1 the role of bioavailability in pharmaceutical product development alwyn pidgen pharmacokinetics...
TRANSCRIPT

1
The Role of BioavailabilityThe Role of Bioavailability
in in
Pharmaceutical Product Pharmaceutical Product DevelopmentDevelopment
Alwyn PidgenAlwyn PidgenPharmacokinetics consultantPharmacokinetics consultant

2
FormulationsFormulations include:- include:-
Tablet, Suspension, Capsule, Solution, PowderTablet, Suspension, Capsule, Solution, Powder Aerosol, Nebulizer, Dry Powder Inhaler (DPI)Aerosol, Nebulizer, Dry Powder Inhaler (DPI) Lotion, Ointment, CreamLotion, Ointment, Cream Suppository, Rectal solution, ImplantsSuppository, Rectal solution, Implants
Different routes Different routes && formulations formulations can impact thecan impact the speed speed andand completeness completeness of drug of drug absorptionabsorption
INTRAVASCULARINTRAVASCULAR IntravenousIntravenous
EXTRAVASCULAREXTRAVASCULAR Intra-muscular; Subcutaneous; Oral; Intra-muscular; Subcutaneous; Oral; Rectal; Topical; Rectal; Topical; Inhalation; Intranasal; Inhalation; Intranasal; Transdermal; Sublingual; BuccalTransdermal; Sublingual; Buccal
3
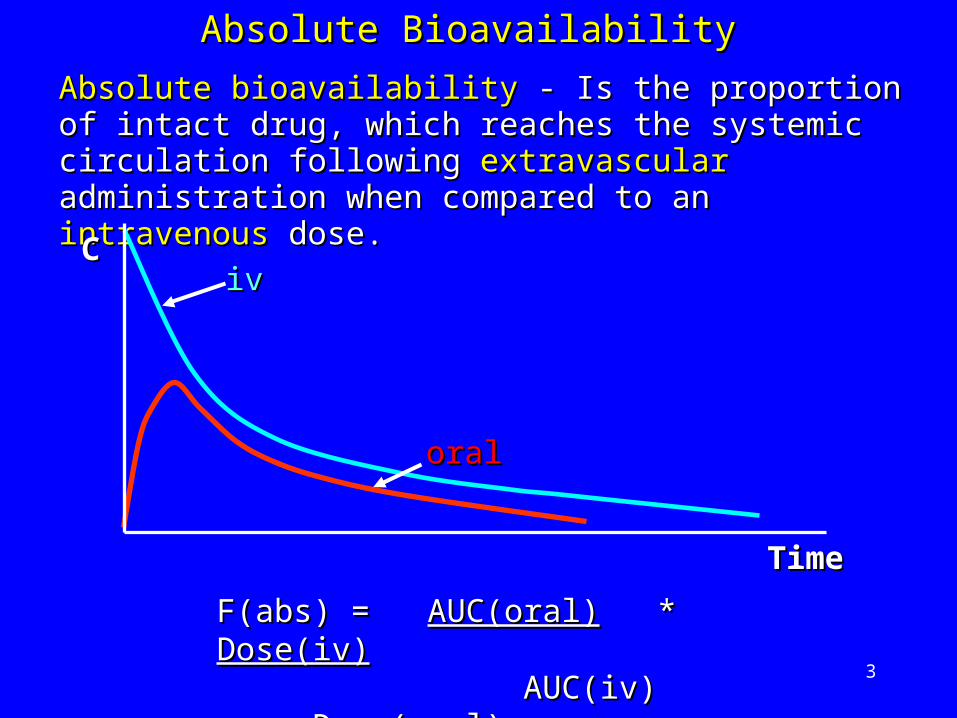
Absolute bioavailabilityAbsolute bioavailability - Is the proportion of intact - Is the proportion of intact drug, which reaches the systemic circulation drug, which reaches the systemic circulation following following extravascular extravascular administration when administration when compared to an compared to an intravenousintravenous dose. dose.
Absolute BioavailabilityAbsolute Bioavailability
F(abs) = F(abs) = AUC(oral)AUC(oral) * * Dose(iv)Dose(iv) AUC(iv) AUC(iv) Dose(oral)Dose(oral)
iviv
oraloral
CC
TimTimee
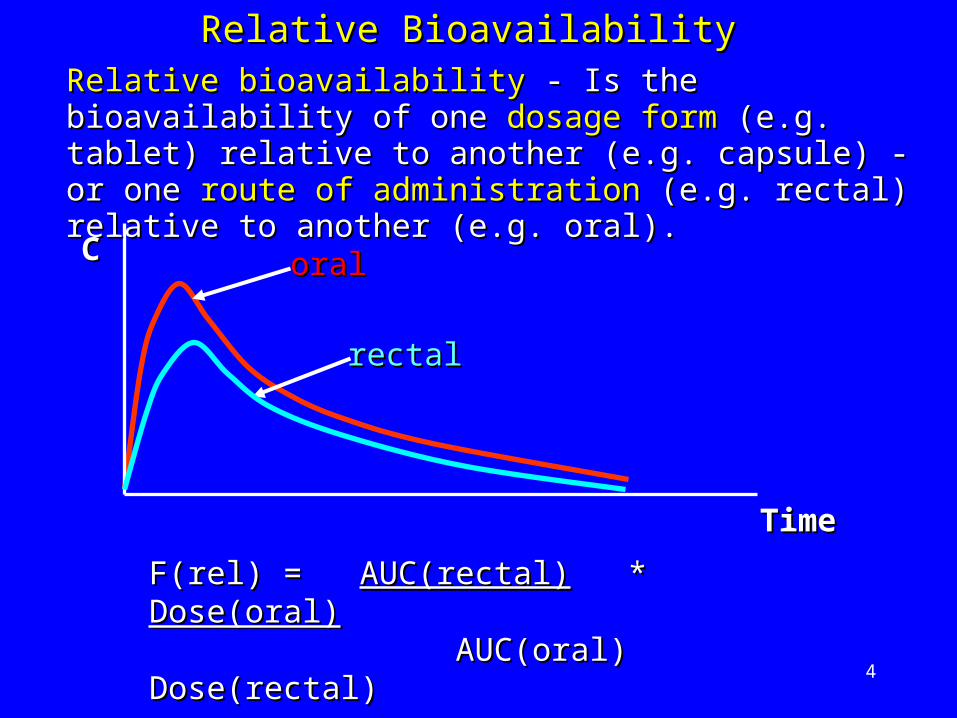
4
Relative BioavailabilityRelative Bioavailability
F(rel) = F(rel) = AUC(rectal)AUC(rectal) * * Dose(oral)Dose(oral) AUC(oral) Dose(rectal)AUC(oral) Dose(rectal)
rectalrectal
oraloral
Relative bioavailabilityRelative bioavailability - Is the bioavailability of one - Is the bioavailability of one dosage formdosage form (e.g. tablet) relative to another (e.g. (e.g. tablet) relative to another (e.g. capsule) - or one capsule) - or one route of administrationroute of administration (e.g. (e.g. rectal) relative to another (e.g. oral).rectal) relative to another (e.g. oral).
CC
TimeTime

5
BioequivalenceBioequivalence
Two medicinal products can be classed Two medicinal products can be classed as as bioequivalent bioequivalent when their when their raterate and and extentextent of of absorption absorption meet strict meet strict regulatory requirements after regulatory requirements after administration of the administration of the same molar dose.same molar dose.

6
Medicinal products are called Medicinal products are called Pharmaceutical Pharmaceutical
equivalentsequivalents if they contain the if they contain the same amountsame amount
of the of the same active substancesame active substance in the in the same same
dosage formdosage form, which meet the same or , which meet the same or
comparable standards.comparable standards.
Pharmaceutical equivalencePharmaceutical equivalence

7
Particle sizeParticle size Physico-chemical factors of drugPhysico-chemical factors of drug SolubilitySolubility Use of different excipientsUse of different excipients Degree of agitationDegree of agitation Change in manufacturing processChange in manufacturing process FoodFood
HOWEVER -HOWEVER - Pharmaceutical equivalence Pharmaceutical equivalence does not does not
automatically assure automatically assure bioequivalencebioequivalence mainly due mainly due
to changes in to changes in dissolutiondissolution, which can be influenced , which can be influenced
by: -by: -
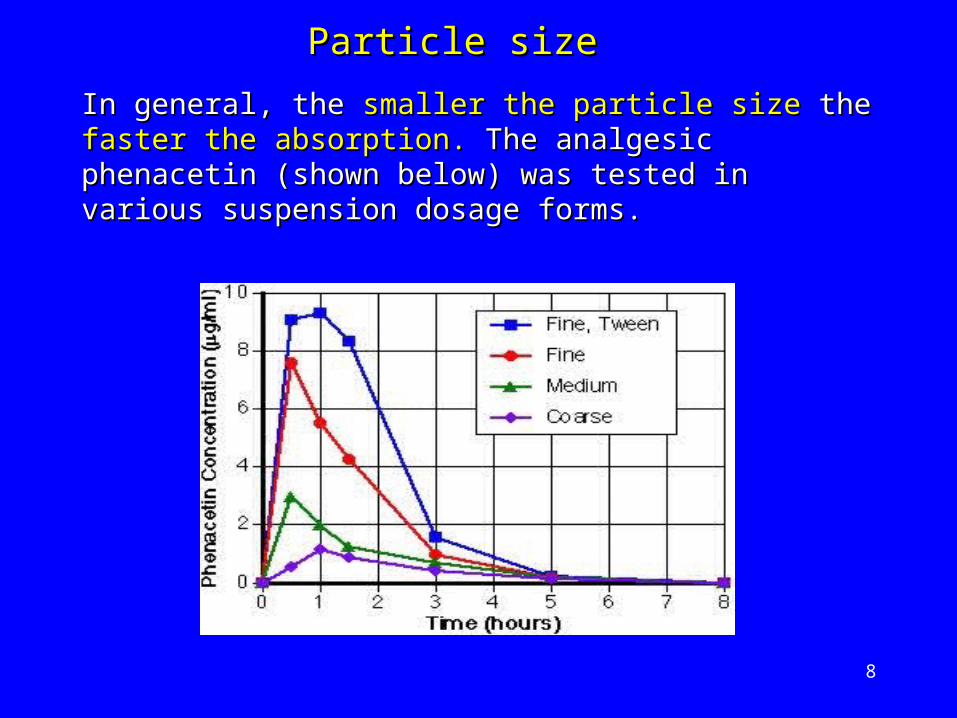
8
In general, the In general, the smaller the particle sizesmaller the particle size the the faster the faster the absorption.absorption. The analgesic phenacetin (shown below) The analgesic phenacetin (shown below) was tested in various suspension dosage forms. was tested in various suspension dosage forms.
Particle sizeParticle size

9
HOWEVERHOWEVER This is replaced by the This is replaced by the indirectindirect approach of a approach of a bioequivalence trialbioequivalence trial based on the principle that:- based on the principle that:- ‘‘Two medicinal products that give rise to Two medicinal products that give rise to ‘essentially ‘essentially equivalent’equivalent’ concentrations of the concentrations of the active speciesactive species in blood in blood (viewed as a profile over time) will give (viewed as a profile over time) will give equivalent equivalent therapeutic effects’therapeutic effects’..
KEY CONCEPT IN BIOEQUIVALENCEKEY CONCEPT IN BIOEQUIVALENCE ‘‘Equal amounts of the same drug administered in Equal amounts of the same drug administered in different products will show equal therapeutic effects’.different products will show equal therapeutic effects’.
A A directdirect demonstration of demonstration of therapeutic equivalencetherapeutic equivalence requires a clinical trialrequires a clinical trial

10
Why perform Bioequivalence studies Why perform Bioequivalence studies ??
To enable clinical trial formulations to be To enable clinical trial formulations to be modified or production ‘scaled up’ modified or production ‘scaled up’ throughout a drug’s development.throughout a drug’s development.
To compare a clinical trial formulation with the To compare a clinical trial formulation with the ‘to be marketed’ product just prior to filing.‘to be marketed’ product just prior to filing.
To compare a generic drug product with a To compare a generic drug product with a corresponding reference drug. corresponding reference drug.
To change the dosage regimen by means of a To change the dosage regimen by means of a change in formulation change in formulation • Immediate release Immediate release Modified-release Modified-release

11
When are bioequivalence studies not normally When are bioequivalence studies not normally
needed ?needed ?
If the product differs only in the If the product differs only in the strength of the strength of the active substanceactive substance it contains and the it contains and the pharmacokinetics are linearpharmacokinetics are linear
If the product has been If the product has been slightly reformulatedslightly reformulated or or the manufacturing method slightly modified by the manufacturing method slightly modified by the original manufacturer in ways that can be the original manufacturer in ways that can be argued to be irrelevant argued to be irrelevant (using in-vitro tests)(using in-vitro tests)
If the product is to be administered If the product is to be administered parenterallyparenterally as a solution and contains the same active as a solution and contains the same active substances and excipients as a medicinal productsubstances and excipients as a medicinal product currently approvedcurrently approved..

12
If the product is a If the product is a liquid oral form in solutionliquid oral form in solution containing the active substance in the same containing the active substance in the same concentration and form as a concentration and form as a currently approved currently approved medicinal productmedicinal product
An An acceptable correlationacceptable correlation between dissolution between dissolution rate in-vivo and in-vitro has been shown (FDA rate in-vivo and in-vitro has been shown (FDA guidance).guidance).
Products intended for Products intended for local uselocal use to act to act without without systemic absorptionsystemic absorption
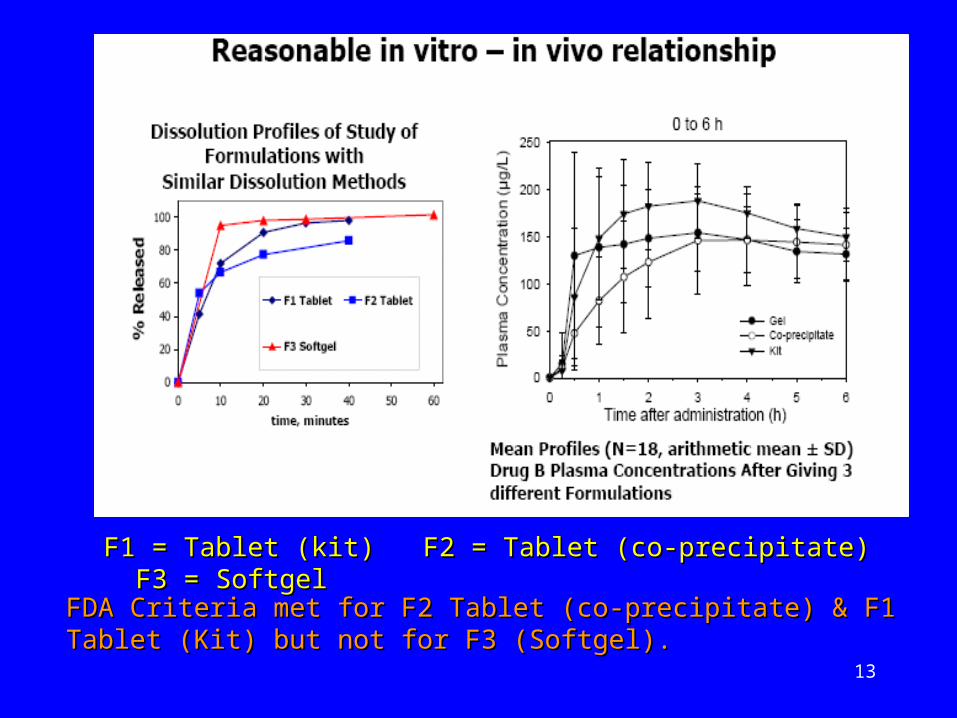
13
F1 = Tablet (kit) F2 = Tablet (co-precipitate) F3 = F1 = Tablet (kit) F2 = Tablet (co-precipitate) F3 = SoftgelSoftgel
FDA Criteria met for F2 Tablet (co-precipitate) & F1 Tablet (Kit) FDA Criteria met for F2 Tablet (co-precipitate) & F1 Tablet (Kit) but not for F3 (Softgel).but not for F3 (Softgel).

14
Healthy volunteersHealthy volunteers
Male subjects or women of non-child bearing Male subjects or women of non-child bearing potentialpotential
Aged between 18 and 55Aged between 18 and 55
Single dose.Single dose.
Crossover designCrossover design – first choice – first choice
Parallel group designParallel group design - for drugs with long half- - for drugs with long half-liveslives
Replicate designReplicate design - for highly variable drugs - for highly variable drugs
Typical Study Design FeaturesTypical Study Design Features

15
Should be appropriately Should be appropriately statistically poweredstatistically powered if study is pivotal for filingif study is pivotal for filing
An estimate of a drug’s An estimate of a drug’s intrinsic variabilityintrinsic variability is is obtained from previous studies or obtained from previous studies or publicationspublications
Should not be smallerShould not be smaller than 12 than 12
SAMPLE SIZESAMPLE SIZE

16
For an inactive For an inactive pro-drugpro-drug the main the main active active metabolitemetabolite should be measured if the plasma should be measured if the plasma levels of the levels of the parentparent are are too lowtoo low for accurate for accurate assay measurement. assay measurement.
What to measureWhat to measure
For BE studies - measurement of For BE studies - measurement of plasma plasma concentrationsconcentrations of the of the parent drugparent drug are are recommended.recommended.
Parent drug is more sensitive to formulation changes Parent drug is more sensitive to formulation changes than any metabolite.than any metabolite.
Examples of prodrugsExamples of prodrugs EnalaprilEnalapril (hypertension) is metabolised to the active (hypertension) is metabolised to the active
form form EnalaprilatEnalaprilat
ValacyclovirValacyclovir (herpes virus) is metabolised to the active (herpes virus) is metabolised to the active form form AcyclovirAcyclovir

17
Enantiomers versus RacematesEnantiomers versus Racemates
For BE studies measurement of the For BE studies measurement of the racemate racemate is is recommended.recommended.
If one enantiomer is pharmacologically active If one enantiomer is pharmacologically active and the other contributes little to activity, it is and the other contributes little to activity, it is sufficient to demonstrate bioequivalence for the sufficient to demonstrate bioequivalence for the active enantiomer only.active enantiomer only.
Measurement of Measurement of individual enantiomersindividual enantiomers is is recommended if:-recommended if:- The enantiomers have different PK or PD The enantiomers have different PK or PD
characteristicscharacteristics The exposure (AUC ratio) of the enantiomers is The exposure (AUC ratio) of the enantiomers is
modified by a difference in the rate of modified by a difference in the rate of absorption.absorption.

18
Endogenous substancesEndogenous substances
Difficult area. A pilot study may be useful to check Difficult area. A pilot study may be useful to check the effect. the effect. Baseline correctionBaseline correction of background of background levels are required to ensure that drug levels levels are required to ensure that drug levels reflect the treatments under test. reflect the treatments under test. A longer A longer washout period may be required.washout period may be required.
Fixed combinationsFixed combinations
A A separate BE analysisseparate BE analysis should be performed on should be performed on each active substanceeach active substance in a fixed combination. This in a fixed combination. This is achieved by considering all other active is achieved by considering all other active substances (in turn) as excipients.substances (in turn) as excipients.

19
Peak exposurePeak exposure
Important parameter - may have potential links to safety Important parameter - may have potential links to safety and/or efficacy.and/or efficacy.
Parameter is Maximum observed drug concentrationParameter is Maximum observed drug concentration (Cmax)(Cmax)
PHARMACOKINETIC PARAMETERSPHARMACOKINETIC PARAMETERS
Early exposureEarly exposure
Only consider if making a claim for Only consider if making a claim for clinically relevant clinically relevant rapid release - and/or - if onset of action is related to rapid release - and/or - if onset of action is related to adverse events.adverse events. Rapid onset of analgesic effectRapid onset of analgesic effect Avoidance of excessive hypotensive actionAvoidance of excessive hypotensive action
Parameters includeParameters include AUC( AUC(0–tmax0–tmax) ) (FDA)(FDA) andand tmax tmax (EMEA)(EMEA)
20
ParametersParameters
AUC to the last measurable time point AUC to the last measurable time point (AUC(AUC0-t0-t)) AUC truncated at 72h AUC truncated at 72h (AUC(AUC0-720-72)) – (EMEA only) – (EMEA only) AUC extrapolated to infinite time AUC extrapolated to infinite time (AUC(AUC0-inf0-inf))
o The elimination rate constant The elimination rate constant (kel)(kel) and terminal half- and terminal half-life life (t(t1/21/2)) should be reported - particularly if should be reported - particularly if AUC(AUC(0-0-
infinf)) is used. is used.
Total exposureTotal exposure
This is the most important BE parameter since AUC is This is the most important BE parameter since AUC is directlydirectly proportional to the amount of drug absorbed.proportional to the amount of drug absorbed.

21
STATISTICAL ANALYSISSTATISTICAL ANALYSIS
Calculate 90% Calculate 90% confidence intervalsconfidence intervals of the ratio of the of the ratio of the treatment means (test/reference) for treatment means (test/reference) for CmaxCmax, and , and AUCAUC..The data are log transformed prior to analysisThe data are log transformed prior to analysis..
If statistical evaluation of If statistical evaluation of tmaxtmax is required then a is required then a non-parametric hypothesis testnon-parametric hypothesis test is performed on the is performed on the untransformed data.untransformed data.

22
Bioequivalence regionBioequivalence region
For Bioequivalence - the ratio of the For Bioequivalence - the ratio of the geometric means geometric means µµTT/µ/µRR of the test and of the test and reference products must bereference products must be ≥ 0.8 ≥ 0.8 andand ≤ 1.25 ≤ 1.25..
0.80.8 1.21.255
BioequivalencBioequivalence e
regionregion
Bio-Bio-inequivalence inequivalence
regionregion
Bio-Bio-inequivalence inequivalence
regionregion
µµTT/µ/µRR

23
tmaxtmax : Only consider : Only consider if clinically relevant rapid release is if clinically relevant rapid release is claimed and/or onset of action is related to adverse events.claimed and/or onset of action is related to adverse events.
AUCAUC : test treatment to be within : test treatment to be within ≥≥ 0.8 and 0.8 and ≤≤ 1.25 of the 1.25 of the reference treatment (FDA and EMEA)reference treatment (FDA and EMEA)
Range to be tightened in the case of a drug with a narrow Range to be tightened in the case of a drug with a narrow therapeutic window (e.g. digoxin, phenytoin) (FDA and therapeutic window (e.g. digoxin, phenytoin) (FDA and EMEA). This may also be applicable to Cmax.EMEA). This may also be applicable to Cmax.
CmaxCmax : : test treatment to be within test treatment to be within ≥≥ 0.8 and 0.8 and ≤≤ 1.25 of the 1.25 of the reference treatmentreference treatment (FDA and EMEA)(FDA and EMEA)
A wider interval may be acceptable (EMEA only). A wider interval may be acceptable (EMEA only). The actual The actual limits vary according to the within-subject variability (%CV) limits vary according to the within-subject variability (%CV) noted in the bioequivalence study. See EMEA guidelines for noted in the bioequivalence study. See EMEA guidelines for details.details.
The Regulatory acceptance criteriaThe Regulatory acceptance criteria

24
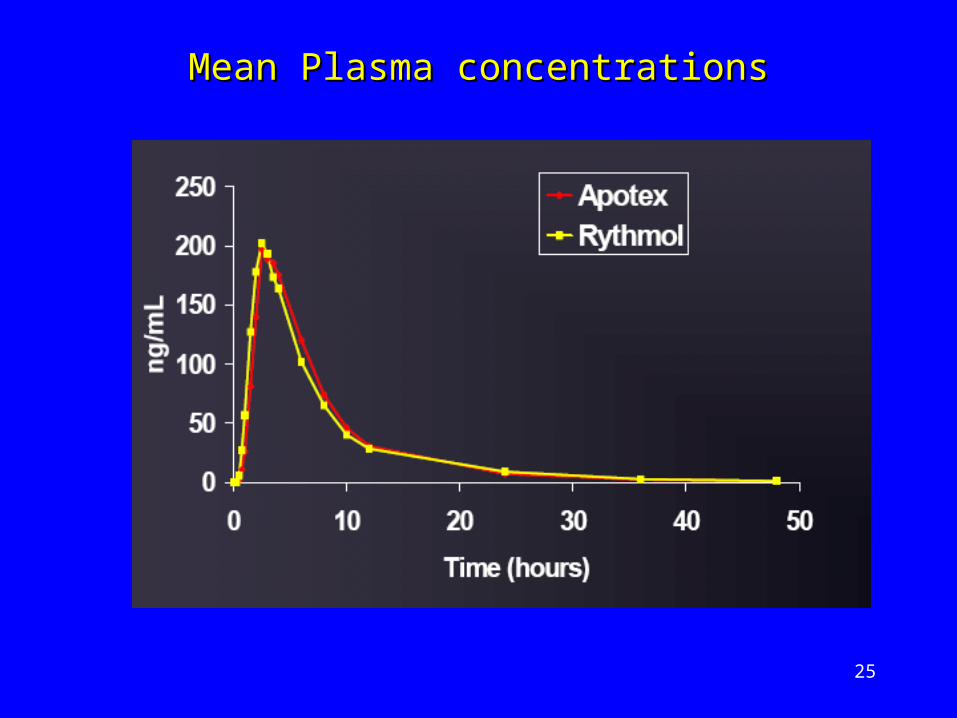
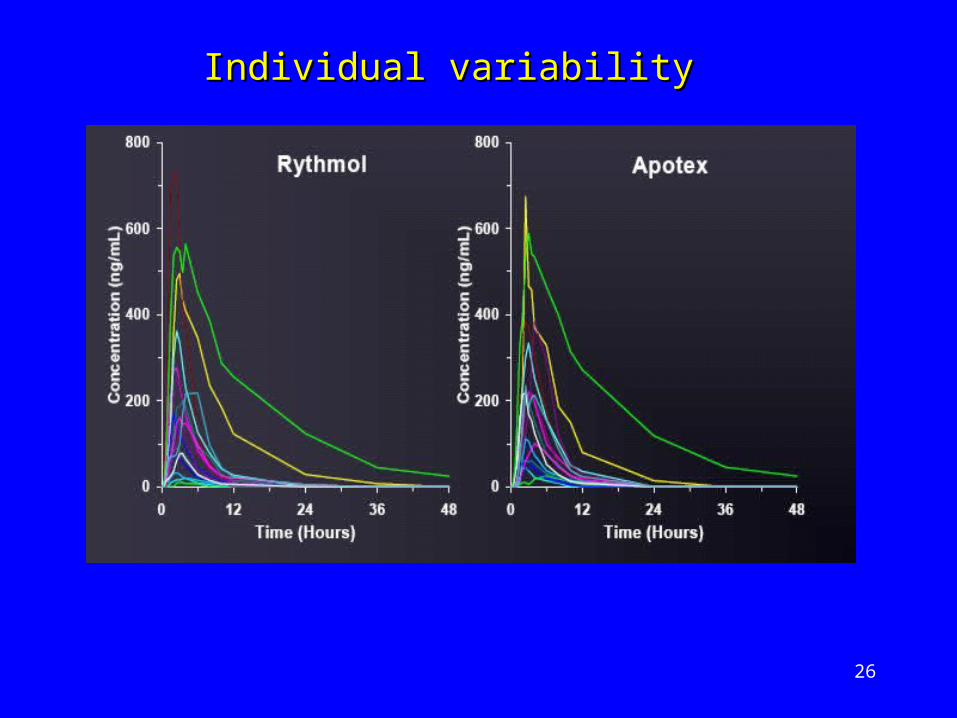
Propafenone – anti-arrhythmic drugPropafenone – anti-arrhythmic drug Undergoes extensive first-pass metabolismUndergoes extensive first-pass metabolism Variable half-lifeVariable half-life
Bioequivalence study undertakenBioequivalence study undertaken
Apotex (Reference) v Rhythmol (Test)Apotex (Reference) v Rhythmol (Test)
300mg tablet 300mg tablet
18 healthy subjects18 healthy subjects
2-way crossover2-way crossover
An exampleAn example
‘‘Highly Variable Drugs:Experience with Propafenone’ -Highly Variable Drugs:Experience with Propafenone’ -Yu Yu Chung Tsang, Radu Pop & Michael SpinoChung Tsang, Radu Pop & Michael Spino http://www.ualberta.ca/~csps/JPPS1(2)/Y.Tsang/Tsang.pdfhttp://www.ualberta.ca/~csps/JPPS1(2)/Y.Tsang/Tsang.pdf
25
Mean Plasma concentrationsMean Plasma concentrations
26
Individual variabilityIndividual variability
27
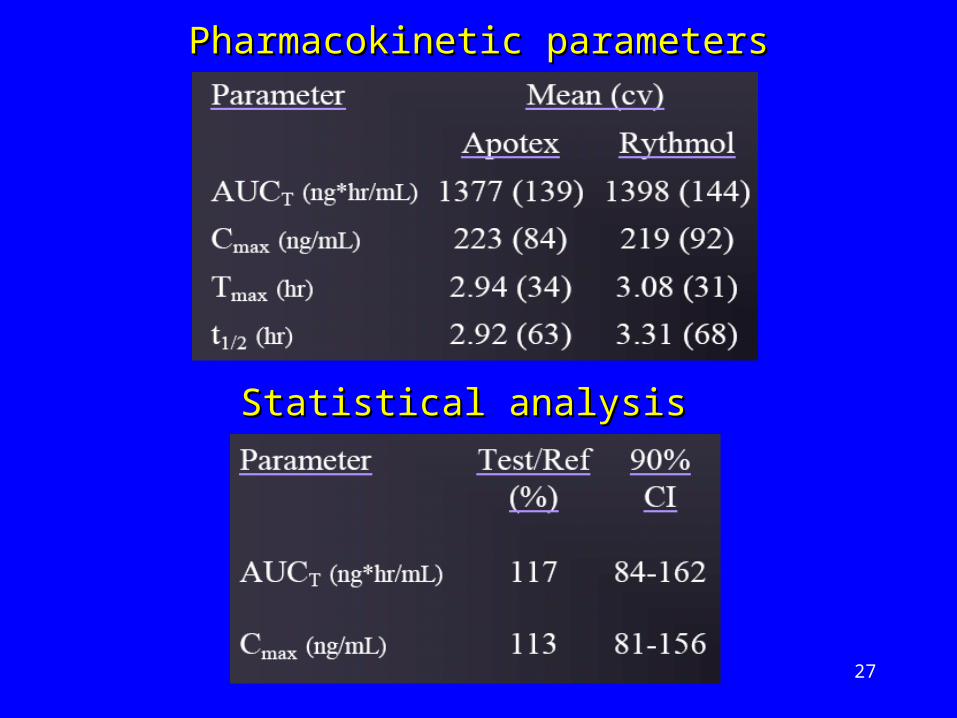
Pharmacokinetic parametersPharmacokinetic parameters
Statistical analysisStatistical analysis

28
Reasons for BE failureReasons for BE failure
Very high Very high inter-subjectinter-subject CV for Cmax & CV for Cmax & AUCAUC
High overall High overall intra-subjectintra-subject variability variability (46%)(46%)
Variability in elimination characteristicsVariability in elimination characteristics
The The metabolismmetabolism of propafenone is of propafenone is influenced by influenced by geneticsgeneticso tt1/21/2 (fast metabolisers) = 2-10h (fast metabolisers) = 2-10ho tt1/21/2 (slow metabolisers) = 10-32h (slow metabolisers) = 10-32ho Drug levels 5 times higher in slow Drug levels 5 times higher in slow
metabolisersmetabolisers

29
Hot topic – large market potential – lot of issuesHot topic – large market potential – lot of issues
More More regulatoryregulatory obstacles than with traditional obstacles than with traditional ‘small molecule’ generics.‘small molecule’ generics. FDA position still under reviewFDA position still under review EMEA guidance availableEMEA guidance available Numerous position papersNumerous position papers
Current ANDA approach Current ANDA approach not considered not considered scientifically appropriatescientifically appropriate for biotechnology for biotechnology productsproducts
Companies asked to show Companies asked to show ‘biosimilarity’‘biosimilarity’ – this – this involves involves clinical trialsclinical trials to demonstrate to demonstrate safety/efficacysafety/efficacy
Biogenerics – current statusBiogenerics – current status

30
Protein productsProtein products have a greater structural have a greater structural complexity, often difficult to characterize and have complexity, often difficult to characterize and have a much higher molecular weight than a much higher molecular weight than ‘small ‘small molecule drugs’molecule drugs’. .
They can be mixtures of They can be mixtures of many molecular speciesmany molecular species and can have and can have unique impurity profilesunique impurity profiles that depend that depend upon the upon the manufacturing processmanufacturing process..
Biogenerics – why the problems ?Biogenerics – why the problems ?

31
Small changesSmall changes in the manufacturing process can in the manufacturing process can lead to lead to big changesbig changes in the drugs safety & in the drugs safety & efficacy. FDA collecting examples to evaluate this efficacy. FDA collecting examples to evaluate this claim.claim.
Improvements in Improvements in protein technologyprotein technology mean mean manufacturers can no longer produce a protein manufacturers can no longer produce a protein that would have identical characteristics to the that would have identical characteristics to the original.original.
ImplicationsImplications - altered PK & PD of the protein - - altered PK & PD of the protein - ultimately leading to ultimately leading to clinical implicationsclinical implications..
Biogenerics - manufactureBiogenerics - manufacture
32
FDA FDA (www.fda.gov/cder/guidance/index.htm)(www.fda.gov/cder/guidance/index.htm) Bioavailability & Bioequivalence studies for orally administered drug Bioavailability & Bioequivalence studies for orally administered drug
products – general considerations – March 2003 products – general considerations – March 2003 Food-effect Bioavailability & Fed Bioequivalence studies – December Food-effect Bioavailability & Fed Bioequivalence studies – December
2002 2002 Modified Release Solid Oral Dosage Forms - Scale-Up and Post-Modified Release Solid Oral Dosage Forms - Scale-Up and Post-
approval Changes: Chemistry, Manufacturing and Controls; In Vitro approval Changes: Chemistry, Manufacturing and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation – Dissolution Testing and In Vivo Bioequivalence Documentation – Sept 1997Sept 1997
Dissolution testing of Immediate release solid oral dosage forms - Dissolution testing of Immediate release solid oral dosage forms - Aug 97Aug 97
Extended release oral dosage forms : Development, Evaluation & Extended release oral dosage forms : Development, Evaluation & Application of In-vitro/In-vivo correlations – Sept 97Application of In-vitro/In-vivo correlations – Sept 97
EMEA (www.emea.europa.eu/index/indexh1.htm)EMEA (www.emea.europa.eu/index/indexh1.htm) Guideline on the Investigation of Bioequivalence - January 2010 - Guideline on the Investigation of Bioequivalence - January 2010 -
Doc. Ref.CPMP/EWP/QWP/1401/98Doc. Ref.CPMP/EWP/QWP/1401/98 Guideline on Similar Biological Medicinal products – November 2004Guideline on Similar Biological Medicinal products – November 2004 Note for guidance on quality of Modified Release products (A Oral Note for guidance on quality of Modified Release products (A Oral
dosage forms; B Transdermal dosage forms) – July 1999dosage forms; B Transdermal dosage forms) – July 1999 Guideline on Similar Biological Medicinal Products – Nov 2004Guideline on Similar Biological Medicinal Products – Nov 2004 Guideline on Similar Biological Medicinal Products containing Guideline on Similar Biological Medicinal Products containing
Biotechnology-derived Proteins as active substance: Quality Issues – Biotechnology-derived Proteins as active substance: Quality Issues – June 2005June 2005
Useful Regulatory Guidance documentsUseful Regulatory Guidance documents