a theoretical investigation of the selective oxidation of methanol
TRANSCRIPT

A Theoretical Investigation of the Selective Oxidation of Methanol to Formaldehyde onIsolated Vanadate Species Supported on Titania
Anthony Goodrow and Alexis T. Bell*Department of Chemical Engineering, UniVersity of California, Berkeley, California 94720-1462
ReceiVed: February 14, 2008; ReVised Manuscript ReceiVed: April 18, 2008
The selective oxidation of methanol to formaldehyde occurring on titania-supported vanadate species hasbeen analyzed theoretically with the aim of understanding why the activity of VOx/TiO2 is ∼103 faster thanthat of VOx/SiO2. The active site was represented by a [(O)3VdO] group located at the corner of a cubicTiOx cluster, a model similar to that used successfully to describe the oxidation of methanol on isolatedvanadate species supported on silica. Density functional theory was used to calculate the geometry, vibrationalfrequencies, and energy of all ground state and transition state structures. The equilibrium constants and ratecoefficients for each elementary reaction step were calculated using statistical mechanics and absolute ratetheory. Methanol oxidation to formaldehyde was taken to proceed via two key steps: the reversible adsorptionof methanol across a V-O-Ti bond followed by the transfer of a hydrogen atom from an adsorbed methoxygroup to a vanadyl O atom. The rate parameters and the apparent first-order rate coefficient determined forVOx/TiO2 were found to be very similar to those reported earlier in a theoretical analysis of VOx/SiO2 [J.Phys. Chem. C 2007, 111, 14753], indicating that the significantly higher rate of reaction seen experimentallyfor VOx/TiO2 is not due to an intrinsic electronic effect of the support on the catalytic properties of the activecenter. Introduction of an O-vacancy adjacent to the vanadate species results in a reduction in the activationbarrier for the rate-limiting step and to close agreement between the rate parameters predicted and thosefound experimentally. The effect of O-vacancies in the support on the rate of methanol on metal oxide-supported vanadate species is further evidenced by a strong correlation between the turnover frequency formethanol oxidation and the energy required to form an O-atom defect on metal oxide supports.
Introduction
The selective oxidation of methanol to formaldehyde cata-lyzed by supported vanadia has been the subject of numerousinvestigations.1–11 For submonolayer coverages of vanadia,support composition has been observed to have a large effecton the specific activity of vanadium. The general consensus isthat activity per vanadium atom decreases with support com-position in the order SiO2 , Al2O3 , TiO2 < ZrO2 < CeO2
and that under identical reaction conditions the turnoverfrequency (TOF) for VOx/CeO2 is roughly 3 orders of magnitudehigher than that that for VOx/SiO2.1,3,8–11 This trend has beenattributed to the effect of the support on the electronic propertiesof the supported vanadate species based on the observation thatthe TOF for formaldehyde formation decreases with increasingSanderson electronegativity of the support cation.3,8,9
Studies of the reaction kinetics have shown that at lowconversions the rate of formaldehyde formation is first-orderin the partial pressure of methanol and zero-order in the partialpressure of oxygen, independent of the support composition.Mechanistic investigations suggest that the reaction occurs intwo steps.3,8,10,11 The first is the reversible adsorption ofmethanol, which occurs by methanol addition across one of theV-O-M bonds anchoring V to the support. The second, rate-limiting step is the transfer of a hydrogen atom from the resultingV-OCH3 species to an O atom associated with the activecenter.10 In the limit of low conversion, the overall rateexpression for the formation of formaldehyde can then be written
as the product of the equilibrium constant for methanoladsorption, Kads, and the rate constant for the rate-limiting step,krls, as shown in eq 1
RCH2O )KadskrlsPMeOH (1)
On the basis of the observed decrease in TOF with increasingSanderson electronegativity of the support cation, it has beenproposed that the electronegativity of the support cation and,hence, reducibility of the support affects the equilibriumadsorption of methanol and the rate-limiting step in which anH atom is abstracted from an adsorbed methoxy group.3,8,9
The mechanism and kinetics of methanol oxidation on isolatedvanadate species supported on different metal oxides have beeninvestigated theoretically by several groups. Khaliullin and Bell12
have analyzed the energetics and kinetics of this reaction onisolated vanadate species supported on SiO2, TiO2, and ZrO2,using a small cluster model comprising a V)O group and threemetal support atoms terminated by hydroxyl groups. Thecalculated TOF was found to be essentially independent ofsupport composition in contradiction to what is observedexperimentally. Zhanpeisov13 has carried out a similar analysis,using a more realistic representation for isolated vanadate speciessupported on rutile. The calculated bond lengths and vibrationalfrequencies for methanol adsorption on VOx/TiO2 were foundto be in close agreement with those reported in previousexperimental11 and theoretical studies.12 Using a large clustermodel of VOx on SiO2, Sauer and co-workers14 have carriedout a detailed analysis of the reaction mechanism for theoxidation of methanol to formaldehyde. These authors reportedan apparent activation energy of 27 kcal/mol, in reasonable
* Author to whom correspondence should be addressed: [email protected].
J. Phys. Chem. C 2008, 112, 13204–1321413204
10.1021/jp801339q CCC: $40.75 2008 American Chemical SocietyPublished on Web 08/01/2008

agreement with that determined from experiments;1,8,10 however,the apparent rate constant was 4 orders of magnitude smallerthan that observed. More recently, Goodrow and Bell15 havereported an analysis of the mechanism and kinetics of methanoloxidation occurring on silica-supported vanadate species, inwhich not only was the pathway to methanol oxidation examinedbut also the pathway for catalyst reoxidation. The equilibriumconstant for methanol adsorption and the apparent first-orderrate coefficient for the reaction determined from first principleswere found to be in very close agreement with the experimentalvalues for these parameters. It was also concluded that reoxi-dation of the catalytically active centers is approximately 4orders of magnitude faster than their rate of reduction, a resultwhich is also in agreement with experimental observation.
The purpose of the present study was to carry out a detailedtheoretical investigation of the mechanism and kinetics ofmethanol oxidation on isolated vanadate species supported onthe anatase phase of TiO2 with the aim of explaining why therate of this reaction is significantly higher on VOx/TiO2 thanon VOx/SiO2. The emphasis on isolated vanadate species wasmade for two reasons. The first is that experimental studiesconducted by Bell and co-workers suggest that the mechanismof methanol oxidation to formaldehyde is the same for isolatedvanadate species supported on silica and titania.10,11 Second,and as noted above, Goodrow and Bell15 have demonstratedthat the kinetics for methanol oxidation on VOx/SiO2 arerepresented with reasonable accuracy by theoretical analysis.These results motivate us to ask whether the higher rate ofmethanol oxidation observed for VOx/TiO2 is due to the effectsof the support on the electronic properties of the supportedvanadate species or to some other cause. The results of thepresent study suggest that the difference in activity of VOx
supported on TiO2 and SiO2 can be attributed to the effects ofO-atom defects present on the surface of TiO2 in proximity tothe active site but is not likely due to effects of the support onthe electronic and, hence, catalytic properties of the supportedvanadate species.
Theoretical Methods
Density functional theory (DFT) was used to computeoptimized geometries, vibrational frequencies, and thermody-namic properties for all species involved in the reactionmechanism. As discussed below, the active center was describedby a cluster that included a single vanadate species and a partof the support. In geometry optimization calculations, all atomsof the cluster were allowed to relax. The B3LYP functionalwas used to describe effects of electron exchange and correla-tion, and the 6-31G* basis set was used for all nonmetal atoms.Both V and Ti atoms were treated using the LANL2DZ basisset within Gaussian 03.16 The calculated frequencies weremultiplied by 0.9614 to compensate for the overestimation ofvibrational frequencies at the B3LYP/6-31G* level of DFT.17
The growing string method18 was used to find an initial estimateof the transition-state geometry, which was then refined byperforming a transition-state search in Gaussian 03. The brokensymmetry approach19,20 was used to calculate the activationenergy for reactions involving multiple spin states, as describedin previous studies.14,15 A more accurate estimate of the energywas obtained by performing single-point calculations on alloptimized ground-state and transition-state structures using the6-311++G** basis set for all nonmetal atoms and augmentingthe LANL2DZ basis set with diffuse and polarization functions(triple-� effective core potential basis sets) for V and Ti.
The Gibbs free energy for all structures was calculated usingthe standard equations of statistical mechanics.21–25 In the
determination of rate parameters, the partition functions fortranslation, rotation, and vibration were computed explicitly.15
Appropriate assumptions were made for all species involvingthe catalyst. For example, adsorbed species were assumed tohave zero degrees of translational and bulk rotational freedom.Scaled frequencies (see above) were used to compute thevibrational partition function for all species. The standard statefor all reported Gibbs free energies, at 450 K, the temperatureat which the selective oxidation reaction was studied experi-mentally,11 is denoted as ∆G °(450). The standard state usedfor gas-phase species was taken to be 1 atm, and a mole fractionof 1 was used for all species involving the active site.
Results and Discussion
Representation of the Support and the Catalytically ActiveSite. Since our experimental studies of methanol oxidationcatalyzed by titania-supported vanadia11 have used the anatasephase of titania as the support, the first objective of the presentwork was to find a suitable representation for the surface ofanatase. The two models shown in Figure 1 were considered.The first of these, model A, is formed by replacing each of theSi atoms in silsesquioxane (Si8O12H8), the model used previouslyto represent silica,14,15 by a Ti atom. The core of this cluster(Ti8O12)isidenticaltothatfoundincrystalline[Ti8O12(H2O)24]8+.26–28
In this model, Ti has a coordination of 4 and O has acoordination of 2. Model B is formed by taking a cluster fromthe energetically preferred (101) surface of anatase.29,30 Thiscluster is terminated by point charges of -2 for O atoms andpseudopotentials for Ti atoms placed at their crystallographicpositions, a method used in previous studies.31,32 In contrast tomodel A, model B exhibits the coordination of Ti and O presentat the surface of anatase: 5-fold coordination for Ti and 2- or3-fold coordination for O. However, while model B is a morerealistic representation of the surface of anatase, the computa-tional cost associated with this model is considerably higherthan that associated with model A. For example, convergenceof geometry optimizations and frequency calculations for modelB (100-125 h of CPU time) took almost five times longer thanfor model A (24-36 h of CPU time). Consequently, it isimportant to establish whether the surface of anatase could berepresented adequately using model A. To address this issue,the geometry, vibrational bands, and heats of adsorption for
Figure 1. (a) Model A: a cubic model of TiO2, analogous to asilsesquioxane model of SiO2. The Ti atoms are terminated with Hatoms. (b) Model B: a cluster representation of the (101) surface ofanatase. The structure is terminated with point charges (where O atomswould be located) and pseudopotentials (where Ti atoms would belocated).
Selective Oxidation of Methanol to Formaldehyde J. Phys. Chem. C, Vol. 112, No. 34, 2008 13205
water and methanol were computed for models A and B, andthe results compared with each other and with experimentalvalues.
Table 1 shows that the Ti-O bond length and Ti-Ti distancecalculated for model A agree very closely with the experimentalvalues for [Ti8O12(H2O)24]8+,28 but do not agree with thecorresponding values for bulk anatase. The calculated Ti-O-Tibond angle is larger than that seen experimentally in the cubictitania complex due to the absence of water coordination inmodel A. The calculated geometry of model B agrees muchmore closely with that of crystalline anatase. The two Ti-Obond distances listed for model B are for O atoms that have acoordination to Ti of 2 or 3, respectively, and the three Ti-Tidistances reflect whether each of the Ti atoms is 5- or 6-foldcoordinated to O. (On the surface, O has a coordination of 2 or3 and Ti has a coordination of 5, whereas in the bulk, O has acoordination of 3 and Ti has a coordination of 6.)
The vibrational frequencies calculated for models A and Bare listed in Table 2 together with the experimental values foranatase.11,33 Both models produce vibrational features that agreereasonably well with those observed for anatase, with thepossible exception of the lowest frequency band. Closerinspection reveals that the calculated frequencies for model Aare in somewhat better agreement with the experimental valuesthan those calculated for model B.
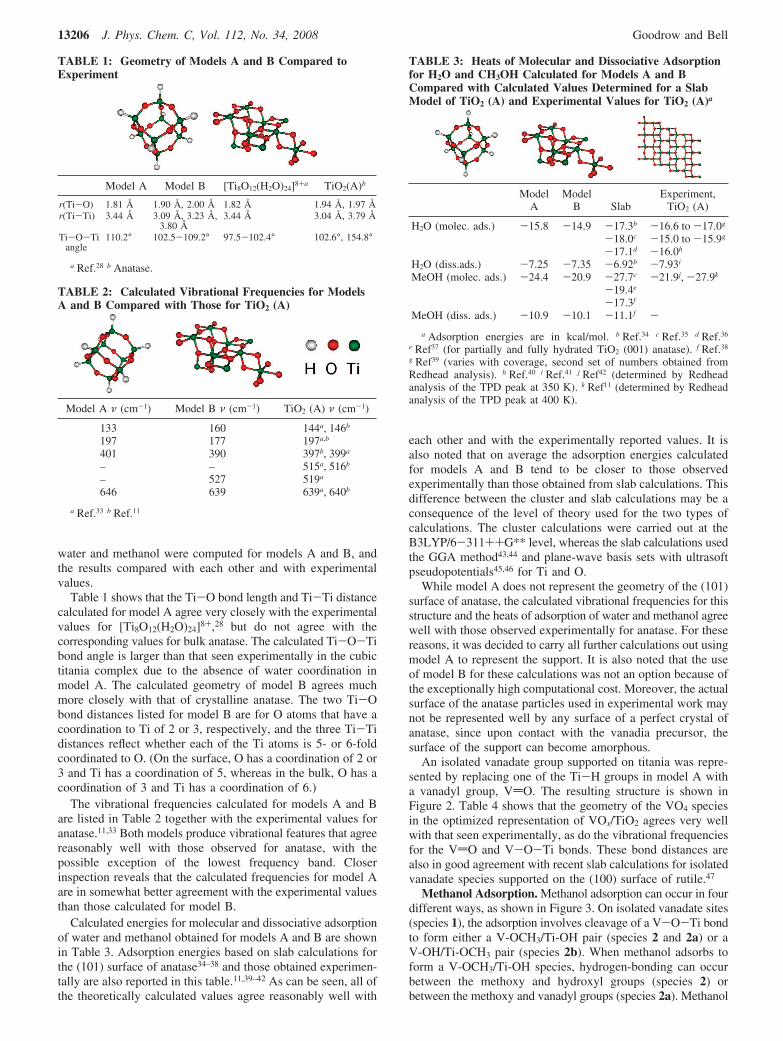
Calculated energies for molecular and dissociative adsorptionof water and methanol obtained for models A and B are shownin Table 3. Adsorption energies based on slab calculations forthe (101) surface of anatase34–38 and those obtained experimen-tally are also reported in this table.11,39–42 As can be seen, all ofthe theoretically calculated values agree reasonably well with
each other and with the experimentally reported values. It isalso noted that on average the adsorption energies calculatedfor models A and B tend to be closer to those observedexperimentally than those obtained from slab calculations. Thisdifference between the cluster and slab calculations may be aconsequence of the level of theory used for the two types ofcalculations. The cluster calculations were carried out at theB3LYP/6-311++G** level, whereas the slab calculations usedthe GGA method43,44 and plane-wave basis sets with ultrasoftpseudopotentials45,46 for Ti and O.
While model A does not represent the geometry of the (101)surface of anatase, the calculated vibrational frequencies for thisstructure and the heats of adsorption of water and methanol agreewell with those observed experimentally for anatase. For thesereasons, it was decided to carry all further calculations out usingmodel A to represent the support. It is also noted that the useof model B for these calculations was not an option because ofthe exceptionally high computational cost. Moreover, the actualsurface of the anatase particles used in experimental work maynot be represented well by any surface of a perfect crystal ofanatase, since upon contact with the vanadia precursor, thesurface of the support can become amorphous.
An isolated vanadate group supported on titania was repre-sented by replacing one of the Ti-H groups in model A witha vanadyl group, VdO. The resulting structure is shown inFigure 2. Table 4 shows that the geometry of the VO4 speciesin the optimized representation of VOx/TiO2 agrees very wellwith that seen experimentally, as do the vibrational frequenciesfor the VdO and V-O-Ti bonds. These bond distances arealso in good agreement with recent slab calculations for isolatedvanadate species supported on the (100) surface of rutile.47
Methanol Adsorption. Methanol adsorption can occur in fourdifferent ways, as shown in Figure 3. On isolated vanadate sites(species 1), the adsorption involves cleavage of a V-O-Ti bondto form either a V-OCH3/Ti-OH pair (species 2 and 2a) or aV-OH/Ti-OCH3 pair (species 2b). When methanol adsorbs toform a V-OCH3/Ti-OH species, hydrogen-bonding can occurbetween the methoxy and hydroxyl groups (species 2) orbetween the methoxy and vanadyl groups (species 2a). Methanol
TABLE 1: Geometry of Models A and B Compared toExperiment
Model A Model B [Ti8O12(H2O)24]8+a TiO2(A)b
r(Ti-O) 1.81 Å 1.90 Å, 2.00 Å 1.82 Å 1.94 Å, 1.97 År(Ti-Ti) 3.44 Å 3.09 Å, 3.23 Å,
3.80 Å3.44 Å 3.04 Å, 3.79 Å
Ti-O-Tiangle
110.2° 102.5-109.2° 97.5-102.4° 102.6°, 154.8°
a Ref.28 b Anatase.
TABLE 2: Calculated Vibrational Frequencies for ModelsA and B Compared with Those for TiO2 (A)
Model A ν (cm-1) Model B ν (cm-1) TiO2 (A) ν (cm-1)
133 160 144a, 146b
197 177 197a,b
401 390 397b, 399a
– – 515a, 516b
– 527 519a
646 639 639a, 640b
a Ref.33 b Ref.11
TABLE 3: Heats of Molecular and Dissociative Adsorptionfor H2O and CH3OH Calculated for Models A and BCompared with Calculated Values Determined for a SlabModel of TiO2 (A) and Experimental Values for TiO2 (A)a
ModelA
ModelB Slab
Experiment,TiO2 (A)
H2O (molec. ads.) -15.8 -14.9 -17.3b -16.6 to -17.0g
-18.0c -15.0 to -15.9g
-17.1d -16.0h
H2O (diss.ads.) -7.25 -7.35 -6.92b -7.93i
MeOH (molec. ads.) -24.4 -20.9 -27.7c -21.9j, -27.9k
-19.4e
-17.3f
MeOH (diss. ads.) -10.9 -10.1 -11.1f -a Adsorption energies are in kcal/mol. b Ref.34 c Ref.35 d Ref.36
e Ref37 (for partially and fully hydrated TiO2 (001) anatase). f Ref.38
g Ref39 (varies with coverage, second set of numbers obtained fromRedhead analysis). h Ref.40 i Ref.41 j Ref42 (determined by Redheadanalysis of the TPD peak at 350 K). k Ref11 (determined by Redheadanalysis of the TPD peak at 400 K).
13206 J. Phys. Chem. C, Vol. 112, No. 34, 2008 Goodrow and Bell

can also adsorb on the titania support to form Ti-OCH3/Ti-OHgroups (species 2c). Figure 3 shows the adsorption energies andGibbs free energies for these four scenarios. The most energeti-cally preferred adsorbed species is species 2a, as this structureallows for the strongest H-bonding. This conclusion is supportedby previous experimental studies, which have shown thatV-OCH3 groups, as shown in species 2a, are necessary for theformation of formaldehyde.1,7,8,10 TPD and TPRx experimentsreported by Bronkema et al.11 show that Ti-OCH3 groups doform formaldehyde but to a significantly lesser extent than doV-OCH3 groups. The results presented in Figure 3 alsodemonstrate that methanol reacts preferentially with V-O-Tibonds than Ti-O-Ti bonds, in agreement with experimentalobservations.11
The calculated and observed frequencies for the bending andstretching modes of CH3 groups in Ti-OCH3 (in species 2c)and V-OCH3 (in species 2a) are shown in Table 5. Thecalculated frequencies for the C-H bending vibration and thosefor symmetric and asymmetric C-H stretching vibrationsassociated with Ti-OCH3 and V-OCH3 species agree with thoseobserved experimentally, which further supports the choice ofmodel A to represent the support.6,11,48,49
Mechanism and Kinetics of Methanol Oxidation. Experi-mental studies suggest that the oxidation of methanol toformaldehyde involves the dissociative adsorption of methanol,followed by the transfer of an H atom from the adsorbedmethoxy group to the vanadyl O of the active site in the rate-limiting step, as illustrated in Figure 4.3,8,10 The reversibleadsorption of methanol by 1 is characterized by ∆E ) -15.7kcal/mol and ∆G °(450) ) -2.7 kcal/mol, and this process isassumed to be quasi-equilibrated. Transfer of one of the threemethoxy H atoms in 2a to form 3 occurs with ∆E ) 36.7 kcal/mol and a ∆G°(450) ) 39.0 kcal/mol. The calculated activationenergy for this step is ∆E‡ ) 38.5 kcal/mol. The brokensymmetry approach was used to determine the activation energyfor this step because the electronic structure of V changes froma singlet in the reactant 2a (formally V5+ with no unpaired d
electrons) to a triplet in the product 3 (formally V3+ with twounpaired d electrons). The weakly bound formaldehyde speciesin 3 desorbs with ∆E ) 16.3 kcal/mol and ∆G °(450) ) -0.6kcal/mol, leaving behind two hydroxyl groups on the surfacein species 4. These groups then react to form water with ∆E )-9.6 kcal/mol and ∆G°(450) ) -24.8 kcal/mol. The activationenergy for the reaction of 4 to 5 is ∆E‡ ) 9.7 kcal/mol.
In the course of the reaction sequence shown in Figure 4,the V atom is reduced from the +5 oxidation state in 1 to the+3 oxidation state in 5. To complete the catalytic cycle, the Vatom in species 5 must be reoxidized. It has been seenexperimentally11 that V remains in the +5 oxidation state underreaction conditions, suggesting that the reoxidation process isvery rapid. Figure 5 shows a possible mechanism for thereoxidation starting from species 5′, which consists of tworeduced V3+ cations in opposite corners of the cubic model.This scheme is identical to that described in a theoretical studyof methanol oxidation on VOx/SiO2.15 In the first step, a peroxidespecies is formed by the adsorption of O2 onto species 5′ with∆E ) -44.8 kcal/mol. An O atom in the resulting peroxidethen migrates through the support until both V atoms arereoxidized to V5+ in species 1′. The largest activation barrierin the reoxidation pathway is ∆E‡ ) 16.2 kcal/mol for thereaction of 7 to 8, which is similar to the barrier for O2 migrationon the surface of anatase, 11.3 kcal/mol.50
The kinetics of methanol oxidation can be represented by eq1 and the apparent first-order rate coefficient can then beexpressed as
kapp )Kadskrls ) kapp0 exp(-∆Eapp
RT ) (2)
∆Eapp )∆Eads +∆Erls‡ (3)
where kapp0 is the apparent pre-exponential factor, ∆Eapp is the
apparent activation energy, ∆Eads is the energy change formethanol adsorption, and ∆Erls
‡ is the activation energy for therate-limiting step.
The equilibrium constant for methanol adsorption, Kads, isdefined as
Kads ) σads
qMeOH•S
qMeOHqSexp(PV
RT ) exp(-∆Eads
RT ) (4)
where σads is the symmetry factor for the adsorption of methanoland qi is the partition function for state i (S ) active site).Methanol was treated as an ideal gas. The rate constant krls forthe rate-limiting step is defined as
krls ) σrlsκ(T)kBT
hq‡
qiexp(-∆Erls
‡
RT ) (5)
where σrls is the symmetry factor for the rate-limiting step andqi and q‡ refer to the partition functions for the reactant andtransition state structures, respectively. The activation energyis denoted by ∆Erls
‡ . The transmission coefficient for tunneling,κ, is a function of temperature and is included because thetransition state involves the transfer of an H atom. The valueof κ at 450 K was determined to be 1.4 using Wigner’sapproximation.51
The rate parameters determined for the oxidation of methanolto formaldehyde on the basis of the mechanism shown in Figure4 for VOx/TiO2 are listed in Table 6 and compared with similarvalues reported earlier for VOx/SiO2.15 The adsorption energyfor methanol, ∆Eads, and the activation energy for the rate-limiting step, ∆Erls
‡ , are essentially the same on both supports.
Figure 2. Model of an isolated vanadate species supported on TiO2.
TABLE 4: Comparison of the Geometry and VibrationalFrequencies for [(O)3VdO] Species Supported on TiO2 (A)Calculated for Species 1 and Observed Experimentally
Theory Experiment, TiO2 (A)a
r(VdO) 1.57 Å 1.58 År(V-O) 1.77 Å 1.79 Åν(VdO) 1035 cm-1 1026 cm-1
ν(V-O-Ti) 977 cm-1 980 cm-1
a Ref.11
Selective Oxidation of Methanol to Formaldehyde J. Phys. Chem. C, Vol. 112, No. 34, 2008 13207

The apparent activation energy on both supports differ by only1.5 kcal/mol and the apparent first-order rate constant is lessthan a factor of 3 larger on TiO2 than SiO2. These results suggestthat the much higher methanol oxidation activity observed fortitania-supported vanadia relative to silica-supported vanadia isnot due to an intrinsic difference in the electronic properties ofthe support, in agreement with the results reported earlier byKhaliullin and Bell.12 Therefore, another explanation must besought for the strong enhancement in the rate of methanoloxidation observed for vanadate species supported on titania.
Titania and silica differ in the concentration of O-vacanciesand their effect on the oxide. These O-vacancies or defects occurin much higher concentration on the surface of titania than that
of silica.52–55 Moreover, the O-atom defects in titania differ fromthose in silica.56 When an O-vacancy is formed in silica byremoval of an O atom from a Si-O-Si bond, the distancebetween the Si atoms decreases. Excess electron density on theSi atoms is localized and leads to the formation of a Si-Si bond.However, when an O-vacancy is formed in titania, the Ti atomsbecome formally Ti3+ and the distance between the Ti atomsincreases. The excess electron density carried on the pair of Tiatoms associated with the O-vacancy shifts to the 3d orbitalsand causes even more electron delocalization in the neighboringatoms.56 A density of states analysis shows that the band gap isdecreased when there is an O-vacancy, making the unoccupiedorbitals in Ti more reactive.
In most studies of O-atom defect formation in titania, defectsare produced by heating the oxide in vacuum. An interestingfinding of such work is that surface O-vacancies are energeticallyfavored compared to those in the bulk.57 This conclusion issupported by theoretical work demonstrating that the diffusionof an O-vacancy from the bulk to the surface of titania issignificantly faster than the reverse process.58 Quantification ofthe surface concentration of O-atom defects is rare; however,STM studies show that upon heating rutile in vacuum to700-1100 K, the (110) surface of rutile can contain 2-10%O-vacancies.42,59–61
An important question is whether O-vacancies can besustained at elevated temperatures when titania is heated inoxygen. Since the energy for O-atom defect formation is 4.0eV for rutile (110) and 4.3 eV for anatase (101),62 O-vacancieswould not be expected to be present on the surface or in thebulk of titania if the process of defect formation and annihilationwere controlled by thermodynamics. However, recent work bySekiya and co-workers has shown that O-atom defects can besustained in anatase at temperatures up to 873 K in the presenceof 1 MPa of O2.63–66 These authors observed the presence ofO-atom defects by polarized absorption spectroscopy and EPR.When an as-grown anatase crystal was heated in O2, a banddeveloped at about 3 eV, which grew in intensity as the
Figure 3. Possible structures for methanol adsorption onto species 1 (left) or TiO2 (right). Values of ∆E and ∆G °(450) are in kcal/mol.
TABLE 5: Comparison of the Vibrational FrequenciesCalculated for Ti-OCH3 (2c) and V-OCH3 (2a) with ThoseObserved Experimentally
AdsorbedSpecies Vibrational Mode
Theory(cm-1)
Experiment(cm-1)a
Ti-OCH3 C-H Bend 1432 1440-1443Symmetric C-H 2832 2819-2833Asymmetric C-H 2919 2921-2932
V-OCH3 C-H Bend 1421 1435-1436Symmetric C-H 2838 2832Asymmetric C-H 2929 2929-2939
a Ref.11
TABLE 6: Comparison of the Energetics and ApparentFirst-Order Rate Coefficient for Methanol Oxidation onIsolated VOx Species Supported on SiO2 and TiO2
VOx/SiO2a VOx/TiO2
∆Eads (kcal/mol) -15.5 -15.7∆E‡
rls (kcal/mol) 39.8 38.5∆E‡
reox (kcal/mol) 28.1 16.2∆Eapp (kcal/mol) 24.3 22.8kapp (1/atm · s) 3.99 × 10-5 9.35 × 10-5
a Ref.15
13208 J. Phys. Chem. C, Vol. 112, No. 34, 2008 Goodrow and Bell

temperature was raised from 473 to 873 K, and the crystalchanged its color from pale blue to yellow.63,65 The EPR signalof the yellow crystal indicated the absence of paramagnetic spinassociated with the conduction band, suggesting that the O trapstwo electrons to form a relatively deep level. When the yellowcrystal was heated in O2 at 1073 K for 60 h the band at 3.0 eVdisappeared completely and the crystal became clear. Thestability of the defects up to 873 K when the yellow crystalwas heated in 1 MPa of O2 led to the conclusion that O-atomdefects in anatase are not in thermodynamic equilibrium withthe gas phase.
When vanadia is supported on titania other changes can occurin addition to the formation of O-vacancies in titania. A numberof authors have shown EPR evidence for the formation ofisolated V4+ cations for vanadia supported on either rutile or amixture of rutile and anatase.67–74 Since g| > g⊥ and A| > A⊥ ,V4+ cations are presumed to have octahedral symmetry and tobe located at the positions of Ti4+ cations in the rutile phase ofthe matrix.70,74 However, estimates of the fraction of thesupported V converted to V4+ for VOx/TiO2 samples containingpredominantly isolated vanadate species show that only about1% of the V is present as V4+.70,74 The incorporation of V4+
Figure 4. Pathway for the selective oxidation of methanol to formaldehyde on isolated vanadate species supported on titania. Values of ∆E, ∆E†,and ∆G°(450) are in kcal/mol.
Figure 5. Pathway for the reoxidation of V from V3+ to V5+ on titania via oxygen migration. Values of ∆E, ∆E†, and ∆G°(450) are in kcal/mol.
Selective Oxidation of Methanol to Formaldehyde J. Phys. Chem. C, Vol. 112, No. 34, 2008 13209

cations into anatase was not considered, since in this case g| <g⊥ .74 For these reasons, it is unlikely that V4+ cations wouldhave an effect on the catalytic activity of isolated vanadatespecies supported on titania.
Direct evidence for the formation of O-vacancies in TiO2 havebeen obtained recently by high-field (14.9 T) EPR carried outon VOx/TiO2 (containing exclusively isolated vanadate species).The catalyst was used for methanol oxidation (T ) 450 K, P )1 atm, MeOH/O2 ) 1: 2), rapidly quenched, and then sealed.75
As shown in the Supporting Information, a sharp peak isobserved at g ) 2.00, corresponding to electrons trapped at theO-vacancies, and a broad peak is observed at g ) 1.96,corresponding to Ti3+. The latter peak is not observed in thesample prior to reaction. Thus, EPR spectroscopy suggests thatduring methanol oxidation O-vacancies are created in thesupport, and that the electrons thus released can form two Ti3+
cations per O-vacancy.O-vacancies present on the surface of VOx/TiO2 can be
associated with either a V or a Ti atom. Previous research hasshown that the migration of surface defects is facilitated in thepresence of gas-phase O2;76 hence, it is reasonable to assumethat the distribution of vacancies is determined by thermody-namics. Equilibrium can be established between an active sitewith an O-vacancy, represented as [V-D] (where V stands fora V atom and D stands for an O-vacancy) and without anO-vacancy, represented as [V-ND] (where ND stands for noO-vacancy). Calculations performed with species 1 show thatthe energetically preferred location for an O-vacancy is next tothe vanadate species, as shown in Figure 6. The stability of anO-vacancy in the vicinity of a vanadate is further supported bythe fact that removal of an O atom from a V-O-Ti bond is
almost 35 kcal/mol higher than removal from a Ti-O-Ti bond.The equilibrium constant for the migration of an O-vacancyfrom titania to a position adjacent to an isolated vanadate speciescan be determined on the basis of the scheme shown in Figure7. Using this scheme, the equilibrium constant for the exchangeof an O-vacancy in titania with an O atom near an isolatedvanadate species (1) is calculated to be 2.65 at 450 K, fromwhich it is concluded that O-vacancies present on the surfaceof VOx/TiO2 prefer to be near vanadate species. Figure 8 showsthe relationship between the fraction of isolated vanadate speciescontaining an adjacent O-vacancy and the fraction of O-vacancies in the titania for the conditions of the experimentsreported by Bronkema et al.11
The change in energy and Gibbs free energy for the reactionmechanism involving an active site with an O-vacancy is shownin Figure 9. This mechanism is analogous to the mechanism inFigure 4 for an active site without an O-vacancy. The adsorptionof methanol onto a defect site, the reaction of 1d to 2ad, occurswith ∆E ) -15.9 kcal/mol and ∆G °(450) ) -2.4 kcal/mol.Both of these values are similar to the energetics seen formethanol adsorption on a site without an O-vacancy, suggestingthat methanol adsorption on VOx/TiO2 is essentially the samefor active sites with and without an O-vacancy. The values forthe change in energy and Gibbs free energy for the rate-limitingstep, the transfer of an H atom from an adsorbed methoxy group,are ∆E ) 19.5 kcal/mol and ∆G°(450) ) 24.7 kcal/mol,respectively. These values are considerably smaller than thosedetermined for the same reaction in the absence of a defect. Aplot of the energy profile as a function of reaction coordinate isshown in Figure 10 highlighting the difference between theH-abstraction step on an active site with and without anO-vacancy. The reason for the smaller energy of the reactionfor 2ad to 3d is that the defect provides the active site withmore flexibility thereby allowing for a larger degree of H-bonding in the product between Ti-OH and V-OH ligands. AMulliken charge analysis shows that the oxidation state of V inthis step is the same as when there is no O-vacancy present.The activation energy for 2ad to 3d is 31.8 kcal/mol, which isalmost 7 kcal/mol lower than that calculated for the case of anactive site without a defect. The decrease in activation energyis a result of H-bond stabilization in the transition state structure.The H-abstraction step is followed by desorption of formalde-hyde from 3d to 4d, with ∆E ) 22.3 kcal/mol and ∆G°(450)) 3.5 kcal/mol. The remaining two hydroxyl groups cometogether to form water and reform a V-O-Ti support bond inthe reaction of 4d to 5d. For this reaction, ∆E ) -8.7 kcal/mol, ∆G°(450) ) -25.0 kcal/mol, and ∆E‡ ) 12.1 kcal/mol.
TABLE 7: Comparison of Theoretical and Experimental Values for the Energetics and Rate Parameters for MethanolOxidation on VOx/TiO2
a
Theory (450 K)No Defect
Theory (450 K)Defect
Theory (450 K) with 1%,2% Defectsb
Experiment(450 K)
Adsorption Step (1,2a), (1d,2ad) ∆Eads (kcal/mol) -15.7 -15.9 -15.9 -14.5c, -13.0d
Kads0 (1/atm) 4.21 × 10-6 6.17 × 10-6 6.17 × 10-6 3.36 × 10-5c, 3.44 × 10-6e
Kads (1/atm) 178 337 337 371c, 301d
Rate-Limiting Step (2a,3), (2ad, 3d) ∆Erls‡ (kcal/mol) 38.5 31.8 31.8 30.5c, 30.7e
krls0 (1/s) 2.63 × 1012 1.75 × 1012 4.03 × 1011, 6.48 × 1011 1.57 × 1011 c, 2.54 × 1012 e
krls (1/s) 5.26 × 10-7 6.34 × 10-4 1.46 × 10-4 Å, 2.35 × 10-4 2.41 × 10-4 c, 3.10 × 10-3 e
Apparent Kinetics ∆Eapp (kcal/mol) 22.8 15.9 15.9 16c, 15.93d
kapp0 (1/atm · s) 1.11 × 107 1.08 × 107 2.48 × 106, 4.00 × 106 5.30 × 106 c, 2.38 × 106 d
kapp (1/atm · s) 9.35 × 10-5 2.14 × 10-1 4.92 × 10-2, 7.92 × 10-2 8.96 × 10-2 c, 4.37 × 10-2 d
a The apparent rate constants are defined on a per active site basis. b Corresponding value in previous column multiplied by 23% (for 1%surface O vacancies) and 37% (for 2% surface O vacancies). Refer to Figure 8. c Ref.11 d Ref.8 e Ref.3
Figure 6. Model of a VOx/TiO2 active site with a defect. The locationof the O-vacancy is circled.
13210 J. Phys. Chem. C, Vol. 112, No. 34, 2008 Goodrow and Bell

The reoxidation mechanism of V on a site with an O-vacancyis achieved in the same manner as in Figure 5 for a site withoutan O-vacancy. Since the surface O-vacancies are mobile on thesurface, an O-vacancy will be rapidly interchanged with an Oin the active site (1 to 1d). The rate of vacancy diffusion hasbeen estimated to be 10-2 to 10-1 s-1 at 250 K and to increasewith temperature and exposure to O2.76 Assuming an Arrheniustemperature dependence and an activation barrier for O migra-tion of 10.4 kcal/mol,77 the rate of vacancy diffusion at 450 Kunder would be 102-103 s-1, which is significantly faster thanthe rate-limiting step in the reaction mechanism, 2ad to 3d.Hence, the rate of reoxidation is rapid and not kineticallyrelevant.
Comparison with Experimental Results. Table 7 lists thecalculated rate parameters for methanol oxidation for an activesite with and without an adjacent O-vacancy at 450 K and theexperimental values reported by Burcham and Wachs3,8 and byBronkema et al.11 The presence or absence an O-vacancy nearthe vanadate center has virtually no effect on the value of theadsorption energy. The calculated values of the adsorptionenergy are in good agreement with the experimental valuereported by Bronkema and Bell11 but somewhat higher than thatreported by Burcham and Wachs.3,8
The corresponding equilibrium adsorption constants at 450K are K1,2a ) 178 atm-1 and K1d,2ad ) 337 atm-1. Both typesof active site show reasonable agreement with experimentalmeasurements of the equilibrium adsorption constant. The betteragreement for the site containing an O-atom defect comesprimarily from the higher pre-exponential factor.
As mentioned previously, the rate-limiting step in the selectiveoxidation of methanol to formaldehyde is abstraction of an Hatom from an adsorbed methoxy group to a vanadyl O. Table7 shows that the difference in activation energy between siteswith and without an O-vacancy is 6.7 kcal/mol. It is also evident
that the value of ∆Erls‡ determined for the site with a O-vacancy
agrees more closely to that seen experimentally by Bronkemaand Bell,11 and Wachs and co-workers.3,8 The value of the pre-exponential factor determined for the site with an O-vacancy isvery similar to that for the site without an O-vacancy. Tocompare the value of the pre-exponential factor observedexperimentally with those determined from experiments, it isnecessary to estimate the fraction of all vanadate sites that areadjacent to an O-vacancy. Unfortunately, this is difficult to do,since there are no experimental data on which to base thisestimate, and there is insufficient knowledge of the kinetics ofO-atom defect formation and annihilation to obtain a theoreticalestimate. Therefore, we have assumed that 1-2% of the O atomspresent at the surface of the support are absent, i.e., are O-atomdefects, and have used Figure 8 to then determine the fractionof surface vanadate groups associated with an O-atom defect.On this basis, we project that 23-37% of the vanadate speciesare associated with an O-vacancy in the support (correspondingto 1-2% O sites with an O-vacancy). Since the pre-exponentialfactors determined from experimental data assume that all ofthe V sites are equally active, we have multiplied the values ofkrls
0 determined for vanadate sites associated with an O-vacancyby 0.23-0.37. Table 7 shows that the corrected value of krls
0
agrees reasonably well with the values of krls0 reported experi-
mentally than does the value of krls0 determined for sites not
associated with an O-vacancy. Likewise, it is seen that the valueof krls determined for vanadate sites associated with O-vacanciesand corrected for the fraction of such sites agrees much moreclosely with the experimentally reported values of this parameter.
Table 7 also lists the apparent pre-exponential factor, apparentactivation energy, and the first-order rate coefficient determinedfrom eq 2 for the cases of isolated vanadate sites that are andare not next to an O-vacancy. The value of ∆Eapp
‡ determinedfor the site which contains an O-vacancy is 6.9 kcal/mol lowerthan that determined for the site that does not have a vacancy,and the value of ∆Eapp
‡ is in excellent agreement with the valuesobserved experimentally. While the presence of O-vacancieshas almost no effect on the calculated values of the apparentpre-exponential factor, kapp
0 , the value of the apparent first-orderrate coefficient, kapp, is roughly 3 orders of magnitude largerwhen an O-atom defect is present near a vanadate site, theprincipal cause of this difference being the lower value of ∆Eapp
‡ .When the values of kapp
0 and kapp are corrected for the estimatedfraction of vanadate species associated with O-vacancies, Table7 shows that the experimental and theoretical values are inexcellent agreement.
The results presented in Tables 6 and 7 suggest that the higheractivity of isolated vanadate species supported on titania relativeto those supported on silica is not due to inherent differencesin the electronic properties of vanadate species caused by thecomposition of the support. Instead, we propose that thesignificantly higher activity of VOx/TiO2 can be attributed to areduction in the activation energy for the rate-limiting step
Figure 7. Scheme for an O-vacancy in TiO2 (TiO2-D) exchanging with an O atom to form an active site with an O-vacancy (1d).
Figure 8. Dependence of the percent of V atoms with an associatedO-vacancy on the percent of O sites with an O-vacancy on the surfaceof titania at 450 K.
Selective Oxidation of Methanol to Formaldehyde J. Phys. Chem. C, Vol. 112, No. 34, 2008 13211

caused by an O-vacancy adjacent to the active center. Thisinterpretation differs from that given by Wachs and co-workerswho proposed that the higher activity of VOx/TiO2 relative toVOx/SiO2 is due to the effect of support composition on theintrinsic electronic properties of the supported vanadate species.This conclusion was based on the observation that the specificactivity of supported vanadate species for methanol oxidationto formaldehyde increases with a decrease in the Sandersonelectronegativity of the metal cation in the support oxide. Whilethe two interpretations for the effects of support compositionappear to be in conflict, in fact, they are not. As noted in Table8, the Sanderson electronegativity of the support cation78 exhibitsa positive correlation with the energy to form an O-vacancy.The net result is a positive correlation between the turnoverfrequency for methanol oxidation to formaldehyde and theenergy of O-vacancy formation. Figure 11 shows a stronginverse correlation between the turnover frequency for methanoloxidation and the energy of O-vacancy formation62,79,80 for dataon different supports reported by Wachs and co-workers7 (R2
value of 0.9486) and Bell and co-workers10,11,81 (R2 value of
0.8974). The lower TOFs reported by Bell and co-workers thanthose reported by Wachs and co-workers may be a consequenceof the difference in the surface coverage of vanadia and, hence,the structure of the vanadate groups present on the supportsurface. In the work of Wachs and co-workers all samples hadhigh vanadia surface coverages, resulting in the presence ofpolyvanadate as well as monovanadate species, whereas in thework of Bell and co-workers all of the sample were prepared
Figure 9. Pathway for the selective oxidation of methanol to formaldehyde on isolated vanadate species supported on titania, with the active sitecontaining an O-vacancy. Values of ∆E, ∆E†, and ∆G°(450) are in kcal/mol.
Figure 10. Energy profile for the H-abstraction step on an active sitewith and without an O-vacancy adjacent to the active site. The reactioncoordinate axis has been normalized.
TABLE 8: Sanderson Electronegativities and Energies forthe Formation of O-Vacancies in Metal Oxide Supports
Metal Oxide Support S (cation)a Ef1/2O2 (eV)b
SiO2 2.138 8.5c
R-Al2O3 1.714 6d
TiO2 (A) 1.5 4.3e
m-ZrO2 0.9 4e
CeO2 0.9 3.3e
a Ref 78. b Ef1/2O2 is defined as the defect formation energy using
half of the total energy of molecular oxygen; see ref 62. c Ref 80.d Ref 79. e Ref 62.
Figure 11. TOF per V atom for methanol oxidation versus theO-vacancy formation energy. a Ref 7. b Refs 10 (SiO2), 11 (TiO2), 81(ZrO2).
13212 J. Phys. Chem. C, Vol. 112, No. 34, 2008 Goodrow and Bell

with predominantly (>90%) monovanadate species. The resultsof the present investigation, therefore, have revealed a new andunexpected effect of O-atom defects on the activity of vanadatespecies for the oxidation of methanol. It is reasonable toanticipate that such defects may have similar effects on otherreactions occurring on dispersed metal oxides.
Conclusions
The oxidation of methanol to formaldehyde catalyzed byisolated sites supported on titania has been examined theoreti-cally. The properties of the active site are well represented bya model consisting of a [(O)3VdO] group positioned at thecorner of a cubic TiOx cluster, a model similar to that usedpreviously to model the oxidation of methanol on isolatedvanadate species supported on silica. Each of the rate parameters,the apparent activation energy, and the apparent rate coefficientdetermined for VOx/TiO2 are very similar to those determinedpreviously for VOx/SiO2.15 This finding indicates that theelectronic properties of isolated vanadate species are not affectedby the composition of the support and, hence, that thesignificantly higher activity of VOx/TiO2 seen experimentallycannot be explained by this means. In contrast to silica,O-vacancies can form on the surface of titania, and such defectscan affect the catalytic properties of species supported on titania.Our calculations show that the introduction of an O-vacancy inthe support at a point adjacent to a vanadate site reduces theapparent activation energy for methanol oxidation from 22.8 to15.9 kcal/mol. If the concentration of O-atom defects on thesurface of the support is taken to be 1-2% of all O atomspresent at the surface, it is found that surface defects concentratepreferentially adjacent to the vanadate species. The pre-exponential factor and activation energy for the rate-limitingstep determined for a model of the active site with an adjacentO-vacancy agree very well with those deduced from experi-ments, as does the apparent activation energy. The role of defectsin facilitating the rate of methanol oxidation is further supportedby the observation that the specific activity of vanadium centersincreases with decreasing energy of defect formation.
Acknowledgment. The authors acknowledge Andrzej Ozarow-ski and Klaus-Peter Dinse, who acquired the EPR data givenin the Supporting Information. This work was supported by theMethane Conversion Cooperative funded by BP.
Supporting Information Available: A zip archive containsminimum energy structures and transition state structures inXYZ format from Gaussian03. README.txt files are includedwithin the archive to guide the reader. EPR spectra of VOx/TiO2 recorded before and after use for methanol oxidation arealso presented. This material is available free of charge via theInternet at http://pubs.acs.org.
References and Notes
(1) Deo, G.; Wachs, I. E. J. Catal. 1994, 146–323.(2) Deo, G.; Wachs, I. E. J. Catal. 1994, 146, 335.(3) Burcham, L. J.; Wachs, I. E. Catal. Today 1999, 49, 467.(4) Lim, S. Y.; Haller, G. L. Appl. Catal., A 1999, 188, 277.(5) Briand, L. E.; Farneth, W. E.; Wachs, I. E. Catal. Today 2000, 62,
219.(6) Baltes, M.; Cassiers, K.; Van Der Voort, P.; Weckhuysen, B. M.;
Schoonheydt, R. A.; Vansant, E. F. J. Catal. 2001, 197, 160.(7) Burcham, L. J.; Briand, L. E.; Wachs, I. E. Langmuir 2001, 17,
6164.(8) Burcham, L. J.; Badlani, M.; Wachs, I. E. J. Catal. 2001, 203, 104.(9) Wachs, I. E. Catal. Today 2005, 100, 79.
(10) Bronkema, J. L.; Bell, A. T. J. Phys. Chem. C 2007, 111, 420.
(11) Bronkema, J. L.; Leo, D. C.; Bell, A. T. J. Phys. Chem. C 2007,111, 14530.
(12) Khaliullin, R. Z.; Bell, A. T. J. Phys. Chem. B 2002, 106, 7832.(13) Zhanpeisov, N. U. Res. Chem. Intermed. 2004, 30, 133.(14) Dobler, J.; Pritzsche, M.; Sauer, J. J. Am. Chem. Soc. 2005, 127,
10861.(15) Goodrow, A.; Bell, A. T. J. Phys. Chem. C 2007, 111, 14753.(16) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G.; E.;
Robb, M. A. C., J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.;;Burant, J. C. M., J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci,B.; Cossi,; M.; Scalmani, G. R., N.; Petersson, G. A.; Nakatsuji, H.; Hada,M.; Ehara, M.;; Toyota, K. F., R.; Hasegawa, J.; Ishida, M.; Nakajima, T.;Honda, Y.; Kitao, O.;; Nakai, H. K., M.; Li, X.; Knox, J. E.; Hratchian,H. P.; Cross, J. B.; Adamo, C.;; Jaramillo, J. G., R.; Stratmann, R. E.;Yazyev, O.; Austin, A. J.; Cammi, R.;; Pomelli, C. O., J. W.; Ayala, P. Y.;Morokuma, K.; Voth, G. A.; Salvador, P.;; Dannenberg, J. J. Z., V. G.;Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas,; O.; Malick, D. K. R.,A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui,; Q.; Baboul,A. G. C., S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.;;Piskorz, P. K., I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.;Peng,; C. Y.; Nanayakkara, A. C., M.; Gill, P. M. W.; Johnson, B.; Chen,W.; Wong,; M. W.; Gonzalez, C. P., J. A.; Gaussian 03; Gaussian, Inc.:Pittsburgh, PA, 2003.
(17) Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502.(18) Peters, B.; Heyden, A.; Bell, A. T.; Chakraborty, A. J. Chem. Phys.
2004, 120, 7877.(19) Caballol, R.; Castell, O.; Illas, F.; Moreira, P. R.; Malrieu, J. P. J.
Phys. Chem. A 1997, 101, 7860.(20) Noodleman, L. J. Chem. Phys. 1981, 74, 5737.(21) Atkins, P.; dePaula, J. Physical Chemistry, 7th ed.; W. H. Freeman
& Co.: New York, 2002.(22) Chandler, D. Introduction to Modern Statistical Mechanics; Oxford
University Press: New York, 1987.(23) Hill, T. L. An Introduction to Statistical Thermodynamics; Dover
Publications, Inc.: New York, 1986.(24) McQuarrie, D. A. Statistical Mechanics; University Science Books,
Sausalito, CA, 2000.(25) Ochterski, J. W. Thermochemistry in Gaussian; Gaussian, Inc.:
Pittsburgh, PA, 2000.(26) Reichmann, M. G.; Bell, A. T. Appl. Catal. 1987, 32, 315.(27) Reichmann, M. G.; Bell, A. T. Langmuir 1987, 3, 111.(28) Reichmann, M. G.; Hollander, F. J.; Bell, A. T. Acta Crystallogr.,
Sect. C: Cryst. Struct. Commun. 1987, 43, 1681.(29) Vittadini, A.; Selloni, A.; Rotzinger, F. P.; Gratzel, M. Phys. ReV.
Lett. 1998, 81, 2954.(30) Hebenstreit, W.; Ruzycki, N.; Herman, G. S.; Gao, Y.; Diebold,
U. Phys. ReV. B 2000, 62, R16334.(31) Stefanovich, E. V.; Truong, T. N. J. Phys. Chem. B 1998, 102,
3018.(32) Stefanovich, E. V.; Truong, T. N. Chem. Phys. Lett. 1999, 299,
623.(33) Zhang, J.; Li, M. J.; Feng, Z. C.; Chen, J.; Li, C. J. Phys. Chem.
B 2006, 110, 927.(34) Selloni, A.; Vittadini, A.; Gratzel, M. Surf. Sci. 1998, 219, 402–
404.(35) Gong, X.-Q.; Selloni, A. J. Catal. 2007, 249, 134.(36) Gong, X. Q.; Selloni, A.; Batzill, M.; Diebold, U. Nat. Mater. 2006,
5, 665.(37) Gong, X. Q.; Selloni, A. J. Phys. Chem. B 2005, 109, 19560.(38) Tilocca, A.; Selloni, A. J. Phys. Chem. B 2004, 108, 19314.(39) Herman, G. S.; Dohnalek, Z.; Ruzycki, N.; Diebold, U. J. Phys.
Chem. B 2003, 107, 2788.(40) Egashira, M.; Kawasumi, S.; Kagawa, S.; Seiyama, T. Bull. Chem.
Soc. Jpn. 1978, 51, 3144.(41) Levchenko, A. A.; Li, G. S.; Boerio-Goates, J.; Woodfield, B. F.;
Navrotsky, A. Chem. Mater. 2006, 18, 6324.(42) Henderson, M. A.; Otero-Tapia, S.; Castro, M. E. Faraday Discuss.
1999, 114, 313.(43) DalCorso, A.; Pasquarello, A.; Baldereschi, A.; Car, R. Phys. ReV.
B 1996, 53, 1180.(44) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.;
Pederson, M. R.; Singh, D. J.; Fiolhais, C. Phys. ReV. B 1992, 46, 6671.(45) Laasonen, K.; Pasquarello, A.; Car, R.; Lee, C.; Vanderblit, D. Phys.
ReV. B 1993, 47, 10142.(46) Vanderbilt, D. Phys. ReV. B 1990, 41, 7892.(47) Calatayud, M.; Mguig, B.; Minot, C. Theor. Chem. Acc. 2005, 114,
29.(48) Burcham, L. J.; Deo, G. T.; Gao, X. T.; Wachs, I. E. Top. Catal.
2000, 11, 85.(49) Calatayud, M.; Minot, C. J. Phys. Chem. C 2007, 111, 6411.(50) Liu, L. M.; McAllister, B.; Ye, H. Q.; Hu, P. J. Am. Chem. Soc.
2006, 128, 4017.
Selective Oxidation of Methanol to Formaldehyde J. Phys. Chem. C, Vol. 112, No. 34, 2008 13213

(51) Masel, R. I. Chemical Kinetics & Catalysis; Wiley-Interscience:New York, 2001.
(52) Zhanpeisov, N. U.; Fukumura, H. J. Phys. Chem. C 2007, 111,16941.
(53) Eder, D.; Kramer, R. Phys. Chem. Chem. Phys. 2003, 5, 1314.(54) Sulimov, V.; Casassa, S.; Pisani, C.; Garapon, J.; Poumellec, B.
Modell. Simul. Mater. Sci. Eng. 2000, 8, 763.(55) Imai, H.; Arai, K.; Imagawa, H.; Hosono, H.; Abe, Y. Phys. ReV.
B 1988, 38, 12772.(56) Laursen, S.; Linic, S. Phys. ReV. Lett. 2006, 97.(57) Sengupta, G.; Chatterjee, R. N.; Maity, G. C.; Ansari, B. J.;
Satyanarayna, C. V. V. J. Colloid Interface Sci. 1995, 170, 215.(58) Jug, K.; Nair, N. N.; Bredow, T. Phys. Chem. Chem. Phys. 2005,
7, 2616.(59) Diebold, U.; Anderson, J. F.; Ng, K. O.; Vanderbilt, D. Phys. ReV.
Lett. 1996, 77, 1322.(60) Diebold, U.; Lehman, J.; Mahmoud, T.; Kuhn, M.; Leonardelli,
G.; Hebenstreit, W.; Schmid, M.; Varga, P. Surf. Sci. 1998, 411, 137.(61) EplingW. S.; Peden, C. H. F.; Henderson, M. A.; Diebold, U. Surf.
Sci. 1998, 333, 412–413.(62) Ganduglia-Pirovano, M. V.; Hofmann, A.; Sauer, J. Surf. Sci. Rep.
2007, 62, 219.(63) Sekiya, T.; Ichimura, K.; Igarashi, M.; Kurita, S. J. Phys. Chem.
Solids 2000, 61, 1237.(64) Sekiya, T.; Tasaki, M.; Wakabayashi, K.; Kurita, S. J. Lumin. 2004,
108, 69.(65) Sekiya, T.; Yagisawa, T.; Kamiya, N.; Das Mulmi, D.; Kurita, S.;
Murakami, Y.; Kodaira, T. J. Phys. Soc. Jpn. 2004, 73, 703.(66) Wakabayashi, K.; Yamaguchi, Y.; Sekiya, T.; Kurita, S. J. Lumin.
2005, 112, 50.
(67) Cavani, F.; Centi, G.; Foresti, E.; Trifiro, F.; Busca, G. J. Chem.Soc., Faraday Trans. 1 1988, 84, 237.
(68) Centi, G.; Giamello, E.; Pinelli, D.; Trifiro, F. J. Catal. 1991, 130,220.
(69) Aboukais, A.; Aissi, C. F.; Dourdin, M.; Courcot, D.; Guelton, M.;Serwicka, E. M.; Giamello, E.; Geobaldo, F.; Zecchina, A.; Foucault, A.;Vedrine, J. C. Catal. Today 1994, 20, 87.
(70) Ciambelli, P.; Lisi, L.; Russo, G.; Volta, J. C. Appl. Catal., B 1995,7, 1.
(71) Alemany, L. J.; Lietti, L.; Ferlazzo, N.; Forzatti, P.; Busca, G.;Giamello, E.; Bregani, F. J. Catal. 1995, 155, 117.
(72) Paganini, M. C.; DallAcqua, L.; Giamello, E.; Lietti, L.; Forzatti,P.; Busca, G. J. Catal. 1997, 166, 195.
(73) Chary, K. V. R.; Kishan, G.; Bhaskar, T.; Sivaraj, H. J. Phys. Chem.B 1998, 102, 6792.
(74) Rodella, C. B.; Franco, R. W. A.; Magon, C. J.; Donoso, J. P.;Nunes, L. A. O.; Saeki, M. J.; Aegerter, M. A.; Sargentelli, V.; Florentino,A. O. J. Sol-Gel Sci. Technol. 2002, 25, 83.
(75) Ozarowki, A.; Dinse, K. P.; National High Magnetic FieldLaboratory, Tallahassee, FL. unpublished work, 2008.
(76) Schaub, R.; Wahlstrom, E.; Ronnau, A.; Laegsgaard, E.; Stensgaard,I.; Besenbacher, F. Science 2003, 299, 377.
(77) Wang, Y.; Pillay, D.; Hwang, G. S. Phys. ReV. B 2004, 70, 193410.(78) Sanderson, R. T. J. Chem. Educ. 1988, 65, 112.(79) Carrasco, J.; Lopez, N.; Sousa, C.; Illas, F. Phys. ReV. B 2005, 72,
54109.(80) Pacchioni, G. Solid State Sci. 2000, 2, 161.(81) Bronkema, J. L.; Bell, A. T. J. Phys. Chem. C 2008, 112, 6404.
JP801339Q
13214 J. Phys. Chem. C, Vol. 112, No. 34, 2008 Goodrow and Bell