a de novo nucleoside synthesis and late-stage
TRANSCRIPT

A De Novo Nucleoside Synthesis and Late-Stage
Heterobenzylic Fluorination Strategy
by
Michael Weiwei Meanwell
M.Sc., University of Victoria, 2015
B.Sc. (Hons.), University of British Columbia, 2014
Thesis Submitted in Partial Fulfillment of the
Requirements for the Degree of
Doctor of Philosophy
in the
Department of Chemistry
Faculty of Science
© Michael W. Meanwell 2020
SIMON FRASER UNIVERSITY
Summer 2020
Copyright in this work rests with the author. Please ensure that any reproduction or re-use is done in accordance with the relevant national copyright legislation.

ii
Approval
Name: Michael Weiwei Meanwell
Degree: Doctor of Philosophy
Title: A De Novo Nucleoside Synthesis and Late-Stage Heterobenzylic Fluorination Strategy
Examining Committee: Chair: Krzysztof Starosta Associate Professor
Robert Britton Senior Supervisor Professor
Roger Linington Supervisor Associate Professor
Robert Young Supervisor Professor
Peter Wilson Internal Examiner Associate Professor
Christopher Vanderwal External Examiner Professor Department of Chemistry University of California, Irvine
Date Defended/Approved: July 30, 2020

iii
Abstract
Nucleoside analogues constitute almost half of today’s major anticancer and antiviral
therapeutics. Despite this, synthetic routes to these valuable molecules have typically
relied on carbohydrate starting materials, which can significantly impair efforts in medicinal
chemistry. Moreover, nucleoside scaffolds with increased complexity (e.g., C2’ or C4’
substitution) often require lengthy syntheses (up to 18 steps). Toward a goal of
streamlining nucleoside synthesis, we have developed a one-pot proline-catalyzed α-
fluorination/aldol reaction that generates enantiomerically enriched fluorohydrins that can
serve as versatile building blocks for the construction of nucleoside analogues. Most
importantly, this process enables access to variously functionalized nucleoside analogues
in only 3 steps from commercial starting materials. The development of this process and
practical application in rapidly accessing C2’- and C4’- modified nucleoside analogues,
locked nucleic acids (LNAs), and iminonucleosides should inspire future efforts in drug
design. Similar challenges also obstruct the synthesis of carbohydrate analogues (CAs),
another important class of molecules to drug discovery efforts. To streamline CA
synthesis, we developed several new proline-catalyzed α-functionalization/aldol reactions
for constructing stereochemically rich and densely functionalized aldol adducts. In only 2
steps, these aldol adducts were then readily converted into a structurally diverse collection
of CAs including iminosugars, annulated furanoses, bicyclic nucleosides, and fluorinated
carbacycles.
Incorporation of a fluorine atom can have several profound effects on a drug’s
physiochemical properties – including metabolic stability, membrane permeability, and
potency. However, the introduction of fluorine into the heterobenzylic position of drug
molecules has remained an unsolved synthetic challenge. Towards this goal, we describe
the first unified platform for the late-stage mono- and difluorination and
trifluoromethylthiolation at heterobenzylic positions. This technology should become a
dynamic tool for drug-lead diversification.
Keywords: proline catalysis, nucleoside analogues, carbohydrate analogues, diversity-
oriented synthesis, late-stage fluorination

iv
Dedication
To my mom for everything that she does for her family

v
Acknowledgements
I am immensely grateful to my supervisor Prof. Rob Britton for the amazing opportunities
and valuable lessons he has given me. I am thankful for his endless patience and positivity
that is constant even in the most challenging of times. His passion and enthusiasm for
chemistry is truly an inspiration and is something I try to emulate everyday.
I am thankful to committee members Prof. Roger Linington and Prof. Robert Young for
their helpful suggestions and kind encouragement over the course of my studies. I want
to thank Prof. Peter Wilson for serving as my internal examiner but also for always showing
a keen interest in my research progress. I am thankful to Prof. Christopher Vanderwal for
taking the time out of his busy schedule to serve as my external examiner.
Some key aspects of this work would not have been possible without the support and help
of Dr. Steven M. Silverman at Merck Process. Thank you for all your efforts and
contributions.
I am thankful to Dr. Rainer E. Martin at Hoffman-La Roche for his support and efforts on
numerous projects.
Thank you to all members of the Britton group both past and present for insightful
discussions and for helping create such a wonderful place to work. To those that I had an
opportunity to collaborate with, it was truly a pleasure to have worked with you. I am
especially grateful to Dr. Matthew Nodwell for mentoring me and helping me through some
of the lows in life. A special thanks goes out to Dr. Milan Bergeron-Brlek, who mentored
me as an undergraduate student, for showing me the ropes in the lab.
I would not be here without the constant love and support of my friends and family. I am
very thankful to my Uncle Nicholas Meanwell for all the advice and help he has given me.
I want to thank my brother, Jason Wang, for believing in me even when I didn’t – you will
always be my best friend, teammate, and opponent. I am grateful to my partner, Nastaran
Yousefi, for her everlasting love and understanding, and for helping me see that there is
more to life than chemistry. I am thankful to my loving parents, Neil Meanwell and Sherrie
Wang, for giving me every opportunity in life to succeed and teaching me there is no
replacement for a strong work ethic.

vi
Table of Contents
Approval .......................................................................................................................... ii
Abstract .......................................................................................................................... iii
Dedication ...................................................................................................................... iv
Acknowledgements ......................................................................................................... v
Table of Contents ........................................................................................................... vi
List of Tables ................................................................................................................. viii
List of Figures................................................................................................................. ix
List of Schemes .............................................................................................................. xi
List of Acronyms ............................................................................................................ xiii
Chapter 1. Introduction .............................................................................................. 1
1.1. Fluorine in medicinal chemistry .............................................................................. 1
1.1.1. Conformational influence ............................................................................... 2
1.1.2. Metabolic stability .......................................................................................... 4
1.1.3. Potency ......................................................................................................... 5
1.1.4. Membrane permeability ................................................................................. 6
1.2. Fluorine as a synthetic handle ............................................................................... 7
1.2.1. Defluorination of C(sp3-F) bonds .................................................................... 7
1.2.2. C(sp2)-F nucleophilic substitution ................................................................. 10
1.2.3. Hydrodefluorination ..................................................................................... 12
1.3. Thesis overview ................................................................................................... 13
Chapter 2. Development of an α-fluorination/aldol reaction and an annulative fluoride displacement for de novo nucleoside analogue synthesis .............. 16
2.1. Nucleoside analogues in drug discovery .............................................................. 17
2.2. Current challenges in NA synthesis ..................................................................... 18
2.3. Proposal for de novo NA synthesis ...................................................................... 20
2.4. Development of an α-fluorination/aldol reaction and an annulative fluoride displacement ................................................................................................................. 21
2.5. Synthesis of C2’-modified nucleoside analogues ................................................. 25
2.6. Experimental ....................................................................................................... 26
General considerations .............................................................................................. 26
Chapter 3. A short de novo synthesis of C4’ nucleosides and locked nucleoside analogues ........................................................................................................... 81
3.1. C4’-modified NAs in drug discovery ..................................................................... 81
3.1.1. Synthetic challenges .................................................................................... 82
3.2. Rapid synthesis of C4’-modified NAs ................................................................... 84
3.2.1. Locked nucleic acids.................................................................................... 87
3.3. Experimental ....................................................................................................... 89
General considerations .............................................................................................. 89

vii
Chapter 4. A platform for diversity-oriented synthesis of carbohydrate analogues 108
4.1. Introduction to carbohydrate analogues ............................................................. 108
4.2. Development of α-functionalization/aldol reactions ............................................ 112
4.3. Rapid synthesis of CAs...................................................................................... 118
4.4. Experimental ..................................................................................................... 121
General Procedures ................................................................................................. 122
Chapter 5. A convenient late-stage flourination of pyridylic C-H bonds ............ 162
5.1. Synthesis of heterobenzylic fluorides ................................................................. 162
5.1.1. Deoxyfluorination ....................................................................................... 163
5.1.2. Halide-exchange reaction .......................................................................... 165
5.1.3. Electrophilic fluorination of heterobenzylic anions ...................................... 166
5.1.4. Late-stage C-H bond fluorination ............................................................... 167
5.1.5. Monofluoromethylation of C(sp2)-H bonds ................................................. 170
5.2. Late-stage fluorination of pyridylic C-H bonds .................................................... 170
5.3. Conclusion......................................................................................................... 177
5.4. Experimental ..................................................................................................... 177
General considerations ............................................................................................ 177
Chapter 6. Direct heterobenzylic monofluorination, difluorination and trifluoromethylthiolation with dibenzenesulfonamide derivatives ............... 188
6.1. Direct functionalization of heterobenzylic C-H bonds ......................................... 188
6.1.1. 18F-fluorination ........................................................................................... 195
6.2. Conclusion......................................................................................................... 195
6.3. Experimental ..................................................................................................... 196
General Considerations ........................................................................................... 196
Chapter 7. Future Work .......................................................................................... 218
7.1. Synthesis of NA and CA Screening-Libraries ..................................................... 218
7.2. Incorporating NAs into Antisense Oligonucleotides ............................................ 219
References ................................................................................................................. 220
Appendix A. Liquid-Chromatography Chromatograms .................................... 242

viii
List of Tables
Table 1.1. Fluorine’s effect on potency and selectivity ..................................................... 5
Table 1.2. Caco-2-permeability of Xa factor inhibitors ..................................................... 6
Table 4.1. α-functionalization/aldol reactions between pentanal and dioxanone (75) ... 113
Table 4.2. Optimization of α-chlorination/aldol with isovaleraldehyde (204) and thiopyranone 206.aketone and aldehyde added at the same time. ........ 114
Table 5.1 Fluorination of 4-alkylpyridines using NFSI. ................................................. 171
Table 6.1 Mono- and difluorination of 4-ethylpyridine (354) and alkyl quinolines 355 and 356 ....................................................................................................... 189

ix
List of Figures
Figure 1.1. Examples of fluorine-containing molecules in medicinal chemistry. ............... 1
Figure 1.2. The effect of fluorine’s on conformation ......................................................... 2
Figure 1.3. Structural conformations of 17 and 18 ........................................................... 3
Figure 1.4. Stragetic use of fluroine to block unproductive metabolsim ........................... 4
Figure 1.5. A) Metabolic precursors of fluoroacetic acid (21). B) Mediating the metabolic profile of KSP inhibitors ............................................................................ 5
Figure 2.1 Nucleoside analogues in drug discovery ...................................................... 17
Figure 2.2. Current challenges in nucleoside synthesis ................................................. 18
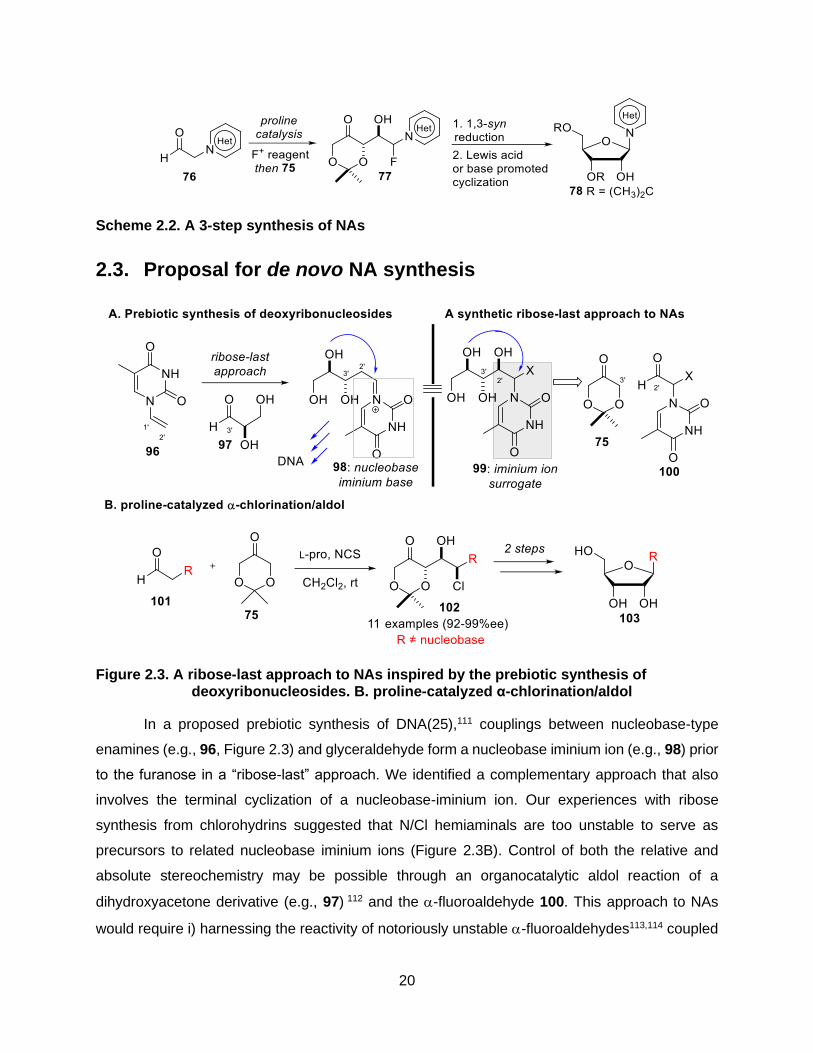
Figure 2.3. A ribose-last approach to NAs inspired by the prebiotic synthesis of deoxyribonucleosides. B. proline-catalyzed α-chlorination/aldol ............. 20
Figure 2.4. Optimization of αFAR and AFD ................................................................... 21
Figure 2.5. Mechanism of cyclization of 107a and 107b ................................................ 22
Figure 2.6. Scope of nucleoside and NA synthesis. ....................................................... 24
Figure 2.7. C3’/C5’ protected nucleosides (R = C(CH3)2) and β-L-nucleosides. ............. 25
Figure 2.8. C2’-modified nucleoside analogues (R = C(CH3)2) ....................................... 26
Figure 3.1. C4’ analogues in medicinal chemistry .......................................................... 81
Figure 3.2. Synthesis of C4’-modified analogues (R = C(CH3)2) .................................... 86
Figure 3.3. Common locked nucleic acid analogues ...................................................... 87
Figure 3.4. Short syntheses of LNAs ............................................................................. 88
Figure 4.1 Carbohydrate analogues in drug discovery ................................................. 108
Figure 4.2. Development of α-functionalization/aldol reactions for drug discovery ....... 111
Figure 4.3. Ketone scope of α-chlorination/aldol reaction ............................................ 115
Figure 4.4. Scope of α-functionalization/aldol reactions. .............................................. 117
Figure 4.5. Scope of α-functionalization/aldol reactions ............................................... 118
Figure 4.6. Platform for rapid diversity-oriented synthesis (253 = 5-(methanesulfonyl)-1-phenyl-1H-tetrazole) ............................................................................. 118
Figure 4.7. Rapid synthesis of CAs.aα:β = 2.5:2.bα:β = 3:1. ......................................... 120
Figure 4.8. Conversion of fluorohydrins 221, 226, and 230 into biologically relevant molecules ............................................................................................. 121
Figure 5.1. Primary sites of metabolism in omeprazole (277) and pioglitazone (279) and the effects of heterocycles and fluorine on physicochemical properties in omarigliptin (278) and gefitinib (280). ................................................... 163
Figure 5.2 Common deoxyfluorination reagents .......................................................... 164
Figure 5.3. Common electrophilic fluorinating N-F reagents ........................................ 166
Figure 5.4 Selective pyridylic fluorination of C-H Bonds .............................................. 174
Figure 6.1. Heterobenzylic fluorides in discovery ......................................................... 188
Figure 6.2. Mono- and difluorination of pyridines, quinolines, pyrimidines, isoquinolines, quinazolines, and purines. .................................................................... 191

x
Figure 6.3. Trifluoromethylthiolation and chlorination of purines and quinazolines ....... 193
Figure 7.1. C2’/C4’-modified NAs for incorporation into oligonucleotides ..................... 219

xi
List of Schemes
Scheme 1.1. Deflourative functionalization. A) Glycosylation of glycosyl fluorides. B) Friedel-Crafts reaction of aliphatic tertiary fluorides .................................. 7
Scheme 1.2. Stereospecific fluoride displacement .......................................................... 9
Scheme 1.3. A) Nucleophilic aromatic substitution for macrocyclization. B. Process scale intramolecular nucleophilic heteroaromatic substitution .......................... 10
Scheme 1.4. Reactivity pathways of gem-difluoroalkenes ............................................. 11
Scheme 1.5. Hydrodefluorination strategies .................................................................. 12
Scheme 1.6. The development of an α-fluorination/aldol reaction (αFAR) and an annulative fluoride displacement (AFD) for nucleoside analogue synthesis ............................................................................................................... 13
Scheme 1.7. Development of new α-functionalization/aldol reactions for carbohydrate analogue synthesis ................................................................................ 14
Scheme 1.8. The direct monofluorination, difluorination, and trifluoromethylthiolation of heterobenzylic C-H bonds ...................................................................... 14
Scheme 2.1. MacMillan’s de novo synthesis of C2’-modified NAs ................................. 19
Scheme 2.2. A 3-step synthesis of NAs......................................................................... 20
Scheme 2.3. Application to the synthesis of MK-3682 penultimate (130) (R = C(CH3)2).26
Scheme 3.1. Common building blocks for the construction of C4’-modified NAs ........... 82
Scheme 3.2. Merck’s syntheses of MK-8591 ................................................................. 83
Scheme 3.3. Synthesis of C4’-allyl C2’-deoxy NA (R = C(CH3)2) ................................... 87
Scheme 4.1. Diversity-oriented synthesis approaches to CAs ..................................... 109
Scheme 4.2. One-pot synthesis of chlorohydrins and DKR of α-chloroaldehydes through proline-catalyzed aldol reactions .......................................................... 112
Scheme 5.1. Deoxyfluorination of quinine led to inversion (282), retention (283), and rearrangement (284) products .............................................................. 164
Scheme 5.2. Late-stage enzymatic oxidation enabled deoxyfluorination ..................... 165
Scheme 5.3 Halide-exchange reaction with silver fluoride ........................................... 165
Scheme 5.4 Halide-exchange reaction for the synthesis of 18F radiotracer (290) ......... 166
Scheme 5.5. Heterobenzylic fluorination of camptothecin ........................................... 167
Scheme 5.6. Manganese-catalyzed C-H fluorination for the generation of 18F radiotracers .......................................................................................... 168
Scheme 5.7. Transition metal-free radical C-H fluorination .......................................... 168
Scheme 5.8. Palladium-catalyzed diastereoselective C-H fluorination ......................... 169
Scheme 5.9 Sanford’s palladium-catalyzed C-H fluorination methods ......................... 169
Scheme 5.10. Fluorination with Selectfluor .................................................................. 170
Scheme 5.11. Fluorination of 4-(cyclopropylmethyl)pyridine (311) and a mechanistic proposal for the formation of 308 and decomposition products of NFSI 172
Scheme 5.12 Direct fluorination of the potent aldosterone synthase inhibitors 333 and 335 ....................................................................................................... 175

xii
Scheme 5.13 Site-selective late-stage fluorination of pyridylic, benzylic, or aliphatic C-H bonds, contrasted with classical α-fluorination ...................................... 176
Scheme 6.1 Late-stage mono- and difluorination, trifluoromethylthiolation of heterocycles ............................................................................................................. 194
Scheme 6.2. 18F-fluorination of heterocycle 407 .......................................................... 195
Scheme 7.1. Synthesis of C2’/C4’-modified NA and ProTide libraries ......................... 218

xiii
List of Acronyms
[α]D specific rotation at the sodium D line (589 nm)
oC Degrees Celsius
Ac acetyl
AcOH acetic acid
AFD annulative fluoride displacement
AIBN azobisisobutyronitrile
BAIB (Diacetoxyiodo)benzene
Bn benzyl
BuLi butyllithium
tBuOK potassium tert-butoxide
CAs carbohydrate analogues
CSA camphor-10-sulfonic acid
D dextrorotatory
DCE 1,2-dichloroethane
DIPEA N,N-diisopropylethylamine
DMF N,N-dimethylforamide
DMSO dimethylsulfoxide
4-DPN-IPN 2,4,5,6-tetrakis(diphenylamino)isophthalonitrile
D-pro D-pro
dr diastereomic ratio
E+ electrophile
EC50 half maximal effective concentration
ee enantiomeric excess
e.g. Exempli grata
Equiv. equivalents
Et ethyl
Et2O diethyl ether
EtOAc ethyl acetate
αFAR α-fluorination-aldol reaction
FDA Food and Drug Administration
18F-DOPA [18F]fluorodeoxyphenylalanine
[18F]FDG 2-deoxy-[18F]fluoroglucose
GABA Gamma Aminobutyric Acid
HAT hydrogen atom transfer
HBTU Hexafluorophosphate Benzotriazole Tetramethyl Uronium

xiv
Het heterocycle
HFIP 1,1,1,3,3,3-Hexafluoro-2-propanol
HMBC heteronuclear multiple bond correlation
HPLC high-performance liquid chromatography
HSQC heteronuclear single quantum coherence
4-HTP 4-hydroxythiophenol
Hz Hertz
i isopropyl
IC50 half maximal inhibitory concentration
i.e. Id est
K2CO3 potassium carbonate
KHMDS potassium bis(trimethylsilyl)amide
KSP kinesin spindle protein
L levorotary
LDA lithium diisopropyl amine
LEDS light-emitting diode
LG leaving group
LNAs locked nucleic acids
L-pro L-proline
M molar
MDG multidrug resistance protein 1 (MDR) ratio
Me methyl
MeCN acetonitrile
MeNO2 nitromethane
MeOH methanol
mmol millimole
mol mole
NaDT sodium decatungstate
NAs nucleoside analogues
n normal (alkyl chain prefix)
NBS N-bromosuccinimide
NCS N-chlorosuccinimide
NFSI N-fluorobenzenesulfonimide
NMP N-methyl-2-pyrrolidine
NMR nuclear magnetic resonance
nOe nuclear Overhauser effect
Nu nucleophile

xv
Pe membrane permeability
PET Positron Emission Tomography
P-gp P-glycoprotein
pKa −logKa
pH −log[H+]
Ph phenyl
PhthNSCF3 N-trifluoromethylthiophthalimide
PMP 1,2,2,6,6-pentamethylpiperidine
PNB p-nitrobenzoate
ppm parts-per-million
PRMT5 Protein arginine methyltransferase 5
rt room temperature
SET single electron transfer
SNAr nucleophilic aromatic substitution
t tertiary
TBS tert-butyldimethylsilyl
TCDI 1,1’-thiocarbonyldiimidazole
TEMPO (2,2,6,6-tetramethylpiperidine-1-yl)oxidanyl
TFA trifluoroacetic acid
THF tetrahydrofuran
TIPS triisopropylsilyl
TMP 2,2,6,6-tetramethylpiperidine
TS transition structure
δ chemical shift (in ppm) from tetramethylsilane

1
Chapter 1. Introduction
1.1. Fluorine in medicinal chemistry
The strategic use of fluorine and fluorine-containing motifs has profoundly impacted drug
design and development.1,2 Remarkably, since the first fluorinated drug (fludrocortisone (1),
Figure 1.1) was released to the market in 1955, over 270 fluorine-containing drugs have been
approved by the U.S. Food and Drug Administration (FDA) and now constitute roughly a third of
all major pharmaceuticals.3 Continued efforts in this vibrant area of chemical research have led
to a greater understanding of the effects of fluorine incorporation and its application in drug design.
Fluorine is often introduced into a drug candidate to enhance its pharmacokinetic and
pharmacodynamic properties such as potency, membrane permeability, lipophilicity,
bioavailability, and metabolic stability. 3–6 However, the influence of fluorination on these
properties can be difficult to predict. In recent years, innovations in chemical synthesis7,8 have
enabled access to novel fluorinated motifs,5,9 thus providing new opportunities to probe the effect
of fluorination on drug properties. Due to its low positron emission energy (0.64 MeV) and
favourable half-life (t1/2 = 109.8 min), novel 18F radiotracers in PET (positron emission
tomography) imaging as disease diagnostics are also highly valuable.10–13 Most notably, 2-deoxy-
[18F]fluoroglucose ([18F]FDG, 2)14 and 18F-DOPA (3)15 are clinical radiotracers used in diagnosing
and monitoring disease progression in cancer and Parkinson’s disease, respectively. Considering
the utility of fluorine in pharmaceuticals and clinical radiotracers, it is expected that the number of
fluorine-containing pharmaceuticals and radiotracers will continue to grow.
Figure 1.1. Examples of fluorine-containing molecules in medicinal chemistry.

2
1.1.1. Conformational influence
Figure 1.2. The effect of fluorine’s on conformation
Fluorine can uniquely affect molecular conformations.1,4,6,16,17 The predominant conformer
is influenced by energic contributions from dipole-dipole interactions, charge-dipole interactions,
and hyperconjugation. Due to the highly polarized nature of C-F bonds, the associated low lying
σ*C-F antibonding orbital can readily accept donating electron density from vicinal C-H bonds. This
form of hyperconjugation is known as the gauche effect and significantly influences molecular
conformations of 1,2-difluoroalkanes to favour the gauche conformer. 4,16,17 For instance, in 1, 2–
difluoroethane the gauche conformer (4a) is the lowest energy conformation in contrast to the
anti-conformer (4b) which is expected to have the least steric hindrance (Figure 1.2A). In the
gauche conformer, the σ*C-F orbital is aligned antiparallel with a vicinal σC-H bonding orbital so that
the σC-H orbital can donate electron density into the vacant σ*C-F orbital. Notably, the gauche effect
is more pronounced in systems featuring F-C-C-O or F-C-C-N (see 6 - 8). Comparatively,
electrostatic interactions (i.e. charge-dipole, dipole-dipole) offer a greater degree of stabilization
than hyperconjugation. In cases where the adjacent substituent bears a positive charge (e.g. 9 –
11), charge-dipole interactions with the partially negative fluorine atoms results in these molecules
adopting the gauche conformer. Likewise, dipole-dipole interactions within the same molecule
can also provide stabilization towards a particular conformation. For example, in α-
fluoroacetamide (12) the fluorine and the carbonyl oxygen are oriented antiperiplanar to minimize
the net dipole of the molecule, thus providing an overall stabilization of 7.5 kcal/mol. A weak

3
hydrogen bonding interaction between the amide N-H and fluorine may also contribute to this
stabilization.
Figure 1.3. Structural conformations of 17 and 18
In drug design, introduction of a fluorine atom is a common tool used for changing
molecular conformations to tune basicity and target-binding affinity. Towards studying
conformation-activity relationships, O’Hagan first synthesized and then evaluated the binding
properties of 3F-GABA enantiomers (17 and 18) against GABAA receptor and GABA
aminotransferase. 18 While both enantiomers were considerably less potent than the natural
substrate GABA, 17 and 18 themselves exhibited similar binding affinities toward GABAA
receptor, however, GABA aminotransferase metabolized 18 10-fold faster compared to 17.
O’Hagan rationalized these observations based on the conformational preferences of each
enantiomer which are summarized in Figure 1.3. As discussed previously, the gauche conformers
(17a/17b and 18b/18c) were assigned as the more stable conformations. Furthermore,
conformers 18a and 17c are disfavoured owing to the antiperiplanar alignment of NH3+ and
fluorine which increases each conformers’ net dipole. The equipotency of the two analogues
towards GABAA receptor suggests that the antiperiplanar alignment of the NH3+ and COO- in 17b
and 18b, both energetically favourable conformations, is important to receptor recognition.
Interestingly, the metabolism of 18 by GABA aminotransferase produced 10-fold more hydrogen
fluoride compared to 17. For elimination of fluoride to occur, fluoride must be positioned
antiperiplanar to a β-hydrogen. Taken together, this points towards conformation 18c possessing

4
the preferred binding orientation of NH3+ and COO- which is an energetically disfavoured
conformation in 17.
1.1.2. Metabolic stability
Many classes of metabolic enzymes, such as cytochrome P450 (CYP) enzymes,
hydrolases, and peroxidases, are involved in drug metabolism.19 The resulting drug metabolites
can provide crucial insight into metabolic pathways and help guide drug design efforts. The short
C(sp3) -F bond (1.40 Å) is the strongest bond (110 kcal/mol) in organic chemistry and, due to
fluorine’s small Van der Waals radius (1.47 Å), it is an effective isostere for a hydrogen atom or a
hydroxyl group. In drug development, strategic fluorine incorporation can be used to block
unproductive metabolism. As a notable example, the aryl fluorides in Ezetimibe (20), a drug used
to reduce cholesterol absorption in patients with hypercholesterolaemia, prevents CYP mediated
aromatic C-H oxidation and O-demethylation that was previously observed in the initial lead
compound 19.20 Ultimately, fluorine replacement, in addition to stereoselective benzylic oxidation
and demethylation, led to a 50-fold increase in activity and an improved pharmacological profile.
Figure 1.4. Stragetic use of fluroine to block unproductive metabolsim
Toxic fluorine-containing metabolites, such as fluoroacetic acid (21), are a recognized
problem when working with fluorinated drugs. Fluoroacetic acid, which can also be generated
from several metabolic precursors (Figure 1.5, 22-27), is lethal to humans at levels ranging from
2 – 10 mg/kg and has encountered toxicological issues during drug discovery campaigns.3 For
instance, to develop an effective treatment for taxane-refractory solid tumors, Merck investigated
piperidine-based kinesin spindle protein (KSP) inhibitors with a specific focus of reducing P-
glycoprotein (P-gp) efflux by tuning the pKa of the piperidine nitrogen to a range of 6.5 to 8.0.21 P-
gp efflux is measured as the ratio of the IC50 evaluated in a cell-line overexpressing P-gp over the

5
IC50 in the parental cell-line that does not express P-gp and is denoted as the multidrug resistance
protein 1 (MDR) ratio.21
Figure 1.5. A) Metabolic precursors of fluoroacetic acid (21). B) Mediating the metabolic profile of KSP inhibitors
As shown in Figure 1.5, the N-2-fluoroethyl analogue 28 had an MDR ratio of 4.5, well
below the predetermined maximum threshold of 10, and an optimal pKa value (7.6). However, in
rats 28 was found to be severely toxic as it undergoes N-dealkylation presumably leading to
fluoroacetic acid via a metabolic precursor 22. Interestingly, fluorine incorporation into the
piperidine ring produced the desired reduction in basicity for compound 30 (MK-0731) while also
avoiding the production of fluoroacetic acid as a primary metabolite.
1.1.3. Potency
compound R R1 17β-HSD1 IC50 (nM)
17β-HSD2 IC50 (nM)
selectivity factor
31 H H 69 1950 28 32 F H 8 940 118 33 F F 56 312 6
Table 1.1. Fluorine’s effect on potency and selectivity

6
The tactical introduction of fluorine into drug candidates is widely used to enhance potency
and target affinity.1,4,6 As mentioned previously, one of the more common strategies is the
substitution of a hydrogen atom for a fluorine atom.9 The effectiveness of this approach largely
depends on the drug-target interactions that are being optimized. For example, Hartmann
explored aryl fluoride substitutions of 31 for the selective inhibition of 17β-hydroxysteroid
dehydrogenase 1 (17β-HSD1), an important target for treating osteoporosis.22,23 32 demonstrated
improved potency and selectivity over 17β-hydroxysteroid dehydrogenase 1 (17β-HSD2), which
catalyzes the reverse reaction (i.e. oxidation of estradiol). Accordingly, it was postulated fluorine
increases the acidity of the phenol moiety allowing for increased hydrogen bonding with Glu282
and His221 in the active site of 17β-HSD1, a key interaction that mimics the enzyme’s natural
substrate (estrogen). Addition of a second fluorine atom reduced potency and selectivity, thus
suggesting the phenol proton has an optimal pKa for this interaction.
1.1.4. Membrane permeability
compd R R1 Caco-2-permeability compd R2 Caco-2-permeability
34 H CH3 1.20 ± 0.09 x 10-6 cm/s 38 H 0.8 x 10-6 cm/s 35 F CH3 3.14 ± 0.10 x 10-6 cm/s 39 F 7.4 x 10-6 cm/s 36 H CF3 3.38 ± 0.08 x 10-6 cm/s 40 CN < 0.1 x 10-6 cm/s 37 F CF3 4.86 ± 0.33 x 10-6 cm/s
Table 1.2. Caco-2-permeability of Xa factor inhibitors
A drug candidate’s cellular membrane permeability (Pe) can significantly affect its
pharmacological and toxicological profiles.4 In regard to passive diffusion across cellular
membranes, Pe decreases with molecular size and increases with lipophilicity. Fluorine
incorporation has been used to increase a molecule’s lipophilicity and, consequently, improve a
molecule’s membrane permeability via passive diffusion by affecting lipophilicity, intramolecular
hydrogen bonding interactions, and amine basicity. As an illustrative example, compounds 34-40

7
were investigated for their Xa factor inhibition for the development of anticoagulant drugs.24,25
Substituting fluorine in for the hydrogen ortho to the anilide N-H resulted in improved membrane
permeability as measured by the Caco-2 permeability assay for 35, 37, and 39 compared to their
hydrogen analogues. Presumably, the ortho anilide N-H is a hydrogen bond donor that interacts
with the ortho fluoride to increase membrane permeability. In contrast, replacement of the
hydrogen with a nitrile instead significantly reduces permeability in 40 as no intramolecular
hydrogen bonding is achievable.
1.2. Fluorine as a synthetic handle
Over the past two decades, C-F bond forming reactions have been explored extensively
to facilitate the growing utility of fluorinated molecules in pharmaceuticals,26 disease
diagnostics,10,11,13 agrochemicals,27 and material science.28 This has also resulted in the
investigation into the unique reactivity of C-F bonds (i.e. C-F bond activation) themselves and the
use of fluorinated molecules as versatile building blocks in chemical synthesis.29,30 To date, C-F
bond activation has been used for glycosylation,31–41 Friedel-Crafts,42–49 nucleophilic
substitution,50–56 dehydrofluorination, 57–60 transition metal cross-coupling,61–64 and halide-
exchange reactions.65,66
1.2.1. Defluorination of C(sp3-F) bonds
Scheme 1.1. Deflourative functionalization. A) Glycosylation of glycosyl fluorides. B) Friedel-Crafts reaction of aliphatic tertiary fluorides

8
Fluoride abstraction with a Lewis acid is a common way to exploit C-F bond reactivity in
defluorinative functionalization reactions. For instance, Lewis acids catalyze the glycosylation of
glycosyl fluorides.32 This has led to the emergence of glycosyl fluorides as an attractive alternative
to traditional glycosyl donors (i.e. glycosyl halides,68 pentenyl glycosides,67
trichloroacetimidates68) in carbohydrate synthesis. The additional strength of the C-F bond affords
glycosyl fluorides improved stability over other glycosyl halides and trichloroacetimidates while
also remaining reactive under mild glycosylation conditions. As depicted in Scheme 1.1A,
Montgomery reported fluoride abstraction with tris(pentafluorophenyl)borane to catalyze the
stereospecific glycosylation of over 35 mono- and disaccharides with a variety of silyl ethers.36
Mechanistically, anchimeric assistance facilitates fluoride abstraction, via oxacarbenium
intermediate 42, and ultimately enables the stereospecific delivery from the silyl ether. Activation
of pyranosyl and furanosyl fluorides with other Lewis acids are well-documented. The use of
C(sp3)-F bond defluorination in Friedel-Crafts alkylations have also been independently reported
by Stephan,45–47 Moran,48 Paquin,44 and Kemnitz.49 Notably, Moran has demonstrated the use of
aliphatic tertiary fluorides in Friedel-Crafts reactions (Scheme 1.1B).48 Here, the
tris(pentafluorophenyl)borane catalyst abstracts fluoride to generate the carbocation species for
reaction with electron-rich (hetero)arenes. While fluorine is generally considered a poor hydrogen-
acceptor, Paquin showed the carbocation species could also be generated from benzylic fluorides
by hydrogen-bonding with HFIP (a strong hydrogen-bond donor) in the synthesis of 1,1-diaryl
methanes.44

9
Scheme 1.2. Stereospecific fluoride displacement
Rare examples of nucleophilic substitutions of aliphatic fluorides under basic or neutral
conditions exist.50–53 Hu reported the intramolecular fluoride displacement of benzylic fluorides
with tertiary and secondary alkoxides for the synthesis of annulated dihydrofurans (Scheme 1.2A)
In this one-pot-two-step sequence, treatment of 47 with n-butyllithium afforded the organolithium
species which then reacts with benzaldehyde to form the alkoxide in situ. Investigations into the
mechanism of cyclization revealed that the loss of fluoride occurred via an SN2 process. From
their work towards bicyclic morpholine and piperidine cores, researchers at Pfizer Inc. explored
nucleophilic fluoride displacement for the construction of the second ring.51 As depicted in
Scheme 1.2B, exposing 51 to KHMDS was unsuccessful due to geometric constraints; however,
under the same reaction conditions, they were able to cyclize compound 52 to compound 54 in
excellent yield. The PNB protecting group allowed epimerization of the α-fluoromethine, thus
enabling backside attack from the tethered alkoxide.

10
1.2.2. C(sp2)-F nucleophilic substitution
Scheme 1.3. A) Nucleophilic aromatic substitution for macrocyclization. B. Process scale intramolecular nucleophilic heteroaromatic substitution
Contrary to reactivity trends observed in nucleophilic substitution reactions of aliphatic
fluorides, aryl fluorides are the most reactive among the aryl halides towards nucleophilic aromatic
substitution (SNAr).29 It is believed SNAr is a step-wise process that proceeds through a transient
reaction intermediate known as the Meisenheimer complex (57), though there is recent evidence
supporting a concerted pathway.69 In SNAr reactions, the rate-determining step (RDS) is the
addition of the nucleophile to the aryl halide. This process kinetically favours aryl fluorides over
other aryl halides due to fluorine being comparatively small and strongly electron-withdrawing.
The preferential reactivity of aryl fluorides is demonstrated in the reaction between 1-bromo-3-
chloro-5-fluorobenzene and cyanoacetate which exclusively displaces fluoride to afford a single
regioisomer (> 30:1).56,70 SNAr reactions have proven to be a highly useful tool for the construction
of macrocycles, as well as for heterocycle synthesis and functionalization in drug discovery. As
shown in Scheme 1.2, reacting 55 with potassium carbonate led to a productive macrocyclization
and afforded the 14-membered ring macrocycle 56 for the synthesis of cycloisodityrocine
derivatives.71 Amgen Inc reported a scalable route to their dual FLT3/CDK4 inhibitor AMG 925

11
(60) for the treatment of acute myeloid leukemia in which the 3-fluoro-pyridyl moiety is a synthetic
handle for base-promoted intramolecular SNAr cyclization to afford the tricyclic intermediate 59.54
Scheme 1.4. Reactivity pathways of gem-difluoroalkenes
The C(sp2)-F bond of fluoroalkenes can undergo nucleophilic substitution through a similar
addition-elimination pathway observed in SNAr reactions. For instance, upon base or acid
treatment gem-difluoroalkenes 61 engage in intramolecular nucleophilic substitutions to generate
a variety of cyclic scaffolds including cyclopentenes, dihydrofurans, dihydropyrroles, and
dihydrothiophenes.55 The double bond of gem-difluoroalkenes is highly polarized as the 13C NMR
indicates an electron deficient difluoromethylene carbon (~ 155 ppm) and an electron rich
methylene carbon (~ 90 ppm).55 Notably, these characteristics engender gem-difluoroalkenes
with two additional modes of reactivity. Rather than eliminating fluoride to regenerate the double
bond, the intermediate carbanion 65 can react with electrophiles to afford 1, 2-addition products.
Alternatively, in the case where the γ-carbon is substituted with a leaving group, an SN2’ process
can lead to difluoromethyl alkene products 66.

12
1.2.3. Hydrodefluorination
Scheme 1.5. Hydrodefluorination strategies
Recently reported protocols for the selective hydrodefluorination of trifluoromethyl
aromatics have provided direct access to the corresponding difluoromethyl products, a valuable
motif in modern drug design. 57–60 While several transition metal cross-coupling and C-H
functionalization reactions have been well-established for introducing difluoromethyl groups into
aromatics and heteroaromatics, 72–88 converting a trifluoromethyl group directly into the
difluoromethyl analogue is a powerful tool for a medicinal chemist. Admittedly, over
hydrodefluorination to the corresponding monofluoromethyl and methyl derivatives is a common
concern in developing these methodologies.57–60 Olah reported the selective hydrodefluorination
of 1,3-bis(trifluoromethyl)arenes using magnesium powder under acidic conditions.58 As
highlighted in Scheme 1.5A, these conditions converted Netupitant (67) into its difluoromethyl
analogue 68 without producing any of the monofluoromethyl side-product. Conversely,

13
subsequent work by Jui58 and Gouverneur59 relied on photoredox approaches to generate a
radical anion intermediate 70. Following loss of fluoride, the resulting difluoromethyl radical 71 is
then trapped via hydrogen atom transfer (HAT) to afford the desired difluoromethyl analogue.
1.3. Thesis overview
In this thesis, the discoveries of 1) novel proline-catalyzed α-functionalization/aldol
reactions for short syntheses of nucleoside and carbohydrate analogues and 2) the late-stage
functionalization of heterobenzylic C-H bonds are presented.
Scheme 1.6. The development of an α-fluorination/aldol reaction (αFAR) and an annulative fluoride displacement (AFD) for nucleoside analogue synthesis
Chapter 2 reports a “ribose-last” approach for the 3-step de novo synthesis of nucleoside
analogues (NAs). NAs are the leading class of antiviral drugs and make up a significant portion
of anticancer therapeutics. Our strategy involves the development of a proline-catalyzed α-
fluorination/aldol reaction (αFAR) to access enantioenriched fluorohydrin intermediates 77 and
unprecedented annulative fluoride displacement (AFD) reaction for cyclization to a protected
nucleoside core 78. The utility of this unique process is highlighted in the synthesis of several C2’-
modified NAs.
Chapter 3 discusses the short syntheses of C4’-modified NAs via 1,2-additions of Grignard
reagents to the aforementioned fluorohydrin intermediates 77 and subsequent AFD. To date,
current syntheses have relied on semisynthetic approaches that are lengthy (~10 steps),
protracted, and not amenable to rapid diversification. Furthermore, we also present short
syntheses of locked nucleic acids (LNAs), a class of conformationally rigid NAs that are
incorporated into oligonucleotides to increase stability and potency of antisense therapeutics.
Ultimately, this process creates opportunities for preparing diversity libraries and will support
future efforts in drug discovery.

14
Scheme 1.7. Development of new α-functionalization/aldol reactions for carbohydrate analogue synthesis
Chapter 4 presents the development of novel asymmetric α-functionalization/aldol reactions for
the synthesis of carbohydrate analogues (CAs). Current syntheses of CAs have relied on
semisynthetic approaches that have limited rapid access to structurally diverse CAs. In contrast,
our strategy readily constructs several unique CA scaffolds from 81, a versatile building block that
can be generated using only proline-catalysis and cheap achiral building blocks in a single step.
This 3-step synthesis enabled access to iminosugars, annulated furanoses, bicyclic nucleosides,
and fluorinated carbacycles.
Scheme 1.8. The direct monofluorination, difluorination, and trifluoromethylthiolation of heterobenzylic C-H bonds
Chapter 5 reports the late-stage monofluorination of pyridylic C-H bonds with N-
fluorobenzenesulfonimide (NFSI). There have been significant advances in the fluorination of
benzylic C-H bonds but these methods have not translated to the fluorination of heterobenzylic
C-H bonds. This strategy is the first method reported for the selective C-H fluorination of 2- and
4-alkyl pyridines, and proceeds via a transient N-sulfonated pyridinium intermediate. Most
importantly, the reaction is tolerant to a wide range of different functional groups present in drug
discovery and we demonstrate its utility for site-selective drug lead fluorination. Considering
nearly 60% of all FDA approved drugs contain at least one heterocycle, this reaction should
become a useful tool for the modulation of drug leads’ physicochemical properties.

15
Chapter 6 reports the improved versatility of the work presented in Chapter 5 and both the C-H
monofluorination and difluorination of pyridines, pyrimidines, quinolines, purines, isoquinolines,
and quinazolines are presented. While advances in the addition of fluoroalkyl radicals to
heterocycles have been made, direct C(sp3)–H heterobenzylic fluorination is comparatively
unexplored. Here we demonstrate both mono- and difluorination of a range of alkyl heterocycles
using a convenient process that relies on transient sulfonylation by the electrophilic fluorinating
agent N-fluorobenzenesulfonimide. We also report heterobenzylic trifluoromethylthiolation and
18F-fluorination, providing a suite of reactions for late-stage C(sp3)–H functionalization of drug
leads and radiotracer discovery.

16
Chapter 2. Development of an α-fluorination/aldol reaction and an annulative fluoride displacement for de novo nucleoside analogue synthesis
The results presented in this chapter have been reported in part, see:
Meanwell, M.; Silverman, S. M.; Lehmann, J.; Adluri, B.; Wang, Y.; Cohen, R.; Campeau, L.-C.;
Britton, R. 2020, Science. Accepted.
Several colleagues contributed to this work. Dr. Steven M. Silverman optimized the route to
compounds 121 and 122. Dr. Johannes Lehmann developed the route to 130. Dr.
Bharanishashank Adluri optimized the AFD reaction for 106 and 108. Dr. Yang Wang obtained
analytical data for compounds 113, 114, and 118. Dr. Ryan Cohen developed the J-based
configurational analysis for the determination of relative configuration.

17
2.1. Nucleoside analogues in drug discovery
Figure 2.1 Nucleoside analogues in drug discovery
As fundamental biomolecules, nucleosides play key roles in diverse cellular processes
ranging from cell signalling to metabolism.89 Not surprisingly, synthetic nucleoside analogues
(NAs), designed to mimic their natural counterparts, are widely exploited in medicinal chemistry90–
95 and used as tool compounds in chemical biology. In fact, NAs have been in use for over half a
century for the treatment of cancer90,92 and represent the largest class of small molecule antivirals
(e.g., 86 – 91, Figure 2.1). 94,95 Mechanistically, NAs operate as toxic antimetabolites that interfere
with nucleic acid synthesis.95 Alternatively, following in vivo phosphorylation, the resulting
nucleotide analogues can inhibit enzymes crucial to cancer cell growth or viral replication (e.g.,
DNA/RNA polymerases, ribonucleotide reductases or nucleoside phosphorylases).90,95 Recently,
NAs have also demonstrated promise as epigenetic modulators, and both decitabine and
azacitidine inhibit DNA methyltransferase and have been approved for cancer therapy.95
The continued discovery and development of anticancer and antiviral NAs builds on
several decades of medicinal chemistry knowledge. 92,96,97 For example, in the early 1980’s it was
found that fluorination at C2' improves metabolic stability (i.e., decreases hydrolysis) and can
influence furanose conformation and enzyme binding (PSI-620698: 86, Figure 2.1).4,94,96 Likewise,

18
replacement of a proton at C2' with a methyl group alters the conformation, and thus C2'-methyl
NAs operate as nonobligate chain terminators.97,99 Modifications at C3' generally interrupt
extension of a growing nucleic acid chain, while C4' functionalization (e.g., methyl, azido, alkynyl)
can beneficially influence furanose conformation or attenuate reactivity of the C3' and C5' alcohols
toward chain extension (MK-8591: 89). 97,100 Modified nucleobases have also been studied to
improve potency, pharmacokinetic and pharmacodynamic properties. For example, NAs
possessing 5-membered ring nucleobases such as ribavirin (90)101 mimic structurally related
intermediates in de novo purine nucleotide biosynthesis and can modulate the activity of enzymes
in this pathway.102 In recent years there has also been increased interest in unnaturally configured
NAs.103,104*For example, the -L-NA Lamivudine (3TC)91 has found widespread use in the
treatment of HIV-1/AIDS and several -D-thymidine analogues have demonstrated promise as
antimalarials. 91
2.2. Current challenges in NA synthesis
Figure 2.2. Current challenges in nucleoside synthesis
Despite decades of NA research, the synthesis of NAs still presents many challenges.105 Firstly,
NAs are often synthesized from naturally occurring carbohydrates, which limits patterns of
substitution and furanose stereochemistry. Secondly, the addition of nucleobases to activated
ribose derivatives (i.e., the Vorbrüggen reaction) often fails or proceeds with poor
diastereoselectivity when NAs are functionalized at C2' or C4'. 106,107 Thirdly, modifications at C2'
often require multistep protecting group manipulations of the C3' and C5' alcohol functions. While
such processes can be accomplished with siloxanes for discovery purposes,108 they are cost
prohibitive on production scale. Lastly, modular and efficient strategies for producing C4' modified
* By convention, -D-NAs are naturally configured (e.g., 86 – 91), while L- denotes the NA is epimeric at
C4' and - or - indicates a trans- or cis-relationship, respectively, between the nucleobase and the C4'-CH2OH group.

19
NAs do not exist, which continues to challenge medicinal and process research chemists. In fact,
a recent summit of key opinion leaders highlighted the synthesis of noncanonical nucleosides as
an “emerging area of high potential impact”.109 While efforts to develop de novo NA syntheses
have aimed to address these challenges, the resulting processes are often lengthy and target-
specific, as highlighted by the 16-step process required to produce the C4'-modified NA MK-8591
(89).107 As a notable example of de novo NA synthesis, MacMillan reported the synthesis of C2'-
modified NAs, including the core of sofosbuvir (86), using a sequence that involved a Mukaiyama
aldol coupling between a ketene acetal (93) and an α-oxyaldehyde (94).110
Scheme 2.1. MacMillan’s de novo synthesis of C2’-modified NAs
Here we disclose a straightforward process for NA synthesis that involves a one-pot,
proline-catalyzed -fluorination-aldol reaction of heteroaryl-substituted acetaldehydes 76
followed by reduction or organometallic addition and annulative fluoride displacement (AFD). This
concise (2-3 step) process addresses several major and longstanding challenges in NA synthesis
by enabling direct access to C3'/C5' protected NAs 78 (and hence C2' modified NAs), providing
flexibility in nucleobase substitution, and offering a direct route to C4' modified NAs. We expect
this strategy will become a powerful tool that enables and inspires drug design.
20
Scheme 2.2. A 3-step synthesis of NAs
2.3. Proposal for de novo NA synthesis
Figure 2.3. A ribose-last approach to NAs inspired by the prebiotic synthesis of deoxyribonucleosides. B. proline-catalyzed α-chlorination/aldol
In a proposed prebiotic synthesis of DNA(25),111 couplings between nucleobase-type
enamines (e.g., 96, Figure 2.3) and glyceraldehyde form a nucleobase iminium ion (e.g., 98) prior
to the furanose in a “ribose-last” approach. We identified a complementary approach that also
involves the terminal cyclization of a nucleobase-iminium ion. Our experiences with ribose
synthesis from chlorohydrins suggested that N/Cl hemiaminals are too unstable to serve as
precursors to related nucleobase iminium ions (Figure 2.3B). Control of both the relative and
absolute stereochemistry may be possible through an organocatalytic aldol reaction of a
dihydroxyacetone derivative (e.g., 97) 112 and the -fluoroaldehyde 100. This approach to NAs
would require i) harnessing the reactivity of notoriously unstable -fluoroaldehydes113,114 coupled

21
with the additional challenge of a nucleobase connected at the same position (e.g., 100), and ii)
the development of an annulative fluoride displacement (AFD) reaction to form the ribose ring in
the last step.
2.4. Development of an α-fluorination/aldol reaction and an annulative fluoride displacement
Figure 2.4. Optimization of αFAR and AFD
To explore this conceptually new approach to NAs, we investigated the -fluorination113 of
-pyrazolyl aldehyde 104 (Figure 2.4) and eventually found that a combination of L-proline and
N-fluorobenzenesulfonimide (NFSI) in DMF115 provided an α-fluorohydrate (not shown) as the
sole product. While we were unable to dehydrate this material, it was eventually found that the
direct addition of dioxanone 75 in MeCN to the reaction mixture afforded the fluorohydrins 105a
and 105b in good yield and enantioselectivity (Figure 2.4, entry 2). As indicated, the fluorohydrins
105a and 105b were formed as a ~1.4:1 mixture of epimers at the pseudo anomeric carbon
(indicated with *) that do not interconvert under the reaction or isolation/purification conditions.
This result suggests that a relatively slow epimerization of -pyrazolyl--fluoroaldehydes
precludes a dynamic kinetic resolution,116 or that the transition structure for the proline catalyzed
aldol reaction between dioxanone 75 and (R)- or (S)--pyrazolyl--fluoroaldehydes are
energetically similar. Notwithstanding, we anticipated that the AFD would proceed via the
formation of a transient azacarbenium cation117 (i.e., an SN1 process), rendering the fluoromethine
configuration inconsequential. To investigate this novel cyclization strategy (i.e., AFD), reduction
of the fluorohydrins 105a and 105b provided a mixture of 1,3-syn diols that was then treated with
one of several Lewis acids in an attempt to promote displacement of the fluoride by the distal

22
alcohol function. After considerable experimentation, an AFD reaction using fluorophilic
Sc(OTf)3118 was realized that afforded the NA 106 in 38% yield as a single -anomer (entry 4).
Additionally, we found that treatment of a mixture of the diols 105a and 105b with base (NaOH)
resulted in the formation of a mixture of - and -anomeric NAs that varied in composition
depending on reaction time and equivalents of base (entries 5 and 6). Using a large excess of
NaOH (10 equiv, entry 6), the -anomer 106 was formed as the exclusive product in excellent
yield (76%).
Figure 2.5. Mechanism of cyclization of 107a and 107b
To further examine the mechanism of cyclization, the intermediate diols 107a and 107b
were separated by flash column chromatography and their relative stereochemistry assigned by
J-based configurational analysis and/or X-ray analysis of derivatives (see experimental section
for full details). Subjecting the purified syn-fluorohydrin 107a to the AFD reaction (NaOH, CH3CN,
Panel C) promoted a clean cyclization to the -anomer 106 via an SN2 process, in contrast to our
expectations. Similarly, the anti-fluorohydrin 107b cyclized to afford the -anomer 108, again via
stereochemical inversion. Fortuitously, under these same reaction conditions the -anomer 108
epimerizes to afford the naturally configured -anomer 106, and thus both fluorohydrin aldol
products 105 can be transformed together into a single naturally configured -D-NA via this
straightforward reaction sequence. It is notable that the enantiomeric purity of the NA 106 (e.r. =
95:5, Figure 2.4 entry 6) represents an average of the enantiomeric purities of the epimeric
fluorohydrin FAR products 105.

23
In order to assess the general utility of this uniquely concise NA synthesis, we prepared a
collection of acetaldehyde derivatives through the straightforward alkylation of several
heterocycles with bromoacetaldehyde diethyl acetal (109) (Figure 2.6). Using either Selectfluor
or NFSI as the electrophilic fluorinating agent (F+), the resulting aldehydes 110 then underwent
proline-catalyzed αFAR with dioxanone 75 to provide a collection of fluorohydrin aldol products
111 functionalized with one of the heterocycles uracil, thymine, triazole, deazadenine, pyrazole,
phthalimide, adenine or 2,6-dichloropyrimidine. These fluorohydrins were generally produced in
good to excellent yield and enantiomeric purity. In the case of the adenine containing fluorohydrin,
the enantiomeric purity was lowered by competing (non-proline) catalysis in the αFAR. Each of
the αFAR products was isolated as a mixture of epimers at the fluoromethine center that
subsequently underwent a 1,3-syn selective carbonyl reduction and AFD promoted by either base
(NaOH, Panel B) or a Lewis acid (Panel C) as indicated. We were pleased to find that several
heterocycles were compatible with this process and that uracil, thymine or adenine-substituted
acetaldehydes could be exploited in short (4 step total) de novo syntheses of the endogenous
ribonucleosides uridine (113), 5-methyluridine (114) and adenosine (A: 120). In general, the
optimal Lewis acids for promoting AFD reactions were InCl3 or Sc(OTf)3, while pyrazole- and
uracil-derived fluorohydrins cyclized using NaOH. With the exception of trifluoromethyluracil 118
and deazaadenines 121 and 122, the NAs were produced as an approximate average of the
enantiomeric purities of the individual fluorohydrin epimers 111. Thus, the majority of NAs
examined in this study undergo epimerization following AFD, providing a straightforward means
to convert the mixture of epimeric aldol products into a single, naturally configured -D-nucleoside
analogue. For the triazole 117, trifluoromethyl uracil 118 and deazaadenines 121 and 122, αFAR
products (e.g., 111) were reduced, separated and treated individually with Sc(OTf)3 or InCl3. As
indicated in Figure 2.6C, for trifluoromethyl uracil, only the anti-fluorohydrin underwent AFD to
form 118, which did not epimerize under the reaction conditions.

24
Figure 2.6. Scope of nucleoside and NA synthesis.
In the case of the deazaadenine, both the syn-fluorohydrin and anti-fluorohydrin
underwent AFD to provide the - and α-anomers 121 and 122, respectively, confirming that these

25
reactions proceed via direct fluoride displacement. To evaluate the practical utility of these
processes, several of the αFARs were demonstrated on >10 g scale (e.g., 113, 114, 118, 119
and 121 (Figure 2.6)) and proceeded without complication, though we noted a small improvement
in diastereoselectivity when reactions were executed on larger scale. We also found that the C-
linked NA 116 could be prepared using this sequence of reactions starting from a
dichloropyrimidine, further extending the utility of this strategy to an additional and important class
of NAs.119 Here, however, the major product of the αFAR cyclizes to an α-D-nucleoside
analogue103 and undergoes a second cyclization event under the reaction conditions to form the
tricycle 116. In addition to naturally configured NAs, this strategy can be easily adapted for the
synthesis of enantiomeric (L-configured) nucleosides and NAs (Figure 2.7) by simply using D-
proline in the αFAR. Thus, L-uridine (ent-113) and the L-configured NA ent-117 were accessed in
this straightforward manner. While crude reaction mixtures were generally treated with aqueous
acid to remove the acetonide protecting group and enable isolation of the targeted NA, eliminating
this step allowed us to isolate C3'/C5'-protected NAs directly (e.g., 123 and 124, Figure 2.7).
Figure 2.7. C3’/C5’ protected nucleosides (R = C(CH3)2) and β-L-nucleosides.
2.5. Synthesis of C2’-modified nucleoside analogues
This modification provides a solution to the challenge of selective C3'/C5' protection that
is required for producing C2'-modified NAs. To demonstrate that these acetonide-protected NAs
can be further derivatized using standard protocols, several C2'-modified NAs were prepared,
including C2'-deoxy (125), C2'-oxo (126), C2'-3º alcohol (127) and C2'-epi (128) (Figure 2.8).

26
Figure 2.8. C2’-modified nucleoside analogues (R = C(CH3)2)
Considering the potential for this process to impact the large-scale production of NAs, we
examined the synthesis of the D-uridine derivative 123 starting with 900 g of uracil. Without
additional optimization, we were able to generate ~380 g of the respective aldol adduct, which
was readily converted into the protected uridine 123 in excellent yield by base-promoted AFD.
Oxidation of the C2'-OH function followed by deprotection and addition of MeMgBr in THF gave
the tertiary alcohol 130 (Scheme 2.3). This later compound is a previously-reported intermediate
in the large-scale production of MK-3682 (Uprifosbuvir: 131),120 an HCV NS5B RNA polymerase
inhibitor developed for the treatment of HCV.
Scheme 2.3. Application to the synthesis of MK-3682 penultimate (130) (R = C(CH3)2).
2.6. Experimental
General considerations
L- and D-proline (99% purity) were purchased from Alfa Aesar. All reactions described were
performed at ambient temperature and atmosphere unless otherwise specified. Column
chromatography was carried out with 230-400 mesh silica gel (E. Merck, Silica Gel 60).
Concentration and removal of trace solvents was done via a Buchi rotary evaporator using
acetone-dry-ice condenser and a Welch vacuum pump.

27
Nuclear magnetic resonance (NMR) spectra were recorded using deuterochloroform (CDCl3),
deuteromethanol (CD3OD), deuteroacetone ((CD3)2CO), deuteroacetonitrile (CD3CN) or
deuterodimethyl sulfoxide (DMSO-d6) as the solvent. Signal positions (δ) are given in parts per
million from tetramethylsilane (δ 0) and were measured relative to the signal of the solvent (1H
NMR: CDCl3: δ 7.26; CD3OD: δ 3.31; (CD3)2CO: δ 2.05; CD3CN: δ 1.96; DMSO-d6: δ 2.50; 13C
NMR: CDCl3: δ 77.16; CD3OD: δ 49.00; (CD3)2CO: δ 29.84; CD3CN: δ 1.32; DMSO-d6: 39.5).
Coupling constants (J values) are given in Hertz (Hz) and are reported to the nearest 0.1 Hz. 1H
NMR spectral data are tabulated in the order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet;
sept, septet; m, multiplet; br broad), coupling constants, number of protons. NMR spectra were
recorded on a Bruker Avance 600 equipped with a QNP or TCI cryoprobe (600 MHz), Bruker 400
(400 MHz) or Bruker 500 (500 MHz). Diastereomeric ratios (dr) are based on analysis of crude
1H-NMR. Assignments of 1H are based on analysis of 1H-1H-COSY and nOe spectra. Assignments
of 13C are based on analysis of HSQC spectra.
High performance liquid chromatography (HPLC) analysis was performed on an Agilent 1100
HPLC, equipped with a variable wavelength UV-Vis detector.
High-resolution mass spectra were performed on an Agilent 6210 TOF LC/MS, Bruker MaXis
Impact TOF LC/MS, or Bruker micrOTOF-II LC mass spectrometer.
Infrared (IR) spectra were recorded neat on a Perkin Elmer Spectrum Two FTIR spectrometer.
Only selected, characteristic absorption data are provided for each compound.
Optical rotation was measured on a Perkin-Elmer Polarimeter 341 at 589 nm.
General Procedure A (one-pot organocatalytic α-fluorination/aldol reaction)
A sample of aldehyde (1.5 equiv.) was added to a stirred suspension of NFSI (1.5 equiv.),
L-proline (1.5 equiv.), and NaHCO3 (1.5 equiv.) in DMF (0.75 M) at 4 °C. When complete
conversion to the α-fluoroaldehyde was observed by 1H-NMR spectroscopic analysis, 2,2-
dimethyl-1,3-dioxan-5-one (75) (1.0 equiv.) in CH2Cl2 or THF or MeCN (1.25*DMF vol.) was then
added and the resulting mixture was allowed to warm to room temperature. After a further 36-72
hours, or when complete consumption of 75 was observed by 1H NMR spectroscopic analysis of
small reaction aliquots, the mixture was diluted with CH2Cl2 and the organic layer was washed
once with saturated sodium bicarbonate solution and once with water. The organic layer was then

28
dried over MgSO4, concentrated under reduced pressure and the crude product was purified by
flash chromatography as indicated.
General Procedure B (syn-reduction)
To a stirred solution of syn- and anti-fluorohydrins (1.0 equiv) in MeCN (0.10 M) at -15˚C was
added tetramethylammoniumtriacetoxyborohydride (5.0 equiv) and acetic acid (10 equiv). The
resulting mixture was stirred 16 hours or until complete consumption of starting material (as
determined by TLC analysis). The reaction mixture was then diluted with a saturated solution of
Rochelle salt and washed three times with CH2Cl2. The organic layer was separated, dried over
MgSO4, concentrated under reduced pressure, and the crude product was purified by flash
chromatography.
General Procedure C (base promoted cyclization)
To a stirred solution of syn-diols, syn- and anti-fluorohydrins (1.0 equiv.) in MeCN (0.10 M) was
added 2 M NaOH (1.5 - 10 equiv.) and the reaction mixture was stirred for 3 hours or until no
starting material remained (as determined by TLC analysis). The reaction mixture was diluted with
CH2Cl2 and washed with saturated ammonium chloride solution. The organic layer was separated,
dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was
purified by flash chromatography.
General Procedure D (Lewis acid promoted cyclization)
To a stirred solution of syn-diols, syn- and anti-fluorohydrin (1.0 equiv.) in MeCN (0.10 M) was
added Sc(OTf)3 or InCl3 (0.10 – 2.5 equiv.) and the reaction mixture was stirred for 6 hours or until
complete consumption of starting material (as determined by TLC analysis). The reaction mixture
was diluted with CH2Cl2 and was washed with saturated sodium bicarbonate solution. The organic
layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The
crude product was purified by flash chromatography.
Preparation and characterization of all compounds
Preparation of S1, aldehyde SM1, aldol adduct A1, diol adducts 107a/107b, and nucleoside
analogues 106, 108, and 124

29
A solution of pyrazole (1.00 g, 14.7 mmol, 1.0 equiv.), bromoacetaldehyde diethyl acetal (2.67
mL, 17.6 mmol, 1.2 equiv.) and K2CO3 (4.06 g, 29.4 mmol, 2.0 equiv.) was stirred in DMF (74 mL)
for 36 hours at 90 °C. The reaction mixture was then filtered and washed with 40 mL of CH2Cl2
and concentrated under reduced pressure. Purification of crude S1 by flash chromatography
(pentane:ethyl acetate – 7:3) afforded S1 (2.43 g, 90 % yield) as a colorless oil. A solution of S1
(0.100g, 0.543, 1.0 equiv.) was heated to 90˚C in 0.5 M HCl (0.54 mL) for 5 hrs. Upon complete
conversion to SM1, the reaction mixture was concentrated under reduced pressure and the
resulting product SM1 was used in the next reaction without purification.
Data for S1: IR (neat): = 2977, 2904, 1516, 1396, 1129, 1063, 751, 621
cm-1; 1H NMR (400 MHz, CDCl3): δ 7.51 (d, J = 1.8 Hz, 1H), 7.46 (d, J = 2.3
Hz, 1H), 6.24 (dd, J = 2.3, 1.8 Hz, 1H), 4.77 (t, J = 5.5 Hz, 2H), 4.22 (d, J =
5.5 Hz, 2H), 3.70 (m, 2H), 3.41 (m, 2H), 1.16 (t, J = 7.1 Hz, 6H); 13C NMR
(125 MHz, CDCl3): δ 139.7, 130.6, 105.6, 101.7, 63.8, 55.2, 15.3
HRMS (EI+) calcd for [C9H17N2O2]+ 185.1285; found 185.1284
α-fluorination/aldol
Following General Procedure A, a solution of SM1 (0.543 mmol), NFSI (0.170 g, 0.543 mmol), L-
proline (0.063 g, 0.543 mmol) and NaHCO3 (0.045 g, 0.543 mmol) was stirred for 12 hours at 4°C
in DMF (0.72 mL). 75 (0.043 mL, 0.362 mmol) in MeCN (0.90 mL) was then added and the
reaction mixture was stirred for 60 hrs at room temperature. Purification of the crude fluorohydrin
A1 by flash chromatography (pentane:Et2O – 25:75) afforded a mixture of syn- and anti-
fluorohydrins A1 (0.060 g, 64 % yield, dr 1.4:1) as a light yellow oil.
Data for syn- and anti-fluorohydrins A1: IR (neat): = 2989, 1749, 1446,
1376, 1091, 1042, 764 cm-1; 1H NMR (600 MHz, CDCl3): δ 7.88, 7.78,
7.63, 6.45, 6.44, 6.39, 6.37, 4.89, 4.50, 4.36, 4.34, 4.31, 4.26, 4.07, 4.04,
1.50, 1.45, 1.45, 1.34; 13C NMR (150 MHz, CDCl3): δ 209.0, 207.4,
141.7, 141.4, 131.5, 131.1, 107.7, 107.5, 101.8, 101.4, 95.0, 94.6, 74.3,
72.4, 71.0, 70.2, 67.0, 66.9, 24.0, 23.7, 23.7, 23.4; 19F NMR (470 MHz, CDCl3): δ -144.9, -154.1
HRMS (EI+) calcd for [C11H16FN2O4]+ 259.1089; found 259.1093

30
Syn-reduction of syn-and anti-fluorohydrins A1
Following General Procedure B, Me4NHB(OAc)3 (0.968 g, 3.68 mmol) and AcOH (0.442 mL, 7.36
mmol) were added to a stirred solution of A1 (0.190 g, 0.736 mmol) at -15 °C in MeCN (7.36 mL)
and the reaction mixture was stirred for 18 hrs. Purification of the crude diols 107a and 107b by
flash chromatography (pentane:ethyl acetate – 1:1) afforded a mixture of 107a and 107b (0.151g,
79% yield, d.r. (syn/anti) = 1:1.2) as a colourless oil.
Data for syn-diol, syn-fluorohydrin 107a: []D20 = +83.2 (c 0.37 in MeCN);
IR (neat): = 3001, 1442, 1375, 1039, 918, 749 cm-1;1H NMR (600 MHz,
CDCl3): δ 7.68 (d, J = 2.4 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 6.38 (dd, J =
2.4, 1.5 Hz, 1H), 6.18 (d, J = 51.2 Hz, 1H), 4.27 (dd, J = 22.4, 8.8 Hz, 1H),
3.95 (dd, J = 11.1, 5.6 Hz, 1H), 3.93 (dd, J = 9.5, 8.0 Hz, 1H), 3.80 (m, 1H), 3.70 (dd, J = 11.2,
11.0 Hz, 1H), 1.52 (s, 3H), 1.39(s, 3H); 13C NMR (150 MHz, CDCl3): δ 141.5, 132.0, 107.2, 99.0,
91.9 (d, J = 211.0 Hz), 72.3 (d, J = 21.8 Hz), 70.6, 67.1, 63.8, 28.7, 19.4; 19F NMR (470 MHz,
CD3CN): δ -150.3
HRMS (EI+) calcd for [C11H18FN2O4]+ 261.1245; found 261.1255
Data for syn-diol, anti-fluorohydrin 107b: []D20 = -10.8 (c 0.91 in MeCN); IR
(neat): = 3646, 3001, 1443, 1375, 1039, 918 cm-1; 1H NMR (600 MHz,
CDCl3): δ 7.70 (d, J = 0.9 Hz, 1H), 7.65 (d, J = 2.5 Hz, 1H), 6.40 (dd, J =
2.5, 0.9 Hz, 1H), 6.29 (dd, J = 48.4, 2.9 Hz, 1H), 4.41 (ddd, J = 8.0, 4.0, 2.9
Hz, 1H), 3.87 (m, 2H), 3.52 (dd, J = 11.3, 2.7 Hz, 1H), 3.17 (dd, J = 8.8, 8.8 Hz, 1H), 1.34 (s, 3H),
1.16 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 142.1, 132.0, 106.9, 98.9, 93.1 (d, J = 207.9 Hz), 76.2
(d, J = 24.7 Hz), 72.2 (d, J = 5.3 Hz), 67.3 (d, J = 4.6 Hz), 63.8, 28.5, 19.3; 19F NMR (470 MHz,
CD3CN): δ -145.9
HRMS (EI+) calcd for [C11H18FN2O4]+ 261.1245 found 261.1262
Cyclization of diols 107a and 107b
Following General Procedure C, a mixture of 107a and 107b (0.025 g, 0.096 mmol, d.r. (syn/anti)
= 1:1) and 2 M NaOH (0.48 mL, 0.962 mmol) was stirred in MeCN (0.96 mL) at 50˚C for 5 hrs.
Purification of the crude 124 by flash chromatography (pentane:ethyl acetate – 65:35) afforded
nucleoside analogue 124 (0.018 g, 76 % yield) as a white solid.

31
Data for nucleoside analogue 124: []D20 = -58.9 (c 2.0 in MeCN); IR (neat):
= 3339, 2926, 1647, 1450, 1397, 1092, 1045, 759 cm-1;1H NMR (400 MHz,
CD3CN): δ 7.70 (d, J = 2.4 Hz, 1H), 7.56 (d, J = 1.6 Hz, 1H), 6.30 (dd, J = 1.6,
2.4 Hz, 1H), 5.70 (s, 1H), 4.47 (d, J = 4.6 Hz, 1H), 4.12 (dd, J = 4.6, 9.6 Hz,
1H), 4.11 (dd, J = 4.6, 9.6 Hz, 1H), 3.91 (dd, J = 9.6, 10.3, 1H), 3.83 (dd, J =
4.6, 9.6 Hz, 1H), 3.72 (br s, 1H), 1.54 (s, 3H), 1.43 (s, 3H); 13C NMR (100 MHz, CD3CN): δ 141.7,
130.1, 106.7, 101.7, 96.1, 74.7, 74.4, 71.8, 65.9, 29.3, 20.1
HRMS (EI+) calcd for [C11H17N2O4]+ 241.1183; found 241.1197
Deprotection of nucleoside analogue 124
124 (0.021g, 0.088 mmol) was dissolved in MeOD (1.0 mL) and two drops of 1 M HCl was added
and the solution was left for 12 hrs at room temperature. Subsequently, the reaction mixture was
concentrated under reduced pressure to afford 106 as a white solid (0.018 g, 100%).
Data for nucleoside analogue 106: []D20 = +70.4 (c 0.48 in MeOH); IR (neat):
= 3325, 2944, 2832, 1449, 1022, 631 cm-1;1H NMR (600 MHz, CD3CN): δ
7.74 (d, J = 2.3 Hz, 1H), 7.58 (d, J = 1.0 Hz, 1H), 6.30 (dd, J = 2.3, 1.0 Hz, 1H),
5.70 (d, J = 4.3 Hz, 1H), 4.51 (m, 1H), 4.33 (m, 1H), 4.08 (br s, 1H), 3.74 (dd, J
= 12.3, 2.8 Hz, 1H), 3.67 (d, J = 5.7 Hz, 1H), 3.59 (dd, J = 12.3, 2.5 Hz, 1H), 3.52 (d, J = 4.3 Hz,
1H); 13C NMR (150 MHz, CD3CN): δ 141.2, 131.1, 106.4, 94.7, 87.2, 76.6, 72.3, 63.4.
HRMS (EI+) calcd for [C8H13N2O4]+ 201.0870; found 201.0870
Cyclization of diol 107b
A solution of 107b (0.043 g, 0.165 mmol) and 2 M NaOH (0.21 mL, 0.443 mmol, 2.5 equiv.) was
stirred for 4 hrs in MeCN (1.65 mL) at 50˚C. Purification of the crude 108 by flash chromatography
(pentane:ethyl acetate – 65:35) afforded nucleoside analogue 108 (0.026 g, 76 % yield) as a white
solid
Data for nucleoside analogue 108: []D20 = +72.2 (c 0.98 in MeCN); IR
(neat): = 3366, 2992, 1306, 1383, 1200, 1076, 754 cm-1,1H NMR (600
MHz, CD3CN): δ 7.76 (d, J = 2.3 Hz, 1H), 7.56 (d, J = 1.2 Hz, 1H), 6.35 (d,
J = 2.3 Hz, 1H), 5.38 (d, J = 0.9 Hz, 1H), 4.12 (dd, J = 0.9, 2.1 Hz, 1H), 3.94
(d, J = 2.1, 9.7 Hz, 1H), 3.81 (dd, J = 5.0, 10.6 Hz, 1H), 3.59 (m, 2H), 3.37 (m), 1.45 (s, 3H), 1.33

32
(s, 3H); 13C NMR (150 MHz, CDCl3): δ 142.1, 131.0, 108.2, 99.9, 71.8, 65.4, 65.2, 64.7, 59.0,
29.1, 19.9
HRMS (EI+) calcd for [C11H17N2O4]+ 241.1183; found 241.1176
Determination of relative stereochemistry for diol 107a
Diol 107a was converted into the bis-p-nitro-benzoyl ester and recrystallized in ethanol. This
allowed for the relative stereochemistry to be assigned using single X-ray crystallography (see X-
ray structures).
Determination of relative stereochemistry for nucleoside analogue 106
Analysis of 2D NOESY of nucleoside analogue 106 supported the indicated
stereochemistry
Determination of relative sterchemistry for nucleoside analogue 108
Analysis of 2D NOESY of nucleoside analogue 108 supported the
indicated stereochemistry

33
Figure S2.1. Cyclization of diols 107a and 107b. Following General Procedure C, diols 107a and
107b were cyclized separately to the same product (106). The α-anomer resulting from an SN2
cyclization from 107b epimerizes following cyclization to the thermodynamically more stable β-
anomer 17 under the reaction conditions. Moreover, taking a 2:1 mixture of products (108:106)
and following General Procedure C affords only the β-anomer 106. Note also the e.r. of 106 (95:5)
represents the average e.r. of 107a (93:7) and 107b (98:2). Such emperizations have been
reported for nucleosides.121
Determination of enantiomeric excess of diol 107a
Following General Procedures A and B, using a 1:1 mixture of L-: D-proline, a racemic sample of
diol 107a was prepared. The enantiomeric diols were separated by chiral HPLC using a Lux® 3µm
Amylose-1 column; flow rate 0.40 mL/min; eluent: hexanes-iPrOH 90:10; detection at 210 nm;
retention time = 6.66 min for (+)-107a; 8.10 min for (-)-107a (see chromatograms).The
enantiomeric ratio of the optically enriched (+)-107a diol was determined using the same method
(93:7 e.r.).
Determination of enantiomeric excess of diol 107b
Following General Procedures A and B, using a 1:1 mixture of L-: D-proline, a racemic sample of
diol 107b was prepared. The enantiomeric diols were separated by chiral HPLC using a Lux® 3µm
Amylose-1 column; flow rate 0.40 mL/min; eluent: hexanes-iPrOH 90:10; detection at 210 nm;
retention time = 6.13 min for (-)-107b; 11.72 min for (+)-107b (see chromatograms).The

34
enantiomeric ratio of the optically enriched (-)-107b diol was determined using the same method
(98:2 e.r.).
Determination of enantiomeric excess of nucleoside analogue 124
Following General Procedures A, B, and C, using a 1:1 mixture of L-: D-proline, a racemic sample
of nucleoside 124 was prepared. The enantiomeric nucleosides were separated by chiral HPLC
using a Lux® 3µm-i-Cellulose-5 column; flow rate 0.10 mL/min; eluent: hexanes-iPrOH 90:10;
detection at 254 nm; retention time = 8.91 min for (-)-124; 13.32 min for (+)-124 (see
chromatograms).The enantiomeric ratio of the optically enriched (-)-124 was determined using
the same method (95:5 e.r.).
Preparation of aldol adduct A2, diol adducts D2, and nucleoside analogues 113, 123, and ent-
113
α-fluorination/aldol
The corresponding starting aldehyde/hydrate SM3 was prepared following literature
procedures.122 Following General Procedure A, a solution of aldehyde (1.32 mmol), NFSI (0.416
g, 1.32 mmol), L-proline (0.152 g, 1.32 mmol) and NaHCO3 (0.111 g, 1.32 mmol) was stirred for
12 hours at 4°C in DMF (1.76 mL). 75 (0.105 mL, 0.880 mmol) in THF (2.64 mL) was then added
and the reaction mixture was stirred for 96 hrs at 4˚C. Purification of the crude fluorohydrin A2 by
flash chromatography (pentane:ethyl acetate – 1:1) afforded an inseparable mixture of syn- and
anti- fluorohydrins A2 (0.159 g, 60 % yield, d.r. 1.2:1) as an off-white solid.
Data for syn- and anti-fluorohydrins A2: IR (neat): = 3432, 2992, 2900,
1692, 1381, 1079 cm-1; 1H NMR (600 MHz, CDCl3): δ 8.87, 8.79, 7.74,
7.68, 6.68, 6.67, 5.80, 5.77, 4.53, 4.40, 4.34, 4.33, 4.30, 4.13, 4.11, 4.06,
3.70, 3.48, 1.52, 1.46, 1.44, 1.44; 13C NMR (150 MHz, CDCl3): δ 211.3,
208.7, 162.8, 162.6, 150.3, 149.8, 141.7, 141.1, 103.2, 102.6, 102.1, 101.9, 90.7, 90.3, 73.3, 71.4,
70.7, 70.5, 66.6, 66.5, 23.7, 23.6, 23.6, 23.3; 19F NMR (470 MHz, CDCl3): δ –162.0, –178.6
HRMS (EI+) calcd for [C12H16FN2O6]+ 303.0987; found 303.0982

35
Syn-reduction of syn-and anti-fluorohydrins A2
Following General Procedure B, Me4NHB(OAc)3 (0.174g, 0.660 mmol) and AcOH (0.076 mL,
1.32 mmol) were added to a stirred solution of A2 (0.040g, 0.130 mmol) at -15 °C in MeCN (1.32
mL) and the reaction mixture was stirred for 24 hrs. Purification of the crude diols D2a and D2b
by flash chromatography (pentane:ethyl acetate – 1:3) afforded diols D2a and D2b (0.020 g, 50
%, d.r. (syn/anti) = 1.2:1) as white solids.
Data for syn-diol, syn-fluorohydrin D2a: 1H NMR (600 MHz, MeOD): δ 7.76
(d, J = 8.0, 1H), 6.46 (dd, J = 44.4, 4.8 Hz, 1H), 5.73 (d, J = 8.0 Hz, 1H),
4.03 (ddd, J = 18.3, 7.0, 5.0 Hz, 1H), 3.82 (dd, J = 11.4, 5.1 Hz, 1H), 3.71
(m, 2H), 3.60 (dd, J = 11.4, 8.1 Hz, 1H), 1.42 (s, 3H), 1.28 (s, 3H); 13C NMR
(150 MHz, MeOD): δ 165.8, 151.7, 143.1 (d, J = 2.6 Hz), 102.9, 100.1, 94.3 (d, J = 208.4 Hz),
74.6 (d, J = 24.6 Hz), 73.7 (d, J = 4.5 Hz), 67.3, 65.3, 28.3, 19.7.
HRMS (EI+) calcd for [C12H18FN2O6]+ 305.1143; found 305.1142
Data for syn-diol, anti-fluorohydrin D2b: 1H NMR (600 MHz, MeOD): δ 7.90
(d, J = 8.1 Hz, 1H), 6.71 (dd, J = 44.2, 6.1 Hz, 1H), 5.74 (d, J = 8.1 Hz, 1H),
4.32 (m, 1H), 3.81 (m, 3H), 3.60 (m, 1H), 1.43 (s, 3H), 1.32 (s, 3H); 13C
NMR (150 MHz, MeOD): δ 165.8, 152.2, 143.0, 103.2 100.2, 92.6 (d, J =
204.4), 75.9 (d, J = 2.8 Hz), 71.5 (d, J = 29.1 Hz), 65.7, 64.5 (d, J = 2.2 Hz), 28.6, 19.4.
HRMS (EI+) calcd for [C12H18FN2O6]+ 305.1143; found 305.1123
Cyclization of diols D2a and D2b
Following General Procedure C, a solution of D2 (0.022 g, 0.072 mmol, d.r. syn/anti = 1.2:1) and
2 M NaOH (0.36 mL, 0.72 mmol) was stirred for 24 hours in MeCN (0.72 mL). Purification of the
crude 123 by flash chromatography (CH2Cl2:MeOH – 92.5:7.5) afforded nucleoside analogue 123
(0.019 g, 95% yield) as a white solid.
Data for nucleoside analogue 123: []D20 = +48.1 (c 0.90 in MeOH); IR (neat):
= 2912, 1436, 1407, 1042, 952, 697 cm-1; 1H NMR (600 MHz, (CD3)2CO): δ
7.71 (d, J = 8.0 Hz, 1H), 5.81 (s, 1H), 5.61 (d, J = 8.0 Hz, 1H), 4.45 (d, J = 4.6
Hz, 1H), 4.20 (dd, J = 9.8, 4.7 Hz, 1H), 4.12 (dd, J = 10.0, 10.0 Hz, 1H), 3.90
(dd, J = 10.0, 4.8 Hz, 1H), 3.86 (ddd, J = 10.0, 10.0, 4.7 Hz, 1H), 1.56 (s, 3H),

36
1.42 (s, 3H); 13C NMR (150 MHz, (CD3)2CO): δ 164.2, 151.8, 142.4, 103.4, 102.3, 94.5, 75.3,
74.6, 72.5, 66.1, 33.1, 22.8
HRMS (EI+) calcd for [C12H17N2O6]+ 285.1081; found 285.1085
Deprotection of nucleoside analogue 123
123 (0.019g, 0.068 mmol) was dissolved in MeOD (0.68 mL) and two drops of 1 M HCl was added
and the solution was left for 12 hrs at room temperature. Subsequently, the reaction mixture was
concentrated under reduced pressure to afford nucleoside 113 as a white solid (0.017 g, 100%).
The spectral data matched previous reports.123
Data for nucleoside 113: [α]D20 = -23 (c = 0.1, MeOH); IR (neat): ν = 3347,
2927, 2857, 1679, 1464, 1381, 1260, 1202, 1104, 1053, 806 cm–1; 1H NMR
(600 MHz, MeOD): δ 8.03 (d, J = 8.1 Hz, 1H), 5.91 (d, J = 4.7 Hz, 1H), 5.70
(d, J = 8.1 Hz, 1H), 4.18 (dd, J = 4.9, 4.9 Hz, 1H), 4.15 (dd, J = 4.9, 4.9 Hz,
1H), 4.00-4.01 (m, 1H), 3.84 (dd, J = 12.2, 2.6 Hz, 1H), 3.74 (dd, J = 12.2, 3.1
Hz, 1H); 13C NMR (150 MHz, MeOD): 166.2, 152.5, 142.7, 102.6, 90.6, 86.4, 75.7, 71.3, 62.3
HRMS (EI+) calcd for [C9H13N2O6]+ 245.0768; found 245.0770
Determination of relative stereochemistry for diol D2a and D2b
Based on J-based configurational analysis of compounds D5a/D5b,
D8a/D8b and XRD analysis of compounds 107a, D7b, D9a a clear trend
was established between the stereochemistry at the fluoromethine center
and the chemical shift of the fluoromethine proton (*). In every case, the
syn-fluorohydrin diol has a lower chemical shift than the diastereomeric anti-fluorohydrin diol.
Here, D2a has a chemical shift of 6.46 ppm while D2b has a chemical shift of 6.71 ppm for the
flouromethine proton. D2a was assigned as the syn-fluorohydrin diol and D2b the anti-fluorohydrin
diol.
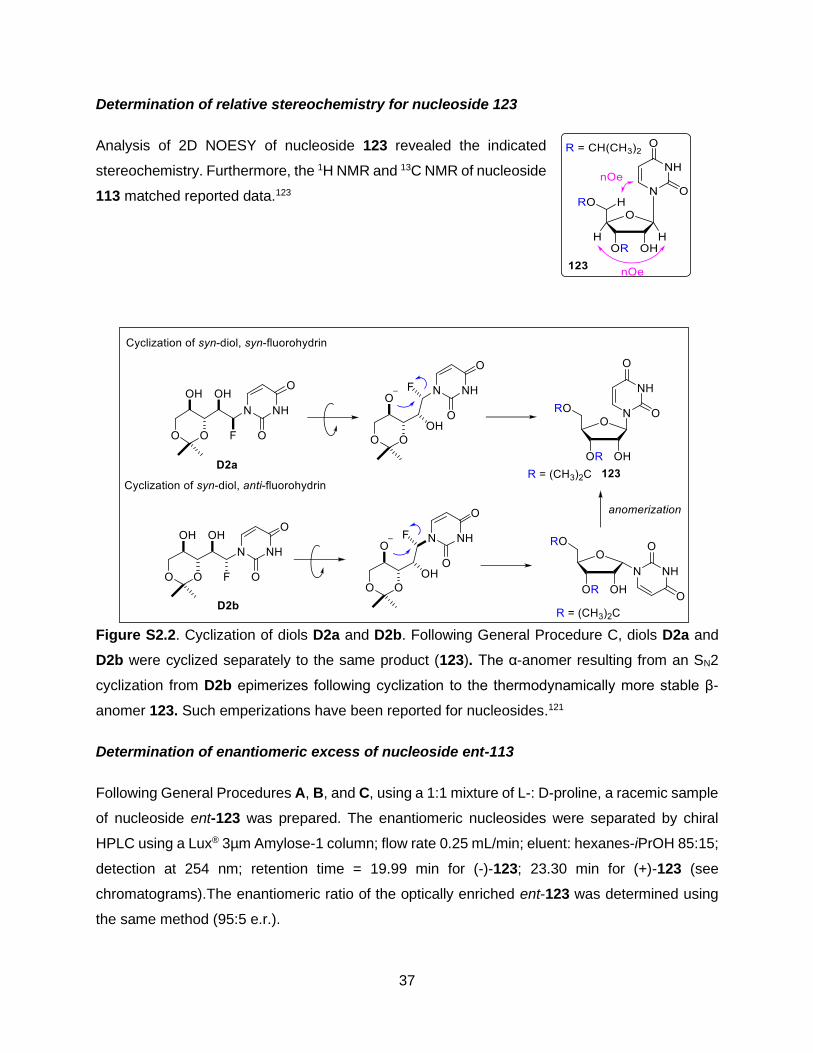
37
Determination of relative stereochemistry for nucleoside 123
Analysis of 2D NOESY of nucleoside 123 revealed the indicated
stereochemistry. Furthermore, the 1H NMR and 13C NMR of nucleoside
113 matched reported data.123
Figure S2.2. Cyclization of diols D2a and D2b. Following General Procedure C, diols D2a and
D2b were cyclized separately to the same product (123). The α-anomer resulting from an SN2
cyclization from D2b epimerizes following cyclization to the thermodynamically more stable β-
anomer 123. Such emperizations have been reported for nucleosides.121
Determination of enantiomeric excess of nucleoside ent-113
Following General Procedures A, B, and C, using a 1:1 mixture of L-: D-proline, a racemic sample
of nucleoside ent-123 was prepared. The enantiomeric nucleosides were separated by chiral
HPLC using a Lux® 3µm Amylose-1 column; flow rate 0.25 mL/min; eluent: hexanes-iPrOH 85:15;
detection at 254 nm; retention time = 19.99 min for (-)-123; 23.30 min for (+)-123 (see
chromatograms).The enantiomeric ratio of the optically enriched ent-123 was determined using
the same method (95:5 e.r.).

38
Preparation of aldol adducts A3, diol adducts D3, and nucleoside analogues NA3 and 114
α-fluorination/aldol
The corresponding starting aldehyde/hydrate SM3 was prepared following literature
procedures.124 Following General Procedure A, a solution of SM3 (0.40 mmol), NFSI (0.126 g,
0.40 mmol), L-proline (0.046 g, 0.40 mmol) and NaHCO3 (0.034 g, 0.40 mmol) was stirred for 14
hours at 4°C in DMF (0.53 mL). Dioxanone 75 (0.032 mL, 0.27 mmol) in CH2Cl2 (0.67 mL) was
then added and the reaction mixture was stirred for 96 hrs at 4°C. Purification of the crude
fluorohydrin A3 by flash chromatography (pentane:ethyl acetate – 3:7) afforded fluorohydrin A3
(0.072 g, 84 % yield, d.r. 1.3:1 ) † as an off-white solid.
Data for syn- and anti-fluorohydrins A3: IR (neat): = 2995, 1696, 1451,
1376, 1087, 1049 cm-1; 1H NMR (600 MHz, CDCl3): δ 8.65, 8.60, 8.52,
7.57, 7.46, 7.41, 7.23, 6.67, 6.66, 6.64, 6.52, 4.59, 4.54, 4.52, 4.40, 4.39,
4.36, 4.35, 4.35, 4.33, 4.33, 4.32, 4.32, 4.12, 4.11, 4.07, 4.06, 3.67, 3.37,
1.97, 1.95, 1.95, 1.94, 1.52, 1.51, 1.51, 1.49, 1.47, 1.46, 1.45, 1.44; 13C NMR (150 MHz, CDCl3):
δ 211.4 208.5, 207.9, 206.4, 163.4, 163.2, 163.2, 163.1, 150.8, 150.5, 149.9, 149.9, 137.2, 136.2,
135.7, 134.6, 112.6, 112.0, 111.9, 111.0, 102.1, 102.1, 101.8, 101.7, 91.9, 90.8, 90.7, 90.1, 73.7,
73.0, 71.5, 70.8, 70.6, 70.5, 68.2, 68.0, 67.1, 66.8, 66.6, 66.5, 24.0, 23.9, 23.7, 23.7, 23.7, 23.6,
23.6, 23.4, 12.7, 12.7, 12.7, 12.7; 19F NMR (470 MHz, CDCl3): δ –159.9, –161.6, –169.6, –177.8
HRMS (EI+) calcd for [C13H18FN2O6]+ 317.1143; found 317.1142
Syn-reduction of syn-fluorohydrin and anti-fluorohydrins A3
Following General Procedure B, Me4NHB(OAc)3 (0.416 g, 1.58 mmol) and AcOH (0.181 mL, 3.16
mmol) were added to a stirred solution of A3 (0.100 g, 0.316 mmol) at -15 °C in MeCN (2.10 mL)
and the reaction mixture was stirred for 18 hrs. Purification of the crude diol D3a by flash
chromatography (pentane:ethyl acetate – 3:7) afforded diols D3a and D3b (0.063 g, 63 % yield,
d.r. (syn:anti) = 1.3:1) as a white solid.
† Mixture of 2 diastereomers and their corresponding tautomers (1:1.1:0.65:0.28). Varying the pH of the solution changes the ratio of these products. Following reduction, only 2 products (d.r. (syn/anti) = 1.3:1) are present in the crude.

39
Data for syn-diol, syn-fluorohydrin D3a: []D20 = -11.8 (c 1.0 in MeOH); IR
(neat): = 3363, 2924, 2858, 1674, 1380, 1209, 1075 cm-1; 1H NMR (600
MHz, CD3CN): δ 7.42 (d, J = 0.90 Hz, 1H), 6.36 (dd, J = 44.9, 5.1 Hz, 1H),
4.04 (ddd, J = 18.1, 6.6, 5.1 Hz, 1H), 3.79 (dd, J = 11.3, 4.5 Hz, 1H), 3.67
(m, 2H), 3.55 (m, 1H), 1.83 (d, J = 0.90 Hz, 3H), 1.39 (s, 3H), 1.24 (s, 3H); 13C NMR (150 MHz,
CD3CN): δ 164.7, 151.5, 137.9, 111.7, 99.9, 94.0 (d, J = 205.9 Hz), 74.8 (d, J = 25.1 Hz), 73.0 (d,
J = 4.3 Hz), 67.1, 65.0, 28.8, 19.9, 12.7; 19F NMR (470 MHz, CD3CN): δ –169.1
1H NMR in MeOD for syn-diol, syn-fluorohydrin D3a for relative stereochemical assignment:1H
NMR (600 MHz, MeOD): δ 7.58 (s, 1H), 6.43 (dd, J = 4.1 Hz, 1H), 4.06 (m, 1H), 3.81 (m 1H), 3.71
(m, 2H), 3.59 (m, 1H), 1.89 (s, 3H), 1.41 (s, 3H), 1.26 (s, 3H).
HRMS (EI+) calcd for [C13H20FN2O6]+ 319.1300; found 319.1329
Data for syn-diol, anti-fluorohydrin D3b: []D20 = +26.2 (c 0.45 in CH3CN);
IR (neat): = 3360, 2922, 2855, 1670, 1380, 1207, 1078 cm-1; 1H NMR
(600 MHz, MeOD): 7.72 (d, J = 1.1 Hz, 1H), 6.71 (dd, J = 44.3, 6.8 Hz, 1H),
4.32 (m, 1H), 3.82 (m, 3H), 3.60 (m, 1H), 1.90 (d, J =1.1 Hz, 3H), 1.44 (s,
3H), 1.32 (s, 3H); 13C NMR (150 MHz, MeOD): δ 166.1, 152.5, 138.3,
112.0, 100.2, 92.6 (d, J = 204.7 Hz), 75.9, 71.3 (d, J = 29.9 Hz), 65.7, 64.4 (d, J = 2.1 Hz), 28.6,
19.5, 12.4. 19F NMR (470 MHz, CD3CN): δ –160.3
HRMS (EI+) calcd for [C13H20FN2O6]+ 319.1300; found 319.1320
Cyclization of diols D3a and D3b
Following General Procedure C, a solution of D3a and D3b (0.100 g, 0.314 mmol, d.r. syn/anti =
1.5:1) and 2 M NaOH (0.236 mL, 0.472 mmol) was stirred for 10 hours in MeCN (3.14 mL).
Purification of the crude nucleoside NA3 by flash chromatography (ethyl acetate) afforded
nucleoside NA3 (0.089 g, 95 % yield) as a white solid.

40
Data for nucleoside NA3: []D20 = +39.4 (c 1.1 in MeCN); IR (neat): ν = 3405,
2993, 1687, 1267, 1138, 845, 734 cm–1; 1H NMR (600 MHz, CD3CN): δ 9.04
(br s, 1H), 7.19 (d, J = 1.1 Hz, 1H), 5.67 (s, 1H), 4.22 (dd, J = 4.8, 3.1 Hz,
1H), 4.15 (dd, J = 9.1, 3.5 Hz, 1H), 4.02 (dd, J =10.1, 9.8 Hz, 1H), 3.70 (m,
2H), 3.55 (m, 1H), 1.85 (d, J = 1.1 Hz, 3H), 1.53 (s, 3H), 1.41 (s, 3H); 13C
NMR (150 MHz, CD3CN): δ 164.9, 151.6, 137.5, 111.8, 102.3, 93.8, 74.7,
74.1, 72.1, 65.6, 29.6, 20.5, 12.7
HRMS (EI+) calcd for [C13H19N2O6]+ 299.1238; found: 299.1277
Deprotection of nucleoside analogue NA3
NA3 (0.010g, 0.034 mmol) was dissolved in MeOD (0.34 mL) and two drops of 1 M HCl was
added and the solution was left for 12 hrs at room temperature. Subsequently, the reaction mixture
was concentrated under reduced pressure to afford 114 as a white solid (8.7 mg, 100%). The
spectral data matched previous reports.125
Data for nucleoside analogue 114: []D20 = -33.0 (c = 0.1 in MeOH); IR (neat):
ν = 3346, 2928, 2867, 1688, 1466, 1378, 1262, 1200, 1104, 1050, 803 cm–1;
1H NMR (600 MHz, MeOD): δ 7.86 (d, J = 1.1 Hz, 1H), 5.91 (d, J = 4.6 Hz,
1H), 4.15-4.18 (m, 2H), 3.98-4.00 (m, 1H), 3.86 (dd, J = 12.2, 2.7 Hz, 1H),
3.75 (dd, J = 12.2, 3.0 Hz, 1H), 1.88 (d, J = 0.9 Hz, 3H); 13C NMR (150 MHz,
MeOD): δ 166.4, 152.7, 138.4, 111.5, 90.3, 86.3, 75.5, 71.3, 62.3, 12.4.
HRMS (EI+) calcd for [C10H15N2O6]+ 259.0925; found: 259.0923
Determination of relative stereochemistry for diol D3a and D3b
Based on J-based configurational analysis of compounds D5a/D5b,
D8a/D8b and XRD analysis of compounds 107a, D7b, D9a a clear trend
was established between the stereochemistry at the fluoromethine center
and the chemical shift of the fluoromethine proton (*). In every case, the
syn-fluorohydrin diol has a lower chemical shift than the diastereomeric anti-fluorohydrin
diol.Here, D3a has a chemical shift of 6.43 ppm while D3b has a chemical shift of 6.69 ppm for
the fluoromethine proton. D3a was assigned as the syn-fluorohydrin diol and D3b the anti-
fluorohydrin diol.

41
Determination of absolute stereochemistry
Comparison of []D20 values of nucleoside 114 with literature values confirmed absolute
stereochemistry.126
Figure S2.3. Cyclization of diols D3a and D3b. Following General Procedure C, diols D3a and
D3b were cyclized separately to the same product, NA3. The α-anomer resulting from an SN2
cyclization from D3b epimerizes following cyclization to the thermodynamically more stable β-
anomer NA3. Such emperizations have been reported for nucleosides.121
Determination of enantiomeric excess of nucleoside NA3
Following General Procedures A, B, and C, using a 1:1 mixture of L-: D-proline, a racemic sample
of nucleoside NA3 was prepared. The enantiomeric nucleosides were separated by chiral HPLC
using a Lux® 3µm Amylose-1 column; flow rate 0.25 mL/min; eluent: hexanes-iPrOH 85:15;
detection at 254 nm; retention time = 5.18 min for (+)-NA3; 12.61 min for (-)-NA3 (see
chromatograms).The enantiomeric ratio of the optically enriched (+)-NA3 was determined using
the same method (91:9 e.r.).
Preparation of aldol adduct A4, diol adducts D4a/D4b, and nucleoside analogue 116
α-fluorination/aldol and syn-reduction of syn-and anti-fluorohydrins
Following General Procedure A, a solution of 2-(4,6-dichloropyrimidin-5-yl)acetaldehyde (0.250
g, 1.31 mmol, 1 equiv.), NFSI (0.413 g, 1.31 mmol, 1 equiv.), L-proline (0.151 g, 1.31 mmol, 1

42
equiv.) and NaHCO3 (0.110 g, 1.31 mmol, 1 equiv.) was stirred for 1 hr at 4°C in DMF (1.19 mL).
Dioxanone 75 (0.521 mL, 4.36 mmol, 3.33 equiv.) was added and the reaction mixture was stirred
for 24 hrs at 4°C. Purification of the crude fluorohydrin A4 by flash chromatography (pentane:ethyl
acetate – 3:7) afforded fluorohydrin A4 (0.301 g, 68 % yield) as an orange oil. Following General
Procedure B, Me4NHB(OAc)3 (2.16 g, 8.21 mmol) and AcOH (0.905 mL, 16.4 mmol) were added
to a stirred solution of A4 (0.555 g, 1.64 mmol) at -15 °C in MeCN (16.4 mL) and the reaction
mixture was stirred for 24 hrs. Purification of the crude diol D4a by flash chromatography
(pentane:ethyl acetate – 4:1) afforded diol D4a (0.295 g, 53 % yield) as an off-white solid.
Data for syn-diol D4a: []D20 = +26.6 (c 5.0 in MeCN); IR (neat): = 3000,
1442, 1375, 1039, 918, cm-1; 1H NMR (600 MHz, CDCl3): δ 8.73 (s, 1H),
6.05 (dd, J = 46.0, 7.9 Hz, 1H), 4.64 (m, 1H), 3.89 (dd, J = 11.5, 5.7 Hz,
1H), 3.80 (m, 1H), 3.73 (dd, J = 9.1, 8.5 Hz, 1H), 3.61 (dd, J = 11.5, 9.5 Hz,
1H), 1.29 (s, 3H), 0.94 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 161.5, 157.4, 127.8, 98.3, 91.1 (d,
J =179.4 Hz), 75.5 (d, J = 21.3 Hz), 71.7 (d, J = 5.5 Hz), 66.6, 63.3, 28.2, 18.7; 19F NMR (470
MHz, CDCl3): δ –193.0
HRMS (EI+) calcd for [C12H16Cl2FN2O4]+ 341.0466; found 341.0425
Cyclization of diol D4a
Following General Procedure C, a solution of D4a (0.014 g, 0.044 mmol, 1 equiv.) and 2 M NaOH
(0.11 mL, 0.22 mmol, 5 equiv.) was stirred for 15 minutes in MeCN (0.30 mL). Purification of the
crude nucleoside 116 by flash chromatography (ethyl acetate:pentane – 50:50) afforded
nucleoside 116 (6.4 mg, 51% yield) as a white solid.
Data for nucleoside analogue 116: []D20 = +51.2 (c 0.34 in CH2Cl2); IR
(neat): = 3363, 2927, 1602, 1598, 1571, 1408, 968 cm-1; 1H NMR (600
MHz, CD3CN): δ 8.66 (s, 1H), 4.19 (dd, J = 10.1, 4.9 Hz, 1H), 3.91 (dd, J
= 10.2, 10.1 Hz, 1H), 3.86 (dd, J = 10.1, 4.7 Hz, 1H), 3.30 (ddd, J = 10.2,
10.1, 4.8 Hz, 1H), 1.56 (s, 3H), 1.51 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 176.6, 160.8, 158.7,
114.6, 101.7, 82.2, 79.1, 75.6, 69.0, 64.7, 28.9, 19.5.
HRMS (EI+) calcd for [C12H14ClN2O4]+ 285.0637; found 285.0644

43
Determination of the relative stereochemistry for nucleoside 116
Analysis of 2D NOESY of nucleoside 116 revealed the indicated
stereochemistry.
Determination of enantiomeric excess of diol D4a
Following General Procedures A and B, using a 1:1 mixture of L-: D-proline, a racemic sample of
diol D4a was prepared. The enantiomeric diols were separated by chiral HPLC using a Lux® 3µm
Amylose-1 column; flow rate 0.25 mL/min; eluent: hexanes-iPrOH 90:10; detection at 254 nm;
retention time = 11.81 min for (-)-D4a; 12.68 min for (+)-D4a (see chromatograms).The
enantiomeric ratio of the optically enriched (+)- D4a diol was determined using the same method
(95:5 e.r.).
Preparation of S5, hydrate SM5, aldol adduct A5, diol adducts D5a and D5b, and nucleoside
analogue 117
A solution of 1,2,3-triazole (1.00 mL, 17.2 mmol, 1.0 equiv.), bromoacetaldehyde diethyl acetal
(3.10 mL, 20.7 mmol, 1.2 equiv.) and K2CO3 (4.75 g, 34.4 mmol, 2.0 equiv.) was stirred for 24
hours at 90 °C in DMF (86 mL). The reaction mixture was then filtered and washed with 40 mL of
CH2Cl2 and concentrated under reduced pressure. Purification of crude S5 by flash
chromatography (pentane:ethyl acetate – 7:3) afforded S5 (2.90 g, 91% yield) as a colorless oil.
A solution of S5 (0.100 g, 0.54 mmol, 1.0 equiv.) was heated to 90˚C in 0.5 M HCl (0.54 mL) for
5 hours. Upon complete conversion to SM5, the reaction mixture was concentrated under reduced
pressure and the resulting product SM5 was used in the reaction without purification.
Data for S5: 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 0.90 Hz, 1H), 7.66
(d, J = 0.90 Hz, 1H), 4.76 (t, J = 5.3 Hz, 1H), 4.48 (d, J = 5.3 Hz, 2H), 3.73
(m, 2H), 3.47 (m, 2H), 1.17 (m, 6H); 13C NMR (125 MHz, CDCl3): δ 133.8,
124.9, 101.1, 64.0, 52.9, 15.3.
HRMS (EI+) calcd for [C8H16N3O2]+ 186.1237; found 186.1233
α-fluorination/aldol

44
Following General Procedure A, a solution of S5 (0.54 mmol), Selectfluor (0.192 g, 0.54 mmol),
L-proline (0.063 g, 0.54 mmol) and NaHCO3 (0.045 g, 0.54 mmol) was stirred for 12 hours at 4°C
in DMF (0.72 mL). Dioxanone 75 (0.043 mL, 0.36 mmol) in MeCN (0.43 mL) was then added and
the reaction mixture was stirred for 72 hrs at room temperature. Purification of the crude
fluorohydrin A5 by flash chromatography (Et2O) afforded fluorohydrin A5 (0.061 g, 65 % yield,
d.r. 1:1) as a light yellow oil.
Data for syn- and anti-fluorohydrins A5: IR (neat): = 3138, 2990, 1749,
1455, 1379, 1224, 1070, 799 cm-1; 1H NMR (600 MHz, CDCl3): δ 8.24
(1H), 8.12 (1H), 7.79 (1H), 7.77 (1H), 6.89 (1H), 6.86 (1H), 4.74 (1H), 4.49
(1H), 4.33 (2H), 4.26 (1H), 4.14 (1H), 4.06 (1H), 3.89 (1H), 1.55 (3H), 1.48
(3H), 1.44 (3H), 1.31 (3H); 13C NMR (150 MHz, CDCl3): δ 210.8, 209.4, 134.5, 134.5, 124.4,
124.4, 102.1, 102.0, 94.5, 93.5, 72.1, 71.3, 70.8, 70.1, 66.5, 66.5, 23.8, 23.5, 23.4, 23.4; 19F NMR
(470 MHz, CDCl3): δ -154.6, -163.8.
HRMS (EI+) calcd for [C10H15FN3O4]+ 260.1041; found 260.1044
Syn-reduction of syn-and anti-fluorohydrins A5
Following General Procedure B, Me4NHB(OAc)3 (0.391 g, 1.49 mmol) and AcOH (0.170 mL, 2.98
mmol) were added to a stirred solution of A5 (0.077 g, 0.30 mmol) at -15 °C in MeCN (3.00 mL)
and the reaction mixture was stirred for 24 hrs. Purification of the crude diols D5a and D5b by
flash chromatography (CH2Cl2:MeOH – 96:4) afforded diols D5a and D5b (0.072 g, 94 % yield,
d.r. (syn/anti) = 1.2:1) as white solids.
Data for syn-diol, syn-fluorohydrin D5a: []D20 = +52.4 (c 0.51 in MeCN);
IR (neat): = 3432, 2997, 2253, 1444, 1375, 1071, 1039 cm-1;1H NMR
(600 MHz, CD3CN): δ 8.17 (d, J = 1.0 Hz, 1H), 7.78 (d, J = 1.0 Hz, 1H),
6.69 (dd, J = 48.1, 4.7 Hz, 1H), 4.36 (ddd, J = 18.4, 5.0, 5.0 Hz, 1H), 3.79
(dd, J = 11.4, 5.0 Hz, 1H), 3.63 (m, 2H), 3.54 (m, 2H), 1.39 (s, 3H), 1.31 (s, 3H); 13C NMR (150
MHz, CD3CN): δ 135.2, 126.2, 100.0, 95.9 (d, J = 206.7 Hz), 74.7 (d, J = 22.7 Hz), 73.1 (d, J =
4.4 Hz), 66.0, 65.2, 28.8, 19.9; 19F NMR (470 MHz, CDCl3): δ -156.0
HRMS (EI+) calcd for [C10H17FN3O4]+ 262.1198; found 262.1209

45
Data for syn-diol, anti-fluorohydrin D5b: []D20 = +40.0 (c 0.37 in MeCN);
IR (neat): = 3000, 1442, 1375, 1039, 918, 740 cm-1;1H NMR (600 MHz,
CD3CN): δ 8.22 (d, J = 1.0 Hz, 1H), 7.79 (d, J = 1.0 Hz, 1H), 6.78 (dd, J =
46.4, 6.0 Hz, 1H), 4.53 (ddd, J = 10.4, 6.0, 4.7 Hz, 1H), 4.09 (br s, 1H),
3.83 (m, 2H), 3.57 (m, 2H), 3.41 (br s, 1H), 1.35 (s, 3H), 1.34 (s, 3H); 13C NMR (150 MHz, CD3CN):
δ 135.3, 125.7, 100.0, 96.5 (d, J = 204.3 Hz), 74.2 (d, J = 2.3 Hz), 72.9 (d, J = 27.2 Hz), 65.4,
65.3 (d, J = 2.0 Hz), 28.9, 19.8; 19F NMR (470 MHz, CDCl3): δ -151.2
HRMS (EI+) calcd for [C10H17FN3O4]+ 262.1198; found 262.1206
Cyclization of diol D5a
Following General Procedure C, a solution of D5a and D5b (0.025 g, 0.096 mmol, 1.0 equiv, d.r.
(syn/anti) = 1.2:1) and Sc(OTf)3 (0.118 g, 0.239 mmol, 2.5 equiv.) was stirred in dry MeCN (1.00
mL). After 12 hours, pyridine (0.50 mL) and acetic anhydride (0.25 mL) were added and the
reaction mixture was left to stir for 3 hrs. Purification of the crude 117 by flash chromatography
(pentane:ethyl acetate – 1:3) afforded nucleoside analogue 117 (0.015 g, 47 % yield) as a clear
colorless oil.
Data for nucleoside analogue 117: []D20 = +1.3 (c 0.60 in CH2Cl2); IR (neat):
= 2926, 1747, 1373, 1227, 1064 cm-1; 1H NMR (600 MHz, CDCl3): δ 7.76 (s,
1H), 7.26 (s, 1H), 6.19 (d, J = 3.7 Hz. 1H), 5.85 (dd, J = 5.0, 3.8 Hz, 1H), 5.63
(dd, J = 5.3, 5.0 Hz, 1H), 4.49 (ddd, J = 5.3, 4.3, 3.0 Hz, 1H), 4.41 (dd, J =
12.4, 3.0 Hz, 1H), 4.22 (dd, J = 12.4, 4.3 Hz, 1H), 2.13 (s, 3H), 2.13 (s, 3H), 2.06 (s, 3H); 13C NMR
(150 MHz, CDCl3): δ 170.5, 169.6, 169.5, 134.3, 122.9, 90.0, 81.0, 74.5, 70.8, 62.9, 20.8, 20.6,
20.6;
HRMS (EI+) calcd for [C13H18N3O7]+ = 328.3005; found 328.3000
Determination of relative stereochemistry for diol D5a
The relative stereochemistry of diol D5a was determined by J-based
configurational analysis. See J-based configurational analysis
section for details.

46
Determination of relative stereochemistry for diol D5b
The relative stereochemistry of diol D5b was determined by J-based
configurational analysis. See J-based configurational analysis section
for details.
Determination of relative stereochemistry for nucleoside 117
Analysis of 2D NOESY of nucleoside 117a supported the indicated
stereochemistry.
Figure S2.5. Cyclization of diol 5a. Following General Procedure D, diol D5a was cyclized
separately to 117 while diol D5b did not cyclize. This suggests the product generated from the
diol mixture comes only from the D5a diol via an SN2 cyclization.
Determination of enantiomeric excess of diol D5a
Following General Procedures A and B, using a 1:1 mixture of L-: D- proline, a racemic sample
of diol D5a was prepared. The enantiomeric diols were separated by chiral HPLC using a Lux®
3µm i-Cellulose-5 column; flow rate 0.20 mL/min; eluent: hexanes-iPrOH 90:10; detection at 210

47
nm; retention time = 4.69 min for (+)-D5a; 5.80 min for (-)-D5a (see chromatograms).The
enantiomeric ratio of the optically enriched (+)-D5a diol was determined using the same method
(93:7 e.r.).
Determination of enantiomeric excess of diol D5b
Following General Procedures A and B, using a 1:1 mixture of L-: D- proline, a racemic sample
of diol D5b was prepared. The enantiomeric diols were separated by chiral HPLC using a Lux®
3µm i-Cellulose-5 column; flow rate 0.20 mL/min; eluent: hexanes-iPrOH 90:10; detection at 210
nm; retention time = 3.94 min for (-)-D5b; 4.95 min for (+)-D5b (see chromatograms).The
enantiomeric ratio of the optically enriched (+)-D5b diol was determined using the same method
(96:4 e.r.).
Determination of enantiomeric excess of diols ent-D5a
Following General Procedures A and B, using a 1:1 mixture of L-: D- proline, a racemic sample
of diol ent-D5a was prepared. The enantiomeric diols were separated by chiral HPLC using a
Lux® 3µm i-Cellulose-5 column; flow rate 0.20 mL/min; eluent: hexanes-iPrOH 90:10; detection
at 210 nm; retention time = 4.69 min for (+)-D5a; 5.80 min for (-)-D5a (see chromatograms).The
enantiomeric ratio of the optically enriched ent-D5a diol was determined using the same method
(95:5 e.r.).
Determination of enantiomeric excess of diols ent-D5b
Following General Procedures A and B, using a 1:1 mixture of L-: D- proline, a racemic sample
of diol ent-D5b was prepared. The enantiomeric diols were separated by chiral HPLC using a
Lux® 3µm i-Cellulose-5 column; flow rate 0.20 mL/min; eluent: hexanes-iPrOH 90:10; detection
at 210 nm; retention time = 3.94 min for (-)-D5b; 4.95 min for (+)-D5b (see chromatograms).The
enantiomeric ratio of the optically enriched ent-D5b diol was determined using the same method
(95:5 e.r.).

48
Preparation of S6, hydrate SM6, aldol adduct A6, diol adducts D6a and D6b, and nucleoside
analogue 118
A solution of trifluoromethyluracil (1.00 g, 5.52 mmol, 1.0 equiv.), bromoacetaldehyde diethyl
acetal (1.66 mL, 11.1 mmol, 2.0 equiv.) and K2CO3 (1.53 g, 11.1 mmol, 2.0 equiv.) was stirred for
24 hours at 90 °C in DMF (27.6 mL). The reaction mixture was then filtered and washed with 40
mL of CH2Cl2 and concentrated under reduced pressure. Purification of crude S6 by flash
chromatography (pentane:ethyl acetate – 7:3) afforded S6 (0.605 g, 37% yield) as a colorless oil.
A solution of S7 (0.100 g, 0.340 mmol, 1.0 equiv.) was heated to 90˚C in 0.5 M HCl (0.34 mL) for
5 hours. Upon complete conversion to aldehyde/hydrate SM6, the reaction mixture was
concentrated under reduced pressure and the resulting aldehyde/hydrate SM6 was used in the
reaction without purification.
Data for S6: IR (neat): = 3430, 2988, 2800, 1109, 1025cm-1; 1H NMR
(600 MHz, CDCl3): δ 8.56 (br s, 1H), 7.82 (s, 1H), 4.61 (t, J = 5.0 Hz),
3.88 (d, J =5.0 Hz), 3.78 (m, 2H), 3.54 (m, 2H), 1.21 (m, 6H); 13C NMR
(150 MHz, CDCl3): δ 158.6, 150.0, 147.0 (q, J = 5.8 Hz), 121.9 (q, J =
270.5 Hz), 104.7 (q, J =33.5 Hz), 100.0, 64.6, 51.0, 15.3
HRMS (EI+) calcd for [C11H16F3N2O4]+ 297.1057; found 297.1056
α-fluorination/aldol
Following General Procedure A, a solution of SM6 (0.340 mmol), NFSI (0.107 g, 0.340 mmol), L-
proline (0.039 g, 0.340 mmol) and NaHCO3 (0.029 g, 0.340 mmol) was stirred for 12 hours at 4°C
in DMF (0.45 mL). Dioxanone 75 (0.027 mL, 0.227 mmol) in CH2Cl2 (0.57 mL) was then added
and the reaction mixture was stirred for 96 hrs at 4°C. Purification of the crude fluorohydrin A6 by
flash chromatography (pentane:ethyl acetate – 65:35 ) afforded fluorohydrin A6 (0.050 g, 60 %
yield) as a light yellow oil.
Data for syn- and anti- fluorohydrins A6: IR (neat): = 2991, 1699, 1450,
1087, 1049 cm-1; 1H NMR (600 MHz, CD3CN): δ 9.53, 9.52, 8.15, 8.11,
6.58, 6.46, 4.62, 4.56, 4.55, 4.43, 4.31, 4.29, 3.98, 3.98, 1.43, 1.40, 1.40,
1.38; 13C NMR (150 MHz, CD3CN): δ 208.4, ,207.9, 159.6, 159.5, 150.6,
150.1, 144.0, 144.0, 123.6, 123.5, 106.6, 106.0, 102.4, 102.3, 95.3, 92.4,

49
76.3, 76.1, 69.9, 69.1, 67.9, 67.8, 24.5, 24.4, 24.2, 23.9; 19F NMR (470 MHz, CD3CN): δ –64.1, –
64.1, –161.4, –169.1
HRMS (EI+) calcd for [C13H14F4N2NaO6]+ 393.0680; found 393.0682
Syn-reduction of syn-and anti-fluorohydrins A6
Following General Procedure B, Me4NHB(OAc)3 (0.355 g, 1.35 mmol) and AcOH (0.155 mL, 2.79
mmol) were added to a stirred solution of A6 (0.100 g, 0.27 mmol, 1 equiv.) at -15 °C in MeCN
(1.80 mL) and the reaction mixture was stirred for 24 hrs. Purification of the crude diols D6a and
D6b by flash chromatography (pentane:ethyl acetate – 4:1) afforded diols D6a (0.040 g, 40%
yield) and D6b (0.019 g, 19% yield) as white solids.
Data for syn-diol, syn-fluorohydrin D6a: []D20 = +18.4 (c 0.50 in CH2Cl2);
IR (neat): = 3426, 2996, 1702, 1463, 1379, 1070 cm-1; 1H NMR (600
MHz, CD3CN): δ 9.42 (br s, 1H), 8.10 (s, 1H), 6.33 (dd, J = 45.1, 5.6 Hz,
1H), 4.28 (dd, J = 14.8, 5.6 Hz, 1H), 3.79 (dd, J = 11.1 5.5 Hz, 1H), 3.70
(m, 2H), 3.60 (dd, J = 9.5, 2.7 Hz, 1H), 3.55 (dd, J = 10.4, 9.5 Hz, 1H), 1.35
(s, 3H), 1.30 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 159.5, 150.1, 144.2 (q, J = 6.3 Hz), 123.5
(q, J = 266.4 Hz), 106.3 (q, J = 32.9 Hz), 99.9, 96.3 (d, J = 210.9 Hz), 73.9 (d, J = 3.8 Hz), 70.5
(d, J = 24.5 Hz), 65.4, 63.0, 29.1, 19.8; 19F NMR (470 MHz, CD3CN): δ –64.1, –168.0.
HRMS (EI+) calcd for [C13H17F4N2NaO6]+ 395.0837; found 395.0836
Data for syn-diol, anti-fluorohydrin D6b: []D20 = -37.2 (c 1.1 in CH2Cl2); IR
(neat): = 3424, 1703, 1466, 1379, 1281, 1138, 1042 cm-1; 1H NMR (600
MHz, CD3CN): δ 8.26 (s, 1H), 6.67 (dd, J = 43.0, 4.9 Hz, 1H), 4.34 (m, 1H),
3.78 (dd, J = 11.2, 5.1 Hz, 1H), 3.72 (m, 2H), 3.54 (dd, J = 11.2, 8.3 Hz,
1H), 1.39 (s, 3H), 1.26 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 159.5,
150.6, 144.2, 123.6 (q, J = 272.9 Hz), 105.9 (q, J = 32.5 Hz), 100.0, 92.5 (d, J = 206.1 Hz), 74.2
(d, J = 4.4 Hz), 72.3 (d, J = 27.7 Hz), 65.4, 64.8, 29.0, 19.7;19F NMR (470 MHz, CD3CN): δ –64.1,
–161.7.
HRMS (EI+) calcd for [[C13H17F4N2NaO6]+ 395.0837; found 395.0838
Cyclization of diols D6a and D6b

50
Following General Procedure D, a solution of D6a and D6b (0.045 g, 0.121 mmol, d.r. (syn/anti)
= 1:2) and Sc(OTf)3 (8.9 mg, 0.018 mmol, 0.15 equiv.) was stirred for 24 hours in dry MeCN (1.21
mL). Purification of the crude 118 by flash chromatography (pentane:ethyl acetate – 3:7) afforded
nucleoside 118 (0.013 g, 45 % yield (from anti-fluorohydrin D6b)) as a colorless oil.
Data for nucleoside analogue 118: []D20 = -16.7 (c 0.49 in CH2Cl2); IR
(neat): = 3405, 2924, 2854, 1702, 1465, 1276 cm-1; 1H NMR (600
MHz, CD3CN): δ 9.33 (br s, 1H), 7.97 (q, J =1.2 Hz, 1H), 6.18 (d, J =
4.1 Hz, 1H), 4.86 (m, 2H), 4.42 (dd, J = 3.6, 2.4 Hz, 1H), 3.67 (m, 2H),
3.21 (dd, J =5.6, 4.4 Hz, 1H), 1.36 (s, 3H), 1.30 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 159.4,
149.9, 143.6 (q, J =6.0 Hz), 123.6 (q, J =269.7 Hz), 113.6, 103.4 (q, J =33.2 Hz), 87.7, 84.7, 82.8,
80.2, 64.0, 25.7, 24.0; 19F NMR (470 MHz, CD3CN): δ –63.8
HRMS (EI+) calcd for [C13H16F3N2O6]+ 353.0955; found 353.0971
Determination of relative stereochemistry for nucleoside 118
Analysis of 2D NOESY of nucleoside 118 supported the indicated
stereochemistry.
Determination of relative stereochemistry for diols D6a and D6b
Based on J-based configurational analysis of compounds D5a/D5b,
D8a/D8b and XRD analysis of compounds 107a, D7b, D9a a clear trend
was established between the stereochemistry at the fluoromethine center
and the chemical shift of the fluoromethine proton (*). In every case, the
syn-fluorohydrin diol has a lower chemical shift than the diastereomeric anti-fluorohydrin diol.
Here, D6a has a chemical shift of 6.33 ppm while D6b has a chemical shift of 6.67 ppm for the
fluoromethine proton. D6a was assigned as the syn-fluorohydrin diol and D6b the anti-fluorohydrin
diol.

51
Figure S2.6. Cyclization of diol D6b. Following General Procedure D, diol D6b was cyclized
separately to 118 while diol D6a did not cyclize. This suggests the product from generated from
the diol mixture comes only from the D6b diol via an SN2 cyclization.
Determination of enantiomeric excess of nucleoside 118
Following General Procedures A, B, and C using a 1:1 mixture of L-:D- proline, a racemic sample
of nucleoside 118 was prepared. The enantiomeric nucleosides were separated by chiral HPLC
using a a Lux® 3µm Amylose-1 column; flow rate 0.25 mL/min; eluent: hexanes-iPrOH 90:10;
detection at 254 nm; retention time = 9.10 min for (+)-118; 13.14 min for (-)-118 (see
chromatograms).The enantiomeric ratio of the optically enriched (-)-118 nucleoside was
determined using the same method (94:6 e.r.).
Preparation of S7, hydrate SM7, aldol adduct A7, diol adducts D7a and D7b, and nucleoside
analogue 119
α-fluorination/aldol and syn-reduction of syn-and anti-fluorohydrins A7
Following General Procedure A, a solution of phthalimidoacetaldehyde (0.100 g, 0.529 mmol, 1.5
equiv.), NFSI (0.167 g, 0.529 mmol, 1.5 equiv.), L-proline (0.061 g, 0.529 mmol, 1.5 equiv.) and
2,6-lutidine (0.061 mL, 0.529 mmol, 1.5 equiv.) was stirred for 12 hours at 4°C in DMF (0.71 mL).
Dioxanone 75 (0.042 mL, 0.353 mmol, 1 equiv.) in CH2Cl2 (0.88 mL) was then added and the
reaction mixture was stirred for 48 hrs at room temperature. Purification of the crude fluorohydrin

52
A7 by flash chromatography (pentane:ethyl acetate – 1:1) afforded fluorohydrin A7 (0.069 g, 58
% yield, d.r. 2.2:1) as a yellow oil. Following General Procedure B, Me4NHB(OAc)3 (0.776 g, 2.95
mmol) and AcOH (0.337 mL, 5.90 mmol) were added to a stirred solution of A7 (0.200 g, 0.59
mmol) at -15 °C in MeCN (5.90 mL) and the reaction mixture was stirred for 24 hrs. Purification
of the crude diols D7a and D7b by flash chromatography (pentane:ethyl acetate – 3:7) afforded
diols D7a and D7b (0.094 g, 47 % yield, d.r. (syn/anti) = 1.5:1) as white solids.
Data for syn-diol, syn-fluorohydrin D7a: []D20 = -11.4 (c 2.0 in CH2Cl2); IR
(neat): = 3442, 2992, 1785, 1724, 1377, 1074, 721 cm-1; 1H NMR (600
MHz, CD3CN): δ 7.93 (m, 2H), 7.89 (m, 2H), 6.07 (dd, J = 48.6, 7.9 Hz,
1H), 4.76 (m, 1H), 4.43 (m, 1H), 3.73 (m, 2H), 3.58 (dd, J = 8.8, 6.0 Hz,
1H), 3.47 (m, 1H), 3.41 (m, 1H), 1.21 (s, 3H), 0.92 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 167.8
(d, J = 1.5 Hz), 136.0, 132.5, 124.6, 99.1, 91.1 (d, J = 202.0 Hz), 73.3 (d, J = 6.6 Hz), 71.8 (d, J
= 25.3 Hz), 65.1, 64.5, 28.1, 19.3; 19F NMR (470 MHz, CD3CN): δ –157.8
HRMS (EI+) calcd for [C16H19FNO6]+ 340.1191; found 340.1190
Data for syn-diol, anti-fluorohydrin D7b: []D20 = -1.0 (c 2.3 in CH2Cl2); IR
(neat): = 3442, 2992, 1784, 1725, 1375, 1070, 723 cm-1; 1H NMR (600
MHz, CD3CN): δ 7.94 (m, 2H), 7.89 (m, 2H), 6.34 (dd, J = 46.0, 9.2 Hz,
1H), 4.80 (m, 1H), 3.92 (ddd, J = 9.5, 1.8, 1.4 Hz, 1H), 3.84 (m, 2H), 3.73
(m, 1H), 3.60 (dd, J = 10.8, 8.7 Hz, 1H), 3.30 (m, 1H), 1.47 (s, 3H), 1.35 (s, 3H); 13C NMR (150
MHz, CD3CN): δ 168.1 (d, J = 1.6 Hz), 136.0, 132.3, 124.6, 99.4, 89.5 (d, J = 202.4 Hz), 75.1,
68.7 (d, J = 31.7 Hz), 65.3, 63.1 (d, J = 3.1 Hz), 28.6, 19.5; 19F NMR (470 MHz, CDCl3): δ –159.8
HRMS (EI+) calcd for [C16H19FNO6]+ 340.1191; found 340.1172
Cyclization of diols D7a and D7b
Following General Procedure C, a solution of D7a and D7b (0.033 g, 0.097 mmol, 1.0 equiv., d.r.
(syn/anti) = 2:1) and Sc(OTf)3 (0.120 g, 0.243 mmol, 2.5 equiv.) was stirred for 6 hours in MeCN
(0.65 mL). 0.25 mL of pyridine and 0.25 mL of acetic anhydride were added and the reaction
mixture was allowed to stir for a further 1.5 hrs. Purification of the crude 119 by flash
chromatography (pentane:ethyl acetate – 7:3) afforded nucleoside analogue 119 (0.027 g, 69 %
yield) as a colourless oil.

53
Data for nucleoside analogue 119: []D20 = -9.0 (c 1.96 in CH2Cl2); IR (neat):
= 2922, 1781, 1744, 1721, 1374, 1222, 1047, 720 cm-1; 1H NMR (500 MHz,
CDCl3): δ 7.88 (m, 2H), 7.77 (m, 2H), 5.94 (dd, J = 6.0, 4.1 Hz, 1H), 5.87 (d,
J = 4.1 Hz, 1H), 5.65 (dd, J = 6.1, 6.0 Hz, 1H), 4.49 (dd, J = 12.1, 3.4 Hz,
1H), 4.29 (ddd, J = 9.5, 5.9, 3.4 Hz, 1H), 4.21 (dd, J = 12.1, 5.9, 1H), 2.12
(s, 3H), 2.11 (s, 3H), 2.09 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 170.9, 169.8, 169.7, 166.9,
134.8, 131.7, 124.0, 82.8, 79.2, 72.0, 70.6, 63.2, 20.9, 20.7, 20.7
HRMS (EI+) calcd for [C19H19NO9 + NH4]+ 423.1398; found 423.1378
Determination of relative stereochemistry for diol D7b
Recyrstallization in ethanol allowed for the relative stereochemistry to
be assigned using single X-ray crystallography (see X-ray structures).
Determination of the relative stereochemistry for nucleoside 119
Analysis of 2D NOESY of nucleoside 119 supported the indicated
stereochemistry.

54
Figure S2.7. Cyclization of diols D7a and D7b. Following General Procedure D, diol D7a was
cyclized separately to 119 while diol D7b cyclized to a mixture of 119 and its corresponding α-
anomer. The diol mixture comes from both diols via an SN2 cyclization and some epimerization of
the α-anomer. Such emperizations have been reported for nucleosides.121
Determination of enantiomeric excess of diol ent-D7a
Following General Procedures A and B, using a 1:1 mixture of L-:D- proline, a racemic sample of
diol D7a was prepared. The enantiomeric nucleosides were separated by chiral HPLC using a a
Lux® 3µm Amylose-1 column; flow rate 0.25 mL/min; eluent: hexanes-iPrOH 90:10; detection at
254 nm; retention time = 9.10 min for (-)-D7a; 13.14 min for (+)-D7a (see chromatograms).The
enantiomeric ratio of the optically enriched (+)-D7a diol was determined using the same method
(95:5 e.r.).
Preparation of SM8, aldol adduct A8, diol adducts D8a/D8b, and nucleoside analogues 121/122
A solution of deazadenine (0.500 g, 1.79 mmol, 1.0 equiv.), bromoacetaldehyde diethyl acetal
(0.323 mL, 2.15 mmol, 1.25 equiv.) and K2CO3 (0.491 g, 3.58 mmol, 2.0 equiv.) was stirred for 24
hours at 90 °C in DMF (9.00 mL). The reaction mixture was then filtered and washed with 10 mL
of CH2Cl2 and concentrated under reduced pressure. Purification of crude S8 by flash
chromatography (pentane:ethyl acetate – 7:3) afforded S8 (0.375 g, 53 % yield) as a white solid.
A solution of S8 (17.0 g, 43.0 mmol, 1.0 equiv.) was heated to 70˚C in 2.0 M HCl (129 mL, 258
mmol, 6.0 equiv.) for 1 hours. The reaction mixture was then cooled to room temperature and
allowed to stir for a further 2 hrs. The reaction mixture was stored overnight at -20 °C and the

55
formed precipitate was then filtered and washed with 1:1 dioxane:water (10 mL x 2). The filtrate
SM8 was dried under reduced pressure and the resulting product SM8 (7.88 g, 54 % yield) was
used in the reaction without purification.
Data for S8: 1H NMR (600 MHz, CDCl3): δ 8.61 (s, 1H), 7.50 (s, 1H), 4.67
(t, J = 5.1 Hz, 1H), 4.35 (d, J = 5.1 Hz, 2H), 3.73 (m, 2H), 3.48 (m, 2H),
1.16 (m, 6H); 13C NMR (150 MHz, CDCl3): δ 152.7, 151.1, 150.8, 136.3,
116.9, 100.7, 63.9, 50.6, 47.7, 15.3
HRMS (EI+) calcd for [C12H16ClIN3O2]+ 395.9970; found 395.9973
α-fluorination/aldol
Following General Procedure A, a solution of SM8 (2.00 g, 5.86 mmol, 1 equiv.), NFSI (1.85 g,
5.86 mmol, 1.0 equiv.), L-proline (0.674 g, 5.86 mmol, 1.0 equiv.) and NaHCO3 (0.984 g, 11.71
mmol, 2.0 equiv.) was stirred for 18 hours at 20°C in DMF (10 mL). Dioxanone 75 (0.762 g, 5.86
mmol, 1.0 equiv.) was then added and the reaction mixture was stirred for 36 hrs at room
temperature. Purification of the crude A8 by flash chromatography (25-75% ethyl acetate in
pentane) afforded syn- and anti-fluorohydrins A8 (1.58 g, 57 % yield, d.r. 1.2:1) as a light yellow
solid.
Data for syn-and anti-fluorohydrins A8: IR (neat): = 3145, 2988, 1747,
1575, 1539, 1444, 1205, 1084, 949, 734 cm-1; 1H NMR (600 MHz, dmso-
d6): δ 8.76, 8.74, 8.39, 8.24, 6.89, 6.85, 6.37, 6.12, 4.98, 4.76, 4.61, 4.32,
4.30, 4.05, 3.95, 3.93, 1.40, 1.34, 1.33, 1.31; 13C NMR (150 MHz, dmso-
d6): δ 206.3, 206.1, 151.6, 151.5, 151.3, 151.2, 151.0, 134.5, 134.1, 116.8,
116.7, 100.4, 100.1, 91.4, 09.4, 76.1, 74.7, 68.7, 68.0, 66.6, 66.4, 55.3, 55.1, 24.6, 24.1, 22.9,
22.7; 19F NMR (470 MHz, dmso-d6): δ –146.0, –152.6
HRMS (EI+) calcd for [C14H15ClFIN3O4]+ 469.9774; found 469.9779
syn-reduction of syn-and anti-fluorohydrins A8
Following General Procedure B, NaHB(OAc)3 (0.316 g, 1.49 mmol, 5 equiv.) and AcOH (0.171
mL, 2.98 mmol, 10 equiv.) were added to a stirred solution of A8 (0.140 g, 0.298 mmol, 1 equiv.)
at 0 °C in MeCN (2.8 mL). The reaction mixture was then stirred at room temperature for 2hrs.

56
Purification of the crude diols D8a and D8b by flash chromatography (pentane:ethyl acetate –
70:30) afforded diols D8a and D8b (0.141 g, 77 % yield, d.r. (syn/anti) = 1.5:1) as a white solid.
Data for syn-diol, syn-fluorohydrin D8a: []D20 = -19.6 (c 2.0 in CH2Cl2); IR
(neat): = 3335, 2989, 2890, 1577, 1540, 1445, 1206, 1076, 951 cm-1;1H
NMR (600 MHz, dmso-d6): δ 8.73 (s, 1H), 8.27 (s, 1H), 6.73 (dd, J = 49.4,
7.0 Hz, 1H), 6.08 (br s, 1H), 4.84 (d, J = 4.1 Hz, 1H), 4.59 (m, 1H), 3.59
(m, 1H), 3.44 (m, 1H), 3.42 (m, 1H), 3.33 (m, 1H), 1.16 (s, 3H), 1.13 (s,
3H); 13C NMR (150 MHz, dmso-d6): δ 151.4, 151.2, 151.1, 134.5, 116.7, 97.8, 92.0 (d, J = 203.3),
73.2 (d, J =5.7 Hz), 71.0 (d, J = 24.2 Hz), 63.8, 62.5, 54.9, 28.0, 19.1; 19F NMR (470 MHz, dmso-
d6): δ –147.1
HRMS (EI+) calcd for [C14H15ClFIN3O4]+ 471.9931; found 471.9940
Data for syn-diol, anti-fluorohydrin D8b: []D20 = -11.6 (c 0.38 in CH2Cl2);
IR (neat): = 3363, 2931, 2890, 1579, 1540, 1444, 1212, 1067, 951 cm-
1; 1H NMR (600 MHz, dmso-d6): δ 8.73 (s, 1H), 8.34 (s, 1H), 6.97 (dd, J
= 46.9, 7.9 Hz, 1H), 5.74 (d, J = 5.7 Hz, 1H), 5.22 (d, J = 5.7 Hz, 1H), 4.61
(m, 1H), 3.84 (m, 1H), 3.72 (m, 1H), 3.52 (dd, J = 11.7, 8.7 Hz, 1H), 1.35 (s, 3H), 1.20 (s, 3H); 13C
NMR (150 MHz, dmso-d6): δ 151.5, 151.4, 151.2, 134.1, 116.6, 97.9, 90.9 (d, J = 203.5 Hz), 74.3,
69.1 (d, J =30.3 Hz), 64.2, 61.4, 54.8, 28.4, 19.0; 19F NMR (470 MHz, , dmso-d6): δ –146.3
HRMS (EI+) calcd for [C14H15ClFIN3O4]+ 471.9931; found 471.9940
Cyclization of diol D8a
Following General Procedure D, a solution of D9a (0.050 g, 0.106 mmol, 1.0 equiv.) and InCl3
(2.3 mg, 0.011 mmol, 0.10 equiv.) was stirred for 16 hrs in dry MeCN (1.00 mL). Purification of
the crude nucleoside 121 by flash chromatography (20-80% ethyl acetate in pentanes) afforded
nucleoside 121 (0.029 g, 61 % yield) as a white solid.
Data for nucleoside analogue 121: []D20 = -23.9 (c 0.46 in CH2Cl2); IR
(neat): = 3339, 3113, 2935, 1576, 1539, 1445, 1207, 1108, 951 cm-1;
1H NMR (600 MHz, dmso-d6): δ 8.69 (s, 1H), 8.23 (s, 1H), 6.34 (d, J = 3.1
Hz, 1H), 5.19 (dd, J = 6.3, 3.1 Hz, 1H), 5.14 (br s, 1H), 4.94 (dd, J = 6.3,
2.9 Hz, 1H), 4.20 (m, 1H), 3.56 (m, 2H), 1.54 (s, 3H), 1.31 (s, 3H); 13C

57
NMR (150 MHz, dmso-d6): δ 151.2, 150.8, 150.4, 133.9, 116.7, 113.2, 89.4, 86.3, 83.9, 80.9,
61.4, 53.7, 27.0, 25.1.
HRMS (EI+) calcd for [C14H16ClIN3O4]+ 451.9869; found 451.9875
Cyclization of diol D8b
Following General Procedure D, a solution of D8b (0.050 g, 0.106 mmol, 1.0 equiv.) and InCl3
(2.3 mg, 0.011 mmol, 0.10 equiv.) was stirred for 16 hrs in dry MeCN (1.00 mL). Purification of
the crude nucleoside 122 by flash chromatography (20-80% ethyl acetate in pentanes) afforded
nucleoside 122 (0.034 g, 70 % yield) as a white solid.
Data for nucleoside analogue 122: []D20 = -47.8 (c 0.51 in CHCl3); 1H
NMR (600 MHz, dmso-d6): δ 8.66 (s, 1H), 7.81 (s, 1H), 6.73 (d, J = 4.3
Hz, 1H), 5.22 (br s, 1H), 4.91 (m, 2H), 4.41 (dd, J = 3.6, 3.1 Hz, 1H),
3.62 (m, 2H), 1.32 (s, 3H), 1.23 (s, 3H); 13C NMR (150 MHz, dmso-d6):
δ 151.0, 150.7, 149.8, 134.6, 116.3, 112.3, 85.6, 83.1, 81.9, 79.4, 62.5,
51.9, 25.2, 23.9.
HRMS (EI+) calcd for [C14H16ClIN3O4]+ 451.9869; found 451.9888
Determination of relative stereochemistry for diol D8a
The relative stereochemistry of diol D8a was determined by J-based
configurational analysis. See J-based configurational analysis section
for details.
Determination of relative stereochemistry for diol D8b
The relative stereochemistry of diol D8b was determined by J-based
configurational analysis. See J-based configurational analysis section
for details

58
Determination of relative stereochemistry for nucleoside 121
Analysis of 2D NOESY of nucleoside 121 supported the indicated
stereochemistry.
Determination of relative stereochemistry for nucleoside 122
Analysis of 2D NOESY of nucleoside 122 supported the indicated
stereochemistry.

59
Figure S2.8. Cylization of diols D8a and D8b. Following General Procedure D, diol D8a was
cyclized separately to 121 while diol D8b cyclized to 122. This supports an SN2 cyclization without
subsequent epimerization.
Determination of enantiomeric excess of diol D8a
Following General Procedures A and B, using a 1:1 mixture of L-:D- proline, a racemic sample of
diol D8a was prepared. The enantiomeric diols were separated by chiral HPLC using an IB
column; eluent: 90:10 (MeCN:water) to 10:90 (MeCN:water); detection at 230 nm; retention time
= 12.23 min for (+)-D8a; 13.39 min for (-)-D8a (see chromatograms).The enantiomeric ratio of the
optically enriched ent-D8a diol was determined using the same method (90:10 e.r.).
Determination of enantiomeric excess of diol D8b
Following General Procedures A and B, using a 1:1 mixture of L-:D- proline, a racemic sample of
diol D8b was prepared. The enantiomeric diols were separated by chiral HPLC using a IG column;
eluent: 90:10 (MeCN:water) to 10:90 (MeCN:water); detection at 230 nm; retention time = 12.35
min for (-)-D8b; 12.56 min for (+)-D8b (see chromatograms).The enantiomeric ratio of the optically
enriched ent-D8b diol was determined using the same method (93:7 e.r.).

60
Preparation of SM9, aldehyde S9, aldol adduct A9, diol adducts D9a/D9b, and nucleoside
analogues SI9/NA9
A solution of iodouracil (2.50 g, 10.5 mmol, 1.0 equiv.), bromoacetaldehyde diethyl acetal (1.91
mL, 12.7 mmol, 1.2 equiv.) and K2CO3 (2.92 g, 21.1 mmol, 2.0 equiv.) was stirred for 16 hours at
90 °C in DMF (70 mL). The reaction mixture was filtered, and the filtrate was diluted with 200 mL
of ethyl acetate. The organic layer was washed 3 times with water, separated, dried over MgSO4,
filtered, and concentrated under reduced pressure. Purification of crude S9 by flash
chromatography (pentane:ethyl acetate – 75:25) afforded S9 (0.301 g, 8% yield) as a white solid.
A solution of S9 (0.142 g, 0.401 mmol, 1.0 equiv.) was heated to 90˚C in 0.5 M HCl (0.40 mL) for
5 hours. Upon complete conversion to aldehyde/hydrate SM9, the reaction mixture was
concentrated under reduced pressure and the resulting aldehyde/hydrate SM9 was used in the
reaction without purification.
Data for S9: IR (neat): = 2975, 1686, 1439, 1121, 1059, 1021 cm-1; 1H
NMR (600 MHz, CDCl3): δ 8.56 (br s, 1H), 7.82 (s, 1H), 4.61 (t, J = 5.0
Hz), 3.88 (d, J =5.0 Hz), 3.78 (m, 2H), 3.54 (m, 2H), 1.21 (m, 6H); 13C
NMR (150 MHz, CDCl3): δ 158.6, 150.0, 147.0 (q, J = 5.8 Hz), 121.9 (q,
J = 270.5 Hz), 104.7 (q, J =33.5 Hz), 100.0, 64.6, 51.0, 15.3
HRMS (EI+) calcd for [C10H16IN2O4]+ 355.0149; found 355.0145
α-fluorination/aldol and syn-reduction of syn- and anti-fluorohydrins A9
Following General Procedure A, a solution of S9 (0.401 mmol), NFSI (0.126 g, 0.401 mmol), L-
proline (0.046 g, 0.401 mmol) and NaHCO3 (0.034 g, 0.401 mmol) was stirred for 12 hours at 4°C
in DMF (0.53 mL). Dioxanone 75 (0.053 mL, 0.270 mmol) in CH2Cl2 (0.67 mL) was then added
and the reaction mixture was stirred for 72 hrs at 4°C. Purification of the crude fluorohydrin A9 by
flash chromatography (pentane-ethyl acetate – 1:1) afforded fluorohydrin A9. as a yellow oil.
Following General Procedure B, Me4NHB(OAc)3 (0.066 g, 0.251 mmol) and AcOH (0.0.30 mL,
0.502 mmol) were added to a stirred solution of A9 (0.021 g, 0.049 mmol) at -15 °C in MeCN
(0.49 mL) and the reaction mixture was stirred for 24 hrs. The crude diols D9a and D9b were
used directly for the cyclization owing to challenges with stability and purification.

61
Cyclization of diols D9a and D9b
Following General Procedure C, a solution of D9a and D9b (16.2 mg, 0.038 mmol, 1 equiv.) and
2 M NaOH (0.038 mL, 0.38 mmol, 10 equiv.) was stirred for 18 hours in MeCN (1.51 mL).
Purification of the crude nucleoside SI9 by flash chromatography (CH2Cl2:MeOH – 90:10) afforded
nucleoside SI9 as a white solid. SI9 (10.3 mg, 0.025 mmol) was dissolved in MeOD (0.25 mL)
and two drops of 1 M HCl was added and the solution was left for 12 hrs at room temperature.
Subsequently, the reaction mixture was concentrated under reduced pressure to afford NA9 as a
white solid. The spectral data matched previous reports.122
Data for nucleoside analogue SI9: 1H NMR (600 MHz, MeOD): δ 7.99 (s,
1H), 5.58 (s, 1H), 4.35 (d, J = 4.5 Hz, 1H), ,4.19 (dd, J =10.0, 4.6 Hz, 1H),
4.08 (dd, J =10.0, 9.7 Hz, 1H), 3.83 (m, 2H), 1.57 (s, 3H), 1.45 (s, 3H); 13C
NMR (150 MHz, MeOD): δ 162.8, 151.7, 147.2, 102.5, 95.7, 74.5, 73.8, 72.5,
68.9, 65.8, 29.3, 20.0
Data for nucleoside NA9: [α]D20 = -41 (c = 0.1, MeOH); IR (neat): ν = 3353,
2929, 1679, 1447, 1262, 1101, 1023, 799 cm–1; 1H NMR (600 MHz, MeOD):
δ 8.61 (s, 1H), 5.86 (d, J = 3.6 Hz, 1H), 4.16-4.17 (m, 2H), 4.02-4.03 (m, 1H),
3.89 (dd, J = 12.2, 2.6 Hz, 1H), 3.76 (dd, J = 12.1, 2.5 Hz, 1H); 13C NMR (150
MHz, MeOD): δ 162.8, 152.2, 147.3, 90.9, 86.3, 76.1, 70.9, 68.3, 61.7.
HRMS (EI+) calcd for [C9H12IN2O6]+ 370.9735; found: 370.9739
Determination of relative stereochemistry for diols D9a
Recyrstallization in ethanol allowed for the relative stereochemistry to be
assigned using single X-ray crystallography (see X-ray structures).

62
Preparation of nucleoside analogue 125
To a solution of nucleoside analogue 125 (0.100 g, 0.352 mmol, 1 equiv.) in THF (3.52 mL) was
added 1, 1’- thiocarbonyldiimidazole (0.125 g, 0.704 mmol, 2 equiv.). The reaction mixture was
stirred for 24 hrs. Subsequently, CH2Cl2 (10 mL) was added to the reaction mixture and washed
with water 3 times. The organic layer was dried over MgSO4, filtered, and concentrated under
reduced pressure to yield crude S125. Purification of crude S125 by flash chromatography (ethyl
acetate) afforded S125 (0.129 g, 96%).
Data for nucleoside analogue S125: []D20 = +25.8 (c 1.2 in MeCN); IR
(neat): = 3000, 1701, 1443, 1375, 1039, 918, 749 cm-1; 1H NMR (600
MHz, CD3CN): δ 9.34 (br s, 1H), 8.38 (s, 1H), 7.73 (s, 1H), 7.43 (d, J = 7.4
Hz, 1H), 7.04 (s, 1H), 6.08 (d, J = 5.2 Hz, 1H), 5.88 (d, J = 5.2 Hz, 1H),
5.69 (d, J = 7.4 Hz, 1H), 4.22 (m, 2H), 4.06 (dd, J = 10.4 Hz, 1H), 3.83
(ddd, J = 10.4, 10.3, 5.0 Hz, 1H), 1.55 (s, 3H), 1.39 (s, 3H); 13C NMR (150
MHz, CD3CN): δ 184.8, 164.1, 151.3, 143.3, 138.4, 132.3, 119.8, 103.8,
102.9, 92.4, 82.7, 73.5, 72.8, 65.5, 29.5, 20.4.
HRMS (EI+) calcd for [C16H19N4O6S]+ 395.1020; found 395.1010
To a solution of nucleoside S125 (0.020 g, 0.045 mmol, 1 equiv.) in dry toluene (3.0 mL) under
nitrogen was added tributyltin hydride (0.024 mL, 0.090 mmol, 2 equiv.) and AIBN (1.8 mgs, 0.011
mmol, 0.25 equiv.). The resulting reaction mixture was purged with nitrogen for 30 minutes.
Subsequently, the reaction mixture was stirred for 16 hrs at 90 °C. The reaction mixture was
diluted with CH2Cl2 (10 mL). The organic layer was washed with water, separated, dried over
MgSO4, filtered, and concentrated under reduced pressure to yield crude 125. Purification of
crude 125 by flash chromatography (ethyl acetate) afforded nucleoside 125 (6.8 mg, 57%) as a
colorless oil.
Data for nucleoside analogue 125: []D20 = +7.8 (c 0.32 in MeOH); 1H NMR
(600 MHz, CD3CN): δ 8.94 (br s, 1H), 7.50 (d, J = 8.2 Hz, 1H), 6.14 (dd, J =
8.7, 2.1 Hz, 1H), 5.63 (d, J = 8.2 Hz, 1H), 4.10 (dd, J = 10.0, 4.6 Hz, 1H),
4.00 (dd, J = 10.3, 10.0 Hz, 1H), 3.94 (m, 1H), 3.35 (ddd, J = 10.3, 10.0, 4.6
Hz, 1H), 2.27 (m, 1H), 2.17 (m, 1H), 1.52 (s, 3H), 1.37 (s, 3H); 13C NMR
(150 MHz, CD3CN): δ 164.1, 151.6, 142.6, 103.3, 102.2, 84.4, 76.3, 72.7,
65.6, 36.4, 29.8, 20.5.

63
HRMS (EI+) calcd for [C12H17N2O5]+ 269.1132; found 269.1111
Preparation of nucleoside analogue 126
To a solution of nucleoside analogue 106 (0.020 g, 0.083 mmol, 1.0 equiv.) in dry CH2Cl2 (0.83
mL) was added TEMPO (1.3 mg, 0.008 mmol, 0.10 equiv.) and (diacetoxyiodo)benzene (0.067
g, 0.208 mmol, 2.5 equiv.). Following 18 hrs or complete consumption of 106 as monitored by 1H
NMR spectroscopy, the reaction mixture was cooled to room temperature and diluted with CH2Cl2.
The organic layer was then washed with saturated sodium bicarbonate solution, dried over
MgSO4, filtered, and concentrated under reduced pressure to yield crude 126. Purification of the
crude nucleoside 126 by flash chromatography (pentane:ethyl acetate – 1:1) afforded nucleoside
126 (0.019 g, 92 % yield) as a white solid.
Data for nucleoside analogue 126: []D20 = -115.6 (c 1.0 in MeCN); IR (neat):
= 3001, 2989, 1694, 1374, 1305, 1088 cm-1; 1H NMR (600 MHz, CD3CN):
δ 7.80 (d, J = 2.4 Hz, 1H), 7.62 (d, J = 1.5 Hz, 1H), 6.36 (dd, J = 2.4, 1.5 Hz,
1H), 5.78 (s, 1H), 4.69 (d, J = 11.1 Hz, 1H), 4.22 (d, J = 10.0, 5.0 Hz, 1H),
4.13 (dd, J = 10.6, 10.6 1H), 3.87 (ddd, J = 11.1, 10.0, 5.0 Hz, 1H), 1.56 (s,
3H), 1.45 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 201.5, 143.3, 133.2, 108.1, 103.5, 86.5, 76.8,
69.4, 66.1, 29.3, 20.0.
HRMS (EI+) calcd for [C11H14N2O4+H3O]+ 257.1132; found 257.1130
Determination of relative stereochemistry for nucleoside 126
Analysis of 2D NOESY of nucleoside 126 supported the indicated
stereochemistry
Preparation of nucleoside analogue 127
To a stirred solution of nucleoside 126 (0.020 g, 0.084 mmol, 1.0 equiv.) in dry THF (0.84 mL)
was added methylmagnesium bromide (0.126 mL, 0.378 mmol, 4.5 equiv.) at -78°C and the
resulting reaction mixture was stirred for 3.5 hrs. The reaction mixture was quenched at -78°C
with 0.50 mL of an ammonium chloride:methanol solution (1:1 – saturated ammonium chloride

64
solution:methanol) and warmed to room temperature. The resulting mixture was diluted with 3 mL
of CH2Cl2 and washed twice with water. The organic layer was dried over MgSO4, filtered, and
concentrated under reduced pressure to give crude 127. Purification of crude 127 by flash
chromatography (ethyl acetate:pentane – 30:70) afforded nucleoside analogue 127 (19.1 mg,
90%) as a white solid.
Data for nucleoside analogue 127: []D20 = -117.7 (c 0.57 in CH2Cl2); IR (neat):
= 3425, 2992, 1398, 1384, 1088, 851cm-1; 1H NMR (600 MHz, CD3CN): δ
7.73 (d, J = 2.3 Hz, 1H), 7.60 (d, J =1.3 Hz, 1H), 6.33 (dd, J = 2.3, 1.3 Hz, 1H),
5.60 (s, 1H), 4.13 (d, J = 10.0 Hz, 1H), 4.06 (dd, J = 9.8, 4.7 Hz, 1H), 3.93 (dd,
J = 10.1, 9.8 Hz, 1H), 3.54 (s, 1H), 3.48 (ddd, J =10.1, 10.0, 4.7 Hz, 1H), 1.53
(s, 3H), 1.41 (s, 3H), 1.36 (s, 3H); 13C NMR (150 MHz, CD3CN): 142.1, 132.5, 107.2, 102.2, 95.1,
80.5, 78.4, 71.6, 66.2, 29.7, 20.6, 20.4.
HRMS (EI+) calcd for [C12H19N2O4]+ 255.1339; found 255.1333
Determination of relative stereochemistry for nucleoside 127
Analysis of 2D NOESY of nucleoside 127 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 128
To a solution of nucleoside analogue 123 (0.025g, 0.088 mmol, 1 equiv.) in CH2Cl2 (0.45 mL) at
0˚C was added dropwise diethylaminosulfur trifluoride (0.058 mL, 0.44 mmol, 5 equiv.). The
reaction mixture was warmed to room temperature and allowed to stir for 1 hr. Subsequently,
ethyl acetate (10 mL) was added and the organic layer was washed 3 times with saturated sodium
bicarbonate solution. The organic layer was then separated, dried, filtered, and concentrated
under reduced pressure. Purification of the crude S128 by flash chromatography (CH2Cl2:MeOH
95:5) afforded 2’,2’-anhydrouridine S128 (0.012 g, 51 % yield) as a white solid. 2’,2’-
anhydrouridine S128 (0.011 g, 0.039 mmol, 1 equiv.) was dissolved in a 1 M HCl:MeOH solution
(0.20mL:0.20mL). The reaction mixture was heated to 50˚C for 24hrs and then concentrated

65
under reduced pressure to yield nucleoside 128 (9.5 mg, 100% yield). The spectral data matched
previous reports.126
Data for nucleoside analogue 128: 1H NMR (600 MHz, dmso-d6): δ 11.28 (d,
J = 2.1 Hz, 1H), 7.62 (d, J = 8.1 Hz, 1H) 5.98 (d, J = 4.5 Hz, 1H), 5.56 (dd, J
= 8.1, 2.1 Hz, 1H), 3.99 (dd, J = 4.4, 3.2 Hz, 1H), 3.89 (dd, J = 3.6, 3.2 Hz,
1H), 3.73 (ddd, J = 5.6, 4.6, 3.6 Hz, 1H), 3.60 (dd, J = 11.6, 4.6 Hz, 1H), 3.56
(dd, J = 11.6, 5.6 Hz, 1H); 13C NMR (150 MHz, dmso-d6): δ 163.4, 150.5,
142.3, 100.0, 85.1, 84.7, 75.5, 75.1, 60.7
HRMS (EI+) calcd for [C9H13N2O6]+ 245.0768; found 245.0777
Preparation of nucleoside analogue 130
To a solution of nucleoside 123 (0.285 g, 1.0 mmol, 1.0 equiv.) in dry dioxane (20 mL) was added
(diacetoxyiodo)benzene (0.805 g, 2.5 mmol, 2.5 equiv.) and TEMPO (0.031 g, 0.20 mmol, 0.2
equiv.). The reaction mixture was stirred for 24 hrs at room temperature until complete
consumption of starting material was detected by TLC analysis. The reaction mixture was
concentrated to 2 mL and purified with flash chromatography (CH2Cl2:Et2O – 75:25) to afford
ketone 129 (0.265 g, 0.94 mmol, 94 % yield) as a white solid. Ketone 129 (0.053 g, 0.19 mmol,
1.0 equiv.) was dissolved in methanol (0.94 mL) and 3 drops of AcCl were added. The solution
was stirred for 12 hrs at room temperature until complete consumption of starting material was
detected by TLC analysis. The reaction mixture was concentrated under reduced pressure to a
white solid S130. The spectral data matched previous reports.127 The crude product was
subsequently dissolved in tetrahydrofuran (4.0 mL) and the resulting solution was cooled to -78°C
and methyl magnesium bromide (3.0 M in THF, 0.38 mL, 1.13 mmol, 6.0 equiv.) was added. The
resulting brown suspension was stirred at -78 °C for 3 hrs. The reaction mixture was quenched at
-78 °C with a solution of methanol:TFA (10:1) and then concentrated under reduced pressure.
The crude product 130 was purified by flash chromatography (CH2Cl2:MeOH – 85:15) to yield
nucleoside analogue (0.024 g, 49 % yield) as a white solid. The spectral data matched previous
reports.128

66
Data for nucleoside analogue 130: 1H NMR (600 MHz, MeOD): δ 7.86 (d, J
= 8.1 Hz, 1H), 5.96 (s, 1H), 5.64 (d, J = 8.1 Hz, 1H), 3.85 (m, 4H), 1.29 (s,
3H).
HRMS (EI+) calcd for [C10H15N2O6]+ 259.0925; found 259.0915
Examples of large-scale preparation of αFAR products
The following scaleup work was carried out by a CRO (WuXi AppTec). No additional optimization
of the reaction conditions was done for large scale synthesis and in most cases only select
chromatographed fractions were included in the final mass.
Large-scale preparation of A2
Three reactions were ran in parallel. To a large reactor was charged DMF (2.1 L) and
uracil (300.0 g, 2.68 mol, 1.0 equiv.) at 15-25°C. Then, the reactor was individually charged with
DBU (807 mL, 5.35 mol, 2.0 equiv.) and 2-bromo-1,1-diethoxy-ethane (483 mL, 3.21 mol, 1.2
equiv.). The reaction mixture was heated to 90°C-100oC for 16 hrs. The reaction mixture cooled
to 25oC and the three batches were combined and concentrated to dryness to give a residue. To
the residue was water (2.5 L) and the pH of the resulting mixture was adjusted with 1M HCl to 6-
7 and extracted with EtOAc (2.0 L x 8). The combined organic layer was dried with Na2SO4, filtered
and the filtrate was concentrated to dryness under reduced pressure to give a residue. The crude
residue was triturated with MBTE (3 L) at 20oC for 60 minutes. The crude residue was purified by
silica gel chromatography (petroleum ether: EtOAc: CH2Cl2 = 10: 2: 1). The alkylated uracil
product (738 g, 3.23 mol, 40.3% yield) was isolated as a white solid.
To a large reactor was charged HCl (1 M, 2.89 L, 1.0 equiv.) and the alkylated thymine
product (660 g, 2.89 mol, 1.0 equiv.) at 15-25°C. The reaction mixture was heated to 90~100°C
and stirred for 3 hours. Following complete consumption of starting material, the reaction mixture
was cooled to 0oC and stirred for 30 minutes. The resulting suspension was filtered, dried, and

67
the crude product was used in the next step without further purification. The aldehyde/hydrate
(425 g, 2.76 mol, 95.4%) was obtained as an off-white solid.
To a large reactor was charged with DMF (2800 mL) and aldehyde (400 g, 2.60 mol, 1.0
eq) and the resulting mixture was cooled to 4°C. Then, the reactor was individually charged with
NFSI (818 g, 2.60 mol, 1.0 equiv.), NaHCO3 (218 g, 2.60 mol, 1.0 equiv.) and L-proline (299 g,
2.60 mol, 1.0 equiv.). The reaction mixture was stirred at 4°C for 18 hrs. HPLC (ET24077-13-
P1A) showed starting material (RT = 0.34) was consumed completely. To the reaction mixture
was added dropwise a solution of dioxanone (226 g, 1.74 mol, 0.67 eq) in CH2Cl2 (1.3 L) at 4°C.
The reaction mixture was stirred at 15~25°C for 18 hrs. HPLC (ET24077-13-P1A) showed starting
material (RT = 1.72 min) showed the α-fluorohydrate was completely consumed. 14.0 L H2O was
added into the reaction mixture and extracted with EtOAc (3.0 L x 8). The organic phase was
dried with Na2SO4, then filtered, and the filtrate was concentrated to dryness under reduced
pressure to give a residue. The residue was purified by flash silica gel chromatography (Eluent of
0~50% ethyl acetate/petroleum ether gradient) to afford A2 as a yellow oil (380 g, 72% yield, d.r.
1:1).
Large-scale preparation of A3
To a large reactor was charged DMF (1.7 L) and thymine (85.0 g, 0.674 mol, 1.0 equiv.) at 15-
25°C. Then, the reactor was individually charged with DBU (203 mL, 1.35 mol, 2.0 equiv.) and 2-
bromo-1,1-diethoxy-ethane (122 mL, 0.809 mol, 1.2 equiv.). The reaction mixture was heated to
90°C for 14.5 hrs. The reaction mixture was concentrated to dryness to give a residue. To the
residue was added EtOAc (1.7 L) and water (1.7 L), the organic layer was separated, the aqueous
layer was extracted with EtOAc (1.7 L x 2). The combined organic layer was washed with brine
(500 mL), dried with Na2SO4, filtered and the filtrate was concentrated to dryness under reduced
pressure to give a residue. The residue was purified by flash silica gel chromatography (ISCO®;
5000 g SepaFlash® Silica Flash Column, Eluent of 30~60% Ethyl acetate/Petroleum ether

68
gradient @ 800mL/min). The alkylated thymine product (80.0 g, 301 mmol, 22.37% yield, 91.3%
purity) was obtained as an off white solid.
To a large reactor was charged HCl (1 M, 330 mL, 1.0 equiv.) and the alkylated thymine product
(80.0 g, 0.330 mol, 1.0 equiv.) at 15-25°C. The reaction mixture was heated to 90~100°C and
stirred for 15 hours. HPLC (ET17680-15-P1A) indicated starting material (RT = 2.77) was
consumed completely. The mixture was concentrated to dryness and the crude product was used
into the next step without further purification. aldehyde and hydrate (63.0 g mixture) was obtained
as an off white solid.
To a large reactor was charged with DMF (190 mL) and aldehyde (0.131 mol, 1.0 eq) and the
resulting mixture was cooled to 4°C. Then, the reactor was individually charged with NFSI (41.3
g, 0.131 mol, 1.0 equiv.), NaHCO3 (11.0 g, 0.131 mol, 1.0 equiv.) and L-proline (15.1 g, 0.131mol,
1.0 equiv.). The reaction mixture was stirred at 4°C for 18.5 hrs. HPLC (ET17918-3-P1A) showed
starting material (RT = 1.99) was consumed completely. 1. To the reaction mixture was added
dropwise a solution of dioxanone (11.4 g, 0.088 mol, 0.67 eq) in CH2Cl2 (200 mL) at 4°C. The
reaction mixture was stirred at 15~25°C for 20.5 hrs. 570 mL CH2Cl2 was added into the mixture,
and the organic phase was washed with water (190 mL x 3). The organic phase was dried with
Na2SO4, then filtered, and the filtrate was concentrated to dryness under reduced pressure to give
a residue. The residue was purified by flash silica gel chromatography (ISCO®; 330 g
SepaFlash® Silica Flash Column, Eluent of 0~100% ethyl acetate/petroleum ether gradient @
200 mL/min) to afford A3 as a yellow oil (21.0 g, 76% yield, d.r. 3:1 (syn:anti)).
Large-scale preparation of A5
This reaction was executed without further optimization. Crude A5 was purified by column
chromatography to afford 16.5 g of A5 (impure fractions were discarded).

69
Large-scale preparation of A6
The reaction was executed without further optimization. The reaction was stopped after only 16
hrs. Crude A6 was purified by prep-HPLC to afford 36.6 g of A6 (impure fractions were discarded).
Large-scale preparation of A8
The reaction was executed without further optimization. Crude A8 was purified by prep-HPLC to
afford 47 g of A8 (impure fractions were discarded).

70
J-based configurational analysis (JBCA)
The fluorine stereoconfigurations of the following compounds were assigned using NMR J-based
configuration analysis, and then the assignments were verified using density functional theory
calculations. The other stereocenters were known based on synthesis.
NMR Spectroscopy
NMR samples were prepared by dissolving several mg in 0.75 mL of DMSO-d6. These solutions
were then transferred to 5-mm NMR tubes. Proton chemical shifts were referenced to residual
DMSO-d5 at 2.50 ppm, and carbon chemical shifts were referenced to DMSO-d6 at 39.52 ppm.
NMR spectra were acquired on either a 600 MHz Bruker AVANCE III HD spectrometer equipped
with a 5-mm triple resonance (HCN) helium cryoprobe or a 500 MHz Bruker AVANCE III HD
spectrometer equipped with a 5-mm inverse Prodigy probe. Data were processed using Mnova,
version 12.0.4. 1H, 13C, COSY, HSQC, and HMBC data were acquired for all compounds to
assign the proton and carbon chemical shifts. Either NOESY or ROESY spectra were acquired
using a 200 ms mixing time to aid in the stereochemical determinations.
DFT calculations
Density functional theory (DFT) calculations of NMR parameters, chemical shifts (, ppm) and
coupling constants (J, Hz), were performed in order to verify the peak assignments and relative
stereoconfiguration. Initially, an ensemble of conformers was generated using a mixed
torsional/low-mode sampling search with the OPLS3e force field, as implemented in
Macromodel.129 The set of conformers less than 5 kcal/mol were then further subjected to DFT
geometry optimizations and frequency determinations (to verify potential energy minima) using
the B3LYP/6-31G(d) model chemistry in Gaussian ’16.130 Isotropic magnetic shielding values, ,
were then calculated starting from the optimized geometries using either WP04/cc-pVDZ or
B97X-D/6-31G(d,p) gauge-including atomic orbital (GIAO) methods for proton and carbon,

71
respectively, with implicit solvent corrections from the polarized continuum model (PCM). Linear
scaling factors [ = intercept – / -slope] were applied to convert the values to chemical shifts,
, in ppm. The scaling factors were previously determined from a large test set of known
structures, curated by Rablen et.al.131 and Lodewyk et.al.132 (1H: intercept = 31.8465, slope = -
0.9976; 13C: intercept = 198.1218, slope = -0.9816). Coupling constants were calculated using
the B3LYP/6-31G(d) model chemistry. Gibbs free energies were calculated using M06-2X/6-
31+G(d,p) with SMD solvation model, and both chemical shifts and coupling constants were
weighted according to the Boltzmann energy distribution.

72
Analysis of syn-diol, syn-fluorohydrin D8a and syn-diol, anti-fluorohydrin D8b
Figure S2.10. DFT calculated conformation of D8b
Figure S2.11. DFT calculated conformation of D8a

73
Table S2.5. Experimental and DFT-calculated coupling constants for compounds D8a and D8b
Table S2.6. Analysis of experimental and DFT-calculated 1H NMR chemical shifts for
compounds D8a and D8b
Analysis of Coupling Constants
Expt Expt DFT DFT
Coupling Atoms Sample 1 Sample 2 R-isomer S-isomer
1J_FC (24,12) 203.4 204.7 -274.5 -285.3
2J_FC (24,13) 29.9 24.5 31.7 23.9
3J_FC (24,14) < 1 5.8 -0.1 4.4
3J_HC (27,2) 3.0 3.7 6.8 6.2
3J_HC (27,8) 5.2 5.2 4.6 4.8
2J_HC (27,13) 2.3 2.4 -3.1 -2.9
3J_HC (27,14) 1.7 1.4 1.8 0.4
2J_FH (27,24) 46.8 49.5 48.6 52.4
3J_HF (28,24) < 1 14.9 1.8 12.6
3J_HH (28,27) 8.0 7.0 9.6 7.8
4J_HF (29,24) 2.0 < 1 3.5 -1.6
3J_HH (29,28) 2.0 3.7 2.5 3.1
Analysis of 1H Chemical Shifts DFT DFT
Expt Expt S R Expt. Diff DFT Diff
atom C# ID Sample 1 Sample 2 ppm ppm ppm ppm
H 4 25 8.75 8.74 8.88 8.90 0.01 -0.02
H 8 26 8.37 8.28 7.51 7.42 0.09 0.09
H 12 27 6.97 6.73 6.49 6.70 0.24 -0.21
H 13 28 4.61 4.60 5.48 5.63 0.01 -0.15
H 14 29 3.85 3.41 3.38 4.19 0.44 -0.81
H 16 31 3.78 3.45 3.98 4.22 0.33 -0.24
H 17 32 3.54 3.33 3.33 3.74 0.21 -0.41
H 17 33 3.73 3.59 3.82 3.79 0.14 0.03
H 21 34,36,35 1.38 1.14 1.20 1.35 0.24 -0.15
H 22 37,39,38 1.23 1.17 1.38 1.49 0.06 -0.11

74
Table S2.7. Analysis of experimental and DFT-calculated 13C NMR chemical shifts for
compounds D8a and D8b
Analysis of 13C Chemical Shifts DFT DFT
Expt Expt S R Expt. Diff DFT Diff
atom C# ID Sample 1 Sample 2 ppm ppm ppm ppm
C 1 1 117.1 116.7 120.5 121.1 0.36 -0.60
C 2 2 152.0 151.1 153.0 152.5 0.90 0.54
C 4 4 151.7 151.2 154.3 153.7 0.45 0.52
C 8 8 134.6 134.4 135.5 135.8 0.18 -0.26
C 12 12 91.3 92.0 99.8 100.6 -0.71 -0.85
C 13 13 69.5 70.9 76.2 74.5 -1.36 1.76
C 14 14 74.7 73.2 77.2 76.2 1.48 0.99
C 16 16 61.8 62.4 64.6 64.8 -0.56 -0.21
C 17 17 64.7 63.9 66.2 66.8 0.80 -0.51
C 19 19 98.4 97.8 102.9 102.8 0.53 0.06
C 21 21 19.5 19.0 21.0 20.8 0.43 0.13
C 22 22 28.9 28.0 30.8 31.3 0.90 -0.50

75
Figure S2.12. Summary of J-based configurational analysis for compounds D8a and D8b

76
Analysis of syn-diol, syn-fluorohydrin D5a and syn-diol, anti-fluorohydrin D5b
Table S2.8. Experimental and DFT-calculated coupling constants for compounds D5a and D5b
Table S2.9. Analysis of experimental and DFT-calculated 1H and 13C NMR chemical shifts for
compounds D5a and D5b
Coupling Constant Analysis
Isomer: 1 2 R S
Coupling Expt Expt DFT DFT3J H10,F 15.4 3.7 0.0 11.4
3J H10,H5 2.7 1.7 -0.2 2.73J H10,H12 7.5 8.6 9.2 8.5
2J H12,F 49.8 47.3 50.0 55.72J F,C10 23.4 28.3 29.4 22.63J F,C5 6.2 < 1 0.1 5.04J F,C6 < 1 2.4 0.3 -0.4
Chemical Shift Analysis
Isomer: 1 2 R S
chemical shifts Expt Expt DFT DFT D-Expt D-DFT
H1' 3.33 3.54 3.75 3.37 -0.21 0.382
H1'' 3.60 3.74 3.83 3.71 -0.14 0.116
H5 3.23 3.83 4.23 3.50 -0.6 0.737
H6 3.43 3.80 4.13 3.83 -0.37 0.303
H8 1.24 1.30 1.36 1.34 -0.06 0.019
H9 1.22 1.41 1.48 1.29 -0.19 0.193
H10 4.48 4.53 5.17 4.96 -0.05 0.207
H12 6.63 6.71 6.75 6.72 -0.08 0.03
H17 7.87 7.86 8.00 7.95 0.01 0.048
H18 8.45 8.56 8.02 7.98 -0.11 0.045
C1 64.0 64.2 67.2 66.6 -0.23 0.51
C3 97.9 98.0 103.1 103.0 -0.14 0.17
C5 73.1 74.0 76.7 76.4 -0.9 0.23
C6 61.4 61.2 65.0 64.7 0.19 0.35
C8 28.2 28.5 31.1 30.9 -0.29 0.14
C9 18.9 19.1 21.1 20.9 -0.11 0.22
C10 71.2 69.4 76.2 76.6 1.7 -0.33
C12 95.0 95.0 99.4 99.4 0 -0.02
C17 133.7 133.8 134.8 134.8 -0.11 -0.06
C18 125.7 124.7 130.7 130.2 1.04 0.53

77
Figure S2.13. Summary of J-based configurational analysis for compounds D5a and D5b

78
Single crystal X-ray diffraction
Suitable crystals were suspended in paratone oil, mounted on a MiTeGen Micro Mount, and
transferred to the X-ray diffractometer, which was set to 150 K using an Oxford Cryosystems
Cryostream. Data was collected at 150 K on a Bruker Smart instrument equipped with an APEX
II CCD area detector fixed at a distance of 5.0 cm from the crystal and a Cu Kα fine focus sealed
tube (λ = 1.54178 Å) operated at 1.5 kW (45 kV, 0.65 mA), filtered with a graphite monochromator.
Data were collected and integrated using the Bruker SAINT software package and were corrected
for absorption effects using the multi-scan technique (SADABS133). The structures were solved
with direct methods (SIR92) and subsequent refinements were performed using SHELXL134 and
ShelXle.135 Hydrogen atoms on carbon atoms were included at geometrically idealized positions
(C–H bond distance 0.95Å) and were not refined. The isotropic thermal parameters of the
hydrogen atoms were fixed at 1.2 times that of the preceding carbon atom. Diagrams were
prepared using Mercury136 and POV-RAY.137 Thermal ellipsoids are shown at the 50% probability
level.
Deposition numbers:
Bis-PNB ester of 107a: 1955427
D7b: 1955420
Figure S2.14. XRD structure of compound D7b

79
Figure S2.15. XRD structure of compound Bis-PNB ester of 107a
The crystal for the diffraction experiment was selected from those provided. The crystal was a
colorless block of dimensions 0.10mm x 0.15 mm x 0.075 mm. All diffraction measurements were
made at approximately at 100 K on a Bruker Apex II diffractometer. The refinement is complete
at an excellent level (R = 2.46%) and the molecular geometry shows no unusual quantities. The
compound has crystallized in the centrosymmetric space group P-1 as an anhydrous racemate
with one molecule in the asymmetric unit.
Figure S2.16. XRD structure of compound D9a

80
Compound Reference Bis-PNB ester of 107a D9a D7b
Chemical Formula C25H23N4O10F C12H16FIN2O6 C16H18O6FN
Formula Mass 558.47 430.1709 339.31
Crystal System
Triclinic
a/Å 16.7932(13) 9.2762(4) 7.9861(18)
b/Å 15.8691(11) 9.6024(4) 8.252(3)
c/Å 19.4773(14) 9.8870(4) 12.936(3)
α/˚ 90 69.8990(10) 79.83(2)
β/˚ 90 64.8030(10) 81.342(19)
γ/˚ 90 87.7980(10) 89.66(2)
Unit cell volume/Å3 5190.6(7) 742.28(5) 829.4(4)
Temperature/K 150(2) 100.15 150(2)
Space group Pbca P-1 P1
Number of formula unit per cell/Z
8 2 2
Radiation type Cu Kα
Cu Kα
Absorption coefficient, μ/mm-1
1.001 17.367 0.951
No. of reflections 4759 18953 4704
Flack parameter - - -0.4 (3)
Rint 0.0309 0.0383 0.0764
Final R1 values (I>2σ(I)) 0.0642 0.0246 0.0693
Final wR(F2) values (I>2σ(I)) 0.1932 0.0632 0.1717
Final R1 values (all data) 0.0711 0.0246 0.0846
Final wR(F2) (all data) 0.2018 0.0632 0.1846
Goodness of fit 1.050 1.116 1.021
Table S2.10. Summary of XRD analysis

81
Chapter 3. A short de novo synthesis of C4’ nucleosides and locked nucleoside analogues
The results presented in this chapter have been reported in part, see:
Meanwell, M.; Silverman, S. M.; Lehmann, J.; Adluri, B.; Wang, Y.; Cohen, R.; Campeau, L.-C.;
Britton, R. 2020, Science. Accepted
Dr. Steven M. Silverman made several contributions to this work. S.M.S. helped develop the
methodology, provided insightful discussions, and synthesized compounds 151-156, 163, and
171-174.
3.1. C4’-modified NAs in drug discovery
Figure 3.1. C4’ analogues in medicinal chemistry
C4’-modified NAs have attracted much attention from drug discovery over the years.
Nucleocidin (132, Figure 3.1), isolated from Streptomyces calvus in 1956, was one of the first C4’-
modified nucleosides reported.138 Its potent antimicrobial properties inspired decades of research
into the effects of different C4’ substituents on the antibiotic, antiviral and chemotherapeutic
properties of NAs. Motivated by the early success of nucleocidin and zidovudine, initial focus on
NAs involved C4’-fluoro- and azido modifications. C4’-methyl and C4’-cyano substitutions have
also been explored and collectively have led to the observation that C4’-substitutions can
profoundly affect nucleoside conformations, thus significantly impacting their binding with target
enzymes and chain termination during replication.96,97 Together, these efforts have informed drug
discovery campaigns. Currently, MK-8591 (also known as islatravir or EFdA), a reverse
transcriptase inhibitor, is in Phase II clinical trials for the treatment of HIV infection.107,139,140 In

82
2011, Balapiravir, a perester prodrug of 4’-azidocytidine (134), advanced to Phase I clinical trials
for the treatment of Dengue fever.141
3.1.1. Synthetic challenges
Scheme 3.1. Common building blocks for the construction of C4’-modified NAs
Synthesis of C4’-modified NAs have largely relied on semisynthetic approaches that are
lengthy and not amenable to rapid library generation to support drug discovery efforts. 135, 138,
and 142 are examples of building blocks used for installing C4’-modifications. 4’,5’-Unsaturated
nucleosides such as 135 have allowed access to C4’-azido and -fluoro substitutions in as few as
7 steps (Scheme 3.1A).142 In contrast, C-C bond formation at C4’ has required more laborious
approaches.143,144 For instance, the C4’-methyl (141) and -ethyl analogues (143) of toyocamycin,

83
an antibiotic, were synthesized in 12 and 13 steps respectively.145 As an additional practical
challenge, separate total synthesis were required to access these two similar analogues (Scheme
3.1C).
Scheme 3.2. Merck’s syntheses of MK-8591
Merck’s recent development of MK-8591 as a HIV treatment has renewed interest in
exploring novel synthetic routes to structurally diverse NAs for both drug discovery and process
scale production.107,139,140 Structurally, MK-8591 presents a unique synthetic challenge as it
contains both C4’- and C2’-modifications. Merck’s first approach featured an enzymatic
desymmetrization of 144 to enable a 16-step enantioselective de novo synthesis of MK-8591.107
Though innovative, this strategy was as long as previously reported semisynthetic routes (12-18
steps).146 Towards a more efficient synthesis, Merck, in collaboration with Codexis, reported a
remarkable biocatalytic cascade for the 3-step synthesis of MK-8591 from racemic 2-
ethynylglycerol in 51% overall yield.139 This short sequence relied on the engineering of five
enzymes, as well as the use of four auxiliary enzymes, to catalyze reactions with non-natural

84
substrates. Here, enzymatic asymmetric oxidization and phosphorylation irreversibly transformed
2-ethynylglycerol (147) into intermediate 148 in excellent yield and enantioselectivity.
Subsequently, in a single pot, three enzymatic reactions (aldol reaction, phosphate transfer, and
glycosylation) reversibly converted 149 into MK-8591 and dihydrogen phosphate. As a
complicating factor, dihydrogen phosphate is a known inhibitor of phosphopentomutase, the
enzyme responsible for phosphate transfer in the aforementioned process. In order to improve
the yield of this one pot process, sucrose phosphorylase (SP) and sucrose were added to the
reaction mixture to convert the generated inorganic phosphate into glucose-1-phosphate, thereby
consuming dihydrogen phosphate and driving the desired reaction to completion. As a critical
aspect, this new enzymatic glycosylation is highly stereoselective and avoids the poor
diastereoselectivity commonly associated with glycosylation of C2’- and C4’-modified furanoses.
In 2020, Merck utilized their enzymatic glycosylation in a stereoselective 9-step route to MK-8591
from 2-deoxyribose.140
3.2. Rapid synthesis of C4’-modified NAs
Having ready access to a range of αFAR products, we anticipated that addition of
organometallic reagents (rather than reduction with hydride) would provide tertiary alcohols
whose subsequent AFD would lead directly to C4'-modified NAs. Toward this goal, we examined
reactions of the deazaadenine-substituted fluorohydrin 150 with a range of organometallic
reagents (e.g., MeMgCl, MeMgBr, Me2Zn, Me3ZnLi, MeLi, Me2Mg, Me3MgLi) in CH2Cl2 or THF at
-78C, 0C or room temperature (Figure 3.2). From this panel of experiments, MeMgX reagents
in CH2Cl2 proved to be most compatible with the densely functionalized fluorohydrin. Executing
the 1,2-addition reaction at -78C proved a necessity, as higher temperatures promoted 1,2-
hydride shift/fluoride displacement as a major degradative pathway. With regards to
stereochemistry, the 1,2-addition reactions generally gave mixtures of tertiary alcohols with a
preference for addition to the re face. 112 Surprisingly, when the reaction was executed in CH2Cl2
and the crude reaction mixture was allowed to warm to room temperature overnight, the
intermediate magnesium alkoxide 151a underwent AFD to provide the C4-modified NA 152
directly. Remarkably, this sequence enables access to enantiomerically enriched C4'-modified
NAs in only 3 steps from simple achiral heterocycles and bromoacetaldehyde diethyl acetal.
Alternatively, quenching the mixture of magnesium alkoxides 151a and 151b with ammonium
chloride followed by a subsequent Lewis acid promoted AFD using InCl3 gave the anomeric -D
NA 153. Thus, in this case, each of the magnesium alkoxides 151a and 151b cyclize selectively

85
using complimentary base- or Lewis acid promoted AFD processes to afford access to -L and
-D configured NAs, both unnaturally configured NAs that are of significant contemporary interest
to medicinal chemists.103,104,147
These results inspired us to examine the reaction of several additional organomagnesium
reagents with fluorohydrin aldol adducts containing triazole, deazaadenine, thymine, pyrazole or
trifluoromethyluracil functions (Figure 3.2). Here, we found the degree of stereoselectivity in 1,2-
addition reactions depended strongly on both the solvent and heterocycle. For example, the
addition of MeMgBr to ketofluorohydrins in THF generally gave mixtures of tertiary alcohols of
different composition to those generated in CH2Cl2. Curiously, the addition of MeMgBr to
ketofluorohydrins substituted with triazole gave predominantly 1,3-syn-diols that underwent AFD
to produce the naturally configured NA β-D-160. Clearly, subtle differences in chelation structures
involving the heterocycle and/or -alkoxide function and organomagnesium reagents play a
significant role in determining the stereochemical outcome of these 1,2-addition reactions and
controlling this aspect of the process will be the subject of future studies. Notwithstanding,
exploiting these straightforward reactions, a collection of deazaadenine-substituted NAs 152 –
156 were readily accessed as both - and -anomers. In general, and as noted above, base
promoted AFD resulted in C3', C5'-protected NAs (e.g., 157 – 159, 161 – 163), while AFD
promoted by Lewis acids resulted in deprotection or protecting group migration (e.g., 153 and
155). As summarized in Figure 3.2, a range of densely functionalized C4'-modified NAs could be
rapidly accessed from the corresponding ketofluorohydrin aldol adducts, including NAs
substituted with methyl, cyclopropyl, aryl and alkynyl groups. From this study, it is clear that much
larger collections of C4'-modified NAs (e.g., focused screening libraries) are now readily available.
It is worth highlighting that each of the C4'-methyl, cyclopropyl, p-methoxyphenyl, p-chlorophenyl,
alkynyl NAs 152 – 163 were prepared in only 3 or 4 steps total, which compares favourably to all
existing syntheses of members of these important classes of NAs.

86
Figure 3.2. Synthesis of C4’-modified analogues (R = C(CH3)2)

87
Scheme 3.3. Synthesis of C4’-allyl C2’-deoxy NA (R = C(CH3)2)
In an effort to demonstrate the advantage of this route for accessing NAs with
modifications at both C2' and C4', we prepared a C4'-modified, C2'-deoxy NA (Scheme 3.3). Here,
C4'-allyl thymine 166 was readily prepared in good yield through addition of allylmagnesium
bromide to the fluorohydrin aldol adduct 164 followed by base-promoted AFD. A Barton-
McCombie deoxygenation then gave the 4'-allyl NA 166 in only 6 steps total from thymine.
3.2.1. Locked nucleic acids
Figure 3.3. Common locked nucleic acid analogues
Locked nucleic acids (LNAs) are rigid nucleoside scaffolds possessing a methylene bridge
between the 2’-oxygen and the 4’-carbon. As depicted in Figure 3.3, structural variations such as
exchanging the oxygen contained within the bridge for a carbon, nitrogen, or sulfur, as well as
modifications to the methylene bridge (e.g. 170) have been reported.148–153 When incorporated
into oligonucleotides, LNAs can enhance oligomer stability and binding affinity towards
complimentary DNA or RNA strands, and thus have shown great promise as DNA-based
diagnostics and antisense therapies. 148

88
Figure 3.4. Short syntheses of LNAs
As a final demonstration of the broader utility of this process for NA synthesis, we aimed
to exploit C4'-functionalization for the preparation of locked nucleic acids (LNAs).148 These
conformationally restricted NAs demonstrate improved stability and their incorporation in
antisense oligonucleotides can lead to significant increases in specificity and potency. However,
much like syntheses of other C4'-modified NAs, the synthesis of LNAs is often protracted.
Towards a unified LNA synthesis, we evaluated the addition of alkynylmagnesium bromide to the
thymine-containing aldol adduct 164 and found the reaction gave two diastereomeric addition
products 171 and 172 in excellent overall yield. Remarkably, the major product was transformed
directly into the unusual LNA 174 by simply reacting with NaOH, which promoted both the AFD
reaction and a subsequent cyclization between the free alcohol function and alkyne in excellent
overall yield (Figure 3.4). It is notable that this 4-step total synthesis compares well with the 23-
step route reported for the analogous uracil LNA 170154 (see Figure 3.2). We were also able to
generate the unusual alkyne-functionalized LNA, a previously unreported scaffold in nucleoside
chemistry, by simply effecting an AFD of the 1,2-addition product 173. From here, formation of
the 2,2'-anhydrothymidine followed by deprotection and treatment with base in warm DMF155 gave
the LNA 175. This unique scaffold is primed for further diversification through standard click or
Sonagashira coupling reactions.

89
3.3. Experimental
General considerations
All reactions described were performed at ambient temperature and atmosphere unless otherwise
specified. Column chromatography was carried out with 230-400 mesh silica gel (E. Merck, Silica
Gel 60). Concentration and removal of trace solvents was done via a Buchi rotary evaporator
using acetone-dry-ice condenser and a Welch vacuum pump.
Nuclear magnetic resonance (NMR) spectra were recorded using deuterochloroform (CDCl3),
deuteromethanol (CD3OD), deuteroacetone ((CD3)2CO), deuteroacetonitrile (CD3CN) or
deuterodimethyl sulfoxide (DMSO-d6) as the solvent. Signal positions (δ) are given in parts per
million from tetramethylsilane (δ 0) and were measured relative to the signal of the solvent (1H
NMR: CDCl3: δ 7.26; CD3OD: δ 3.31; (CD3)2CO: δ 2.05; CD3CN: δ 1.96; DMSO-d6: δ 2.50; 13C
NMR: CDCl3: δ 77.16; CD3OD: δ 49.00; (CD3)2CO: δ 29.84; CD3CN: δ 1.32; DMSO-d6: 39.5).
Coupling constants (J values) are given in Hertz (Hz) and are reported to the nearest 0.1 Hz. 1H
NMR spectral data are tabulated in the order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet;
sept, septet; m, multiplet; br broad), coupling constants, number of protons. NMR spectra were
recorded on a Bruker Avance 600 equipped with a QNP or TCI cryoprobe (600 MHz), Bruker 400
(400 MHz) or Bruker 500 (500 MHz). Diastereomeric ratios (dr) are based on analysis of crude
1H NMR. Assignments of 1H are based on analysis of 1H-1H-COSY and nOe spectra. Assignments
of 13C are based on analysis of HSQC spectra.
High performance liquid chromatography (HPLC) analysis was performed on an Agilent 1100
HPLC, equipped with a variable wavelength UV-Vis detector.
High-resolution mass spectra were performed on an Agilent 6210 TOF LC/MS, Bruker MaXis
Impact TOF LC/MS, or Bruker micrOTOF-II LC mass spectrometer.
Infrared (IR) spectra were recorded neat on a Perkin Elmer Spectrum Two FTIR spectrometer.
Only selected, characteristic absorption data are provided for each compound.
Optical rotation was measured on a Perkin-Elmer Polarimeter 341 at 589 nm.
General Procedure A (Grignard additions)

90
A stirred solution of fluorohydrin aldol adduct (1 equiv.) in CH2Cl2 (0.025 M) was cooled to -78°C.
Organomagnesium reagent (2.2 – 5 equiv.) was added dropwise and the resulting reaction
mixture was stirred for 5 hrs. The reaction mixture was quenched at -78°C with an ammonium
chloride:methanol solution (1:1 – saturated ammonium chloride solution:methanol) and warmed
to room temperature. The resulting mixture was diluted with CH2Cl2 and washed twice with water.
The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure to
give crude product. The crude product was either purified by flash chromatography or used
directly for cyclization.
General Procedure B (base promoted cyclization)
To a stirred solution of syn-diols, syn- and anti-fluorohydrins (1.0 equiv.) in MeCN (0.10 M) was
added 2 M NaOH (1.5 - 10 equiv.) and the reaction mixture was stirred for 3 hours or until no
starting material remained (as determined by TLC analysis). The reaction mixture was diluted with
CH2Cl2 and washed with saturated ammonium chloride solution. The organic layer was separated,
dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was
purified by flash chromatography.
General Procedure C (Lewis acid promoted cyclization)
To a stirred solution of syn-diols, syn- and anti-fluorohydrin (1.0 equiv.) in MeCN (0.10 M) was
added Sc(OTf)3 or InCl3 (0.10 – 2.5 equiv.) and the reaction mixture was stirred for 6 hours or until
complete consumption of starting material (as determined by TLC analysis). The reaction mixture
was diluted with CH2Cl2 and was washed with saturated sodium bicarbonate solution. The organic
layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The
crude product was purified by flash chromatography.
Preparation of nucleoside analogue 152
Methylmagnesium chloride (3.0 M in THF, 1.49 mL, 4.47 mmol, 2.1 equiv.) was added dropwise
to a solution of 150 (syn-/anti-fluorohydrin = 3:1, 1.00 g, 2.13 mmol, 1.0 equiv.) at -78°C in CH2Cl2
(10 mL). The reaction mixture was stirred at this temperature for 2 hrs and then allowed to warm
gradually to room temperature and stirred for 12 hrs. The reaction mixture was quenched with
saturated ammonium chloride solution and diluted with ethyl acetate. The organic layer was
separated, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification of

91
crude product 152 by flash chromatography (0-10% MeOH in CH2Cl2) afforded nucleoside 152
(0.418 g, 42%) as a white solid.
Data for nucleoside analogue 152: []D20 = -13.6 (c 0.28 in CH2Cl2); IR
(neat): = 3443, 2250, 1661, 1053, 1005, 821 cm-1; 1H NMR (600 MHz,
CDCl3): δ 8.64 (s, 1H), 7.55 (s, 1H), 6.28 (d, J = 7.6 Hz, 1H), 4.92 (ddd, J =
9.8, 7.5, 4.4 Hz, 1H), 4.21 (d, J = 4.5 Hz, 1H), 3.83 (d, J = 12.6 Hz, 1H),
3.74 (d, J = 12.6 Hz, 1H), 3.40 (d, J = 9.8 Hz, 1H), 1.53 (s, 3H), 1.49 (s,
3H), 1.42 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 153.2, 151.0, 151.0, 132.6, 118.1, 99.2, 89.9,
79.1, 75.5, 73.9, 66.2, 52.8, 27.4, 23.0, 20.8
HRMS (EI+) calcd for [C15H18ClIN3O4]+ 466.0025; found 466.0054
Determination of relative stereochemistry for nucleoside 152
Analysis of 2D NOESY of nucleoside 152 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 153
Methylmagnesium chloride (3.0 M in THF, 1.56 mL, 4.68 mmol, 2.2 equiv.) was added dropwise
to a solution of 150 (syn-/anti-fluorohydrin = 3:1, 1.00 g, 2.13 mmol, 1.0 equiv.) at -78°C in CH2Cl2
(20.0 mL). The resulting reaction mixture was stirred at -78°C for 5 hrs. The reaction mixture was
quenched with an ammonium chloride:methanol solution (1:1 – saturated ammonium chloride
solution:methanol) and warmed to room temperature. The reaction mixture was diluted with
CH2Cl2 (50 mL) and the organic layer was separated, dried over MgSO4, filtered, and
concentrated under reduced pressure. Purification of crude product 151b by flash
chromatography (pentane:ethyl acetate – 65:35) afforded 151b (0.498 g, 48%) as an off-white
solid.

92
Data for 151b: []D20 = -17.7 (c 1.8 in CH2Cl2); IR (neat): = 3316, 2991,
1206, 1086, 863, 736 cm-1; 1H NMR (600 MHz, dmso-d6): δ 8.76 (s,
1H), 8.28 (s, 1H), 6.92 (dd, J = 45.8, 3.3 Hz, 1H), 6.23 (d, J = 5.0 Hz,
1H), 4.65 (s, 1H), 4.45 (m, 1H), 3.44 (d, J =11.1 Hz, 1H), 3.28 (d, J =
8.0 Hz, 1H), 3.23 (d, J = 11.1, 1H), 1.28 (s, 3H), 1.13 (s, 3H), 0.75 (s,
3H);13C NMR (150 MHz, dmso-d6): δ 151.5, 151.4, 151.2, 134.3, 116.0, 98.3, 90.2 (d, J = 202.7
Hz), 74.1 (d, J = 4.5 Hz), 70.1 (d, J = 25.1 Hz), 70.0, 66.7, 55.2, 28.4, 19.7, 18.1; 19F NMR (470
MHz, dmso-d6): δ –151.1
HRMS (EI+) calcd for [C15H19ClFIN3O4]+ 486.0087; found 486.0080
To a stirred solution of 151b (0.100 g, 0.206 mmol, 1.0 equiv.) in dry MeCN (2.0 mL) was added
InCl3 (0.046 g, 0.206 mmol, 1.0 equiv.). The resulting reaction mixture was heated to 50°C for 2
hrs. 2,2-dimethoxypropane (0.214 mg, 2.06 mmol, 10.0 equiv.) and camphorsulfonic acid (9.6
mg, 0.041 mmol, 0.20 equiv.) were added and the reaction mixture was stirred for a further 1 hr
at 50 °C. The reaction mixture was then concentrated and purified by flash chromatography (0-
10% MeOH in CH2Cl2) to afford nucleoside 153 (0.049 g, 51%) as a white solid.
Data for nucleoside analogue 153: []D20 = +1.4 (c 0.84 in MeOD); 1H
NMR (600 MHz, CDCl3): δ 8.58 (s, 1H), 7.68 (s, 1H), 6.83 (d, J = 4.5
Hz. 1H), 5.01 (dd, J = 6.0, 4.7 Hz, 1H), 4.77 (d, J = 6.0 Hz, 1H), 3.79
(dd, J = 10.9, 5.2 Hz, 1H), 3.74 (dd, J = 10.9, 3.6 Hz, 1H), 2.02 (dd, J
= 5.2, 3.6 Hz, 1H), 1.48 (s, 3H), 1.41 (s, 3H), 1.31 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 152.6,
150.8, 150.3, 134.5, 117.4, 113.2, 85.1, 85.0, 83.0, 81.1, 69.5, 50.8, 25.6, 24.1, 17.4.
HRMS (EI+) calcd for [C15H18ClIN3O4]+ 466.0025; found 466.0000
Determination of relative stereochemistry for nucleoside 153
Analysis of 2D NOESY of nucleoside 153 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 154

93
Ethynylmagnesium chloride (0.5 M in THF, 8.94 mL, 4.47 mmol, 2.1 equiv.) was added dropwise
to a solution of 150 (syn-/anti-fluorohydrin = 3:1, 1.00 g, 2.13 mmol, 1.0 equiv.) at -78°C in CH2Cl2
(10 mL). The reaction mixture was stirred at this temperature for 2 hrs and then allowed to warm
gradually to room temperature and stirred for 12 hrs. The reaction mixture was quenched with
saturated ammonium chloride solution and diluted with ethyl acetate. The organic layer was
separated, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification of
crude product 154 by flash chromatography (0-10% MeOH in CH2Cl2) afforded nucleoside 154
(0.415 g, 41%) as a white solid.
Data for nucleoside analogue 154: []D20 = -29.5 (c 0.58 in MeOH); IR
(neat): = 3291, 2924, 1446, 1201, 1023, 600 cm-1; 1H NMR (600 MHz,
dmso-d6): δ 8.72 (s, 1H), 8.02 (s, 1H), 6.44 (d, J = 8.1 Hz, 1H), 5.05 (dd,
J = 8.1, 3.6 Hz, 1H), 4.44 (d, J = 3.6 Hz, 1H), 4.16 (s, 1H), 4.01 (d, J =
13.2 Hz, 1H), 3.82 (d, J = 13.2 Hz, 1H), 3.44 (br s, 1H), 1.49 (s, 3H), 1.43
(s, 3H);13C NMR (150 MHz, dmso-d6): δ 151.7, 151.4, 151.1, 132.8, 116.6, 97.5, 86.5, 81.1, 80.5,
75.0, 74.1, 72.3, 64.2, 53.0, 28.5, 18.9
HRMS (EI+) calcd for [C16H16ClIN3O4]+ 475.9869; found 475.9849
Determination of relative stereochemistry for nucleoside 154
The relative stereochemistry was assigned based on comparison of the chemical shift of the
anomeric proton with compounds 152 and 156.
Preparation of nucleoside analogue 155
Ethynylmagnesium chloride (0.5 M in THF, 8.94 mL, 4.47 mmol, 2.1 equiv.) was added dropwise
to a solution of 150 (syn-/anti-fluorohydrin = 3:1, 1.00 g, 2.13 mmol, 1.0 equiv.) at -78°C in CH2Cl2
(20 mL). The resulting reaction mixture was stirred at -78°C for 1 hr. The reaction mixture was
quenched with an ammonium chloride:methanol solution (1:1 – saturated ammonium chloride
solution:methanol) and warmed to room temperature. The reaction mixture was diluted with
CH2Cl2 (50 mL) and the organic layer was separated, dried over MgSO4, filtered, and
concentrated under reduced pressure. Purification of crude product S155 by flash
chromatography (pentane:ethyl acetate – 65:35) afforded S155 (0.720 g, 68%, 1:1 mixture of
diastereomers) as an off-white solid.

94
To a stirred solution of S155 (0.050 g, 0.101 mmol, 1.0 equiv.) in dry MeCN (2.0 mL) was added
InCl3 (0.022 g, 0.101 mmol, 1.0 equiv.). The resulting reaction mixture was heated to 50°C for 2
hrs. 2,2-dimethoxypropane (0.124 mL, 1.01 mmol, 10.0 equiv.) and camphorsulfonic acid (4.7
mg, 0.020 mmol, 0.20 equiv.) were added and the reaction mixture was stirred for a further 1 hr
at 50 °C. The reaction mixture was then concentrated and purified by flash chromatography (0-
10% MeOH in CH2Cl2) to afford nucleoside 155 (0.029 g, 60%) as a white solid.
Data for nucleoside analogue 155: []D20 = +6.3 (c 2.0 in CH2Cl2); 1H
NMR (600 MHz, CDCl3): δ 8.59 (s, 1H), 7.82 (s, 1H), 6.85 (d, J = 4.6
Hz, 1H), 5.03 (dd, J = 6.0, 4.9 Hz, 1H), 4.98 (d, J =6.0 Hz, 1H), 3.97
(dd, J =11.5, 4.4 Hz, 1H), 3.92 (dd, J =11.5, 3.5 Hz, 1H), 2.82 (s, 1H),
2.18 (dd, J = 4.4, 3.5 Hz, 1H), 1.53 (s, 3H), 1.34 (s, 3H); 13C NMR
(150 MHz, CDCl3): δ 152.7, 150.9, 1505., 134.6, 117.4, 114.6, 85.3, 83.0, 82.9, 80.6, 78.2, 77.8,
68.7, 51.4, 25.7, 24.5.
HRMS (EI+) calcd for [C16H16ClIN3O4]+ 475.9869; found 475.9885
Determination of relative stereochemistry for nucleoside 155
Analysis of 2D NOESY of nucleoside 155 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 156
Phenylmagnesium chloride (2.0 M in THF, 2.24 mL, 4.47 mmol, 2.1 equiv.) was added dropwise
to a solution of 150 (syn-/anti-fluorohydrin = 3:1, 1.00 g, 2.13 mmol, 1.0 equiv.) at -78°C in CH2Cl2
(10 mL). The reaction mixture was stirred at this temperature for 2 hrs and then allowed to
gradually warm to room temperature and stirred for 12 hrs. The reaction mixture was quenched
with saturated ammonium chloride solution and diluted with ethyl acetate. The organic layer was
separated, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification of
crude product by flash chromatography (0-10% MeOH in CH2Cl2) afforded nucleoside 156 (0.496
g, 45%) as a white solid.

95
Data for nucleoside analogue 156: []D20 = -23.6 (c 1.7 in CH2Cl2); IR
(neat): = 3309, 2990, 2938, 1575, 1538, 1445, 1200 cm-1; 1H NMR (600
MHz, dmso-d6): δ 8.70 (s, 1H), 7.63 (s, 1H), 7.43 (m, 5H), 6.55 (d, J = 8.3
Hz, 1H), 5.55 (d, J = 6.9 Hz, 1H), 4.77 (d, J = 3.8 Hz, ,1H), 4.67 (ddd, J
=8.3, 6.9, 3.8 Hz, 1H), 3.81 (d, J = 12.9 Hz, 1H), 3.68 (d, J =12.9 Hz, 1H),
1.62 (s, 3H), 1.50 (s, 3H); 13C NMR (150 MHz, dmso-d6): δ 152.0, 151.3, 151.0, 140.4, 133.4,
128.5, 128.0, 125.3, 111.8, 97.4, 86.1, 80.8, 73.9, 72.5, 67.0, 54.3, 28.3, 20.2
HRMS (EI+) calcd for [C20H20ClIN3O4]+ 528.0182; found 528.0206
Determination of relative stereochemistry for nucleoside 156
Analysis of 2D NOESY of nucleoside 156 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 157
Following General Procedure A, cyclopropylmagnesium bromide (1.0 M in 2-methylTHF, 0.79
mL, 0.79 mmol, 5 equiv.) was added to a solution of 164 (0.050 g, 0.158 mmol, 1 equiv.) in CH2Cl2
(6.30 mL) at -78 °C. The reaction mixture was stirred for 5 hrs. Without further purification, crude
S157 was dissolved in MeCN (1.60 mL) and 2 M NaOH (0.193 mL, 0.395 mmol) was added and
the reaction mixture was stirred for 4 hrs at 50 °C. Purification of crude product S157 by flash
chromatography (pentane:ethyl acetate – 30:70) afforded nucleoside 157 (0.021 g, 40 % yield)
as an off-white solid.
Data for nucleoside analogue 157: []D20 = -32.6 (c 0.47 in CH2Cl2); IR
(neat): = 3500, 3251 2997, 2175, 1690, 1088, 888 cm-1; 1H NMR (600
MHz, CDCl3): δ 7.10 (s, 1H), 6.04 (d, J = 7.9 Hz, 1H), 4.25 (dd, J = 7.9,
5.1 Hz. 1H), 4.08 (d, J = 5.1 Hz, 1H), 3.70 (d, J = 11.9 Hz, 1H), 3.63 (d, J
= 11.9 Hz, 1H), 3.15 (br s, 1H), 1.93 (s, 3H), 1.44 (s, 3H), 1.43 (s, 3H),
1.21 (m, 1H), 0.63 (m, 1H), 0.55 (m, 1H), 0.46 (m, 1H), 0.42 (m, 1H); 13C

96
NMR (150 MHz, CDCl3): δ 163.3, 151.0, 134.9, 111.9, 100.1, 87.5, 81.2, 74.0, 72.5, 64.3, 25.9,
25.6, 16.2, 12.9, 1.31, 0.50.
HRMS (EI+) calcd for [C16H22N2O6]+ 339.1551; found 339.1575
Determination of relative stereochemistry for nucleoside 157
Analysis of 2D NOESY of nucleoside 157 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 158
Following General Procedure A, p-tolylmagnesium bromide (1.0 M in THF, 0.712 mL, 0.71 mmol)
was added to a solution of 164 (0.050 g, 0.158 mmol) in CH2Cl2 (6.30 mL) at -78 °C. The reaction
mixture was stirred for 4.5 hrs. Without further purification, crude S158 was dissolved in MeCN
(1.58 mL) and 2 M NaOH (0.198 mL, 0.395 mmol) was added and the reaction mixture was heated
to 50°C for 4 hrs. Purification of crude product 158 by flash chromatography (pentane:ethyl
acetate – 35:65) afforded nucleoside 158 (0.024 g, 39 % yield over two steps) as colorless oil.
Data for nucleoside analogue 158: []D20 = -56.5 (c 0.4 in MeOH); IR
(neat): = 3432, 2939, 1700, 1466, 1378, 1129, 1051 cm-1; 1H NMR (600
MHz, CD3CN): δ 8.96 (br s, 1H), 7.38 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 8.1
Hz, 2H), 6.78 (d, J = 0.90 Hz, 1H), 6.24 (d, J = 8.2 Hz, 1H), 4.76 (d, J =
3.8 Hz, 1H), 4.19 (s, 1H), 3.80 (d, J = 13.2 Hz, 1H), 3.73 (d, J = 13.2 Hz,
1H), 3.48 (br s), 2.35 (s, 3H), 1.68 (d, J = 0.90 Hz, 3H), 1.60 (s, 3H), 1.49
(s, 3H); 13C NMR (150 MHz, CD3CN): δ 164.6, 152.8, 139.6, 138.7, 137.4, 130.6, 126.7, 112.0,
99.2, 88.9, 81.6, 74.9, 74.3, 68.7, 29.0, 21.4, 20.8, 12.8.
HRMS (EI+) calcd for [C20H25N2O6]+ 389.1707; found 389.1707

97
Determination of relative stereochemistry for nucleoside 158
Analysis of 2D NOESY of nucleoside 158 supported the indicated
stereochemistry
Preparation of nucleoside analogue 159
Following General Procedure A, p-methoxyphenylmagnesium bromide (0.5 M in THF, 1.58 mL,
0.79 mmol, 5 equiv.) was added to a solution of 164 (0.050 g, 0.158 mmol, 1 equiv.) in CH2Cl2
(6.30 mL) at -78 °C. The reaction mixture was stirred for 5 hrs. Without further purification, crude
S159 was dissolved in MeCN (1.60 mL) and 2 M NaOH (0.193 mL, 0.395 mmol) was added and
the reaction mixture was stirred for 4 hrs at 50 °C. Purification of crude product 159 by flash
chromatography (pentane:ethyl acetate – 30:70) afforded nucleoside 159 (0.026 g, 41 % yield)
as a white solid.
Data for nucleoside analogue 159: []D20 = -52.8 (c 1.0 in CH2Cl2); IR
(neat): = 3197, 2990, 1693, 1252, 1036, 834 cm-1; 1H NMR (600 MHz,
CDCl3): δ 7.38 (d, J = 8.7 Hz, 2H), 6.96 (d, J = 8.7 Hz, 2H), 6.78 (s, 1H),
6.37 (d, J = 7.9 Hz, 1H), 4.75 (d, J = 4.1 Hz, 1H), 4.16 (m, 1H), 3.87 (d, J
= 13.1 Hz, 1H), 3.84 (s, 3H), 3.79 (d, J = 13.1, 1H), 2.99 (br s, 1H), 1.63
(s, 3H), 1.56 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 163.2, 159.9, 151.1,
135.8, 131.8, 126.5, 114.5, 111.7, 98.7, 88.7, 80.6, 74.8, 73.2, 67.7, 55.6, 28.1, 20.4, 12.7.HRMS
(EI+) calcd for [C20H25N2O7]+ 405.1656; found 405.1650
Determination of relative stereochemistry for nucleoside analogue 159
Analysis of 2D NOESY of nucleoside 159 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 160

98
Methylmagnesium iodide (3.0 M in THF, 0.39 mL, 1.16mmol, 3 equiv.) was added dropwise to a
solution of A5 (0.100 g, 0.388 mmol, 1 equiv.) at -78°C in CH2Cl2. The resulting reaction mixture
was gradually warmed to -10°C and allowed to stir for 2 hours. Following completion of the
reaction as monitored by TLC analysis, the reaction mixture was quenched with saturated
ammonium chloride solution and diluted with CH2Cl2. The organic layer was subsequently washed
twice with water and once with brine. The organic layer was then dried over MgSO4, filtered, and
concentrated under reduced pressure. Purification of crude product S160 by flash
chromatography (pentane:ethyl acetate – 25:75) afforded S160 (0.089 g, 84%) as a light yellow
oil.
Data for S160: 1H NMR (600 MHz, CDCl3): δ 8.16, 8.02, 7.76, 7.76, 6.80,
6.58, 4.62, 4.52, 4.40, 4.31, 4.07, 3.81, 3.59, 3.55, 3.45, 3.25, 3.13, 3.10,
1.52, 1.47, 1.45, 1.40, 1.38, 1.17; 13C NMR (150 MHz, CDCl3): δ 134.2,
134.1, 124.9, 124.3, 99.8, 99.8, 95.6, 93.4, 72.4, 72.4, 71.9, 71.8, 70.2,
70.0, 67.9, 67.8, 28.8, 28.7, 20.0, 19.8, 19.2, 18.5; 19F NMR (470 MHz, CDCl3): δ –157.8, -162.8
HRMS (EI+) calcd for [C11H19FN3O4]+ 276.1354; found 276.1366
To a solution of S160 (0.060 g, 0.218 mmol, 1 equiv.) in dry MeCN (2.18 mL) was added Sc(OTf)3
(0.268 g, 0.545 mmol, 2.5 equiv.). After stirring the reaction mixture for 16 hrs, 0.50 mL of acetic
anhydride and 0.50 mL of pyridine were added to the reaction mixture. The reaction mixture was
stirred for a further 4 hrs and then diluted with CH2Cl2. The organic layer was washed with twice
with 1 M HCl and once with water, dried over sodium sulfate, filtered, and concentrated under
reduced pressure to yield crude 160. Purification of crude product 160 by flash chromatography
(pentane:ethyl acetate – 60:40) afforded 160 (0.024 g, 32 % yield).
Data for nucleoside analogue 160: []D20 = +18.4 (c 1.46 in CH2Cl2); IR (neat):
= 2925, 1744, 1374, 1215, 1049 cm-1; 1H NMR (600 MHz, CDCl3): δ 7.76
(d, J = 0.60 Hz, 1H), 7.75 (d, J = 0.60 Hz, 1H) 6.19 (d, J = 4.7 Hz, 1H), 6.02
(dd, J = 5.4, 4.7 Hz, 1H), 5.67 (d, J = 5.4 Hz, 1H), 4.17 (d, J = 12.0 Hz, 1H),
4.08 (d, J = 12.0 Hz, 1H), 2.15 (s, 3H), 2.09 (s, 3H), 2.03 (s, 3H), 1.37 (s, 3H);
13C NMR (150 MHz, CDCl3): δ 170.3, 169.3, 169.2, 134.4, 122.7, 89.4, 85.6, 75.0, 71.9, 67.9,
20.8, 20.5, 20.5, 19.3.
HRMS (EI+) calcd for [C14H20N3O7]+ 342.1296; found 342.1312

99
Determination of relative stereochemistry for nucleoside analogue 160
Analysis of 2D NOESY of nucleoside 160 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 161
Following General Procedure A, methylmagnesium bromide (3.0 M in THF, 0.258 mL, 0.78 mmol,
4 equiv.) was added to a solution of A1 (0.050 g, 0.194 mmol, 1 equiv.) in CH2Cl2 (3.90 mL) at -
78 °C. The reaction mixture was stirred for 6 hrs. Crude S161 was purified by flash
chromatography (ethyl acetate-pentane – 6:4) to yield S161 (0.026 g, 49 % yield). S161 (0.030 g,
0.109 mmol) was dissolved in MeCN (1.09 mL) and 2 M NaOH (0.545 mL, 1.09 mmol, 10 equiv.)
was added and the reaction mixture was stirred for 5 hrs at 50 °C. Purification of crude nucleoside
analogue 161 by flash chromatography (pentane:ethyl acetate – 25:75) afforded 161 (0.017 g, 61
% yield) as a light yellow oil.
Data for nucleoside analogue 161: []D20 = +11.3 (c 0.38 in CH2Cl2); );
IR (neat): = 3383, 2992, 2922, 1382, 1199, 1090, 908 cm-1 1H NMR
(600 MHz, CDCl3): δ 7.60 (d, J = 2.4 Hz, 1H), 7.59 (d, J = 1.6 Hz, 1H),
6.35 (dd, J = 2.4, 1.6 Hz, 1H), 5.29 (d, J = 1.3 Hz, 1H), 4.12 (dd, J =
3.0, 1.3 Hz, 1H), 3.98 (d, J = 3.0 Hz, 1H), 3.76 (d, J = 11.3 Hz, 1H), 3.52 (d, J = 11.3 Hz, 1H),
1.47 (s, 3H), 1.44 (s, 3H), 1.41 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 141.3, 129.3, 107.4, 99.6,
72.6, 70.3, 67.3, 64.9, 57.0, 28.8, 20.5, 19.0.
HRMS (EI+) calcd for [C12H19N2O4]+ 255.1339; found 255.1320

100
Determination of relative stereochemistry for nucleoside analogue 161
Analysis of 2D NOESY of nucleoside 161 supported the indicated
stereochemistry
Preparation of nucleoside analogue 162
Following General Procedure A, p-methoxyphenylmagnesium bromide (0.5 M in THF, 4.66 mL,
2.33 mmol, 3 equiv.) was added to a solution of A1 (0.200 g, 0.775 mmol, 1 equiv.) in CH2Cl2
(7.75 mL) at -78 °C. The reaction mixture was stirred for 6 hrs. Crude S162 was purified by flash
chromatography (ethyl acetate-pentane – 4:6) to yield S162 (0.157 g, 55 % yield). S162 (0.155 g,
0.423 mmol, 1 equiv.) was dissolved in MeCN (2.82 mL) and 2 M NaOH (0.53 mL, 1.06 mmol,
2.5 equiv.) was added and the reaction mixture was stirred for 5 hrs at 50 °C. Purification of crude
nucleoside analogue 162 by flash chromatography (pentane:ethyl acetate – 40:60) afforded 162
(0.085 g, 58 % yield) as a light orange oil.
Data for nucleoside analogue 162: []D20 = -14.8 (c 1.4 in CH2Cl2); IR
(neat): = 3418, 2991, 1611, 1512, 1250, 1032, 759 cm-1; 1H NMR (600
MHz, CD3CN): δ 7.69 (d, J = 2.7 Hz, 1H), 7.56 (d, J = 1.4 Hz, 1H), 7.39
(d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 6.35 (dd, J = 2.7, 1.4 Hz,
1H), 5.99 (d, J = 7.9 Hz, 1H), 4.73 (dd, J = 7.9, 3.7 Hz, 1H), 4.59 (d, J =
3.7 Hz, 1H), 3.92 (d, J = 13.3 Hz, 1H), 3.78 (s, 3H), 3.68 (d, J = 13.3 Hz,
1H), 1.62 (s, 3H), 1.51 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 160.6, 141.5, 133.7, 132.1, 128.4,
114.8, 107.6, 99.1, 93.9, 82.0, 75.9, 75.1, 68.9, 56.3, 29.0, 21.2.
HRMS (EI+) calcd for [C18H23N2O5]+ 347.1601; found 347.1610

101
Determination of relative stereochemistry for nucleoside 162
Analysis of 2D NOESY of nucleoside 162 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 163
p-Chlorophenylmagnesium bromide (1.0 M in diethyl ether, 4.32 mL, 4.32 mmol, 3.2 equiv.) was
added dropwise to a stirred solution of fluorohydrin aldol adduct A6 (0.500 g, 1.35 mmol, 1 equiv.)
in THF (10.0 mL) at 0°C. The resulting reaction mixture was stirred for 14 hrs at room temperature
and for a further 8 hrs at 40°C. The reaction mixture was then diluted with ethyl acetate (100 mL)
and washed once with water (100 mL) and once with brine (50 mL). The organic layer was
separated, dried over MgSO4, filtered, and concentrated under reduced pressure to give crude
163. Purification of crude nucleoside analogue 163 by flash chromatography (pentane:ethyl
acetate – 50:50) afforded 163 (0.289 g, 46%).
Data for nucleoside 163: []D20 = +10.5 (c 0.8 in CH2Cl2); IR (neat):
= 3087, 2996, 1699, 1467, 1283, 1129, 1085 cm-1; 1H NMR (600
MHz, dmso-d6): δ 11.94 (br s, 1H), 8.74 (s, 1H), 7.57 (d, J = 8.7 Hz,
2H), 7.50 (d, J = 8.7 Hz, 2H), 6.13 (d, J = 7.2 Hz, 1H), 5.67 (br s,
1H), 4.66 (d, J =4.3 Hz, 1H), 4.17 (dd, J =6.8, 4.3 Hz, 1H), 3.98 (d,
J =13.4 Hz, 1H), 3.88 (d, J =13.4 Hz, 1H), 1.63 (s, 3H), 1.40 9s, 3H);
13C NMR (150 MHz, CD3CN): δ 159.9, 150.5, 144.3 (q, J = 5.9 Hz), 138.0, 134.9, 129.9, 128.1,
124.1 (q, J = 269.0 Hz), 104.0 (q, J = 32.0), 99.6, 84.7, 81.6, 73.6, 73.6, 67.7, 28.6, 19.9; 19F NMR
(470 MHz, CD3CN): δ –62.9
HRMS (EI+) calcd for [C19H19ClF3N2O6]+ 463.0878; found 463.0875

102
Determination of relative stereochemistry for nucleoside 163
Analysis of 2D NOESY of nucleoside 163 supported the indicated
stereochemistry.
Preparation of nucleoside analogue 165
Following General Procedure A, allylmagnesium bromide (1.0 M in diethyl ether, 1.42 mL, 1.42
mmol, 4.5 equiv.) was added to a solution of 164 (0.100 g, 0.316 mmol, 1 equiv.) in CH2Cl2 (12.6
mL) at -78 °C. The reaction mixture was stirred for 5 hrs. Without further purification, crude S165
was dissolved in MeCN (3.16 mL) and 2 M NaOH (0.395 mL, 0.79 mmol, 2.5 equiv.) was added
and the reaction mixture was stirred for 4 hrs at 50 °C. Purification of crude 165 by flash
chromatography (CH2Cl2:MeOH – 4:96) afforded nucleoside analogue 165 (0.050 g, 47 % yield)
as a dark orange oil.
Data for nucleoside analogue 166: []D20 = -6.0 (c 0.4 in MeOH); IR
(neat): = 3340, 2992, 1670, 1376, 1044 cm-1; 1H NMR (600 MHz,
CD3CN): δ 8.95 (br s, 1H), 7.27 (s, 1H), 6.02 (d, J = 8.3 Hz, 1H), 5.87 (m,
1H), 5.22 (d, J = 17.7 Hz, 1H), 5.20 (d, J = 10.1 Hz, 1H), 4.31 (ddd, J =
9.3, 8.3, 4.9 Hz, 1H), 4.11 (d, J = 4.9 Hz, 1H), 3.68 (d, J = 12.2 Hz, 1H),
3.64 (d, J = 12.2 Hz, 1H), 3.41 (d, J = 9.3 Hz, 1H), 2.50 (dd, J =14.2, 6.7
Hz, 1H), 2.41 (dd, J = 14.2 , 8.1 Hz, 1H), 1.85 (s, 3H), 1.40 (s, 3H), 1.39 (s, 3H); 13C NMR (150
MHz, CD3CN): δ 164.6, 152.3, 136.8, 133.7, 120.4, 112.3, 100.3, 88.4, 81.7, 73.9, 73.3, 65.3,
41.7, 26.9, 22.4, 12.8.
HRMS (EI+) calcd for [C16H22N2O6]+ 339.1551; found 339.1556

103
Determination of relative stereochemistry for nucleoside 165
Analysis of 2D NOESY of nucleoside 165 supported the indicated
stereochemistry
Preparation of nucleoside analogue 166
To a solution of nucleoside 165 (0.022 g, 0.061 mmol, 1 equiv.) in dry THF (0.61 mL) was added
1, 1’- thiocarbonyldiimidazole (0.022 g, 0.122 mmol, 2 equiv.). The reaction mixture was stirred
for 18 hrs. Subsequently, CH2Cl2 (5 mL) was added to the reaction mixture and washed with
water 3 times. The organic layer was dried over MgSO4, filtered, and concentrated under reduced
pressure to yield crude S166. Purification of crude S166 by flash chromatography (pentane:ethyl
acetate – 40:60) afforded S166 (0.018 g, 66% yield). To a solution of nucleoside S166 (0.014 g,
0.031 mmol, 1 equiv.) in dry toluene (4.45 mL) under nitrogen was added tributyltin hydride (8.35
µL, 0.031 mmol, 1 equiv.) and AIBN (5.1 mgs, 0.031 mmol, 1.0 equiv.). The resulting reaction
mixture was purged with nitrogen for 30 minutes. Subsequently, the reaction mixture was stirred
for 16 hrs at 90 °C. Upon competition, CH2Cl2 was added to reaction mixture and the washed with
water. The organic layer was dried over MgSO4, filtered, and concentrated under reduced
pressure to yield crude 166. Purification of crude 166 by flash chromatography (ethyl acetate)
afforded nucleoside analogue 166 (6.0 mg, 61%) as a white solid.
Data for nucleoside analogue 166: []D20 = +13.3 (c 0.46 in CH2Cl2); IR
(neat): = 2924, 1690, 1467, 1375, 1263, 1226, 1053 cm-1; 1H NMR (600
MHz, CDCl3): δ 8.26 (s, 1H), 7.31 (d, J = 1.1 Hz, 1H), 6.38 (dd, J = 9.6, 4.8
Hz, 1H), 5.86 (m, 1H), 5.25-5.27 (m, 2H), 4.22 (d, J = 5.2 Hz, 1H), 3.69 (d,
J = 12.0 Hz, 1H), 3.64 (d, J = 12.0 Hz, 1H), 2.50 (m, 2H), 2.41 (dd, J =
13.5, 4.8 Hz, 1H), 2.00 (dd, J = 13.5, 9.6, 5.2 Hz, 1H), 1.92 (s, 3H), 1.37
(s, 6H); 13C NMR (150 MHz, CDCl3): δ 163.3, 150.0, 135.0, 131.9, 120.2, 111.1, 99.5, 85.8, 84.0,
73.9, 63.9, 40.9, 37.8, 25.6, 22.5, 12.7
HRMS (EI+) calcd for [C16H23N2O5]+ 323.1601; found 323.1580

104
Preparation of fluorohydrins 171 and 172
Following General Procedure A, ethynylmagnesium chloride (0.5 M in THF, 3.5 mL, 1.75 mmol,
3.5 equiv.) was added to a solution of 164 (0.160 g, 0.50 mmol, 1 equiv.) in CH2Cl2 (25.0 mL) at
-78 °C. The reaction mixture was stirred for 4 hrs. The crude products 171 and 172 were purified
by flash chromatography (ethyl acetate:hexane – 70:30) to afford 171 (0.058 g, 38 % yield) and
172 (0.072 g, 42 % yield) as white solids.
Data for fluorohydrin 171: []D20 = -38.0 (c 1.2 in MeOH); IR (neat): = IR
(neat): = 3395, 2994, 1694, 1468, 1381, 1282, 1043 cm-1; 1H NMR (600
MHz, CD3CN): δ 9.29 (br s, 1H), 7.41 (s, 1H), 6.40 (dd, J = 43.4, 4.6 Hz,
1H), 4.54 (m, 1H), 4.27 (m, 1H), 4.22 (m, 1H), 3.82 (d, J = 9.5 Hz, 1H),
3.79 (d, J = 11.5 Hz, 1H), 3.75 (d, J = 11.5 Hz, 1H), 2.81 (s, 1H), 1.85 (s,
3H), 1.41 (s, 3H), 1.28 (s, 3H); 13C NMR (150 MHz, CD3CN): δ 164.8, 151.4, 137.7, 111.5, 100.8,
94.1 (d, J = 206.9 Hz), 84.4, 75.7, 73.6 (d, J = 3.8 Hz), 73.4 (d, J = 24.7 Hz), 69.3, 67.3, 28.8,
19.4, 12.8; 19F NMR (470 MHz, CD3CN): δ –175.5
HRMS (EI+) calcd for [C15H20N2O6]+ 343.1300; found 343.1305
Data for fluorohydrin 172: []D20 = -60.8 (c 0.4 in MeOH); IR (neat): =
3320, 2944, 2832, 1670, 1449, 1022, 638 cm-1; 1H NMR (600 MHz, dmso-
d6): δ 11.47 (br s, 1H), 7.56 (s, 1H), 6.36 (dd, J = 43.7, 4.1 Hz, 1H), 6.21 (d,
J = 5.3 Hz, 1H), 5.37 (br s, 1H), 4.14 (m, 1H), 3.71 (d, J = 8.7 Hz, 1H), 3.68
(br s, 1H), 3.42 (s, 1H), 3.16 (d, J = 5.0 Hz, 1H), 1.78 (s, 3H), 1.33 (s, 3H), 1.21 (s, 3H); 13C NMR
(150 MHz, dmso-d6): δ 163.6, 150.0, 136.7, 109.2, 98.8, 92.7 (d, J = 206.6 Hz), 83.7, 76.2, 72.8
(d, J = 2.8 Hz), 71.2 (d, J = 24.6 Hz), 68.2, 65.7, 27.8, 18.7, 12.1;19F NMR (470 MHz, dmso-d6):
δ –170.5
HRMS (EI+) calcd for [C15H20N2O6]+ 343.1300; found 343.1298
Preparation of nucleoside analogue 174
Following General Procedure B, a solution of 172 (0.100 g, 0.292 mmol, 1.0 equiv.) and NaOH
(29.2 mg, 0.73 mmol, 2.5 equiv.) in MeCN (2.0 mL) was heated to 50 °C for 36 hrs. Purification
of the crude 174 by flash chromatography (0-10% MeOH in dichloromethane) afforded nucleoside
analogue 174 (58.6 mg, 62 % yield) as a white powder.

105
Data for nucleoside analogue 174: []D20 = -8.7 (c 0.6 in CH2Cl2); IR (neat):
= 2994, 1748, 1690, 1270, 1043 cm-1; 1H NMR (600 MHz, dmso-d6): δ
11.42 (s, 1H), 7.61 (d, J = 1.3 Hz, 1H), 5.46 (s, 1H), 4.86 (s, 1H), 4.63 (d, J
= 11.2 Hz, 1H), 4.45 (d, J = 2.6 Hz, 1H), 4.37 (d, J = 2.6 Hz, 1H), 4.23 (d, J
= 11.2 Hz, 1H), 3.91 (s, 1H), 1.84 (s, 3H), 1.51 (s, 3H), 1.30 (s, 3H); 13C
NMR (150 MHz, dmso-d6): δ 163.8, 158.8, 150.0, 135.0, 109.3, 100.4, 87.2,
83.0, 78.4, 76.5, 71.9, 58.5, 28.7, 19.5, 12.0
HRMS (EI+) calcd for [C15H19N2O6]+ 323.1238; found 323.1235
Preparation of nucleoside analogue 173
Following General Procedure C, a solution 171 (0.220 g, 0.64 mmol, 1 equiv.) and 2M NaOH
(0.640 mL, 1.28 mmol, 2.0 equiv.) was heated to 50°C and stirred for 24 hours in MeCN (6.4 mL).
Purification of the crude 173 by flash chromatography (MeOH:CH2Cl2 – 3:97) afforded nucleoside
analogue 173 (0.144 mg, 70 % yield) as a white powder.
Data for nucleoside analogue 173: []D20 = +30.8 (c 1.66 in CH2Cl2); 1H
NMR (600 MHz, CD3CN): δ 9.06 (br s, 1H), 7.48 (s, 1H), 6.16 (d, J = 8.2
Hz, 1H), 4.61 (ddd, J = 8.4, 8.2, 3.7 Hz, 1H), 4.41 (d, J = 3.7 Hz, 1H), 4.06
(d, J = 13.3 Hz, 1H), 3.88 (d, J = 13.3 Hz, 1H), 3.64 (d, J = 8.4 Hz, 1H), 3.29
(s, 1H) 1.86 (s, 3H), 1.48 (s, 3H), 1.43 (s, 3H); 13C NMR (150 MHz, CD3CN):
δ 164.7, 152.5, 136.9, 112.7, 99.3, 89.4, 81.2, 80.8, 76.5, 75.5, 73.9, 65.9, 29.1, 19.7, 13.1
HRMS (EI+) calcd for [C15H19N2O6]+ 323.1238; found 323.1245
Determination of relative stereochemistry for nucleoside 173
Analysis of 2D NOESY of nucleoside 173 supported the indicated
stereochemistry
A solution of 173 (0.050 g, 0.155 mmol, 1 equiv.) in dry CH2Cl2 (0.78 mL) was cooled to 0 °C and
diethylaminosulfur trifluoride (0.102 mL, 0.776 mmol, 5 equiv.) was added dropwise over 5

106
minutes. The resulting reaction mixture was slowly warmed to room temperature over 3 hrs.
Following completion of the reaction, as monitored by TLC analysis, the reaction mixture was
diluted with 5 mL of ethyl acetate and washed with 3 mL of H2O (3x). Subsequently, the organic
layer was dried over MgSO4, filtered, and concentrated under reduced pressure. Purification of
the crude product by flash chromatography (ethyl acetate) afforded nucleoside analogue S173
(0.043g, 91%) as a white solid.
Data for nucleoside analogue S173: []D20 = -47.5 (c 1.1 in MeCN); IR
(neat): = 3284, 3002, 1626, 1554, 1497, 1134, 1066, 1030 cm-1; 1H NMR
(600 MHz, CD3CN): δ 7.46 (s, 1H), 6.32 (d, J = 5.3 Hz, 1H), 5.13 (d, J =
5.3 Hz, 1H), 4.74 (s, 1H), 4.10 (d, J = 13.7 Hz, 1H), 4.00 (d, J = 13.7 Hz,
1H), 2.87 (s, 1H), 1.87 (s, 3H), 1.47 (s, 3H), 1.34 (s, 3H); 13C NMR (150
MHz, CD3CN): δ 173.1, 161.5, 132.3, 119.6, 99.7, 91.8, 87.4, 79.8, 79.1, 77.8, 74.6, 64.9, 29.0,
19.3, 14.4
HRMS (EI+) calcd for [C15H17N2O5]+ 305.1132; found 305.1108
To a solution of S173 (0.042 g, 0.138 mmol, 1 equiv.) in wet MeCN (2.76 mL) was added InCl3
(0.122g, 0.553 mmol, 4 equiv.). The resulting reaction mixture was heated to 50 °C and was
stirred for 16 hrs or until the reaction was complete as monitored by TLC. The reaction mixture
was concentrated under reduced pressure and purified by flash chromatography (MeOH:CH2Cl2
– 7.5:92.5) to afford S175 (0.038 g, 96%). To a solution of S175 (0.038 g, 0.133 mmol, 1 equiv.)
in DMF (1.73 mL) was added K2CO3 (0.096g, 0.69 mmol, 5 equiv.). The resulting reaction mixture
was heated to 90 °C and stirred for 7 days or until the reaction was complete as monitored by 1H
NMR spectroscopy. Subsequently, the reaction mixture was filtered, concentrated under reduced
pressure, and the crude product was purified by flash column chromatography (MeOH:CH2Cl2 –
10:90) to afford 175 (0.027g, 71%) as a white solid.
Data for nucleoside analogue 175: []D20 = +16.9 (c 1.0 in MeOH); IR (neat):
= 3261, 2988, 1686, 1272, 1203, 1047, 799 cm-1; 1H NMR (600 MHz, CD3CN):
δ 9.43 (br s, 1H), 7.31 (d, J = 1.1 Hz, 1H), 5.48 (s, 1H), 4.27 (s, 1H), 4.15 (s,
1H), 4.03 (d, J = 8.0 Hz, 1H), 3.93 (d, J = 8.0 Hz, 1H), 3.16 (s, 1H), 1.85 (d, J
= 1.1 Hz, 3H); 13C NMR (150 MHz, CD3CN): δ 165.1, 151.4, 135.6, 111.0, 88.6,
80.9, 80.3, 80.2, 75.8, 75.2, 75.1, 13.0
HRMS (EI+) calcd for [C12H13N2O5]+ 265.0819; found 265.0813

107
10-20g scale preparation of 164
39.0 g of A3 was dissolved in 240 mL of ethyl acetates and repurified by prep-HLPC to give 18.0
g of product. The 18.0 g product was dissolved in 240 mL of CH2Cl2 and concentrated under
reduced pressure to give 17.5 g of 164. The 17.5 g of 164 was freeze-dried to obtain 15.8 g of
164 as a white solid (94.3 % purity).
Data for syn-fluorohydrin 164: []D20 = -89.4 (c 1.1 in MeOH); IR (neat):
= 2993, 1694, 1450, 1369, 1082, 1045 cm-1; 1H NMR (400 MHz,
CDCl3): δ 8.30 (br s, 1H), 7.57 (dd, J = 1.3, 1.2 Hz, 1H), 6.66 (ddd, J =
42.7, 2.3, 1.3 Hz, 1H), 4.40 (dd, J = 8.9, 1.4 Hz, 1H), 4.33 (dd, J = 17.7,
1.4 Hz, 1H), 4.12 (d, J = 17.7 Hz, 1H), 4.10 (ddd, J = 15.4, 3.1, 2.3 Hz,
1H), 3.64 (d, J = 3.0 Hz, 1H), 1.95 (d, J = 1.2 Hz, 3H), 1.52 (s, 3H), 1.46 (s, 3H); 13C NMR (100
MHz, CDCl3): δ 211.2, 163.2, 149.9, 137.1 (d, J = 4.0 Hz), 111.0, 102.1, 90.2 (d, J = 207.8 Hz),
71.6 (d, J = 2.3 Hz), 70.9 (d, J = 23.4 Hz), 66.5, 23.8, 23.4, 12.6; 19F NMR (470 MHz, CDCl3): δ –
177.8
HRMS (EI+) calcd for [C13H18FN2O6]+ 317.2929; found 317.1142

108
Chapter 4. A platform for diversity-oriented synthesis of carbohydrate analogues
Several colleagues contributed to this work. Gaelen Fehr developed the methodology for α-
chlorination/aldol reactions and synthesized compounds 207-212, 231-233, 235, 236, 256, 257,
and 265-269. Dr. Weiwu Ren synthesized compounds 221, 230, 274, and 276. Dr.
Bharanishashank Adluri synthesized compounds 231, 234, and 270-272.
4.1. Introduction to carbohydrate analogues
Figure 4.1 Carbohydrate analogues in drug discovery
Carbohydrates are essential biomolecules that play critical roles in cell signaling and
metabolism.156 Consequently, structural mimics of carbohydrates (carbohydrate analogues
(CAs)), have found widespread use in drug discovery and chemical biology and have had a
profound impact in therapeutic areas such as diabetes,157 virology158 and cancer.159,160 As a
notable example, Oseltamivir (Tamiflu, 176 (Figure 4.1)), a neuraminidase inhibitor, is a WHO
essential medicine for treating influenza and was the frontline drug used during the H1N1
pandemic in 2009.161 Likewise, Miglitol (178) is an antidiabetic drug that functions by inhibiting α-
glucosidases and decreasing the carbohydrate metabolism in patients with type II diabetes.157
From a mechanistic perspective, therapeutic CAs often inhibit enzymes involved in carbohydrate
processing (glycoside hydrolases and transferases), and thus mitigate the effects of dysregulated
metabolism. As selective probe molecules, CAs are often used to gain insight into enzyme

109
function and disease pathology.162 Importantly, carbohydrates and CAs are also core structural
units found in several approved drugs (e.g., doxorubicin, vancomycin) and play a vital role in their
therapeutic efficacy.
Scheme 4.1. Diversity-oriented synthesis approaches to CAs
Unsurprisingly, CAs display a high degree of structural diversity. The canonical endocyclic
oxygen is frequently exchanged for a nitrogen,163 carbon,164 or sulfur,165 and the ring-size varied,
with 5- and 6-membered rings being the most common. While the structural landscape of CAs is
vast, a smaller collection of scaffolds has dominated drug discovery efforts owing to their
predictive properties. For example, iminosugars are CAs in which the endocyclic oxygen is
replaced with a nitrogen that is protonated at physiological pH, mimicking the oxacarbenium ion
transition state traversed during hydrolysis by glycoside hydrolases (GHs).163,166 The
corresponding sulfur analogues or thiosugars possess distinct geometries, conformations, and
electronic properties that can profoundly influence their physiochemical properties (e.g.,
metabolic stability, bioavailability, lipophilicity, potency).165 Carbasugars or cyclitols have shown
promise as transition-state analogues and inhibitors of α-glycosidases.164 While diversity-oriented
synthesis (DOS) approaches are useful for the discovery of new CAs and exploring their
associated chemical space, these methods often rely on the derivatization of naturally occurring
carbohydrates, leading to lengthy synthetic sequences (> 10 steps) that can limit structural
diversity and drug discovery efforts.167 For example, several DOS strategies rely on single step
transformations of stereochemically rich CA cores to generate screening libraries. Wong has
reported a library of potent and selective α-fucosidase inhibitors derived from 1-aminomethyl-

110
fuconojirimycin (182) (Figure 1).168 Here, the advantage of late-stage diversification using a robust
reaction (i.e. amide coupling) is challenged by the 12-step route required to access the key
scaffold 182. DOS approaches that provide access to stereochemically and structurally diverse
scaffolds have also been reported. For example, Marcaurelle demonstrated the use of 2,3-
unsaturated C-glycoside scaffolds to synthesize several new bicyclic carbohydrates in as few as
6 total steps.169 Recently, Loh reported a robust strategy for the diversification of carbohydrates
using hydrogen- and halogen-bond catalyzed strain release glycosylation to produce complex
O,N-glycoside analogues.170 The synthesis of CA building blocks for incorporation into
oligosaccharides has also attracted interest, and new platforms for producing CA-containing
oligosaccharides have been exploited in the generation of tobramycin analogues that target
pathogenic RNA. 171

111
Figure 4.2. Development of α-functionalization/aldol reactions for drug discovery
As an alternative de novo approach, we have described a one-pot process that involves
the proline-catalyzed α-chlorination of aliphatic aldehydes followed by a L-proline-catalyzed aldol
reaction with dioxanone 75 to produce syn-chlorohydrins of general structure 188 in good yield,
diastereoselectivity, and enantioselectivity.172 Chlorohydrin scaffolds produced via this -
chlorination/aldol reaction (CAR) have proven to be versatile building blocks for the construction
of CAs including carbasugars (e.g., 190)173 and iminosugars (e.g., 191).116,174 For example, we
have exploited this process in the discovery of inhibitors of OGlcNAcase174 and -
galactosidases173,175 (i.e. 190 and 191). As detailed in Chapters 2 and 3, we have reported a
complimentary proline-catalyzed α-fluorination/aldol reaction (FAR) that supports the rapid
synthesis of nucleoside analogues (NAs, e.g., 192).176 Critical to this NA synthesis is the stability
of heteroaryl-C-F functions (compared to heteroaryl-C-Cl), which enabled the production and
isolation of fluorohydrin aldol adducts (e.g., 189: R = heteroaryl). A subsequent intramolecular

112
displacement of the fluoride results in stereospecific formation of the carbohydrate core of
nucleosides and NAs. While these unique CARs and FARs provide a convenient means to
construct CAs and NAs, all efforts here have relied on the well-established reactivity of dioxanone
75 as the aldol coupling partner and thus limited our exploration of more unusual carbohydrate
scaffolds. We envisioned new -functionalization/aldol reactions, involving a larger variety of
electrophiles and carbonyl compounds, would support the rapid construction diverse collections
of CAs. Here, we describe the development of these reactions and ultimately demonstrate their
broad utility for synthesizing CAs.
4.2. Development of α-functionalization/aldol reactions
Scheme 4.2. One-pot synthesis of chlorohydrins and DKR of α-chloroaldehydes through proline-catalyzed aldol reactions
Asymmetric enamine catalysis utilizing chiral amines has been widely exploited in the
enantioselective α-functionalization of aldehydes, including α-chloro-,177 α-fluoro,115 α-hydroxy,178
α-amino,179 α-thio,180 and α-trifluoromethyl aldehydes.181 Despite intense efforts, many of these
aldehydes remain underutilized as chiral building blocks owing in part to their instability, volatility,
propensity to epimerize and/or challenges with their purification.115 For these reasons, processes
that avoid isolation of α-functionalized aldehydes but still take advantage of their synthetic
potential as chiral building blocks are highly sought. We previously demonstrated that proline
catalyzes the racemic -chlorination of aldehydes, the interconversion of the resulting racemic -
chloroaldehydes and finally their aldol reactions with dioxanone 75.172 As depicted in Scheme 4.2,
we proposed that electrostatic repulsion between the Cl and O atoms in the transition structure

113
leading to anti-chlorohydrins served as the key diastereodiscriminating interaction in this dynamic
kinetic resolution (DKR). The high levels of enantioselectivity then derive from H-bonding, which
directs the facial approach of the correctly configured -chloroaldehyde to the proline-derived
enamine through a Houk-List-type transition structure TS1.182
entry X+ source solvent syn:anti %ee yield
1 NCS CH2Cl2 6:1 94 72
2 NCS CH2Cl2:DMF 2.2:1 ND ND
3 NFSI CH2Cl2:DMF 15:1 96 61
4 PhthN-SCF3 CH2Cl2:DMSO 6:1 91 52
5 NBS CH2Cl2:DMF 1.5:1 ND 21
6 CbzNNCbz MeNO2 3:1 98 45
Table 4.1. α-functionalization/aldol reactions between pentanal and dioxanone (75)
To explore the broader utility of this strategy, we prepared a series of α-functionalized
pentanal derivatives and evaluated their reactivity in proline-catalyzed aldol reactions with
dioxanone 75. Here, α-functionalization/aldol reactions were performed as two-step-one-pot
sequences (see experimental section for details). Specifically, we examined the L-proline
catalyzed α-functionalization of pentanal using a series of electrophilic reagents (e.g., N-
chlorosuccinimide (NCS), N-fluorobenzenesulfonimide (NFSI), N-trifluoromethylthiophthalimide
(PhthN-SCF3)), and subsequently added dioxanone 75 and examined the production of aldol
adducts 198, 200 – 203. As shown in Table 4.1, this process preferentially affords syn-
fluorohydrin 200, -trifluoromethylthiohydrin 201, -bromohydrin 202, and -aminohydrin 203 in
variable diastereoselectivity and excellent enantioselectivity. Notably, the observed
diastereoselectivity for the formation of the series of halohydrins (198 (d.r. 15:1), 200 (d.r. 2.2:1),
and 202 (d.r. 1.5:1)) correlated well with increasing electronegativity of the halogen atom. Thus,
despite the smaller van der Waals radius for F (F = 1.47 Å; Cl = 1.75 Å; Br = 1.85 Å) and shorter
bond length (C(sp3)-F = 1.40 Å; C(sp3)-Cl = 1.79 Å; C(sp3)-Br = 1.97 Å), the reactions of
enantiomeric α-fluoroaldehydes were more greatly differentiated based on the increased charge
density on F (ENF = 3.98; ENCl = 3.16; ENBr = 2.96). These results are in accord with our original
proposal172 that electrostatic repulsion between the O and X atoms in TS-2 disfavour production
of anti-halohydrins 199. To gauge the effect of solvent polarity, the αCAR was repeated in CH2Cl2

114
(rather than 9:1 CH2Cl2-DMF) and we observed a substantial increase in diastereoselectivity
(2.2:1 to 6:1; see Table 4.1 entries 1 and 2). This observation further supports the role of
electrostatic interactions in the diastereodifferentiating step. As summarized in Table 4.1 (entry
4), the -trifluoromethylthioaldehyde derived from pentanal also underwent a highly diastero- and
enantioslective aldol reaction with dioxanone 75. Considering that both the organocatalyzed -
fluorination115 and -trifluoromethylthiolation183 of aldehydes proceed with almost no
enantioselctivity (ee’s <15%), the high level of stereoselectivity in these processes is attributed to
DKR of the intermediate -fluoro- and -trifluoromethylthioaldehydes. Thus, proline-catalyzed
racemization is more facile than the subsequent aldol reactions (i.e., krac > k(D) or k(L)). In the case
of the α-aza aldehyde generated from the reaction of pentanal and dibenzyl azodicarboxylate
(entry 6), steric hindrance precludes formation of a proline enamine required for racemization.
Furthermore, in a separate experiment the proline-catalyzed aldol reaction between (S)-2-Cbz-
aminopentanal and dioxanone yielded the corresponding anti-aminohydrin as the sole product,
confirming that these α-aminoaldehydes do not racemize under the reaction conditions (krac <
k(L)). Likewise, the L-proline catalyzed aldol of 2-phenylpropanal and dioxanone affords an equal
mixture of syn- and anti- diastereomers (see experimental section).
Table 4.2. Optimization of α-chlorination/aldol with isovaleraldehyde (204) and thiopyranone 206.aketone and aldehyde added at the same time.
Having demonstrated the use of dioxanone (75) in several α-functionalization/aldol
reactions, we were intrigued to expand the scope of compatible ketones. As summarized in Table
4.2, we began by examining the L-proline catalyzed αCAR of tetrahydro-4H-thiopyranone (206)
entry additive temp. (°C) solvent A solvent B 207:208 yield
1a None RT CH2Cl2 None N.D. <5%
2a None RT DMSO None N.D. 0%
3a None 0 CH2Cl2 None N.D. <5%
4 None 0 CH2Cl2 DMSO 10:1 38%
5 H2O 0 CH2Cl2 DMSO 10:1 45%

115
with isovaleraldehyde. Carrying out this reaction using our previously reported one-step-one-pot
procedure in a variety of solvents afforded the desired syn-chlorohydrin, albeit in low yield (See
Table 4.2, entries 1-3). Suspecting that competitive chlorination of thiopyranone 206 was
complicating this process, we adopted a two-step-one-pot sequence. Satisfyingly, when the L-
proline catalyzed α-chlorination of isovaleraldehyde was first carried out in CH2Cl2 at 0 °C and
then followed by direct addition of thiopyranone 206 in DMSO, the desired 207 was produced in
good yield and excellent diastereo-and enantioselectivity (entry 4). Addition of catalytic amounts
of water with the thiopyranone led to modest improvements in yield while catalytic amounts of
TFA had minimal effect (Table 4.2, entry 5). Using this straightforward process, several additional
ketones (O-TBS-hydroxyacetone, cyclohexanone, and tetrahydro-4H-pyran-4-one) were
engaged in αCARs giving syn-chlorohydrins 207 and 209 – 211 in good yield and excellent
diastereo- and enantioselectivity. Unfortunately, simple aliphatic ketones such as acetone,
cyclopentanone, and 3-pentanone, were incompatible (e.g., 212-217).
Figure 4.3. Ketone scope of α-chlorination/aldol reaction
With several new α-functionalization/aldol reactions in hand, we were eager to
demonstrate the broad applicability of this technology to synthesize CA building blocks through
the use of a panel of functionally diverse aldehydes. Towards this goal, we synthesized a
collection of fluorohydrins (218 – 230 and 237 – 242), chlorohydrins (231-234),
trifluoromethylthiohydrins (243-247), and aminohydrins (235 and 236) in excellent enantio- and
diastereoselectivity. As shown in Figure 4.3, the functional group compatibility of the αFAR is
highlighted in the reactions of TIPS-protected 3-hydroxypropanal and Cbz-protected 3-
aminopropanal, which delivered the unusual fluorohydrins 226 and 230 bearing a heteroatom at

116
each position in the carbon chain. Overall, the αFAR was tolerant to alkyl-, alkene-, alkyne-and
(hetero)aryl substituents. Furthermore, simply using D-proline enables access to enantiomeric
syn-fluorohydrins 221 and 226. Gratifyingly, adding 206 and cyclohexanone neat to reaction
mixtures of α-fluoroaldehydes (generated from pentanal, pentenal, and phthalimidoacetaldehyde)
successfully engaged in α-FARs to afford anti-aldol-syn-fluorohydrin products (237 – 242) in good
to excellent yield. We were particularly encouraged by the ability to engage α-fluoro-α-
phthalimidyl-acetaldehyde in productive aldol reactions to afford 241 and 242 and have reported
similar heteroaryl-C-F containing aldol intermediates for NAs synthesis. Considering the SCF3
group is commonly used to increase lipophilicity (Hansch hydrophobicity parameter π = 1.44) and
modulate pKa,184 the utility of this process was further highlighted in the production of syn-
trifluoromethylthiohydrins containing an alkyl (243 and 245), alkene (244), aryl (246), or heteroaryl
(247) in excellent diastereo- and enantioselectivity. To the best of our knowledge, these are the
first examples of an asymmetric reaction involving α-(trifluoromethylthio)aldehyde. Finally, we
explored the compatibility of a small collection of aldehydes in αCARs with OTBS-
hydroxyacetone, which afford chlorohydrins 231-234, and demonstrated that α-amination/aldol
reactions deliver enantiomerically enriched syn-aminohydrins (235 and 236).

117
Figure 4.4. Scope of α-functionalization/aldol reactions.

118
Figure 4.5. Scope of α-functionalization/aldol reactions
4.3. Rapid synthesis of CAs
Figure 4.6. Platform for rapid diversity-oriented synthesis (253 = 5-(methanesulfonyl)-1-phenyl-1H-tetrazole)
To demonstrate the utility of these aldol adducts for rapidly producing diverse scaffolds for
medicinal chemistry, several readily available chlorohydrins, fluorohydrins,

119
trifluoromethylthiohydrins, and aminohydrins were converted into a diverse collection of
iminosugars, bicyclic nucleoside analogues, annulated furanoses, and fluorinated CAs (Figure
4.6). Hydrogenation of syn-aminohydrins allowed access to cyclic hydrazones 256 and 257 in
excellent yield over 2 total steps. This compares favourably with syntheses of azafagomines 254
and 255, a class of α-fucosidase inhibitors, that required 17 step syntheses.185 The recent use of
258 as potent and selective PRMT5 inhibitors for cancer treatment highlights the importance of
bicyclic nucleoside analogues, a relatively unexplored NA scaffold.186 In attempt to develop a
more efficient approach to this class of compounds, we prepared bicyclic NAs 259 and 260 in 3
total steps via a 1,3-syn reduction and indium chloride mediated cyclization from fluorohydrins
259 and 260. Presumably, under cyclization conditions, epimerization occurs to give a mixture of
α- and β- anomers. Given the synthetic challenges currently associated with NA synthesis, this
short sequence should provide new opportunities for medicinal chemistry. We also evaluated the
utility of fluoro- and trifluoromethylthiohydrins as building blocks for the synthesis of fluorinated
carbacycles. As depicted in Figure 4.7, Julia-Kocienski olefination and subsequent RCM of 223,
239, 240 and 244 afforded bicyclic carbacycles 261 – 264 in only 3 total steps. 1,3-syn reduction
with sodium borohydride and subsequent microwave cyclization of syn-chlorohydrins 198, 207,
209, 211, and 233 afforded uniquely annulated tetrahydrofuran (THF) products 265 – 269,
including scaffolds that have not yet been reported 265 and 266. Alternatively, 1,3-syn reductive
amination with benzyl amine followed by reflux in toluene under basic conditions of 231, 232 and
234 gave access to selectively protected 5’-deoxy-iminosugars 270 – 272. 1,3-syn or 1,3-anti
selective reduction of the vicinal fluorohydrin 230 followed by removal of the silyl protecting group
and oxidation of the resultant primary alcohol delivered the epimeric 2-deoxy-2-fluoropyranoses
273 and 274. This short sequence avoids the iterative alcohol protection-deprotection steps
commonly required in fluorosugar synthesis. Hydrogenation of the Cbz-protected amino
fluorohydrin 226 (made using D-proline catalysis) gave directly and in excellent yield a previously
undescribed fluorinated analogue 275 of the drug migalastat (galafold),187 a pharmacological
chaperone used to treat Fabry disease. As an additional target of interest, we also prepared a
fluorinated analogue 276 of D-ribo-phytosphingosine, a precursor to the potent natural killer T cell
stimulator α-galactosylceramide,188 in a straightforward manner through the reductive amination
of the fluorohydrin 221 with benzyl amine, followed by hydrogenolysis.

120
Figure 4.7. Rapid synthesis of CAs.aα:β = 2.5:2.bα:β = 3:1.

121
Figure 4.8. Conversion of fluorohydrins 221, 226, and 230 into biologically relevant molecules
In summary, we have developed a DOS approach that relies on achiral and readily
accessible starting materials to access an array of highly relevant CA scaffolds for drug discovery.
These processes are enabled by an expanding family of α-functionalization/aldol reactions that,
in a single step, can afford stereochemically rich and densely functionalized aldol adducts in good
yield and excellent diastereo- and enantioselectivity. These versatile building blocks were rapidly
derivatized, through a suite of established procedures, into a variety of CAs in 1-2 steps.
4.4. Experimental
Data for compounds 207-212, 231-236, 256, 257, and 265-269 were reported in the thesis of G.
Fehr.189
L- and D-proline (99% purity) were purchased from Alfa Aesar. All reactions described were
performed at ambient temperature and atmosphere unless otherwise specified. Column
chromatography was carried out with 230-400 mesh silica gel (E. Merck, Silica Gel 60).
Concentration and removal of trace solvents was done via a Buchi rotary evaporator using
acetone-dry-ice condenser and a Welch vacuum pump.
Nuclear magnetic resonance (NMR) spectra were recorded using deuterochloroform (CDCl3),
deuteromethanol (CD3OD) or deuterodimethyl sulfoxide (DMSO-d6) as the solvent. Signal
positions (δ) are given in parts per million from tetramethylsilane (δ 0) and were measured relative

122
to the signal of the solvent (1H NMR: CDCl3: δ 7.26; CD3OD: δ 3.31; DMSO-d6: δ 2.50; 13C NMR:
CDCl3: δ 77.16; CD3OD: δ 49.0; DMSO-d6: 39.5). Coupling constants (J values) are given in Hertz
(Hz) and are reported to the nearest 0.1 Hz. 1H NMR spectral data are tabulated in the order:
multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; sept, septet; m, multiplet; br broad), coupling
constants, number of protons. NMR spectra were recorded on a Bruker Avance 600 equipped
with a QNP or TCI cryoprobe (600 MHz), Bruker 400 (400 MHz) or Bruker 500 (500 MHz).
Diastereomeric ratios (dr) are based on analysis of crude 1H-NMR. Assignments of 1H are based
on analysis of 1H-1H-COSY and nOe spectra. Assignments of 13C are based on analysis of HSQC
spectra.
High performance liquid chromatography (HPLC) analysis was performed on an Agilent 1100
HPLC, equipped with a variable wavelength UV-Vis detector and Chiralcel OD-H chiral column
(0.46 cm x 25 cm). Enantiomeric excess (ee) has been determined using corresponding bis-p-
nitrobenzoate of all syn-fluorohydrins except for 228, 238, 241, and 242 which required no
derivatization.
High-resolution mass spectra were performed on an Agilent 6210 TOF LC/MS, Bruker MaXis
Impact TOF LC/MS, or Bruker micrOTOF-II LC mass spectrometer.
Infrared (IR) spectra were recorded neat on a Perkin Elmer Spectrum Two FTIR spectrometer.
Only selected, characteristic absorption data are provided for each compound.
Optical rotation was measured on a Perkin-Elmer Polarimeter 341 at 589 nm.
General Procedures
General Procedure A (α-fluorination/aldol reaction with dioxanone)
A sample of aldehyde (1.5 equiv.) was added to a stirred suspension of NFSI (1.5 equiv.),
L-proline (1.5 equiv.), and NaHCO3 (1.5 equiv.) in DMF (0.75 M) at -10 °C. When complete
conversion to the α-fluoroaldehyde was observed by 1H NMR spectroscopic analysis, 2,2-
Dimethyl-1,3-dioxan-5-one (75) (1.0 equiv.) in CH2Cl2 (0.055 – 0.10 M) was then added and the
resulting mixture was allowed to warm gradually to room temperature. After a total of 72 hrs, or
when complete consumption of the 75 was observed by 1H NMR spectroscopic analysis of small
reaction aliquots, the mixture was diluted with Et2O and the organic layer was washed twice with

123
water and once with brine. The organic layer was then dried over MgSO4, concentrated under
reduced pressure and the crude product was purified by flash chromatography as indicated.
General Procedure B (α-fluorination/aldol reaction with cyclohexanone/thiopyranone 206)
A sample of aldehyde (1.0 equiv.) was added to a stirred suspension of NFSI (1.5 equiv.),
L-proline (1.0 equiv.), and NaHCO3 (1.0 equiv.) in DMF (0.75 M) at -10 °C. When complete
conversion to the α-fluoroaldehyde was observed by 1H NMR spectroscopic analysis,
cyclohexanone or thiopyranone 206 (5.0 – 10.0 equiv.) was then added and the resulting mixture
was allowed to warm gradually to room temperature. After a total of 18 hrs, the mixture was diluted
with Et2O and the organic layer was washed twice with water and once with brine. The organic
layer was then dried over MgSO4, concentrated under reduced pressure and the crude product
was purified by flash chromatography as indicated.
General Procedure C (α-trifluoromethylthiolation/aldol reaction)
A sample of aldehyde (2.0 equiv.) was added to a stirred suspension of NFSI (1.5 equiv.),
L-proline (2.0 equiv.), and NaHCO3 (2.0 equiv.) in DMSO (0.75 M) at room temperature. When
complete consumption of aldehyde was observed by 1H NMR spectroscopic analysis, 2,2-
Dimethyl-1,3-dioxan-5-one (75) (1.0 equiv.) in CH2Cl2 (5 x vol. of DMSO) was then added and the
resulting mixture was stirred for a further 48 – 72 hrs. When complete consumption of the 75 was
observed by 1H NMR spectroscopic analysis of small reaction aliquots, the mixture was diluted
with Et2O and the organic layer was washed twice with water and once with brine. The organic
layer was then dried over MgSO4, concentrated under reduced pressure and the crude product
was purified by flash chromatography as indicated.
General Procedure D (olefination/ring-closing metathesis)
To a stirred solution of 5-(methanesulfonyl)-1-phenyl-1H-tetrazole (2 – 2.2 equiv.) in dry THF (0.80
M) at -78oC was added dropwise a 1 M LiHMDS (2 – 2.2 equiv.) and the resulting reaction mixture
was stirred for 30 minutes. A solution of fluorohydrin (or trifluoromethylthiohydrin) (1.0 equiv.) in
dry THF (0.30 – 0.50 M) was then added dropwise and the reaction mixture was allowed to stir
for 5 hrs at -78°C. Following complete consumption of fluorohydrin (or trifluoromethylthiohydrin),
as monitored by TLC analysis, the reaction mixture was quenched with saturated ammonium
chloride solution and diluted with CH2Cl2. The organic layer was washed twice with water,

124
separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude
alkene was purified by flash chromatography as indicated. A mixture Grubbs II catalyst (0.05
equiv.) and alkene (1.0 equiv.) in dry toluene (0.025 M) was purged with N2 for 45 minutes in a
sealed reaction vessel and subsequently heated to 80 - 90°C for 6 -12 hrs. The reaction mixture
was then diluted with CH2Cl2 and washed twice with water. The organic layer was dried with
MgSO4, filtered, and concentrated under reduced pressure. The crude carbacycle was purified by
flash chromatography as indicated.
General Procedure E (reduction and benzoylation)
To a stirred solution of racemic or optically enriched syn-fluorohydrin (1.0 equiv) in MeOH (0.15
M) was added sodium borohydride (1.5 equiv), and the resulting mixture was stirred at room
temperature for 1 hour or until no starting material remained (as determined by TLC analysis).
The reaction mixture was then diluted with CH2Cl2 and washed with H2O. The organic layer was
removed, dried over MgSO4, concentrated under reduced pressure, and the crude product was
purified by flash chromatography. To a solution of purified diol in CH2Cl2 (0.10 M) was added
triethylamine (6.0 equiv.), either p-nitro benzoyl chloride (3.0 equiv.) or p-bromo benzoyl chloride
(3.0 equiv.), and 4-dimethylaminopyridine (cat.), and left to stir for 1 hour or until no starting
material remained (as determined by TLC analysis). The reaction mixture was diluted with CH2Cl2
and washed with NaHCO3. The organic layer was removed, dried over MgSO4, concentrated
under reduced pressure, and the crude product was purified by flash chromatography.
General Procedure F (hydrogenation)
Through a solution of fluorohydrin (1.0 equiv) and Pd/C (50 % by weight) in MeOH (0.10 M) was
bubbled H2 gas for 1 hr. The reaction vial was then sealed and left over night. The reaction mixture
was then filtered, concentrated under reduced pressure, and the crude product was purified by
flash chromatography.
Preparation and characterization of all compounds
Preparation of aldol adduct 200
Following General Procedure A, a solution of pentanal (0.050 mL, 0.470 mmol), NFSI (0.149 g,
0.470 mmol), L-proline (0.054 g, 0.470 mmol), and NaHCO3 (0.039 g, 0.470 mmol) was stirred for
45 minutes at -10 °C in 0.65 mL of DMF. Dioxanone 75 (0.0380 mL, 0.314 mmol) in CH2Cl2 (5.6
mL) was added and the reaction mixture was left to stir at room temperature for 72 hrs. Purification

125
of the crude fluorohydrin 200 by flash chromatography (pentane:EtOAc – 9:1) afforded
fluorohydrin 200 (0.045 g, 61 % yield) as a colourless oil.
Data for syn-fluorohydrin 200: []D20 = -6.2 (c 2.27 in CHCl3); IR (neat):
= 3429, 2990, 1742, 1376, 1225, 1091, 864 cm-1; 1H NMR (600 MHz,
CDCl3): δ 4.69 (dddd, J = 47.3, 9.0, 4.0, 1.6 Hz, 1H), 4.39 (dd, J = 8.5,
1.1 Hz, 1H), 4.30 (dd, J = 17.7, 1.5 Hz, 1H), 4.08 (d, J = 17.5 Hz, 1H),
3.80 (ddd, J = 27.2, 8.5, 1.5 Hz), 3.29 (s, 1H), 1.94 (m, 1H), 1.61 (m,
1H), 1.52 (m, 1H), 1.50 (s, 3H), 1.43 (s, 3H), 1.42 (m, 1H), 0.97 (t, J = 7.0 Hz, 3H); 13C NMR (150
MHz, CDCl3): δ 212.6, 101.8, 91.4 (d, J = 175.3), 71.9 (d, J = 5.1 Hz), 71.4 (d, J = 18.0 Hz), 66.9,
32.6 (d, J = 20.8 Hz), 23.8, 23.8, 18.5 (d, J = 5.4 Hz), 14.3; 19F NMR (470 MHz, CDCl3): δ –202.4
HRMS (EI+) calcd for [C11H20FO4]+ 235.1340; found 235.1354
Determination of the absolute stereochemistry for fluorohydrin 200
Following General Procedure E, the fluorohydrin 200 (0.105 g, 0.449 mmol) was converted into
the corresponding bis-p-bromobenzoate 200-XRD. Recyrstallization in dichloromethane and
ethanol (1:1) allowed for the absolute stereochemistry to be assigned using single X-ray
crystallography (see X-ray structures).
Determination of enantiomeric excess of fluorohydrin 200
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 200 was prepared. Following General Procedure E, optically enriched and racemic
samples of 200 (0.015 g, 0.064 mmol) were converted into the bis-p-nitrobenzoate derivative. The
enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection at 260
nm; retention time = 17.27 min for (-)-200-Bis-PNB; 22.06 min for (+)-200-Bis-PNB (see
chromatograms).The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (96% ee).
Preparation of aldol adduct 219
Following General Procedure A, a solution of propanal (0.050 mL, 0.687 mmol), NFSI (0.217 g,
0.687 mmol), L-proline (0.079 g, 0.687 mmol) and NaHCO3 (0.058 g, 0.687 mmol) was stirred for
45 minutes at -10 °C in DMF (0.92 mL). Dioxanone 75 (0.055 mL, 0.458 mmol) in CH2Cl2 (8.3 mL)

126
was then added and the reaction mixture was stirred for 48 hrs. Purification of the crude
fluorohydrin 219 by flash chromatography (pentane-EtOAc 4:1) afforded fluorohydrin 219 (0.053
g, 56 % yield) as a yellow oil.
Data for syn-fluorohydrin 219: []D20 = -13.5 (c 3.42 in CHCl3); IR (neat): =
3428, 2990, 1742, 1378, 1225, 1091, 864 cm-1;1H NMR (600 MHz, CDCl3): δ
4.90 (dq, J = 47.0 Hz, 6.5 Hz, 1H), 4.38 (dd, J = 8.5, 1.4 Hz, 1H), 4.30 (dd, J =
17.6, 1.5 Hz, 1H), 4.08 (d, J = 17.6, 1H), 3.75 (ddd, J = 26.1, 2.5, 2.5, 1H), 3.29
(d, J = 2.7), 1.50 (s, 3H), 1.43 (s, 3H), 1.43 (dd, J = 24.0, 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3):
δ 212.1, 101.6, 88.0 (d, J = 171.0 Hz) , 72.3 (d, J = 17.7 Hz), 72.0 (d, J = 5.1 Hz), 66.8, 23.7, 23.7,
16.4 (d, J = 22.9 Hz); 19F NMR (470 MHz, CDCl3): δ -195.5
HRMS (EI+) calcd for [C9H16FO4]+ 207.1027; found 207.1054
Determination of relative stereochemistry for fluorohydrin 219
Following General Procedure E, the fluorohydrin 219 (0.105 g, 0.449 mmol) was converted into
the corresponding p-nitrobenzoyl diester (219-XRD). Recyrstallization in ethanol allowed for the
relative stereochemistry to be assigned using single X-ray crystallography (see X-ray structures).
Determination of enantiomeric excess of fluorohydrin 219
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 219 was prepared. Following General Procedure E, optically enriched and racemic
samples of 219 (0.040 g, 0.19 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a
DIACEL CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection
at 260 nm; retention time = 28.1 min for (-)-219-Bis-PNB; 37.4 min for (+)-219-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (95% ee).
Preparation of aldol adduct 220
Following General Procedure A, a solution of 3-methylbutanal (0.050 mL, 0.465 mmol), NFSI
(0.147 g, 0.465 mmol), L-proline (0.053 g, 0.465 mmol), and NaHCO3 (0.039 g, 0.465 mmol) was
stirred for 45 minutes at -10 °C in 0.60 mL of DMF. Dioxanone 75 (0.037 mL, 0.31 mmol) in CH2Cl2

127
(5.4 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 220 by flash chromatography
(pentane-EtOAc 4:1) afforded fluorohydrin 220 (0.049 g, 67 % yield) as a colorless oil.
Data for syn-fluorohydrin 220: []D20 = -117 (c 2.6 in CHCl3); IR (neat): =
3526, 2968, 1740, 1377, 864;1H NMR (600 MHz, CDCl3): δ 4.39 (d, J = 8.7
Hz, 1H), 4.30 (d, J = 17.8 Hz, 1H), 4.21 (dd, J = 46.5, 9.6 Hz, 1H), 4.08 (d, J
= 17.6 Hz, 1H), 3.95 (dd, J = 28.6, 8.8 Hz, 1H), 3.26 (d, J = 2.5 Hz, 1H), 2.26
(m, 1H), 1.50 (s, 3H), 1.43 (s, 3H), 1.07 (d, J = 6.6 Hz, 3H), 0.92 (d, J =6.6
Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 212.8, 101.6, 96.1 (d, J = 178.1 Hz), 71.5 (d, J = 6.2 Hz),
69.2 (d, J = 18.4 Hz), 66.7, 28.1 (d, J = 20.0 Hz), 23.6, 23.6, 19.1 (d, J = 4.8 Hz), 18.1 (d, J = 9.3
Hz); 19F NMR (470 MHz, CDCl3): δ –203.3
HRMS (EI+) calcd for [C11H20FO4]+ 235.1340; found 235.1334
Determination of enantiomeric excess of fluorohydrin 220
Following General Procedure A, using a 1:1 mixture of L-: D-proline, a racemic sample of the
fluorohydrin 220 was prepared. Following General Procedure E, optically enriched and racemic
samples of 220 (0.035 g, 0.15 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric fluorohydrin were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 93.5:6.5; detection at 260
nm; retention time = 24.94 min for (+)-220-Bis-PNB; 27.31 min for (-)-220-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically pure bis-p-nitrobenzoate derivative
was determined using the same method (95 % ee).
Preparation of aldol adduct 221
Following General Procedure A, a solution of pentadecanal (0.453 g, 2.0 mmol), NFSI (0.731 g,
2.0 mmol), D-proline (0.23 g, 2.0 mmol) and NaHCO3 (0.168 g, 2.0 mmol) was stirred for 3 hrs at
-10 °C in DMF (2.7 mL). Dioxanone 75 (0.287 mL, 1.33 mmol) in CH2Cl2 (24 mL) was stirred for
48 hours. Purification of the crude fluorohydrin by flash chromatography (pentane-EtOAc 30:1)
afforded fluorohydrin 221 (0.239 g, 48% yield) as a clear oil.

128
Data for syn-fluorohydrin 221: []D20 = -83.0 (c 1.2 in CHCl3); IR (neat):
= 3530, 2924, 2854, 1740, 1337, 1224, 1090, 865 cm-1; 1H NMR (600
MHz, CDCl3): δ 4.66 (ddd, J = 47.3, 8.9, 4.9 Hz, 1H), 4.38 (dd, J = 8.7,
1.4 Hz, 1H), 4.30 (dd, J = 17.5, 1.4 Hz, 1H), 4.07 (d, J = 17.5 Hz, 1H),
3.79 (dddd, J = 27.1, 8.5, 3.1, 1.7 Hz, 2H), 3.31 (d, J = 3.1 Hz, 1H), 1.92 (m, 1H), 1.25-1.63 (26
H), 0.88 (dd, J =6.6, 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 212.3, 101.6, 91.5 (d, J = 174.9
Hz) , 71.7 (d, J = 5.3 Hz), 71.2 (d, J = 18.5 Hz), 66.7, 32.1, 30.4 (d, J = 21.3 Hz), 29.8, 29.8, 29.8,
29.8, 29.6, 29.6, 29.5, 25.4, 25.3, 23.6, 23.6, 22.8, 14.2; 19F NMR (470 MHz, CDCl3): δ –201.9
HRMS (EI+) calcd for [C21H39FNaO4]+ 397.2725; found 397.2755
Determination of relative stereochemistry for fluorohydrin 221
Analysis of 1H-NMR of fluorohydrins 200 and 221 revealed identical signals between 1.60 and
4.70 ppm indicating the two compounds share the same relative stereochemistry.
Determination of enantiomeric excess of fluorohydrin 221
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 221 was prepared. Following General Procedure E, optically enriched and racemic
samples of 221 (0.050 g, 0.13 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric p-nitrobenzoyl esters were separated by chiral HPLC using a
DIACEL CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection
at 260 nm; retention time = 8.76 min for (+)-221-Bis-PNB; 12.06 min for (-)-221-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (91% ee).
Preparation of aldol adduct 222
Following General Procedure A, a solution of 4-methyl-4-nitropentanal (0.050 mL, 0.379 mmol),
NFSI (0.119 g, 0.379 mmol), L-proline (0.044 g, 0.379 mmol), and NaHCO3 (0.032 g, 0.379 mmol)
was stirred for 120 minutes at -10 °C in 0.50 mL of DMF. Dioxanone 75 (0.030 mL, 0.253 mmol)
in CH2Cl2 (5.5 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 222 by flash
chromatography (pentane-EtOAc 3:1) afforded fluorohydrin 222 (0.034 g, 46 % yield) as a
colorless oil.

129
Data for syn-fluorohydrin 222: []D20 = -71.6 (c 1.9 in CHCl3); IR (neat):
= 3515, 2989, 1741, 1540, 1376, 861, cm-1;1H NMR (500 MHz, CDCl3): δ
4.77 (ddd, J = 48.7, 10.0, 1.0 Hz, 1H), 4.35 (dd, J = 9.0, 1.3 Hz, 1H), 4.30
(dd, J = 17.7, 1.4 Hz, 1H), 4.08 (d, J = 17.7 Hz, 1H), 3.76 (dddd, J = 27.4,
8.9, 2.3, 2.3 Hz, 1H), 3.43 (d, J = 2.3 Hz, 1H), 2.57 (m, 1H), 2.30 (m, 1H), 1.69 (s, 3H), 1.67 (s,
3H), 1.48 (s, 3H), 1.40 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 212.5, 101.8, 87.9 (d, J = 176.9
Hz), 87.1, 72.2 (d, J = 18.6 Hz), 71.2 (d, J = 5.5 Hz) 66.5, 41.3 (d, J = 20.5 Hz), 28.0, 25.4, 23.6,
23.5; 19F NMR (470 MHz, CDCl3): δ –201.4
HRMS (EI+) calcd for [C12H21FNO6]+ 294.1347; found 294.1359
Determination of enantiomeric excess of fluorohydrin 222
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 222 was prepared. Following General Procedure E, optically enriched and racemic
samples of 222 (0.050 g, 0.171 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric fluorohydrin were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.5 mL/min; eluent: hexanes-iPrOH 80:20; detection at 260
nm; retention time = 34.51 min for (-)-222-Bis-PNB; 37.98 min for (+)-222-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically pure bis-p-nitrobenzoate derivative
was determined using the same method (95% ee).
Preparation of aldol adduct 223
Following General Procedure A, a solution of pentenal (0.050 g, 0.60 mmol), NFSI (0.189 g, 0.60
mmol), L-proline (0.069 g, 0.60 mmol) and NaHCO3 (0.055 g, 0.60 mmol) was stirred at -10 °C in
DMF (0.80 mL) for 1 hr. Dioxanone 75 (0.048 mL, 0.40 mmol) in CH2Cl2 (7.2 mL) was stirred for
72 hours. Purification of the crude fluorohydrin 223 by flash chromatography (pentane-EtOAc
85:15) afforded fluorohydrin 223 (0.059 g, 64 % yield) as a light yellow oil.
Data for syn-fluorohydrin 223: []D20 = -115.8 (c 2.46 in CHCl3); IR (neat):
= 3509, 2989, 1740, 1643, 1422, 1377, 1089, 863 cm-1; 1H NMR (600 MHz,
CDCl3): δ 5.83 (m, 1H), 5.20 (d, J = 17.4 Hz, 1H), 5.14 (d, J = 9.9, 1.0 Hz,
1H), 4.73 (dddd, J = 47.0, 7.0, 7.0, 1.3 Hz, 1H), 4.39 (dd, J = 8.9, 1.0 Hz,
1H), 4.30 (dd, J = 17.7, 1.0 Hz), 4.08 (d, J = 17.6), 2.69 (m, 1H), 2.48 (m, 1H) 1.49 (s, 3H), 1.43
(s, 3H);13C NMR (150 MHz, CDCl3): δ 212.4, 133.0 (d, J = 7.6 Hz), 118.5, 101.6, 90.5 (d, J =

130
178.0 Hz), 71.5 (d, J = 5.3 Hz), 70.5 (d, J = 18.2 Hz), 66.6, 34.9 (d, J = 22.2 Hz), 23.6, 23.6; 19F
NMR (470 MHz, CDCl3): δ –201.6
HRMS (EI+) calcd for [C11H18FO4]+ 233.1184; found 233.1202
Determination of relative stereochemistry for fluorohydrin 223
Following General Procedure F, the fluorohydrin 223 (0.105 g, 0.45 mmol) was converted to the
fluorohydrin 200. Comparison of 1H and 19F NMR with fluorohydrin 200 confirmed relative
stereochemistry.
Determination of the absolute stereochemistry for fluorohydrin 223
Following General Procedure F, the fluorohydrin 223 (0.105 g, 0.45 mmol) was converted to the
fluorohydrin 200. Comparison of []D values with fluorohydrin 200 confirmed absolute
stereochemistry
Determination of enantiomeric excess of fluorohydrin 223
Following General Procedure F, the optically enriched sample of 223 (0.105 g, 0.45 mmol) was
converted into fluorohydrin 200. Following General Procedure E, the optically enriched and
racemic samples of fluorohydrin 200 were converted into their corresponding bis-p-nitrobenzoate
derivative. The enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a
DIACEL CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection
at 260 nm; retention time = 17.27 min for (-)-200-Bis-PNB; 22.06 min for (+)-200-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (93 % ee).
Preparation of aldol adduct 224
Following General Procedure A, a solution of 3-(4-methoxyphenyl)propanal (0.050 mL, 0.317
mmol), NFSI (0.100 g, 0.317mmol), L-proline (0.037 g, 0.317 mmol), and NaHCO3 (0.027 g, 0.317
mmol) was stirred for 90 minutes at -10 °C in 0.43 mL of DMF. Dioxanone 75 (0.025 mL, 0.211
mmol) in CH2Cl2 (3.8 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 224 by flash
chromatography (pentane-EtOAc 4:1) afforded fluorohydrin 224 (0.034 g, 51 % yield) as a white
solid.

131
Data for syn-fluorohydrin 224: []D20 = -233.4 (c 3.0 in CHCl3); 1H
NMR (600 MHz, CDCl3): δ 7.20 (d, J = 8.8 Hz, 2H), 6.86 (d, J =
8.8Hz, 2H) 4.81 (dddd, J = 46.8, 7.3, 7.3 Hz, 0.8 Hz, 1H), 4.40 (d,
J = 8.8 Hz, 1H), 4.26 (d, J = 17.8 Hz, 1H), 4.07 (d, J = 17.8 Hz,
1H), 3.79 (s, 3H), 3.77 (d, J = 24.1, 8.8 Hz, 1H), 3.39 (br s, 1H), 3.15 (m, 1H), 2.99 (m, 1H), 1.46
(s, 3H), 1.39 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 212.7, 158.6, 130.6, 128.9 (d, J = 8.0 Hz),
114.1, 101.7, 91.9 (d, J = 179.2 Hz), 71.4 (d, J = 5.1 Hz), 70.1 (d, J =18.1 Hz), 66.6, 55.4, 35.8
(d, J = 22.4 Hz), 23.6, 23.5; 19F NMR (470 MHz, CDCl3): δ –200.1
HRMS (EI+) calcd for [C16H22FO5]+ 313.1446; found 313.1450
Determination of enantiomeric excess of fluorohydrin 224
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 224 was prepared. Following General Procedure E, optically enriched and racemic
samples of 224 (0.040 g, 0.128 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric fluorohydrin were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.5 mL/min; eluent: hexanes-iPrOH 93.5:6.5; detection at 260
nm; retention time = 74.8 min for (+)-224-Bis-PNB; 110.4 min for (-)-224-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically pure bis-p-nitrobenzoate derivative
was determined using the same method (95% ee).
Preparation of aldol adduct 225
Following General Procedure A, a solution of pentynal (0.050 g, 0.61 mmol), NFSI (0.192 g, 0.61
mmol), L-proline (0.070 g, 0.61 mmol) and NaHCO3 (0.051 g, 0.61 mmol) was stirred for 1.5 hrs
at -10°C in DMF (0.81 mL). Dioxanone 75 (0.049 mL, 0.41 mmol) in CH2Cl2 (7.3 mL) was stirred
for 48 hours. Purification of the crude fluorohydrin by flash chromatography (pentane-EtOAc; 9:1)
afforded fluorohydrin 225 (0.052 g, 55 % yield) as a light yellow oil.
Data for syn-fluorohydrin 225: []D20 = -62.9 (c 3.42 in CHCl3); IR (neat):
= 3512, 3293, 2993, 1743, 1378, 1224, 1033 cm-1; 1H NMR (500 MHz,
CDCl3): δ 4.86 (dddd, J = 46.4, 7.3, 7.3, 1.5 Hz, 1H), 4.38 (dd, J = 8.9, 1.5
Hz, 1H), 4.31 (dd, J = 17.6, 1.6 Hz, 1H), 4.10 (d, J = 17.7 Hz, 1H), 4.00
(ddd, J = 27.9, 9.1, 1.5 Hz, 1H), 2.76 (m, 2H), 2.04 (t, J = 2.8 Hz, 1H), 1.49 (s, 3H), 1.43 (s, 3H);
13C NMR (150 MHz, CDCl3): δ 212.2, 101.7, 89.0 (d, J = 180.0 Hz), 79.0 (d, J = 15.4 Hz), 71.3 (d,

132
J = 5.1 Hz), 70.8, 69.7 (d, J = 17.4 Hz), 66.6, 23.7, 23.6, 20.4 (d, J = 28.9 Hz); 19F NMR (470
MHz, CDCl3): δ –200.1
HRMS (EI+) calcd for [C11H16FO4]+ 231.1027; found 231.1042
Determination of relative stereochemistry for fluorohydrin 225
Following General Procedure F, the fluorohydrin 225 (0.10 g, 0.43 mmol) was converted to the
fluorohydrin 200. Comparison of 1H and 19F NMR with fluorohydrin 200 confirmed relative
stereochemistry.
Determination of the absolute stereochemistry for fluorohydrin 225
Following General Procedure F, the fluorohydrin 225 (0.10 g, 0.43 mmol) was converted to the
fluorohydrin 200. Comparison of []D values with fluorohydrin 200 confirmed absolute
stereochemistry.
Determination of enantiomeric excess of fluorohydrin 225
Following General Procedure F, the optically enriched sample of 225 (0.10 g, 0.43 mmol) was
converted into fluorohydrin 200. Following General Procedure E, the optically enriched and
racemic samples of fluorohydrin 200 were converted into their corresponding bis-p-nitrobenzoate
derivative. The enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a
DIACEL CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection
at 260 nm; retention time = 17.27 min for (-)-225-Bis-PNB; 22.06 min for (+)-225-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (92% ee).
Preparation of aldol adduct 227
Following General Procedure A, a solution of 3-(5-methylfuran-2-yl)propanal (0.050 mL, 0.376
mmol), NFSI (0.119 g, 0.376 mmol), L-proline (0.029 g, 0.376 mmol), and NaHCO3 (0.032 g, 0.376
mmol) was stirred for 90 minutes at -10 °C in 0.50 mL of DMF. Dioxanone 75 (0.030 mL, 0.251
mmol) in CH2Cl2 (4.5 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 227 by flash
chromatography (pentane-EtOAc 5:1) afforded fluorohydrin 227 (0.038 g, 53 % yield) as a
colorless oil.

133
Data for syn-fluorohydrin 227: []D20 = 16.4 (c 2.5 in CHCl3); 1H NMR
(600 MHz, CDCl3): δ 6.02 (d, J = 3.1 Hz, 1H), 5.88 (d, J = 3.1 Hz, 1H),
4.95 (ddd, J = 46.8, 6.9, 6.9 Hz, 1H), 4.41 (d, J = 9.0 Hz, 1H), 4.29 (d,
J = 17.6 Hz, 1H), 4.08 (d, J = 17.6 Hz, 1H), 3.82 (dd, J = 27.4, 8.8 Hz,
1H), 3.34 (d, J = 1.5 Hz, 1H), 3.18 (m, 1H), 3.06 (m, 1H), 2.26 (s, 3H),
1.49 (s, 3H), 1.42 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 212.4, 151.4, 149.0 (d, J = 9.5 Hz),
108.3, 106.4, 101.7, 89.4 (d, J = 179.3 Hz) , 71.4 (d, J = 5.1 Hz), 70.4 (d, J = 17.7 Hz), 66.6, 29.5
(d, J = 25.2 Hz), 23.6, 23.6, 13.7; 19F NMR (470 MHz, CDCl3): δ –201.2
HRMS (EI+) calcd for [C14H20FO5]+ 287.1289; found 287.1289
Determination of enantiomeric excess of fluorohydrin 227
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 227 was prepared. Following General Procedure E, optically enriched and racemic
samples of 227 (0.043 g, 0.15 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric fluorohydrin were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.5 mL/min; eluent: hexanes-iPrOH 93.5:6.5; detection at 260
nm; retention time = 30.00 min for (-)-227-Bis-PNB; 40.48 min for (+)-227-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically pure bis-p-nitrobenzoate derivative
was determined using the same method (95% ee).
Preparation of aldol adduct 228
Following General Procedure A, a solution of hydrocinnamaldehyde (0.050 mL, 0.38 mmol), NFSI
(0.120 g, 0.38 mmol), L-proline (0.044 g, 0.38 mmol), and NaHCO3 (0.032 g, 0.38 mmol) was
stirred for 75 minutes at -10 °C in 0.50 mL of DMF. Dioxanone 75 (0.036 mL, 0.30 mmol) in CH2Cl2
(4.5 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 228 by flash chromatography
(pentane-EtOAc 4:1) afforded fluorohydrin 228 (0.053 g, 62 % yield) as a colorless oil.
Data for syn-fluorohydrin 228: []D20 = -3.1 (c 2.1 in CHCl3); IR (neat):
= 3511, 2923, 1739, 1705, 1650, 1585, 1453, 863, 698 cm-1;1H NMR
(600 MHz, CDCl3): δ 7.30 (m, 5H), 4.87 (dd
d, J = 46.5, 7.0, 7.0 Hz, 1H), 4.42 (dd, J = 9.0, 0.9 Hz, 1H), 4.27 (dd, J = 17.7, 1.0 Hz, 1H), 4.08
(d, J = 17.6 Hz, 1H), 3.79 (dd, J = 27.6, 9.0 Hz, 1H), 3.42 (br s, 1H), 3.24 (m, 1H), 3.05 (m, 1H),

134
1.48 (s, 3H), 1.40 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 212.6, 136.9 (d, J = 8.1 Hz), 129.6,
128.7, 126.8, 101.7, 91.7 (d, J = 177.8 Hz) , 71.4 (d, J = 5.2 Hz), 70.2 (d, J = 18.1 Hz), 66.6, 36.7
(d, J = 22.4 Hz), 23.6, 23.5; 19F NMR (470 MHz, CDCl3): δ –200.1
HRMS (EI+) calcd for [C15H20FO4]+ 283.1340; found 283.1365
Determination of relative stereochemistry for fluorohydrin 228
Reduction with sodium borohydride of the fluorohydrin 228 (0.105 g, 0.37 mmol) in methanol
allowed for conversion to the corresponding syn-diol 228-XRD. Recyrstallization in ethanol (1:1)
allowed for the relative stereochemistry to be assigned using single X-ray crystallography (see X-
ray structures)
Determination of enantiomeric excess of fluorohydrin 228
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 228 was prepared. The enantiomeric fluorohydrin were separated by chiral HPLC
using a DIACEL CHIRALCEL-OD column; flow rate 1.5 mL/min; eluent: hexanes-iPrOH 97:3;
detection at 260 nm; retention time = 12.80 min for (-)-228; 13.64 min for (+)-228 (see
chromatograms). The enantiomeric excess of the optically pure 228 was determined using the
same method (98% ee).
Preparation of aldol adduct 229
Following General Procedure A, a solution of 3-(4-bromophenyl)propanal (0.050 g, 0.236 mmol),
NFSI (0.074 g, 0.236 mmol), L-proline (0.028 g, 0.236 mmol), and NaHCO3 (0.020 g, 0.236 mmol)
was stirred for 120 minutes at -10 °C in 0.30 mL of DMF. Dioxanone 75 (0.022 mL, 0.157 mmol)
in CH2Cl2 (2.8 mL) was stirred for 72 hrs. Purification of the crude fluorohydrin 229 by flash
chromatography (pentane-EtOAc 4:1) afforded fluorohydrin 229 (0.034 g, 61 % yield) as a white
solid.
Data for syn-fluorohydrin 229: []D20 = -76.4 (c 0.8 in CHCl3); IR (neat):
= 3513, 2989, 1740, 1490, 1377, 864cm-1;1H NMR (600 MHz, CDCl3):
δ 7.44 (d, J = 8.2 Hz), 7.16 (d, J = 8.2 Hz), 4.80 (ddd, J = 46.8, 7.2, 7.2,
0.9 Hz, 1H), 4.40 (d, J = 18.9 Hz, 1H), 4.27 (d, J = 17.7 Hz, 1H), 4.08 (d,
J = 17.7 Hz, 1H), 3.75 (dd, J = 27.6, 9.0 Hz, 1H), 3.42 (br s, 1H), 3.18 (m, 1H), 2.97 (m, 1H), 1.46
(s, 3H), 1.39 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 212.6, 136.0 (d, J = 7.2 Hz), 131.8, 131.4,

135
120.8, 101.7, 91.4 (d, J = 179.9 Hz) , 71.3 (d, J = 5.0 Hz), 70.2 (d, J = 17.1 Hz), 66.6, 36.2 (d, J
= 22.4 Hz), 23.6, 23.5; 19F NMR (470 MHz, CDCl3): δ –200.6
HRMS (EI+) calcd for [C15H19BrFO4]+ 361.0445; found 361.0434
Determination of enantiomeric excess of fluorohydrin 229
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 229 was prepared. Following General Procedure E, optically enriched and racemic
samples of 229 (0.037 g, 0.103 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric fluorohydrin were separated by chiral HPLC using a DIACEL
CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection at 260
nm; retention time = 52.3 min for (+)-229-Bis-PNB; 63.5 min for (-)-229-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically pure bis-p-nitrobenzoate derivative
was determined using the same method (95% ee).
Preparation of aldol adduct 230
A solution of 3-OTIPS-propanal (1.152 g, 5.0 mmol, 1.5 equiv.), Selectfluor (1.77 g, 5.0 mmol, 1.5
equiv.), and L-proline (0.576 g, 5.0 mmol, 1.5 equiv.) were dissolved in 50 mL DMF (0.1 M) and
stirred at 4 °C for 3 hrs or until the reaction was complete as determined by TLC analysis. The
reaction mixture was diluted with 500 mL of diethyl ether and washed 3 x H2O (100 mL). The
organic layer was removed, dried over MgSO4, and concentrated under reduced pressure.
Dioxanone 75 (0.434 g, 3.33 mmol, 1.0 equiv.) and L-proline (0.306 g, 2.7 mmol, 0.8 equiv.) were
added to the crude α-fluoroaldehyde in 25 mL of CH2Cl2 (0.2 M). After 48 hours, the reaction
mixture was diluted with CH2Cl2 and washed with H2O. Purification of the crude fluorohydrin 230
by flash chromatography (pentane-EtOAc; 20:1) afforded fluorohydrin 230 (0.692 g, 55 % yield)
as a colorless oil.
Data for syn-fluorohydrin 230: []D20 = -25.6 (c 5.0 in CHCl3); IR (neat): =
3503, 2994, 2867, 1741, 1224, 1094, 883 cm-1; 1H NMR (600 MHz, CDCl3):
δ 4.75 (dddd, J = 47.1, 5.7, 5.7, 1.9 Hz, 1H), 4.41 (dd, J = 8.5, 1.4 Hz, 1H),
4.31 (dd, J = 17.5, 1.4 Hz, 1H), 4.08 (d, J = 17.6 Hz, 1H), 4.05 (ddd, J =
27.4, 9.4, 1.7 Hz, 2H), 4.02 (dd, J = 18.5, 5.5 Hz, 1H), 3.33 (br s, 1H), 1.50 (s, 3H), 1.44 (s, 3H),

136
1.07 (m, 21H); 13C NMR (150 MHz, CDCl3): δ 211.7, 101.6, 91.0 (d, J = 178.1 Hz) , 71.7 (d, J =
5.2 Hz), 69.6 (d, J = 18.2 Hz), 66.8, 62.4 (d, J = 27.4 Hz), 23.7, 23.7, 18.1, 12.1; 19F NMR (470
MHz, CDCl3): δ –209.0
HRMS (EI+) calcd for [C18H36FO5Si]+ 379.2311; found 379.2343
Determination of the absolute stereochemistry for fluorohydrin 230
Following General Procedure E, the fluorohydrin 230 (0.050 g, 0.13 mmol) was converted into the
corresponding bis-p-bromobenzoate derivative 230-XRD. Recyrstallization in dichloromethane
and ethanol (1:1) allowed for the absolute stereochemistry to be assigned using single X-ray
crystallography (see X-ray structures).
Determination of enantiomeric excess of fluorohydrin 230
Following General Procedure A, using a 1:1 mixture of L-:D- proline, a racemic sample of the
fluorohydrin 230 was prepared. Following General Procedure E, optically enriched and racemic
samples of 230 (0.050 g, 0.13 mmol) were converted into the corresponding bis-p-nitrobenzoate
derivative. The enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a
DIACEL CHIRALCEL-OD column; flow rate 1.0 mL/min; eluent: hexanes-iPrOH 80:20; detection
at 260 nm; retention time = 6.26 min for (-)-230-Bis-PNB; 8.33 min for (+)-230-Bis-PNB (see
chromatograms). The enantiomeric excess of the optically enriched bis-p-nitrobenzoate derivative
was determined using the same method (99 % ee).
Preparation of aldol adduct 237
Following General Procedure B, a solution of pentanal (0.050 mL, 0.47 mmol), NFSI (0.148 g,
0.47 mmol), L-proline (0.054 g, 0.47 mmol) and NaHCO3 (0.039 g, 0.47 mmol) was stirred at -10
°C in DMF (0.63 mL) for 75 minutes. Cyclohexanone (0.488 mL, 4.70 mmol) was added and the
reaction mixture was stirred for 18 hours. Purification of crude fluorohydrin 237 by flash
chromatography (pentane:EtOAc – 95:5 → 90:10) afforded fluorohydrin 237 (0.051 g, dr = 10:1,
54% yield) as an off-white solid.

137
Data for syn-fluorohydrin 237: []D20 = -16.4 (c 1.65 in CH2Cl2); IR (neat): =
3498, 2957, 2938, 2863, 1698, 1450, cm-1; 1H NMR (600 MHz, CDCl3): δ 4.55
(ddd, J = 47.8, 8.3, 4.2 Hz, 1H), 3.77 (dddd, J = 29.0, 8.3, 4.2, 2.1 Hz, 1H),
3.55 (d, J = 4.2 Hz, 1H), 2.45 (m, 1H), 2.36 (dddd, J = 13.4, 13.4, 6.2, 1.1
Hz, 1H), 2.23 (m, 1H), 2.12 (m, 1H), 1.85 – 1.97 (2H), 1.55 – 1.78 (3H), 1.36 – 1.55 (3H), 0.96
(dd, J = 7.4, 7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 215.7, 92.7 (d, J = 174.1 Hz), 72.2 (d, J
= 19.3 Hz), 52.8 (d, J = 3.5 Hz), 42.9, 32.8 (d, J = 21.2), 30.1, 27.8, 24.8; 19F NMR (470 MHz,
CDCl3): δ –199.3
HRMS (EI+) calcd for [C11H20FO2]+ 203.1442; found 203.1421
Determination of enantiomeric excess of fluorohydrin 237
Following General Procedure E, the optically enriched and racemic samples of fluorohydrin 237
were converted into their corresponding p-nitrobenzoyl diesters. The enantiomeric p-nitrobenzoyl
diesters were separated by chiral HPLC using a Lux® 3µm Amylose-1 column; flow rate 0.40
mL/min; eluent: hexanes-iPrOH 90:10; detection at 254 nm; retention times = 8.96 min and 10.37
min (see chromatograms). The enantiomeric excess of the optically enriched p-nitrobenzoyl
diesters was determined using the same method (94 % ee).
Preparation of aldol adduct 238
Following General Procedure B, a solution of pentanal (0.0.50 mL, 0.47 mmol), NFSI (0.148 g,
0.47 mmol), L-proline (0.054 g, 0.47 mmol) and NaHCO3 (0.039 g, 0.47 mmol) was stirred for 75
minutes at -10 °C in DMF (0.63 mL). Thiopyranone 206 (0.546 g, 4.70 mmol) was added and the
reaction mixture was stirred for 24 hours at 4°C. Purification of crude fluorohydrin 238 by flash
chromatography (pentane-Et2O 4:1) afforded syn-fluorohydrin 238 (0.049 g, 47% yield) as a white
solid.
Data for syn-fluorohydrin 238: []D20 = -16.4° (c 1.65 in CH2Cl2); IR (neat):
= 3353, 2952, 1706, 1428, 510 cm-1; 1H NMR (600 MHz, CDCl3): δ 4.58
(ddd, J = 48.0, 8.8, 2.3 Hz, 1H), 3.95 (ddd, J = 26.7, 6.6, 2.3 Hz, 1H), 3.07
(m, 1H), 3.06 (m, 1H), 2.94 – 3.02 (3H), 2.72 – 2.86 (3H), 1.88 (m, 1H), 1.64
(m, 1H), 1.60 (m, 1H), 1.51 (m, 1H), 1.42 (m 1H), 0.97 (dd, J = 7.4, 7.4 Hz, 3H);13C NMR (150
MHz, CDCl3): δ 211.7, 92.8 (d, J = 173.6 Hz), 71.8 (d, J = 20.8 Hz), 55.2 (d, J = 3.1 Hz), 44.7,

138
32.9 (d, J = 20.9 Hz), 32.5 (d, J = 1.2 Hz), 30.9, 18.7 (d, J = 4.9 Hz), 14.0; 19F NMR (470 MHz,
CDCl3): δ –197.6
HRMS (EI+) calcd for [C10H18FO2S]+ 221.1006; found 221.0999
Determination of enantiomeric excess of fluorohydrin 238
Using a 1:1 mixture of L-: D-proline, a racemic sample of fluorohydrin 238 was prepared. The
enantiomeric fluorohydrins were separated by chiral HPLC using a Lux® 3µm Amylose-1 column;
flow rate 0.40 mL/min; eluent: hexanes-iPrOH 90:10; detection at 254 nm; retention time = 7.14
min for (+)-238; 8.89 min for (-)-238 (see chromatograms). The enantiomeric excess of the
optically enriched fluorohydrin 238 was determined using the same method (84% ee).
Preparation of aldol adduct 239
Following General Procedure B, a solution of pentenal (0.050 g, 0.595 mmol), NFSI (0.188 g,
0.595 mmol), L-proline (0.069 g, 0.595 mmol) and NaHCO3 (0.050 g, 0.595 mmol) was stirred for
1.5 hrs at -10°C in DMF (0.79 mL). Cyclohexanone (0.62 mL, 5.95 mmol) was added and the
reaction mixture stirred for 16 hours. Purification of crude fluorohydrin 239 by flash
chromatography (pentane:EtOAc – 95:5 → 90:10) afforded fluorohydrin 239 (0.059 g, 50% yield)
as a white solid.
Data for syn-fluorohydrin 239: []D20 = +22.2 (c 0.60 in CH2Cl2); IR (neat):
= 3513, 2937, 2863, 1698, 1449, 1132 cm-1; 1H NMR (600 MHz, CDCl3):
δ 5.83 (dddd, J = 17.2, 10.3, 7.2, 7.0 Hz, 1H), 5.19 (dd, J = 17.2, 1.5 Hz,
1H), 5.13 (d, J = 10.3 Hz, 1H), 4.57 (ddd, J = 47.7, 8.3, 4.7 Hz, 1H), 3.80
(dddd, J = 29.7, 8.3, 4.1, 2.0 Hz, 1H), 3.58 (d, J = 4.1 Hz, 1H), 2.74 (m, 1H), 2.67 (m, 1H), 2.42 –
2.55 (2H), 2.36 (dddd, J = 13.4, 13.4, 6.1, 1.1 Hz, 1H), 2.22 (m, 1H), 2.12 (m, 1H), 1.93 (m, 1H),
1.73 (ddddd, J = 13.0, 13.0, 13.0, 3.6, 3.6 Hz, 1H), 1.67 (ddddd, J = 13.0, 13.0, 13.0, 3.6, 3.6 Hz,
1H), 1.42 (dddd, J = 12.7, 12.7, 12.7, 3.6, 3.6 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 215.6, 133.3
(d, J = 7.5 Hz), 118.4, 92.0 (d, J = 177.6 Hz), 71.6 (d, J = 18.8 Hz), 52.7 (d, J = 3.6 Hz), 42.8, 35.4
(d, J = 23.4 Hz), 30.0, 27.8 24.8; 19F NMR (470 MHz, CDCl3): δ –198.7
HRMS (EI+) calcd for [C11H18FO2]+ 201.1285; found 201.1260
Determination of relative stereochemistry for fluorohydrin 239

139
Following General Procedure D, the fluorohydrin 239 was converted to carbacycle 263. NOE
analysis of carbacycle 263 confirmed relative stereochemistry of fluorohydrin 239.
Determination of enantiomeric excess of fluorohydrin 239
Following General Procedure E, optically enriched and racemic samples of fluorohydrin 239 were
converted into their corresponding p-nitrobenzoyl diesters. The enantiomeric p-nitrobenzoyl
diesters were separated by chiral HPLC using a Lux® 3µm i-Cellulose-3 column; flow rate 0.20
mL/min; eluent: hexanes-iPrOH 85:15; detection at 254 nm; retention times = 4.32 min and 6.09
min (see chromatograms). The enantiomeric excess of the optically enriched p-nitrobenzoyl
diester was determined using the same method (96% ee).
Preparation of aldol adduct 240
Following General Procedure B, a solution of pentenal (0.100 mL, 1.02 mmol), NFSI (0.319 g,
1.02 mmol), L-proline (0.118 g, 1.02 mmol) and NaHCO3 (0.086 g, 1.02 mmol) was stirred for 60
minutes at -10 °C in DMF (1.35 mL). Thiopyranone 206 (1.19 g, 10.2 mmol) was then added and
the reaction mixture was stirred for 24 hrs at 4°C. Purification of the crude fluorohydrin 240 by
flash chromatography (pentane:Et2O – 4:1) afforded syn-fluorohydrin 240 (0.069 g, dr = 6:1, 31
% yield) as a waxy white solid.
Data for syn-fluorohydrin 240: []D20 = -11.0 (c 0.30 in CH2Cl2); IR (neat):
= 3455, 2929, 1703, 1428, 1117, 923 cm-1;1H NMR (600 MHz, CDCl3): δ
5.82 (dddd, J = 17.1, 10.3, 7.0, 6.9 Hz, 1H), 5.21 (dd, J = 17.1, 1.4 Hz, 1H),
5.15 (d, J = 10.3, 1H), 4.61 (ddd, J = 47.7, 5.9, 2.0 Hz, 1H), 3.97 (ddd, J =
27.5, 6.4, 2.0 Hz, 1H), 3.09 (m, 1H), 3.05 (m, 1H), 3.04 (m, 1H), 2.99 (m, 1H), 2.97 (m, 1H), 2.81
(m, 2H), 2.76 (m, 1H), 2.64 (m, 1H), 2.51 (m, 1H) ; 13C NMR (150 MHz, CDCl3): δ 211.7, 132.8
(d, J = 7.3 Hz), 118.7, 92.1 (d, J = 176.3 Hz), 71.2 (d, J = 19.3 Hz), 55.1 (d, J = 3.0 Hz), 44.7, 35.4
(d, J = 22.2 Hz), 32.4, 30.8; 19F NMR (470 MHz, CDCl3): δ -197.0
HRMS (EI+) calcd for [C10H16FO2S]+ 219.0850; 219.0833
Determination of relative stereochemistry for fluorohydrin 240

140
Following General Procedure D, the fluorohydrin 240 was converted to carbacycle 264. NOE
analysis of carbacycle 264 confirmed relative stereochemistry of fluorohydrin 240.
Preparation of aldol adduct 241
Following General Procedure B, a solution of phthalimidoacetaldehyde (0.050 g, 0.265 mmol),
NFSI (0.84 g, 0.265 mmol), L-proline (0.031 g, 0.265 mmol) and 2,6-lutidine (0.031 mL, 0.265
mmol) was stirred at 4°C in DMF (0.35 mL) for 16 hrs. cyclohexanone (0.275 mL, 2.65 mmol)
was added and the reaction mixture was stirred for 18 hours. Purification of crude fluorohydrin
241 by flash chromatography (pentane:EtOAc – 60:40) afforded fluorohydrin 241 (0.068 g, d.r. =
5:1, 84% yield) as a white solid.
Data for fluorohydrin 241: 1H NMR (600 MHz, CDCl3): δ 7.92, 7.91, 7.78,
7.78, 6.29, 6.07, 5.37, 4.63, 3.51, 2.93, 2.92, 2.89, 2.80, 2.44, 2.41, 2.30,
2.25, 2.16, 2.01. 1.99, 1.87, 1.78, 1.71; 13C NMR (150 MHz, CDCl3): δ
215.9, 213.5, 167.1, 167.1, 134.9, 134.8, 131.7, 131.6, 124.1, 124.1, 89.9,
88.3, 69.9, 65.5, 51.8, 51.0, 43.3, 42.7, 32.4, 28.3, 27.8, 26.1, 25.4, 24.8; 19F NMR (470 MHz,
CDCl3): δ –156.0, –160.7
HRMS (EI+) calcd for [C16H17FNO4]+ 306.1136; observed 306.1135
Determination of enantiomeric excess of fluorohydrin 241
Using a 1:1 mixture of L-: D-proline, a racemic sample of fluorohydrin 241 was prepared. The
enantiomeric fluorohydrins were separated by chiral HPLC using a Lux® 3µm Amylose-1 column;
flow rate 0.40 mL/min; eluent: hexanes-iPrOH 90:10; detection at 254 nm; retention times = 26.70
min and 28.10 min (see chromatograms). The enantiomeric excess of the optically enriched
fluorohydrin 241 was determined using the same method (92% ee).
Preparation of aldol adduct 242
Following General Procedure B, a solution of phthalimidoacetaldehyde (0.050 g, 0.265 mmol),
NFSI (0.84 g, 0.265 mmol), L-proline (0.031 g, 0.265 mmol) and 2,6-lutidine (0.031 mL, 0.265
mmol) was stirred at 4°C in DMF (0.35 mL) for 15 hrs. Thiopyranone 206 (0.307 g, 2.65 mmol)
was added and the reaction mixture was stirred for 18 hours. Purification of crude fluorohydrin
242 by flash chromatography (pentane:EtOAc – 60:40) afforded fluorohydrin 242 (0.075 g, d.r. =
5:1, 87% yield) as a white solid.

141
Data for fluorohydrin 242: 1H NMR (600 MHz, CDCl3): δ 7.93, 7.92, 7.79,
7.79, 6.26, 6.11, 5.37, 4.78, 3.44, 3.25, 3.24, 3.16, 3.11, 3.09, 3.03, 2.99,
2.98, 2.85, 2.80, 2.79; 13C NMR (150 MHz, CDCl3): δ 212.8, 210.2, 167.1,
167.1, 135.1, 134.9, 131.6, 131.5, 124.3, 124.2, 89.6, 88.3, 70.1, 66.1,
54.6, 53.6, 45.7, 44.9, 34.6, 31.3, 30.7, 30.1; 19F NMR (470 MHz, CDCl3): δ –155.5, –158.5
HRMS (EI+) calcd for [C15H14FNO4S + NH4]+ 341.0966; observed 341.0938
Determination of enantiomeric excess of fluorohydrin 242
Using a 1:1 mixture of L-: D-proline, a racemic sample of fluorohydrin 242 was prepared. The
enantiomeric fluorohydrins were separated by chiral HPLC using a Lux® 3µm Amylose-1 column;
flow rate 0.70 mL/min; eluent: hexanes-iPrOH 80:20; detection at 254 nm; retention times = 15.99
min and 17.07 min (see chromatograms). The enantiomeric excess of the optically enriched
fluorohydrin 242 was determined using the same method (90% ee).
Preparation of aldol adduct 243
Following General Procedure C, a solution of pentanal (0.100 mL, 0.941 mmol), N(SCF3)Phth
(0.232 g, 0.941 mmol), L-proline (0.108 g, 0.941 mmol), and NaHCO3 (0.078 g, 0.941 mmol) was
stirred for 50 minutes at RT in DMSO (1.30 mL). Dioxanone 75 (0.044 mL, 0.270 mmol) in CH2Cl2
(5.6 mL) was stirred for 60 hrs. Purification of crude trifluoromethylthiohydrin 243 by flash
chromatography (pentane:Et2O – 9:1) afforded trifluoromethylthiohydrin 243 (0.082 g, d.r. = 6:1,
55 % yield) as a light yellow oil.
Data for syn- trifluoromethylthiohydrin 243: []D20 = -111.1° (c 3.0 in
CH2Cl2); IR (neat): = 3508, 2961, 1735, 1741, 1377, 1110, 861, 735 cm-
1; 1H NMR (600 MHz, CDCl3): δ 4.42 (dd, J = 9.1, 1.5 Hz, 1H), 4.30 (dd, J
= 17.6, 1.5 Hz, 1H), 4.08 (d, J = 17.6 Hz, 1H), 4.08 (m, 1H), 3.70 (dd, J =
2.2, 1.7 Hz, 1H), 3.46 (m, 1H), 2.00 (m, 1H), 1.87 (m, 1H), 1.48 (s, 3H), 1.42 (s, 3H), 0.95 (dd, J
= 7.4, 7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 213.5, 131.6 (q, J = 306.6 Hz), 101.7, 72.4,
71.4, 66.5, 47.0, 36.7, 23.8, 23.7, 20.3, 13.8; 19F NMR (470 MHz, CDCl3): δ –39.4
HRMS (EI+) calcd for [C12H19F3O4S + NH4]+ 334.1294; found 334.1303
Determination of relative stereochemistry for trifluoromethylthiohydrin 243

142
Following General Procedure F, trifluoromethylthiohydrin 244 was converted to
trifluoromethylthiohydrin 243. 1H NMR analysis revealed identical signals with 243 synthesized
using General Procedure C.
Determination of enantiomeric excess of fluorohydrin 243
Following General Procedure E, optically enriched and racemic samples of
trifluoromethylthiohydrin 243 were converted into their corresponding p-nitrobenzoyl diesters. The
enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a Lux® 3µm Amylose-
1 column; flow rate 0.50 mL/min; eluent: hexanes-iPrOH 92.5:7.5; detection at 254 nm; retention
time = 6.20 min and 9.35 min (see chromatograms). The enantiomeric excess of the optically
enriched p-nitrobenzoyl diester was determined using the same method (90% ee).
Preparation of aldol adduct 244
Following General Procedure C, a solution of pentenal (0.100 mL, 1.01 mmol), N(SCF3)Phth
(0.250 g, 1.01 mmol), L-proline (0.116 g, 1.01 mmol), and NaHCO3 (0.085 g, 1.01 mmol) was
stirred for 50 minutes at RT in DMSO (1.35 mL). Dioxanone XX (0.061 mL, 0.506 mmol) in CH2Cl2
(6.7 mL) was added and the reaction mixture was stirred for 60 hrs. Purification of the crude
trifluoromethylthiohydrin 244 by flash chromatography (pentane-Et2O 4:1) afforded
trifluoromethylthiohydrin 244 (0.103 g, d.r. = 6:1, 65 % yield) as a light yellow oil.
Data for syn-trifluoromethylthiohydrin 244: []D20 = -100.5° (c 1.28 in
CH2Cl2); IR (neat): = 3650, 3150, 1737, 1377, 1224, 1109 cm-1;1H NMR
(600 MHz, CDCl3): δ 5.79 (m, 1H), 5.18 (d, J = 17.0 Hz, 1H), 5.14 (d, J =
10.3 Hz, 1H), 4.42 (d, J = 9.0 Hz, 1H), 4.29 (d, J = 17.8 Hz, 1H), 4.12 (d, J
= 9.0 Hz, 1H), 4.07 (d, J = 17.8 Hz, 1H), 3.63 (s, 1H), 3.51 (dd, J = 10.1, 4.8 Hz, 1H), 2.78 (ddd,
J = 14.3, 9.9, 7.9 Hz, 1H), 2.68 (ddd, J = 14.3, 6.6, 5.1 Hz, 1H), 1.48 (s, 3H), 1.42 (s, 3H); 13C
NMR (150 MHz, CDCl3): δ 213.3, 134.4, 131.6 (q, J = 303.5 Hz), 118.7, 101.8, 72.3, 70.4, 66.4,
46.3, 38.9, 23.8, 23.6; 19F NMR (470 MHz, CDCl3): δ –39.7
HRMS (EI+) calcd for [C12H17F3O4S + NH4]+ 332.1138; found 332.1110
Determination of relative stereochemistry for trifluoromethylthiohydrin 244

143
Following General Procedure D, the trifluoromethylthiohydrin 244 was converted to carbacycle
262. NOE analysis of carbacycle 262 confirmed relative stereochemistry of
trifluoromethylthiohydrin 244.
Determination of enantiomeric excess of trifluoromethylthiohydrin 244
Following General Procedure F, trifluoromethylthiohydrin 244 was converted to
trifluoromethylthiohydrin 243. Following General Procedure E, optically enriched and racemic
samples of trifluoromethylthiohydrin 243 were converted into their corresponding p-nitrobenzoyl
diesters. The enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a Lux®
3µm Amylose-1 column; flow rate 0.50 mL/min; eluent: hexanes-iPrOH 92.5:7.5; detection at 254
nm; retention time = 6.92 min and 10.52 min (see chromatograms). The enantiomeric excess of
the optically enriched p-nitrobenzoyl diester was determined using the same method (91% ee).
Preparation of aldol adduct 245
Following General Procedure C, a solution of isovaleraldehyde (0.050 mL, 0.456 mmol),
N(SCF3)Phth (0.113 g, 0.456 mmol), L-proline (0.053 g, 0.456 mmol), and NaHCO3 (0.038 g,
0.456 mmol) was stirred for 50 minutes at RT in DMSO (0.61 mL). Dioxanone XX (0.027 mL,
0.228 mmol) in CH2Cl2 (3.04 mL) was added and the reaction mixture was stirred for 60 hrs.
Purification of the crude trifluoromethylthiohydrin 245 by flash chromatography (pentane:Et2O –
88:12) afforded trifluoromethylthiohydrin 245 (0.060 g, d.r. = 10:1, 42 % yield) as a colorless oil.
Data for syn-trifluoromethylthiohydrin 245: []D20 = -106.0° (c 2.15 in CH2Cl2);
IR (neat): = 3508, 2967, 173, 1101, 865 cm-1;1H NMR (600 MHz, CDCl3): δ
4.41 (dd, J = 9.0, 1.3 Hz, 1H), 4.30 (dd, J = 17.6, 1.5 Hz, 1H), 4.19 (d, J = 9.0
Hz, 1H), 4.08 (d, J = 17.6 Hz, 1H), 3.73 (dd, J = 2.3, 1.3 Hz, 1H), 3.35 (d, J =
5.5 Hz, 1H), 2.18 (m, 1H), 1.47 (s, 3H), 1.43 (s, 3H), 1.10 (dd, J = 5.8, 5.7 Hz, 3H); 13C NMR (150
MHz, CDCl3): δ 213.6, 131.6 (q, J = 305.0 Hz), 101.7, 72.2, 70.4, 66.5, 53.5, 33.1, 23.8, 23.7,
20.8, 19; 19F NMR (470 MHz, CDCl3): δ –38.6
HRMS (EI+) calcd for [C12H19F3O4S + NH4]+ 334.1294; found 334.1266
Preparation of aldol adduct 246
Following General Procedure C, a solution of 3-(4-methoxyphenyl)propanal (0.050 mL, 0.317
mmol), PhthN(SCF3) (0.078 g, 0.317 mmol), L-proline (0.037 g, 0.317 mmol), and NaHCO3 (0.027

144
g, 0.317 mmol) was stirred for 50 minutes at RT in DMSO (0.42 mL). Dioxanone 75 (0.019 mL,
0.228 mmol) in CH2Cl2 (2.11 mL) was added and the reaction mixture stirred for 60 hrs.
Purification of crude trifluoromethylthiohydrin 246 by flash chromatography (pentane:Et2O –
80:20) afforded trifluoromethylthiohydrin 246 (0.032 g, d.r. = 10:1, 56 % yield) as a yellow oil.
Data for syn-trifluoromethylthiohydrin 246: []D20 = -61.4° (c 1.6 in
CH2Cl2); IR (neat): = 3518, 1736, 1512, 1108, 863 cm-1;1H NMR (600
MHz, CDCl3): δ 7.15 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 4.40
(dd, J = 9.1, 1.3 Hz, 1H), 4.20 (dd, J = 17.6, 1.5 Hz, 1H), 4.01 (d, J =
17.6 Hz, 1H), 3.89 (d, J = 9.1 Hz, 1H), 3.80 (s, 3H), 3.58-3.62 (2H), 3.26 (dd, J = 13.7, 11.0 Hz,
1H), 3.21 (dd, J = 13.7, 5.5 Hz, 1H), 1.46 (s, 3H), 1.34 (s, 3H;13C NMR (150 MHz, CDCl3): δ 213.6,
158.6, 131.6 (q, J = 306.0 Hz), 130.5, 130.0, 114.1, 101.7, 72.4, 69.2, 66.4, 55.4, 48.2, 39.2, 23.8,
23.6; 19F NMR (470 MHz, CDCl3): δ –39.7
HRMS (EI+) calcd for [C17H21F3O5S + NH4]+ 412.1400.1340; found 412.1369
Determination of enantiomeric excess of trifluoromethylthiohydrin 246
Using sodium borohydride in methanol, optically enriched and racemic samples of
trifluoromethylthiohydrin 246 were converted into their corresponding diols. The enantiomeric
diols were separated by chiral HPLC using a Lux® 3µm Amylose-1 column; flow rate 0.50 mL/min;
eluent: hexanes-iPrOH 95:5; detection at 254 nm; retention time = 6.01 min and 8.49 min (see
chromatograms). The enantiomeric excess of the optically enriched diol was determined using
the same method (93% ee).
Preparation of aldol adduct 247
Following General Procedure C, a solution of 3-(5-methylfuran-2-yl)propanal (0.050 mL, 0.376
mmol), N(SCF3)Phth (0.093 g, 0.376 mmol), L-proline (0.043 g, 0.376 mmol), and NaHCO3 (0.032
g, 0.376 mmol) was stirred for 50 minutes at RT in DMSO (0.50 mL). Dioxanone 75 (0.023 mL,
0.188 mmol) in CH2Cl2 (2.50 mL) was added and the reaction mixture stirred for 60 hrs.
Purification of crude trifluoromethylthiohydrin 247 by flash chromatography (pentane:Et2O –
85:15) afforded trifluoromethylthiohydrin 247 (0.032 g, d.r. > 10:1, 46 % yield) as a colorless oil.

145
Data for syn-trifluoromethylthiohydrin 247: []D20 = -101.7° (c 1.0 in
CH2Cl2); IR (neat): = 3517, 2995, 1736, 1386, 1113, 737 cm-1;1H NMR
(600 MHz, CDCl3): δ 6.00 (d, J = 2.8 Hz, 1H), 5.86 (d, J = 2.8 Hz, 1H),
4.42 (dd, J = 9.0, 1.3 Hz, 1H), 4.26 (dd, J = 17.6, 1.3 Hz, 1H), 4.05 (d,
J = 17.6 Hz, 1H), 4.03 (d, J = 9.0 Hz, 1H), 3.76 (dd, J = 10.6, 5.1 Hz, 1H), 3.62 (dd, J = 2.3, 1.3
Hz, 1H), 3.31 (dd, J = 15.1, 10.6 Hz, 1H), 3.18 (dd, J = 15.1, 5.1 Hz, 1H), 2.26 (s, 3H), 1.48 (s,
3H), 1.40 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 213.6, 151.6, 149.9, 131.4 (q, J = 306.0 Hz),
108.6, 106.2, 101.7, 72.4, 70.3, 66.4, 45.8, 33.2, 23.8, 23.6, 13; 19F NMR (470 MHz, CDCl3): δ –
39.8
HRMS (EI+) calcd for [C15H19F3O5S + NH4]+ 386.1244; found 386.1227
Determination of enantiomeric excess of trifluoromethylthiohydrin 247
Following General Procedure E, optically enriched and racemic samples of
trifluoromethylthiohydrin 247 were converted into their corresponding p-nitrobenzoyl diesters. The
enantiomeric p-nitrobenzoyl diesters were separated by chiral HPLC using a Lux® 3µm Amylose-
1 column; flow rate 0.50 mL/min; eluent: hexanes-iPrOH 95:5; detection at 254 nm; retention time
= 20.83 min and 22.46 min (see chromatograms). The enantiomeric excess of the optically
enriched p-nitrobenzoyl diester was determined using the same method (93% ee).
Preparation of nucleoside analogue 259
To a stirred solution of fluorohydrins 241 (0.105 g, 0.344 mmol, 1.0 equiv) in MeCN (3.00 mL) at
-15˚C was added tetramethylammoniumtriacetoxyborohydride (0.453 g, 1.72 mmol, 5.0 equiv)
and acetic acid (0.190 mL, 3.44 mmol, 10 equiv). The resulting mixture was stirred 16 hours. The
reaction mixture was then diluted with a saturated solution of Rochelle salt and washed three
times with CH2Cl2. The organic layer was separated, dried over MgSO4, filtered and concentrated
under reduced pressure. The crude product S260 was purified by flash chromatography
(EtOAc:pentane – 70:30) to afford S259 as a white solid (0.076 g, 72%)
To a stirred solution of syn-diol-fluorohydrins S259 (0.076, 0.248 mmol, 1.0 equiv.) in MeCN (2.50
mL) was added InCl3 (0.014 g, 0.062 mmol, 0.25 equiv.) and the reaction mixture was stirred for
24 hours. The reaction mixture was diluted with CH2Cl2 and was washed with saturated sodium
bicarbonate solution. The organic layer was separated, dried over MgSO4, filtered, and

146
concentrated under reduced pressure. The crude product 259 was purified by flash
chromatography (EtOAc:pentane – 25:75) to afford 259 as a colorless oil (42.7 mg, 60%)
Data for nucleoside analogue 259: []D20 = +46.6 (c 0.38 in CH2Cl2); IR
(neat): = 3475, 2935, 1708, 1370, 720 cm-1; 1H NMR (600 MHz, CDCl3):
δ 7.88 (m, 2H), 7.77 (m, 2H), 6.13 (d, J = 5.0 Hz, 1H), 4.40 (ddd, J = 11.8,
5.0, 4.8 Hz, 1H), 4.03 (ddd, J = 10.6, 10.6, 4.1 Hz, 1H), 3.13 (d, J =11.9
Hz, 1H), 2.22 (m, 1H), 1.94 (m, 1H), 1.85 (m, 2H), 1.62 (dddd, J = 11.9, 11.9, 4.6, 3.2 Hz, 1H),
1.51 (m, 1H), 1.23 – 1.40(3H); 13C NMR (150 MHz, CDCl3): δ 169.1, 134.6, 132.1, 123.8, 84.4,
81.1, 75.3, 51.4, 31.7, 25.4, 24.0, 24.0
HRMS (EI+) calcd for C16H18NO4 [M + H+] 288.1230; found 288.1246
Determination of relative stereochemistry for nucleoside 259
Analysis of 2D NOESY of nucleoside 259 supported the indicated
stereochemistry
Preparation of nucleoside analogue 260
To a stirred solution of fluorohydrins 242 (0.097 g, 0.30 mmol, 1.0 equiv) in MeCN (3.00 mL) at -
15˚C was added tetramethylammoniumtriacetoxyborohydride (0.395 g, 1.50 mmol, 5.0 equiv) and
acetic acid (0.172 mL, 1.50 mmol, 10 equiv). The resulting mixture was stirred 16 hours. The
reaction mixture was then diluted with a saturated solution of Rochelle salt and washed three
times with CH2Cl2. The organic layer was separated, dried over MgSO4, filtered and concentrated
under reduced pressure. The crude product S260 was purified by flash chromatography
(EtOAc:pentane – 70:30) to afford S260 as a white solid (0.068 g, 70%)
To a stirred solution of syn-diol-fluorohydrins S260 (0.047, 0.143 mmol, 1.0 equiv.) in MeCN (1.43
mL) was added InCl3 (7.9 mg, 0.036 mmol, 0.25 equiv.) and the reaction mixture was stirred for
24 hours. The reaction mixture was diluted with CH2Cl2 and was washed with saturated sodium
bicarbonate solution. The organic layer was separated, dried over MgSO4, filtered, and
concentrated under reduced pressure. The crude product 260 was purified by flash
chromatography (EtOAc:pentane – 40:60) to afford 260 as a colorless oil (23.7 mg, 73%)

147
Data for nucleoside analogue 260: []D20 = +18.6 (c 2.37 in CH2Cl2); IR
(neat): = 3475, 2923, 1774, 1709, 1373, 719 cm-1; 1H NMR (600 MHz,
CDCl3): δ 7.88 (m, 2H), 7.77 (m, 2H), 6.13 (d, J = 4.9 Hz, 1H), 4.40 (ddd,
J = 11.5, 4.7, 4.7 Hz, 1H), 4.03 (ddd, J = 11.2, 11.2, 3.6 Hz, 1H), 3.35 (d,
J = 11.9 Hz, 1H), 2.98 (dd, J = 13.1 11.9 Hz, 1H), 2.82 (m ,2H), 2.69 (m, 1H), 2.50 (m, 1H), 2.10
(m ,1H), 1.74 (m , 1H); 13C NMR (150 MHz, CDCl3): δ 169.2, 134.8, 131.9, 124.0, 83.0, 80.2, 75.2,
51.3, 33.5, 27.6, 27.4
HRMS (EI+) calcd for C15H19N2O4S [M + NH4+] 323.1060; found 323.1037
Determination of relative stereochemistry for nucleoside 260
Analysis of 2D NOESY of nucleoside 260 supported the indicated
stereochemistry
Preparation of carbocycle 261
Following General Procedure D, to a stirred solution of 5-(methanesulfonyl)-1-phenyl-1H-tetrazole
(0.126 g, 0.560 mmol) in dry THF (0.70 mL) at -78oC was added dropwise a 1 M LiHMDS (0.560
mL, 0.560 mmol) and the resulting reaction mixture was stirred for 30 minutes. A solution of
fluorohydrin 223 (0.052 g, 0.224 mmol) in dry THF (0.45 mL) was then added dropwise and the
reaction mixture was allowed to stir for 5 hrs at -78°C. Purification of crude alkene S261 by flash
chromatography (pentane:EtOAc – 80:20) afforded alkene S261 (0.034 g, 65 % yield) as a
colorless oil. A mixture Grubbs II catalyst (2.9 mg) and alkene S261 (0.034 g, 0.148 mmol) in dry
toluene (5.91 mL) was purged with N2 for 45 minutes in a sealed reaction vessel and subsequently
heated to 80°C for 6 hrs. Purification of crude carbacycle 261 by flash chromatography
(pentane:EtOAc – 75:25) afforded carbacycle 261 (0.019 g, 63 % yield) as a white solid.
Data for carbacycle 261: []D20 = -32.8 (c 0.50 in CH2Cl2); 1H NMR (600 MHz,
CDCl3): δ 5.45 (s, 1H), 4.96 (dd, J = 46.8, 5.6 Hz, 1H), 4.61 (s, 1H), 4.51 (d, J =
13.5 Hz, 1H), 4.13 (m, 1H), 4.10 (d, J = 13.5 Hz, 1H), 2.71 (dd, J = 3.8, 1.5 Hz,
1H), 2.62 (ddd, J = 19.2, 5.6, 2.6 Hz, 1H), 2.31 (dd, J = 21.2, 19.2 Hz, 1H), 1.57
(s, 3H), 1.44 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 128.2, 117.8, 99.4, 88.5 (d, J
= 166.5 Hz), 66.1 (d, J = 2.7 Hz), 65.9 (d, J = 27.7 Hz), 63.7, 29.0, 27.4 (d, J = 22.1), 20.0.

148
HRMS (EI+) calcd for [C10H16FO3]+ 203.1078; found 203.1058
Preparation of carbocycle 262
Following General Procedure D, to a stirred solution of 5-(methanesulfonyl)-1-phenyl-1H-tetrazole
(0.066 g, 0.295 mmol) in dry THF (0.37 mL) at -78oC was added dropwise a 1 M LiHMDS (0.295
mL, 0.295 mmol) and the resulting reaction mixture was stirred for 30 minutes. A solution of
fluorohydrin 244 (0.042 g, 0.134 mmol) in dry THF (0.54 mL) was then added dropwise and the
reaction mixture was allowed to stir for 3 hrs at -78°C. Purification of the crude alkene S262 by
flash chromatography (pentane:Et2O – 80:20) afforded alkene S262 (0.023 g, 56 % yield) as a
colorless oil. A mixture Grubbs II catalyst (2.9 mg) and alkene S262 (0.021 g, 0.067 mmol) in dry
toluene (2.70 mL) was purged with N2 for 30 minutes in a sealed reaction vessel and subsequently
heated to 90°C for 6 hrs. Purification of the crude carbacycle 262 by flash chromatography
(pentane:EtOAc – 80:20) afforded carbacycle 262 (0.013 g, 74 % yield) as a white solid.
Data for carbacycle 262: []D20 = -54.3 (c 0.83 in CH2Cl2); IR (neat): = 3470,
1430, 1111, 879, cm-1;1H NMR (600 MHz, CDCl3): δ 5.52 (m, 1H), 4.62 (m, 1H),
4.48 (dd, J = 13.4, 2.6 Hz, 1H), 4.11 (m, 1H), 4.06 (d, J = 13.4 Hz, 1H), 3.75 (m,
1H), 3.00 (d, J = 18.8 Hz, 1H), 2.94 (d, J = 1.4 Hz, 1H), 2.21 (ddd, J = 18.8, 4.4,
2.1 Hz, 1H), 1.58 (s, 3H), 1.43 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 130.9 (q, J
= 307.7 Hz), 128.8, 119.6, 99.6, 67.6, 65.7, 63.8, 41.5 (q, J = 1.6 Hz), 29.0, 27.4, 20.1; 19F NMR
(470 MHz, CDCl3): δ –39.9
HRMS (EI+) calcd for [C11H16F3O3S]+ 285.0767; found 285.0780
Determination of relative stereochemistry for carbacycle 262
Analysis of 2D NOESY of carbacycle 262 supported the indicated
stereochemistry
Preparation of carbacycle 263
Following General Procedure D, to a stirred solution of 5-(methanesulfonyl)-1-phenyl-1H-tetrazole
(0.134 g, 0.60 mmol) in dry THF (0.75 mL) at -78oC was added dropwise a 1 M LiHMDS (0.60
mL, 0.60 mmol) and the resulting reaction mixture was stirred for 30 minutes. A solution of

149
fluorohydrin 239 (0.060 g, 0.30 mmol) in dry THF (1.20 mL) was then added dropwise and the
reaction mixture was allowed to stir for 5 hrs at -78°C. Purification of the crude alkene S263 by
flash chromatography (pentane:EtOAc – 90:10) afforded alkene S263 (0.031 g, 52 % yield) as a
white solid. A mixture Grubbs II catalyst (5.5 mg, 0.05 equiv.) and alkene S263 (0.026 g, 0.13
mmol) in dry toluene (5.30 mL) was purged with N2 for 30 minutes in a sealed reaction vessel and
subsequently heated to 80°C for 8 hrs. Purification of the crude carbacycle 263 by flash
chromatography (pentane:Et2O – 85:15) afforded carbacycle 263 (15.9 mg, 72 % yield) as a
colorless oil.
Data for carbacycle 263: []D20 = -111.3 (c 0.3 in CH2Cl2); IR (neat): = 3418,
2925, 2853, 1447, 1003 cm-1;1H NMR (600 MHz, CDCl3): δ 5.17 (m, 1H), 4.68
(dddd, J = 52.2, 13.8, 8.2, 5.6 Hz, 1H), 3.99 (m, 1H), 2.50 (m, 1H), 2.44 (m, 1H),
2.26 (m, 1H), 2.23 (m, 1H), 2.09 (d, J = 3.7 Hz), 2.06 (m, 1H), 1.95 (m, 1H), 1.87
(m ,1H), 1.81 (m ,1H), 1.39 (ddd, J = 13.2, 3.8, 3.8 Hz, 1H), 1.24 (ddd, J = 13.0,
3.8, 3.8 Hz, 1H), 1.13 (dd, J = 12.8, 3.7 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 136.9 (d, J = 1.8
Hz), 113.0 (d, J = 8.8 Hz), 90.6 (d, J = 171.2 Hz) , 71.2 (d, J = 19.2 Hz), 42.4 (d, J = 4.8 Hz), 35.4,
29.8 (d, J = 19.8 Hz), 28.7, 28.3; 19F NMR (470 MHz, CDCl3): δ –191.8
HRMS (EI+) calcd for C10H16FO4 171.1180; found 171.1154
Determination of relative stereochemistry for carbacycle 263
Analysis of 2D NOESY of carbacycle 263 supported the indicated
stereochemistry
Preparation of carbocycle 264
Following General Procedure D, to a stirred solution of 5-(methanesulfonyl)-1-phenyl-1H-tetrazole
(0.099 g, 0.44 mmol) in dry THF (0.55 mL) at -78oC was added dropwise a 1 M LiHMDS (0.440
mL, 0.440 mmol) and the resulting reaction mixture was stirred for 30 minutes. A solution of
fluorohydrin 240 (0.048 g, 0.220 mmol) in dry THF (2.20 mL) was then added dropwise and the
reaction mixture was allowed to stir for 3 hrs at -78°C. Purification of the crude alkene S264 by
flash chromatography (pentane:EtOAc – 85:15) afforded alkene S264 (0.029 g, 61 % yield) as a

150
off-white solid. A mixture Grela catalyst (5.9 mg, 0.10 equiv.) and alkene S264 (0.019 g, 0.088
mmol) in dry toluene (3.52 mL) was purged with N2 for 30 minutes in a sealed reaction vessel and
subsequently heated to 80°C for 8 hrs. Purification of the crude carbacycle 264 by flash
chromatography (pentane:EtOAc – 90:10) afforded carbacycle 264 (0.013 g, 71 % yield) as a
white solid.
Data for carbacycle 264: []D20 = -77.2 (c 0.53 in CH2Cl2); IR (neat): = 3424,
2924, 1426, 1290, 1088, 1057, 1021 cm-1;1H NMR (600 MHz, CDCl3): δ 5.27 (m,
1H), 4.64 (dddd, J = 52.7, 15.5, 9.3, 6.3 Hz, 1H), 4.00 (ddd, J = 16.3, 9.3, 7.1 Hz,
1H), 3.01 (dd, J = 12.9, 2.9 Hz, 1H), 2.85 (m, 1H), 2.63 (m, 2H), 2.55 (m, 1H),
2.53 (m, 1H), 2.43 (dd, J = 12.8, 12.4 Hz, 1H), 2.35 (m, 1H), 2.32 (m, 1H), 2.27
(m, 1H); 13C NMR (150 MHz, CDCl3): δ 138.9, 116.2 (d, J = 10.4 Hz), 90.3 (d, J = 172.2 Hz), 71.5
(d, J = 17.8 Hz), 45.3 (d, J = 5.6 Hz), 31.8, 30.8, 30.3 (d, J = 20.0 Hz); 19F NMR (470 MHz, CDCl3):
δ –191.4
HRMS (EI+) calcd for [C9H14FOS]+ 189.0744; found 189.0757
Determination of relative stereochemistry for carbacycle 264
Analysis of 2D NOESY of carbacycle 264 supported the indicated
stereochemistry
Synthesis of 2-fluoro-2-deoxy- D-altrose acetonide 273
The fluorohydrin 230 (0.150 g, 0.397 mmol, 1 equiv.) in MeCN (0.45 mL, 0.9 M) was added to a
stirred solution of Me4NBH(OAc)3 (0.520 g, 1.98 mmol, 5 equiv.) and AcOH (0.23 mL, 3.97 mmol,
10 equiv.) in MeCN (2.0 mL, 0.2 M) at -25°C and the resulting mixture was stirred for 24 hours.
The reaction mixture was then quenched by addition of a saturated aqueous solution of sodium
tartrate. The aqueous layer was removed and extracted four times with CH2Cl2, and the combined
organic layers were dried over MgSO4 and concentrated under reduced pressure. Purification of
the crude product by flash chromatography (pentane-EtOAc; 75:25) afforded the 1,3 syn-
fluorodiol (0.114 g, 76 %).

151
To a cold solution (0 °C) of the 1,3-syn-fluorodiol (0.060 g, 0.16 mmol) in THF (1.6 mL) was added
a solution of tetrabutylammonium fluoride in THF (1 M, 0.18 mL, 0.18 mmol), and the reaction
mixture was stirred for 30 minutes. The reaction mixture was then diluted with Et2O (2 mL) and
was washed with a solution of saturated aqueous ammonium chloride. The organic layer was
dried over MgSO4 and concentrated under reduced pressure. Purification of the crude product by
flash chromatography (CH2Cl2-MeOH 95:5) afforded the deprotected 1,3-syn-fluorotriol (32 mg,
91% yield). To a cold (0°C) solution of bis-(acetoxy)iodobenzene (35 mg, 0.108 mmol) and the
1,3-syn-fluorotriol (23 mg, 0.103 mmol), in CH2Cl2 (1.1 mL), was added 2,2,6,6-
tetramethylpiperidinyloxy (1 mg, cat.) and the reaction mixture was allowed to gradually warm to
room temperature and stirred for 5 hours and the reaction mixture was concentrated under
reduced pressure. Purification of the crude fluorohydrin 273 (dr 1:1) by flash chromatography
(pentane-EtOAc 5:5) afforded 273 (15 mg, 65% yield) as a clear oil.
Data for 2-fluoro-2-deoxy- L-altrose acetonide (273) []D20 = + 2.5 (c 0.83 in
CHCl3); IR (neat): = 3413, 2918, 1078, 1043 cm-1; 1H-NMR (600 MHz,
CDCl3): 5.12 (2H), 4.68 (2H), 4.38 (1H), 4.27 (2H), 4.16 (1H), 3.91 (7H),
3.27 (1H), 2.68 (1H), 2.30 (1H), 1.49 (12H); 13C-NMR (150 MHz, CDCl3): =
100.4, 100.3, 92.9, 91.8, 89.2, 86.5, 69.0, 68.7, 67.7, 67.6, 64.4, 62.6, 62.3, 59.4, 29.2, 29.1, 19.5,
19.4 19F-NMR (470 MHz, CDCl3): δ -194.9, -216.5 HRMS (ESI) m/z calcd for C9H16FO5 [M + H]+
223.0976, found 223.0965
Synthesis of 2-fluoro-2-deoxy-L-galactose (274)
To a cold (0 °C), stirred solution of fluorohydrin 230 (0.189 g, 0.500 mmol, 1.0 equiv.) in dry THF
(0.1 M) was added a solution of catechol borane in THF (1.1 mL, 1.0 M, 2.2 equiv.). The resulting
mixture was allowed to warm gradually to room temperature and was then stirred for an additional
45 minutes or until complete consumption of starting chlorohydrin was observed by TLC analysis.
The mixture was then diluted with MeOH (to 0.05 M) and a solution of saturated aqueous sodium
tartrate was added. The biphasic mixture was stirred vigorously for 2 hours, after which time the
aqueous layer was removed and extracted three times with Et2O. The combined organic layers
were dried over MgSO4, concentrated under reduced pressure, and the crude product was
purified by flash chromatography (pentane- EtOAc; 2:1) to yield the 1,3-anti-fluorodiol (0.056 g,
82 %)

152
To a cold solution (0 °C) of the 1,3-anti-fluorodiol (0.076 g, 0.20 mmol, 1 equiv.) in THF (2.0 mL)
was added a solution of tetrabutylammonium fluoride in THF (1 M, 0.22 mL, 0.22 mmol, 1.1
equiv.), and the reaction mixture was stirred for 4 hours. The reaction mixture was then diluted
with Et2O (2 mL) and was washed with a solution of saturated aqueous ammonium chloride. The
organic layer was dried over MgSO4 and concentrated under reduced pressure. Purification of
the crude product by flash chromatography (CH2Cl2-MeOH 95:5) afforded the deprotected 1,3-
anti-fluorotriol (41 mg, 91% yield) (See Pre274-XRD for X-ray). To a cold (0°C) solution of bis-
(acetoxy)iodobenzene (12.9 mg, 0.040 mmol) and the 1,3-anti-fluorotriol (9 mg, 0.04 mmol), in
CH2Cl2 (0.40 mL), was added 2,2,6,6-tetramethylpiperidinyloxy (1 mg, cat.) and the reaction
mixture was allowed to gradually warm to room temperature and stirred for 8 hours and the
reaction mixture was concentrated under reduced pressure. Purification of the crude fluorohydrin
(dr 1:0.1.0) by flash chromatography (pentane-EtOAc 5:5) afforded (4.6 mg, 51 % yield) as a clear
oil. The purified product (4.6 mg, 0.020 mmol) was then dissolved in CH2Cl2 (0.20 mL) and and
0.05 mL of TFA was added. The reaction mixture was left to stir for 24 hrs and the solvent was
removed under reduced pressure to give pure 274 (4.3 mg, 100%) as a colorless oil. The data for
274 matched those of previously reported for 2-fluoro-2-deoxy-D-galactose.190
Synthesis of fluorohydrin 226 and 2-fluoro-2-deoxy migalastat (275)
Following General Procedure A, a solution of 3-N-Cbz-aminopropanal (0.10 g, 0.483 mmol), NFSI
(0.152 g, 0.483 mmol), (R)-proline (0.056 g, 0.483 mmol) and NaHCO3 (0.041 g, 0.483 mmol)
was stirred for 3 hours at -10 °C in DMF (0.65 mL). Dioxanone 2 (0.039 mL, 0.322 mmol) in CH2Cl2
(5.8 mL) was stirred for 24 hours. Purification of the crude fluorohydrin 226 by flash
chromatography (pentane-EtOAc 3:1) afforded fluorohydrin 226 (0.056 g, 49 % yield) as a yellow
oil. 1H-NMR spectroscopic analysis of this material indicated that it exists as a complicated 1:1
mixture of fluorohydrin 226:hemiminals (1:1 mixture of diastereomers).
Following General Procedure C, the fluorohydrin 226 (0.051 g, 0.144 mmol) and Pd/C powder
were stirred in MeOH (1.4 mL) with bubbling in H2 gas for 18 hrs. The Pd/C was filtered off, the
solvent was removed under reduced pressure, and the crude product (dr 7:1)was purified with

153
flash chromatography (EtOAc: pentanes; 40:60) to give a white powder (0.024 g, 83 %). The
purified product (0.028 g, 0.139 mmol) was then dissolved in MeOH (1.4 mL) and 0.5 mL of 1 M
HCl was added. The reaction mixture was left to stir for 24 hrs and the solvent was removed under
reduced pressure to give pure 275(0.022 g, 97%).
Data for 2-fluoro-2-deoxy migalastat (275): []D20 = + 10.7 (c 1.88 in MeOH);
IR (neat): = 3307, 2952, 2464, 1406, 1111, 1055 cm-1; 1H NMR (500 MHz,
MeOD): 5.25 (dddd, J = 49.2, 10.8, 9.3, 5.6 Hz, 1H), 4.06 (m, 1H), 3.80 (m,
3H), 3.57 (ddd, J = 12.4, 5.6, 2.3 Hz, 1H), 3.40 (dd, J = 8.1, 5.3 Hz, 1H), 3.08
(m, 1H); 13C NMR (150 MHz, MeOD): 88.5 (J = 175.5 Hz), 73.1 (d, J = 17.7 Hz), 68.9 (d, J =
10.1 Hz), 62.0, 60.4, 45.0 (d, J = 32.6); 19F NMR (470 MHz, MeOD):δ -204.5
HRMS (ESI) m/z calcd for C6H13FNO3+ [M + H]+ 166.0874, found 166.0893
Determination of enantiomeric excess of fluorohydrin 226
Following General Procedure A, using a 1:1 mixture of L: D - proline, a racemic sample of the
fluorohydrin 226 was prepared. Following General Procedure B, the optically enriched and
racemic samples of fluorohydrin 226 (0.055 g, 0.155 mmol) were converted into their
corresponding cyclized products. These were then diacylated with R)-(+)-MTPA-OH (3 equiv.),
DIC (6 equiv.), pyridine (3 equiv.), and 4-dimethylaminopyridine (cat.) in CH2Cl2 (0.10 M). By
analysis of 19F NMR it was determined that the enantiomeric excess was 92 %.
Synthesis of (5R)-5-D-ribo-fluorophytosphingosine (276)
To a stirred solution of the fluorohydrin 221 (0.187 g, 0.50 mmol, 1.0 equiv.) in 5.0 mL of THF (0.1
M) was added to benzylamine (0.137 mL, 1.25 mmol, 2.5 equiv.) and glacial acetic acid (0.030 g,
0.50 mmol, 1.0 equiv.), and the resulting mixture was stirred at 20°C for 2 hours or until complete
conversion into the corresponding imine was accomplished (as determined by 1H-NMR
spectroscopic analysis of small samples removed from the reaction mixture). NaCNBH3 (0.080 g,
1.25 mmol, 2.5 equiv.) was then added and the mixture was stirred for a further 1 hour. The
reaction mixture was then diluted with CH2Cl2 to a concentration of 0.05 M and treated with water.
The layers were separated and the organic layer was washed with brine, dried (MgSO4), and
concentrated under reduced pressure. The crude product was purified by flash chromatography

154
(CH2Cl2-MeOH; 15:1) to afford the reductive amination product (0.206 g, 88 % yield). Pd/C (2 mg)
was added to a stirred solution of purified product (9.3 mg, 0.02 mmol, 1.0 equiv.) in 0.20 mL of
MeOH (0.1 M) under a H2 atmosphere. After 24 hrs the reaction was filtered, concentrated under
reduced pressure, and purified by flash column chromatography (CH2Cl2-MeOH; 20:1) to give the
debenzylated product (7 mg, 93 %). A solution of the debenzylated product (9 mg, 0.024 mmol)
in 0.25 mL MeOH (0.1 M) was added 0.05 mL of 1 M HCl and left for 24 hrs. The reaction mixture
was then concentrated under reduced pressure to afford pure 276 (8.7 mg, 98 %).
Data for (5R)-5-D-ribo-fluorophytosphingosine (276): []D20
= -4.5 (c 0.75 in DMSO-d6); IR (neat): = 3425, 2924, 1025,
1005, 822, 760, 614 cm-1; 1H NMR (600 MHz, DMSO-d6):
= 7.84 (br s), 4.69 (ddd, J = 47.5, 8.6, 4.7 Hz, 1H), 3.80 (dd,
J = 9.8, 2.7 Hz, 1H), 3.75 (dd, J = 11.2, 3.8 Hz, 1H), 3.58
(dd, J = 11.0, 9.4 Hz, 1H), 3.28 (dd, J = 29.6, 9.8 Hz, 1H), 1.77 (m, 1H), 1.56 (m, 1H), 1.26 (m, 26
H), 0.87 (dd, J = 6.8, 6.8 Hz, 3H); 13C NMR (150 MHz, DMSO-d6): = 91.8 (J = 173.1 Hz), 71.1
(d, J = 18.1 Hz), 67.8 (d, J = 5.0 Hz), 56.9, 54.5, 31.3, 30.4 (d, J = 21.4 Hz), 29.0, 29.0, 29.0, 29.0,
28.9, 28.9, 28.7, 24.8, 24.8, 22.1, 13.9; 19F NMR (470 MHz, DMSO-d6):δ -201.0
HRMS (ESI) m/z calcd for C18H39FNO3+ [M + H]+ 336.2908, found 336.2920

155
Figure S4.1. XRD structure of compound 219-XRD

156
Figure S4.2. XRD structure of compound 200-XRD

157
Figure S4.3. XRD structure of compound 228-XRD

158
Figure S4.4. XRD structure of compound 230-XRD

159
Figure S4.5. XRD structure of compound Pre274-XRD
160
Compound Reference 219-XRD 200-XRD 228-XRD 230-XRD Pre274-
XRD
Chemical Formula C23H23N2O10F C25H27O6Br2F C15H21O4F C32H43Br2FO7Si C9H17FO5
FW 506.43 602.28 284.32 746.57 224.22
Crystal System Orthorhombic Orthorhombic Orthorhombic Monoclinic Triclinic
Space group P212121 P212121 P212121 P21 P1
a/Å 7.8910(2) 5.5445(4) 5.60440(10) 12.6483(4) 9.3287(6)
b/Å 11.8417(3) 18.6627(13) 13.0932(3) 11.3201(4) 9.3568(5)
c/Å 24.9605(6) 24.3453(19) 20.2022(4) 13.4868(4) 25.3029(1
5)
α/˚ 90 90 90 90 91.911(2)
β/˚ 90 90 90 115.6810(10) 97.536(2)
γ/˚ 90 90 90 90 90.195(3)
Unit cell volume/Å3 2332.38(10) 2519.1(3) 1482.43(5) 1740.29(10) 2188.2(2)
Z 4 4 4 2 8
Temperature/K 150(2) 150(2) 296(2) 150(2) 150(2)
Radiation type Cu Kα Cu Kα Cu Kα Cu Kα Cu Kα
Absorption
coefficient, μ/mm-1
1.023 4.476 0.83 3.689 1.038

161
All Reflections 16537 16215 9652 25308 60788
Unique Reflections 4273 4631 2610 6336 14635
Flack parameter 0.03(4) 0.002(8) 0.07(5) 0.001(5) 0.04(3)
Rint 0.0337 0.0389 0.0205 0.0305 0.0532
Final R1 values
(I>2σ(I))
0.0281 0.0251 0.0368 0.0351 0.0436
Final wR(F2) values
(I>2σ(I))
0.0735 0.0642 0.1023 0.0916 0.1146
Final R1 values (all
data)
0.0286 0.0258 0.0388 0.0361 0.0437
Final wR(F2) (all data) 0.074 0.0648 0.1048 0.0927 0.1147
Goodness of fit 1.049 1.034 1.085 1.031 1.044

162
Chapter 5. A convenient late-stage flourination of pyridylic C-H bonds
The results presented in this chapter have been reported in part, see:
Meanwell, M.; Nodwell, M.; Martin, R. E.; Britton. R. Angew. Chem. Int. Ed. 2016, 55, 13244-
13248 and Meanwell, M.; Britton, R. Synthesis 2018, 50, 1228-1236.
Dr. Matthew Nodwell synthesized compound 343 and contributed insightful discussions to this
work.
5.1. Synthesis of heterobenzylic fluorides
Nitrogen-containing heteroaromatics are privileged scaffolds in both pharmaceutical and
agrochemical research.191–195 In fact, roughly 60% of FDA approved drugs incorporate a nitrogen-
containing heterocycle, of which pyridines, thiazoles, and imidazoles are among the most
common.191 The prevalence of these heterocycles in approved pharmaceuticals has inspired
significant advances in both their synthesis196,197 and functionalization198 that enable the fine-
tuning of potency and physicochemical properties of drug leads. Thus, through the careful choice
and positioning of substituents, features such as basicity (Figure 5.1; 277199,200), inter- and
intramolecular hydrogen bonding (Figure 5.1; 278201), and π-stacking interactions (Figure 5.1;
278201) can be optimized for ligand-target binding.202 Notably, owing to the small size of fluorine
atoms, the polarized nature of carbon-fluorine bonds and consequent impact on compound
lipophilicity, hydrofluorocarbon substituents (e.g., CF3 and CH2F) can have profound effects on
biological activity.1,4,6,9,203–205 In addition, fluorine is also an isostere for both hydrogen and
hydroxyl groups, and strategic fluorination at metabolically labile sites is a common tactic
employed to mediate enzymatic degradation and adjust pharmacokinetic properties.1,4,6,9,203–205
For example, installation of the aryl fluoride in the anti-cancer drug gefitinib (280) markedly
prevents metabolism at this position, resulting in an increased in vivo half-life (Figure 5.1).206
Owing to their relatively weak C-H bond strength, heterobenzylic C-H bonds are also prone to
metabolism (see 277 and 279207 Figure 5.1). Thus, strategic fluorination at these centers provides
unique opportunities to modulate basicity and metabolism. However, heterobenzylic fluorination,
especially at a late-stage in a synthesis or on structurally complex and functional group-rich drug
leads, remains a significant synthetic challenge.204,205

163
Figure 5.1. Primary sites of metabolism in omeprazole (277) and pioglitazone (279) and the effects of heterocycles and fluorine on physicochemical properties in omarigliptin (278) and gefitinib (280).
Over the past decade several late-stage C-H fluorination strategies have been reported
that enable the direct fluorination of benzylic C(sp3)–H bonds.208–212 These strategies are
particularly useful tools for lead optimization and also present opportunities for the 18F-labelling of
ligands for positron-emission tomography (PET) imaging.213 In contrast, however, there are very
few examples of heterobenzylic fluorination. In fact, rarely have C(sp3)–H fluorination reactions
been demonstrated on molecules that include a heterocycle. While this may relate to fundamental
incompatibilities between electrophilic fluorinating agents and nucleophilic heteroaromatics, there
is a clear need for robust reactions that engender the synthesis of heterobenzylic fluorides.
Previous reviews on late-stage C-H fluorination8,204,205,214 have included examples of
heterobenzylic monofluorination, however, there is no focused review on the topic. Here, we will
provide a survey of methods available for the synthesis of heterobenzylic fluorides, summarize
recent advances in this area and identify limitations that we hope will inspire further investigation.
5.1.1. Deoxyfluorination
The most common strategy to access heterobenzylic fluorides is through
deoxyfluorination.215–226 While these processes are not late-stage transformations and require
prior synthesis of a heterobenzylic alcohol, they have proven to be a valuable resource for
medicinal chemists. Here, reagents such as DAST,217–221 Deoxofluor,216 and Xtalfluor227 (Figure

164
5.2), have enabled transformation of a broad range of heterobenzylic alcohols into the
corresponding heterobenzylic fluorides. Mechanistically, these reagents function by activation of
the hydroxyl group followed by nucleophilic displacement by fluoride.
Figure 5.2 Common deoxyfluorination reagents
Deoxyfluorination at the heterobenzylic position in quinoline-,215 pyrazole-,216,217
pyrimidine-,218,219 thiophene-,220 imidazole-,221 thiazole-,222 pyridine-,223 and purine-containing226
heterocycles, among others224,225 have been described. Though deoxyfluorination is a robust and
widely used transformation, it is fundamentally limited by a reliance on the prior formation of a
heterobenzylic alcohol and can be complicated by the formation of by-products derived from
elimination or isomerization processes.214 In 2009, Gilmour and colleagues reported the
deoxyfluorination of quinine alkaloids using DAST in THF at -20 °C as part of a broader medicinal
chemistry campaign (Scheme 5.1).215 Here, products derived from both stereochemical inversion
282 and retention 283 were produced in low to modest yield. In addition, the ring-expanded
azepane 284 was produced via formation of an aziridinium intermediate.
Scheme 5.1. Deoxyfluorination of quinine led to inversion (282), retention (283), and rearrangement (284) products
A recent and particularly interesting example of this transformation was reported by Huisman and
co-workers, who introduced a heterobenzylic alcohol via the selective late-stage oxidation of 285
with microcytochrome P450 monooxygenase followed by deoxyfluorination, which provided access
to the fluoromethyl imidazole 286 (Scheme 5.2).228

165
Scheme 5.2. Late-stage enzymatic oxidation enabled deoxyfluorination
5.1.2. Halide-exchange reaction
Halide exchange reactions have also proven useful for the synthesis of heterobenzylic
fluorides, and have been described for purines,226 imidazoles,229 pyridines,230 quinolines,231–233
indoles,234 benzofurans,235 thiazoles236 and other heterocycles.237 However, as with
deoxyfluorination, the requirement for prior installation of a heterobenzylic halide limits the utility
of these processes and their suitability as late-stage modifications for lead optimization.
Scheme 5.3 Halide-exchange reaction with silver fluoride
An excellent example of halide exchange was reported as part of an investigation into the
cytostatic activity of 6-(fluoromethyl)purine nucleoside analogues. Here, Hocek and co-workers
converted the protected 6-(iodomethyl)purine nucleoside 287 into its fluoromethyl derivative 288
using silver fluoride in THF (Scheme 5.3).226 Silver fluoride, tetrabutylammonium fluoride (TBAF),
potassium fluoride, cesium fluoride, and hydrogen pyridinium fluoride (Olah reagent) are common
fluoride sources used in halide exchange reactions, and their efficient preparation as 18F isotopes
has provided opportunities for the synthesis of 18F-labelled radiotracers for PET imaging.232,236,237
For example, Sutherland and colleagues have reported a radiotracer for imaging of the
translocator protein. Here, K18F in MeCN under moderate heating rapidly converted 289 into its
18F-fluorinated derivative 290 in 38% radiochemical yield (Scheme 5.4).232

166
Scheme 5.4 Halide-exchange reaction for the synthesis of 18F radiotracer (290)
Recently, Yan and co-workers reported a late-stage iodination of 2-alkyl quinolines with iodine
and triphenylphosphine in the presence of sodium bicarbonate. Coupling this process with a
subsequent halide exchange reaction using silver(II) fluoride provided a means to access 2-
fluoroalkyl quinolines in excellent yield.233
5.1.3. Electrophilic fluorination of heterobenzylic anions
Figure 5.3. Common electrophilic fluorinating N-F reagents
The deprotonation of a heterobenzylic methyl or methylene by strong base, followed by
reaction with an electrophilic fluorinating agent (e.g., Figure 5.3) has also provided access to
heterobenzylic fluorides. Here, however, the substrate scope is often limited to molecules with
little additional functionality or relatively acidic heterobenzylic protons. Thus, to facilitate
deprotonation, the heterobenzylic position is often adjacent to a carbonyl,238 nitro,239 or sulfonate
group.240 Notably, this strategy can also provide access to difluorinated adducts by simply
employing an excess of base and fluorinating reagent.241 While much less common, fluorination
of unfunctionalized alkyl heterocycles have also been reported. For example, in 1991, Anders and
co-workers investigated the deprotonation of 4-alkyl pyridines using LDA followed by reaction with
various electrophiles, including NFSI, a process that delivered the corresponding pyridylic fluoride
in modest yield.242

167
Scheme 5.5. Heterobenzylic fluorination of camptothecin
As a notable additional example, Varchi and co-workers utilized this sequence in their fluorination
of the TES-protected natural product camptothecin (291, Scheme 5.5).243 Here, deprotonatation
with LiHMDS at -78 ˚C in THF, and subsequent addition of NFSI, afforded 292 in excellent yield.
5.1.4. Late-stage C-H bond fluorination
Following Groves and co-workers pioneering report on the fluorination of unactivated C(sp3)-H
bonds in 2012,244 several complimentary C-H fluorination strategies have been developed that
provide access to aliphatic,244,245 allyl,246 or benzylfluorides.208–212 Many of these processes take
advantage of the observation made by Sammis and Paquin that electrophilic fluorinating agents
such as NFSI and Selectfluor are capable of transferring a fluorine atom to an intermediate
carbon-centered radical owing to their low N-F bond dissociation energies (Selectfluor BDENF =
62.2 kcal/mole in H2O; NFSI BDENF = 63.5 kcal/mole in H2O).247 Unfortunately, as pointed out
earlier by Crugeiras,248 these reagents are often incompatible with basic amines. Thus, the
preponderance of reports on C-H fluorination that employ electrophilic fluorinating reagents lack
examples of nitrogen-containing heterocycles. Uniquely, the C(sp3)-H fluorination reaction
described by Groves relies instead on the in situ formation of a Mn(IV) species that is a competent
fluorine transfer agent. It has also been demonstrated that this process is amenable to 18F-
fluorination of benzylic C-H bonds213 including the two heterobenzylic C-H 18F-fluorination
reactions depicted in Scheme 5.6 (i.e., 294 and 295).

168
Scheme 5.6. Manganese-catalyzed C-H fluorination for the generation of 18F radiotracers
In 2015, Yi and co-workers described a transition metal-free radical benzylic fluorination using
potassium persulfate in combination with Selectfluor. It was also demonstrated that 8-
(fluoromethyl)quinoline (297) could be produced in 80% yield using this process (Scheme 5.7).212
The authors proposed that thermal decomposition of persulfate generates a sulfate radical that
abstracts a benzylic hydrogen atom. Subsequent fluorine atom transfer from Selectfluor249
provides the fluorinated adduct. Interestingly, an additional 1.5 equiv. of both potassium persulfate
and Selectfluor led to selective difluorination. The authors also noted a competitive benzylic
oxidation reaction that predominated at lower temperatures.
Scheme 5.7. Transition metal-free radical C-H fluorination
Transition metal catalyzed stereocontrolled fluorinations have also been employed for the
synthesis of enantiopure heterobenzylic fluorides.250–252 The Pd-catalyzed β-C(sp3)-H directed
fluorination reported by Yu and co-workers for the synthesis of enantiopure anti-β-fluoro-α-amino
acids provided 299 in 43 % yield and excellent diastereoselectivity (Scheme 5.8).250 Here, it was
proposed that the active catalyst (PdIILn) is formed in situ from the quinoline ligand coordinating
to Pd(TFA)2. A trans-substituted 5-membered palladacycle derived from C(sp3)-H activation

169
undergoes oxidative addition with Selectfluor to generate a Pd(IV) fluoride intermediate.
Reductive elimination then affords the pyridylic fluoride 299.
Scheme 5.8. Palladium-catalyzed diastereoselective C-H fluorination
In 2006, the palladium-catalyzed directed C-H fluorination of both heterobenzylic and aryl C-H
bonds was reported by Sanford and co-workers and represents the first example of metal-
catalyzed heterobenzylic fluorination (Scheme 5.9).253 Here, following generation of an
intermediate 5-membered palladacycle, oxidation by N-fluoropyridinium triflate to Pd(IV) and a
subsequent reductive elimination generates a new C-F bond. In 2012, Sanford and co-workers
described an important advance by demonstrating that the palladium-catalyzed C-H fluorination
could also be effected using nucleophilic fluoride (Scheme 5.9).254 Here, a hypervalent iodine
source is responsible for oxidation of Pd(II) to Pd(IV), which upon ligand exchange with silver
fluoride and subsequent reductive elimination generates 8-(fluoromethyl)quinolines 303.
Scheme 5.9 Sanford’s palladium-catalyzed C-H fluorination methods
In the same year, Freeze and collegues described the heterobenzylic fluorination of quinazoline
304 with Selectfluor in their synthesis of nicotinamide phosphoribosyltransferase inhibitors

170
(Scheme 5.10; 305).58 Given these mild conditions, this simple reaction may well prove useful for
the the late-stage fluorination of other heterobenzylic C-H bonds.
Scheme 5.10. Fluorination with Selectfluor
5.1.5. Monofluoromethylation of C(sp2)-H bonds
In 2012, Baran and co-workers reported an efficient and complementary synthesis of
heterobenzylic fluorides by demonstrating that these compounds could be accessed through the
direct functionalization of heteroaromatic C(sp2)–H bonds.88 Here, it was shown that a zinc
monofluoromethane sulphinate, in the presence of an oxidant, effected the direct
monofluoromethylation of xanthines, pyridines, quinoxalines , and pyrroles. Notably, in cases
where multiple monofluoromethylation events are possible, single products were observed with
excellent regioselectivity. Mechanistically, it was proposed that the reaction involves a Minisci-like
radical process, whereby a zinc monofluoromethane sulphinate generates a nucleophilic CH2F
radical. Through the use of alternative zinc sulphinate salts it was also shown that heterocycles
could be readily modified by addition of CF3, CF2H, and CH2CF3.
5.2. Late-stage fluorination of pyridylic C-H bonds
In 2014, we reported the direct fluorination of unactivated C-H bonds using the unique
combination of a decatungstate photocatalyst and N-fluorobenzenesulfonimide (NFSI).211,245,255
Considering our prior success in the fluorination of aliphatic,245,255 acyl,245 and benzylic211 C – H
bonds, we endeavoured to expand the reaction scope to include the fluorination of relatively weak
pyridylic C –H bonds (4-picoline BDE (CH3) 87 kcalmol-1 vs. toluene BDE (CH3) 90 kcalmol-1).256
As a significant complicating factor, however, Crugeiras has reported248 that several amine bases
react with NFSI at the sulfonyl group (and not the electrophilic fluorine) and decompose this
reagent.

171
entry conditions (pyridine)
solvent additive (1 equiv.)
t (h) Product (%yield)
1 A (306) MeCN HCl 24 308 (0) 2 A (306) MeCN none 24 308 (0) 3 A (306) MeCN-H2O HCl 24 308 (0) 4 A (307) MeCN-H2O TFA 36 309 (38) 5 A (306) MeCN AlF3 18 308 (26) 6 B (306) MeCN AlF3 18 308 (32) 7 B (306) MeCN none 18 308 (29) 8 C (306) MeCN none 18 308 (79) 9 D (306) MeCN none 15 308 (87)
10 D (306) benzene none 18 308 (81) 11 D (306) EtOAc none 18 308 (90) 12 D (307) MeCN none 18 310 (61)
Table 5.1 Fluorination of 4-alkylpyridines using NFSI.
In an effort to prevent such undesired sulfonyl transfer reactions, we initiated our investigation by
exploring the fluorination of N-oxides and various Brønsted acid salts of 4-ethylpyridine (306) and
4-isobutylpyridine (307) under our standard decatungstate/NFSI reaction conditions.245 As
summarized in Table 5.1 (entries 1–4), while we were unable to effect the fluorination of salts of
4-ethylpyridine, 4-isobutylpyridine·TFA was fluorinated selectively at the branched aliphatic
position (entry 4). While encouraging, this result suggested that protonation disfavours formation
of the intermediate pyridylic radical. The addition of Lewis acids of varying strength was also
explored and we were delighted and surprised to find that pyridylic fluorination of 4-ethylpyridine
occurred only in the presence of AlF3 (entry 5), a relatively weak and largely insoluble Lewis acid.
To assess the individual roles of the various reagents now present, the reaction was repeated
without the decatungstate or photoirradiation, and a similar outcome was observed (entry 6), thus
clearly indicating that the photocatalyst was not a participant. Furthermore, removal of AlF3 also
had no effect on the reaction outcome (entry 7). In fact, simply stirring 4-ethylpyridine with an
excess of NFSI in MeCN led cleanly to 4-(1-fluoroethyl)pyridine (308). Remarkably, only a single
related example of this operationally straightforward process has been reported; in 1996
DesMarteau found257 that 2- and 4-picoline react with the highly reactive fluorinating agent

172
(CF3SO2)2NF (fluorine plus detachment (FPD) = 200.5 kcalmol-1 in CH3CN vs. 229.6 kcalmol-1 for
NFSI)258 and base in CH2Cl2 to provide fluoromethyl-picolines, along with lesser amounts of
difluoromethyl and fluoropyridine products. Quite distinctly, in the present reaction, the addition of
base (NaHCO3 or Li2CO3) had little effect on the outcome (entries 8 and 9), and the reaction
proceeds equally well in C6H6 and EtOAc (entries 10 and 11) but gives poorer results in CH2Cl2
and THF. Optimally, simply heating a mixture of 4-ethylpyridine and NFSI in MeCN at 60°C
afforded 4-(1-fluoroethyl)pyridine (308) in excellent yield (87%, entry 9). Gratifyingly, reaction of
4isobutylpyridine (307) under the same conditions afforded the pyridylic fluorination product 310
(entry 12), with selectivity complimentary to that observed in the photocatalytic C-H fluorination of
the same substrate (entry 4).
Scheme 5.11. Fluorination of 4-(cyclopropylmethyl)pyridine (311) and a mechanistic proposal for the formation of 308 and decomposition products of NFSI
Our previous studies211 have shown that benzylic fluorination can involve radical
propagation by NFSI, and we questioned whether radical intermediates are involved in the
present pyridylic fluorination. When the reaction of 4-ethylpyridine with NFSI (Table 5.1, entry 9)
was repeated with addition of the galvinoxyl free-radical trap,259 the yield of 4-(1-
fluoroethyl)pyidine (306) was not affected. Moreover, fluorination of 4-(cyclopropylmethyl)pyridine
(311)260 with NFSI delivered only the pyridylic fluoride 312 in excellent yield (Scheme 5.11). The
absence of rearrangement products 313 or 314 suggests this reaction does not proceed via a
pyridylic radical or cation generated through a SET process.247 Further investigation of the
fluorination of 4-ethylpyridine provided additional insight. For example, when NFSI was replaced

173
with N-fluoropyridinium triflate, an equally reactive fluorinating agent,258 only unreacted starting
materials were recovered, thus suggesting that NFSI plays a critical role. Moreover, the major
byproducts produced in the 4-ethylpyridine fluorination were found to be benzenesulfonamide,
phenylsulfonyl fluoride, dibenzenesulfonimide and, to a lesser extent, products apparently derived
from phenylsulfonyl nitrene261 (Scheme 5.11). Based on these findings and the propensity of
nitrogen nucleophiles to react with NFSI at the sulfur,248 we posit that this pyridylic fluorination
involves a transient reaction between pyridine and NFSI to generate N-sulfonylpyridinium salt 315
and a nitrene.261 Loss of an α-methylene proton from the pyridinium then affords the resonance
stabilized tautomer 316, which can subsequently react with NFSI to provide the fluoroalkylpyridine
308. Phenylsulfonyl fluoride, which is generated in this reaction, may also play a role and serve
as the key sulfonylating reagent. While it is conceivable that this process could be initiated by
transient fluorination of the pyridine nitrogen, when a mixture of pyridine and NFSI was heated in
MeCN-d3, the expected N-fluoropyridinium salt was not detected by 1H or 19F NMR spectroscopy.
A corollary of this mechanistic proposal is that pyridylic fluorination is restricted to 2- and 4-
substituted pyridines, where deprotonation results in a resonance-stabilized carbanion. As
detailed below, 3-substituted pyridines fail to engage in productive reactions with NFSI.

174
Figure 5.4 Selective pyridylic fluorination of C-H Bonds
In order to evaluate the regioselectivity and scope of this reaction, we investigated the
fluorination of a structurally diverse collection of alkylpyridines (Figure 5.4). In general, the direct
C-H fluorination of 2- and 4-alkyl pyridines and annulated pyridines proceeds in good to excellent
yield. Notably, the reaction is highly tolerant to common functional groups, including amides,
esters, ketones, imidazolidinones, tertiary alcohols, and sulfonamides. While conceptually,
fluoroalkyl pyridines could be accessed from the deprotonation of alkyl pyridines using a strong
base, followed by reaction with NFSI, the reaction of 4-ethylpyridine with LDA followed by NFSI
optimally provided the fluoroethyl pyridine 308 in inferior yield (57%). Moreover, the reaction of

175
5,6,7,8-tetrahydroisoquinoline with LDA followed by NFSI delivered none of the fluorinated adduct
324, and these conditions only effected α-fluorination of carbonyl-containing substrates (see the
Supporting Information). Conversely, fluorination of 5,6,7,8-tetrahydroisoquinoline using our
optimized method (Table 5.1, entry 9) provided 324 in good yield and with complete selectivity for
fluorination at C5. Likewise, fluorination of 6,7-dihydro-5H-cyclopenta[b]pyridine262 afforded the
7-fluoro adduct 323 exclusively. This selectivity is consistent with the mechanistic proposal
outlined above and the fact that other 3-alkylpyridines (e.g., 321, 322, and 326) failed to undergo
fluorination. Importantly, this predictive selectivity creates opportunities for site-selective
fluorination in more complex or annulated pyridines263–268 (e.g., 323 – 325, 331, and 332), thus
making this method a potentially powerful tool for the late-stage modification of drug leads.
Substrates containing acid-sensitive functionalities were also well tolerated under the mild
reaction conditions, which enabled access to the tertiary alcohols 327 and 329. Interestingly,
pyridylic fluorination occurs in preference to fluorination at the α-carbon of ketones, amides, and
esters (e.g., 325, 331, and 332), which further highlights the enhanced acidity of the pyridylic
proton following activation (Figure 5.4) and the distinctness of this process from classical
deprotonation/fluorination strategies.
Scheme 5.12 Direct fluorination of the potent aldosterone synthase inhibitors 333 and 335
With an interest in exploring the reliability of this transformation for medicinal chemistry
purposes, the potent (IC50 < 10 nm) aldosterone synthase (CYP11B2) inhibitors 333 and 335 268
were also reacted with NFSI to afford the fluorinated analogues 334 and 336 in good yield and
with complete regioselectivity for the position indicated. It is notable that this simple modification
should have a measurable influence on both the acidity (ΔpKa ≈ 1.4)269 and metabolic stability of
these drug leads.

176
Scheme 5.13 Site-selective late-stage fluorination of pyridylic, benzylic, or aliphatic C-H bonds, contrasted with classical α-fluorination
To demonstrate the utility of this advance within the context of site-selective late-stage C-
H fluorination, we explored the fluorination of esters derived from ibuprofen and leucine (Scheme
5.13). When employing our photocatalytic decatungstate reaction conditions245 with the TFA salts
of 337 and 341, we observed complete selectivity for the expected211,245,255 benzylic or aliphatic
fluorination products 340 and 343, respectively. Conversely, simply heating the esters 337 and
341 with NFSI in MeCN delivered the corresponding pyridylic fluorides 338 and 342 in good yield.
Notably, treatment of 337 with LDA followed by NFSI provided complimentary selectivity and
afforded the α-fluoroester 339 as the exclusive product.

177
5.3. Conclusion
In summary, we have developed a convenient and regioselective fluorination reaction that
enables the late-stage fluorination of pyridylic C-H bonds. This reaction tolerates a wide variety
of functional groups and offers selectivity complementary to decatungstate-catalyzed C-H
fluorination. Importantly, this process provides a means to directly modulate basicity, improve
lipophilicity, and alter the metabolic profile of 2- and 4-alkylpyridines. Considering that pyridines
are prominent scaffolds in small-molecule drugs, this simple reaction may well serve as an
enabling tool in medicinal chemistry. Further optimization aimed at decreasing reaction times
(e.g., in continuous flow) may expand the utility of this process to include radiotracer synthesis
using [18F]NFSI270 (18F t1/2 = 110 min).
5.4. Experimental
General considerations
All reactions were carried out with commercial solvents and reagents that were used as
received. For extended NaDT photochemical reactions, degassing of the solvent was carried out
via several freeze/pump/thaw cycles. Flash chromatography was carried out with Geduran®
Si60 silica gel (Merck). Concentration and removal of trace solvents was done via a Büchi rotary
evaporator using dry ice/acetone condenser, and vacuum applied from an aspirator or Büchi V-
500 pump. All reagents and starting materials were purchased from Sigma Aldrich, Alfa Aesar,
TCI America, and/or Strem, and were used without further purification. All solvents were
purchased from Sigma Aldrich, EMD, Anachemia, Caledon, Fisher, or ACP and used without
further purification, unless otherwise specified.
Nuclear magnetic resonance (NMR) spectra were recorded using chloroform-d (CDCl3),
acetonitrile-d3 (CD3CN), or methanol-d4 (MeOD). Signal positions (δ) are given in parts per million
from tetramethylsilane (δ 0) and were measured relative to the signal of the solvent (1H NMR:
CDCl3: δ 7.26, CD3CN: δ 1.96, MeOD: δ 3.31; 13C NMR: CDCl3: δ 77.16, CD3CN: δ 118.26,
MeOD: δ 49.00). Coupling constants (J values) are given in Hertz (Hz) and are reported to the
nearest 0.1 Hz. 1H NMR spectral data are tabulated in the order: multiplicity (s, singlet; d, doublet;
t, triplet; q, quartet; quint, quintet; m, multiplet), coupling constants, number of protons. NMR
spectra were recorded on a Bruker Avance 600 equipped with a QNP or TCI cryoprobe (600
MHz), Bruker 500 (500 MHz), or Bruker 400 (400 MHz). Assignments of 1H and 13C NMR spectra

178
are based on analysis of 1H-1H COSY, HSQC, and HMBC spectra, where applicable. Methyl
propiolate was added to the crude reaction mixtures and used as an internal standard. Yields
were then calculated following analysis of 1H NMR spectra. phenylsulfonylfluoride,
benzenesulfonamide, and dibenzenesulfonimide were identified by comparison of their spectral
data to that reported previously.
High-resolution mass spectra were performed on an Agilent 6210 TOF LC/MS, Bruker
MaXis Impact TOF LC/MS, or Bruker micrOTOF-II LC mass spectrometer.
Preparative RP –HPLC was performed on an Agilent series 1200 instrument with a
Phenomenex Gemini-NX C18 preparative column (5 um 110 Å 50 x 30 mm, flow rate 15 mL/min).
General Procedure 1: pyridylic fluorination
To a solution of substrate in CH3CN (0.1 M substrate) was added N-fluorobenzenesulfonimide
(NFSI) (3.0 eq) and Li2CO3 (1.1 eq). The resulting reaction mixture was then heated to 60 °C for
18-24 h. The reaction mixture was cooled, diluted with CH2Cl2 and washed with saturated
NaHCO3 solution. The organic layer was dried (MgSO4), concentrated, and the crude reaction
product was purified by column chromatography on silica gel (as indicated).
Preparation of compound 308
Prepared following General Procedure 1: 4-ethylpyridine (50.0 mg, 0.466 mmol), NFSI (0.441 g,
1.40 mmol, 3.0 eq.), Li2CO3 (38.2 mg, 0.517 mmol, 1.1 eq.). Purified by flash chromatography
using ethyl acetate – pentane (50:50) as the eluent. Yield determined by analysis of 1H NMR
spectra of crude reaction product using an internal standard (methyl propiolate): 87 %
1H NMR (400 MHz, CD3CN): δ 8.74 (d, J = 6.6 Hz, 2H), 7.98 (d, J = 6.4 Hz, 2H),
5.99 (dq, J = 47.5, 6.7 Hz, 1H), 1.69 (dd, J = 24.5, 6.9 Hz, 3H); 13C NMR (150
MHz, CD3CN): δ 162.7 (d, J = 20.6 Hz), 142.3, 124.1 (d, J = 9.0 Hz) , 89.8 (d, J
= 171.8 Hz), 22.3 (d, J = 23.0 Hz); 19F NMR (470 MHz, CD3CN): –180.8
HRMS (EI+) calcd for [C7H9NF]+ 126.0714, found 126.0717
Preparation of compound 312

179
Prepared following General Procedure 1: 4-(cyclopropylmethyl)pyridine (30.0 mg, 0.225 mmol),
NFSI (0.213 g, 0.676 mmol, 3.0 eq.), Li2CO3 (18.3 mg, 0.248 mmol, 1.1 eq.). Purified by flash
chromatography using ethyl acetate – pentane (50:50) as the eluent. Yield determined by the
analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 73 %
1H NMR (500 MHz, CD3CN): δ 8.63 (d, J = 5.6 Hz, 2H), 7.42 (d, J = 5.5 Hz, 2H),
4.89 (dd, J = 47.8, 9.3 Hz, 1H), 1.31 (m, 1H), 0.73 (m, 2H), 0.64 (m, 1H), 0.58
(m, 1H); 13C NMR (150 MHz, CD3CN): δ 150.9, 150.0 (d, J = 23.0 Hz), 121.3 (d,
J = 7.4 Hz), 97.6 (d, J = 171.8 Hz), 17.0 (d, J = 28.2 Hz), 4.07 (d, J = 2.2 Hz),
2.92 (d, J = 9.1 Hz); 19F NMR (470 MHz, CD3CN) –174.2.
HRMS (EI+) calcd for [C9H11FN]+ 152.0870, found 152.0866
Preparation of compound 318
Prepared following General Procedure 1: 4-propylpyridine (28.0 mg, 0.231 mmol), NFSI (0.219 g,
0.693 mmol, 3.0 eq.), Li2CO3 (18.7 mg, 0.254 mmol, 1.1 eq.). Purified by flash chromatography
using ethyl acetate – pentane (50:50) as the eluent. Yield determined analysis of 1H NMR spectra
of crude reaction product using an internal standard (methyl propiolate): 86 %
1H NMR (500 MHz, CDCl3): δ 8.74 (d, J = 6.0 Hz, 2H), 7.73 (d, J = 6.0 Hz, 2H),
5.62 (ddd, J = 47.8, 7.6, 4.4 Hz, 1H), 1.99 (m, 2H), 1.06 (t, J = 7.5 Hz, 3H) 13C
NMR (150 MHz, CDCl3): δ 159.6 (d, J = 22.5 Hz), 140.8, 122.4 (d, J = 9.1 Hz),
91.9 (d, J = 180.3 Hz), 29.3 (d, J = 22.2 Hz), 8.24 19F NMR (470 MHz, CDCl3):
–186.5
HRMS (ESI+) calcd for [C8H11NF]+ 140.0870, found 140.0868
Preparation of compound 323
Prepared following General Procedure 1: 6,7-dihydro-5H-cyclopenta[b]pyridine (25.0 mg, 0.201
mmol), NFSI (0.199 g, 0.603 mmol, 3.0 eq.), Li2CO3 (16.3 mg, 0.221 mmol, 1.1 eq.). Purified by
flash chromatography using ethyl acetate – pentane (40:60) as the eluent. Yield determined by
analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 52 %

180
1H NMR (500 MHz, CDCl3): δ 8.55 (d, J = 4.8 Hz, 1H), 7.66 (d, J = 7.7 Hz, 1H),
7.26 (dd, J = 7.7, 4.9 Hz, 1H), 5.95 (ddd, J = 56.6, 6.4, 2.5 Hz, 1H), 3.18 (m, 1H),
2.90 (m, 1H), 2.41 (m, 2H) ; 13C NMR (150 MHz, CDCl3): δ 159.2 (d, J = 16.8 Hz),
148.4 (d, J = 2.3 Hz), 137.0 (d, J = 4.3 Hz), 133.2 (d, J = 1.6 Hz) 123.7(d, J = 3.3
Hz), 95.0 (d, J = 176.0 Hz), 30.3 (d, J = 23.3 Hz), 27.4; 19F NMR (470 MHz, CDCl3): –167.6
HRMS (EI+) calcd for [C8H9NF]+ 138.0714, found 138.0719
Preparation of compound 324
Prepared following General Procedure 1: 5,6,7,8-tetrahydroisoquinoline (50.0 mg, 0.375 mmol),
NFSI (0.356 g, 1.13 mmol, 3.0 eq.), Li2CO3 (30.5 mg, 0.413 mmol, 1.1 eq.). Purified by flash
chromatography using ethyl acetate – pentane (30:70) as the eluent. Yield determined by
analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 68 %
1H NMR (600 MHz, MeOD): δ 8.41 (s, 1H), 8.40 (d, J = 5.3 Hz, 1H), 7.47 (d, J =
5.3 Hz, 1H), 5.64 (ddd, J = 49.7, 5.3, 5.3 Hz, 1H), 2.90 (m, 1H), 2.79 (m, 1H),
2.14 (m, 1H), 2.00 (m, 1H), 1.89 (m, 1H); 13C NMR (150 MHz, MeOD): δ 149.0,
145.6 (d, J = 1.4 Hz), 143.4 (d, J = 18.0 Hz), 133.2 (d, J = 3.3 Hz), 122.7 (d, J =
5.0 Hz), 86.4 (d, J = 170.0 Hz), 28.2 (d, J = 20.0), 24.7, 17.3 (d, J = 5.2 Hz); 19F
NMR (CDCl3): –166.5.0
HRMS (EI+) calcd for [C9H11FN]+ 152.0870, found 152.0871
Preparation of compound 325
Prepared following General Procedure 1: 6,7-dihydroisoquinolin-8(5H)-one (30.0 mg, 0.204
mmol), NFSI (0.193 g, 0.612 mmol, 3.0 eq.), Li2CO3 (16.6 mg, 0.224 mmol, 1.1 eq.). Purified by
flash chromatography using ethyl acetate – pentane (50:50) as the eluent. Yield determined by
analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 51 %

181
1H NMR (600 MHz, CDCl3): δ 9.21 (s, 1H), 8.85 (d, J = 5.1 Hz, 1H), 7.57 (d, J
= 5.2 Hz, 1H), 5.75 (ddd, J = 48.8, 9.0, 4.2 Hz, 1H), 2.96 (ddd, J = 17.8, 6.9,
4.2 Hz), 1H), 2.67 (ddd, J = 17.5, 11.0, 4.9 Hz, 1H), 2.58 (m, 1H), 2.43 (m, 1H);
13C NMR (150 MHz, CDCl3) δ 195.0 (d, J = 1.7 Hz), 153.6, 149.6 (d, J = 18.9
Hz), 148.8, 126.0 (d, J = 4.3 Hz), 121.4 (d, J = 6.8 Hz), 86.7 (d, J = 178.7 Hz),
34.9 (d, J = 8.7 Hz), 29.1 (d, J = 20.7 Hz); 19F NMR (470 MHz, CDCl3): –178.7
HRMS (EI+) calcd for C9H9FNO+ 166.0663, found 166.0660
Preparation of compound 332
Prepared following General Procedure 1: ethyl 2-(6,7-dihydro-5H-cyclopenta[c]pyridine-6-
carboxamido)-4-methylpentanoate (20.0 mg, 0.0657 mmol), NFSI (0.062 g, 0.20 mmol, 3.0 eq.),
Li2CO3 (5.0 mg, 0.072 mmol, 1.1 eq.). Purified by flash chromatography using ethyl acetate –
pentane (60:40) as eluent. Yield determined by analysis of 1H NMR spectra of crude reaction
product using an internal standard (methyl propiolate): 69 %. Characterization data shown for one
diastereomer.
1H NMR (600 MHz, MeOD): δ 8.58 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 7.55 (d,
J = 5.0 Hz, 1H), 6.28 (d, J = 54.4, 5.7 Hz, 1H), 4.52 (t, J = 7.2 Hz, 1H), 4.21
(m, 2H), 3.49 (m, 2H) , 3.18 (m, 1H), 1.79 (m, 1H), 1.70 (m, 2H), 1.31 (t, J
= 7.4 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H), 1.00 (d, J = 6.6 Hz, 3H); 13C NMR
(150 MHz, MeOD) δ 174.3 (d, J = 2.8), 174.0, 150.4 (d, J = 18.2 Hz), 148.8,
147.3, 139.0 (d, J = 4.4 Hz), 121.2, 98.8 (d, J = 184.3 Hz), 62.4, 53.2 (d, J
= 20.2 Hz), 52.6, 41.3, 33.2 (d, J = 3.3 Hz), 26.1, 23.3, 21.8, 14.5; 19F NMR
(470 MHz, MeOD): –175.0
HRMS (EI+) calcd for [C17H24FN2O3]+ 323.1771, found 323.1778
Preparation of compound 327
Prepared following General Procedure 1: 8-vinyl-5,6,7,8-tetrahydroisoquinolin-8-ol (14.0 mg,
0.0799 mmol), NFSI (0.076 g, 0.24 mmol, 3.0 eq.), Li2CO3 (7.0 mg, 0.088 mmol, 1.1 eq.). Purified
by flash chromatography using ethyl acetate – pentane (80:20) as the eluent. Yield determined
by analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 58 %. Characterization data shown for one diastereomer.

182
1H NMR (500 MHz, CDCl3): δ 9.00 (s, 1H), 8.57 (d, J = 5.7 Hz, 1H), 7.48 (d, J
= 5.8, 1H), 6.04 (dd, J = 17.3, 10.6 Hz, 1H), 5.59 (ddd, J = 48.5, 8.4, 6.0 Hz,
1H), 5.39 (d, J = 10.6 Hz, 1H), 5.33 (d, J = 17.2 Hz, 1H), 2.47 (m, 1H), 2.31
(m, 2H), 2.08 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 153.1 (d, J = 19.2 Hz),
143.2, 141.1, 141.0 (d, J = 3.5 Hz), 138.9, 124.6 (d, J = 7.2 Hz), 117.1, 86.6
(d, J = 179.4 Hz), 71.4, 33.0 (d, J = 7.6 Hz), 25.2 (d, J = 19.3 Hz); 19F NMR (CDCl3) –175.0
HRMS (ESI+) calcd for [C11H13FNO]+ 194.0976, found 194.1006
Preparation of compound 329
Prepared following General Procedure 1: 8-(1-(phenylsulfonyl)-1H-indol-2-yl)-5,6,7,8-
tetrahydroisoquinolin-8-ol (95.0 mg, 0.235 mmol), NFSI (0.222 g, 0.705 mmol, 3.0 eq.), Li2CO3
(19.1 mg, 0.259 mmol, 1.1 eq.). Purified by flash chromatography using ethyl acetate – pentane
(65:35) as the eluent.. Yield determined by analysis of 1H NMR spectra of crude reaction product
using an internal standard (methyl propiolate): 34 %
1H NMR (600 MHz, MeOD): δ 8.47 (d, J = 5.4 Hz, 1H), 8.18 (s, 1H),
8.07 (d, J = 8.2 Hz 1H), 7.60 (m, 2H) 7.53, (t, J = 7.61, 1H), 7.31 (m,
5H), 7.11 (m, 2H), 5.85 (ddd, J = 50.3, 10.3, 5.6 Hz, 1H), 3.30 (m,
1H), 2.35 (m, 2H), 2.21 (m, 1H); 13C NMR (150 MHz, MeOD): δ
150.0, 149.3, 148.9, 147.9, 140.4, 140.2, 137.3, 135.1, 130.2, 130.2
(d, J = 17.4 Hz), 127.6, 127.0, 126.2, 125.3, 122.4, 121.3, 116.5, 89.1
(d, J = 172.5 Hz), 72.6, 36.2 (d, J = 11.6 Hz) , 27.2 (d, J = 18.8 Hz); 19F NMR (470 MHz, CD3CN)
–179.2.
HRMS (EI+) calcd for [C23H20FN2O3S]+ 423.1173, found 423.1170
Preparation of compound 328
Prepared following General Procedure 1: 3-(pyridin-4-yl)propyl benzoate (62.0 mg, 0.254 mmol),
NFSI (0.240 g, 0.762 mmol, 3.0 eq.), Li2CO3 (20.6 mg, 0.279 mmol, 1.1 eq.). Purified by flash
chromatography using ethyl acetate – pentane (50:50) as the eluent. Yield determined by
analysis 1H NMR spectra of crude reaction product using an internal standard (methyl propiolate):
70 %

183
1H NMR (600 MHz, CDCl3): δ 8.66 (d, J = 5.2 Hz, 2H), 7.98 (dd, J =
8.5, 1.3 Hz, 2H), 7.58 (tt, J = 7.6, 1.2 Hz, 1H), 7.45 (dd, J = 7.7, 7.7
Hz, 2H), 7.36 (d, J = 5.2 Hz, 2H), 5.72 (ddd, J = 47.9, 6.2, 6.2 Hz,
1H), 4.52 (m, 2H), 2.37 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 166.4,
149.8 (d, J = 21.9 Hz), 149.3, 133.4, 129.9, 129.7, 128.6, 120.3 (d,
J = 7.4 Hz), 89.8 (d, J = 176.3 Hz), 60.4 (d, J = 4.5 Hz), 36.2 (d, J = 22.8 Hz); 19F NMR (470 MHz,
CDCl3) –187.6
HRMS (EI+) calcd for [C15H15FNO2]+ 260.1081, found 260.1109
Preparation of compound 330
Prepared following General Procedure 1: 3-(pyridin-4-yl)propyl 4-nitrobenzoate (80.0 mg, 0.280
mmol), NFSI (0.265 g, 0.840 mmol, 3.0 eq.), Li2CO3 (22.7 mg, 0.308 mmol, 1.1 eq.). Purified by
flash chromatography using ethyl acetate – pentanes (50:50) as the eluent. Yield determined by
analysis of 1H NMR spectra of crude reaction product using an internal standard (methyl
propiolate): 64 %
1H NMR (500 MHz, MeOD): δ 8.54 (d, J = 6.0 Hz, 2H), 8.32 (d, J =
8.9 Hz, 2H), 8.14 (d, J = 8.9 Hz 2H), 7.49 (d, J = 9.0 Hz, 2H) 5.84
(ddd, J = 47.8, 7.7, 4.4 Hz, 1H), 4.56 (dd, J = 6.2, 6.2 Hz, 2 H),
2.45 (m, 2H); 13C NMR (150 MHz, MeOD): δ 165.8, 151.6 (d, J =
21.0 Hz), 150.4, 136.7, 131.8, 124.6, 121.7 (d, J = 8.5 Hz), 91.2
(d, J = 174.2 Hz), 62.4 (d, J = 4.9), 36.6 (d, J = 22.5); 19F NMR (470 MHz, MeOD) –188.6
HRMS (EI+) calcd for [C15H14FN2O4]+ 305.0932, found 305.0957
Preparation of compound 331
Prepared by following General Procedure 1: 3-(pyridin-4-yl)propyl 6-((4R,5S)-5-methyl-2-
oxoimidazolidin-4-yl)hexanoate (86.0 mg, 0.255 mmol), NFSI (0.241 g, 0.765 mmol, 3.0 eq.),
Li2CO3 (20.7 mg, 0.281 mmol, 1.1 eq.). Purified by flash chromatography using dichloromethane
– methanol (95:5) as the eluent. Yield determined by analysis of 1H NMR spectra of crude reaction
product using an internal standard (methyl propiolate): 63 %.

184
1H NMR (500 MHz, MeOD): δ 8.59 (d, J = 6.0 Hz, 2H), 7.46
(d, J = 6.0 Hz, 2H), 5.73 (ddd, J = 48.1, 7.5, 4.6 Hz 1H), 4.27
(m, 2H) 3.84 (m, 1H), 3.72 (m, 1H), 2.36, 2.30 (m, 3H), 2.25
(m, 1H), 1.64 (quint., J = 7.4 Hz, 2H), 1.52 (m, 2H), 1.40 (m,
2H), 1.13 (d, J = 6.5 Hz, 3H) ; 13C NMR (150 MHz, MeOD): δ
175.3, 166.2, 151.7 (d, J = 20.5 Hz), 150.5, 121.7 (d, J = 7.9
Hz), 91.1 (d, J = 174.0 Hz), 61.0 (d, J = 5.3 Hz), 57.4, 52.7, 36.7 (d, J = 22.3 Hz), 34.8, 30.7,
30.1, 27.1, 25.8, 15.6; 19F NMR (470 MHz, MeOD) –188.8
HRMS (EI+) calcd for [C18H27FN3O3]+ 352.2031, found 352.2059
Preparation of compound 334
Prepared following General Procedure 1: 6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile
(25.0 mg, 0.173 mmol), NFSI (0.164 g, 0.520 mmol, 3.0 eq.), Li2CO3 (14.0 mg, 0.190 mmol, 1.1
eq.). Purified by flash chromatography using ethyl acetate –pentane (40:60) as the eluent. Yield
determined by analysis of 1H NMR spectra of crude reaction product using an internal standard
(methyl propiolate): 60 %
1H NMR (600 MHz, CDCl3): δ 8.82 (s, 1H), 8.81 (s, 1H), 6.2 (ddd, J = 54.8, 6.7,
3.3 Hz, 1H), 3.28 (m, 1H), 3.03 (m, 1H), 2.51 (m, 2H); 13C NMR (150 MHz,
CDCl3) δ 150.5 (d, J = 16.8 Hz), 150.1, 150.0, 140.2, 114.1, 106.5, 93.5 (d, J
= 178.1 Hz), 31.3 (d, J = 22.7 Hz), 27.6; 19F NMR (CDCl3) –169.2
HRMS (ESI+) calcd for [C9H8FN2]+ 163.0666, found 165.0662
Preparation of compound 336
Prepared following General Procedure 1: 4-(2-fluoro-4-(trifluoromethyl)phenyl)-6,7-dihydro-5H-
cyclopenta[c]pyridine (30.0 mg, 0.107 mmol), NFSI (0.101 g, 0.321 mmol, 3.0 eq.), Li2CO3 (9.0
mg, 0.12 mmol, 1.1 eq.). Purified by flash chromatography using ethyl acetate – pentane (50:50)
as the eluent. Yield determined by analysis of 1H NMR spectra of crude reaction product using
an internal standard (methyl propiolate): 78 %

185
1H NMR (500 MHz, CDCl3): δ 8.71 (s, 1H), 8.57 (s, 1H), 7.61 (dd, J = 7.8,
1H) 1H), 7.56 (d, J = 7.8, 1H), 7.50 (d, J = 10.1, 1H), 5.97 (ddd, J = 54.1
Hz, 1H), 3.32 (m, 1H), 3.06 (m, 1H), 2.43 (m, 2H); 13C NMR (150 MHz,
CDCl3) δ 159.4 (d, J = 250.4 Hz), 148.5 (d, J = 17.2 Hz), 147.4, 146.0,
141.0 (d, J = 3.4 Hz), 133.0 (qd, J = 32.8, 8.0 Hz), 132.5(dq, J = 3.1, 1.5
Hz), 128.4, 127.3 (d, J = 15.4 Hz), 123.2 (qd, J = 272.6, 2.8 Hz), 121.6 (dq, J = 7.5, 3.9 Hz), 113.7
(dq, J = 25.8, 3.7 Hz), 94.9 (dd, J = 176.1, 2.9 Hz), 32.5 (d, J = 23.2 Hz), 28.1; 19F NMR (CDCl3)
–62.8, –113.3, –165.5
HRMS (EI+) calcd for [C15H11F5N]+ 300.0806, found 300.0802
Preparation of compound 338
Prepared following General Procedure 1: 3-(pyridin-4-yl)propyl 2-(4-isobutylphenyl)propanoate
(35.0 mg, 0.108 mmol), NFSI (0.102 g, 0.323 mmol, 3.0 eq.), Li2CO3 (9.0 mg, 0.12 mmol, 1.1 eq.).
Purified by flash chromatography using ethyl acetate – pentane (50:50) as the eluent. Yield
determined by analysis of 1H NMR spectra of crude reaction product using an internal standard
(methyl propiolate): 54 %.
1H NMR (500 MHz, MeOD): δ 8.53 (m, 2H), 7.27 (m, 4H), 7.15
(m, 2H), 5.38 (m, 1H), 4.23 (m, 2H), 3.73 (m, 1H), 2.46 (m, 2H),
2.20 (m, 2H), 1.83 (m, 1H), 1.47 (m, 3H), 0.88 (m, 6H); 13C NMR
(150 MHz, MeOD): δ 174.2, 149.7, 148.3, 140.1, 137.5, 128.6,
126.4, 119.8, 88.8, 59.3, 44.4, 44.1, 34.9, 29.6, 20.8, 16.7. 19F
NMR (470 MHz, MeOD) –188.8, –188.8.
HRMS (EI+) calcd for [C21H27FNO2]+ 344.2020, found 344.2021
Preparation of compound 340
To a solution of 337 ·TFA (25 mg, 0.057 mmol) in CH3CN (0.45 ml) and H2O (0.05 ml) was added
NFSI (0.054 g, 0.17 mmol, 3.0 eq) and NaDT (7.0 mg, 0.0029 mmol, 0.05 eq.) and the resulting
mixture was degassed via 3 x freeze/pump/thaw cycles. The reaction mixture was then irradiated
with long wave UV light (365 nm) for 36 h and monitored by 1H NMR spectroscopy. After this
time, the resulting suspension was diluted with CH2Cl2 and washed with saturated NaHCO3
solution. The organic layer was dried (MgSO4), concentrated, and the crude reaction product was

186
purified by column chromatography on silica gel using ethyl acetate – pentane (50:50) as the
eluent. Yield determined by analysis of 1H NMR spectra of crude reaction product using an internal
standard (methyl propiolate): 57 %.
1H NMR (600 MHz, CD3CN): δ 8.43 (d, J = 5.5 Hz, 2H), 7.37
(d, J = 7.8 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.07 (d, J = 5.3 Hz,
2H), 5.2 (dd, J = 47.6, 7.1 Hz, 1H), 4.06 (m, 2 H), 3.80 (q, J =
7.4 Hz, 1H), 2.55 (dd, J = 7.5, 7.5 Hz, 2H), 1.89 (m, 2H), 1.48
(d, J = 7.9 Hz, 3H), 1.31 (m, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.81
(d, J = 6.8 Hz, 3H); 13C NMR (150 MHz, CD3CN): δ 175.1, 151.9, 150.3, 142.2, 139.4 (d, J = 21.7
Hz), 128.5, 127.6 (d, J = 6.6 Hz), 125.0, 100.0 (d, J = 170.1 Hz), 64.3, 46.0, 34.9 (d, J = 22.6 Hz),
31.8, 29.9, 18.6, 18.5 (d, J = 6.5 Hz), 17.9 (m) ; 19F NMR (470 MHz, CD3CN) –178.9, –179.0.
HRMS (EI+) calcd for [C21H27FNO2]+ 344.2020, found 344.2019
Preparation of compound 342
Prepared following General Procedure 1: 341 (140. mg, 0.227 mmol), NFSI (0.215 g, 0.682 mmol,
3.0 eq.), Li2CO3 (18.4 mg, 0.250 mmol, 1.1 eq.). Purified by flash chromatography using ethyl
acetate – pentane (50:50) as the eluent. Yield determined by analysis of 1H NMR spectra of crude
reaction product using an internal standard (methyl propiolate): 73%.
1H NMR (500 MHz, MeOD): δ 8.51 (m, 2H), 7.81 (m, 2H), 7.68 (m, 2H),
7.39 (m, 4H) 7.31 (m, 2H), 5.68 (m, 1H), 4.39 (m, 2H), 4.31 (m, 2H),
4.22 (m, 2H), 2.23 (m, 2H), 1.73 (m, 1H), 1.61 (m, 2H), 0.97 (m, 6H);
13C NMR (150 MHz, MeOD): δ 174.6, 174.5, 158.7, 150.4, 145.2, 142.6,
128.8, 128.2, 126.2, 121.6, 120.9, 90.8, 67.9, 61.8, 54.0, 41.1, 36.9,
30.4, 25.9, 23.3, 21.7.; 19F NMR (470 MHz, MeOD) –188.5, 189.1
HRMS (EI+) calcd for [C29H32FN2O4]+ 491.2341, found 491.2358
Preparation of compound 343
To a solution of 341·2TFA (0.20 g, 0.42 mmol, 1.0 eq) in CH3CN (3.4 ml) and H2O (0.8 ml) was
added NFSI (0.40 g, 1.26 mmol,, 3.0 eq) and NaDT (51 mg, 0.021 mmol, 0.05 eq. ), and the
resulting mixture was degassed via 3 x freeze/pump/thaw cycles. The reaction mixture was
irradiated with long wave UV (365 nm) for 48 h. After this time, the resulting suspension was

187
diluted with CH2Cl2 and washed with 3 x 1.0 M NaOH. The organic layer was dried (MgSO4),
concentrated, and the crude reaction product was purified by preparative HPLC eluting with
solvent (A: 0.1 % TFA in H2O B: 0.1 % TFA in ACN) on a gradient of 2 % → 30 % solvent B over
15 minutes. Yield determined by analysis of 1H NMR spectra of crude reaction product using an
internal standard (methyl propiolate): 41 %.
1H NMR (600 MHz, D2O): δ 8.67 (d, J = 6.0 Hz, 2H), 7.96 (d, J =
6.1 Hz, 2H), 4.44 (dd, J = 8.9, 4.4 Hz 1H), 4.35 (t, J = 6.6 Hz, 2H)
3.08 (t, J = 7.9 Hz, 2H), 2.40 (ddd, J = 31.0, 15.8, 4.3 Hz, 1 H),
2.30 (ddd, J = 30.7 15.6, 9.0 Hz, 1H), 2.19 (m, 2H), 1.50 (dd, J =
22.5, 8.0 Hz, 6H) ; 13C NMR (150 MHz, D2O): δ 170.2, 163.8,
140.5, 127.3, 96.7, 66.2, 50.0, 40.1 (d, J = 21.4 Hz), 31.9, 27.5,
26.8 (d, J = 23.7 Hz), 24.7 (d, J = 24.0 Hz); 19F NMR (470 MHz, D2O) –138.7.
HRMS (EI+) calcd for [C14H22FN2O2]+ 269.1660, found 269.1649

188
Chapter 6. Direct heterobenzylic monofluorination, difluorination and trifluoromethylthiolation with dibenzenesulfonamide derivatives
The results presented in this chapter have been reported in part, see:
Meanwell, M.; Adluri, B.; Yuan, Z.; Newton, J.; Prevost, P.; Nodwell, M.; Friesen, C. M.;
Schaffer, P.; Martin, R. E.; Britton. R. Chem. Sci. 2018, 9, 5608-5613.
Other colleagues contributed to this work. Dr. Bharanishashank Adluri synthesized compounds
377, 378, 380, and 405. Dr. Zheliang Yuan developed the 18F procedure.
6.1. Direct functionalization of heterobenzylic C-H bonds
Figure 6.1. Heterobenzylic fluorides in discovery
The development of synthetic strategies that provide access to heterobenzylic fluorides is
of particular interest to medicinal chemistry and much success has been realized in
trifluoromethylation of heterocycles.271 However, introduction of heterobenzylic monofluoroalkyl
or difluoroalkyl groups remains largely reliant on cross coupling reactions80,81,84,272–277 or
deoxyfluorination of heterobenzylic alcohols278 and carbonyls,114,279 processes that require prior

189
functionalization. As a notable exception, Baran has reported innate C(sp2)–H functionalization of
heterocycles as a means to add each of the CHF2,87 CH2F88 and CF2CH3280
groups (e.g., 348 –
350) by employing the corresponding zinc sulphinate salts in Minisci-like radical addition
processes. Likewise, the introduction of difluoroacetates 351,76,281–285 difluoroacetamides 352286–
288 and difluorophosphonates 353289 has been accomplished via transition metal catalysis or
radical processes.290 Unfortunately, despite considerable advances in C(sp3)–H benzylic mono-
and difluorination, heterobenzylic C(sp3)–H fluorination278 or difluorination are largely unexplored
owing to fundamental incompatibilities between common fluorine transfer reagents (e.g., N-
fluorobenzensulfonimide (NFSI)) and nucleophilic heterocycles.248 Here, we demonstrate that
activation by transient sulfonylation is general for a range of alkylheterocycles and can be
extended to heterobenzylic difluorination and trifluoromethylthiolation. Collectively, these
convenient processes provide a platform for late-stage functionalization of drug leads and enable
direct 18F-fluorination of alkylheterocycles for the purpose of radiotracer synthesis for positron
emission tomography (PET) imaging.
entry hetero aromatic
solvent (conc. (M))
NFSI (equiv.)
temp. (°C)
product (ratio)
yield
1 354 MeCN (0.1) 3 60 357:360 (>20:1) 87 2 354 MeCN (0.5) 10 75 357:360 (1:1) 74 3 354 EtOAc (0.5) 6 75 357:360 (2:3) 82 4 355 MeCN (0.1) 3 65 358:361 (>20:1) 71 5 355 EtOAc (0.5) 10 75 358:361 (10:1) 81 6 356 MeCN( 0.1) 3 65 359:362 (1:3) 30 7 356 MeCN (0.3) 4 75 359:362 (1:8) 61 8 356 MeCN (0.5) 5 75 359:362 (1:10) 74
Table 6.1 Mono- and difluorination of 4-ethylpyridine (354) and alkyl quinolines 355 and 356
While examining the scope of the pyridylic fluorination reaction,291 we found that at
elevated temperatures (>65 C) small amounts of the corresponding difluoroalkyl derivatives were
formed and could be identified by a characteristic resonances at ~ -95 ppm in 19F NMR spectra
recorded on crude reaction mixtures. These observations prompted us to investigate the pyridylic

190
difluorination reaction as a complimentary process. As summarized in Table 6.1, heating a
solution of 4-ethylpyridine in MeCN with an excess of NFSI afforded exclusively the
monofluorinated adduct 357 at 60 C (entry 1). Increasing the reaction temperature above 80 C
(in a microwave) provided a complex mixture of products that included the corresponding
acetamide derived from displacement of fluoride by solvent (MeCN).211 However, when the
reaction was repeated at 75 C with a further increase in equivalents of NFSI, a ~1:1 mixture of
the mono- and difluorinated ethylpyridines 357 and 360 were produced in good yield (74%, entry
2) and were readily separable by flash column chromatography. Notably, for difluorination,
sequential activation by sulfonylation consumes 2 equivalents of NFSI and a further 2 equivalents
are required for fluorination. The additional excess of NFSI is required to offset its slow
decomposition over the course of the reaction (48 h). Several alternative solvents were evaluated
and a modest increase in yield was realized in EtOAc (entry 3). The fluorination of 4-ethylquinoline
(355) was also examined and we were pleased to find that heterobenzylic fluorination of this
alkylquinoline provided the monofluoroethyl product 358 in good yield (entry 4). However, despite
considerable effort, this substrate proved reluctant to undergo difluorination. Under more forcing
conditions (e.g., >90 C, microwave) decomposition occurred, and after 36 h at 75 C with a large
excess of NFSI only ~7% of the difluoroethyl quinoline 361 was produced (entry 5). Considering
the importance of both the mono- and difluoromethyl groups as bioisosteres,9 we also investigated
the fluorination of 4-methyl quinoline (356) and were surprised to find that difluorination
predominated even at low conversion, suggesting that here the second fluorination event is a
more facile process (entry 6). Increasing the equivalents of NFSI and reaction temperature (entry
7) as well as concentration (entry 8) ultimately provided the difluoromethyl quinoline 362 in
excellent yield. In both the mono- and difluorination of alkylquinolines 355 and 356, phenylsulfonyl
fluoride was observed as a by-product, suggesting that these reactions rely on activation of
quinoline through transient sulfonylation by NFSI.291 It is notable that this approach to
heterobenzylic fluorination is complimentary to the Minisci-like radical reactions described by
Baran, which favour trifluoromethylation at C7 or difluoromethylation at C2 of quinolines.87,88,280

191
Figure 6.2. Mono- and difluorination of pyridines, quinolines, pyrimidines, isoquinolines, quinazolines, and purines.
Encouraged by the susceptibility of 4-alkylquinolines 355 and 356 to undergo mono- or
difluorination, we explored the scope of these reactions with a broader range of heterocycles
including pyridines, isoquinolines, pyrimidines, quinazolines and purines. As summarized in

192
Figure 6.2, by simply modifying the equivalents of reagent and temperature, in several cases
mono- or difluorination could be effected selectively. For example, both mono- and difluoroalkyl
pyridines, quinazolines and purines could be produced in good yield following this straightforward
procedure (e.g., 357/383, 371/384, 372/385 and 373/386). As noted above, alkylquinolines were
reluctant to difluorinate but were monofluorinated in excellent yield providing 363 and 364.
Conversely, a series of methylquinolines were transformed directly into the corresponding
difluoromethylquinolines 374 – 380 in good yield. In addition to the obvious compatibility with
azaheterocycles, substituted aromatics (e.g., 377 – 382), esters (e.g., 364) and amides (e.g., 363)
were well tolerated. It is notable that 2,4-dimethylquinoline and 1-propyl-3,4-dimethlyisoquinoline
did not undergo fluorination using our standard reaction conditions. Here, we postulate that steric
hindrance from the adjacent alkyl group(s) prevents sulfonylation of the heterocycle by NFSI and
thus precludes fluorination. In several cases, complete separation of mono- and difluorinated
products by flash column chromatography proved challenging. Thus, while purified product could
be isolated this way, yields for these reactions were determined by analysis of NMR spectroscopic
data using an internal standard.

193
Figure 6.3. Trifluoromethylthiolation and chlorination of purines and quinazolines
Considering that sulfonyl transfer from NFSI is a key feature of this process (e.g., 389,
Figure 6.3),291 we examined a small collection of dibenzensulfonamide derivatives to explore their
potential in the direct heterobenzylic functionalization of alkylquinazolines and purines. As
depicted in Figure 6.3, we found that both trifluoromethylthiolation (e.g., 390 – 392 and 394 – 396)
and chlorination (e.g., 393) were facile processes. For example, 2- and 4-alkylquinazolines and
6-ethylpurine underwent heterobenzylic trifluoromethylthiolation using
N-trifluoromethylthiodibenzenesulfonimide (N(SCF3)SI).292 Surprisingly, we observed no
competing heteroaryl trifluoromethylthiolation292184 of quinazolines and purines, and attempts to
effect the equivalent transformation using trifluoromethylthiophthalimide, an electrophilic trifluoro-
methylthiolation reagent,184 delivered none of the expected trifluoromethylthiolated products. This
later result provides support for a mechanism involving activation by transient sulfonylation with
dibenzenesulfamide derivatives. Again, 2,4-disubstituted quinazolines failed to provide any
trifluoromethylthiolated product (e.g., 397 or 398) presumably due to steric hindrance impeding
sulfonylation of the heterocycle by N(SCF3)SI. Notably, this heterobenzylic

194
trifluoromethylthiolation293 reaction offers a unique opportunity to significantly alter lipophilicity
(Hansch hydrophobicity parameter = 1.44)184 and pKa of a drug lead.
Scheme 6.1 Late-stage mono- and difluorination, trifluoromethylthiolation of heterocycles
In an effort to further demonstrate the utility of this suite of transformations, we explored
the monofluorination, difluorination and trifluoromethylthiolation of quazodine (399),294 a cardiac
stimulant. As depicted in Scheme 6.1, each of these transformations proceeded smoothly and
provided access to the unique quazodine derivatives 400 – 402 in good to excellent yield. To
gauge the impact of heterobenzylic functionalization on relevant physiochemical properties, the
pKa, distribution coefficient (logD) at pH 7.4 and aqueous solubility of each compound was
measured. As summarized in Scheme 6.1, these transformations significantly affected each
property and provide a straightforward means to modulate lipophilicity and basicity. Likewise, the

195
peracetate 404 of the cytotoxic purine nucleoside analogue 403295 could be mono- or
difluorinated, affording the analogues 405 or 406, respectively, in good yield.
6.1.1. 18F-fluorination
Scheme 6.2. 18F-fluorination of heterocycle 407
Finally, we explored the direct 18F-fluorination of the annulated pyridine 407 to
demonstrate the additional utility of this transformation for rapidly generating radiotracers for
positron emission tomography (PET) imaging. We have previously exploited [18F]NFSI270 in the
direct radiofluorination of branched aliphatic amino acids296 and were pleased to find that simply
heating a solution of the annulated pyridine 407 and [18F]NFSI in MeCN at 75 C for 40 min
provided the 18F-labelled derivative 408 in good radiochemical conversion (RCC) and yield (RCY).
This streamlined heterobenzylic 18F-fluorination does not rely on prior functionalization or
sensitive reagents and thus offers certain advantages for the rapid generation of radiotracers for
PET imaging.
6.2. Conclusion
In summary, we demonstrate that transient sulfonylation of a range of nitrogen-containing
heterocycles enables direct heterobenzylic mono or difluorination using the bench stable
electrophilic fluorinating agent NFSI or radiofluorination with [18F]NFSI. Taking advantage of this
heterocycle activation process, both trifluoromethylthiolation and chlorination could also be
achieved using the corresponding dibenzenesulfonamide derivatives. This collection of late-stage
transformations should enable the rapid tuning of pKa and lipophilicty of heterocycle-containing
drug leads and provides a complimentary means to incorporate pharmaceutically relevant
bioisosteres (e.g., -CHF2, -CF2R and –CH(SCF3)R) as well as a method to rapidly generate 18F-
labelled imaging agents for PET imaging.

196
6.3. Experimental
General Considerations
All reactions were carried out with commercial solvents and reagents that were used as received.
Flash chromatography was carried out with Geduran® Si60 silica gel (Merck). Concentration and
removal of trace solvents was done via a Büchi rotary evaporator using dry ice/acetone
condenser, and vacuum applied from an aspirator or Büchi V-500 pump. All reagents and starting
materials were purchased from Sigma Aldrich, Alfa Aesar, TCI America, and/or Strem, and were
used without further purification. All solvents were purchased from Sigma Aldrich, EMD,
Anachemia, Caledon, Fisher, or ACP and used without further purification, unless otherwise
specified.
Nuclear magnetic resonance (NMR) spectra were recorded using chloroform-d (CDCl3) or
acetonitrile-d3 (CD3CN). Signal positions (δ) are given in parts per million from tetramethylsilane
(δ 0) and were measured relative to the signal of the solvent (1H NMR: CDCl3: δ 7.26, CD3CN:
δ 1.96; 13C NMR: CDCl3: δ 77.16, CD3CN: δ 118.26). Coupling constants (J values) are given in
Hertz (Hz) and are reported to the nearest 0.1 Hz. 1H NMR spectral data are tabulated in the
order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet), coupling
constants, number of protons. NMR spectra were recorded on a Bruker Avance 600 equipped
with a QNP or TCI cryoprobe (600 MHz), Bruker 500 (500 MHz), or Bruker 400 (400 MHz).
Assignments of 1H and 13C NMR spectra are based on analysis of 1H-1H COSY, HSQC, and
HMBC spectra, where applicable. Methyl propiolate or 4-fluorotoluene was added to the crude
reaction mixtures and used as an internal standard. Yields were then calculated following
analysis of 1H NMR spectra.
High-resolution mass spectra were performed on an Agilent 6210 TOF LC/MS, Bruker MaXis
Impact TOF LC/MS, or Bruker micrOTOF-II LC mass spectrometer.
General Procedure A: Heterobenzylic monofluorination
To a solution of substrate in CH3CN (0.1-0.25 M substrate) was added N-
fluorobenzenesulfonimide (NFSI) (3.0 equiv.) and Li2CO3 (1.1 equiv.). The resulting reaction
mixture was then heated to 65 °C and maintained at this temperature for 18-24 h. The reaction
mixture was cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic

197
layer was dried (MgSO4), concentrated and the crude reaction product was purified by column
chromatography on silica gel.
General Procedure B: Heterobenzylic difluorination
To a solution of substrate in CH3CN (0.25-0.50 M substrate) was added N-
fluorobenzenesulfonimide (NFSI) (5.0 equiv.) and Li2CO3 (5.0 equiv.). The resulting reaction
mixture was then either heated to 75 °C and maintained at this temperature for 48 h or heated to
125 °C and maintained at this temperature for 1 h in a microwave reactor. The reaction mixture
was cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer
was dried (MgSO4), concentrated and the crude reaction product was purified by column
chromatography on silica gel.
General Procedure C: Heterobenzylic trifluoromethylthiolation
To a solution of substrate in CH3CN (0.20-0.50 M substrate) was added N-
trifluoromethylthiobenzenesulfonimide (2.4 equiv.) and Li2CO3 (1.1 equiv.). The resulting reaction
mixture was then either heated to 75 °C and maintained at this temperature for 48 h or heated to
125 °C and maintained at this temperature for 1 h in a microwave reactor. The reaction mixture
was cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer
was dried (MgSO4), concentrated and the crude reaction product was purified by column
chromatography on silica gel.
Preparation of compound 363
Following General Procedure A, to a solution of N-pentyl-3-(quinolin-4-yl)propenamide (0.0485 g,
0.179 mmol) in 1.2 mL of CH3CN (0.15 M substrate) was added NFSI (0.170 g, 0.538 mmol, 3.0
equiv.) and Li2CO3 (0.015 g, 0.197 mmol, 1.1 equiv.). The resulting reaction mixture was then
heated to 65 °C and maintained at this temperature for 24 h. Purification of the crude 363 by flash
chromatography (pentane-ethyl acetate 4:6) afforded 363 (32.5 mg, 63%)
1H NMR (600 MHz, CDCl3): δ 8.95 (d, J = 4.5 Hz, 1H), 8.22 (d, J =
8.4 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.78 (dd, J = 8.4, 7.1 Hz, 1H),
7.63 ( dd, J = 8.4, 7.1 Hz, 1H), 7.55 (d, J = 4.5 Hz, 1H), 6.73 (ddd, J
= 46.8, 9.1, 2.6 Hz, 1H), 5.63 (br s, 1H), 3.32 (m, 2H), 2.90 (ddd, J
= 37.1, 15.4, 2.8 Hz, 1H), 2.78 (ddd, J = 17.5, 15.3, 9.1, 4.4 Hz 1H),
1.52 (m, 2H), 1.31 (m, 4H), 0.90 (dd, J = 7.3, 7.3 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 168.2,

198
149.8, 130.9, 130.2, 130.1, 129.9, 127.7, 124.4 (d, J = 5.1 Hz), 122.9, 116.9 (d, J = 11.4 Hz), 88.5
(d, J = 175.5 Hz), 44.5 (d, J = 24.8 Hz), 40.1, 29.3, 29.1, 22.5, 14.1; 19F NMR (470 MHz, CDCl3):δ
–183.6
HRMS (EI+) calcd for [C17H22FN2O]+ 289.1711, found 289.1724.
Preparation of compound 364
Following General Procedure A, to a solution of substrate (0.040 g, 0.070 mmol) in 0.35 mL of
CH3CN (0.20 M substrate) was added NFSI (0.066 g, 0.21 mmol, 3.0 equiv.) and Li2CO3 (6.0 mg,
0.077 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 65 °C and maintained
at this temperature for 24 h. Purification of the crude 364 by flash chromatography (pentane-ethyl
acetate 5:5) afforded 364 as a 1:1 mixture of diastereomers (37.0 mg, 90%).
IR (neat): = 2939, 2868, 1724, 1178, 905, 730 cm-
1; 1H NMR (600 MHz, CDCl3): δ 8.96 (1H), 8.17 (1H),
7.93 (1H), 7.76 (1H), 7.62 (1H), 7.53 (1H), 6.66 (1H),
4.84 (1H), 3.66 (3H), 2.99(1H), 2.34 (1H), 2.21 (1H),
1.96 (1H), 1.91-0.98 (25H), 0.94 (3H), 0.91 (3H),
0.65 (3H); 13C NMR (150 MHz, CDCl3): δ 174.9,
169.0, 150.5, 148.5, 144.1, 130.8, 129.7, 127.4, 124.5, 122.7, 117.3, 87.8, 75.8, 56.6, 56.2, 51.6,
42.9, 42.4, 42.1, 42.1, 40.6, 40.3, 36.0, 35.5, 35.1, 34.7, 32.4, 32.3, 31.2, 31.2, 28.3, 27.1, 26.8,
26.7, 26.5, 24.3, 23.5, 21.0, 18.4, 12.2; 19F NMR (470 MHz, CDCl3):δ –182.9, –182.9.
HRMS (EI+) calcd for [C37H51FNO4]+ 592.3797, found 592.3793.
Preparation of compound 365
Following General Procedure A, to a solution of 4-ethylquinoline (0.025 g, 0.159 mmol) in 1.60
mL of CH3CN (0.10 M substrate) was added NFSI (0.150 g, 0.478 mmol, 3.0 equiv.) and Li2CO3
(13.0 mg, 0.175 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 65 °C and
maintained at this temperature for 24 h. Purification of the crude 365 by flash chromatography
(pentane-EtOAc 7:3) afforded 365 (17.3 mg, 62%).

199
1H NMR (600 MHz, CDCl3): δ 8.95 (d, J = 4.5 Hz, 1H), 8.17 (d, J = 8.5 Hz, 1H), 7.90
(d, J = 8.5 Hz, 1H), 7.74 (dd, J = 8.2, 8.2 Hz, 1H), 7.59 (dd, J = 7.9, 7.9 Hz, 1H),
7.51 (d, J = 4.4 Hz, 1H), 6.33 (dq, J = 46.8, 6.5 Hz, 1H), 1.81 (dd, J = 24.3, 6.5 Hz,
3H) 13C NMR (150 MHz, CDCl3): δ 150.6 (d, J = 22.5 Hz), 148.4, 146.9 (d, J = 19.1
Hz), 130.7, 129.4, 127.0, 124.7 (d, J = 4.8 Hz), 122.9, 116.6 (d, J = 10.9 Hz) 87.8 (d, J = 172.3
Hz), 22.7 (d, J = 25.1 Hz); 19F NMR (470 MHz, CDCl3): δ –176.7
HRMS (ESI+) calcd for [C11H11FN]+ 176.0870, found 176.0841
Preparation of compound 366
A solution of 5-bromo-4-ethylpyrimidine (35.0 mg, 0.187 mmol, 1.0 eq.), NFSI (0.177 g, 0.56
mmol, 3.0 eq.), and Li2CO3 (15.2 mg, 0.206 mmol, 1.1 eq.) in 1.2 mL CH3CN was added to a
microwave vial. The vial was sealed in a CEM Discover LabMate microwave reactor and the
resulting mixture was heated to 120 °C (as monitored by a vertically focused infrared temperature
sensor) for 75 min. The reaction was concentrated under reduced pressure and the crude product
366 was purified by column chromatography on silica gel using ethyl acetate – pentanes (5:95)
as the eluent. Yield determined by analysis of 1H NMR spectra of crude reaction product using an
internal standard (methyl propiolate): 59 %.
1H NMR (500 MHz, CDCl3): δ 9.18 (s, 1H), 8.85 (s, 1H), 5.96 (dq, J = 47.0, 6.5
Hz, 1H), 1.73 (dd, J = 24.1, 6.5 Hz, 3H); 13C NMR (150 MHz, SO(CD3)2) δ 163.1
(d, J = 18.8 Hz), 160.0, 157.0, 119.2 (d, J = 2.5 Hz), 88.2 (d, J = 171.3 Hz), 18.6
(d, J = 24.6 Hz); 19F NMR (470 MHz, CDCl3) δ –179.5
HRMS (ESI+) calcd for [C6H7BrFN2]+ 204.9771, found 204.9798
Preparation of compound 367
A solution of 2-chloro-4-ethylpyrimidine (35.0 mg, 0.245 mmol, 1.0 eq.), NFSI (0.233 g, 0.74
mmol, 3.0 eq.), and Li2CO3 (20.0 mg, 0.270 mmol, 1.1 eq.) in 1.0 mL CH3CN was added to a
microwave vial. The vial was sealed in a CEM Discover LabMate microwave reactor and the
resulting mixture was heated to 150 °C (as monitored by a vertically focused infrared temperature
sensor) for 75 min. The reaction was concentrated under reduced pressure and the crude product
367 was purified by column chromatography on silica gel using ethyl acetate – pentanes (5:95)
as the eluent. Yield determined by analysis of 1H NMR spectra of crude reaction product using an
internal standard (methyl propiolate): 44 %.

200
1H NMR (500 MHz, CDCl3): δ 8.69 (d, J = 4.9 Hz, 1H), 7.48 (d, J = 5.0, 1H), 5.6 (dq,
J = 48.1, 6.8 Hz, 1H), 1.71 (dd, J = 24.6, 6.8 Hz, 3H); 13C NMR (150 MHz, SO(CD3)2)
δ 171.8 (d, J = 23.7 Hz), 161.7, 159.8 (d, J = 2.6 Hz), 115.9 (d, J = 6.8 Hz), 89.2 (d,
J = 170.6 Hz), 20.3 (d, J = 25.0); 19F NMR (470 MHz, CDCl3) δ –184.0
HRMS (ESI+) calcd for [C6H7ClFN2]+ 161.0276, found 161.0299
Preparation of compound 368
Following General Procedure A, to a solution of 4-(3-phenylpropyl)pyrimidine (0.025 g, 0.126
mmol) in 1.25 mL of CH3CN (0.10 M substrate) was added NFSI (0.238 g, 0.756 mmol, 6.0 equiv.)
and Li2CO3 (10.0 mg, 0.139 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to
125 °C and maintained at this temperature for 25 minutes in a microwave reactor. The yield for
368 (48%) was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-
fluorotoluene as an internal standard. Purification of the crude material by flash column
chromatography (pentane-ethyl acetate; 6:4) provided an analytical sample of 368.
IR (neat): = 2829, 1582, 1350, 1056, 750 cm-1;1H NMR (600 MHz, CDCl3): δ
9.16 (s, 1H), 8.78 (d, J = 4.8 Hz, 1H), 7.52 (d, J = 4.9 Hz, 1H), 7.29 (dd, J = 7.4,
7.4 Hz, 1H), 7.22 (d, J = 7.4 Hz, 1H), 7.20 (dd, J = 7.4, 7.4 Hz, 1H), 5.49 (ddd,
J = 48.4, 8.6, 2.3 Hz, 1H), 2.84 (dd, J = 8.3, 8.3 Hz, 2H), 2.39 (m, 1H), 2.22 (m,
1H); 13C NMR (150 MHz, CDCl3) δ 168.8 (d, J = 26.0 Hz), 158.1 (d, J = 2.9 Hz), 157.5 (d, J = 1.6
Hz), 140.6, 128.7, 128.6, 126.4, 117.0 (d, J = 8.3 Hz), 92.5 (d, J = 175.9 Hz), 36.9 (d, J = 21.7
Hz), 31.0 (d, J = 3.2 Hz); 19F NMR (470 MHz, CDCl3): δ –193.4
HRMS (EI+) calcd for [C13H14FN2]+ 217.1136, found 217.1128
Preparation of compound 369
Following General Procedure A, to a solution of 1-ethylisoquinoline (0.025 g, 0.159 mmol) in 1.60
mL of CH3CN (0.25 M substrate) was added NFSI (0.150 g, 0.478 mmol, 3.0 equiv.) and Li2CO3
(13.0 mg, 0.175 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 75 °C and
maintained at this temperature for 24 h. Purification of the crude 369 by flash chromatography
(pentane-EtOAc 85:15) afforded 369 (19.3 mg, 69%).

201
IR (neat): = 3055, 2989, 1624, 1586, 1378, 1055, 827 cm-1;1H NMR (600 MHz,
CDCl3): δ 8.52 (d, J = 5.7 Hz, 1H), 8.38 (d, J = 8.6 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H),
7.71 (dd, J = 7.8, 7.8 Hz, 1H), 7.65 (d, J = 5.5 Hz, 1H), 7.64 (dd, J = 7.6 Hz, 1H),
6.35 (dq, J = 48.0, 6.6 Hz, 1H), 1.93 (dd, J = 24.0 Hz, 6.6 Hz, 3H); 13C NMR (150
MHz, CDCl3) δ 157.8 (d, J = 19.7 Hz), 141.5, 136.9, 130.2, 127.6 (d, J = 1.7 Hz), 127.6, 126.2,
125.3 (d, J = 5.8 Hz), 121.6 (d, J = 1.7 Hz), 91.2 (d, J = 168.2 Hz), 20.5 (d, J = 23.6 Hz); 19F NMR
(470 MHz, CDCl3): δ –169.5
HRMS (EI+) calcd for [C11H11FN]+ 176.0870, found 176.0887
Preparation of compound 370
Following General Procedure A, to a solution of 1-propylisoquinoline (0.025 g, 0.146 mmol) in
0.60 mL of CH3CN (0.25 M substrate) was added NFSI (0.138 g, 0.438 mmol, 3.0 equiv.) and
Li2CO3 (12.0 mg, 0.161 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 75
°C and maintained at this temperature for 24 h. The yield for 28 (78%) was determined by analysis
of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard.
Purification of the crude material by flash chromatography (pentane-EtOAc 85:15) provided an
analytical sample of 370.
IR (neat): = 2957, 1604, 1499, 1119, 900 cm-1;1H NMR (600 MHz, CDCl3): δ
8.51 (d, J = 5.5 Hz, 1H), 8.37 (d, J = 8.3 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.70
(dd, J = 7.4, 7.4 Hz, 1H), 7.64 (d, J = 5.5 Hz, 1H), 7.62 (dd, J = 7.4, 7.4 Hz, 1H),
6.04 (ddd, J = 48.1, 8.6, 5.2 Hz, 1H), 2.35 (m, 1H), 2.20 (m, 1H), 1.12 (dd, J = 7.4,
7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 157.4 (d, J = 19.8 Hz), 141.5, 136.8, 130.1, 127.5,
127.4 (d, J = 1.4 Hz), 126.2, 125.2 (d, J = 6.2 Hz), 121.3 (d, J = 1.4 Hz), 96.2 (d, J = 173.4 Hz),
28.2 (d, J = 22.7 Hz), 9.9 (d, J = 5.7 Hz); 19F NMR (470 MHz, CDCl3): δ –178.0
HRMS (EI+) calcd for [C12H13FN]+ 190.1027, found 190.1027
Preparation of compound 371
Following General Procedure A, to a solution of 2-ethylquinazoline (0.025 g, 0.158 mmol) in 0.60
mL of CH3CN (0.25 M substrate) was added NFSI (0.100 g, 0.50 mmol, 3.0 equiv.) and Li2CO3
(0.026 g, 0.348 mmol, 2.2 equiv.). The resulting reaction mixture was then heated to 125 °C and
maintained at this temperature for 60 minutes in a microwave reactor. The yield for 371 (72%)

202
was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as
an internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 7.5:2.5) provided an analytical sample of 371.
IR (neat): = 2959, 1620, 1585, 1490, 1379, 1077, 765 cm-1;1H NMR (600
MHz, CDCl3): δ 9.45 (s, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.96 (m, 2H), 7.69 (dd, J
= 7.6, 7.6 Hz, 1H), 5.91 (dq, J = 48.5, 6.6 Hz), 1H), 1.85 (dd, J = 24.1, 6.7 Hz,
3H); 13C NMR (150 MHz, CDCl3): δ 164.5 (d, J = 19.6 Hz), 161.1, 150.2, 134.7,
128.7, 128.2, 127.4, 124.2, 91.2 (d, J = 173.9 Hz), 20.9 (d, J = 24.1 Hz); 19F NMR (470 MHz,
CDCl3): δ –177.1
HRMS (EI+) calcd for [C10H10FN2]+ 177.0823, found 177.0817
Preparation of compound 372
Following General Procedure A, to a solution of 6-bromo-4-ethylquinazoline (0.025 g, 0.106
mmol) in 1.1 mL of CH3CN (0.10 M substrate) was added NFSI (0.040 g, 0.127 mmol, 1.2 equiv.)
and Li2CO3 (9.0 mg, 0.117 mmol, 1.1 equiv.). The resulting reaction mixture was then left room
temperature and maintained at this temperature for 96 hours. Purification of the crude 372 by
flash chromatography (pentane-ethyl acetate; 85:15) afforded 372 (24.0 mg, 91%).
IR (neat): = 3975, 2254, 1498, 903, 726 cm-1;1H NMR (600 MHz, CDCl3): δ
9.32 (s, 1H), 8.01 (dd, J = 9.0, 2.0 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H), 6.18 (dq,
J = 48.1, 6.9 Hz, 1H), 1.91 (dd, J = 24.2, 6.6 Hz, 3H); 13C NMR (150 MHz,
CDCl3): δ 166.3 (d, J = 21.3 Hz), 154.5, 149.8, 137.7, 131.2, 127.6 (d, J = 9.2
Hz), 123.3, 122.1, 91.2 (d, J = 172.1 Hz), 20.7 (d, J = 23.1 Hz); 19F NMR (470 MHz, CDCl3): δ –
174.2
HRMS (EI+) calcd for [C10H9BrFN2]+ 254.9928, found 254.9949
Preparation of compound 373
Following General Procedure A, to a solution of 6-ethyl-9-methyl-9H-purine (0.015 g, 0.093 mmol)
in 0.40 mL of CH3CN (0.25 M substrate) was added NFSI (0.096 g, 0.303 mmol, 3.3 equiv.) and
Li2CO3 (8.2 mg, 0.111 mmol, 1.2 equiv.). The resulting reaction mixture was then heated to 75
°C and maintained at this temperature for 36 hours. Purification of the crude 373 by flash
chromatography (CH2Cl2-MeOH 96:4) afforded 373 (11.1 mg, 70%).

203
IR (neat): = 2978, 2262, 1039, 832 cm-1;1H NMR (600 MHz, CD3CN): δ 8.92
(s, 1H), 8.21 (s, 1H), 6.18 (dq, J = 47.3, 6.6 Hz, 1H), 1.81 (dd, J = 24.7, 6.7 Hz,
3H); 13C NMR (150 MHz, CD3CN): δ 157.5 (d, J = 19.4 Hz), 153.7, 152.8, 147.8,
131.7, 88.8 (d, J = 168.8 Hz), 30.2, 20.2 (d, J = 24.4 Hz); 19F NMR (470 MHz,
CD3CN): δ –176.6
HRMS (EI+) calcd for [C8H10FN4]+ 181.0884, found 181.0890
Preparation of compound 374
Following General Procedure B, to a solution of 4-methylquinoline (0.030 mL, 0.226 mmol) in 0.45
mL of CH3CN (0.50 M substrate) was added NFSI (0.357 g, 1.13 mmol, 5.0 equiv.) and Li2CO3
(0.084 g, 1.13 mmol, 5.0 equiv.). The resulting reaction mixture was then heated to 75 °C and
maintained at this temperature for 48 hours. The yield for 374 (68%) was determined by analysis
of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard.
Purification of the crude material by flash column chromatography (pentane-ethyl acetate; 6:4)
provided an analytical sample of 374.
1H NMR (600 MHz, CDCl3): δ 9.03 (d, J = 4.3 Hz, 1H), 8.21 (d, J = 8.5 Hz, 1H),
8.10 (d, J = 8.5 Hz, 1H), 7.80 (dd, J = 7.5, 7.5 Hz, 1H), 7.67 (dd, J = 7.5 Hz, 1H).
7.60 (d, J = 4.3 Hz, 1H), 7.17 (t, J = 54.6 Hz, 1H) ; 13C NMR (150 MHz, CDCl3): δ
150.1, 148.8, 138.0 (t, J = 21.8 Hz), 130.6, 130.1, 128.0, 124.3 (t, J = 3.1 Hz),
123.4, 118.1 (t, J = 7.7 Hz), 113.4 (t, J = 241.0 Hz); 19F NMR (470 MHz, CDCl3): δ –115.1
HRMS (EI+) calcd for [C10H8F2N]+ 180.0619, found 180.0600.
Preparation of compound 375
Following General Procedure B, to a solution of 6-bromo-4-methylquinoline (0.030 g, 0.136 mmol)
in 0.30 mL of CH3CN (0.50 M substrate) was added NFSI (0.214 g, 0.679 mmol, 5.0 equiv.) and
Li2CO3 (0.050 g, 0.679 mmol, 5.0 equiv.). The resulting reaction mixture was then heated to 75
°C and maintained at this temperature for 48 hours. The yield for 375 (64%) was determined by
analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard.
Purification of the crude material by flash column chromatography (pentane-ethyl acetate; 6:4)
provided an analytical sample of 375.

204
1H NMR (600 MHz, CDCl3): δ 9.03 (d, J = 4.3 Hz, 1H), 8.26 (s, 1H), 8.08 (d, J =
, 9.0 Hz, 1H), 7.88 (dd, J = 9.1, 1.8 Hz, 1H), 7.61 (d, J = 4.5 Hz, 1H), 7.10 (t, J
= 54.4 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 150.4, 147.4, 137.2 (t, J = 22.4
Hz), 133.7, 132.2, 126.0, 125.3 (t, J = 2.7 Hz), 122.4, 119.0 (t, J = 7.5 Hz), 113.2
(t, J = 241.1 Hz); 19F NMR (470 MHz, CDCl3):δ –114.9
HRMS (EI+) calcd for [C10H7BrF2N]+ 257.9724, found 257.9711
Preparation of compound 376
Following General Procedure B, to a solution of 4-methyl-6-phenylquinoline (0.046 g, 0.210 mmol)
in 0.42 mL of CH3CN (0.50 M substrate) was added NFSI (0.331 g, 1.05 mmol, 5.0 equiv.) and
Li2CO3 (0.078 g, 1.05 mmol, 5.0 equiv.). The resulting reaction mixture was then heated to 75 °C
and maintained at this temperature for 48 hours. The yield for 376 (41%) was determined by
analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard.
Purification of the crude material by flash column chromatography (pentane-ethyl acetate; 1:1)
provided an analytical sample of 376.
1H NMR (600 MHz, CDCl3): δ 9.06 (d, J = 4.4 Hz, 1H), 8.31 (d, J = 8.8 Hz, 1H),
8.29 (s, 1H), 8.10 (dd, J = 8.8, 1.9 Hz, 1H), 7.77 (d, J = 7.7 Hz, 2H), 7.66 (d, J =
4.3 Hz, 1H), 7.57 (dd, J = 7.7, 7.7 Hz, 2H), 7.48 (dd, J = 7.7, 7.7 Hz, 1H), 7.25
(t, J = 54.6 Hz, 1H) ; 13C NMR (150 MHz, CDCl3): δ 150.0, 148.1, 140.8, 140.3,
138.0 (t, J = 21.6 Hz), 130.9, 129.9, 129.2, 128.3, 127.8, 124.5 (t, J = 2.9 Hz), 121.2, 118.5 (t, J
= 7.8 Hz), 113.5 (t, J = 240.8 Hz); 19F NMR (470 MHz, CDCl3):δ –115.0
HRMS (EI+) calcd for [C16H12F2N]+ 256.0932, found 256.0930
Preparation of compound 377
Following General Procedure B, to a solution of 4-(methyl)-6-(4-fluorophenyl)quinoline (0.035 g,
0.147 mmol) in 0.29 mL of CH3CN (0.50 M substrate) was added NFSI (0.232 g, 0.735 mmol, 5.0
equiv.) and Li2CO3 (0.059 g, 0.735 mmol, 5.0 equiv.). The resulting reaction mixture was then
heated to 75 °C and maintained at this temperature for 48 hours. The yield for 377 (63%) was
determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an
internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 1:1) provided an analytical sample of 377.

205
1H NMR (600 MHz, CDCl3): δ 9.02 (s, 1H), 8.27 (d, J = 8.8 Hz, 1H), 8.19
(s, 1H), 8.00 (dd, J = 8.8, 1.9 Hz, 1H), 7.68 (m, 2H), 7.62 (d, J = 3.9 Hz,
1H), 7.57 (dd, J = 7.7, 7.7 Hz, 2H), 7.21 (dd, J = 8.7, 8.7 Hz, 1H), 7.19 (t, J
= 54.6 Hz, 1H) ; 13C NMR (150 MHz, CDCl3): δ 163.1 (d, J = 248.7 Hz),
150.0, 148.0, 139.8, 138.0 (t, J = 21.7 Hz), 136.4 (d, J = 3.2 Hz), 131.0, 129.8, 129.4 (d, J = 8.2
Hz) 124.5, 121.1, 118.7 (t, J = 7.0 Hz), 116.2 (d, J = 21.1 Hz), 113.6 (t, J = 241.3 Hz); 19F NMR
(470 MHz, CDCl3):δ –114.2, –114.7
HRMS (EI+) calcd for [C16H11F3N]+ 274.0838, found 274.0813
Melting point: 111-114 ˚C
Preparation of compound 378
Following General Procedure B, to a solution of 4-methyl-6-(4-(trifluoromethyl)phenyl)quinoline
(0.035 g, 0.122 mmol) in 0.24 mL of CH3CN (0.50 M substrate) was added NFSI (0.192 g, 0.610
mmol, 5.0 equiv.) and Li2CO3 (0.045 g, 0.610 mmol, 5.0 equiv.). The resulting reaction mixture
was then heated to 75 °C and maintained at this temprature for 48 hours. The yield for 378 (60%)
was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as
an internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 1:1) provided an analytical sample of 378.
IR (neat): = 2948, 1680, 1173, 722 cm-1; 1H NMR (600 MHz, CDCl3): δ
9.08 (s, 1H), 8.35 (d, J = 8.8 Hz, 1H), 8.29 (s, 1H), 8.06 (dd, J = 8.8, 1.8 Hz,
1H), 7.83 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 3.7 Hz,
1H), 7.20 (t, J = 54.7 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 150.1, 147.8,
143.7, 139.6, 138.7 (t, J = 22.5 Hz), 130.8, 130.5 (q, J = 32.6 Hz), 129.9, 128.2, 126.2 (q, J = 3.7
Hz), 124.6, 124.2 (q, J = 272.4 Hz), 122.0, 119.0, 113.5 (t, J = 241.2 Hz) 121.1, 118.7 (t, J = 7.0
Hz), 116.2 (d, J = 21.1 Hz), 113.6 (t, J = 241.3 Hz); 19F NMR (470 MHz, CDCl3):δ –62.5, –114.6
HRMS (EI+) calcd for [C17H11F5N]+ 324.0806, found 324.0813

206
Preparation of compound 379
Following General Procedure B, to a solution of 7-bromo-4-methylquinoline (0.025 g, 0.113 mmol)
in 0.22 mL of CH3CN (0.50 M substrate) was added NFSI (0.178 g, 0.566 mmol, 5.0 equiv.) and
Li2CO3 (0.042 g, 0.566 mmol, 5.0 equiv.). The resulting reaction mixture was then heated to 75
°C and maintained at this temperature for 48 hours. The yield for 379 (68%) was determined by
analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard.
Purification of the crude material by flash column chromatography (pentane-ethyl acetate; 1:1)
provided an analytical sample of 379.
IR (neat): = 2971, 2254, 1604, 902, 724 cm-1; 1H NMR (600 MHz, CDCl3): δ 9.03
(d, J = 4.3 Hz, 1H), 8.41 (d, J = 2.0 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H), 7.76 (dd, J
= 9.0 Hz, 1H) 7.61 (d, J = 4.3 Hz, 1H), 7.12 (t, J = 54.2 Hz, 1 H); 13C NMR (150
MHz, CDCl3): δ 151.1, 149.4, 138.2 (t, J = 22.4 Hz), 132.9, 131.5, 124.9, 124.4,
118.5 (t, J = 7.6 Hz), 113.3 (t, J = 241.3 Hz); 19F NMR (470 MHz, CDCl3):δ –114.6
HRMS (EI+) calcd for [C10H7BrF2N]+ 257.9724, found 257.9739
Melting point: 68-72˚C
Preparation of compound 380
Following General Procedure B, to a solution of 4-methyl-7-(4-(trifluoromethyl)phenyl)quinoline
(0.035 g, 0.122 mmol) in 0.24 mL of CH3CN (0.50 M substrate) was added NFSI (0.192 g, 0.61
mmol, 5.0 equiv.) and Li2CO3 (0.045 g, 0.61 mmol, 5.0 equiv.). The resulting reaction mixture
was then heated to 75 °C and maintained at this temperature for 48 hours. The yield for 380 (46%)
was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as
an internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 1:1) provided an analytical sample of 380.
IR (neat): = 2925, 1617, 1326, 1119, 1071, 729 cm-1; 1H NMR (600
MHz, CDCl3): δ 9.07 (d, J = 4.2 Hz, 1H), 8.44 (d, J = 1.7 Hz, 1H), 8.21
(d, J = 8.6 Hz, 1H), 7.92 (dd, J = 8.8, 1.8 Hz, 1H) 7.87 (d, J = 8.2 Hz,
2H), 7.87 (d, J = 8.2 Hz, 2H), 7.63 (ddd, J = 4.2 Hz, 1H), 7.18 (t, J =
54.5 Hz, 1H); 13C NMR (150 MHz, CDCl3): δ 150.9, 148.9, 143.3, 141.3,
138.1 (t, J = 22.7 Hz), 136.0, 130.5 (q, J = 32.7), 128.5, 128.0, 127.3, 126.2 (q, J = 3.7 Hz), 124.4,

207
123.8 (t, J = 3.1 Hz), 118.5 (t, J = 7.9 Hz), 113.4 (t, J = 241.3 Hz); 19F NMR (470 MHz, CDCl3):δ
–62.5, –114.8
HRMS (EI+) calcd for [C17H11F5N]+ 324.0806, found 324.0806
Preparation of compound 381
Following General Procedure B, to a solution of 3-(pyridin-4-yl)propyl 4-nitrobenzoate (0.025 g,
0.087 mmol) in 0.17 mL of ethyl acetate (0.50 M substrate) was added NFSI (0.274 g, 0.87 mmol,
10 equiv.) and Li2CO3 (0.033 g, 0.44 mmol, 5.0 equiv.). The resulting reaction mixture was then
heated to 75 °C and maintained at this temperature for 48 hours. Purification of the crude 381 by
flash chromatography (pentanes: ethyl acetate 1:1) afforded 381 (13.1 mg, 49%).
1H NMR (600 MHz, CDCl3): δ 8.80 (d, J = 3.8 Hz, 1H), 8.28 (d, J = 8.5
Hz, 2H), 8.05 (d, J = 8.5, 1H), 7.64 (d, J = 3.8 Hz, 2H), 4.60 (dd, J = 6.3,
6.3 Hz, 2H), 2.69 (m, J = 3.7 Hz, 2H); 13C NMR (150 MHz, CDCl3): δ
164.4, 150.9, 149.4, 149.3, 135.0, 130.8, 123.8, 120.3, 120.2, 59.3,
37.8 (t, J = 26.7 Hz); 19F NMR (470 MHz, CDCl3):δ –97.9.
HRMS (EI+) calcd for [C15H13F2N2O4]+ 323.0838, found 323.0845.
Preparation of compound 382
Following General Procedure B, to a solution of 3-(pyridin-4-yl)propyl 4-bromobenzoate (0.025 g,
0.078 mmol) in 0.16 mL of ethyl acetate (0.50 M substrate) was added NFSI (0.247 g, 0.78 mmol,
10 equiv.) and Li2CO3 (0.029 g, 0.39 mmol, 5.0 equiv.). The resulting reaction mixture was then
heated to 75 °C and maintained at this temperature for 48 hours. Purification of the crude 382 by
flash chromatography (pentanes: ethyl acetate 1:1) afforded 382 (12.1 mg, 47%).
IR (neat): = 2973, 1723, 1272, 1118, 732 cm-1;1H NMR (600 MHz,
CDCl3): δ 8.73 (d, J = 3.8 Hz, 2H), 7.68 (d, J = 7.8 Hz, 2H), 7.56 (d, J =
7.8 Hz, 2H), 7.42 (d, J = 3.8 Hz, 2H), 4.51 (t, J = 5.9 Hz, 2H), 2.66 (tt, J
= 15.7, 6.2 Hz, 2H); 13C NMR (150 MHz, CDCl3): δ 165.5, 150.6, 144.9
(t, J = 26.8 Hz), 131.9, 131.1, 128.6, 128.5, 120.5 (t, J = 242.9 Hz),
119.6 (t, J = 6.3 Hz), 58.8 (t, J = 5.4 Hz), 37.9 (t, J = 27.2 Hz); 19F NMR (470 MHz, CDCl3):δ –
97.6

208
HRMS (EI+) calcd for [C15H13BrF2NO2]+ 356.0092, found 356.0091
Preparation of compound 383
Following General Procedure B, to a solution of 4-ethylpyridine (0.025 mL, 0.22 mmol) in 0.45 mL
of ethyl acetate (0.50 M substrate) was added NFSI (0.694 g, 2.20 mmol, 10 equiv.) and Li2CO3
(0.081 g, 1.10 mmol, 5 equiv.). The resulting reaction mixture was then heated to 75 °C and
maintained at this temperature for 48 hours. The yield for 383 (43%) was determined by analysis
of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard. TFA
was added to the reaction mixture prior to concentration under reduced pressure. Purification of
the crude material by flash column chromatography (pentane-ethyl acetate; 1:1) provided an
analytical sample of 383.297
IR (neat): = 3085, 2923, 1666, 1293, 1150, 720 cm-1;1H NMR (600 MHz, CDCl3): δ
11.11 (br s, 1H), 9.02 (d, J = 6.2 Hz, 2H), 7.95 (d, J = 6.3 Hz, 2H), 2.00 (t, J = 18.4
Hz, 3H); 19F NMR (470 MHz, CDCl3):δ –75.9, –92.2
HRMS (EI+) calcd for [C7H8F2N]+ 144.0619, found 144.0623
Melting point: 86-89˚C
Preparation of compound 384
Following General Procedure B, to a solution of 2-ethylquinazoline (0.025 g, 0.158 mmol) in 0.65
mL of CH3CN (0.25 M substrate) was added NFSI (0.249 g, 0.791 mmol, 5.0 equiv.) and Li2CO3
(0.026 g, 0.348 mmol, 2.2 equiv.). The resulting reaction mixture was then heated to 125 °C and
maintained at this temperature in a microwave reactor for 1 hour. The yield for 384 (86%) was
determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an
internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 4:6) provided an analytical sample of 384.
IR (neat): = 2977, 1620, 1584, 905, 729 cm-1; 1H NMR (600 MHz, CDCl3): δ
9.52 (s, 1H), 8.17 (d, J = 8.7 Hz, 1H), 8.01 (m, 2H), 7.76 (dd, J = 7.3 Hz, 1H),
2.19 (t, J = 18.7 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 161.4, 159.3 (t, J = 28.1
Hz), 149.9, 135.0, 129.2, 129.2, 127.3, 124.7, 119.8 (t, J = 239.7 Hz), 23.4 (t,
J = 26.8 Hz); 19F NMR (CDCl3): δ –92.8

209
HRMS (EI+) calcd for [C10H9F2N2]+ 195.0728, found 195.0725
Preparation of compound 385
Following General Procedure B, to a solution of 6-bromo-4-ethylquinazoline (0.025 g, 0.106
mmol) in 0.22 mL of CH3CN (0.50 M substrate) was added NFSI (0.100 g, 0.318 mmol, 3.0 equiv.)
and Li2CO3 (9.0 mg, 0.117 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to
125 °C and maintained at this temperature in a microwave reactor for 1 hour. The yield for 385
(74%) was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-
fluorotoluene as an internal standard. Purification of the crude material by flash column
chromatography (pentane-ethyl acetate; 8:2) provided an analytical sample of 385.
IR (neat): = 3059, 2976, 1394, 1117, 1036, 759 cm-1; 1H NMR (600 MHz,
CDCl3): δ 9.36 (s, 1H), 8.66 (d, J = 1.2 Hz, 1H), 8.04 (dd, J = 9.0, 1.8 Hz, 1H)
1H), 8.02 (d, J = 9.0, 1H), 2.23 (t, J = 19.4, 1H); 13C NMR (150 MHz, CDCl3) δ
160.6 (d, J = 32.2 Hz), 153.8, 150.7, 138.1, 131.1, 128.5 (t, J = 6.1 Hz), 123.0 (t,
J = 240.7 Hz), 123.0, 122.2, 22.7 (t, J = 25.7 Hz); 19F NMR (CDCl3): δ –84.4
HRMS (EI+) calcd for [C10H8BrF2N2]+ 272.9833, found 272.9833
Preparation of compound 386
Following General Procedure B, to a solution of 6-ethyl-9-methyl-9H-purine (0.025 g, 0.169 mmol)
in 0.68 mL of CH3CN (0.25 M substrate) was added NFSI (0.266 g, 0.843 mmol, 5.0 equiv.) and
Li2CO3 (0.027 g, 0.372 mmol, 2.2 equiv.). The resulting reaction mixture was then heated to 125
°C and maintained at this temperature in a microwave reactor for 1 hour. The yield for 386 (69%)
was determined by analysis of a 1H NMR spectrum (500 MHz, CD3CN) using 4-fluorotoluene as
an internal standard. Purification of the crude material by flash column chromatography (pentane-
ethyl acetate; 1:9) provided an analytical sample of 386.
IR (neat): = 2975, 1594, 1328, 1119, 907, 731 cm-1; 1H NMR (600 MHz, CDCl3):
δ 9.05 (s, 1H), 8.19 (s, 1H), 3.96 (s, 3H) 1H), 2.20 (t, J = 19.4, 3H); 13C NMR (150
MHz, CDCl3) δ 153.7, 152.9 (t, J = 30.1 Hz), 152.1, 146.7, 130.1, 120.4 (t, J = 240.2
Hz), 30.1, 23.6 (t, J = 26.4 Hz); 19F NMR (CDCl3): δ –90.8
HRMS (EI+) calcd for [C8H9F2N4]+ 199.0790, found 199.0784

210
Preparation of compound 390
Following General Procedure C, to a solution of 6-bromo-4-ethylquinazoline (0.025 g, 0.106
mmol) in 0.53 mL of CH3CN (0.20 M substrate) was added N-
trifluoromethylthiobenzenesulfonimide (0.100 g, 0.252 mmol, 2.4 equiv.) and Li2CO3 (9.0 mg,
0.117 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 75 °C and maintained
at this temperature for 48 hours. Purification of the crude 390 by flash chromatography (pentanes:
ethyl acetate; 85:15) afforded 390 (33.0 mg, 92%).
IR (neat): = 2977, 1557, 1485, 1115, 731 cm-1;1H NMR (600 MHz, CDCl3): δ
9.30 (s, 1H), 8.28 (d, J = 1.8 Hz, 1H), 8.02 (dd, J = 9.0, 2.0 Hz, 1H), 8.00 (d, J
= 9.0 Hz, 1H) 5.25 (q, J = 7.1 Hz, 1H), 1.94 (d, J = 7.1 Hz, 3H); 13C NMR (150
MHz, CDCl3): δ 167.9, 154.7, 149.4, 138.0, 131.6, 130.9 (q, J = 307.8 Hz),
125.9, 122.7, 122.6, 41.4, 22.9; 19F NMR (470 MHz, CDCl3):δ –40.3
HRMS (EI+) calcd for [C11H9BrF3N2S]+ 336.9616, found 336.9621
Preparation of compound 391
Following General Procedure C, to a solution of 6-ethyl-9-methyl-9H-purine (0.025 g, 0.169 mmol)
in 0.68 mL of CH3CN (0.25 M substrate) was added N-trifluoromethylthiobenzenesulfonimide
(0.147 g, 0.372 mmol, 2.2 equiv.) and Li2CO3 (0.015 g, 0.20 mmol, 1.1 equiv.). The resulting
reaction mixture was then heated to 125 °C and maintained at this temperature in a microwave
reactor for 50 minutes. The yield for 391 (56%) was determined by analysis of a 1H NMR spectrum
(500 MHz, CD3CN) using 4-fluorotoluene as an internal standard. Purification of the crude material
by flash column chromatography (pentane-ethyl acetate; 1:1) provided an analytical sample of
391.
IR (neat): = 2953, 1750, 1591, 1331, 1223, 1114 cm-1; 1H NMR (600 MHz, CDCl3):
δ 8.96 (s, 1H), 8.08 (s, 1H), 5.28 (q, J = 7.1 Hz, 1H), 3.93 (s, 3H), 1.90 (d, J = 7.4,
1H); 13C NMR (150 MHz, CDCl3) δ 159.9, 152.8, 152.2, 145.4, 130.9 (q, J = 304.2
Hz), 40.7 (q, J = 1.8 Hz), 30.1, 21.7; 19F NMR (CDCl3): δ –40.4
HRMS (EI+) calcd for [C9H10F3N4S]+ 263.0573, found 263.0573

211
Preparation of compound 392
Following General Procedure C, to a solution of 2-propylquinazoline (0.025 g, 0.158 mmol) in 0.65
mL of CH3CN (0.25 M substrate) was added N-trifluoromethylthiobenzenesulfonimide (0.249 g,
0.791 mmol, 5.0 equiv.) and Li2CO3 (0.026 g, 0.348 mmol, 2.2 equiv.). The resulting reaction
mixture was then heated to 125 °C and maintained at this temperature in a microwave reactor for
1 hour. The yield for 392 (74%) was determined by analysis of a 1H NMR spectrum (500 MHz,
CD3CN) using 4-fluorotoluene as an internal standard. Purification of the crude material by flash
column chromatography (pentane-ethyl acetate; 1:1) provided an analytical sample of 392.
IR (neat): = 2973, 1624, 1113, 905, 731 cm-1; 1H NMR (600 MHz, CDCl3): δ
8.52 (d, J = 5.7 Hz, 1H), 8.17 (d, J = 8.7 Hz, 1H), 7.88 (dd, J = 7.3, 7.3 Hz, 1H),
7.67 (dd, J = 7.3, 7.3 Hz, 1H), 7.62 (d, J = 5.5 Hz, 1H), 5.25 (dd, J = 5.9, 5.7
Hz, 1H), 2.42 (m, 1H), 2.32 (m, 1H), 0.91 (t, J = 7.6 Hz, 3H); 13C NMR (150
MHz, CDCl3) δ 158.7, 141.9, 136.7, 131.3 (q, J = 307.7 Hz), 130.6, 128.0, 126.0, 124.2, 120.8,
47.7, 30.0, 11.4; 19F NMR (CDCl3): δ –40.3
HRMS (EI+) calcd for [C12H12F3N2S]+ 273.0668, found 273.0688
Preparation of compound 393
To a solution of 6-bromo-4-ethylquinazoline (0.020 g, 0.085 mmol) in 0.35 mL of CH3CN (0.25 M
substrate) was added N-chlorobenzenesulfonimide (0.034 g, 0.102 mmol, 1.2 equiv.) and Li2CO3
(6.9 mg, 0.093 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to and
maintained at 75 °C for 48 hours. Purification of the crude 393 by flash chromatography
(pentanes: diethyl ether; 6:4) afforded 393 (17.7mg, 78%).
IR (neat): = 2925, 1558, 1484, 905, 837, 729 cm-1; 1H NMR (600 MHz,
CDCl3): δ 9.35 (s, 1H), 8.44 (d, J = 1.9 Hz, 1H), 8.01 (dd, J = 8.9, 1.9 Hz, 1H),
7.99 (d, J = 8.9 Hz, 1H) 5.75 (q, J = 6.7 Hz, 1H), 2.08 (d, J = 6.7 Hz, 3 H); 13C
NMR (150 MHz, CDCl3): δ 166.4, 154.8, 149.7, 137.7, 131.4, 126.7, 123.4,
122.3, 52.7, 22.1
Melting point: 107-108˚C
HRMS (EI+) calcd for [C10H9BrClN2]+ 270.9632, found 270.9642.

212
Preparation of compound 394
To a solution of 4-ethyl-6-phenylquinazoline (0.030 g, 0.128 mmol) in 0.60 mL of CH3CN (0.20 M
substrate) was added N-trifluoromethylthiobenzenesulfonimide (0.102 g, 0.256 mmol, 2.0 equiv.)
and Li2CO3 (10.0 mg, 0.141 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to
and maintained at 75 °C for 36 hours. Purification of the crude 394 by flash chromatography
(pentanes: diethyl ether; 6:4) afforded 394 (20.9 mg, 49%).
1H NMR (500 MHz, CDCl3): δ 9.29 (s, 1H), 8.26 (d, J = 1.5 Hz, 1H), 8.21
(dd, J = 8.7, 1.5 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H) 5.42 (q, J = 7.0 Hz, 1H),
1.98 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 168.7, 154.4, 150.1,
141.6, 139.7, 134.2, 131.2 (d, J = 307.6 Hz), 130.3, 129.4, 128.7, 127.7,
121.9, 121.1, 41.4, 22.9; 19F NMR (470 MHz, CDCl3): δ -40.3
HRMS (EI+) calcd for [C17H14F3N2S]+ 335.0824, found 335.0844
Preparation of compound 395
To a solution of 4-ethyl-6-(4-fluorophenyl)quinazoline (0.030 g, 0.119 mmol) in 0.60 mL of CH3CN
(0.20 M substrate) was added N-trifluoromethylthiobenzenesulfonimide (0.095 g, 0.240 mmol, 2.0
equiv.) and Li2CO3 (9.7 mg, 0.13 mmol, 1.1 equiv.). The resulting reaction mixture was then
heated to and maintained at 75 °C for 36 hours. Purification of the crude 395 by flash
chromatography (pentanes: diethyl ether; 6:4) afforded 395 (33.3 mg, 79%).
1H NMR (500 MHz, CDCl3): δ 9.28 (s, 1H), 8.21 (d, J = 1.5 Hz, 1H), 8.19
(d, J = 8.6 Hz, 1H), 8.14 (dd, J = 8.6, 1.8 Hz, 1H), 7.67 (m, 2H), 7.24 (m,
2H), 5.40 (q, J = 7.0 Hz, 1H), 1.98 (d, J = 7.0 Hz, 3 H); 13C NMR (125
MHz, CDCl3): δ 168.8, 160.3 (d, J = 248.5 Hz), 154.5, 150.1, 140.5, 135.8
(d, J = 2.8 Hz), 134.0, 131.1 (q, J = 307.2), 130.4, 129.4 (d, J = 8.0 Hz), 121.9, 120.9, 116.4 (d, J
= 21.7 Hz), 41.3, 22.9; 19F NMR (470 MHz, CDCl3): δ -40.3, -113.4
HRMS (EI+) calcd for [C17H13F4N2S]+ 353.0730, found 353.0711.

213
Preparation of compound 396
To a solution of 4-ethyl-6-(4-ethylphenyl)quinazoline (0.030 g, 0.115 mmol) in 0.55 mL of CH3CN
(0.20 M substrate) was added N-trifluoromethylthiobenzenesulfonimide (0.091 g, 0.230 mmol, 2.0
equiv.) and Li2CO3 (9.3 mg, 0.270 mmol, 1.1 equiv.). The resulting reaction mixture was then
heated to and maintained at 75 °C for 48 hours. Purification of the crude 396 by flash
chromatography (pentanes: diethyl ether; 6:4) afforded 396 (28.3 mg, 68%).
1H NMR (500 MHz, CDCl3): δ 9.27 (s, 1H), 8.23 (d, J = 1.6 Hz, 1H),
8.20 (dd, J = 9.1, 1.8 Hz, 1H), 8.16 (d, J = 9.1 Hz, 1H), 7.63 (d, J = 8.2
Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 5.41 (q, J = 7.0 Hz, 1H), 2.75 (q, J =
7.6 Hz, 2H), 1.98 (d, J = 7.0 Hz, 3H), 1.31 (t, J = 7.6Hz, 3H); 13C NMR
(150 MHz, CDCl3): δ 168.2, 154.3, 150.0, 145.1, 141.5, 137.0, 134.2,
131.1 (q, J = 309.0 Hz), 130.2, 129.0, 127.6, 122.0, 120.7, 41.5, 28.7, 23.0, 15.7; 19F NMR (470
MHz, CDCl3): δ -40.3.
HRMS (EI+) calcd for [C19H18F3N2S]+ 363.1137, found 363.1119.
Preparation of compound 400
Following General Procedure A, to a solution of quazodine (0.025 g, 0.115 mmol) in 1.2 mL of
CH3CN (0.10 M substrate) was added NFSI (0.044 g, 0.138 mmol, 1.2 equiv.) and Li2CO3 (10.0
mg, 0.127 mmol, 1.1 equiv.). The resulting reaction mixture was then left room temperature and
maintained at this temperature for 120 hours. Purification of the crude 400 by flash
chromatography (pentane-ethyl acetates; 1:1) afforded 400 (14.0 mg, 50%).
IR (neat): = 2957, 1617, 1501, 1233, 1133, 905, 729 cm-1; 1H NMR (600
MHz, CDCl3): δ 9.12 (s, 1H), 7.56 (s, 1H), 7.38 (s, 1H), 6.15 (dq, J = 48.1, 6.6
Hz, 1H), 4.08 (s, 3H), 4.06 (s, 3H), 1.90 (dd, J = 24.2, 6.7 Hz, 3H); 13C NMR
(150 MHz, CDCl3) δ 163.6 (d, J = 20.8 Hz), 156.1, 153.0, 150.5, 149.5, 118.2,
107.3, 102.2 (d, J = 9.2 Hz), 91.7 (d, J = 171.5 Hz), 56.6, 56.4, 20.5 (d, J = 23.2 Hz); 19F NMR
(470 MHz, CDCl3): δ –174.1
HRMS (EI+) calcd for [C12H14FN2O2]+ 237.1034, found 237.1042
Melting point: 111-114˚C

214
Preparation of compound 401
Following General Procedure B, to a solution of quazodine (0.025 g, 0.115 mmol) in 0.23 mL of
CH3CN (0.50 M substrate) was added NFSI (0.108 g, 0.344 mmol, 3.0 equiv.) and Li2CO3 (9.0
mg, 0.127 mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 125 °C in a
microwave reactor and maintained at this temperature for 1 hour. Purification of the crude 401 by
flash chromatography (pentanes: ethyl acetate; 7:3) afforded 401 (17.8 mg, 62%).
1H NMR (600 MHz, CDCl3): δ 9.14 (s, 1H), 7.68 (s, 1H), 7.39 (s, 1H), 4.09 (s,
1H), 4.07 (s, 1H), 2.22 (t, J = 19.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ
158.0 (t, J = 30.3 Hz), 156.4, 152.3, 150.9, 150.3, 123.5 (t, J = 239.9 Hz),
117.2, 107.1, 103.0, 56.6, 56.4, 22.7 (t, J = 25.5 Hz); 19F NMR (470 MHz,
CDCl3): δ –85.3
HRMS (ESI+) calcd for [C12H15F2N2O2]+ 255.0940, found 255.0949
Preparation of compound 402
Following General Procedure C, to a solution of quazodine (0.025 g, 0.115 mmol) in 0.60 mL of
CH3CN (0.20 M substrate) was added N-trifluoromethylthiobenzenesulfonimide (0.108 g, 0.275
mmol, 2.4 equiv.) and Li2CO3 (9.0 mg, 0.127 mmol, 1.1 equiv.). The resulting reaction mixture
was then heated to 75 °C and maintained at this temperature for 48h. Purification of the crude
402 by flash chromatography (pentanes: ethyl acetate; 1:1) afforded 402 (34.0 mg, 94%).
1H NMR (600 MHz, CDCl3): δ 9.12 (s, 1H), 8.18 (s, 1H), 7.38 (s, 1H), 7.25 (s,
1H), 5.20, (q, J = 7.0, 1H), 4.08 (s, 3H), 4.08 (s, 3H), 1.96 (d, J = 6.9 Hz, 3H);
13C NMR (150 MHz, MeOD): δ 164.7, 156.4, 153.4, 151.1, 149.0, 131.2 (q, J
= 307.7 Hz), 117.5, 107.7, 100.7, 56.7, 56.4, 41.4, 22.4; 19F NMR (470 MHz,
CDCl3):δ –40.4
HRMS (EI+) calcd for [C13H14F3N2O2S]+ 319.0723, found 319.0725
Melting point: 104-107˚C
Preparation of compound 405
Following General Procedure A, to a solution of 404 (0.020 g, 0.049 mmol) in 0.20 mL of CH3CN
(0.25 M substrate) was added NFSI (0.047 g, 0.148 mmol, 3.0 equiv.) and Li2CO3 (4.0 mg, 0.054

215
mmol, 1.1 equiv.). The resulting reaction mixture was then heated to 75 °C and maintained at
this temperature for 48 hours. The yield for 405 (69%) was determined by analysis of a 1H NMR
spectrum (500 MHz, CD3CN) using 4-fluorotoluene as an internal standard. Purification of the
crude material by flash column chromatography (pentane-ethyl acetate; 25:75) provided an
analytical sample of 405 as a 1:1 mixture of diastereomers.
IR (neat): = 2971, 1748, 1592, 1223, 1057 cm-1; 1H NMR (600 MHz,
CDCl3): δ 9.00 (1H), 8.26 (1H), 6.25 (1H), 6.19 (1H), 5.98 (1H), 5.68
(1H), 4.48 (1H), 4.46 (1H), 4.39 (1H), 2.16 (3H), 2.12 (3H), 2.09 (3H),
1.88 (3H); 13C NMR (150 MHz, CDCl3): δ 170.4, 169.7, 169.5, 159.2,
152.8, 151.7, 143.4, 131.5, 88.4, 86.7, 80.6, 73.2, 70.7, 63.1, 20.9, 20.7,
20.7, 20.5; 19F NMR (470 MHz, CDCl3):δ –180.2, 180.2.
HRMS (EI+) calcd for [C18H22FN4O7]+ 425.1467, found 425.1459
Preparation of compound 406
Following General Procedure B, to a solution of 404 (0.010 g, 0.025 mmol) in 0.10 mL of CH3CN
(0.25 M substrate) was added NFSI (0.039 g, 0.123 mmol, 5.0 equiv.) and Li2CO3 (9.1 mg, 0.123
mmol, 5.0 equiv.). The resulting reaction mixture was then heated to 75 °C and maintained at
this temperature for 48 hours. Purification of the crude 406 by flash chromatography (pentanes:
ethyl acetate 25:75) afforded 406 (6.3 mg, 57%).
IR (neat): = 2952, 1749, 1590, 1224, 1133 cm-1; 1H NMR (600 MHz,
CDCl3): δ 9.05 (s, 1H), 8.35 (s, 1H), 6.28 (d, J = 5.1 Hz 1H), 5.98 (t,
J = 5.2, 5.2 Hz, 1H), 5.66 (dd, J = 5.1, 5.1 Hz, 1H), 4.49 (m, 1H), 4.46
(dd, J = 13.1, 1.8 Hz, 1H), 4.40 (dd, J = 13.1 Hz, 4.1 Hz, 1H), 2.20 (t,
J = 18.8 Hz, 3H), 2.17 (s, 3H), 2.13 (s, 3H), 2.09 (s, 3H); 13C NMR
(150 MHz, CDCl3): δ 170.3, 169.6, 169.3, 153.4 (t, J = 30.9 Hz), 152.7, 152.2, 144.3, 130.8, 120.0
(t, J = 239.6 Hz), 86.7, 80.5, 73.1, 70.6, 62.9, 23.4 (t, J = 25.9 Hz), 20.8, 20.5, 20.4; 19F NMR (470
MHz, CDCl3):δ –90.9.
HRMS (EI+) calcd for [C18H21F2N4O7]+ 443.1373, found 443.1370

216
Radiochemistry
Production of [18F]F2 gas
[18F]F2 gas was produced on TRIUMF’s TR13 cyclotron via the 18O(p,n)18F nuclear reaction in an
aluminum-body target using two proton irradiations. First [18O]O2 was loaded into the target to
~270 psi and irradiated with 25 A of 13 MeV protons for 5-10 minutes. The gas was removed
under reduced pressure and cryogenically trapped for recycling. F2 gas (3 % in Ar) was filled into
the target to 14 psi and topped with Ar to 290 psi. The target was then irradiated for 2-5 min with
20 A of 13 MeV protons. The target was emptied to the chemistry lab carried by Ar.
Synthesis of [18F]N-fluorodibenzenesulfonamide ([18F]NFSI)
Sodium dibenzenesulfonamide (40 mg, 125 µmol) was dissolved in 1 mL of 4:1 CH3CN:H2O and
placed in a conical vial. [18F]F2 produced in the cyclotron target was then passed through the
solution over a period of ~10 min. The waste gas was trapped by saturated KI solution. Typically
3-4 GBq was trapped in the reaction mixture. The resulting solution was then passed through a
SepPak (Waters tC18 SepPak Plus Long Cartridge). The cartridge was washed with 10 mL H2O
followed by 600 µL CH3CN. [18F]NFSI was then eluted from the SepPak cartridge in 2.4 mL
CH3CN. Typically, 50 ± 8 µmol of purified NFSI with an activity of 0.3-0.5 GBq is produced from
this process. The amount of NFSI generated in each reaction was calculated following HPLC
analysis of the reaction mixture and comparison with a calibration curve prepared from NFSI.
Synthesis of [18F] 408
The [18F]NFSI solution was concentrated under vacuum at 75 oC, then 360 µL CH3CN was added.
The mixture of 407 (6.70 mg, 50 µmol), Li2CO3 (4.2 mg, 57 µmol) and [18F]NFSI (180 µL CH3CN
solution) were place in a 5 mL conical vial and reacted at 75 oC for 40 min. After this time, a
fraction of the resulting mixture was subjected to HPLC analysis to get the radiochemical
conversion (RCC). Analytical HPLC was carried out on a Phenomenex Luna C18 (4.6 x 100 mm,
1 mL/min) using a gradient of 100% solvent A (0.1% TFA in H2O) to 100% solvent B (0.1% TFA
in CH3CN) over 15 min. A fraction of 15 µL reaction mixture was used for the purification, the
mixture was subjected to semi-preparatory HPLC purification. Semi-preparatory HPLC condition:
Phenomenex Luna C18 (4.6 x 100 mm, 1 mL/min) using a gradient of 100% solvent A (0.1% TFA
in H2O) to 100% solvent B (0.1% TFA in CH3CN) over 15 min. The radiochemical yield (RCY) is

217
reported as a percentage and represents the total activity present in the purified 18F-labeled 408
divided by the total activity present in the purified [18F]NFSI x 100 (decay corrected).
Figure 6.1: HPLC radio trace of purified [18F]NFSI (top), HPLC radio trace of crude
(middle) and HPLC radio trace of purified 408 (bottom, blue line) overlaid with HPLC UV trace
(220 nm) of authentic reference (bottom, red line).
HPLC radio trace of purified
[18F]NFSI
HPLC radio trace of
crude 408
Blue line: HPLC radio
trace of
Purified 408
Red line: HPLC UV
trace of
authentic reference

218
Chapter 7. Future Work
7.1. Synthesis of NA and CA Screening-Libraries
High-throughput screening of chemical libraries represents a powerful tool for drug-lead
identification.298 However, these libraries often suffer from poor structural and stereochemical
diversity thus limiting their effectiveness in drug discovery.298 The chemical methods established
in Chapters 2-4 of this thesis provide a platform for the efficient synthesis of a broad variety of
different nucleoside and carbohydrate scaffolds and thus should facilitate future efforts in
generating NAs and CAs for screening libraries.
Scheme 7.1. Synthesis of C2’/C4’-modified NA and ProTide libraries
Libraries of carbacycles, iminosugars, fluorosugars, and numerous NA scaffolds can
readily be accessed for screening against targets associated with cancer, viral and bacterial
infections, and other diseases. Of particular interest to medicinal chemistry, would be a library
consisting of NAs that incorporate modifications at both the C2’ and C4’-positions (Scheme 7.1A),
a subclass of antiviral NAs whose syntheses were previously lengthy and unamenable to rapid
diversification.107 Furthermore, utilizing chemistry developed at Merck, NAs 409 can be readily

219
converted into their corresponding phosphoramidate prodrugs (ProTide) 411 which are also used
as antiviral and anticancer therapies.299
7.2. Incorporating NAs into Antisense Oligonucleotides
Antisense Oligonucleotides (ASO) consist of single stranded DNA or RNA and function by
targeting genes that are linked to disease.300 Incorporation of nucleoside analogues into an
oligonucleotide can prevent its degradation by nucleases and improve its potency.300 For
example, Damha reported the synthesis of 4’-modified-2’-deoxy-2’-fluorouridine nucleoside
analogues where he demonstrated 4’-methoxy analogues significantly improved oligomer stability
against nucleases.301 As mentioned previously, our NA synthesis allows for the rapid synthesis of
a variety of novel NAs. Observing the effects of these NAs on oligomer stability and potency may
provide new structural insights for the further development of ASO therapies. As shown in Figure
7.1, concurrent modifications to the phosphate ester backbone, such as a phosphorothiolate
linkage (413 and 414), may also be investigate to improve nuclease resistance.
Figure 7.1. C2’/C4’-modified NAs for incorporation into oligonucleotides

220
References
(1) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317 (5846), 1881–1886.
(2) Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.;
Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114 (4), 2432–2506.
(3) Johnson, B. M.; Shu, Y. Z.; Zhuo, X.; Meanwell, N. A. J. Med. Chem. 2020.
(4) Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem.
2015, 58 (21), 8315–8359.
(5) Meanwell, N. A. J. Med. Chem. 2018, 61 (14), 5822–5880.
(6) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37 (2), 320–
330.
(7) Szpera, R.; Moseley, D. F. J.; Smith, L. B.; Sterling, A. J.; Gouverneur, V. Angew. Chem.
Int. Ed. 2019, 58 (42), 14824–14848.
(8) Champagne, P. A.; Desroches, J.; Hamel, J. D.; Vandamme, M.; Paquin, J. F. Chem. Rev.
2015, 115 (17), 9073–9174.
(9) Meanwell, N. A. J. Med. Chem. 2011, 54 (8), 2529–2591.
(10) Preshlock, S.; Tredwell, M.; Gouverneur, V. Chem. Rev. 2016, 116 (2), 719–766..
(11) Campbell, M. G.; Mercier, J.; Genicot, C.; Gouverneur, V.; Hooker, J. M.; Ritter, T. Nature
Chemistry 2016, 9 (1), 1–3.
(12) Yuan, Z.; Nodwell, M. B.; Yang, H.; Malik, N.; Merkens, H.; Bénard, F.; Martin, R. E.;
Schaffer, P.; Britton, R.; Campbell, M. G.; Mercier, J.; Genicot, C.; Gouverneur, V.;
Hooker, J. M.; Ritter, T. Angew. Chem. Int. Ed. 2016, 57 (1), 1–3.
(13) Tredwell, M.; Gouverneur, V. 18F Labeling of Arenes. Angew. Chem. Int. Ed. 2012, 51 (46),
11426–11437.

221
(14) Som, P.; Atkins, H. L.; Bandoypadhyay, D.; Fowler, J. S.; Macgregor, A. R.; Matsui, K.;
Oster, Z. H.; Sacker, D. F.; Shiue, C. Y.; Turner, H.; Wan, C. N.; Wolf, A. P.; Zabinski, S.
V. J. Nuc. Med. 1980, 21 (7), 670-675
(15) Rubello, D.; Nanni, C.; Fanti, S. J. Nuc. Med. 2007, 48 (10), 1577–1579.
(16) O’Hagan, D. Chem. Soc. Rev. 2008, 37 (2), 308–319.
(17) Hunter, L. Beilstein J.Org. Chem. 2010, 6, 1–14.
(18) Deniau, G.; Slawin, A. M. Z.; Lebl, T.; Chorki, F.; Issberner, J. P.; van Mourik, T.; Heygate,
J. M.; Lambert, J. J.; Etherington, L. A.; Sillar, K. T.; O’Hagan, D. ChemBioChem 2007, 8
(18), 2265–2274.
(19) Kirchmair, J.; Göller, A. H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I. D.; Glen, R. C.;
Schneider, G. Nat. Rev. Drug. Discov. 2015, 14 (6), 387-404
(20) Sharma, G.; Kaur, K. Ezetimibe. Indian Journal of Pharmacology 2003, 35 (6), 406.
(21) Cox, C. D.; Coleman, P. J.; Breslin, M. J.; Whitman, D. B.; Garbaccio, R. M.; Fraley, M. E.;
Buser, C. A.; Walsh, E. S.; Hamilton, K.; Schaber, M. D.; Lobell, R. B.; Tao, W.; Davide,
J. P.; Diehl, R. E.; Abrams, M. T.; South, V. J.; Huber, H. E.; Torrent, M.; Prueksaritanont,
T.; Li, C.; Slaughter, D. E.; Mahan, E.; Fernandez-Metzler, C.; Yan, Y. 2008, 51 (14),
4239–4252.
(22) Bey, E.; Marchais-Oberwinkler, S.; Werth, R.; Negri, M.; Al-Soud, Y. A.; Kruchten, P.;
Oster, A.; Frotscher, M.; Birk, B.; Hartmann, R. W. J. Med. Chem. 2008, 51 (21), 6725–
6739.
(23) Bey, E.; Marchais-Oberwinkler, S.; Negri, M.; Kruchten, P.; Oster, A.; Klein, T.; Spadaro,
A.; Werth, R.; Frotscher, M.; Birk, B.; Hartmann, R. W. J. Med. Chem. 2009, 52 (21),
6724–6743.
(24) Pinto, D. J. P.; Orwat, M. J.; Wang, S.; Fevig, J. M.; Quan, M. L.; Amparo, E.; Cacciola, J.;
Rossi, K. A.; Alexander, R. S.; Smallwood, A. M.; Luettgen, J. M.; Liang, L.; Aungst, B. J.;
Wright, M. R.; Knabb, R. M.; Wong, P. C.; Wexler, R. R.; Lam, P. Y. S. J. Med. Chem.
2001, 44 (4), 566–578.

222
(25) Quan, M. L.; Lam, P. Y. S.; Han, Q.; Pinto, D. J. P.; He, M. Y.; Li, R.; Ellis, C. D.; Clark, C.
G.; Teleha, C. A.; Sun, J. H.; Alexander, R. S.; Bai, S.; Luettgen, J. M.; Knabb, R. M.;
Wong, P. C.; Wexler, R. R.; Orwat, M. J.; Wang, S.; Fevig, J. M.; Quan, M. L.; Amparo,
E.; Cacciola, J.; Rossi, K. A.; Alexander, R. S.; Smallwood, A. M.; Luettgen, J. M.; Liang,
L.; Aungst, B. J.; Wright, M. R.; Knabb, R. M.; Wong, P. C.; Wexler, R. R.; Lam, P. Y. S.
J. Med. Chem. 2005, 48 (4), 566–578.
(26) Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acenã, J. L.; Soloshonok, V. A.; Izawa, K.;
Liu, H. Chem. Rev. 2016, 116 (2), 422–518.
(27) Fujiwara, T.; O’Hagan, D. J. Fluor. Chem. 2014, 167, 16–29.
(28) Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40
(7), 3496–3508.
(29) Amii, H.; Uneyama, K. Chem. Rev. 2009, 109 (5), 2119–2183.
(30) Stahl, T.; Klare, H. F. T.; Oestreich, M. ACS Catal. 2013, 3 (7), 1578–1587.
(31) Fügedi, P.; Garegg, P. J.; Lönn, H.; Norberg, T. Glycoconjugate Journal 1987, 4 (2), 97–
108.
(32) Shimizu, M.; Togo, H.; Yokoyama, M. Synthesis 1998, No. 6, 799–822.
(33) Williams, S. J.; Withers, S. G. Carbohydrate Research 2000, 327 (1–2), 27–46.
(34) Toshima, K. Glycosyl Fluorides in Glycosidations. Carbohydrate Research 2000, 327 (1–
2), 15–26.
(35) Pelletier, G.; Zwicker, A.; Allen, C. L.; Schepartz, A.; Miller, S. J. J. Am. Chem. Soc. 2016,
138 (9), 3175–3182.
(36) Sati, G. C.; Martin, J. L.; Xu, Y.; Malakar, T.; Zimmerman, P. M.; Montgomery, J. J. Am.
Chem. Soc. 2020, 142 (15), 7235–7242.
(37) Nielsen, M. M.; Stougaard, B. A.; Bols, M.; Glibstrup, E.; Pedersen, C. M. Eur. J. Org.
Chem. 2017, 2017 (9), 1281–1284.
(38) Kanie, O.; Ito, Y.; Ogawa, T. J. Am. Chem. Soc. 1994, 116 (26), 12073–12074.

223
(39) Mukaiyama, T.; Takeuchi, K.; Jona, H.; Maeshima, H.; Saitoh, T. Helv. Chim. Acta 2000,
83 (8), 1901–1918.
(40) Czernecki, S.; Ville, G. J. Org. Chem. 1989, 54 (3), 610–612.
(41) Yadav, L. D. S.; Rai, V. K. Nucleosides, Nucleotides and Nucleic Acids 2008, 27 (12),
1227–1237.
(42) Olah, G. A.; Olah, J. A.; Ohyama, T. J. Am. Chem. Soc. 1984, 106 (18), 5284–5290.
(43) Olah, G. A.; Farooq, O.; Farnia, S. M. F.; Olah, J. A. J. Am. Chem. Soc. 1988, 110 (8),
2560–2565.
(44) Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J. F. Angew. Chem. Int. Ed.
2014, 53 (50), 13835–13839.
(45) Guo, J.; Bamford, K. L.; Stephan, D. W. Org. Biomol. Chem. 2019, 17 (21), 5258–5261.
(46) Bamford, K. L.; Chitnis, S. S.; Qu, Z. wang; Stephan, D. W. Chem. Eur. J. 2018, 24 (60),
16014–16018.
(47) Zhu, J.; Pérez, M.; Caputo, C. B.; Stephan, D. W. Angew. Chem. Int. Ed. 2016, 55 (4),
1417–1421.
(48) Dryzhakov, M.; Moran, J. ACS Catal. 2016, 6 (6), 3670–3673.
(49) Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem. Int. Ed. 2013, 52 (20),
5328–5332.
(50) Champagne, P. A.; Pomarole, J.; Thérien, M. È.; Benhassine, Y.; Beaulieu, S.; Legault, C.
Y.; Paquin, J. F. Org. Lett. 2013, 15 (9), 2210–2213.
(51) Choi, C.; Nuhant, P.; Mousseau, J. J.; Yang, X.; Gerstenberger, B. S.; Williams, J. M.;
Wright, S. W. Org. Lett. 2016, 18 (21), 5748–5751.
(52) Haufe, G.; Suzuki, S.; Yasui, H.; Terada, C.; Kitayama, T.; Shiro, M.; Shibata, N. Angew.
Chem. Int. Ed. 2012, 51 (49), 12275–12279.
(53) Zhang, L.; Zhang, W.; Liu, J.; Hu, J. J. Org. Chem. 2009, 74 (7), 2850–2853.

224
(54) Affouard, C.; Crockett, R. D.; Diker, K.; Farrell, R. P.; Gorins, G.; Huckins, J. R.; Caille, S.
Org. Process Res. Dev. 2015, 19 (3), 476–485.
(55) Ichikawa, J.; Wada, Y.; Fujiwara, M.; Sakoda, K. Synthesis 2002, No. 13, 1917–1936.
(56) Grecian, S. A.; Hadida, S.; Warren, S. D. Tet. Lett. 2005, 46 (27), 4683–4685.
(57) Munoz, S. B.; Ni, C.; Zhang, Z.; Wang, F.; Shao, N.; Mathew, T.; Olah, G. A.; Prakash, G.
K. S. Eur. J. Org. Chem. 2017, 2017 (16), 2322–2326.
(58) Vogt, D. B.; Seath, C. P.; Wang, H.; Jui, N. T. J. Am. Chem. Soc. 2019, 141 (33), 13203–
13211.
(59) Sap, J. B. I.; Straathof, N. J. W.; Knauber, T.; Meyer, C. F.; Medebielle, M.; Buglioni, L.;
Genicot, C.; Trabanco, A. A.; Noel, T.; am Ende, C. W.; Gouverneur, V. J. Am. Chem.
Soc. 2020,142 (20), 9181-9187
(60) Dang, H.; Whittaker, A. M.; Lalic, G. Chem. Sci. 2016, 7 (1), 505–509.
(61) Blessley, G.; Holden, P.; Walker, M.; Brown, J. M.; Gouverneur, V. Org. Lett. 2012, 14
(11), 2754–2757.
(62) Zhu, F.; Wang, Z. X. J. Org. Chem. 2014, 79 (10), 4285–4292.
(63) Yoshikai, N.; Mashima, H.; Nakamura, E. J. Am. Chem. Soc. 2005, 127 (51), 17978–
17979.
(64) Luo, H.; Wu, G.; Xu, S.; Wang, K.; Wu, C.; Zhang, Y.; Wang, J. Chem. Commun. 2015,
51 (68), 13321–13323.
(65) Mizukami, Y.; Song, Z.; Takahashi, T. Org. Lett. 2015, 17 (24), 5942–5945.
(66) Namavari, M.; Satyamurthy, N.; Barrio, J. R. J. Fluor. Chem. 1995, 72 (1), 89–93.
(67) Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988,
110 (16), 5583–5584.
(68) Handbook of Chemical Glycosylation: Advances in Stereo- selectivity and Therapeutic
Relevance. Demchenko, A. V., Ed. Wiley: Weinheim, 2008.

225
(69) Lennox, A. J. J. Angew. Chem. Int. Ed. 2018, 57 (45), 14686–14688.
(70) Kowalczyk, B. A. Synthesis 1997, 1411–1414.
(71) Bigot, A.; Zhu, J. Tet. Lett. 1998, 39 (7), 551–554.
(72) Ruan, Z.; Zhang, S. K.; Zhu, C.; Ruth, P. N.; Stalke, D.; Ackermann, L. Angew. Chem. Int.
Ed. 2017, 56 (8), 2045–2049.
(73) Hu, J.; Zhang, W.; Wang, F. Chemical Commun. 2009, No. 48, 7465–7478.
(74) Li, Z. Y.; Li, L.; Li, Q. L.; Jing, K.; Xu, H.; Wang, G. W. Chem. Eur. J.l 2017, 23 (14),
3285–3290.
(75) Xu, P.; Guo, S.; Wang, L.; Tang, P. Synlett 2015, 26 (1), 36–39.
(76) Fujikawa, K.; Fujioka, Y.; Kobayashi, A.; Amii, H. Org. Lett. 2011, 13 (20), 5560–5563.
(77) Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J. Tet. Lett. 2007, 48 (18), 3251–3254.
(78) Nagura, H.; Murakami, S.; Fuchigami, T. Tetrahedron 2007, 63 (41), 10237–10245.
(79) Hu, J.; Zhang, W.; Wang, F. Chem. Commun. 2009, No. 48, 7465–7478.
(80) Jiang, X. L.; Chen, Z. H.; Xu, X. H.; Qing, F. L. Org. Chem. Front. 2014, 1 (7), 774–776.
(81) Feng, Z.; Min, Q. Q.; Fu, X. P.; An, L.; Zhang, X. Nat. Chem. 2017, 9 (9), 918–923.
(82) Xu, L.; Vicic, D. A. J. Am. Chem. Soc. 2016, 138 (8), 2536–2539.
(83) Gu, Y.; Leng, X.; Shen, Q. Nat. Commun. 2014, 5, 1–7.
(84) Fier, P. S.; Hartwig, J. F. J. Am. Chem. Soc. 2012, 134 (12), 5524–5527.
(85) Rong, J.; Ni, C.; Hu, J. Asian J. Org. Chem. 2017, 6 (2), 139–152.
(86) Fujikawa, K.; Kobayashi, A.; Amii, H. Synthesis 2012, 44 (I), 15–18.
(87) Fujiwara, Y.; Dixon, J. a; Rodriguez, R. a; Baxter, R. D.; Dixon, D. D.; Collins, M. R.;
Blackmond, D. G.; Baran, P. S.; Baxter, R. D. J. Am. Chem. Soc. 2012, 134 (3), 1494–
1497.

226
(88) Fujiwara, Y.; Dixon, J. A.; O’hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.;
Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012,
492 (7427), 95–99.
(89) Johnson, C. S.; Galmarini, C. M.; Brown, T.; Huryn, D. M.; Okabe, M.; Shelton, J.; Lu, X.;
Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 1993, 116 (1),
1745–1768.
(90) Galmarini, C. M. J. Oncol. 2002, 3 (1), 22–32.
(91) Huryn, D. M.; Okabe, M. Chem. Rev. 1992, 92 (8), 1745–1768.
(92) Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem.
Rev. 2016, 116 (23), 14379–14455.
(93) Ewald, B.; Sampath, D.; Plunkett, W. Oncogene 2008, 27 (50), 6522–6537.
(94) De Clercq, E. Med. Res. Rev. 2013, 33, 1215–1248.
(95) Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discov. 2013, 12
(6), 447–464.
(96) Seley-Radtke, K. L.; Yates, M. K. Antivir. Res. 2018, 154, 66–86.
(97) Yates, M. K.; Seley-Radtke, K. L. Antivir. Res 2019, 162, 5–21.
(98) Ma, H.; Jiang, W. R.; Robledo, N.; Leveque, V.; Ali, S.; Lara-Jaime, T.; Masjedizadeh, M.;
Smith, D. B.; Cammack, N.; Klumpp, K.; Symons, J. J. Biol. Chem. 2007, 282 (41),
29812–29820.
(99) Deval, J.; Powdrill, M. H.; D’Abramo, C. M.; Cellai, L.; Götte, M. Antimicrob. Agents
Chemother. 2007, 51 (8), 2920–2928.
(100) Ohrui, H. Chem. Rec. 2006, 6 (3), 133–143.
(101) Witkowski, J. T.; Robins, R. K.; Sidwell, R. W.; Simon, L. N. J. Med. Chem. 15 (11), 1150.
(102) Zeidler, J.; Baraniak, D.; Ostrowski, T. Eur. J. Med. Chem. 97, 409–418.

227
(103) Ni, G.; Du, Y.; Tang, F.; Liu, J.; Zhao, H.; Chen, Q. RSC Adv. 2019, 9 (25), 14302–14320.
(104) Gumina, G.; Song, G. Y.; Chu, C. K. FEMS Microbiol. Let. 2001, 202 (1), 9–15.
(105) Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chem. Rev. 110 (6), 3337–3370.
(106) Brodszki, M.; Bäckström, B.; Horvath, K.; Larsson, T.; Malmgren, H.; Pelcman, M.;
Wähling, H.; Wallberg, H.; Wennerberg, J. Org. Process Res. Dev. 2011, 15 (5), 1027–
1032.
(107) Mclaughlin, M.; Kong, J.; Belyk, K. M.; Chen, B.; Gibson, A. W.; Keen, S. P.; Lieberman,
D. R.; Milczek, E. M.; Moore, J. C.; Murray, D.; Peng, F.; Qi, J.; Reamer, R. A.; Song, Z.
J.; Tan, L.; Wang, L.; Williams, M. J. Org. Lett. 2017, 19 (4), 926.
(108) Markiewicz, W. T.; Wiewiórowski, M. Nucleic Acids Res. 1978, 5 (suppl_1), s185–s190.
(109) Campos, K.; Coleman, P.; Alvarez, J.; Dreher, S.; Garbaccio, R.; Terrett, N.; Tillyer, R.;
Truppo, M.; Parmee, E. Science 2019, 363 (6424), 244.
(110) Peifer, M.; Berger, R.; Shurtleff, V. W.; Conrad, J. C.; Macmillan, D. W. C. J. Am. Chem.
Soc. 2014, 136 (16), 5900–5903.
(111) Teichert, J. S.; Kruse, F. M.; Trapp, O. Angew. Chem. Int. Ed. 2019, 58 (29), 9944–9947.
(112) Grondal, C.; Enders, D. Adv. Synth. Catal. 349 (4–5), 694–702.
(113) Davis, F.; Kasu, P.; Sundarababu, G.; Qi, H. J. Org. Chem. 1995, 62 (22), 7546–7547.
(114) Middleton, W. J.; Bingham E. M. J. Org. Chem. 1980, 45 (14), 2883–2887.
(115) Steiner, D. D.; Mase, N.; Barbas, C. F. Angew. Chem. Int. Ed. 2005, 44 (24), 3706.
(116) Bergeron-Brlek, M.; Meanwell, M.; Britton, R. Nat. Commun. 2015, 6.
(117) Sánchez-Fernández, E. M.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.
M.; Nieto, P. M.; Angulo, J. Chemistry 2012, 18 (27), 8527.
(118) Huang, W.; Diaconescu, P. Organometallics 36 (1), 89–96.
(119) De Clercq, E. J. Med. Chem. 2016, 59 (6), 2301–2311.

228
(120) Hyde, A. M.; Calabria, R.; Arvary, R.; Wang, X.; Klapars, A. Org. Process Res. Dev.
2019, 23 (9), 1860–1871.
(121) T. C. Britton, M. E. LeTourneau. Process for Anomerizing Nucleosides. 5,420,266, 1995.
Eli Lilly and Company
(122) Downey, A. M.; Richter, C.; Pohl, R.; Mahrwald, R.; Hocek, M. Org. Lett. 2015, 17 (18),
4604–4607.
(123) Xu, Z.-Q.; Qiu, Y.-L.; Chokekijchai, S.; Mitsuya, H.; Zemlicka, J. J. Med. Chem. 1995, 38
(6), 875–882.
(124) Moyroud, E.; Biala, E.; Strazewski, P. Tetrahedron 2000, 56 (11), 1475–1484.
(125) Hadj-Bouazza, A.; Zerrouki, R.; Krausz, P.; Laumond, G.; Aubertin, A. M.; Champavier,
Y. Nucleosides, Nucleotides and Nucleic Acids. 2005, 24 (8), 1249–1263.
(126) Mehellou, Y.; Valente, R.; Mottram, H.; Walsby, E.; Mills, K. I.; Balzarini, J.; Mcguigan, C.
Bioorg. Med. Chem. 2010, 18 (7), 2439.
(127) Cook, A. Frederick.; Moffatt, J. G. J. Am. Chem. Soc. 1967, 89 (11), 2697–2705.
(128) Jenkinson, S.; Jones, N.; Moussa, A.; Stewart, A.; Heinz, T.; Fleet, G. Tet. Lett. 2007, 48
(25), 4441–4444.
(129) Schrödinger Release 2018-3: MacroModel, Schrödinger, LLC, New York, NY (2019).
(130) Gaussian 16, Revision C.01, M. J. Frisch et al. Gaussian, Inc., Wallingford CT (2016).
(131) Rablen, P.; Pearlman, S.; Finkbiner, J.; J. Phys. Chem. A. 1999 103 (36), 7357–7363.
(132) Lodewyk, M.; Siebert, M.; Tantillo, D. Chem. Rev. 2012, 112 (3), 1839–1862.
(133) Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, WI (2016).
(134) Sheldrick, G. M. Acta crystallographica. Section C, Structural chemistry 71 (Pt 1), 3.
(135) C. B. Hübschle, G. M. Sheldrick, B. Dittrich. J. Appl. Crystallogr. 2011, 44, 1281-1284

229
(136) Macrae, C.; Bruno, I.; Chisholm, J.; Edgington, P.; Mccabe, P.; Pidcock, E.; Rodriguez-
Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. J. Appl. Crystallogr. 2008, 41 (2), 466–
470.
(137) Fenn, T.; Ringe, D.; Petsko, G. J. Appl. Crystallogr. 2003, 36 (2), 944–947.
(138) Waller, C. W.; Patrick, J. B.; Fulmor, W.; Meyer, W. E. J. Am. Chem. Soc. 1961, 79 (4),
1011–1012.
(139) Huffman, M. A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K. R.; Canada,
K. A.; Devine, P. N.; Duan, D.; Forstater, J. H.; Grosser, S. T.; Halsey, H. M.; Hughes, G.
J.; Jo, J.; Joyce, L. A.; Kolev, J. N.; Liang, J.; Maloney, K. M.; Mann, B. F.; Marshall, N.
M.; Mclaughlin, M.; Moore, J. C.; Murphy, G. S.; Nawrat, C. C.; Nazor, J.; Novick, S.;
Patel, N. R.; Rodriguez-Granillo, A.; Robaire, S. A.; Sherer, E. C.; Truppo, M. D.;
Whittaker, A. M.; Verma, D.; Xiao, L.; Xu, Y.; Yang, H. Science, 2019, 366 (6470), 1255.
(140) Nawrat, C. C.; Whittaker, A. M.; Huffman, M. A.; Mclaughlin, M.; Cohen, R. D.; Andreani,
T.; Ding, B.; Li, H.; Weisel, M.; Tschaen, D. M. Org. Lett. 2020, 22 (6), 2167.
(141) Nguyen, N. M.; Tran, C. N. B.; Phung, L. K.; Duong, K. T. H.; Huynh, H. L. A.; Farrar, J.;
Nguyen, Q. T. H.; Tran, H. T.; Nguyen, C. V. V.; Merson, L.; Hoang, L. T.; Hibberd, M. L.;
Aw, P. P. K.; Wilm, A.; Nagarajan, N.; Nguyen, D. T.; Pham, M. P.; Nguyen, T. T.;
Javanbakht, H.; Klumpp, K.; Hammond, J.; Petric, R.; Wolbers, M.; Nguyen, C. T.;
Simmons, C. P. J. Infect. Dis. 2013, 207 (9), 1442–1450.
(142) Smith, D. B.; Martin, J. A.; Klumpp, K.; Baker, S. J.; Blomgren, P. A.; Devos, R.;
Granycome, C.; Hang, J.; Hobbs, C. J.; Jiang, W. R.; Laxton, C.; Pogam, S. le; Leveque,
V.; Ma, H.; Maile, G.; Merrett, J. H.; Pichota, A.; Sarma, K.; Smith, M.; Swallow, S.;
Symons, J.; Vesey, D.; Najera, I.; Cammack, N. Bioorg. Med. Chem. Lett. 2007, 17 (9),
2570–2576.
(143) Griffon, J. F.; Dukhan, D.; Pierra, C.; Benzaria, S.; Loi, A. G.; la Colla, P.; Sommadossi,
J. P.; Gosselin, G. Nucleosides, Nucleotides and Nucleic Acids 2003, 22 (5–8), 707–709.
(144) Kozak, J.; Johnson, C. R. Nucleosides and Nucleotides 1998, 17 (12), 2221–2239.

230
(145) Gunic, E.; Girardet, J. L.; Pietrzkowski, Z.; Esler, C.; Wang, G. Bioorg. Med. Chem. 2001,
9 (1), 163–170.
(146) Kageyama, M.; Nagasawa, T.; Yoshida, M.; Ohrui, H.; Kuwahara, S. Org. Lett. 2011,13
(19), 5264.
(147) Cui, H.; Carrero-Lérida, J.; Silva, A. P. G.; Whittingham, J. L.; Brannigan, J. A.; Ruiz-
Pérez, L. M.; Read, K. D.; Wilson, K. S.; González-Pacanowska, D.; Gilbert, I. H. J. Med.
Chem. 2012, 55 (24), 10948.
(148) Campbell, M. A.; Wengel, J. Chem. Soc. Rev. 2011, 40 (12), 5680–5689.
(149) Madsen, A. S.; Jorgensen, A. S.; Jensen, T. B.; Wengel, J. J. Org. Chem. 2012, 77 (23),
10718–10728.
(150) Madsen, A. S.; Hrdlicka, P. J.; Kumar, T. S.; Wengel, J. Carbohydrate Research 2006,
341 (10), 1398–1407.
(151) Wengel, J.; Koshkin, A.; Singh, S. K.; Nielsen, P.; Meldgaard, M.; Rajwanshi, V. K.;
Kumar, R.; Skouv, J.; Nielsen, C. B.; Jacobsen, J. P.; Jacobsen, N.; Olsen, C. E.
Nucleosides and Nucleotides 1999, 18 (6–7), 1365–1370.
(152) Li, Q.; Yuan, F.; Zhou, C.; Plashkevych, O.; Chattopadhyaya, J. J. Org. Chem. 2010, 75
(18), 6122–6140.
(153) Xu, J.; Liu, Y.; Dupouy, C.; Chattopadhyaya, J. J. Org. Chem. 2009, 74 (17), 6534–6554.
(154) Seth, Punit P. Swayze, E. E. 6-Disubstituted or Unsaturated Bicyclic Nucleic Acid
Analogs. US 8278283 B2, 2008.
(155) Yamaguchi, T.; Horiba, M.; Obika, S. Chem. Commun. 2015, 51 (47), 9737–9740.
(156) W. G. Overend. Nature 1973, 242 (5392), 71.
(157) Scott, L. J.; Spencer, C. M. Drugs 2000, 59 (3), 521–549.
(158) Pascolutti, M.; von Itzstein, M. Design and Synthesis of Carbohydrates and
Carbohydrate Mimetics as Anti-Influenza Agents John Wiley & Sons, Ltd, 2016; pp 455–
481.

231
(159) Hossain, F.; Andreana, P. R. Pharmaceuticals 2019, 12 (2), 84 –102
(160) Goss, P. E.; Baker, M.; Dennis, J. W.; Carver, J. Clin. Cancer Res. 1995, 1, 935–944.
(161) Wang, S. Q.; Du, Q. S.; Huang, R. B.; Zhang, D. W.; Chou, K. C. Biochem. Biophys.
Res. Commun. 2009, 386 (3), 432–436.
(162) Cecioni, S.; Vocadlo, D. J. J. Am. Chem. Soc. 2017, 139 (25), 8392.
(163) Compain, Philippe.; Martin, O. R. Iminosugars : From Synthesis to Therapeutic
Applications / Editors, Philippe Compain and Olivier R. Martin.; Compain, P., Martin, O.
R., Eds.; John Wiley and Sons, 2007.
(164) Arjona, O.; Gómez, A. M.; López, J. C.; Plumet, J. Chem. Rev. 2007, 107 (5), 1919.
(165) Witczak, Z. J.; Culhane, J. M. Appl. Microbiol. Biotechnol. 2005, 69 (3), 237–244.
(166) Horne, G.; Wilson, F.; Tinsley, J.; Williams, D.; Storer, R. Drug Discov. today 2011, 16,
107–118.
(167) Lenci, E.; Menchi, G.; Trabocchi, A. Org. Biomol. Chem. 2016, 14 (3), 808–825.
(168) Wu, C. Y.; Chang, C. F.; Chen, J. S. Y.; Wong, C. H.; Lin, C. H. Angew. Chem. Int. Ed.
2003, 42 (38), 4661–4664.
(169) Gerard, B.; Lee, M. D.; Dandapani, S.; Duvall, J. R.; Fitzgerald, M. E.; Kesavan, S.;
Lowe, J. T.; Marié, J. C.; Pandya, B. A.; Suh, B. C.; O’Shea, M. W.; Dombrowski, M.;
Hamann, D.; Lemercier, B.; Murillo, T.; Akella, L. B.; Foley, M. A.; Marcaurelle, L. A. J.
Org. Chem. 2013, 78 (11), 5160–5171.
(170) Xu, C.; Loh, C. C. J. J. Am. Chem. Soc. 2019. 141 (13), 5381.
(171) Liang, F. S.; Wang, S. K.; Nakatani, T.; Wong, C. H. Angew. Chem. Int. Ed. 2004, 43
(47), 6496–6500.
(172) Bergeron-Brlek, M.; Teoh, T.; Britton, R. Org. Lett. 2013, 15 (14), 3554.

232
(173) Adamson, C.; Pengelly, R. J.; Shamsi Kazem Abadi, S.; Chakladar, S.; Draper, J.;
Britton, R.; Gloster, T. M.; Bennet, A. J. Angew. Chem. Int. Ed. 2016, 55 (48), 14978–
14982.
(174) Bergeron-Brlek, M.; Goodwin-Tindall, J.; Cekic, N.; Roth, C.; Zandberg, W. F.; Shan, X.;
Varghese, V.; Chan, S.; Davies, G. J.; Vocadlo, D. J.; Britton, R. Angew. Chem. Int. Ed.
2016, 54 (51), 15429.
(175) Ren, W.; Pengelly, R.; Farren-Dai, M.; Shamsi Kazem Abadi, S.; Oehler, V.; Akintola,
O.; Draper, J.; Meanwell, M.; Chakladar, S.; Świderek, K.; Moliner, V.; Britton, R.; Gloster,
T. M.; Bennet, A. J. Nat. Commun. 2018, 9 (1), 1–12.
(176) Meanwell, M.; Silverman, S. M. Lehmann, J.; Adluri, B.; Yang, W.; Cohen, R.;
Campeau, L.-C.; Britton, R. Accepted. Science 2020.
(177) Brochu, M. P.; Brown, S. P.; MacMillan, D. W. C. J. Am. Chem. Soc. 2004, 126 (13),
4108–4109.
(178) Zhong, G. Angew. Chem. Int. Ed. 2003, 42 (35), 4247–4250.
(179) List, B. J. Am. Chem. Soc. 2002, 124 (20), 5656.
(180) Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Angew. Chem. Int. Ed.
2005, 44 (5), 794–797.
(181) Nagib, D. A.; Scott, M. E.; Macmillan, D. W. C. J. Am. Chem. Soc. 131 (31), 10875–
10877.
(182) Bahmanyar, S.; Houk, K. N.; Martin, H. J.; List, B. J. Am. Chem. Soc. 125 (9), 2475–
2479.
(183) Hu, L.; Wu, M.; Wan, H.; Wang, J.; Wang, G.; Guo, H.; Sun, S. New J. Chem. 2016, 40
(8), 6550–6553.
(184) Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. Ang. Chem. Int. Ed. 2013,
52 (49), 12856–12859.

233
(185) Moreno-Clavijo, E.; Carmona, A. T.; Moreno-Vargas, A. J.; Rodríguez-Carvajal, M. A.;
Robina, I. Bioorg. Med. Chem. 2010, 18 (13), 4648–4660.
(186) Machacek, M.; Witter, D.; Gibeau, C.; Huang, C.; Kawamura, S.; Sloman, D. L.;
Siliphaivanh, P.; Quiroz, R.; Wan, M.; Schneider, S.; Yeung, C.; Reutershan, M. H.;
Henderson, T. J. Paparin, J.-L.; Rahali, H.; Hughes, J. M. E.; Sanyal, S.; Ye, Y.; Candito,
D. A.; P.S. Fier, Silverman, S. M. WO 2020/033288 A1, 2020, Merck Sharpe & Dohme
Corp.
(187) Kato, A.; Kato, N.; Kano, E.; Adachi, I.; Ikeda, K.; Yu, L.; Okamoto, T.; Banba, Y.;
Ouchi, H.; Takahata, H.; Asano, N. J. Med. Chem. 2005, 48 (6), 2036–2044.
(188) Gao, Y.; He, X.; Ding, F.; Zhang, Y. Synthesis 2016, 48 (23), 4017–4037.
(189) Fehr, G. Expanding the organocatalyzed α-chlorination/aldol reaction, 2018. MSc
(190) Barlow, J. N.; Blanchard, J. S. Carbohydrate Research, 2000, 328, 473
(191) Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57 (24), 10257–10274.
(192) Guan, A.-Y.; Liu, C.-L.; Sun, X.-F.; Xie, Y.; Wang, M.-A. Bioorg. Med. Chem. 2016, 24
(3), 342–353.
(193) Meanwell, N. A. A Synopsis of the Properties and Applications of Heteroaromatic
Rings in Medicinal Chemistry; 2017; Vol. 123.
(194) Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57 (14), 5845–
5859.
(195) Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi,
O. Org. Bio. Chem. 2016, 14 (28), 6611–6637.
(196) Martins, M. A. P.; Frizzo, C. P.; Moreira, D. N.; Buriol, L.; Machado, P. Chem. Rev.
2009, 109 (9), 4140–4182.
(197) Zhang, B.; Studer, A. Chem. Soc. Rev. 2015, 44 (11), 3505–3521.
(198) Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Acc. Chem. Res. 2016, 49 (9), 1911–
1923.

234
(199) Abelo, A.; Andersson, T. B.; Antonsson, M.; Naudot, A. K.; Skanberg, I.; Weidolf, L.
Drug Metab. Dispos. 2000, 28 (8), 966–972.
(200) Olbe, L.; Carlsson, E.; Lindberg, P. Nat. Rev. Drug. Discov. 2003, 2 (2), 132–139.
(201) Biftu, T.; Sinha-Roy, R.; Chen, P.; Qian, X.; Feng, D.; Kuethe, J. T.; Scapin, G.; Gao,
Y. D.; Tan, Y.; Krueger, D.; Bak, A.; Eiermann, G.; He, J.; Cox, J.; Hicks, J.; Lyons, K.;
He, H.; Salituro, G.; Tong, S.; Patel, S.; Doss, G.; Petrov, A.; Wu, J.; Xu, S. S.; Sewall,
C.; Zhang, X.; Zhang, B.; Thornberry, N. A.; Weber, A. E. J. Med. Chem. 2014, 57 (8),
3205–3212.
(202) Meanwell, N. A. A Synopsis of the Properties and Applications of Heteroaromatic
Rings in Medicinal Chemistry. Advances in Heterocyclic Chemistry, VOL 123; Scriven,
EFV and Ramsden, CA, Ed. 2017; Vol. 123, pp 245–361.
(203) Bergstrom, C. G.; Nicholson, R. T.; Dodson, R. M. J. Org. Chem. 1963, 28 (10), 2633–
2640.
(204) Neumann, C. N.; Ritter, T. Angew. Chem. Int. Ed. 2015, 54 (11, SI), 3216–3221.
(205) Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Bio. Chem. 2016, 14 (36), 8398–8427.
(206) Trinks, U.; Buchdunger, E.; Furet, P.; Kump, W.; Mett, H.; Meyer, T.; Müller, M.;
Regenass, U.; Rihs, G.; Lydon, N.; Traxler, P. J. Med. Chem. 1994, 37 (7), 1015–1027.
(207) Jaakkola, T.; Laitila, J.; Neuvonen, P. J.; Backman, J. T. Basic Clin. Pharm. 2006, 99
(1), 44–51.
(208) Liu, W.; Groves, J. T. Angew. Chem. Int. Ed. 2013, 52 (23), 6024–6027.
(209) Bloom, S.; McCann, M.; Lectka, T. Org. Lett. 2014, 16 (24), 6338–6341.
(210) Bloom, S.; Pitts, C. R.; Woltornist, R.; Griswold, A.; Holl, M. G.; Lectka, T. Org. Lett.
2013, 15 (7), 1722–1724.
(211) Nodwell, M. B.; Bagai, A.; Halperin, S. D.; Martin, R. E.; Knust, H.; Britton, R. Chem.
Commun. 51 (59), 11783–11786.
(212) Ma, J.-J.; Yi, W.-B.; Lu, G.-P.; Cai, C. Org. Biomol. Chem. 13 (10), 2890–2894.

235
(213) Huang, X.; Liu, W.; Ren, H.; Neelamegam, R.; Hooker, J. M.; Groves, J. T. J. Am.
Chem. Soc. 2014, 136 (19), 6842–6845.
(214) Koperniku, A.; Liu, H.; Hurley, P. B. Eur. J. Org. Chem. 2016, 2016 (5), 871–886.
(215) Bucher, C.; Sparr, C.; Schweizer, W. B.; Gilmour, R. Chem. Eur. J. 2009, 15 (31),
7637–7647.
(216) Fustero, S.; Roman, R.; Sanz-Cervera, J. F.; Simon-Fuentes, A.; Bueno, J.; Villanova,
S. J. Org. Chem. 2008, 73 (21), 8545–8552.
(217) Lee, S. H.; Seo, H. J.; Lee, S.-H.; Jung, M. E.; Park, J.-H.; Park, H.-J.; Yoo, J.; Yun, H.;
Na, J.; Kang, S. Y.; Song, K.-S.; Yim, M.; Chang, C.-H.; Kim, J.; Lee, J. J. Med. Chem.
2008, 51 (22), 7216–7233.
(218) Loksha, Y. M.; Pedersen, E. B.; Loddo, R.; Sanna, G.; Collu, G.; Giliberti, G.; la Colla,
P. J. Med. Chem. 2014, 57 (12), 5169–5178.
(219) Radi, M.; Angeli, L.; Franchi, L.; Contemori, L.; Maga, G.; Samuele, A.; Zanoli, S.;
Armand-Ugon, M.; Gonzalez, E.; Llano, A.; Este, J. A.; Botta, M. Bioorg. Med. Chem.
Lett. 2008, 18 (21), 5777–5780.
(220) Anilkumar, R.; Burton, D. J. J. Fluor. Chem. 2005, 126 (8), 1174–1184.
(221) Minakawa, N.; Kojima, N.; Matsuda, A. J. Org. Chem. 1999, 64 (19), 7158–7172.
(222) Stump, B.; Kohler, P. C.; Schweizer, W. B.; Diederich, F. Heterocycles 2007, 72, 293–
326.
(223) McElroy, W. T.; Tan, Z.; Basu, K.; Yang, S.-W.; Smotryski, J.; Ho, G. D.; Tulshian, D.;
Greenlee, W. J.; Mullins, D.; Guzzi, M.; Zhang, X.; Bleickardt, C.; Hodgson, R. Bioorg.
Med. Chem. Lett. 2012, 22 (3), 1335–1339.
(224) Ostrowski, T.; Januszczyk, P.; Cieslak, M.; Kazmierczak-Baranska, J.; Nawrot, B.;
Bartoszak-Adamska, E.; Zeidler, J. Bioorg. Med. Chem. 2011, 19 (14), 4386–4398.
(225) Blessley, G.; Holden, P.; Walker, M.; Brown, J. M.; Gouverneur, V. Org. Lett. 2012, 14
(11), 2754–2757.

236
(226) Silhar, P.; Pohl, R.; Votruba, I.; Hocek, M. Org. Biomol. Chem. 2005, 3 (16), 3001–
3007.
(227) Bugarolas Brufau, P.; Popko, B.; Appelbaum, D. (University of Chicago)
WO2013173746 A2, 2013
(228) Huisman, J. W.; Hubbs, J. L.; Zhang, X.; Osborne, R. WO2016033304 A1, 2016
(229) Lange, J. H. M.; van Stuivenberg, H. H.; Coolen, H.; Adolfs, T. J. P.; McCreary, A. C.;
Keizer, H. G.; Wals, H. C.; Veerman, W.; Borst, A. J. M.; de Looff, W.; Verveer, P. C.;
Kruse, C. G. J. Med. Chem. 2005, 48 (6), 1823–1838.
(230) Mobinikhaledi, A.; Foroughifar, N. Phosphorus Sulfur 2006, 181 (2), 405–412.
(231) Cappelli, A.; Matarrese, M.; Moresco, R. M.; Valenti, S.; Anzini, M.; Vomero, S.;
Turolla, E. A.; Belloli, S.; Simonelli, P.; Filannino, M. A.; Lecchi, M.; Fazio, F. Bioorg. Med.
Chem. 2006, 14 (12), 4055–4066.
(232) Blair, A.; Zmuda, F.; Malviya, G.; Tavares, A. A. S.; Tamagnan, G. D.; Chalmers, A. J.;
Dewar, D.; Pimlott, S. L.; Sutherland, A. Chem. Sci. 2015, 6 (8), 4772–4777.
(233) Pang, X.; Xiang, L.; Ma, J.; Yang, X.; Yan, R. RSC Adv. 2016, 6 (113), 111713–
111717.
(234) Woolridge, E. M.; Rokita, S. E. Tet. Lett., 1991, 30 (45), 6117–6120.
(235) Brooke, G. M.; Wallias, D. I. J. Chem. Soc. Perk. Trans I. 1981, No. 5, 1417–1420.
(236) Simeon, F. G.; Brown, A. K.; Zoghbi, S. S.; Patterson, V. M.; Innis, R. B.; Pike, V. W. J.
Med. Chem. 2007, 50 (14), 3256–3266..
(237) Perrone, M. G.; Malerba, P.; Uddin, Md. J.; Vitale, P.; Panella, A.; Crews, B. C.; Daniel,
C. K.; Ghebreselasie, K.; Nickels, M.; Tantawy, M. N.; Manning, H. C.; Marnett, L. J.;
Scilimati, A. Eur. J. Med. Chem. 2014, 80, 562–568.
(238) Gonzalez, A. Z.; Li, Z.; Beck, H. P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.;
Eksterowicz, J.; Fox, B. M.; Fu, J.; Huang, X.; Houze, J.; Jin, L.; Li, Y.; Ling, Y.; Lo, M.-C.;
Long, A. M.; McGee, L. R.; McIntosh, J.; Oliner, J. D.; Osgood, T.; Rew, Y.; Saiki, A. Y.;

237
Shaffer, P.; Wortman, S.; Yakowec, P.; Yan, X.; Ye, Q.; Yu, D.; Zhao, X.; Zhou, J.; Olson,
S. H.; Sun, D.; Medina, J. C. J. Med. Chem. 2014, 57 (7), 2963–2988.
(239) Vara, B. A.; Johnston, J. N. J. Am. Chem. Soc. 2016, 138 (42), 13794–13797.
(240) Oslob, J.; Aubele, D.; Kim, J.; McDowell, R.; Song, Y.; Sran, A.; Zhong, M (MyoKardia
Inc.) US Patent 2016243100 A1, 2016
(241) Boehmer, J. E.; Mclachlan, M. M. W. (Syngenta Ltd.) WO2007096576 A1, 2007
(242) Anders, E.; Opitz, A.; Bauer W. Synthesis 1991, No. 12, 1221–1227.
(243) Samori, C.; Guerrini, A.; Varchi, G.; Fontana, G.; Bombardelli, E.; Tinelli, S.; Beretta,
G. L.; Basili, S.; Moro, S.; Zunino, F.; Battaglia, A. J. Med. Chem. 2009, 52 (4), 1029–
1039.
(244) Liu, W.; Huang, X.; Cheng, M.-J.; Nielsen, R. J.; Goddard III, W. A.; Groves, J. T.
Science 2012, 337 (6100), 1322–1325.
(245) Halperin, S. D.; Fan, H.; Chang, S.; Martin, R. E.; Britton, R. Angew. Chem. Int. Ed.
2014, 53 (18), 4690–4693.
(246) Braun, M.-G.; Doyle, A. G. J. Am. Chem. Soc. 2013, 135 (35), 12990–12993.
(247) Rueda-Becerril, M.; Chatalova Sazepin, C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl,
P.; Paquin, J. F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134 (9), 4026–4029.
(248) Antelo, J. M.; Crugeiras, J.; Leis, J. R.; Rios, A. J. Chem. Soc. Perk. Trans. 2 2000,
No. 10, 2071–2076.
(249) Rueda-Becerril, M.; Sazepin, C. C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.;
Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134 (9), 4026–4029.
(250) Zhu, R.-Y.; Tanaka, K.; Li, G.-C.; He, J.; Fu, H.-Y.; Li, S.-H.; Yu, J.-Q. J. Chem. Am.
Soc. 2015, 137 (22), 7067–7070.
(251) Reddy, D. S.; Shibata, N.; Horikawa, T.; Suzuki, S.; Nakamura, S.; Toru, T.; Shiro, M.
Chem. Asian J. 2009, 4 (9), 1411–1415.

238
(252) Paull, D. H.; Scerba, M. T.; Alden-Danforth, E.; Widger, L. R.; Lectka, T. J. Am. Chem.
Soc. 2008, 130 (51), 17260.
(253) Hull, K. L.; Anani, W. Q.; Sanford, M. S. J. Am. Chem. Soc. 2006, 128 (22), 7134–
7135.
(254) McMurtrey, K. B.; Racowski, J. M.; Sanford, M. S. Org. Lett. 2012, 14 (16), 4094–4097.
(255) Halperin, S. D.; Kwon, D.; Holmes, M.; Regalado, E. L.; Campeau, L.-C.; DiRocco, D.
A.; Britton, R. Org. Lett. 2015, 17 (21), 5200–5203.
(256) Luo, Y.; Kerr, J. Bond Dissociation Energies. CRC Handbook of Chemistry and
Physics 2007, 65–98.
(257) Ying, W. W.; DesMarteau, D. D.; Gotoh, Y. Tetrahedron 1996, 52 (1), 15–22.
(258) Xue, X.-S.; Wang, Y.; Li, M.; Cheng, J.-P. J. Org. Chem. 2016, 81 (10), 4280–4289.
(259) Albeniz, A. C.; Espinet, P.; Lopez-Fernandez, R.; Sen, A. J. Am. Chem. Soc. 2002,
124 (38), 11278–11279.
(260) E. Umemura et al. (Meiji Seika Kaisha Ltd.), EP 2166 015 A1, 2010.
(261) Roy, A.; Schneller, S. W. Org. Lett. 2005, 7 (18), 3889–3891.
(262) Martin, R. E.; Morawitz, F.; Kuratli, C.; Alker, A. M.; Alanine, A. I. Eur. J. Org. Chem.
2012, 2012 (1), 47–52.
(263) Lehmann, J.; Alzieu, T.; Martin, R. E.; Britton, R. Org. Lett. 2013, 15 (14), 3550–3553.
(264) Goetz, A. E.; Garg, N. K. Nat. Chem. 2013, 5 (1), 54–60.
(265) Xi, L.-Y.; Zhang, R.-Y.; Liang, S.; Chen, S.-Y.; Yu, X.-Q. Org. Lett. 2014, 16 (20),
5269–5271.
(266) Wei, Y.; Yoshikai, N. J. Am. Chem Soc. 2013, 135 (10), 3756–3759.
(267) Gati, W.; Rammah, M. M.; Rammah, M. B.; Couty, F.; Evano, G. J. Am. Chem. Soc.
2012, 134 (22), 9078–9081.

239
(268) Martin, R. E.; Lehmann, J.; Alzieu, T.; Lenz, M.; Corrales, M. A. C.; Aebi, J. D.; Marki,
H. P.; Kuhn, B.; Amrein, K.; Mayweg, A. V.; Britton, R. Org. Bio. Chem. 2016, 14 (25),
5922–5927.
(269) Perrin, D. D.; Dempsey, B.; Serjeant, E. P. pKa Prediction for Organic Acids and
Bases, Chapman and Hall, London, 1981.
(270) Teare, H.; Robins, E. G.; Arstad, E.; Luthra, S. K.; Gouverneur, V. Chem. Commun.
2007, No. 23, 2330–2332.
(271) Studer, A. A. Angew. Chem. Int. Ed. 2012, 51 (36), 8950–8958.
(272) Prakash, G. K. S.; Ganesh, S. K.; Jones, J.-P.; Kulkarni, A.; Masood, K.; Swabeck, J.
K.; Olah, G. A. Angew. Chem. Int. Ed. 2012, 51 (48), 12090–12094.
(273) Gu, Y.; Leng, X.; Shen, Q. Nat. Commun. 2014, 5.
(274) Matheis, C.; Jouvin, K.; Goossen, L. J. Sandmeyer. Org. Lett. 2014, 16 (22), 5984–
5987.
(275) Feng, Z.; Min, Q.-Q.; Zhang, X. Org. Lett. 2016, 18 (1), 44–47.
(276) Xu, L.; Vicic, D. A. J. Am. Chem. Soc. 2016, 138 (8), 2536–2539.
(277) Bour, J. R.; Kariofillis, S. K.; Sanford, M. S. Organometallics 2017, 36 (7), 1220–1223.
(278) Meanwell, M.; Britton, R. Synthesis 2018, 50 (6), 1228–1236.
(279) Deng, W. P.; Nam, G.; Fan, J. F.; Kirk, K. L. J. Org. Chem. 2003, 68 (7), 2798–2802.
(280) Zhou, Q.; Ruffoni, A.; Gianatassio, R.; Fujiwara, Y.; Sella, E.; Shabat, D.; Baran, P. S.
Angew. Chem. Int. Ed. 2013, 52 (14), 3949–3952.
(281) Taguchi, T.; Kitagawa, O.; Morikawa, T.; Nishiwaki, T.; Uehara, H.; Endo, H.;
Kobayashi, Y. Tet. Lett. 1986, 27 (50), 6103–6106.
(282) Murakami, S.; Ishii, H.; Tajima, T.; Fuchigami, T. Tetrahedron 2006, 62 (15), 3761–
3769.

240
(283) Jung, J.; Kim, E.; You, Y.; Cho, E. J. Adv. Synth. Catal. 2014, 356 (13), 2741–2748.
(284) Ohtsuka, Y.; Yamakawa, T. Tetrahedron 2011, 67 (12), 2323–2331.
(285) Shao, C.; Shi, G.; Zhang, Y.; Pan, S.; Guan, X. Org. Lett. 2015, 17 (11), 2652–2655.
(286) Chen, H.; Li, P.; Wang, M.; Wang, L. Org. Lett. 2016, 18 (19), 4794–4797.
(287) Wang, L.; Wei, X.-J.; Jia, W.-L.; Zhong, J.-J.; Wu, L.-Z.; Liu, Q. Org. Lett. 2014, 16
(22), 5842–5845.
(288) Ge, S.; Arlow, S. I.; Mormino, M. G.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136 (41),
14401–14404.
(289) Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Chem. Eur. J. 2015, 21
(37), 12836–12865.
(290) Douglas, J. J.; Albright, H.; Sevrin, M. J.; Cole, K. P.; Stephenson, C. R. J. Angew.
Chem. Int. Ed. 2015, 54 (49), 14898–14902.
(291) Meanwell, M.; Nodwell, M. B.; Martin, R. E.; Britton, R. Angew. Chem. Int. Ed. 2016, 55
(42), 13244–13248.
(292) Zhang, P.; Li, M.; Xue, X.-S.; Xu, C.; Zhao, Q.; Liu, Y.; Wang, H.; Guo, Y.; Lu, L.; Shen,
Q. J. Org. Chem. 2016, 81 (17), 7486–7509.
(293) Lefebvre, Q.; Fava, E.; Nikolaienko, P.; Rueping, M. Chem. Commun. 2014, 50 (50),
6617–6619.
(294) Parratt, J. R.; Winslow, E. Br. J. Pharamcol. 1971, 43 (3), 612.
(295) Hassan, A. E. A.; Abou-Elkhair, R. A. I.; Riordan, J. M.; Allan, P. W.; Parker, W. B.;
Khare, R.; Waud, W. R.; Montgomery, J. A.; Secrist III, J. A. Eur. J. Med. Chem. 2012,
47, 167–174.
(296) Nodwell, M. B.; Yang, H.; Colovic, M.; Yuan, Z.; Merkens, H.; Martin, R. E.; Benard, F.;
Schaffer, P.; Britton, R. J. Am. Chem. Soc. 2017, 139 (10), 3595–3598.
(297) Haas, A.; Spitzer M.; Lieb, M. Chem. Ber. 1988, 121, 1329-1340

241
(298) Krier, M.; Bret, G.; Rognan, D. J. Chem. Inf. Model. 2006, 46, 512-524
(299) DiRocco, D. A.; Ji, Y.; Sherer, E. C.; Klapars, A.; Reibarkh, M. et al. Science 2017,
356, 426-430
(300) DeVos, S. L.; Miller, T. M. Neurotherapeutics 2013, 10, 486-497
(301) Malek-Adamian, E.; Guenther, D. C.; Matsuda, S.; Martínez-Montero, S. et al. J. Am.
Chem. Soc. 2017, 139, 14542-14555

242
Appendix A. Liquid-Chromatography Chromatograms

243

244

245

246

247

248

249

250

251

252

253

254

255

256

257
(+/-)-224-Bis-PNB
(-)-224-Bis-PNB

258
(+/-)-227-Bis-PNB
(+)-227-Bis-PNB

259
(+/-)-229-Bis-PNB
(-)-229-Bis-PNB

260
(+/-)-220-Bis-PNB
(-)-220-Bis-PNB

261
(+/-)-222-Bis-PNB
(-)-222-Bis-PNB

262
(+/-)-200-Bis-PNB
(-)-200-Bis-PNB

263
(+/-)-200-Bis-PNB
(-)-200-Bis-PNB derived from 223

264
(+/-)-200-Bis-PNB
(-)-200-Bis-PNB derived from 225

265
(+/-)-221-Bis-PNB
(-)-221-Bis-PNB

266
(+/-)-219-Bis-PNB
(-)-219-Bis-PNB

267
(+/-)-230-Bis-PNB
(-)-230-Bis-PNB
268
(+/-)-228
(-)-228

269
(+/-)-237-Bis-PNB
(+/-)-237-Bis-PNB

270
(+/-)-238
238
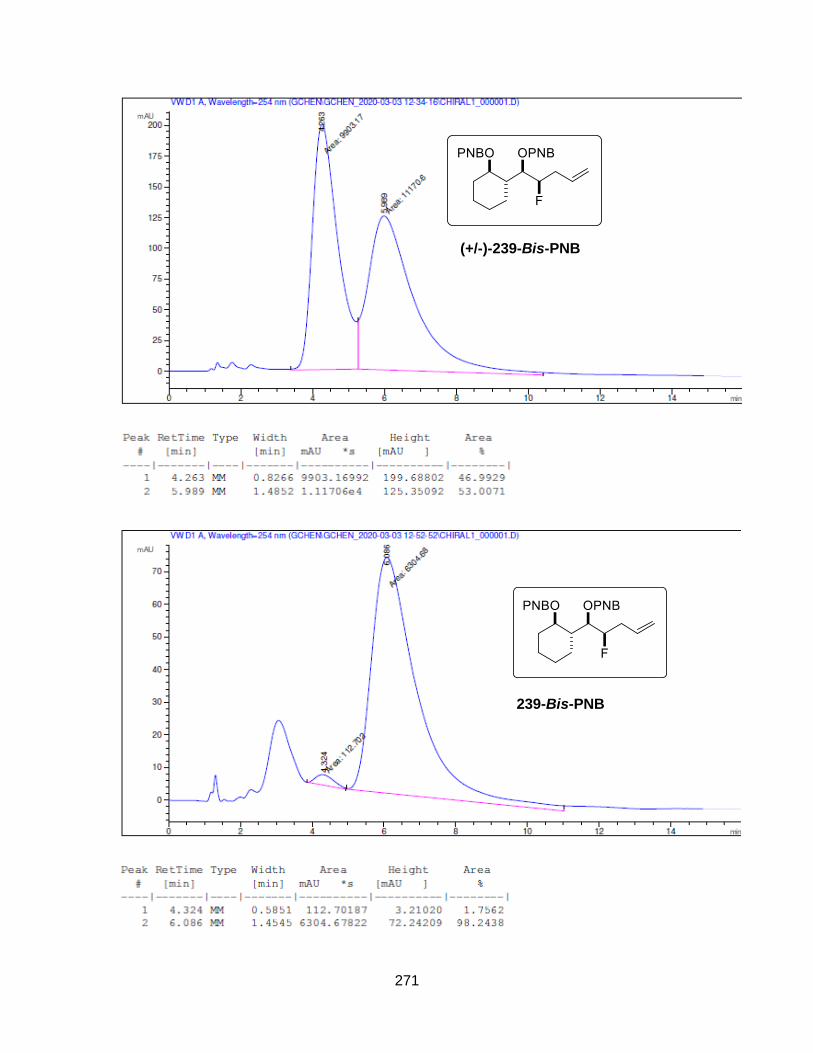
271
(+/-)-239-Bis-PNB
239-Bis-PNB

272
(+/-)-241
241

273
(+/-)-242
242

274
(+/-)-243-Bis-PNB
243-Bis-PNB

275
(+/-)-243-Bis-PNB
243-Bis-PNB derived
from 244

276
(+/-)-246-diol
246-diol

277
(+/-)-247-Bis-PNB
247-Bis-PNB