© 2001 nature publishing group ......letter nature genetics • volume 29 • december 2001 465...
TRANSCRIPT

letter
nature genetics • volume 29 • december 2001 465
Mutations in PTPN11, encoding the protein tyrosinephosphatase SHP-2, cause Noonan syndrome
Marco Tartaglia1,4, Ernest L. Mehler2, Rosalie Goldberg5, Giuseppe Zampino6, Han G. Brunner7, HannieKremer7, Ineke van der Burgt7, Andrew H. Crosby8, Andra Ion8, Steve Jeffery8, Kamini Kalidas8, Michael A.Patton8, Raju S. Kucherlapati5 & Bruce D. Gelb1,3
Departments of 1Pediatrics, 2Physiology and Biophysics and 3Human Genetics, Mount Sinai School of Medicine, New York, New York 10029, USA.4Laboratorio di Metabolismo e Biochimica Patologica, Istituto Superiore di Sanità, Rome, Italy. 5Department of Molecular Genetics, Albert Einstein Collegeof Medicine, Bronx, New York 10461, USA. 6Istituto di Clinica Pediatrica, Università Cattolica del Sacro Cuore, Rome, Italy. 7Department of HumanGenetics, University Medical Centre, Nijmegen, The Netherlands. 8Department of Medical Genetics, St George’s Hospital Medical School, London, UK.Correspondence should be addressed to M.T. (e-mail: [email protected]).
Noonan syndrome (MIM 163950) is an autosomal dominant dis-order characterized by dysmorphic facial features, proportion-ate short stature and heart disease (most commonly pulmonicstenosis and hypertrophic cardiomyopathy)1,2. Webbed neck,chest deformity, cryptorchidism, mental retardation and bleed-ing diatheses also are frequently associated with this disease.This syndrome is relatively common, with an estimated inci-dence of 1 in 1,000–2,500 live births. It has been mapped to a 5-cM region (N-SH2) on chromosome 12q24.1, and geneticheterogeneity has also been documented3–6. Here we showthat missense mutations in PTPN11 (MIM 176876)—a geneencoding the nonreceptor protein tyrosine phosphatase SHP-2,which contains two Src homology 2 (SH2) domains—causeNoonan syndrome and account for more than 50% of the casesthat we examined. All PTPN11 missense mutations cluster ininteracting portions of the amino N-SH2 domain and the phos-photyrosine phosphatase domains, which are involved inswitching the protein between its inactive and active confor-mations. An energetics-based structural analysis of two N-SH2mutants indicates that in these mutants there may be a signifi-cant shift of the equilibrium favoring the active conformation.This implies that they are gain-of-function changes and thatthe pathogenesis of Noonan syndrome arises from excessiveSHP-2 activity.We considered that PTPN11 might be a candidate gene for NS1because it maps to chromosome 12q24.1 between D12S84 andD12S79 and because its protein product, SHP-2, is essential inseveral intracellular signal transduction pathways that control
diverse developmental processes, including cardiac semilunarvalvulogenesis7–9. We established the gene organization andintron boundary sequence of PTPN11 using complementaryDNA and genomic sequences (Fig. 1). Mutation screening wasinitially conducted with two moderate-sized families (NS-C andNS-L) in which the Noonan syndrome (NS) phenotype cosegre-gates with haplotypes defined by D12S84, D12S105, D12S354and D12S2070.
Bi-directional sequencing of the 15 PTPN11 exons and theirintron boundaries for family NS-C shows that there is a G→Ttransversion at position 214 in exon 3 (Fig. 2a), predicting theAla72Ser substitution in the N-SH2(N-SH2) domain. We con-firmed that this sequence change was present in all affected fam-ily members but absent in unaffected ones by a PCR-basedrestriction fragment–length polymorphism assay (Fig. 2b). Wedid not observe this change in over 200 control individuals.Sequence comparison of SHP-2 with its orthologs and otherclosely related protein tyrosine phosphatases shows that there iscomplete conservation of Ala 72 (data not shown).
Analysis of family NS-L reveals an A→G transition at position236 in exon 3 (Fig. 2a). This change predicts the Gln79Arg sub-stitution, which affects another highly conserved residue in theN-SH2 domain. This sequence change was confirmed in allaffected individuals in this family (Fig. 2b), but was absent inunaffected family members and control individuals. The findingsin the two families establish that PTPN11 is the gene on NS1causing NS.
Because NS is genetically heterogeneous, we considered the rel-ative importance of PTPN11 defects in the epidemiology of NS.We carried out mutation screening of 22 unrelated individualswith Noonan syndrome who represented either sporadic cases orsmall kindreds for which linkage analysis would not have beeninformative. Missense mutations in PTPN11 are present in 11 ofthese individuals (Table 1). Among the five additional mutationsaffecting the N-SH2 domain, individuals N-17 and N-30 haveA→G transitions at nt 182 and 188, respectively, leading toAsp61Gly and Tyr63Cys substitutions. In family NS-U, Ala 72 isaltered by a second mutation, a C→G transversion at nt 215 thatpredicts its substitution with a glycine (Ala72Gly). In individualN-34 (sporadic case), there is a G→C transversion at nt 228, pre-dicting the Gly76Asp substitution. The Gln79Arg mutation isrecurrent in individual NS-D (sporadic case).
Three missense mutations affect the phosphotyrosine phos-phatase (PTP) domain, of which one is recurrent. An A→G
Published online: 12 November 2001, DOI: 10.1038/ng772
Fig. 1 PTPN11 organization and SHP-2 domain structure. The coding exons areshown at the top as numbered filled boxes, and the positions of the ATG andTGA codons are indicated. The functional domains of the SHP-2 protein, com-prising two tandemly arranged SH2 domains at the N terminus (N-SH2 and C-SH2) followed by a protein tyrosine phosphatase (PTP) domain, are shownbelow. Numbers below the domain structure indicate the amino-acid bound-aries of those domains.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com

letter
466 nature genetics • volume 29 • december 2001
transition at nt 922 in exon 8 is found in families NS-A, NS-Vand N-7, as well as in individual NS-B (sporadic case). Thischange predicts the substitution Asn308Asp. Sporadic casesNS-F and NS-N have A→G transitions at nt 844 and 1510,respectively, leading to the substitutions of Ile282Val andMet504Val, respectively.
All amino-acid substitutions affect highly conserved residuesof SHP-2. For familial cases, mutations cosegregate with the NSphenotype, and no mutations are present in more than 200 con-trol individuals. Combining the findings from sporadic caseswith those from the two chromosome-12-linked families,PTPN11 mutations account for slightly over 50% of NS in thissmall sample. Because we did not screen for certain types ofmutations, such as promoter and enhancer changes, however,this might be an underestimate.
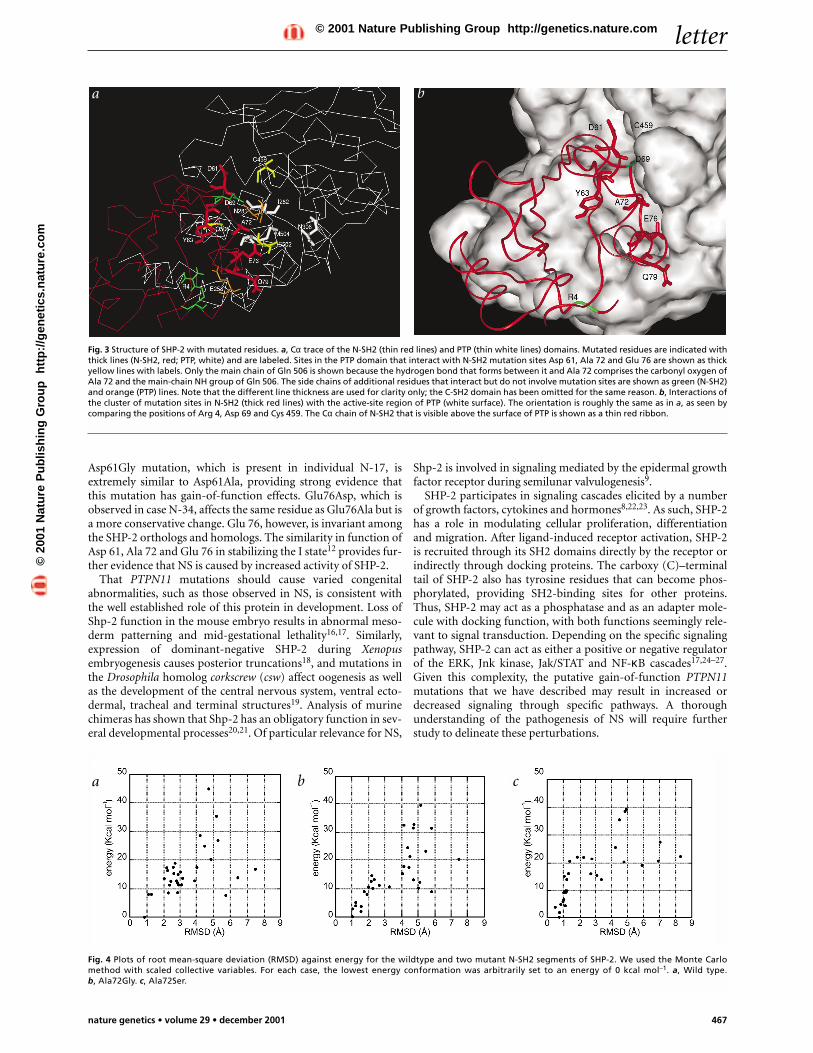
To explore the molecular consequences of the amino-acid sub-stitutions, we examined the crystallographic structures of SHP-2(refs 10–12). SHP-2 exists in an inactive (I) or an active (A) con-formation, with the N-SH2 domain acting as a molecular switch.In the I state, N-SH2 assumes a conformation that blocks the PTPactive site and disrupts its own, separate phosphopeptide-bindingcleft. On binding of phosphopeptide, the N-SH2 domain assumesthe A conformation, which disrupts its PTP recognition surface.In the three-dimensional structure of SHP-2 in the I conforma-tion, the eight residues affected by NS-causing mutations are alllocated in and around the interactive surfaces of the N-SH2 andPTP domains (Fig. 3a,b). Most notably, Ala 72, which is affectedby two mutations, is located in the loop between β-sheet F andand α-helix B and interacts directly with PTP in the I conforma-tion by forming a hydrogen bond with the PTP domain residueGln 506. These observations indicate that the Ala72Ser andAla72Gly mutations may affect the intrinsic structural properties
of the loop segment around Ala 72, shifting the equilibriumbetween the A and I states of SHP-2.
We determined the energetically accessible conformations ofthe Gly68–Glu76 segment in the context of the N-SH2 domain inthe A conformation using Monte Carlo simulations13 with scaledcollective variables14 (MC–SCV). More than 30 structures for thewildtype and both mutants were analyzed by this procedure torelate their structure and energy11.
For the wild type (Fig. 4a), only three low-energy structureshave a root-mean square deviation (r.m.s.d.) of 2 Å or less for theCα chain from the native structure. The lowest-energy structure(r.m.s.d. 0.86 Å) is about 8 kcal mol–1 below the other two stateswith small r.m.s.d., which supports the observed stability of theactive conformation of isolated N-SH2 (ref. 10). In addition tothe two low r.m.s.d. states lying at about 8 kcal mol–1 above thelowest energy conformation, several conformations are found atthis energy with considerably larger r.m.s.d. values. Therefore,the shuttling of the wildtype SHP-2 between the A and I confor-mations becomes energetically plausible if, on interacting withthe PTP domain, the energy gap between the lowest energy con-formation and the nearby states decreases. This would makethose conformations accessible and confer the requisite flexibilityto this segment of N-SH2.
By contrast, both the Ala72Gly and Ala72Ser mutations havelarge populations with small r.m.s.d. values and low energies(Fig. 4b,c). Even if the energies of these states were to shift relativeto each other after binding the PTP domain, there would still beseveral low-energy conformations available that were close to theA conformation, but none that would confer the flexibilityrequired for transition to the I state. Consequently, we predictthat the ability of these two mutants to shuttle between the A andI states is impaired, which would imply that the equilibrium isshifted toward the A state relative to the wildtype protein.
Functional data support the conclusions of these energetics-based structural analyses. Two SHP-2 mutants, D61A and E76A,were predicted to be gain-of-function changes, as both are N-SH2 residues that interact directly with the PTP domain15. Thesemutants were created and characterized, documenting an
increase in basal phosphataseactivity (Glu76Ala > Asp61Ala> wildtype) and the retentionof normal phosphopeptide-binding properties. Whenexpressed in Xenopus ectoder-mal explants, both mutantsinduce changes that mimicsome aspects of developmentthat are induced by fibroblastgrowth factor, indicating thebasal stimulation of some sig-naling cascades in vivo. The
Fig. 2 PTPN11 mutations in Noonan syndrome. a, Sequence analysis of ampli-fied DNA containing PTPN11 exon 3 and flanking intronic boundaries revealsheterozygous changes in the NS-C and NS-L families. Left, a G→T transver-sion at nt 214 in an affected individual from NS-C. Right, an A→G transitionat nt 236 in an individual from NS-L. Wildtype exonic sequences and theamino-acid residues that they encode are shown below the electrophero-grams. Mutated bases are shown in bold with arrows indicating the positionof the heterozygous sequences. b, Segregation of the G214T (left) andA236G (right) mutations in NS-C and NS-L families, respectively. Family treesfor these two families are shown above. A single BglI site is present in theproduct amplified from the wild type, and so this digestion results in 164-and 374-bp products. The G214T change obliterates that BglI site so that theheterozygotes also have an undigested 538-bp product. The A236G mutationintroduces a BsaWI restriction site, resulting in 177- and 361-bp products,whereas wildtype alleles remain undigested at 538 bp.
Table 1 • PTPN11 mutations among familial and sporadic cases of Noonan syndrome
Family ID Nucleotide substitution Exon Amino-acid substitution Functional domainN-17 A182G 3 Asp61Gly N-SH2N-30 A188G 3 Tyr63Cys N-SH2NS-C G214T 3 Ala72Ser N-SH2NS-U C215G 3 Ala72Gly N-SH2N-34 G228C 3 Glu76Asp N-SH2NS-L, NS-D A236G 3 Gln79Arg N-SH2NS-F A844G 7 Ile282Val PTPNS-A, NS-B, NS-V, N-7 A922G 8 Asn308Asp PTPNS-N A1510G 13 Met504Val PTP
a
b
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com
letter
nature genetics • volume 29 • december 2001 467
Asp61Gly mutation, which is present in individual N-17, isextremely similar to Asp61Ala, providing strong evidence thatthis mutation has gain-of-function effects. Glu76Asp, which isobserved in case N-34, affects the same residue as Glu76Ala but isa more conservative change. Glu 76, however, is invariant amongthe SHP-2 orthologs and homologs. The similarity in function ofAsp 61, Ala 72 and Glu 76 in stabilizing the I state12 provides fur-ther evidence that NS is caused by increased activity of SHP-2.
That PTPN11 mutations should cause varied congenitalabnormalities, such as those observed in NS, is consistent withthe well established role of this protein in development. Loss ofShp-2 function in the mouse embryo results in abnormal meso-derm patterning and mid-gestational lethality16,17. Similarly,expression of dominant-negative SHP-2 during Xenopusembryogenesis causes posterior truncations18, and mutations inthe Drosophila homolog corkscrew (csw) affect oogenesis as wellas the development of the central nervous system, ventral ecto-dermal, tracheal and terminal structures19. Analysis of murinechimeras has shown that Shp-2 has an obligatory function in sev-eral developmental processes20,21. Of particular relevance for NS,
Shp-2 is involved in signaling mediated by the epidermal growthfactor receptor during semilunar valvulogenesis9.
SHP-2 participates in signaling cascades elicited by a numberof growth factors, cytokines and hormones8,22,23. As such, SHP-2has a role in modulating cellular proliferation, differentiationand migration. After ligand-induced receptor activation, SHP-2is recruited through its SH2 domains directly by the receptor orindirectly through docking proteins. The carboxy (C)–terminaltail of SHP-2 also has tyrosine residues that can become phos-phorylated, providing SH2-binding sites for other proteins.Thus, SHP-2 may act as a phosphatase and as an adapter mole-cule with docking function, with both functions seemingly rele-vant to signal transduction. Depending on the specific signalingpathway, SHP-2 can act as either a positive or negative regulatorof the ERK, Jnk kinase, Jak/STAT and NF-κB cascades17,24–27.Given this complexity, the putative gain-of-function PTPN11mutations that we have described may result in increased ordecreased signaling through specific pathways. A thoroughunderstanding of the pathogenesis of NS will require furtherstudy to delineate these perturbations.
Fig. 3 Structure of SHP-2 with mutated residues. a, Cα trace of the N-SH2 (thin red lines) and PTP (thin white lines) domains. Mutated residues are indicated withthick lines (N-SH2, red; PTP, white) and are labeled. Sites in the PTP domain that interact with N-SH2 mutation sites Asp 61, Ala 72 and Glu 76 are shown as thickyellow lines with labels. Only the main chain of Gln 506 is shown because the hydrogen bond that forms between it and Ala 72 comprises the carbonyl oxygen ofAla 72 and the main-chain NH group of Gln 506. The side chains of additional residues that interact but do not involve mutation sites are shown as green (N-SH2)and orange (PTP) lines. Note that the different line thickness are used for clarity only; the C-SH2 domain has been omitted for the same reason. b, Interactions ofthe cluster of mutation sites in N-SH2 (thick red lines) with the active-site region of PTP (white surface). The orientation is roughly the same as in a, as seen bycomparing the positions of Arg 4, Asp 69 and Cys 459. The Cα chain of N-SH2 that is visible above the surface of PTP is shown as a thin red ribbon.
Fig. 4 Plots of root mean-square deviation (RMSD) against energy for the wildtype and two mutant N-SH2 segments of SHP-2. We used the Monte Carlomethod with scaled collective variables. For each case, the lowest energy conformation was arbitrarily set to an energy of 0 kcal mol–1. a, Wild type.b, Ala72Gly. c, Ala72Ser.
a b
a b c
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com

letter
468 nature genetics • volume 29 • december 2001
MethodsSubject recruitment. We recruited people with NS, primarily through aGeneTests listing (http://www.genetests.org/), and obtained informed con-sent from all subjects. NS was diagnosed on the basis of clinical evaluationsfrom the referring clinical geneticists. Affected individuals with NS wereAmericans, predominantly of European descent, and control individualswere obtained from a mixed American group comprising predominantlyindividuals of European descent.
Molecular analyses. We isolated genomic DNA from peripheral bloodlymphocytes (Gentra). Genotyping was carried out using dye-labeled sim-ple tandem repeat markers, D12S84, D12S105, D12S354 and D12S2070(Research Genetics). We resolved PCR products using a ABI Prism 377DNA Sequencer (Perkin Elmer) and made genotype determinations usingGeneScan v.3.1 and Genotyper v.2.1 software packages (Perkin Elmer).
We compared genomic and cDNA sequences for PTPN11 using DNAanalysis software (MacVector) and made predictions of the gene organiza-tion using a NIX software package (UK HGMRP). We carried out muta-tional screening of PTPN11 by direct sequencing of purified PCR productsbi-directionally, using an ABI BigDye terminator Sequencing Kit (PerkinElmer) and an ABI 3700 Capillary Array Sequencer (Perkin Elmer). Primerpairs and PCR conditions for amplification of PTPN11 exons 1–15 areavailable on request. We analyzed sequences using Sequencing Analysisv.3.6.1 and AutoAssembler v.1.4.0 software packages (Perkin Elmer). Toconfirm sequence changes in affected individuals and exclude them fromunaffected family members and controls, we amplified the relevant exonsby PCR and then either digested them with restriction endonucleases (BglIfor G214T and C215G, BsaWI for A236G, and EcoRV for A922G) accord-ing to the manufacturer’s instructions (New England Biolabs) or analyzedby denaturing high-performance liquid cromatography (DHPLC)(A182G, A188G, G228C, A844G and A1510G) using the Wave DNA Frag-ment Analysis System (Transgenomics) at column temperatures recom-mended by the WaveMaker v.4.0.28 software (Transgenomics).
Computational methods. A two-step protocol has been devised for calcu-lating the (unknown) structure of segments connecting known elements ofsecondary structure in proteins. Monte Carlo–simulated annealing is firstcarried out to allow the segment to find all structures that are intrinsic toits sequence while it remains tethered to the protein only at its N terminus.Then MC–SCV14 is used to drive the C terminus to its attachment point.The entire procedure has been programed into the CHARMM suite of pro-grams28 using the PAR22 all-atom force field29. The method is described infull in ref. 30.
We modified the first step of the general procedure for our calcula-tions. Starting from the native conformation, the segment was cutbetween Glu 76 and Leu 77, and a long MC–SCV simulation was carriedout on the nine-residue segment Gly68–Glu76 at a temperature of 310 K.In this step, only a few fixed residues at the N and C termini of the vari-able segment were included in the calculation. This step allows the seg-ment to relax and explore conformation space to find conformationsintrinsic to its sequence, and results in several conformations that pro-vide the starting structures for the second step. To reduce the calculation,we first clustered31 the structures on the basis of structural similarity andthen used a representative from each class (n=31).
For the second step, we used MC–SCV14 to drive the segment back to itsattachment point at Leu 77. This was achieved by attaching a dummyresidue at the C terminus identical with the target residue (Leu 77) towhich the segment will attach. This dummy residue had no role in theenergetics but served as a geometric reference for adding a harmonic term,Σk(ri– ri
o)2, to the energy that drove the segment to its target; ri and rio are
the coordinates of atom i in the dummy and target residues, respectively,and the sum runs over the backbone and Cβ atoms. Starting with k=0, wecarried out an MC–SCV simulation at 310 K on each coordinate setobtained from the first step to relax it in the field of the tertiary structure ofthe protein that had now been added to the system, as well as the aqueoussolvent represented by the screened Coulomb potential-implicit solventmodel32. We subsequently increased k in successive steps, with anMC–SCV simulation carried out at each step, to facilitate the movement ofthe C terminus to its attachment point. We used a power schedule ofki=10ki–1, starting from ko=10–6. We found that ten steps were sufficient toachieve closure.
GenBank accession numbers. Human PTPN11, NM_002834; BAC cloneRP3-329E11, AC004086.
AcknowledgmentsWe thank the individuals with Noonan syndrome and their families whoparticipated in this study, the physicians who referred the subjects, X. Songfor technical assistance, H. Weinstein for insightful suggestions about thestructural analysis, S. Hassan for providing algorithms and programs to carryout the Montre Carlo calculations and for discussions and G. Diaz and Y.Ioannou for reading the manuscript. This work was supported in part bygrants from the NIH (to B.D.G., E.L.M. and R.K.) and from the HumanGenetics Program at the Albert Einstein College of Medicine (to R.K.).
Received 1 August; accepted 17 October 2001.
1. Noonan, J.A. Hypertelorism with Turner phenotype. A new syndrome withassociated congenital heart disease. Am. J. Dis. Child. 116, 373–380 (1968).
2. Allanson, J.E. Noonan syndrome. J. Med. Genet. 24, 9–13 (1987).3. Jamieson, C.R. et al. Mapping a gene for Noonan syndrome to the long arm of
chromosome 12. Nature Genet. 8, 357–360 (1994).4. Brady, A.F. et al. Further delineation of the critical region for Noonan syndrome
on the long arm of chromosome 12. Eur. J. Hum. Genet. 5, 336–337 (1997).5. Legius, E., Schollen, E., Matthijs, G. & Fryns, J.P. Fine mapping of Noonan/cardio-
facio cutaneous syndrome in a large family. Eur. J. Hum. Genet. 6, 32–37 (1998).6. van Der Burgt, I. & Brunner, H. Genetic heterogeneity in Noonan syndrome:
evidence for an autosomal recessive form. Am. J. Med. Genet. 94, 46–51 (2000).7. Dechert, U. et al. Protein-tyrosine phosphatase SH-PTP2 (PTPN11) is localized to
12q24.1-24.3. Hum. Genet. 96, 609–615 (1995).8. Feng, G.-S. Shp-2 tyrosine phosphatase: signaling one cell or many. Exp. Cell Res.
253, 47–54 (1999).9. Chen, B. et al. Mice mutant for Egfr and Shp2 have defective cardiac semilunar
valvulogenesis. Nature Genet. 24, 296–299 (2000).10. Lee, C.H. et al. Crystal structures of peptide complexes of the amino-terminal SH2
domain of the Syp tyrosine phosphatase. Structure 2, 423–438 (1994).11. Eck, M.J., Pluskey, S., Trub, T., Harrison, S.C. & Shoelson, S.E. Spatial constraints on
the recognition of phosphoproteins by the tandem SH2 domains of thephosphatase SH-PTP2. Nature 379, 277–280 (1996).
12. Hof, P., Pluskey, S., Dhe-Paganon, S., Eck, M.J. & Shoelson, S.E. Crystal structure ofthe tyrosine phosphatase SHP-2. Cell 92, 441–450 (1998).
13. Allen, M.P. & Tildesley. Computer Simulation of Liquids (Clarendon, Oxford,1987).
14. Noguti, T. & Go, N. Efficient Monte Carlo method for simulation of fluctuatingconformations of native proteins. Biopolymers 24, 527–546 (1985).
15. O’Reilly, A.M., Pluskey, S., Shoelson, S.E. & Neel, B.G. Activated mutants of SHP-2preferentially induce elongation of Xenopus animal caps. Mol. Cell. Biol. 20,299–311 (2000).
16. Arrandale, J.M. et al. Insulin signaling in mice expressing reduced levels of Syp. J.Biol. Chem. 271, 21353–21358 (1996).
17. Saxton, T.M. et al. Abnormal mesoderm patterning in mouse embryos mutant forthe SH2 tyrosine phosphatase Shp-2. EMBO J. 16, 2352–2364 (1997).
18. Tang, T.L., Freeman, R.M. Jr, O’Reilly, A.M., Neel, B.G. & Sokol, S.Y. The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAPkinase for early Xenopus development. Cell 80, 473–483 (1995).
19. Perkins, L.A., Johnson, M.R., Melnick, M.B. & Perrimon, N. The nonreceptorprotein tyrosine phosphatase corkscrew functions in multiple receptor tyrosinekinase pathways in Drosophila. Dev. Biol. 180, 63–81 (1996).
20. Qu, C.K. et al. Biased suppression of hematopoiesis and multiple developmentaldefects in chimeric mice containing Shp-2 mutant cells. Mol. Cell. Biol. 18,6075–6082 (1998).
21. Saxton, T.M. et al. The SH2 tyrosine phosphatase shp2 is required for mammalianlimb development. Nature Genet. 24, 420–423 (2000).
22. Stein-Gerlach, M., Wallasch, C. & Ullrich, A. SHP-2, SH2-containing proteintyrosine phosphatase-2. Int. J. Biochem. Cell. Biol. 30, 559–566 (1998).
23. Tamir, I., Dal Porto, J.M. & Cambier, J.C. Cytoplasmic protein tyrosine phosphataseSHP-1 and SHP-2: regulators of B cell signal transduction. Curr. Opin. Immunol.12, 307–315 (2000).
24. Shi, Z.Q., Lu, W. & Feng, G.S. The Shp-2 tyrosine phosphatase has opposite effectsin mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases. J. Biol. Chem. 273, 4904–4908(1998).
25. You, M., Yu, D.H. & Feng, G.S. Shp-2 tyrosine phosphatase functions as a negativeregulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 19,2416–2424 (1999).
26. Maroun, C.R., Naujokas, M.A., Holgado-Madruga, M., Wong, A.J. & Park, M. Thetyrosine phosphatase SHP-2 is required for sustained activation of extracellularsignal-regulated kinase and epithelial morphogenesis downstream from the metreceptor tyrosine kinase. Mol. Cell. Biol. 20, 8513–8525 (2000).
27. You, M., Flick, L.M., Yu, D. & Feng, G.S. Modulation of the nuclear factor κBpathway by Shp-2 tyrosine phosphatase in mediating the induction of interleukin(IL)-6 by IL-1 or tumor necrosis factor. J. Exp. Med. 193, 101–110 (2001).
28. Brooks, B.R. et al. CHARMM — a program for macromolecular energy,minimization and dynamics calculations. J. Comput. Chem. 4, 165–175 (1983).
29. MacKerell, A.D. et al. All-atom empirical potential for molecular modeling anddynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
30. Hassan, S.A., Mehler, E.L. & Weinstein, H. in Lecture Notes Series inComputational Science (ed. Schlick, T.) (Springer, New York, in press).
31. Shenkin, P.S. & McDonald, D.Q. Cluster analysis of molecular conformations. J.Comput. Chem. 15, 899–916 (1994).
32. Hassan, S.A., Guarnieri, F. & Mehler, E.L. A general treatment of solvent effectsbased on screened Coulomb potentials. J. Phys. Chem. B 104, 6478–6489 (2000).
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://g
enet
ics.
nat
ure
.co
m© 2001 Nature Publishing Group http://genetics.nature.com