4. recrystallization and melting pointscourses.chem.psu.edu/chem36/experiments/recrstal.pdf · 4....
TRANSCRIPT

39
4. Recrystallization and Melting PointsPreLab - All PreLabs must be completed before coming to lab. You cannot start any
experimental work until the PreLab white pages are turned in! There are no exceptions tothis rule!
This first experiment is quite lengthy because it involves two techniques, recrystallization andmelting point. Therefore, be forewarned that the PreLab is quite extensive and because it’s yourfirst one, will probably take about two hours to do. First, read through the Introduction and allthe Experimental sections given in this chapter. Then, read through sections 1 and 2 of Chapter3 describing the PreLab writing format with examples. Finally, in your lab notebook, usingcarbonless copies, write an introductory PreLab section for the experiments to be run including:
1. A brief Summary of what will be done and why it will be done.
2. Four Learning Objectives or Goals for the experiment.
3. Diagrams of Apparatus. There are no chemical reactions or equations in this experiment.
4. Complete Chemical Data Tables including ALL possible solvents and unknowns used inProcedures 1 to 5, but not Procedure 6. Fill in the data on the Common Shelf ChemicalData Table for the ten solvents designated Chapter 4 in column 3 of that table (includingwater). On the blank Chemical Data Table, fill in data for the 6 possible organic solidunknowns, phthalic acid, and methylene blue (0.01% solution in water).
5. Do the PreLab Exercise given here.a) If cooling fails to produce crystals, give two methods for inducing crystallization and
describe how they work.b) Define immiscible and give one common household example of this phenomenon.c) When choosing a solvent for recrystallization, what factors should be considered? (List
at least four criteria for choosing a solvent and explain where appropriate.)d) What is decantation and when should you employ this technique?
The PreLab and Final Report for this first experiment will be graded on a ✔, ✔+ , ✔- basis.This means that the report will not be graded in the strict sense, but errors will be indicated andcomments for improvement will be given by your TA. Almost everyone will receive a ✔ or 100points each on the PreLab and Final Report if they do a reasonable job. A few may receive a ✔-or 90 points if blatantly sloppy or inferior work is turned in. Also, a few may receive a ✔+ or 110points if their work is truly exceptional. The intent here is to use these first lab reports as feedbackto help your TA communicate to you what he or she is looking for in a typical PreLab and FinalReport. It is obviously very important for you to look over your TA’s analysis and comments anddiscuss any areas you don’t understand so that you are better prepared to do a good job in writingsubsequent PreLabs and Final Reports which will be graded more rigorously.
The white pages of this section including the Chemical Data Table will be collected by yourTA at the beginning of the lab period. Tear out the appropriate Grading Sheet from the back ofthe Lab Guide in (Appendix A.10) and attach it to the front of the PreLab. The point assignmentsfor each portion of the PreLab are shown below.
Before coming toLab
PreLab Exercise
Grading
GRADING FOR RECRYSTALLIZATION/MELTING POINT EXPERIMENT:Possible
PreLab: PointsDate, Name, Desk #, Experiment # & Title (abbreviated after 1st page), Section & TA Name 8Summary 16Goals 16Reactions and/or Diagrams of Special Apparatus 20Common Shelf Chemical Data Table and experiment Chemical Data Table 20PreLab Exercise 20Total for PreLab 100

40
Solubility
Introduction
Italian salad dressing is a classic example of aneveryday solubility problem. It is clear fromobserving the interaction between vegetable oiland water, they are not compatible and do not mixwell. This phenomenon can be understood by thesimple principle “like-dissolves-like”. Onlymolecules of similar structure will be soluble ineach other and mix easily. Water (a small polarmolecule) and vegetable oil (a long saturated alkanechain) are very different compounds and thus donot mix well. However, methanol (CH3OH) orethanol (CH3CH2OH) which are small organicmolecules containing a functional group that canH-bond, are readily soluble in water. We wouldfind that the vegetable oil, which contains moleculeswith large hydrocarbon chains are readily solublein hydrocarbon solvents such as ligroine or paintthinner. These are examples of like-dissolves-like.
Table 4.1: List of common solvents bydecreasing polarity.
Solubility depends on the polarities and interactions between the solute molecules (the solidto be purified) and the solvent molecules. For recrystallization, a solvent must be picked with apolarity similar to the polarity of the compound you are attempting to purify. In general, the moreheteroatoms, such as nitrogen or oxygen, a molecule contains, the more polar the molecule. Table4.1 lists some common organic solvents in order of polarity. This table can be used to selectsolvents with polarities closely matching the polarities of your solute. It is quite unlikey that asolvent with polarity that is very different from your solute will work at all, since you need to havea least partial solubility in a solvent for it to be a possible recrytallizing solvent.
Chemicals arefound everywhere!!
Like-dissolves-like! Most PolarWater (H20)Acetic Acid (CH3COOH)Methanol (CH3OH)Ethanol (CH3CH2OH)Acetone (CH3COCH3)Dichloromethane (CH2Cl2)Chloroform (CH3Cl)Ether (CH3CH2OCH2CH3)Benzene (C6H6)Toluene (C6H5CH3)Hexane (CH3(CH2)4CH3)Ligroin (mix of hydrocarbons)
Least Polar
Most of chemistry deals with the production of PURE chemicals for use in products wepurchase and use everyday. As you look around your kitchen or bathroom you see many of theseproducts. Can you tell which are pure compounds and which are mixtures? You should concludethat most of the foods, cosmetics, cleaning agents or drugs are mixtures. Very likely, the only puresingle-compound materials in the kitchen are water (H2O), salt (NaCl), baking soda (Na2HCO3),and sugar (sucrose, C12H22O11). In the bathroom you might have some crystals of Epsom salts(MgSO4) and some liquid rubbing alcohol (2-propanol), but these are about the only commonexamples. Pills and capsules are all mixtures of a pharmaceutical compound and an inert bindersuch as starch or gelatin. Many materials are really solutions, for example vinegar (5% acetic acidin water) or hydrogen peroxide (usually 3% in water). Even though most things are mixtures,many are formulated from pure compounds. For example, Anacin is a mixture of pure crystallineaspirin and pure crystalline caffeine mixed with starch and pressed into tablets. The Food andDrug Administration (FDA) sets very rigorous standards for purity of all the chemical componentsthat go into these mixtures or “formulations” sold as consumer products.
How do drug firms or the FDA measure the purity of organic compounds used in drugs? Moreimportantly, how do they purify compounds to meet these purity standards? This experiment willanswer these questions by demonstrating two common techniques: recrystallization for purifyingorganic solids and melting point determination for testing the purity of organic solids. Masteringthese techniques will be critical to the success of your synthetic experiments in the second halfof this course. As with all techniques, mastery means not just learning the motions, but alsounderstanding the underlying principles involved. For this experiment, the principles are:solubility (like-dissolves-like) including its variation with temperature and the melting behaviorof mixtures versus pure compounds.

41
The second factor affecting solubility is temperature. The temperature of the solvent directlyrelates to the amount of material that can be dissolved. One example is making rock candy fromsugar. In order to obtain the saturated sugar solution needed to make rock candy, the water isbrought to a vigorous boil. More sugar will dissolve in the water when the temperature isincreased. This is a very important factor in recrystallization because these differences insolubility at different temperatures will be used to our advantage.
The best solvent for any particular recrystallization should only sparingly dissolve thecompound at room temperature and easily dissolve the complex at higher temperatures. Figure4.1 shows the solubility of a typical organic compound in three different solvents as a functionof temperature.
Temperature (°C)
A
B
C
Sol
ubili
ty(w
eigh
t in
gram
s of
sol
id d
isso
lved
per
vol
ume
of s
olve
nt)
Figure 4.1: Solubility differences of a solid in three solvents, A, B & C verses temperature.
At any given temperature, the compound has a very low solubility in solvent B. Solvent C isexactly the opposite, its solubility is too great at all the temperatures. However, solvent A seemsto be a good choice because it has a low solubility at moderate temperatures, and the solubilityof the compound changes significantly as the temperature increases. This difference in solubilitywith small changes in temperature is an ideal solvent for recrystallization.
By understanding the solubility principles and the temperature factors involved, recrystallizationcan be a useful method for purifying solid organic compounds in the laboratory. This easy,convenient, and inexpensive purification technique can be used to remove small amounts ofimpurities from solid compounds. Essentially the process of recrystallization breaks down intofive simple steps:
• Choosing a suitable solvent• Dissolving the compound in a minimum amount of solvent• Removing insoluble and/or colored impurities• Crystallization• Collecting and washing the collected solids• Drying the crystals.
1. Choosing a Suitable Recrystallization SolventThe success of a recrystallization is based on the amount of pure solid crystals that can be
obtained using this technique. Ideally one would like to obtain 100% of the original weight ofmaterial. This is rarely possible, but recovery of 80 to 90 % of the original weight can be obtainedif: 1) the “correct” solvent is chosen and 2) the recrystallizing technique is carried out correctly.
Selecting an appropriate recrystallization solvent is the first and normally the most difficultstep in the purification process. A successful recrystallization depends on the solubilitydifferences between the substance and the impurity. Recrystallizing a known substance makes
Six easy steps torecrystallization.
Temperature playsa large role inrecrystallization.

42
choosing a solvent of similar polarity easier. Reference textbooks, such as the Handbook ofChemistry and Physics, can be used to find information on the solubility of organic compounds.If the material you will be recrystallizing is unknown, than the process of choosing a solvent isnot as straight-forward and is mainly trail and error. Dissolve the unknown compound in a varietyof solvents, heat and observe the results.
Based on the principle of “like-dissolves-like”, which solvent, benzene or water, would be abetter candidate solvent for recrystallizing naphthalene? Which solvent would be better forbenzoic acid? *
Solids:
Solvents:
Benzene
OHH
Water
Napthalene
C
O
OH
Benzoic Acid
* Benzene would be a better choice for recrystallizing naphthalene. Both of these compoundsare nonpolar organic molecules and the naphthalene should dissolve readily in hot benzene.Water would be a better solvent for organic molecules having polar substituents such as alcoholgroups (sugar) or carboxylic acid groups (benzoic acid).
A Problem!
There are also several practical concerns when choosing a recrystallization solvent. Forobvious reasons, the solvent should not react with the solute. The solvent must have a boiling pointlower than the melting point of the compound. If the boiling point of the solvent is higher thanthe melting point of the solid it will tend to “melt” instead of dissolve. This will lead to a very poorcrystallization. Chemists use the term “oiling out” to describe when a solid turns to an oil insteadof well-defined crystals. Oils are hard to handle and often difficult to crystallize. By choosinga different recrystallization solvent, oiling out can normally be avoided. In addition, a non-toxic,inexpensive, volatile and nonflammable solvent is the best choice. However, since most solventsdo not fit all these requirements, as the chemist you will have to find the best compromise for yourspecific recrystallization.
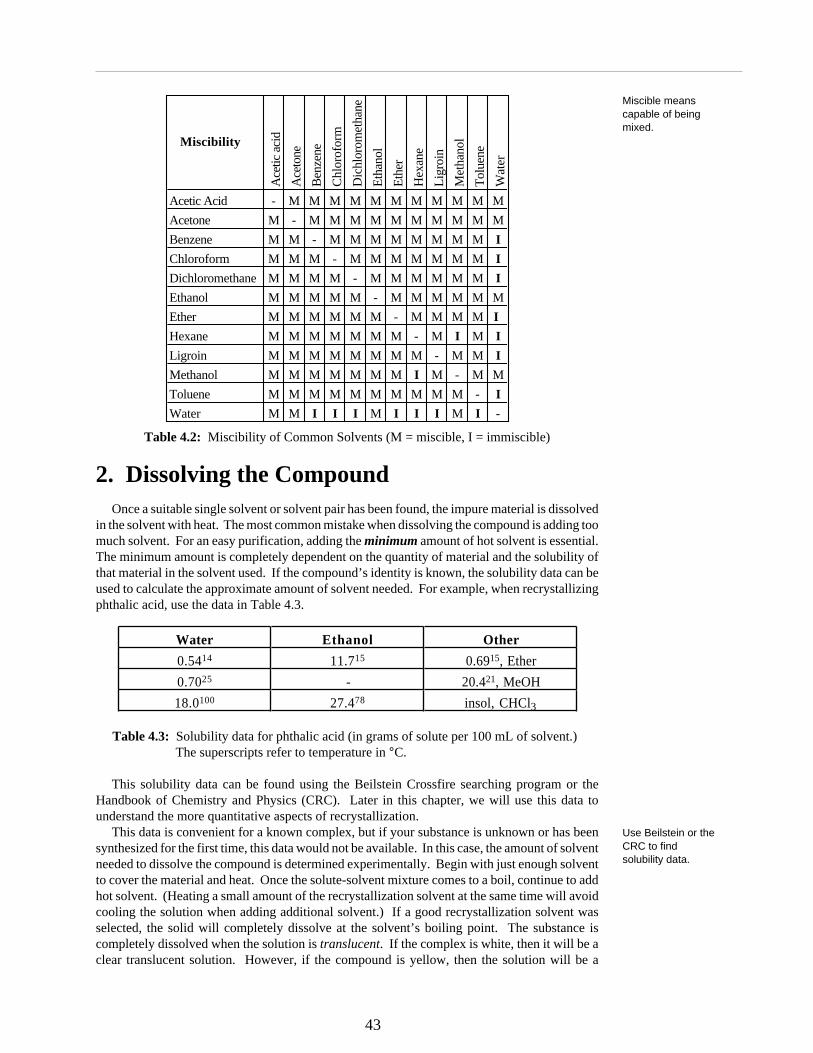
In an ideal system, the compound should be completely soluble in the hot solvent and quiteinsoluble in the cold solvent and the impurities should be soluble at all temperatures. This idealsituation is never obtainable. A single solvent system is always the best for recrytallization, butsometimes this cannot be found and therefore a two solvent system may be necessary. Whenchoosing a two solvent system, the two solvents must be miscible. Miscible means capable ofbeing mixed in all proportions. Table 4.2 summarizes the miscibility of some common solvents.Notice that with only one exception (can you find it?) organic solvents are miscible with eachother, but many are not miscible with water.
Some typical two solvent systems include; ethanol-water, methanol-water, acetone-ligroin,ethanol-toluene and acetic acid-water. (Notice that each of these combinations are miscible.)When using two solvents, it is best to find one solvent that will completely dissolve the compoundto be recrystallized at high temperatures and the second that dissolves it poorly at hightemperatures. Dissolve the compound in a minimum amount of the good solvent (dissolvescompound well at the boiling point), then add the bad solvent (dissolves the compound poorly)dropwise until the solution becomes slightly cloudy. Reheat the solution until clear and then coolslowly. Upon cooling, the compound should crystallize. If you are having problems crystallizingthe compound, reheat and add more of the poor solvent. The complex should eventuallyprecipitate, although this can often lead to the production of an oily non-crystalline mass.
The Answer!
43
Miscibility
Ace
tic a
cid
Ace
tone
Ben
zene
Chl
orof
orm
Dic
hlor
omet
hane
Eth
anol
Eth
er
Hex
ane
Ligr
oin
Met
hano
l
Tol
uene
Wat
er
Acetic Acid - M M M M M M M M M M M
Acetone M - M M M M M M M M M M
Benzene M M - M M M M M M M M I
Chloroform M M M - M M M M M M M I
Dichloromethane M M M M - M M M M M M I
Ethanol M M M M M - M M M M M M
Ether M M M M M M - M M M M I
Hexane M M M M M M M - M I M I
Ligroin M M M M M M M M - M M I
Methanol M M M M M M M I M - M M
Toluene M M M M M M M M M M - I
Water M M I I I M I I I M I -
Table 4.2: Miscibility of Common Solvents (M = miscible, I = immiscible)
2. Dissolving the Compound
Miscible meanscapable of beingmixed.
Once a suitable single solvent or solvent pair has been found, the impure material is dissolvedin the solvent with heat. The most common mistake when dissolving the compound is adding toomuch solvent. For an easy purification, adding the minimum amount of hot solvent is essential.The minimum amount is completely dependent on the quantity of material and the solubility ofthat material in the solvent used. If the compound’s identity is known, the solubility data can beused to calculate the approximate amount of solvent needed. For example, when recrystallizingphthalic acid, use the data in Table 4.3.
Water Ethanol Other
0.5414 11.715 0.6915, Ether
0.7025 - 20.421, MeOH
18.0100 27.478 insol, CHCl3
Table 4.3: Solubility data for phthalic acid (in grams of solute per 100 mL of solvent.) The superscripts refer to temperature in °C.
This solubility data can be found using the Beilstein Crossfire searching program or theHandbook of Chemistry and Physics (CRC). Later in this chapter, we will use this data tounderstand the more quantitative aspects of recrystallization.
This data is convenient for a known complex, but if your substance is unknown or has beensynthesized for the first time, this data would not be available. In this case, the amount of solventneeded to dissolve the compound is determined experimentally. Begin with just enough solventto cover the material and heat. Once the solute-solvent mixture comes to a boil, continue to addhot solvent. (Heating a small amount of the recrystallization solvent at the same time will avoidcooling the solution when adding additional solvent.) If a good recrystallization solvent wasselected, the solid will completely dissolve at the solvent’s boiling point. The substance iscompletely dissolved when the solution is translucent. If the complex is white, then it will be aclear translucent solution. However, if the compound is yellow, then the solution will be a
Use Beilstein or theCRC to findsolubility data.

44
translucent yellow solution. Recognizing when the solid just dissolves completely is veryimportant. DO NOT add too much solvent. This will cause problems when attempting to collectthe solid. The goal when heating this mixture is to create a saturated solution in hot solvent. (Justlike our rock candy example.) By creating a completely saturated solution, the less solublecompound will crystallize upon cooling. If an appropriate solvent was selected, your compoundwill crystallize with ease. Too much solvent will hold more solid on cooling and it may notcrystallize. However, if too much solvent is added, there is a simple solution. Heat the solutionto the boiling point and evaporate some of the solvent to reduce the volume of the solution. It isvery easy to add too much solvent while learning to recrystallize organic compounds. This is away to correct your mistakes without starting over or wasting material.
One practical aspect of dissolving the compound in heated solvent is how to avoid aphenomenon called “superheating” or “bumping” . Superheating is when the temperature of thesolvent reaches a temperature greater than that of the boiling point of the solvent. When thisoccurs, the solvent will very rapidly and sometimes violently bump or splash. Bumping is thesudden explosive boiling of the liquid near the bottom of the flask. Avoid bumping by using aTeflon boiling chip (small white chips found on the Common Shelf) or a wooden boiling stick (alsofound on the Common Shelf). These two methods allow the solution to heat evenly and boilsmoothly.
3. Removing Insoluble and Colored ImpuritiesOnce a hot saturated solution is obtained, there may still be some remaining particles, including
dirt, sand, or insoluble impurities. If these particles are dense and settle to the bottom, the liquidcan often be poured off or decanted from the solid. If they don’t settle to the bottom, there areseveral methods to remove these insoluble materials. When working on small scale, pipetfiltration is a good method. To make a filter pipet, carefully stuff a small portion of cotton(Common Shelf) into a disposable glass pipet as shown in Figure 4.2. Some chemists also addabout 1/2” of silica or alumina.
Figure 4.2: Pipet filtrationfor insoluble materials.
Transfer the hot solution into the filter pipet with another pipet. The insoluble materials collectin the glasswool. Be careful though; since the filter pipet cools the solution, crystallization canoccur inside the pipet. One method to avoid precipitation inside the pipet is to heat some purerecrystallization solvent to the boiling point and run this hot solution through the pipet first. Thiswill increase the temperature of the pipet and glasswool decreasing the chance of precipitation.Another way to insure that the compound will not crystallize inside the pipet is to use extra solventand create a more dilute solution. This will decrease the chance of crystallization at coolertemperatures. After collecting the dilute solution, you will have to evaporate the extra solvent toinitiate crystallization.
If you are working on a larger scale, use the hot filtration apparatus shown in Figure 4.3. Asin the microscale pipet filtration, it is essential to have the entire flask and funnel heated. This canbe accomplished by heating a small amount of recrystallization solvent in the bottom of theErlenmeyer flask. This allows the vapor to rise warming the flask, funnel, and filter paper. If the
Use boiling chips ora boing stick toavoid bumping!!
Pipet filtration ofinsoluble particles.
Reaction tube to c olle ct mother liquor
Pour in dilute solu tion
Cotton used to collect insoluble materials
Silica gel or alumina plug or layer on top of cotton

45
Figure 4.3: Hot filtration set-up
4. Crystallization
Once you have obtained a clear saturated solution in hot solvent free of insoluble or coloredimpurities, it is time to crystallize your solid product. Crystallization can sometimes occur withinseconds and other times it may take days. A perfect crystal needed for X-ray crystallographyrequires very slow crystallization in order to obtain a crystal large enough to analyze. It shouldalso be mentioned that crystallization is different from precipitation. Precipitation is defined asthe formation of a solid product from a reaction that separates from solution. Precipitationnormally occurs very rapidly and results in very fine crystals. Crystallization is similar toprecipitation in that you separate a solid from the remaining solution; however, no chemicalreaction occurs and crystallization normally refers to the slow growth of crystals. Slow crystalgrowth yields purer crystalline formations. There are three ways to initiate crystallization;cooling, scratching and seeding.
Slow cooling is the easiest method and works in most cases. Simply allow the hot saturatedsolution to cool slowly without agitating or disturbing the solution in any way. Allow the hotsolution to cool to room temperature by placing the flask on a book or paper towel, then move itto the cooler bench top and finally put it in a beaker of ice. Setting a beaker on an insulating surfacesuch as a book or placing a test tube in a beaker of glasswool will slow the heat losses and thecooling process. The slower the solution cools, the more likely it will form perfect crystals whichcan exclude the impurities since these will not fit in the crystal structure. Alternatively, you maycool the solution slowly to room temperature and then place the solution in the refrigerator until
entire apparatus is not warm, the product will crystallize along the inside of the filter paper.These filtration methods will remove insoluble materials from the solution. However, if you
have soluble colored impurities, Norit® pellets can be used to remove them. Norit® is thecommercial name for activated carbon. When mixed with the solution, the colored materialsphysically absorb onto the Norit® pellets. You may have to heat the mixture and repeat theprocedure more than once. (Note that even very small quantities of colored impurities can producehighly colored solutions.) The benefit of using the Norit® pellet form of activated carbon insteadof finely powdered charcoal when removing colored impurities is that the pellets can then beeasily removed by pipet or gravity filtration as described above. In other words, the solublecolored impurities stuck to the Norit® pellets are now an easily removable insoluble impurity.Remember, however, that some of your product will be absorbed so don’t use too much Norit.
Obviously filtration and/or treatment with activated carbon are only necessary when there areinsoluble or colored impurities present. Most recrystallizations will not require either step.
Heating the entirehot filtrationapparatus isessential to theprocess.
Slow colling leadsto the best crystals.

46
the next lab period. (Note: All solutions placed in the refrigerator must be in capped and labeledcontainers!) This slow cooling process normally yields beautiful crystals.
Sometimes it is difficult to obtain crystals even after the solution is cooled in an ice bath. Inthis situation crystallization can be induced by scratching the inside of the beaker or flask witha glass stirring rod. This will produce microscopic fragments of glass that may act as surfaces onwhich crystal growth can begin.
If slow cooling and scratching does not produce crystals, crystallization may be induced byseeding. Seeding is accomplished by taking a small crystal from the original solid and droppingit into the solution. This will provide a nucleation site for similar crystals to grow. This methodworks well if you are having problems with a solid that separates without crystallization or “oils”out. However, by adding some of the original material, you are inevitably increasing the amountof impurity in the resulting solid. Seeding is normally used only as a last resort.
One crystallization method that is not encouraged is evaporation. Naturally, if you are havingtrouble crystallizing a solid, evaporating the solvent completely will recover the material.However this method has defeated the purification process. Not only have you recovered the purematerial, but also the impurities. When this is done, the recrystallization procedure must becarried out again.
5. Collecting and Washing the CrystalsThe filtration method used to separate the crystals from the remaining solution and “collect”
the solid depends largely on the amount of crystals and solution. For recrystallizations involving1 g to 100 g or more, gravity filtration through filter paper can be used. For recrystallizationsinvolving at least 0.1 g up to 100 g or more, organic chemists use a fast and effective filtrationmethod called vacuum filtration. But for the small quantities of 10 - 100 mg in a few mL ofsolution, pipet filtration is quick and effective.
The first method, gravity filtration is used when there is at least a half a gram of large, well-defined crystals. A glass funnel fitted with fluted filter paper is used to collect the crystals. Withvery large amounts of solid, this process can consume a lot of time.
A quicker and more efficient method for this same quantity of material is vacuum filtration.Vacuum filtration is probably the most common filtration type used by chemists. The funnel usedfor this method called a Hirsch funnel if small or a Buchner funnel if large. These funnels havea large flat plate inside which can either be made from ceramic materials that contains lots of smallholes through it or from a porous polyethylene disk.
Figure 4.4: Vacuum filtrationapparatus using a Hirsch funnel.
To use these funnels, a round piece of filter paper ofa matching diameter is placed over the perforated orporous plate. The Hirsch or Buchner funnel is put intoa vacuum filter flask with a rubber adapter in between toassure a vacuum tight seal. The filter flask has aconnecting tube or “side arm” which is connected to thehouse vacuum or water aspirator. Wet the filter paperwith a small amount of solvent or recrystallizationsolution to prohibit loss of product under the filter paper.Once the vacuum is turned on, the filter paper is suckeddown on the plate so that it seals uniformly. The crystalsand solution are swirled or agitated and quickly pouredonto the filter funnel. The vacuum quickly draws theliquid through the filter paper disk, leaving the crystalsbehind. Any crystals remaining behind in the beaker arescraped out with a spatula and/or washed out with someof the recrystallizing solution from the filter flask or withsmall amount of cold recrystallizing solvent. This steprequires great care. Using too much solvent that is notcold enough can dissolve a lot of solid product, thusdecreasing your recovery and yield significantly.
The filtrationmethod useddepends largely onthe amount ofcrystals collected.
Wash yourcrystals with care!

47
One variation on a traditional vacuum filtration apparatus is shown in Figure 4.5. The motherliquor is the filtrate or liquid left behind after removing solids from a solution. In this set-up, themother liquor is collected in a clean beaker and can be used again. Often during a recrystallization,the first collection process will not crystallize all the material from the solution. By collecting thefiltrate, the remaining crystals can be retrieved. After the first filtration, concentrate the motherliquor by evaporating the solvent to approximately half the original volume. Upon cooling,additional crystals, called a second crop, may appear.
Figure 4.5: Bell Jar with vacuum filtration used to collect the mother liquor.
To use this set-up, first apply a light coating of stopcock grease to the bottom edge of the belljar. Seat the Hirsch funnel in a rubber filter adapter and place in the top of the bell jar. Connectthe side arm of the bell jar to the house vacuum using heavy-walled vacuum tubing. Pour thesolution carefully onto the filter. Once you have collected the solid, turn off the vacuum and allowair to leak slowly into the jar. Carefully disconnect the tubing being careful not spill the contentsof the filter apparatus or beaker. If there is not sufficient time to concentrate the mother liquorby evaporation, than simply setting the beaker in the back of the hood for slow evaporation. This
Figure 4.6: Pipet Filtration of Solid Product.
will often yield crystals without effort. Ifcrystals are obtained from this secondcrop, the crystals may not be as pure asthe first crop because there were a higherpercentage of impurities present duringthe second attempt. A melting-pointdetermination can be used to determinethe purity of both crops.
For small quantities of materials, pipetfiltration is the preferred method. Figure4.6 shows the pipet filtration method forcollecting solid products.
When you have solid material left-over in a reaction tube or small test tube,there will be less product loss if youremove the solvent and leave the solidbehind. This can be easily achieved bypipet filtration. Select a pipet with aperfectly square tip (when looked at fromthe side), and without chips on the tip.Push out all the air by squeezing the pipetbulb. Slowly lower the pipet into thesolution and ease your way to the very
Filtration methodused to obtain asecond crop ofcrystals.
Square tip pipet
Pipet filtration of asolid product.

48
bottom of the reaction tube. Once the pipet tip is squarely and firmly pressed against the bottomof the tube, slowly release the pipet bulb while keeping the pipet tip pressed against the bottomof the tube. The solvent will be slowly drawn up into the pipet and the product should remainbehind. Initially you may see a small amount of product move up the pipet. Don’t panic! Thisis a normal loss for this technique. This technique is a little trickier than vacuum filtration andnormally requires some practice. Practice with some inert materials from the Common Shelf, suchas sodium sulfate and hexane. The separation is easier if you have big, chunky crystals. Very finecrystals will be difficult to separate using this method.
After the crystals have been successfully separated from the solvent, washing the crystals withcold recrystallization solvent will put the finishing touches on your purification. This will washaway any last traces of soluble impurity that have been left on the surface of the crystals. It isessential to use cold solvent; hot or room temperature solvent will dissolve too many of yourcrystals. If you used gravity filtration or vacuum filtration to collect the crystals, drop the coldliquid directly onto the crystals with a pipet. If you performed a pipet filtration, add the coldsolvent directly to the tube while immersing it in ice, stir the solvent and then remove the solventusing pipet filtration as you did before. Careful pipet filtration will increase your percent recoveryof the product.
6. Drying the CrystalsThe easiest way to dry the collected crystals is to be patient and allow nature to take its course.
Leave the crystals in a container open to the air in your desk. Over a period of a few days,depending on the boiling point of the solvent, they will dry. If there is not sufficient time to allowthis slow drying process or if you are using test tubes, reaction tubes or solvents with higher boilingpoints, such as toluene or water, there are several other methods including vacuum drying or usinga stream of nitrogen. Pressing the solid between pieces of filter paper or paper towel is a simpleway to remove a major proportion of a solvent.
Vacuum drying can be easily accomplished with the desk equipment provided. If your productis in a reaction tube from a pipet filtration, you can connect the yellow thermometer adapter to thereaction tube and then on to a piece of thick-walled rubber tubing using the cut off barrel from a1-mL plastic syringe. Connect the tubing to the house vacuum and allow it to dry. (Figure 4.7)Alternatively, the reaction tube can be pushed directly into the opening of the thick-walled tubing.Eveaporation is speeded up by clamping the reaction tube in a beaker of warm water. This methodshould only be used to dry residual solvent. Using a vacuum to remove large amounts of solventwill be slow and could eventually destroy the vacuum pumps.
For volatile or low boiling solvents, placing your product under a stream of nitrogen may besufficient to dry your product. Each hood has its own nitrogen line. The outlet should be fit witha plastic pipet attached to the nitrogen line by a piece of Tygon tubing. Simply place the end ofthe plastic pipet over the solid to be dried or directly into the reaction tube. Be careful not to “blow”all the product away with a very fast flow rate. Drying your product under a stream of nitrogenworks very well for very volatile solvents such as ether and dichloromethane.
Pipet filtration takespractice andpatience.
Air Drying
Vacuum Drying
Nitrogen
Figure 4.7 Vacuumdrying solids in areaction tube.

49
Melting Behavior of Mixtures vs. Pure CompoundsOnce you have purified the solid by recrystallization, its purity can be verified by determining
its melting temperature which is commonly called a melting point, but is really a melting range.To better understand this behavior, the theory of melting point determination must be examined.
Butter is a mixture of many different types of fat or triglyceride molecules. When stored at 5°Cin a refrigerator, it is fairly solid. On warming to room temperature (~ 25°C) it becomes soft andif allowed to stand in the warm sun (40°C), it will turn to a liquid. Thus, this mixture of organiccompounds “melts” over a very broad range of temperatures. Pure water containing only one typeof molecule, H2O, freezes (if we remove heat from the liquid) or melts (if we add heat to the solid)at a single temperature, 0°C, which is defined as the melting point (mp) of pure water. Until allthe solid ice is melted to liquid water, the temperature will remain at 0°C no matter how hard weheat the container. What would happen if we add an impurity, such as salt, to water? Everydayexperience with the effects of spreading salt on highways and sidewalks tells us that the meltingtemperature of a water-salt mixture is lower than 0°C causing the ice to melt as it dissolves thesalt. This behavior is called melting point depression. Hopefully you are familiar with this fromgeneral chemistry.
Of the 18 million organic compounds known today, only a few thousand are liquid or gaseousorganic compounds. The remainder are all solids, many of which will melt from just above roomtemperature to about 350°C. Above this temperature, organic compounds just decompose andchar rather than melt. (Also, the glass in a thermometer starts to get red hot and soften.) Solidswith normal melting behavior are in the molecular weight range from 59 (e.g., acetamideCH3CONH2, mp 79-81°C) to roughly 1500 ( e.g. the antibiotic bacitracin, C66H103N17O18S, mp221-225°C). Notice that while we talk about melting point and use the abbreviation mp for it, wealmost always report a melting temperature range of a few degrees as seen in the examples justcited. This is an anachronism that chemists have perpetuated and we will continue this peculiarhabit.
As with water, we should expect that adding impurities to organic compounds would lowertheir melting point also. In addition we observe that the melting range is broadened. Even smallamounts of impurities, ~1%, will lower and broaden the melting temperature range. Thisbehavior of solids is consistent and can therefore be used as a test of whether we have a purecompound or one mixed with some impurities. Thus, a melting point (really melting range)determination is a quick and simple test of a compounds purity and drug companies will use thisand other analyses to assure the purity of any drug compounds they produce. For example, thetrue melting point range for caffeine is 234 - 237 ° C. If the caffeine is obtained directly from teaand is not purified, the observed melting point is much lower. In fact, crude caffeine extractedfrom tea bags melts as low 180 - 220 ° C. This melting range is significantly different from thetrue melting point and can be used to determine that it is impure.
Melting Point (Range) DeterminationThe melting point or freezing point of a compound is the physical state change from a solid to
a liquid or a liquid to a solid respectively. Each pure organic compound has a characteristicmelting point or range. This determination can be used to correctly identify and/or demonstratethe purity of the compound.
In addition to traditional melting points, compounds can sometimes decompose beforeactually melting. Instead of a solid clearly turning into a liquid, the solid may turn gray, brownor black at a certain temperature. This does not mean that you have obtained the wrong compound,just that this particular compound is not stable at the observed temperatures. Saccharin forexample does not melt, but decomposes at 229°C. This is symbolized as 229 d. This may happento some of the compounds you purify throughout the semester.
There are over 18million organiccompounds knowntoday.
Not all compoundsmelt.

50
Purity can be determined by evaluating the melting point range. When an impurity is solublein the compound, the melting point will be depressed. Figure 4.8 is a Melting Point CompositionDiagram that shows how the melting temperatures are affected in a two component mixture.Either component may be regarded as an impurity in the other component.
Mole percent of AMole percent of B
100 50 0 0 50 100
MP in °C of pure A
MP in °C of pure B
Ep
Et
Increasing MeltingTemperature
Figure 4.8: Melting Point Composition Diagram of a Two Component Mixture.
The left and right y-axis intercept the known melting temperatures for pure compound A andpure compound B respectively. However, upon mixing A with B, the melting temperature of themixture is decreased. The amount the melting point is depressed is a function of the molar percentof each component and can be read directly from the diagram. There are two special points onthis diagram. The first is the Eutectic temperature, designated by Et. This is the lowesttemperature at which the mixture will begin to melt. The Eutectic point, Ep is the mole percentagewhere the mixture of two compounds are dissolved equally in each other. At this particular point,even though the mixture is not pure, it will appear to have a sharp melting point. As discussedearlier, a sharp melting point indicates a pure material. A broad melting point is normally amixture of compounds. The Eutectic point is an exception to this rule and can lead tomisidentification of the sample. It should also be noted that insoluble impurities, although veryundesirable, will not affect the melting point. One example of an insoluble impurity would be apiece of sand. The sand will have not affect on the melting point.
If you are determining the melting point of a known compound, the results can be comparedto literature values. Aldrich catalog has an enormous amount of physical data for organiccompounds including melting points. Use these and other sources to verify your melting points.However, if the compound is unknown or has been synthesized for the first time, the melting rangemay still help in determining the purity of the substance. The melting point range is thetemperature at which the solid begins to melt to the temperature when the solid has completelybeen converted into a liquid. Melting points should always be recorded as a range. As previouslymentioned, the purer the compound is the narrower the melting point range. In contrast, the moreimpure the compound is the broader the melting point range. Melting point ranges can vary, buta good range is typically 1-3 °C. Poor ranges can be as broad as 20°C.
One of the biggest and most common mistakes when obtaining a melting point, is not allowingthe sample to dry completely. Traces of solvent in the sample act just as any other impurity. Itwill depress the melting point and broaden its range. This will lead to incorrect identification ofthe unknowns you will be recrystallizing in this chapter. Another common mistake is to heat thesample too fast. In general it is best to heat the sample slowly, no faster than 2°C per min in therange of the melting point. Use this rate to expedite the use of the few melting point units we have.
Melting PointDepression.
Solids must be freeof solvent in orderto obtain anaccurate meltingpoint.

51
In our laboratory, we use two different types of melting point apparatuses: Mel-Temps, locatedat the end of every other laboratory bench, and Thomas Hoover’s located at the North end of Lab205. Both are electrically heated melting point apparati. The directions for the Thomas-Hooverand Mel-Temp Melting Point Apparati can be found in Step 3 of the experiment found in thischapter. Since both the Mel-Temp and the Thomas Hoover machine use thermometers, it is veryimportant to calibrate the thermometers. Obviously, the identification of your unknown will beincorrect if the thermometer reading is wrong. This is a very common and serious mistake duringthe determination of unknown samples. Please check to see that the thermometer is givingaccurate temperature readings by recording the temperatures of boiling water and ice. It is asimple and straight-forward test you must complete before the melting point determination.
One advantage of a melting point analysis is the very small amount of sample needed to obtaingood results. Each machine uses glass capillary tubes to obtain the melting point. To load tocapillary, take a few milligrams of the material to be analyzed and push the solid together on awatch glass or other clean surface. Use the open end of the capillary tube (found in your deskequipment) and push the open end onto the pile of solid material. Once a small portion of thecompound is in the tube, turn the capillary over and rap the tube gently on the desk top to packthe sample in the botton. Be careful not to break the tube accidentally. Even though the tubes aresmall, the glass can be very dangerous and puncture the skin very easily. While rapping on thedesktop, the compound (if thoroughly dry) will move to the closed end of the tube. If this doesnot occur, drop the mp capillary through a 3 foot long plastic tube held vertically on the benchtop.These plastic tubes can be found clamped to the side shelves. You only need a few millimetersof sample in the end of the tube. Too much sample will lead to uneven melting and possiblyincrease the range of your melting point. Insert the tube, closed-end down, into the Mel-Tempor Thomas Hoover, heat it slowly, and observe the melting point range.
Although melting points can be used as an identification tool, there are many compounds thathave coincident melting points. There may be an occasion when you have two compounds thathave the same melting point. They could be the same compound or it may be a coincidence. Amixed-melting point determination can verify whether they are the same compound or twodifferent compounds. Mix equal amounts of the two solids and determine the melting point of thismixture. If the mixed-melting point is the same for both the mixed materials and the individualcompounds, they are the same compounds. However, since mixtures of different materials meltat lower than either compound, if the combined melting point is depressed compared to theindividual samples, then the materials are not the same. This simple method can easily verify ifthese compounds with the same melting point are the same.
Mixed-meltingpoints.
Mel-temps &Thomas Hoovers
only 1 to 2mm of samplein mp capillary

52

53
Procedures 1,2, and 3 provide challenging tests of your experimental and observational skills.You will be given a small amount of an impure unknown organic compound whose identity canbe determined from its melting point. However, since its melting point is depressed by impurities,you will need to purify the compound by recrystallization before the melting point determination.Procedure 4 involves a more quantitative analysis of solubility in cold and hot solutions.Procedure 5 demonstrates how colored impurities might be removed from an organic solid.Procedure 6 shows how the melting point can be used to help identify an unknown solid.
All students should complete Procedures 1, 2 & 4 in the first lab. Procedure 3,determination of unknown melting points and Procedure 5, decolorization can be done inthe following lab period. Procedure 6 may be done in subsequent labs or in the instrumentroom. In order to use your time efficiently, whenever possible try to do more than oneoperation or experiment at a time.
CH3
O
O
O HC
NH2
CO H
CH2CO H
CH3
O
O
Benzamidem.p. 127-130°C
654
321o-Toluic Acid
m.p. 104-107°Cm-Toluic Acid
m.p. 109-113°CFluorene
m.p. 114-116°C
E-Cinnamic Acidm.p. 133-134°C
E-Stilbenem.p. 122-124°C
Procedure 1 • Finding a Suitable Recrystallization Solvent for your Unknown
You will use a sand bath, not a Bunsen burner or steam bath, to heat solvents. As soon as youarrive and while you’re waiting for your TA to collect your PRELAB pages, fill your heatingmantle half full of sand, mount it to a ring stand in your hood, and plug it into the heating controller(often called a varistat or powerstat) which is mounted on the outside of the hood. Plug thecontroller into the electrical socket above the bench and set the dial to 50.
To effect smooth boiling and avoid explosive boiling or bumping of the solvent when heatingit, be sure to use boiling sticks (wooden applicator sticks). Boiling sticks also can serve as stirrers.
Your instructor will provide you with approximately 100 mg of an impure unknown organicsolid (record the number of your unknown immediately into your lab notebook) which is one ofthe six compounds shown below:
UnknownCompounds
NOTE: Set up andheat your sand bathas soon you arrivein lab.
Recrystallization Experiments
Solubility Test
You will test the solubility of your unknown in differentsolvents as follows:
Weigh out between 8 and 13 mg (0.008 to 0.013g) of yourunknown and transfer it to a tared reaction tube or micro test tube.(This is about the amount that can be picked up on the last half inchof the sharply pointed end of your stainless steel micro-spatula.)Using a Pasteur pipet, add to this solid about 0.25 mL of distilledwater. You can use the graduations on the side of the reactiontubes to estimate the 0.25 mL of solvent.
GraduatedReaction Tube

54
Always makecarefulobservations.
Cleaning Up
Alternatively, use your 1 mL x 1/100 mL pipette to measure out liquid volumes less than 1 mL.To draw liquids into the pipet in a controllable manner, attached your 2.5-mL plastic syringe tothe pipet with a small rubber connector available on the Common Shelf. (Ask your TA for ademonstration if you are unsure of how to use the rubber connector.) Volumes can be approximatefor the solubility tests; for 0.25 mL of water, for example, you can use a volume between 0.20and 0.30 mL.
Agitate the tube vigorously for 10-30 sec by hitting or tapping the bottom with your fingers,or by crushing the large crystals with a stirring rod. If the solid dissolves, the unknown is toosoluble in the solvent and the solvent can be eliminated as suitable for recrystallization. If the soliddoes not dissolve after mixing for a short period, place a boiling stick (wooden applicator) in thetube and push the bottom of the tube part-way down into the hot sand of your sand bath. Thesolvent should start to boil within 10-30 seconds. If the boiling becomes too vigorous, pull thetube partially out of the sandbath. Continue gently boiling the solvent for about a minute, notinghow the vapors condense on the tube walls and run back down into the liquid. This is calledrefluxing .
Carefully observe the mass of crystals while you’re refluxing the liquid. If the solid dissolves,place the tube in a small beaker of ice water for one or two minutes to cool the solution. If youobserve solid crystals coming out of solution you have found a suitable solvent for recrystallization.Go directly to Step 2 and recrystallize your unknown according to the instructions there.
Note: Be alert to the difference between melting and dissolving! Compounds that melt justabove the boiling point of water when pure may melt just below the boiling point of water whensmall amounts of impurities are present (the maximum solvent temperature in these experimentsis approximately that of the boiling point of the solvent). Solids that melt but do not dissolve willform two immiscible liquids in the tube; upon crystallization, little impurities will stay in thesolvent, so the degree of purification of the solid from this step will be rather small.
If refluxing in the solvent leads to an observable decrease in the mass of the solid, but notcomplete dissolution, try adding more solvent, a few drops at a time, heating the solution to boiling(refluxing) for 30 to 45 seconds after each addition to see whether all of the solid dissolves. If itfinally does, place the tube in an small ice-filled beaker for one or two minutes to cool the solution.If you observe solid crystals coming out of solution you have found a suitable solvent forrecrystallization. Go directly to Step 2 and recrystallize your unknown according to theinstructions there.
If the mass of solid remains unchanged after one or two minutes in the refluxing solvent, thenthe unknown is obviously insoluble in both cold and hot solvent, and the solvent is unsuitable forrecrystallization. Set the tube aside. With a clean tube, carry out the solubility test procedure asdescribed above with another 8 to 13 mg sample of your unknown and a different solvent. Trythe other extreme in solvent polarity, for the second test, ligroin (ligroin is the common name fora mixture of hexane and cyclohexane isomers with boiling points between 60 and 90°C). If thisdoesn’t work, try a solvent with polarities intermediate between water and ligroin such asmethanol. If you don’t find a suitable recrystallization solvent in 3 or 4 trials, talk with yourinstructor.
Cleaning Up Place organic solvents and solutions of the compounds in the NonhalogenatedOrganics Disposal container. Dilute the aqueous solutions with water and flush down the drain.
Procedure 2 •• Microscale Recrystallization of your Unknown
Having found a suitable solvent for recrystallization in Step 1 above, recrystallize theremainder of your unknown as follows:
Refluxing

55
Obtain approximately 80 mg of your unknown solid by weighing your unknown on theelectronic balance. Transfer the weighed unknown into a tared reaction tube. (See the Check-in instructions in the back of the laboratory notebook for information on how to permanently marktare weights on your reaction tubes, if they haven’t been marked already.) Make sure you haveabout 5 mg of the impure solid left to be used in a melting point determination in Step 3. Use theplastic funnel from your red glassware kit can be used as a powder funnel to make the transfer ofthe solid into the narrow neck of the reaction tube easier. If you do not have 80 mg, you’ll haveto obtain more from the stockroom. Please be sure to tell the stockroom personnel your unknownnumber if you need more.
To the solid in the tube, add about 1.0 mL of the solvent you’ve found to be suitable forrecrystallization, place a boiling stick in the tube and heat to refluxing in the sand bath. If the solidhas not completely dissolved after a minute of boiling, add additional solvent, 5 or 6 drops at atime, bringing the solution back to a gentle reflux for a brief period after each addition. Be sureto allow time for as much undissolved solid to go into solution as possible at the boiling point ofthe solvent. After all the solid has dissolved, set the tube into a test tube rack and allow the solutionto cool slowly. This will lead to slower crystal growth, yielding larger, more perfect, and purercrystals. While this is cooling, put approx. 0.5 mL fresh recrystallization solvent in a small testtube and cool in an ice bath. You will use this later to wash your purified crystals.
After about five to ten minutes of slow cooling, crystals should have started to form. If theyhaven’t scratch the inside wall of the tube with a glass stirring rod. When you are sure that thesolution is close to room temperature, cool it further by placing the tube in a small beaker of ice.After a few minutes in ice, if necessary, break up any solid crystalline mass that may have formedand rap the reaction tube bottom on a book or paper towel to force the liquid to the bottom. Removethe solvent using the pipet filtration method described in Figure 4.6 and demonstrated to you byyour instructor. Add 6 to 10 drops of previously-cooled pure solvent to the crystals in the ice bathand agitate to wash the crystals. Remove this wash solvent using the pipet filtration method.
If your recrystallization solvent was water, you should remove a small clump (about 5 mg, justenough to fit on the tip of the spatula) of crystals and place them on an open watch glass in yourlocker so that they can dry thoroughly by the next lab period. They will be used to obtain andaccurate melting point. If it was ligroin or other more volatile solvent, the solids can be left in theopen reaction tubes in your locker until the next lab period and weighed directly in the tube(subtracting the tare weight of the tube). This will reduce losses in of solid during transfers whichcan be a problem at microscale. Alternatively, you can speed up the removal of the residualsolvent adhering to the solids by applying vacuum to the reaction tube while warming in a beakerof warm water as shown in Figure 4.7.
When dry, simply weigh the tube containing the sample and subtract the reaction tube tareweight as determined previously. Calculate and report the percent recovery in your final report.A small portion of this solid will be used to determine the melting point of your purified unknownin Step 3.
Cleaning Up Place organic solvents and solutions in the halogenated or nonhalogenatedorganics waste container, whichever is appropriate. Dilute the aqueous solutions with water andflush down the drain. Carry your sandbath to the side shelf and dump the HOT! sand back intothe sand supply bowl. Do NOT dump it in the waste bins.
Scratch or Seed
Your compoundmust be dry todetermine thecorrect meltingpoint.
Cleaning Up
Procedure 3 • Melting Points of Your Impure and Recrystallized Unknown
In this experiment, you will determine the melting temperature ranges of both your impureunknown and your recrystallized, pure unknown. These will be determined using melting pointcapillary tubes (found in your locker) and one of the “Mel-Temp” electrically heated meltingpoint apparati found on the shelf above your desk (1 per 4 students) or “Thomas-Hoover” typefound on the side shelves. (Please don’t move the ones on the side shelves.) Two students can
Obtain the weight ofyour unknown.

56
use one Thomas-Hoover apparatus at the same time, since 4 melting point tubes can be placed inthe heating oil bath of this apparatus, and the melting points determined for all 4 samples byheating from low temperature, say 70°C, to high temperature, say over 130°C, in one pass. Itshould take 20-30 min for each temperature “scan”, including the time necessary for the apparatusto cool down. All students should be able to determine their mp’s in one three-hour lab.
Turn the power on, but turn the heating rate controldial all the way counter-clockwise to zero. If thetemperature of the aluminum block is more than 70°Cas read on the thermometer, wait for it to cool see thetemperature on the thermometer rise. At this point youneed to keep an eye on both the mp tubes (to watch forany signs of melting) and the thermometer (to monitorthe rate at which the temperature is rising). [It helps tohave chameleon eyes!] You may need to adjust theheating rate control dial so that the temperature does notrise too rapidly.
It is best to heat the samples slowly, no faster than1°C per min. However, it is possible to get useable mp’sheating at 2 to 3°C per minute, and to expedite the useof the few mp units we have, try this faster rate. Watchthe samples, and when you start to see any one of themstart to melt, record the temperature. When the solid in
Mel-Temp
It is of utmost importance that your sample be thoroughly dry and free from all traces of solventbefore trying to determine its melting point. Otherwise, the solvent, like any other impurity, willcause the melting point to be depressed, and you won’t obtain an accurate melting point.Normally, most solvents evaporate completely from crystals in an open reaction tube in the twoto five days between labs, but higher boiling solvents such as water (bp 100°C) and toluene (bp110°C) may require drying on an open watch glass for a day. If your solid still seems wet andsticky after this period of time, you may have to allow additional time for drying by leaving it openin your locker for another few days, or you may have to speed up the evaporation process byapplying vacuum. Assuming your crystals are dry, proceed as follows:
Remove a few mg of the purified unknown and place it on a watch glass. If it is lumpy, grindit into a fine powder using the flat end of a spatula. Push the crystals into a small pile and thenpush the open end of a melting point capillary tube into the crystals so that some solid is forcedinto the opening of the tube. The sample is then shaken down into the closed end of the capillaryeither by: 1) wrapping the capillary tube with a paper towel and rapping sharply onto a hardsurface; or by: 2) dropping it down a 2 to 3-ft length of 5-10 mm I.D. tube onto a hard surface.(there are a few of these tubes on the side shelves) If necessary, push more crystals into the openend and pack it down into the closed end so that you have no more than 2-3 mm column of solidat the bottom of the mp. tube.
In a second melting point capillary, load a small amount of the unrecrystallized and impureunknown that you retained from Step 2 in the snap-top plastic vial. Mark the tubes so they canbe differentiated.
When a melting point apparatus is available, check that it has cooled to below 70°C. Thenyou (and another student in the case of the Thomas Hoover units) can insert two samples and taketheir melting points using one of the following procedures, depending on the melting pointapparatus. Be sure to bring your notebooks with you to the mp apparatus so that you can recordthe mp data
Loading the meltingpoint capillary.
Your unknown mustbe solvent free.
Figure 4.12: Mel-Temp Apparatus
Mel-Temp Melting Point Apparatus Procedure
only 1 to 2mm of samplein mp capillary

57
Thomas-Hoover Melting Point Apparatus Procedure
Turn the power on, if it isn’t already. Turn the heating rate control dial all the way counter-clockwise to zero. Make sure the stirrer speed control dial is set at a little more than half of fullspeed. If the temperature of the oil bath is more than 70°C as read on the thermometer, wait forit to cool below this. Once below 70°C, you and another student can place your two mp tubesgently into the holes above the oil bath. Now start heating the oil by turning the heating rate controldial to about 3. After a moment, you should start to see the temperature on the thermometer rise.At this point you need to keep an eye on both the mp tubes (to watch for any signs of melting) andthe thermometer (to monitor the rate at which the temperature of the oil bath is rising). [It helpsto have chameleon eyes!] You may need to adjust the heating rate control dial so that thetemperature does not rise too rapidly.
Thomas-Hoover
Figure 4.13: Thomas Hoover Apparatus
It is best to heat the oil bath slowly, no fasterthan 1°C per min. However, it is possible toget useable mp’s heating at 2 to 3°C perminute, and to expedite the use of the few mpunits we have, try this faster rate. Watch all thesamples, and when you start to see any one ofthem start to melt, you or your co-workershould record the temperature and the positionof the tube, for example #3 (from right) - 86.5°.If that is one of your partner’s tubes, it wouldbe nice if you could let them watch the tubeswhile you keep tabs on the temperature andrate of heating and then change places againwhen yours starts to melt. When the solid in aparticular tube has completely melted, recordthis temperature also (87-88°C, for example).Once all samples have melted, you shouldhave recorded melting ranges for each. Turnthe heating rate control dial to zero, but do notturn off the power or stirrer, since stirringhelps to cool the oil bath down faster.
a particular tube has completely melted, record this temperature also (87-88°C, for example).Once both samples have melted, you should have recorded melting ranges for each. Turn theheating rate control dial to zero and turn off the power. Cooling of the aluminum block can beexpedited by holding a moist (not sopping wet!) paper towel on it, but be careful not to burn yourfingers.
Proof that an unknown compound is identical to known compound can be obtained by takinga mixed melting point. If two organic solids are thoroughly mixed together, two possibilities areobserved:
1. If they are identical, the mp will be unaffected.2. If they are different, each acts as an impurity in the other, and the mp will be considerably
depressed. For example, you could verify that your unknown is fluorene by grinding it together with an
authentic sample of fluorene using a glass rod on a watch glass. If the melting point is still 114-116°C, your unknown is indeed fluorene; if lower, it must be something else. Occasionally, themelting point range of your purified unknown is still more than a few degrees and in between twopossibilities. If this is the case, ask you TA to provide you with small amounts of pure samplesof each of the possibilities, grind a small amount of your unknown together with each, and takethe mp’s. Your unknown is then identical to the authentic compound that doesn’t effect the mpof your unknown when mixed with it.
Mixed Melting Points If your mp dataisn’t definitive, trythis!

58
Once cooled close to room temperature, cool the flask in an ice bath with occasional swirling.If the crystals have formed a solid mass, break this up with a spatula or boiling stick. Swirl to makesure everything is in suspension and then pour the solid-liquid slurry into the Hirsch funnel. Thewater solution should be rapidly sucked through the filter. Rinse out any crystals remaining inthe flask into the Hirsch funnel with about 1 mL of ice water. Press down on the crystals in theHirsch funnel with a spatula to squeeze out as much water as possible. Using a stainless steelspatula, scrape the crystals and filter paper disc out of the funnel onto a tared beaker or onto a taredwatchglass. Remove the 1.3-cm filter paper disc and discard it. Allow the crystals to dry in yourlocker until the next lab period and then weigh them. For your Final Report, compare thecalculated volume of water with the volume of water actually used to dissolve the acid. Also,calculate the percent recovery of dry, recrystallized phthalic acid.
Collecting thepurified crystals.
The large difference in solubility in water as a function of temperature suggests this as thesolvent of choice. The solubility in alcohol is too high at room temperature. Ether is difficult touse because it has such a low boiling point and boils away rapidly; the compound is insoluble inchloroform (insol. CHCl3).
Crystallize 1.0 g of phthalic acid from the minimum volume of water, using the above data tocalculate the required volume. (This calculation should appear in your Observation and DataSection.) Add the solid to a 25-mL Erlenmeyer flask and then, using a Pasteur pipet, add theamount of water calculated above to the flask from a full 10-mL graduated cylinder. A boilingstick (a wooden applicator stick) facilitates even boiling and will prevent bumping. After a portionof the water has been added, gently heat the solution to boiling on a shallow sand bath or a hot plate.(A hot plate can be shared between 3 or four people.) As soon as boiling begins, continue to addwater in small portions, heating to boiling each time, until all the solid just dissolves. Record thetotal amount of water used to do this.
Place the flask on a book or other insulator and allow it to cool undisturbed to roomtemperature, during which time the crystallization process can be observed. Slow cooling favorslarger and purer crystals. While this is taking place, set up for suction filtration by clamping your25 mL filter flask to a ring stand, and connecting the vacuum hose from the house vacuum outletto the side arm of the filter flask. Place the white plastic Hirsch funnel into the filter flask and coverthe porous disc with a 1.3 cm filter paper disc from the Common Shelf (see Figure 4.4) The filterpaper keeps the porous plastic frit from getting clogged with small particles. Open the vacuumvalve and place the palm of your hand firmly over the top of the Hirsch funnel to see if you canfeel a vacuum. If you don’t, there may be an air leak or the vacuum system isn’t working properly.Consult your TA if you can’t figure out the problem. An alternative filtration method is alsodemonstrated above in Figure 4.5.
Don’t forget tocollect a secondcrop of crystals.
Water Ethanol Other0.5414 11.715 0.6915, Ether
0.7025 - 20.421, MeOH
18.0100 27.478 insol, CHCl3
Table 4.3: Solubility data for phthalic acid (in grams of solute per 100 mL of solvent). The superscripts refer to temperature in °C.
Procedure 4 • Macroscale Recrystallization of Phthalic Acid
The more quantitative aspects of recrystallization will be studied using phthalic acid. Usingthe Beilstein Crossfire as a reference the following solubility data (in grams of solute per 100 mLof solvent) can be summarized. The superscripts refer to temperature in °C:
Quantative aspectsof recrystallization.

59
More phthalic acid crystals (a second crop) can be obtained from the filtrate (mother liquor)by concentrating the solution. Pour the filtrate from the first crystallization above into a 100-mLbeaker and either let the solution evaporate in your desk until the next laboratory period or boilthe solution to at least half the volume. Collect the second crop of crystals by suction filtration,wash the crystals with about 1 mL of ice water, and air dry the crystals on tared watch glass untilthe next lab period. Weigh the watchglass and determine the yield and percent recovery basedon the original sample of phthalic acid.
The technique used for recovering the second crop of crystals is a general approach in syntheticchemistry. Occasionally, you may find that you’ve formed an unsaturated solution that is sodilute, that you have to boil away a lot of solvent before it becomes concentrated enough thatcooling gives a supersaturated solution. You may also find that upon crystallization, there is agreat loss of product; as long as you do not throw out your product, the solvent can always beremoved and repeat the crystallization procedure using less solvent.
Cleaning Up Phthalic acid crystals can be recycled in the specially marked recycle containerson the hooded side shelves. Please don’t put filter paper in the recycling bottle. The watersolutions from the phthalic acid recrystallization can go down the drain. Phthalic acid is notconsidered toxic to the environment. Carry your sandbath to the side shelf and dump the HOTsand back into the sand supply bowl. Do NOT dump it in the waste bins. (Why is this a bad idea?)
Cleaning Up
Into a reaction tube place 1.0 mL of a 0.01% solution of methylene blue dye. Add to the tubea few pieces (8 or 10) of pelletized Norit decolorizing charcoal. Shake, and observe the color overa period of a minute or two. Add a boiling stick, heat the contents of the tube to boiling (reflux)and observe the color by holding the tube in front of a piece of white paper from time to time. Howrapidly is the color removed at room temperature versus heating to higher temperature? Ifthe color is not removed after heating for a minute or so, add more pelletized Norit charcoal, a fewpieces at a time, until the color does disappear.
Cleaning Up Place the Norit in the waste bin (the nonhazardous solid waste container.) Thewater solutions can go down the drain.
Procedure 5 • Removing Colored Impurities Using Activated Carbon
Cleaning Up
Decolorization
Obtaining a secondcrop.
Procedure 6 • Melting Point Determination of your Spectral Unknown
Obtain the melting point of your spectral unknown. Assume that the sample is pure and doesnot need to be recrystallized. You do not necessarily need to do this during the two lab periodsscheduled for this recrystallization/melting point technique experiment. If the melting pointapparati are tied up, you can take your melting point during the distillation technique lab periodsor in the instrument room any time it is open. If you plan to do it outside your regularly scheduledlab time, put your unknown sample tube in a labeled ziplock bag (available on the Common Shelf)and place it by the melting point apparatus in the instrument room or carry it with you.
Use only as much sample as necessary. Remember that you need most of your sample for NMRand IR analysis.
Once you have determined the melting point, look at the Unknown Possibilities by MeltingPoint list located on the Web at:
http://courses.chem.psu.edu/chem36/HTML/Experiments/Unknown_Lists/Unknown.html
In you Final Report, give the name and draw the structures for all compounds that havemelting points within plus or minus 5 °C of your spectral unknown’s melting point.
Spectral Unknownmelting point

60
Summary of In-Lab and Post-Lab Points for Final Report
OBSERVATION /DATA - Accuracy and completeness 8
Solubility test data; Weighing data; Melting point data, Calculated water volume forPhthalic Acid Recrystallization, Decolorizing Ex data and observations
10
RESULTS /DISCUSSION - Overall organization, readability, completeness. 8
Identification of Unknown with explanation 12
Yield and % recovery for unknown 8
Yield and % recovery of phthalic acid (first crop) 8
Yield and % recovery of phthalic acid (second crop) 4
Calculate solubility of phthalic acid in hot water from your data and Table 4.3 8
Discussion of the volume of water used vs calculated 8
Solution decolorization results and discussion 6
Spectral Unknown melting point and possible compounds' names and structures 8
PostLab Questions 12
Grading
Follow the guidelines given below in the Summary for In-Lab and Post-Lab Points to write athorough discussion of recrystallization, solubility, and melting point determination. Include allin-lab observations and data and calculations or answers to questions shown in bold to the aboveexperiments. Answer the PostLab Questions below.
PostLab Questions:1) Under what circumstances might it be necessary to use a mixture of solvents to carry out
crystallization? What characteristics should these two solvents have?2) What is the effect of an insoluble impurity, such as sodium sulfate, on the observed melting
point of a compound?3) Strictly speaking, why is it incorrect to speak of a melting point?
Final Report
A copy of the In-Lab and Post-Lab portions of the Recrystallization/Melting Point GradingSheet is givenbelow
PostLab Questions