1 analysis and displays associated with the qt studies · 1 analysis and displays associated with...
TRANSCRIPT

Version 1.0
1
1 Analysis and Displays associated with the QT studies
Version 1.0 Draft for Broad Review Created 08 September 2015
A White Paper by the PhUSE CSS Development of Standard Scripts for Analysis and Programming Working Group
Disclaimer: The opinions expressed in this document are those of the authors and do not necessarily represent the opinions of PhUSE, the members’ respective companies or organizations, or regulatory authorities. The content in this document should not be interpreted as a data standard and/or information required by regulatory authorities.

Version 1.0
2
2 Table of Contents 1 Analysis and Displays associated with the QT studies .......................................................................................... 1 2 Table of contents .................................................................................................................................................... 2 3 Purpose ................................................................................................................................................................... 3 4 Introduction ............................................................................................................................................................ 4 5 ECG background .................................................................................................................................................... 5 6 Pre-analytical issues ............................................................................................................................................... 7
6.1 Correction of the QT-interval for heart rate ................................................................................................. 7 6.1.1 Historical Population-Based Formula from a Historical Population ....................................................... 7 6.1.2 Population-Based Formula from the Population under Study ................................................................. 9 6.1.3 Individual-Based Formula (QTcI) ............................................................................................................ 9
6.2 Thorough QT (TQT) Study Design ............................................................................................................ 10 6.2.1 Brief Background ................................................................................................................................... 10 6.2.2 Specific designs ...................................................................................................................................... 14
6.3 Baseline and Treatment Difference (Drug Effect) ...................................................................................... 16 6.3.1 Time-Matched Lead-in Day Baseline; Double-Delta Treatment Difference ......................................... 16 6.3.2 Time-Averaged Lead-in Day Baseline; Double-Delta Treatment Difference ....................................... 16 6.3.3 Predose Averaged Baseline; Double-Delta Treatment Difference ........................................................ 17
7 Analysis ................................................................................................................................................................ 18 7.1 Primary analysis ......................................................................................................................................... 18
7.1.1 Testing of QT prolongation .................................................................................................................... 18 7.1.2 Assay Sensitivity .................................................................................................................................... 19 7.1.3 Categorical Analyses .............................................................................................................................. 20 7.1.4 Morphological (Qualitative) Analyses ................................................................................................... 21
7.2 Concentration-Response Relationship (CRR) ............................................................................................ 21 7.3 P-values and Confidence Intervals ............................................................................................................ 23
8 List of outputs ....................................................................................................................................................... 24 9 Outputs shells ....................................................................................................................................................... 25 10 References ....................................................................................................................................................... 46

Version 1.0
3
3 Purpose Under CDISC, standards have been defined for data collection (Clinical Data Acquisition Standards Harmonization - CDASH), tabulation (Study Data Tabulation Model - SDTM), and analysis (Analysis Data Model - ADaM) datasets. The next step is to develop standard tables, figures, and listings. The Development of Standard Scripts for Analysis and Programming Working Group is leading an effort to create several White Papers providing recommended analyses and displays for common measurements, and has developed a Script Repository as a place to store shared code.
The purpose of this White Paper is to provide advice on displaying, summarizing, and analyzing Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs (henceforth referred to as TQT study). The intent is to begin the process of developing industry standards with respect to analyses and reporting for these trials. In particular, this White Paper provides recommended processes for:
• Pre-analytical issues: Study design, QT interval corrections, and Baseline adjustments • Analytical issues: Testing for QT prolongation, Assay sensitivity, Outlier analysis / Categorical analysis,
Morphological (Qualitative) abnormalities, and PK/PD analysis
This paper attempts to give recommendations for difficult decisions related to the analysis of difficult topics such as QT interval correction, baseline, and PK/PD analysis. Since there are on-going discussions regarding these topics the recommendations made here are mainly based on the authors experience with these trials and submission to regulatory bodies (and ICH-E14 guidelines and Q&A at the time this White Paper was written).
The content of this document can be used when developing the analysis plan for individual clinical trials for Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs.
Development of standard Tables, Figures, and Listings (TFLs) and associated analyses will lead to improved standardization from collection through data storage, as it is necessary to determine how the results should be reported and analyzed before finalizing how to collect and store the data. The development of standard TFLs will also lead to improved product lifecycle management by ensuring reviewers receive the desired analyses for consistent and efficient evaluation of patient safety. Although having standard TFLs is an ultimate goal, this White Paper reflects recommendations only and should not be interpreted as “required” by any regulatory agency.
Detailed specifications for TFL or dataset development are considered out-of-scope for this White Paper. However, the hope is that specifications and code (utilizing SDTM and ADaM structures) will be developed consistent with the concepts outlined in this White Paper, and placed in the publicly available Standard Scripts Repository.

Version 1.0
4
4 Introduction Industry standards have evolved over time for data collection (CDASH), observed data (SDTM), and analysis datasets (ADaM). There is now recognition that the next step would be to develop standard TFLs for common measurements across clinical trials and therapeutic areas. Having industry standards for data collection and analysis datasets provides a good basis for creating standard TFLs.
The beginning of the effort leading to this white paper came from the initiation of the FDA/PhUSE Computational Science Collaboration, a yearly conference and ongoing working groups to support addressing computational needs of the industry. The FDA identified key priorities and teamed up with the PhUSE to tackle various challenges using collaboration, crowd sourcing, and innovation (Rosario LA, 2012). The FDA and PhUSE created several Computational Science (CS) working groups to address several of these challenges. The working group, titled “Development of Standard Scripts for Analysis and Programming,” has led the development of this white paper, along with the development of a platform for storing shared code.
Several existing documents contain suggested TFLs for common measurements. Some of the documents are now relatively outdated, and generally lack sufficient detail to be used as support for the entire standardization effort. Nevertheless, these documents were used as a starting point in the development of this White Paper. The documents include:
• ICH E3: Structure and Content of Clinical Study Reports • Guideline for Industry: Structure and Content of Clinical Study Reports • Guidance for Industry: Premarketing Risk Assessment • Reviewer Guidance. Conducting a Clinical Safety Review of a New Product Application and Preparing a
report on the Review. • ICH M4E: Common Technical Document for the Registration of Pharmaceuticals for Human Use –
Efficacy • ICH E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential For Non-
Antiarrhythmic Drugs • ICH E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-
antiarrhythmic drugs Questions and Answers R1. • FDA Guidance for Industry: ICH E14 Clinical Evaluation of QT/QTc. Interval Prolongation and
Proarrhythmic Potential for Non-Antiarrhythmic Drugs. • QT Studies Therapeutic Area Data Standards User Guide (TAUG) V1. CDISC.
The ICH E14 guidelines, FDA Guidance for Industry and TAUG are considered key documents. They do not provide, however, detailed information that would enable standardization of all analysis and presentation of TQT studies.

Version 1.0
5
5 ECG background Some basic understanding of ECGs can be helpful in planning and completing analyses for Thorough QT (TQT) studies. The ECG is a graphical representation of the electrical depolarization and repolarization of the heart’s cells that initiates and spreads through the heart in an organized manner and causes contraction of the heart muscle that results in the pumping of blood. In 1895, Einthoven established the five primary topographic features of the ECG tracing (P, Q, R, S, and T waves; discussed in more detail below) and in 1912 defined the now standard ECG leads (the waveform of potential difference over time between two sets of one or more electrodes attached to the body) I, II, and III. Additional standard leads were established in 1938 (V1 – V6) and in 1942 (aVR, aVL, and aVF). Therefore, the standard ECG records this activity at the body surface for 12 leads (I, II, III, aVR, aVL, aVF, V1, V2, V3, V4, V5, V6). A continuous waveform (positive and negative changes over time) of electrical activity is recorded for each lead. A standard ECG is a 10-sec recording, but ECG data can be recorded and stored digitally for any amount of time (limited only by storage media capacity). A standard paper ECG displays 3 1/3 seconds of each lead (4 sets of 3 leads) and all 10 seconds of 1 lead as illustrated in Figure 1.
Figure 5-1: Standard 10-sec ECG
The waveforms are a series of complexes that represent the sequential depolarization and repolarization electrical activity that spreads through the heart. These complexes have parts, briefly noted above, that are named as shown in Figure 2. Note that a single complex contains a P-wave, a QRS-complex (that consists of a Q-wave [sometimes absent], an R-wave, and an S-wave; the R- and S-waves can have opposite polarities across leads), a T-wave, and sometimes a U-wave. Each of these complexes represents a complete depolarization and repolarization of the heart. There is an isoelectric gap (no electrical activity) between complexes. The RR-interval, not represented in Figure 2, is the time between successive R-waves (and, therefore, the time between complexes). Analyses in TQT studies will focus on The QT-interval and the RR-interval, but secondary analyses will also be conducted on the PR-interval and the QRS-complex. The width of the waves, and intervals, including the RR-interval, represent time and are most commonly expressed in millisecond (msec) units. Heart rate (HR) which is the number of complexes per minute, usually expressed as beats per minute (bpm). Therefore, the RR-interval measurement, in msec, and HR, in bpm, have the following relationship:

Version 1.0
6
• RR = (1/(HR/60)*1000) • HR = 60,000/RR
Figure 5-2: A single ECG waveform complex and its parts

Version 1.0
7
6 Pre-analytical issues 6.1 Correction of the QT-interval for heart rate The QT-interval is a measure or biomarker for the time of ventricular depolarization and repolarization to occur but is practically used as a biomarker for the time of ventricular repolarization. The QT-interval changes in inverse relationship to HR for appropriate physiological coordination of the pumping of blood by the heart. Therefore, because subjects’ heart rates are not constant throughout participation in a TQT study (or when evaluated clinically) it is necessary to correct the QT-interval for HR in order to make comparisons of the QT interval recorded at different HRs at different times. Complicating the situation a bit more is the fact that the QT-interval does not change instantaneously with a change in HR. The change in QT-interval is delayed; its change is subject to hysteresis. Hysteresis is generally ignored in the analysis of TQT studies, but one researcher (Malik, 2008) has developed methods for evaluating hysteresis patterns on an individual basis and incorporating them into QT correction. Discussion of this topic is beyond the scope of this White Paper. The ideal corrected QT interval, QTc, would be uncorrelated with HR or the RR-interval. In other words if QTc were plotted against either RR or HR and the data were fit to a linear model, the correlation would be “0” and the slope of the regression line would be “0”. Essentially, QT correction for HR attempts to adjust the individual subject’s QT-interval, at any HR, to a value that would be expected if the subject’s HR were constant. In the majority of QT correction formulas, RR is used rather than HR because RR-interval is measured and expressed in the same units as the QT-interval, msec, while HR is measured and expressed in bpm as illustrated above. In general, there are three basic methods to adjust or correct the QT- interval for HR (RR-interval). The methods are:
1. Historical population-based formulas derived from historical populations 2. Study population-based formulas derived from the populations under study 3. Individual-based formulas derived for each individual in the population under study
All three methods are based on exploring the mathematical relationship between the QT-interval and the RR-interval, but they use different populations for finding this relationship. The exploration of this mathematical relationship amounts to finding a function and its numerical coefficients or finding the specific numerical coefficient(s) for either a prespecified function or best fitting mathematical function (linear or nonlinear) from among a number functions that models the relationship between the QT-interval and the RR-interval for a set of ECGs from a population of multiple individuals (or from one individual in the case of Individual-based formulas). The mathematical function is then translated into a correction formula using the numerical coefficient that was found in the data fitting process. The same formula is then applied to all ECGs for which a QTc is being computed. Therefore, for example, a set of QT-interval measurements and associated RR-interval measurements could be fitted to the mathematical function:
QT = β * RRα
The value of the coefficient α that is found to give the best fit for the data might be 0.25. Then the correction formula for QTc would be: QTc = QT / RR0.25.
6.1.1 Historical Population-Based Formula from a Historical Population In a historical population, this would be a group of normal, healthy persons, generally with 1 ECG from each person. Due to the normal variance between different populations, multiple researchers using different groups of subjects have derived different formulas even when fitting their respective data to the same mathematical function. The most commonly used historical population-based correction formulae were proposed in 1920 by Bazett and separately by Fridericia. Unfortunately, each formula can lead to bias for some clinically relevant values of HR as will be illustrated below. For an extensive list of 31 such historical correction formulas, including those listed

Version 1.0
8
below, based on multiple mathematical functions, see a manuscript by Malik (2002). As indicated above, each of these formulas could be expressed using HR, where RRmiliseconds = ((1/(HRbeats-per-minute/60))*1000)
(i) Bazett: QTc = QT/ RR1/2
(ii) Fridericia: QTc=QT/RR1/3 (iii) Framingham: QTc=QT+(0.154*(1-RR))
(iv) Van de Water: QTc=QT–((0.087*(1-RR))
It is reasonably well known that the Bazett formula under-corrects at faster heart rates (over 60 bpm) and conversely over-corrects at slower heart rates. That is, at faster HRs (smaller R- intervals), the computed QTc is ‘larger than it should be’ and at slower HRs (larger RR-intervals), the computed QTc is ‘smaller than it should be’. When Bazett corrected QTc is plotted against RR interval and a regression line is plotted, the slope is negative (Figure 3; with a perfect correction, the slope of the regression line would be “0” as described above). In spite of this, Bazett’s formula is still the most widely used for clinical correction of QT intervals. However, it is becoming more acceptable in regulatory documents to use the Fridericia formula correction, without use of the Bazett formula (ICH, 2012; Question 11), along with additional correction results as described below.
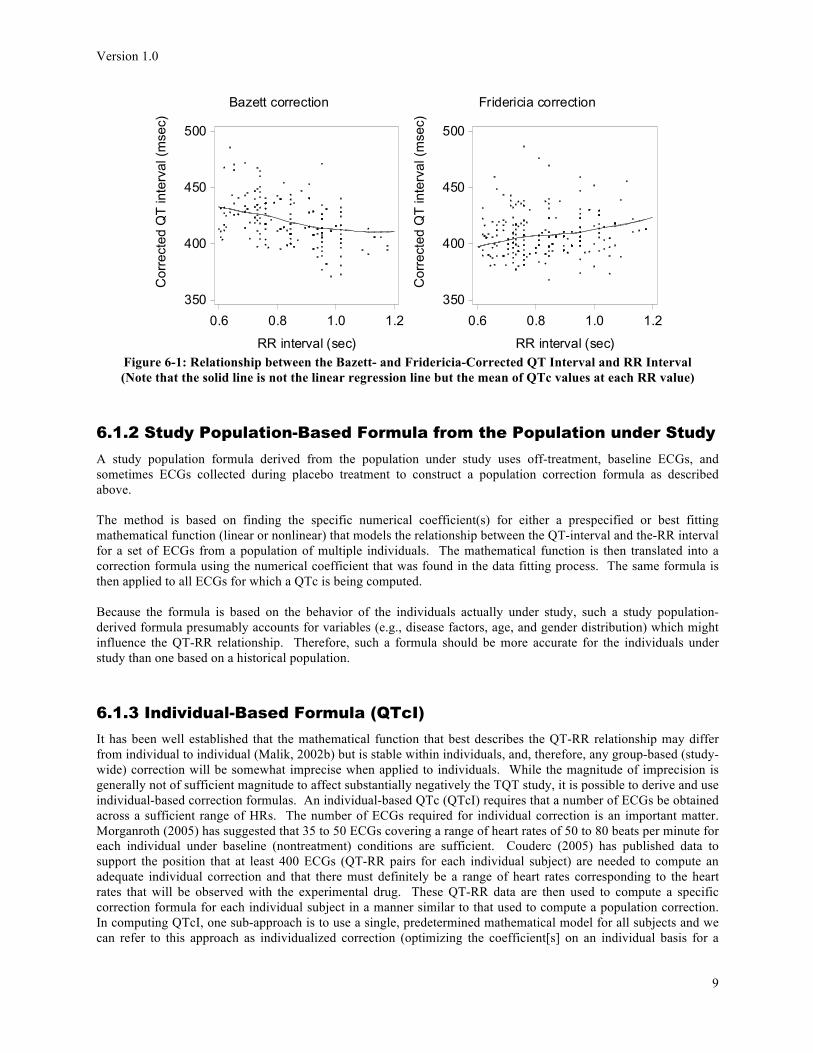
Version 1.0
9
Figure 6-1: Relationship between the Bazett- and Fridericia-Corrected QT Interval and RR Interval (Note that the solid line is not the linear regression line but the mean of QTc values at each RR value)
6.1.2 Study Population-Based Formula from the Population under Study A study population formula derived from the population under study uses off-treatment, baseline ECGs, and sometimes ECGs collected during placebo treatment to construct a population correction formula as described above. The method is based on finding the specific numerical coefficient(s) for either a prespecified or best fitting mathematical function (linear or nonlinear) that models the relationship between the QT-interval and the-RR interval for a set of ECGs from a population of multiple individuals. The mathematical function is then translated into a correction formula using the numerical coefficient that was found in the data fitting process. The same formula is then applied to all ECGs for which a QTc is being computed. Because the formula is based on the behavior of the individuals actually under study, such a study population-derived formula presumably accounts for variables (e.g., disease factors, age, and gender distribution) which might influence the QT-RR relationship. Therefore, such a formula should be more accurate for the individuals under study than one based on a historical population.
6.1.3 Individual-Based Formula (QTcI) It has been well established that the mathematical function that best describes the QT-RR relationship may differ from individual to individual (Malik, 2002b) but is stable within individuals, and, therefore, any group-based (study-wide) correction will be somewhat imprecise when applied to individuals. While the magnitude of imprecision is generally not of sufficient magnitude to affect substantially negatively the TQT study, it is possible to derive and use individual-based correction formulas. An individual-based QTc (QTcI) requires that a number of ECGs be obtained across a sufficient range of HRs. The number of ECGs required for individual correction is an important matter. Morganroth (2005) has suggested that 35 to 50 ECGs covering a range of heart rates of 50 to 80 beats per minute for each individual under baseline (nontreatment) conditions are sufficient. Couderc (2005) has published data to support the position that at least 400 ECGs (QT-RR pairs for each individual subject) are needed to compute an adequate individual correction and that there must definitely be a range of heart rates corresponding to the heart rates that will be observed with the experimental drug. These QT-RR data are then used to compute a specific correction formula for each individual subject in a manner similar to that used to compute a population correction. In computing QTcI, one sub-approach is to use a single, predetermined mathematical model for all subjects and we can refer to this approach as individualized correction (optimizing the coefficient[s] on an individual basis for a
Bazett correction
Cor
rect
ed Q
T in
terv
al (m
sec)
350
400
450
500
RR interval (sec)
0.6 0.8 1.0 1.2
Fridericia correction
Cor
rect
ed Q
T in
terv
al (m
sec)
350
400
450
500
RR interval (sec)
0.6 0.8 1.0 1.2

Version 1.0
10
single correction formula). An alternative sub-approach is to fit the individual subject’s data to several preselected mathematical models and use the best mathematical model for each individual subject (model with the best fit to the data and that results in flattest regression line after correction (QTcI vs. RR)) and we can refer to this approach as individualized individual correction (optimizing the actual correction formula and its coefficient[s] on an individual basis). Malik et al. (2004) have described 12 mathematical models that could be considered when finding an individual best-fit model for a given subject. As such, this latter method for computing QTcI, individualized individual correction, is probably the best. However, either type of individual correction formula computation is also very labor intensive and costly to use. Some researchers have developed methods of assessing changes in ventricular repolarization based on the QT interval that do not rely on an explicit correction of the QT- interval for HR (the RR-interval). These methods are particularly important when the experimental drug results in marked changes in autonomic nervous system tone and HR. These changes can be so large that it will be difficult to obtain ECG data at heart rates that will be observed during treatment with the experimental drug, which would raise concerns about the validity of any correction factor. Discussion of these alternatives beyond the introduction of the concept is outside the scope of this document but can be reviewed in the manuscript by Garnett et al. (2012). These methods would generally rely on continuous recording data.
6.2 Thorough QT (TQT) Study Design
6.2.1 Brief Background 6.2.1.1 Historical Reason for the TQT Study
Jervell and Lange-Nielsen (1957) described correlations between hereditary long QT intervals and sudden death. Smirk and Palmer (1960) noted that initiation of ventricular depolarization (R waves) prematurely occurring before the complete repolarization of the ventricle following the preceding depolarization (during the T waves – referred to as “R-on-T Pattern”) increase the risk of ventricular arrhythmia. Torsade de Pointe (TdP), a specific type of ventricular tachyarrhythmia (fast arrhythmia), was first described in a publication by Dessertenne (1966). Although some drugs that had been developed as anti-arrhythmic agents also altered ventricular repolarization as evidenced by prolonged QTc, it was not widely appreciated that non-cardiac drugs could also have this property. The use of non-sedating antihistamines, e.g. terfenadine and astemizole, from 1985 to 1999 provided an important case study of the public health issues with the widespread use of non-cardiac drugs with such cardiac effects. Initial reports of cardiac arrhythmias, including TdP, were predominately associated with high blood concentrations of these antihistamines subsequent to overdose. Given the metabolic pathway of these drugs, arrhythmias were eventually reported subsequent to co-administration with drugs and substances that slowed the metabolism of terfenadine and astemizole, including grapefruit juice (also resulting in high blood concentrations). Despite warning letters to physicians and restricted product labeling in 1992, inappropriate medications continued to be co-administered with these drugs. Both drugs were withdrawn in 1999 from use in the U.S. after safer alternatives were developed. The high visibility of the association between non-sedating antihistamines and fatal ventricular arrhythmias prompted extensive research into the mechanisms by which drugs cause these cardiac arrhythmias. Although many details remain unknown, current research suggests that most drugs with strong arrhythmic potential interfere with a specific potassium channel in cardiac muscle fiber that functions to repolarize the muscle fiber cells. Partially or completely blocking the potassium channel results in delayed repolarization of the muscle fiber cells. Delayed repolarization increases the time required to restore the normal resting potential prior to the next depolarization for the next muscle contraction. Arrhythmias such as TdP are possibly triggered by the initiation of the R-waves (beginning the depolarization of the ventricles) during the period of delayed repolarization while the ventricles are still partially depolarized. In summary, drugs that delay ventricular repolarization might place a person at increased risk of a fatal ventricular arrhythmia. Again, delayed ventricular repolarization is manifested on the ECG tracing as a prolonged QTc. QTc is clearly recognized as an imperfect biomarker for increased risk of fatal arrhythmia because it can be increased by a number

Version 1.0
11
of drugs that are not associated with a significant incidence of such arrhythmias. None-the-less, an increase in QTc is considered an important risk factor and any drug-induced increase is considered important to assess and quantify. On an individual basis, the increase in QTc generally needs to be substantial to place the patient at risk, but for a potential new drug, even a slight mean increase1 in QTc can be clinically meaningful, in that some degree of risk cannot be excluded in a small number of individuals in a large population that will receive the drug during its use in clinical medicine. The TQT study is considered the most precise way of studying the potential drug effect on QTc in human subjects.
6.2.1.2 Study Design Background Considerations
Clinical studies to detect QTc mean increases as small as 5 msec face significant challenges because of the substantial variability in QT intervals. The first source of variability is the process of acquiring and measuring the QT interval. Placement of ECG electrodes, choice of lead(s) to be measured, standardization of ECG machines, choice of media (paper vs. digital), and variability in expert measurement of the QT interval comprise critical components of the process. QT intervals are characterized by substantial inter- and intra-subject variability apart from that engendered by acquisition and measurement. Sources of inter-subject variability can include a genetic predisposition to long QT intervals, electrolyte concentrations, autonomic activity, age, and sex. Intra-subject variability is strongly influenced by diurnal rhythms (transitioning to sleep from wakefulness and vice versa) that influence autonomic tone and heart rate. Dose selection, duration of dosing, timing of ECG measurements, patient population, and control of factors influencing variability will need to be addressed in any study designed to evaluate QT interval. While the TQT study is considered the most definitive study of the potential influence of a drug on QT interval, it might suffer from limitations due to sample size, the health of the subject population, and many other factors that cause the drug administration in the study to be different from how the drug will be used in broad clinical practice. This brief background provided below is informed primarily by the May 2005 ICH-E14 document [ICH, 2005], which describes the basic conduct, purpose, and expected analyses of the TQT study as well as its update in a subsequent Q&A document (ICH, 2012) The purpose of a TQT study is to evaluate the potential for an experimental drug to delay cardiac ventricular repolarization, which the study does through evaluation of changes in QTc during drug treatment; and also to demonstrate that the study is capable of detecting differences in the variability that can be observed during placebo treatment (random variability; approximately 5 msec), so as to confirm that any lack of detected change is due to actual lack of change rather than lack of assay sensitivity. These TQT studies are generally conducted in healthy volunteers, highly screened for normal cardiac electrical activity, for ease of precise measurement of the QT- interval, and to avoid additional confounding factors. The TQT study designs can be a crossover design or a parallel design discussed in more detail below. In general, the treatments are:
1. A dose of the experimental drug that is several times higher, if possible, than the intended maximum dose, in order to account for drug-drug interactions and/or genetic metabolic enzyme deficiencies that might lead to greater exposure to the experimental drug than otherwise intended with a given dose during routine clinical use
2. Placebo 3. A positive control for purpose of demonstration of assay sensitivity (most often moxifloxacin, usually oral
but sometimes intravenous) 4. Optionally, a dose of the experimental drug that is within the intended therapeutic range (generally the
maximum intended therapeutic dose)
1 >5 milliseconds (msec) would be considered to exceed random variability (Malik, 2001) and a mean increase of ≥10 msec could be of
regulatory interest (ICH, 2005).

Version 1.0
12
The administration of the active control has been allowed by regulators to be open-label, but the administrations of the experimental drug dose(s) and placebo are double-blind, and ECG measurements and readings are performed by persons completely blinded to associated treatments, subject details, and date/time of the ECG.
6.2.1.3 Days of ECG Collection and Time points of ECG Collection on the Days of Collection
ECGs are collected as a set of replicates (in close temporal proximity, e.g., 3 ECGs collected at 1-minute intervals) of 10 seconds in duration and utilizing all 12 leads. In analyses, the QTc values of the replicates will be averaged before analysis of differences in changes in QTc to reduce the signal-to-noise ratio and improve the accuracy of the measurement. When discussing the collection of ECGs below, “ECG” will refer to the set of replicate ECGs. ECGs can be collected as conventional ECGs, or they can be extracted from a continuous high fidelity ECG recording. Experimental drug and metabolite concentrations are often collected for assay immediately after the time of ECG collection for PK/PD analysis, which can be a useful secondary analysis pertinent to the potential influence of the experimental drug on ventricular repolarization. The timing and collection of replicate ECGs are guided by the known properties (e.g., PK) of the drug and its metabolites.
6.2.1.3.1 Baseline ECGs
In general, in the analyses of QTc, baseline QTc values are subtracted from on-treatment QTc values to create a “single ∆” value that is “change in QTc” and this “change in QTc” is compared between treatments. Several alternative baselines exist as will be further discussed in Section 4 below that describes alternative analyses that are in large part influenced by the definition of baseline. Baseline ECGs are collected:
• On the day (or for multiple days) preceding the day of first dose administration of each treatment; § If this type of baseline is used, ECGs are collected at multiple time points that match the
time points at which ECGs are collected on-treatment § If ECGs are collected on multiple days, then QTc values from those days can be averaged
for the baseline value used in analyses; this multiple baseline day collection is rarely done • The averaging can be for each time point when a time-matched baseline is being
used (time-matched) or across all time points (time averaged), if a time averaged baseline is being used (see Section 4. Baseline and Treatment Difference below for a more detailed description of baseline alternatives)
§ Baseline day(s) and time points are the same for each treatment to maintain blind § Although consideration might be given to using a single, common baseline for each
treatment in a crossover study, either before the first treatment period for all subjects or with a subset of subjects assigned by random allocation before each of the treatment periods (Section 3.2 discusses study design in more detail), this is not done
§ This baseline that collects multiple ECGs at the same time points as the ECGs will be collected while on treatment on at least one day that precedes the first administration of test is necessary for parallel studies (allows time-matched baseline)
and/or
• Immediately preceding the first dose administration of each treatment § ECGs would be collected at several time points shortly before first dose administration
such as 60 minutes, 45 minutes, 30 minutes, 15 minutes, and immediately before treatment administration
§ This baseline is generally used for crossover studies and not allowed by regulators for parallel design studies
§ This baseline ECG collection can be combined with the ECG collection on the day or days preceding treatment administration, resulting in complex baseline definitions and treatment difference definitions

Version 1.0
13
6.2.1.3.2 On-treatment ECGs
The days on which ECGs are collected and the time points of collection are determined by the PK characteristics of the test drug. The intent of the study is to measure QTc at that time at which a maximum increase in QTc would occur if the drug, or relevant metabolites, does increase QTc. In crossover studies, it is often the case that the drug is sufficiently well tolerated that desired supratherapeutic exposure could be achieved with a single dose, so only a single dose of treatments is given. Sometimes in crossover studies, it is necessary to titrate the drug up to intended exposure with multiple doses over multiple days. In parallel studies, dosing is often extended over multiple days before intended exposure is reached. For single-dose studies, ECGs are collected on the day of treatment administration at a time point shortly before the time of the maximum drug concentration (Tmax), around Tmax, and should continue even after Tmax to evaluate any delayed effects of the drug or its metabolites on cardiac repolarization. Depending on the PK of drug and metabolites, the ECG collection might continue for one or more days following the day of drug administration. For multiday dose studies, ECGs are collected according to the schedule described in the paragraph above but beginning on the day that the drug reaches steady state or intended exposure has been achieved. In some multiday dose studies, ECGs will also be collected following the first dose at identical times. To demonstrate assay sensitivity, ECGs should also be collected close to the Tmax of the positive control. Replicate ECGs should be collected on the same days and at the same time points in all treatment groups to ensure that blinding is maintained. The diagrams below show how ECG data are organized within 10-second ECGs, and how those 10-second ECGs are organized within and across time points. Although analysis methods that use all the data from continuous monitoring over a long period (e.g., 24 hours) have been developed, the analysis usually assumes that data is organized by time points. ECGs should be recorded (or extracted from continuous recordings) in triplicate as noted above (replicates, number can vary but will generally be 3 and can be more), 30-120 seconds apart, to account for inherent variability; each recording lasting 10 seconds (these 10-sec ECGs are either recorded as 10-sec ECGs or extracted from continuous recording of the ECG record that is digitally stored for later processing, typically in 24-hour increments). Figure 5 is illustrating the on-treatment collection of triplicate ECGs, as an example of the replicate collection, on a single day of ECG collection following treatment administration.
Figure 6-2: Illustration of 1 of 12 Leads of Continuous ECG recording from which a 10-sec ECG can be extracted Each cycle, in a normal ECG obtained from a healthy person, consists of a P-QRS-T complex and the subsequent isoelectric activity before the next P-QRS-T complex as described above in Section 1.
24 hours
Beat 1 (7:59:00.00)
Beat 2 (7:59:01.30)
Beat 3 (7:59:02.15)
Beat 12 (7:59:10.05)
Beat 100,800
Extracted 10 - second ECG ≈ 12 P - QRS - T
… …

Version 1.0
14
Figure 6-3: Illustration of the concepts of recording multiple replicate ECGs at multiple time points subsequent to treatment administration (recording would also occur at baseline) ECGs are taken after subjects have rested, but not sleeping, for at least 5 to 10 minutes in the supine position (in an attempt to obtain a stable heart rate under similar physiological conditions at each time of collection). If the ECGs are to be extracted from a continuous recording, then the subjects rest as they would for actual 10-second ECG recordings.
6.2.2 Specific designs The examples of study designs presented below illustrate specific TQT study designs. A typical TQT study is designed as double-blind (partial double-blind as in some cases the investigator might not be blind to administration of the active control), placebo- and positive-controlled to determine whether the test treatment fails to prolong the QTc (primary statistical test is noninferiority) and to demonstrate the assay sensitivity using the positive control treatment in the study population. Traditional TQT studies employ parallel or crossover designs, are generally designed with equal study duration, and sample size for the different treatment arms or periods.
6.2.2.1 Parallel Studies
Under certain circumstances (related to the PK characteristics of the test drug), a parallel design may be preferred for a TQT study. Such circumstances include (ICH, 2005):
• Drugs with long elimination half-lives for which lengthy time intervals would be required to achieve steady-state and complete washout
• If carryover effects are prominent for other reasons, such as irreversible receptor binding or long-lived active metabolites
• If multiple doses of the investigational drug are required to evaluate the effect on QT/QTc intervals

Version 1.0
15
Example of TQT - Parallel Study Below is the study schema diagram for a parallel study. This study has 4 treatment arms (placebo, positive control, therapeutic study drug dose, and supratherapeutic study drug dose), which correspond to the 4 possible left-to-right "paths" through the study. Moxifloxacin has become the standard positive control with a well characterized (peak effect and time course), expected influence on QTc in healthy subjects with a mean increase in QTc in the range of 10 – 15 msec. Other positive control compounds are possible (e.g., low dose ibutilide).
Figure 6-4: Parallel Study Design Schema for Example TQT Study 1 T = Therapeutic Dose (DRUG A 1 MG), ST = Supratherapeutic Dose (DRUG A 100 MG)
6.2.2.2 Crossover Studies
In comparison to parallel studies, crossover studies have at least two potential advantages: • A smaller number of subjects are typically required. Subjects serve as their own controls, resulting in
reduced variability of differences related to inter-subject variability. • Heart rate correction approaches based on individual subject data may be more feasible (as baseline ECGs
are collected before each treatment period; therefore, more ECGs are available for each subject for computation).
Example of TQT – Crossover Study Below is the study schema diagram for a crossover study. In this example, subjects were screened for eligibility and then randomized in a 1:1:1:1 ratio to receive 1 of 4 treatment sequences (Williams design). As with the parallel design, the therapeutic dose is optional. If the test drug is sufficiently well tolerated such that the necessary supratherapeutic exposure can be achieved with a single dose and washout is not lengthy, then these crossover studies often involve administration of a single dose of drug. If the drug must be titrated to reach required exposure but that titration period is not too lengthy, and washout is not lengthy then the crossover design can be used. When that titration or washout is lengthy, the parallel design is used. Sponsors make the decision regarding whether a study should be crossover or parallel based on the required titration and or washout time. As in most crossover studies, the treatment arms are distinguished by the order of treatments, with all treatments present in each arm.
Figure 6-5: Crossover Study Design Schema for Example TQT Study 2 T = Therapeutic Dose (DRUG A 1 mg), ST = Supratherapeutic Dose (DRUG A 100 mg) A washout period sufficient to clear all drug exposure would be present between treatment periods.
Note: Moxifloxacin is one example of a positive control.
Note: This is an optional arm.

Version 1.0
16
6.2.2.3 Non-standard designs
A design has been used for a parallel TQT study that required lengthy treatment periods in which the positive control treatment was embedded in the placebo treatment arm (Malik, 2008b). Discussion of this design alternative is beyond the scope of this White Paper, but the reader can review the cited manuscript. When both a therapeutic dose and a supratherapeutic dose are studied, they might be contained in a single arm of a parallel study with the supratherapeutic dose following the therapeutic dose (dose escalation) or the supratherapeutic dose can follow the therapeutic dose (dose escalation) in a crossover study. When such designs are employed, the supratherapeutic dose clearly does not have the same design characteristics as the other treatments and questions regarding potential bias can arise. Discussion of such design alternatives is beyond the scope of this White Paper.
6.3 Baseline and Treatment Difference (Drug Effect) In this section, three different baseline definition alternatives are described. For each baseline definition, the resulting definition of treatment differences is described. Note that this is not an exhaustive list of possibilities. For example, triple-delta (∆∆∆QTc) treatment difference definitions are possible where both lead-in day ECGs are collected at matched time points to the time points of collection on the treatment day and one or more ECGs are collected immediately before treatment administration (and at the same time point on the lead-in days), essentially combining 6.3.1 and 6.3.3 below. Multiple lead-in days could be used to create averaged lead-in day values to be used for a time-matched baseline. Potentially, on-treatment QTc values could be compared without any baseline difference comparison, especially in crossover studies where each subject is acting as his/her own control (single-delta - ∆QTc).
6.3.1 Time-Matched Lead-in Day Baseline; Double-Delta Treatment Difference
For time-matched baseline, the baseline for each period is the average of the replicate set values at a time point on the lead-in (baseline) day (Day -1) that corresponds to the post-dose time point. ECGs are collected or extracted from continuous recording in replicate sets (usually 3 replicates about a minute or so apart) at each bj and Xij. The average of the replicates is used for analysis. With the original ICH E-14 guidance, this was the standard baseline definition for both crossover and parallel studies. With the publication of the ICH E-14 Q&A (ICH, 2012; Question 6), the requirement for this baseline definition for crossover studies was relaxed (See Section 6.3.3 below)
For crossover design, ΔΔQTcij is computed for each subject: ΔΔQTcij = X!" − b! !"#$ !
− X!" − b! !"#$%&' where
i=1, 2, … d, j=1, 2, … n; d=days postdose and n=time point. ΔΔQTcij is the difference between drug and placebo in the change from baseline (time-matched) in QTc at each time point for each day of treatment on an individual subject basis. For a parallel design, (Xij – bj) would be averaged across subjects: ΔΔQTc!" = X!" − b! !!"# !
− X!" − b! !"#$%&'
ΔΔQTcij is the difference between drug and placebo in the average across subjects of the change from baseline (time-matched) in QTc at each time point for each day of treatment.
6.3.2 Time-Averaged Lead-in Day Baseline; Double-Delta Treatment Difference
For time-averaged baseline from a lead-in (baseline) day, the baseline for each period is the average of all values for all the replicate sets of ECGs on the baseline day (e.g. Day -1, 1 hr., 2 hr., 3 hr., 4 hr., etc.). ECGs are collected or

Version 1.0
17
extracted from continuous recording in replicate sets (usually 3 replicates about a minute or so apart) at each bj and Xij. The average of the replicates is used for analysis. Several statistical manuscripts have advocated this baseline definition over the time-matched lead-in day baseline for parallel studies (Meng, 2010) and both crossover and parallel studies (Sethuraman, 2008) but this has not become a regulatory standard.
For crossover design, ΔΔQTcij is computed for each subject: ∆∆QTc!" = X!" − b!"# !"#$ !
− X!" − b!"# !"#$%&'
where b!"# = b!/n; i=1, 2, … d, j=1, 2, … n; d = days postdose and n = time point. ΔΔQTcij is the difference between drug and placebo in the change from baseline (time-averaged) in QTc at each time point for each day of treatment on an individual subject basis. For a parallel design, (Xij – bavg) would be averaged across subjects: ΔΔQTc!" = X!" − b!"# !"#$ !
− X!" − b!"# !"#$%&' ΔΔQTcij is the difference between drug and placebo in the
average across subjects of the change from baseline (time-averaged) in QTc at each time point for each day of treatment.
6.3.3 Predose Averaged Baseline; Double-Delta Treatment Difference For predose averaged baseline, ECGs are collected or extracted as replicate sets (usually 3 replicates about a minute or less apart) at predose in close temporal proximity to treatment administration (e.g., 15 min intervals and immediately before treatment administration on the same day of treatment administration) and as replicate sets (usually 3 replicates about a minute or so apart) at each Xij post dose. The average of all the replicates collected predose is used as the baseline for analysis. This baseline definition has the advantage of eliminating the necessity of an inpatient lead-in day from the experimental design with all the monetary and operational expense for each treatment period. This baseline definition is now accepted for crossover studies that are both single-dose and multiple-dose administered over multiple days (ICH, 2012; Question 6).
For crossover design, ΔΔQTcij is computed for each subject: ∆∆QTc!" = X!" − b! !"#$ !
− X!" − b! !"#$%&' where
b! = b!/k; i=1, 2, … d, j=1, 2, … n; d = days postdose, k = number of predose replicates, n = time point. ΔΔQTcij is the difference between drug and placebo in the change from baseline (predose-matched) in QTc at each time point for each day of treatment on an individual subject basis. For a parallel design, the (Xij – bj)’s would be averaged across subjects: 𝛥𝛥𝑄𝑇𝑐!" = 𝑋!" − 𝑏! !"#$ !
− 𝑋!" − 𝑏! !"#$%&' ΔΔQTcij is the difference between drug and placebo in the
average across subjects of the change from baseline (predose-averaged) in QTc at each time point for each day of treatment.

Version 1.0
18
7 Analysis 7.1 Primary analysis There are two hypothesis tests to be performed in a thorough QT/QTc studies:
1. The hypothesis test to confirm no study drug effect that results in prolongation of the QT/QTc as compared to the placebo group;
2. The study is capable of detecting differences in QT/QTc, however, small it may be (to establish the assay sensitivity) by demonstrating the QT/QTc effects of the active control that are already known.
7.1.1 Testing of QT prolongation The primary endpoint should be the time-matched mean difference between the drug and placebo after baseline adjustment at each time point. According to the ICH E14 (Section 2.2.4), the test drug is classified as negative (lack of evidence of QT/QTc prolongation) if the upper bound of the one-sided 95% confidence interval for the largest time matched mean difference between the drug and placebo excludes 10 msec. This definition was chosen to provide reasonable assurance that the mean effect of the study drug on the QT/QTc is not greater than around 5 ms. When the largest time-matched difference exceeds the threshold, the study is termed “positive,” (lack of prolongation effect cannot be established) and additional electrocardiogram safety evaluation in subsequent clinical studies should be performed. The QT intervals (means of replicates) are usually measured at multiple time points to provide reasonable assurance that the mean difference between study drug and placebo on the QT/QTc interval is not greater than the pre-defined threshold. In practice, an intersection-union test (IUT) is applied for its practicality, ease of implementation, and conservatism with respect to assessing QT/QTc prolongation. It is the uniformly most powerful unbiased test (Berger and Hsu, 1996). The hypothesis is specified as follows:
0 drug( ) ( ): {( ) 10}, 1,2,...,i i
T Tt placebo tH i nµ µ− ≥ =U , versus
1 drug( ) ( ): {( ) 10}, 1,2,...,n i i
T Tt placebo tH iµ µ− < =I
where drug( )iT
tµ and )( itplaceboµ are the mean change from baseline of QT for drug and placebo respectively, at time
point ti. The statistical model for estimating the treatment effects and the confidence intervals depend on the study design and other factors. An analysis of covariance model (ANCOVA) or repeated measures mixed effects model is usually used to estimate the treatment effect and the confidence intervals. For crossover designs, the ANCOVA model usually includes treatment, time, period, sequence, and the time-by-treatment interactions as fixed effects, and “pre-Dose averaged” baseline as a covariate. For parallel designs, the model usually includes treatment, time as fixed effects, and “Time-Matched” baseline as a covariate. The ANCOVA model using day-averaged (time-averaged; 6.3.2 above) baseline is recommended for the analysis of parallel-group thorough QT/QTc studies (Sun and Quan etc. 2012). Other covariates should only be added in an exploratory fashion (the simpler model being the primary analysis) only if there are excellent clinical reasons for including them.
7.1.1.1 Multiplicity Issues
For the test drug to placebo comparison, as noted above, an intersection-union test (IUT) method has been proposed and most frequently used as the primary method of analyzing the through QT/QTc study. The IUT method controls the Type I error. Specifically, the comparison between the test drug and placebo requires no adjustment for multiplicity and thus the standard one-sided 95% confidence intervals are used at all post-dose

Version 1.0
19
time points. However, the IUT method may potentially lead to false positive trial results (failing to reject inferiority [not finding non-inferiority] in this specific case of TQT study analysis). The false positive rates depend on several factors including variability of the study, sample size, the number of time points, and the true mean difference to be detected. The probability of incorrectly not being able to reject a potentially clinically meaningful QT/QTc effect increases (or statistical power decreases) with the number of post-dose time points (Patterson et al., 2005).
7.1.2 Assay Sensitivity The confidence in the ability of the study to detect QT/QTc prolongation can be greatly enhanced by the use of a concurrent positive control group to establish assay sensitivity. The positive control should have an effect on the mean QT/QTc interval of about 5 ms (i.e., an effect that is close to the QT/QTc effect that represents the threshold of regulatory concern, around 5 ms). However, as moxifloxacin is the accepted regulatory positive control standard, an effect in the 10-15 ms range for the positive control is acceptable. In the ICH E14 Question and Answers in 2012 [1], FDA clarified how to access the adequacy of the positive control
in the QTc study. There are two conditions required for ensuring assay sensitivity:
1. The positive control should show a significant increase in QTc; i.e., the lower bound of the one-sided 95% confidence interval (CI) must be above 0 ms. This result shows that the trial is capable of detecting an increase in QTc, a conclusion that is essential to concluding that a negative finding for the test drug is meaningful.
2. The study should be able to detect an effect of about 5 ms (the QTc threshold of regulatory concern). Therefore, the size of the effect of the positive control is of particular relevance. It determines the threshold of the lower bound.
a. If a positive control has a known effect of greater than 5 ms (e.g., 10 ms), assay sensitivity will be
established if the lower bound of the one-sided 95% confidence interval for the mean treatment difference between the positive control and placebo is above 5 ms. This approach has proven to be useful in many regulatory cases. However, if the positive control has too large an effect, the study’s ability to detect a 5 ms QTc prolongation might be questioned. 𝐻! : 𝜇!"#$%& !! − 𝜇!"#$%&' !! ≤ 5!∈! , versus 𝐻! : 𝜇!"#$%& !! − 𝜇!"#$%&' !! > 5!∈! .
Where R is a subset of a pre-selected subset of time points; 𝜇𝑎𝑐𝑡𝑖𝑣𝑒 𝑡𝑖 and 𝜇𝑝𝑙𝑎𝑐𝑒𝑏𝑜 𝑡𝑖
are mean changes from baseline of QT for active drug and placebo respectively, at time point ti. The authors note that if moxifloxacin is used, then this criterion implicitly requires that the experimental group respond to moxifloxacin in the same manner as historical control groups. Even if the point estimate of the difference is only 5 ms, the study will be declared as not having demonstrated assay sensitivity because moxifloxacin is “known” to produce a certain effect. The authors recommend, based on their experience with oral moxifloxacin, to reduce risk of failing to establish assay sensitivity by using IV moxifloxacin to avoid potential issues with other factors such as food effect.
b. If a positive control has a known effect close to 5 ms, assay sensitivity can be demonstrated if the point estimate of the maximum mean difference with placebo is close to 5 ms, and the lower bound of the one-sided 95% confidence interval for the mean treatment difference between the positive control and placebo is above 0 ms. 𝐻! : 𝜇!"#$%& !! − 𝜇!"#$%&' !! ≤ 0!∈! , versus 𝐻! : 𝜇!"#$%& !! − 𝜇!"#$%&' !! > 0!∈! .

Version 1.0
20
Where R is a subset of a pre-selected subset of time points; 𝜇𝑎𝑐𝑡𝑖𝑣𝑒 𝑡𝑖 and 𝜇𝑝𝑙𝑎𝑐𝑒𝑏𝑜 𝑡𝑖
are mean changes from baseline of QT for active drug and placebo respectively, at time point ti.
The analyses model of the positive control compared to placebo is similar to the analyses of the test drug compared to placebo. 7.1.2.1 Multiplicity Issues
Assay sensitivity is usually defined in terms of the statistically significant difference between the positive control and placebo at one or more post-dose time points. Due to the multiple comparisons, the probability of demonstrating assay sensitivity is inflated. To avoid the inflation, the clinical trial sponsor can consider the following options:
• Perform the assay sensitivity analysis at fewer post-dose time points. Since the effect of a positive control on QTc interval is generally well understood, it is reasonable to restrict the positive control versus placebo comparisons to the number of time points when the QTc effect of the positive control is most pronounced. For example, if moxifloxacin 400 mg serves as the positive control, significant QT interval prolongation is likely to occur during the 2-4-hour window after the dose and the sponsor can consider excluding the post-dose ECG recordings collected after 10 hours post-dose from the assay sensitivity analysis.
• Perform a multiplicity adjustment. When performing this adjustment, it is important to utilize a multiple testing procedure that takes into account correlations among the estimated treatment differences at post-dose time points (e.g., resampling-based multiplicity adjustments, Westfall and Young, 1993). Basic multiple tests such as the Bonferroni test may be avoided because they tend to be very conservative in multiplicity problems. The choice of which multiplicity adjustment method to use must be pre-specified for a specific study.
7.1.3 Categorical Analyses
Categorical (or outlier) analyses are often performed to gain an impression of the proportion of study participants who exceed predefined upper reference limit values. Outlier reference limits can be defined in terms of absolute values, change from baseline values or a combination of change from baseline and absolute value. The following thresholds are often used (but alternative limits may be used): Absolute QTc interval prolongation:
• QTc interval >450 msec
• QTc interval >480 msec
• QTc interval >500 msec
Change from baseline measurement in QTc interval:
• QTc interval increase >30 msec
• QTc interval increase >60 msec

Version 1.0
21
It has to be noted that the limits above were selected based on the experience of the writers of this white paper and ICH E14 guidance. As these limits have their basis in QTcB where QTcF is most commonly used, it is strongly recommended for the reader to investigate recent literature from the regulators before defining their analysis, as these recommended limits may change in the future. Change limits should be put in raw numbers or can be percentage adjusted if empirically derived percentage limits are available. All outliers should be summarized for each treatment group on at each time point and overall basis. The outlier summary tables should include counts of subjects (at each time point and overall). Therefore, if a subject experienced more than one subject of a particular outlier event, the subject should be countered only once for that event. Statistical analyses comparing treatments may be performed but is considered out of the scope of this White Paper.
7.1.4 Morphological (Qualitative) Analyses
Morphological (qualitative) abnormal findings (e.g., rhythm; axis; conduction; evidence of ischemia, injury, or infarction; evidence of hypertrophy; other ST abnormalities; other T-wave abnormalities; U-wave abnormalities; findings consistent with pericarditis, electrolyte abnormalities, COPD, etc.) in the ECG waveform should be described and the data presented in terms of the number and percentage of subjects in each treatment arm who had changes from baseline that represented the appearance or worsening of the morphological abnormality (e.g., tables of the incidences of the observed treatment emergent abnormalities by specific abnormal finding, not just by category of findings). Special attention can be directed at abnormalities and/or changes in the appearance of the T-wave/U-wave that might be indicative of delayed repolarization, such as double humps ("notched" T wave), indistinct terminations (TU complex), delayed inscription (prolonged isoelectric ST segment), widening, flattening, and inversion. T wave alternans (beat-to-beat variability in the amplitude, vector, and/or morphology of the T wave), is considered to be a harbinger of ventricular arrhythmias and might receive special attention with respect to occurrence of any of these findings. Several of these T-wave/U-wave findings can be numerically quantified and analyzed, but this is not a routine expectation in TQT study analyses. While the predictive value of morphological analyses is not well characterized (even if the drug does have an effect on the ECG, these abnormal morphological findings will be observed with low frequency if at all in a TQT study), differences in the incidence of abnormalities between treatment arms, if observed, have proved to be informative. Statistical analyses comparing treatments may be performed but is considered out of the scope of this White Paper.
7.2 Concentration-Response Relationship (CRR) Why a. TQT study is negative (non-inferiority is supported by study results) When the primary analysis shows evidence of lack of meaningful QT/QTc changes, there still may be small QTc changes taking place upon administration of the investigational drug at supra-therapeutic doses below the threshold of regulatory concern. A CRR analysis can clarify whether this is the case or not and inform drug development (e.g. predict the QTc changes at doses and in subpopulations/factors that were not studied directly). It can also help in increasing confidence in regards to the timepoints chosen for the primary analysis by investigating possible delayed effects. b. TQT study is positive (cannot reject inferiority based on study results) When the primary analysis does not support lack of QT/QTc prolongation, CRR analysis is an excellent tool to inform further sponsors and regulators not only about the magnitude of the possible QTc prolongation but also:
− help predict the QT effects of doses, dosing regimens, routes of administration, or formulations that were not studied directly. Interpolation within the range of concentrations studied is considered more reliable than extrapolation above the range;

Version 1.0
22
− inform dose selection for later studies; − inform whether the QTc change occurs simultaneously with the peak concentration (Cmax) or delayed
(e.g., effect-compartment or turnover models); − may assist and clarify the interpretation of equivocal data (on occasion, a TQT study can yield ambiguous
results); − analyses of CRR by sex can be helpful for studying the effect of the drug on QT/QTc interval in cases
where there is evidence or mechanistic theory for a gender difference; − can help predict the effects of intrinsic (e.g., Cytochrome P450 isoenzyme status) or extrinsic (e.g., drug-
drug PK interactions) factors, possibly affecting inclusion criteria or dosing adjustments in later phase studies;
− if the results for the study drug are ambiguous (e.g., possible QT prolongation at lower dose but no prolongation at higher dose or QTc prolongation at a single isolated time point), CRR analysis can help interpret the data.
When a. TQT study is negative If the TQT is negative a PK-QTc analysis is not required by authorities; however when a small drug effect is expected (based on pre-clinical info, such as hERG test, animal data, etc.) it is a ‘nice to have’. b. TQT study is positive As mentioned earlier, the primary IUT analysis is very conservative (the false-positive rate reported in literature [ICH,2014] is around 20%) and a CRR analysis can either confirm the ICH E14 results as well as provide a non-biased characterization of the drug effect or point towards further investigation being needed. c. Assay sensitivity is not demonstrated CCR might demonstrate that the PK-PD relationship for the positive control is as expected based on historical control and that failure to demonstrate assay sensitivity was likely due to inadequate positive control exposure due to one or more of several factors (e.g., delayed absorption of an oral formulation and failure to reach an expected Cmax due to a food effect when a meal was given shortly before the positive control). Furthermore, it might be possible to demonstrate that assay sensitivity would have likely been demonstrated if sufficient, and expected, exposure to the positive control had been achieved. How In all situations, it is important that the modeling assumptions, criteria for model selection, and rationale for model components be specified prior to analysis to limit bias as models with different underlying assumptions on the same data can produce discordant results. For the same reason pre-specification of model characteristics (e.g., structural model, objective criteria, goodness of fit) based on knowledge of the pharmacology is recommended whenever possible. Mixed effects model can be used to describe the CRR with (Δ)ΔQTc as response (the (Δ)ΔQTc notation will be used to show that the equation/statement applies both for the ΔQTc and ΔΔQTc subject to the study design). The following model definition can be considered: (Δ)ΔQTci (t) = Intercept(i) + drugEffect + eta(i) + eps, for subject i where eta(i) stands for subjects i inter-individual variability and eps stands for the residual variability. The drug effect is given by (i) in linear effect models

Version 1.0
23
drugEffect = Concentration * Slope where Slope = drug effect slope (ii) in power models
drugEffect = Concentrationb where b = drug effect power and (iii) in Emax models drugEffect = Emax * Concentration / (EC50 + Concentration) where Emax = maximal effect of the drug on QTc changes and EC50 = the concentration at which half of the maximal drug effect is reached. If a time delay is observed between peak concentration and peak QT effect, other models will need to be considered. These models are considered out-of-scope for this White Paper. For crossover designs, the ΔΔQTc should be used. For parallel designs, the ΔQTc is used. There are different opinions for parallel designs whether Placebo observations should be included in the analysis as having zero concentration; as no formal guidance exists at the time of writing the authors leave it at the readers person experience but they recommend for the reader to investigate recent literature from the regulators in case such guidance is issued. The baselines recommended are the same as in the Primary analysis i.e. for crossover designs “pre-Dose averaged” baseline and for parallel designs “Time-Matched” baseline. Other considerations for PK/PD If assay sensitivity is in question based on the results of the primary analysis and PK/PD analysis of the active control data can be performed to bring confidence in the assay sensitivity claim. The models recommended here are the same as the ones for the other PK/PD analysis. If Moxifloxacin is to be used then based on Tornøe et al. (2011) and Florian et al. (2011), we recommend model (i) from the models above. Finally, the authors stress that a CRR analysis is credible only when the data are well behaved with respect to the regression line along its entire observed length.
7.3 P-values and Confidence Intervals There has been an ongoing debate on the value or lack of value for the inclusion of p-values and/or confidence intervals in safety assessments (Crowe, et. al. 2009). This White Paper does not attempt to resolve this debate. As noted in the Reviewer Guidance, p-values or confidence intervals can provide some evidence of the strength of the finding, but unless the trials are designed for hypothesis testing, these should be thought of as descriptive. Throughout this White Paper, p-values and measures of spread are included in several places. Where these are included, they should not be considered as hypothesis testing. If a company or compound team decides that these are not helpful as a tool for reviewing the data, they can be excluded from the display. Some teams may find p-values and/or confidence intervals useful to facilitate focus, but have concerns that lack of “statistical significance” provides unwarranted dismissal of a potential signal. Conversely, there are concerns that due to multiplicity issues, there could be over-interpretation of p-values adding potential concern for too many outcomes. Similarly, there are concerns that the lower- or upper-bound of confidence intervals will be over-interpreted. A mean change can be as high as x causing undue alarm. It is important for the users of these TFLs to be educated on these issues if p-values and/or confidence intervals are included in the TFLs.

Version 1.0
24
8 List of outputs In TQT studies the following list of outputs are commonly produced (for the baseline definitions for Parallel and Crossover studies, please refer to Section 6.3): Type Title Figure Individual QT vs. RR plot and QTcF-RR plot Figure Box plots of change from baseline in QTc by time-point for
each treatment Figure Estimated mean difference in comparison to placebo and
90% CI for change from baseline in QTc (ddQTc) for treatment
Figure Estimated mean difference in comparison to placebo and 90% CI for change from baseline in QTc (ddQTc) for active control
Figure Mean (+/-SE) change from baseline in QT, QTc and HR by treatment
Figure Concentration response for change from baseline in QTc for active control (assay-sensitivity)
Figure Mean (+/-SE) QT and QTc intervals by treatment Figure Mean (+/-SE) HR by treatment Figure Concentration response for change from baseline in QTc
for treatment Table Treatment comparisons of change from baseline in QTc
intervals by time for treatment Table Treatment comparisons of change from baseline in QTc
intervals by time for active control Table Treatment comparisons of change from baseline to all time
points in ECG parameters (HR, PR, QRS) by time for treatment
Table Summary of values and changes from baseline to all time points in ECG parameters by time and treatment
Table Number and percentage of subjects meeting or exceeding clinically noteworthy QT and QTc interval changes by time point and overall
Table Number and percentage of subjects meeting or exceeding clinically noteworthy PR, QRS and HR interval changes by time point and overall
Table Number and percentage of subjects with abnormal morphological/qualitative ECG findings
Listing ECG intervals (average over repeated measurements) Listing Change from baseline in ECG intervals(average over
repeated measurements) Listing ECG intervals (each replicate) Listing ECG interpretation Listing ECG findings