22.10.2017 muskel trauma und mehr! … aus radiologischer ... · 07.11.2017 12. bremer...
TRANSCRIPT

07.11.2017
www.klinikum-bochum.de
12. Bremer MR-Symposium, 20. – 22.10.2017
Muskel – Trauma und mehr! … aus radiologischer Perspektive Christoph M. Heyer Institut für Kinderradiologie, Ruhr-Universität Bochum MVZ Radiologie JosefCarrée
Matthias Vorgerd Neurologische Universitätsklinik, Heimer Institut für Muskelforschung, Univ.-Klinikum Bergmannsheil, Bochum

Klassifikation der Myopathien
Erworbene Myopathien - Toxische Myopathien
(Statine u.a.)
- Immunogene Myositiden
- Erregerbedingte Myositiden
- Traumatische Muskelläsionen
Erbliche Myopathien - Muskeldystrophien
- X-chromosomale Muskeldystrophien
- Gliedergürtelmuskel- dystrophien (LGMD) - Kongenitale
Muskeldystrophien
- Myofibrilläre Myopathien
- Metabolische Myopathien Mittlerweile sind >150 verschiedene Krankheitsgene beschrieben worden

Symptome von Myopathien
- Katarakt - Skoliose - Gelenkkontrakturen - Hautveränderungen
(z.B. Hyperlaxizität, Gesichts-erythem, Teleangiektasien der Nagelfalz)
- Kardiomyopathie - ZNS-Beteiligung - Gastrointestinale Symptome
Minus-Symptome Muskelschwächen/Muskelatrophien - Generalisiert oder fokal? - Proximal oder distal betont? - Symmetrisch oder asymmetrisch? - Mitbeteiligung der Augen-,
Gesichts-, Schluckmuskulatur? - Mitbeteiligung der
Atemmuskulatur?
Plus-Symptome - Muskelschmerzen, Krampi - Myoglobinurie - Muskelsteifigkeit
Hinweise auf multisystemische Beteiligung
Skelettmuskel-assoziierte Symptome

Stufendiagnostik von Myopathien
Diagnostik Stufe 1 („Allgemeine Muskeldiagnostik“) CK (bei Myopathien häufig erhöht, nl CK schließt Myopathie nicht aus), Schilddrüsen-Funktion, EMG, Neurographie
Basisuntersuchungen: Anamnese, klinischer Befund, Familienanamnese, multisystemische Hinweise?
Diagnostik Stufe 3 („Molekulare und invasive Diagnostik“) Genetik (DMD-, BMD-, FSHD-, LGMD-, DM1-, DM2-Verdacht) Muskelbiopsie (bei LGMD-, Pompe-, Myositis-, MFM-Verdacht)
Diagnostik Stufe 2 („Spezielle Muskeldiagnostik“) MRT Myositis-spezifische AK, Auto-AK-Diagnostik Nicht-Ischämischer Arbeitstest, Fahrradbelastungs-Test

•5
MRT: Strukturdarstellung der Muskel-Makroskopie (Muskel-Bündel-Faser)
Stoffwechseltests: Metabolite im Blut durch Stoffwechsel vieler Muskeln
EMG: Funktionszustand von Muskelfasergruppen (in motorischen Einheiten)
Muskelbiopsie: Strukturdarstellung von Bündeln und Fasern
EM: subzelluläre Strukturen in Muskelfasern
Ca. 300 Muskeln, 35 – 45 % der Körpermasse
Aufbau: Muskel-Bündel-Faser-Sarkomer
Durchmesser der Muskelfasern: 30 bis 80 µm, Länge: 5 bis 35 cm

X-chromosomale Muskeldystrophie Duchenne / Becker
ein Beispiel aus der großen, klinisch und
genetisch heterogenen Gruppe der Muskeldystrophien

Dystrophinopathien: Typ Duchenne (DMD)
Häufigste Muskeldystrophie im Kindesalter Inzidenz 1/3.500 männliche Neugeborene nur Jungen betroffen
Klinik
• progrediente, proximal betonte Paresen • Entwicklungsverzögerung • Watschelgang, Gower-Zeichen • Wadenhypertrophie, Hyperlordose
• Beginn meist vor dem 5. LJ • meist vor dem 12. LJ rollstuhlpflichtig • CK bis 30.000 U/L • Lebenserwartung 18-25 Jahre, Kyphoskoliose,
Gelenkkontrakturen, Kardiomyopathie, Ateminsuffizienz

Dystrophinopathien: Typ Becker (BMD) „der Duchenne im Zeitlupentempo“
mildere Verlaufsform (Inzidenz 1/18.500)
Klinik
• Beginn meist nach dem 7. LJ
• langsam progrediente Paresen
(proximal > distal)
• Myokrampi können im Vordergrund stehen
• Kardiomyopathie kann führend sein
• Gehverlust mit 16 – 80 Jahren
• eingeschränkte Lebenserwartung
(im Mittel 40 J.)

Molekulargenetik der DMD / BMD
Dystrophin-Gen:
Xp21.2, 2500 kB, enthält 75-80 Exone; Dystrophin 427 kDa
Mutationen:
70% Deletionen im Dystrophin-Gen (out of frame bei DMD, in frame bei
BMD), ca. 5% Duplikationen; übrige: Punktmutationen

Wie wirkt sich eine Muskeldystrophie am Skelettmuskel aus? • Muskelbioptische Befunde
• Grundlage für MRT-Veränderungen

07.11.2017
www.klinikum-bochum.de
Warum Muskel-MRT?
Differenzialdiagnostische Abklärung
Planung von Muskelbiopsien
Therapiemonitoring
Prognoseabschätzung
Beurteilung von Aktivität / Chronizität
Evaluation von Läsionslokalisation und -ausmaß

07.11.2017
www.klinikum-bochum.de
Tejvir S et al., Nature Reviews Drug Discovery 2003
Dystrophinopathien

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien
Dystrophinopathie Typ Duchenne (DMD)

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien
Dystrophinopathie Typ Duchenne (DMD)

07.11.2017
www.klinikum-bochum.de
34 J
Dystrophinopathien
Dystrophinopathie Typ Becker (BMD)

07.11.2017
www.klinikum-bochum.de
34 J
Dystrophinopathien
Dystrophinopathie Typ Becker (BMD)

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien
Leung DG, J Neurol 2016

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien – Spektroskopie
Forbes SC et al., PLoS ONE 2014
Magnetic Resonance Imaging and Spectroscopy
Assessment of Lower Extremity Skeletal
Muscles in Boys with Duchenne Muscular
Dystrophy: A Multicenter Cross Sectional Study

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien – Kardio-MRT
Myocardial Delayed Enhancement by
Magnetic Resonance Imaging
in Patients With Muscular Dystrophy
Silva MC et al., J Am Coll Cardiol. 2007
19 J
Dystrophinopathie Typ Duchenne, zunehmende Herzinsuffizienz
19 J

07.11.2017
www.klinikum-bochum.de
Dystrophinopathien – Kardio-MRT
Myokarditis Sarkoidose Chagas-Krankheit Amyloidose
Pulmonale Hypertonie Muskeldystrophie Chloroquin-induzierte Kardiomyopathie

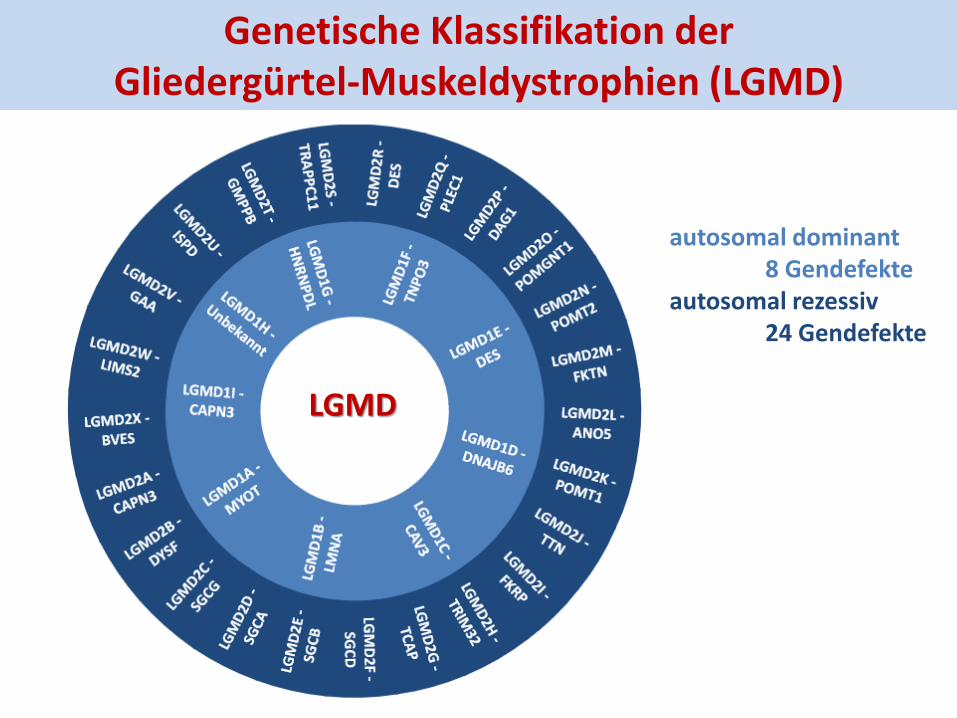
Definition und Historie der LGMD
Klinische Merkmale einer LGMD proximale Paresen und Atrophien, oft der UE CK-Erhöhung dystrophe Gewebsveränderungen selten: Prävalenz 4-7/100.000 Einwohner
Historie 1954 Erstbeschreibung der Krankheitsentität LGMD (Abgrenzung zu den X-chromosomalen Dystrophinopathien) Brain 1954;77:169-231
1995 genetische Erstbeschreibung der LGMD: Calpainopathie (LGMD2A) Cell 1995:81:27-40
LGMD
autosomal dominant 8 Gendefekte autosomal rezessiv 24 Gendefekte
Genetische Klassifikation der Gliedergürtel-Muskeldystrophien (LGMD)

Die Vielfalt der Gendefekte bedingt heterogene Pathomechanismen der LGMD
Sarkomer-Defekte • Aktin- / Myosin-System Plectin, Myotilin, Desmin Titin
Zellkern-Defekte Lamin A/C Transportin-3
Sarkolemm-Defekte • α-Dystroglykan FKRP, POMPT1, POMPT2 POMGNT1 • Dystrophin-Glykoprotein- komplex Sarkoglykane, Caveolin-3 • Membran-Reparatur ANO5, Dysferlin

Diagnostik-Algorithmus der LGMD
Typische LGMD - Klinik
MRT Muskel
Genetische Panel – Diagnostik
Muskel- biopsie
LGMD MFM andere erbliche
Myopathie
unauffällig
keine Myositis
vereinbar mit MD
Myositis möglich (auffällige Ödemkomponente)
unklare Myopathie
Myositis
LGMD – untypische Klinik
distaler Schwer- punkt
rasche Progredienz
proximal - distale
Verteilung
wegweisende klinische Stigmata
Beevor Sign, Polyhill Sign
PIRC, Rippling
Ptose, Dysphagie, Familien- anamnese
Trias, proximale
Parese, paradoxe Atmung,
CK erhöht
Einzelgenanalyse
FSHD Caveolinopathie OPMD Pompe
unauffällig
erbliche Myopathie (LGMD, MFM, usw.)

07.11.2017
www.klinikum-bochum.de
Tejvir S et al., Nature Reviews Drug Discovery 2003
Gliedergürtelmuskeldystrophien (LGMD)

07.11.2017
www.klinikum-bochum.de
LGMD Typ 1a (Myotilin)
33 J
Gliedergürtelmuskeldystrophien (LGMD)

07.11.2017
www.klinikum-bochum.de
55 J
Gliedergürtelmuskeldystrophien (LGMD)
LGMD Typ 2a (Calpain 3)
07.11.2017
www.klinikum-bochum.de
39 J
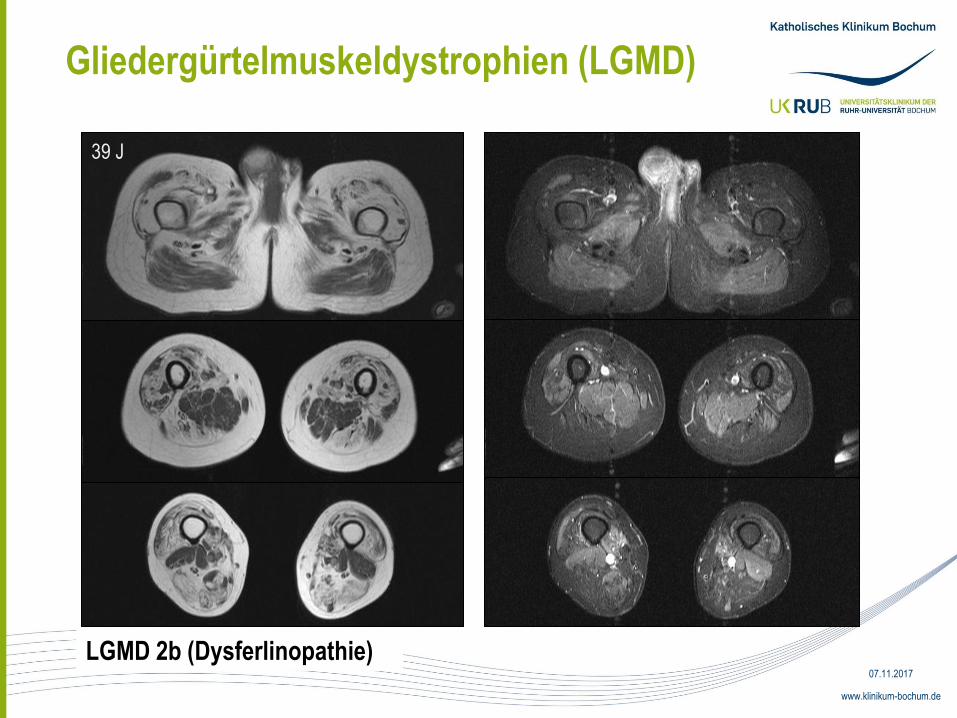
Gliedergürtelmuskeldystrophien (LGMD)
LGMD 2b (Dysferlinopathie)

07.11.2017
www.klinikum-bochum.de
39 J
Gliedergürtelmuskeldystrophien (LGMD)
LGMD 2b (Dysferlinopathie)

07.11.2017
www.klinikum-bochum.de
Dystrophie versus Entzündung
Dystrophie Entzündung
T1
T2

07.11.2017
www.klinikum-bochum.de
Dystrophie versus neurogene Myopathie
Dystrophie Neurogene Myopathie
T1 T1
07.11.2017
www.klinikum-bochum.de
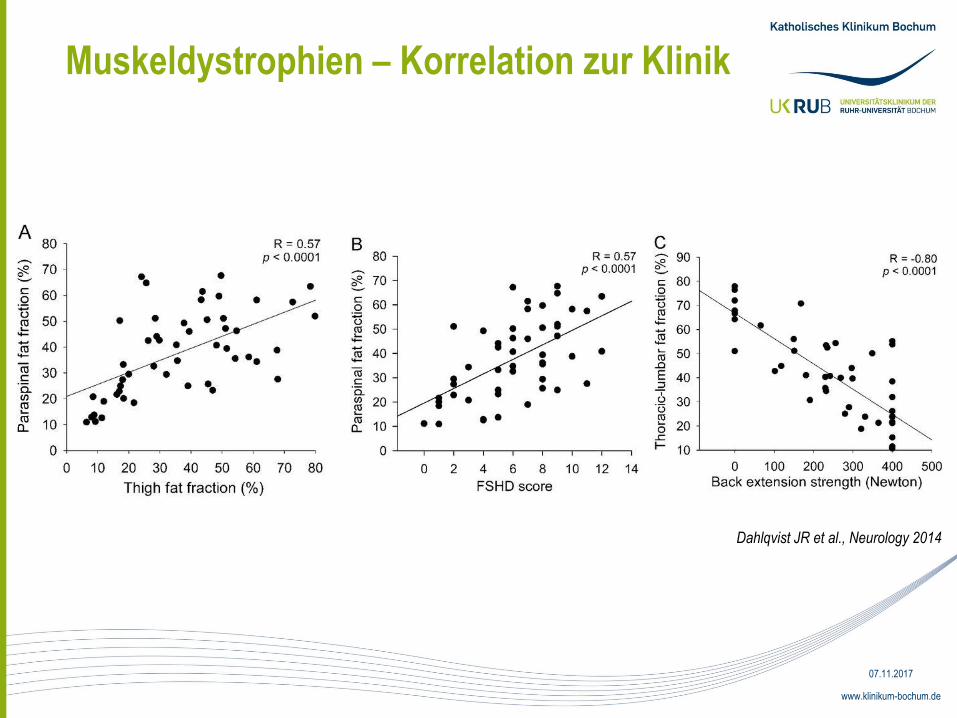
Muskeldystrophien – Korrelation zur Klinik
Dahlqvist JR et al., Neurology 2014

07.11.2017
www.klinikum-bochum.de
Muskeldystrophien – Therapiemonitoring
Cross-sectional comparisons of MRI T2 between corticosteroid-treated (CS) and corticosteroid-naive boys (CS-naive) with Duchenne muscular dystrophy
Arpan I et al., Neurology 2014

Aktueller Therapiestandard der Muskeldystrophien
Glukokortikoide
Duchenne: Deflazacort (0,9 mg/kg KG) Goldstandard Lancet 2009
Translarna (PTC124, Ataluren) bei Stop-Mutationen (mind. 5 J., gehfähig)
LGMD2B: Deflazacort unwirksam Orphanet J Rare Dis 2013
(BMBF MD-NET-Studie in D, n=35, placebo-kontrolliert)
Physiotherapie Aerobes Training wirksam bei LGMD2A, 2I, 2L Neurology 2007;68:59-61;
Muscle Nerve 2014;50:119-23; Muscle Nerve 2013;47:163-69
Orthopädische Hilfsmittelversorgung, operative Behandlung Kontrakturen, Skoliose Immobilität
Kardiomyopathie früher Therapiebeginn mit β-Blockern / ACE-Hemmern
Restriktive Ventilationsstörung nicht-invasive Beatmung, Tracheostoma

Myofibrilläre Myopathien (MFM) Klinische Phänotypen und bislang bekannte Gendefekte
Myotilinopathie
Olive et al. Brain 2005
FHL1-Myopathie
Schessl et al. Brain 2009
Filaminopathie
Kley et al. Brain 2007, Fürst et al. Acta Neuropathol 2013
BAG3-Myopathie
Konersman et al. NMD 2015 Kley et al. Curr Opin Neurol 2016
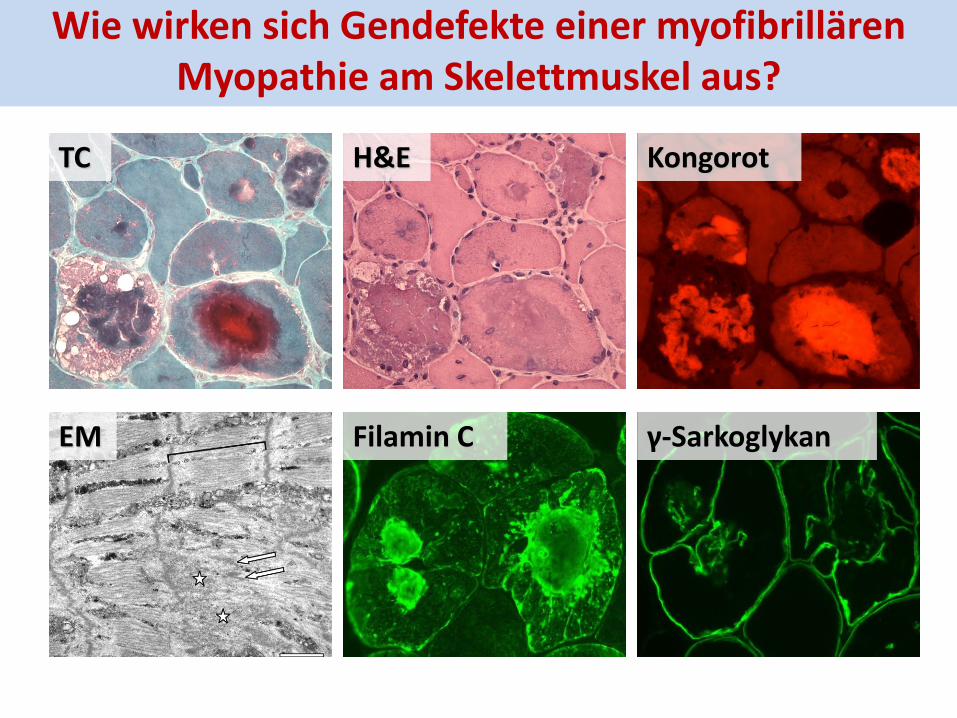
H&E TC Kongorot
Filamin C γ-Sarkoglykan EM
Wie wirken sich Gendefekte einer myofibrillären Myopathie am Skelettmuskel aus?

07.11.2017
www.klinikum-bochum.de
Myofibrilläre Myopathien
53 J
Myofibrilläre Myopathie Typ I (Desminopathie)

07.11.2017
www.klinikum-bochum.de
Myofibrilläre Myopathien
Myofibrilläre Myopathie Typ I (Desminopathie)
53 J

07.11.2017
www.klinikum-bochum.de
Myofibrilläre Myopathien

07.11.2017
www.klinikum-bochum.de
Tejvir S et al., Nature Reviews Drug Discovery 2003
Kongenitale Muskeldystrophien (CMD)

07.11.2017
www.klinikum-bochum.de
Kongenitale Muskeldystrophien (CMD)
• Gruppe seltener hereditärer Myopathien, Inzidenz 2,6 x 10-5 – 4,56 x 10-5
• gekennzeichnet durch muskuläre Hypotonie, verzögerte motorische
Entwicklung, frühzeitiges Einsetzen von Muskelschwäche und dystrophen
Gewebsveränderungen
• Herzmuskel nur selten betroffen

07.11.2017
www.klinikum-bochum.de
Kongenitale Muskeldystrophien (CMD)
Muntoni F, Eur J Ped Neurol 2002

07.11.2017
www.klinikum-bochum.de
Kongenitale Muskeldystrophien (CMD)
• Gruppe seltener hereditärer Myopathien, Inzidenz 2,6 x 10-5 – 4,56 x 10-5
• gekennzeichnet durch muskuläre Hypotonie, verzögerte motorische
Entwicklung, frühzeitiges Einsetzen von Muskelschwäche und dystrophen
Gewebsveränderungen
• Herzmuskel nur selten betroffen
• 30 – 40%: Laminin-Alpha2-Mangel (MDC1A) = Merosinopathie
• Typische Symptome: schwere Muskelschwäche und Atrophien,
Gelenkkontrakturen, Gesichtsdysmorphien, CK-Erhöhung,
in der Regel keine Beeinträchtigung kognitiver Funktionen
• Einzelne Subtypen der CMD mit Hirn- und Augenbeteiligung bekannt

07.11.2017
www.klinikum-bochum.de
Kongenitale Muskeldystrophien (CMD)
Muntoni F, Eur J Ped Neurol 2002
kortikale Migrationsstörungen, okzipitale Agyrie, verplumpte Gyri in
den übrigen Abschnitten („cobblestone cortex“), Leukenzephalopathie,
Vermis- und Hirnstammhypoplasie, Hydrozephalus
Lissenzephalie Typ II, zerebelläre Polymikrogyrie, zerebrale/zerebelläre
Leukenzephalopathie, Balkenhypoplasie, Pons- und Vermishypoplasie,
Dandy-Walker-Malformation, Hydrozephalus, Enzephalozelen

07.11.2017
www.klinikum-bochum.de
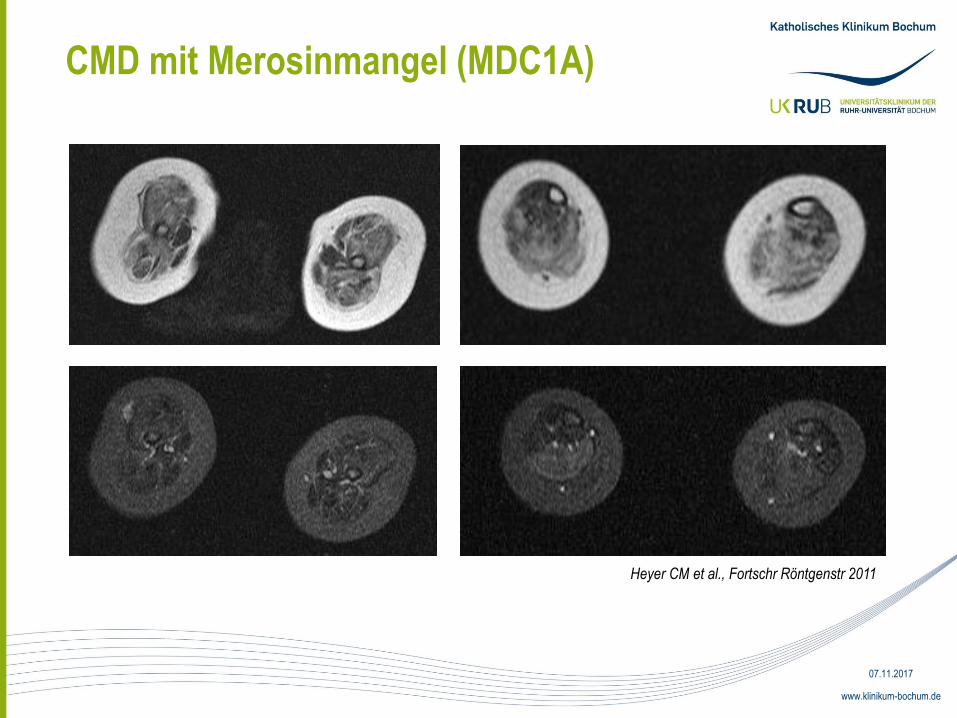
CMD mit Merosinmangel (MDC1A)
• 5 Monate alter deutscher Junge, Eltern nicht konsanguin
• Ausgeprägte postpartale muskuläre Hypotonie, keine Trinkschwäche
• CK 2604 U/l
• Genanalyse: zwei heterozygote Mutationen am LAMA2-Gen
(Exon 32:c.4645C>T, p,Arg1549X, Exon 4:c.498G>A p,Trp166X)
• Beide Elternteile heterozygote Träger je einer Mutation
• Muskel- und Schädel-MRT im Alter von 23 Monaten
07.11.2017
www.klinikum-bochum.de
CMD mit Merosinmangel (MDC1A)
Heyer CM et al., Fortschr Röntgenstr 2011

07.11.2017
www.klinikum-bochum.de
CMD mit Merosinmangel (MDC1A)
Heyer CM et al., Fortschr Röntgenstr 2011

07.11.2017
www.klinikum-bochum.de
CMD mit Merosinmangel (MDC1A)
• Typischer MRT-Befund der MDC: ausgedehnte Veränderungen der
supratentoriellen weißen Hirnsubstanz (Leukenzephalopathie)
• Mögliche Ätiologie: Störungen der Bluthirn-Schranke wegen erhöhter
Kapillarpermeabilität; LAMA2 Bestandteil der Basalmembran zerebraler
Gefäße (Villanova M, 1997)
• Erhebliche Befundvariabilität in den ersten zwei Lebensjahren
• Neuronale Migrationsdefekte bei MDC möglich (Polymikrogyrien, fokale-
kortikale Dysplasien, meist okzipital)
• Mögliche Ätiologie: LAMA2 beeinflusst periphere Myelogenese
Schwann´scher Zellen im Mausmodell (Matsumura K, 1997)

Metabolische Myopathien Erbliche Stoffwechselerkrankungen mit einem breiten klinischen
Spektrum (Belastungsintoleranz – permanente Muskelschwächen)

Wie wirken sich Gendefekte einer metabolischen Myopathie am Skelettmuskel aus?
• Muskelbioptische Befunde • Histologische Grundlagen für MRT-Veränderungen
H&E
PAS
Ölrot
Trichrom

Klinisches Spektrum der Pompe-Erkrankung
Typische klinische Trias: Proximale Paresen + CK-Erhöhung
Rumpf-Paresen (Hyperlordose, Skoliose) Atemmuskelschwäche mit “paradoxer Atmung”
Muscle Nerve 2012;47:594-600
EMG: myotone Entladungsserien DD: DM1, PROMM
Gefäßveränderungen: Dolichobasilaris Aneurysmen
Herzbeteiligung: Sinusarrhythmie Sick Sinus-Syndrom WPW-Syndrom Leitungsblockierungen
Rigid Spine
- seltene, progrediente, autosomal-rezessive Glykogenspeicherkrankheit - Primärdefekt: lysosomale saure α-Glukosidase (GAA)

07.11.2017
www.klinikum-bochum.de
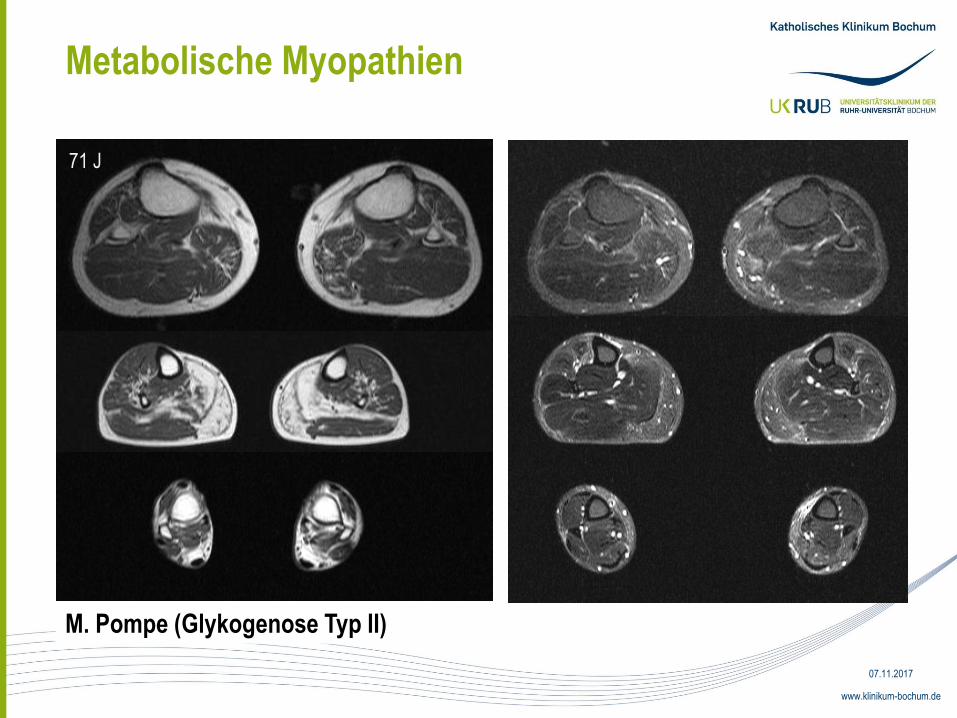
Metabolische Myopathien
71 J
M. Pompe (Glykogenose Typ II)
07.11.2017
www.klinikum-bochum.de
Metabolische Myopathien
71 J
M. Pompe (Glykogenose Typ II)

07.11.2017
www.klinikum-bochum.de
Metabolische Myopathien
The results demonstrate that fatty muscle
degeneration can occur before clinical
manifestation of muscle weakness and
suggest that mildly affected muscles may
respond better to ERT treatment than
severely involved muscles (...)
It should be discussed if muscle
alterations detected by muscle MRI may
be an objective sign of disease
manifestation justifying an early start of
ERT in clinically asymptomatic patients in
order to improve the long-term outcome.

N Engl J Med 2010;362:1396-406
Therapie der Pompe-Erkrankung
Zugelassene Behandlung: Enzym-Ersatztherapie mit rekombinanter α-Glukosidase (Myozyme®)
• Dosierung: 20 mg/kg KG (i.v.) • Intervall: 14 Tage • Infusionsdauer: ca. 4 Stunden • Therapiedauer: lebenslang • Kosten: ca. 7000 €/a pro kg KG
Effektivität wurde mittlerweile in mehreren Studien nachgewiesen

Klinische Symptomatik
Trockenblut-Test (GAA-Aktivität < 50% in Leukozyten)
GAA-Enzymdiagnostik in Skelettmuskulatur oder Fibroblasten
oder GAA-Mutationsanalyse
Enzym-Ersatztherapie (alle Kriterien müssen erfüllt sein): - gesicherte Diagnose - symptomatisch (Skelettmuskel und / oder respiratorische Einschränkungen) - residuelle Muskelkraft und respiratorische Funktion - keine lebensbegrenzende Komorbidität - Einhalten des Therapieregimes (Patienten, Behandler)
andere Myopathie (z.B. LGMD, FSHD,
Strukturmyopathie, Myositis
Diagnostik-Algorithmus der Pompe-Erkrankung
ja
ja
nein
nein

Einteilung der immunogenen Myositiden (nicht erreger-bedingt)
Merkmale Polymyositis
(PM) Dermatomyositis
(DM) Nekrotisierende Myopathie (NM)
Einschlußkörper-myositis (IBM)
Frauen : Männer 2:1 2:1 1:1 1:3
Erkrankungsalter > 18 Jahre 1-15 und 45-65 Jahre > 18 Jahre > 45 Jahre
Verlauf akut – subakut akut – subakut akut – subakut chronisch > 12 Monate
Hautveränderungen Nein ja Nein Nein
Paresen proximal > distal symmetrisch
proximal > distal symmetrisch
proximal > distal symmetrisch
proximal = distal asymmetrisch, Prädilektion: Kniestrecker ≥ Hüftbeuger und/oder Fingerbeuger ≥ Schulterabduktoren
Muskelschmerzen (+) + + (+)
Muskelatrophien + (+) + ++
EMG Myopathisch Myopathisch Myopathisch myopathisch und neurogen
CK bis 50x normal bis 50x bis 50x normal bis < 15x

Diagnostik – Algorithmus der Myositiden
Muskel-MRT
proximale Paresen an UE
- Hautveränderungen - hohe CK - akuter - subakuter
Verlauf
- hohe CK - akuter - subakuter
Verlauf
- hohe CK - Statin-Exposition - akuter - subakuter
Verlauf
- normal - leichte CK - proximal-distal - langsamer Verlauf
Myositis - spezifische Antikörper +
Muskelbiopsie
Mi-2 TIF1 NXP2 SAE
MUP-44
DM PM NM IBM
Tumor - Suche
engmaschiges Tumor Follow - Up
HMGCoA-R SRP
TIF1 NXP2
seronegativ HMGCoA-R
07.11.2017
www.klinikum-bochum.de
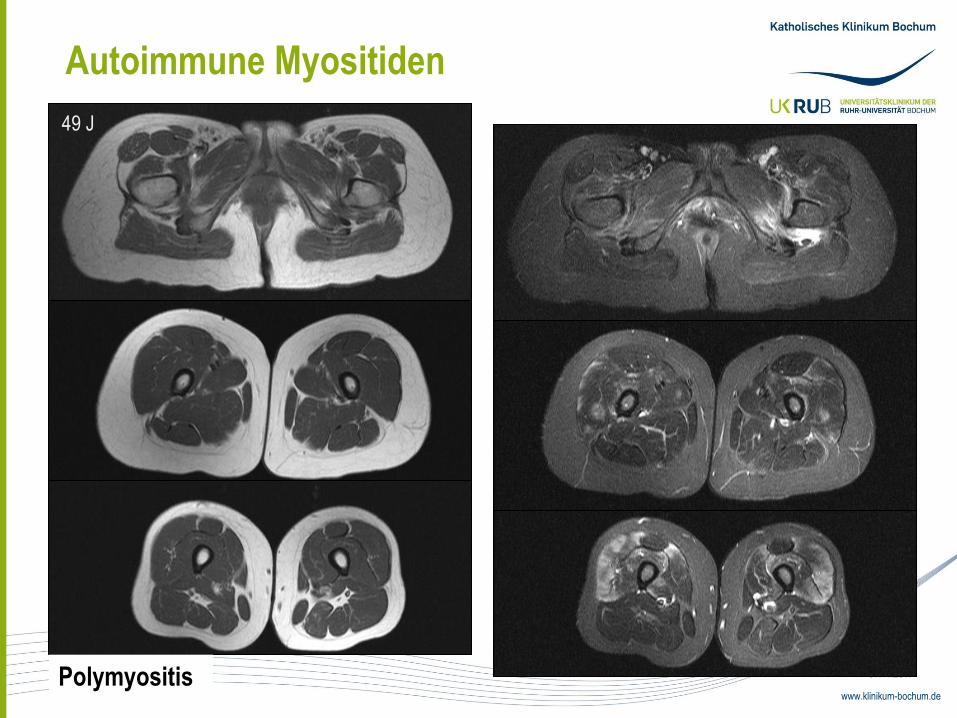
Autoimmune Myositiden
49 J
Polymyositis

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
49 J
Polymyositis
07.11.2017
www.klinikum-bochum.de
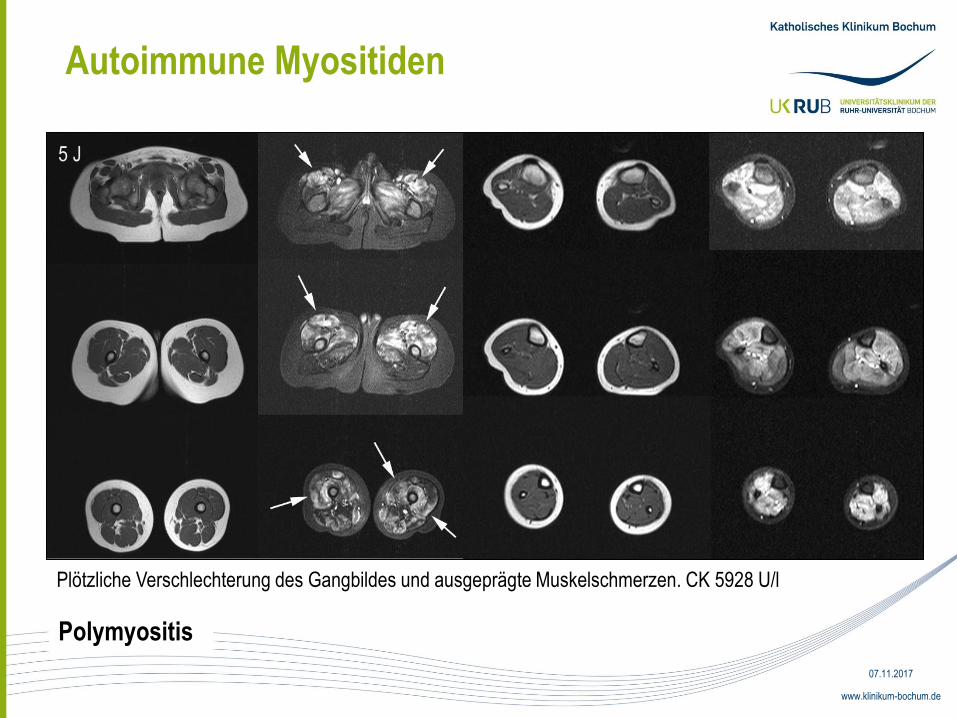
Autoimmune Myositiden
5 J
Plötzliche Verschlechterung des Gangbildes und ausgeprägte Muskelschmerzen. CK 5928 U/l
Polymyositis

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
5 J
Polymyositis

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
Chronische Polymyositis mit Atrophien
51 J

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
HE CD 20
bekannte Dermatomyositis, Muskelschmerzen und -schwäche nach initialer Remission
7 J

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden – DM
Hoch-florides Rezidiv einer Dermatomyositis
7 J

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
57 J
Einschlusskörpermyositis (IBM)

07.11.2017
www.klinikum-bochum.de
Autoimmune Myositiden
57 J
Einschlusskörpermyositis (IBM)

Aktueller Therapiestandard der Myositis
aus: Akt Rheuma- tologie 2016

Toxische Myopathien
grundsätzlich: toxische Myopathien bisher bei etwa 120 Substanzen beschrieben häufigste toxische Myopathien: Alkohol, Steroide, Statine entweder direkte toxische Wirkung auf den Muskel oder indirekt
(durch metabolische oder immunologische Mechanismen)
Äthyltoxische Myopathien
Klinik: akute schmerzhafte Rhabdomyolyse (deutliche CK- Erhöhung) subakute hypokaliämische vakuoläre Myopathie chronische schmerzlose Myopathie mit Atrophie
in 50%, >100 g Alkohol/d >10 J. geringe CK, Typ2-Atrophie, tubuläre Aggregate

Toxische Myopathien
Steroid-assoziierte Myopathie
grundsätzlich: kumulative Gesamtdosis relevant
fluorierte Steroide (Dexamethason, Triamcinolon) toxischer (10-60% der Hirn-Tu-Patient entwickeln nach 2-wöchiger Dexamethason-Therapie eine Steroidmyopathie)
selten endokrin bei M. Cushing (ACTH-HVL-Tu, NNR-Tu)
Klinik: akute schwere Tetraparese mit Atemmuskelschwäche hohe CK, zahlreiche Nekrosen
chronische schmerzlose Steroidmyopathie geringe CK, Typ2-Faseratrophie

Statin-assoziierte Myopathien
grundsätzlich:
in D erhalten ca. 4,6 Mio. Menschen Statine (1,7 Milliarden Statin-Tagesdosen 2013) - Indikation: Schlaganfallprophylaxe, MS, Demenz - Statine sind HMG-CoA-Reduktasehemmer - Statine bewirken eine Senkung der Low-Density-Lipoproteine (LDL) - NW meist bis 6 Wochen nach Therapie-beginn, bei 7-29% der Statin-behandelten
Patienten - Risikofaktoren: Alter Begleitmedikation (u.a. Gemfibrozil, Makrolide, Inhibitoren von CYP450) Komorbiditäten (u.a. Diabetes, Hypothyreose, Vit D- Mangel) genetische Disposition (SNP in Cytochrom P450)
Einteilung der Muskelschädigungen:
asymptomatische CK-Erhöhung Myalgien ± CK-Erhöhung (5-10% der Patienten)
Paresen ± CK-Erhöhung (sog. Statin-Myopathie, 0,1% der Pat.)
Rhabdomyolyse (0,01% der Pat.)
persistierende Symptomatik nach Absetzen der Lipidsenker (Demaskierung einer Myopathie / nekrotisierende Myopathie durch HMGCoA-R-AK)
JAMA 2003;289:1681-1690, Cardiovasc Drugs Ther 2004;17:459-465, J Neurol Neurosurg Psychiatry 2009;80:832-38

Pathogenese der Statin-assoziierten Myopathie (die einer MRT-Veränderung zugrunde liegen können)
Statin-induzierte Muskelläsionen: - mitochondriale Alterationen
(COX-Minderung, Ragged Red-Fasern, vermehrter Lipidgehalt)
- Verminderung der mtDNA - verminderte Aktivitäten von Complex I, II,
III, IV - verminderte oxidative Phosphorylierung
J Neurol Sci 2013;325:142-7; Arch Neurol 2005;62:1709-12; Eur Heart J 2015
Mitochondrienpathologie
Ragged Red Fasern
Ragged Blue Fasern

07.11.2017
www.klinikum-bochum.de
Toxische Myopathien
Statin-Therapie seit 3 Wochen, ausgeprägte Muskelschmerzen
41 J

07.11.2017
www.klinikum-bochum.de
Toxische Myopathien
Statin-assoziierte toxische Myopathie
41 J

07.11.2017
www.klinikum-bochum.de
Statin-assoziierte toxische Myopathie
• Untersuchte Patienten: n=21, 16 Männer (76%)
• Medikationsdauer Δ 41 Monate
• CKmax 6356 U/l
• Myalgien 71%, Paresen 19%
• MRT: Muskelödeme 62%, lipomatöse Veränderungen 29%
• Kein spezifisches Befallsmuster, aber vornehmlich Oberschenkel-Flexoren und
oberflächliche Unterschenkel-Flexoren betroffen
• Signifikante Korrelationen zwischen
- radiologischer Diagnose (+) und CKmax (p=0,012)
- Muskelödem und CKmax (p=0,012)
- lipomatösen Veränderungen und CKmax (negativ, p=0,019)

Pragmatische Vorgehensweise (Empfehlungen des Am. College of Cardiology, DGN-S1 Leitlinie 2012)
● CK-↑ < 10-fach ● blande Myalgien → Statine weiter (evtl. niedrigere Dosierung oder alternatives Statin, CK-Kontrollen)
● CK-↑ > 10-fach (w. > 1.460 U/L; m > 1.720 U/L) ● Myalgien ± CK-↑ → Pausieren der Statine → danach Umstellung auf alternative Statin-Therapie oder andere lipidsenkende Therapie
● progrediente Paresen ± CK-↑ ● Rhabdomyolyse → HMGCoA-R-AK, MRT, ggf. Muskelbiopsie → Absetzen der Statine, andere lipidsenkende Therapie (Ezetimib, Fibrate, PCSK9-AK, CETP-Hemmer)
07.11.2017
www.klinikum-bochum.de
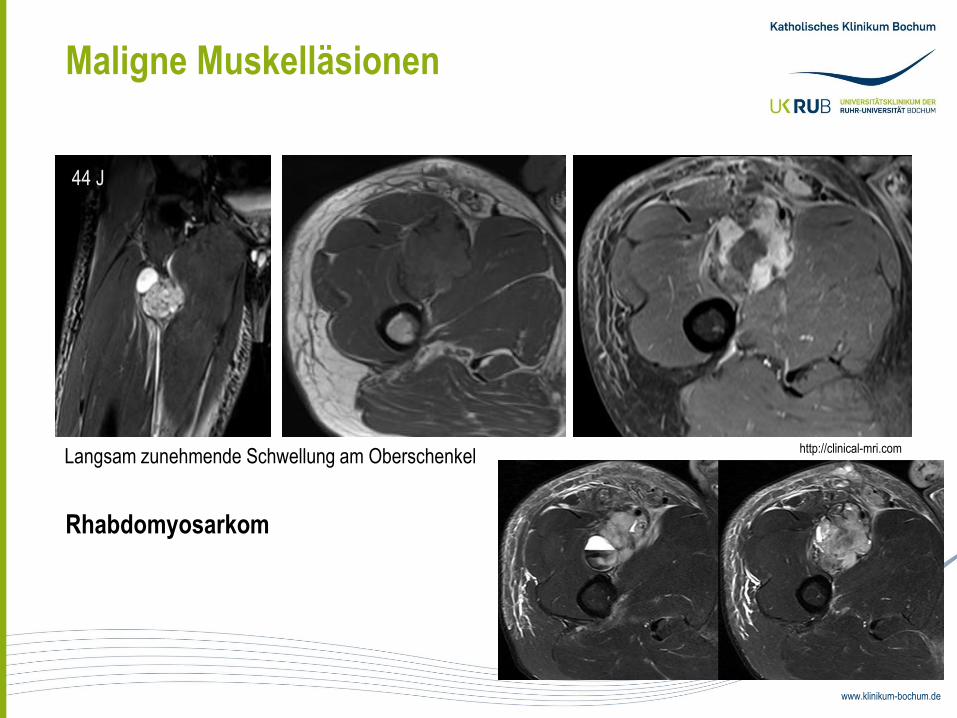
Maligne Muskelläsionen
Langsam zunehmende Schwellung am Oberschenkel
Rhabdomyosarkom
http://clinical-mri.com
44 J

07.11.2017
www.klinikum-bochum.de
Maligne Muskelläsionen
Langsam zunehmende, schmerzlose Schwellung am dorsalen Oberschenkel
Tumorzellen SMA-, EMA- und Desmin-positiv, S100, SOX10, CD34, Myogenin negativ
Myxofibrosarkom
60 J

07.11.2017
www.klinikum-bochum.de
Maligne Muskelläsionen
Generalisierte Muskelschmerzen
55 J

07.11.2017
www.klinikum-bochum.de
Maligne Muskelläsionen
Muskelmetastasen eines
Ösophagusadenokarzinoms
Heyer CM et al., Scand J Gastroenterol 2005
55 J

07.11.2017
www.klinikum-bochum.de
Tumorähnliche Muskelläsionen
Z.n. Hohlhand-OP, zunehmende Schwellung
Myositis ossificans
36 J

07.11.2017
www.klinikum-bochum.de
Traumatische Muskelerkrankungen
Müller-Wohlfahrt HW et al., Br J Sports Med. 2013

07.11.2017
www.klinikum-bochum.de
Traumatische Muskelerkrankungen
Basisprotokoll koronare T1wSE/TSE, koronare STIR, transversale STIR, transversale T2wTSE Schichtdicke 3 – 5 mm, In-Plane-Auflösung 0,5 mm
Vereinfachte Klassifikation myotendinöser Verletzungen (nach Wörtler)
Grad 1 – mikroskopische Verletzung ohne Funktionsverlust („Muskelzerrung“)
Grad 2 – partieller Muskelriss mit variablem Funktionsdefizit („Muskelfaserriss“)
Grad 3 – kompletter Muskelriss oder Sehnenavulsion mit vollständigem Funktionsausfall
Bevorzugte Lokalisation
- Muskeln mit hohem Typ-II-Faseranteil
- mehrköpfige Muskeln (M. quadriceps femoris, M. gastrocnemius)
- Muskeln, die zwei Gelenke überschreiten (ischiokrurale Muskulatur)
- Myotendinöser Übergang, Aponeurosen
07.11.2017
www.klinikum-bochum.de
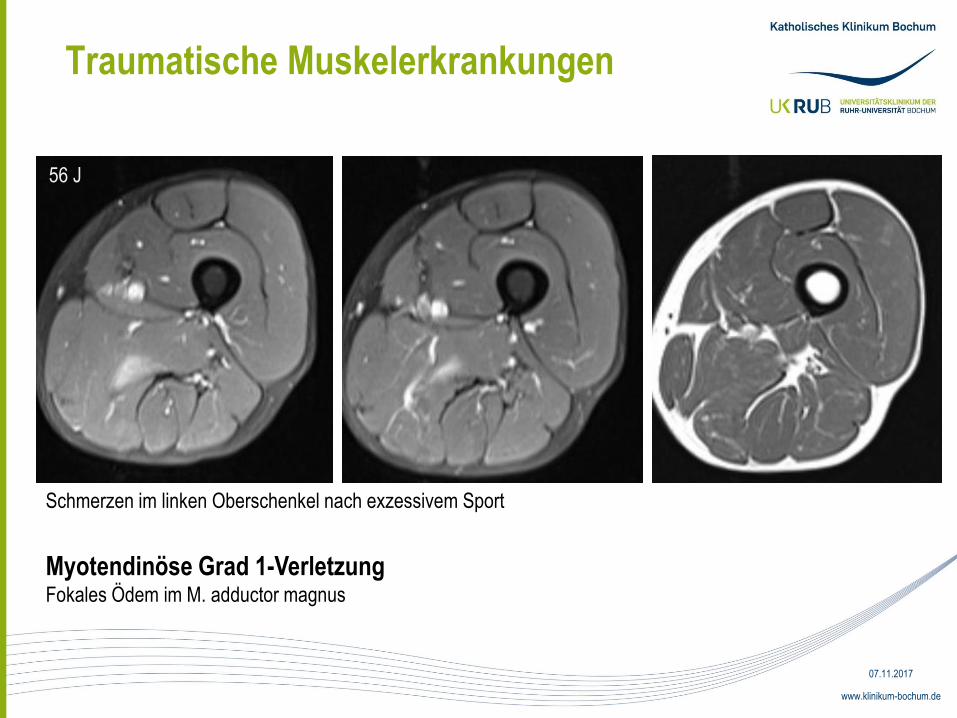
Traumatische Muskelerkrankungen
Schmerzen im linken Oberschenkel nach exzessivem Sport
Myotendinöse Grad 1-Verletzung Fokales Ödem im M. adductor magnus
56 J

07.11.2017
www.klinikum-bochum.de
Traumatische Muskelerkrankungen
Wadenschmerzen nach Sport Myotendinöse Grad 2-Verletzung Ausgedehnte Partialruptur des
M. gastrocnemius mit intramuskulärem Hämatom
56 J

07.11.2017
www.klinikum-bochum.de
Traumatische Muskelerkrankungen
Hüftschmerzen nach Tanzsport
Myotendinöse Grad 3-
Verletzung Vollständiger Abriss der
ischiokruralen Muskulatur vom
Tuber ischiadicum
(„Hamstring“-Läsion)
16 J

07.11.2017
www.klinikum-bochum.de
Traumatische Muskelerkrankungen
Relativ seltene Ursache chronischer Muskelschmerzen (ca. 200 case reports)
Konstitutionell oder posttraumatisch bzw. postoperativ
Häufigste Lokalisation: untere Extremität (M. tibialis anterior)
Bildgebende Methode der Wahl: dynamische Sonographie / MRT
Kleine Muskelhernie am M. vastus lateralis
Schmerzen am rechten Oberschenkel lateral, Z.n. Femurfraktur
34 J

07.11.2017
www.klinikum-bochum.de
Zusammenfassung
• Visualisierung pathologischer Muskelveränderungen durch MRT
• Lipomatose (T1/T2) → Atrophie/Dystrophie Ödem (T2)→ Inflammation
• Weitere Indikationen: Biopsieplanung, Verlaufskontrolle, Therapiemonitoring
• Herzbildgebung: Late Enhancement, kardiale Beteiligung bei Myopathien
• Analyse der MR-tomographischen Muster → Differenzialdiagnose

07.11.2017
www.klinikum-bochum.de
47 J, w
„Muskelatrophien, CK unauffällig. Muskelerkrankung?“

07.11.2017
www.klinikum-bochum.de
47 J, w
„Muskelatrophien, CK unauffällig. Muskelerkrankung?“
Relativ ausgedehnte, flächige Lipomatosen mit zum Teil
deutlichen Atrophien
Asymmetrische Verteilung (≠ Dystrophie, IBM, PM)
Distale Befundbetonung (≠ LGMD)
Kaum Muskelödeme (≠ Myositis, toxische Myopathie)
Chronischer Prozess ohne Aktivierung
Am ehesten chronisch-neurogener Schaden
Motoneuronerkrankung
Spastische Spinalparalyse (1. MN)
Spinale Muskelatrophien (2. MN)
Amyotrophische Lateralsklerose (1.und 2. MN)
Poliomyelitis und Postpolio-Syndrom (2. MN)