4 1.electronic excitation energy transfer a förster energy transfer demonstration experiment
TRANSCRIPT
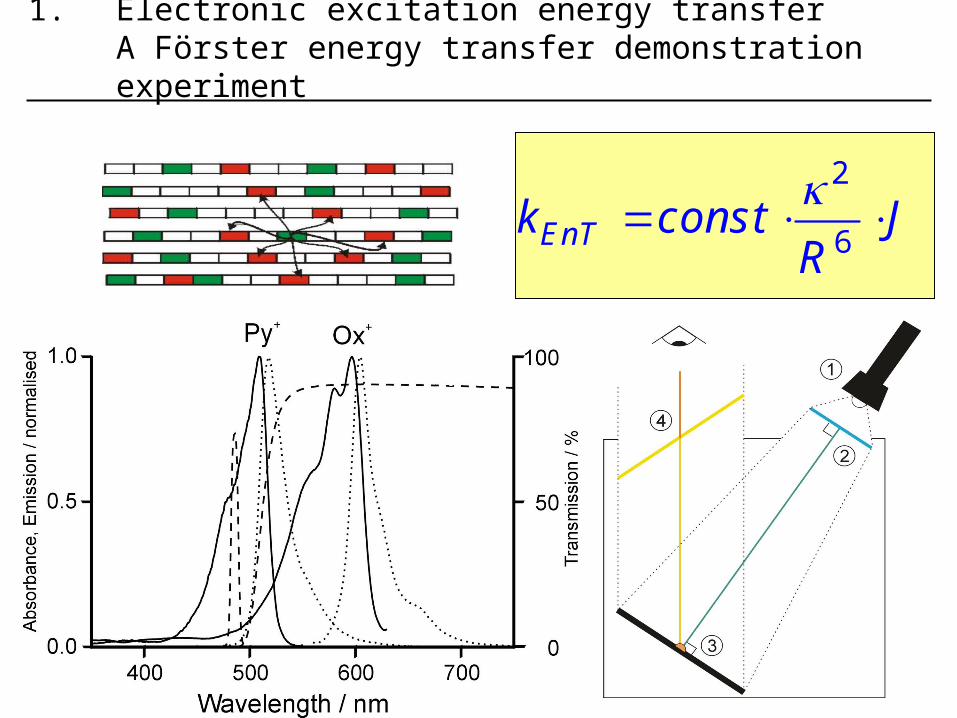
4
2
6EnTk const JR
1. Electronic excitation energy transferA Förster energy transfer demonstration experiment
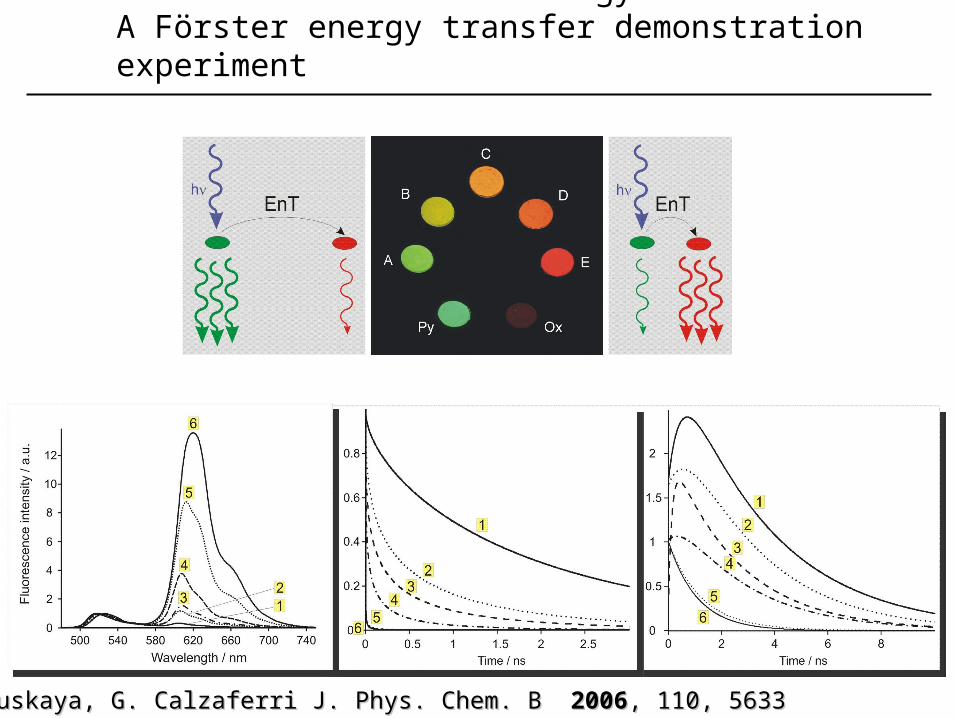
1. Electronc excitation energy transferA Förster energy transfer demonstration experiment
K. K. Lutkouskaya, G. Calzaferri J. Phys. Chem. BLutkouskaya, G. Calzaferri J. Phys. Chem. B 20062006, , 110, 5633110, 5633
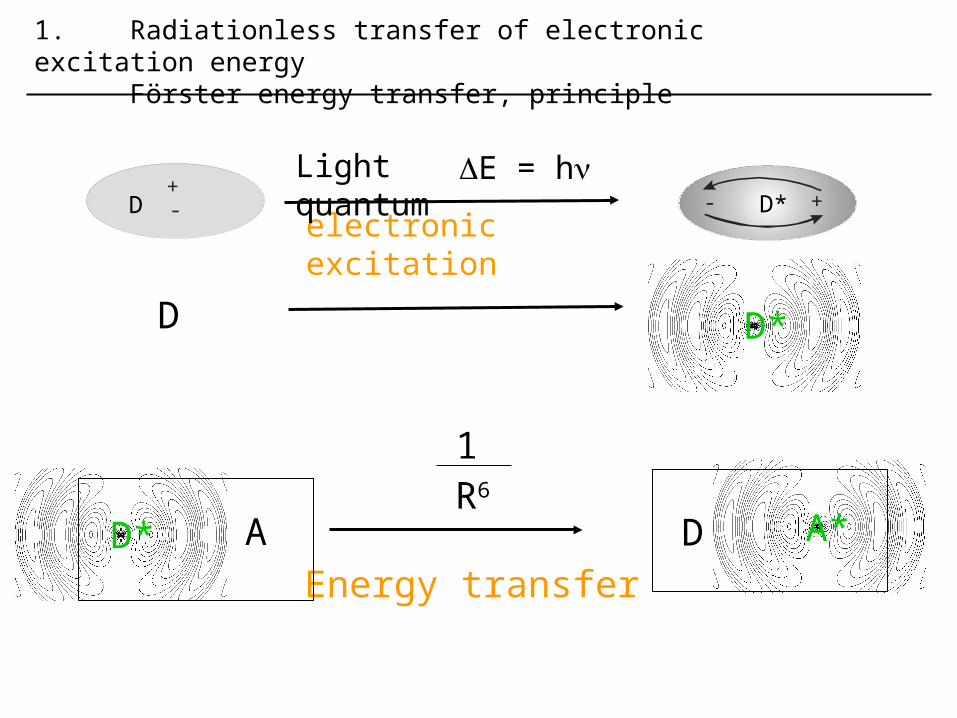
D
A*D
1R6
Energy transfer
1. Radiationless transfer of electronic excitation energy Förster energy transfer, principle
+-D +- D*
electronic excitation
Light quantum E = h
D* A
D*

VDi-Di = the first term in a Taylor's series expansion of the electrostatic interaction between two neutral molecules.
VDi-Di = interaction energy between two dipoles 1=l1q and 2=l2q
2 Comment on the dipole-dipole interaction and on the
orientation factor
3
0
3( )1
4Di DiVR
μ μ n μ n μ
-
+
-
+
1 2R
l2
l1
To do this, we assume two fixed positive charges ea and eb at distance R, each of them compensated by a negative charge –ea and –eb.
We express VDi-Di between two dipoles in polar coordinates. We introduce the factor to describe the angle dependence VDi-Di = VDi-Di (R,1,2,12).
10 1 2 104 1.11265 10 J C m
n = unit vector in direction of R.

2 2
11 022 1 2
1 1 1 1 1
4a ba b
a bb a
e e
r rV e e
R r r r
Hence, the interaction energy Vdd between two dipoles can be expressed as:
The first 4 terms represent the mutual interaction of two dipoles.
0 12 1 2
1 1 1 1
4a b
ddb a
e eV
R r r r
Potential energy:
Expansion in a series along the Cartesian coordinates:
-41 2 1 2 1 23
0
12 Terms in R ...
4a b
dde e
V x x y y z zR
Approximate expression for this interaction by assuming:
This means, that the term indicated in blue color is of constant value (does not depend on R and also not on r12, r2a , r1b). This is the condition for a
dipole-dipole interaction.
(b) the distances between ea and –ea, and also between eb and –eb, are very short with respect to the distance R between the objects 1 and 2 (R >> r1a and R >> r2b).
(a) R is constant (R changes only slowly with respect to the movements of the electrons)

Neglecting higher terms, we get:
1 2 1 2 1 230
12
4a b
dde e
V x x y y z zR
z2 = l2 cos(2)z1 = l1 cos(1)
y2 = l2 sin(2) sin(2)
x2 = l2 sin(2) cos(2)
y1 = l1 sin(1) sin(1)
x1 = l1 sin(1) cos(1)
This equation is equivalent to:
30
3( )1
4Di DiVR
n n
x1 x2 + y1y 2 -2 z1 z2 = l1l2 {sin(1) sin(2)[cos(1) cos(2)+ sin(1) sin(2)]-2
cos(1) cos(2)}
cos12=cos(1-2)cos()
1 21
012 1 223
sin s cosin 2cos cos4a b
dde e l l
VR
21
30
12
4a be e l l
R
We now express the dependence of Vdd on the coordinates (R,1,2,12):
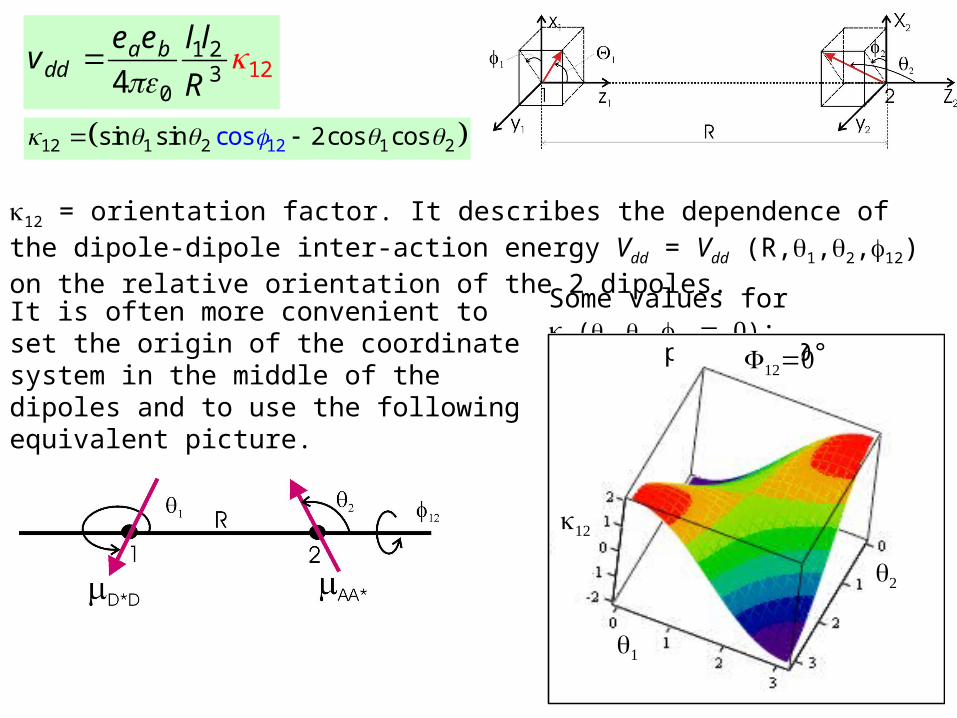
1 23 2
014
a bdd
e e l lv
R
1212 1 2 1 2cossin sin 2cos cos
12 = orientation factor. It describes the dependence of the dipole-dipole inter-action energy Vdd = Vdd (R,1,2,12) on the relative orientation of the 2 dipoles. Some values for
(1,2,): It is often more convenient to set the origin of the coordinate system in the middle of the dipoles and to use the following equivalent picture.
phi12 = 0°
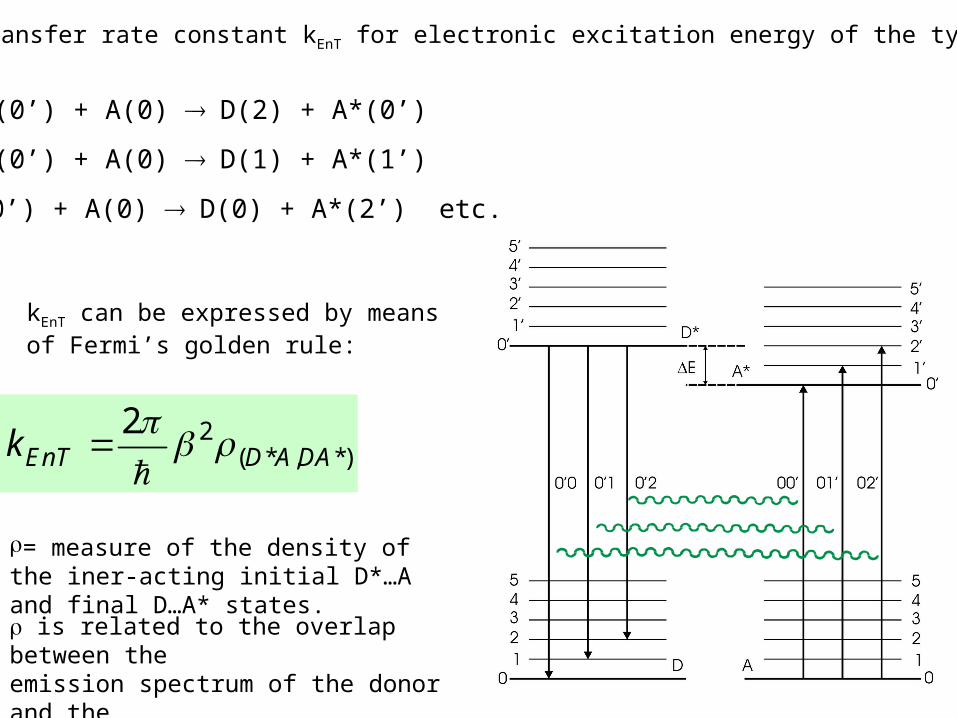
D*(0’) + A(0) D(1) + A*(1’)
D*(0’) + A(0) D(0) + A*(2’) etc.
D*(0’) + A(0) D(2) + A*(0’)
The energy transfer rate constant kEnT for electronic excitation energy of the type:
kEnT can be expressed by means of Fermi’s golden rule:
2( * , *)
2EnT D ADAk
is related to the overlap between theemission spectrum of the donor and theabsorption spectrum of the acceptor.
= measure of the density of the iner-acting initial D*…A and final D…A* states.

5
*
4 6
2
0*
40 4
9000 ln(10)
128
1 ( )( )D A
L DA
EnT
D
AD
nN Rk c S d
0* * *D D D
The dimension of S() is equal to that of -1. Hence, expressing the spectral overlap integralin wave numbers, and using , we get:
0c
* 4
( )( ) A
DD A S dJ
5
*
4 6
*4
*
29000 ln(10)
128
( )( )D A
DAL
EnTD A
DDnN R
k S d
*
4 65
*
*
2
*9 ln(10)
128
D A
DAL
EnTD
D ADn RN
k J
The spectral overlap integral is usually abbreviated with the symbol J:
The above formula is correct if the dimension of [J] is chosen to be cm6mole-1For chemists the more natural way to choose the dimension of the spectral overlap integral is: [J] = [cm3M-1], [M] =[mol L-1].
kEnT for energy transfer must then be expressed as follows: