˘ ˇˆ - diva portal311806/fulltext01.pdf · don't underestimate the value of doing nothing,...
TRANSCRIPT

������������������� ���������� �
����
���������� �������������� ������������������� ������� ����������������������������
���������� ���������������������������������������������������������������������������
��� !�"#!$%�&
���%'��'(�)*����%+,-(+'(��.(,-',(+���/�0�/��/��/��1(')2*�2

����������������������� ������� �������������������������������������������������������������������������� ������ !!"�#���"���������$���%&��%�'������&(')�*��� ��������*������*�! ���� ��+,�������*�$������-.�/ �����������0����������������"0���� .
��������1������$.�%�'�.�2����������3�����������"��������4*���������50����������0�� ������������ ������2��������2�����.������ ������������ ���������.����������� � ���� ������ ���������������� ������������� ������������ ����� �)6).�7)���. ������.�4"58�&79:&':))�:79'7:&.
/ ���� ���������������������������������0�� ��*����������0�����������+45�-������������������� +22-.� 1�� ������������ ������� ���� ��� ��� ����� ��� ����������� ��� /:��������������� ���*����������������������������������������0 �� ���� ������������������*���������� ��*������+,-�������*������*������������������������������� ����������������./ ����0��������������� ����������������+32!-�������� ���������;�+3!;-����� ������ �������������������������+$!<-���������������+2-=��������������� ��������+2��-��2 �������5�+2�5-���������������+"8-����������*���332������������������ ���������������������.1�� *��� � ��� �� ��������� ,2� ������ ��� ������ ��� �� ��������� ������� *�� �������*��
�������������������0�� �45������������������� �� �,2������������*��� ������������+����-.�,��� �������,2����,:$!<������������������������� ��,:3!;�������������������45�.�1������������,:32!��,:3!;��,:$!<����,2������������*���������������������������������������������0�� �22�+��� ���-������������� ���,:32!�0���� ������������������*�����������������22.�8���������,:32!����,:3!;�����������������������*��������*����������. 1��� 0���� ���� ���*����������22����� ����������������������������� ��������� ���� ������������� ������������+��� ��-.�/:������ �������0���������*������������������22�� �����������������.�������������������������� ����������������*����� ������/:������������������0�� ������������������������.�/ ��*������������� ����������������� � ��� � ������ ���� ��������������� ���� �� � ����� � �������*�22.�$������� �� ����������*�,:2����,:2�5����,:"8�0����*�������������0�� �22�� ����� ���0�� 45�����������+��� ��-������������������������*��� ��������������:���������������� ����� �������*����������������.
� �������2�����������������*����������0�������������������������������2� >����������*������������������� ����/:�������32!��3!;��$!<��������������*�0���������� ����������� �������5����������������������
����� ������ � �� ���� ������ ��������� �� � �!"�� ��"���#�"��� � ������������� ����� �$%&'()'�������� ��� � �
?�$�� ����1�����%�'�
4""8�'6)':6%�64"58�&79:&':))�:79'7:&��(�(��(��(����:'%@�)@�+ ���(AA��.��.��A������B��C��(�(��(��(����:'%@�)@-

Don't underestimate the value of Doing Nothing, of just going along, listening to all the things you can't hear, and not bothering.”
Winnie the Pooh (A A. Milnes)

Supervisors:
Marie Carlson, Associate professor Department of medical Sciences Gastroenterology Research Group Uppsala University Uppsala Sweden
Maria Lampinen, Ph D Department of medical Sciences Gastroenterology Research Group Uppsala University Uppsala Sweden
Per Sangfelt, Associate Professor Department of medical Sciences Gastroenterology Research Group Uppsala University Uppsala Sweden

List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Wagner M, Peterson CGB , Ridefelt P, Sangfelt P, Carlson M. Fecal markers of inflammation used as surrogate markers for treatment outcome in relapsing IBD.
World J Gastroenterol 2008;14:5584-9.
II Wagner M, Lampinen M, Sangfelt P, Agnarsdottir M, Carlson M. Budesonide treatment of patients with collagenous colitis restores normal eosinophil and T-cell activity in the colon. Inflammatory bowel diseases 2009. Dec 21. Epub ahead of print.
III Wagner M, Peterson CGB, Stolt I, Sangfelt P, Agnarsdottir M, Lampinen M, Carlson M.
Fecal eosinophil cationic protein as a marker of active disease and treatment outcome in collagenous colitis.
Submitted for publication.
IV Wagner M, Stridsberg M, Peterson CGB. Lampinen M, Sangfelt P, Carlson M.
Elevated fecal levels of chromogranin A, chromogranin B and secretoneurin in patients with collagenous colitis.
In manuscript
Reprints were made with permission from the respective publishers. Faculty opponent: Associate Professor Erik Hertervig

Cover: Immunohistochemical identification of eosinophils by using the eosinophil marker eosinophil peroxidase in colonic biopsy specimens from a patient with active collagenous colitis.( Published with kindly permission of Alkwin Wanders).

Contents
Introduction...................................................................................................11 Background ..............................................................................................11
Ulcerative colitis..................................................................................11 Crohn´s disease....................................................................................12 Collagenous colitis...............................................................................13 Treatment.............................................................................................14 Pathogenesis in IBD ............................................................................15 Pathogenesis in collagenous colitis .....................................................16
Inflammatory cells involved in the mucosal inflammation......................17 Neutrophil granulocytes.......................................................................17 Eosinophil granulocytes.......................................................................18 T-cells ..................................................................................................19
Mucosal inflammation and the enteric neuro-endocrine system..............20 Mucosal inflammation and flow cytometry .............................................21 Mucosal inflammation and fecal markers ................................................21 Faecal markers in IBD..............................................................................21 Faecal markers in collagenous colitis.......................................................22
Aims..............................................................................................................23 Subjects .........................................................................................................24
Study I ......................................................................................................24 Study II.....................................................................................................25 Study III ...................................................................................................26 Study IV ...................................................................................................26
Methods ........................................................................................................27 Study design - studies I and IV ................................................................27 Study design - study II .............................................................................28 Study design - studies III and IV..............................................................28 Faecal markers of inflammation – study I................................................29 Collection and preparation of biopsy samples – study II .........................30 Antibodies for flow cytometry – study II .................................................30 Flow cytometry assay – study II...............................................................30 Histopathology – studies II and III...........................................................33 Immuno-histochemistry – studies II and III .............................................33 Ethics........................................................................................................34 Statistics ...................................................................................................34

Results...........................................................................................................35 Study I: Faecal markers of inflammation used as surrogate markers for treatment outcome in relapsing IBD ........................................................35 Study II: Budesonide treatment of patients with collagenous colitis restores normal eosinophil and T-cell activity in the colon .....................37 Study III: Faecal Eosinophil Cationic Protein as a Marker of Active Disease and Treatment Outcome in Collagenous Colitis .........................40
Eosinophil markers of inflammation ...................................................41 Neutrophil markers of inflammation ...................................................42
Study IV: Increased Fecal Levels of Chromogranin A, Chromogranin B and Secretoneurine in Collagenous Colitis ..............................................43
Discussion.....................................................................................................45 Faecal markers of inflammation in inflammatory bowel disease.............45 Faecal markers of inflammation in collagenous colitis ............................47 Mucosal inflammation in collagenous colitis...........................................48
Eosinophil granulocytes.......................................................................48 T-cells ..................................................................................................50
Markers of neuroendocrine activity in CC and IBD ................................50 Clinical implications and future perspectives ...............................................52 Conclusions...................................................................................................53 Svensk sammanfattning ................................................................................54
Introduktion..............................................................................................54 Bakgrund ..................................................................................................54 Studie I .....................................................................................................55 Studie II ....................................................................................................56 Studie III...................................................................................................56 Studie IV ..................................................................................................57 Slutsatser och egna kommentarer.............................................................58
Acknowledgements.......................................................................................59 References.....................................................................................................62

Abbreviations
5-ASA 5-aminosalicylic acid APC Allophycocyanin BSA Bovine serum albumin CARD Caspace activation and recruitment domain CC Collagenous colitis CD Crohn´s disease CgA Chromogranin A CgB Chromogranin B CTAB N-cetyl-N,N,N-trimetylammonium bromide DAB 3,3’-diaminobenzidine ECP Eosinophil cationic protein EEC Entero-endocrine ELISA Enzyme-linked immunosorbent assay ENS Enteric nervous system EPO Eosinophil peroxidase EPX Eosinophil protein X FACS Flourescence activated cell sorting FC Fecal calprotectin FITC Flourescein isothiocyanate HBI Harvey-Bradshaw´s clinical activity index HC Healthy control subject HLA Human leucocyte antigen HNL Human neutrophil lipocalin IBD Inflammatory bowel disease IFN Interferon IL Interleukin IBD Inflammatory bowel disease MBP Major basic protein MFI Mean fluorescence intensity MHC Major histocompatibility complex MPO Myeloperoxidase NOD Nucleotide binding oligomerisation domain NPV Negative predictive value NSAID Non-steroid anti-inflammatory drugs PBS Phosphate buffered saline PE Phycoerythrin

PerCP Peridinin chlorophyll protein PPV Positive predictive value RIA Radioimmuno assay SN Secretoneurin TGF Transforming growth factor TNF Tumor necrosis factor UC Ulcerative colitis ULN Upper limit of normal VEGF Vascular endothelial growth factor

11
Introduction
Ulcerative colitis (UC) and Crohn´s disease (CD) are chronic, relapsing, immunologically mediated disorders that are collectively referred to as inflammatory bowel disease (IBD). Ulcerative colitis and CD are considered as two separate conditions with distinguishing clinical, endoscopical and histological features, but they also have overlapping features and may even represent several different diseases with similar characteristics (1).
Collagenous colitis (CC) is, together with lymphocytic colitis (LC), a subtype of microscopic colitis and was first described by the Swedish pathologist Lindström (2-4). Lymphocytic and collagenous colitis have indistinguishable clinical features but different histopathological characteristics. Conversion of CC to LC and vice versa occurs, but is not frequentl (5). This together with the observed difference in sex ratio and HLA pattern gives reason to consider CC and LC as related but separated conditions (6). An interchange between UC or CD and microscopic colitis has been reported occasionally (7, 8). Wheter this is due to a common genetic predisposition or shared immunological pathways, or is simply a chance association remains unknown so far.
Background Ulcerative colitis Ulcerative colitis is characterised by diffuse, non-transmural mucosal inflammation restricted to the colon, usually involving the rectum it may extend proximally in a contiguous pattern. If the involvement is limited to the rectum (ie distal to the rectosigmoid junction) it is classified as proctitis; left sided colitis (distal colitis) is limited to the proportion of the colon distal to the splenic flexure; and extensive colitis (pancolitis) extends proximal to the splenic flexure (9). However, the extent of the disease shows instability over time and both regression and progression may occur (10).
Patients typically present with bloody diarrhoea, passage of pus and/or mucus, and abdominal cramping during bowel movements (11).
Numerous extraintestinal manifestations can appear, such as oral ulcerations (10%), arthritis (5-10%), primary sclerosing colangitis (3%),

12
uveitis (0.5-3%), pyoderma gangrenosum (0.5-3%) and thrombo-embolic disease (0.2%) (12).
Ulcerative colitis is a clinical diagnosis confirmed by endoscopy, and shows characteristic changes including loss of the typical vascular pattern, friability, exudates, ulcerations and granularity in a continuous, circumferential pattern. In addition, mucosal biopsy for histological examination reveals marked architectural distortion, cryptitis and crypt abscesses as well as goblet cell depletion. Furthermore, a mixed inflammatory cell infiltrate, including neutrophils, eosinophils, lymphocytes, plasma cells, and macrophages, is present in the lamina propria (13, 14).
The incidence of UC in northern Europe ranges from 3.2 – 20.3 per 100 000 individuals per year and in Sweden it is 15 per 100 000. The onset most commonly occurs between 15 and 40 years of age, with a second peak in incidence between 50 and 80 years (15). Men and women are affected at similar rates.
A positive family history is the main independent risk factor for the disease. Among UC patients 5-15% have a first-degree relative with UC and 6 – 16% have a first-degre relative with IBD. The estimated lifetime risk that a first-degree relative of a UC proband will develop IBD is 1.5 -5 % (16) and the relative risk is more than 15 (17). The concordances for monozygotic and dizygotic twins are 16 – 18% and 2 – 4.5%, respectively (18-20) Cigarette smokers have a lower risk of developing UC than do non-smokers, but compared with those who have never smoked, former smokers are more likely to develop the disease (21). Furthermore, cessation of smoking seems to have a negative impact on the course of UC (21, 22). Appendectomy at a young age appears to be protective against the development of UC (23, 24). Patients with extensive UC are at higher risk in developing colorectal cancer than healthy individuals, but the magnitude of this is debated (25).
Crohn´s disease Crohn´s disease is a relapsing, transmural inflammatory disease of the gastrointestinal mucosa with discontinuous involvement of various portions of the gastrointestinal tract which can affect any part from the mouth to the anus. At diagnosis, the disease has been found to be located in the terminal ileum in 20-30 %, the colon in 20-50 %, the ileo-colon in 10-40 % and the upper gastrointestinal tract in 3-30 % (26, 27). The disease may be classified regarding behaviour as non-stricturing non-penetrating in 35-60 %, stricturing in 20-30% and penetrating (fistulas and/or abscesses) in 10-40 % (26). The anatomical location of CD is fairly stable but the behaviour of the disease over time is unstable and both regression and progression may occur (28).
The clinical presentation, which is largely dependent on the disease location and the development of complications including strictures,

13
abscesses or fistulas, may include diarrhoea, abdominal pain, fever, clinical signs of bowel obstruction and passage of blood and/or mucus. Numerous extraintestinal manifestations may occur and they are mainly the same as for UC.
The diagnosis in CD is made on the basis of the patient history and physical examination, with the addition of objective findings from endoscopic, radiological, laboratory and histological studies (29). The endoscopic appearance in CD may vary from small superficial aphthous ulceration, loss of vascular pattern, friability, exudates and granularity, to linear and serpiginous deep ulceration and patchy inflammation ultimately resulting in a cobblestone pattern and occasionally fibrosis and stenosis interspersed by normal mucosa.
Histological features in CD include focal inflammation with skip lesions (ie aphthous erosion or ulceration occurring in a background of normal mucosa), submucosal involvement, cryptitis and crypt abscesses, granulomas and goblet cell preservation. The degree of crypt architectural distortion may be less pronounced than in UC (13, 14).
The incidence of CD in northern Europe ranges from 3.6 – 9.8 per 100 000 per year, and in Sweden it is 6 -7 per 100 000. The onset is most common between the ages of 15 and 30 years (15). CD affects men and women at similar rates.
As for UC, a positive family history is the main independent risk factor for the disease. Among CD patients, 2-16% have a first-degree relative with CD and 5 – 22% with IBD. The estimated lifetime risk that a first-degree relative of a CD proband will develop IBD is 5%– 8 % (16) and the relative risk is more than 35 (17). The concordances for monozygotic and dizygotic twins are 22 – 64% and 3 - 4%, respectively, for CD (18-20). Cigarette smokers are at higher risk of developing CD than are non-smokers (30), and smoking has a negative impact on the clinical course (31-33). On a hopeful note, patients with CD who stop smoking have fewer exacerbations and require less corticosteroid and immunosuppressive treatment to control symptoms compared with patients who continue to smoke (34). Appendicectomy seems to be associated with a future risk of CD, especially when performed after 10 years of age, but the relation is weaker than in UC (35, 36).
Similary to patients with extensive UC, patients with extensive Crohn colitis are at higher risk in developing colorectal cancer than healthy individuals, but the magnitude of this is debated.
Collagenous colitis Clinically CC is characterized by chronic watery diarrhoea and occasionally abdominal pain, distension and weight loss (37, 38). Fatigue, nausea and faecal incontinence are other associated symptoms and the disease may

14
impair the quality of life (39). The clinical course is often chronically relapsing and benign, and even if some patients suffer from severe diarrhoea, serious dehydration is rare (40).
Patients with CC often have concomitant autoimmune diseases, most commonly thyroid disorders, coeliac disease, diabetes mellitus and rheumatoid arthritis (37). Macroscopically the colonic mucosa appears normal or almost normal, whereas microscopic examination of mucosal biopsies reveals characteristic histopathological changes, with abnormal thickening of the subepithelial collagen layer (> 10 μm) and lymphocytic infiltration of the epithelium and the lamina propria. Epithelial detachment and loss may occur (41). Cryptitis and/or Paneth cell metaplasia does not rule out a diagnosis of CC (42). The abnormal thickening of the subepithelial collagen layer is most pronounced in the proximal part of the colon and may be absent from the sigmoid colon and rectum (43). This is of importance when diagnosing CC.
Previously CC was considered to be rare, but it has recently emerged as a common cause of chronic diarrhoea with an incidence ranging from 5 - 6 per 100 000 individuals per year. CC mainly affects middle-aged women, with a peak incidence of 25 per 100 000 individuals of about 65 years of age. The female/male ratio is about 7/1 (37, 38, 44).
Treatment The treatment of UC and CD aims to induce and maintain remission of the disease (1). The medical treatment of UC and CD includes aminosalicylates, corticosteroids, antimicrobials, immuno-modulators and biological therapies (29, 45). Surgical treatment is reserved for patients in whom medical treatment fails or who develop severe intestinal bleeding, perforation or cancer. In CD specific indications for surgery include formation of fibrotic strictures leading to partial or complete bowel obstruction, internal fistulas complicated by abdominal abscess, and enterovesical and enterocutaneous fistulas(46-48).
Recommendations for treatment in CC were formerly based mainly on retrospective and uncontrolled data and generally a “step up” regime was proposed (6). In patients with mild symptoms, loperamide or cholestyramine may be used, the latter preferably in patients with bile acid malabsorption (5). Bismuth subsalicylate, prednisolone and mesalamine, with or without cholestyramine, may be effective, but at present budesonide has the best documented efficacy in treating CC (49) although there is a high relapse rate when this medication is tapered or ceased (50). In patients with unresponsive or steroid-resistant CC immunosuppressive therapy may be used, although the evidence for this is limited (51, 52). Owing to the improvement of medical therapy, the indications for surgery are narrow, but it may be considered in severe unresponsive CC (53).

15
Pathogenesis in IBD The pathogenesis of IBD remains obscure. The most widely held hypothesis is that an overly aggressive acquired immune response to a subset of commensal enteric bacteria develops in genetically susceptible hosts, and environmental factors precipitate the onset or reactivation of the disease (fig. 1). In the healthy intestinal mucosa there is a low-level physiological inflammation, a condition of “controlled inflammation”, representing a state of preparedness to deal with potentially harmful agents. Innate immune cells (granulocytes, macrophages, mast cells, dendritic cells and natural killer cells), adaptive immune cells (T-cells and B-cells) and non-immune cells (including epithelial and endothelial cells, cells of the entero-endocrine and enteric nervous system and fibroblasts) engage in complex interactions. However, there is a high proportion of anti-inflammatory and regulatory cytokine responses and the inflammation is kept in check through an active process of tolerance (54, 55). In IBD, the balance between mucosal responsiveness and tolerance towards antigens is disturbed, and there is an exaggerated immune response to the commensal flora and/or other luminal agents, culminating in the elaboration of pro-inflammatory mediators that overwhelm the homeostatic defences of the intestine and injure the intestinal epithelium.
Several genes have been associated with the pathogenesis of UC and CD. The first to be associated with CD was CARD15, formerly known as NOD2 (56, 57). The genes linked with the pathogenesis of IBD regulate innate immune responses, mucosal barrier function and bacterial killing (54, 58).

16
Figure 1: Factors that precipitate the onset or reactivation of IBD.
Pathogenesis in collagenous colitis Data on mucosal inflammation in CC are limited and its pathophysiology is poorly understood, but at present, as for UC and CD, the disease is considered to represent specific mucosal responses to various noxious luminal agents in predisposed individuals. Several mechanisms have been discussed, such as autoimmunity (59, 60), bile acid malabsorption (61),drug induced injury (62), infections (63-65), nitric oxide (66-69), and luminal agents of unknown origin (53). Recently an increased bacterial uptake has been proposed (70).
Familial occurrence of CC has been reported, but the role of genetic factors in this disease remains largely unknown (71-73). An association between CC and HLA-DQ2 and between CC and HLA-DR3-DQ2 has been found, irrespective of the presence of concomitant coeliac disease (74, 75). Madisch et al demonstrated polymorphism in the matrix metallo-proteinase-9 gene (collagen degrading enzyme, collagenase IVb) associated with CC (76). However, in contrast to CD, no association between CARD15 polymorphism and CC has been observed (77).
Luminal factors/antigen
Immune response
Environmental factors
Genetic predisposition IBD

17
Inflammatory cells involved in the mucosal inflammation Neutrophil granulocytes A prominent feature in mucosal biopsy samples from patients with active IBD is the infiltration of neutrophil granulocytes (78). Neutrophils constitute about 60-70% of the leucocytes in the circulation. If not activated, they are short lived and die within 4-10 hours. The mature cell is typically characterised by the multilobed nucleus, and the cytoplasm contains several types of granules. The two major types of granules are the azurophilic (primary) granules and the specific (secondary) granule. The azurophilic granules contain myeloperoxidase (MPO), lysozymes, proteinases and antibiotic proteins (defensins) (79). The specific granules contain metalloproteinases (collagenase and gelatinase) and antimicrobial proteins (HNL, lactoferrin and cathelicidin)(79, 80). Neutrophils constitute the first line defence against invading micro-organisms and they also play an important role in tissue healing (79, 81). When activated, neutrophils immediately migrate to the site of inflammation by responding to successive combinations of chemoattractant gradients. Chemoattractants are released by endothelial cells and by activated stromal cells such as macrophages and epithelial cells, and by the inflammatory targets (bacteria, dying cells). At the site of inflammation neutrophils can act as professional phagocytes or they can release their granule constituents, including radical oxygen species, proteinases, bactericidal proteins and cytokines, which either alone or in concert, may interact in the regulation of inflammatory processes.
18
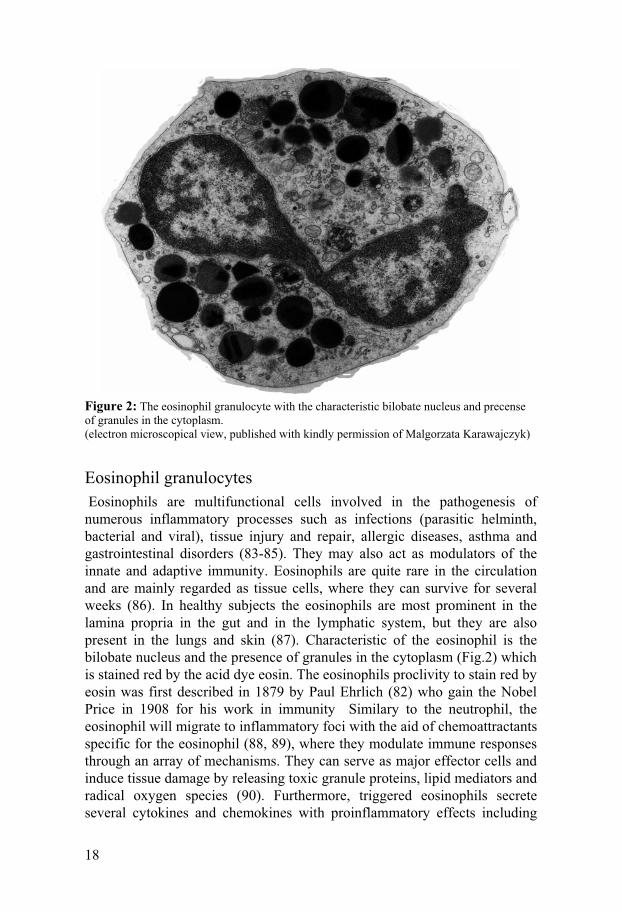
Figure 2: The eosinophil granulocyte with the characteristic bilobate nucleus and precense of granules in the cytoplasm. (electron microscopical view, published with kindly permission of Malgorzata Karawajczyk)
Eosinophil granulocytes Eosinophils are multifunctional cells involved in the pathogenesis of numerous inflammatory processes such as infections (parasitic helminth, bacterial and viral), tissue injury and repair, allergic diseases, asthma and gastrointestinal disorders (83-85). They may also act as modulators of the innate and adaptive immunity. Eosinophils are quite rare in the circulation and are mainly regarded as tissue cells, where they can survive for several weeks (86). In healthy subjects the eosinophils are most prominent in the lamina propria in the gut and in the lymphatic system, but they are also present in the lungs and skin (87). Characteristic of the eosinophil is the bilobate nucleus and the presence of granules in the cytoplasm (Fig.2) which is stained red by the acid dye eosin. The eosinophils proclivity to stain red by eosin was first described in 1879 by Paul Ehrlich (82) who gain the Nobel Price in 1908 for his work in immunity Similary to the neutrophil, the eosinophil will migrate to inflammatory foci with the aid of chemoattractants specific for the eosinophil (88, 89), where they modulate immune responses through an array of mechanisms. They can serve as major effector cells and induce tissue damage by releasing toxic granule proteins, lipid mediators and radical oxygen species (90). Furthermore, triggered eosinophils secrete several cytokines and chemokines with proinflammatory effects including

19
upregulation of adhesion systems, modulation of cellular trafficking, regulation of vascular permeability, mucus secretion and smooth muscle contraction. (87). In addition they may act as antigen presenting cells that stimulate T-cell proliferation and activation (91).
Activated eosinophils release four cationic proteins that are thought to be specific for eosinophils: eosinophil cationic protein (ECP)(92), eosinophil protein X (EPX) (93), eosinophil peroxidase (EPO) (94) and major basic protein (MBP) (95). ECP has a bactericidal function, is toxic to helminthic parasites, promotes mast cell degranulation and has ribonuclease activity (96-100). EPX is weakly toxic to parasites and mammalian cells and possesses antiviral activity in respiratory infection (96, 101). EPO is cytotoxic, degradative to connective tissue, bactericidal and able to induce oxidative damage to DNA and RNA (102-106). MBP has several properties, such as cytotoxicity and toxicity against helminthic parasites (95).
T-cells T-cells are developed in the bone marrow and mature in the thymus, were they undergo so called positive and negative selection through presentation of dendritic (antigen presenting) cells and mature to form helper T-cells (Th-cells) or cytotoxic T-cells (CTL). Th-cells express CD4 and recognise class II major histocompatibility complex (MHC II). There are different kinds of Th-cells: Th-1 cells participate in cell mediated immunity and inflammation and activate macrophages, natural killer cells and CTLs via cytokines (such as interferon-�, interleukin-2 and 12). Th-2 cells participate in antibody mediated immunity and activate B-cells via cytokines (such as interleukin-4, 5 and 13); they can also activate eosinophils and mast cells. (107) Th-17 cells are predominantly found in epithelial surfaces that interact with the external environment such as the gastrointestinal tract. Here they are positioned to attack bacteria by secreting defensins and to recruit neutrophils (108). There is also a subset of CD4+ T-cells (Th-3 cells, TR-1 cells and CD4+CD25+ regulatory T-cells) that have regulatory functions and act as immunosuppressive and anti-inflammatory cells (227).
Cytotoxic T-cells express CD8 and recognise MHC I. They act as professional killer cells and can kill other “target” cells, which may include cells infected with virus, intracellular bacteria or parasites, allografts and cancer cells. They are also active in autoimmune disorders. When activated by antigen presenting cells they enter the cell cycle and a “clonal expansion” takes place, followed by differentiation to killer cells. Killing is achieved by perforins and granzymes released from CTL at a so called “immunological synapse” with a target cell. Perforins form a pore in the cell membrane of the target cell that enables granzymes to enter the cell and cause the cell to self-destruct by apoptosis. Another mechanism of killing is via a transmembrane protein expressed on CTL, called Fas-L, which binds to a receptor on the

20
target cell and leads to its death by apoptosis. Most of the CTLs will die of apoptosis when they have completed their task, but some will become memory cells; ie long-lived cells (several years) that are prepared to respond to the antigen if it should reappear (107).
Mucosal inflammation and the enteric neuro-endocrine system Several studies suggest that interactions between the enteric nervous system and the immune system play an important role in the pathophysiology of IBD (109-112). Involved in these interactions is the secretion of chemical messengers that carry signals, occasionally bidirectional, between enteric neurones and immunological cells. Close anatomical associations in the gut wall between the terminal axons of enteric neurons, entero-endocrine (EEC) cells and inflammatory cells facilitate the neuroimmune communication (113, 114). Among others, neuropeptides have been shown to act as chemical messengers (110) and can also directly influence immunoglobulin production, lymphocyte proliferation, chemotaxis, phagocytosis, release of granular proteins from neutrophils, and also the migration and homing patterns of lymphocytes (110, 115, 116). Secretoneurin (SN), a neuropeptide derived from secretogranin II (chromogranin C) (117) has been shown to be a major peptide within the human enteric neuro-endocrine system (118). SN can act as a chemoattractant for blood eosinophils (119) and may increase the spontaneous locomotion of neutrophils (120). Chromogranin A is a EEC cell marker and has been reported to be elevated in plasma and serum from patients with IBD (121-123). Moreover, neutrophil granulocytes have emerged as a significant source of intact and processed forms of CgA (124, 125). Chromogranin B and its related peptides have been identified in both humans and animals and some of these have been shown to possess biological activity (126, 127). In most neuroendocrine cells, CgB co-exists together with CgA and it has been shown that CgB can be used as a complement to CgA measurements as an important marker for neuroendocrine toumors (128-130).
Entero-endocrine cells are scattered amongst the epithelial cells lining the intestinal mucosa and react to luminal stimulants such as antigens, nutrients, irritants, enteric secretions, bulk, and mechanical distortion (131). In response to a stimulus EEC cells release proteins and peptides, which may act locally (on nerve endings, on epithelial cells and on immune cells) or at remote sites by entering the circulation (131). Furthermore, studies in rats suggest that CgA and serotonin could be released in to the intestinal lumen (132, 133).

21
Mucosal inflammation and flow cytometry Measurements of cell products in intestinal perfusion fluid and faeces in patients with UC, CD and CC may provide a good idea of eosinophil and neutrophil activity in intestinal disease. However, to look more closely at the different cells at the site of inflammation, we have established a method for studying the presence and activity of intestinal granulocytes and lymphocytes by flow cytometry, using intestinal biopsiy samples (134). By means of this method we can evaluate the number of activated versus resting granulocytes and lymphocytes as well as the degree of activation of individual cells. It may lead to a better understanding of the roles of granulocytes and lymphocytes in different stages of CC.
Mucosal inflammation and fecal markers Classical acute phase proteins such as C-reactive protein (CRP) and orosomucoid, together with the erythrocyte sedimentation rate (ESR), have been used as a complement to clinical indices when evaluating patients with IBD (135). However, it is generally agreed that these markers are insensitive in detecting disease activity (136-138). Currently, the most reliable method for assessing intestinal inflammation is endoscopy with biopsy sampling. These techniques however, are costly, invasive, time consuming and unpopular with patients. Furthermore, the site of inflammation is not always reached by endoscopy, as in the case of small bowel involvement in CD. Simple, inexpensive and objective tools for assessment of mucosal inflammation are therefore desirable. Proteins released by inflammatory cells, involved in the mucosal inflammation in the gut, may leak into the bowel lumen and appear in the stools. Hence, these proteins in faeces could be markers of local inflammation in the gut and have the advantage of not being elevated in extra-intestinal processes (139-146). Previous studies have indicated that faecal markers may be used in differentiation of IBD against functional gastrointestinal disorders, but the usefulness of these markers in monitoring effects of treatment in patients with active relapse of IBD needs further evaluation (136, 147-149) .
Faecal markers in IBD Calprotectin is a major protein of neutrophil granulocytes and macrophages, accounting for about 60% of the cytosol in these cells (150). It is a calcium-binding protein with antibacterial, antiproliferative and immunomodulating effects (151-153). Several authors have proposed that elevated FC correlates to inflammation in the intestinal tract in both adults and children (141, 144,

22
154) and has the ability to predict relapse in IBD (228,229)Moreover, calprotectin is stable in the stool, and samples are easily collected (140, 145, 147, 155-157).
Other faecal proteins of interest in monitoring IBD are MPO and EPX. MPO is mainly derived from neutrophil granulocytes and its release has been observed both in mucosa (146, 158, 159) and in gut lavage, with the potential of monitoring treatment outcome (160). EPX is a granulae protein released upon activation of the eosinophil granulocyte, which has been shown to be abundant in the mucosa during active IBD (161-164). In adition to this, we have reported elevated faecal levels of EPX and MPO with the potential of monitoring therapy in UC (146).
Faecal markers in collagenous colitis Besides the characteristic lymphocytic infiltration of the epithelium and the lamina propria in CC, infiltrates of eosinophil and neutrophil granulocytes can be observed in the mucosa (165-168). Taha et al reported increased luminal levels of ECP, in perfusion fluids from colon (173) and a reduction in rectal release of ECP was noted after oral prednisolone treatment of active CC (169). Increased levels of ECP and EPX in faeces from patients with CC have been demonstrated (142, 170). Neutrophil recruitment and activation, with release of MPO, is a dominant feature in IBD, but CC patients only have a slightly increased number of neutrophils in the mucosa (171, 172) Moreover, levels of MPO have been found to be low in perfusion fluids from colon in patients with active CC (173) implying low activity of the recruited neutrophils. Nevertheless, elevated levels of calprotectin (174) and MPO (142, 170) in faeces from patients with CC have been reported.

23
Aims
The general aim of this investigation was to study inflammatory processes in the intestinal mucosa in patients with inflammatory bowel disease and collagenous colitis, and to determine whether these processes are reflected in the faecal content of specific proteins secreted by cells in the intestinal mucosa.
Specific aims of the studies described in this thesis were:
• To assess the value of faecal calprotectin (FC) as a surrogate marker of the outcome of relapse treatment in patients with UC and CD compared to standard criteria of response in the clinical setting and to compare FC with fecal MPO and EPX regarding their applicability in monitoring treatment outcome (study I).
• To quantify and assess the activity of eosinophil and neutrophil
granulocytes in patients with active CC before and after budesonide treatment compared to healthy individuals with aim to elucidate there role in the mucosal inflammation. The second aim was to investigate the activity of CD4+ and CD8+ T-cells in the same patients (study II).
• To examine inflammatory processes as indicated by excretion of
faecal ECP and EPX, FC, and fecal MPO in active CC before and after budesonide treatment and to evaluate the ability of these markers to monitor disease activity in CC (study III).
• To determine whether secretoneurin, chromogranin A and
chromogranin B are detectable in faeces from patients with CC and to compare the levels with patients with UC, CD and healthy control subjects. A further aim was to find out if there were any dynamic changes in faecal SN, faecal CgA and faecal CgB during treatment in the same patients (study IV).

24
Subjects
Study I Thirty-eight adult patients with symptoms of a relapse of a previously known diagnosis of IBD ( UC, n=27; CD, n=11), seeking medical advice at the Section of Gastroenterology, University Hospital, Uppsala, Sweden between October 2002 and April 2003, completed study. On inclusion in the study, at the first visit to the clinic, all patients had mild to moderate clinical activity and endoscopical findings consistent with active disease. Exclusion criteria were as follows: pregnant or lactating women, enteritis due to infections, or intestinal biopsies performed within three days before inclusion. For background data, treatment on inclusion and the extent of the disease study population, see Table 1. Faecal samples obtained from 44 healthy adult individuals (median age 43.5 years, range 18–73) were used as samples from healthy controls. A questionnaire including health status was filled in by each control individual. Individuals who were considered healthy were, within the scope of the questionnaire, not suffering from thyroid disease, heart and vascular disease, tumors, joint disease, diabetes, liver disease, lung disease, allergic disease, dermatitis or eczema, food allergy, IBD or other gastrointestinal disease, frequent urinary infection, or other recent infection and not receiving anti-inflammatory treatment.

25
Table 1. Clinical data of the study population of study I UC CD
Number of subjects 27 11
Gender (female/male) 15/12 3/8
Agea 42.5 (21 – 66) 34.6 (21 – 70)
Disease durationa 9.3 (0 – 38) 5.6 (0 – 18)
Extent of disease:
Colon 9b 8
Left sided colitis 14 -
Proctitis 4 -
Ileocolitis - 3
Prior surgery 1 1
Treatment on inclusion:
No treatment 7 4
5-ASA (topical/systemic) 19 4
Azatioprine 1 1
Prednisone (topical/systemic) 5 -
Metronidazol - 1 CD = Crohn´s disease, UC = ulcerative colitis, 5-ASA= 5-aminosalicylic acid, a Years, mean (min-max), b Extensive colitis = Proximal to the splenic flexure
Study II
Eleven adult patients with previously diagnosed collagenous colitis and ongoing clinically active disease, defined as a stool frequency > four per day, seeking medical advise at the Section of Gastroenterology, University Hospital, Uppsala, Sweden, participated in study II. (For baseline characteristics of the study population, see Table 2) The diagnostic criteria for collagenous colitis were histological findings of a subepithelial collagen layer thicker than 10 μm, at least focally, and lymphocytic infiltration of the epithelium and the lamina propria (41). Patients treated with anti-inflammatory drugs (5-ASA, corticosteroids, NSAID, azathioprine, antibiotics) within the last four weeks were excluded. Additional exclusion criteria were coeliac disease, gastrointestinal infection, previous colonic resection and pregnant or breast feeding women.

26
Controls were recruited among patients examined for anaemia due to gastrointestinal bleeding, or were healthy volunteers (n=10, mean age 38 years, range 23-72).
Table 2. Baseline characteristics of patients in Study II Patients
Sex (F/M) 8/3
Age (years)* 59 (22 – 79)
Duration of symptoms (weeks)* 36 (3 – 104)
Watery stools/day* 9 (5 – 17) *Mean (min-max)
Study III Twelve patients (all 11 patients in study II and in addition a female patient aged 51 years with a symptom duration of 12 and an average of 12 watery stools/day) participated in study III. The healthy controls who provided faecal samples were the same as in study I. In addition five healthy volunteers (mean age 58 years, range 30 – 78), who served as controls, underwent one colonoscopy with biopsy sampling
Study IV Study IV comprised all patients who participated in study I and study III. The healthy controls who provided faecal samples were the same as in study I.

27
Methods
Study design - studies I and IV Patients were examined according to the study protocol on inclusion and after four and eight weeks of treatment, see Table 3. In patients with UC a semi quantitative four-graded (normal, mild, moderate and severe) scale was used for clinical and endoscopical scoring (175). In patients with CD the Harvey-Bradshaw clinical activity index (HBI) was used (176). The histopathological findings were graded as active or inactive inflammation based on the basis of the number of neutrophil granulocytes in the mucosa; the grading was done by an experienced pathologist according to accepted criteria adapted from Truelove and Whitts (177). Stool samples were analysed for calprotectin by enzyme-linked immunosorbent assay (ELISA) and for F-MPO and F-EPX by radioimmunoassay (RIA) (study I). In study IV stool samples were analysed for F-CgA, F-CgB and F-SN by RIA. Treatment was individualised, according to standard recommendations for the management of IBD, and included 5-ASA, prednisone, azathioprine and methotrexate. Topical and/or systemic 5-ASA was administered to 26/27 UC patients and 9/11 CD patients. Topical and/or systemic prednisone was prescribed in 22/27 UC patients and in 7/11 CD patients. Azathioprine was used in 3/27 UC patients and in 3/11 CD patients. One CD patient received methotrexate.
In UC patients response to treatment was defined as complete if the clinical and endoscopical scores decreased to normal. A partial response was defined as a decrease in both clinical and endoscopical score but not to normal. Non-response was defined as a decrease in only the clinical or endoscopical score or as an unchanged or increased clinical and/or endoscopical score. In CD patients treatment response was defined as complete if the HBI score decreased to ≤ 5 points, and as partial if the clinical score decreased but not < 6 points; finally, non-response was defined as an unchanged or increased HBI score.

28
Table 3. Study protocol (studies I + IV) Procedure On inclusion At 4Weeks At 8 Weeks
Stool sample Done Done Done
Clinical score Done Done Done
Endoscopy and biopsy Donea Doneb Donec
Histopathology Donea Doneb Donec
a: not done in 2 Crohn´s disease (CD) patients, b: not done in 4 CD patients, c: not done in 2 CD patients.
Study design - study II On inclusion, the patient´s demographic data and medical history were recorded. Before treatment (on inclusion) and after eight weeks of treatment with budesonide 9 mg once daily (Entocort® 3 mg Astra-Zeneca), clinical symptoms and the stool frequency and consistency were recorded and an ileo-colonoscopy with biopsy sampling from each of seven different locations (terminal ileum,caecum, right and left flexures of the colon, descending colon, sigmoid colon and rectum) was performed. The intestinal biopsy samples from patients and controls were analysed by flow cytometry, histopathology and immuno-histochemistry. Peripheral blood was also collected from all patients before and after treatment, and from control subjects, and analysed by flow cytometry.
Study design - studies III and IV On inclusion, patient demographic data and the medical history were recorded. Before (on inclusion) and after 3, 7, 28 and 56 days of treatment with budesonide 9 mg daily (Entocort® 3 mg Astra-Zeneca) clinical symptoms and stool frequency and consistency were recorded and stool samples were collected. Stool samples from patients and controls were analysed for ECP by UniCAP and for EPX, MPO and calprotectin by ELISA (study III). In study IV stool samples were analysed for F-CgA, F-CgB and F-SN by RIA. Colonoscopy with biopsy sampling from each of six different locations (caecum, right and left flexures of the colon, descending colon, sigmoid colon and rectum) was performed on inclusion and on day 56 after treatment in patients and controls, on one occasion. The biopsy samples were analysed by histopathology and immuno-histochemistry.
.

29
Faecal markers of inflammation – study I Stool samples were collected in screw-capped plastic containers on inclusion, and after four and eight weeks of treatment. Stool samples were kept at +4 °C for up to 2 days before freezing at –70 °C. Calprotectin in faecal extracts was measured with the calprotectin ELISA according to the manufacturer´s instructions (Calprest;Eurospital SpA, Trieste, Italy). Before testing, the supernatants were thawed, diluted 1:50 with assay buffer and then measured with Calprest. Calprotectin was expressed as microgram per gram of faeces. The manufacturer suggests considering a calprotectin level of > 50 μg/g as pathological. The concentration of faecal calprotectin was determined in samples from 44 apparently healthy adults and the normal range was calculated to be 9.2 – 94.5 μg/g (5 th – 95th percentile). When extracts of stool samples were prepared for measurement of EPX and MPO, approximately 0.1–1 g of faeces was weighed and diluted five times by adding 4 volumes (vol/wt) of an extraction buffer consisting of phosphate-buffered saline (PBS), pH 7.4, supplemented with 10 mmol/L ethylenediaminetretraacetic acid, 0.2% N-cetyl-N,N,N-trimethylammonium bromide (CTAB), 20% glycerol, 0.05% Tween 20, and 1% bovine serum albumin (BSA). The mixture was homogenised using a Polytron PT1200 CL mixer (Kinematica, Lucerne, Switzerland) for 5–90 s until a homogeneous solution was obtained. An aliquot of 0.5 ml of the homogenate was then further diluted 20 times in extraction buffer. After incubation at 6°C for 30 min and mixing, the homogenate was centrifuged at 20,800 × g for 30 min at 5°C. The particle-free supernatant was thereafter transferred to three tubes and frozen at -70°C for later analysis. Two separate tubes containing 1500 μl × l of the 1:5 diluted homogenate were weighed and centrifuged at 20,800 × g for 30 min at 5°C. By weighing the pellet obtained after discarding the supernatant, a measure of semi-dry weight was obtained. EPX and MPO were determined using radioimmunoassay (Pharmacia Diagnostics, Uppsala, Sweden). Concentrations of fecal markers in healthy adults (n = 44, median age 44 years, range 18 - 73) for respectively fecal MPO (1.3 - 8.8 μg/g, 5 - 95th percentile) and fecal EPX (0.2 - 1.7 μg/g, 5 - 95th percentile) has recently been described (142).
In study III ELISA assays for measurement of calprotectin were obtained from Bühlmann Laboratories AG (Basel, Switzerland). ECP was measured with UniCAP (Phadia AB, Uppsala, Sweden), whereas EPX and MPO were measured by ELISA obtained from Diagnostics Development AB (Uppsala, Sweden). Marker concentrations in faeces were adjusted for faecal water content as described in study I and expressed as �g/g semi-dry faeces.
In study IV faecal CgA, F-CgB and F-SN were measured by specific radioimmunoassay as described before (EuRIA CGA, Eurodiagnostica, and (129, 178). The intra- and inter-assay variations were less than 10% for all assays used in study I, III and IV.

30
Collection and preparation of biopsy samples – study II During ileo-colonoscopy, four adjacent biopsy samples were taken from each of seven different locations in all patients and control subjects: the terminal ileum, caecum, right and left flexures of the colon, descending colon, sigmoid colon and rectum. Two of the samples from each location were sent for histological analysis. The remaining two samples were immediately transferred into tubes filled with physiological saline solution at room temperature, and were further processed within one hour.
Single-cell suspensions of biopsy cells were obtained, using a loosely fit glass homogenizer, and the cells were then washed twice with a buffer assigned for fluorescence activated cell sorting (FACS) containing 0.05% NaN3 , 0.1% BSA and 0.4% trisodium citrate dihydrate in PBS. Heparinised peripheral blood from the same individuals was haemolysed with a 0.83 % ammonium chloride solution and washed twice in the FACS buffer to obtain a suspension of blood leucocytes. Both types of cell suspensions were incubated with fluorochrome-conjugated monoclonal antibodies (mAbs) for 30 minutes at room temperature in the dark. After a final wash, the cells were suspended in 500 �L of the FACS buffer and analysed.
Antibodies for flow cytometry – study II Mouse-anti-human mAbs conjugated to fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin-chlorophyll protein (PerCP) or allophycocyanin (APC) were used for all antigens. Isotype-matched control labelling was also performed, using fluorochrome-conjugated mouse anti-human IgM� and IgG2b� as controls for non-specific staining. All antibodies used for flow cytometry were purchased from Becton Dickinson (BD) Biosciences/ Pharmingen, San Diego, USA.
Flow cytometry assay – study II The flow cytometry assay was performed on a two laser FACS Calibur cytometer (BD Immunocytometry systems, San José, Ca, USA). Three types of fluorophores were used to enable analysis of three different antigens in the same sample: FITC, emitting 519 nm green colour in FL1; PE emitting 578 nm yellow-green colour in FL2; and PerCP emitting 675 nm red colour in FL3. Fluorescence measurements were collected using a logarithmic amplifier; forward and side scatter was studied with a linear amplifier. Ten thousand cells were counted and analysed in each sample. For data analyses, Cell Quest Pro software from Becton Dickinson was used.

31
Eosinophil and neutrophil granulocytes from peripheral blood or biopsy samples were gated by their forward and side scatter properties and further identified by surface markers as described in Figure 3.
CD9 has previously been used as a marker for eosinophils (179), but in our setting the specificity needed to be further increased. We therefore used CD9 combined with anti-CDw125, which is expressed on eosinophils and basophils (180). CD44, the receptor for hyaluronic acid, was used as a marker of eosinophil activation (181). Another indicator of eosinophil activity is mean fluorescence intensity (MFI) of CD9, which is high on resting cells and decreases on activation, probably because of shedding (182). CD15 is expressed on both eosinophils and neutrophils, but the MFI of this molecule is 10–100 times higher on neutrophils (183), and the neutrophil population is therefore easily distinguished from eosinophils. CD66b is stored in the secondary granules of neutrophils and is mobilised to the surface on activation. Accordingly, the MFI of this molecule was used as a measure of neutrophil activation (184, 185).
Lymphocytes were gated by their forward and side scatter properties and further identified by CD4 and CD8 antibodies. The MFI of CD69 was used as a measure of lymphocytic activation.

32
Figure 3. Representative plots describing the procedure of cell identification by flow cytometry. Cell populations are selected for further analysis by drawing gates (numbered R1, R2, etc) around the cells of interest in the forward/side scatter plot. Gated cells are then transferred to new plots for evaluation of their expression of specific markers. (a) Peripheral blood leucocytes: gating of eosinophil (R1) and neutrophil (R2) granulocytes.
0 200 400 600 800 1000FSC-H
R1
R2
a)
10 0 10 1 10 2 10 3 10 4FL2-H
CD15 PE
CD66b FITC
Gate R5
c)
100 101 102 103 104FL1-H
R3
CD9 FITC
CDw125 PE
Gate R4
e)
10 0 10 1 10 2 10 3 10 4FL3-H
M1
Gate R4 and R3
f)
Isotype control/ CD44 PerCP
100 101 102 103 10 4FL2-H
Gate R5
d)
Isotype kontrol FITC
isotype control PE
0 200 400 600 800 1000FSC-H
R4
R5
b)

33
(b) Intestinal biopsy: gating of eosinophil (R4) and neutrophil (R5) granulocytes. (c) Intestinal neutrophils gated by R5. The upper right quadrant contains CD66b positive neutrophils. (d) Intestinal neutrophils gated by R5. Isotype matched control monoclonal antibodies (mAbs) for CD66b and CD15 (mouse antihuman IgMk fluorescein isothiocyanate (FITC) and phycoerythrin (PE), respectively). (e) Intestinal eosinophils gated by R4. Cells in the upper right quadrant expressing CD9 and CDw125 are gated (R3) for evaluation of CD44. (f) Expression of CD44 on eosinophils gated by R4 and R3 compared with isotype matched control mAb (mouse anti-human IgG2bk peridinin-chlorophyll protein (PerCP)). A gate (M1) is set on the eosinophil population with expression of CD44 exceeding isotype control staining.
Histopathology – studies II and III
The biopsy samples were fixed in 4% formaldehyde and embedded in paraffin. Three sections were cut at different levels and stained with haematoxylin-eosin and van Gieson. A four-graded scale was employed to evaluate the inflammatory response: 0 = no inflammation; 1 = light inflammation through the whole thickness of the lamina propria, or light inflammation only involving the luminal part of the lamina propria; 2 = heavy inflammation only involving the luminal surface of the lamina propria; 3 = heavy inflammation through the whole thickness of the lamina propria. Areas containing the thickest collagen band were measured with a calibrated ocular microscale and the maximum thickness and corresponding location were recorded. The same pathologist reviewed the biopsies.
Immuno-histochemistry – studies II and III Immuno-histochemical analyses were performed on biopsy samples from 11 (12 in study III) CC patients before and after eight weeks of budesonide treatment and from five control subjects. A monoclonal antibody to EPO (Dept. of Medical Sciences, Clinical Chemistry, University of Uppsala, Sweden) was used to identify eosinophil granulocytes.
Sections cut from wax-embedded blocks (prepared for routine histological analysis) were de-paraffinised in xylene, rehydrated through decreasing concentrations of alcohol, and finally rinsed in distilled water. To expose antigenic sites and reduce background, the sections were heated in a citrate buffer using a pressure cooker. The slides were subsequently placed in an automated slide processing system (AutostainerPlus, Dako Cytomation, Glostrup, Denmark), where sections were blocked in hydrogen peroxide/methanol, washed, and stained in several steps. The antigen-antibody complex was visualized with 3,3'-diaminobenzidine (DAB), using a commercial Envision kit (Dako Cytomation, Glostrup, Denmark)

34
according to the instructions given in the manual. The samples were then counterstained with Mayer’s haematoxylin (Histolab Products AB, Gothenburg, Sweden), dehydrated, and mounted. The sections were examined by the same examiner with an Olympus BH2-MDO microscope (Olympus Optical Co. LTD., Tokyo, Japan) and the examiner was blinded to patient data and treatment outcome. The result was expressed as mean values of numbers of eosinophils identified in 10 high power field (x340 magnification).
Ethics The project was approved by the Ethical Committee of the Medical Faculty, Uppsala University and all participitants provided written informed consent.
Statistics The non-parametric tests Kruskal-Wallis ANOVA and the Mann-Whitney U-test were used for unpaired comparisons. For paired analyses we used Friedman ANOVA and the Wilcoxon matched pairs test. Spearman rank order correlations were used to express relationship between variables. A p value of < 0.05 was adopted as significant. All calculations were performed on a personal computer by means of the statistical software Statistica (Statsoft Inc, Tulsa, Oklahoma USA).

35
Results
Study I: Faecal markers of inflammation used as surrogate markers for treatment outcome in relapsing IBD On inclusion in the study 37 of the 38 patients displayed elevated FC levels. The FC levels on inclusion and during the study period are shown in Table 4.
Table 4. Faecal calprotectin (FC) levels (μg/g, mean, 10th - 90th percentile) in patients with a relapse of IBD on inclusion and after four and eight weeks of treatment. FC inclusion FC four weeks FC eight weeks
All patients 5430 (151– 4170) 1920 (61–4150) 1720 (38–3390) p<0.01*
UC 5600 (210-14170) 1730 (61–2620) 1820 (40–3390) p<0.01*
CD 5010 (151–9180) 2440 (124–7910) 1460 (36–5030) p=ns*
UC = ulcerative colitis, CD = Crohn´s disease, * Friedman´s test for 3 variables
In UC patients a correlation between FC level and clinical score was seen after four weeks of treatment (R =0.42, p < 0.01), but not on inclusion or after eight weeks. Correlation between FC levels and endoscopic score was noted after four (R = 0.50, p < 0.01) and eight weeks (R = 0.78, p < 0.01) of treatment but not on inclusion. In CD patients the FC levels was correlated to clinical score after eight weeks of treatment (R = 0.78, p < 0.01) but not on inclusion or after four weeks. Patients with UC and histological signs of active inflammation had higher levels of FC than UC patients with inactive inflammation after four and eight weeks of treatment (p < 0.05). In CD patients no such difference was found during the study.
Among the 27 patients with UC a complete response was observed in 14 (52%) and 21 (78%) after four and eight weeks of treatment, respectively. Two patients were classified as partial responders and four patients as non-responders after eight weeks of treatment. In complete responders there was a decline in FC ( p < 0.01) that was not seen in partial or non-responders and which was already significant after four weeks of treatment (p < 0.01) (fig 2). Among the 11 CD patients a complete response was noted in 9 (81%) and 10 (91%) after four and eight weeks of treatment, respectively. A tendency to a decline in FC levels was observed in CD patients with a complete response during the study period (p=0.13) (fig. 4).

36
Figure 4. Faecal calprotectin levels (box shows the median and the 25th - 75th
percentiles and the lines 10th - 90th percentile with circles displaying outliers) in patients of study I with ulcerative colitis (UC) and Crohn´s disease (CD) treated for eight weeks for a relapse of their respective disease, and treatment outcome in terms of complete and non-complete response.
FC correlated to MPO and EPX at all visits in the whole group of patients as well as in sub-group analyses in UC and in CD patients.
With the aim of predicting the treatment outcome a positive predictive value (PPV) and negative predictive value (NPV) were calculated after eight weeks of treatment. All patients with a normalised FC value after eight weeks of treatment fulfilled the predefined criteria of a complete response whether their diagnosis was UC or CD. However, an elevated FC value was noted in 10/21 UC patients (48%) and 6/9 CD patients (67%) who fulfilled complete response criteria after eight weeks of treatment. Regarding MPO, the same pattern was found among both UC and CD patients, but for EPX this was only seen in CD patients (Table 5).

37
Table 5. Positive predictive values (PPV) and negative predictive values (NPV) of a complete response after eight weeks of treatment given for a relapse of ulcerative colitis (UC) or Crohn´s disease (CD), calculated by using faecal markers values above the 95th percentile as cut-off values. Faecal marker Patients PPV(95%CI) NPV(95%CI)
FC All n = 37a 30 (13-53) 100 (77-100)
FC UC n = 27 38 (13-65) 100 (71-100)
FC CD n = 10a 14 (2-58) 100 (30-100)
MPO All n = 37a 23 (10-42) 100 (59-100)
MPO UC n = 27 27 (10-50) 100 (48-100)
MPO CD n = 10a 12 (2-53) 100 (19-100)
EPX All n = 37a 22 (8-42) 90 (56 -98)
EPX UC n = 27 25 (8-49) 85 (42-98)
EPX CD n = 10a 14 (3 -58) 100 (30-100)
Faecal calprotectin (FC) cut-off value 94.5 μg/g; Myeloperoxidase (MPO) cut-off value 8.8 μg/g; eosinophil protein X (EPX) cut-off value 1.7μg/g, a one missing value.
Study II: Budesonide treatment of patients with collagenous colitis restores normal eosinophil and T-cell activity in the colon Clinical remission (stool frequency � three per day) was observed in ten out of 11 patients after eight weeks of treatment. In one patient the stool frequency decreased from 17 to eight but the clinical remission criteria were not fulfilled.
Immunohistochemical staining for the eosinophil specific protein EPO revealed increased numbers of eosinophils in the lamina propria in patients with active CC, most markedly in the right flexure of the colon, compared to control subjects (p<0.05). After eight weeks of treatment the number of eosinophils had decreased (p<0.05) but had not reached the control level.
We found increased eosinophil activation, assessed as decreased MFI of CD9, in biopsy samples from untreated CC patients compared to control subjects. After eight weeks of treatment, the CD9 expression increased indicating attenuated eosinophil activity (Fig. 5).

38
0
200
400
600
800
1000
1200
1400
p=0.06
MFI
of C
D9
CC inclusion (n=11) CC day 56 (n=11) Control subjects (n=10)
p=0.04
p=0.03
p=0.01p=0.02
Terminal Caecum Right Left Descending Sigmoid Rectum ileum flexure flexure colon colon
Figure 5
Figure 5. Mean fluorescense intensity (MFI) of CD9 on eosinophils from patients with active collagenous colitis (CC) before treatment (CC inclusion), and after 8 weeks of budesonide treatment (CC day 56), and from control subjects. P-values, assessed by a Wilcoxon matched pairs test (inclusion and day56) and the Mann-Whitney U test (inclusion and control subjects), are indicated. The results are expressed as mean±SEM.
No significant difference in the proportion of activated eosinophils, measured as high expression of CD44, was seen between patients with active or treated CC and control subjects, although there was a tendency towards increased CD44 in patients with active CC. The percentage of CD44 high
eosinophils was decreased after eight weeks of treatment, indicating reduced eosinophil activity (fig. 6).

39
0
10
20
30p=0.06
% C
D44
high
eos
inop
hils
CC Inclusion (n=11) CC day 56 (n=11) Control subjects (n=10)
Terminal Caecum Right Left Descending Sigmoid Rectum ileum flexure flexure colon colon
p=0.04
p=0.01 p=0.04Figure 6
Figure 6. Percentage numbers of activated (CD44+high) eosinophils in patients with active collagenous colitis (CC) before treatment (CC inclusion), after 8 weeks of budesonide treatment (CC day 56) and in control subjects. P-values, assessed by the Wilcoxon matched pairs test, are indicated. The results are expressed as mean±SEM.
No difference in neutrophil activity was found between patients with active CC and control subjects. After eight weeks of treatment the neutrophil activity was diminished (p < 0.05) in the right flexure of colon and in the descending colon, where the expression was even lower than in control subjects (p < 0.05) (not shown).
There was a tendency towards lower CD69 expression (MFI) on CD4+ T-cells from patients with active CC compared to controls (p = 0.09 in the caecum and the right flexure of colon), indicating suppressed activation of CD4+ T-cells in active CC. After eight weeks of treatment, the expression of CD69 increased to the control level (not shown).
The MFI of CD69 on CD8+ T-cells was lower in patients with active CC than in control subjects. An increased MFI of CD69 on CD8+ T-cells was observed in patients after eight weeks of treatment (fig. 7).

40
0
20
40
60
80
100
p=0.07
p=0.06 p=0.06
p=0.03 p=0.05
Terminal Caecum Right Left Descending Sigmoid Rectum ileum flexure flexure colon colon
MFI
of C
D69
on
CD
8+ T
-cel
ls
CC Inclusion (n=11) CC day 56 (n=11) Control subjects (n=10)
p=0.03
p=0.03
Figure 7
Figure 7. Mean fluorescense intensity (MFI) of CD69 on CD8+ T-cells from patients with active collagenous colitis (CC) before treatment (CC inclusion) and after 8 weeks of budesonide treatment (CC day 56), and from control subjects. p-values, assessed by the Wilcoxon matched pairs test (inclusion and day 56) and the Mann-Whitney U Test (inclusion and control subjects), are indicated. The results are expressed as mean±SEM.
The grade of mucosal inflammation, assessed from histological findings, was reduced (p < 0.05) after eight weeks of treatment in all parts of the colon but only occasionally was it normalised. There was no change in the collagen layer thickness after eight weeks of treatment.
No significant difference in markers of eosinophil, neutrophil or T-cell activation in the peripheral blood was observed after eight weeks of treatment compared with values on inclusion or with control subjects.
Study III: Faecal Eosinophil Cationic Protein as a Marker of Active Disease and Treatment Outcome in Collagenous Colitis As in study II all but one patient fulfilled the remission criteria at the end of the study period.. Regarding the immunohistochemical staining for the eosinophil specific protein EPO and the histological findings before and after treatment the same pattern was found as was found in study II.
The values of the studied faecal markers on inclusion and at the end of the study period are displayed in Table 6.

41
Table 6. Levels of faecal (F) inflammatory markers (μg/g, median, min-max) in 12 patients with collagenous colitis before (on inclusion) and after 56 days of budesonide treatment. Marker On inclusion Day 56 Friedman ANOVA
F-ECP 11.1 (1.9-31.1) 1.4 (0.12-3.5) p < 0.001
F-EPX 8.1 (0.71-72.9) 0.52 (0.03-2.3) p < 0.001
F-MPO 12.4 (2.7-202) 3.0 (0.22-34.2) p < 0.001
FC 235 (21.0-1948) 40.5 (7.0-308) P < 0.001
ECP: eosinophil cationic protein, EPX: Eosinophil protein X, MPO: myeloperoxidase, FC: faecal calprotectin
Eosinophil markers of inflammation On inclusion 11/12 patients (92%) displayed F-ECP values above the upper limit of normal (ULN). During the study period there was a decline in F-ECP, which was significant after only three days of treatment (Wilcoxon matched pairs test p<0.01) (Fig. 8), when the values were below ULN in 7/12 patients (58%). After 28 and 56 days of treatment all patients had F-ECP values below ULN. On inclusion 8 of the 12 patients (67%) displayed F-EPX values above ULN. During the study period F-EPX declined, and the decrease was significant after only three days of treatment (Wilcoxon matched pairs test p<0.01) when 8/12 patients (67%) showed F-EPX values below ULN. After 56 days of treatment 11/12 patients (92%) had F-EPX values below ULN. The patient with partial remission had normalised F-ECP and F-EPX at the end of the study.

42
0
5
10
15
20
25
F-E
CP
μg/g
Inclusion Day 3 Day 7 Day 28 Day 56
Figure 8
** **
**
**
Figure 8. Levels of faecal eosinophil cationic protein (F-ECP) in 12 patients with collagenous colitis (CC) before (on inclusion) and after 3, 7, 28 and 56 days of budesonide treatment. The boxes display the median and the 25th -75th percentiles and whiskers 10th -90th percentiles. The horizontal dotted line indicates the upper limit of normal (95th percentile=5.81 μg/g). Values significantly different from those in active CC (on inclusion) are indicated above the respective box. (**p<0.01). The Wilcoxon matched pairs test was used for the statistical evaluation.
Neutrophil markers of inflammation On inclusion 8 out of the 12 patients (67%) displayed F-MPO values above ULN. During the study period there was a decrease which was significant after seven days of treatment (Wilcoxon matched pairs test p<0.01) when 9/12 patients (75%) showed F-MPO values below ULN. At the end of the study 10/12 patients (83%) had F-MPO values below ULN. On inclusion 9 out of the 12 patients (75%) had FC values above ULN. During the study period there was a decrease in FC, which was significant after 7 days of treatment (Wilcoxon matched pairs test p<0.05) when 6/12 patients (50%) showed FC values below ULN. At the end of the study 9/12 patients (75%) had FC values below ULN. The patient with partial remission showed normalised level of F-MPO and FC during the study period.

43
Study IV: Increased Fecal Levels of Chromogranin A, Chromogranin B and Secretoneurine in Collagenous Colitis In the group of CC patients 11/12 (92%) achieved remission (stool frequency � three per day). One patient had a partial response and decreased from 17 to eight stools per day. In the group of UC patients a complete response (clinical and endoscopical score decreased to normal) was observed in 21/27 patients (78%). Two UC patients were classified as partial responders and four as non responders. Ten CD patients (91%) had a complete response (clinical score decreased to normal) and one a partial response.
The levels of F-CgA, F-CgB and F-SN in patients and controls on inclusion are displayed in fig. 9.
0,01
0,1
1
10
100
1000
nmol
/g lo
g
F-CgA F-CgB F-SN
Figure 9 Healthy controls n=43
Collagenous colitis n=12
Ulcerive colitis n=27
Crohn´s disease n=11
*****
******
***
******
*****
******
***
Figure 9. Faecal levels of chromogranin A (F-CgA), chromogranin B (F-CgB) and secretoneurine (F-SN) in healthy controls and in patients with collagenous colitis, ulcerative colitis and Chrohn´s diseas before treatment. The boxes display median and the 25th - 75th percentiles and the whiskers 10th - 90th percentiles. The Mann-Whitney U test was used for the statistical evaluation. ** p<0.01, *** p<0.001.
In patients with CC who achieved remission after 56 days of treatment, no significant reduction in F-CgA or F-CgB was noted (Friedman ANOVA p=0.14 and p=0.52, respectively). A decrease in F-SN (Friedman ANOVA p=0.0061) was found, which was significant after only three days of treatment (Wilcoxon matched pairs test p<0.05) (Fig. 10). The F-SN decreased to levels found in healthy controls. The F-SN levels in patients

44
with CD and UC on inclusion were significantly lower than in controls, and F-SN in CC patients was still markedly higher than in these patients at the end of the study. In patients with UC or CD no change was observed in F-CgA or F-SN levels during the study period.
A significant decrease in F-CgB was found in UC patients with a complete response to treatment (Friedman ANOVA p<0.05) even if the level was within the 5th - 95th percentile of the level found in healthy controls. In UC patients with a non-complete response or in CD patients no difference was found in F-CgB levels during treatment.
0
5
10
F-S
N n
mol
/g
Inclusion Day 3 Day 7 Day 28 Day 56
** ** **
Figure 10
Figure 10. Levels of faecal secretoneurin (F-SN) in 11 patients with collagenous colitis (CC) before (on inclusion) and after 3, 7, 28 and 56 days of budesonide treatment. The boxes display median and the 25th - 75th percentiles and the whiskers 10th - 90th percentiles. The horizontal dotted line indicate upper limit of healthy control subjects (95th percentile) =2.14 nmol/g. Friedman ANOVA p=0.0061. Values significantly different from those in active CC (on inclusion) are indicated above the respective box. *p<0.05, **p<0.01. The Wilcoxon matched pairs test was used for the statistical evaluation.

45
Discussion
This thesis describes studies on the inflammatory processes in the intestinal mucosa of patients with inflammatory bowel disease and collagenous colitis. We have particularly focused on the roles of eosinophil and neutrophil granulocytes and T-cells. We have also conducted studies aimed at determining whether these inflammatory processes are reflected in the faecal content of specific proteins secreted by cells in the intestinal mucosa. For this purpose we have measured the eosinophil derived ECP and EPX and the neutrophil derived MPO and calprotectin; in addition we have measured CgA, CgB and secretoneurin derived from EEC cells and cells in the enteric nervous system.
Faecal markers of inflammation in inflammatory bowel disease In the first study of this doctoral research (study I) we assessed the faecal conten of calprotectin (FC) as a surrogate marker of the treatment outcome in patients with IBD. The levels of FC were monitored before and after four and eight weeks of treatment in patients with a relapse of UC or CD. Patients who had normalised levels of FC after eight weeks of treatment also fulfilled the predefined criterion of a complete response: i.e., when FC values below ULN were used as a negative predictor of active disease, they had a negative predictive value of 100%. This is in line with the previously reported conclusion that normalisation of FC predicts mucosal healing in patients with IBD (139, 148). However, elevated levels of FC after eight weeks of treatment did not rule out responses to treatment: i.e., use of an FC value above ULN as a positive predictor of ongoing active disease (treatment failure), led to a positive predictive value of 30% (38% in UC patients and 14 % in patients with CD). Only few studies on serial determinations of FC levels in patients being treated for IBD have been performed. Roseth et al followed up a single patient treated for UC and found that the FC level decreased and corresponded with clinical, endoscopical and histological healing (148). Others have demonstrated a transient decrease in FC levels in two IBD patients after treatment corresponding to improvement in clinical disease activity (186). Kolho and co-workers followed up the FC level in 15

46
children treated for IBD and concluded that this level declined along with clinical improvement but seldom fell within the normal range (187). It is known that although patients with IBD achieve clinical remission during therapy, complete remission at the microscopic level is rare. In the UC patients in study I who had a complete response, 7 out of 21 were graded as having active inflammation on histological examination after eight weeks of treatment. These patients expressed higher FC values than patients with inactive inflammation. This suggests that the elevated FC levels in some of our patients who responded to treatment reflect ongoing inflammation in a clinically silent disease. There is growing evidence that mucosal healing (i.e. a normal or near normal endoscopical appearance of the mucosa) is a relevant outcome parameter when treating patients with IBD (188). It has been reported that FC correlates more closely with histological than with endoscopical findings, suggesting that this marker is more sensitive than endoscopy in evaluating activity in IBD (189, 190).
In patients with CD, ten out of eleven were classified as complete responders, but only three of these ten showed normalised FC levels. The treatment outcome in CD patients was evaluated only on the basis of clinical indications. The Harvey-Bradshaw activity index improved, mainly on account of a reduced frequency of loose stools and abatement of abdominal pain. In previous studies only a weak correlation has been found between clinical indices and intestinal inflammation as detected by endoscopy in CD patients (191-193). Thus, evaluation of CD patients on the grounds of clinical indices alone seems to underestimate the inflammatory activity. Our results may indicate that although the clinical activity index decreased to normal, the intestinal inflammation was not resolved.
It is possible that in the patients who responded to therapy in study I, but continued to have elevated FC levels, the FC levels would have been normalised if the study had been prolonged. On the other hand, elevated FC levels have been reported to predict relapse of IBD, so another explanation is that the patients with persistently elevated levels of FC in study I may have relapsed again soon after termination of the study. After the completion of study I we followed up the studied patients retrospectively (194) and found that 14 out of the 38 patients had a relapse within a year after study I was ended. Among these 14 patients six were classified as non-complete responders at the end of study I. Of the eight patients who were classified as responders at the end of study I and had a relapse, three (38%) expressed FC > 100 μg/g at the end of that study. In the group of patients without a relapse, 13 out of 22 eligible patients (60%) expressed FC > 100 μg/g. Using FC values > 100 μg/g to predict a relapse within a year, PPV was 19% (95%CI 4.3-45.7) and use of FC > 1000 μg/g gives a PPV 25% (95%CI 3.4-64.9). Thus, at present there are not sufficient data to conclude that patients with elevated FC levels may benefit from more aggressive anti-inflammatory treatment.

47
In study I we also measured F-MPO and F-EPX and compared them with the FC levels, in order to monitor the treatment outcome, and we noted close correlations between these three variables and the treatment outcome. Nevertheless, we found that FC and F-MPO discriminated the treatment outcome better than F-EPX in patients with UC. Interestingly, normalised levels of EPX indicated a complete response, especially in patients with CD.
Faecal markers of inflammation in collagenous colitis
FC is used as a marker for intestinal inflammation in IBD, but no reliable marker for CC has been identified so far (170, 174). In study II we demonstrated that the mucosal inflammation in CC is characterised by enhanced eosinophil activation. In study III we therefore evaluated the eosinophil markers F-ECP and F-EPX in comparison with the neutrophil markers F-MPO and FC in patients with CC. Although the number of subjects included in study III was small, our results indicate that F-ECP provides the best discrimination in detecting active disease before treatment (all but one patient (92%) had elevated levels of F-ECP on inclusion). The only previously published report on F-ECP in CC is from our group, on a study including three patients with active CC,of whom two had increased F-ECP levels (142). Others have studied F-EPX, F-MPO (170) and FC (174) in patients with CC and concluded that these proteins are not uniform markers of the disease, which is in line with our findings.
As demonstrated in study II, the enhanced eosinophil activity found in CC is restored to normal during budesonide treatment. All of the evaluated inflammatory markers in study III declined during budesonide treatment but this effect was most pronounced in the eosinophil markers, and all but one patient displayed values below ULN at the end of the study. After only three days of treatment the levels of the eosinophil markers had declined. This may indicate that the onset of the restoration to normal eosinophil activity begins soon after the initiation of treatment. Furthermore, the decline in eosinophil markers isaccompanied by clinical improvement and this could indicate an essential role for the eosinophils participating in the pathophysiology in CC.
In study II we demonstrated that the mucosal neutrophils are not activated in active CC. In spite of this, we found elevated levels of F-MPO and FC in more than half of the studied patients before treatment in study III. However, the level found in active CC was markedly lower than those in patients with active IBD in study I. Thus, there might be a low grade of neutrophil activity in active CC, but it should be noted that MPO and calprotectin are not unique to the neutrophil. These proteins are also released from monocytes and macrophages (150, 195) and increased numbers of macrophages have been found in the colonic mucosa in CC (196). It may

48
therefore be speculated that this cell population contributes to the enhanced levels of F-MPO and FC found in active CC. It is known that faecal markers of neutrophils increase in gastrointestinal infections and in inflammation caused by the use of NSAIDs (157, 197), but these conditions were ruled out in our patients included in study III.
Mucosal inflammation in collagenous colitis Eosinophil granulocytes Data on the mucosal inflammation in CC are limited. As mentioned, eosinophil infiltration in the colonic mucosa is considered to be a histological feature of CC. Increased luminal levels of eosinophil granular protein such as ECP in perfusion fluids from the colon (173) and increased degranulation of MBP in colonic tissues from patients with CC have been found (198). In study II we confirmed these data by means of histochemical staining for the eosinophil specific granule protein EPO in biopsy samples from the colon in patients with active CC. Furthermore, we found functionally activated colonic eosinophils in these patients by using flow cytometry. For the first time, we also demonstrated that the increased eosinophil activity (demonstrated by flow cytometry as increased expression of CD9 and a decreased percentage of CD44high
eosinophils) decreased to control levels after 8 weeks of budesonide treatment, with concomitant clinical improvement. In study III we found normalised levels of faecal eosinophil markers during treatment of patients with active CC and our findings in study II suggest that down-regulation of eosinophil activity may have contributed to this.
There are a number of reasons to belive that the eosinophil is involved in the pathogenesis of CC. The eosinophil is a multifunctional cell that is capable of generating a) epithelial cell injury (199), b) subepithelial collagen deposition (200-205), c) secretory diarrhoea (206-208), d) a type 1 cytokine profile (91) and e) chemokines that may attract T-cells to the mucosa (209) (Fig. 11). All these characteristics are found in CC.

49
Helminths
Cytok
Allergens
Neuro-mediators:Substance P
VIP
Chemokines:Chemokines:Eotaxin,RANTES,Mip-1α Cyto
IL-2, IL-3, ILIL-8, IL-12, IL
IFN-γ, T N
Viral infec�ons
ines
Tissue injury
Viral infec�ons
Granule proteins:ECP, EPO, MBP,
EDN/EPX
Lipid mediators:Lipid mediators:Platelet ac�va�ng factor,Leukotrienes
kines:L-4, IL-5, IL-6,L-18, GM-CSF,
NF- α, TGF-β
Figure 11. The multifunctional eosinophil granulocyte
Secretoneurin, a potent chemoatractant for blood eosinophils (119), has been shown to be a major peptide within the human enteric neuroendocrine system (118). In study IV we aimed to elucidate neuroendocrine proteins and found elevated levels of F-SN in patients with active CC, with a subsequent reduction during treatment. Elevated levels of SN have also been found in nasal lavage fluid from patients with seasonal allergic rhinitis (210) and in sputum from histamine challenged healthy humans (211). Just as in CC, eosinophil inflammation is a key feature in allergic rhinitis and asthma. In patients with asthma, the eosinophils and their granule proteins have been reported to cluster around airway nerves, suggesting a possible interaction with nerve endings in promoting disease symptom (212). Similarly to the airways, there is a close anatomical association between enteric neurones and inflammatory cells in the intestinal mucosa (113, 114), where the eosinophils are also positioned for interactions with nerve endings (213, 214). These observations, together with our findings of elevated F-SN levels in active CC, suggest a possible role of SN as an important participator in the pathophysiology of CC. Speculatively SN may promote the migration of eosinophils in the colonic mucosa. Considering these findings, CC may well be “the allergic rhinitis or asthma of the colon”.

50
T-cells The mucosal lymphocytes in CC mainly consist of CD8+ T-cells in the epithelium and CD4+ T-cells in the lamina propria (215). Bonderup et al reported a reduction of the lymphocytic infiltrate in budesonide treated patients (216, 217). Our findings in study II confirmed that budesonide treatment in CC reduces the mucosal lymphocyte infiltration. In addition, our data suggest that this reduction is most apparent for CD4+ T-cells. Similarly to the finding of a residual infiltration of eosinophils, there is a remaining infiltration of T-cells in CC. As shown by Munch et al, there is an increased mucosal uptake of E.coli12 in CC that persists after budesonide treatment, suggesting an underlying barrier dysfunction as an explanation (70). To the best of our knowledge, study II is the first systematic investigation of lymphocyte activity in the colonic mucosa of patients with CC. Surprisingly, we observed lower CD69 expression on both CD4+ and CD8+ T-cells in active CC compared to controls, indicating suppressed activation of CD4+ and CD8+ T-cells in active CC. The reason for this remains obscure, but this finding along with the absence of neutrophil activity implies that there is a smouldering rather than a highly active inflammation in the mucosa in active CC. During budesonide treatment, the expression of CD69 in CC patients increased to the levels found in controls; this was most obvious in CD4+ T-cells but was also detected in CD8+ T-cells. In an in vitro study of the immunosuppressive effect of budesonide on human lamina propria lymphocytes it was concluded that there is an inhibitory effect on proliferation together with decreased TNF-� secretion, suggesting a down-regulation of the activity (218). The latter is in contrast to our findings and it may be asked if there is a suppressing factor influencing T-cell activity in active CC which is altered by budesonide treatment.
Markers of neuroendocrine activity in CC and IBD It is suggested that interactions between the enteric nervous system and the immune system may play an important role in the pathophysiology of IBD (109-112). Close anatomical associations in the gut wall between the terminal axons of enteric neurones, EEC cells and inflammatory cells facilitate the neuroimmune communication (113, 114). In the normal intestinal mucosa EEC cells are scattered amongst the epithelial cells. In response to a stimulus they release proteins and peptides, which may act locally (on nerve endings, on epithelial cells and on immune cells) or act at remote sites by entering the circulation (131). Moreover, animal studies suggest that CgA, a general marker of EEC cells, and serotonin could be released into the intestinal lumen (132, 133).

51
For the first time we have demonstrated higher faecal levels of CgA and CgB in patients with active CC than in patients with active IBD and healthy controls (study IV). Furthermore, patients with UC expressed elevated levels of F-CgA compared to healthy controls. No obvious reduction was noted in the levels of F-CgA or F-CgB during treatment in any of the studied groups. There are no published data on CgA levels in patients with CC, but increased levels of circulating CgA have been reported in patients with UC (121-123). These findings together with our data on elevated F-CgA and F-CgB levels may indicate an up-regulation of the intestinal EEC system in patients with CC or UC. In studies regarding CgA positive cells in the colon in patients with UC, different results have been presented. Some studies have shown unchanged numbers of CgA positive cells in both active and quiescent disease (123, 219), while others have demonstrated an increase in CgA positive cells (220). Moreover, hyperplasia of colonic neuroendocrine cells has been observed in patients with UC (219, 221-224).
Intact and processed forms of CgA have been found in neutrophils (124, 125), and neutrophil recruitment and activation are dominant features of UC (225, 226). In study I we demonstrated high faecal levels of the neutrophil markers MPO and calprotectin in patients with active IBD. However, no correlation between the F-CgA level and F-MPO or FC could be found at any time (data not shown). In study II we concluded that there was no enhancement of neutrophil activity in patients with active CC. Taken together, these observations indicate that the neutrophil is not the source of the F-CgA levels found in IBD and CC.
There are no available data concerning CgB in patients with CC or IBD. In most neuroendocrine cells CgB co-exists with CgA and could be measured as a complement to CgA assays (128-130). In line with this we found a correlation between F-CgA and F-CgB before and during treatment in study IV.
Patients with IBD expressed markedly lower amounts of F-SN compared to patients with CC and healthy controls, and during the study the levels were unchanged. In IBD neural involvement is evident from colonic nerve damage, extensive axonal degeneration and infiltration of the myenteric plexus with inflammatory cells, and this may explain the low levels of F-SN found in IBD (109).

52
Clinical implications and future perspectives
It has been claimed that faecal calprotectin has good diagnostic precision for separating organic and functional intestinal disease but that it is not specific for IBD. Thus, a high concentration of FC in patients without a known gastrointestinal disease provides a strong reason for carrying out a colonoscopy. We have demonstrated that in patients treated for a relapse of IBD, a normalised level of FC, F-MPO or F-EPX could be used as a marker for a successful treatment outcome. However, the significance of persistent elevation of FC, F-MPO or F-EPX in patients with IBD needs further evaluation. More studies are needed to elucidate the capacity of the faecal markers to predict IBD relapse. The mucosal inflammation in active CC is characterised by activated eosinophils, but there is no neutrophil activity. Hence, it would be preferable to use eosinophil markers of inflammation to detect patients with CC. Increased F-ECP could be used as a marker for active disease in CC, and normalised levels of F-ECP and F-EPX could indicate a successful treatment outcome. However, further longitudinal studies are needed to confirm the utility of F-ECP and F-EPX in the clinical setting concerning patients with CC. In patients investigated for gastrointestinal symptoms such as diarrhoea or abdominal cramping it would be desirable to collect faecal samples for analyse not only of FC but also of ECP and/or EPX. In summary, together with good clinical judgement a panel of faecal markers of inflammation, including both markers of eosinophils and neutrophils, could be of value when diagnosing patients with gastrointestinal symptoms, and elevated levels should be a strong reason for performing a colonoscopy.
In future studies of CC it would be of interest to further classify the subgroups of T-cells involved in the mucosal inflammation in CC and to look more closely at the mast cell, which may also be implicated. In addition, it would be of great importance to study the involvement of the neuro-endocrine system in the pathogenesis of CC.

53
Conclusions
A normalised FC level has the potential to be used as a surrogate marker for a successful treatment outcome in patients with UC and CD. However, patients with persistently increased FC levels need further evaluation. FC and F-MPO provide better discrimination than F-EPX in predicting the treatment outcome in IBD.
The mucosal inflammation in CC is characterised by activated eosinophils, but there is no neutrophil activity. There are increased numbers of CD4+ and CD8+ T cells in active CC, but they express a lower grade of activity than in control subjects. Budesonide treatment restores the normal activation of eosinophils and T-cells, at the same time leading to clinical remission. These results suggest that the eosinophil has a central role in the pathophysiology of CC.
Increased F-ECP may be used as a surrogate marker for active CC. Faecal ECP and F-EPX allows a better assessment of the treatment outcome than F-MPO and FC. Activation of mucosal eosinophil granulocytes and the subsequent increase in the faecal excretion of ECP, and to a lesser extent EPX, are part of the inflammatory processes in patients with active CC. During budesonide treatment there is a rapid decrease in F-ECP and F-EPX, accompanied by clinical improvement, and this may indicate an essential role for the eosinophil in the pathophysiology of CC.
Chromogranin A, CgB and SN are detectable in faeces from patients with CC, UC and CD and from healthy controls. Patients with active CC display markedly elevated levels of F-CgA, F-CgB and F-SN compared to healthy controls and patients with UC or CD. Patients with UC have higher levels of F-CgA than healthy controls. During treatment no appreciable change was observed in the level of F-CgA or F-CgB in any of the studied groups and only in patients with CC was a rapid decrease of F-SN noted. The markedly high levels of F-CgA, F-CgB and F-SN found in collagenous colitis indicate a role for the intestinal neuro-endocrine system in the pathogenesis of this disease.

54
Svensk sammanfattning
Introduktion Studierna i avhandlingsarbetet, som är utförda på patienter med inflammatorisk tarmsjukdom och kollagen kolit (KK), med särskild tonvikt på KK, är gjorda ur både klinisk och experimentell synvinkel. Det övergripande syftet var att studera inflammatoriska processer i tarmslemhinnan och om dessa processer avspeglar sig i halten av specifika proteiner i avföringen, utsöndrade av celler i tarmslemhinnan. Celler som vi har undersökt tillhör de vita blodkropparna: eosinofila och neutrofila granulocyter samt T-celler. Vi har analyserat eosinofil cationic protein (F-ECP) och eosinofil protein X (F-EPX) som markör för den eosinofila granulocyten, och myeloperoxidas (F-MPO) och calprotektin (FC) som netrofil markörer. Därtill har vi analyserat förstadier till hormoner (chromogranin A (CgA) och chromogranin B (CgB)) och en nervsignalsubstans (secretoneurin (SN)), som utsöndras från endokrina (hormonproducerande) celler i tarmen respektive celler i tarmkanalens nervsystem.
Bakgrund Begreppet inflammatorisk tarmsjukdom (IBD) omfattar sjukdomarna ulcerös kolit (UC) och Crohn´s sjukdom (CD). De är kroniska inflammatoriska sjukdomar i magtarmkanalen vars orsak är okänd. En förklaringsmodell som ofta anges är att ett överaktivt immunförsvar reagerar på någonting i tarminnehållet t.ex normalt förekommande tarmbakterier, och att omgivningsfaktorer så som infektioner kan utlösa reaktionerna. Dessutom är genetiska faktorer av betydelse. IBD förlöper i skov som karaktäriseras av diarréer, ofta blodiga, ibland också magknip och buksmärtor. Sjukdomarna är vanliga, i Sverige drabbas ca 15 per 100 000 individer per år av UC och ca 6 – 7 per 100 000 individer per år insjuknar i CD. Alla åldrar drabbas men vanligaste insjuknandeåldern är mellan 15 och 40 år, kvinnor och män drabbas lika. Ulcerös kolit är lokaliserad till tjocktarmen, vanligen engagerar den ändtarmen och sträcker sig på ett kontinuerligt sätt upp i tjocktarmen med varierande utbredning från fall till fall. Crohn´s sjukdom kan lokaliseras till alla delar av magtarmkanalen, från mun till ändtarm, men oftast sitter den

55
i tunntarmen eller tjocktarmen eller i båda samtidigt. Symptomen vid CD kan likna dem vid UC men sjukdomen har många ”ansikten” och symptomen är beroende av var i magtarmkanalen sjukdomen är lokaliserad. Dessutom kan det bildas besvärande fistlar (vanligen kring ändtarmen), varansamlingar i buken och förträngningar i tarmen.
Diagnosen vid IBD ställs med hjälp av den kliniska presentationen tillsammans med endoskopisk undersökning (tarmkikarundersökning) av tarmen, som ser svullen och sårig ut, kompletterat med mikroskopisk värdering av vävnadsprover från tarmslemhinnan. Behandlingen, som i första hand är medicinsk, syftar till att få inflammationen att läka ut (framkalla remmission) och att förhindra återfall. Om inte medicinsk behandling har effekt eller om komplikationer utvecklas kan kirurgisk behandling hjälpa.
Kollagen kolit tillhör gruppen mikroskopiska koliter och beskrevs första gången 1976 av den svenska patologen Clas Lindström. Sjukdomen, som förlöper i skov, karaktäriseras av frekventa vattentunna, oblodiga diarree´r, ibland upp till 15 – 20 per dygn, stundtals åtföljt av buksmärtor, uppspändhet och viktnedgång. Kollagen kolit är lokaliserad till tjocktarmen och drabbar framförallt kvinnor i medelåldern, med en topp på 25 per 100 000 individer per år vid 65 års ålder. Orsaken till sjukdomen är okänd. Diagnostiken baseras på den kliniska presentationen tillsammans med endoskopisk undersökning där provtagning från slemhinnan är nödvändig för att säkerställa diagnosen. Makroskopiskt ser tarmslemhinnan frisk ut, men vid mikroskopisk granskning av vävnadsprov från tarmslemhinnan framträder karaktäristiska förändringar, som utgörs av ett förtjockat bindvävslager och ett ökat antal vita blodkroppar. Sjukdomen har oftast ett ofarligt förlopp men orsakar stort lidande eftersom de frekventa tarmtömningarna hos den drabbade kan vara invalidiserande. Behandlingen är i lindriga fall symptomatisk med stoppande mediciner men den bäst dokumenterade behandlingen utgörs idag av budesonid, ett kortisonpreparat i kapselberedning som har sin största effekt lokalt i tarmen. Återfall är dock vanligt när behandlingen trappas ner eller avslutas.
Studie I Sjukhistoria och klinisk undersökning i kombination med udersöknigar med tarmkikare (endoskopi) utgör hörnstenarna i att utvärdera behandling vid IBD. Tarmkikarundersökning av tjocktarmen (koloskopi) är besvärliga för patienterna och dessutom tidskrävande och kostsamma. Syftet med studien var att undersöka om fekalt calprotektin (FC) kan användas för värdering av behandlingsutfallet vid skov av UC och CD. Dessutom jämfördes FC med F-MPO och F-EPX. Totalt 38 patienter med tidigare diagnostiserad IBD (27

56
med UC respektive 11 med CD) studerades före och efter fyra respektive åtta veckors behandling. Tjugoen av de 27 UC patienterna och 10 av 11 CD patienter hade ett komplett behandlingssvar (normal avföring och normal eller nästan normal tarmkikarundersökning). Resultaten visar att förhöjda FC värden kunde verifiera skov av sjukdomen. Normaliserade nivåer av FC och/eller F-MPO efter åtta veckors behandling i UC gruppen indikerade komplett behandlingssvar, dessa patienter torde kunna besparas ny endoskopisk undersökning. Normaliserade nivåer av FC och/eller F-MPO respektive F-EPX efter åtta veckors behandling i CD gruppen indikerade komplett behandlingssvar. Däremot var kvarstående förhöjda nivåer av FC, F-MPO respektive F-EPX inte liktydigt med uteblivet behandlingssvar hos vare sig UC eller CD, utan bör värderas närmare.
Studie II Sjukdomsmekanismerna vid KK är okänd. Tidigare studier på tarmsköljvätska från tjocktarmen hos patienter med KK har visat en ökad halt av ECP tydande på ökad aktivitet av eosinofila granulocyter. Syftet var att på cellnivå undersöka eosinofilers, neutrofilers och T-cellers fördelning och aktivitet i tarmslemhinnan hos patienter med aktiv KK före och efter åtta veckors budesonidbehandling. Elva patienter med aktiv KK genomgick tarmkikarundersökning av tjocktarmen där vävnadsprov togs från sju olika stationer före och efter behandling. Vävnadsproven analyserades med flödescytometri, en metod som kan klassificera och sortera celler efter sort och aktivitet med hjälp av specifika antikroppar. Efter åtta veckors behandling hade 10 patienter ett komplett behandlingssvar (� tre lösa avföringar per dygn) och hos en minskade antalet diarréer från 17 till 8. Resultaten visar att vid aktiv KK karaktäriseras inflammationen i tarmslemhinnan av en ökad eosinofil aktivitet, men det finns ingen neutrofil aktivering. Antalet T-celler är ökade vid aktiv KK, men aktiviteten är lägre än hos kontroller. Budesonidbehandling återställer aktiviteten hos eosinofiler och T-celler, åtföljt av klinisk förbättring. Fynden talar för att eosinofil aktivitet har betydelse för sjukdomsmekanismerna vid KK
Studie III Fekalt calprotectin kan användas som markör för intestinal inflammation vid UC och CD men för KK har man inte påvisat någon tillförlitlig markör. I studie II påvisade vi en ökad eosinofil aktivitet i tarmslemhinnan vid aktiv KK. Syftet i studie III var därför att undersöka om eosinofilmarkörerna F-ECP och F-EPX kan användas som markör för sjukdomsaktivitet och behandlingsutfall vid KK i jämförelse med FC och F-MPO. Tolv patienter

57
med aktiv KK studerades före och efter 3, 7, 28 och 56 dagars behandling med budesonid. Elva av det tolv studerade patienterna hade ett komplett behandlingssvar (� tre lösa avföringar per dygn).
Resultaten visar att F-ECP hade bästa förmågan att påvisa aktiv KK. Normaliserade nivåer av F-ECP och F-EPX efter 56 dagars behandling indikerade ett komplett behandlingssvar. Vid budesonidbehandling av KK sjunker nivåerna av F-ECP och F-EPX tidigt efter insatt behandling, åtföljt av klinisk förbättring. Fynden talar för att eosinofilen spelar en viktig roll vid sjukdomsmekanismerna vid KK.
Studie IV Interaktioner mellan nervceller, hormonproducerande celler och celler i immunsystemet i tarmkanalen har föreslagits spela en viktig roll när det gäller sjukdomsmekanismer vid IBD.
Syftet var att undersöka om CgA, CgB och SN kan analyseras i feces från patienter med aktiv KK och jämföra resultaten från patienter med UC, CD och med friska kontroller. Vi ville också undersöka om det var någon förändring av fekalt CgA (F-CgA), F-CgB och F-SN under behandling av dessa patienter. Tolv patienter med KK undersöktes före och efter 3, 7, 28 och 56 dagars behandling. Tjugosju patienter med UC och elva med CD undersöktes före och efter 28 och 56 dagars behandling. Kliniska data, avföringsfrekvens och konsistens registrerades och avföringsprover togs vid varje tillfälle.
Resultaten visar att det finns mätbara nivåer av CgA, CgB och SN i feces. Patienter med aktiv KK uppvisar markant högre nivåer av F-CgB, F-CgA och F-SN jämfört med patienter med UC eller CD, liksom med friska kontroller. Vid behandling av KK sjunker F-SN nivåerna snabbt men ingen förändring noterades vid behandling av patienter med UC eller CD. Patienter med UC hade förhöjda F-CgA jämfört med kontroller. Ingen större förändring observerades för nivåerna av F-CgA eller F-CgB vid behandling i någon av de undersökta grupperna. De markant förhöjda värdena av F-CgA, F-CgB och F-SN vid KK indikerar att interaktioner mellan nervceller, hormonproducerande celler och celler i immunsystemet i tarmen kan ha betydelse vad gäller sjukdomsmekanismerna vid kollagen kolit.

58
Slutsatser och egna kommentarer Det har i tidigare studier visats att fekalt calprotectin kan vara användbart för att skilja sjukdomar i magtarmkanalen med organisk bakomliggande orsak från sjukdomar med funktionell orsak (irritabel tarm, IBS), men FC är inte specifikt för IBD. Förhöjda värden av FC är därför ett starkt skäl att utföra en tarmkikarundersökning för vidare bedömning. Vi har visat, att hos patienter som behandlas för ett skov av IBD kan normaliserade värden av FC, F-MPO och F-EPX användas för att bekräfta ett lyckat behandlings-resultat. Kvarstående förhöjda värden av FC, F-MPO och F-EPX hos patienter med IBD kräver dock närmare utvärdering. Vidare studier, som också sträcker sig över längre tid, krävs för att studera dugligheten av fekala inflammationsmarkörer vid behandling och utvärdering av patienter med IBD.
Vid aktiv KK karaktäriseras inflammationen i tarmslemhinnan av en ökad eosinofil aktivitet men det finns ingen neutrofil aktivitet. Därför kan det vara fördelaktigt att använda eosinofila markörer för att upptäcka patienter med KK. Förhöjda värden av F-ECP är användbara som markör för aktiv KK, och normaliserade värden av F-ECP och F-EPX indikerar ett lyckat behandlingsresultat. Ytterligare studier är dock nödvändiga för att bekräfta användbarheten av F-ECP och F-EPX i klinisk praxis när det gäller patienter med KK.
Sammantaget; en god klinisk bedömning tillsammans med en panel av fekala inflammationsmarkörer, inkluderande både eosinofila och neutrofila markörer, kan vara värdefulla vid utredning av patienter med diarré och/eller återkommande buksmärtor/magknip. Förhöjda markörnivåer utgör ett starkt skäl att gå vidare med utredningen och i första hand utföra en tarmkikarundersökning.
De markant förhöjda värdena av F-CgA, F-CgB och F-SN påvisade vid KK är mycket intressanta fynd, som talar för att interaktioner mellan nervceller, hormonproducerande celler och celler i immunsystemet i tarmkanalen spelar en viktig roll när det gäller sjukdomsmekanismer vid KK. Det är angeläget att i framtida studier undersöka dessa interaktioner närmare och även att kartlägga vilka undergrupper av T-celler och andra typer av vita blodkroppar som är involverade i sjukdomsmekanismerna och inflammationen vid kollagen kolit.

59
Tack! (Acknowledgements)
Det är många som på olika sätt har bidragit till att genomföra detta avhandlingsarbete, vissa med kunskap i ämnet och andra med glada tillrop och omtanke när det varit motlut på färden. Tack till alla er alla! Det är dock några som jag speciellt vill nämna:
Marie Carlson, min huvudhandledare och kollega, tänd på eosinofiler, och fäst vid sina pennor, (ssk den röda som kan sätta , och .) har sakta men säkert fått även mig att förstå att eosinofiler minsann inte går av för hackor; nej! de går av för många andra faktorer (fig 11). Med din stora kunskap, entusiasm, fantasi, noggrannhet, tillgänglighet, generositet, uppmuntran, och rimliga krav har du drivit mig framåt genom det här arbetet så att jag slutligen kunnat gå i mål.
Mia Lampinen, bihandledare, biomedicinare och också eosinofil-fan, för att du med hjälp av hi-tech maskiner (FACS), men också med enkla mikroskop, tagit mig med ner på cellnivå och visat vad man kan hitta down under och hur man hittar det. För allt arbete på lab och för att du guidat mig genom submissionlabyrinten, rätat ut engelskan, gjort mig skeptisk till korrelationer, alltid lämnat användbara svar på mobil och mail (tom från Argentina) så att jag kommit vidare i arbetet.
Pelle Sangfelt, bihandledare, fd sektionschef, kollega, karriärcoach och trofast vän som vill att allt ska gå ärligt och rätt till här i livet. Tur att jag har starka nerver för det var stark läsning när peken reviderats hos dig men du har ju ofta (alltid?) rätt. Du har gett mig mantrat som skapat ordning, bollat frågor och med fria associationer med din omfångsrika kunskap, både klinikiskt och vetenskapligt, bidragit till att föra mig framåt på många plan. För deltagande i sorger och alla skratten och uppmuntrande mail med mer eller mindre vetenskapliga attachments.
Ingrid Stolt, medförfattare, och galant laborant, för erfaren, skicklig och noggrann provhantering och laborering och för att du visat mig hur det skall gå till för att det skall bli perfekt

60
Christer GB Peterson, medförfattare och avföringsmarkörmetodmakare, för kloka synpunkter och omvandlade och ovandlade värden i diverse tabeller med mikro, pico, och nanonivåer.
Margret Agnarsdottir, medförfattare, för rask och noggrann gradering av den mikroskopiska inflammationen. Alkwin Wanders för hjälp med foto av EPO-färgade snitt i studie II, varav ett pryder omslaget omslaget).
Mats Stridsberg, medförfattare, för att ha introducerat mig för familjen Granin.
Lena, Ulla, Ulrika, Mona och Ann-Chatrin på skopi-mottagningen för att med entusiasm, glatt humör och god vilja alltid klämma in mina forskningsskopier på xtratider och med fast handlag och glada tillrop sett till så att vi kom hela vägen runt och dessutom hållit ordning på biopsierna. Inger, Katarina och Gunilla på mottagningen för att ni med ert goda humör hållit mina pat. på gott humör och sett till att de blivit omhändertagna under tiden jag har varit avhållsam med det kliniska arbetet. Inger, för att du fångade upp patienter till studie I. Cattha, Helena och Lasse och all övrig personal på avd 30C för att ni fortfarande känner igen mig trots att det varit långt mellan besöken.
Mari Thörn, kollega, nyligen tillträdd sektionschef och fd schemaläggare, för att du skapat utrymme för min forskning med dina schemaläggartalanger, och för att du ansträngt dig för att få det hela att gå ihop. Mina Kollegor på gastrosektionen Fredrik Rorsman, Anders Rönnblom, Fredrik Eriksson Linder, Lars Åkerberg, Ronald Malcher, för att frustande ha tagit i lite extra med det redan innan tunga kliniska arbetet utan att gruffa, så att jag har kunnat ägna mig åt denna bok. Professor Per Hellström för att du tog tag i BUC-studien som jag intet själv mäktat med under denna tid.
Bernice Wiberg och Maria Liden för tips och uppmuntran i hela den här komplicerade proceduren kring själva boken. Erik Torell, partisan i antifeodalstyrkan och operasångare, för att då och då plötsligt dyka upp för att kolla läget och Marie Raiend för ssk omtanke om min familj.
Maud Marsden, for your excellent British English, som har lyft språket i denna avhandling till hög höjd och för att du läst den med sådan inlevelse trots att tiden var knapp.
Mina rumskamrater Jarl Hellman (JH) och Per Anton Westerberg (PA); för att ni inte alltid har varit alldeles för högljudda utan har gett mig arbetsro för att slutföra mitt avhandlingsarbete; för möjlighet till omedelbar debreefing när något gått åt skogen och för att ni har delat stort och smått bakom lykta

61
dörrar; till JH för hjälp med PP, PCn. och ovärderlig information om universitetsvärlden; till PA för differentialdiagnostiska övervägande vad gäller armen och för att vara en pålitlig maltwhiskeyleverantör.
Personal och kollegor på klinisk kemis forskningsavdelning för vänligt bemötande och kaffe och kaka på fredagar inte minst. Inger Olsson för analyser av Graniner. Mikael Åberg som på något magiskt sätt blixtsnabbt förvandlade en fig. från ett format till något annat så att submission för artikel II kunde fullföljas. Malgorzata Karawajczyk för den fina elektromikroskopibilden (Fig.2)
Patienter och kontrollpersoner för att ni har ställt upp och följt studieprotokollen, trots de besvär som detta inneburit.
Inst för medicinska vetenskaper under ledning av Prefekt Eva Tiensuu Jansson för att jag har fått möjlighet att göra min avhandling vid inst.
Följande Fonder och Stiftelser Vetenskapsrådet – Medicin. FoU-medel ALF, lokala och centrala, vid Akademiska sjukhuset, Uppsala. Lisa & Johan Grönbergs Stiftelse. Ruth and Richard Juhlin´s Foundation Research Grant (Karolinska Institutets Stiftelser) Petrus och Augusta Hedlunds Stiftelse.
Erling och Lise, mina framlidna föräldrar, för att ni gav mig liv, trygghet, glädje och möjligheter och många, många goda minnen.
Edda Ferm för att du är en sådan fantastisk Mormor och raskt ställer upp när vi kallar
Mina barn Lisa, David, Linn och Linus, det bästa jag någonsin varit med om att åstadkomma, för att ni ger livet mening och innehåll och gör mig till en otroligt stolt far.
Sist men mest, Helene, min älskade och älskande hustru, livskamrat, kollega och sparringpartner, för kärlek, omtanke, utrymme och tålamod och för att du står ut med min oförmåga till att planera och ha framförhållning; för att du låter mig komma först i mål på våra joggingrundor i skogen, det håller liksom stämningen uppe. Snart är vi där igen
Återigen, Stort Tack till er alla!

62
References
1. Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflammatory bowel diseases 2006;12 Suppl 1:S3-9.
2. Lindstrom CG. 'Collagenous colitis' with watery diarrhoea--a new entity? Pathologia Europaea 1976;11:87-9.
3. Flejou JF, Bogomoletz WV. [Microscopic colitis: collagenous colitis and lymphocytic colitis. A single concept?]. Gastroenterologie clinique et biologique 1993;17:T28-32.
4. Levison DA, Lazenby AJ, Yardley JH. Microscopic colitis cases revisited. Gastroenterology 1993;105:1594-6.
5. Tysk C, Bohr J, Nyhlin N, et al. Diagnosis and management of microscopic colitis. World J Gastroenterol 2008;14:7280-8.
6. Nyhlin N, Bohr J, Eriksson S, et al. Systematic review: microscopic colitis. Alimentary pharmacology & therapeutics 2006;23:1525-34.
7. Pokorny CS, Kneale KL, Henderson CJ. Progression of collagenous colitis to ulcerative colitis. Journal of clinical gastroenterology 2001;32:435-8.
8. Aqel B, Bishop M, Krishna M, et al. Collagenous colitis evolving into ulcerative colitis: a case report and review of the literature. Digestive diseases and sciences 2003;48:2323-7.
9. Satsangi J, Silverberg MS, Vermeire S, et al. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut 2006;55:749-53.
10. Langholz E, Munkholm P, Davidsen M, et al. Changes in extent of ulcerative colitis: a study on the course and prognostic factors. Scandinavian journal of gastroenterology 1996;31:260-6.
11. Kornbluth A, Sachar DB. Ulcerative colitis practice guidelines in adults (update): American College of Gastroenterology, Practice Parameters Committee. The American journal of gastroenterology 2004;99:1371-85.
12. Rothfuss KS, Stange EF, Herrlinger KR. Extraintestinal manifestations and complications in inflammatory bowel diseases. World J Gastroenterol 2006;12:4819-31.
13. Garland CF, Lilienfeld AM, Mendeloff AI, et al. Incidence rates of ulcerative colitis and Crohn's disease in fifteen areas of the United States. Gastroenterology 1981;81:1115-24.
14. Tanaka M, Riddell RH, Saito H, et al. Morphologic criteria applicable to biopsy specimens for effective distinction of inflammatory bowel disease from other forms of colitis and of Crohn's disease from ulcerative colitis. Scandinavian journal of gastroenterology 1999;34:55-67.
15. Loftus EV, Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004;126:1504-17.

63
16. Russell RK, Satsangi J. IBD: a family affair. Best Pract Res Clin Gastroenterol 2004;18:525-39.
17. Van Limbergen J, Russell RK, Nimmo ER, et al. The genetics of inflammatory bowel disease. The American journal of gastroenterology 2007;102:2820-31.
18. Halfvarson J, Bresso F, D'Amato M, et al. CARD15/NOD2 polymorphisms do not explain concordance of Crohn's disease in Swedish monozygotic twins. Dig Liver Dis 2005;37:768-72.
19. Vind I, Jespersgaard C, Hougs L, et al. Genetic and environmental factors in monozygotic twins with Crohn's disease and their first-degree relatives: a case report. Digestion 2005;71:262-5.
20. Spehlmann ME, Begun AZ, Burghardt J, et al. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflammatory bowel diseases 2008;14:968-76.
21. Boyko EJ, Koepsell TD, Perera DR, et al. Risk of ulcerative colitis among former and current cigarette smokers. The New England journal of medicine 1987;316:707-10.
22. Beaugerie L, Massot N, Carbonnel F, et al. Impact of cessation of smoking on the course of ulcerative colitis. The American journal of gastroenterology 2001;96:2113-6.
23. Andersson RE, Olaison G, Tysk C, et al. Appendectomy and protection against ulcerative colitis. The New England journal of medicine 2001;344:808-14.
24. Koutroubakis IE, Vlachonikolis IG, Kouroumalis EA. Role of appendicitis and appendectomy in the pathogenesis of ulcerative colitis: a critical review. Inflammatory bowel diseases 2002;8:277-86.
25. Hertervig E, Befrits R, Ekbom A, et al. [Colitis cancer--myth or reality?]. Lakartidningen 2009;106:3000-2.
26. Gasche C, Scholmerich J, Brynskov J, et al. A simple classification of Crohn's disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflammatory bowel diseases 2000;6:8-15.
27. Lapidus A. Crohn's disease in Stockholm County during 1990-2001: an epidemiological update. World J Gastroenterol 2006;12:75-81.
28. Louis E, Collard A, Oger AF, et al. Behaviour of Crohn's disease according to the Vienna classification: changing pattern over the course of the disease. Gut 2001;49:777-82.
29. Carter MJ, Lobo AJ, Travis SP. Guidelines for the management of inflammatory bowel disease in adults. Gut 2004;53 Suppl 5:V1-16.
30. Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Digestive diseases and sciences 1989;34:1841-54.
31. Sutherland LR, Ramcharan S, Bryant H, et al. Effect of cigarette smoking on recurrence of Crohn's disease. Gastroenterology 1990;98:1123-8.
32. Cosnes J, Carbonnel F, Carrat F, et al. Effects of current and former cigarette smoking on the clinical course of Crohn's disease. Alimentary pharmacology & therapeutics 1999;13:1403-11.
33. Picco MF, Bayless TM. Tobacco consumption and disease duration are associated with fistulizing and stricturing behaviors in the first 8 years of Crohn's disease. The American journal of gastroenterology 2003;98:363-8.
34. Cosnes J, Beaugerie L, Carbonnel F, et al. Smoking cessation and the course of Crohn's disease: an intervention study. Gastroenterology 2001;120:1093-9.

64
35. Andersson RE, Olaison G, Tysk C, et al. Appendectomy is followed by increased risk of Crohn's disease. Gastroenterology 2003;124:40-6.
36. Radford-Smith GL, Edwards JE, Purdie DM, et al. Protective role of appendicectomy on onset and severity of ulcerative colitis and Crohn's disease. Gut 2002;51:808-13.
37. Bohr J, Tysk C, Eriksson S, et al. Collagenous colitis: a retrospective study of clinical presentation and treatment in 163 patients. Gut 1996;39:846-51.
38. Olesen M, Eriksson S, Bohr J, et al. Microscopic colitis: a common diarrhoeal disease. An epidemiological study in Orebro, Sweden, 1993-1998. Gut 2004;53:346-50.
39. Madisch A, Heymer P, Voss C, et al. Oral budesonide therapy improves quality of life in patients with collagenous colitis. International journal of colorectal disease 2005;20:312-6.
40. Kingham JG, Levison DA, Morson BC, et al. Collagenous colitis. Gut 1986;27:570-7.
41. Warren BF, Edwards CM, Travis SP. 'Microscopic colitis': classification and terminology. Histopathology 2002;40:374-6.
42. Ayata G, Ithamukkala S, Sapp H, et al. Prevalence and significance of inflammatory bowel disease-like morphologic features in collagenous and lymphocytic colitis. The American journal of surgical pathology 2002;26:1414-23.
43. Tanaka M, Mazzoleni G, Riddell RH. Distribution of collagenous colitis: utility of flexible sigmoidoscopy. Gut 1992;33:65-70.
44. Pardi DS, Loftus EV, Jr., Smyrk TC, et al. The epidemiology of microscopic colitis: a population based study in Olmsted County, Minnesota. Gut 2007;56:504-8.
45. Travis SP, Stange EF, Lemann M, et al. European evidence based consensus on the diagnosis and management of Crohn's disease: current management. Gut 2006;55 Suppl 1:i16-35.
46. Berg DF, Bahadursingh AM, Kaminski DL, et al. Acute surgical emergencies in inflammatory bowel disease. American journal of surgery 2002;184:45-51.
47. Larson DW, Pemberton JH. Current concepts and controversies in surgery for IBD. Gastroenterology 2004;126:1611-9.
48. Itzkowitz SH, Present DH. Consensus conference: Colorectal cancer screening and surveillance in inflammatory bowel disease. Inflammatory bowel diseases 2005;11:314-21.
49. Chande N, MacDonald JK, McDonald JW. Interventions for treating microscopic colitis: a Cochrane Inflammatory Bowel Disease and Functional Bowel Disorders Review Group systematic review of randomized trials. The American journal of gastroenterology 2009;104:235-41; quiz 4, 42.
50. Miehlke S, Madisch A, Voss C, et al. Long-term follow-up of collagenous colitis after induction of clinical remission with budesonide. Alimentary pharmacology & therapeutics 2005;22:1115-9.
51. Pardi DS, Loftus EV, Jr., Tremaine WJ, et al. Treatment of refractory microscopic colitis with azathioprine and 6-mercaptopurine. Gastroenterology 2001;120:1483-4.
52. Riddell J, Hillman L, Chiragakis L, et al. Collagenous colitis: oral low-dose methotrexate for patients with difficult symptoms: long-term outcomes. Journal of gastroenterology and hepatology 2007;22:1589-93.

65
53. Jarnerot G, Tysk C, Bohr J, et al. Collagenous colitis and fecal stream diversion. Gastroenterology 1995;109:449-55.
54. Coombes JL, Robinson NJ, Maloy KJ, et al. Regulatory T cells and intestinal homeostasis. Immunological reviews 2005;204:184-94.
55. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet 2007;369:1627-40.
56. Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001;411:599-603.
57. Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001;411:603-6.
58. Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nature clinical practice 2006;3:390-407.
59. Bohr J, Tysk C, Yang P, et al. Autoantibodies and immunoglobulins in collagenous colitis. Gut 1996;39:73-6.
60. Freeman HJ. Perinuclear antineutrophil cytoplasmic antibodies in collagenous or lymphocytic colitis with or without celiac disease. Canadian journal of gastroenterology = Journal canadien de gastroenterologie 1997;11:417-20.
61. Ung KA, Gillberg R, Kilander A, et al. Role of bile acids and bile acid binding agents in patients with collagenous colitis. Gut 2000;46:170-5.
62. Beaugerie L, Pardi DS. Review article: drug-induced microscopic colitis - proposal for a scoring system and review of the literature. Alimentary pharmacology & therapeutics 2005;22:277-84.
63. Makinen M, Niemela S, Lehtola J, et al. Collagenous colitis and Yersinia enterocolitica infection. Digestive diseases and sciences 1998;43:1341-6.
64. Bohr J, Nordfelth R, Jarnerot G, et al. Yersinia species in collagenous colitis: a serologic study. Scandinavian journal of gastroenterology 2002;37:711-4.
65. Erim T, Alazmi WM, O'Loughlin CJ, et al. Collagenous colitis associated with Clostridium difficile: a cause effect? Digestive diseases and sciences 2003;48:1374-5.
66. Lundberg JO, Herulf M, Olesen M, et al. Increased nitric oxide production in collagenous and lymphocytic colitis. European journal of clinical investigation 1997;27:869-71.
67. Perner A, Andresen L, Normark M, et al. Expression of nitric oxide synthases and effects of L-arginine and L-NMMA on nitric oxide production and fluid transport in collagenous colitis. Gut 2001;49:387-94.
68. Perner A, Nordgaard I, Matzen P, et al. Colonic production of nitric oxide gas in ulcerative colitis, collagenous colitis and uninflamed bowel. Scandinavian journal of gastroenterology 2002;37:183-8.
69. Olesen M, Middelveld R, Bohr J, et al. Luminal nitric oxide and epithelial expression of inducible and endothelial nitric oxide synthase in collagenous and lymphocytic colitis. Scandinavian journal of gastroenterology 2003;38:66-72.
70. Munch A, Soderholm JD, Ost A, et al. Increased transmucosal uptake of E. coli K12 in collagenous colitis persists after budesonide treatment. The American journal of gastroenterology 2009;104:679-85.
71. van Tilburg AJ, Lam HG, Seldenrijk CA, et al. Familial occurrence of collagenous colitis. A report of two families. Journal of clinical gastroenterology 1990;12:279-85.

66
72. Abdo AA, Zetler PJ, Halparin LS. Familial microscopic colitis. Canadian journal of gastroenterology = Journal canadien de gastroenterologie 2001;15:341-3.
73. Jarnerot G, Hertervig E, Granno C, et al. Familial occurrence of microscopic colitis: a report on five families. Scandinavian journal of gastroenterology 2001;36:959-62.
74. Fine KD, Do K, Schulte K, et al. High prevalence of celiac sprue-like HLA-DQ genes and enteropathy in patients with the microscopic colitis syndrome. The American journal of gastroenterology 2000;95:1974-82.
75. Koskela RM, Karttunen TJ, Niemela SE, et al. Human leucocyte antigen and TNFalpha polymorphism association in microscopic colitis. European journal of gastroenterology & hepatology 2008;20:276-82.
76. Madisch A MSea. Matrix metalloproteinase-9 gene polymorphism is associated with collagenous colitis. Gut 2006; Supplement nov Vol 55:A113.
77. Madisch A, Hellmig S, Schreiber S, et al. NOD2/CARD15 gene polymorphisms are not associated with collagenous colitis. International journal of colorectal disease 2007;22:425-8.
78. Saverymuttu SH, Camilleri M, Rees H, et al. Indium 111-granulocyte scanning in the assessment of disease extent and disease activity in inflammatory bowel disease. A comparison with colonoscopy, histology, and fecal indium 111-granulocyte excretion. Gastroenterology 1986;90:1121-8.
79. Xu SY, Carlson M, Engstrom A, et al. Purification and characterization of a human neutrophil lipocalin (HNL) from the secondary granules of human neutrophils. Scandinavian journal of clinical and laboratory investigation 1994;54:365-76.
80. Witko-Sarsat V, Rieu P, Descamps-Latscha B, et al. Neutrophils: molecules, functions and pathophysiological aspects. Laboratory investigation; a journal of technical methods and pathology 2000;80:617-53.
81. Nathan C. Neutrophils and immunity: challenges and opportunities. Nature reviews 2006;6:173-82.
82. Mahmoud AAF AK, Simon AS., ed. The eosinophil in health and disease. New York, London, Toronto, Sydney, San Fransisco: Grune & Stratton, 1980.
83. Gleich GJ, Loegering DA. Immunobiology of eosinophils. Annual review of immunology 1984;2:429-59.
84. Rothenberg ME. Eosinophilia. The New England journal of medicine 1998;338:1592-600.
85. Weller PF. Eosinophils: structure and functions. Current opinion in immunology 1994;6:85-90.
86. Rothenberg ME, Owen WF, Jr., Silberstein DS, et al. Eosinophils cocultured with endothelial cells have increased survival and functional properties. Science (New York, NY 1987;237:645-7.
87. Rothenberg ME, Hogan SP. The eosinophil. Annual review of immunology 2006;24:147-74.
88. Gutierrez-Ramos JC, Lloyd C, Gonzalo JA. Eotaxin: from an eosinophilic chemokine to a major regulator of allergic reactions. Immunology today 1999;20:500-4.
89. Teran LM, Noso N, Carroll M, et al. Eosinophil recruitment following allergen challenge is associated with the release of the chemokine RANTES into asthmatic airways. J Immunol 1996;157:1806-12.

67
90. Gleich GJ, Adolphson CR. The eosinophilic leukocyte: structure and function. Advances in immunology 1986;39:177-253.
91. Woerly G, Roger N, Loiseau S, et al. Expression of CD28 and CD86 by human eosinophils and role in the secretion of type 1 cytokines (interleukin 2 and interferon gamma): inhibition by immunoglobulin a complexes. The Journal of experimental medicine 1999;190:487-95.
92. Venge P, Stromberg A, Braconier JH, et al. Neutrophil and eosinophil granulocytes in bacterial infection: sequential studies of cellular and serum levels of granule proteins. British journal of haematology 1978;38:475-83.
93. Peterson CG, Venge P. Purification and characterization of a new cationic protein--eosinophil protein-X (EPX)--from granules of human eosinophils. Immunology 1983;50:19-26.
94. Carlson MG, Peterson CG, Venge P. Human eosinophil peroxidase: purification and characterization. J Immunol 1985;134:1875-9.
95. Hogan SP, Rosenberg HF, Moqbel R, et al. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy 2008;38:709-50.
96. Ackerman SJ, Gleich GJ, Loegering DA, et al. Comparative toxicity of purified human eosinophil granule cationic proteins for schistosomula of Schistosoma mansoni. The American journal of tropical medicine and hygiene 1985;34:735-45.
97. Lehrer RI, Szklarek D, Barton A, et al. Antibacterial properties of eosinophil major basic protein and eosinophil cationic protein. J Immunol 1989;142:4428-34.
98. McLaren DJ, McKean JR, Olsson I, et al. Morphological studies on the killing of schistosomula of Schistosoma mansoni by human eosinophil and neutrophil cationic proteins in vitro. Parasite immunology 1981;3:359-73.
99. Slifman NR, Loegering DA, McKean DJ, et al. Ribonuclease activity associated with human eosinophil-derived neurotoxin and eosinophil cationic protein. J Immunol 1986;137:2913-7.
100. Zheutlin LM, Ackerman SJ, Gleich GJ, et al. Stimulation of basophil and rat mast cell histamine release by eosinophil granule-derived cationic proteins. J Immunol 1984;133:2180-5.
101. Rosenberg HF, Domachowske JB. Eosinophils, eosinophil ribonucleases, and their role in host defense against respiratory virus pathogens. Journal of leukocyte biology 2001;70:691-8.
102. Slungaard A, Mahoney JR, Jr. Thiocyanate is the major substrate for eosinophil peroxidase in physiologic fluids. Implications for cytotoxicity. The Journal of biological chemistry 1991;266:4903-10.
103. Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry 2000;39:5474-82.
104. Henderson JP, Byun J, Williams MV, et al. Bromination of deoxycytidine by eosinophil peroxidase: a mechanism for mutagenesis by oxidative damage of nucleotide precursors. Proceedings of the National Academy of Sciences of the United States of America 2001;98:1631-6.
105. Pegorier S, Wagner LA, Gleich GJ, et al. Eosinophil-derived cationic proteins activate the synthesis of remodeling factors by airway epithelial cells. J Immunol 2006;177:4861-9.

68
106. Wang J, Slungaard A. Role of eosinophil peroxidase in host defense and disease pathology. Archives of biochemistry and biophysics 2006;445:256-60.
107. Delves P MS, Burton D, Roitt I, ed. Roitt's Essential Immunology, 11: Wiley-Blackwell, 2006.
108. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nature medicine 2007;13:139-45.
109. Goyal RK, Hirano I. The enteric nervous system. The New England journal of medicine 1996;334:1106-15.
110. Collins SM. The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology 1996;111:1683-99.
111. Levite M, Chowers Y. Nerve-driven immunity: neuropeptides regulate cytokine secretion of T cells and intestinal epithelial cells in a direct, powerful and contextual manner. Ann Oncol 2001;12 Suppl 2:S19-25.
112. Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflammatory bowel diseases 2007;13:918-32.
114. Liu L, Shang F, Markus I, et al. Roles of substance P receptors in human colon circular muscle: alterations in diverticular disease. The Journal of pharmacology and experimental therapeutics 2002;302:627-35.
113. Castagliuolo I, Keates AC, Qiu B, et al. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. Proceedings of the National Academy of Sciences of the United States of America 1997;94:4788-93.
115. Mantyh CR, Gates TS, Zimmerman RP, et al. Receptor binding sites for substance P, but not substance K or neuromedin K, are expressed in high concentrations by arterioles, venules, and lymph nodules in surgical specimens obtained from patients with ulcerative colitis and Crohn disease. Proceedings of the National Academy of Sciences of the United States of America 1988;85:3235-9.
116. Renzi D, Pellegrini B, Tonelli F, et al. Substance P (neurokinin-1) and neurokinin A (neurokinin-2) receptor gene and protein expression in the healthy and inflamed human intestine. The American journal of pathology 2000;157:1511-22.
117. Reinisch N, Kirchmair R, Kahler CM, et al. Attraction of human monocytes by the neuropeptide secretoneurin. FEBS letters 1993;334:41-4.
118. Schurmann G, Bishop AE, Facer P, et al. Secretoneurin: a new peptide in the human enteric nervous system. Histochemistry and cell biology 1995;104:11-9.
119. Dunzendorfer S, Schratzberger P, Reinisch N, et al. Secretoneurin, a novel neuropeptide, is a potent chemoattractant for human eosinophils. Blood 1998;91:1527-32.
120. Schratzberger P, Reinisch N, Kahler CM, et al. Deactivation of chemotaxis of human neutrophils by priming with secretogranin II-derived secretoneurin. Regulatory peptides 1996;63:65-71.
121. Granberg D, Stridsberg M, Seensalu R, et al. Plasma chromogranin A in patients with multiple endocrine neoplasia type 1. The Journal of clinical endocrinology and metabolism 1999;84:2712-7.

69
122. Spadaro A, Ajello A, Morace C, et al. Serum chromogranin-A in hepatocellular carcinoma: diagnostic utility and limits. World J Gastroenterol 2005;11:1987-90.
123. Sciola V, Massironi S, Conte D, et al. Plasma chromogranin a in patients with inflammatory bowel disease. Inflammatory bowel diseases 2009;15:867-71.
124. Lugardon K, Raffner R, Goumon Y, et al. Antibacterial and antifungal activities of vasostatin-1, the N-terminal fragment of chromogranin A. The Journal of biological chemistry 2000;275:10745-53.
125. Briolat J, Wu SD, Mahata SK, et al. New antimicrobial activity for the catecholamine release-inhibitory peptide from chromogranin A. Cell Mol Life Sci 2005;62:377-85.
126. Russell J, Gee P, Liu SM, et al. Inhibition of parathyroid hormone secretion by amino-terminal chromogranin peptides. Endocrinology 1994;135:337-42.
127. Strub JM, Garcia-Sablone P, Lonning K, et al. Processing of chromogranin B in bovine adrenal medulla. Identification of secretolytin, the endogenous C-terminal fragment of residues 614-626 with antibacterial activity. European journal of biochemistry / FEBS 1995;229:356-68.
128. Stridsberg M, Oberg K, Li Q, et al. Measurements of chromogranin A, chromogranin B (secretogranin I), chromogranin C (secretogranin II) and pancreastatin in plasma and urine from patients with carcinoid tumours and endocrine pancreatic tumours. The Journal of endocrinology 1995;144:49-59.
129. Stridsberg M, Husebye ES. Chromogranin A and chromogranin B are sensitive circulating markers for phaeochromocytoma. European journal of endocrinology / European Federation of Endocrine Societies 1997;136:67-73.
130. Stridsberg M, Eriksson B, Fellstrom B, et al. Measurements of chromogranin B can serve as a complement to chromogranin A. Regulatory peptides 2007;139:80-3.
131. Furness JB, Jones C, Nurgali K, et al. Intrinsic primary afferent neurons and nerve circuits within the intestine. Progress in neurobiology 2004;72:143-64.
132. Fujimiya M, Okumiya K, Kuwahara A. Immunoelectron microscopic study of the luminal release of serotonin from rat enterochromaffin cells induced by high intraluminal pressure. Histochemistry and cell biology 1997;108:105-13.
133. Okumiya K, Fujimiya M. Immunoelectron microscopic study of the luminal release of chromogranin A from rat enterochromaffin cells. Histochemistry and cell biology 1999;111:253-7.
134. Lampinen M, Ronnblom A, Amin K, et al. Eosinophil granulocytes are activated during the remission phase of ulcerative colitis. Gut 2005;54:1714-20.
135. Nielsen OH, Vainer B, Madsen SM, et al. Established and emerging biological activity markers of inflammatory bowel disease. The American journal of gastroenterology 2000;95:359-67.
136. Vermeire S, Van Assche G, Rutgeerts P. Laboratory markers in IBD: useful, magic, or unnecessary toys? Gut 2006;55:426-31.
137. Desai D, Faubion WA, Sandborn WJ. Review article: biological activity markers in inflammatory bowel disease. Alimentary pharmacology & therapeutics 2007;25:247-55.
138. Gisbert JP, Gonzalez-Lama Y, Mate J. [Role of biological markers in inflammatory bowel disease]. Gastroenterologia y hepatologia 2007;30:117-29.

70
139. Roseth AG, Fagerhol MK, Aadland E, et al. Assessment of the neutrophil dominating protein calprotectin in feces. A methodologic study. Scandinavian journal of gastroenterology 1992;27:793-8.
140. Sugi K, Saitoh O, Hirata I, et al. Fecal lactoferrin as a marker for disease activity in inflammatory bowel disease: comparison with other neutrophil-derived proteins. The American journal of gastroenterology 1996;91:927-34.
141. Roseth AG, Aadland E, Jahnsen J, et al. Assessment of disease activity in ulcerative colitis by faecal calprotectin, a novel granulocyte marker protein. Digestion 1997;58:176-80.
142. Peterson CG, Eklund E, Taha Y, et al. A new method for the quantification of neutrophil and eosinophil cationic proteins in feces: establishment of normal levels and clinical application in patients with inflammatory bowel disease. The American journal of gastroenterology 2002;97:1755-62.
143. Langhorst J, Elsenbruch S, Mueller T, et al. Comparison of 4 neutrophil-derived proteins in feces as indicators of disease activity in ulcerative colitis. Inflammatory bowel diseases 2005;11:1085-91.
144. D'Inca R, Dal Pont E, Di Leo V, et al. Calprotectin and lactoferrin in the assessment of intestinal inflammation and organic disease. International journal of colorectal disease 2006.
145. Fagerberg UL, Loof L, Lindholm J, et al. Fecal calprotectin: a quantitative marker of colonic inflammation in children with inflammatory bowel disease. Journal of pediatric gastroenterology and nutrition 2007;45:414-20.
146. Peterson CG, Sangfelt P, Wagner M, et al. Fecal levels of leukocyte markers reflect disease activity in patients with ulcerative colitis. Scandinavian journal of clinical and laboratory investigation 2007;67:810-20.
147. Carroccio A, Iacono G, Cottone M, et al. Diagnostic accuracy of fecal calprotectin assay in distinguishing organic causes of chronic diarrhea from irritable bowel syndrome: a prospective study in adults and children. Clinical chemistry 2003;49:861-7.
148. Roseth AG, Aadland E, Grzyb K. Normalization of faecal calprotectin: a predictor of mucosal healing in patients with inflammatory bowel disease. Scandinavian journal of gastroenterology 2004;39:1017-20.
149. von Roon AC, Karamountzos L, Purkayastha S, et al. Diagnostic precision of fecal calprotectin for inflammatory bowel disease and colorectal malignancy. The American journal of gastroenterology 2007;102:803-13.
150. Johne B, Fagerhol MK, Lyberg T, et al. Functional and clinical aspects of the myelomonocyte protein calprotectin. Mol Pathol 1997;50:113-23.
151. Brandtzaeg P, Gabrielsen TO, Dale I, et al. The leucocyte protein L1 (calprotectin): a putative nonspecific defence factor at epithelial surfaces. Advances in experimental medicine and biology 1995;371A:201-6.
152. Brun JG, Ulvestad E, Fagerhol MK, et al. Effects of human calprotectin (L1) on in vitro immunoglobulin synthesis. Scandinavian journal of immunology 1994;40:675-80.
153. Steinbakk M, Naess-Andresen CF, Lingaas E, et al. Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 1990;336:763-5.
154. Tibble J, Teahon K, Thjodleifsson B, et al. A simple method for assessing intestinal inflammation in Crohn's disease. Gut 2000;47:506-13.
155. Dale I, Brandtzaeg P, Fagerhol MK, et al. Distribution of a new myelomonocytic antigen (L1) in human peripheral blood leukocytes.

71
Immunofluorescence and immunoperoxidase staining features in comparison with lysozyme and lactoferrin. American journal of clinical pathology 1985;84:24-34.
156. Roseth AG, Schmidt PN, Fagerhol MK. Correlation between faecal excretion of indium-111-labelled granulocytes and calprotectin, a granulocyte marker protein, in patients with inflammatory bowel disease. Scandinavian journal of gastroenterology 1999;34:50-4.
157. Summerton CB, Longlands MG, Wiener K, et al. Faecal calprotectin: a marker of inflammation throughout the intestinal tract. European journal of gastroenterology & hepatology 2002;14:841-5.
158. Izzo RS, Witkon K, Chen AI, et al. Interleukin-8 and neutrophil markers in colonic mucosa from patients with ulcerative colitis. The American journal of gastroenterology 1992;87:1447-52.
159. Carlson M, Raab Y, Seveus L, et al. Human neutrophil lipocalin is a unique marker of neutrophil inflammation in ulcerative colitis and proctitis. Gut 2002;50:501-6.
160. Sangfelt P, Carlson M, Thorn M, et al. Neutrophil and eosinophil granule proteins as markers of response to local prednisolone treatment in distal ulcerative colitis and proctitis. The American journal of gastroenterology 2001;96:1085-90.
161. Bischoff SC, Wedemeyer J, Herrmann A, et al. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology 1996;28:1-13.
162. Levy AM, Gleich GJ, Sandborn WJ, et al. Increased eosinophil granule proteins in gut lavage fluid from patients with inflammatory bowel disease. Mayo Clinic proceedings 1997;72:117-23.
163. Raab Y, Fredens K, Gerdin B, et al. Eosinophil activation in ulcerative colitis: studies on mucosal release and localization of eosinophil granule constituents. Digestive diseases and sciences 1998;43:1061-70.
164. Carlson M, Raab Y, Peterson C, et al. Increased intraluminal release of eosinophil granule proteins EPO, ECP, EPX, and cytokines in ulcerative colitis and proctitis in segmental perfusion. The American journal of gastroenterology 1999;94:1876-83.
165. Jessurun J, Yardley JH, Giardiello FM, et al. Chronic colitis with thickening of the subepithelial collagen layer (collagenous colitis): histopathologic findings in 15 patients. Human pathology 1987;18:839-48.
166. Lazenby AJ, Yardley JH, Giardiello FM, et al. Pitfalls in the diagnosis of collagenous colitis: experience with 75 cases from a registry of collagenous colitis at the Johns Hopkins Hospital. Human pathology 1990;21:905-10.
167. Offner FA, Jao RV, Lewin KJ, et al. Collagenous colitis: a study of the distribution of morphological abnormalities and their histological detection. Human pathology 1999;30:451-7.
168. Lampinen M, Carlson M, Sangfelt P, et al. IL-5 and TNF-alpha participate in recruitment of eosinophils to intestinal mucosa in ulcerative colitis. Digestive diseases and sciences 2001;46:2004-9.
169. Taha Y, Raab Y, Carlson M, et al. Steroids reduce local inflammatory mediator secretion and mucosal permeability in collagenous colitis patients. World J Gastroenterol 2006;12:7012-8.

72
170. Lettesjo H, Hansson T, Peterson C, et al. Detection of inflammatory markers in stools from patients with irritable bowel syndrome and collagenous colitis. Scandinavian journal of gastroenterology 2006;41:54-9.
171. Gubbins GP, Dekovich AA, Ma CK, et al. Collagenous colitis: report of nine cases and review of the literature. Southern medical journal 1991;84:33-7.
172. Zins BJ, Tremaine WJ, Carpenter HA. Collagenous colitis: mucosal biopsies and association with fecal leukocytes. Mayo Clinic proceedings 1995;70:430-3.
173. Taha Y, Carlson M, Thorn M, et al. Evidence of local eosinophil activation and altered mucosal permeability in collagenous colitis. Digestive diseases and sciences 2001;46:888-97.
174. Wildt S, Nordgaard-Lassen I, Bendtsen F, et al. Metabolic and inflammatory faecal markers in collagenous colitis. European journal of gastroenterology & hepatology 2007;19:567-74.
175. Binder V. A comparison between clinical state, macroscopic and microscopic appearances of rectal mucosa, and cytologic picture of mucosal exudate in ulcerative colitis. Scandinavian journal of gastroenterology 1970;5:627-32.
176. Harvey RF, Bradshaw JM. A simple index of Crohn's-disease activity. Lancet 1980;1:514.
177. Truelove SC, Richards WC. Biopsy studies in ulcerative colitis. British medical journal 1956;1:1315-8.
178. Stridsberg M, Eriksson B, Janson ET. Measurements of secretogranins II, III, V and proconvertases 1/3 and 2 in plasma from patients with neuroendocrine tumours. Regulatory peptides 2008;148:95-8.
179. Fernvik E, Hallden G, Hed J, et al. Intracellular and surface distribution of CD9 in human eosinophils. Apmis 1995;103:699-706.
180. Upham JW, Sehmi R, Hayes LM, et al. Retinoic acid modulates IL-5 receptor expression and selectively inhibits eosinophil-basophil differentiation of hemopoietic progenitor cells. The Journal of allergy and clinical immunology 2002;109:307-13.
181. Matsumoto K, Appiah-Pippim J, Schleimer RP, et al. CD44 and CD69 represent different types of cell-surface activation markers for human eosinophils. American journal of respiratory cell and molecular biology 1998;18:860-6.
182. Fernvik E, Lundahl J, Hallden G. The impact of eotaxin- and IL-5-induced adhesion and transmigration on eosinophil activity markers. Inflammation 2000;24:73-87.
183. Bochner BS, Sterbinsky SA, Bickel CA, et al. Differences between human eosinophils and neutrophils in the function and expression of sialic acid-containing counterligands for E-selectin. J Immunol 1994;152:774-82.
184. Stocks SC, Kerr MA, Haslett C, et al. CD66-dependent neutrophil activation: a possible mechanism for vascular selectin-mediated regulation of neutrophil adhesion. Journal of leukocyte biology 1995;58:40-8.
185. Torsteinsdottir I, Arvidson NG, Hallgren R, et al. Enhanced expression of integrins and CD66b on peripheral blood neutrophils and eosinophils in patients with rheumatoid arthritis, and the effect of glucocorticoids. Scandinavian journal of immunology 1999;50:433-9.
186. Aadland E, Fagerhol MK. Faecal calprotectin: a marker of inflammation throughout the intestinal tract. European journal of gastroenterology & hepatology 2002;14:823-5.

73
187. Kolho KL, Raivio T, Lindahl H, et al. Fecal calprotectin remains high during glucocorticoid therapy in children with inflammatory bowel disease. Scandinavian journal of gastroenterology 2006;41:720-5.
188. Froslie KF, Jahnsen J, Moum BA, et al. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology 2007;133:412-22.
189. Limburg PJ, Ahlquist DA, Sandborn WJ, et al. Fecal calprotectin levels predict colorectal inflammation among patients with chronic diarrhea referred for colonoscopy. The American journal of gastroenterology 2000;95:2831-7.
190. Bunn SK, Bisset WM, Main MJ, et al. Fecal calprotectin: validation as a noninvasive measure of bowel inflammation in childhood inflammatory bowel disease. Journal of pediatric gastroenterology and nutrition 2001;33:14-22.
191. Gomes P, du Boulay C, Smith CL, et al. Relationship between disease activity indices and colonoscopic findings in patients with colonic inflammatory bowel disease. Gut 1986;27:92-5.
192. Korelitz BI. An indicator of Crohn's disease activity. A need unfulfilled. Journal of clinical gastroenterology 1986;8:220-2.
193. Sipponen T, Karkkainen P, Savilahti E, et al. Correlation of faecal calprotectin and lactoferrin with an endoscopic score for Crohn's disease and histological findings. Alimentary pharmacology & therapeutics 2008;28:1221-9.
194. Eklund G. Monitorering av fekala markörproteiner vid IBD-skov-vilken patientnytta? Uppsala: Faculty of Medicin, Uppsala University, 2010.
195. Olofsson T, Olsson I, Venge P, et al. Serum myeloperoxidase and lactoferrin in neutropenia. Scandinavian journal of haematology 1977;18:73-80.
196. Wildt S, Rumessen JJ, Csillag C, et al. Cyclooxygenase-2 immunoreactivity in collagenous colitis. Apmis 2009;117:500-6.
197. Tibble JA, Sigthorsson G, Foster R, et al. High prevalence of NSAID enteropathy as shown by a simple faecal test. Gut 1999;45:362-6.
198. Levy AM, Yamazaki K, Van Keulen VP, et al. Increased eosinophil infiltration and degranulation in colonic tissue from patients with collagenous colitis. The American journal of gastroenterology 2001;96:1522-8.
199. Gleich GJ, Frigas E, Loegering DA, et al. Cytotoxic properties of the eosinophil major basic protein. J Immunol 1979;123:2925-7.
200. Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. The Journal of biological chemistry 1986;261:4337-45.
201. Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proceedings of the National Academy of Sciences of the United States of America 1986;83:4167-71.
202. Edwards DR, Murphy G, Reynolds JJ, et al. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. The EMBO journal 1987;6:1899-904.
203. Pincus SH, Ramesh KS, Wyler DJ. Eosinophils stimulate fibroblast DNA synthesis. Blood 1987;70:572-4.
204. Birkland TP, Cheavens MD, Pincus SH. Human eosinophils stimulate DNA synthesis and matrix production in dermal fibroblasts. Archives of dermatological research 1994;286:312-8.

74
205. Stahle-Backdahl M, Maim J, Veress B, et al. Increased presence of eosinophilic granulocytes expressing transforming growth factor-beta1 in collagenous colitis. Scandinavian journal of gastroenterology 2000;35:742-6.
206. Jacoby DB, Ueki IF, Widdicombe JH, et al. Effect of human eosinophil major basic protein on ion transport in dog tracheal epithelium. The American review of respiratory disease 1988;137:13-6.
207. Metwali A, Blum AM, Ferraris L, et al. Eosinophils within the healthy or inflamed human intestine produce substance P and vasoactive intestinal peptide. Journal of neuroimmunology 1994;52:69-78.
208. Minnicozzi M, Duran WN, Gleich GJ, et al. Eosinophil granule proteins increase microvascular macromolecular transport in the hamster cheek pouch. J Immunol 1994;153:2664-70.
209. Dajotoy T, Andersson P, Bjartell A, et al. Human eosinophils produce the T cell-attracting chemokines MIG and IP-10 upon stimulation with IFN-gamma. Journal of leukocyte biology 2004;76:685-91.
210. Korsgren M, Erjefalt JS, Hinterholzl J, et al. Neural expression and increased lavage fluid levels of secretoneurin in seasonal allergic rhinitis. American journal of respiratory and critical care medicine 2003;167:1504-8.
211. Korsgren M, Fischer-Colbrie R, Andersson M, et al. Secretoneurin is released into human airways by topical histamine but not capsaicin. Allergy 2005;60:459-63.
212. Costello RW, Schofield BH, Kephart GM, et al. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. The American journal of physiology 1997;273:L93-103.
213. Stead RH. Innervation of mucosal immune cells in the gastrointestinal tract. Regional immunology 1992;4:91-9.
214. O'Brien LM, Fitzpatrick E, Baird AW, et al. Eosinophil-nerve interactions and neuronal plasticity in rat gut associated lymphoid tissue (GALT) in response to enteric parasitism. Journal of neuroimmunology 2008;197:1-9.
215. Mosnier JF, Larvol L, Barge J, et al. Lymphocytic and collagenous colitis: an immunohistochemical study. The American journal of gastroenterology 1996;91:709-13.
216. Bonderup OK, Hansen JB, Birket-Smith L, et al. Budesonide treatment of collagenous colitis: a randomised, double blind, placebo controlled trial with morphometric analysis. Gut 2003;52:248-51.
217. Bonderup OK, Hansen JB, Teglbjaerg PS, et al. Long-term budesonide treatment of collagenous colitis: a randomised, double-blind, placebo-controlled trial. Gut 2009;58:68-72.
218. Elitsur Y, Lichtman SN, Neace C, et al. Immunosuppressive effect of budesonide on human lamina propria lymphocytes. Immunopharmacology 1998;38:279-85.
219. Aoki S, Watanabe M, Hasegawa H, et al. Rectal carcinoid arising in ulcerative colitis associated with rectal adenocarcinoma. Journal of gastroenterology 2004;39:697-8.
220. El-Salhy M, Danielsson A, Stenling R, et al. Colonic endocrine cells in inflammatory bowel disease. Journal of internal medicine 1997;242:413-9.
221. Matsumoto T, Jo Y, Mibu R, et al. Multiple microcarcinoids in a patient with long standing ulcerative colitis. Journal of clinical pathology 2003;56:963-5.

75
222. Miller RR, Sumner HW. Argyrophilic cell hyperplasia and an atypical carcinoid tumor in chronic ulcerative colitis. Cancer 1982;50:2920-5.
223. Rybakova MG, Botina AV, Solov'eva OI. [Immunomorphological characteristics of mucosal and endocrine cells of the colon in patients with chronic ulcerative colitis]. Arkhiv patologii 2005;67:30-3.
224. Stewart CJ, Matsumoto T, Jo Y, et al. Multifocal microcarcinoid tumours in ulcerative colitis. Journal of clinical pathology 2005;58:111-2; author reply 1112.
225. Elliott SN, Wallace JL. Neutrophil-mediated gastrointestinal injury. Canadian journal of gastroenterology = Journal canadien de gastroenterologie 1998;12:559-68.
226. Panes J, Granger DN. Leukocyte-endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology 1998;114:1066-90.
227. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nature reviews 2003;3:331-41.
228. Tibble JA, Sigthorsson G, Bridger S, et al. Surrogate markers of intestinal inflammation are predictive of relapse in patients with inflammatory bowel disease. Gastroenterology 2000;119:15-22.
229. Costa F, Mumolo MG, Ceccarelli L, et al. Calprotectin is a stronger predictive marker of relapse in ulcerative colitis than in Crohn's disease. Gut 2005;54:364-8.

!��3��1��������3������������������� �������������� ������������������� ������� ����������������������������
������/4����������5���������������
!�������������������������5���������������63����3��1������6�������������������0���������7!�����������������������������������8�����9������������������0�����6��������������������������0��������������������������������������������������1�����������3�����������������������5���������������7:;������<����6)**�6�������������0�������������������=�����������1�����������3�����������������������5���������������>7?
������0�����/��0��������7��7�����/�0�/��/��/��1(')2*�2
������������������� ���������� �
����