1 june 2015dimacs - machine learning in bioinformatics 1 machine learning as applied to structural...
Post on 18-Dec-2015
216 views
TRANSCRIPT

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
1
Machine Learning as Applied to Structural Bioinformatics: Results and Challenges
Philip E. Bourne
University of California San Diego

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
2
The Current Situation
• Structure contributes greatly to our understanding of living systems
• We are locked into thinking about structure in specific ways which limits our view– All too often we consider
structure as a static entity
– The view at left is not how another protein or a small molecule ligand sees PKA
• We are still not very good at certain problems …

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
3
Example Unsolved Problems that Machine Learning Can Address
• Predicting flexibility and disorder in protein structure• Predicting sites of protein-protein and protein-ligand
interaction• Predicting protein function• Defining domain boundaries from sequence• Predicting secondary, tertiary and quaternary
structure• Predicting what will crystallize

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
4
Example Unsolved Problems that Machine Learning Can Address
• Predicting flexibility and disorder in protein structure• Predicting sites of protein-protein and protein-ligand
interaction• Predicting protein function• Defining domain boundaries from sequence• Predicting secondary, tertiary and quaternary
structure• Predicting what will crystallize
* Will talk about this* Will offer as a challenge

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
5
The Current Situation: The Potential “Training Set” is Growing Quickly
• High level of redundancy as measured by sequence or structure
• Structure space is clearly very finite, but not clear how much is covered
• Increase in functionally uncharacterized structures
• Complexity is increasing, but still lack complexes
• Structures predominantly 1 and 2 domains
• Lack membrane proteins
• In summary the training set is still not truly representative but structural genomics will improve this situation

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
6
Predicting Functional Flexibility
Jenny Gu
Gu, Gribskov & Bourne PLoS Computational Biology 2006 Early On-line Release

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
7
Spectrum of Protein Order and Disorder
OrderedStructures
DisorderedStructures
If we believe that the 3-dimensional structure of a protein is defined by its 1-dimensional sequence then why not its
flexibility?
April 18, 2023 DIMACS - Machine Learning in Bioinformatics
8
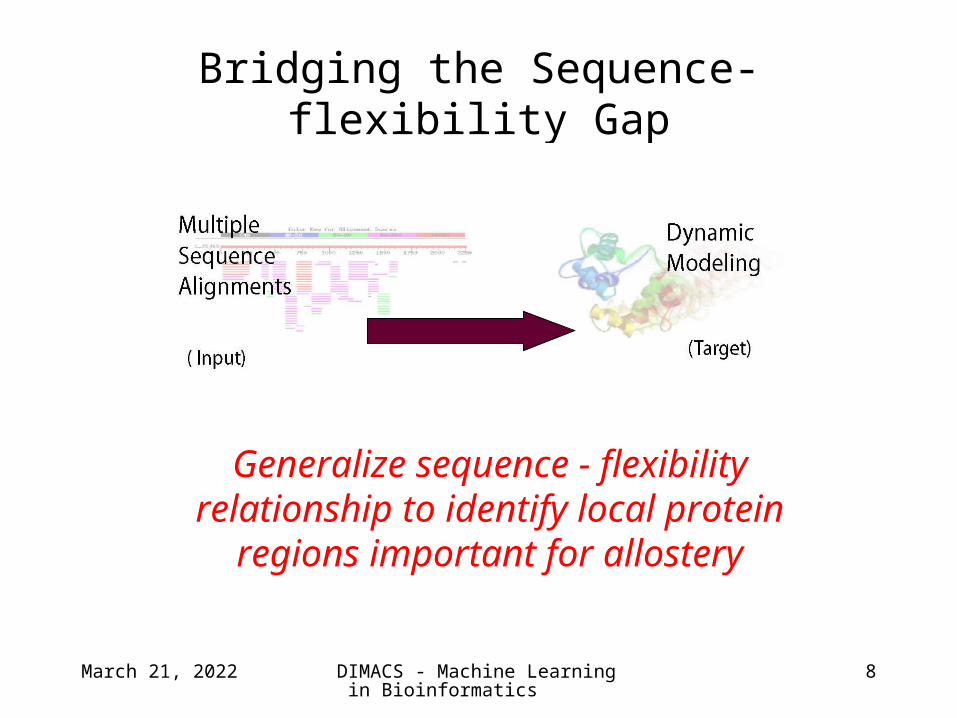
Bridging the Sequence-flexibility Gap
Generalize sequence - flexibility relationship to identify local protein
regions important for allostery

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
9
The Training Dataset
The dataset contains the following qualities:
• Non-redundant sequences– training set with sequences containing ≤ 10% identity.
• With good quality structures– R-factor < 0.30
• At high resolution– Resolution < 2.0 Å.
Total number of proteins in dataset: 1277 sequences

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
10
Obtaining Protein Dynamic Information
Protein structures treated as a 3-D elastic network.
Bahar, I., A.R. Atilgan, and B. Erman
Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential.
Folding & Design, 1997. 2(3): p. 173-181.

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
11
Defining the Target Features
Gaussian Network Model:
• Models protein structure as a 3-D elastic network.
– Each Ca is a node in the network.
– Each node undergoes Gaussian-distributed fluctuations influenced by neighboring interactions within a given cutoff distance. (7Å)
• Decompose protein fluctuation into a summation of different modes.
Bahar, I., A.R. Atilgan, and B. Erman
Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential.
Folding & Design, 1997. 2(3): p. 173-181.

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
12
Side Note: Gaussian Network Model vs Molecular Dynamics
• GNM relatively cause grained
• GNM fast to compute vs MD– Look over larger time scales– Suitable for high throughput

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
13
Functional Flexibility Score
• Utilize correlated movements to help define regional flexibility with functional importance.
Functionally Flexible Score
For each residue:
1. Find Maximum and Minimum Correlation
2. Use to scale normalized fluctuation to determine functional importance

Example: Identifying Functional Flexible Regions (FFR) in HIV Protease
Gu, Gribskov & BournePLoS Comp. Biol.. 2006 Early Release
Correlated modes (yellow)Anti-correlated (blue)
Normalized scores – single chain
April 18, 2023 DIMACS - Machine Learning in Bioinformatics
15
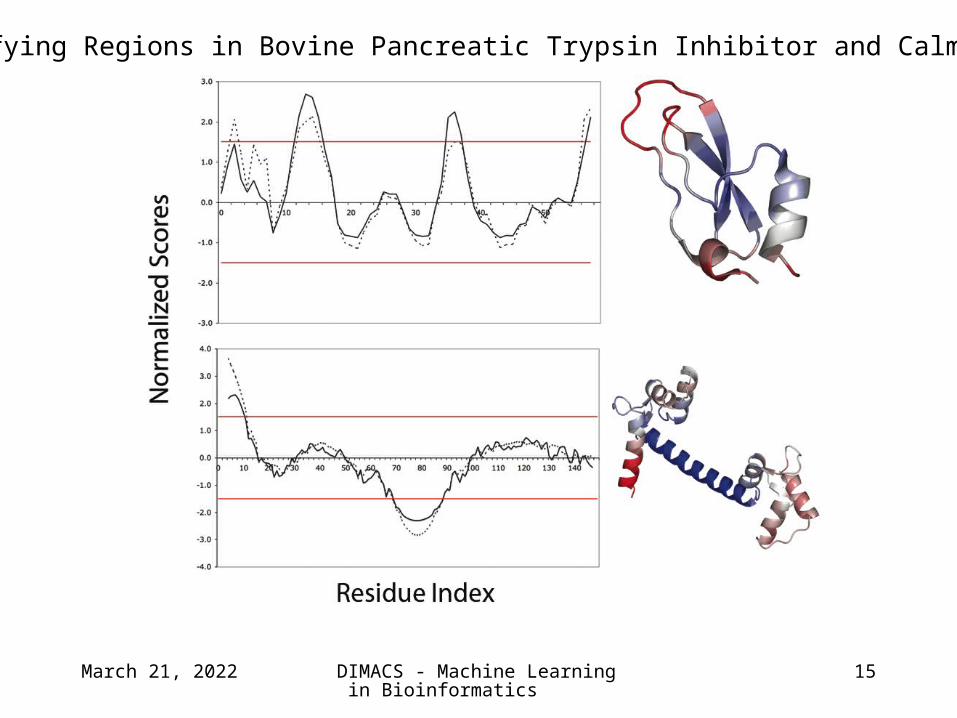
Identifying Regions in Bovine Pancreatic Trypsin Inhibitor and Calmodulin

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
16
How to Represent the Protein Sequence?
• Residues characterized as FFs or not – approx 20% of residues with lengths typically 9+/-11
• The longer the protein the longer the FFR• We use hidden Markov models to represent each
protein sequence in the training dataset.• Hidden Markov models captures evolutionary
information along with the probability of finding one of the 20 amino acids in each position of the sequence.
• Use probability states as input features in the first layer of an architecture containing two SVM layers.

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
17
Architecture of Wiggle
CapturesEvolutionary
Effects
CapturesLocal
Effects(smoothing)
9*29 featuresused for each residue

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
18
Generating Additional Input FeaturesModified Bootstrapping – for Tripeptides – Accounts for Nearest Neighbors Effects
Calculate Z score and P value for each pattern
with respective null models
Sample with replacement44645 times
Pooled Patterns(window size : 3)
Null Model* for FFR Regions
Null Model* for Non-FFR Regions
Sample with replacement199515 times
* Generate 10,000 Null Models

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
19
Architecture of Wiggle
CapturesEvolutionary
Effects
CapturesLocal
Effects(smoothing)
9*29 featuresused for each residue
April 18, 2023 DIMACS - Machine Learning in Bioinformatics
20
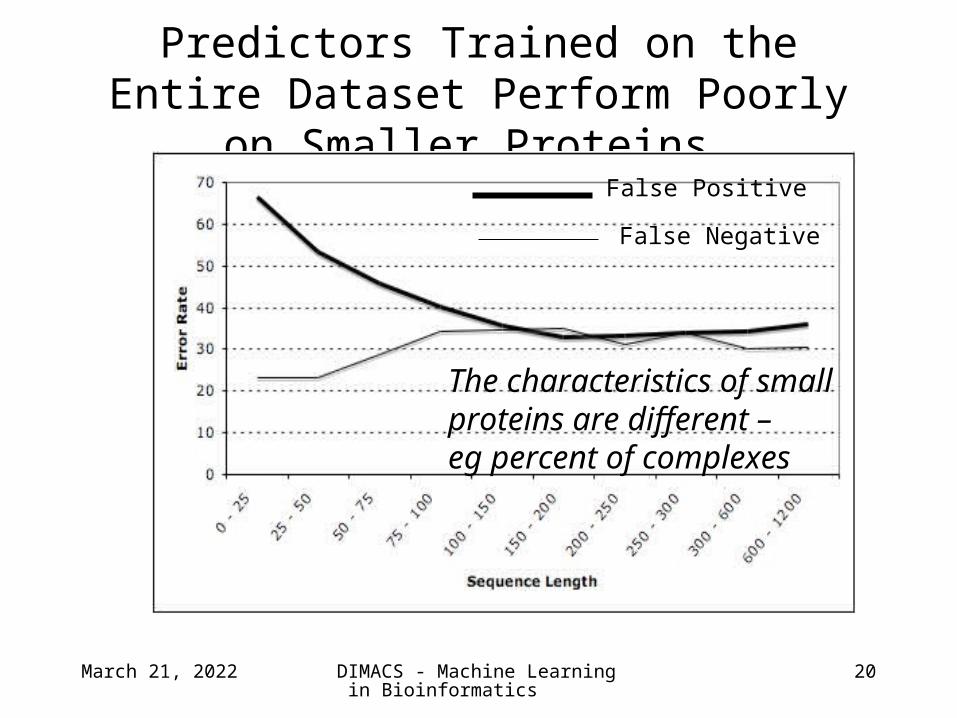
Predictors Trained on the Entire Dataset Perform Poorly on Smaller Proteins.
False Positive
False Negative
The characteristics of small proteins are different – eg percent of complexes

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
21
Partition Training Set Based on Sequence Length
• Prediction performance of SVM trained on a partitioned dataset (solid lines) is compared to that was trained on the entire dataset (dashed line).
• Prediction quality improved when dataset is partitioned. Most notably for proteins up to 200 amino acid residues long. Slight improvements observed for proteins longer than 200 residues.
<200 AA Long >200 AA Long

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
22
Performance of Wiggle Predictors
Wiggle
Accuracy: 66.01%
Precision: 37.11%
Recall: 70.49%
Wiggle 200
Accuracy: 76.46%
Precision: 48.99%
Recall: 78.27%

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
23
Case Study: PvuII Endonuclease
FF SCORE
(homodimer for DNA specific cleavage)
Wiggle 200
• Identify known loop for minor grove recognition • Identify hinge residues not previously seen • Important result for mutagenesis studies

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
24
Conclusions for Wiggle
• FFRs can be measured from structure• With some empirical effort these data can be used as
input to an SVM to predict FFRs from sequence alone
• Useful for:– Improving docking studies– Better understand protein function– Engineer more or less stable proteins– ……
Gu, Gribskov & Bourne 2006PLoS Comp. Biol.. 2006 Early Release

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
25
Exploiting Sequence and Structure Homologs to Identify
Protein-Protein Binding Sites
JoLan Chung
Chung, Wang & Bourne 2006 Proteins: Structure, Function and Bioinformatics, 62(3)
630-640

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
26
Methods to Identify Protein-protein Binding Sites
• Docking• Threading and homology modeling• Evolutionary tracing• Correlated mutations• Properties of patches• Hydrophobicity• Neural networks and support vector machines
(SVM)

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
27
• None of the above methods consider the residues which are spatially conserved on the surfaces of structure homologs
• These residues are reported to correspond to the energy hot spots on protein interfaces and can be derived from multiple structure alignments
Structurally Conserved Surface Residues?

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
28
Method: Incorporate Structural Conservation to Predict the Interface Residue Using SVM
Support vector machine
Sequence + structure information
Binding site location

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
29
Derive the Structurally Conserved Residues
• The structural conservation scores were derived from multiple structural alignments and weighted by the normalized B-factors to consider the structure flexibility that will result in a bad alignment (could use FFRs in the future)
• Each position in the alignment has a structural conservation score, which represents the conservation in 3D space
• A position has a high conservation score if the aligned residues are spatially conserved

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
30
Structurally Conserved Residues and Interface Residues
E.g. Residues with the top 20% of structure conservation scores (red) mapped to adrenodoxin (Adx, PDB code 1E6E:B) and known to bind adrenodoxin reductase (AR, blue).

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
31
Training Dataset
• 274 non-redundant chains of heterocomplexes (<30% sequence identity) extracted from the PDB
• Each of these chains was accompanied with a structure alignment with at least 4 members

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
32
SVM Training
A surface residue
↓
Sequence profile + ASA + Structural conservation score
in a window of 13 residues
(The residue to be predicted and 12 spatially nearest surface residues)
↓
Support vector machine classifier
↓
Interface or non-interface residue ?

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
33
SVM Training
• Each residue was encoded as a feature vector with 13×21 dimensions: (the surface residue to be predicted + 12 nearest neighbors) x (20 amino acids + accessible surface area)
• Implemented using SVMlight with the radial basis function as a kernel. (γ = 0.01, regularization parameter C =10)
• A set of non-interface surface residues was randomly selected to make the ratio of positive and negative data 1:1
• 3 fold cross-validation was performed

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
34
Predictor 1: Sequence profile + ASA.Predictor 2: Sequence profile + ASA + structural conservation scorePredictor 3: Sequence profile + ASA + raw structural conservation score without weighted by the normalized B-factor Predictor 4: Sequence profile + ASA+ normalized B-factor
The Performance of Various Predictors

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
35
Precise prediction: at least 70% interface residues were identifiedPrecise prediction: at least 70% interface residues were identifiedCorrect prediction: at least 50 % interface residues were identifiedCorrect prediction: at least 50 % interface residues were identifiedPartial prediction: some but less than 50 % interface residues were Partial prediction: some but less than 50 % interface residues were identifiedidentifiedWrong prediction: no interface residues were identifiedWrong prediction: no interface residues were identified
The Performances of the Predictors

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
36
Predicted Binding Sites - Example 1
Protein : domain 1 of the human coxsackie and adenovirus receptor (CAR D1)• Mediate adenoviruses and coxsackie virus B infection• CAR is an integral membrane protein expressed in a broad range of human and
murine cell type. CAR D1 is one of its two extracellular domains
Binding partner: knob domain of the adenoviruses serotype 12 (Ad12)

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
37
Predicted Binding Sites - Example 2
Protein : adrendoxin (Adx) • In mitochondria of the adrenal cortex, the steroid hydroxylating system requires the
transfer of electrons from the membrane-attached flavoprotein AR via the soluble Adx to the membrane-integrated cytochrome P450 of the CYP 11 family
Binding partner: adrenodoxin reductase (AR)

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
38
Predicted Binding Sites - Example 3
Protein : fibroblast growth factor receptor 2 (FGFR2) Ser252Trp Mutant
• Apert syndrome (AS) is caused by substitution of one of two adjacent residues, Ser252Trp or Pro253Arg
Binding partner: fibroblast growth factor (FGF2)

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
39
Conclusions – Protein-protein Binding Sites
• Incorporating the structural conservation score improved the prediction performance of SVM significantly
• This study is an initial trial that exploits multiple structure alignment for the large scale prediction of functional regions
• We need better algorithms for multiple structure alignment (we have one benchmark for anyone interested)
• This method can be used to guide experiments, such as site-specific mutagenesis, or combined with docking procedures to limit the search space

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
40
General Conclusions
• Using known features of protein structure these can be mapped to the corresponding sequences and used to train an SVM
• Having evaluated the SVM in a cross validation tests the performance can be determined
• Good performance is shown in training for both flexibility and sites of protein-protein interaction
• These predictors are currently being used to solve real biological problems
• Can this approach be applied to other aspects of structure?

1ytf
PUU: 1 Experts: 2
1d0gt
PUU: 1Experts: 3
1dgk
PUU: 6 Experts: 4
1aoga
PUU: 4 Experts: 3
1fohb
PUU: 2 Experts: 3
A. B.
C.D.
E.
Consider Domain Definitions:Holland et al. 2006 JMB Early Release
Veretnik et al. 2004 JMB 339(3), 647-678

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
42
Challenge – Defining Domain Boundaries from Sequence
• A domain is the unit of currency of proteins – domain structures define function, indicate evolutionary relationships etc…
• Domain prediction from structure easier than from sequence, but still not a solved problem
• Recently developed an accurate test set of domain definitions and boundaries: http://pdomains.sdsc.edu
• Good luck!
Benchmark Data Available See:Holland et al 2006 JMB Early Release

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
43
Acknowledgements
• Functional Flexibility– Jenny Gu & Michael
Gribskov
• Protein-protein Interactions– JoLan Chung & Wei Wang
• Domain Definitions– Stella Veretnik, Tim Holland,
Ilya Shindalov, Nick Alexandrov, Abdur Sikur
• Funding, NSF, NIH

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
44
The structural conservation score
• Raw structural conservation score
where
if a is not gap and b is not gap otherwise
where N is the total number of aligned structures, si(x) is the amino acid at position x in the ith structure in the alignment, m is a modified PET substitution matrix calculated by Valdar et al.
N
i
N
ij
ji xsxsLNN
xC ))(),(()1(
2)(
))(),(()))(),((exp())(),(( xsxsMxsxsdxsxsL jijiji
)min()max(
)min(),(
0))(),(( mm
mbam
ji xsxsM

April 18, 2023 DIMACS - Machine Learning in Bioinformatics
45
The structure conservation score
• The B-factors determined by X-ray crystallographic experiments provide an indication of the degree of mobility and disorder of an atom in a protein structure
• Raw structural conservation scores were weighted by the normalized B-factors (Bnorm, i) to consider the structure flexibility
where
)()()( xrweighted xCxC
)exp()( , inormBxr