boletinspars.es/wp-content/uploads/2015/10/bol-spars-2010-vol-40-n2.pdf · 40 linfadenitis...
TRANSCRIPT

BBOOLLEETTIINNSociedad de Pediatría de
ARAGÓN, LA RIOJA Y SORIA
ARTÍCULO ESPECIALProtocolo de actuación ante un síncope en urgencias de pediatríaI. Galé Ansó, E. Sancho Gracia, O. Gómez Cámara, A. Aldana Tabuenca, A. Manero Oteiza,M.D. Gracia de la Calzada
CASOS CLÍNICOSHistiocitosis de células de Langerhans congénitaC. Guerrero Laleona, M. Guiu Campos, C. Calvo Escribano, I. Marquina Ibáñez,A.M. Morales Callaghan
Linfadenitis tuberculosa por Mycobacterium bovisF. de Juan Martín
Transmisión congénita de Trypanosoma cruzi (enfermedad de Chagas):a propósito de un casoF. de Juan Martín, L. Roc Alfaro, E. Lomba Fuentes
SESIONES DE LA SOCIEDADResúmenes de las Sesiones de Comunicaciones Libres celebradasel 13 de mayo y el 17 de junio de 2010 en ZaragozaAlcalosis metabólica hipoclorémica: presentación de fibrosis quísticaen un lactanteConvulsiones precoces en el recién nacido a término.«Causa poco frecuente»Síndrome de Dandy-Walker. Comunicación de dos casos y revisiónPaciente con deleción terminal del brazo largo del cromosoma 10.Caso clínico¿Qué papel tiene la enfermería de manera autónomaen el programa del niño sano?Intumescencia mamaria neonatal: ¿un cuadro benigno?Esofagitis esofinofílica: patología emergenteInfección perinatalManejo respiratorio de la hernia diafragmáticaCaso clínico: Síndrome de WolframHomocistinemia clásica detectada por cribado neonatal ampliado.Revisión de nuestra casuística¿Qué niños se han vacunado de la gripe A y por qué?Insuficiencia cardiaca como consecuencia de anemia graveSíndrome de Bloch-sulzberger neonatalEvolución clínica y analítica en paciente con hipoaldosteronismocongénito primarioUrticaria y parasitosis intestinalAgenesia del pectoral mayor en la infancia.Complejo malformativo Poland.A propósito de un casoDiarrea oleosa con heces anaranjadasPolaquiuria primariaSíndrome de JobImportancia de la educación en el control del niño con TDAH.Visión desde atención primaria
mayo agosto 2010
volumen 40
número 2 SUM
AR
IO

BBOOLLEETTIINNSociedad de Pediatría de
ARAGÓN, LA RIOJA Y SORIAÓrgano de expresión fundamental de la Sociedad de Pediatría de Aragón, La Rioja y Soria
Con la colaboración de
Junta directiva:Presidente:Juan Elías Pollina
Vicepresidente 1.º:Manuel Domínguez Cunchillos
Vicepresidente 2.º:Javier Membrado Granizo
Secretario General:Javier F. Sierra Sirvent
Secretaria de Actas:Isabel Lostal Gracia
Tesorero:Héctor Colán Villacorta
Bibliotecaria y Directora del Boletín:M.ª Pilar Samper Villagrasa
Vocal por Huesca:M.ª Jesús Oliván del Cacho
Vocal por La Rioja:Juan Antonio Pérez Marrodán
Vocal por Soria:Olga García Bodega
Vocal por Teruel:María Cristina de Miguel Pardo
Vocal por Zaragoza:Juan Ángel Giménez Olivas
Vocal de PediatríaExtrahospitalaria:Javier Sanz Aguareles
Vocal MIR:Alberto Aldana Tabuenca
Fundador:Luis Boné Sandoval
Edita:Sociedad de Pediatríade Aragón, La Rioja y SoriaPaseo de Ruiseñores, 250006 Zaragoza
Dep. legal:M. 21. 402-1970
I.S.S.N.:1.696-358-X
Imprime:TIPOLINEA, S.A.
Publicación autorizada por el Ministerio de Sanidad como Soporte Válido Ref. n.º 393
Publicación cuatrimestral(3 números al año)
Dirección:M.ª Pilar Samper Villagrasa
Secretario de redacción:Gerardo Rodríguez MartínezJuan Carlos I, 43, 12.º A50009 Zaragozacorreo: [email protected]
Consejo de redacción:Directora:M.ª Pilar Samper Villagrasa
Secretario de Redacción:Gerardo Rodríguez Martínez
Consejo de Redacción:L. Alonso TomásC. Baselga AsensioF. Cucalón ManzanosF. De Juan MartínJ. Fleta ZaragozanoM.V. Labay y MatíasA. Lacasa ArreguiA. Lázaro AlmarzaC. Loris PabloJ.L. Olivares LópezI. Pastor MourónV. Pérez-ChólizL. Ros MarF. Valle Sánchez
Presidentes de honor:E. Casado de FríasM.A. Soláns CastroA. Sarría ChuecaA. Baldellou VázquezM. Bueno SánchezM. Adán PérezA. Ferrández Longás
Sociedad de Pediatría de Aragón, La Rioja y Soriahttp://www.comz.org/spars/spars.html
REVISTA INCLUIDA EN EL ÍNDICE MÉDICO ESPAÑOL

Sumario
BBOOLLEETTIINNSociedad de Pediatría de
ARAGÓN, LA RIOJA Y SORIA
mayoagosto2010volumen 40número 2
SUMARIO ARTÍCULO ESPECIAL33 Protocolo de actuación ante un síncope en urgencias de pediatría
I.Galé Ansó, E. Sancho Gracia,O.Gómez Cámara,A.Aldana Tabuenca,A.Manero Oteiza,M.D.Gracia de la Calzada
CASOS CLÍNICOS37 Histiocitosis de células de Langerhans congénita
C. Guerrero Laleona, M. Guiu Campos, C. Calvo Escribano, I. Marquina Ibáñez, A.M. Morales Callaghan
40 Linfadenitis tuberculosa por Mycobacterium bovisF. de Juan Martín
43 Transmisión congénita de Trypanosoma cruzi (enfermedad de Chagas):a propósito de un casoF. de Juan Martín, L. Roc Alfaro, E. Lomba Fuentes
SESIONES DE LA SOCIEDADResúmenes de las Sesiones de Comunicaciones Libres celebradas el 13 de mayoy el 17 de junio de 2010 en Zaragoza
45 Alcalosis metabólica hipoclorémica: presentación de fibrosis quística en un lactante45 Convulsiones precoces en el recién nacido a término. «Causa poco frecuente»46 Síndrome de Dandy-Walker. Comunicación de dos casos y revisión46 Paciente con deleción terminal del brazo largo del cromosoma 10. Caso clínico47 ¿Qué papel tiene la enfermería de manera autónoma en el programa del niño sano?47 Intumescencia mamaria neonatal: ¿un cuadro benigno?48 Esofagitis esofinofílica: patología emergente48 Infección perinatal49 Manejo respiratorio de la hernia diafragmática49 Caso clínico: Síndrome de Wolfram50 Homocistinemia clásica detectada por cribado neonatal ampliado.
Revisión de nuestra casuística50 ¿Qué niños se han vacunado de la gripe A y por qué?51 Insuficiencia cardiaca como consecuencia de anemia grave51 Síndrome de Bloch-sulzberger neonatal52 Evolución clínica y analítica en paciente con hipoaldosteronismo congénito primario52 Urticaria y parasitosis intestinal53 Agenesia del pectoral mayor en la infancia. Complejo malformativo Poland.
A propósito de un caso53 Diarrea oleosa con heces anaranjadas54 Polaquiuria primaria54 Síndrome de Job55 Importancia de la educación en el control del niño con TDAH.
Visión desde atención primaria

BBOOLLEETTIINNSociedad de Pediatría de
ARAGÓN, LA RIOJA Y SORIA
MayAugust2010volume 40number 2
CONTENTS SPECIAL ARTICLE33 Protocol to action in emergency syncope pediatrics
I. Galé Ansó, E. Sancho Gracia, O. Gómez Cámara, A. Aldana Tabuenca, A. Manero Oteiza,M.D. Gracia de la Calzada
CLINICAL CASES37 Langerhans cell histiocytosis congenital
C. Guerrero Laleona, M. Guiu Campos, C. Calvo Escribano, I. Marquina Ibáñez, A.M. Morales Callaghan
40 Cervical lymphadenitis due to Mycobacterium bovisF. de Juan Martín
43 Vertical transmission of Trypanosoma cruzi infection (Chagas’ disease):a case reportF. de Juan Martín, L. Roc Alfaro, E. Lomba Fuentes
SOCIETY SESSIONS

33VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Protocolo de actuación ante un síncopeen urgencias de pediatría
I. Galé Ansó, E. Sancho Gracia, O. Gómez Cámara, A. Aldana Tabuenca, A. Manero Oteiza, M.D. Gracia de la Calzada
Hospital Infantil Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 33-36]
Correspondencia: Inés Galé AnsóHospital Infantil Miguel Servet. Zaragozae-mail: [email protected]: junio de 2010. Aceptado: junio de 2010
RESUMENEl síncope es una patología frecuente generalmente benigna que, sin embargo, genera gran ansiedad en pacientes yfamiliares. Se ha descrito que hasta un 50% de los niños habrá presentado al menos un síncope o un presíncope antesde llegar a la adolescencia y que puede ser la causa de hasta un 3% de las visitas a urgencias pediátricas. Presentamosla actualización del protocolo de actuación en urgencias de pediatría empleado en nuestro hospital ante pacientespediátricos con clínica de síncope y la revisión del cumplimiento del protocolo anterior.
PALABRAS CLAVESíncope, protocolo de actuación, niños.
Protocol to action in emergency syncope pediatricsABSTRACTSyncope is a usually benign frequent pathology, however, generates great anxiety in patients and relatives. Described that upto 50% of children will be submitted at least a syncope or a presyncope reach adolescence and can be up to 3% of pediatricemergency visits.We introduce the employee in our hospital protocol update in pediatric patients with syncope clinic and therevision of the fulfillment of the previous protocol.
KEY WORDSSyncope, protocolization, children.
INTRODUCCIÓNEn nuestro hospital se está trabajando en un plan de mejo-ra de la calidad asistencial de las urgencias pediátricasmediante la protocolización de la actuación en diferentespatologías, como el síncope.
DEFINICIÓNSe define un síncope como la pérdida de conciencia y deltono postural cuya recuperación es espontánea y rápiday que está producida por un mecanismo de déficit brus-co del flujo cerebral.
El síncope se puede encuadrar en una de estas cua-tro categorías: neurocardiogénico, cardiológico, neuroló-gico y otros(1-5). En el paciente pediátrico la mayoría de losepisodios serán debidos a un síncope neurocardiogénico,pero es muy importante diferenciar e identificar aquellosinfrecuentes casos de síncopes debidos a causas poten-cialmente graves(1-5). Una clasificación podría ser :
1.º Síncope neurocardiogénico y afines. Neurocar-diogénico o vasovagal (que supone el 85% de los episo-dios en pediatría), espasmo del sollozo, síncope situacio-nal (miccional, tusígeno, relacionado con el peinado, contos paroxística), hipotensión ortostática.
Artículo Especial

Boletín de la Sociedad de Pediatría de Aragón, La Rioja y Soria
34 I. Galé Ansó, E. Sancho Gracia, O. Gómez Cámara, A. Aldana Tabuenca, A. Manero Oteiza, M.D. Gracia de la Calzada
2.º Síncope cardiaco
–Mecánico: Por obstrucción en los tractos de salidaventriculares (Estenosis aórtica, Miocardiopatíahipertrófica, Estenosis pulmonar severa…) o porafectación miocárdica (miocardiopatía, miocarditis,anomalías arterias coronarias).
–Eléctrico: Arritmias (bloqueo AV, QT largo,Taquicardia ventricular, taquicardia supraventricular-Wolf-Parkinson-White).
3.º Síncope neurológico: Epilepsia, migraña, ACV nocardiogénico.
4.º Otros: Hipoglucemia, histeria, vértigo…
POBLACIÓN DIANA
Este protocolo se aplicará a los niños que acudan aurgencias de pediatría con sintomatología compatiblecon un síncope.
PAUTA DE ACTUACIÓN EN URGENCIASANTE NEFRITIS AGUDA
Anamnesis
Una anamnesis detallada es lo más importante, haciendohincapié en:
• Antecedentes familiares: Muerte súbita, cardiopatías,enfermedades metabólicas, enfermedades neurológi-cas, historia familiar de migrañas y episodios sincopales.
• Antecedentes personales: Síncopes previos, patologíacardiaca, enfermedades metabólicas, enfermedadesneurológicas, tratamientos farmacológicos y/o drogas(abuso).
• Descripción del episodio: Sobre todo si existe rela-ción con algún factor precipitante, si existen otros pro-cesos intercurrentes (ayuno, fiebre…) o si han existidosíntomas/signos presincopales. Duración del episodio ydatos acompañantes (movimientos de EE, revulsiónocular, pérdida control esfínteres…).
Exploración
Exploración cardiovascular y neurológica, orientada a labúsqueda de alteraciones. Destacar que el fondo de ojose considera parte constituyente de la exploración neu-rológica y se debe intentar siempre, sobre todo en niñosmayores de 4 años y colaboradores.
Pruebas complementarias
• 1.º Constantes: Tensión arterial, temperatura, satura-ción oxígeno y prueba de ortostatismo.
• 2.º Glucemia capilar: Excepto si el desencadenante hasido un pinchazo.
• 3.º ECG: De acuerdo con el servicio de cardiología elmédico que atienda el síncope se lo guardará y acudi-rá al día siguiente a cardiología para confirmar que notiene alteraciones.
–Una anamnesis, exploración física y ECG normalesdescartan, en un 95% de probabilidad, la existencia deuna patología cardiaca.
• 4.º Valorar RX TÓRAX en síncope inexplicado o sos-pecha de síncope cardiaco.
Actitud
• Alta y control por su pediatra: Niños que no cumplencriterios de ingreso, con exploración cardiológica yneurológica,TA y ECG normales.
• Hospital de día: Nos lo plantearemos cuando el niñoviene en ambulancia o existe angustia familiar.
• Ingreso: Ante síncopes de esfuerzo, valorar ante sínco-pes tras sustos o ruidos fuertes, si existen alteracionesen la exploración y/o ECG o antecedentes familiaresde muerte súbita precoz, si la duración es prolongaday ante un síncope en un cardiópata conocido.Ademásse valorará ante síncope con historia atípica o sínco-pes recurrentes no estudiados o ante dudas de lasituación, angustia o presión familiar y/o distocia social.
EVALUACIÓN DEL ANTERIOR PROTOCOLO
Se realizó un estudio descriptivo retrospectivo, mediantela revisión de informes de urgencias de los niños con diag-nóstico de síncope/presíncope en el período comprendi-do entre enero y noviembre de 2009 y así poder estudiarlos datos epidemiológicos de esta población, y se revisa-ron los registros de urgencias de 34 pacientes selecciona-dos al azar para analizar el cumplimiento de los indicado-res de calidad del protocolo (la constancia de factor de-sencadenante, puntuación en la escala de Glasgow, ten-sión arterial, glucemia y el electrocardiograma).
En este período se atendieron 143 niños con diag-nóstico de síncope/presíncope, lo que supone el 0,3% deltotal de urgencias pediátricas. El 56% eran niñas, supo-niendo el grupo de entre 12 y 15 años el 39%. Hasta un37,7% fue catalogado como síncope vasovagal, destacan-do el 4% de síncopes convulsivos; el 22% fueron diagnos-ticados de presíncopes. El 12,5% de los niños ingresaronen planta y el 86,8% fueron dados de alta domiciliariahabiendo estado previamente en la unidad de observa-ción el 24,8% de las altas. Se realizaron 2 pruebas de neu-roimagen de carácter urgente resultando normales y nin-guno de los ECG realizados en urgencias presentó alte-raciones. De los niños que fueron ingresados destacan losdiagnósticos definitivos de una epilepsia, una bradicardiasinusal en reposo, una crisis generalizada del despertar,una migraña y un trastorno de conducta alimentaria.

35
Protocolo de actuación ante un síncope en urgencias de pediatría
VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
sentar otras alteraciones en el resto de exploración neu-rológica que se había registrado en el 90% de los pacien-tes. La medición de TA y su valor constaba en un 91,1%de los informes revisados, estando dentro de rangos denormalidad según peso y talla en el 100% de los casos.Asimismo el valor de glucemia capilar se reflejaba en el91,1%, siendo normal en todos ellos. La realización deECG y su interpretación aparecía en un 67,6%, sin encon-trar alteraciones en ninguno de los pacientes.
En los informes de urgencias revisados de los 34casos con diagnóstico de síncope se puede observar unpredominio del sexo femenino (64,7%), con una media-na de edad de 10 años, y una moda de 12 años. La cons-tancia del factor desencadenante se cumplía en un 79,5%de los casos, siendo el dolor el más frecuente y desta-cando dos en relación con hiperextensión del cuello(peinarse). La puntuación en la escala de Glasgow cons-taba en un 67,4%, siendo 15 en todos los casos y sin pre-
ALGORITMO DE ACTUACIÓN
EPISODIO SINCOPAL
Anamnesis:Antecedentes personalesAntecedentes familiaresDescripción episodio
Exploración:CardiólogicaNeurológica (Glasgow, FO)Constantes (FC,TA,Tª)ECG (2 copias)Glucemia
• Ritmo sinusal • Frecuencia cardiaca • Amplitud QRS• Intervalo PR, QT, QTc • Anomalías segmento ST• No existencia de arritmias
ALTA Y CONTROL POR SU PEDIATRA
Niños con exploración neurológica y cardiológicanormales y con ECG y TA normales sin signos dealarma.
INGRESAN
–Síncopes de esfuerzo.–Valorar ante síncopes tras sustos o ruidos fuertes.–Alteraciones en la exploración y/o ECG.–Duración prolongada.–Antecedentes familiares de muerte súbita precoz.–Síncope en un cardiópata conocido.–Alteraciones del estado de conciencia o de la exploraciónneurológica.
–Valorar ante síncope con historia atípica o síncopes recu-rrentes no estudiados.
–Valorar ante dudas de la situación, angustia o presión fami-liar, distocia social.
** Uso Hospital de Día:No necesario en principio y su uso se limitará a:• Niño que viene en ambulancia.• Angustia familiar.• Niño que no se recupera bien (ver encefalo-
patía aguda) o alteraciones en exploración.
4. Ruiz JA,Montero R,Hernández N,et al. Síncope en Publicaciónde libros médicos, S.L.U. editores. Manual de diagnóstico yterapéutica en pediatría. 4ª ed. Madrid: Cofás, 2003; pp. 81-84.
5. Campo F, Navarro A. Síncope en pediatría. Protocolos de lasociedad española de cardiología pediátrica 2005. [Consul-tado 19.10.05]. Disponible en:http://www.secardioped.org/protocolos/protocolos/Capítulo_28.pdf.
BIBLIOGRAFÍA
1. Johnston MV. Procesos que simulan crisis epilépticas. En:Nelson.Tratado de pediatría. 17 ed. Madrid: Elsevier EspañaS.A. 2004; pp. 2010-2012.
2. Toral E, Quintillá JM, Luaces C. Síncope. En: Urgencias enpediatría. 3ª ed. Madrid: Ergón España, 2002; pp. 592-597.
3. Park MK. Síncope. En: Cardiología pediátrica. 3ª ed. Madrid:Elsevier España S.A., 2003; pp. 219-223.

Boletín de la Sociedad de Pediatría de Aragón, La Rioja y Soria
36 I. Galé Ansó, E. Sancho Gracia, O. Gómez Cámara, A. Aldana Tabuenca, A. Manero Oteiza, M.D. Gracia de la Calzada
9. Prieto ML, Pérez AM. Protocolos de Cardiología: Síncopes.Bol Pediatr 2006; 46: 281-285.
10. Rueda F, Rodríguez A. Síncope. En: Cardiología Pediátrica enAtención Primaria.
11. Bo I, Carano N, Agnetti A, et al. Syncope in children andadolescents: a two-year experience at the Department ofPaediatrics in Parma. Acta Biomed 2009; 80: 36-41.
12. Alonso MT, Palomino A.Trastornos paroxísticos no epilép-ticos. Protocolos Diagnóstico Terapéuticos de la AEP:Urgencias Pediátricas 2008.
6. Brignole M,Alboni P, Benditt DG, et al. Guidelines on mana-gement (diagnosis and treatment) of syncope. Update2004. Executive summary. Rev esp cardiol. 2005; 58: 175-193.
7. Benito FJ, Mintegi S.Trastornos paroxísticos no epilépticos. En:Diagnóstico y tratamiento de Urgencias pediátricas 4ª ed.2006; pp. 478-481.
8. Gary R. Fleisher, Stephen Ludwig, Fred M. Henretig.Syncope.Text book of Pediatric Emergency Medicine 5ª ed.2006; pp. 649-655.

37VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Histiocitosis de células de Langerhans congénita
Casos clínicos
C. Guerrero Laleona(1), M. Guiu Campos(2), C. Calvo Escribano(3), I. Marquina Ibáñez(4), A.M. Morales Callaghan(5)
(1)Servicio de Pediatría. Hospital de Alcañiz (Teruel). (2)Medicina Familiar y Comunitaria del área de Alcañiz (Teruel)(3)Servicio de Oncología. Hospital Infantil Universitario Miguel Servet. Zaragoza
(4)Servicio de Anatomía Patológica del Hospital de Alcañiz (Teruel). (5)Servicio de Dermatología. Hospital de Alcañiz (Teruel)
[Bol Pediatr Arag Rioj Sor, 2010; 40: 37-39]
Correspondencia: Carmelo Guerrero LaleonaServicio de Pediatría. Hospital de AlcañizDoctor Repollés, 2. 44600 Alcañiz (Teruel)e-mail: [email protected]: julio de 2010. Aceptado: julio de 2010
RESUMENDescribimos el caso de un lactante varón con diagnóstico de Histiocitosis de células de Langerhans (HCL) conafectación localizada en piel y mucosas de inicio neonatal. Si la Histiocitosis de células de Langerhans es unaenfermedad muy infrecuente, todavía lo es más la forma congénita o neonatal también llamada enfermedad deHashimoto-Pritzker que se da con una frecuencia de 1-2/1.000.000 de recién nacidos.
PALABRAS CLAVEHistiocitosis, Langerhans, congénita.
Langerhans cell histiocytosis congenitalABSTRACTWe describe a male newborn with the diagnosis of Langerhans cell histiocytosis who had skin and mucous membranesinvolvement. If Langerhans cell histiocytosis is a very rare disease, the congenital or newborn variety (Hashimoto-Pritzkerdisease) is even more 1,2, which has a frequence of 1-2/1000000 newborns.
KEY WORDSCongenital, Langerhans, histiocytosis.
INTRODUCCIÓN
La Histiocitosis de células de Langerhans congénita oenfermedad de Hashimoto-Pritzker es una rara enfer-medad que se da en recién nacidos, caracterizada por lapresencia de pápulas y/o nódulos generalizados.Presentamos el caso de un varón con afectación de piely mucosas de inicio neonatal (1,2).
CASO CLÍNICO
Presentamos el caso de un lactante varón de 3 meses deedad, nacido a término y sin antecedentes familiares de
interés, que desde el nacimiento ha presentado variosbrotes intercurrentes de lesiones maculosas y papulove-siculosas, generalizadas, diagnosticadas en un comienzode pustulosis del recién nacido y posteriormente de pio-dermitis, sin respuesta a tratamiento. En el último brotedichas lesiones se acompañaron además de erosiones enmucosa oral y conjuntivitis.
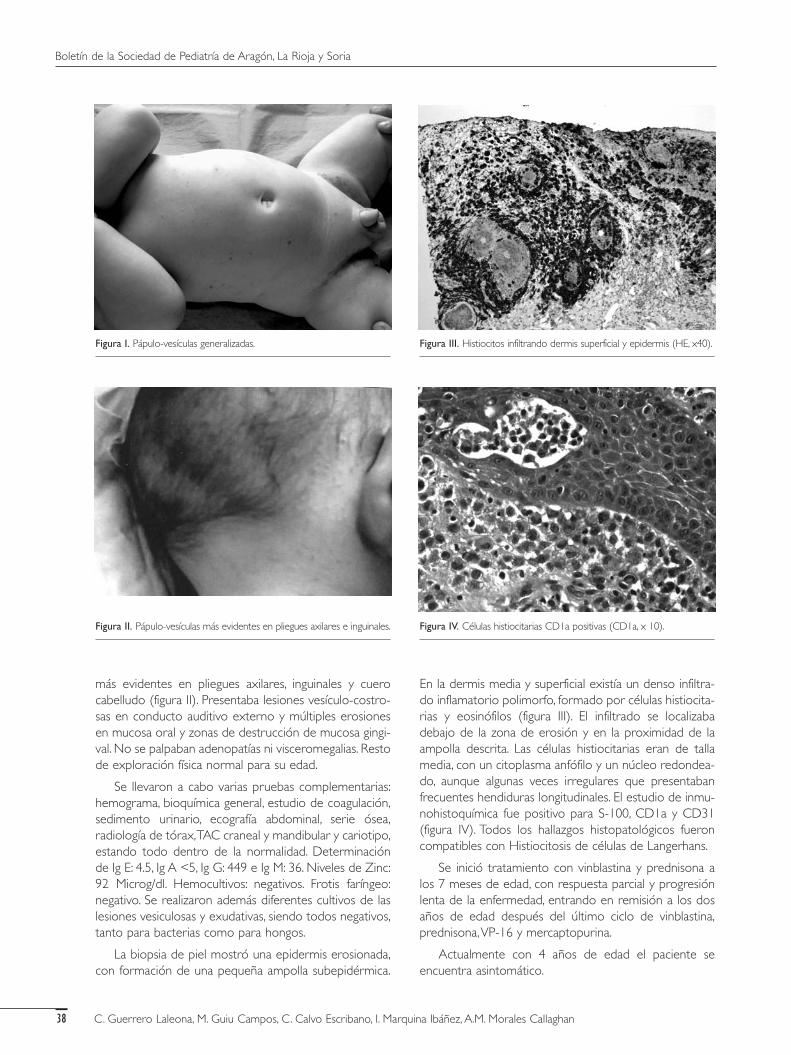
En la exploración física general el lactante presentabaun peso de 6.500 gramos (P75), talla de 63 centímetros(P75) y perímetro craneal de 41,5 centímetros (P50). Encuanto a la exploración cutánea se observaban lesionesmaculosas y papulovesiculosas generalizadas (figura 1),
Boletín de la Sociedad de Pediatría de Aragón, La Rioja y Soria
38 C. Guerrero Laleona, M. Guiu Campos, C. Calvo Escribano, I. Marquina Ibáñez, A.M. Morales Callaghan
más evidentes en pliegues axilares, inguinales y cuerocabelludo (figura II). Presentaba lesiones vesículo-costro-sas en conducto auditivo externo y múltiples erosionesen mucosa oral y zonas de destrucción de mucosa gingi-val. No se palpaban adenopatías ni visceromegalias. Restode exploración física normal para su edad.
Se llevaron a cabo varias pruebas complementarias:hemograma, bioquímica general, estudio de coagulación,sedimento urinario, ecografía abdominal, serie ósea,radiología de tórax,TAC craneal y mandibular y cariotipo,estando todo dentro de la normalidad. Determinaciónde Ig E: 4.5, Ig A <5, Ig G: 449 e Ig M: 36. Niveles de Zinc:92 Microg/dl. Hemocultivos: negativos. Frotis faríngeo:negativo. Se realizaron además diferentes cultivos de laslesiones vesiculosas y exudativas, siendo todos negativos,tanto para bacterias como para hongos.
La biopsia de piel mostró una epidermis erosionada,con formación de una pequeña ampolla subepidérmica.
En la dermis media y superficial existía un denso infiltra-do inflamatorio polimorfo, formado por células histiocita-rias y eosinófilos (figura III). El infiltrado se localizabadebajo de la zona de erosión y en la proximidad de laampolla descrita. Las células histiocitarias eran de tallamedia, con un citoplasma anfófilo y un núcleo redondea-do, aunque algunas veces irregulares que presentabanfrecuentes hendiduras longitudinales. El estudio de inmu-nohistoquímica fue positivo para S-100, CD1a y CD31(figura IV). Todos los hallazgos histopatológicos fueroncompatibles con Histiocitosis de células de Langerhans.
Se inició tratamiento con vinblastina y prednisona alos 7 meses de edad, con respuesta parcial y progresiónlenta de la enfermedad, entrando en remisión a los dosaños de edad después del último ciclo de vinblastina,prednisona,VP-16 y mercaptopurina.
Actualmente con 4 años de edad el paciente seencuentra asintomático.
Figura I. Pápulo-vesículas generalizadas. Figura III. Histiocitos infiltrando dermis superficial y epidermis (HE, x40).
Figura IV. Células histiocitarias CD1a positivas (CD1a, x 10).Figura II. Pápulo-vesículas más evidentes en pliegues axilares e inguinales.

39
Histiocitosis de células de Langerhans congénita
VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
DISCUSIÓN
La Histiocitosis es un grupo de enfermedades caracteri-zadas por la proliferación de macrófagos en diferentesórganos y sistemas. La proliferación puede ser localizada(en piel o hueso) o generalizada, invasiva o no invasiva,de elementos maduros o inmaduros.Todo ello determi-na el compromiso de unos u otros órganos y vísceras delsistema mononuclear-fagocítico, y el tipo de afectación,benigna o maligna en cada caso.
La gran diversidad de posibilidades clínicas ha dadolugar a múltiples cuadros, la mayoría con expresión cutá-nea específica y precoz. Desde 1987 se clasifican en tresgrandes grupos: Histiocitosis tipo I o Histiocitosis de célu-las de Langerhans; Histiocitosis tipo II o Histiocitosis decélulas no Langerhans e Histiocitosis tipo III o malignas.
Su etiología es desconocida. Se cree que existe unabase genética por su mayor frecuencia en gemelos, laexistencia de agrupaciones familiares de casos, la asocia-ción con otras neoplasias y la mayor frecuencia de ines-tabilidad cromosómica en los pacientes con HCL (si bienno se ha demostrado ninguna lesión cromosómica espe-cífica). Es posible que cambios en genes reguladores pre-dispongan a un control inadecuado de la respuesta inmu-nológica que lleve a una proliferación, clonal o no, decélulas de Langerhans o a alteraciones de sus mecanis-mos de apoptosis. Dicha proliferación estaría potenciadapor la producción de citocinas. Las formas localizadas seobservan más frecuentemente en varones, mientras queno hay predominio sexual en las generalizadas.
Creemos que nuestro caso es la forma neonatal, aun-que para definirla como tal es necesario hacer el diag-nóstico antes de los 28 días de vida, en la mayoría de loscasos éste se retrasa por error diagnóstico y sólo el 20%de los casos se diagnostica dentro de este período (3). Esnecesario establecer diagnóstico diferencial con otrasenfermedades cutáneas como varicela congénita, herpes,melanosis pustulosa, impétigo, penfigoide gestacionalneonatal, eritema tóxico neonatal y candidiasis neonatalo congénita (4-5). Nuestro paciente debutó con lesionescutáneas generalizadas (figura I) con predominio encuero cabelludo (figura II), conducto auditivo, plieguesaxilar e inguinal y posteriormente aparecieron lesionesdestructivas en mucosa gingival, acompañado de erup-ción dental temprana. Aunque en un principio se pensa-ba que la forma neonatal cutánea autoinvoluciona deforma espontánea (2, 4, 6, 7), actualmente cada vez son máslas publicaciones que hablan de un claro predominiomultisistémico en su evolución (3-8, 9). Sin embargo, no estáclaro cuál es el momento idóneo para iniciar el trata-miento para evitar yatrogenia (3, 9, 10).
En nuestro caso decidimos iniciar tratamiento a los 7meses de edad con vinblastina y prednisona por la per-sistencia de lesiones cutáneas, otitis recidivante y presen-cia de lesiones en mucosa oral, a pesar de lo cual noentró en remisión. Actualmente, con 2 años de vida, seencuentra asintomático aunque sigue en control estrictopor parte de los Servicios de Pediatría, Oncología yDermatología.
BIBLIOGRAFÍA1. Hashimoto K, Pritzker MS. Electron microcopic study of reti-
culohistiocytoma. An unusual case of congenital self healingreticulohistiocytosis. Arch Dermatol 1973; 107: 263-270.
2. Pavloviç MD, Miniç A, Zolotarevski L,Vesiç S. Disseminatedcrusted papules in a newborn. Vojnosanit Pregl 2006; 63:681-683.
3. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocy-tosis in neonates. Pediatr Blood Cancer 2005; 45: 802-807.
4. Guillermo N, Hernández-Machín B, Borrego L. Pápulas cos-trosas generalizadas en una recién nacida. Actas Dermosi-filiogr 2008; 99: 567-568.
5. Fuentes MA, Montahud C, Belenguer MJ, Pérez M,Vargas F.Presentación congénita de una histiocitosis de células deLangerhans no autoinvolutiva. An Pediatr (Barc) 2006; 65:634-635.
6. Walia M, Paul P, Mishra S, Mehta R. Congenital Langerhanscell histiocytosis: the self-healing variety. J Pediatr HematolOncol 2004; 26: 398-402.
7. Isaacs H Jr. Fetal and neonatal histiocytosis. Pediatr BloodCancer 2006; 47: 123-129.
8. Querings K, Starz H, Balda BR. Clinical spectrum of cutane-ous Langerhans’ cell histiocytosis mimicking various diseases.Acta Derm Venereol 2006; 86: 39-43.
9. Lau L, Krafchik B, Trebo MM, Weitzman S. Cutaneous Lan-gerhans cell histiocytosis in children under one year. PediatrBlood Cancer 2006; 46: 66-71.
10. Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cellHistiocytosis presenting in the neonatal period. A retros-pective case series. Arch Pediatr Adolesc Med 2001; 155:778-783.

40
Linfadenitis tuberculosa por Mycobacterium bovisFernando de Juan Martín
Servicio de Infecciosas. Hopsital Infantil Universitario Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 40-42]
RESUMENLa tuberculosis por Mycobacterium bovis (M. bovis) es una zoonosis que afecta a la especie humana en todo el mundo.Infecta al ganado vacuno que puede transmitir este agente a la especie humana al consumir leche no pasteurizada,también puede ser transmitido entre humanos por vía inhalatoria. M. bovis puede causar linfadenitis cervical sinenfermedad pulmonar. A su vez produce una pirazinamidasa que inactiva la acción terapéutica de la pirazinamida,por lo que es necesario su identificación ya que la pirazinamida no debe ser incluida en la pauta de tratamiento.Presentamos el caso de un niño de 2 años de edad, nacido en Marruecos pero viviendo en Soria (España), quepresenta una linfadenitis cervical derecha en la que, en el examen del aspirado realizando punción con aguja fina(PAAF), se observaron BAAR, que se identificaron como M. bovis.
PALABRAS CLAVELinfadenitis cervical,Tuberculosis bovina.
Cervical lymphadenitis due to Mycobacterium bovisABSTRACTTuberculosis due to M. bovis is a zoonosis that affects humans all around the world. Primarily it infects cattle which cantransmit the agent to humans, and can also be transmitted among humans. M. bovis may cause acute cervical lymphadenitiswithout pulmonary disease. It produces a pyrazinamidase and its identification is required because pyrazinamide should notbe included in the treatment regimen. Accurate speciation is important to select the antibiotic regimen.We introduce here a2-year-old baby, who was born in Morocco but lives in Soria (Spain). He had a right cervical lymph node. Examination ofaspirate revealed bacilli identified as M. bovis.
KEY WORDSCervical lynphadenitis, Bovine tuberculosis.
Correspondencia: Fernando de Juan MartínHospital Infantil Universitario Miguel Servet. Sección de Enfermedades InfecciosasIsabel la Católica 1-3. 5008-Zaragozae-mail: [email protected]: junio de 2010. Aceptado: junio de 2010
INTRODUCCIÓN
M. bovis es el agente causal de la tuberculosis en anima-les salvajes y domésticos y en ocasiones también en laespecie humana. El ganado bovino es el principal reser-vorio de esta especie. El contagio a la especie humana serealiza generalmente por la ingesta de leche no pasteuri-zada y se observa principalmente en inmigrantes prove-nientes de países con tasas elevadas de infección del
ganado. La enfermedad se suele manifestar más frecuen-temente en forma de linfoadenitis cervical y afectaciónabdominal (1-3).
Varón de 2 años de edad, hijo de padres inmigrantesprocedentes de Marruecos, donde viaja periódicamentesiendo el último viaje hace 4 meses. A un primo herma-no se le diagnóstico 2 meses antes de linfadenitis cervi-cal por M. bovis.
Fernando de Juan Martín
Casos clínicos

41
Linfadenitis tuberculosa por Mycobacterium bovis
VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
El paciente es controlado en el Hospital «Virgen delMirón» de Soria, donde es derivado desde su Centro deSalud por presentar una adenopatía cervical derecha devarios meses de evolución y de 4 x 4 cm de tamaño, noadherida a planos profundos.
La ecografía mostró un nódulo de 4,2 x 2,7 x 2 cmhipoecoica con un centro graso. En el TAC cervical seobservó una masa cervical derecha que parecía estar for-mada por dos grandes adenopatías, de unos 4 cm de diá-metro la adenopatía externa y de 2,2 cm la adenopatíainterna parafaríngea. Ambas presentaban múltiples zonasde hipocaptación compatibles con necrosis ganglionar.
El Mantoux fue de 15 mm y la PAAF realizada mostróuna necrosis caseosa compatible con etiología tuberculosa.
Se inició tratamiento con isoniazida, rifampicina ypirazinamida.
En las muestras de jugo gástrico se aisló M. bovis resis-tente a pirazinamida, observándose al mismo tiempo unaumento significativo del tamaño de la adenopatía, por loque se remitió al Hospital Infantil Universitario MiguelServet.
Al Laboratorio de Microbiología del Hospital Uni-versitario Miguel Servet se remitieron tres muestras deaspirado gástrico y dos de PAAF; en las baciloscopias rea-lizadas en estas muestras se observaron escasos BAARalterados morfológicamente.
Las muestras recibidas fueron procesadas por elmétodo de descontaminación de N-acetil-L-cisteína yconcentradas tras su procesamiento, se inocularon en losfrascos de cultivo (BacT/ALERT® MP, bioMerieux) y elsedimento restante se guardó congelado para poder rea-lizar otras determinaciones por genética molecular, comoidentificación y detección de genes de resistencia paraisoniazida y rifampicina.
Todos los cultivos fueron negativos, probablementeporque los BAAR observados no estuvieran viables,debido al tratamiento recibido y a que M. bovis presentamayor dificultad de crecimiento en los medios de cultivohabituales, aunque los utilizados en nuestro laboratorioestán preparados para ello.
Ante los resultados negativos de los cultivos, realiza-mos del sedimento congelado la detección de RNA deM. tuberculosis complex con el GenoType ®MycobacteriaDirect (HAIN LIFESCENCE) con resultado positivo paraM. tuberculosis complex. Seguidamente realizamos elGenoType ®MTBC que está diseñado para diferenciarentre las especies del complejo tuberculoso, en cultivopositivo. Puesto que se trataba de un sedimento conge-lado, y no de un cultivo positivo, aumentamos en núme-ro de ciclos en el programa del termociclador recomen-dado por el fabricante, hasta un total de 40 ciclos, y con-seguimos una identificación como M. bovis. Por último,con el test GenoType ®MTBDRplus que detecta lasresistencias a isoniazida y rifampicina por las mutaciones
más frecuentes en los genes que las codifican: katG einhA para isoniazida y rpoB para rifampicina, obtuvimosun resultado de sensibilidad a ambos fármacos, al nodetectar ninguna mutación, ni ninguna ausencia del geno-tipo salvaje para los genes testados. No se pudo realizarla sensibilidad a pirazinamida por no disponer de ese test,tampoco se pudo realizar el antibiograma al no conseguircrecimiento de la micobacteria. Una vez conocida que laetiología era debida a M. bovis, se modificó el tratamien-to y se continuó con isoniazida, rimfampicina, etambutoly amikacina. Posteriormente se prolongó el tratamientocon isoniazida más rifampicina.
A los 3 meses la adenopatía se mantenía con untamaño parecido y fistulizó y se procedió a su extirpa-ción quirúrgica (figura I).
El estudio anatomopatológico mostró una linfadenitisgranulomatosa necrotizante con trayecto fistuloso a piel.
En el estudio microbiológico, se observaron escasosBAAR alterados morfológicamente, cultivo para mico-bacterias negativo, y la detección de RNA para M. tuber-culosis complex resultó positiva, los mismos resultadosque anteriormente.
La evolución a los 3 meses de la exéresis es satisfac-toria, observándose solamente una cicatriz ligeramenteantiestética, por lo que es remitida para control al servi-cio de cirugía estética.
DISCUSIÓN
Dentro del complejo Mycobacterium tuberculosis se agru-pan diferentes micobacterias que causan enfermedad enel hombre y en los animales. Este complejo se componede las especies M. tuberculosis, M. africanum, M. bovis, M.microti, M. pinnipedii y M. canettii (4). La especie M. bovis hasido subdividida recientemente en M. bovis subsp. bovis,resistente a pirazinamida (PZA) y M. bovis subsp. caprae,sensible a PZA9-11.
Figura I. Linfadenitis cervical fistulizada.

Boletín de la Sociedad de Pediatría de Aragón, La Rioja y Soria
42
La vacuna BCG está elaborada a partir de una cepa deM. bovis que por sucesivos pases ha perdido poder pató-geno y conserva su capacidad inmunógena. En la actuali-dad es la única vacuna existente contra la tuberculosis.
El M. bovis es el más frecuente de este grupo en nues-tro medio. En el año 2006 sobre un total de 2.164 cepasde M. tuberculosis estudiadas en Laboratorio deReferencia de Micobacterias del Centro Nacional deMicrobiología del Instituto de Salud Carlos III, se identifi-caron: M. africanus 3, M. bovis 5 y M. tuberculosis 2.156.
M. bovis se caracteriza por ser resistente a pirazina-mida debido a un cambio del nucleótido guanina porcitosina, lo que origina la sustitución del aminoácido his-tidina por ácido aspártico, lo que da lugar a la inactivaciónde la pirazinamidasa: enzima necesaria para convertir lapirazinamida en su principio activo (5).
La tuberculosis bovina es un problema importante enla economía y en la sanidad veterinaria. La ComunidadEconómica Europea clasifica la tuberculosis bovina comode categoría I «Enfermedad animal con importanciasobre la salud pública».
El programa de lucha en España se inicia de forma sis-temática a partir de 1986 tras la incorporación de nues-tro país a la CEE. El avance conseguido con el ProgramaNacional de Erradicación de la Tuberculosis Bovina a lolargo de los años ha supuesto beneficios importantes nosólo para la economía y la sanidad animal, sino tambiénpara la seguridad alimentaria y la salud pública. Se ha inte-grado con los programas de inspección en el controlsanitario de la carne y el tratamiento térmico de la lechey productos lácteos (2,3).
En España, estas campañas están cofinanciadas por laComunidad Europea y se basan en la prueba de la tuber-culina con PPD bovino y el sacrificio de los animalesinfectados. Estas campañas han supuesto una disminuciónimportante de la enfermedad en el ganado bovino y de
los casos de enfermedad en humanos, prácticamente lamisma que en el resto de países desarrollados. En Españala prevalencia de tuberculin-positivos en la cabaña vacu-na era de 1,38% en el año 1996 y descendió a 0,49 enel año 2007. La incidencia sigue disminuyendo pero toda-vía se considera muy elevada cuando se compara con lamayoría de los países europeos (6,7).
La tuberculosis por M. bovis en la especie humana escaracterística de los países en vías de desarrollo. En nues-tro medio es muy infrecuente y la mayoría de los casosocurren en niños inmigrantes procedentes de paísesdonde todavía existe una alta prevalencia de tuberculo-sis bovina y no se realiza la pasteurización de la leche (8,9).
La enfermedad suele presentarse como tuberculosisextrapulmonar, especialmente como linfadenitis cervicaly menos frecuente como tuberculosis abdominal queincluye la infección del peritoneo, los ganglios linfáticos, elhígado, el bazo y el páncreas (10-11).
El tratamiento de la linfadenitis cervical es el mismo, enlíneas generales, que el de la tuberculosis pulmonar. Debeiniciarse empíricamente con cuatro fármacos, isoniazida,rifampicina, pirazinamida y etambutol o un aminoglucósido.
Una vez aislada la micobacteria, si se identifica comoM. bovis, debido a su resistencia a pirazinamida debe sus-penderse este fármaco y continuar con los tres restanteshasta completar dos meses de tratamiento, para conti-nuar con isoniazida y rifampicina durante 4 meses más (12).Cuando la evolución no es favorable y persiste la adeno-patía, hay que recurrir a la exéresis quirúrgica.
En nuestro caso se inició tratamiento con isoniazida,rifampicina y pirazinamida que se modificó por isoniazida,rinfampicina, etambutol y amikacina al identificarse la mico-bacteria como M. bovis, hasta completar 2 meses, y se con-tinuó con isoniazida más rifampicina durante 4 meses más.La evolución en nuestro caso fue tórpida con fistulizaciónde la misma, necesitando la extirpación de la adenopatía.
Fernando de Juan Martín
BIBLIOGRAFÍA1. Scorpio A, Collins D, Whipple D et al. Rapid differentiation
of bovine and human tubercle bacilli based on a characte-ristic mutation in bovine pyrazinamidase gene. J ClinMicrobiol 1997; 35: 106-110.
2. Thoen C, LoBue P, Kantor I. The importance of Mycobac-terium bovis as a zoonosis.Vet Microbiol 2006; 112: 339-345.
3. O’Reilly LM, Daborn CJ. The epidemiology of Mycobac-terium bovis infections in animals and man: a review.TuberLung Dis 1995; 76: S1-46.
4. Herrera-León L, Pozuelo-Díaz R, Molina T, et al. Aplicaciónde métodos moleculares para la identificación de las espe-cies del complejo Mycobacterium tuberculosis. EnfermInfecc Microbiol Clin 2009; 27: 496-502.
5. Grange JM. Mycobacterium bovis infection in human beings.Tuberculosis 2001; 81: 71-77.
6. Programa de erradicación de la tuberculosis bovina presen-tado por España para los años 2009-2010. DirecciónGeneral de Recursos Agrícolas y Ganaderos. SubdirecciónGeneral de Producción Ganadera. Ref: PN TB 2009-2010.
7. Benito J, March P, Balfagón P, Cayla J.Tuberculosis bovina enEspaña. Med Clin (Barc) 2005; 125: 475-479.
8. Alfayate S, Piñero J, Montero MT, Mula JA, Paredes P, ZarauzJM. Enfermedad tuberculosa por Mycobacterium bovis en laregión de Murcia. An Pediatr (Barc) 2009; 71: 327-330.
9. Rodríguez E, Sánchez LP, Pérez S, et al. Human tuberculosisdue to Mycobacterium bovis and M. caprae in Spain, 2004-2007. Int J Tuberc Lung dis 2009; 13: 1536-1541.
10. Lavas M, Moonan P, Cowan L, et al. Human tuberculosis dueMycobacterium bovis in the the United States, 1995-2005.Clin Infect Dis 2008; 47: 168-175.
11. Yellin L. Mycobacterium bovis versus Mycobacterium tuber-culosis as a cause of acute cervical lymphadenitis withoutpulmonary disease. Pediatr Infect Dis J 2004; 23: 590-591.
12. Ruiz-Manzano JR, Blanquer R, Calpe JL, Caminero JA, Cayla Jet al. Normativa SEPAR. Diagnóstico y tratamiento de latuberculosis. Arch Bronconeumol 2008; 44: 551-566.

43VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Transmisión congénitade Trypanosoma cruzi (enfermedad de Chagas):a propósito de un caso
Casos clínicos
F. de Juan Martín(1), L. Roc Alfaro(2), E. Lomba Fuentes(2)
(1)Sección Enfermedades Infecciosas. Hospital Infantil Universitario Miguel Servet. Zaragoza(2)Servicio Microbiología. Hospital General Universitario Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 43-44]
Correspondencia: Fernando de Juan MartínHospital Infantil Universitario Miguel Servet. Sección de Enfermedades InfecciosasIsabel la Católica 1-3. 5008-Zaragozae-mail: [email protected]: junio de 2010. Aceptado: junio de 2010
RESUMENComunicamos un caso de transmisión vertical de Trypanosoma cruzi de una madre boliviana que vive en España y afectade enfermedad de Chagas asintomática. La serología a T. cruzi era positiva en la madre. La reacción en cadena de lapolimerasa (PCR) y el examen directo del parásito fueron positivos en sangre del recién nacido. Recibió tratamiento conbenznidazole 10 mg/kg/día durante 90 días.La PCR,el examen directo y los test serológicos a T.cruzi se hicieron negativos.
PALABRAS CLAVETrypanosoma cruzi,Transmisión vertical.
Vertical transmisión of Trypanosoma cruzi infection (Chagas’disease): a case reportABSTRACTWe report a case of congenital transmission of Trypanosoma cruzi from a Bolivian mother with asymtomatic Chagas diseaseliving in Spain.The serology to T. cruzi were positive for the mother. Nested PCR and direct examination were positive in theblood of the neonate. Received treatment with benznidazole 10 mg/kg/day for 90 days. Nested PCR, examination direct andserologicol test to T. cruzy were negative.
KEY WORDSTrypanosoma cruzi,Vertical transmission.
INTRODUCCIÓN
La enfermedad de Chagas representa un problemaimportante de salud pública en América Latina. Se calculaque en áreas endémicas hay alrededor de 13 millones deinfectados y la prevalencia de infección materna puedesuperar el 50% (1). En España, así como en otros paíseseuropeos, el incremento de inmigrantes latinoamerica-nos con enfermedad de Chagas ha aumentado el riesgode trasmisión por vía vertical de esta enfermedad y porconsiguiente la aparición de formas congénitas (2-5).Comunicamos un caso de transmisión congénita deTrypanosoma cruzi de una madre boliviana con enferme-dad de Chagas con residencia en España.
CASO CLÍNICO
Recién nacido, primer hijo de una madre de 23 años deedad, producto de un embarazo y parto normal. Pesó alnacer 3.740 g. La madre presentó anticuerpos frente a T.cruzi durante el embarazo. En el recién nacido la deter-minación de anticuerpos IgG anti-T. cruzi fueron positivospor el método ELISA y títulos >1/160 por inmunofluo-rescencia (IFI). El estudio parasitológico en fresco, portécnica de gota gruesa y microhematocrito de sangrecapilar, permitió identificar el parásito. La reacción encadena de la polimerasa (PCR) a T. cruzi fue tambiénpositiva.

Boletín de la Sociedad de Pediatría de Aragón, La Rioja y Soria
44 Fernando de Juan Martín, Lourdes Roc Alfaro, Elena Lomba Fuentes
La exploración clínica del niño fue siempre normal,no presentó signos de distrés respiratorio, no se obser-varon visceromegalias, la exploración cardiológica fuenormal, no se apreciaron alteraciones neurológicas y pre-sentó una buena ganancia ponderoestatural.
Se estableció el diagnóstico de enfermedad de Chagascongénita asintomática y se inició tratamiento a los 2meses de vida con benznidazol a la dosis de 10 mg/kg/díadurante 90 días. En el control realizado al mes del trata-miento, la parasitemia había desaparecido y la PCR se hizonegativa. Los niveles de anticuerpos IgG anti-T. cruzi fuerondisminuyendo hasta desaparecer a los 6 meses de vida.
DISCUSIÓNLa enfermedad de Chagas o tripanosomiasis americanaes producida por el protozoo flagelado Trypanosoma cruziy representa un problema importante de salud públicaen América Latina. La transmisión del parásito se realizaen áreas endémicas fundamentalmente por la picadurade un vector (hemípteros redúvidos de la subfamiliaTriatominae). Otros mecanismos de infección son latransmisión vertical vía madre infectada, la transfusión desangre y el transplante de órganos. En áreas no endémi-cas, donde el vector no está presente, estos dos últimosmecanismos son los más importantes.
La prevalencia de esta infección en embarazadas lati-noamericanas en España oscila entre el 3,4% y el 4,64%,llegando a ser en bolivianas del 17,5% y la tasa de trans-misión vertical supera el 7,3% (6,7).
La enfermedad de Chagas congénita se puede pre-sentar de forma sintomática o asintomática. La forma sin-tomática oscila entre el 2-10% de los casos y aparece deforma precoz durante el primer mes de vida o de formatardía si lo hace más tarde. Pueden presentar manifesta-ciones graves como distrés respiratorio, hepatoespleno-megalia, miocarditis y meningoencefalitis que a vecespone en peligro la vida del niño. Después de la fase aguda,sea sintomática o asintomática, si no son tratados entra-rán en la fase crónica y entre el 25-35% desarrollarán enla edad adulta complicaciones cardiacas, digestivas o neu-rológicas. El tratamiento suele ser eficaz en más del 90%de los niños pero su eficacia disminuye con la edad (1).
Los hijos de embarazadas infectadas presentan riesgode infección. Como los test serológicos no pueden dife-renciar si los anticuerpos anti-T. cruzi proceden de lamadre o del recién nacido, deben ser sometidos a exa-men directo de la sangre, para buscar la presencia delparásito, y a pruebas de PCR. La positividad de cualquie-ra de las dos pruebas junto con el antecedente de sero-logía positiva en la madre sirve para establecer el diag-nóstico de enfermedad de Chagas congénita e iniciar eltratamiento. Si la parasitemia y la PCR son negativas sedeben repetir antes del mes de vida y si siguen siendonegativas se realizarán estudios serológicos hasta los 9-10meses, cuando se supone que el nivel de anticuerposmaternos tiene que haber disminuido. Las técnicas utili-zadas para determinar IgM específicas dan resultados fal-sos con mucha frecuencia (8, 9).
En nuestro caso, la madre presentó anticuerpos anti-T. cruzi en el estudio serológico, se observó la presenciade parásitos en examen directo de sangre y la PCR a T.cruzi fue positiva.
Una vez establecido el diagnóstico de la enfermedadhay que comenzar, tan pronto como sea posible, el tra-tamiento con benznidazol a la dosis de 10 mg/kg/díadurante un período de 30-60 días. La eficacia del trata-miento en las formas congénitas es superior al 90% cuan-do son tratados durante el primer año de vida. La res-puesta al tratamiento se valora por la desaparición de laparasitemia y la negativización de la PCR (6).
Los enfermos tratados de enfermedad de Chagascongénita deben ser controlados posteriormente median-te la evolución de la tasa de anticuerpos específicos(ELISA e IFI) y la determinación de la PCR. Este controlestá indicado a los 3 meses de terminar el tratamiento yposteriormente de forma anual durante 3-5 años.
En nuestro caso, la parasitemia desapareció a los 90días de tratamiento y se negativizó la PCR. Los anticuer-pos fueron indetectables a los 6 meses de vida.
Debido a la aparición de casos de enfermedad deChagas congénita en nuestro medio es aconsejable el criba-do de esta enfermedad en las mujeres embarazadas proce-dentes de países endémicos, especialmente de Bolivia (4-6, 9).
BIBLIOGRAFÍA1. Bern C, Montgomery S, Herwald B et al. Evaluation and tre-
atment of Chagas Disease in the United States. A systema-tic review. JAMA 2007; 298: 2171-2181.
2. Paricio JM, Benlloch MJ, Collar JI et al.Vigilancia epidemioló-gica de la transmisión vertical de la enfermedad de Chagasen tres maternidades de la Comunidad Valenciana. EnfermInfecc Microbiol Clin 2008; 26: 609-613.
3. Muñoz J, Coll O, Juncosa T et al. Prevalence and verticaltransmission of Tripanosoma cruzi infection among pregnantlatin american women attending 2 maternity clinics inBarcelona, Spain. Clin Infect Dis J 2009; 48: 1736-1740.
4. Riera C, Guarro A, Kassab HE et al. Congenital transmission ofTripanosoma cruzi in Europe (Spain): a case report. Am J TropMed Hyg 2006; 75: 1078-1081.
5. Flores-Chavez M, Faez Y, Olalla J et al. Fatal congenital Chagasdisease in a non-endemic area: a case report. Cases Journal.2008; I: 302.
6. Carrilero B, Quesada J, Alfayate S, Segovia M. Enfermedadde Chagas congénita en recién nacido de madre de origenboliviano. Enferm Infecc Microbiol Clin 2008; 27: 487-488.
7. Jackson Y, Myers C, Diana A et al. Congenital transmission ofChagas disease in latin american immigrants in Switzerland.Emerg Infect Dis 2009; 15: 601-603.
8. Gascón J. Diagnóstico y tratamiento de la Enfermedad deChagas importada. Conferencia de Consenso. Med Clin(Barc) 2005; 125: 230-235.
9. Gascón J, Pinazo MJ. Control de la transmisión vertical deTripanosoma cruzi en España: principal reto de la patologíaimportada. Enferm Infecc Microbiol Clin 2008; 26: 607-608.

45VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
Alcalosis metabólica hipoclorémica:presentación de fibrosis quística en un lactante
P. Huerta, L. Cuadrón, F. Fuertes, S. Ortiz, E. Muñoz, A. Lázaro, J.L. Olivares
Servicio de Pediatría. Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 45]
INTRODUCCIÓN
La fibrosis quística (FQ) es la enfermedad autosómica recesiva gravemás frecuente de la raza caucásica. El defecto fundamental es la funciónreducida o ausente de la proteína reguladora de la conductancia trans-membrana (CFTR). La presentación clínica clásica se caracteriza porenfermedad pulmonar crónica, deficiencia pancreática y concentracio-nes altas de electrolitos en sudor. La deshidratación con alcalosis meta-bólica hipoclorémica es una manifestación conocida de la FQ, peroinfrecuente como forma de presentación. Se describe un caso de unalactante con diagnóstico en período neonatal de FQ homozigota AF508que presentó un cuadro de alcalosis metabólica hipoclorémica e hipo-natrémica.
CASO CLÍNICO
Lactante de 6 meses con antecedente de diagnóstico en período neo-natal de FQ que ingresa por presentar en el último mes estancamien-to ponderal y anorexia. En la exploración física destaca un peso de
5,735 kg (<P3). Regular estado nutricional con disminución de la tur-gencia y signo del pliegue negativo. Fontanela anterior levementedeprimida. Resto de exploración por aparatos normal. En los exáme-nes complementarios iniciales destaca pH 7,67, pCO3 43 mmHg,CO3H 43,8 mmol/L, EB 21,3 mmol/L, Na 132,9 mEq/L, K 3,69 mEq/L,cloro 74 mEq/L. En orina las cifras de sodio y cloro son inferiores dellímite de detección. A su ingreso se procedió a reponer el déficithidroelectrolítico, precisando nutrición enteral total a débito continuoen los primeros días de ingreso, siendo la evolución favorable en losdías posteriores.
COMENTARIOS
Aunque en nuestro caso el diagnóstico de FQ estaba establecido, sedebe considerar la FQ en pacientes de cualquier edad, especialmenteniños pequeños con cuadro clínico de anorexia, vómitos, detención delpeso y episodios recurrentes de deshidratación con alcalosis metabóli-ca hipoclorémica, sin otra causa que lo justifique, aunque no presentensíntomas respiratorios, digestivos o mal progreso de peso.
Convulsiones precoces en el recién nacido a término.«Causa poco frecuente»
S. Ortiz, F. Fuertes, J. Morales,T. Pérez, P. Collado, O. Bueno, P. Ventura
Servicio de Pediatría. Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 45]
INTRODUCCIÓN
La presencia de convulsiones en el período neonatal suele ser la mani-festación de una disfunción neurológica aguda como consecuencia deuna agresión cerebral. La cronología de aparición de las mismas puedeayudar en el establecimiento de la etiología; así la aparición en las pri-meras 24 horas de vida, probablemente se relacione con encefalopatíahipóxico-isquémica.
CASO CLÍNICO
Recién nacido a término, parto eutócico, aguas meconiales claras,Apgar7/8, remitido desde Hospital Comarcal por presentar desde el períodoneonatal inmediato episodios de apnea. AP: sin interés. Evolución: a sullegada se objetiva la existencia de «movimientos de boxeo» de extre-midad superior derecha, que no ceden a pesar de contención manual,iniciando tratamiento con fenobarbital IV, cediendo completamente. Alas 24 horas presenta de nuevo convulsiones generalizadas, comenzan-do tratamiento con fenitoína, con respuesta parcial. Se induce coma
barbitúrico, cediendo las convulsiones completamente, tras la retiradadel mismo se inicia tratamiento con ácido valproico, no presentandonuevos episodios. Pruebas complementarias: Ecografía transfontanelar :área hiperecogénica desde región cápsulo-talámica izquierda afectandoa todo hemisferio cerebral de ese lado y ligero desplazamiento de lalínea media. EEG: aplanamiento izquierdo. TAC cerebral: infarto isqué-mico izquierdo con afectación del territorio de la cerebral media yanterior izquierda. Estudio de trombofilias: madre portadora homoci-goto mutación MHTFR A1298C. En el momento del alta presenta dis-creta hipertonía de hemicuerpo derecho con exaltación de reflejososteotendionosos.
COMENTARIOS
La incidencia de accidentes cerebro-vasculares isquémicos en el reciénnacido no es bien conocida. Los factores predisponentes, etiología, asícomo la respuesta del cerebro a la agresión van a ser diferentes a otrasedades de la vida, dada la plasticidad neuronal, es por ello que precisanun enfoque diagnóstico-terapéutico individualizado.

46
Sesiones de la sociedad
Síndrome de Dandy-Walker.Comunicación de dos casos y revisión
N. Clavero,V. Giménez, D. Royo, M.Vara, A. Manero,V. Rebage
Servicio de Pediatría. Hospital Universitario Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 46]
INTRODUCCIÓN
Las malformaciones quísticas de fosa posterior en el recién nacido sonde gran interés, en particular el espectro malformativo de Dandy-Walker. El síndrome de Dandy-Walker (SDW) se caracteriza por la dila-tación quística del cuarto ventrículo e hipoplasia del vermis cerebelosoasociada a hidrocefalia congénita. La etiología es heterogénea. Las mani-festaciones clínicas suelen evidenciarse desde la infancia y puedenacompañarse de otras malformaciones congénitas externas y del siste-ma nervioso central.
CASOS CLÍNICOS
Recién nacido que ingresa en Neonatal por diagnóstico prenatal dequiste de fosa posterior. Como antecedentes familiares, hermana de 9años con microcefalia y retraso cognitivo. Peso 3.450 g (>p95), talla:53 cm (p90), perímetro craneal: 36 cm (p90-97), Apgar 9/9 y explora-ción neurológica normal. En ecografía transfontanelar al nacimiento seapreciaba dilatación del cuarto ventrículo, y en TAC y RM hallazgos com-patibles con SDW. Resto de exámenes complementarios normales. Elpaciente se controla periódicamente en la Policlínica llevando hasta el
momento una evolución favorable, con normalidad del desarrollo neu-rológico y del crecimiento craneal.
Varón de 21 días, remitido para valoración de hidrocefalia congéni-ta. Madre DGA2, Apgar 9/10, peso 4.115 g (>p97), talla: 55 cm (>p97),PC: 38 cm (>p97). Al ingreso presenta macrocefalia, occipucio promi-nente y PC: 42,3 cm, fontanela prominente, ojos en sol poniente con nis-tagmo vertical intermitente, hipotonía axial y depresión sensorial. Laneuroimagen mostró gran dilatación triventricular con atrofia cerebelo-sa y quiste de fosa posterior compatible con un SDW.Valorado por elServicio de Neurocirugía, se programó derivación ventricular con buenaevolución posterior.
COMENTARIOS
Nuestros dos casos corresponden a una malformación quística de lafosa posterior por un síndrome de Dandy-Walker con dos formas clíni-cas muy distintas. Destacar el antecedente materno de DGA2 en elsegundo caso, que en la literatura aparece como factor predisponente.Incidir en la importancia que tiene el perímetro cefálico para la detec-ción de patología intracraneal en el recién nacido que curse con altera-ción del tamaño y crecimiento del cráneo.
Paciente con deleción terminaldel brazo largo del cromosoma 10. Caso clínico
S.T. Jiménez, C. Cristóbal, A.I. Fernández, M.T. Llorente
Servicio de Pediatría. Fundación Hospital Calahorra
[Bol Pediatr Arag Rioj Sor, 2010; 40: 46]
CASO CLÍNICO
Se trata de una niña de 4 años fruto del segundo embarazo de padresañosos no consanguíneos, con un hijo varón sano. Amniocentesis 46XX. Parto a las 38 semanas de gestación, por cesárea riesgo de pérdi-da de bienestar fetal. Peso recién nacido: 3.600 g. Apgar 3/7/10.Reanimación tipo II. Fenotipo peculiar con facies triangular, asimetríafacial con tortícolis congénito, megalotriquiasis, filtrum largo, labiosuperior fino, paladar ojival, mamilas separadas e hipotonía generaliza-da. Desde el nacimiento presentó cardiopatía tipo comunicación inter-auricular ostium secundum más ductus arterioso persistente, orificiolacrimal imperforado bilateral, estrabismo convergente bilateral y limi-tación a la mirada superior, displasia acetabular bilateral con subluxa-ción de cadera izquierda y reflujo vesico-ureteral grado IV bilateral. Seintervino en dos ocasiones del reflujo, se canalizaron los lacrimalestambién en dos ocasiones, llevó férula ortopédica en caderas durante6 meses, ha sido intervenida del estrabismo y la comunicación inter-auricular cerró de manera espontánea. Durante la evolución ha pre-sentado retraso psicomotor y del lenguaje importante con microcefa-
lia progresiva. Ha requerido tratamiento rehabilitador multidisciplinarconsiguiendo la sedestación a los 9 meses, deambulación y bisílabos alos 2 años, control de esfínteres diurno a los 3 años y medio, mastica-ción a los 4 años y todavía precisa logopedia. Ha sido escolarizada enintegración y presenta importantes dificultades para el aprendizaje ycarácter sociable pero impulsivo. Las pruebas de imagen cerebralesfueron normales. Ante la evolución se realizó cariotipo donde sedetectó deleción terminal del brazo largo de uno de los cromosomasdel par 10 a nivel de la banda q26.10 (46XX del (10) (q.26.1)(15). Elcariotipo de los padres fue normal, por lo que la deleción se conside-ró de novo.
COMENTARIOS
Las deleciones terminales del cromosoma 10 a nivel de la banda q26.1son poco frecuentes (16 casos descritos en la literatura) pero se tratade una cromosomopatía a buscar en el cariotipo para filiar un retrasopsicomotor, a pesar de amniocentesis normal, como en este caso, sobretodo si presenta las malformaciones asociadas típicas.

47VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
¿Qué papel tiene la enfermería de manera autónomaen el programa del niño sano?
M. Biosca, M.E. Doménech, M. Cunillera, E. Mateus, L. Olivart
ABS Les Borges Blanques. Lleida
[Bol Pediatr Arag Rioj Sor, 2010; 40: 47]
INTRODUCCIÓN
En Cataluña se han establecido una serie de revisiones del niño sano enlas que se ha obviado el papel del pediatra y la idea es que en un futu-ro próximo la enfermería se encargue en exclusiva de este programa. Secree que la enfermera de pediatría se puede entrenar para la realizaciónincluso de la exploración física, siendo capaz de detectar los problemasmás frecuentes y derivar a pediatra sólo si lo considera necesario.
OBJETIVO
Determinar qué alteraciones detecta la enfermera en las revisiones querealiza de manera autónoma (7-15 días, 8 meses, 15 meses i 6 años).
MATERIAL Y MÉTODOS
Recogida de datos de 236 revisiones realizadas de manera autónomaen una consulta de enfermería pediátrica (perdidos=20) durante 7meses (junio-diciembre de 2009).
RESULTADOS
N = 216 (59,25% niños y 40,75% niñas). Alteraciones detectadas poraparatos:
Piel: manchas (16,20%), dermatitis (13,88%), nevus (12,96%), 1 tiñacapitis y 2 eritemas solares.
Genitourinario: fimosis (7,87%), enuresis (1,85%), 4 circuncisiones y4 sinequias labios menores.
Digestivo: cólico lactante (5,09%), estreñimiento (1,38%), 2 vegeta-rianos.
Boca: caries (8,33%), maloclusión (6,01%).ORL: impactación cerumen (3,70%).Cabeza: microcefalia (2,31%), plagiocefalia (1,85%).Locomotor: pie plano (2,77%), genu valgo (2,31%), dismetría EEII
(1,85%).
COMENTARIOS
La enfermera de pediatría bien entrenada es capaz de detectar muchasde las alteraciones más frecuentes que se detectan en las revisiones delniño sano. Queda reservado al pediatra la exploración de los órganosinternos (soplos, visceromegalias, etc.). Difícilmente se puede obviar elpediatra para la realización de todas las revisiones del niño sano. Es unabuena opción la coordinación de revisiones entre los distintosprofesionales para llegar al objetivo: la educación sanitaria para la pre-vención y promoción de la salud y detección de alteraciones físicas, psí-quicas y sensoriales.
Intumescencia mamaria neonatal:¿un cuadro benigno?
P. Murillo, G. Herráiz, G. González,V. Rosel, E. Elías, G. Rodríguez
Servicio de Pediatría. Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 47]
INTRODUCCIÓN
Hasta 1/3 de los recién nacidos presentan tumefacción mamaria induci-da por estímulos hormonales procedentes de la madre que es másevidente al tercer y cuarto día tras el nacimiento y se prolonga 2-3semanas, desapareciendo espontáneamente. Durante el período demáxima tumefacción pueden secretar unas gotas de líquido serosoconocidas como «leche de brujas».
CASO CLÍNICO
Varón de 20 días de vida de origen árabe que acude a urgencias poraumento del tamaño de ambas mamas. A la exploración se objetivaimportante tumefacción mamaria bilateral, eritema de la zona y secre-ción blanquecina por ambos pezones. No parece doloroso a la palpa-ción. Afebril, orexia conservada y buena ganancia ponderal. Se plantea
diagnóstico diferencial entre ingurgitación mamaria neonatal transitoriay mastitis. Hemograma y reactantes de fase aguda dentro de la norma-lidad, por lo que se decide alta y control evolutivo en UrgenciasPediátricas en 5 días, donde se aprecia mejoría evidente del cuadro.
COMENTARIOS
La intumescencia mamaria neonatal es un motivo frecuente de consul-ta en urgencias que genera gran ansiedad en los padres. En otras cultu-ras la extracción de la «leche de brujas» es una práctica común. Esimportante informar a la familia de la benignidad del cuadro y evitar lamanipulación de las mamas y la extracción de la secreción por el ries-go de infección. Aunque es un cuadro benigno, en ocasiones, debido asu intensidad puede plantearse el diagnóstico diferencial con cuadrosinfecciosos como la mastitis, siendo necesaria la realización de pruebascomplementarias.

48
Sesiones de la sociedad
Esofagitis esofinofílica: patología emergenteM. Arqued, G. Herráiz,V. Rosel, P. Meléndez, J.L. Olivares, A. Lázaro
Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 48]
INTRODUCCIÓN
La esofagitis eosinofílica se puede sospechar ante una clínica sugestivade reflujo gastroesofágico que no responde al tratamiento intensivo coninhibidores de la bomba de protones. La confirmación se obtiene porendoscopia con toma de biopsias.
OBJETIVO
Revisión de los pacientes con diagnóstico de esofagitis eosinofílica aten-didos en la consulta de Gastroenterología Pediátrica de nuestro hospi-tal en los últimos dos años.
MATERIAL Y MÉTODOS
Estudio retrospectivo: revisión de las historias de los pacientes con eso-fagitis eosinofílica: se detectan 6 casos. El 100% son varones, de entre 7y 12 años, exceptuando uno de ellos de 21 meses.
RESULTADOS
Dos pacientes consultaron en el Servicio de Urgencias por disfagia, elresto fueron remitidos por su pediatra, en 2 casos por diarrea crónica yen otros 2 por epigastralgia y pirosis. Respecto a las pruebas comple-mentarias, en el 83% se encontró elevación de la IgE y eosinofilia peri-
férica, en un 50% se detectaron sensibilizaciones, en 2 casos a alergenosambientales y en 1 caso a alergenos alimenticios, en 5 casos se realizóendoscopia con biopsias confirmando el diagnóstico. Individualizando eltratamiento, un caso fue tratado exclusivamente con dieta hipoalergé-nica, 5 casos notaron mejoría con corticoide deglutido, complementadocon inhibidores de la bomba de protones, siendo posible la suspensióndel tratamiento en un caso. En uno de los casos se detectó HelicobacterPylori, precisando tratamiento erradicador. En un tercio se añadió mon-telukast al corticoide. Uno de los casos que consultó por disfagia preci-só tratamiento endoscópico para extracción de resto alimenticio. El100% ha evolucionado favorablemente con el tratamiento, continuandoen seguimiento en nuestra consulta.
COMENTARIOS
La esofagitis eosinofílica, a veces de diagnóstico tardío, tiene morbilidadconsiderable que evoluciona bien con el tratamiento adecuado.Dependiendo de los resultados de las pruebas de alergia tenemos dife-rentes actitudes terapéuticas: eliminar dicho alergeno de la dieta o, si nose encuentra alergeno específico, considerar una dieta de eliminación abase de una fórmula basada en aminoácidos. El tratamiento farmacoló-gico se basa en los corticoides, tanto deglutidos, el propionato de fluti-casona, como vía oral. Otras opciones son el montelukast y el mepoli-zumab, este último en fase de ensayo.
Infección perinatalS. Ortiz, J. Morales, P. Collado, S.Valle, G. Rodríguez, M.P. Samper, P. Ventura
Servicio de Pediatría. Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 48]
INTRODUCCIÓN
La infección neonatal por virus herpes simple puede llegar a tener gra-ves complicaciones a pesar de un tratamiento etiológico correcto. Lasmadres portadoras de una primoinfección por virus herpes genitaldurante el embarazo o el parto tienen un elevado riesgo de transmi-sión vertical, este riesgo es mucho menor si se trata de recidivas. Lainfección localizada en piel, boca u ojos, rara vez presenta graves com-plicaciones; por el contrario cuando se trata de una infección disemina-da o con afectación del sistema nervioso central, la mortalidad es supe-rior al 80%, a pesar de un tratamiento adecuado, y los que sobrevivensuelen presentar graves secuelas neurológicas.
CASO CLÍNICO
Neonato de 30 semanas de edad gestacional, nacido por cesárea urgen-te debido a prolapso de cordón. Apgar 3/6, P: 1.510 g, L: 41 cm, conexploración física compatible con la normalidad para la edad gestacio-nal. A los 6 días de vida presenta lesión eritematosa de 1,5 cm de diá-
metro en párpado inferior y ala de nariz, que en días sucesivos comien-za con vesículas de contenido transparente que al romper dejan costrassangrantes. Ante la sospecha de infección por virus herpes se extraecontenido de la lesión para análisis de PCR confirmando la infección porVHS-2, serología VHS en LCR: negativa. Se inicia tratamiento con aciclo-vir intravenoso durante 3 semanas, en ese tiempo el niño presenta unempeoramiento progresivo precisando ventilación mecánica. Al finalizartratamiento el estado general es bueno sin precisar soporte respirato-rio, no obstante a los 6 días se produce empeoramiento brusco conconvulsiones y bradicardia que no remonta a pesar de maniobras dereanimación avanzada, presentando finalmente una parada cardiorrespi-ratoria. El estudio anatomopatológico es compatible con el diagnósticode encefalitis herpética.
COMENTARIOS
La morbilidad en los pacientes que ha sufrido una encefalitis por virusherpes es elevada, tan sólo un tercio de los niños van a tener un de-sarrollo posterior normal.

49VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
Manejo respiratorio de la hernia diafragmáticaG. Sierra, L. Portero, C. Alonso, C. Pallás
Servicio de Neonatología. Hospital Universitario 12 de Octubre. Madrid
[Bol Pediatr Arag Rioj Sor, 2010; 40: 49]
INTRODUCCIÓN
La hernia diafragmática es una entidad de fisiopatología compleja, en laque coexisten anomalías en el desarrollo del parénquima y de los vasospulmonares que dificultan el manejo respiratorio de estos pacientes.Actualmente la asistencia preoperatoria va dirigida a evitar el daño pul-monar con el empleo de ventilación permisiva. Se trata de un grupo enel que la respuesta al óxido nítrico es pobre.
OBJETIVO
Se presenta el caso de una hernia diafragmática congénita y se discuteel manejo respiratorio de ésta, en una unidad de cuidados intensivosneonatal.
CASO CLÍNICO
Se trata de un embarazo controlado desde la semana 24, con ecografíasprenatales normales.Al nacimiento el neonato está hipotónico, cianóticoy sin esfuerzo respiratorio con latido cardiaco desplazado a la derecha,
requiriendo reanimación tipo IV en paritorio. La radiografía muestra her-nia diafragmática con asas intestinales en hemitórax izquierdo. La ventila-ción y oxigenación del paciente resulta difícil, requiriéndose ventilaciónmecánica convencional y alta frecuencia, así como óxido nítrico. La inter-vención quirúrgica se realiza en dos pasos de manera programada, conun quilotórax como complicación secundaria. Este caso clínico nosrecuerda que la hernia diafragmática es una entidad de fisiopatologíacompleja, en la que coexisten anomalías en el desarrollo del parénquimay de los vasos pulmonares que dificultan el manejo respiratorio de estospacientes. Actualmente la asistencia preoperatoria va dirigida a evitar eldaño pulmonar con el empleo de ventilación permisiva. Se trata de ungrupo en el que la respuesta al óxido nítrico es pobre.
COMENTARIOS
La forma óptima de ventilar a estos pacientes no está definida aún.Existen estudios controlados sobre la repercusión perjudicial que elbarotrauma tiene sobre los pulmones de la hernia diafragmática, perono los hay sobre las consecuencias que la ventilación permisiva puedatener en la supervivencia y en el neurodesarrollo de estos pacientes.
Caso clínico: Síndrome de WolframV. Giménez, N. Clavero, L. Zandueta, A. Campos,Y. Armendáriz, G.M. Lou, M. Rodríguez
Servicio de Diabetes Infantil. Hospital Universitario Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 49]
INTRODUCCIÓN
El Síndrome de Wolfram (SW), también denominado DIDMOAD, esun trastorno autosómico recesivo constituido por la asociación de dia-betes insípida (DI), diabetes mellitus (DM), atrofia óptica (OA) y sor-dera (D). El gen responsable es el WFS1, situado en la región 4p16.1,que sintetiza una proteína transmembrana denominada wolframina.
CASO CLÍNICO
Paciente mujer de 14 años, hija de padres sanos consanguíneos.Antecedente personal de comunicación interventricular, comunicacióninterauricular e hipertensión pulmonar en período neonatal con reso-lución espontánea. A los 5,6 años inicia poliuria, polidipsia, polifagia ypérdida de peso de 1 mes de evolución mostrando la analítica una glu-cemia de 217 mg/dl, por lo que se diagnostica de diabetes mellitus insu-linodependiente con Anti-GAD negativos. A los 6 años es remitida aoftalmología por disminución de agudeza visual de ojo derecho, objeti-vándose palidez de papila derecha y lentificación de potenciales evoca-dos visuales (PEV) compatible con atrofia óptica.Ante sospecha de SWse solicita estudio genético que confirma el diagnóstico de presunciónapareciendo la mutación G736A en homocigosis, siendo ambos padresheterocigotos para la misma. Pasados 5 años comienza nuevamente apresentar poliuria, siendo catalogada de diabetes insípida y 2 años mástarde se diagnostica de vejiga neurógena. Por presentar incontinencia
urinaria con abundante residuo postmiccional, se le realiza derivaciónurinaria continente según técnica de Mitrofanoff con apéndice cecal.Actualmente recibe tratamiento con insulina, vasopresina y precisa deautosondajes vesicales varias veces al día.
COMENTARIOS
El SW presenta una morbilidad elevada con gran deterioro en la calidadde vida, así como alta mortalidad. El seguimiento de esta patología escomplejo puesto que precisa de un enfoque multidisciplinario, tantopara el diagnóstico precoz como para su tratamiento. En la actualidad elestudio genético permite el diagnóstico antes de que aparezcan lasmanifestaciones clínicas, pudiéndose realizar incluso diagnóstico prenatal.
BIBLIOGRAFÍA1. Vilan A, Faria O, Campos MM.Wolfram síndrome. Rev Neurol. 2009; 49:
221-222.2. Zmysl/owska A, Bodalski J, Ml/ynarski W. The rare syndromic forms of
monogenic diabetes in childhood. Pediatr Endocrinol Diabetes Metab.2008; 14: 41-43.
3. Doménech E, Gómez-Zaera M, Nunes V.Wolfram/DIDMOAD syndro-me, a heterogenic and molecularly complex neurodegenerative disease.Pediatr Endocrinol Rev. 2006; 3: 249-257.

50
Sesiones de la sociedad
¿Qué niños se han vacunado de la gripe A y por qué?M. Biosca, M. Cunillera, M.E. Doménech, L. Olivart, M. Guillén
ABS Les Borges Blanques. Lleida
[Bol Pediatr Arag Rioj Sor, 2010; 40: 50]
INTRODUCCIÓN
La gripe A (GA) ha tenido gran repercusión mediática. Se han genera-do dudas entre la población sobre la conveniencia de optar o no por lavacunación.
OBJETIVO
Medir la incidencia de vacunación contra la GA y comparación con lavacunación de gripe estacional (GE).
Determinar los motivos de vacunación o no vacunación.
MATERIAL Y MÉTODOS
Estudio descriptivo de la cobertura de vacunación de GA en una UBAde pediatría (Total 1.116 niños).
RESULTADOS77 pacientes con indicación de vacuna de GA según programa infor-mático e-cap (6,9%).Acuden 55 pacientes (71,4%): 34,5% niñas y 65,5%
niños. Un 28,6% con indicación de vacuna de GA no acude a consulta.De los que acuden, un 20% se vacuna de GA: 16,4% vacunadosGE+GA, 3,6%, sólo vacuna GA, 56,4 % sólo vacuna GE y 23,6% no seadministran ninguna vacuna. Cobertura de vacunación de GA de nues-tra población de riesgo 14,3% y cobertura vacunación GE 56%.Indicaciones de los vacunados GA: asma 87,3%, cardiopatía 5,5%, diabe-tes mellitus 1,8%, enfermedad hepática 1,8%, obesidad mórbida 1,8%.Motivos de vacunación GA: considerarse de riesgo 81,8%, recomenda-ción del médico 9,1% y motivo no especificado 9,1%. Motivos de novacunación: sentimiento de conejillo de indias 50%, por las noticias de laprensa 25%, porque creen que la han pasado 4,5% y motivo no especi-ficado 20%. Existe un 0,2% de nuestra población que se ha vacunado deGA sin tener factores de riesgo.
COMENTARIOS
Cobertura de vacunación de GA de la población de riesgo baja. Másaceptación de la vacuna de GE que de GA. La nueva vacuna ha gene-rado dudas de seguridad.
Homocistinemia clásicadetectada por cribado neonatal ampliado.Revisión de nuestra casuística
L. Gil, M. Odriozola, S. Beltrán, M.ªP. Ruiz-Echarri, M.C. García
Servicio de Pediatría y de Enfermedades metabólicas. Hospital Infantil Universitario Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 50]
INTRODUCCIÓN
Las homocistinurias abarcan un grupo de errores congénitos del meta-bolismo de los aminoácidos azufrados que, con una gran heterogenei-dad etiológica y clínica, poseen una serie de características comunes. Lahomocistinemia clásica es la causa más frecuente de homocistinuria. Sudetección mediante cribado neonatal ampliado permite el tratamientoprecoz de estos pacientes y evita las complicaciones a largo plazo, algu-nas de ellas irreversibles.
CASO CLÍNICO
Presentamos el caso de recién nacido varón en cuyo cribado neonatalampliado se detecta una elevación importante de la metionina. Se com-pleta el estudio, que muestra un perfil bioquímico compatible con homo-cistinuria clásica, por lo que se inicia tratamiento dietético y farmacológico.
Se realiza, además, una revisión de los casos de homocistinemiacontrolados en nuestro centro. La determinación de la homocisteínaplasmática total se incluyó para el estudio etiológico de los trastornosneurológicos de causa no identificada, accidentes vasculares de carác-ter tromboembólico y anemias megaloblásticas, diagnosticándose 12
pacientes de hiperhomocistinemia, de los cuales 5 eran homocistine-mias clásicas.
COMENTARIOS
La homocistinemia clásica es una patología más frecuente de lo que sepensaba hasta ahora; además existen en la actualidad posibilidades tera-péuticas, efectivas siempre y cuando se apliquen de forma precoz, antesde que se hayan producido lesiones orgánicas irreversibles. La impor-tancia del cribado neonatal ampliado en esta patología por tanto esindiscutible.
BIBLIOGRAFÍA
1. Couce Picó ML, Fernández Lorenzo JR, Fraga Bermúdez JM.Trastornosdel metabolismo de los aminoácidos azufrado. En Sanjurjo P, Baldellou A,ed. Diagnóstico y Tratamiento de las Enfermedades MetabólicasHereditarias, 3ª ed. Ergon SA, Madrid, 2010; 465-474.
2. García Jiménez MC, Baldellou Vázquez A. Homocistinuria: ¿Una grandesconocida? Claves para el diagnóstico en Atención Primaria. ActaPediatr Esp. 2009; 67: 535-541.

51VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
Síndrome de Bloch-sulzberger neonatalI. Galé, S. Beltrán, O. Gómez, E. Sancho, M. Domínguez, S.Torres
Servicio de Neonatología. Hospital Infantil Miguel Servet Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 51]
INTRODUCCIÓN
El síndrome de Bloch-sulzberger o incontinencia pigmenti (IP) es untrastorno neuroectodérmico de escasa incidencia, que afecta principal-mente a la piel aunque puede asociar afectación de otros órganos (sis-tema nervioso central, ojos…). Las lesiones cutáneas evolucionan encuatro estadios: vesicular, verrugoso, hiperpigmentario y atrófico.
CASO CLÍNICO
Recién nacida mujer, cesárea electiva a las 37 semanas, embarazo geme-lar. Apgar 9/10. PRN: 2.200 g (BPEG); ingresa a los 17 días de vida porpresentar lesiones hiperqueratósicas, de base eritematosa con algunapequeña pústula en su superficie, distribuidas de forma segmentaria enlas cuatro extremidades, afectando a manos y pies. Destaca una faciestriangular, frente amplia, hendidura palpebral pequeña y casi ausencia depestañas. En la analítica presenta una leucocitosis con marcada eosinofi-lia (49%), proteína C reactiva negativa, los cultivos de las lesiones yhemocultivo fueron negativos. Fondo de ojo, Rx tórax, serie ósea y eco-cardiograma normales. En la ecografía transfontanelar se observa unadilatación de III ventrículo, y en la RNM cerebral una atrofia generaliza-da de predominio bifrontal y lesiones de isquemia en núcleos de la basey sustancia blanca del hemisferio derecho. Se realizó una biopsia de laslesiones con una discreta espongiosis en la epidermis y moderada can-
tidad de eosinófilos en dermis y epidermis. El estudio genético muestrauna deleción de los exones 4 al 10 del gen IKKG (NEMO). Presentómarcada hiporexia con dificultades en la succión, ingresando de nuevo alos dos meses de vida por una neumonitis intersticial difusa. Actual-mente es controlada por el servicio de dermatología, con buena evolu-ción de sus lesiones y neuropediatría, por retraso psicomotor, hipotoníaaxial y hemiparesia izquierda.
COMENTARIOS
La IP es una genodermatosis de transmisión dominante ligada al X, letalen varones, de expresividad variable. La mutación del gen NEMO apa-rece en el 80% de los pacientes, que permite realizar un consejo gené-tico a la familia. El pronóstico está marcado por la afectación ocular yneurológica.
BIBLIOGRAFÍA
1. Emre S, Firat Y, Güngör S et al. Incontinentia pigmenti: a case report andliterature review.The Turkish Journal of Pediatrics 2009; 51: 190-194.
2. Ehrenreich M, Mordechai M, Godlewska-janusz MD et al. Incontinenciapigmenti (Bloch-Sulzberger syndrome): a sistemic disorder. Pediatric der-matology cutis 2007; 79: 355-362.
Insuficiencia cardiacacomo consecuencia de anemia grave
I. Ruiz(1), A. Miralles(1), G. González(1), E. Elías(1), S. Ortiz Madinaveitia(2), A. Ayerza(1), E. Muñoz(1)
(1)Hospital Clínico Universitario Lozano Blesa. Zaragoza. (2)Hospital San Jorge. Huesca
[Bol Pediatr Arag Rioj Sor, 2010; 40: 51]
INTRODUCCIÓN
La insuficiencia cardiaca es una entidad poco frecuente en la infancia. Suetiología puede ser diversa, siendo la anemia una de sus posibles causas.
CASO CLÍNICO
Niño de 2 años que acude a Urgencias por presentar tos productiva yfiebre de hasta 37,5 ºC de 24 horas de evolución. Los padres refierenastenia, palidez y orexia disminuida desde hace una semana. A la explo-ración presenta regular estado general con palidez de piel y de muco-sas.Adenopatías 0-0-1.Auscultación cardiaca: soplo sistólico II/VI y taqui-cardia (150 lpm). Auscultación pulmonar: subcrepitantes en bases,taquipnea y discretos signos de dificultad respiratoria. Hepatomegalia de1 cm y esplenomegalia de 3 cm bajo el reborde costal. Resto de laexploración compatible con la normalidad. Pruebas complementarias:Hemograma: 1,6 mill/mm3 de hematíes, Hb: 4,5 g/dl, Hematocrito: 13%,neutrófilos: 1,2 %, linfocitos: 93,6%, plaquetas: 58 mil/mm3. Bioquímica ygasometría venosa normales. PCR: 0,67. Análisis serológico: IgG anti-Toxoplasma, IgM anti-VCA VEB e IgM anti-CMV negativos. Radiografía
de tórax: engrosamiento peribroncovascular con ligera elevación dehemidiafragma izquierdo.
Se ingresa por presentar anemia sintomática con sospecha de pro-ceso linfoproliferativo y clínica compatible con insuficiencia cardiaca. Eltratamiento consiste en transfusión de concentrado de hematíes, esta-bilizándose al paciente desde el punto de vista clínico y hemodinámico.En el estudio hematológico se aprecia bicitopenia con un 98% de linfo-citos de pequeño tamaño, sin nucleolos evidentes con alta relaciónnúcleo-citoplasma, indentaciones nucleares e irregularidades núcleo-citoplasmáticas. Se decide traslado a hospital de referencia por sospe-cha de leucemia linfoblástica aguda tipo L1.
COMENTARIOS
La leucemia linfoblástica aguda (LLA) supone el 80% de las leucemias yes la neoplasia más frecuente en niños (22,8% en nuestro medio), con unpico de incidencia entre los 1 y los 4 años. La LLA debuta frecuentementecon sintomatología de anemia grave que en ocasiones puede evolucionarhacia un cuadro de insuficiencia cardiaca como en nuestro caso.

52
Sesiones de la sociedad
Evolución clínica y analíticaen paciente con hipoaldosteronismo congénito primario
G. Herráiz, D. Clavero, L. Escartín, O. Bueno(1), G. Rodríguez(1), J.M. Garagorri(2), G. Bueno(2), J.L. Olivares
(1)Servicio de Neonatos. (2)Servicio de Endocrinología Pediátrica. Hospital Clínico Universitario Lozano Blesa. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 52]
INTRODUCCIÓN
El hipoaldosteronismo congénito primario es un defecto selectivo de labiosíntesis de la aldosterona determinado por mutaciones del genCYP11B2.
CASO CLÍNICO
Recién nacido varón que ingresa en unidad neonatal al décimo día devida por rechazo al alimento y pérdida de peso. Presenta deshidrataciónmoderada, y genitales masculinos normoconfigurados sin virilización nimacrogenitosomía. Los datos analíticos confirman pérdida salina (Sodio122 mEq/L, Potasio 7,2 mEq/L), iniciándose fluidoterapia intravenosa connecesidades de sodio de hasta 8 mEq/kg/día, para mantener natremiasentre 125-129 mEq/L. Los resultados hormonales iniciales no sugierenun déficit clásico de 21 hidroxilasa (ACTH, Cortisol y 17-OH-progeste-rona en niveles de normalidad), sospechándose un defecto limitado a lazona glomerular. Tras realizar extracciones para estudio del metabolis-mo de la aldosterona, se inicia tratamiento con fludrocortisona a0,15mg/día y suplementación oral de ClNa a 3 g/día, normalizándose lanatremia. El estudio bioquímico es compatibe con defecto CMO tipo IIde la aldosterona sintasa (aldosterona descendida con elevación de susprecursores: Renina, DOC, Corticosterona y 18 OH-Corticosterona). Elestudio genético confirma tres alteraciones en homocigosis del gen
CYP11B2 (E198D,V386A y R173K). Durante los tres primeros años devida recibe tratamiento sustitutivo con fludrocortisona sin descompen-saciones ante procesos intercurrentes, por lo que se decide suspendergradualmente. Actualmente los niveles de sodio y potasio en plasma yorina son normales. Su curva pondoestatural, con y sin tratamiento,transcurre en el percentil 10.
COMENTARIOS
El hipoaldosteronismo congénito primario debe ser sospechado antetodo neonato o lactante con retraso ponderoestarural y clínica de pér-dida salina. Estos pacientes necesitan un seguimiento estrecho. El trata-miento con fludrocortisona puede reducirse e incluso suspenderse endependencia del balance del sodio y del crecimiento.
BIBLIOGRAFÍA
1. Lopes LA, Dubuis JM,Valloton MB. Should we monitor more closelythe dosage of 9 alpha-fluorhydrocortisone in salt-losing congenitaladrenal hyperplasia? J Pediatra Endocrinol (Copenh) 1998; 73: 417-426.
2. Stapenhorst L. 9 alpha-fluorohydrocortisone therapy in aldosteronesynthase deficiency. Pediatr Nephrol 2005; 20: 839.
Urticaria y parasitosis intestinalR. Manso, A. Manero, O. Gómez, M.P. Sanz de Miguel, C. García Vera,T. Cenarro
Centro de Salud Sagasta. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 52]
INTRODUCCIÓN
La urticaria es un motivo de consulta habitual. La etiología alimentariaes la más frecuente en el caso de urticaria agua en los niños, de urtica-ria crónica (duración mayor a 6 semanas) son idiopáticos hasta en un75%, siendo la parasitosis una causa poco frecuente (alrededor de un5%). Comentamos dos casos de urticaria en los que la única pruebapositiva fue el test de Graham para Enterobius vermicularis (en una delas pacientes de forma recurrente), y que se resolvieron con tratamien-to antiparasitario.
CASO CLÍNICO
Presentación de dos pacientes mujeres de 18 y 30 meses respectiva-mente, con antecedentes familiares de atopia y como único anteceden-te personal a destacar el de dermatitis atópica. Presentan clínica de urti-caria de repetición tratada con antihistamínicos y corticoides orales sinresolución de los cuadros. Entre las pruebas complementarias realizadasse encuentran analítica de sangre, tres coprocultivos y pruebas cutáneasde alergia que resultaron normales. En ambos casos se realizaron 3 testde Graham, uno de los cuales resultó positivo para Enterobius vermi-
cularis, por lo que se pautó tratamiento con mebendazol con posteriordesaparición de las lesiones cutáneas y negativización de los test deGraham. En el caso de la paciente de 30 meses, tras el tratamiento per-maneció libre se síntomas 18 meses, tras los cuales presenta nuevoepisodio de urticaria con test de Graham nuevamente positivo aEnterobius y con buena respuesta al tratamiento (mebendazol) hasta elmomento actual.
COMENTARIOS
Con la presentación de estos casos queremos recordar la importanciade pensar en la posible asociación de urticaria con parasitosis, inclusoen los casos sin clínica gastrointestinal y con antecedentes de atopia.Yremarcar la utilidad de realizar 3 pruebas de Graham para el diagnósti-co debido a la gran cantidad de falsos negativos del test.
BIBLIOGRAFÍA
1. Jirapongsananiruk O, Pongpreuksa S, Sangacharoenkit P. Identification ofthe etilogogies of cronocurticaria in children: A prospective study of 94patients. Peditri Allergy Immunol 2010; 21: 508-514.

53VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
Diarrea oleosa con heces anaranjadasJ. Caro, M. Cosculluela, F. Beltrán
Centro de Salud Oliver. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 53]
INTRODUCCIÓN
La diarrea oleosa con heces anaranjadas ha sido objeto de estudio porparte de los Centros de Vigilancia Epidemiológica al haberse presenta-do brotes, pero también podemos encontrar casos aislados y está rela-cionada con el consumo de pescado.
CASO CLÍNICO
Niña de 5 años que consulta por haber expulsado, al orinar, un líquidocomo gotas de aceite, de color naranja, en gran cantidad, dos vecesseguidas.Acudió a urgencias donde se realizó tira de orina que fue nor-mal. Al día siguiente acudió a consulta, no se había repetido, se solicitóserología celiaca que fue negativa.
Niño de 8 años que presentó un episodio de incontinencia fecalmanchando la ropa y posteriormente deposición diarreica, las heces ibanacompañadas de abundante líquido graso anaranjado que la madre des-cribía como caldo de mejillones en lata. Acudió a urgencias y posterior-mente a consulta, no se repitió el episodio y se solicitó estudio: copro-cultivo, parásitos, análisis de heces, digestión de principios inmediatos, san-gre oculta y serología celiaca, siendo todos los resultados normales.
A posteriori se constató que ambos niños habían cenado la nocheanterior mero.
COMENTARIOS
En nuestra comunidad los casos descritos se relacionan con el consumode «escolar negro» (Lepidocybium flavobrunneum). La legislación europeareglamenta su comercialización con unas normas estrictas y, sin embar-go, podemos encontrarlo en las pescaderías como «mero». El mecanis-mo de producción se debe a un éster ceroso que contienen sus múscu-los. El cuadro clínico parece estar causado por la incorrecta digestión deléster ceroso. El conocimiento de esta entidad y preguntar por la inges-tión de mero nos permitirá un diagnóstico inmediato y evitar exámenescomplementarios innecesarios. El control sanitario requiere la notifica-ción a Higiene Alimentaria.
BIBLIOGRAFÍA
1. Martín Granado A, Varela Martínez MC, Martínez Sánchez EVet al. Interés de la identificación de la especie de pescado en brotesde diarrea oleosa con heces anaranjadas. Boletín EpidemiológicoSemanal. Centro Nacional de Epidemiología. 2007; 15: 25-36.
2. Ling KH, Nichols PD, But PP. Fish-induced keriorrhea.Adv Food Nutr Res2009; 57: 1-52.
Agenesia del pectoral mayor en la infancia.Complejo malformativo Poland.A propósito de un caso
A. Manero(1,2), L. Gracia Torralba(2), B. Chapí (2), R. Manso(1,2), I. Galé(2), C. García Vera(1)
(1)EAP Centro de Salud José Ramón Muñoz Fernández. Zaragoza. (2)Servicio de Pediatría. Hospital Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 53]
INTRODUCCIÓN
La anomalía muscular congénita más frecuente es la agenesia o hipo-plasia del pectoral mayor. Su frecuencia es de 1/4.000 a 1/30.000 reciénnacidos. Suelen ser esporádicos, aunque se han descrito casos familiares,son excepcionales. Puede darse de forma aislada, o asociar malforma-ciones de la mano homolateral (Síndrome de Poland), en ambos casosforma parte del complejo malformativo Poland, ya que pueden acom-pañarse de malformaciones a distancia, como dextrocardia, hipoplasiapulmonar, malformaciones renales, hipospadias, síndrome de Moebius.La etiología no está aclarada, la hipótesis más extendida es la de este-nosis temporal de la arteria subclavia en período embrionario, en fun-ción de su gravedad y duración causaría defectos de la pared torácicacon o sin afectación de la extremidad.
CASO CLÍNICO
Varón de 12 meses al que en revisión del niño sano se detecta una asi-metría de pared torácica. El esternón es más prominente en el ladoizquierdo con aplanamiento en región pectoral, ausencia del pliegue
anterior axilar, mamila izquierda desplazada lateralmente, con menorrelieve que la contralateral. No anomalías en cintura escapular ni manohomolateral. Resto de exploración sin hallazgos. Se realiza radiografía detórax, encontrando asimetría de partes blandas de la pared torácica, sinalteraciones de parrilla costal. Tiene controles ecográficos prenatalesnormales. En época neonatal persistencia del ductus, resuelta en eco-cardiograma realizado al mes de vida. Su madre presenta defecto simi-lar de pared torácica.
Se consulta con neuropediatría y traumatología pediátrica que apo-yan el diagnóstico de agenesia aislada del músculo pectoral izquierdo.Pendiente de confirmar con resonancia magnética para definir el defec-to de la pared torácica.
COMENTARIOS
El complejo malformativo Poland comprende un amplio abanico demanifestaciones clínicas con diverso grado de afectación. La etiologíaaún no está aclarada. En caso de agenesia aislada del pectoral, comopodría ser el nuestro, si no hay limitación funcional se reduce a un pro-blema estético.

54
Sesiones de la sociedad
Polaquiuria primariaA. Aldana(1), O. Gómez(1), E. Sancho(1),V. Caballero(1), M.P. Sanz(1),Y. Romero(2)
(1)Servicio de Pediatría. (2)Servicio de Nefrología. Hospital Miguel Servet. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 54]
INTRODUCCIÓN
La polaquiuria primaria o psicógena, también denominada «falsa infec-ción de tracto urinario», se define como la realización de al menos 1micción/hora con pequeño volumen urinario, durante el día, en niñoscontinentes (>3a) y con comportamiento vesical nocturno normal. Suetiología es desconocida aunque se conocen varios factores desenca-denantes. El diagnóstico es fundamentalmente clínico, tras descartarotros cuadros poliúricos, siendo su tratamiento principal el soporteemocional.
CASO CLÍNICO
Presentamos una niña con antecedentes familiares de uropatía (herma-no con válvulas de uretra posterior y abuela materna con duplicidadureteral), que es controlada en la consulta de nefrología tras su ingresoa los 19 meses por primera ITU febril, donde se le detectó ectasia pié-lica izquierda de 13 x 14 mm sin dilatación ureteral, mínima hipofuncióndel riñón izquierdo en la Gammagrafía y reflujo vesicoureteral izquierdogrado III en la CUMS. A los 5 años de edad inicia cuadro de polaquiuriade 2 meses de evolución, exclusivamente diurna, con sensación inter-mitente de disuria, sin presentar escapes ni otra sintomatología. En laanamnesis destaca la separación de los padres hace 4 meses. Se realizahemograma, bioquímica en sangre y orina, que muestran función renal,
hepática y capacidad de concentración urinaria normal. Sedimento deorina normal y Urocultivo negativo. Se plantea el diagnóstico de pola-quiuria primaria que se confirma con la mejoría clínica objetivada en laevolución.
COMENTARIOS
El interés de esta patología radica en su frecuencia, similar a la de lainfección de tracto urinario bajo, y por tanto está infradiagnosticada ennuestro medio, quizás también por su escasa documentación bibliográ-fica. Por ello debemos tenerla presente para obviar la realización deexámenes complementarios innecesarios, algunos de ellos a vecescruentos y evitar también así el gasto económico que conllevan.
BIBLIOGRAFÍA
1. Berfmann M, Corigliano T, Ataia I et al. Chilhood extraordinary dayti-me urinary frequency – a case series and a systematic literaturereview. Pediatr Nephrol 2009; 24: 789-795.
2. Corigliano T, Renella R, Robbiani A, Riavis M, Bianchetti MG. Isolatedextraordinary daytime urinary frequency of childhood: a case of 26 chil-dren in Switzerland.Acta Paediatr 2007; 96: 1347-1349.
Síndrome de JobI. Cordeiro, J. Martin, S. Sumsky, M. Domínguez, B. Baquero
Servico de Pediatría. Hospital Obispo Polanco.Teruel
[Bol Pediatr Arag Rioj Sor, 2010; 40: 54]
INTRODUCCIÓN
Síndrome de Job, también conocido como Síndrome de hiperinmuno-globulina E o Síndrome de Buckley, es una rara enfermedad de inmu-nodeficiencia primaria autosómica dominante, causada por una muta-ción genética (gen STAT3 en el cromosoma 4). Caracterizada por lapresencia de abscesos cutáneos e infecciones de las vías aéreas conniveles elevados de inmunoglobulina E.
CASO CLÍNICO
Paciente femenino de 12 años de edad. Natural de Ecuador, referidodel centro de salud para control pediátrico. Diagnosticada de Síndromede Job a pocos meses de nacer, tras un marcado retraso psicomotor,pondo-estatural, múltiples abscesos cutáneos y neumonías de repeti-ción. Sin antecedentes familiares neonatales o alimentarios. Esquema devacunación incompleta. Múltiples ingresos por neumonías, absceso pul-monar derecho y múltiples abscesos en piel por Estaphilococus áurease infecciones de herpes. Examen físico: hipodesarrollo pondo-estatural(P <3 en peso y talla), facies tosca, cuello corto. Piel xerótica, hiper-
queratosis en codos, rodillas y tobillos. Auscultación cardiopulmonar :normal. Abdomen: prominente, blando, no visceromegalias, herniaumbilical reducible. Manos alargadas, pies planos. Columna dorsal conescoliosis derecha. Retraso psicomotor marcado, torpeza motora, con-trol de esfínteres reciente, lenguaje verbal de difícil comprensión.Hemograma, bioquímica coagulación, Factor Reumatoide, HormonasTiroideas: normales. IgE: 3.512 UI/ml, otras Inmunoglobulinas: normales.Rast alérgico positivo: Polvo doméstico, pelo gato, leche de vaca, gra-míneas. Cariotipo 46XX. Fondo de ojo, Rx de Tórax, TAC de cráneo:normales. Rx Columna dorsal: escoliosis.
COMENTARIOS
No existe tratamiento específico, indicado el manejo de procesosinfecciosos cuando se presentan, resección quirúrgica de abscesos yquistes pulmonares. Extracciones dentarias en caso de retraso delrecambio de dentición primaria, seguimiento de escoliosis y fracturasde repetición, aplicar medidas de soporte: higiene ambiental, alimen-tación adecuada.

55VOL. 40 - Nº 2 • MAYO-AGOSTO 2010
Sesiones de la sociedad
Importancia de la educaciónen el control del niño con TDAH.Visión desde atención primaria
G. Herráiz(1), F.J. Lozano(2), M. Marco(3), E. Atance(3), M.J. Blasco(3)
(1)Hospital Clínico Lozano Blesa. (2)Colegio San Valero. (3)C.S.Valdefierro. Zaragoza
[Bol Pediatr Arag Rioj Sor, 2010; 40: 55]
INTRODUCCIÓN
El Trastorno por Déficit de Atención e Hiperactividad (TDAH) consti-tuye un patrón de comportamiento inadaptado de base neurobiológicacon una prevalencia en población infanto-juvenil del 6%. La mediaciónes efectiva en un 80% de los niños. La mejor respuesta combina medi-cación y métodos conductuales.
OBJETIVO
Conocer la situación del TDAH en la población pediátrica del Centrode Salud de Valdefierro. Sensibilizar a los padres de que el tratamientoconsiste en el binomio medicación-educación y constatar qué impor-tancia le conceden.
MATERIAL Y MÉTODOS
Estadística del centro. Reunión informativa con padres y educadores. Aposteriori, entrega de un cuestionario.
RESULTADOS
El 2,53% de los niños del C.S. de Valdefierro son controlados por TDAHy todos están medicados. Acudieron a la reunión 52 adultos.Respondieron a la encuesta 20, el 85% eran madres. La edad media erade 10,7 años, un 65% acuden a colegios concertados. El 65% entresemana dedican más de 4 horas/día a estar con su hijo y diez horas /día
el fin de semana. El 40% de los niños acude a actividades extraescola-res. Un 60% ayudan a sus hijos con los deberes y el 45% cuenta con unprofesor en casa.
El 70% creen que con ayuda el niño puede mejorar, el 25% que notiene solución y el 5% que es algo pasajero.
Para el rendimiento y comportamiento del niño, la institución másvalorada es la familia, en el 60%.Al colegio y al médico le conceden el 45%y 40% respectivamente. A los fármacos les otorgan 0 puntos en el 25%.
COMENTARIOS
Todos los niños TDAH controlados en el C.S. de Valdefierro están medi-cados. La mayoría de los padres dedican un tiempo razonable a estarcon sus hijos. En gran proporción confían en que el rendimiento y com-portamiento de su hijo pueda mejorar con apoyos. El orden de con-fianza es en primer lugar la familia, seguida del colegio y el médico y enúltimo lugar los fármacos. Sería conveniente que el control de estosniños recayera en mayor medida en la familia y la escuela, siempre coor-dinados, y menos en la medicación.
BIBLIOGRAFÍA
1. Curso de TDAH para Pediatras.Vaquerizo-Madrid 2008.2. American Academy of Pediatric. Clinical practice guideline: diagnosis and
evaluation of the child with attention-deficit/hyperactivity disorder.Pediatrics 2000; 105: 1158-1170.
