a 1,3-dipolar cycloaddition approach to the synthesis...
TRANSCRIPT

A 1,3-DIPOLAR CYCLOADDITION APPROACH TO THE SYNTHESIS OF RESINIFERATOXIN
by
Jennifer A. Loyer-Drew
B.S., Western Washington University, 2003
Submitted to the Graduate Faculty of
Arts and Sciences in partial fulfillment
of the requirements for the degree of
Master of Science
University of Pittsburgh
2008

UNIVERSITY OF PITTSBURGH
ARTS AND SCIENCES
This thesis was presented
by
Jennifer A. Loyer-Drew
It was defended on
July 31, 2008
and approved by
Professor Tara Y. Meyer
Professor Dennis P. Curran
Thesis Advisor: Professor Kay M. Brummond
ii

A 1,3-DIPOLAR CYCLOADDITION APPROACH TO THE SYNTHESIS OF
RESINIFERATOXIN
Jennifer A. Loyer-Drew, M.S.
University of Pittsburgh, 2008
The Rh(I)-catalyzed allenic cyclocarbonylation reaction is a formal [2 + 2 + 1] cycloaddition
process that has been used to gain access to 4-alkylidenecyclopentenones. Incorporation of a
six-membered ring on the tether between the allene and the alkyne components allows access to
a variety of [6-7-5] ring structures featured in the skeletons of various natural products, including
resiniferatoxin. This thesis describes the development of two systems, each with a future
synthesis of resiniferatoxin in mind. First, a model system was designed to demonstrate the
compatibility of the isoxazoline moiety with the Rh(I)-catalyzed cyclocarbonylation reaction.
The second investigation involved the synthesis of an asymmetrically functionalized 2-
cyclohexenone in order to attempt a stereoselective 1,3-dipolar cycloaddition. The first model
system successfully led to the synthesis of the unfunctionalized [6-7-5] core of resiniferatoxin
via cyclocarbonylation of an isoxazoline-containing allene-yne. Unfortunately, under numerous
conditions, the functionalized cyclohexenone synthesized for the second study failed to undergo
1,3-dipolar cycloaddition with a nitrile oxide.
iii

TABLE OF CONTENTS
ABBREVIATIONS ....................................................................................................................... x
1.0 INTRODUCTION................................................................................................................ 1
1.1 RESINIFERATOXIN ................................................................................................. 1
1.2 STRUCTURALLY-RELATED NATURAL PRODUCTS ...................................... 5
2.0 PREVIOUS APPROACHES TO THE SYNTHESIS OF RESINIFERATOXIN AND
RELATED COMPOUNDS .......................................................................................................... 7
3.0 RETROSYNTHETIC ANALYSIS OF RESINIFERATOXIN ..................................... 11
3.1 ACCESSING THE A AND B RINGS VIA A Rh(I)-CATALYZED ALLENIC
CYCLOCARBONYLATION REACTION ..................................................................... 11
3.2 THE MASKED ALDOL STRATEGY: 1,3-DIPOLAR CYCLOADDITION TO
FORM AN ISOXAZOLINE .............................................................................................. 13
4.0 RESULTS AND DISCUSSION ........................................................................................ 18
4.1 INTRODUCTION ..................................................................................................... 18
4.2 MODEL SYSTEM CONTAINING AN UNFUNCTIONALIZED C RING ........ 19
4.3 SYNTHESIS OF AN ASYMMETRICALLY-FUNCTIONALIZED C RING .... 26
4.3.1 Conjugate addition to cyclohexadienone 62 to form enone 60 ................... 27
4.3.2 Elaboration of enone 60 ................................................................................. 32
5.0 CONCLUSION .................................................................................................................. 34
iv

6.0 EXPERIMENTAL ............................................................................................................. 35
6.1 GENERAL ................................................................................................................. 35
6.2 SYNTHESIS AND CHARACTERIZATION ......................................................... 36
APPENDIX A .............................................................................................................................. 49
BIBLIOGRAPHY ....................................................................................................................... 74
v

LIST OF TABLES
Table 1. Magnesium alkoxide direction in 1,3-dipolar cycloaddition (Kanemasa) ..................... 15
Table 2. Hydrogen bond direction in 1,3-dipolar cycloaddition (Choi) ....................................... 15
Table 3. Propargylation of aldehyde 50 ........................................................................................ 24
Table 4. Literature examples of conjugate addition to quinone monoketal 62 ............................. 28
Table 5. Product distribution in copper-catalyzed conjugate addition to cyclohexadienone 62 .. 30
Table 6. Attempted stereoselective cycloaddition of enone 26 .................................................... 33
vi

LIST OF FIGURES
Figure 1. Structures of resiniferatoxin and capsaicin ...................................................................... 1
Figure 2. Phorbol and prostratin .................................................................................................... 4
Figure 3. Daphnane natural products .............................................................................................. 6
Figure 4. Molecular modeling of cyclocarbonylation products .................................................... 17
Figure 5. 1H NMR verification of regioselectivity ...................................................................... 21
Figure 6. Minimum energy conformations and dihedral angles for 59α (left) and 59β (right) ... 26
Figure 7. Chiral phosphoramidite ligands ..................................................................................... 28
vii

LIST OF SCHEMES
Scheme 1. Wender's synthesis of resiniferatoxin ............................................................................ 8
Scheme 2. Phenol p-alkylation to yield spirocycle 11 .................................................................... 9
Scheme 3. Carreira's approach to resiniferatoxin ........................................................................... 9
Scheme 4. Cha's synthesis of phorbol ........................................................................................... 10
Scheme 5. Cyclocarbonylation of a tethered allene-yne ............................................................... 11
Scheme 6. Access of [6-7-5] skeletons via allenic cyclocarbonylation reaction ......................... 12
Scheme 7. Retrosynthetic analysis of resiniferatoxin ................................................................... 13
Scheme 8. Steric direction of nitrile oxide cycloaddition (Martin) .............................................. 16
Scheme 9. Synthesis of model system ......................................................................................... 19
Scheme 10. Dependence of chemical shift on regioselectivity in nitrile oxide cycloaddition
(Grünanger) ................................................................................................................................... 20
Scheme 11. Propargylation of aldehyde 50 .................................................................................. 22
Scheme 12. Regioselectivity in propargylation reactions ............................................................. 23
Scheme 13. Cyclocarbonylation of the model system .................................................................. 25
Scheme 14. Proposed synthesis of functionalized C ring ............................................................. 27
Scheme 15. Synthesis of racemic phosphoramidite ligand 63 ...................................................... 29
Scheme 16. Improved synthesis of phosphoramidite (S,R,R)-64 ................................................. 31
Scheme 17. Synthesis of enone 28 ............................................................................................... 32
viii

Thank you, Erech. Go Team Venture!
ix

x
ABBREVIATIONS
Ac2O Acetic anhydride
9-BBN 9-Borabicyclo[3.3.1]nonane
BINOL 1,1’-Bi(2-naphthol)
DCM Dichloromethane
DIPA Diisopropylamine
DMAP 4-N,N-Dimethylaminopyridine
DMF N,N-Dimethylformamide
DMSO Dimethylsulfoxide
EtOAc Ethyl acetate
NMR Nuclear magnetic resonance
PAA para-Anisaldehyde
PhH Benzene
TBS tert-Butyldimethylsilyl
TEA Triethylamine
THF Tetrahydrofuran
TLC Thin layer chromatography
TMEDA N,N,N’,N’-Tetramethylethylenediamine
TMS Trimethylsilyl

1.0 INTRODUCTION
1.1 RESINIFERATOXIN
The therapeutic benefits of euphorbium—the dried latex of plants of the genus Euphorbia—have
been appreciated for several centuries; in his posthumously published Tractatus de Materia
Medica, 18th century French chemist and physician Etiénne-François Geoffroy cites euphorbium
as an effective remedy for bone cavities and nerve pains.1 Although euphorbium disappeared
from the documented pharmacopoeia in the 1800s, interest in its active constituent,
resiniferatoxin (1, Figure 1), has been renewed, as resiniferatoxin shows potential in treating,
among other ailments, chronic pain and bladder incontinence.
OO
O9
13
41
OOH
H
Ph
20
OO
OH
OMe
1
AB
COMe8
NHOH
O
( )4
2
Figure 1. Structures of resiniferatoxin and capsaicin
Resiniferatoxin was isolated in 1975 from the latex of Euphorbia resinifera and related
plants by Hecker and colleagues.2 The high irritant activity of the compound was immediately
recognized as novel and, seven years later, Hecker published a revised structure of the natural
product along with the results of preliminary structure-activity studies.3 Although the
1

pharmacophore has not been determined, the phenol and orthoester moieties have been deemed
essential for potency.
Resiniferatoxin is a known agonist of the transient receptor potential vanilloid (TRPV1).
Analogous physiological effects and structural similarities to capsaicin (2)—also an irritant that
binds to a TRPV1—suggest a common mode of action for the two compounds. Both natural
products induce pain, neurogenic edema and hypothermia in rats.4
Vanilloid receptors are ligand-gated cation channels located in the membranes of
nociceptive sensory nerves. Activation of the proteins by chemical agonists or heat causes the
channel to open, leading to an influx of intracellular calcium cations; this depolarization
generates an action potential that is perceived by the central nervous system and leads to a
burning sensation. In 1997, Julius and coworkers5 cloned TRPV1 and characterized the protein
as containing six transmembrane domains. The vanilloid binding pocket was later located
through the use of radiolabeled resiniferatoxin, and was shown to exist between domains three
and four.6
Although capsaicin and resiniferatoxin cause initial irritation at the site of application,
tachyphylaxis is demonstrated upon subsequent exposure. This phenomenon is much more
prominant with the use of resiniferatoxin than with capsaicin, although the initial irritation
caused by resiniferatoxin is only marginally more severe. The exact mechanism of
desensitization remains elusive, but summaries of thoughtful speculation can be found in several
reviews.7-9
Not only are nociceptors exposed to resiniferatoxin densensitized to further treatment with
resiniferatoxin, but they often show diminished responses to other stimuli as well, including
capsaicin, heat and other exogenous inflammatory agents. Thus, resiniferatoxin is viewed as a
2

potentially valuable analgesic for those suffering from chronic neurogenic pain and other
phenomena that result from hyperactive nociceptors.
In particular, some subjects exhibiting symptoms of overactive bladder and incontinence
have been reported to have an abnormally high density of nociceptive neurons present in their
bladder tissues. Introduced intravesically, low concentration solutions of resiniferatoxin have
been shown to lead to a decreased frequency in incontinent episodes in some of these patients;
the effects of a single treatment can last up to three months and the initial discomfort is
minimal.10, 11
Current investigation of clinical applications of resiniferatoxin seems to be taking place
predominantly in the academic arena. This may be due, in part, to a 2004 press release issued by
ICOS stating their findings that resiniferatoxin did not pass Phase II trials for treatment of
interstitial cystitis12 and that ICOS’s interest in resiniferatoxin had ceased.13 Since this statement
by ICOS, however, additional contradictory reports14 demonstrating the efficacy of
resiniferatoxin in treating bladder pain syndrome and interstitial cystitis have appeared in the
literature.
Some scientists have expressed reservations with respect to treating humans with
resiniferatoxin based on the similarities between its structure and those of the tumor-promoting
phorbol esters (12,13-diesters of 3, R1 = OH, R2 = H, Figure 2);15 however, these concerns are
not corroborated by experimental evidence. Notably, prostratin (3, R1 = H, R2 = Ac, Figure 2), a
12-deoxytigliane recently synthesized by Wender and coworkers16 from phorbol, was shown to
be a non-tumor-promoting potential anti-HIV therapeutic. Prostratin is currently in preclinical
development.
3

12 13H
R2OR1
OHH
OHO
OH
3phorbol R1 = OH, R2 = H
prostratin R1 = H, R2 = Ac
Figure 2. Phorbol and prostratin
The FDA designated resiniferatoxin as an orphan drug in 2003 for the treatment of
“intractable pain at end-stage disease.”17 This classification seems especially befitting in light of
a 2005 publication from the School of Veterinary Medicine at the University of Pennsylvania.
Administered intrathecally, resiniferatoxin appeared to largely diminish the pain associated with
bone cancer as experienced by a group of canine companion animals.18 When the dogs entered
the study, many were not deemed to be achieving adequate pain relief through the use of
conventional analgesics. After treatment with resiniferatoxin, the comfort level of most of the
dogs seemed to improve so drastically that analgesic use was tapered or in some cases
discontinued completely. No lasting ill effects of resiniferatoxin were observed, pre- or
postmortem.
TRPV1 has become a popular target for development of analgesics; a number of TRPV1
antagonists are in clinical development to address painful phenomena ranging from migraine
headaches to HIV neuropathy-associated pain.19, 20 Capsaicin is available over-the-counter as a
topical ointment for the treatment of arthritis pain. Recent publications regarding the therapeutic
possibilities of resiniferatoxin include a study of its use as a long-lasting local anasthetic21 and a
patent application alleging the efficacy of injections of resiniferatoxin in treating joint pain.22
4

The isolation of resiniferatoxin from euphorbium continues to be a materials-intensive
and fairly noxious process. Fattorusso23 has published an improved procedure for the isolation
of resiniferatoxin, but only managed a 0.0020% yield from the fresh latex of the E. resinifera
plant. One of the altruistic goals of the synthesis of resiniferatoxin is to provide a vehicle for
synthesis and testing of analogs that are more accessible or that perhaps possess more optimal
biological activity than the natural product itself.
1.2 STRUCTURALLY-RELATED NATURAL PRODUCTS
While it is our objective to carry out a synthesis of resiniferatoxin, our expectation is that the
methods that we utilize will find application in the synthesis of structurally-similar molecules.
Many daphnane diterpenoids have been isolated from natural sources; they collectively exhibit a
wide variety of fascinating and potentially useful biological activities.24 Several of these natural
products are depicted in Figure 3 with their corresponding biological activities.25-31 Phorbol (3,
R1 = OH, R2 = H, Figure 2), a tigliane diterpene, is also notable not only for the biological
activity of its 12,13-diesters, but also for the inspirational amount of creative chemistry that has
been developed in pursuit of its synthesis.
5

OO
OH
OH
Gnidimacrin26, 27
anti-tumor agent, PKC activator
O
OHOHOBz
HO
( )4
R
OO
O
OOH
H
OH
Maprouneacin28
exhibits antihyperglycemic activity
O
O
O
O
O
O
( )3R =
Ph
OO
OH
OH
Rediocide A30
potent anti-flea compound
OH
O
OHOH
H
O
O
O
O
Ph
OO
OH
Genkwanine D31
inhibitor of endothelium cell proliferationand cytotoxic towards tumor cells
OHOHOBz
OHOH
OH
OO
OH
OH
Kirkinine B29
possesses neutrophic and antitumor activity
O
OHOH
( )8
O
OO
OH
OH
Huratoxin25
potent piscicidal activity
O
OHOHO
( )7
C
AB
Daphnane skeleton
Figure 3. Daphnane natural products
6

2.0 PREVIOUS APPROACHES TO THE SYNTHESIS OF RESINIFERATOXIN AND
RELATED COMPOUNDS
To date, there has been only one total synthesis of resiniferatoxin, published by Wender in
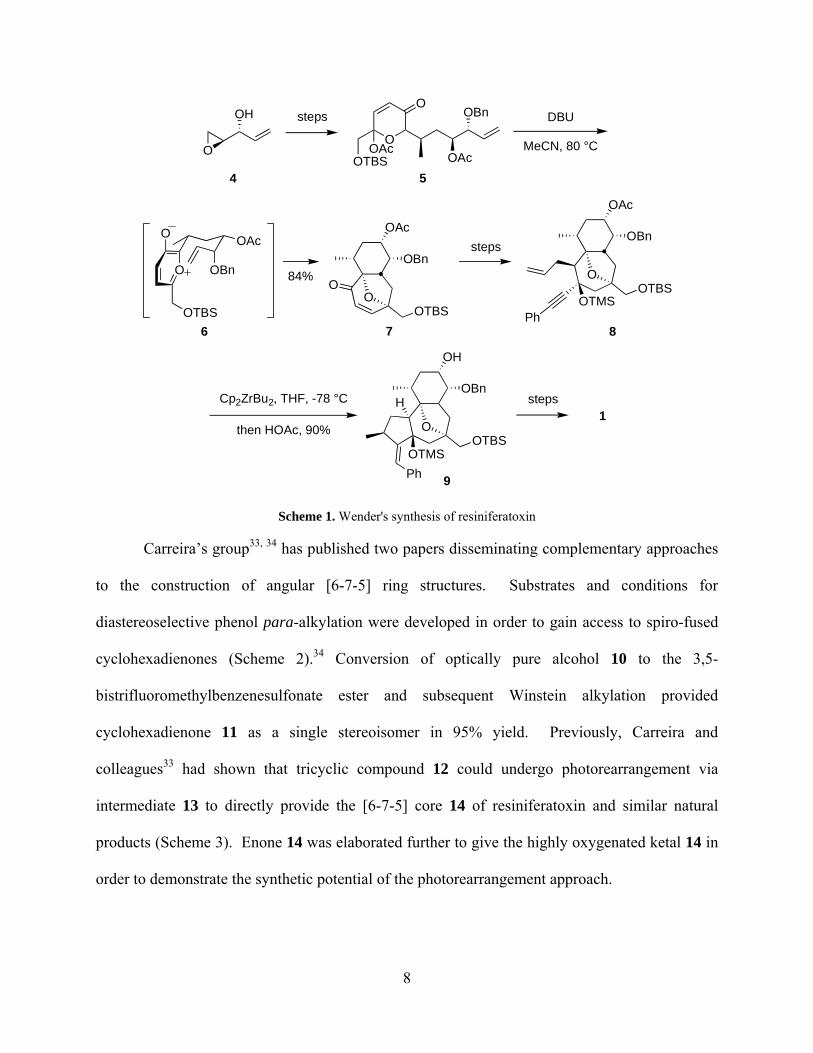
1997.32 The absolute stereochemistry was set in the first step: known epoxide 4 (Scheme 1) was
synthesized from 1,4-pentadien-3-ol using Sharpless’ asymmetric epoxidation conditions.
Subsequent steps proceeded stereoselectively, affording intermediate 5. The key step in
Wender’s synthesis was a [5 + 2] intramolecular cycloaddition of oxidopyrylium 6, forming the
B and C rings simultaneously. An advantage of this tactic was the resulting rigidity of the
tricyclic ring system. The presence of the bridging ether in 7 lent bias to the formation of
subsequent stereocenters. A zirconium-mediated cyclization of enyne 8 closed the A ring of
resiniferatoxin and further elaboration of 9 led to completion of the natural product.
Esterification at C20 and construction of the orthoester were performed towards the end of the
synthesis, since these moieties have been shown to be essential for potent irritant activity.
7
OH
O
OBn
OAcO
O
OAcOTBS
DBU
MeCN, 80 °C
OAc
OBnO
O
OTBS
OAc
OBn
OO
OTBS
84%
6
4 5
steps
7
OAc
OBn
OOTBS
8
OTMSPh
Cp2ZrBu2, THF, -78 °C
then HOAc, 90%
OH
OBn
OOTBS
9
OTMSPh
steps
steps1
H
Scheme 1. Wender's synthesis of resiniferatoxin
Carreira’s group33, 34 has published two papers disseminating complementary approaches
to the construction of angular [6-7-5] ring structures. Substrates and conditions for
diastereoselective phenol para-alkylation were developed in order to gain access to spiro-fused
cyclohexadienones (Scheme 2).34 Conversion of optically pure alcohol 10 to the 3,5-
bistrifluoromethylbenzenesulfonate ester and subsequent Winstein alkylation provided
cyclohexadienone 11 as a single stereoisomer in 95% yield. Previously, Carreira and
colleagues33 had shown that tricyclic compound 12 could undergo photorearrangement via
intermediate 13 to directly provide the [6-7-5] core 14 of resiniferatoxin and similar natural
products (Scheme 3). Enone 14 was elaborated further to give the highly oxygenated ketal 14 in
order to demonstrate the synthetic potential of the photorearrangement approach.
8

OEtBzO
OO
H
PhOH
OBz F3C
CF3
SO2Cl
DMAP, DCM
i,
ii, K2CO3, EtOH/i-PrOH95% OEt
O
O
O PhHO
H
10 11
Scheme 2. Phenol p-alkylation to yield spirocycle 11
OEtO
OMe
HOH
OH
hν
OH
OHH
OEtO
OMe
TFA/pentane
O
EtO
O OHH
HOMe
82%
HH
OMe
OHO
HO
OHMeO OMe
12
14 15
13
Scheme 3. Carreira's approach to resiniferatoxin
The Wender group has applied their [5 + 2] cycloaddition chemistry several times to the
synthesis of phorbol; their most recent synthesis was asymmetric.35-38 Wender39 recently
published preliminary results on the application of this methodology towards the synthesis of
gnidimacrin.
The only other complete synthesis of phorbol to date was published by Lee and Cha40 in
2001. Like Wender, Lee and Cha employed a cycloaddition as a key step. [4 + 3] Cycloaddition
of furan 16 (Scheme 4) and the oxyallyl generated from 1,1,3-trichloroacetone (17), followed by
dechlorination with zinc, afforded meso compound 18 (R = TBS). Desymmeterization of the
corresponding acetate (18, R = Ac) was performed enzymatically to give alcohol 19 in 90% yield
9

and 80% ee. Subsequent steps proceeded diastereoselectively to give substrate 20 which
underwent an intramolecular Heck reaction to close the C ring. Lee and Cha later intersected
with Wender’s synthesis of phorbol.
There have been numerous innovative approaches to the construction of a suitably
functionalized angular [6-7-5] tricyclic core of phorbol; these strategies include: Diels–Alder
cyclization,41-43 an intramolecular 1,3-dipolar cycloaddition,44, 45 a diyl-trapping cycloaddition,46
and an anionic oxy-Cope rearrangement.47 The obvious challenges of constructing the polycyclic
core of resiniferatoxin and related natural products that possess opportunely located functionality
have inspired many chemists in the synthetic community, including the Brummond group.
O
OTBS
OTBS
Cl
Cl
OCl
OR
OR
O O
OAc
OH
O Oi, TEA, TFE
ii, Zn, MeOH+
Candida rugosa
OTBS
O
I
3
16 17 18 19
20
TMSO
Ph
steps steps
Scheme 4. Cha's synthesis of phorbol
10

3.0 RETROSYNTHETIC ANALYSIS OF RESINIFERATOXIN
3.1 ACCESSING THE A AND B RINGS VIA A Rh(I)-CATALYZED ALLENIC
CYCLOCARBONYLATION REACTION
The Brummond group has been preeminent in applying transition metal catalysis to the creation
of a diverse array of polycyclic scaffolds via the Pauson–Khand-type cyclocarbonylation of
allenes.48-55 Former group members have demonstrated the utility of the molybdenum-mediated
and rhodium-catalyzed methodologies in their application to natural product synthesis.48, 50-52 A
unique feature of the rhodium-catalyzed system is its selectivity for reaction with the distal
double bond of the allene;49 in this way, our group has been able to selectively access a variety of
4-alkylidene cyclopentenones by varying the length of the tether between the allene and alkyne
moieties (Scheme 5).
•O
[Rh(CO)2Cl]2, CO
Scheme 5. Cyclocarbonylation of a tethered allene-yne
Incorporation of a six-membered ring on the tether between the allene and alkyne has
allowed access to a variety of angular and linear [6-7-5] tricyclic skeletons present in several
natural products (Scheme 6).48, 50 The angular [6-7-5] substructure present in resiniferatoxin
seems specially suited to showcase our group’s cyclocarbonylation methodology.48 The
11

unsaturation present in the cyclocarbonylation product is conveniently situated at sites requiring
oxidation. Further, the rhodium-catalyzed reaction has demonstrated excellent functional group
compatibility. In the case of resiniferatoxin, this tolerance would permit us to submit an allene-
yne tethered by a functionalized C ring to the cyclocarbonylation reaction, yielding an advanced
synthetic intermediate.
HO
•
R
( )n
( )m
R
O
R
O
R
O
RO
OO
O
OOH
H
Ph
OR
1
OHC
OAc
O
HO HOOH
HOHO OH
H H
H
OH
Guanacastepene A Grayanotoxin III Rippertene
Scheme 6. Access of [6-7-5] skeletons via allenic cyclocarbonylation reaction
We envision a formal synthesis of resiniferatoxin, intersecting Wender’s total synthesis at
intermediate alcohol 21 (Scheme 7). We intend to gain access to this intermediate via functional
group manipulation of 22. A key premise of our proposal is the use of the isoxazoline moiety as
a masked aldol equivalent (see section 3.2); thus, opening of the isoxazoline ring in 23 will yield
β-hydroxy ketone 22. Isoxazoline 23 may be obtained by performing selective oxidations and
12
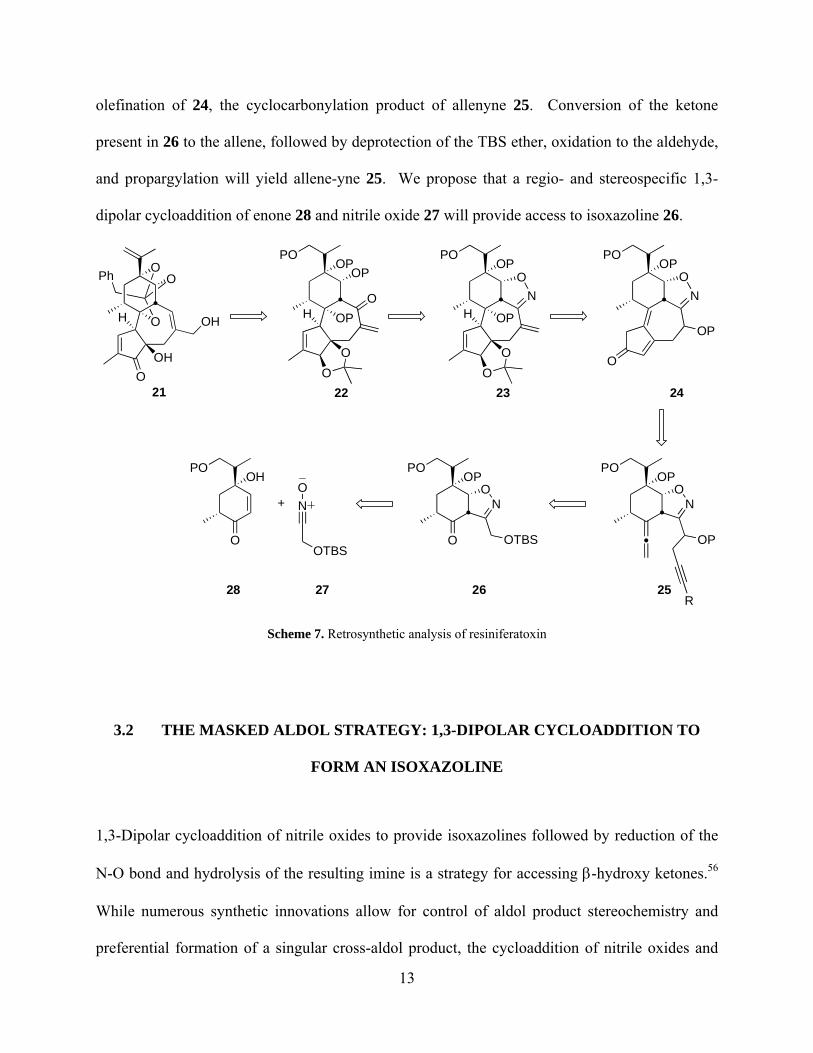
olefination of 24, the cyclocarbonylation product of allenyne 25. Conversion of the ketone
present in 26 to the allene, followed by deprotection of the TBS ether, oxidation to the aldehyde,
and propargylation will yield allene-yne 25. We propose that a regio- and stereospecific 1,3-
dipolar cycloaddition of enone 28 and nitrile oxide 27 will provide access to isoxazoline 26.
OO
O OH
OOH
H
Ph
21
O
OPOP
OO
OPH
OP
OO
OPHN
O
22 23
OP
OP
O
NO
•
NO
OP
R
OP
O
OP
NO
OTBSO
OHON
OTBS
24
+
25262728
PO PO
PO PO PO
PO
Scheme 7. Retrosynthetic analysis of resiniferatoxin
3.2 THE MASKED ALDOL STRATEGY: 1,3-DIPOLAR CYCLOADDITION TO
FORM AN ISOXAZOLINE
1,3-Dipolar cycloaddition of nitrile oxides to provide isoxazolines followed by reduction of the
N-O bond and hydrolysis of the resulting imine is a strategy for accessing β-hydroxy ketones.56
While numerous synthetic innovations allow for control of aldol product stereochemistry and
preferential formation of a singular cross-aldol product, the cycloaddition of nitrile oxides and
13

olefins continues to offer an elegant alternative to the aldol reaction.57-61 The characteristics that
attracted us to the nitrile oxide cycloaddition approach were:
(1) The potential for selectively establishing two stereocenters and latent
functionality in a single step;
(2) The ability of the isoxazoline to act as a protecting group for functionality to be
unmasked late in the synthesis; and
(3) Rigidification of the cyclocarbonylation product to provide a clear facial
preference for subsequent oxidations.
We anticipate that the regioselectivity of the cycloaddition will be influenced by the
presence of the enone. High regioselectivity has been reported for the reaction of nitrile oxides
with cycloalkenones, largely due to steric effects.62 We anticipate that the hydroxyl group may
aid in guiding the nitrile oxide to a syn approach, as well as in reinforcing the regioselectivity;
although, this type of assistance is not well supported in the literature. While hydrogen-bonding
has not been observed to be a strong enough force to consistently direct the 1,3-dipolar
cycloaddition of nitrile oxides,63-65 supplanting the proton of the alcohol with a stronger Lewis
acid has been demonstrated to lead to high regio- and stereoselectivity.66-70
Kanemasa and colleagues70 found that conversion of allylic alcohols to the corresponding
magnesium alkoxide (e.g., 30, X = MgBr, Table 1) and subsequent exposure to benzonitrile
oxide (generated through dehydrohalogenation of hydroximoyl chloride 29) gave the
corresponding isoxazolines in a highly regio- and stereoselective fashion (Table 1, entry 2). It
was noted, however, that the reaction of the magnesium alkoxide generated from 2-cyclohexenol
“led to the formation of a complex mixture of many products.”
14

Table 1. Magnesium alkoxide direction in 1,3-dipolar cycloaddition (Kanemasa)
NOH
ClPh
baseOX
Ph
N O
OHPh
N O
OHPh
N O
OH
+ +
31 32 3329
30
Base X Yield (%) 31 : 32 : 33
TEA H 28 55 : 27 : 18
EtMgBr MgBr 53 94 : 6 : 0
A related result was realized in a study of the directing abilities of 2° amides conducted
by a member of the Curran group. After exposure to conditions that led to satisfactory formation
of the syn-cycloadduct 36 of the corresponding cyclopentenyl amide (35, n = 0, Table 2, entry 1),
unreacted cyclohexenyl amide (35, n = 1) was recovered, even when the reaction was heated to
80 °C for nearly 10 days (Table 2, entries 2 and 3).68 Choi suggested this result was due to the
relatively low reactivity of cyclohexene-derived substrates towards cycloaddition and to the
inability of such substrates to adopt the necessary hydrogen-bonded transition state.
Table 2. Hydrogen bond direction in 1,3-dipolar cycloaddition (Choi)
N Ph
OH
TEA N Ph
OH
ON
t-Bu
+NOH
Clt-Bu
N Ph
OH
ON
t-Bu
N Ph
OH
NO
t-Bu+ +
34 35 36 37 38
( )n ( )n ( )n ( )n
Entry n Time (days) T (°C) Yield (%) 36 : 37 : 38
1 0 4.5 25 92 85 : 1 : 14
2 1 4.5 25 NR NA
3 1 9.5 80 NR NA
15

It is possible that the facial selectivity of the 1,3-dipolar cycloaddition will be controlled
by sterics alone. In studies directed toward the synthesis of breynolide, the Martin group71, 72
observed a single cycloadduct 41 resulting from addition of the nitrile oxide generated from
hydroximoyl chloride 40 anti to a large substituent on a cyclohexenone substrate 39 (Scheme 8).
O
ON
SBnO
H
H
O
SBnO
H
H
+
NOH
ClO O
TEA, Et2O
37%
O
O
39 40 41
Scheme 8. Steric direction of nitrile oxide cycloaddition (Martin)
The presence of the isoxazoline ring will impose considerable conformational constraints
on our synthetic intermediates. Molecular modeling of the two possible diastereomers (resulting
from the propargylation step) of the cyclocarbonylation product using Spartan shows that each
molecule possesses a convex and a concave face (Figure 4). In both cases, the more sterically
accessible convex face is the face on which we hope to perform selective oxidations of the
enone.
16

O
TMSO
NO OMe
O
TMSO
NO OMe
Figure 4. Molecular modeling of cyclocarbonylation products
17

4.0 RESULTS AND DISCUSSION
4.1 INTRODUCTION
Prior to committing to the synthesis of resiniferatoxin, our retrosynthetic plan required that we
address three questions:
(1) Is the isoxazoline moiety tolerated by the reaction conditions we wish to
employ, namely the Rh(I)-catalyzed cyclocarbonylation protocol?
(2) Is selective oxidation of the resulting alkylidene cyclopentenone feasible?
(3) Are we able to carry out a stereoselective [3 + 2] cycloaddition?
In order to satisfy these queries, we embarked on two separate investigations. We utilized a
model system to demonstrate the compatibility of the isoxazoline moiety with the rhodium-
catalyzed cyclocarbonylation reaction (section 4.2). Also, this model system will allow us to
ascertain the utility of the cyclocarbonylation product in subsequent selective oxidation.
Concurrently, an asymmetrically functionalized C ring was prepared to probe the possibility of
obtaining selectivity in the 1,3-dipolar cycloaddition (section 4.3).
18

4.2 MODEL SYSTEM CONTAINING AN UNFUNCTIONALIZED C RING
OHO2N
TBSCl, imidazole
DMFOTBS
O2N
43
OCN
NCO
TEA, PhH, refluxO
NO
OTBS45
NO
OTBS
46
THF, 0 °C
MgBr
HO DCM, 0 °C
NO
OTBS
47
AcO
Ac2O, TEA, DMAP
toluene, H2O
[(Ph3P)CuH]6
NO
OTBS
48
•THF, 0 °C
TBAF NO
OH
49
•TEA, DCM, -65 °C
(COCl)2, DMSO NO
O
50
•
92%
2-cyclohexenone (44)
43%
84% 74% 87%
89% 75%
42
Scheme 9. Synthesis of model system
Under my direction, undergraduate researcher Darla Seifried explored the steps shown in
Scheme 9.73 I have since repeated the reactions several times to investigate the propargylation
and subsequent cyclocarbonylation steps. All yields reported are from my syntheses.
Nitroethanol (42) was protected as the TBS ether 43, which then underwent in situ dehydration
and 1,3-dipolar cycloaddition with 2-cyclohexenone (44) to give bicycle 45 as a single
regioisomer. Initially, phenylisocyanate was used as the dehydrating agent;74 however, the
diphenyl urea byproduct generated during the reaction was extremely difficult to remove from
the desired product via either multiple precipitations (using ethyl acetate, hexanes, ether, or
water) of the urea or column chromatography.73 Kurth and colleagues75 have reported a clever
solution to this frequently observed problem: the use of 1,4-phenylene diisocyanate generates a
19

polyurea that is insoluble in organic solvents (THF, methylene chloride and benzene) and can
therefore be removed by filtration. We found Kurth’s method to be satisfactory, although a
single filtration is rarely sufficient to remove the significant quantities of polymeric urea present
in the reaction mixture.
The regioselectivity of the cycloaddition was confirmed by 1H NMR. In isoxazoline 45,
H5 gives a doublet of triplets resonance at δ 4.95 ppm and H4 appears as a doublet at δ 3.86 ppm
(Figure 5). This is consistent with the findings of Grünanger and colleagues62 in their studies of
the relationship between regioisomers and chemical shift in similar systems. In the addition of
benzonitrile oxide to 2-cyclohexenone (44), the multiplet resonance for H5 appears significantly
downfield from the H4 doublet in the major product 51 (Scheme 10). The ΔδH4,H5 is much
smaller for the regioisomer 52, and the more deshielded proton—H5—is alpha to the ketone and
therefore only split by H4 (i.e., appears as a doublet). Grünanger observed only cis-addition to
cycloalkenones, the products exhibiting a characteristically large 3JH4,H5 (8.8 to 12.0 Hz). The 3J
coupling (9.7 Hz) exhibited by H4 and H5 in isoxazoline 45 falls neatly within this range.76, 77
45 N
O
Ph
H
H
O
OPh
NOH
Cl
TEA
Et2O, rt, 12 h85%
3 : 1 (51 : 52)
+ +
2944 51H4: δ 4.23 (d)H5: δ 5.12 (m)
52H4: δ 4.20 (m)H5: δ 4.68 (d)
45 N
O
Ph
H
HO
Scheme 10. Dependence of chemical shift on regioselectivity in nitrile oxide cycloaddition (Grünanger)
20

45 N
OH
HO
45OTBS
H4
H5
Figure 5. 1H NMR verification of regioselectivity
Addition of ethynylmagnesium bromide to the ketone 45, followed by acylation of the
tertiary alcohol 46, gave propargyl acetate 47 as a single diastereomer. As has been experienced
by other group members,48, 51 Stryker’s reagent ([(Ph3P)CuH]6) was suitable for the reduction of
the propargyl acetate 47 to the corresponding 1,1-disubstituted allene 48. Initial yields obtained
for the reduction of the propargyl acetate 47 using commercial samples of Stryker’s reagent were
around 45%. Others78-80 have remarked on the variable (and generally inferior) quality of
commercial [(Ph3P)CuH]681 as compared to Stryker’s reagent that has been prepared in the
laboratory. Consequently, the copper hydride species was prepared according to the literature82
and, using the fresh reagent, the yield of allene 48 nearly doubled to 87%. Deprotection of the
TBS ether 48 followed by Swern oxidation83 of the primary alcohol 49 gave aldehyde 50.
21

The addition of nucleophiles to 3-formyl-Δ2-isoxazolines is precedented,84 although
propargylation has not been reported. A zinc Barbier reaction of the aldehyde 50 and propargyl
bromide in aqueous ammonium chloride produced the desired homopropargyl alcohol 53 in
variable yield as a 3:1 mixture of diastereomers (Scheme 11). However, subjecting this substrate
to the [Rh(CO)2Cl]2 cyclocarbonylation conditions led mostly to decomposition. Other group
members have observed similar behavior in the case of terminal alkynes.48 Attempts at protecting
the terminal alkyne 53 with a trimethylsilyl group proved unsuccessful and efforts to force the
formation of 54 by exposure of the protected alcohol to excess base led primarily to
decomposition.
NO
O•
Br
Zn, sat NH4Cl, THF
NO
OH•54-70%
533:1 dr
NO
OR•
TMS5450
Scheme 11. Propargylation of aldehyde 50
In reexamining the propargylation step, it seemed necessary to install the propargyl group
and to protect the terminal alkyne in a single step. There are relatively few methods of
accomplishing this. Most propargylation conditions rely upon the favorable equilibrium of the
propargylic and allenic organometallic species, 56 and 57, respectively (Scheme 12), the latter
leading to the α-addition product 58 via an SE2’ mechanism.85 Sterically demanding groups on
the terminus of the alkyne (e.g., R1 = TMS) may shift this equilibrium to favor the propargyl
organometallic species 56, giving the allenic alcohol 55 preferentially. The product distribution
has, however, been found to be highly dependent on the metal used, the nature of the substituent
on the alkyne (R1), the electrophile, and reaction solvent.
22

R1M
•R1
M
α β
γ R2CHO
R2
OH R1
R2
OH•
R1
R2CHO
55 56 57 58
α βγ
Scheme 12. Regioselectivity in propargylation reactions
Paquette and Daniels86 have published an often-cited study that exemplifies the
capricious behavior of propargylation reactions. Treatment of aldehydes and ketones with
trimethylsilylpropargylzinc bromide in THF led to selective formation of the homopropargylic
alcohol, while treatment of the same substrates with the analogous aluminum reagent yielded
allenyl alcohols preferentially. Additionally, increasing the coordinating ability of the solvent
was highly influential in the product distribution of protonolysis of the aluminum reagent; in
diethyl ether, 1-trimethylsilylpropyne was obtained after quenching the organometallic species,
while in THF or diglyme the regioselectivity was reversed.
Unfortunately, Paquette and Daniels’87 zinc amalgam protocol failed to produce the
desired homopropargyl alcohol 54 (Table 3, entry 1). The majority of the starting material 50
was recovered. Propargylation of aldehyde 50 was attempted using conditions developed by Loh
and Lin,88 who have shown that simple aldehydes undergo regioselective propargylation with
trialkylsilyl propargyl bromides in the presence of indium and catalytic indium trifluoride. Lin
and Loh attribute the high selectivity for the α-alkylation product to the probable coordination of
the halogen and the silicon in the allenic organometallic species 57. This protocol proved
ineffective in the propargylation of our aldehyde 50, however (entry 2). The majority of the
starting material was recovered.
23

Table 3. Propargylation of aldehyde 50
NO
O•
Br
conditions
NO
OR2•
5450TMS
R1
Entry R1 Conditions R2 Yield of 54 (%)
1 TMS Zn, HgCl2, cat I2 NA NR
2 TMS In, InF3, THF, reflux NA NR
3 H 2 equiv n-BuLi, TMEDA, then TMSCl TMS 21 (2:1 dr)
Cabezas89, 90 has also presented an approach to the problem of regioselective addition, in
which 1,3-dilithiopropyne is prepared from propargyl bromide and added to the aldehyde.
Quenching of the reaction with TMSCl protects both the newly formed alcohol and the alkyne
terminus. Employment of this method gave the desired bis-protected homopropargyl alcohol 54
(R2 = TMS), albeit in 21% yield as a 2:1 mixture of diastereomers by 1H NMR (Table 3, entry
3).
Gratifyingly, when subjected to [Rh(CO)2Cl]2 in the presence of CO, allene-yne 54
smoothly underwent cyclocarbonylation to give the desired product 59 in 91% yield after 3 hours
(Scheme 13). Following cyclocarbonylation, the two diastereomers (originating in the
propargylation step) were easily separated by column chromatography, yielding 10 mg (73%) of
the major diastereomer, and 3 mg (18%) of the minor isomer.
24

NO
OTMS
54
•
TMS
OTMS
O
NO
30 mol% [Rh(CO)2Cl]2
CO, toluene, 60 °C91%
TMS59
Scheme 13. Cyclocarbonylation of the model system
There is a minor discrepancy between the isolated yields of the diastereomers of 59 and
the previously observed 2:1 dr observed in the 1H NMR of the homopropargyl silyl ether 54.
Were the remaining 9% of the material not isolated as product comprised solely of the minor
diastereomer of 54, the diastereomeric ratio observed in the cyclocarbonylation reaction
(potentially 73:27) still would not be equivalent to 2:1. This lack of agreement may be attributed
to two things. First, the 2:1 dr is based on the integration of methine protons that are not fully
resolved in the 1H NMR spectrum (see Appendix A). Second, the margin of error in weighing
milligram quantities is presumably large enough to account for the observed discrepancy.
In an effort to gain insight into the relative stereochemistry of the cyclocarbonylation
products, the two epimers of 59 were modeled using CAChe. The global minimum
conformation for each isomer was found using CONFLEX/MM3 parameters (Figure 6).91 The
minimum energy conformer for the epimer with the α-TMS ether (59α) exhibited dihedral
angles of 70.2 and 44.3° between Ha and Hb, and Ha and Hc, respectively. The measured dihedral
angles for 59β are 176.2 and 60.9°. Based on these calculations, one would expect to observe a
doublet of doublets with two medium-to-small 3J couplings for the resonance of Ha in 59α. In
contrast, the calculated dihedral angles for 59β predict a doublet of doublets with one large and
one small vicinal coupling. Indeed, the resonance for Ha in the 1H NMR spectrum of the major
diastereomer of 59 is a triplet at δ 5.01 ppm with J = 3.6 Hz (see Appendix A). The
25

corresponding signal in the 1H NMR spectrum of the minor diastereomer is a doublet of doublets
at δ 4.76 ppm with J = 10.8 and 5.4 Hz. Therefore, the major and minor diastereomers can
tentatively be assigned as 59α and 59β, respectively.
O
NO
TMS59α
OTMSHa
HcHb O
NO
TMS59β
Ha
OTMS
HcHb
Ha-C-C-Hb ∠ 70.2°Ha-C-C-Hc ∠ 44.3°
observed for Ha: δ 5.01 (t, J = 3.6 Hz)
Ha-C-C-Hb ∠ 176.2°Ha-C-C-Hc ∠ 60.9°
observed for Ha: δ 4.75 (dd, J = 10.8, 5.4 Hz)
Figure 6. Minimum energy conformations and dihedral angles for 59α (left) and 59β (right)
4.3 SYNTHESIS OF AN ASYMMETRICALLY-FUNCTIONALIZED C RING
Our strategy for the synthesis of a functionalized C ring was to add isopropenyl Grignard to
known enone 60 (Scheme 14), hydrate the exocyclic olefin of 62 and cleave the ketal to access
functionalized enone 28.
26

O
MeO OMe MeO OMe
OH OHPO
O60 61 28
Scheme 14. Proposed synthesis of functionalized C ring
4.3.1 Conjugate addition to cyclohexadienone 62 to form enone 60
There are only a few literature examples of conjugate addition of a methyl group to quinone
monoketal 62. Addition of lithium dimethylcopper to quinone monoketal 6292 is known to lead
to reductive aromatization rather than conjugate addition (Table 4, entry 1).93 Complexation of
quinone monoketals with an organoaluminum reagent followed by addition of either an
alkyllithium or Grignard reagent has been shown to yield the corresponding 1,4-addition
products in many cases; however, in the case of the addition of methyllithium, only a 24% yield
of the desired product 60 was achieved (entry 2).94 Fortunately, Feringa and colleagues95 have
developed an enantioselective copper-catalyzed conjugate addition protocol that was reported to
give the desired enone 60 in 76% yield and 99% ee from quinone monoketal 62 (entry 3).
27

Table 4. Literature examples of conjugate addition to quinone monoketal 62
O
MeO OMe62
conditionsO
MeO OMe60
Entry Conditions Yield (%) Ref.
1 LiMe2Cu —a 93
2 MAD,b MeLi 24 94
3 Cu(OTf)2, (S,R,R)-64, Me2Zn 76 (99% ee) 95 ap-Methoxyphenol obtained exclusively; bMAD =
methylaluminum bis(2,6)-di-tert-butyl-4-methylphenoxide)
The enantioselectivity of Feringa’s protocol arises from the use of chiral phosphoramidite
ligand 64 (Figure 7). The Feringa group has published the use of a variety of phosphoramidite
ligands synthesized from BINOL, PCl3, and 2° amines.96 Because the price of the amines varies
widely, we sought to synthesize an effective phosphoramidite ligand while minimizing cost. We
were encouraged to see that both ligand (S)-63 and (S,R,R)-64 were effective in the copper-
catalyzed addition of diethylzinc to 2-cyclohexenone, each giving 3-ethylcyclohexanone in
>75% yield and 83% ee and >98% ee, respectively.96 Initially less concerned with the
enantiopurity of the enone than with obtaining the desired material, we opted to synthesize
racemic phosphoramidite 63 from racemic BINOL (rac-65), PCl3, and diisopropylamine
(Scheme 15).
OP
ON
OP
ON
Ph
Ph
(S,R,R)-64(S)-63
Figure 7. Chiral phosphoramidite ligands
28

OHOH
rac-65
PCl3, TEA
toluene, -60 °C OP
OCl
rac-66
DIPA, TEA
toluene, -40 °C → rt19% O
PO
N
rac-63
Scheme 15. Synthesis of racemic phosphoramidite ligand 63
Unfortunately, exposure of quinone monoketal 62 to Cu(OTf)2, ligand rac-63, and Me2Zn
in toluene at –25 °C failed to produce the desired product 60 in a reasonable yield (Table 5,
entries 1 and 2). The reaction did not go to completion, even when the mixture was allowed to
stir at -25 °C for several days. The lack of desired reactivity may have been due, at least in part,
to the poor solubility of the catalyst complex in toluene at the reaction temperature. Upon
cooling the premixed solution of Cu(OTf)2 and ligand from room temperature to -25 °C,
significant precipitation was observed.
Enantiomerically pure phosphoramidite (S,R,R)-64 was synthesized in 26% yield in a
fashion analogous to the previous phosphoramidite synthesis.96 Cu(OTf)2 was premixed with the
phosphoramidite ligand and was then cooled to -25 °C; the quinone monoketal was added
followed by a solution of Me2Zn. Within approximately six hours, all cyclohexadienone 62
starting material was consumed (this is in contrast to the 16 hours the reaction is reputed95 to
require). Surprisingly, a significant amount of the diaddition product 67 was observed in the
product mixture (Table 5, entry 3). Protonation of the Zn-enolate intermediate prior to
quenching of the catalyst is requisite in order for a second 1,4-addition to occur. TLC indicated
a dramatic difference in the relative amounts of mono- and diaddition products (60 and 67,
respectively) before and after quenching. The final ratio of mono- to diaddition products has
been found to be somewhat contingent on the method of quenching. This trend is summarized in
Table 5 (entries 3-5), although it is important to note that exact ratios were not reproducible.
29

Following the published procedure,95 the addition of saturated aqueous NH4Cl to the reaction
mixture led to greater relative amounts of the diaddition product 67 in the crude 1H NMR than
did a basic quench (entries 3 and 4), which was prescribed by Rosalinde Imbos in her Ph.D.
thesis.97 The ratio of mono- to diaddition product (60 : 67) was further improved by performing a
reverse basic quench (entry 5).
Table 5. Product distribution in copper-catalyzed conjugate addition to cyclohexadienone 62
O
MeO OMe
2.5 mol% Cu(OTf)25 mol% ligand
toluene, -25 °C
62
O
MeO OMe60
O
MeO OMe67
*+ + unreacted
62
Entry Ligand Equiv Me2Zn
Time (h) Quench 60 : 67 : 62a Yield of 60
(%)
1 rac-63 1.6 16 direct, sat NH4Cl — 17
2 rac-63 1.6 + 1b 48 direct, sat NH4Cl — 20
3 (S,R,R)-64 1.6 6 direct, sat NH4Cl 18 : 82 : 0 —c
4 (S,R,R)-64 1.6 16 direct, 1 M NaOH 60 : 40 : 0 33
5 (S,R,R)-64 1.6 6 reverse, 1 M NaOH 71 : 29 : 0 —c
6 (S,R,R)-64 1.6 4d reverse, 1.5 M NaOH 75 : 0 : 25 50
7 (S,R,R)-64e 1.6 16 direct, 1 M NaOH 100 : 0 : 0 21f adetermined by integration of crude 1H NMR; badditional equivalent Me2Zn added after 45 h; cyield not determined; dreaction followed closely by TLC and quenched at first faint sign of diaddition product; eligand of high purity; flow yield due to volatility of product
The presence of the diaddition product proved to be problematic since separation of the
mono- and diaddition products by column chromatography was not possible. Two methods of
minimizing the formation of the diaddition product were discovered. The first was to monitor
the reaction closely and quench the reaction as soon as there was TLC evidence of diaddition
product forming (Table 5, entry 6). An early quench of the reaction allowed for the isolation of
an easily separable 3:1 mixture (by 1H NMR) of desired product 60 to unreacted starting
30

material. (Fortuitously, ketone 67 stains very darkly with PAA and although the diaddition
product was evident by TLC, it often was only faintly—if at all—visible in the crude 1H NMR.)
The use of phosphoramidite ligand of a higher purity also suppressed the formation of the
unwanted diaddition product.98 Unsatisfied with the previously low-yielding synthesis of
phosphoramidite 64, we developed a revised synthesis based on methods reported in Ate
Duursma’s Ph.D. thesis.99 (S)-BINOL (65) was refluxed in PCl3 (Scheme 16). Removal of the
excess PCl3 under vacuum furnished phosphochloridite 66, which was dissolved in toluene and
added to a solution of deprotonated amine (R,R)-68. Not only did this method drastically
improve the isolated yield of the ligand 64 (77% versus 26%), but also the ligand was
significantly more pure by 1H NMR.100 Use of this ligand in the copper-catalyzed conjugate
addition led only to the desired monoaddition product (Table 5, entry 7). Unfortunately,
repetition of the improved ligand synthesis procedure led to ligand of inferior purity that, in turn,
led to significant quantities of diaddition product 67 produced in the copper-catalyzed 1,4-
addition reaction.
OHOH
(S)-65
reflux OP
OCl
(S)-66
toluene/THF, -40 °C → rt77% O
PO
N
(S,R,R)-64
Ph
Ph
PCl3
NPh
PhLi
(R,R)-68
Scheme 16. Improved synthesis of phosphoramidite (S,R,R)-64
31

4.3.2 Elaboration of enone 60
Isopropenylmagnesium bromide was added to the ketone 60, affording the tertiary alcohol 61 in
74% yield (Scheme 17). Hydroboration of the 1,1-disubstituted olefin with BH3·DMS followed
by oxidation101 gave the corresponding primary alcohol as a mixture of diastereomers. Attempts
to improve the diastereoselectivity by using bulky 9-BBN led to no appreciable hydroboration, as
observed by the 1H NMR spectra of aliquots of the reaction mixture. The mixture of
diastereomers was taken on crude due to the lability of the ketal on silica gel, even when the
column was pretreated with TEA. The primary alcohol was protected as the TBS ether102 prior
to cleavage of the ketal by Montmorillonite K10 clay,103 giving the enone 28 in 21% yield from
61 as a 2:1 mixture of diastereomers by 1H NMR.
O
MeO OMe60
*
MgBr
THFMeO OMe
61
OH 1. BH3·DMS, then H2O, NaOH, H2O2
28
OH
2. TBSCl, DMAP, TEA3. Montmorillonite-K10
TBSO
O21%, 3 steps74%
Scheme 17. Synthesis of enone 28
A variety of 1,3-dipolar cycloaddition conditions were surveyed, all of which were
designed to generate the nitrile oxide very gradually to minimize homodimerization of the nitrile
oxide.56 Unfortunately, enone 28 failed to undergo 1,3-dipolar cycloaddition and, in all cases,
most of the starting material was recovered (Table 6). A very small amount of a mixture of
unknown products was isolated from the reaction of enone 28 with nitroalkane 43 when Kurth’s
diisocyanate methodology75 was used (entry 6). Since selectivity was not exhibited,
investigation was discontinued.
32

Table 6. Attempted stereoselective cycloaddition of enone 26
OHTBSO
O
OHTBSO
O
NO
R28 26
Ph Cl
NOH
NO2TBSO
29 43
or
conditions
Entry 29 or 43 Conditions Scale (mg of 28)
Yield of 26 (%)
1 29 EtMgBr, DCM70 10 —
2 29 TEA, Et2O71 10 —
3 29 KHCO3, DME104 10 —
4 29 KHCO3, DME, Δ 10 —
5 29 4 Å mol sieves, DCM105 21 —
6 43 1,4-phenylene diisocyanate, TEA, PhH, reflux75 21 —a aMost of starting material was unreacted; however, a small amount of a mixture of unknown products was obtained.
33

5.0 CONCLUSION
Our proposed plan for the synthesis of resiniferatoxin required that we addressed several
uncertainties, namely whether an isoxazoline moiety would be tolerated by the Rh(I)-catalyzed
cyclocarbonylation reaction, whether selective oxidations of the resulting alkylidene
cyclopentenone could be carried out, and whether a stereoselective 1,3-dipolar cycloaddition was
feasible. We synthesized a simple isoxazoline-containing model system, which underwent
Rh(I)-catalyzed allenic cyclocarbonylation to give the unfunctionalized angular [6-7-5] core of
resiniferatoxin in 91% yield. The cyclopentenone scaffold produced by the cyclocarbonylation
reaction was an appropriate substrate for probing conditions for selective olefin oxidation,
although selective oxidation was not pursued due to the disappointing results obtained in the area
of stereoselective 1,3-dipolar cycloaddition. An asymmetrically functionalized enone,
corresponding to the C ring of resiniferatoxin, was synthesized utilizing Feringa’s Cu-catalyzed
asymmetric conjugate addition protocol. Unfortunately, the functionalized 2-cyclohexenone
failed to undergo stereoselective 1,3-dipolar cycloaddition under numerous conditions tested.
34

6.0 EXPERIMENTAL
6.1 GENERAL
Unless otherwise specified, all nonaqueous reactions were performed in flame- or oven-dried
glassware under N2 atmosphere using the appropriate syringe, cannula, and septum techniques.
All solvents and reagents were purchased commercially and used as received unless otherwise
noted. Toluene, acetonitrile (MeCN), triethylamine (TEA), N,N,N’,N’-
tetramethylethylendiamine (TMEDA), and chlorotrimethylsilane (TMSCl) were distilled from
CaH2 prior to use. Dimethyl sulfoxide (DMSO) was distilled from CaH2 and stored over
activated 4 Å mol sieves. Tetrahydrofuran (THF) and diethyl ether (Et2O) were dried and
deoxygenated by sequential passage through activated alumina and Q5 columns in a Sol-Tek ST-
002 solvent purification system; dichloromethane (DCM) was passed through an activated
alumina column in the same system. 2-Cyclohexenone was distilled (47 °C at 9 mm Hg) and
stored at -20 °C. Acetic anhydride (Ac2O) was distilled (30 °C at 20 mm Hg) from P2O5 and
stored in a desiccator. Cu(OTf)2 and [(Ph3P)CuH]6 were stored in a glove box and dispensed
using standard glove box techniques. All solvents used in the preparation and reactions of air-
sensitive reagents were degassed by bubbling N2 for 20-30 min before use.
Column chromatography was performed using silica gel (32-63 μm particle size, 60 Å
pore size) purchased from Scientific Adsorbents, Inc. following the guidelines described in the
35

seminal publication on flash chromatography by Still and colleagues.106 Ethyl acetate (EtOAc)
and hexanes used for chromatography were distilled prior to use. TLC was performed using
silica gel plates (60 F254, 250 μm thickness).
1H and 13C NMR spectra were obtained on Varian 300 MHz instruments. All chemical
shifts (δ) are reported in ppm. 1H NMR spectra were calibrated to the residual CHCl3 peak at δ
7.27; 13C NMR spectra were referenced to the CDCl3 resonance at δ 77.0. The following
abbreviations are used to denote the indicated splitting pattern 1H NMR spectra: s = singlet, d =
doublet, t = triplet, q = quartet; abbreviations are used in combination to indicate more complex
splitting (e.g., dtd = doublet of triplets of doublets). Infrared spectra were obtained on a Nicolet
Avatar E. S. P. 360 FT-IR.
6.2 SYNTHESIS AND CHARACTERIZATION
OTBSO2N
43
OCN
NCO
TEA, PhH, refluxO
NO
OTBS45
2-cyclohexenone (44)
3-((tert-Butyldimethylsilyloxy)methyl)-5,6,7,7a-tetrahydrobenzo[d]isoxazol-4(3aH)-one
(45). A mixture of cyclohexenone (44) (1.80 mL, 18.59 mmol, 1.0 equiv), nitroalkane 43 (3.75
g, 18.28 mmol, 1.0 equiv), 1,4-phenylenediisocyanate (9.08 g, 56.69 mmol, 3.1 equiv) and TEA
(0.50 mL, 3.59 mmol, 0.2 equiv) in benzene (180 mL) was refluxed. After approximately 16 h,
the turbid yellow mixture was cooled to rt; water (9 mL) was added and the mixture stirred 1 h.
The polymeric urea was removed via vacuum filtration through a pad of celite; the filtrate was
36

dried (MgSO4) and concentrated under reduced pressure. Benzene was added and the filtration
step was repeated. The filtrate was concentrated onto silica gel, which was dry-loaded onto a
silica gel column for purification. Gradient elution with EtOAc/hexanes (1:9 to 1:6) yielded
isoxazoline 45 (2.212 g, 43%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 4.95 (dt, J =
9.7, 4.5 Hz, 1H), 4.56 (A of ABq, J = 12.5 Hz, 1H), 4.44 (B of ABq, J = 12.5 Hz, 1H), 3.86 (d, J
= 9.7 Hz, 1H), 2.54-2.45 (m, 1H), 2.37-2.26 (m, 1H), 2.16-1.79 (m, 4H), 0.89 (s, 9H), 0.10 (s,
3H), 0.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 205.3, 157.4, 81.5, 58.7, 58.5, 39.9, 26.4, 25.8,
18.4, 18.3, -5.5, -5.6; IR (neat, NaCl): 2930, 2857, 1713, 1088, 837 cm-1; MS m/z (%): 283 (9),
268 (73), 226 (90), 74 (100); EI-HRMS calcd for C14H25NO3Si [M]+ m/z: 283.1604, found:
283.1610.
NO
OTBS
46
THF, 0 °C
MgBr
HO84%
O
NO
OTBS
45
3-((tert-Butyldimethylsilyloxy)methyl)-4-ethynyl-3a,4,5,6,7,7a-hexahydrobenzo[d]isoxazol-
4-ol (46). To a solution of the ketone 45 (0.511 g, 1.802 mmol, 1 equiv) in THF (18 mL) at 0 °C
was added ethynylmagnesium bromide (14 mL of a 0.5 M soln in THF, 7 mmol, 4 equiv) over
25 min via syringe pump. The mixture stirred at 0 °C and reaction progress was monitored by
TLC. Complete consumption of the starting material was observed ~30 min after the addition
was finished; the reaction was quenched by pouring the mixture into a flask containing sat
NH4Cl solution. The aqueous layer was extracted with Et2O (4×) and the combined organic
layers were washed with brine, dried (MgSO4), filtered, and concentrated under reduced
pressure. The residue was purified via column chromatography, using EtOAc/hexanes (1:6) as
the eluent, to give pure propargyl alcohol 46 (0.470 g, 84%) as a colorless oil. 1H NMR (300
37

MHz, CDCl3): δ 4.67-4.61 (m, 1H), 4.62 (A of ABq, J = 11.7 Hz, 1H), 4.45 (B of ABq, J = 11.7
Hz, 1H), 3.53 (d, J = 9.0 Hz, 1H), 2.48 (s, 1H), 1.97-1.71 (m, 4H), 1.61-1.50 (m, 1H), 0.93 (s,
9H), 0.16 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 157.6, 87.2, 80.2, 71.6, 65.4, 58.6, 57.9, 36.6,
25.8, 25.0, 18.2, 16.1, -5.5; IR (neat, NaCl): 3416, 3310, 2930, 2090, 1257, 1081, 838 cm-1; MS
m/z (%): 309 (9), 294 (69), 252 (82), 105 (74), 74 (100); EI-HRMS calcd for C16H27NO3Si [M]+
m/z: 309.1760, found: 309.1762.
NO
OTBS
46
HO DCM, 0 °C
NO
OTBS
47
AcO
Ac2O, TEA, DMAP
74%
3-((tert-Butyldimethylsilyloxy)methyl)-4-ethynyl-3a,4,5,6,7,7a-hexahydrobenzo[d]-isoxazol-
4-yl acetate (47). To a soln of the propargyl alcohol 46 (0.560 g, 1.81 mmol, 1 equiv) and
DMAP (0.230 g, 1.88 mmol, 1 equiv) in DCM (18 mL) at 0 °C were added Ac2O (1.7 mL, 18.00
mmol, 10 equiv) and TEA (2.5 mL, 17.94 mmol, 10 equiv). The mixture stirred at 0 °C and
reaction progress was monitored by TLC. After approximately 12 h, the reaction was quenched
by addition of saturated NaHCO3 soln and the aqueous layer was extracted with DCM (3×). The
combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated
under reduced pressure. The residue was purified by column chromatography, eluting with
EtOAc/hexanes (1:6), to give pure propargyl acetate 47 (0.504 g, 74%) as a colorless oil. 1H
NMR (300 MHz, CDCl3): δ 4.58 (A of ABq, J = 12.7 Hz, 1H), 4.52 (B of ABq, J = 12.7 Hz,
1H), 4.54-4.50 (m, 1H), 3.58 (d, J = 8.4 Hz, 1H), 2.72-2.66 (m, 1H), 2.68 (s, 1H), 2.12-2.04 (m,
1H), 2.03 (s, 3H), 1.87-1.57 (m, 5H), 0.91 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H); 13C NMR (75 MHz,
CDCl3): δ 168.7, 159.8, 82.8, 79.0, 74.9, 70.8, 58.7, 54.3, 32.4, 25.8, 24.2, 21.8, 18.2, 15.3, -5.2,
-5.4; IR (neat, NaCl): 3250, 2931, 2100, 1748, 1221, 1076, 838 cm-1; MS m/z (%) 294 (85), 234
38

(100), 156 (20), 117 (41), 105 (24); EI-HRMS calcd for C17H26NO4Si [M]+ m/z: 336.1631,
found: 336.1631.
NO
OTBS
47
AcO toluene, H2O
[(Ph3P)CuH]6
87%
NO
OTBS
48
•
3-((tert-Butyldimethylsilyloxy)methyl)-4-vinylidene-3a,4,5,6,7,7a-
hexahydrobenzo[d]isoxazole (48). To a dark red suspension of [(Ph3P)CuH]682 (1.68 g, 0.86
mmol, 0.50 equiv) in toluene (8.5 mL) was added a mixture of the propargyl acetate 47 (0.607 g,
1.727 mmol, 1 equiv), toluene (17 mL) and water (0.08 mL, 4.44 mmol, 2.57 equiv) via cannula
over 20 min. The mixture gradually turned brown (i.e., appeared quenched) approximately 1 h
after the addition. The septum was removed from the flask and the contents stirred open to the
air for 20 min before being diluted with Et2O and filtered through a plug of silica gel. After
concentration under reduced pressure, the filtrate was purified by column chromatography using
gradient elution with hexanes/EtOAc (19:1, 9:1). A second purification using 19:1
hexanes/EtOAc was required to remove remaining PPh3. Allene 48 (0.305 g, 87%) was obtained
as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 4.72-4.60 (m, 3H), 4.46 (A of ABq, J = 13.2
Hz, 1H), 4.42 (B of ABq, J = 13.2 Hz, 1H), 3.86 (br d, J = 9.3 Hz, 1H), 2.17-2.12 (m, 2H), 1.79-
1.62 (m, 4H), 1.53-1.45 (m, 1H), 0.87 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (75 MHz,
CDCl3): δ 205.6, 160.2, 95.3, 79.7, 75.2, 58.0, 49.0, 27.2, 26.1, 25.7, 19.3, 18.1, -5.5, -5.6; IR
(neat, NaCl): 2953, 2830, 1960, 1255, 1088, 839 cm-1; MS m/z (%): 278 (41), 239 (33), 238 (96),
237 (73), 236 (100), 156 (13); EI-HRMS calcd for C15H24NO2Si [M]+ m/z: 278.1576, found:
278.1576.
39

NO
OTBS
48
•THF, 0 °C
TBAF NO
OH
49
•89%
(4-Vinylidene-3a,4,5,6,7,7a-hexahydrobenzo[d]isoxazol-3-yl)methanol (49). To a soln of the
TBS ether 48 (0.166 g, 0.565 mmol, 1 equiv) in THF (5.7 mL) at 0 °C was added TBAF (0.85
mL of a 1.0 M soln in THF, 0.85 mmol, 1.5 equiv) dropwise. Mixture stirred at 0 °C and the
reaction progress was monitored by TLC. Approximately 20 min after the addition was
complete, the reaction was quenched by the addition of sat. NH4Cl soln. The aqueous layer was
extracted with Et2O (4×). The combined organic layers were washed with brine, dried (MgSO4),
filtered, and concentrated under reduced pressure. The crude residue was purified via column
chromatography, using EtOAc/hexanes (3:7), to give alcohol 49 (0.075 g, 75%) as a pale yellow
oil. 1H NMR (300 MHz, CDCl3): δ 4.72-4.71 (m, 2H), 4.64 (dt, J = 9.1, 5.1 Hz, 1H), 4.42 (A of
ABq, J = 14.7 Hz, 1H), 4.34 (B of ABq, J = 14.7 Hz, 1H), 3.84 (br d, J = 9.1 Hz, 1H), 3.25 (br s,
1H), 2.20-2.10 (m, 2H), 1.78-1.58 (m, 3H), 1.53-1.40 (m, 1H); 13C NMR (75 MHz, CDCl3): δ
205.4, 161.0, 95.0, 79.7, 75.6, 57.6, 49.1, 27.1, 26.1, 19.4; IR (neat, NaCl): 3392, 2941, 1959,
1610, 1442, 1037 cm-1.
NO
OH
49
•TEA, DCM, -65 °C
(COCl)2, DMSO NO
O
50
•75%
4-Vinylidene-3a,4,5,6,7,7a-hexahydrobenzo[d]isoxazole-3-carbaldehyde (50). To a soln of
oxalyl chloride (0.08 mL, 0.932 mmol, 2.1 equiv) in DCM (3 mL) at –65 °C was added a soln of
DMSO (0.14 mL, 1.971 mmol, 4.4 equiv) in DCM (1.5 mL) dropwise over 10 min. The mixture
stirred 10 min before a soln of the alcohol 49 (0.080 g, 0.446 mmol, 1 equiv) in DCM (1.5 mL)
40

was added dropwise over 10 min. The mixture became cloudy while stirring for approximately
20 min at –65 °C. TEA was added dropwise over 5 min and the mixture was allowed to warm to
room temperature over 30 min. The reaction mixture was washed with sat NH4Cl soln and
water. The combined aqueous washings were back-extracted with DCM (3×). The combined
organic layers were washed with brine, dried (MgSO4), filtered, and concentrated under reduced
pressure. The crude residue was purified via column chromatography, eluting with
hexanes/EtOAc (6:1), to give aldehyde 50 (0.059 g, 75%) as a colorless oil. 1H NMR (300 MHz,
CDCl3): δ 9.90 (s, 1H), 4.75-4.64 (m, 2H), 3.94 (dt, J = 8.7, 3.6 Hz, 1H), 2.30-2.01 (m, 3H),
1.88-1.77 (m, 1H), 1.71-1.60 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 204.9, 185.4, 162.2, 95.4,
84.5, 76.9, 44.4, 26.9, 24.7, 19.4; IR (neat, NaCl): 2946, 2843, 1960, 1696, 1160 cm-1; MS m/z
(%): 177 (65), 160 (82), 106 (87), 105 (100), 104 (70); EI-HRMS calcd for C10H11NO2 [M]+ m/z:
177.0790, found: 177.0781.
NO
O•
Br
Zn, sat NH4Cl, THF
NO
OH•54-70%
5350
1-(4-Vinylidene-3a,4,5,6,7,7a-hexahydrobenzo[d]isoxazol-3-yl)but-3-yn-1-ol (53). To a slurry
of Zn dust (0.046 g, 0.701 mmol, 5 equiv) in THF (0.2 mL) at 0 °C, a soln of the aldehyde 50
(0.025 g, 0.140 mmol, 1 equiv) in THF (0.2 mL) and propargyl bromide (0.04 mL of an 80 wt%
soln in toluene, 0.359 mmol, 2.5 equiv) were added, followed by the addition of sat NH4Cl soln
(0.16 mL) dropwise over 45 min. The reaction mixture was allowed to slowly warm to room
temperature and stirred overnight. After approximately 20 hours, TLC indicated complete
consumption of starting material and the reaction mixture was filtered through a pad of Celite.
41

The filtrate was washed with sat NH4Cl soln and brine, dried (Na2SO4), filtered, and
concentrated under reduced pressure. The residue was purified via column chromatography
using hexanes/EtOAc (3:1) as the eluent. The homopropargyl alcohol 53 (0.021 g, 70%) was
achieved as a pale yellow crystalline solid and an approximate 3:1 mixture of diastereomers (1H
NMR). 1H NMR (300 MHz, CDCl3): δ 4.78-4.75 (m, 3H), 4.68-4.66 (m, 1H), 4.00 (br d, J =
9.3, 0.2 H),* 3.89 (br d, J = 9.0, 0.8 H),** 2.96 (br d, J = 5.7, 1H), 2.75 (A of ABqdd, J = 17.1,
5.4, 2.7, 1H), 2.65 (B of ABqdd, J = 17.1, 5.7, 2.7, 1H), 2.26-2.13 (m, 2H), 2.08 (t, J = 2.7, 1H),
1.86-1.48 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 205.8, 161.1, 95.3, 80.6, 78.9, 75.8, 71.6,
66.8, 49.8, 27.2, 26.3, 25.6, 19.5; IR (neat, NaCl): 3390, 3292, 2946, 2120, 1958, 1059. (*Minor
diastereomer; **major diastereomer.)
NO
O•
Br
2 equiv n-BuLi, TMEDA, then TMSCl21%, 2:1 dr
NO
OTMS•
5450TMS
3-(4-(Trimethylsilyl)-1-(trimethylsilyloxy)but-3-ynyl)-4-vinylidene-3a,4,5,6,7,7a-
hexahydrobenzo[d]isoxazole (54). n-BuLi (0.5 mL of a 1.6 M soln in hexanes, 0.8 mmol, 4.2
equiv) was added to a mixture of Et2O (0.7 mL) and hexanes (0.3 mL) at -78 °C. TMEDA (30
μL, 0.2 mmol, 1.05 equiv) was added dropwise followed by slow dropwise addition of propargyl
bromide (45 μL of an 80 wt% soln in toluene, 0.5 mmol, 2.1 equiv). A white precipitate
developed upon stirring at –78 °C 20 min. A soln of the aldehyde 50 (0.034 g, 0.192 mmol, 1
equiv) in Et2O (0.25 mL) was added dropwise and the mixture continued to stir at –78 °C until
complete consumption of starting material was observed by TLC. The reaction was then
quenched by rapid addition of TMSCl (0.10 mL, 0.79 mmol, 4 equiv). The mixture was allowed
42

to slowly approach room temperature until no additional progress was observed by TLC, at
which point the mixture was poured into an Erlenmeyer flask containing ~10 mL H2O. The
aqueous layer was extracted with Et2O (4×). The combined organic portions were dried
(Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by
column chromatography, using hexanes/EtOAc (9:1) as the eluent, to give allene-yne 54 (0.014
g, 21%) as a 2:1 mixture of diastereomers (1H NMR). 1H NMR (300 MHz, CDCl3): δ 4.74-4.67
(m, 4H), 3.98 (d, 9.9 Hz, 0.3H),* 3.92 (d, J = 9.6 Hz, 0.7H),** 2.78 (A of ABqd, J = 16.8, 5.7
Hz, 1H), 2.65 (B of ABqd, J = 16.8, 7.8 Hz, 1H), 2.28-2.20 (m, 2H), 1.80-1.35 (m, 4H), 0.20 (s,
9H), 0.15 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 206.1, 160.2, 103.2, 95.8, 86.8, 80.2, 75.3,
67.4, 49.6, 27.0, 26.5, 26.4, 19.3, 0.3, 0.0; IR (neat, NaCl): cm-1; TOF-HRMS calcd for
C19H31NO2Si2 [M+H]+ m/z: 362.1972, found: 362.1955. (*Minor diastereomer; **major
diastereomer.)
NO
OTMS
54
•
TMS
OTMS
O
NO
30 mol% [Rh(CO)2Cl]2
CO, toluene, 60 °C91%
TMS59
Cyclopentenone 59. To a test tube containing the allene-yne 54 (0.013 g, 0.036 mmol) and a stir
bar was added toluene (0.4 mL). The test tube was evacuated and flushed with CO (3×).
[Rh(CO)2Cl]2 (0.005 g, 0.012 mmol, 0.33 equiv) was added in one portion and the test tube was
evacuated and flushed with CO (3×) before being placed under CO (1 atm) and immersed in a 60
°C bath. TLC analysis indicated near complete consumption of starting material after 3.5 h, at
which point the reaction mixture was flushed through a plug of silica gel with hexanes/EtOAc
(1:1) to remove the Rh catalyst. The collected eluent was concentrated under reduced pressure
43

and the residue was purified via column chromatography using hexanes/EtOAc (3:1) as the
eluent, giving the major diastereomer of cyclopentenone 59 (0.010 g, 73%) followed by the
minor diastereomer (0.003 g, 21%). Both were white crystalline solids. Major diastereomer: 1H
NMR (300 MHz, CDCl3): δ 5.01 (t, J = 3.6 Hz, 1H), 4.55 (dt, J = 8.2, 3.0 Hz, 1H), 3.81 (br d, J
= 8.2 Hz, 1H), 3.39 (dd, J = 14.4, 3.6 Hz, 1H), 2.95 (A of ABq, J = 20.4 Hz, 1H), 2.88 (B of
ABq, J = 20.4, 1H), 2.85 (dd, J = 14.4, 3.6 Hz, 1H), 2.58-2.53 (m, 1H), 2.28-2.24 (m, 1H), 1.87-
1.67 (m, 4H), 0.28 (s, 9H), 0.20 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 208.2, 173.4, 162.7,
135.8, 132.8, 80.4, 66.3, 49.3, 41.6, 37.3, 31.0, 25.1, 19.2, 0.2, 0.1; IR (neat, NaCl): 2950, 2880,
1676, 1549, 1246, 839 cm-1; MS m/z (%): 389 (11), 374 (61), 147 (29), 75 (74), 73 (100); HR-
EIMS calcd for C20H31NO3Si [M]+ m/z: 389.1843, found: 389.1839. Minor diastereomer: 1H
NMR (300 MHz, CDCl3): δ 4.76 (dd, J = 10.8, 5.4 Hz, 1H), 4.55-4.53 (m, 1H), 3.92 (d, J = 6.9
Hz, 1H), 3.35 (dd, J = 13.5, 5.4 Hz, 1H), 2.99-2.78 (m, 3H), 2.56-2.51 (m, 1H), 2.35-2.31 (m,
1H), 2.04-1.69 (m, 4H), 0.29 (s, 9H), 0.21 (s, 9H).
OHOH
(S)-65
reflux OP
OCl
(S)-66
toluene/THF, -40 °C → rt77% O
PO
N
(S,R,R)-64
Ph
Ph
PCl3
NPh
PhLi
(R,R)-68
O,O’-(S)-(1,1’-Dinaphthyl-2,2’-diyl)-N,N’-di-(R,R)-1-phenylethylphosphoramidite (64). In
the glove box, a 25 mL schlenk flask was charged with (S)-BINOL (65, 1.063 g, 3.714 mmol, 1
equiv) and PCl3 (4 mL, 46 mmol, 12 equiv). The flask was removed from the glove box and the
suspension was heated to reflux under Ar atmosphere. After refluxing 16 h, the mixture had
become a homogeneous solution. After an additional 8 h, the excess PCl3 was removed under
vacuum at 40 °C. The foamy residue was dissolved in toluene (3 mL) and the solvent was
44

removed under vacuum. This step was repeated two more times (2 × 3 mL toluene). The residue
remained under vacuum for 16 h after the final rinse. In a separate flask, n-BuLi (2.6 mL of a
1.6 M soln in hexanes, 4.2 mmol, 1.1 equiv) was added dropwise via syringe to a soln of bis((R)-
phenylethyl)amine (0.85 mL, 3.72 mmol, 1 equiv) in THF (13 mL) at -78 °C. The fuschia-
colored soln was allowed to warm to -40 °C. The phosphochloridite 66 was dissolved in toluene
(20 mL) and the soln was added to the deprotonated amine 68 via cannula. The amber-colored
soln was allowed to warm from -40 °C to rt and stirred 16 h. The resulting cloudy mixture was
filtered through a pad of celite and the filtrate was concentrated under reduced pressure. The
residue was purified by column chromatography using hexanes/EtOAc (14:1) as the eluent to
give pure phosphoramidite (S,R,R)-64 (1.770 g, 77%) as a white foamy solid. The 1H NMR
spectrum is in agreement with published values.96
O
MeO OMe
1 mol% Cu(OTf)22.4 mol% (S,R,R)-64
toluene, -25 °C50%
62
O
MeO OMe60
*
4,4-Dimethoxy-5-methyl-2-cyclohexenone (60). To a two-neck roundbottom flask fitted with a
thermometer adapter and containing a suspension of Cu(OTf)2 (0.059 g, 0.162 mmol, 0.010
equiv) in toluene (25 mL) was added a soln of the phosphoramidite ligand (S,R,R)-64 (0.201 g,
0.373 mmol, 0.024 equiv) in toluene (6 mL). The pale orange mixture stirred 1 h before being
cooled to -25 °C in a cryocool bath. The cyclohexadienone 6292 (2.390 g, 15.51 mmol, 1 equiv)
was added. Me2Zn (21 mL of a 1.2 M soln in toluene, 25 mmol, 1.6 equiv) was added so that the
internal temperature ≤-20 °C. The bright yellow reaction mixture stirred at -25 °C and the
reaction progress was monitored every 30 min by TLC. After 4 h, the slightly less polar
diaddition product was visible above the monoaddition product spot on the TLC plate. The
45

reaction was quenched by pouring the mixture into an Erlenmeyer flask containing rapidly
stirring 1.5 M NaOH. The aqueous layer was extracted with Et2O (3×). The combined organic
layers were washed with 1.5 N NaOH and brine, dried (Na2SO4), filtered and carefully
concentrated under reduced pressure to remove most of the Et2O. The remaining 3:1 mixture (by
1H NMR) of enone 60 and unreacted sm in toluene was applied to a silica gel column and eluted
with pentane/Et2O (3:1 to 0:1) to separate the toluene from the product mixture. Fractions
containing the product were combined and carefully concentrated under reduced pressure. The
residue was purified by column chromatography, eluting with pentane/Et2O (3:1) to give pure
cyclohexenone 60 (1.321 g, 50%) as a colorless oil. The 1H NMR spectrum is in agreement with
literature values.95
O
MeO OMe60
*
MgBr
THFMeO OMe
61
OH 1. BH3·DMS, then H2O, NaOH, H2O2
28
OH
2. TBSCl, DMAP, TEA3. Montmorillonite-K10
TBSO
O21%, 3 steps74%
4-(1-(tert-Butyldimethylsilyloxy)propan-2-yl)-4-hydroxy-6-methyl-2-cyclohexenone (28).
To the enone 60 (0.557 g, 3.28 mmol, 1 equiv) in THF (16 mL) at 0 °C was added
isopropenylmagnesium bromide (20 mL of a 0.5 M soln in THF, 10 mmol, 3 equiv) via syringe
pump over 20 min. The reaction was monitored by TLC and quenched by pouring the reaction
mixture into a flask containing saturated aqueous NH4Cl. The aqueous layer was extracted with
Et2O (4×). The combined organic portions were washed with brine, dried (MgSO4), filtered, and
concentrated under reduced pressure. The crude residue was flushed through a plug of pretreated
(TEA) silica gel with hexanes/EtOAc (4:1) to remove major impurities. Tertiary alcohol 61
(0.514 g, 74%) was obtained as an amber oil. 1H NMR (300 MHz, CDCl3): δ 5.80 (A of ABq, J
= 10.2 Hz, 1H), 5.74 (B of ABq, J = 10.2 Hz, 1H), 5.02 (s, 1H), 4.88 (quintet, J = 1.5 Hz, 1H),
46

3.28 (s, 3H), 3.22 (s, 3H), 2.18-2.12 (m, 2H), 1.77 (s, 3H), 1.68 (dd, J = 15.6, 6.6 Hz, 1H), 1.05
(d, J = 6.9 Hz, 3H).
To a soln of the olefin 61 (0.271 g, 1.276 mmol, 1 equiv) in THF (2 mL) was added
BH3·DMS (0.64 mL of a 2 M soln in THF, 1.28 mmol, 1 equiv) dropwise via syringe. The
reaction was monitored by 1H NMR analysis of aliquots. The reaction stirred 16 h, at which
point the flask was placed in an ambient temperature water bath and H2O (0.11 mL, 6.1 mmol, 5
equiv) was added followed by dropwise addition of NaOH (5 mL of a 3 M aqueous soln, 15
mmol, 12 equiv) and H2O2 (2.2 mL of a 33 wt% aqueous soln, 23.5 mmol, 18 equiv). The
mixture stirred 3 h and was diluted with Et2O. The aqueous layer was extracted with Et2O (4×).
The combined organic portions were washed with brine, dried (Na2SO4), decanted, and
concentrated under reduced pressure to give the crude primary alcohol (0.207 g, 71%).
To a soln of DMAP (11 mg, 0.09 mmol, 0.1 equiv) and the primary alcohol (0.207 g,
0.900 mmol, 1 equiv) in DCM (1 mL) was added TEA (0.31 mL, 2.22 mmol, 2.5 equiv)
followed by dropwise addition of a soln of TBSCl (0.163 g, 1.08 mmol, 1.2 equiv) in DCM (0.5
mL). Reaction progress was monitored by TLC. After 2 h, the mixture was diluted in H2O and
the aqueous layer was extracted with DCM (3×). The combined organic layers were washed
with brine, dried (Na2SO4), decanted, and concentrated under reduced pressure, giving the crude
silyl ether (0.310 g, 84%).
To a soln of the ketal (0.259 g, 0.752 mmol) in DCM (15 mL) was added
Montmorillonite K10 clay (0.378 g). Reaction progress was monitored by TLC. After 10 min,
additional clay was added (0.376 g). When no starting material was observable by TLC, the
reaction mixture was filtered through a pad of celite and the filtrate was concentrated under
reduced pressure. The crude residue, containing a 2:1 mix of diastereomers of 28 by 1H NMR,
47

was purified by column chromatography using 4:1 hexanes/EtOAc as the eluent to give the
desired enone 28 (0.082 g, 36% from 57). Partial separation of the diastereomers was achieved
during column chromatography. Major diastereomer: 1H NMR (300 MHz, CDCl3): δ 6.87 (dd, J
= 10.3, 2.1 Hz, 1H), 5.92 (d, J = 10.3 Hz, 1H), 4.43 (s, 1H), 4.07 (dd, J = 10.5, 3.3 Hz, 1H), 3.68
(dd, J = 10.5, 4.5 Hz, 1H), 2.48-2.36 (m, 2H), 2.08-1.94 (m, 2H), 1.17 (d, J = 6.3 Hz, 3H), 1.14
(d, J = 6.9 Hz, 3H), 0.93 (s, 9H), 0.13 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 201.2, 154.7,
127.9, 73.6, 66.9, 43.7, 40.2, 38.8, 18.0, 15.8, 12.7, -5.7, -5.8; IR (neat, NaCl): 3463, 2931, 2858,
1684, 1462, 1377, 1073, 836 cm-1; MS m/z (%): 321 (12), 281 (16), 251 (80), 191 (13), 121 (29);
TOF-HRMS calcd for C16H30O3NaSi [M + Na]+ m/z: 321.1862, found: 321.1858; Minor
diastereomer: 1H NMR (300 MHz, CDCl3): δ 6.96 (dd, J = 10.2, 2.1 Hz, 1H), 5.92 (d, J = 10.2
Hz, 1H), 4.39 (s, 1H), 4.05 (dd, J = 10.2, 3.0 Hz, 1H), 3.69 (dd, J = 10.2, 2.7 Hz, 1H), 2.39 (dqd,
J = 13.5, 6.6, 4.7 Hz, 1H), 2.27 (ddd, J = 13.5, 4.7, 2.1 Hz, 1H), 1.99 (qt, J = 7.2, 3.0 Hz, 1H),
1.76 (t, J = 13.5 Hz, 1H), 1.19 (d, J = 7.2 Hz, 3H), 1.18 (d, J = 6.6 Hz, 3H), 0.94 (s, 9 H), 0.13 (s,
3H), 0.12 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.3, 156.3, 127.3, 73.8, 67.1, 41.2, 39.1,
38.4, 25.8, 18.0, 15.7, 12.6, -5.7, -5.8.
48

APPENDIX A
1H AND 13C NMR SPECTRA FOR NEW COMPOUNDS
49

50
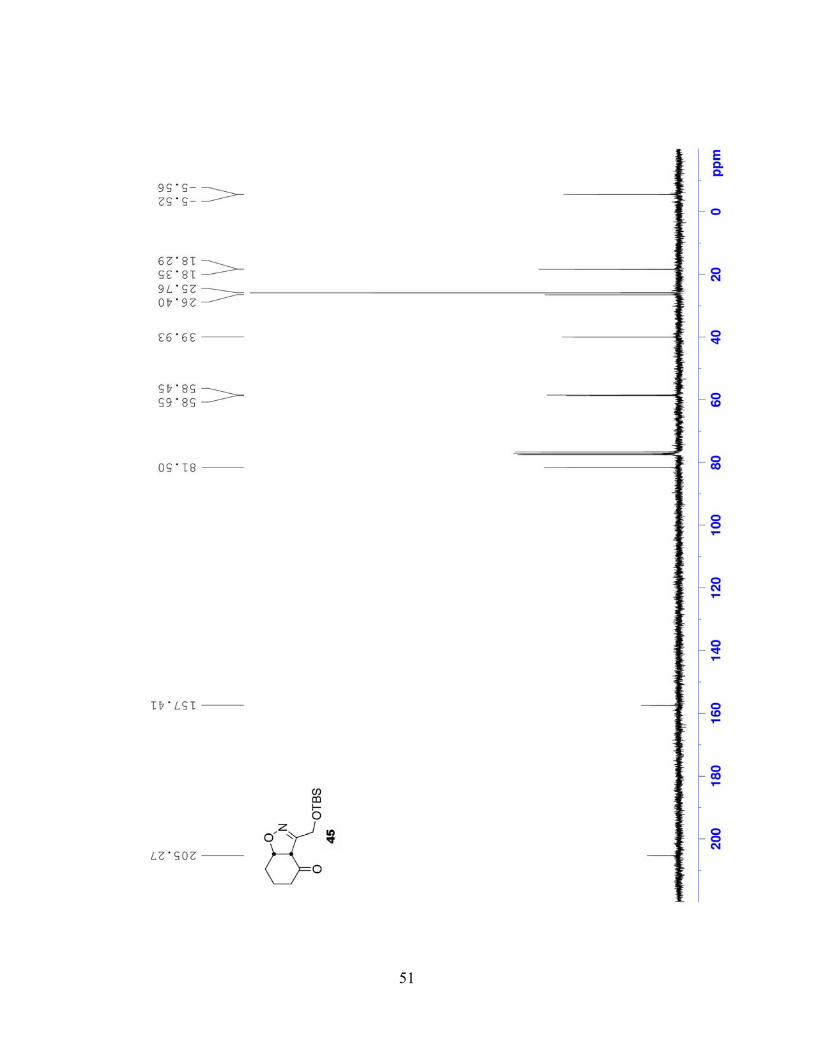
51
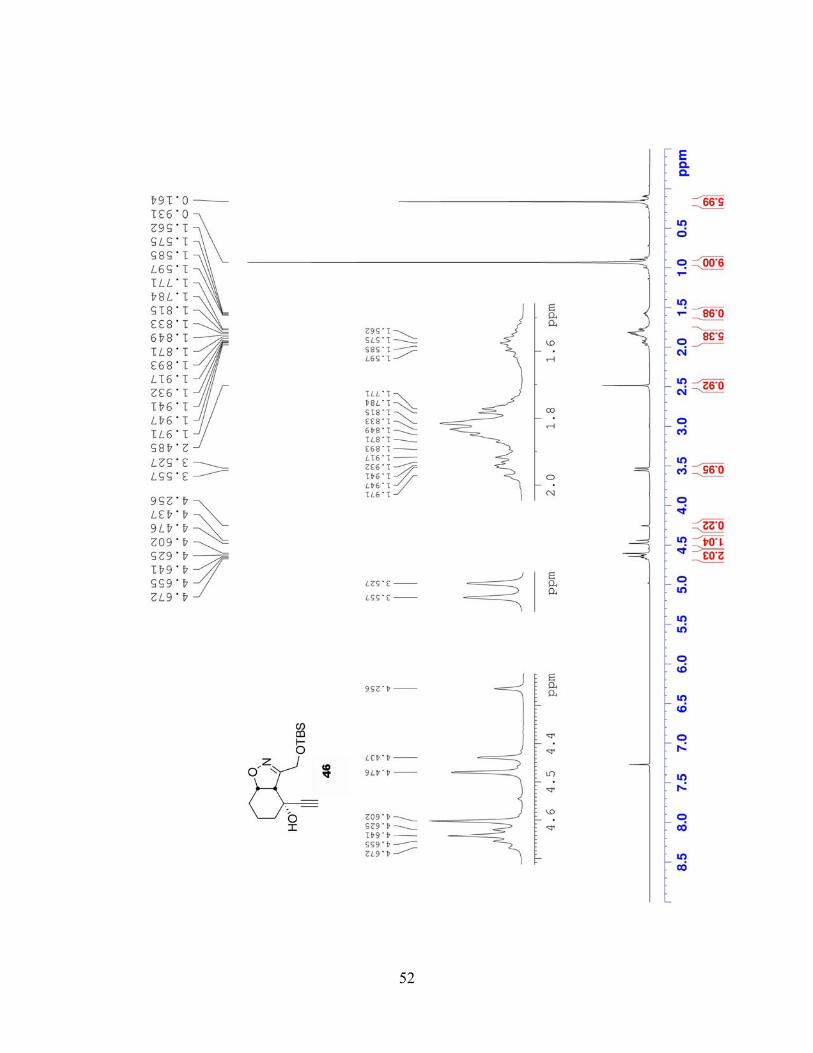
52

53

54

55
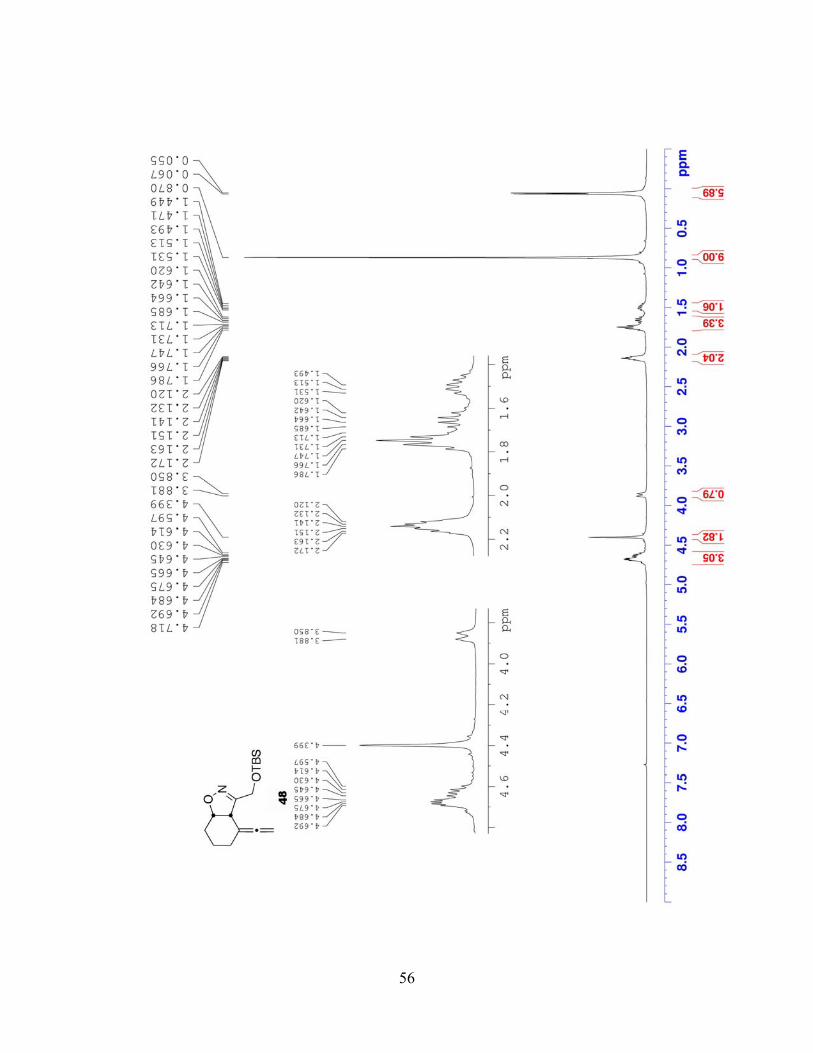
56

57

58
59

60

61

62

63

64

65

66

67

68

69

70

71

72

73

BIBLIOGRAPHY
1. Appendino, G.; Szallasi, A. Euphorbium: Modern Research on its Active Principle, Resiniferatoxin, Revives an Ancient Medicine. Life Sci. 1997, 60, 681-696.
2. Hergenhahn, M.; Adolf, W.; Hecker, E. Resiniferatoxin and Other Esters of Novel Polyfunctional Diterpenes from Euphorbia Resinifera and Unispina. Tetrahedron Lett. 1975, 1975, 1595-1598.
3. Adolf, W.; Sorg, B.; Hergenhahn, M.; Hecker, E. Structure-Activity Relations of Polyfunctional Diterpenes of the Daphnane Type. I. Revised Structure for Resiniferatoxin and Structure-Activity Relations of Resiniferonol and Some of Its Esters. J. Nat. Prod. 1982, 45, 347-354.
4. Szallasi, A.; Blumberg, P. M. Resiniferatoxin, a Phorbol-Related Diterpene, Acts as an Ultrapotent Analog of Capsaicin, the Irritant Constituent in Red Pepper. Neuroscience 1989, 30, 515-520.
5. Caterina, M. J.; Schumacher, M. A.; Tominaga, M.; Rosen, T. A.; Levine, J. D.; Julius, D. The Capsaicin Receptor: A Heat-Activated Ion Channel in the Pain Pathway. Nature 1997, 389, 816-824.
6. Chou, M. Z.; Mtui, T.; Gao, Y.-D.; Kohler, M.; Middleton, R. E. Resiniferatoxin Binds to the Capsaicin Receptor (TRPV1) Near the Extracellular Side of the S4 Transmembrane Domain. Biochemistry 2004, 43, 2501-2511.
7. Szallasi, A.; Blumberg, P. M. Vanilloid Receptors: New Insights Enhance Potential as a Therapeutic Target. Pain 1996, 68, 195-208.
8. Szallasi, A.; Blumberg, P. M. Vanilloid (Capsaicin) Receptors and Mechanisms. Pharmacological Rev. 1999, 51, 159-211.
9. Immke, D. C.; Gavva, N. R. The TRPV1 Receptor and Nociception. Semin. Cell Dev. Biol. 2006, 17, 582-591.
74

10. Cruz, F.; Guimaraes, M. Suppression of Bladder Hyperreflexia by Intravesical Resiniferatoxin. Lancet 1997, 350, 640-641.
11. Lazzeri, M.; Beneforti, P.; Turini, D. Urodynamic Effects of Intravesical Resiniferatoxin in Humans: Preliminary Results in Stable and Unstable Detrusor. J. Urol. 1997, 158, 2093-2096.
12. Payne, C. K.; Mosbaugh, P. G.; Forrest, J. B.; Evans, R. J.; Whitmore, K. E.; Antoci, J. P.; Perez-Marrero, R.; Jacoby, K.; Yu, A. S.; Frumkin, L. R. Intravesical Resiniferatoxin for the Treatment of Interstitial Cystitis: A Randomized, Double-Blind, Placebo Controlled Trial. J. Urol. 2005, 173, 1590-1594.
13. ICOS Corporation February 3, 2004 Press Release. http://media.corporate-ir.net/media_files/nsd/icos/news/PR_Q403_ICOS.pdf (October 2006).
14. Hanno, P.; Nordling, J.; van Ophoven, A. What is New in Bladder Pain Syndrome/Interstitial Cystitis? Curr. Opin. Urol. 2008, 18, 353-358.
15. Blumberg, P. M. Protein Kinase C as the Receptor for the Phorbol Ester Tumor Promoters: Sixth Rhoads Memorial Award Lecture. Cancer Res. 1988, 48, 1-8.
16. Wender, P. A.; Kee, J.-M.; Warrington, J. M. Practical Synthesis of Prostratin, DPP, and Their Analogs, Adjuvant Leads Against Latent HIV. Science 2008, 320, 649-652.
17. United States Food and Drug Administration List of Orphan Designations and Approvals. http://ww.fda.gov/orphan/designat/list.htm (November 2006).
18. Brown, D. C.; Iadarola, M. J.; Perkowski, S. Z.; Erin, H.; Shofer, F.; Laszlo, K. J.; Olah, Z.; Mannes, A. J. Physiologic and Antinociceptive Effects of Intrathecal Resiniferatoxin in a Canine Bone Cancer Model. Anesthesiology 2005, 103, 1052-1059.
19. Szallasi, A.; Cortright, D. N.; Blum, C. A.; Eid, S. R. The Vanilloid Receptor TRPV1: 10 Years From Channel Cloning to Antagonist Proof-of-Concept. Nat. Rev. Drug Discov. 2007, 6, 357-372.
20. Szallasi, A.; Cruz, F.; Geppetti, P. TRPV1: A Therapeutic Target for Novel Analgesic Drugs? Trends Mol. Med. 2006, 12, 545-554.
21. Kissin, I. Vanilloid-Induced Conduction Analgesia: Selective, Dose-Dependent, Long-Lasting, with a Low Level of Potential Neurotoxicity. Anesth. Analg. 2008, 107, 271-281.
22. Meyer, D. Use of Resiniferatoxin (RTX) for Producing an Agent for Treating Joint Pains and Method for Applying Said Agent. USPTO Application # 20080139641, 2008.
75

23. Fattorusso, E.; Lanzotti, V.; Taglialatela-Scafati, O.; Tron, G. C.; Appendino, G. Bisnorsesquiterpenoids from Euphorbia resinifera Berg. and an Expeditious Procedure to Obtain Resiniferatoxin from Its Fresh Latex. Eur. J. Org. Chem. 2002, 71-78.
24. Stanoeva, E.; He, W.; De Kimpe, N. Natural and Synthetic Cage Compounds Incorporating the 2,9,10-Trioxatricyclo[4.3.1.03,8]decane Type Moiety. Bioorg. Med. Chem. 2005, 13, 17-28.
25. Sakata, K.; Kawazu, K.; Mitsui, T.; Masaki, M. Structure and Stereochemistry of Huratoxin, a Piscicidal Constituent of Hura crepitans. Tetrahedron Lett. 1971, 1141-1144.
26. Yoshida, M.; Yokokura, H.; Hidaka, H.; Ikekawa, T.; Saijo, N. Mechanism of Antitumor Action of PKC Activator, Gnidimacrin. Int. J. Cancer 1998, 77, 243-250.
27. Kupchan, S. M.; Shizuri, Y.; Murae, T.; Sweeny, J. G.; Haynes, H. R.; Shen, M.-S.; Barrick, J. C.; Bryan, R. F.; van der Helm, D.; Wu, K. K. Gnidimacrin and Gnidimacrin 20-Palmitate, Novel Macrocyclic Antileukemic Diterpenoid Esters from Gnidia subcordata. J. Am. Chem. Soc. 1976, 98, 5719-5720.
28. Carney, J. R.; Krenisky, J. M.; Williamson, R. T.; Luo, J.; Carlson, T. J.; Hsu, V. L.; Moswa, J. L. Maprouneacin, a New Daphnane Diterpenoid with Potent Antihyperglycemic Activity from Maprounea africana. J. Nat. Prod. 1999, 62, 345-347.
29. He, W.; Cik, M.; Van Puyvelde, L.; Van Dun, J.; Appendino, G.; Lesage, A.; Van der Lindin, I.; Leysen, J. E.; Wouters, W.; Mathenge, S. G.; Mudida, F. P.; De Kimpe, N. Neurotrophic and Antileukemic Daphnane Diterpenoids from Synaptolepsis kirkii. Bioorg. Med. Chem. 2002, 10, 3245-3255.
30. Jayasuriya, H.; Zink, D. L.; Singh, S. B.; Borris, R. P.; Nanokorn, W.; Beck, H. T.; Balick, M. J.; Goetz, M. A.; Slayton, L.; Gregory, L.; Zakson-Aiken, M.; Shoop, W.; Singh, S. B. Structure and Stereochemistry of Rediocide A, a Highly Modified Daphnane from Trigonostemon reidioides Exhibiting Potent Insecticidal Activity. J. Am. Chem. Soc. 2000, 122, 4998-4999.
31. Zhan, Z.-J.; Fan, C.-Q.; Ding, J.; Yue, J.-M. Novel Diterpenoids with Potent Inhibitory Activity Against Endothelium Cell HMEC and Cytotoxic Activities from a Well-known TCM Plant Daphne genkwa. Bioorg. Med. Chem. 2005, 13, 645-655.
32. Wender, P. A.; Jesudason, C. D.; Nakahira, H.; Tamura, N.; Tebbe, A. L.; Ueno, Y. The First Synthesis of a Daphnane Diterpene: The Enantiocontrolled Total Synthesis of (+)-Resiniferatoxin. J. Am. Chem. Soc. 1997, 119, 12976-12977.
76

33. Jackson, S. R.; Johnson, M. G.; Mikami, M.; Shiokawa, S.; Carreira, E. M. Rearrangement of a Tricyclic 2,5-Cyclohexadienone: Towards a General Synthetic Route to the Daphnanes and (+)-Resiniferatoxin. Angew. Chem. Int. Ed. 2001, 40, 2694-2697.
34. Ritter, T.; Zarotti, P.; Carreira, E. M. Diastereoselective Phenol para-Alkylation: Access to a Cross-Conjugated Cyclohexadienone en Route to Resiniferatoxin. Org. Lett. 2004, 6, 4371-4374.
35. Wender, P. A.; Kogen, H.; Lee, H. Y.; Munger, J. D., Jr.; Wilhelm, R. S.; Williams, P. D. Studies on Tumor Promoters. 8. The Synthesis of Phorbol. J. Am. Chem. Soc. 1989, 111, 8957-8958.
36. Wender, P. A.; Lee, H. Y.; Wilhelm, R. S.; Williams, P. D. Studies on Tumor Promoters. 7. The Synthesis of a Potentially General Precursor of the Tiglianes, Daphnanes, and Ingenanes. J. Am. Chem. Soc. 1989, 111, 8954-8957.
37. Wender, P. A.; McDonald, F. E. Studies on Tumor Promoters. 9. A Second-Generation Synthesis of Phorbol. J. Am. Chem. Soc. 1990, 112, 4956-4958.
38. Wender, P. A.; Rice, K. D.; Schnute, M. E. The First Formal Asymmetric Synthesis of Phorbol. J. Am. Chem. Soc. 1997, 119, 7897-7898.
39. Wender, P. A.; D'Angelo, N.; Elitzin, V. I.; Ernst, M.; Jackson-Ugueto, E. E.; Kowalski, J. A.; McKendry, S.; Rehfeuter, M.; Sun, R.; Voigtlaender, D. Function-Oriented Synthesis: Studies Aimed at the Synthesis and Mode of Action of 1α-Alkyldaphnane Analogues. Org. Lett. 2007, 9, 1829-1832.
40. Lee, K.; Cha, J. K. Formal Synthesis of (+)-Phorbol. J. Am. Chem. Soc. 2001, 123, 5590-5591.
41. Brickwood, A. C.; Drew, M. G. B.; Harwood, L. M.; Ishikawa, T.; Marais, P.; Morisson, V. Synthetic Approaches Towards Phorbols via the Ultra-High-Pressure Mediated Intramolecular Diels-Alder Reaction of Furans (IMDAF): Effect of Furan Substitution. J. Chem. Soc., Perkin Trans. 1 1999, 913-921.
42. Page, P. C. B.; Hayman, C. M.; McFarland, H. L.; Willcock, D. J.; Galea, N. M. An IMDA Approach to Tigliane and Daphnane Diterpenoids: Generation of Rings A, B and C Incorporating C-18. Synlett 2002, 583-587.
43. Rigby, J. H.; Kierkus, P. C.; Head, D. Studies on the Stereoselective Construction of the Tigliane Ring System. Tetrahedron Lett. 1989, 30, 5073-5076.
77

44. Shigeno, K.; Sasai, H.; Shibasaki, M. Synthetic Studies Towards Phorbols: A Stereocontrolled Synthesis of the Phorbol Skeleton in the Naturally Occurring Form. Tetrahedron Lett. 1992, 33, 4937-4940.
45. Sugita, K.; Shigeno, K.; Neville, C. F.; Sasai, H.; Shibasaki, M. Synthetic Studies Towards Phorbols: Synthesis of B or C Ring Substituted Phorbol Skeletons in the Naturally Occurring Form. Synlett 1994, 325-329.
46. McLoughlin, J. I.; Brahma, R.; Campopiano, O.; Little, R. D. Stereoselectivity in Intramolecular Diyl Trapping Reactions. Model Studies Directed Toward the Phorbols. Tetrahedron Lett. 1990, 31, 1377-1380.
47. Paquette, L. A.; Sauer, D. R.; Edmondson, S. D.; Friedrich, D. A Concise Route to the Tetracyclic Core of Phorbol. Tetrahedron Lett. 1994, 50, 4071-4086.
48. Brummond, K. M.; Chen, D.; Davis, M. M. A General Synthetic Route to Differentially Functionalized Angularly and Linearly Fused [6-7-5] Ring Systems: A Rh(I)-Catalyzed Cyclocarbonylation Reaction. J. Org. Chem. 2008, 73, 5064-5068.
49. Brummond, K. M.; Chen, H.; Fisher, K. D.; Kerekes, A. D.; Rickards, B.; Sill, P. C.; Geib, S. J. An Allenic Pauson-Khand-Type Reaction: A Reversal in π-Bond Selectivity and the Formation of Seven-Membered Rings. Org. Lett. 2002, 4, 1931-1934.
50. Brummond, K. M.; Gao, D. Unique Strategy for the Assembly of the Carbon Skeleton of Guanacastapene A Using an Allenic Pauson-Khand-Type Reaction. Org. Lett. 2003, 5, 3491-3494.
51. Brummond, K. M.; Lu, J. A Short Synthesis of the Potent Antitumor Agent (±)-Hydroxymethylacylfulvene Using an Allenic Pauson-Khand Type Cycloaddition. J. Am. Chem. Soc. 1999, 121, 5087-5088.
52. Brummond, K. M.; Lu, J.; Petersen, J. A Rapid Synthesis of Hydroxymethylacylfulvene (HMAF) Using the Allenic Pauson-Khand Reaction. A Synthetic Approach to Either Enantiomer of This Illudane Structure. J. Am. Chem. Soc. 2000, 122, 4915-4920.
53. Brummond, K. M.; Wan, H. The Allenic Pauson-Khand Cycloaddition. Dependence on π-Bond Selectivity on Substrate Structure. Tetrahedron Lett. 1998, 39, 931-934.
54. Brummond, K. M.; Wan, H.; Kent, J. L. An Intramolecular Allenic [2 + 2 + 1] Cycloaddition. J. Org. Chem. 1998, 63, 6535-6545.
78

55. Kent, J. L.; Wan, H.; Brummond, K. M. A New Allenic Pauson-Khand Cycloaddition for the Preparation of α-Methylene Cyclopentenones. Tetrahedron Lett. 1995, 36, 2407-2410.
56. Curran, D. P. The Cycloadditive Approach to β-Hydroxy Carbonyls: An Emerging Alternative to the Aldol Strategy. In Advances in Cycloaddition; Curran, D. P., Ed.; JAI Press Inc.: Greenwich, CT, 1988; Vol. 1, pp 129-189.
57. Jäger, V.; Colinas, P. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A., Pearson, W. H., Eds.; John Wiley & Sons: New York, 2002; pp 361-472.
58. Belen'kii, L. I. Nitrile Oxides. In Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis: Novel Strategies in Synthesis; 2nd ed.; Feuer, H., Ed.; John Wiley & Sons: Hoboken, NJ, 2008; pp 1-127.
59. Caramella, P.; Grünanger, P. Nitrile Oxides and Imines. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984; Vol. 1, pp 291-392.
60. Grundmann, C.; Grünanger, P. The Nitrile Oxides: Versatile Tools of Theoretical and Preparative Chemistry,Springer-Verlag: New York, 1971.
61. Torssell, K. B. G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis: Novel Strategies in Synthesis,1st ed.; VCH Publishers, Inc.: New York, 1988.
62. Bianchi, G.; De Micheli, C.; Gandolfi, R.; Grünanger, P.; Finzi, P. V.; de Pava, O. V. Isoxazoline Derivatives. Part VI. Regioselectivity in the 1,3-Dipolar Cycloaddition of Nitrile Oxides to α,β-Unsaturated Ketones. J. Chem. Soc., Perkin Trans. 1 1973, 1148-1155.
63. Caramella, P.; Cellerino, G. Selectivity in Cycloadditions - II. Polar and Steric Control in the 1,3-Dipolar Cycloaddition of Benzonitrile Oxide to Some 3-Substituted Cyclopentenes. Tetrahedron Lett. 1974, 229-232.
64. Houk, K. N.; Moses, S. R.; Wu, Y.-D.; Rondan, N. G.; Jäger, V.; Schohe, R.; Fronczek, F. R. Stereoselective Nitrile Oxide Cycloadditions to Chiral Allyl Ethers and Alcohols. The "Inside Alkoxy" Effect. J. Am. Chem. Soc. 1984, 106, 3880-3882.
65. Curran, D. P.; Choi, S.-M.; Gothe, S. A.; Lin, F.-T. Directed Nitrile Oxide Cycloaddition Reactions. The Use of Hydrogen Bonding to Direct Regio- and Stereochemistry in Nitrile Oxide Cycloadditions with Cyclopentenylamides. J. Org. Chem. 1990, 55, 3710-3712.
79

66. Becker, N.; Carreira, E. M. Hydroxyl-Directed Nitrile Oxide Cycloaddition Reactions with Cyclic Allylic Alcohols. Org. Lett. 2007, 9, 3857-3858.
67. Bode, J. W.; Fraefel, N.; Muri, D.; Carreira, E. M. A General Solution to the Modular Synthesis of Polyketide Building Blocks by Kanemasa Hydroxy-Directed Nitrile Oxide Cycloadditions. Angew. Chem. Int. Ed. 2001, 40, 2082-2085.
68. Choi, S.-M. Part 1. Hydrogen-Bond Directing Nitrile Oxide Cycloadditions Part II. 1,2-Asymmetric Induction by Radical and Ionic Reductions. Ph.D. Thesis, University of Pittsburgh, 1992.
69. Kim, H. R.; Song, J. H.; Rhie, S. Y.; Ryu, E. K. Regioselective and Stereoselective 1,3-Dipolar Cycloadditions of Nitrile Oxide with Allylic Alcohols Prepared in Situ from α,β-Unsaturated Carbonyl Compounds with Grignard Reagents. Synth. Commun. 1995, 25, 1801-1807.
70. Kanemasa, S.; Nishiuchi, M.; Kamimura, A.; Hori, K. First Successful Metal Coordination Control in 1,3-Dipolar Cycloadditions. High-Rate Acceleration and Regio- and Stereocontrol of Nitrile Oxide Cycloadditions to the Magnesium Alkoxides of Allylic and Homoallylic Alcohols. J. Am. Chem. Soc. 1994, 116, 2324-2339.
71. Martin, S. F.; Anderson, B. G.; Daniel, D.; Gaucher, A. A Cycloaddition Approach to Breynolide. Tetrahedron 1997, 53, 8997-9006.
72. Martin, S. F.; Daniel, D. A Novel Approach to Breynolide. Tetrahedron Lett. 1993, 34, 4281-4284.
73. Seifried, D. unpublished results.
74. Mukaiyama, T.; Hoshino, T. The Reactions of Primary Nitroparaffins with Isocyanates. J. Am. Chem. Soc. 1960, 82, 5339-5342.
75. Kantorowski, E. J.; Brown, S. P.; Kurth, M. J. Use of Diisocyanates for in Situ Preparation of Nitrile Oxides: Preparation of Isoxazoles and Isoxazolines. J. Org. Chem. 1998, 63, 5272-5274.
76. 3.7-6.6 Hz is a typical 3JH4,H5 coupling for trans-isoxazolines; see ref 77.
77. Aversa, M. C.; Cum, G.; Crisafulli, M. Spettri NMR di Δ2-Isossazoline. - Nota II. Isomeria cis-trans di Δ2-Isossazoline 4,5-Bisostituite. Gazz. Chim. Ital. 1968, 98, 42-47.
78. Chiu, P.; Li, Z.; Fung, K. C. M. An Expedient Preparation of Stryker's Reagent. Tetrahedron Lett. 2003, 44, 455-457.
80

79. Kamenecka, T. M.; Overman, L. E.; Ly Sakata, S. K. Construction of Substituted Cyclohexenones by Reductive Cyclization of 7-Oxo-2,8-alkadienyl Esters. Org. Lett. 2002, 4, 79-82.
80. Lipshutz, B. H.; Chrisman, W.; Noson, K.; Papa, P.; Sclafani, J. A.; Vivian, R. W.; Keith, J. M. Copper Hydride-Catalyzed Tandem 1,4-Reduction/Alkylation Reactions. Tetrahedron 2000, 56, 2779-2788.
81. Lipshutz observes significant quantities of impurities in two samples of Stryker's reagent obtained frou Aldrich (see ref 80). We generally purchased our commercial Stryker's reagent from Acros. The quality of Stryker's reagent can be judged to some degree by the appearance of the copper hydride. Freshly prepared Stryker's reagent is bright red to deep red and crystalline, whereas quenched Stryker's reagent is dark brown. The typical sample we received from Acros ranged from brick red to brownish-red and often appeared somewhat heterogeneous.
82. Lee, D.-W.; Yun, J. Direct Synthesis of Stryker's Reagent From a Cu(II) Salt. Tetrahedron Lett. 2005, 46, 2037-2039.
83. Caldirola, P.; De Amici, M.; De Micheli, C.; Wade, P. A.; Price, D. T.; Bereznak, J. F. Metal-Hydride Reduction of Isoxazoline-3-Carboxylate Esters. Tetrahedron 1986, 42, 5267-5272.
84. Kamimura, A.; Kakehi, A.; Hori, K. An Experimental and Theoretical Study on Stereoselective Addition to 3-Formyl-Δ2-isoxazolines. Part 1. 1,3-Antiselectivity Induced by BF3�OEt2. Tetrahedron 1993, 49, 7637-7648.
85. Yamamoto, H. Propargyl and Allenyl Organometallics. In Comprehensive Organic Synthesis; Trost, B., Fleming, I., Eds.; Pergamon Press: Elmsford, NY, 1991; Vol. 2, pp 81-98.
86. Daniels, R. G.; Paquette, L. A. Silanes in Organic Synthesis. II. Regiocontrolled Synthesis of α-Hydroxymethylated (Trimethylsilyl)allenes. Tetrahedron Lett. 1981, 22, 1579-1582.
87. Daniels, R. G. Regiochemical and Stereochemical Studies of Organosilanes. Ph.D. Thesis, Ohio State University, Columbus, OH, 1982.
88. Lin, M.-J.; Loh, T.-P. Indium-Mediated Reaction of Trialkylsilyl Propargyl Bromide with Aldehydes: Highly Regioselective Synthesis of Allenic and Homopropargylic Alcohols. J. Am. Chem. Soc. 2003, 125, 13042-13043.
81

89. Cabezas, J. A.; Alvarez, L. X. Propargylation of Carbonyl Compounds: An Efficient Method for the Synthesis of Homopropargyl Alcohols. Tetrahedron Lett. 1998, 39, 3935-3938.
90. Cabezas, J. A.; Pereira, A. R.; Amey, A. A New Method for the Preparation of 1,3-Dilithiopropyne: An Efficient Synthesis of Homopropargyl Alcohols. Tetrahedron Lett. 2001, 42, 6819-6822.
91. Gotō, H.; Ōsawa, E. An Efficient Algorithm for Searching Low-energy Conformers of Cyclic and Acyclic Molecules. J. Chem. Soc., Perkin Trans. 2 1993, 187-198.
92. Pelter, A.; Elgendy, S. M. A. Phenolic Oxidations with Phenyliodonium Diacetate. J. Chem. Soc., Perkin Trans. 1 1993, 1891-1896.
93. Nilsson, A.; Ronlán, A. A Novel Synthesis of 4-Chloro-4-Methylcyclohexa-2,5-dienone and 4,4-Dimethoxycyclohexa-2,5-dienone. Tetrahedron Lett. 1975, 1107-1110.
94. Stern, A. J.; Rohde, J. J.; Swenton, J. S. Oxygenophilic Organoaluminum-Mediated Conjugate Addition of Alkyllithium and Grignard Reagents to Quinone Monoketals and Quinol Ethers. The Directing Effect of a Methoxy Group on the 1,4-Addition Process. J. Org. Chem. 1989, 54, 4413-4419.
95. Imbos, R.; Brilman, M. H. G.; Pineschi, M.; Feringa, B. L. Highly Enantioselective Catalytic Conjugate Additions to Cyclohexadienones. Org. Lett. 1999, 1, 623-625.
96. Arnold, L. A.; Imbos, R.; Mandoli, A.; de Vries, A. H. M.; Naasz, R.; Feringa, B. L. Enantioselective Catalytic Conjugate Addition of Dialkylzinc Reagents Using Copper-Phosphoramidite Complexes; Ligand Variation and Non-linear Effects. Tetrahedron 2000, 56, 2865-2878.
97. Imbos, R. Catalytic Asymmetric Conjugate Additions and Heck Reactions. Ph.D. Thesis, University of Groningen, Groningen, Netherlands, 2002.
98. The Feringa group has commented on the somewhat variable purity of phosphoramidite ligands. See ref 97.
99. Duursma, A. Asymmetric Catalysis with Chiral Monodentate Phosphoramidite Ligands. Ph.D. Thesis, University of Groningen, Groningen, Netherlands, 2004.
100. The identities of the ligand impurities are unknown, although evidence of their existence can be seen by both 1H and 31P NMR. Extraneous peaks are visible in the olefinic region of the 1H NMR spectrum of impure ligand. In addition to the expected resonance at δ 146.0 ppm in the 31P NMR, impure samples of phosphoramidite ligand 64 also gave
82

83
small peaks at δ 139.7 and 138.1 ppm. The impurities are not visible by TLC and were not removed by either column chromatography or recrystallization.
101. Ley, S. V.; Anthony, N. J.; Armstrong, A.; Brasca, M. G.; Clarke, T.; Culshaw, D.; Greck, C.; Grice, P.; Jones, A. B.; Lygo, B.; Madin, A.; Sheppard, R. N.; Slawin, A. M. Z.; Williams, D. J. A Highly Convergent Total Synthesis of the Spiroacetal Macrolide (+)-Milbemycin β1. Tetrahedron 1989, 45, 7161-7194.
102. Paquette, L. A.; Earle, M. J.; Smith, G. F. (4R)-(+)-tert-Butyldimethylsiloxy-2-cyclopenten-1-one. In Organic Syntheses; Wiley & Sons: New York, 1998; Collect. Vol. No. 9, pp 132-136.
103. Gautier, E. C. L.; Graham, A. E.; McKillop, A.; Standen, S. P.; Taylor, R. J. K. Acetal and ketal Deprotection using Montmorillonite K10: The First Synthesis of syn-4,8-Dioxatricyclo[5.1.0.03,5]-2,6-octanedione. Tetrahedron Lett. 1997, 38, 1881-1884.
104. Moore, J. E.; Davies, M. W.; Goodenough, K. M.; Wybrow, R. A. J.; York, M.; Johnson, C. N.; Harrity, J. P. A. Investigation of the Scope of a [3 + 2] Cycloaddition Approach to Isoxazole Boronic Esters. Tetrahedron 2005, 61, 6707-6714.
105. Kim, J. N.; Ryu, E. K. 1,3-Dipolar Cycloaddition: Molecular Sieves Assisted Generation of Nitrile Oxides from Hydroximoyl Chlorides. Heterocycles 1990, 31, 1693-1697.
106. Still, W. C.; Kahn, M.; Mitra, A. Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J. Org. Chem. 1978, 43, 2923-2925.