1 stn 125085/91 bevacizumab (avastin®) plus paclitaxel for 1 st line metastatic breast cancer odac...
TRANSCRIPT

1
STN 125085/91Bevacizumab (Avastin®) plus Paclitaxel
for 1st line Metastatic Breast Cancer
ODAC MeetingDecember 5th, 2007
Clinical Review - Lee Pai-Scherf, M.D.Statistical Review - Hong Lu, Ph.D.
CDER/FDA

2
Proposed Indication
Avastin®, in combination with paclitaxel, is indicated for the treatment of patients who have not received chemotherapy for their locally recurrent or metastatic breast cancer.

3
Overview
• Regulatory Background
• Clinical studies
– Pivotal study E2100 (Phase 3)
– AVF2119g (Phase 3)
• Summary of Findings
• Questions to ODAC

4
Regulatory Background

5
Regulatory Background
Bevacizumab is approved by FDA for:
• 1st (2004) and 2nd line (2006) metastatic colorectal cancer in combination with 5-FU–based chemotherapy. Approval endpoint: OS
• 1st line unresectable, or metastatic non-squamous non-SCLC in combination with carboplatin and paclitaxel (2006) . Approval endpoint: OS

6
Regulatory Background – AVF2119g
• July, 2000
Genentech and FDA discussed study AVF2119g design [RCT of capecitabine +/- bevacizumab for 2nd and 3rd line therapy of MBC] intended to support Avastin approval
• November, 2000 – March 2002
Accrual period
• March, 2002
Genentech and FDA discussed BLA based on this trial
• September, 2002
AVF2119g failed to meet primary endpoint of PFS

7
Regulatory Background – E2100• October, 2001
– NCI submitted protocol E2100 [RCT of paclitaxel +/- bevacizumab for 1st line therapy of MBC]. Study not identified by NCI as intended to support drug approval.*
– Study opened for accrual December 2001
• May, 2002– Genentech identified E2100 as additional study to
support drug approval– FDA provided comments to NCI; noted that SAP was
extremely deficient– Key issues:
• SAP did not clearly identify primary & important secondary efficacy endpoints
• Primary analysis methods for primary & important secondary endpoints not described

8
Regulatory Background – E2100
October, 2002 (2nd letter to NCI re: E2100)– FDA reiterated that study identified by Genentech as
to support Avastin approval – NCI did not request meeting to discuss adequacy of
the trial design and analysis plan
– FDA asked NCI for additional clarification regarding SAP
– FDA stated it was crucial that primary endpoint and statistical plan be adequate if study to serve as basis for drug approval

9
Regulatory Background – E2100
• May 2004– E2100 completed patient accrual
• October 2004 – Genentech submitted SAP addressing FDA’s letters
to NCI and request for meeting re: adequacy of E2100 to support Avastin label expansion
– FDA noted E2100 may not be adequate to support licensure due to 1) non-blinded nature of study and 2) lack of pre-specified, detailed and objective radiological and clinical parameters for determination of disease progression

10
Regulatory Background – E2100
October 2004 (cont.)– FDA noted Genentech must provide overall
survival data for regular approval of the proposed indication. In reviewing the results of E2100, FDA will consider data from AVF2119g (negative phase 3 study)
– Genentech asked if PFS would be an adequate endpoint for full approval. FDA replied it depends on the overall robustness and magnitude of PFS and results of survival data at time of PFS analysis

11
Regulatory Background – E2100• April, 2005
– 1st interim efficacy analysis by ECOG DMC: Improved PFS (6.1 vs. 10.9 mos) in favor of the bevacizumab/paclitaxel arm (HR 0.49, log rank p < 0.001). Unplanned survival analysis reported HR 0.67, log rank test p=0.01
– Trial “stopped” based on these findings
– Genentech made results public April 14, 2005 and ASCO (May 2005) meeting

12
Regulatory Background
• September 2005 (pre-sBLA meeting)
– FDA agreed E2100 could form basis of sBLA
– FDA stated PFS would support accelerated approval and final overall survival necessary for regular approval

13
Regulatory Background
May, 2006– Genentech submitted sBLA for labeling
expansion of Avastin
September 8, 2006– FDA issued a Complete Review Letter

14
Complete Review Letter – Key issues
1. Data set incomplete, without data cut-off date for efficacy and safety. Per Genentech, data collection and clean-up was still ongoing
Why do we need a clean data set with clear data cut-off date?
Data “cut-off”
date
No. of PFS events per ECOG
ECOG 1st interim analysis (4/05) 2/9/05 260
Data presented at ASCO (5/05) 2/9/05 355
1st sBLA submission (5/06) 4/14/05 395
2nd sBLA submission (8/07) 2/9/05 445

15
Complete Review Letter – Key issues
2. FDA reiterated need for independent radiology review of progression events in at least a subset of patients, given subjective nature of PFS endpoint and open-label design of E2100
3. Submission incomplete in regards to documentation of eligibility, baseline tumor description, study violations, drug exposure, and treatment delays/discontinuation due to toxicity
Data submitted did not allow full evaluation of efficacy and safety

16
Regulatory Background
• November 2006 – March 2007
– Agreement reached regarding the data cut off dates for efficacy and safety
– Genentech to submit a “cleaned” dataset– Genentech to conduct independent, blinded review of
all patients enrolled in E2100 study to verify efficacy results
– The primary regulatory endpoint to be PFS adjudicated by independent review facility (IRF)
– Genentech to submit updated survival data
• August 2007– sBLA resubmitted for labeling expansion of Avastin

17
E2100

18
E2100 Study Design
• Recurrent or metastatic adenocarcinoma of the breast • No prior chemo for recurrent or metastatic disease• HER2 neu negative
Arm A
Paclitaxel 90mg/m2 q wk x 3 Bevacizumab 10mg/kg wks 1 and 3
4 week cycles
Arm B
Paclitaxel 90mg/m2 q wk x 3
4 week cycles
Stratificationdisease-free interval (≤ 24, > 24 months), number of metastatic sites (<3, ≥ 3), prior adjuvant chemotherapy (yes, no) and ER status (positive, negative, and unknown)

19
Study Plan
• Treatment continued until disease progression or unacceptable toxicity
• Crossover not allowed
• Tumor assessment every 12 weeks– Protocol required “x-rays and scans”
• Follow up every 3 months if < 2 years and every 6 months (2-5 years)

20
Efficacy Endpoints
Primary• PFS adjudicated by blinded independent
radiographic facility (IRF)
Secondary • Survival• RR and duration • QOL (FACT-B questionnaire)

21
E2100 Enrollment
• December 21, 2001 – May 26, 2004
• N = 722 (368/354)
• 258 centers from ECOG, CALGB, SWOG, NSABP, NCCTG, RTOG, GOG and EPP (NCI’s Expanded Participation Project)

22
Patient and Disease Characteristics
Demographic/Tumor Characteristics Total (N=722)
Female 99.2
Age 55 (27-85)
Post Menopausal 55.3
Metastatic disease 98.3
No. of involved sites < 3 ≥ 3
54.345.7
Common sites of involvement Bone Liver Lung
54.541.741.5
ER status Negative 61.8
No measurable disease at baseline 27.3 (23% vs. 32%)

23
Prior Cancer Therapy
Prior Cancer Therapy Total N= 722)
Hormonal therapy Adjuvant or Metastatic setting 61%
Adjuvant Chemo 66%
Prior taxane 20%
Prior anthracycline 50%

24
E2100 Protocol Deviation
Protocol DeviationTotal N=722
Treated beyond progression6 %
(4 vs. 7)
Stratification error* (ER status, adj. chemo) 7%
Initiation of Non-Protocol anti-cancer therapy (NPT) prior to documented PD
16%

25
E2100 Efficacy Results

26
PFS as the Primary Efficacy Endpoint
• Application rests solely on evidence of an improvement on PFS in a single study
• A 5.5 month improvement in PFS is claimed by Genentech.

27
PFS as the Primary Efficacy Endpoint
In considering Genentech’s claim, the
FDA needs to verify:
1. Robustness (i.e., is there an effect?)
2. Magnitude (i.e., is the 5.5 month
improvement in PFS reliable ?)

28
Outline
• Summary of issues
• Results in PFS
• Confidence in PFS measurement– IRF radiologist results – IRF vs. ECOG results– Sensitivity analyses for PFS
• Results in OS
• Results in Objective Response

29
Summary of Issues
• Confidence in PFS results– Incomplete data– Loss-to-follow-up– Lack of consistent scan readings
• No effect on overall survival

30
E2100: Results in PFS

31
PFS Results based on IRF Assessments (data cutoff date 2/9/2005)
PAC PAC/BV
No. of patients 354 368
No. of patients with an event (%)
184 (52%) 173 (47%)
Median (month) 5.8 11.3
HR (relative to PAC) 0.48
p-valued < 0.0001

32
Type of Progression by IRF
PFS eventsPAC
N=184
PAC/BEV
N=173
Radiologic Progression 79% 76%
Clinical Progression 11% 15%
Death 10% 9%

33
Cumulative Incidence
0. 0
0. 1
0. 2
0. 3
0. 4
0. 5
0. 6
0. 7
0. 8
0. 9
1. 0
PFS Ti me ( mont hs ) ( I RF)
0 6 12 18 24 30 36
PAC:
PAC/ BEV:
# at r i sk
Progression-Free Survival (IRF)
34
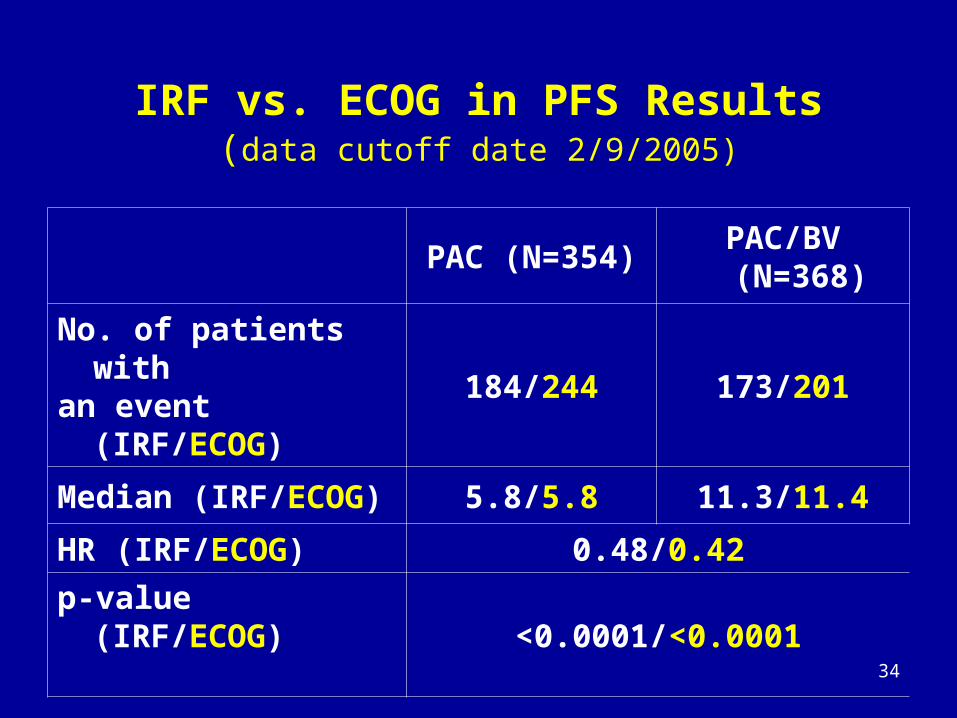
IRF vs. ECOG in PFS Results(data cutoff date 2/9/2005)
PAC (N=354)PAC/BV
(N=368)
No. of patients with an event (IRF/ECOG)
184/244 173/201
Median (IRF/ECOG) 5.8/5.8 11.3/11.4
HR (IRF/ECOG) 0.48/0.42
p-value (IRF/ECOG)<0.0001/<0.0001

35
Confidence in PFS Results

36
Completeness of Tumor assessment
• Missing Radiographic Information: In retrospective collection, Genentech unable to obtain scans in 73 (10%) patients
• Loss-to-follow up: 247 (34%) patients not
followed until IRF-PFS event or end of study

37
• Two radiologists assigned to read all films for each patient
• Readings to be performed independently
• If readings are discordant, 3rd radiologist performed adjudication of radiology results
• An oncologist reviewed all pertinent clinical information
IRF assessment procedure

38
Lack of consistency between the IRF Radiologists in Scan Reading
N No. Patients Adjudicated for Discordant Response or PD Status/Date
Patients with Scan 649 328 (50.5%)
PAC 319 158 (49.5%)
PAC/BEV 330 170 (51.5%)

39
Lack of Consistency between IRF Radiologists for PD Status or Date
NNo. of Patients with Discordant
PD Status or Date (%)
Patients with Scan 649 222 (34.2%)
Radiographic PD 278 131 (47.1%)
No Radiographic PD 371 91 (24.5%)

40
Lack of Consistency between IRF and ECOG for PFS Event Status or Date
Discordance in PFS status (%)Discordance in PFS date when PFS status are agreed upon (%)
IRF PDECOG no PD
IRF no PDECOG PD
Total N =722
43 (6%) 131 (18%) 194 (27%)
51%

41
Lack of consistency between IRF and ECOG
for PFS Event Status
Treatment
No. of discordance (%)
IRF PDECOG no PD
IRF no PDECOG PD
PAC 12 (3.4%) 72 (20.3%)
PAC/BV 31 (8.4%) 59 (16.0%)

42
Lack of Consistency between IRF and ECOG
for PFS Event Status
Treatment
No. of discordance (%)
IRF PDECOG no PD
IRF no PDECOG PD
PAC 12 (3.4%) 72 (20.3%)
PAC/BV 31 (8.4%) 59 (16.0%)

43
Additional PFS Analyses (1)
Median (month)Hazard
RatioPAC PAC/BEV
Primary analysis-IRF 5.8 11.3 0.48
Time to IRF-PFS event, NPT or early discontinuation
4.2 8.1 0.49
Time to IRF-PFS event without censoring for NPT
6.1 11.2 0.57

44
Additional PFS Analyses (2)
Median (month)
Hazard RatioPAC PAC/BEV
NPT and early discontinuation as events in PAC/BEV arm only
5.8 8.1 0.78
Time to earliest PFS event(IRF or ECOG)
4.9 9.0 0.46
Time to earliest PFS event (IRF or ECOG), NPT or early discontinuation
4.1 8.1 0.49
45
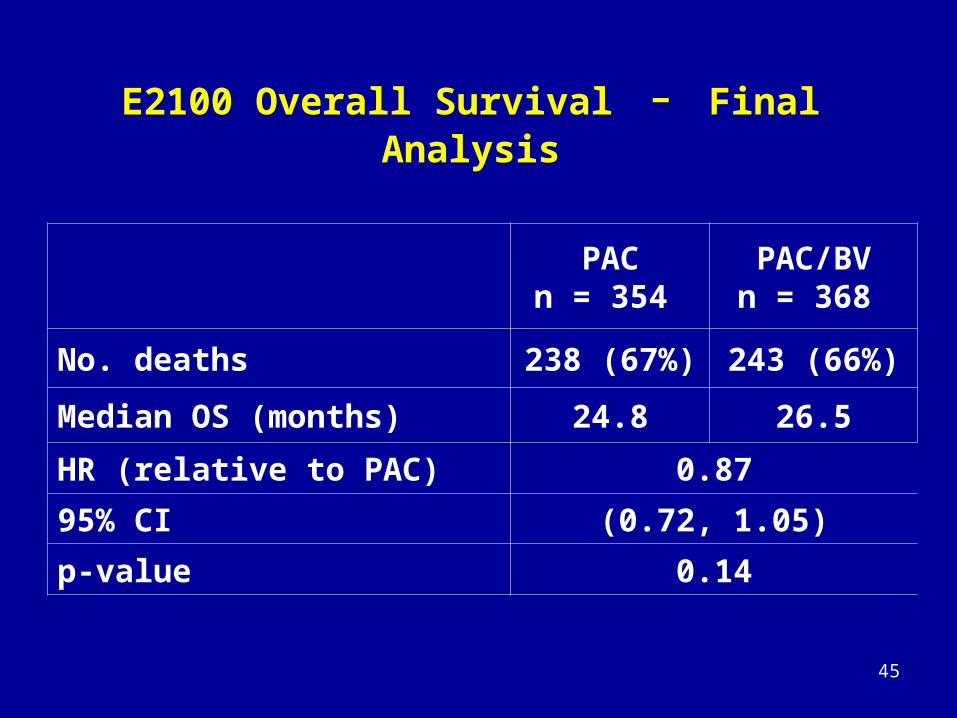
E2100 Overall Survival - Final Analysis
PACn = 354
PAC/BVn = 368
No. deaths 238 (67%) 243 (66%)
Median OS (months) 24.8 26.5
HR (relative to PAC) 0.87
95% CI (0.72, 1.05)
p-value 0.14

46
Cumulative Incidence
0. 0
0. 1
0. 2
0. 3
0. 4
0. 5
0. 6
0. 7
0. 8
0. 9
1. 0
Dur at i on of Sur vi val ( Mont hs )
0 6 12 18 24 30 36 42 48 54 60
PAC
PAC/ BEV
# at r i sk
E2100 Overall Survival – Final Analysis

47
PACn = 243
PAC/BVn = 229
No. with objective response 54 (22%) 112 (49%)
(PAC/BV − PAC) 27%
p-value < 0.0001
Median duration of response (month) 9.7 9.4
Objective Response (IRF)

48
Summary of Issues
• Confidence in PFS results – Genentech unable to obtain scans
retrospectively for 10% of patients– 34% patients not followed until IRF-PFS event
or end of study – 34% discordance between IRF radiologists in
PFS status or date – 51% discordance between IRF and ECOG in
PFS status or date
• No effect on overall survival

49
E2100 Safety Results

50
Drug Exposure (Estimated)
PAC PAC/BV
PAC BEV
Duration of treatment 5 months 9 months
No. of cycles 6 10
Total cumulative dose 1440 mg/m2 1926 mg/m2 180 mg/kg
Relative dose intensity 95 % 85 % 93 %
51
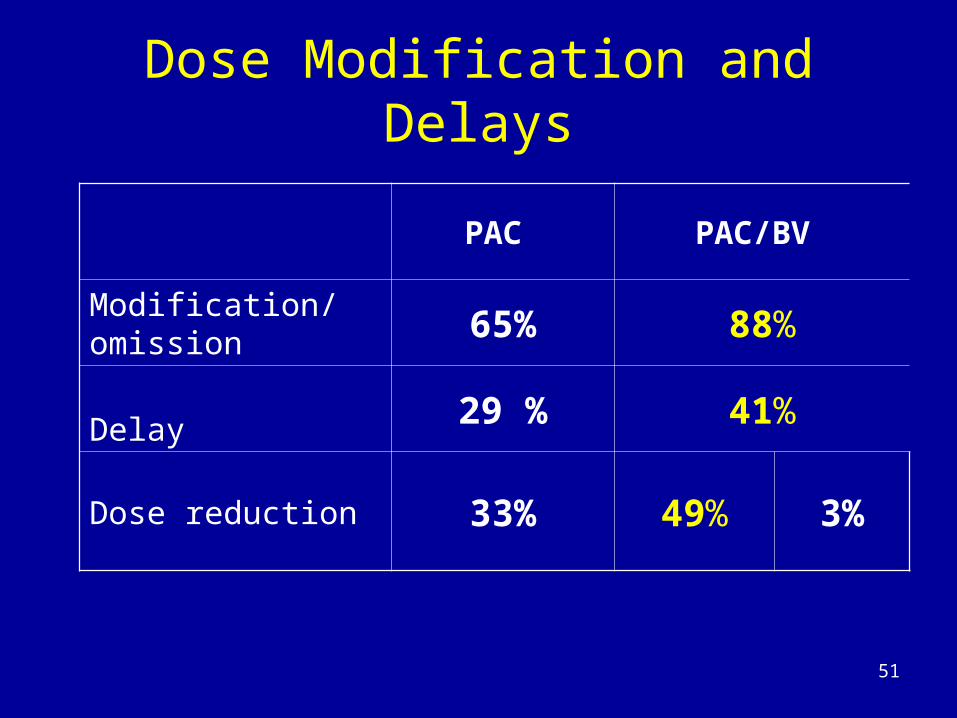
Dose Modification and Delays
PAC PAC/BV
Modification/omission 65% 88%
Delay 29 % 41%
Dose reduction 33% 49% 3%

52
Treatment Discontinuation due to Toxicity
• 142 patients (19.6%) discontinued therapy due to SAE– 70 in PAC arm (20%), – 72 in PAC/BV arm (19.8%)
• Specific adverse event(s) leading to treatment discontinuation not collected in the E2100 study
• Based on temporal association:– PAC: neuropathy and allergic reactions– PAC/BV: neuropathy, thrombosis, proteinuria,
hypertension, arterial thromboembolic event, fatigue, left ventricular dysfunction

53
ECOG Safety Data Collection
• AEs collected once every 3 cycles (12
weeks)
• Date of onset and resolution of AE not
collected
• Only grade 3-5 non-hematologic AEs and
grade 4-5 hematologic AEs collected

54
ECOG Safety Data Collection
• NCI/AdEERS collected serious events
from only PAC/BEV arm
• Laboratory data not collected

55
Grade 3, 4 and 5 Toxicity
PAC N=348 (%)
PAC/BV
N=363 (%)
Total 176 (51) 257 (71)
Grade 5 7 (2) 11 (3)
Grade 4 32 (9) 44 (12)
Grade 3 137 (39) 202 (56)

56
Grade 3, 4 and 5 AEs Known to Occur with Bevacizumab
PAC
N=348 (%)
PAC/BV
N=363 (%)
Hypertension 5 (1.4) 57 (15.7)
Proteinuria 0 10 (2.8)
Arterial Thromboembolic Events
Cerebrovascular ischemia
Cardiac ischemia
0
0
0
10 (2.8)
7 (1.9)
3 (0.8)
Venous Thromboembolic Events 15 (4.3) 9 (2.5)
Bleeding/Hemorrhage 1 (0.3) 6 (1.7)
Congestive Heart Failure 1 (0.3) 5 (1.4)
GI perforation/fistula 0 3 (0.9)
Neutropenia/infection 28 (8) 62 (17.1)

57
Additional Treatment Emergent Grade 3-4 AEs
PAC
% (N=348)
PAC/BV
% (N=363)
Sensory neuropathy 18 24
Vomiting
Diarrhea
Dehydration
2
1
1
6
5
3
Fatigue 5 11
Pain 10 16

58
Deaths on Study per Genentech
Primary cause of death PAC
N = 348 (%)PAC/BV
N = 363 (%)
All deaths* 256 (74) 255 (70)
Due to this disease 241 (69) 243 (67)
Due to protocol treatment
1 (0.3) 0 (0)
Due to other cause 7 (2) 9 (2.5)
Unknown 7 (2) 3 (0.8)
Adapted from Genentech’s CSR, Table 14.3/26* Data from ECOG CRFs. All deaths included. No cut off date.

59
Deaths on Study/within 30 days of End of Study Genentech and FDA’s Attribution of the Cause of Death
PAC PAC/BV
Cause of deathApplicant FDA Applicant FDA
Death on study/within 30d 7 7 12 12
Due to treatment Definite Probable
00
00
00 2
3
Due to this disease 4 3 8 4
Due to other cause 2 1 4 1Unknown 1 2 0 2Insuf. information 0 1 0 0

60
Deaths on Study Possibly/Definitively Related to Protocol Therapy per FDA
• ID 21010: 79 yo, after 6 cycles PAC/BEV, developed severe diarrhea, fatigue, muscle weakness and lethargy and death 11 days after last dose of protocol (Applicant’s attribution: death due to breast cancer)
• ID 21258: 64 yo, after 6 cycles PAC/BEV, developed abdominal pain with gastrointestinal perforation, neutropenia, sepsis and death. (Applicant’s attribution: death due to breast cancer)
• ID 21314: 84 yo, after 3 cycles of PAC/BEV, developed acute abdomen with gastrointestinal perforation, sepsis, respiratory failure and death. (Applicant’s attribution: death due to other cause)

61
• ID 21403: 73 yo, 22 days after BEV/PAC developed progressive fatigue, pneumonitis, and fatal cardiac ischemia/infarction and LV dysfunction. (Applicant’s attribution: death due to other cause)
• ID 26004: 66 yo, after 11 cycles of PAC/BEV was admitted with severe diarrhea, black tarry stool and abdominal pain. Symptoms were attributed to diverticulitis and PAC/BEV. 22 days later became hypotensive, bradycardic and died. (Applicant attribution: death due to other cause)
• ID 21390: 69 yo, discontinued protocol after 3 cycles of BEV/PAC due to grade 4 proteinuria (nephrotic syndrome). Patient had a fatal acute myocardial infarction 7 weeks after being discontinued from protocol (Applicant’s attribution: death due to other cause)

62
Cumulative Incidence
0. 0
0. 1
0. 2
0. 3
0. 4
0. 5
0. 6
0. 7
0. 8
0. 9
1. 0
Dur at i on of Sur vi val ( Mont hs )
0 6 12 18 24 30 36 42 48 54 60
PAC
PAC/ BEV
# at r i sk
E2100 - Overall Survival

63
AVF2119g

64
AVF2119g
N = 462• Progressive metastatic breast cancer• Previously treated with anthracycline and taxane
Capecitabine 2500 mg/m2 d 1-14
Cycles q 3 wks
Capecitabine 2500 mg/m2 d1-14
Bevacizumab 15 mg/kg q 3 wks
Cycles q 3 wks
StratificationECOG PS (0 or ≥ 1)N of prior chemo for MBC (0 or ≥ 1)

65
Primary Endpoint - PFS
CAP
N=230 (%)
CAP + AVF
N=232 (%)
Median (months)4.17
(3.71, 5.13)
4.86
(4.17, 5.52)
HR 0.98 (0.77, 1.25)
P-value 0.857

66Tr eat ment Gr oup ( Char ) CAP Al one CAP+AVF
Cumulative Incidence
0. 2
0. 3
0. 4
0. 5
0. 6
0. 7
0. 8
0. 9
1. 0
Dur at i on of Sur vi val ( mont hs )
0 2 4 6 8 10 12 14 16 18 20
CAP CAP+AVF
Median (month) 14.5 15.1
HR 1.08
P-value 0.63
AVF2119g Overall Survival

67
AVF2119g Response Rate
CAP
N=230 (%)
CAP+BEV
N=232 (%)
ORR 21 (9.1) 46 (19.8)
95% CI 4.3%, 17.0%
P-value 0.001
Duration of response
7.5 months 4.9 months

68
AVF2119g - AEs
• Common AEs in both arms: asthenia, pain, diarrhea, nausea, vomiting, hand-foot syndrome
• Common AE in CAP/Bev arm: headache, hypertension, epistaxis and proteinuria
CAP N=215 (%)
CAP + BEV N=229 (%)
Grade 3-4 All grades Grade 3-4 All grades
Any AE 124 (57.7) 211 (98.1) 165 (72.1) 229 (100)

69
Grade 3-4 AEs Known to Occur with Bevacizumab
CAPN=215 (%)
CAP + BEVN=229 (%)
Hypertension 0.5 20.1
Thromboembolism 3.7 6.1
CHF/Cardiomyopathy 1.0 3.5
Proteinuria 0 0.9
Bleeding 0.5 0.4

70
Summary of Findings

71
E2100 Efficacy
• Estimated 5.5 months improvement in PFS by independent review
• PFS improvement is similar to the ECOG investigators’ findings
• No survival advantage
objective response

72
E2100 Efficacy Robustness of effect: YES
Magnitude of effect: No
Factors affecting confidence in magnitude of PFS finding:
– Missing scans(10%) – 34 % patients not followed until IRF-PFS event or
end of study – Lack of reliability in determination of radiologic
disease progression and date of progression between independent radiologists and investigators and independent radiologists.

73
E2100 - Safety
• Incomplete assessment of toxicity profile:– Grade 1-2 not collected– Laboratory data not collected
• 20.2% increase in grade 3-5 toxicity
• 1.7 % treatment related death in the bevacizumab plus paclitaxel arm

74
AVF2119g
• Did not increase PFS in MBC patients • No survival advantage
objective response (short duration)
• 14.4% in grade 3-4 toxicity

75
Thank you

76
Questions to ODAC

77
Question 1 (non-voting)
In the E2100 study, PFS is not a surrogateendpoint for overall survival (OS) in first-line breast cancer.
Please discuss whether PFS alone without a demonstrated survival advantage should be considered a measure of direct clinical benefit in the initial treatment of metastatic breast cancer.

78
Questions 2Summary results:
• Estimated 5.5 month improvement in median PFS
claimed by Genentech• No improvement in OS • Increased toxicity/toxic death • No effect on PFS or OS in 2nd and 3rd line MBC
Are the data provided sufficient to establish a favorable risk/benefit analysis for the use of bevacizumab plus paclitaxel for first-line treatment of patients with metastatic
breast cancer ?