a eukaryotic translation initiation factor 4e-binding protein promotes mrna decapping and is
TRANSCRIPT

A Eukaryotic Translation Initiation Factor 4E-Binding ProteinPromotes mRNA Decapping and Is Required for PUF Repression
Nathan H. Blewetta,b and Aaron C. Goldstrohma,b
Cellular and Molecular Biology Training Programa and Department of Biological Chemistry,b University of Michigan Medical School, Ann Arbor, Michigan, USA
PUF proteins are eukaryotic RNA-binding proteins that repress specific mRNAs. The mechanisms and corepressors involved inPUF repression remain to be fully identified. Here, we investigated the mode of repression by Saccharomyces cerevisiae Puf5pand Puf4p and found that Puf5p specifically requires Eap1p to repress mRNAs, whereas Puf4p does not. Surprisingly, we ob-served that Eap1p, which is a member of the eukaryotic translation initiation factor 4E (eIF4E)-binding protein (4E-BP) class oftranslational inhibitors, does not inhibit the efficient polyribosome association of a Puf5p target mRNA. Rather, we found thatEap1p accelerates mRNA degradation by promoting decapping, and the ability of Eap1p to interact with eIF4E facilitates thisactivity. Deletion of EAP1 dramatically reduces decapping, resulting in accumulation of deadenylated, capped mRNA. In sup-port of this phenotype, Eap1p associates both with Puf5p and the Dhh1p decapping factor. Furthermore, recruitment of Eap1pto downregulated mRNA is mediated by Puf5p. On the basis of these results, we propose that Puf5p promotes decapping by re-cruiting Eap1p and associated decapping factors to mRNAs. The implication of these findings is that a 4E-BP can repress proteinexpression by promoting specific mRNA degradation steps in addition to or in lieu of inhibiting translation initiation.
Precise regulatory mechanisms are crucial for the execution ofgene expression programs and integration of signals. As inter-
mediaries between genes and proteins, mRNAs are an importantnexus for regulation. Posttranscriptional control of mRNA stabil-ity and translation is achieved through the concerted action ofRNA binding factors, RNA decay enzymes, and the translationmachinery (53, 62). Specific mRNAs are targeted for regulation byRNA binding factors that recognize sequences often found in the3= untranslated region (3=UTR). PUF proteins (Pumilio andFem-3 binding factor) are one class of regulators (47, 73). PUFproteins are defined by a conserved RNA binding domain thatmediates high-affinity binding to specific, 8- to 10-nucleotide, sin-gle-stranded RNA sequences (41, 48, 70, 76, 77). PUFs controldiverse biological processes, including cell proliferation, develop-ment, fertility, and neurological functions (3, 7, 16, 17, 20, 36, 38,39, 46, 52, 59, 75, 77). At the root of these functions lies the abilityof PUFs to repress protein production from target mRNAs (47).The preponderance of evidence indicates that the major mecha-nism of PUF-mediated repression is by enhancing mRNA degra-dation (23, 47, 51, 71). In several cases, PUFs were shown to ac-celerate mRNA decay by removal of the 3= polyadenosine tail (6,21, 23, 24, 28, 51). PUFs were also reported to inhibit translation(9, 10, 26, 63, 72). A remaining challenge is to discover the core-pressors and mechanisms of PUF-mediated repression.
Saccharomyces cerevisiae possess six PUFs, each of which bind adistinct set of mRNAs, dictated by their unique RNA bindingspecificities (22, 73). Puf4p and Puf5p/Mpt5p bind multiplemRNAs (22, 60) and share at least one well-characterized target,the mRNA encoding HO endonuclease, which catalyzes switchingof the mating type (23, 28, 65). Puf4p and Puf5p were previouslyshown to bind specific sites in the HO mRNA 3=UTR and acceler-ate deadenylation and degradation of the message (23, 28). Dead-enylation is essential for repression by Puf4p (28); however, Puf5pcan still repress HO mRNA when deadenylation was blocked bydeletion of the CCR4 gene (23), which encodes the major dead-enylase (24, 67, 68). This finding indicated that Puf5p can repress
by a second, deadenylation-independent mechanism (28). An ad-ditional corepressor(s) may be necessary for Puf5p activity.
We report here that a eukaryotic translation initiation factor4E (eIF4E)-binding protein (4E-BP), Eap1p, serves as an essentialcorepressor for Puf5p. 4E-BPs are found throughout eukaryotesand are thought to inhibit translation by binding to the 5= cap-bound initiation factor eIF4E, thereby blocking interaction withinitiation factor eIF4G (62). 4E-BPs possess a conserved eIF4Ebinding motif, YXXXXL� (� indicates a hydrophobic aminoacid, and X indicates any residue) (27, 42). 4E-BPs might globallyreduce cap-dependent translation; however, specific examplesdemonstrate more-specialized roles (62). Two 4E-BPs have beenidentified in S. cerevisiae, Caf20p and Eap1p (1, 14). Both containan eIF4E binding motif but are otherwise unrelated. Neither pro-tein is essential for growth under standard conditions, but severalmutant phenotypes have been described (8, 14, 32, 44, 45, 61).Eap1p was originally identified based on its ability to bind to eIF4Eand was shown to compete with eIF4G (14). Therefore, like other4E-BPs, Eap1p was proposed to repress by inhibiting translationinitiation.
In this work, we show that Eap1p is required for Puf5p-medi-ated mRNA repression but is not necessary for Puf4p function. Incontrast, Caf20p is fully dispensable for regulation by both Puf4pand Puf5p. Translational analysis demonstrates that Eap1p doesnot affect global translation nor does it inhibit polyribosome as-sociation of a Puf5p-targeted mRNA. Instead, we identify a novelactivity of Eap1p to promote degradation of specific mRNAs, in-
Received 11 April 2012 Returned for modification 4 May 2012Accepted 7 August 2012
Published ahead of print 13 August 2012
Address correspondence to Aaron C. Goldstrohm, [email protected].
Supplemental material for this article may be found at http://mcb.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/MCB.00483-12
October 2012 Volume 32 Number 20 Molecular and Cellular Biology p. 4181–4194 mcb.asm.org 4181
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

cluding a Puf5p target mRNA. Intriguingly, this activity is facili-tated by the interaction of Eap1p with eIF4E. We find that deletionof the EAP1 gene causes deadenylated, capped mRNA to accumu-late to substantial levels, indicating that Eap1p functions to pro-mote removal of the mRNA’s 5= 7-methylguanosine cap. In accor-dance, coimmunoprecipitation experiments indicate that Eap1passociates with Puf5p and the decapping factor Dhh1p. Together,these results provide a new regulatory mechanism for a member ofthe diverse class of eIF4E-binding proteins, enhancement ofmRNA decapping.
MATERIALS AND METHODSYeast strains. Yeast strains were obtained from Open Biosystems unlessotherwise noted. The yeast strains used in this study were AGY111(BY4741 MATa his3�1 leu2�0 met15�0 ura3�0), AGY153 (BY4741MATa eap1::KanR), AGY152 (BY4741 MATa caf20::KanR), AGY150(BY4741 MATa puf5::KanR), AGY109 (BY4741 MATa puf4::KanR), andAGY151 (BY4741 MATa dhh1::KanR).
Plasmids. Plasmid ACG858 (YCp33 HIS3 HO 3=UTR) was previouslydescribed by Goldstrohm et al. (23). Plasmid ACG399 (YCp33 LacZ HO3=UTR) was derived from ACG858 by replacing the HIS3 open readingframe (ORF) with the coding sequence for �-galactosidase (�-Gal). Plas-mid ACG441 (YEp181 PUF5-T7) was previously described by Gold-strohm et al. (23). Plasmid ACG705 (p415 GPD PUF4-T7) was previouslydescribed by Hook et al. (28). Plasmid ACG137 (pACG1 NTB) containsthe ADH1 promoter and 3=UTR. The ACG137 plasmid has a 2� origin ofreplication and zeocin selectable marker. Plasmid ACG693 (pACG1 NTB-EAP1) was created by inserting the EAP1 open reading frame into KpnIand NotI sites in pACG1 NTB. To construct plasmid NB1 (YEp181 EAP1-FLAG), EAP1 was PCR amplified from S288C genomic DNA and clonedinto XmaI sites of YEp181. The C-terminal FLAG epitope was added byinverse PCR using primers NB85/86. To construct plasmid NB2 (YEp181EAP1 mt Y109A/L114A-FLAG), QuikChange PCR (Stratagene) was per-formed on plasmid NB1 with primers AG787/788 to mutate Y109A andL114A. To construct plasmid NB3 (pACG1-eIF4E-T7), the CDC33 ORF,encoding eIF4E, was PCR amplified from S288C genomic DNA andcloned into the KpnI and NotI sites of pACG1 plasmid. All plasmids wereverified by restriction digestion and DNA sequencing.
Oligonucleotides. Synthetic oligonucleotides were purchased from In-tegrated DNA Technologies. The synthetic oligonucleotides used in this studywere as follows: NB64 (5=-TCCATTCCCGGGGTTTTAATGTATTGAAAATCACTTAGTTGTATATAGCC-3=), NB65 (5=-GCAAGGCCCGGGGCTTTCAGGCGCAGAAAACCTGAAAA-3=), NB85 (5=-GATGCGTAACGAGCGAGTACTTGACAGG-3=), NB86 (5=-TCACTTGTCATCGTCATCCTTGTAATCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCTTTTATATTCTTTTTAGAG-3=), AG787 (5=-GCAATACAAGACATATGCAGCGTCCATGAATGAAGCGTATCATTTGAAACCATCTTTGGC-3=), AG788 (5=-GCCAAAGATGGTTTCAAATGATACGCTTCATTCATGGACGCTGCATATGTCTTGTATTGC-3=), AG1029(5=-GCACGGTACCTATGTCCGTTGAAGAAGTTAGCAAG-3=), andAG1030 (5=-GCACGCGGCCGCTTACAAGGTGATTGATGGTTGAGGG-3=). The HO ORF riboprobe T7 transcription template PCR primersused were NB22 (5=-GGATCCTAATACGACTCACTATAGGGAGAACCTGCGTTGTTACCACAACTCTTATGA-3=) and NB23 (5=-AAGTGGTCACAAAACAAGAGAAGTTCCG-3=). The HO 3=UTR riboprobe T7transcription template PCR primers used were NB92 (5=-GGATCCTAATACGACTCACTATAGGGAGACATCCAAAATATTAAATTTTACTTTTATTAC-3=) and NB98 (5=-GCAAGTATGTACCAGAAGCACGTGAA-3=). The LacZ 3=UTR riboprobe T7 transcription template PCR primerswere NB92 (5=-GGATCCTAATACGACTCACTATAGGGAGACATCCAAAATATTAAATTTTACTTTTATTAC-3=) and NB115 (5=-CATCAGCCGCTACAGTCAACAGCAA-3=). The oligonucleotide used in HO 3=UTRRNase H cleavage was NB99 (5=-ATACAGTGATGACCGCTG-3=). Theoligonucleotide used in LacZ 3=UTR RNase H cleavage was NB116 (5=-AACTGGAAGTCGCCGCGCCAC-3=). The RNR1 probes were pNB5 (5=-
AAAGCACATTCCTTCAAGGTGTCGTAAATCCCCTCGATAGAGTCCTCCTTC-3=) and pNB6 (5=-AAGAGGACATTTGAGGTTTTGGAGTACCGGCATTGAACAACGTTGGAGAGG-3=). The RPL41A probe waspNB76 (5=-CCGCTTATTTGGATCTGGCTCTCACCTTCCGTCTCTTTCTCTTAAG-3=). The SCR1 probe was pNB79 (5=-CGCCTCCATCACGGGTCACCTTTGCTGAC-3=). The HO quantitative reverse transcrip-tion-PCR (qRT-PCR) primers were NB36 (5=-CCTCATAAGCAGCAATCAATTCTATCTAT-3=) and NB90 (5=-TTTAATTTCACCGTTAGCCATCAGAA-3=). The 18S rRNA qRT-PCR primers were NB69 (5=-ACGGAAGGGCACCACCA-3=) and NB70 (5=-CCACCCACAAAATCAAGAAAGAGCTCTC-3=).
PUF repression assay. Yeast growth assays were performed to detectrepression by Puf4p and Puf5p as described by Goldstrohm et al. (23) withthe following modifications. Wild-type yeast strain BY4741 or gene-spe-cific deletion strains were transformed with the reporter gene YCp33 HOpHIS3-HO 3=UTR and either empty vector YEp181 or the PUF5 expressionplasmid YEp181 PUF5 or p415 GPD PUF4. Colonies were isolated andgrown to mid-log phase at 30°C, and the indicated number of cells wasspotted onto selective minimal medium with or without histidine. TheHis3p competitive inhibitor 3-aminotriazole was added at a final concen-tration of 1 mM to increase stringency. For assays presented in Fig. 1C, themedium was supplemented with 300 �g/ml zeocin to select for pACG1NTB EAP1 or the negative-control plasmid pACG1.
�-Galactosidase reporter assay. Wild-type or gene-specific deletionstrains were transformed with the YCp33 LacZ-HO3=UTR reporter gene.Each strain was then grown to mid-log phase, and 8.9 � 107 cells (3 opticaldensity at 600 nm [OD600] units) from each sample were harvested andresuspended in 100 �l of fresh medium. An equal volume of room tem-perature Beta-Glo (Promega) reagent was added to each tube. Sampleswere transferred to 96-well plate and incubated for 1 h at 25°C. Lumines-cence measurements were made using a GloMax Multi� detection system(Promega). Specific signals were 2 orders of magnitude above the back-ground level in wells measured with Beta-Glo reagent or medium or inempty wells. Each assay was performed with five biological replicates, anddata are plotted as the mean value of relative light units with standarderror of the mean.
Polyribosome fractionation. Yeast cultures were seeded in 250 ml ofthe appropriate medium at an optical density at 600 nm (OD600) of 0.2and grown to an OD600 of 0.8. The cells were rapidly harvested at 4°C, andall subsequent steps were carried out in a cold room. When indicated,cycloheximide (60 �g/ml) or EDTA (50 mM) was added to cultures,which were immediately poured into cold centrifuge bottles with one-third volume of crushed ice. The cells were pelleted by centrifugation at3,200 � g for 5 min. The medium was decanted, and the cells were washedwith 10 ml of ice-cold 50 mM Tris-Cl (pH 6.8), 100 mM NaCl, 30 mMMgCl2, and 50 �g/ml cycloheximide (or when indicated, 50 mM EDTAwas added in lieu of cycloheximide). The cells were pelleted again andresuspended in 650 �l of ice-cold 50 mM Tris-Cl (pH 6.8), 100 mM NaCl,30 mM MgCl2, and 50 �g/ml cycloheximide (or 50 mM EDTA was addedin lieu of cycloheximide) with 20 U/ml RNasin, and 2� protease inhibi-tors (2 mM phenylmethylsulfonyl fluoride [PMSF], 100 �g/ml aprotinin,100 �g/ml pepstatin, 100 �g/ml leupeptin) in 1.5-ml tubes containing 650�l glass beads. Cells were lysed in a FastPrep (MP Biomedicals) 2 times for60 s each time at 6.5 m/s. Cell debris and beads were removed from theextract by centrifugation for 5 min at 1,400 � g. The extracts were diluted1/200, and the absorbance at 260 nm (A260) was measured with a Nano-Drop spectrophotometer (Thermo Scientific). Twenty A260 units of ex-tract were loaded onto each sucrose gradient. Ribosome runoff was per-formed as described above, except that cycloheximide was omitted fromall steps.
To fractionate ribosomes and polyribosomes, 7 to 47% sucrose gradi-ents were prepared using a Gradient Master (BioComp). Samples wereapplied to the top of each gradient. In Fig. 2, the gradients were centri-fuged 2 h and 30 min at 28,000 rpm in an SW41 Ti rotor at 4°C. In Fig. 3,the gradients were centrifuged 4 h to resolve 40S, 60S, 80S, and polyribo-
Blewett and Goldstrohm
4182 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

some peaks. The gradients were fractionated using a Biologic DuoFlowsystem with an Econo Gradient peristaltic pump (Bio-Rad) at a rateof 1.75 ml/min while collecting 500-�l fractions. Gradient tubes werepierced with a gradient fractionation device (Brandel), and gradientswere pumped from the bottom of the tube using Fluorinert. A260 readingswere made continuously during fractionation using a Bio-Rad Quadtecspectrophotometer. Samples were stored at �80°C after fractionation.RNA was extracted from a total of 460 �l of each gradient fraction usingthe Maxwell RNA purification system and 16 cell LEV RNA purificationkit (Promega). The percentage of each mRNA in each fraction was calcu-lated relative to the total detected by Northern blotting in all fractions.Values from each fraction were represented as the mean value of multiplereplicates. For Western blot analysis of fractions, 45 �l of each fractionwas boiled 5 min in 1� SDS-PAGE loading buffer and then separated in a4 to 12% SDS-polyacrylamide gel.
Northern blotting. RNAs were separated on 1.4% agarose-formalde-hyde denaturing gels with 1� morpholinepropanesulfonic acid (MOPS)running buffer and transferred to Immobilon NY� membranes (Milli-pore) using a downward transfer method. The membranes were UVcross-linked with a UVP CL1000. Membrane blocking and hybridizationwere performed using oligo-hyb or ultra-hyb hybridization buffers (Am-bion). End-labeled oligonucleotide probes were hybridized in oligo-hybbuffer overnight at 42°C and washed two times for 30 min each time in 2�SSC (1� SSC is 0.15 M NaCl plus 0.015 M sodium citrate) with 0.5% SDSat 42°C. Body-labeled riboprobes for HO mRNA were hybridized in ultra-hyb buffer at 68°C and washed two times for 5 min each time in 2� SSCwith 0.1% SDS, and then washed two times for 15 min each time in 0.1�SSC with 0.1% SDS at 68°C. The membranes were exposed to phosphorscreens and scanned using a Typhoon Trio phosphorimager (GeneralElectric).
Measurement of mRNA half-life. Transcription shutoff using thiolu-tin (Pfizer) and RNA purification was performed as described previously(5). Specific Northern blot bands were quantitated on a Typhoon phos-phorimager using ImageQuant TL software. Each time point was normal-ized to the SCR1 RNA in the same sample, and decay was calculatedrelative to the time of drug addition. mRNA half-lives were calculatedwith GraphPad Prism software using nonlinear regression and one-phasedecay analysis of biological replicate samples.
Polyadenosine tail length analysis. Forty micrograms of total RNAwas first treated with Turbo DNase (Ambion). RNA was precipitated with1/10 volume of 3 M sodium acetate and 3 volumes of 100% ethanol at�20°C. The pellets were washed with 70% ethanol and resuspended in 24�l buffer A (20 mM KCl, 1 mM EDTA), 20 pmol of cleavage oligonucle-otide NB99, and either 5 �l of water or 5 �l of oligo(dT) (500 ng/�l). Thesamples were heated for 5 min at 90°C, cooled for 5 min at 65°C, and thencooled for 20 min at room temperature. To each sample, 30 �l of buffer B(40 mM Tris-Cl [pH 8.0], 56 mM MgCl2) was added, along with 1 unit ofRNase H (NEB), and 1 unit of RNasin Plus (Promega). The samples wereincubated for 1 h at 37°C. RNA was then precipitated with 1/10 volume of3 M sodium acetate and then with 3 volumes of ethanol at �20°C. RNApellets were washed with 70% ethanol and resuspended in denaturingpolyacrylamide Northern loading buffer. After heating for 5 min at 95°C,the RNA was separated on a 6% polyacrylamide, 7 M urea denaturing geland transferred to Immobilon NY� with a transblotter (Bio-Rad). North-ern blotting was performed as described above.
Xrn1 sensitivity assay. Twenty micrograms of total RNA extractedfrom wild-type and eap1 strains was used for each reaction. Where indi-cated, control samples were treated with 15 units of tobacco acid pyro-phosphatase (TAP) (Epicentre) to remove 5= cap structures using thesupplied TAP reaction buffer for 1 h at 37°C. RNA was precipitated, pel-leted, and washed with 70% ethanol. RNA was then digested with 10 unitsof Xrn1 (NEB) in reaction buffer with 1 unit of RNase Inhibitor Plus for 1h at 37°C. The RNA was precipitated, washed with 70% ethanol, andresuspended in Northern loading buffer.
Protein coimmunoprecipitation. Yeast cells expressing FLAG-EAP1,or empty vector (mock) as bait, and T7-tagged prey were grown overnightin the appropriate medium. On the next day, yeast cells were seeded to anoptical density, OD600, of 0.2 in 1 liter of medium and then grown to anOD600 of 0.8. The cells were then harvested, washed with TNMT250 (50mM Tris-Cl [pH 8.0], 250 mM NaCl, 2 mM MgCl2, 0.1% Tween 20),pelleted, and stored at �80°C. Anti-FLAG M2 agarose (Sigma) and a50-�l bed volume of beads were preequilibrated in 10 ml TNMT250 with300 �g/ml denatured salmon sperm DNA and 500 �g/ml bovine serumalbumin (BSA) for 1 h at 4°C. The beads were then washed twice for 10min each time with TNMT250. The cell pellets were thawed on ice, sus-pended in an equal volume of TNMT250 with 10 �g RNase A (Fermen-tas), 100 units of RNase One (Promega), and 2� protease inhibitors. Thecells were transferred to 15-ml tubes containing 700 �l acid-washed glassbeads. Lysis was performed with a FastPrep (MP) with three 60-s pulses at6.5 m/s. Cell debris was pelleted by centrifugation at 6,000 � g for 10 min,and the supernatant was transferred to a fresh tube and pelleted again bycentrifugation at 12,000 � g for 10 min. The resulting lysate was pre-cleared by incubation with IgG-agarose to remove nonspecific interac-tions. After preclearing, RNase-treated lysates were applied to anti-FLAGM2 affinity agarose (Sigma) and incubated for 2 h at 4°C. The beads werepelleted at 1,000 � g for 5 min, the supernatant was removed, and beadswere washed 5 times for 15 min each time with 10 ml TNMT250. After thefinal wash, beads were transferred to a microcentrifuge tube. The beadswere resuspended in 100 �l of TNMT250 and 150 ng of FLAG peptide(Sigma). Elution was performed 30 min at 4°C with end-over-end rota-tion. After incubation, the supernatant was passed through a Bio-Radmini-spin column to remove beads. Eluates were then separated by 4 to12% SDS-PAGE gels and probed with anti-FLAG antibody (Sigma) andanti-mouse horseradish peroxidase (HRP)-conjugated monoclonal anti-body (Thermo Scientific), anti-T7 monoclonal antibody linked to HRP(Novagen), or antiactin monoclonal antibody (MP Biomedical).
RNA coimmunoprecipitation. Coimmunoprecipitation of mRNAwith Eap1p was performed as described above with the following altera-tions. Yeast cells were lysed in TKNM140 buffer (40 mM Tris-Cl [pH 8.0],140 mM KCl, 0.1% NP-40, 2 mM MgCl2, 40 units/ml RNasin Plus with2� protease inhibitors). The beads were washed five times for 10 min eachtime in TKNM140 buffer, and then protein and RNA were eluted as de-scribed above with 150 ng of FLAG peptide.
Quantitative PCR detection of HO mRNA immunoprecipitation.Input and elution samples were first treated with 4 units of Turbo DNase(Ambion) in 1� Turbo buffer at 37°C for 30 min. RNA was precipitatedwith 1/10 volume sodium acetate and 3 volumes ethanol for 1 h at �20°C,and the pellets were washed with 70% ethanol. qRT-PCR was carried outas described previously (5). Briefly, HO cDNA was generated withGoScript reverse transcriptase (Promega) using 20 pmol of primer NB90.Amplification of PCR products was measured using GoTaq qPCR mastermix (Promega) with 200 nM each primer in 50-�l reactions. A Bio-RadCFX 96 C1000 real-time PCR instrument was used for all assays. Eachimmunoprecipitation was performed in triplicate, and the elution thresh-old cycle (CT) was normalized to HO input CT for each sample. Foldenrichment was then calculated relative to the mock immunoprecipita-tion samples using the ��CT method (40, 57).
RESULTSThe eIF4E-binding protein, Eap1p, is essential for Puf5p-medi-ated repression. To identify corepressors necessary for PUF-mediated repression, we undertook a genetic approach using areporter gene that is repressed by Puf4p and Puf5p (23, 28). TheHIS3-HO 3=UTR reporter gene was created by replacing the openreading frame of HO with the auxotrophic marker gene HIS3 (Fig.1A) (23). In wild-type cells, wherein Puf4p and Puf5p levels do notfully silence the reporter, introduction of the reporter confers his-tidine biosynthesis and thus growth on medium lacking histidine(Fig. 1B, wild-type [WT] strain). As previously demonstrated (23,
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4183
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

28), increased expression of Puf4p or Puf5p repressed HIS3-HOexpression, thereby abrogating growth in the absence of histidine(Fig. 1B, wild-type strain with PUF5 and PUF4 expression plas-mids). Importantly, repression depends on the PUF binding sitesand the RNA binding activity of each PUF (23, 28).
If a corepressor is required for PUF repression, deletion of itsgene will result in loss of repression and thus growth on mediumlacking histidine. We tested candidate genes with known roles inmRNA degradation and translational control. Deletion of onegene encoding a 4E-BP, EAP1, abrogated Puf5p repression buthad no effect on Puf4p repression (Fig. 1B, �eap1 strain withPUF5 versus PUF4). Therefore, EAP1 is necessary for Puf5p re-pression and dispensable for Puf4p function, indicating a uniquemode of repression by Puf5p. Because Eap1p binds to eIF4E (14),our data suggest that Puf5p may repress translation using Eap1p as
a corepressor. We also tested the second known yeast 4E-BP,CAF20. In contrast to Eap1p, Caf20p is not necessary for repres-sion by either PUF4 or PUF5, as revealed by the ability of each PUFto repress in cells lacking the CAF20 gene (Fig. 1B, �caf20 strain).This indicates that Puf4p represses through a separate mechanismthat does not require a 4E-BP.
If Eap1p is a Puf5p corepressor, then overexpression of Eap1pmay also repress the HIS3-HO reporter. Indeed, when Eap1p wasoverexpressed by introducing an Eap1p expression plasmid intowild-type cells, the HIS3-HO reporter was repressed (Fig. 1C).Eap1p inhibitory activity was dependent on Puf5p, as Eap1p nolonger repressed in a puf5 deletion strain (Fig. 1C). Together, thesedata demonstrate that Eap1p and Puf5p function together.
To further measure the contribution of Eap1p to Puf5p to re-pression, we created a reporter gene encoding �-galactosidase (�-
FIG 1 EAP1 is required for Puf5p-mediated repression. (A) The HIS3-HO 3=UTR reporter contains the HIS3 open reading frame (ORF) with the HO 3=untranslated region (3=UTR) containing the Puf4p binding site (Puf4BS) and Puf5p binding site (PUF5BS). (B) Growth assays measure repression of HIS3-HOreporter in the wild-type (WT) strain and in eap1 (�eap1) or caf20 (�caf20) deletion strains transformed with plasmids expressing PUF4 or PUF5 or with controlplasmid. The indicated numbers of cells (Cell #) were spotted onto medium with histidine (Control) or without histidine (� Histidine). Repression by each genewas scored by no growth on medium lacking histidine (�) or growth (�). (C) Growth assay for repression by PUF5 and EAP1 in wild-type and puf5 deletion(�puf5) cells. (D) Diagram of the LacZ-HO 3=UTR reporter mRNA. The LacZ HO 3=UTR mt reporter was created by mutating the Puf4p and Puf5p binding sites(Puf4BS mt and Puf5BS mt [mt stands for mutant]). (E) Graph of �-galactosidase (�-gal) activity from wild-type or mutant (mt) LacZ reporters measured fromequal number of wild-type, �puf5, or �eap1 cells. The fold change in relative light unit values is plotted relative to wild-type reporter in wild-type cells. (F) Graphof fold change in LacZ HO (WT or mt) mRNA levels, as measured by Northern blotting (see Fig. S1 in the supplemental material) and calculated relative to thewild-type reporter in wild-type cells. (G) Graph of fold change in the ratio of �-galactosidase activity in panel E to LacZ mRNA level in panel F. In each graph,mean values are plotted with standard errors (error bars) from multiple biological replicates.
Blewett and Goldstrohm
4184 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

Gal) controlled by the HO 3=UTR (LacZ-HO 3=UTR [Fig. 1D]).Protein expression from LacZ-HO was measured from an identi-cal number of cells using a luminescence-based �-Gal activity as-say in three genetic backgrounds: wild type, eap1 deletion, andpuf5 deletion. If Eap1p and Puf5p function together to repressHO, then �-Gal activity should increase when each is absent. In-deed, deletion of PUF5 resulted in a 6.5-fold increase in �-Galactivity relative to the wild type (Fig. 1E), and deletion of EAP1caused a 14.8-fold fold increase (Fig. 1E). For a positive control forcomparison, mutation of both PUF binding sites (LacZ-HO3=UTR mt [Fig. 1D]) resulted in a 6.2-fold increase (Fig. 1E) (28).These results indicate that Eap1p represses �-Gal synthesis or pro-motes its decay. To gain additional insight, the steady-state level ofLacZ mRNA was measured by Northern blotting (see Fig. S1 in thesupplemental material). Deletion of EAP1 increased LacZ-HOmRNA by 2.5-fold (Fig. 1F), suggesting that Eap1p represses LacZmRNA synthesis or promotes mRNA degradation. Consistentwith their role in promoting decay of HO mRNA, PUF bindingsite mutations or deletion of PUF5 caused a 2.3-fold increase inthe reporter mRNA (Fig. 1F). Deletion of EAP1 increased the ratioof �-Gal activity to LacZ mRNA by 5.6-fold relative to wild-typecells (Fig. 1G), suggesting that the amount of protein synthesizedper mRNA may increase. Together, our data demonstrate thatboth Puf5p and Eap1p repress a target mRNA and that Puf5pactivity is dependent on Eap1p. The results show that Eap1p re-duces both protein and mRNA expression, and given that 4E-BPsare generally thought to inhibit translation, we next investigatedthe effect of Eap1p on HO translation.
Eap1p does not inhibit polyribosome association of a Puf5p-regulated mRNA. 4E-BPs are proposed to inhibit translation byblocking interaction of eIF4E and eIF4G during initiation (62). Inthis context, we hypothesized that Puf5p may utilize Eap1p toinhibit HO translation. To measure the effect of Eap1p on thetranslation state of HO mRNA, we performed sucrose gradientultracentrifugation to separate ribosome-bound and -unboundmRNAs. If Eap1p inhibits initiation, Eap1p should decrease thepercentage of HO mRNA bound to ribosomes (ribosome occu-pancy) and reduce the number of ribosomes bound to HO mRNA.Therefore, deletion of EAP1 should increase the ribosome occu-pancy and density of HO mRNA.
To test these predictions, cell extracts from wild-type and eap1deletion strains were separated on 7 to 47% sucrose gradients.Each gradient was fractionated while monitoring UV absorptionto show separation of 80S ribosomes and polyribosomes (Fig. 2Aand B), corroborated by ethidium bromide staining of rRNAs(Fig. 2C). The chromatograms of wild-type and eap1 deletionstrains were highly similar, indicating that Eap1p does not sub-stantially alter global translation (compare Fig. 2A and B showingthe WT and �eap1 strains, respectively).
We next detected HO mRNA in the gradient fractions fromthree biological replicates to quantitate the HO translation state.In wild-type cells, 97% of total HO mRNA was in polyribosome-bound fractions (Fig. 2D and 2E, WT, fractions 7 to 19). For com-parison, the average ribosome occupancy for mRNAs in S. cerevi-siae is 71% (2). HO mRNA was predominantly detected infractions containing 3 or more ribosomes, steadily increasing inthe fractions containing 7 or more ribosomes (Fig. 2D, WT frac-tions 9 to 19). Less than 1% of HO mRNA was found in ribosome-free fractions (Fig. 2D, fractions 1 to 3), and only 2% was presentin fractions containing monoribosomes (Fig. 2D, lanes 1 to 9). For
a control, polyribosomes were dissociated with EDTA. This treat-ment caused HO mRNA to shift from the bottom to the top of thegradient (see Fig. S2 in the supplemental material), consistentwith HO being polyribosome associated. These results indicatethat HO mRNA efficiently engages with ribosomes in wild-typecells, contradicting the prediction that Eap1p inhibits translationinitiation of HO mRNA.
Next, the effect of Eap1p on HO translation state was investi-gated. Deletion of EAP1 did not alter the ribosome occupancy ofHO mRNA; 98% associated with polyribosomes, nearly identicalto the wild type (Fig. 2D and E, �eap1 strain, fractions 7 to 19).Like the wild type, less than 1% of HO mRNA was present in theribosome-free fractions (Fig. 2D, �eap1 strain, fractions 1 to 3).The ribosome density of HO mRNA actually decreased slightly inthe eap1 deletion strain, with the peak density shifting from poly-ribosome fraction 16 in wild-type cells to the less-dense fraction15 in eap1 deletion cells (Fig. 2D and E, �eap1 strain). Theseobservations contradict the prediction that deletion of EAP1would increase ribosome occupancy and density of HO mRNA.
The major difference observed between wild-type and eap1deletion strains is a change in the abundance of HO mRNA, whichincreased by 1.7-fold in the eap1 deletion strain (Fig. 2D and E,�eap1 strain). We conclude that Eap1p does not inhibit transla-tion initiation of HO mRNA but instead decreases the abundanceof HO mRNA, suggesting an effect on HO mRNA synthesis orstability.
We also investigated the effect of Eap1p on the translation stateof two mRNAs that are not PUF targets: the ribonucleotide reduc-tase mRNA, RNR1, and the large ribosomal subunit protein L47mRNA, RPL41A (22, 23, 60). Like HO, RNR1 is a low-abundance,cell cycle-regulated mRNA with a large ORF of 2,667 nucleotides(compared to HO ORF, which is 1,761 nt). The distribution ofRNR1 mRNA was not altered by Eap1p (Fig. 2F). All RNR1 asso-ciated with polyribosomes in wild-type and eap1 deletion cells(Fig. 2F. compare fractions 12 to 19 from the WT and �eap1strains). RPL41A is an abundant mRNA with a 78-nucleotide ORFthat engages, on average, one ribosome in wild-type cells (Fig. 2G,WT strain, peak fractions 4 to 6) (2). The ribosome association ofRPL41A mRNA was not altered by deletion of EAP1 (Fig. 2G,�eap1 strain). Therefore, EAP1 does not change the ribosomeassociation of two mRNAs that are not targeted by PUFs.
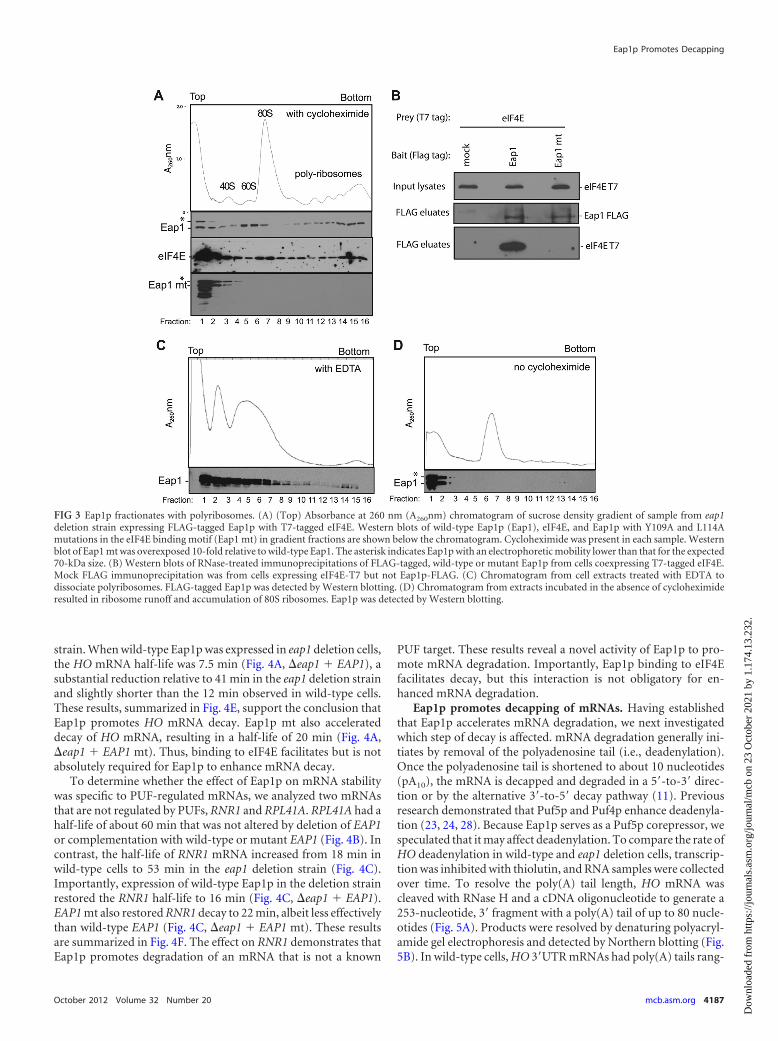
Eap1p associates with polyribosomes. We next evaluatedwhether Eap1p associates with ribosomes. If Eap1p blocks trans-lation initiation, then Eap1p would be expected to be found ex-clusively in translationally inactive, ribosome-free fractions of thesucrose gradient. Eap1p with a FLAG tag was expressed in the eap1deletion strain, and extracts were fractionated by sucrose densitygradients using conditions that separate ribosomal subunits,mono-, and polyribosomes (Fig. 3A). Three peaks of Eap1p wereobserved in the gradient fractions. At the top of the gradient, apeak was present in the ribosome-free fractions (Fig. 3A, fractions1 and 2). A second peak cofractionated with 60S ribosomal sub-unit (Fig. 3A, fraction 5 and 6). A third major peak of Eap1pfractionated with polyribosomes (Fig. 3A, fractions 14 to 16). In-terestingly, a slower-migrating Eap1p species was observed in thefirst two gradient fractions, perhaps the result of a posttransla-tional modification(s).
Eap1p binds eIF4E, and this interaction may mediate Eap1passociation with polyribosomes. First, we assessed the distributionof eIF4E in the gradient fractions. Western blotting of eIF4E re-
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4185
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

vealed that the protein was distributed throughout the gradient,with a major peak at the top of the gradient (Fig. 3A, middle panel,fractions 1 to 4) and a secondary peak corresponding to the 80Smonoribosome (Fig. 3A, fractions 6 to 8). Like Eap1p, eIF4E wasalso present in polyribosome fractions (Fig. 3A, fractions 9 to 16).To test whether Eap1p binding to eIF4E is necessary for polyribo-some association, the eIF4E binding motif, Y109XXXXL114, wasmutated by introducing two alanine substitutions, Y109A andL114A, to create Eap1p mt (mt stands for mutant) (14). This mu-tant was expressed in the eap1 deletion strain, and then extractfrom these cells was fractionated on a sucrose gradient. The Eap1pmutant was detected only in the first three fractions at the top ofthe gradient (Fig. 3A, Eap1 mt, fractions 1 to 3), indicating that theassociation of Eap1p with polyribosomes is dependent on bindingto eIF4E. To confirm that Eap1p mt no longer bound to eIF4E,wild-type Eap1p or Eap1p mt were immunoprecipitated. eIF4Ewas detected only in the wild-type Eap1p immunoprecipitate (Fig.3B); therefore, the mutations disrupted interaction with eIF4E.
Two controls were performed to verify that Eap1p associatedwith polyribosomes. First, extracts were treated with EDTA, re-sulting in collapse of polyribosomes into 40S and 60S peaks (Fig.3C) and causing Eap1p to fractionate predominantly at the top ofthe gradient (Fig. 3C, fractions 1 to 7). Importantly, the portion ofEap1p that fractionated with polyribosomes in wild-type cells(Fig. 3A, fractions 9 to 16) was greatly diminished (Fig. 3C, frac-tions 9 to 16). Second, ribosomes were allowed to elongate and“run off” by omitting cycloheximide (37), resulting in accumula-tion of 80S particles (Fig. 3D, fractions 6 and 7). If Eap1p wereengaged in translating polyribosomes, then runoff should causeEap1p to shift into lighter fractions. Indeed, Eap1p was detectedonly at the top of the gradient (Fig. 3D, fractions 1 and 2). Collec-tively, these data indicate that Eap1p associates with polyribo-somes. This finding was unexpected and is not consistent with themodel that Eap1p inhibits translation initiation. Instead, our find-ings indicate that Eap1p may promote a distinct mode of repres-sion that remained to be discovered.
Eap1p accelerates mRNA decay. Puf5p promotes degradationof the mRNAs it targets (23, 24, 28, 60). Because Eap1p serves as acorepressor for Puf5p and HO (Fig. 2D) and LacZ-HO 3=UTR(Fig. 1F) mRNA levels increased in the eap1 deletion strain, wereasoned that Eap1p may affect mRNA decay. To test this hypoth-esis, mRNA decay rates were measured in the presence or absenceof Eap1p. Cells were treated with thiolutin to inhibit transcription,and RNA samples were collected over time. Next, specific mRNAswere detected by Northern blotting (Fig. 4). In wild-type cells, thehalf-life of HO mRNA was 12 min (Fig. 4A), which is consistentwith past measurements (23, 28). Deletion of EAP1 dramaticallystabilized HO, increasing the half-life to 41 min (Fig. 4A). Theseresults represent the first demonstration that EAP1 affects the rateof mRNA decay.
We next tested whether the interaction of Eap1p with eIF4Ewas required for acceleration of mRNA decay. To do so, the eap1deletion strain was complemented with plasmid expressing eitherwild-type Eap1p (EAP1) or eIF4E binding-defective mutant(EAP1 mt). The HO mRNA half-life was then measured in each
FIG 2 Eap1p does not inhibit polyribosome association of a Puf5p-regulatedmRNA. Ribosome profiles of sucrose density gradients from wild-type (WT)cells (A) and eap1 deletion (�eap1) cells (B). UV absorbance at 260 nm(A260nm) was measured during collection of fractions to generate the chro-matograms. “Top” and “Bottom” refer to the relative position in the gradienttube. Ribosome species are indicated within the chromatogram. (C) Theethidium bromide-stained gel shows the rRNA (26S and 18S rRNA) content ofeach fraction from WT cells. (D) Northern blots of HO mRNA in gradientfractions from WT and �eap1 strains. (E) Quantitation of HO mRNA profileacross gradient fractions from three biological replicate samples of wild-type
and �eap1 strains. Mean values are plotted with standard errors (error bars).(F and G) Northern blots of RNR1 (F) and RPL41A (G) mRNAs in gradientfractions from WT and �eap1 strains.
Blewett and Goldstrohm
4186 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.
strain. When wild-type Eap1p was expressed in eap1 deletion cells,the HO mRNA half-life was 7.5 min (Fig. 4A, �eap1 � EAP1), asubstantial reduction relative to 41 min in the eap1 deletion strainand slightly shorter than the 12 min observed in wild-type cells.These results, summarized in Fig. 4E, support the conclusion thatEap1p promotes HO mRNA decay. Eap1p mt also accelerateddecay of HO mRNA, resulting in a half-life of 20 min (Fig. 4A,�eap1 � EAP1 mt). Thus, binding to eIF4E facilitates but is notabsolutely required for Eap1p to enhance mRNA decay.
To determine whether the effect of Eap1p on mRNA stabilitywas specific to PUF-regulated mRNAs, we analyzed two mRNAsthat are not regulated by PUFs, RNR1 and RPL41A. RPL41A had ahalf-life of about 60 min that was not altered by deletion of EAP1or complementation with wild-type or mutant EAP1 (Fig. 4B). Incontrast, the half-life of RNR1 mRNA increased from 18 min inwild-type cells to 53 min in the eap1 deletion strain (Fig. 4C).Importantly, expression of wild-type Eap1p in the deletion strainrestored the RNR1 half-life to 16 min (Fig. 4C, �eap1 � EAP1).EAP1 mt also restored RNR1 decay to 22 min, albeit less effectivelythan wild-type EAP1 (Fig. 4C, �eap1 � EAP1 mt). These resultsare summarized in Fig. 4F. The effect on RNR1 demonstrates thatEap1p promotes degradation of an mRNA that is not a known
PUF target. These results reveal a novel activity of Eap1p to pro-mote mRNA degradation. Importantly, Eap1p binding to eIF4Efacilitates decay, but this interaction is not obligatory for en-hanced mRNA degradation.
Eap1p promotes decapping of mRNAs. Having establishedthat Eap1p accelerates mRNA degradation, we next investigatedwhich step of decay is affected. mRNA degradation generally ini-tiates by removal of the polyadenosine tail (i.e., deadenylation).Once the polyadenosine tail is shortened to about 10 nucleotides(pA10), the mRNA is decapped and degraded in a 5=-to-3= direc-tion or by the alternative 3=-to-5= decay pathway (11). Previousresearch demonstrated that Puf5p and Puf4p enhance deadenyla-tion (23, 24, 28). Because Eap1p serves as a Puf5p corepressor, wespeculated that it may affect deadenylation. To compare the rate ofHO deadenylation in wild-type and eap1 deletion cells, transcrip-tion was inhibited with thiolutin, and RNA samples were collectedover time. To resolve the poly(A) tail length, HO mRNA wascleaved with RNase H and a cDNA oligonucleotide to generate a253-nucleotide, 3= fragment with a poly(A) tail of up to 80 nucle-otides (Fig. 5A). Products were resolved by denaturing polyacryl-amide gel electrophoresis and detected by Northern blotting (Fig.5B). In wild-type cells, HO 3=UTR mRNAs had poly(A) tails rang-
FIG 3 Eap1p fractionates with polyribosomes. (A) (Top) Absorbance at 260 nm (A260nm) chromatogram of sucrose density gradient of sample from eap1deletion strain expressing FLAG-tagged Eap1p with T7-tagged eIF4E. Western blots of wild-type Eap1p (Eap1), eIF4E, and Eap1p with Y109A and L114Amutations in the eIF4E binding motif (Eap1 mt) in gradient fractions are shown below the chromatogram. Cycloheximide was present in each sample. Westernblot of Eap1 mt was overexposed 10-fold relative to wild-type Eap1. The asterisk indicates Eap1p with an electrophoretic mobility lower than that for the expected70-kDa size. (B) Western blots of RNase-treated immunoprecipitations of FLAG-tagged, wild-type or mutant Eap1p from cells coexpressing T7-tagged eIF4E.Mock FLAG immunoprecipitation was from cells expressing eIF4E-T7 but not Eap1p-FLAG. (C) Chromatogram from cell extracts treated with EDTA todissociate polyribosomes. FLAG-tagged Eap1p was detected by Western blotting. (D) Chromatogram from extracts incubated in the absence of cycloheximideresulted in ribosome runoff and accumulation of 80S ribosomes. Eap1p was detected by Western blotting.
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4187
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.
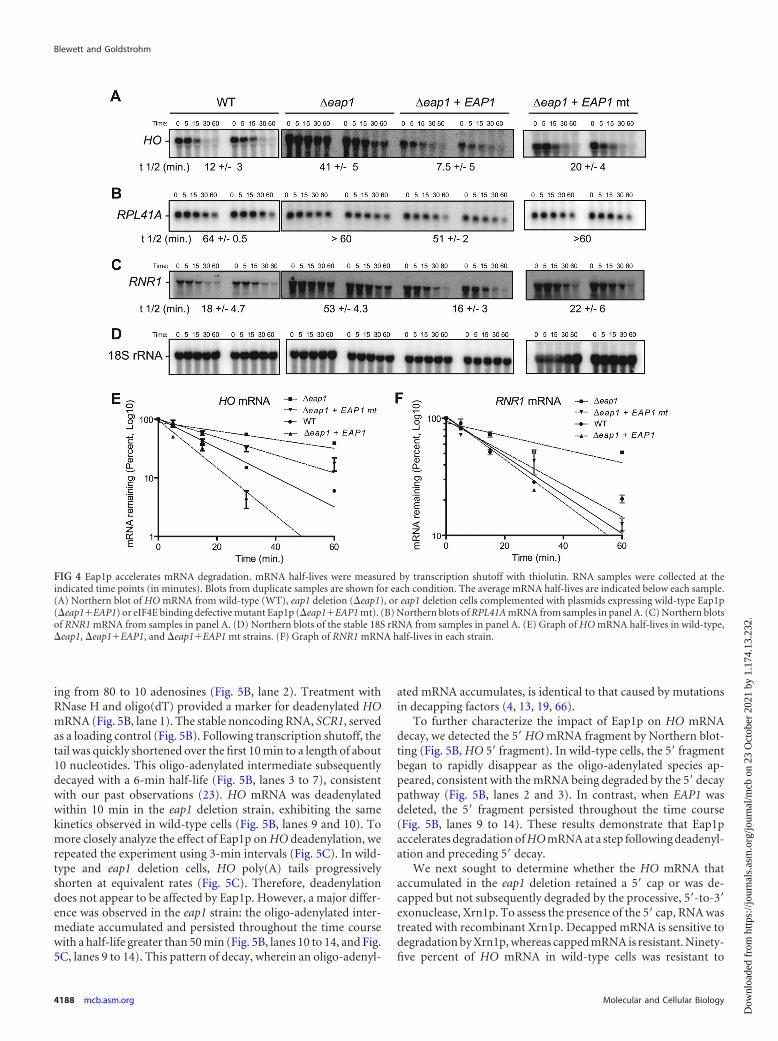
ing from 80 to 10 adenosines (Fig. 5B, lane 2). Treatment withRNase H and oligo(dT) provided a marker for deadenylated HOmRNA (Fig. 5B, lane 1). The stable noncoding RNA, SCR1, servedas a loading control (Fig. 5B). Following transcription shutoff, thetail was quickly shortened over the first 10 min to a length of about10 nucleotides. This oligo-adenylated intermediate subsequentlydecayed with a 6-min half-life (Fig. 5B, lanes 3 to 7), consistentwith our past observations (23). HO mRNA was deadenylatedwithin 10 min in the eap1 deletion strain, exhibiting the samekinetics observed in wild-type cells (Fig. 5B, lanes 9 and 10). Tomore closely analyze the effect of Eap1p on HO deadenylation, werepeated the experiment using 3-min intervals (Fig. 5C). In wild-type and eap1 deletion cells, HO poly(A) tails progressivelyshorten at equivalent rates (Fig. 5C). Therefore, deadenylationdoes not appear to be affected by Eap1p. However, a major differ-ence was observed in the eap1 strain: the oligo-adenylated inter-mediate accumulated and persisted throughout the time coursewith a half-life greater than 50 min (Fig. 5B, lanes 10 to 14, and Fig.5C, lanes 9 to 14). This pattern of decay, wherein an oligo-adenyl-
ated mRNA accumulates, is identical to that caused by mutationsin decapping factors (4, 13, 19, 66).
To further characterize the impact of Eap1p on HO mRNAdecay, we detected the 5= HO mRNA fragment by Northern blot-ting (Fig. 5B, HO 5= fragment). In wild-type cells, the 5= fragmentbegan to rapidly disappear as the oligo-adenylated species ap-peared, consistent with the mRNA being degraded by the 5= decaypathway (Fig. 5B, lanes 2 and 3). In contrast, when EAP1 wasdeleted, the 5= fragment persisted throughout the time course(Fig. 5B, lanes 9 to 14). These results demonstrate that Eap1paccelerates degradation of HO mRNA at a step following deadenyl-ation and preceding 5= decay.
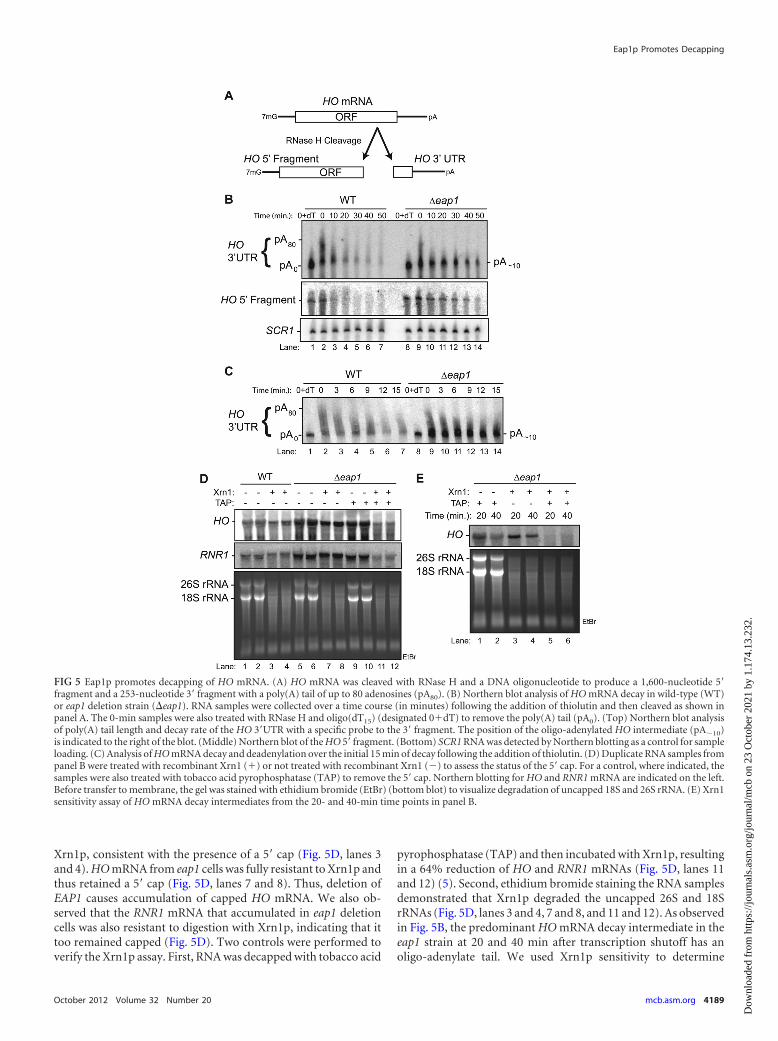
We next sought to determine whether the HO mRNA thataccumulated in the eap1 deletion retained a 5= cap or was de-capped but not subsequently degraded by the processive, 5=-to-3=exonuclease, Xrn1p. To assess the presence of the 5= cap, RNA wastreated with recombinant Xrn1p. Decapped mRNA is sensitive todegradation by Xrn1p, whereas capped mRNA is resistant. Ninety-five percent of HO mRNA in wild-type cells was resistant to
FIG 4 Eap1p accelerates mRNA degradation. mRNA half-lives were measured by transcription shutoff with thiolutin. RNA samples were collected at theindicated time points (in minutes). Blots from duplicate samples are shown for each condition. The average mRNA half-lives are indicated below each sample.(A) Northern blot of HO mRNA from wild-type (WT), eap1 deletion (�eap1), or eap1 deletion cells complemented with plasmids expressing wild-type Eap1p(�eap1�EAP1) or eIF4E binding defective mutant Eap1p (�eap1�EAP1 mt). (B) Northern blots of RPL41A mRNA from samples in panel A. (C) Northern blotsof RNR1 mRNA from samples in panel A. (D) Northern blots of the stable 18S rRNA from samples in panel A. (E) Graph of HO mRNA half-lives in wild-type,�eap1, �eap1�EAP1, and �eap1�EAP1 mt strains. (F) Graph of RNR1 mRNA half-lives in each strain.
Blewett and Goldstrohm
4188 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.
Xrn1p, consistent with the presence of a 5= cap (Fig. 5D, lanes 3and 4). HO mRNA from eap1 cells was fully resistant to Xrn1p andthus retained a 5= cap (Fig. 5D, lanes 7 and 8). Thus, deletion ofEAP1 causes accumulation of capped HO mRNA. We also ob-served that the RNR1 mRNA that accumulated in eap1 deletioncells was also resistant to digestion with Xrn1p, indicating that ittoo remained capped (Fig. 5D). Two controls were performed toverify the Xrn1p assay. First, RNA was decapped with tobacco acid
pyrophosphatase (TAP) and then incubated with Xrn1p, resultingin a 64% reduction of HO and RNR1 mRNAs (Fig. 5D, lanes 11and 12) (5). Second, ethidium bromide staining the RNA samplesdemonstrated that Xrn1p degraded the uncapped 26S and 18SrRNAs (Fig. 5D, lanes 3 and 4, 7 and 8, and 11 and 12). As observedin Fig. 5B, the predominant HO mRNA decay intermediate in theeap1 strain at 20 and 40 min after transcription shutoff has anoligo-adenylate tail. We used Xrn1p sensitivity to determine
FIG 5 Eap1p promotes decapping of HO mRNA. (A) HO mRNA was cleaved with RNase H and a DNA oligonucleotide to produce a 1,600-nucleotide 5=fragment and a 253-nucleotide 3= fragment with a poly(A) tail of up to 80 adenosines (pA80). (B) Northern blot analysis of HO mRNA decay in wild-type (WT)or eap1 deletion strain (�eap1). RNA samples were collected over a time course (in minutes) following the addition of thiolutin and then cleaved as shown inpanel A. The 0-min samples were also treated with RNase H and oligo(dT15) (designated 0�dT) to remove the poly(A) tail (pA0). (Top) Northern blot analysisof poly(A) tail length and decay rate of the HO 3=UTR with a specific probe to the 3= fragment. The position of the oligo-adenylated HO intermediate (pA�10)is indicated to the right of the blot. (Middle) Northern blot of the HO 5= fragment. (Bottom) SCR1 RNA was detected by Northern blotting as a control for sampleloading. (C) Analysis of HO mRNA decay and deadenylation over the initial 15 min of decay following the addition of thiolutin. (D) Duplicate RNA samples frompanel B were treated with recombinant Xrn1 (�) or not treated with recombinant Xrn1 (�) to assess the status of the 5= cap. For a control, where indicated, thesamples were also treated with tobacco acid pyrophosphatase (TAP) to remove the 5= cap. Northern blotting for HO and RNR1 mRNA are indicated on the left.Before transfer to membrane, the gel was stained with ethidium bromide (EtBr) (bottom blot) to visualize degradation of uncapped 18S and 26S rRNA. (E) Xrn1sensitivity assay of HO mRNA decay intermediates from the 20- and 40-min time points in panel B.
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4189
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

whether this mRNA species possesses a 5= cap. At 20 min, 76% ofthe HO intermediate was resistant to Xrn1p. At the 40-min timepoint, the HO intermediate was fully resistant to Xrn1p digestion(Fig. 5E, lanes 3 and 4). Removal of the 5= cap with TAP and thendigestion with Xrn1p resulted in decapping and destruction of95% of the oligo-adenylated HO intermediate (Fig. 5E, lanes 5 and6). Collectively, these results support the conclusion that deletionof EAP1 causes the accumulation of capped, oligo-adenylated HOmRNA. We conclude that Eap1p promotes decapping, a novelfunction for a 4E-BP.
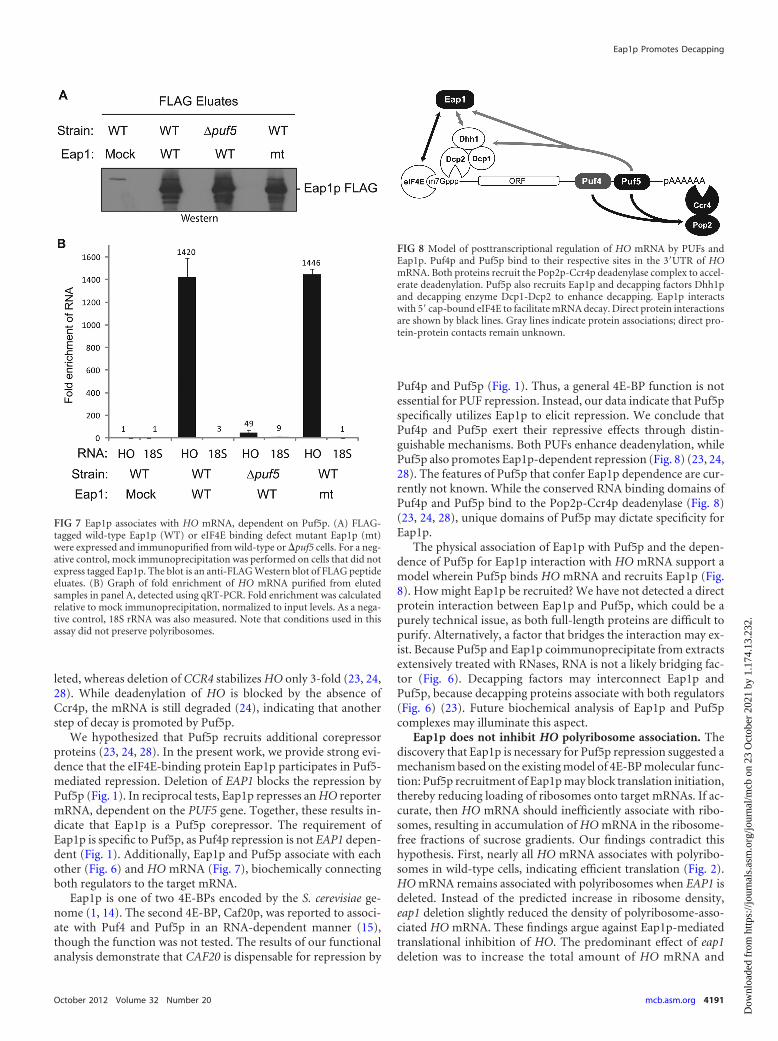
Eap1p associates with Puf5p and Dhh1p. We next askedwhether Puf5p associates with Eap1p. FLAG-tagged Eap1p (Eap1-FLAG) was coexpressed with T7-tagged Puf5p (Puf5-T7) in wild-type cells. Eap1p was then immunoprecipitated with FLAG anti-body resin, washed extensively, and specifically eluted with FLAGpeptide. Eluates were then analyzed by Western blotting (Fig. 6A).As a negative control, a mock FLAG immunoprecipitation wasperformed on cells expressing only Puf5p-T7 (Fig. 6A). Puf5p wasdetected in the Eap1p immunoprecipitate (Fig. 6A). As a negativecontrol, the actin protein, which is abundant in the input extracts,was not detected in the FLAG eluates (Fig. 6A). Because theseextracts were extensively treated with both RNase A and RNaseOne to degrade RNA prior to immunoprecipitation (Fig. 6B), we
conclude that Puf5p likely associates with Eap1p via protein inter-actions, not by a bridging RNA.
The observation that Eap1p enhanced decapping suggestedthat it may physically associate with the decapping machinery.The Dhh1 protein, a DEXD/H box helicase, is a well-known acti-vator of decapping (11). T7-tagged Dhh1p coimmunoprecipi-tated with Eap1p-FLAG but was not present in mock immunopre-cipitate (Fig. 6C). These extracts were also RNase treated prior toimmunoprecipitation; therefore, association of Eap1p and Dhh1pis not dependent on RNA. This result provides a physical linkbetween Eap1p and the decapping machinery. In addition to thisphysical interaction, the mRNA decay phenotypes caused bydeletion of EAP1 (Fig. 5A) and DHH1 (see Fig. S3 in the supple-mental material) (13) were remarkably similar; HO mRNA wasstabilized (half-life [t1/2] of 50 min) and accumulated as anoligo-adenylated species.
Eap1p associates with HO mRNA in a Puf5p-dependentmanner. Our data show that Eap1p participates in Puf5p-medi-ated degradation of HO mRNA. Puf5p binds directly to the HO3=UTR and may recruit Eap1p to the message. Alternatively,Eap1p may associate with HO mRNA by binding to eIF4E. To testthese models, we expressed FLAG-tagged Eap1p or the eIF4Ebinding-defective Eap1p mt and asked whether HO mRNA coim-munoprecipitated with each protein. To assess the role of Puf5p inrecruiting Eap1p, wild-type Eap1p-FLAG was immunoprecipi-tated from the puf5 deletion strain. As a negative control, a mockimmunoprecipitation was performed using extract from wild-type cells. The FLAG eluates were analyzed by Western blotting toconfirm purification of Eap1p and Eap1 mt (Fig. 7A). HO mRNAwas measured in the eluates by reverse transcription and quanti-tative PCR. HO mRNA was nearly undetectable in mock eluates,whereas it was enriched 1,400-fold in the Eap1p FLAG eluates(Fig. 7B). This demonstrates that Eap1p associates with HOmRNA.
Purification of Eap1p from a puf5 deletion strain reduced itsassociation with HO mRNA by 35-fold relative to the wild-typestrain (Fig. 7B, 49-fold enrichment); therefore, Puf5p facilitatesEap1p association with HO mRNA. We next tested the contribu-tion of Eap1p interaction with eIF4E. No significant change wasobserved relative to wild-type Eap1p; HO mRNA was enriched1,400-fold in the Eap1 mt FLAG eluates (Fig. 7B). As a negativecontrol, the noncoding 18S rRNA was not enriched in these im-munoprecipitates. These findings demonstrate that Eap1p associ-ates with HO mRNA, mediated by Puf5p.
DISCUSSIONEap1p is required for Puf5p-mediated repression. Yeast PUFproteins have a well-documented role in accelerating mRNA deg-radation (23, 24, 28, 29, 34, 51, 60, 69), and deadenylation plays animportant role. Both Puf4p and Puf5p enhance deadenylation ofHO mRNA in vivo and in vitro (23, 24, 28). Puf4p repressiondepends on both POP2 and CCR4 genes, which encode subunits ofthe Ccr4-Not deadenylase complex, and the catalytic activity ofCcr4p deadenylase (24, 28). That said, several clues indicate thatadditional mechanisms are utilized by specific PUFs. First, Puf5prepresses a target mRNA even when deadenylation is geneticallyblocked by removal of the CCR4 gene (23). Further evidence of adeadenylation-independent mechanism was revealed from analy-sis of HO mRNA half-lives in different genetic backgrounds. HOmRNA is stabilized 10-fold when both PUF4 and PUF5 are de-
FIG 6 Eap1p associates with Puf5p and Dhh1p. (A) T7-tagged Puf5p coim-munoprecipitates with FLAG-tagged Eap1p from cell extracts (input) treatedwith RNases A and One. Western blot detection of input lysates and FLAGpeptide eluates from mock or Eap1-FLAG immunoprecipitations. (B) RNase-mediated destruction of RNA in extracts from panel A was confirmed byethidium bromide (EtBr) staining of nucleic acids. The migration positions (inthousands of nucleotides or kilonucleotides [knt]) of RNA size markers (laneM) are shown to the left of the blot. (C) T7-tagged Dhh1p coimmunoprecipi-tates with Eap1-FLAG from RNase-treated cell lysates (input).
Blewett and Goldstrohm
4190 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.
leted, whereas deletion of CCR4 stabilizes HO only 3-fold (23, 24,28). While deadenylation of HO is blocked by the absence ofCcr4p, the mRNA is still degraded (24), indicating that anotherstep of decay is promoted by Puf5p.
We hypothesized that Puf5p recruits additional corepressorproteins (23, 24, 28). In the present work, we provide strong evi-dence that the eIF4E-binding protein Eap1p participates in Puf5-mediated repression. Deletion of EAP1 blocks the repression byPuf5p (Fig. 1). In reciprocal tests, Eap1p represses an HO reportermRNA, dependent on the PUF5 gene. Together, these results in-dicate that Eap1p is a Puf5p corepressor. The requirement ofEap1p is specific to Puf5p, as Puf4p repression is not EAP1 depen-dent (Fig. 1). Additionally, Eap1p and Puf5p associate with eachother (Fig. 6) and HO mRNA (Fig. 7), biochemically connectingboth regulators to the target mRNA.
Eap1p is one of two 4E-BPs encoded by the S. cerevisiae ge-nome (1, 14). The second 4E-BP, Caf20p, was reported to associ-ate with Puf4 and Puf5p in an RNA-dependent manner (15),though the function was not tested. The results of our functionalanalysis demonstrate that CAF20 is dispensable for repression by
Puf4p and Puf5p (Fig. 1). Thus, a general 4E-BP function is notessential for PUF repression. Instead, our data indicate that Puf5pspecifically utilizes Eap1p to elicit repression. We conclude thatPuf4p and Puf5p exert their repressive effects through distin-guishable mechanisms. Both PUFs enhance deadenylation, whilePuf5p also promotes Eap1p-dependent repression (Fig. 8) (23, 24,28). The features of Puf5p that confer Eap1p dependence are cur-rently not known. While the conserved RNA binding domains ofPuf4p and Puf5p bind to the Pop2p-Ccr4p deadenylase (Fig. 8)(23, 24, 28), unique domains of Puf5p may dictate specificity forEap1p.
The physical association of Eap1p with Puf5p and the depen-dence of Puf5p for Eap1p interaction with HO mRNA support amodel wherein Puf5p binds HO mRNA and recruits Eap1p (Fig.8). How might Eap1p be recruited? We have not detected a directprotein interaction between Eap1p and Puf5p, which could be apurely technical issue, as both full-length proteins are difficult topurify. Alternatively, a factor that bridges the interaction may ex-ist. Because Puf5p and Eap1p coimmunoprecipitate from extractsextensively treated with RNases, RNA is not a likely bridging fac-tor (Fig. 6). Decapping factors may interconnect Eap1p andPuf5p, because decapping proteins associate with both regulators(Fig. 6) (23). Future biochemical analysis of Eap1p and Puf5pcomplexes may illuminate this aspect.
Eap1p does not inhibit HO polyribosome association. Thediscovery that Eap1p is necessary for Puf5p repression suggested amechanism based on the existing model of 4E-BP molecular func-tion: Puf5p recruitment of Eap1p may block translation initiation,thereby reducing loading of ribosomes onto target mRNAs. If ac-curate, then HO mRNA should inefficiently associate with ribo-somes, resulting in accumulation of HO mRNA in the ribosome-free fractions of sucrose gradients. Our findings contradict thishypothesis. First, nearly all HO mRNA associates with polyribo-somes in wild-type cells, indicating efficient translation (Fig. 2).HO mRNA remains associated with polyribosomes when EAP1 isdeleted. Instead of the predicted increase in ribosome density,eap1 deletion slightly reduced the density of polyribosome-asso-ciated HO mRNA. These findings argue against Eap1p-mediatedtranslational inhibition of HO. The predominant effect of eap1deletion was to increase the total amount of HO mRNA and
FIG 7 Eap1p associates with HO mRNA, dependent on Puf5p. (A) FLAG-tagged wild-type Eap1p (WT) or eIF4E binding defect mutant Eap1p (mt)were expressed and immunopurified from wild-type or �puf5 cells. For a neg-ative control, mock immunoprecipitation was performed on cells that did notexpress tagged Eap1p. The blot is an anti-FLAG Western blot of FLAG peptideeluates. (B) Graph of fold enrichment of HO mRNA purified from elutedsamples in panel A, detected using qRT-PCR. Fold enrichment was calculatedrelative to mock immunoprecipitation, normalized to input levels. As a nega-tive control, 18S rRNA was also measured. Note that conditions used in thisassay did not preserve polyribosomes.
FIG 8 Model of posttranscriptional regulation of HO mRNA by PUFs andEap1p. Puf4p and Puf5p bind to their respective sites in the 3=UTR of HOmRNA. Both proteins recruit the Pop2p-Ccr4p deadenylase complex to accel-erate deadenylation. Puf5p also recruits Eap1p and decapping factors Dhh1pand decapping enzyme Dcp1-Dcp2 to enhance decapping. Eap1p interactswith 5= cap-bound eIF4E to facilitate mRNA decay. Direct protein interactionsare shown by black lines. Gray lines indicate protein associations; direct pro-tein-protein contacts remain unknown.
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4191
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

increase its half-life, pointing toward a role of Eap1p in pro-moting mRNA decay. The small reduction of HO ribosomedensity caused by the absence of Eap1p may reflect the accu-mulation of HO mRNA with short poly(A) tails in the eap1deletion strain (Fig. 5).
If Eap1p inhibits translation initiation, then Eap1p would bepredicted to be found in ribosome-free fractions at the top of thesucrose gradient. In contrast to this, a significant portion of Eap1passociates with polyribosomes (Fig. 3). Because polyribosomalmRNAs have undergone multiple rounds of initiation, polyribo-some-associated Eap1p cannot have blocked initiation. Our dataindicate that interaction of Eap1p with eIF4E mediates its associ-ation with polyribosomes. This conclusion is supported by theobservation that eIF4E was distributed across the gradient, includ-ing polyribosome fractions (Fig. 3). Furthermore, mutations inEap1p that disrupt binding to eIF4E cause a complete loss ofEap1p polyribosome association (Fig. 3). At this time, the func-tional significance of polyribosomal Eap1p remains unknown.
Eap1p is also present in two other peaks of the sucrose gradient.One peak, in the ribosome-free fractions at the top of the gradient,increases upon EDTA treatment, ribosome runoff, and mutationof the eIF4E binding motif (Fig. 3). These observations indicatethat this pool of Eap1p is not bound to ribosomes or eIF4E. AnEap1p species with lower electrophoretic mobility is present inthis peak, suggesting that Eap1p may be modified posttranslation-ally. This idea is supported by proteomic identification of multiplephosphorylation sites in Eap1p (56). Eap1p phosphorylationcould block its interaction with eIF4E in a manner similar to other4E-BPs (62). A third peak of Eap1p cofractionates with 60S ribo-somal subunits. This peak shifts to the ribosome-free fractionsupon ribosome runoff and mutation of the eIF4E binding motif;therefore, it might represent an intermediate in translation. Fu-ture biochemical analysis of Eap1p complexes will be necessary tounderstand their composition and functions.
Deletion of EAP1 does not alter the global translation state, asdeduced from the identical chromatograms of polyribosomesfrom wild-type and eap1 deletion strains (Fig. 2) (32). This con-clusion is supported by analysis of ribosome association of RNR1and RPL41A mRNAs, which like HO, were not inhibited byEap1p. That said, on the basis of the available data, we do notexclude the prospect that specific mRNAs may be translationallyinhibited by Eap1p. In the study that identified Eap1p, an in vitrotranslation assay indicated that Eap1p inhibits translation (14);however, the role of eIF4E binding and impact on mRNA stabilitywere not addressed. More recently, a microarray-based studyfound that deletion of EAP1 changed the translation state of 329mRNAs by greater than 1.8-fold, as measured by the ratio ofmRNA in polyribosomes to monoribosomes (15). Of these, theratios of 176 mRNAs increased, suggesting that Eap1p inhibitstheir translation. In our analysis of HO mRNA, deletion of EAP1caused a 1.6-fold increase in the polyribosome-to-monoribosomeratio; however, this change reflects the 1.7-fold increase in HOabundance. Moreover, eap1 deletion did not increase HO ribo-some occupancy and density (Fig. 2). It remains to be investigatedwhether Eap1p and Puf5p function together to regulate additionalmRNAs. The mRNAs affected by eap1 deletion did not correlatewith those that coimmunoprecipitate with Puf5p (15). Cridge etal. also reported that deletion of EAP1 altered steady-state levels of99 mRNAs more than 2-fold, and of these, 56 increased, hintingthat Eap1p may affect the stability of additional mRNAs (15).
Whether the observed effects of eap1 deletion on translation stateand mRNA levels are direct remains to be established. Germane tothe challenge of discerning direct Eap1p effects from indirect ef-fects, our demonstration that an Eap1p-regulated mRNA (i.e.,HO) coimmunoprecipitates with Eap1p provides proof of princi-ple for future ribonomic approaches to identify Eap1p targetmRNAs.
In summary, our findings do not support a role of Eap1p ininhibition of translation initiation of Puf5p-targeted HO mRNA.Instead, our results argue that Eap1p elicits a mode of repressionthat is divergent from the canonical 4E-BP mechanism of transla-tion inhibition.
Eap1p accelerates mRNA decay. We discovered a novel func-tion for Eap1p in promoting mRNA degradation. Deletion ofEAP1 stabilizes HO mRNA more than 3-fold. Conversely, overex-pression of Eap1p enhances decay of HO mRNA beyond that ob-served in wild-type cells. Targeting of HO by Eap1p is likely di-rected by Puf5p, a hypothesis supported by their mutualinterdependence for repression (Fig. 1) and the finding that asso-ciation of Eap1p with HO mRNA depends on Puf5p (Fig. 7).
Enhancement of mRNA decay by Eap1p is not likely to besolely dependent on Puf5p, because we also observed an effect onRNR1 mRNA, which is not regulated by Puf5p (23, 60) and is notknown to associate with other PUF proteins (22). The factors thatcontrol RNR1 mRNA remain to be discovered in future work.Other mRNAs, such as RPL41A, are unaffected by Eap1p, suggest-ing that Eap1p-enhanced mRNA decay may be restricted to spe-cific messages.
Is the eIF4E binding activity of Eap1p necessary for mRNAdegradation? An Eap1p mutant that cannot bind eIF4E exhibitshalf of the mRNA decay activity of the wild-type protein (Fig. 4);therefore, we conclude that binding to eIF4E facilitates, but is notessential for, Eap1p mRNA decay activity. Binding to eIF4E couldpromote decay by displacing eIF4G (14), destabilizing theeIF4E-5= cap interaction and facilitating access of mRNA degra-dation enzymes (Fig. 8). This idea is supported by data showingthat mutations in translation initiation factors, including eIF4Eand eIF4G, increase mRNA decay (58). However, our data alsoindicate that this explanation in itself is not sufficient. First, theEap1p mutant does not bind eIF4E (Fig. 3), nor does it associatewith polyribosomes (Fig. 3), yet it still stimulates mRNA decay(Fig. 4), albeit with reduced efficiency. We interpret this as evi-dence that Eap1p promotes mRNA decay by another means, per-haps by affecting decapping (Fig. 8), as discussed below.
Eap1p promotes decapping. Degradation of yeast mRNAstypically initiates by shortening of the poly(A) tail to an oligo-adenylated length, followed by decapping (11, 25). DecappedmRNA is then rapidly degraded by Xrn1p. As Puf5p enhancesdeadenylation of HO mRNA, we examined the effect of Eap1p.Loss of Eap1p does not affect poly(A) removal, which occurs rap-idly in both wild-type and eap1 deletion cells (Fig. 5). Instead,Eap1p dramatically affects the fate of the oligo-adenylated inter-mediate. In wild-type cells, this species is rapidly degraded, coin-cident with disappearance of the 5= end of the mRNA (Fig. 5).When EAP1 is deleted, the oligo-adenylated mRNA is highly sta-bilized, as is the 5= end of HO (Fig. 5). The oligo-adenylated HOthat accumulates is resistant to Xrn1p and thus remains capped(Fig. 5). We conclude that Eap1p promotes decapping of HOmRNA. Our data support that HO is degraded by the 5= decappingpathway; deletion of decapping factor genes greatly stabilizes HO
Blewett and Goldstrohm
4192 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

mRNA, including the �pat1 (t1/2 55 min, and �dcp2 (t1/2 60min) strains (unpublished data) and the �dhh1 strain (t1/2 50min) (see Fig. S3 in the supplemental material). Additionally, ac-cumulation of deadenylated, capped mRNA species is identical tothe effect of mutations in decapping factors, including Dhh1p (seeFig. S3), Pat1p, and the Lsm1-7p complex (4, 13, 19, 66). We notethat deletion of genes encoding the 3=-to-5= exosome complex didnot increase HO mRNA levels (see Fig. S4 in the supplementalmaterial) or abrogate Puf5p repression (23); therefore, the 3= de-cay pathway does not impact HO mRNA degradation.
Eap1p associates with the decapping factor Dhh1p. This find-ing suggests that Eap1p recruits decapping factors or alters theiractivity (Fig. 8). Puf5p also associates with decapping factors, in-cluding Dhh1p and Dcp1p (23). These associations do not dependon RNA, indicating that the proteins were not simply tethered tothe same mRNA, though the direct protein contacts remain to bedelineated.
We propose a model of posttranscriptional regulation of HOmRNA that integrates past and new data (Fig. 8). Both Puf4p andPuf5p bind to their respective recognition sequence in the 3=UTRof HO and recruit the Ccr4-Not complex via direct contact withthe Pop2p subunit, thereby enhancing deadenylation. Puf5p alsorecruits Eap1p and decapping factor Dhh1p and decapping en-zyme Dcp2-Dcp1 to promote removal of the 5= cap. Eap1p bind-ing to eIF4E facilitates decay. This model provides a useful frame-work for future research on PUF repression.
Recent evidence indicates that mRNA decay can occur cotrans-lationally on polyribosomes (30, 31). It is tempting to speculatethat decay of HO mRNA, promoted by Eap1p, might occur onpolyribosomes; however, because eIF4E binding-defective Eap1pretains partial activity but does not associate with polyribosomes,ribosome association is unlikely to be an essential feature.
Our analysis does not exclude the possibility that PUFs blocktranslation by additional mechanisms. Chritton et al. reportedthat the Puf5p inhibits translation of a capped, polyadenylatedreporter mRNA in vitro (10). Eap1-mediated decapping may ac-count for the observed regulation. Alternatively, Puf5p may havean independent direct effect on translation. Dhh1p can inhibittranslation (12); therefore, Puf5p recruitment of Dhh1p may im-pact translation.
Do other 4E-BPs affect mRNA decay? Multiple 4E-BPs havebeen identified in eukaryotes (1, 14, 18, 35, 42, 43, 49, 50, 54, 55,64, 74). The finding that Eap1p is required for repression by Puf5pis reminiscent of other examples wherein an RNA-binding proteinutilizes a 4E-BP to repress an mRNA (49, 50, 62, 64, 74). In thesecases, translation inhibition is thought to be the means of repres-sion. Our results showing that Eap1p enhanced decay and decap-ping were therefore unanticipated in the context of current under-standing of 4E-BP repression. Furthermore, Puf5p may not be theonly RNA binding factor to use Eap1p as a corepressor, as the yeastVts1p protein employs Eap1p to enhance mRNA decay (C. Smib-ert, University of Toronto, personal communication).
Additional clues are emerging that implicate specific 4E-BPs asversatile regulators that can influence steps of gene expressionother than translation initiation. The human eIF4E transporterwas implicated in the destabilization of adenine uracil-rich ele-ment (ARE)-containing mRNAs (18). A recent analysis of theDrosophila 4E-BP, Cup, found that it represses mRNAs by specif-ically enhancing deadenylation (33). Interestingly, Cup subse-quently stabilizes the deadenylated mRNA by blocking decapping
(33). These findings suggest the exciting possibility that each4E-BP has unique activities to control translation, localization,and degradation of distinct groups of mRNAs.
ACKNOWLEDGMENTS
A.C.G. gratefully acknowledges support by Marvin Wickens, Universityof Wisconsin, during the initial phase of this research. We appreciateadvice from Trista Schagat, Jamie Van Etten, Chase Weidmann, NathanRaynard, and Joel Hrit. We thank Brad Hook, Daniel Seay, Jeff Coller, andMay Tsoi for technical assistance and Eric Wagner for comments on themanuscript. Pfizer generously supplied thiolutin.
Nathan Blewett was supported by NIH Cellular and Molecular Biologytraining grant T32-GM007315.
REFERENCES1. Altmann M, Schmitz N, Berset C, Trachsel H. 1997. A novel inhibitor of
cap-dependent translation initiation in yeast: p20 competes with eIF4Gfor binding to eIF4E. EMBO J. 16:1114 –1121.
2. Arava Y, et al. 2003. Genome-wide analysis of mRNA translation profiles inSaccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 100:3889–3894.
3. Asaoka-Taguchi M, Yamada M, Nakamura A, Hanyu K, Kobayashi S.1999. Maternal Pumilio acts together with Nanos in germline develop-ment in Drosophila embryos. Nat. Cell Biol. 1:431– 437.
4. Beelman CA, et al. 1996. An essential component of the decapping en-zyme required for normal rates of mRNA turnover. Nature 382:642– 646.
5. Blewett N, Coller J, Goldstrohm A. 2011. A quantitative assay for mea-suring mRNA decapping by splinted ligation reverse transcription poly-merase chain reaction: qSL-RT-PCR. RNA 17:535–543.
6. Chagnovich D, Lehmann R. 2001. Poly(A)-independent regulation ofmaternal hunchback translation in the Drosophila embryo. Proc. Natl.Acad. Sci. U. S. A. 98:11359 –11364.
7. Chen D, et al. 2012. Pumilio 1 suppresses multiple activators of p53 tosafeguard spermatogenesis. Curr. Biol. 22:420 – 425.
8. Chial HJ, Stemm-Wolf AJ, McBratney S, Winey M. 2000. Yeast Eap1p,an eIF4E-associated protein, has a separate function involving geneticstability. Curr. Biol. 10:1519 –1522.
9. Cho PF, et al. 2006. Cap-dependent translational inhibition establishestwo opposing morphogen gradients in Drosophila embryos. Curr. Biol.16:2035–2041.
10. Chritton JJ, Wickens M. 2010. Translational repression by PUF proteinsin vitro. RNA 16:1217–1225.
11. Coller J, Parker R. 2004. Eukaryotic mRNA decapping. Annu. Rev.Biochem. 73:861– 890.
12. Coller J, Parker R. 2005. General translational repression by activators ofmRNA decapping. Cell 122:875– 886.
13. Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. 2001. TheDEAD box helicase, Dhh1p, functions in mRNA decapping and interacts withboth the decapping and deadenylase complexes. RNA 7:1717–1727.
14. Cosentino GP, et al. 2000. Eap1p, a novel eukaryotic translation initiationfactor 4E-associated protein in Saccharomyces cerevisiae. Mol. Cell. Biol.20:4604 – 4613.
15. Cridge AG, et al. 2010. Identifying eIF4E-binding protein translationallycontrolled transcripts reveals links to mRNAs bound by specific PUF pro-teins. Nucleic Acids Res. 38:8039 – 8050.
16. Crittenden SL, et al. 2002. A conserved RNA-binding protein controlsgermline stem cells in Caenorhabditis elegans. Nature 417:660 – 663.
17. Dubnau J, et al. 2003. The staufen/pumilio pathway is involved in Dro-sophila long-term memory. Curr. Biol. 13:286 –296.
18. Ferraiuolo MA, et al. 2005. A role for the eIF4E-binding protein 4E-T inP-body formation and mRNA decay. J. Cell Biol. 170:913–924.
19. Fischer N, Weis K. 2002. The DEAD box protein Dhh1 stimulates thedecapping enzyme Dcp1. EMBO J. 21:2788 –2797.
20. Forbes A, Lehmann R. 1998. Nanos and Pumilio have critical roles in thedevelopment and function of Drosophila germline stem cells. Develop-ment 125:679 – 690.
21. Gamberi C, Peterson DS, He L, Gottlieb E. 2002. An anterior functionfor the Drosophila posterior determinant Pumilio. Development 129:2699 –2710.
22. Gerber AP, Herschlag D, Brown PO. 2004. Extensive association of func-tionally and cytotopically related mRNAs with Puf family RNA-binding pro-teins in yeast. PLoS Biol. 2:E79. doi:10.1371/journal.pbio.0020079.
Eap1p Promotes Decapping
October 2012 Volume 32 Number 20 mcb.asm.org 4193
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.

23. Goldstrohm AC, Hook BA, Seay DJ, Wickens M. 2006. PUF proteins bindPop2p to regulate messenger RNAs. Nat. Struct. Mol. Biol. 13:533–539.
24. Goldstrohm AC, Seay DJ, Hook BA, Wickens M. 2007. PUF protein-mediated deadenylation is catalyzed by Ccr4p. J. Biol. Chem. 282:109–114.
25. Goldstrohm AC, Wickens M. 2008. Multifunctional deadenylase com-plexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 9:337–344.
26. Gu W, Deng Y, Zenklusen D, Singer RH. 2004. A new yeast PUF familyprotein, Puf6p, represses ASH1 mRNA translation and is required for itslocalization. Genes Dev. 18:1452–1465.
27. Haghighat A, Mader S, Pause A, Sonenberg N. 1995. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 forbinding to eukaryotic initiation factor-4E. EMBO J. 14:5701–5709.
28. Hook BA, Goldstrohm AC, Seay DJ, Wickens M. 2007. Two yeast PUFproteins negatively regulate a single mRNA. J. Biol. Chem. 282:15430 –15438.
29. Houshmandi SS, Olivas WM. 2005. Yeast Puf3 mutants reveal the com-plexity of Puf-RNA binding and identify a loop required for regulation ofmRNA decay. RNA 11:1655–1666.
30. Hu W, Petzold C, Coller J, Baker KE. 2010. Nonsense-mediated mRNAdecapping occurs on polyribosomes in Saccharomyces cerevisiae. Nat.Struct. Mol. Biol. 17:244 –247.
31. Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. 2009. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461:225–229.
32. Ibrahimo S, Holmes LE, Ashe MP. 2006. Regulation of translation ini-tiation by the yeast eIF4E binding proteins is required for the pseudohy-phal response. Yeast 23:1075–1088.
33. Igreja C, Izaurralde E. 2011. CUP promotes deadenylation and inhibitsdecapping of mRNA targets. Genes Dev. 25:1955–1967.
34. Jackson JS, Jr, Houshmandi SS, Lopez Leban F, Olivas WM. 2004.Recruitment of the Puf3 protein to its mRNA target for regulation ofmRNA decay in yeast. RNA 10:1625–1636.
35. Jung MY, Lorenz L, Richter JD. 2006. Translational control by neurogui-din, a eukaryotic initiation factor 4E and CPEB binding protein. Mol. Cell.Biol. 26:4277– 4287.
36. Kedde M, et al. 2010. A Pumilio-induced RNA structure switch in p27-3=UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 12:1014–1020.
37. Lee B, Udagawa T, Singh CR, Asano K. 2007. Yeast phenotypic assays ontranslational control. Methods Enzymol. 429:105–137.
38. Lehmann R, Nusslein-Volhard C. 1987. Involvement of the pumilio genein the transport of an abdominal signal in the Drosophila embryo. Nature329:167–170.
39. Lin H, Spradling AC. 1997. A novel group of pumilio mutations affectsthe asymmetric division of germline stem cells in the Drosophila ovary.Development 124:2463–2476.
40. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(-Delta Delta C(T)) method.Methods 25:402– 408.
41. Lu G, Dolgner SJ, Hall TM. 2009. Understanding and engineering RNAsequence specificity of PUF proteins. Curr. Opin. Struct. Biol. 19:110–115.
42. Mader S, Lee H, Pause A, Sonenberg N. 1995. The translation initiationfactor eIF-4E binds to a common motif shared by the translation factoreIF-4 gamma and the translational repressors 4E-binding proteins. Mol.Cell. Biol. 15:4990 – 4997.
43. Matsuo Y, et al. 2004. Stacking of molecules possessing a fullerene apexand a cup-shaped cavity connected by a silicon connection. J. Am. Chem.Soc. 126:432– 433.
44. Meier KD, Deloche O, Kajiwara K, Funato K, Riezman H. 2006.Sphingoid base is required for translation initiation during heat stress inSaccharomyces cerevisiae. Mol. Biol. Cell 17:1164 –1175.
45. Mendelsohn BA, et al. 2003. Genetic and biochemical interactions be-tween SCP160 and EAP1 in yeast. Nucleic Acids Res. 31:5838 –5847.
46. Menon KP, et al. 2004. The translational repressor Pumilio regulatespresynaptic morphology and controls postsynaptic accumulation oftranslation factor eIF-4E. Neuron 44:663– 676.
47. Miller MA, Olivas WM. 2011. Roles of Puf proteins in mRNA degrada-tion and translation. Wiley Interdiscip. Rev. RNA 2:471– 492.
48. Murata Y, Wharton RP. 1995. Binding of pumilio to maternal hunchbackmRNA is required for posterior patterning in Drosophila embryos. Cell80:747–756.
49. Nakamura A, Sato K, Hanyu-Nakamura K. 2004. Drosophila cup is aneIF4E binding protein that associates with Bruno and regulates oskarmRNA translation in oogenesis. Dev. Cell 6:69 –78.
50. Nelson MR, Leidal AM, Smibert CA. 2004. Drosophila Cup is an eIF4E-binding protein that functions in Smaug-mediated translational repres-sion. EMBO J. 23:150 –159.
51. Olivas W, Parker R. 2000. The Puf3 protein is a transcript-specific regu-lator of mRNA degradation in yeast. EMBO J. 19:6602– 6611.
52. Parisi M, Lin H. 1999. The Drosophila pumilio gene encodes two func-tional protein isoforms that play multiple roles in germline development,gonadogenesis, oogenesis and embryogenesis. Genetics 153:235–250.
53. Parker R, Song H. 2004. The enzymes and control of eukaryotic mRNAturnover. Nat. Struct. Mol. Biol. 11:121–127.
54. Pause A, et al. 1994. Insulin-dependent stimulation of protein synthesis byphosphorylation of a regulator of 5=-cap function. Nature 371:762–767.
55. Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. 1998.4E-BP3, a new member of the eukaryotic initiation factor 4E-bindingprotein family. J. Biol. Chem. 273:14002–14007.
56. Ptacek J, et al. 2005. Global analysis of protein phosphorylation in yeast.Nature 438:679 – 684.
57. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by thecomparative C(T) method. Nat. Protoc. 3:1101–1108.
58. Schwartz DC, Parker R. 1999. Mutations in translation initiation factorslead to increased rates of deadenylation and decapping of mRNAs in Sac-charomyces cerevisiae. Mol. Cell. Biol. 19:5247–5256.
59. Schweers BA, Walters KJ, Stern M. 2002. The Drosophila melanogastertranslational repressor pumilio regulates neuronal excitability. Genetics161:1177–1185.
60. Seay D, Hook B, Evans K, Wickens M. 2006. A three-hybrid screenidentifies mRNAs controlled by a regulatory protein. RNA 12:1594 –1600.
61. Sezen B, Seedorf M, Schiebel E. 2009. The SESA network links duplica-tion of the yeast centrosome with the protein translation machinery.Genes Dev. 23:1559 –1570.
62. Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiationin eukaryotes: mechanisms and biological targets. Cell 136:731–745.
63. Sonoda J, Wharton RP. 2001. Drosophila brain tumor is a translationalrepressor. Genes Dev. 15:762–773.
64. Stebbins-Boaz B, Cao Q, de Moor CH, Mendez R, Richter JD. 1999.Maskin is a CPEB-associated factor that transiently interacts with elF-4E.Mol. Cell 4:1017–1027.
65. Tadauchi T, Matsumoto K, Herskowitz I, Irie K. 2001. Post-transcriptional regulation through the HO 3=-UTR by Mpt5, a yeast ho-molog of Pumilio and FBF. EMBO J. 20:552–561.
66. Tharun S, et al. 2000. Yeast Sm-like proteins function in mRNA decap-ping and decay. Nature 404:515–518.
67. Tucker M, Staples RR, Valencia-Sanchez MA, Muhlrad D, Parker R.2002. Ccr4p is the catalytic subunit of a Ccr4p/Pop2p/Notp mRNA dead-enylase complex in Saccharomyces cerevisiae. EMBO J. 21:1427–1436.
68. Tucker M, et al. 2001. The transcription factor associated Ccr4 and Caf1proteins are components of the major cytoplasmic mRNA deadenylase inSaccharomyces cerevisiae. Cell 104:377–386.
69. Ulbricht RJ, Olivas WM. 2008. Puf1p acts in combination with otheryeast Puf proteins to control mRNA stability. RNA 14:246 –262.
70. Wang X, McLachlan J, Zamore PD, Hall TM. 2002. Modular recognitionof RNA by a human pumilio-homology domain. Cell 110:501–512.
71. Weidmann CA, Goldstrohm AC. 2012. Drosophila Pumilio protein con-tains multiple autonomous repression domains that regulate mRNAs in-dependently of Nanos and brain tumor. Mol. Cell. Biol. 32:527–540.
72. Wharton RP, Sonoda J, Lee T, Patterson M, Murata Y. 1998. ThePumilio RNA-binding domain is also a translational regulator. Mol. Cell1:863– 872.
73. Wickens M, Bernstein DS, Kimble J, Parker R. 2002. A PUF familyportrait: 3=UTR regulation as a way of life. Trends Genet. 18:150 –157.
74. Wilhelm JE, Hilton M, Amos Q, Henzel WJ. 2003. Cup is an eIF4Ebinding protein required for both the translational repression of oskar andthe recruitment of Barentsz. J. Cell Biol. 163:1197–1204.
75. Wreden C, Verrotti AC, Schisa JA, Lieberfarb ME, Strickland S. 1997.Nanos and pumilio establish embryonic polarity in Drosophila by pro-moting posterior deadenylation of hunchback mRNA. Development 124:3015–3023.
76. Zamore PD, Williamson JR, Lehmann R. 1997. The Pumilio proteinbinds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3:1421–1433.
77. Zhang B, et al. 1997. A conserved RNA-binding protein that regulatessexual fates in the C. elegans hermaphrodite germ line. Nature 390:477–484.
Blewett and Goldstrohm
4194 mcb.asm.org Molecular and Cellular Biology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 23
Oct
ober
202
1 by
1.1
74.1
3.23
2.