1 (c) mark gerstein, 1999, yale, bioinfo.mbb.yale.edu several motifs ( -sheet, beta-alpha-beta,...
Post on 21-Dec-2015
220 views
TRANSCRIPT

1
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
•Several motifs (-sheet, beta-alpha-beta, helix-loop-helix) combine to form a compact globular structure termed a domain or tertiary structure
•A domain is defined as a polypeptide chain or part of a chain that can independently fold into a stable tertiary structure
•Domains are also units of function (DNA binding domain, antigen binding domain, ATPase domain, etc.)
•Another example of the helix-loop-helix motif is seen within several DNA binding domains including the homeobox proteins which are the master regulators of development
Modules
(Figures from Branden & Tooze)
HMMs, Profiles, Motifs, and Multiple Alignments used to define modules
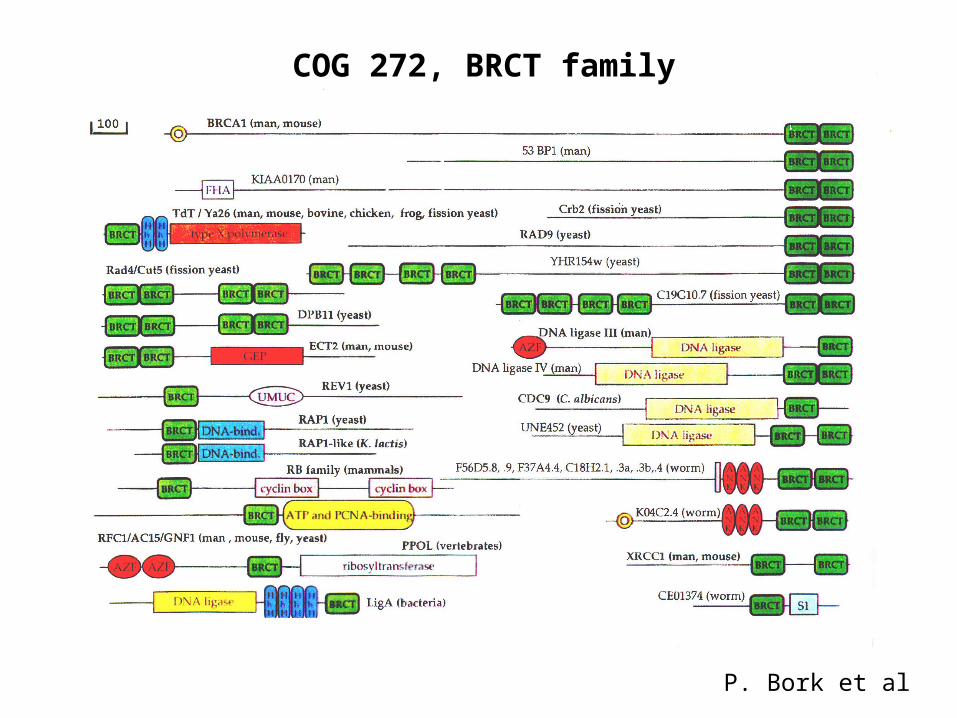
COG 272, BRCT family
P. Bork et al

Five Principal Fold Classes
All folds
All folds
+ folds
/ folds
small irregular folds

SCOP - Protein Fold Hierarchy
Class - 5Fold - ~500
Superfamily - ~ 700Family ~ 1000
Family - domains with common evolutionary origin

Sequence Similarity May Miss Functional Homologies Which Can Be Detected by
3D Structural Analysis
Residues Aligned
% S
equ
ence
Id
enti
ty
Homologous 3D Structure
Non-homologous3D Structure
Adapted from Chris Sander
} “Twilight zone”

Structural Validation Structural Validation of Homologyof Homology
Adenylate Kinase Guanylate Kinase
19% Seq ID
Z = 12.2
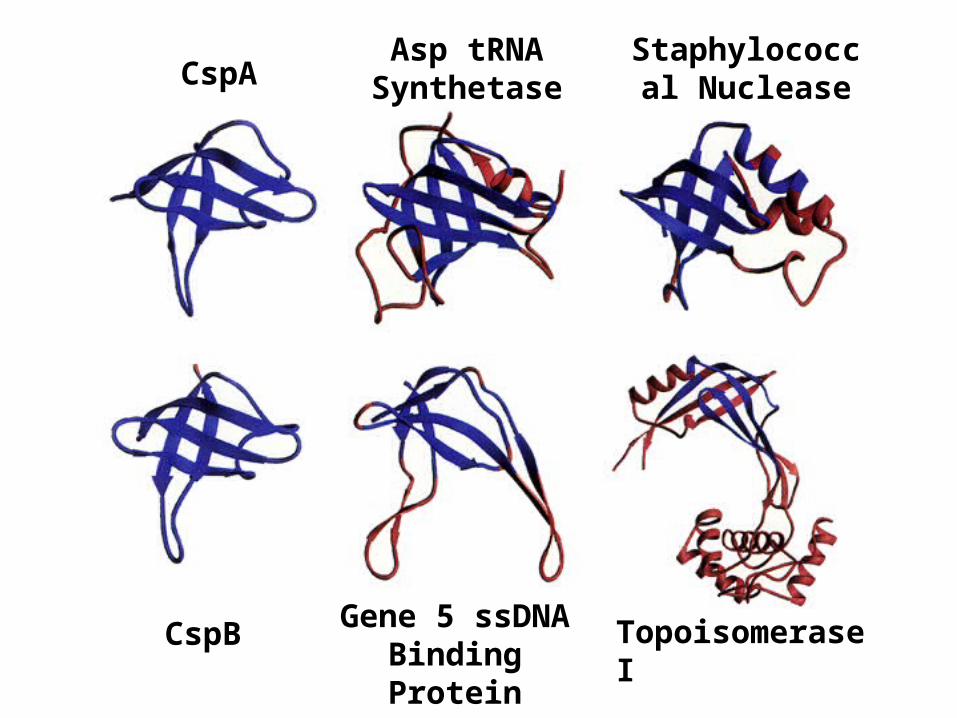
CspA
CspB Topoisomerase I
Asp tRNA Synthetase
Staphylococcal Nuclease
Gene 5 ssDNA Binding Protein

8
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
What is Protein Geometry?
• Coordinates (X, Y, Z’s)
• Dihedral Angles Assumes standard bond
lengths and bond angles

9
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Other Aspects of Structure, Besides just Comparing Atom Positions
Atom Position, XYZ triplets Lines, Axes,
AnglesSurfaces, Volumes

10
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Depicting Protein
Structure:Sperm Whale
Myoglobin

11
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Sperm Whale Myoglobin

12
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Structural Alignment of Two Globins

13
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Automatic Alignment to Build Fold LibraryHb
Mb
Alignment of Individual Structures
Fusing into a Single Fold “Template”
Hb VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAV ||| .. | |.|| | . | . | | | | | | | .| .| || | || . Mb VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASEDLKKHGVTVLTALGAIL
Hb AHVD-DMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR------ | | . || | .. . .| .. | |..| . . | | . ||.Mb KK-KGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRHPGDFGADAQGAMNKALELFRKDIAAKYKELGYQG
Structure Sequence Core Core
2hhb HAHU - D - - - M P N A L S A L S D L H A H K L - F - - R V D P V N K L L S H C L L V T L A A H <
HADG - D - - - L P G A L S A L S D L H A Y K L - F - - R V D P V N K L L S H C L L V T L A C H
HATS - D - - - L P T A L S A L S D L H A H K L - F - - R V D P A N K L L S H C I L V T L A C H
HABOKA - D - - - L P G A L S D L S D L H A H K L - F - - R V D P V N K L L S H S L L V T L A S H
HTOR - D - - - L P H A L S A L S H L H A C Q L - F - - R V D P A S Q L L G H C L L V T L A R H
HBA_CAIMO - D - - - I A G A L S K L S D L H A Q K L - F - - R V D P V N K F L G H C F L V V V A I H
HBAT_HO - E - - - L P R A L S A L R H R H V R E L - L - - R V D P A S Q L L G H C L L V T P A R H
1ecd GGICE3 P - - - N I E A D V N T F V A S H K P R G - L - N - - T H D Q N N F R A G F V S Y M K A H <
CTTEE P - - - N I G K H V D A L V A T H K P R G - F - N - - T H A Q N N F R A A F I A Y L K G H
GGICE1 P - - - T I L A K A K D F G K S H K S R A - L - T - - S P A Q D N F R K S L V V Y L K G A
1mbd MYWHP - K - G H H E A E L K P L A Q S H A T K H - L - H K I P I K Y E F I S E A I I H V L H S R <
MYG_CASFI - K - G H H E A E I K P L A Q S H A T K H - L - H K I P I K Y E F I S E A I I H V L Q S K
MYHU - K - G H H E A E I K P L A Q S H A T K H - L - H K I P V K Y E F I S E C I I Q V L Q S K
MYBAO - K - G H H E A E I K P L A Q S H A T K H - L - H K I P V K Y E L I S E S I I Q V L Q S K
Consensus Profile- c - - d L P A E h p A h p h ? H A ? K h - h - d c h p h c Y p h h S ? C h L V v L h p p <
Elements: Domain definitions; Aligned structures, collecting together Non-homologous Sequences; Core annotation
Previous work: Remington, Matthews ‘80; Taylor, Orengo ‘89, ‘94; Artymiuk, Rice, Willett ‘89; Sali, Blundell, ‘90; Vriend, Sander ‘91; Russell, Barton ‘92; Holm, Sander ‘93; Godzik, Skolnick ‘94; Gibrat, Madej, Bryant ‘96; Falicov, F Cohen, ‘96; Feng, Sippl ‘96; G Cohen ‘97; Singh & Brutlag, ‘98

Explain Concept of Distance Matrix on Blackboard
N x N distance matrix
N dimensional space
Metric matrixMij = Dij
2 - Dio2 - Djo
2
Eigenvectors of metric matrix
Principal component analysis

15
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Automatically Comparing Protein Structures
Given 2 Structures (A & B),
2 Basic Comparison Operations1 Given an alignment optimally
SUPERIMPOSE A onto BFind Best R & T to move A onto B
2 Find an Alignment between A and B based on their 3D coordinates

16
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
RMS Superposition (1)
A
B

17
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
RMS Superposition (2):Distance Between
an Atom in 2 Structures

18
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
RMS Superposition (3):RMS Distance Between
Aligned Atoms in 2 Structures

19
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
RMS Superposition (4):Rigid-Body Rotation and Translation
of One Structure (B)

20
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
RMS Superposition (5):Optimal Movement of One Structure
to Minimize the RMS
Methods of Solution:
springs(F ~ kx)
SVD
Kabsch

21
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Alignment (1) Make a Similarity Matrix
(Like Dot Plot)
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 1 1
C 1 1 1
K
C 1 1 1
R 1 1
B 1
P 1

22
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Structural Alignment (1b) Make a Similarity Matrix
(Generalized Similarity Matrix)• PAM(A,V) = 0.5
Applies at every position
• S(aa @ i, aa @ J) Specific Matrix for each pair of
residues i in protein 1 and J in protein 2
Example is Y near N-term. matches any C-term. residue (Y at J=2)
• S(i,J) Doesn’t need to depend on a.a.
identities at all! Just need to make up a score
for matching residue i in protein 1 with residue J in protein 2
1 2 3 4 5 6 7 8 9 10 11 12 13
A B C N Y R Q C L C R P M1 A 12 Y 1 5 5 5 5 5 53 C 1 1 14 Y 15 N 16 R 1 17 C 1 1 18 K9 C 1 1 110 R 1 111 B 112 P 1
J
i

23
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Seq. Alignment, Struc. Alignment, Threading

24
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Structural Alignment (1c*)Similarity Matrix
for Structural Alignment• Structural Alignment
Similarity Matrix S(i,J) depends on the 3D coordinates of residues i and J
Distance between CA of i and J
M(i,j) = 100 / (5 + d2)
• Threading S(i,J) depends on the how well
the amino acid at position i in protein 1 fits into the 3D structural environment at position J of protein 2
222 )()()( JiJiJi zzyyxxd −+−+−=

25
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Alignment (2): Dynamic Programming,Start Computing the Sum Matrixnew_value_cell(R,C) <= cell(R,C) { Old value, either 1 or 0 } + Max[ cell (R+1, C+1), { Diagonally Down, no gaps } cells(R+1, C+2 to C_max),{ Down a row, making col. gap } cells(R+2 to R_max, C+2) { Down a col., making row gap } ]
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 1 1
C 1 1 1
K
C 1 1 1
R 1 2 0 0
B 1 2 1 1 1 1 1 1 1 1 1 0 0
P 0 0 0 0 0 0 0 0 0 0 0 1 0
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 1 1
C 1 1 1
K
C 1 1 1
R 1 1
B 1
P 1

26
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Alignment (3):Dynamic Programming, Keep Going
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 5 4 3 3 2 2 0 0
C 3 3 4 3 3 3 3 4 3 3 1 0 0
K 3 3 3 3 3 3 3 3 3 2 1 0 0
C 2 2 3 2 2 2 2 3 2 3 1 0 0
R 2 1 1 1 1 2 1 1 1 1 2 0 0
B 1 2 1 1 1 1 1 1 1 1 1 0 0
P 0 0 0 0 0 0 0 0 0 0 0 1 0
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 1 1
C 1 1 1
K
C 1 1 1
R 1 2 0 0
B 1 2 1 1 1 1 1 1 1 1 1 0 0
P 0 0 0 0 0 0 0 0 0 0 0 1 0

27
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Alignment (4): Dynamic Programming, Sum Matrix All Done
A B C N Y R Q C L C R P MA 8 7 6 6 5 4 4 3 3 2 1 0 0
Y 7 7 6 6 6 4 4 3 3 2 1 0 0C 6 6 7 6 5 4 4 4 3 3 1 0 0Y 6 6 6 5 6 4 4 3 3 2 1 0 0N 5 5 5 6 5 4 4 3 3 2 1 0 0R 4 4 4 4 4 5 4 3 3 2 2 0 0C 3 3 4 3 3 3 3 4 3 3 1 0 0K 3 3 3 3 3 3 3 3 3 2 1 0 0C 2 2 3 2 2 2 2 3 2 3 1 0 0R 2 1 1 1 1 2 1 1 1 1 2 0 0B 1 2 1 1 1 1 1 1 1 1 1 0 0P 0 0 0 0 0 0 0 0 0 0 0 1 0
A B C N Y R Q C L C R P M
A 1
Y 1
C 1 1 1
Y 1
N 1
R 5 4 3 3 2 2 0 0
C 3 3 4 3 3 3 3 4 3 3 1 0 0
K 3 3 3 3 3 3 3 3 3 2 1 0 0
C 2 2 3 2 2 2 2 3 2 3 1 0 0
R 2 1 1 1 1 2 1 1 1 1 2 0 0
B 1 2 1 1 1 1 1 1 1 1 1 0 0
P 0 0 0 0 0 0 0 0 0 0 0 1 0

28
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Alignment (5): TracebackFind Best Score (8) and Trace BackA B C N Y - R Q C L C R - P MA Y C - Y N R - C K C R B P
A B C N Y R Q C L C R P M
A 8 7 6 6 5 4 4 3 3 2 1 0 0
Y 7 7 6 6 6 4 4 3 3 2 1 0 0
C 6 6 7 6 5 4 4 4 3 3 1 0 0
Y 6 6 6 5 6 4 4 3 3 2 1 0 0
N 5 5 5 6 5 4 4 3 3 2 1 0 0
R 4 4 4 4 4 5 4 3 3 2 2 0 0
C 3 3 4 3 3 3 3 4 3 3 1 0 0
K 3 3 3 3 3 3 3 3 3 2 1 0 0
C 2 2 3 2 2 2 2 3 2 3 1 0 0
R 2 1 1 1 1 2 1 1 1 1 2 0 0
B 1 2 1 1 1 1 1 1 1 1 1 0 0
P 0 0 0 0 0 0 0 0 0 0 0 1 0

29
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
In Structural Alignment, Not Yet Done (Step 6*)
• Use Alignment to LSQ Fit Structure B onto Structure A However, movement of B will now
change the Similarity Matrix
• This Violates Fundamental Premise of Dynamic Programming Way Residue at i is aligned can
now affect previously optimal alignment of residues(from 1 to i-1)
ACSQRP--LRV-SH -R SENCVA-SNKPQLVKLMTH VK DFCV-

30
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
How central idea of dynamic programming is violated in structural
alignment

31
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Structural Alignment (7*), Iterate Until Convergence
1 Compute Sim. Matrix
2 Align via Dyn. Prog.
3 RMS Fit Based on Alignment
4 Move Structure B
5 Re-compute Sim. Matrix
6 If changed from #1, GOTO #2

32
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Some Similarities are Readily Apparent others are more Subtle
Easy:Globins
125 res., ~1.5 Å
Tricky:Ig C & V
85 res., ~3 Å
Very Subtle: G3P-dehydro-genase, C-term. Domain >5 Å

33
(c)
Mar
k G
erst
ein
, 19
99,
Yal
e, b
ioin
fo.m
bb
.yal
e.ed
u
Some Similarities are Readily Apparent others are more Subtle
Easy:Globins
125 res.,
~1.5 Å
Tricky:Ig C & V
85 res., ~3 Å
Very Subtle: G3P-dehydro-genase, C-term. Domain >5 Å

DALI: Protein Structure Comparison by Alignment of Distance Matrices
L. Holm and C. Sander J. Mol. Biol. 233: 123 (1993)
• Generate C-C distance matrix for each protein A and B• Decompose into elementary contact patterns; e.g. hexapeptide-
hexapeptide submatrices• Systematic comparisons of all elementary contact patterns in the
2 distance matrices; similar contact patterns are stored in a “pair list”
• Assemble pairs of contact patterns into larger consistent sets of pairs (alignments), maximizing the similarity score between these local structures
• A Monte-Carlo algorithm is used to deal with the combinatorial complexity of building up alignments from contact patterns
• Dali Z score - number of standard deviations away from mean pairwise similarity value

Structural Validation Structural Validation of Homologyof Homology
Adenylate Kinase Guanylate Kinase
19% Seq ID
Z = 12.2

Dali Domain DictionaryDeitman, Park, Notredame, Heger, Lappe, and Holm
Nucleic Acids Res. 29: 5557 (2001)
• Dali Domain Dictionary is a numerical taxonomy of all known domain structures in the PDB
• Evolves from Dali / FSSP Database Holm & Sander, Nucl. Acid Res. 25: 231-234 (1997)
• Dali Domain Dictionary Sept 2000 10,532 PDB enteries 17,101 protein chains 5 supersecondary structure motifs (attractors) 1375 fold types 2582 functional families 3724 domain sequence families

SCOP the Structural Classification Of Proteins database
This contains all proteins, and protein domains,
of known structure classified in terms of
their structure and evolutionary relationships.
SUPERFAMILY This database contains:
(a) hidden Markov models (HMMs) of all the
proteins and protein domains in SCOP
(b) a list of the matches made by these HMMs to
the sequences of 56 genomes classsified by family.
http://stash.mrc-lmb.cam.ac.uk/SUPERFAMILY/
courtesy of C. Chothia

Most proteins in biology have been
produced by the duplication,
divergence and recombination of
the members of a small number of
protein families.
courtesy of C. Chothia

SUPERFAMILY matches to genome sequences
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
hs at ce dm mk sc pa eo ec mu bs bh mb vc cc cs dr ss xf sa af ll nn ph hb nm pm mt tm pb mj hi sq cj ml hp aa tv hq ta cq cp cr tp cm ct bb rp mq mp uu bn mg
Genomes
% Coverage
GenesGenome
courtesy of C. Chothia

SUPERFAMILY Results forBuchnera and Human Genome Sequences
Buchnera HumansNumber of sequences 564 23867Sequences matched by SUPERFAMILY 410 12616Coverage of genome 61% 41%Number of matched domains 609 22548Number of families 277 648Mean family size 2.2 35Number of large families that form
half the matched domains 37 17
courtesy of C. Chothia

SUPERFAMILY Results forBuchnera and Human Genome Sequences:
Top Five Domain Families
BuchneraP-loop containing nucleotide triphosphate hydrolasesNucleic acid binding proteinsNAD-binding Rossman domainsNucleotidylyl transferasesClass II aaRS synthetases
HumansClassic zinc fingersImmunoglobulin superfamilyP-loop containing nucleotide triphosphate hydrolasesEGF/LamininCadherin
courtesy of C. Chothia

Eukaryotes
0
5000
10000
15000
20000
25000
30000
sc at dm ce hs
Other families
dm+ce+hs: 45 families
at+dm+ce+hs: 56 families
All: 381 families
courtesy of C. Chothia

CDH-4
CDH-3
HMR-1b
CDH-6
CDH-10
CDH-8
CDH-5
CDH-11
CDH-9
T01D3.1
Y37E11A.94.a
Cadherin Proteins in Caenorhabditis elegans
233
95
120
143
56
64
156
113
82
4307
3343
2920
HMR-1a120
1223
2610
2302
1671
1507
984
1156
623
3191
411
S
S
S
S
S
S
S
S
S
S
S
S
3 2 2 3 1 2 3
3 3 3 3 2 3 3
3 3 2 1 2 1 3 1 3 3 3 3 0 3 3 3
0 3 3 21 1 0 3 3 0
0 3 3 3 3 3
3 0 1
3 3 0 0
33 1 1 3
3 2
2
33 3 3
22 2 1
3 3 3 3 3 3 3 2
3 2
1 3 2 0 0 3 1 2 3 0 3 1 2 3 3 1 3 3 1 3 3 3 3 2
CDH-7 76S
111 2 2 3 3
CDH-1 2072775S
3 3 0 3 0 3 0 3 3 3 0 1 0 2 0 1 3 0 3 3 1 3 0 2
CDH-12 1243017S
332 1 0 10 0 3 1 3 3 1 3 0 0 3
Fat
CG7749
Stan
Ds
CadN
CG14900
CG3389
CG15511/CG7805
CG6445
CG7527
Shg
CG4655/CG4509
CG6977
CG11059
CG10421
Cadherin Proteins in Drosophila melanogaster538
320
613
437
158
332
132
286
43
227
158
968
80
189
5147
4643
3666
3503
3097
2204
2005
1821
1820
1783
1507
1963
1346
978
678
S
S
S
S
S
S
S
S
S
S
3 3 3 3
2? 3 3 3
3 3 0 3 3 3 2 3 3 3 1 3 3 3 3 3 3 3 3 2 3 3 3 1 1 3 3 3 3
3 3 3 3 3 3 1 3 3 3 3 3 3 3 1 3 3 3 1
1 3 2 3 0 3 3 3 3 1 3 3 3 3 3 3 3
3 3 3 3
3
3 3 3 33 2
3 3 3 1 3 3 2
3 2 1 3 3 1 3 3 3 3 2 3 3 3 3 3 3 33 3 1 3 3 3 3 3 3 2 1 3 2 1 1
3 3 2 3 3 3 3 2 3 3 3
3 3 3 3 3 3 3 3 3 3 3 3 32
3 2 3 3 3 3 2 3 3
3
3 ?3
3 3 3 3 3 3 3
2? 3 3 3 3 3 3
1 3
3 0 3 3 3 3 3
3 3 3
33 3
2
3 3 1 3 2 3 3 1 33
CG10244/HD-14 503838S
Ret 5181120
S Signal peptide
Cadherin
EGF
EGF_CA
Laminin G
HormR
GPS
Transmembrane Helix
7 Pass Transmembrane Domain
Classic cytoplasmic domain
Tyrosine Kinase cytoplasmic domain
Type 1 Cytoplasmic domain
Type 2 Cytoplasmic domain
Other Cytoplasmic domainMerge Position
Cadherins
courtesy of C. Chothia

Domain Combinations in Genome Sequences
In bacteria close to1/3 of proteins consist of one domain and2/3 consist of two or more domains.
In eukaryotes close to1/4 of proteins consist of one domain and3/4 consist of two or more domains.
courtesy of C. Chothia

Bacteria
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
mk pa eo ec bs bh mb mu cc vc sm ca cs dr au ss sa af ll pm av xf st sr tm hb nn nm mt hi ml aa sq pb ph cj mj ap ta tv hq hp tp cp cr cq cm ct rp bb bn mq mp uu mg
Genome
TotalEC +BS
courtesy of C. Chothia

A Global Representation of Protein Fold Space Hou, Sims, Zhang, Kim, PNAS 100: 2386 - 2390 (2003)
Database of 498 SCOP “Folds” or “Superfamilies”
The overall pair-wise comparisons of 498 folds lead to a 498 x 498 matrix of similarity scores Sijs, where Sij is the alignment score between the ith and jth folds.
An appropriate method for handling such data matrices as a whole is metric matrix distance geometry . We first convert the similarity score matrix [S ij] to a distance matrix [Dij] by using Dij = Smax - Sij, where Smax is the maximum similarity score among all pairs of folds.
We then transform the distance matrix to a metric (or Gram) matrix [M ij] by using Mij = Dij
2 - Dio2 - Djo
2
where Di0, the distance between the ith fold and the geometric centroid of all N = 498 folds. The eigen values of the metric matrix define an orthogonal system of axes, called factors. These axes pass through the geometric centroid of the points representing all observed folds and correspond to a decreasing order of the amount of information each factor represents.

A Global Representation of Protein Fold Space Hou, Sims, Zhang, Kim, PNAS 100: 2386 - 2390 (2003)