activation of receptor activator of nf-b ligand gene expression by
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, Sept. 2006, p. 6469–6486 Vol. 26, No. 170270-7306/06/$08.00�0 doi:10.1128/MCB.00353-06Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Activation of Receptor Activator of NF-�B Ligand Gene Expressionby 1,25-Dihydroxyvitamin D3 Is Mediated through Multiple
Long-Range Enhancers†Sungtae Kim, Miwa Yamazaki, Lee A. Zella, Nirupama K. Shevde, and J. Wesley Pike*
Department of Biochemistry, University of Wisconsin—Madison, Madison, Wisconsin 53706
Received 27 February 2006/Returned for modification 4 May 2006/Accepted 12 June 2006
RANKL is a tumor necrosis factor (TNF)-like factor secreted by mesenchymal cells, osteoblast derivatives,and T cells that is essential for osteoclastogenesis. In osteoblasts, RANKL expression is regulated by two majorcalcemic hormones, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] and parathyroid hormone (PTH), as well as byseveral inflammatory/osteoclastogenic cytokines; the molecular mechanisms for this regulation are unclear. Toidentify such mechanisms, we screened a DNA microarray which tiled across the entire mouse RankL genelocus at a 50-bp resolution using chromatin immunoprecipitation (ChIP)-derived DNA precipitated withantibodies to the vitamin D receptor (VDR) and the retinoid X receptor (RXR). Five sites of dimer interactionwere observed on the RankL gene centered at 16, 22, 60, 69, and 76 kb upstream of the TSS. These regionscontained binding sites for not only VDR and RXR, but also the glucocorticoid receptor (GR). The most distantof these regions, termed the distal control region (RL-DCR), conferred both VDR-dependent 1,25(OH)2D3 andGR-dependent glucocorticoid (GC) responses. We mapped these activities to an unusual but functionally activevitamin D response element and to several potential GC response elements located over a more extensiveregion within the RL-DCR. An evolutionarily conserved region within the human RANKL gene contained asimilar vitamin D response element and exhibited an equivalent behavior. Importantly, hormonal activation ofthe RankL gene was also associated with chromatin modification and RNA polymerase II recruitment. Ourstudies demonstrate that regulation of RankL gene expression by 1,25(OH)2D3 is complex and mediated by atleast five distal regions, one of which contains a specific element capable of mediating direct transcriptionalactivation.
Skeletal remodeling in adults occurs through the coupledactions of bone-forming osteoblasts and bone-resorbing osteo-clasts (17). The latter are terminally differentiated, multinu-cleated cells of the monocyte-macrophage lineage (63). Theprocess of osteoclastogenesis is highly complex and is orches-trated by a number of growth factors, steroidal components,and cytokines, all of which exert their actions in a highly tem-poral fashion (57). Many of these regulatory factors are pro-duced and secreted by adjacent support cells that includestroma, B and T cells, and cells of the osteoblast lineage. Theproduction of such factors by osteoblasts highlights the extrin-sic role that these cells play in the process of bone resorption.Importantly, while many of these secreted components arecritical for normal adult bone remodeling, their aberrant se-cretion can be pathological and lead to either focal or systemicbone disease (45).
Although many factors participate in osteoclastogenesis, themolecule that is now considered to be both necessary andsufficient in vivo and in vitro is the receptor activator of NF-�Bligand (RankL). RankL is a tumor necrosis factor (TNF)-likefactor that is produced by stromal cells and osteoblasts as wellas a variety of other cell types (38). This factor not only actively
promotes the process of osteoclast differentiation, but also isrequired for the cell’s bone-resorbing activity and for its sur-vival (22). The interaction of RankL with receptor activator ofNF-�B (Rank), an integral receptor protein located on thesurface of osteoclast precursors, triggers a number of signalingcascades that include the IKK/IK�/NF-�B transduction path-way and the mitogen-activated protein kinase (MAPK), Src,and phosphatidylinositol 3-kinase (PI3K)/AKT pathways aswell (64). A novel calcium oscillation pathway that involvesITAM coreceptors is also involved in the downstream effects ofRankL (35). Importantly, stimulation of these pathways culmi-nates in the activation of multiple transcription factors, includ-ing c-fos, NF-�B, and NFATc1, all of which play strategic rolesin the differentiation process at the genetic level (44, 61).Overall, the timely activation of these transcription factorsinitiates growth arrest and promotes osteoclast differentiation,fusion, activation, and survival (63). The evidence that sup-ports the essentiality of both RankL and its receptor in osteo-clast formation is most strongly supported by the skeletal phe-notypes of both RankL- and Rank-null mice, neither of whichare capable of producing osteoclasts in vivo and thus are phe-notypically osteopetrotic (20, 37).
RankL is synthesized and expressed on the surface of regu-latory cells in response to a myriad of both local and systemicfactors, many of which are essential to physiologic bone turn-over. These include the two hormones integral to calciumhomeostasis, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] (60)and parathyroid hormone (PTH) (36, 41). RankL expression
* Corresponding author. Mailing address: Department of Biochem-istry, University of Wisconsin—Madison, 433 Babcock Dr., Madison,WI 53706. Phone: (608) 262-8229. Fax: (608) 263-7609. E-mail: [email protected].
† Supplemental material for this article may be found at http://mcb.asm.org/.
6469
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

can also be influenced by the glucocorticoid (GC) stress hor-mones (18, 54), inflammatory cytokines such as TNF-� andinterleukin-1 (IL-1) (23), the gp130-activating cytokines IL-6and IL-11 (51, 62), certain prostaglandins (66), and transform-ing growth factor � (TGF-�) (28). While these factors canfunction in a physiologic setting, their activities are often man-ifested during disease. Thus, RankL overproduction can in anumber of circumstances lead to the pathological bone resorp-tion associated with age-related and postmenopausal bonedisease, rheumatoid arthritis and osteoarthritis, multiple mye-lomas, diabetic neuropathy, metastatic cancer, general hyper-calcemia of malignancy, and a variety of other syndromes thatimpact the skeleton (19).
The calciotropic hormones 1,25(OH)2D3 and PTH, as wellas certain growth factors, cytokines, and prostaglandins, allregulate the expression of RankL from stromal cells and os-teoblasts (63). Interestingly, the molecular mechanisms re-sponsible for activation by these regulators remain largely un-known. With respect to vitamin D, 1,25(OH)2D3 is known toinduce RankL upregulation primarily through actions initiatedby the vitamin D receptor (VDR) (29). Kitazawa and col-leagues (34) reported a 1,25(OH)2D3 response in the mouseRankL gene promoter and mapped this activity to a 16-bpvitamin D response element (VDRE) that was located 935 bpupstream of the transcriptional start site (TSS). Inducibilitywas modest, however, and subsequent studies by a number oflaboratories have failed to confirm this finding (11, 48). Morerecently, Kabe et al. (24) have explored the ability of a poten-tial VDRE located immediately downstream of the RankL TSSto mediate 1,25(OH)2D3 activity. Although this element issimilar to that of a consensus VDRE, it is not conservedwithin RankL genes of other species and is capable of onlya modest activity in the context of the RankL proximalpromoter. Thus, it remains unclear at this stage whether orhow these two elements contribute to the RankL gene’sregulation by 1,25(OH)2D3.
PTH also regulates RankL expression (40), although themechanism through which this peptide acts has remainedequally elusive. Recent studies by O’Brien and coworkers (12)have established that PTH is capable of both stabilizing RankLmRNA and inducing its expression, the latter via stimulation ofthe protein kinase A (PKA) pathway and activation of theCREB transcription factor. It seems likely that activation ofthis transcription factor may underlie the ability of the prosta-glandin PGE2 to stimulate RankL expression as well (25).Despite these insights, however, the mechanism by whichCREB induces transcriptional activation at the level of theRankL gene promoter remains undefined. The accompanyingarticle by Fu, Manolagas, and O’Brien delineates the molecu-lar mechanism whereby PTH stimulates RankL gene expres-sion (13).
Finally, the actions of GCs on the skeleton are highly com-plex. Enhanced exposure to these hormones, however, gener-ally results in osteoporosis (42). At the level of the osteoblast,GCs reduce both the functional capacity of these cells to formbone and the period of time during which this functionalityoccurs (43, 67). GCs also influence the production of osteo-clasts (18, 54). These actions occur through a direct activity onthe osteoblast primarily to suppress the expression of osteo-protegerin, a decoy receptor which blocks RankL activity, but
also to increase RankL gene expression. Although a role forthe glucocorticoid receptor (GR) in RankL induction is likely,the mechanism whereby the stress hormone induces RankLexpression remains unexplored.
As outlined above, the mechanism of action of 1,25(OH)2D3
involves a ligand-initiated interaction between the VDR andthe regulatory regions of target genes wherein VDR functionstogether with its retinoid X receptor (RXR) partner to recruitcoregulatory complexes that are essential for modulation oftranscriptional output (46, 58). Binding sites for the VDR havebeen found in a number of genes and are generally, althoughnot always, comprised of two hexanucleotide half-sites sepa-rated by a short spacer and frequently located within the firstkilobase or two upstream of the transcriptional start site (50).The absence of bona fide target sites for VDR action withinthe first 8 kb of the RankL gene, as reported by us and others(11, 48), prompted a more expansive approach to delineateRankL regulatory regions. We therefore used contemporarychromatin immunoprecipitation (ChIP)/chip analysis (as de-scribed below) in this endeavor. We discovered five sites ofaction located at increasing distances from the mouse RankLgene TSS, the furthest approximately 76 kb upstream. Thelatter region, which we termed the RankL distal control region(RL-DCR), was transcriptionally active in transfection studiesand contained an unusual VDRE sequence. Our studies, andthe results described in the accompanying article by O’Brienand colleagues (13), provide essential clues as to how1,25(OH)2D3 and PTH regulate RankL induction.
MATERIALS AND METHODS
Reagents. General biochemicals were obtained from Fisher Scientific (Pitts-burgh, PA) and Sigma Chemical Co. (St. Louis, MO). 1,25(OH)2D3 was obtainedfrom Solvay (da Weesp, The Netherlands) and Tetrionics (Madison, WI).ZK159222 was kindly provided by Andreas Steinmeyer and Ekkehard May ofSchering AG (Berlin, Germany). Alpha minimum essential medium-Earle’s me-dium (�-MEM) was purchased from Mediatech (Herndon, VA), and minimumessential medium alpha (MEM-�) was obtained from Invitrogen Corporation(Carlsbad, CA). Oligonucleotide primers were obtained from IDT (Coralville,IA). Anti-VDR (Sc-1008), -RXR (Sc-774), -C/EBP� (Sc-150), and -GR (Sc-1004) antibodies were obtained from Santa Cruz Biotechnology, Inc. (SantaCruz, CA). Anti-acetyl H4 antibody (06-866) was obtained from Upstate (Char-lottesville, VA), and anti-RNA polymerase II antibody (8WG16) was obtainedfrom Berkeley Antibody Company (Richmond, CA). Lipofectamine Plus wasobtained from Invitrogen Corporation (Carlsbad, CA). [�-32P]dATP was ob-tained from NEN Life Science Products, Inc. (Boston, MA). Dexamethasone(D4902), anti-rat immunoglobulin G (IgG; R5128), and RU486 (M8046) werepurchased from Sigma Chemical Co. (St. Louis, MO).
Cell culture. Mouse MC3T3-E1 and ST2 osteoblastic cells were cultured in�-MEM and MEM-�, respectively. Primary calvarial osteoblasts (mOBs) wereobtained as previously described (53) and cultured in �-MEM. Human osteo-sarcoma MG63 cells were grown in Dulbecco’s modified Eagle’s medium(DMEM) supplemented with 1% nonessential amino acids. COS-7 fibroblastswere also cultured in DMEM. Each medium was supplemented with 10% fetalbovine serum (FBS) obtained from HyClone (Logan, UT), 100 U/ml penicillin,and 100 �g/ml streptomycin. All ligands were added in ethanol (0.1% maximumfinal concentration) or dimethyl sulfoxide.
RNA isolation and analysis. Total RNA was isolated from cells using Triazolreagent obtained from MRC (Cincinnati, OH). The isolated RNA was reversetranscribed using the SuperScript III RNase H reverse transcriptase kit fromInvitrogen (Carlsbad, CA) and then subjected to PCR analysis using standardPCR methods. Primers used include those for the mouse �-actin gene (m�-actin)(forward, TGTTTGAGACCTTCAACACCC; reverse, CGTTGCCAATAGTGATGACCT), mCyp24a1 (forward, GTGCGGATTTCCTTTGTGATA; reverse,GGTAGCGTGTATTCACCCAGA), mVDR (forward, TCACTGATGTCTCCAGAGCTGGGC; reverse, TGGATAGGCGGTCCTGAATGGC), and mOpn(forward, CTAACTACGACCATGAGATTGGCAG; reverse, CTTTAGTTGA
6470 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

CCTCAGAAGATGAA) and mRankL (forward, GAATCCTGAGACTCCATGAAAACGC; reverse, CCATGAGCCTTCCATCATAGCTGG). Primers forthe human genes used included those for the human �-actin gene (h�-actin)(forward, TTAGTTGCGTTACACCCTTTC; reverse, GTCACCTTCACCGTTCCAGTT), hCYP24A1 (forward, CTTTGCTTCCTTTTCCCAGAAT; reverse,CGCCGTAGATGTCACCAGTC), and hRANKL (forward, AACAGGCCTTTCAAGGAGCTGTGC; reverse, AAGAGGACAGACTCACTTTATGGG).
siRNA studies. All small interfering RNA (siRNA) duplexes were obtainedfrom Dharmacon RNA Technologies (Lafayette, CO). ST2 cells were seededinto six-well plates at a concentration of 1.5 � 105 cells/well and transfectedapproximately 24 h later using Lipofectamine Plus in serum and antibiotic-freemedium. A 20 nM concentration of mVDR siRNA (D-058923-01), nontargetingsiRNA pool (D-001206-13), or cyclophilin B siRNA (D-001136-01) was used fortransfection. After transfection, the cells were cultured in medium supplementedwith 10% FBS for 48 h before they were treated with a routine concentration of10�7 M 1,25(OH)2D3 for 6 h. RNA isolation and standard PCR analysis werecarried out using the primers listed above. Western blot analysis confirmeddepletion of VDR protein expression in ST2 cells (data not shown).
ChIP assay. Chromatin immunoprecipitation assays were performed as pre-viously described (30). Primer sets used for amplifying mouse and humanCyp24a1, osteopontin (Opn), and RankL gene regions of interest are all listed inTable 1. Densitometric analysis was carried out using Kodak ID Image Analysis(software version 3.5).
Tiled oligonucleotide microarray analysis. ChIP/chip analysis was carried outas described by others (31–33, 47). In brief, DNA was isolated by specificimmunoprecipitation using the ChIP methodology described above and thensubjected to ligation-mediated PCR as described by Oberley et al. (47). ChIP-purified DNA was blunt-ended using T4 polymerase, ligated to linkers with thesequences 5 GCGGTGACCCGGGAGATCTGAATTC 3 and 5 GAATTCA
GATC 3, and subjected to repeated, low-cycle PCR amplification. The resulting500-bp amplicons were then labeled with Cy3 or Cy5 dyes using an indirectlabeling protocol. In this method, biotinylated dUTP is first incorporated into theindividual amplicons by standard procedures and the conjugated DNA is labeledsubsequently with either Cy5- or Cy3-conjugated streptavidin. Cy3- and Cy5-labeled DNA samples are then mixed in the presence of CoT-1 DNA, denatured,and cohybridized to custom oligonucleotide microarrays (Nimblegen SystemsInc., Madison, WI) as described. The microarrays are washed extensively andscanned using an Axon 4000B scanner at the appropriate wavelengths.
Custom oligonucleotide arrays were synthesized by Nimblegen Systems, Inc(Madison, WI). The microarray probes consisted of maskless-array, in situ-synthesized 50-mer oligonucleotides at 2-bp intervals representing a screen ofover 300 kb of the mouse RANKL gene locus from 200 kb upstream of the gene’sTSS to 100 kb downstream of the final 3 noncoding exon. The tiled arrays weresynthesized in duplicate in both the forward as well as reverse directions, pro-viding four independent measurements at each site within the gene. In addition,each analysis was carried out using two independently derived ChIP DNA sam-ples. Both the Cyp24a1 and Opn genes (as well as other candidate VDR targetgenes that are not discussed) were tiled in a similar fashion. A series of com-parisons were made between (i) IgG in the presence or absence of hormone, (ii)VDR in the presence or absence of hormone, (iii) VDR in the presence ofhormone versus input DNA, (iv) RXR in the presence or absence of hormone,and (v) RXR in the presence of hormone versus input DNA. After samplecohybridization, the logarithmic enrichment ratios of Cy5 to Cy3 hybridizationintensities (log2) were plotted as a function of chromosome nucleotide position.While all of the peaks representative of enhanced VDR or RXR binding eitherin the presence of 1,25(OH)2D3 or as compared to input DNA are presented asthe raw data, a peak-finding algorithm was utilized to score the relative levels ofbinding between the three regions identified (14).
Plasmids. Full-length hVDR and hRXR� were cloned into the pET-29bvector obtained from Novagen (Darmstadt, Germany) and expressed with C-terminal His6 tags. The pCH110-�-galactosidase (pCH110-�gal) reporter plas-mid and the pcDNA-hVDR vector or a mutant (hVDR L417A/E420A) version[pcDNA-hVDR(m)] and pRSV-GR� were previously described (70). The parentthymidine kinase (TK) and luciferase (luc) vectors pTK-luc and pGL3-luc wereutilized in subsequent cloning efforts. mRLD1 (�16.4 to �15.2), mRLD2 (�23.1to �21.5), mRLD3 (�60.4 to �59.3), mRLD4 (�69.0 to �68.1), and mRLD5(�76045 to �74973) were amplified using primers that contained HindIII,HindIII/SalI, or HindIII/BamHI restriction ends and then cloned into the cor-responding sites within the pTK-luc vector. pTK-mRLD5 fragments pTK-mRLD5-5/1 (�75724 to �74973), pTK-mRLD5-5/2 (�75475 to �74973), pTK-mRLD5-5/3 (�75228 to �74973), pTK-mRLD5-3/1 (�75724 to �75228), andpTK-mRLD5-3/2 (�75724 to �75475) were subcloned similarly into the pTK-lucvector using HindIII/SalI restriction sites. The hRLD5 region of the humanRANKL gene (�96903 to �95805) was amplified from genomic DNA andcloned into the HindIII/BamHI sites of the TK-luc vector to produce pTK-hRLD5. mRL-VDRE (�75620 to �75590) and the hRL-VDRE (�96467 to�96431) as well as mRL-VDRE1 and mRL-VDRE2, each containing severaloverhanging 5 and 3 nucleotides, were synthesized, annealed, and similarlycloned into the HindIII/BamHI sites of pTK-luc. Triplet mutations in the mRL-VDRE half-sites and in the hRL-VDRE half-sites in the context of pTK-mRLD5and pTK-hRLD5 were created using the Quikchange mutagenesis kit fromStratagene (San Diego, CA). mRL-VDRE segments containing mutations withineach half-site were also synthesized and cloned into the HindIII/BamHI sites ofpTK-luc. The pmRL(100) vector was prepared by introducing an amplifiedsegment of the mouse RankL gene promoter (�101 to �54 relative to the RankLTSS) into the pGL3-luc expression vector at the XhoI/HindIII sites. Each of themRLD regions, mRLD1 (�16.4 to �15.2), mRLD2 (�23.1 to �21.5), mRLD3(�60.4 to �59.3), mRLD4 (�69.0 to �68.1), and mRLD5 (�76045 to �74973),as well as mRL-VDRE (�75620 to �75590), was then amplified and clonedupstream of pmRL(100) using the MluI/XhoI restriction sites. All plasmid con-structs were sequenced to verify successful cloning.
Transfection assays. MC3T3-E1 and/or ST2 cells were seeded into 24-wellplates at appropriate densities and cultured in �-MEM or MEM-� containing10% FBS. Cells were transfected 24 h later with Lipofectamine Plus in serum andantibiotic-free medium. Individual wells were transfected with 250 ng of a lucif-erase reporter vector, 50 ng of pCH110-�gal, and 50 ng of pcDNA-hVDR (whichwas routinely transfected with all luciferase reporters unless otherwise indicated)or 50 ng phRSV-GR� (added only when specifically indicated). Nontargeting,cyclophilin B or VDR siRNA pools (50 nM) were also transfected where indi-cated. After transfection, the cells were cultured first for 48 h in a mediumsupplemented with 20% FBS and subsequently for an additional 24 h with orwithout 1,25(OH)2D3. Cells were then harvested, and the lysates were assayed
TABLE 1. Primers used for ChIP analysis
Primer andspecies Sequence
MouseCyp24a1 .......................5 GGTTATCTCCGGGGTGGAGT
3 AGTGGCCAATGAGCACGCOpn ..............................5 ACCACCTCTTCTGCTCTATATGGC
3 TTGACACTTGAACTATGCAGCCGCIS7 ................................5 CTGAAGCCAAGAGGCAGATT
3 CGCACATCCTTTCAGGTGCTIS6 ................................5 GGTACCACATGTGCACATTA
3 CAGGTGTTGTTTTAAGCTACIS5 ................................5 GCTCAGAAAGCAGGACCTCC
3 CACTTTCTCTTAGAACAGTGIS4 ................................5 CCTCCTATCTGTTTTACTGACGTT
3 CACATAGGCAGAAAGTTGAAAAGCIS2 ................................5 GCTATCATTTATACCTTGGA
3 CCTGAATTTCTGATCTTCCTIS1 ................................5 CATGAGTATATGTGTGGAGT
3 CCTTCATAATGTTTGAGACCD5.................................5 GGTCAAGAGGGGCCTGACTT
3 GCAGTGTGTAAACAAAGAGAD4.................................5 GTGCTGTGAAGCAAAAATTC
3 GCACTACAAATGTGAGGGAAD3.................................5 GAGCTGTGTCCTAGAAGAAT
3 CCCTGCACATAGTAAACAAAD2.................................5 TAAGGCTCTGACCATAGGAA
3 CTGGATTTAAGTGAAACAGTD1.................................5 GAGAAAGCCAAGTCCTGGGT
3 CATTCTGGGGCCGGAACAAAP1 (TSS) ......................5 CAGAAACCAACCACTGGACCCAA
3 CAGGAACATGGAGCGGGAGG
HumanCYP24A1 ....................5 CGAAGCACACCCGGTGAACT
3 CCAATGAGCACGCAGAGGAGD5-1 .............................5 CAAGTTTCTTTGCTGTCATC
3 CAGCCACATAAAGCAGTGAGD5-2 .............................5 GCTCAAGCCTTATGTTTCGG
3 TAAGCTAGTGTGACCCCTGA
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6471
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

for luciferase and �-galactosidase activities as previously described (70). Lucif-erase activity was normalized to �-galactosidase activity in all cases.
Protein purification. Human VDR and RXR� proteins were produced usingthe bacterial expression vectors pET-hVDR and pET-hRXR� in BL21(DE3)codon Plus RIL cells obtained from Stratagene (San Diego, CA). Soluble full-length hVDR and hRXR� proteins were purified to homogeneity using sequen-tial Ni-nitrilotriacetic acid (NTA) and SP-Sepharose column chromatography(70). Two forms of RXR� were present due to redundant start sites.
DNA band shift analysis. The duplex oligonucleotide probes comprised ofthe mouse osteopontin VDRE, mRL-VDRE, mRL-VDRE1m (M1), mRL-mVDRE2m (M2), and hRL-VDRE, as documented in Table 2, were end labeledusing [�-32P]dATP. Probes were incubated at room temperature with the indi-cated concentrations of hVDR and hRXR� in 10 mM HEPES, pH 7.4, 1 mMEDTA, 5 mM MgCl2, 10% glycerol, 0.5 mM dithiothreitol, 0.7 mM phenylmethyl-sulfonyl fluoride, and 50 or 150 mM KCl in the absence or presence of1,25(OH)2D3 for 30 min. Complexes were resolved on nondenaturing 6% poly-acrylamide gels, dried, and then visualized using autoradiography. Densitometricanalyses of complex 1 and complex 2 were carried out using Kodak ID ImageAnalysis (software version 3.5).
Statistical analyses. All values are expressed as the mean � standard error ofthe mean. All statistical calculations were performed with the GraphPad PRISMversion 4 statistical software package (GraphPad Software Inc., San Diego, CA).We evaluated differences between groups through one-way analysis of varianceor Student’s two-tailed t test. Significance was determined at P � 0.05.
RESULTS
1,25(OH)2D3 induces RankL gene expression in osteoblast-like ST2 cells. Earlier studies indicated that 1,25(OH)2D3 in-duces RankL expression in a variety of osteoblast-like cells,including the mouse ST2 cell line (34). To explore this processfurther and to assess the potential effects of GCs on this in-duction, we treated ST2 cells with a maximal dose of either1,25(OH)2D3, dexamethasone (DEX), or the combination andevaluated the level of RankL transcripts produced. As can beseen in Fig. 1A (see Fig. S1 in the supplemental material),1,25(OH)2D3 was capable of inducing RankL mRNA levelswith a time course consistent with that observed for several1,25(OH)2D3 target genes, including both Opn and Cyp24a1.Interestingly, while DEX was generally inactive on its own, itappeared to potentiate the activity of 1,25(OH)2D3 when usedin combination, suggesting a potentially additive or synergisticeffect on RankL expression. DEX of course had no effect oneither Cyp24a1 or Opn mRNA levels. To establish that theinduction of RankL by 1,25(OH)2D3 was mediated via theVDR, we employed an interference assay using VDR siRNAand assessed the effect of this action on RankL expression. ST2
cells were mock transfected or transfected with either nontar-geted or cyclophilin B control siRNA or a VDR siRNA pooland treated at 48 h for an additional 6 h with either vehicle or1,25(OH)2D3. The isolated RNA was then analyzed for targetgene expression. As noted in Fig. 1B, exposure to VDR siRNAeffectively reduced basal expression of VDR mRNA; it alsoprevented the well-established upregulation of VDR mRNAseen in response to 1,25(OH)2D3 (as shown in reference 71and confirmed here) when control siRNAs were administered.Most importantly, knockdown of VDR mRNA with VDRsiRNA but not control siRNAs effectively blocked the abilityof 1,25(OH)2D3 to stimulate RankL expression. Opn andCyp24a1 induction was suppressed as well. These experimentsdemonstrate that 1,25(OH)2D3 is able to induce RankL ex-pression in the ST2 cells in a fashion mediated by the VDR andfacilitated in some way by GCs.
ChIP/chip analysis of the RankL locus identifies potentialVDR binding sites. Despite earlier studies (24, 34), regulatorysites within the RankL gene that mediate the actions of1,25(OH)2D3 remain unclear (11, 48). As a result, we electedto utilize a chromatin immunoprecipitation method combinedwith DNA microarray analysis (ChIP/chip analysis) to scan the
FIG. 1. Induction of mRankL mRNA by 1,25(OH)2D3 in ST2 cellsis enhanced by DEX and mediated by the VDR. (A) Induction ofRankL expression levels by 1,25(OH)2D3 and DEX in vitro. ST2 cellswere treated for periods of up to 24 h with either 1,25(OH)2D3 (10�7
M), DEX (10�7 M), or both (at 10�7 M). Total RNA was isolated andsubjected to reverse transcription-PCR (RT-PCR) analysis using prim-ers specific to mouse Cyp24a1 (30 cycles), osteopontin (Opn) (15cycles), RankL (30 cycles), or �-actin (20 cycles) as documented inMaterials and Methods. The results are typical of multiple similarexperiments. (B) Effects of mVDR siRNA on 1,25(OH)2D3-inducedRankL expression levels in ST2 cells. ST2 cells were transfected with 20nM nontargeting siRNA, cyclophilin B (Cyclo B) siRNA, or mVDRsiRNA. After 48 h, the cells were treated for an additional 6 h witheither vehicle or 1,25(OH)2D3 (10�7 M). Total RNA was isolated andsubjected to standard RT-PCR analysis using the primers identified inpanel A above. The numbers of cycles for amplification of each tran-script are as follows: VDR, 20 cycles; Cyp24a1, 25 cycles; Opn, 15cycles, and �-actin, 15 cycles. These results are typical of several in-dependent experiments.
TABLE 2. DNA sequences for EMSA
DNA typeand species Sequencea
MouseOpn VDRE ...............5 AACAAGGTTCACGAGGTTCACGTCTRankL VDRE ...........5 GGGTGAACTCAGGCAACCAACAAC
TCGGTGACTCTGGRankL VDRE1m......5 GGGTGAACTCAGGCAACCAACAgga
CGGTGAggaTGGRankL VDRE2m......5 GGGTGAggaCAGGCAggaAACAACTC
GGTGACTCTGG
HumanRankL VDRE ...........5 GGGTGAACTCAGACAACCGACAAC
TTGGTGACTTCGA
a VDRE sequences are underlined. Mutated sequences are indicated in lower-case letters.
6472 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
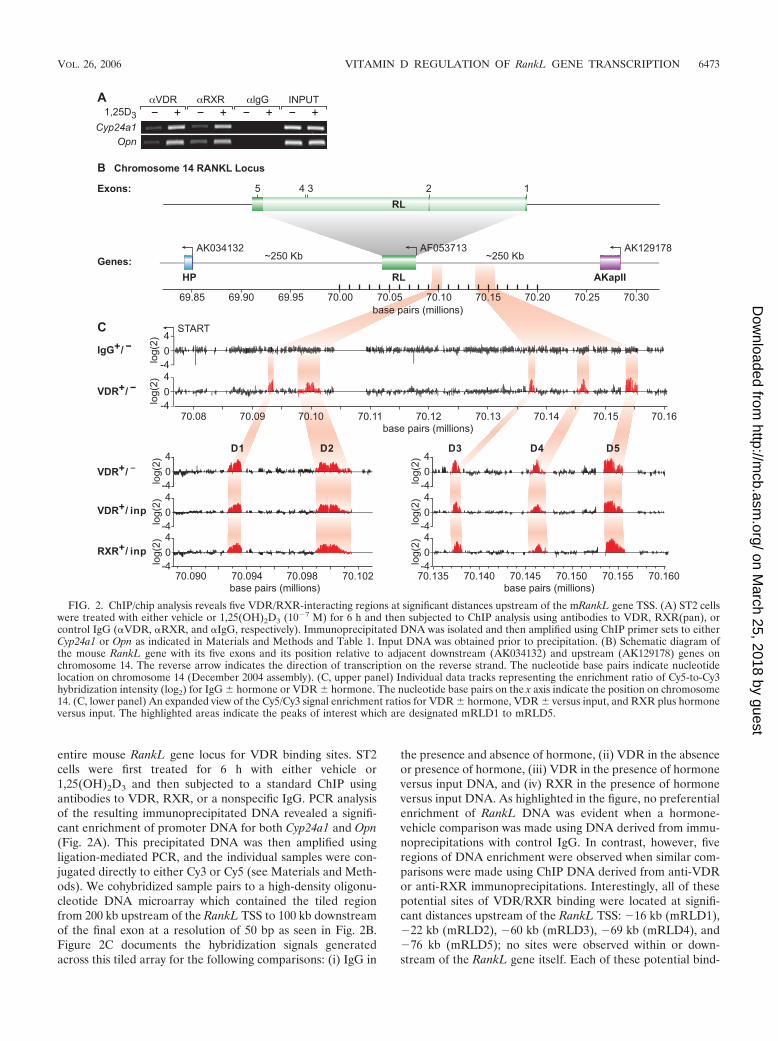
entire mouse RankL gene locus for VDR binding sites. ST2cells were first treated for 6 h with either vehicle or1,25(OH)2D3 and then subjected to a standard ChIP usingantibodies to VDR, RXR, or a nonspecific IgG. PCR analysisof the resulting immunoprecipitated DNA revealed a signifi-cant enrichment of promoter DNA for both Cyp24a1 and Opn(Fig. 2A). This precipitated DNA was then amplified usingligation-mediated PCR, and the individual samples were con-jugated directly to either Cy3 or Cy5 (see Materials and Meth-ods). We cohybridized sample pairs to a high-density oligonu-cleotide DNA microarray which contained the tiled regionfrom 200 kb upstream of the RankL TSS to 100 kb downstreamof the final exon at a resolution of 50 bp as seen in Fig. 2B.Figure 2C documents the hybridization signals generatedacross this tiled array for the following comparisons: (i) IgG in
the presence and absence of hormone, (ii) VDR in the absenceor presence of hormone, (iii) VDR in the presence of hormoneversus input DNA, and (iv) RXR in the presence of hormoneversus input DNA. As highlighted in the figure, no preferentialenrichment of RankL DNA was evident when a hormone-vehicle comparison was made using DNA derived from immu-noprecipitations with control IgG. In contrast, however, fiveregions of DNA enrichment were observed when similar com-parisons were made using ChIP DNA derived from anti-VDRor anti-RXR immunoprecipitations. Interestingly, all of thesepotential sites of VDR/RXR binding were located at signifi-cant distances upstream of the RankL TSS: �16 kb (mRLD1),�22 kb (mRLD2), �60 kb (mRLD3), �69 kb (mRLD4), and�76 kb (mRLD5); no sites were observed within or down-stream of the RankL gene itself. Each of these potential bind-
FIG. 2. ChIP/chip analysis reveals five VDR/RXR-interacting regions at significant distances upstream of the mRankL gene TSS. (A) ST2 cellswere treated with either vehicle or 1,25(OH)2D3 (10�7 M) for 6 h and then subjected to ChIP analysis using antibodies to VDR, RXR(pan), orcontrol IgG (�VDR, �RXR, and �IgG, respectively). Immunoprecipitated DNA was isolated and then amplified using ChIP primer sets to eitherCyp24a1 or Opn as indicated in Materials and Methods and Table 1. Input DNA was obtained prior to precipitation. (B) Schematic diagram ofthe mouse RankL gene with its five exons and its position relative to adjacent downstream (AK034132) and upstream (AK129178) genes onchromosome 14. The reverse arrow indicates the direction of transcription on the reverse strand. The nucleotide base pairs indicate nucleotidelocation on chromosome 14 (December 2004 assembly). (C, upper panel) Individual data tracks representing the enrichment ratio of Cy5-to-Cy3hybridization intensity (log2) for IgG � hormone or VDR � hormone. The nucleotide base pairs on the x axis indicate the position on chromosome14. (C, lower panel) An expanded view of the Cy5/Cy3 signal enrichment ratios for VDR � hormone, VDR � versus input, and RXR plus hormoneversus input. The highlighted areas indicate the peaks of interest which are designated mRLD1 to mRLD5.
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6473
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
ing sites is intergenic in view of the location of AkapII, theannotated gene lying approximately 250 kb upstream. While allof these peaks were obvious to visual inspection, a peak-findinganalysis of these data (47) indicated that binding of the VDRand RXR to the mRLD5 region was two- to fivefold morerobust than that observed for mRLD1 to mRLD4; mRLD4generated the weakest signal. These data suggest the locationsof at least five potential VDR/RXR binding sites situated longdistances upstream of the RankL gene.
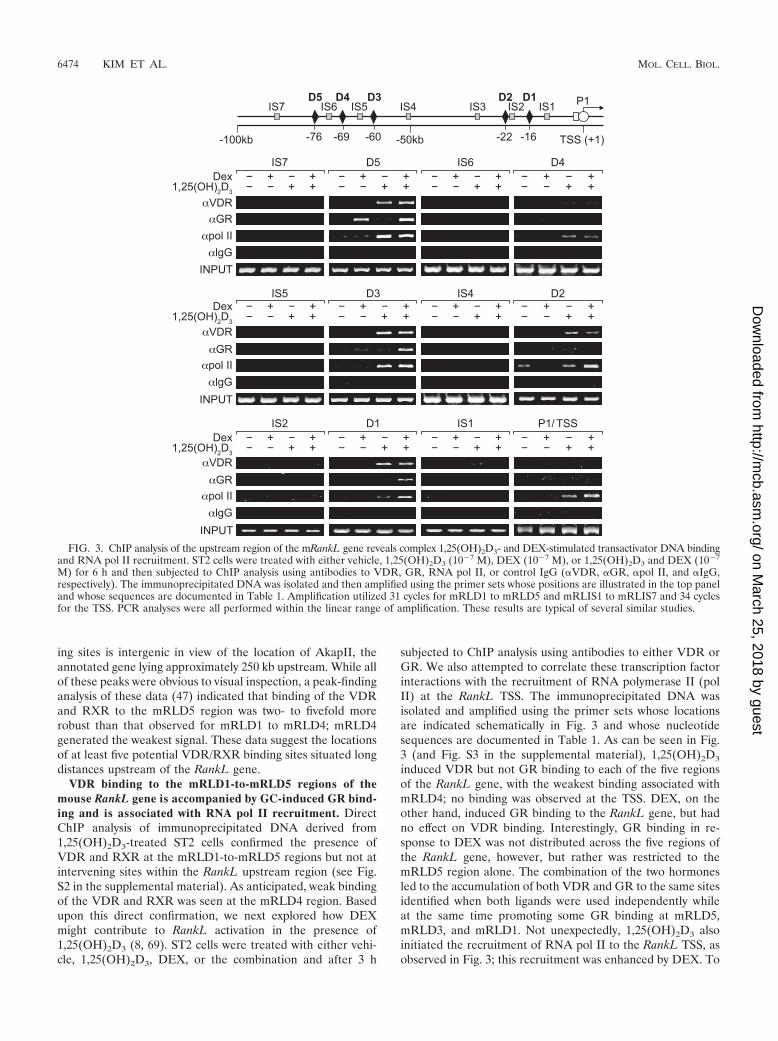
VDR binding to the mRLD1-to-mRLD5 regions of themouse RankL gene is accompanied by GC-induced GR bind-ing and is associated with RNA pol II recruitment. DirectChIP analysis of immunoprecipitated DNA derived from1,25(OH)2D3-treated ST2 cells confirmed the presence ofVDR and RXR at the mRLD1-to-mRLD5 regions but not atintervening sites within the RankL upstream region (see Fig.S2 in the supplemental material). As anticipated, weak bindingof the VDR and RXR was seen at the mRLD4 region. Basedupon this direct confirmation, we next explored how DEXmight contribute to RankL activation in the presence of1,25(OH)2D3 (8, 69). ST2 cells were treated with either vehi-cle, 1,25(OH)2D3, DEX, or the combination and after 3 h
subjected to ChIP analysis using antibodies to either VDR orGR. We also attempted to correlate these transcription factorinteractions with the recruitment of RNA polymerase II (polII) at the RankL TSS. The immunoprecipitated DNA wasisolated and amplified using the primer sets whose locationsare indicated schematically in Fig. 3 and whose nucleotidesequences are documented in Table 1. As can be seen in Fig.3 (and Fig. S3 in the supplemental material), 1,25(OH)2D3
induced VDR but not GR binding to each of the five regionsof the RankL gene, with the weakest binding associated withmRLD4; no binding was observed at the TSS. DEX, on theother hand, induced GR binding to the RankL gene, but hadno effect on VDR binding. Interestingly, GR binding in re-sponse to DEX was not distributed across the five regions ofthe RankL gene, however, but rather was restricted to themRLD5 region alone. The combination of the two hormonesled to the accumulation of both VDR and GR to the same sitesidentified when both ligands were used independently whileat the same time promoting some GR binding at mRLD5,mRLD3, and mRLD1. Not unexpectedly, 1,25(OH)2D3 alsoinitiated the recruitment of RNA pol II to the RankL TSS, asobserved in Fig. 3; this recruitment was enhanced by DEX. To
FIG. 3. ChIP analysis of the upstream region of the mRankL gene reveals complex 1,25(OH)2D3- and DEX-stimulated transactivator DNA bindingand RNA pol II recruitment. ST2 cells were treated with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M), or 1,25(OH)2D3 and DEX (10�7
M) for 6 h and then subjected to ChIP analysis using antibodies to VDR, GR, RNA pol II, or control IgG (�VDR, �GR, �pol II, and �IgG,respectively). The immunoprecipitated DNA was isolated and then amplified using the primer sets whose positions are illustrated in the top paneland whose sequences are documented in Table 1. Amplification utilized 31 cycles for mRLD1 to mRLD5 and mRLIS1 to mRLIS7 and 34 cyclesfor the TSS. PCR analyses were all performed within the linear range of amplification. These results are typical of several similar studies.
6474 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
our surprise, however, 1,25(OH)2D3 and the combination ofboth 1,25(OH)2D3 and DEX also promoted the recruitment ofRNA pol II to the upstream mRLD1-to-mRLD5 regions of theRankL gene. This unanticipated finding suggests that theRankL enhancer regions we have identified may function asrecruitment centers for RNA pol II as well (59). Despite thisspeculation, the results of this experiment provide additionalevidence that mRLD1 to mRLD5 may represent importantenhancer modules essential to the regulation of RankL geneexpression. It is unclear, however, how VDR and GR mightcollaborate to sensitize the RankL gene to stimulation by bothhormones.
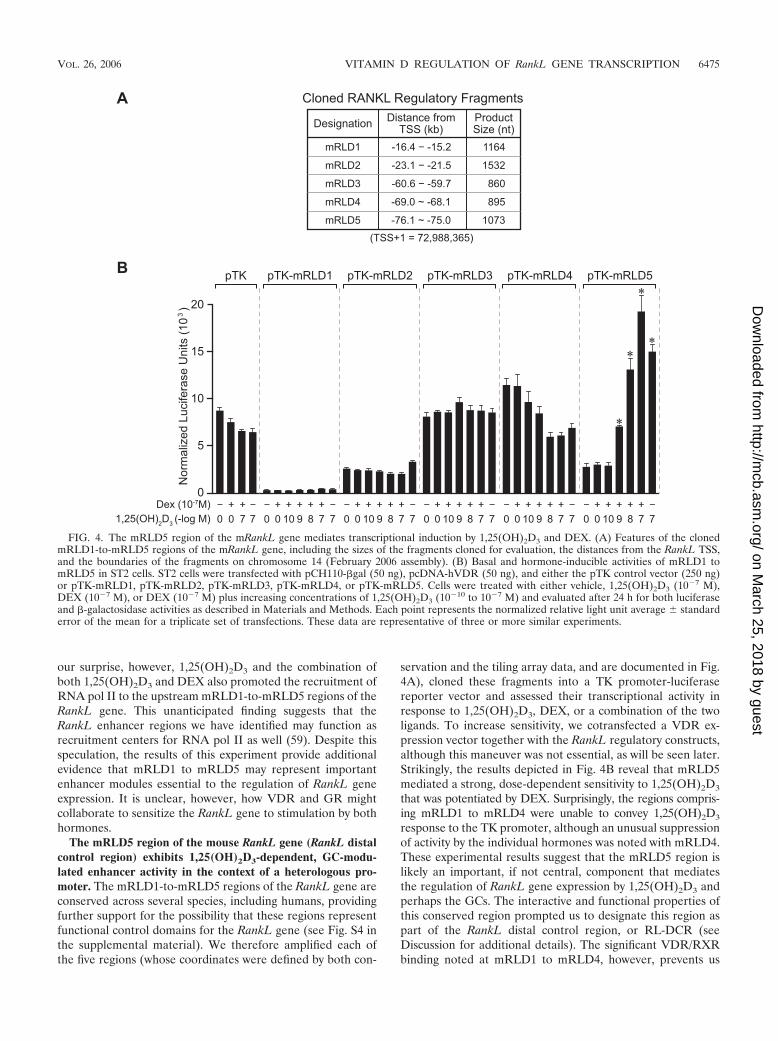
The mRLD5 region of the mouse RankL gene (RankL distalcontrol region) exhibits 1,25(OH)2D3-dependent, GC-modu-lated enhancer activity in the context of a heterologous pro-moter. The mRLD1-to-mRLD5 regions of the RankL gene areconserved across several species, including humans, providingfurther support for the possibility that these regions representfunctional control domains for the RankL gene (see Fig. S4 inthe supplemental material). We therefore amplified each ofthe five regions (whose coordinates were defined by both con-
servation and the tiling array data, and are documented in Fig.4A), cloned these fragments into a TK promoter-luciferasereporter vector and assessed their transcriptional activity inresponse to 1,25(OH)2D3, DEX, or a combination of the twoligands. To increase sensitivity, we cotransfected a VDR ex-pression vector together with the RankL regulatory constructs,although this maneuver was not essential, as will be seen later.Strikingly, the results depicted in Fig. 4B reveal that mRLD5mediated a strong, dose-dependent sensitivity to 1,25(OH)2D3
that was potentiated by DEX. Surprisingly, the regions compris-ing mRLD1 to mRLD4 were unable to convey 1,25(OH)2D3
response to the TK promoter, although an unusual suppressionof activity by the individual hormones was noted with mRLD4.These experimental results suggest that the mRLD5 region islikely an important, if not central, component that mediatesthe regulation of RankL gene expression by 1,25(OH)2D3 andperhaps the GCs. The interactive and functional properties ofthis conserved region prompted us to designate this region aspart of the RankL distal control region, or RL-DCR (seeDiscussion for additional details). The significant VDR/RXRbinding noted at mRLD1 to mRLD4, however, prevents us
FIG. 4. The mRLD5 region of the mRankL gene mediates transcriptional induction by 1,25(OH)2D3 and DEX. (A) Features of the clonedmRLD1-to-mRLD5 regions of the mRankL gene, including the sizes of the fragments cloned for evaluation, the distances from the RankL TSS,and the boundaries of the fragments on chromosome 14 (February 2006 assembly). (B) Basal and hormone-inducible activities of mRLD1 tomRLD5 in ST2 cells. ST2 cells were transfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK control vector (250 ng)or pTK-mRLD1, pTK-mRLD2, pTK-mRLD3, pTK-mRLD4, or pTK-mRLD5. Cells were treated with either vehicle, 1,25(OH)2D3 (10�7 M),DEX (10�7 M), or DEX (10�7 M) plus increasing concentrations of 1,25(OH)2D3 (10�10 to 10�7 M) and evaluated after 24 h for both luciferaseand �-galactosidase activities as described in Materials and Methods. Each point represents the normalized relative light unit average � standarderror of the mean for a triplicate set of transfections. These data are representative of three or more similar experiments.
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6475
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
from excluding these regions as potential regulators of RankLgene expression.
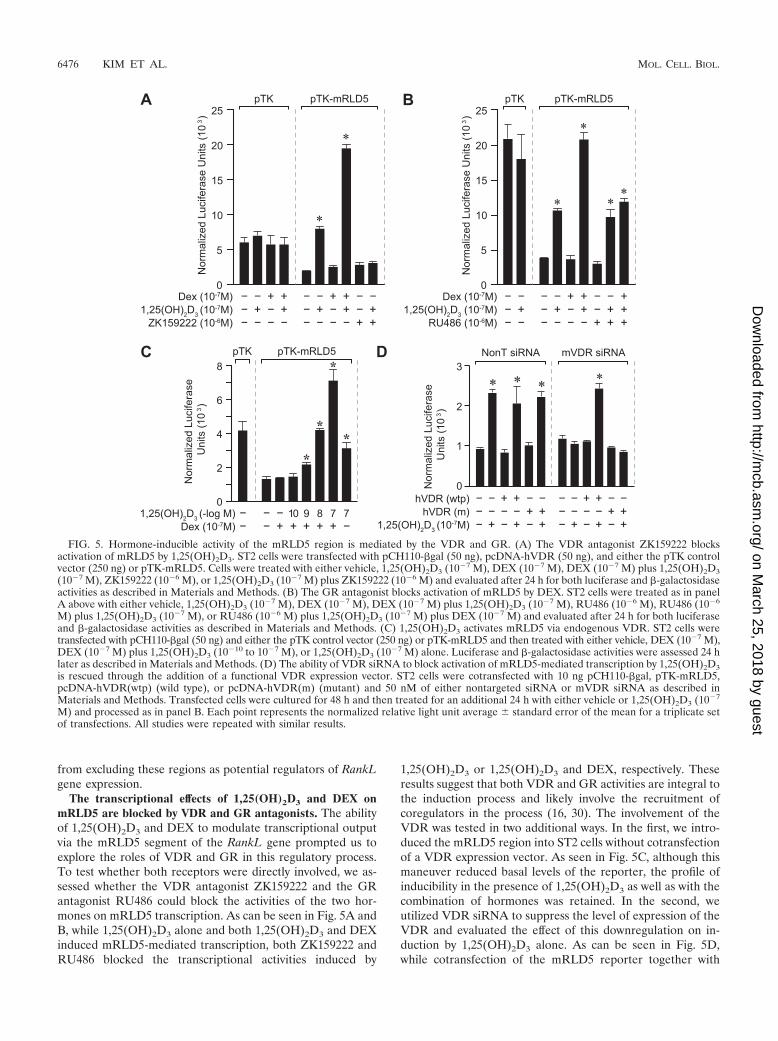
The transcriptional effects of 1,25(OH)2D3 and DEX onmRLD5 are blocked by VDR and GR antagonists. The abilityof 1,25(OH)2D3 and DEX to modulate transcriptional outputvia the mRLD5 segment of the RankL gene prompted us toexplore the roles of VDR and GR in this regulatory process.To test whether both receptors were directly involved, we as-sessed whether the VDR antagonist ZK159222 and the GRantagonist RU486 could block the activities of the two hor-mones on mRLD5 transcription. As can be seen in Fig. 5A andB, while 1,25(OH)2D3 alone and both 1,25(OH)2D3 and DEXinduced mRLD5-mediated transcription, both ZK159222 andRU486 blocked the transcriptional activities induced by
1,25(OH)2D3 or 1,25(OH)2D3 and DEX, respectively. Theseresults suggest that both VDR and GR activities are integral tothe induction process and likely involve the recruitment ofcoregulators in the process (16, 30). The involvement of theVDR was tested in two additional ways. In the first, we intro-duced the mRLD5 region into ST2 cells without cotransfectionof a VDR expression vector. As seen in Fig. 5C, although thismaneuver reduced basal levels of the reporter, the profile ofinducibility in the presence of 1,25(OH)2D3 as well as with thecombination of hormones was retained. In the second, weutilized VDR siRNA to suppress the level of expression of theVDR and evaluated the effect of this downregulation on in-duction by 1,25(OH)2D3 alone. As can be seen in Fig. 5D,while cotransfection of the mRLD5 reporter together with
FIG. 5. Hormone-inducible activity of the mRLD5 region is mediated by the VDR and GR. (A) The VDR antagonist ZK159222 blocksactivation of mRLD5 by 1,25(OH)2D3. ST2 cells were transfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK controlvector (250 ng) or pTK-mRLD5. Cells were treated with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M), DEX (10�7 M) plus 1,25(OH)2D3(10�7 M), ZK159222 (10�6 M), or 1,25(OH)2D3 (10�7 M) plus ZK159222 (10�6 M) and evaluated after 24 h for both luciferase and �-galactosidaseactivities as described in Materials and Methods. (B) The GR antagonist blocks activation of mRLD5 by DEX. ST2 cells were treated as in panelA above with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M), DEX (10�7 M) plus 1,25(OH)2D3 (10�7 M), RU486 (10�6 M), RU486 (10�6
M) plus 1,25(OH)2D3 (10�7 M), or RU486 (10�6 M) plus 1,25(OH)2D3 (10�7 M) plus DEX (10�7 M) and evaluated after 24 h for both luciferaseand �-galactosidase activities as described in Materials and Methods. (C) 1,25(OH)2D3 activates mRLD5 via endogenous VDR. ST2 cells weretransfected with pCH110-�gal (50 ng) and either the pTK control vector (250 ng) or pTK-mRLD5 and then treated with either vehicle, DEX (10�7 M),DEX (10�7 M) plus 1,25(OH)2D3 (10�10 to 10�7 M), or 1,25(OH)2D3 (10�7 M) alone. Luciferase and �-galactosidase activities were assessed 24 hlater as described in Materials and Methods. (D) The ability of VDR siRNA to block activation of mRLD5-mediated transcription by 1,25(OH)2D3is rescued through the addition of a functional VDR expression vector. ST2 cells were cotransfected with 10 ng pCH110-�gal, pTK-mRLD5,pcDNA-hVDR(wtp) (wild type), or pcDNA-hVDR(m) (mutant) and 50 nM of either nontargeted siRNA or mVDR siRNA as described inMaterials and Methods. Transfected cells were cultured for 48 h and then treated for an additional 24 h with either vehicle or 1,25(OH)2D3 (10�7
M) and processed as in panel B. Each point represents the normalized relative light unit average � standard error of the mean for a triplicate setof transfections. All studies were repeated with similar results.
6476 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

control siRNA had no effect on the ability of 1,25(OH)2D3 toinduce this DNA construct, the addition of VDR siRNA fullyblocked that induction. Importantly, this loss of inducibilitycould be rescued by cotransfecting a wild-type human VDRexpression vector but not a vector expressing a transcription-ally inactive form of the VDR which contained mutations inthe transactivating domain (1). Our results support the ideathat both the VDR and GR are required for the hormone-inducible activity mediated by the mRLD5 region.
Mapping the VDR and GR regulatory regions located withinthe mRLD5 fragment. The above ChIP assays together withthe transfection studies suggest that the mRLD5 region con-tains regulatory elements capable of binding both VDR andGR and mediating the activity of 1,25(OH)2D3 and DEX. Wetherefore created a series of 5 and 3 deletion constructs ofthis region as indicated in Fig. 6A, introduced them togetherwith a VDR expression vector into ST2 cells, and mapped theactivity of the two hormones. The results in Fig. 6B reveal thatwhile changes in basal activity are evident within the deletionconstructs, the activity of 1,25(OH)2D3 maps directly to a cen-tral core fragment of 256 bp (mRLD5-3/2) which is locatedwithin mRLD5. The synergistic response to DEX, in contrast,maps to an activity that appears to span two fragments desig-nated mRLD5-3/2 and mRLD5-5/2. Thus, while neither ofthese fragments is responsive to DEX synergistically, an over-lapping fragment termed mRLD5-3/1 that contained theboundary between the above two fragments manifested strik-ing DEX induction. This interpretation is further supported bydirect analysis of DEX activity wherein the addition of cotrans-fected GR provides a direct readout of this receptor’s activityon mRLD5 subfragments. These results suggest that inducibleactivity of mRLD5 is potentially governed by a single regula-tory element for 1,25(OH)2D3 and perhaps via several ele-ments for DEX.
Identification of the mRL-VDRE within the mRLD5 region.An evaluation of the DNA sequence within the 1,25(OH)2D3-inducible fragment of the mRLD5 region (mRLD5-3/2) usingthe CONSITE (http://mordor.cgb.ki.se/cgi-bin/CONSITE/consite)algorithm revealed several nuclear receptor-like regulatory el-ements. One such region, however, contained two highly con-served VDRE-like sequences separated by a single base pair(see Fig. S5 in the supplemental material). To test whether thisunusual element as seen in Fig. 7A might mediate the actionsof 1,25(OH)2D3, a set of 3-bp mutations was introduced in thecontext of the mRLD5 fragment into each of the four half-sitescomprising the putative VDRE. The constructs were thentransfected into ST2 cells, and their activities were evaluated inresponse to 1,25(OH)2D3. As can be seen in Fig. 7B, mutationsin three of the four half-sites completely abolished hormonalresponse; activity derived from a fourth construct was compro-mised. These results suggest that this interesting element doesindeed mediate the actions of 1,25(OH)2D3 in the context ofmRLD5. We also examined whether the VDR and its RXRpartner could bind to this VDRE, particularly as a complexcomprised of two heterodimers. To this end, we carried out anelectrophoretic mobility shift assay (EMSA) using duplex oli-gonucleotides of either the Opn-VDRE or the putative mRL-VDRE and purified VDR and RXR� proteins. As can be seenin Fig. 7C, the VDR/RXR heterodimer bound to both theOpn- and the mRL-VDREs in a salt-sensitive, hormone-de-
pendent fashion. Similar binding of endogenous VDR andRXR was also observed in the presence of ST2 nuclear extracts(data not shown). Competition studies suggest that the relativebinding affinities of the VDR/RXR heterodimer for these twoVDREs were similar (see Fig. S6A in the supplemental mate-rial). Interestingly, the results in Fig. 7C also show that incu-bation of VDR/RXR with the mRL-VDRE but not the Opn-VDRE results in the appearance of a second, more slowlymigrating species, suggestive of a specific complex comprisedof two VDR/RXR heterodimers. Support for this contention isprovided by the observation that the higher-order complex failsto form when purified VDR/RXR is incubated with mRL-VDREs that contain mutations in either VDRE1 or VDRE2(see Fig. S6B in the supplemental material). Taken together,these results suggest that the element located at �75620 to�75590 upstream of the mouse RankL TSS is capable of bothbinding two VDR/RXR heterodimers and mediating the trans-activation potential of this complex in transfected cells.
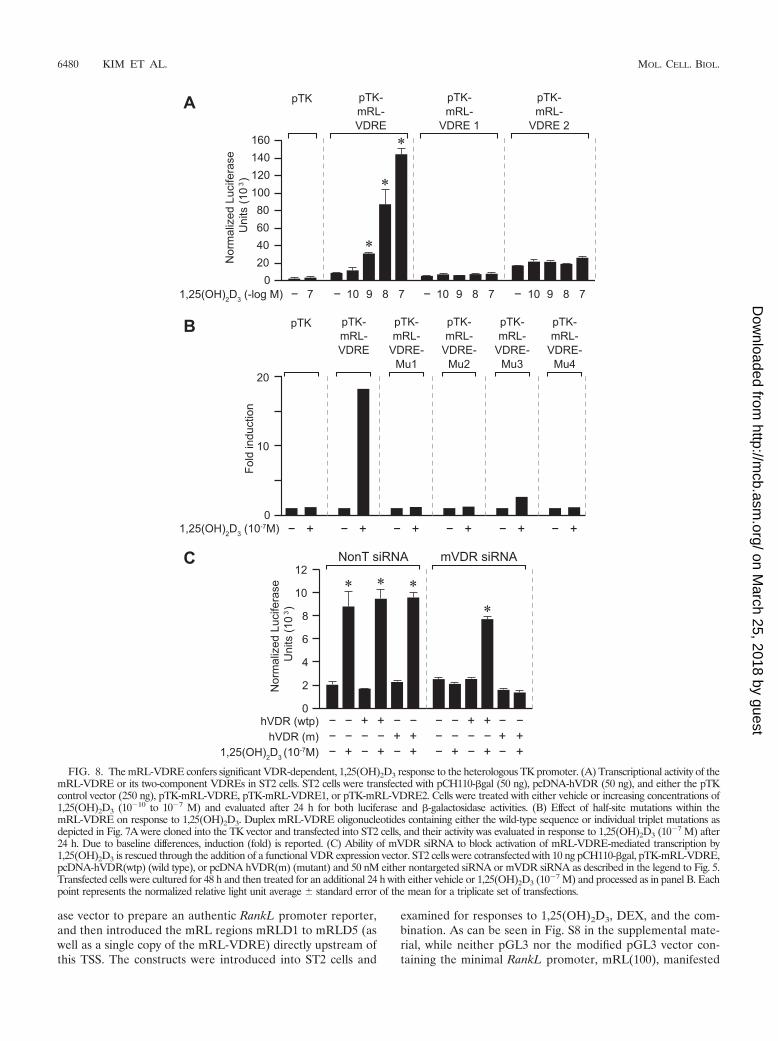
The mRL-VDRE alone confers strong 1,25(OH)2D3 respon-siveness to a heterologous promoter. In a final set of experi-ments aimed at characterizing the mRL-VDRE, we cloned thisDNA sequence as well as each of the half-VDREs that com-prise the element as single copies upstream of a TK promoterand examined their ability to mediate 1,25(OH)2D3 responseindependent of the mRLD5 environment. Constructs were in-troduced into ST2 cells together with a VDR expression vec-tor, and their activities were assessed in response to increasingconcentrations of 1,25(OH)2D3. As can be seen in Fig. 8A, themRL-VDRE was strongly induced by 1,25(OH)2D3 in a dose-dependent fashion. Interestingly, neither of the two individualVDREs that comprise the mRL-VDRE was capable of medi-ating any significant response to 1,25(OH)2D3. Further analy-sis of the mRL-VDRE indicated that the introduction of a 3-bpchange in any one of the four mRL-VDRE half-sites (as doc-umented in Fig. 7) fully compromised the response to1,25(OH)2D3 (Fig. 8B). Only the mutation at the third half-siteretained some small activity. The role of the VDR in thisinduction was further confirmed by a VDR siRNA knockdownand human VDR rescue experiment. Accordingly, while co-treatment of ST2 cells with control siRNA had no effect on theability of 1,25(OH)2D3 to induce the mRL-VDRE, as docu-mented in Fig. 8C, the introduction of VDR siRNA fully com-promised this upregulation. The induction was fully rescued bythe addition of wild-type human VDR, although not with atranscriptionally inactive human VDR allele containing muta-tions in its activation domain as described previously. Impor-tantly, as shown in Fig. S7 in the supplemental material, thismRL-VDRE was also able to mediate 1,25(OH)2D3 inducibil-ity in other mouse osteoblastic cell types, including primarymouse calvarial osteoblasts, as well as in primate cells. Our studiestherefore define a functional VDRE within the mRLD5 regionthat is structurally unique and that likely plays a significant rolein mediating the ability of 1,25(OH)2D3 to induce RankL geneexpression.
1,25(OH)2D3 stimulates and DEX potentiates the activity ofmRLD5 when cloned upstream of the authentic RankL genepromoter. The studies described above made use of a heterol-ogous viral promoter known to contain elements potentiallyresponsive to glucocorticoids. As a result, we cloned a RankLminimal promoter fragment, mRL(100), into a pGL3 lucifer-
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6477
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
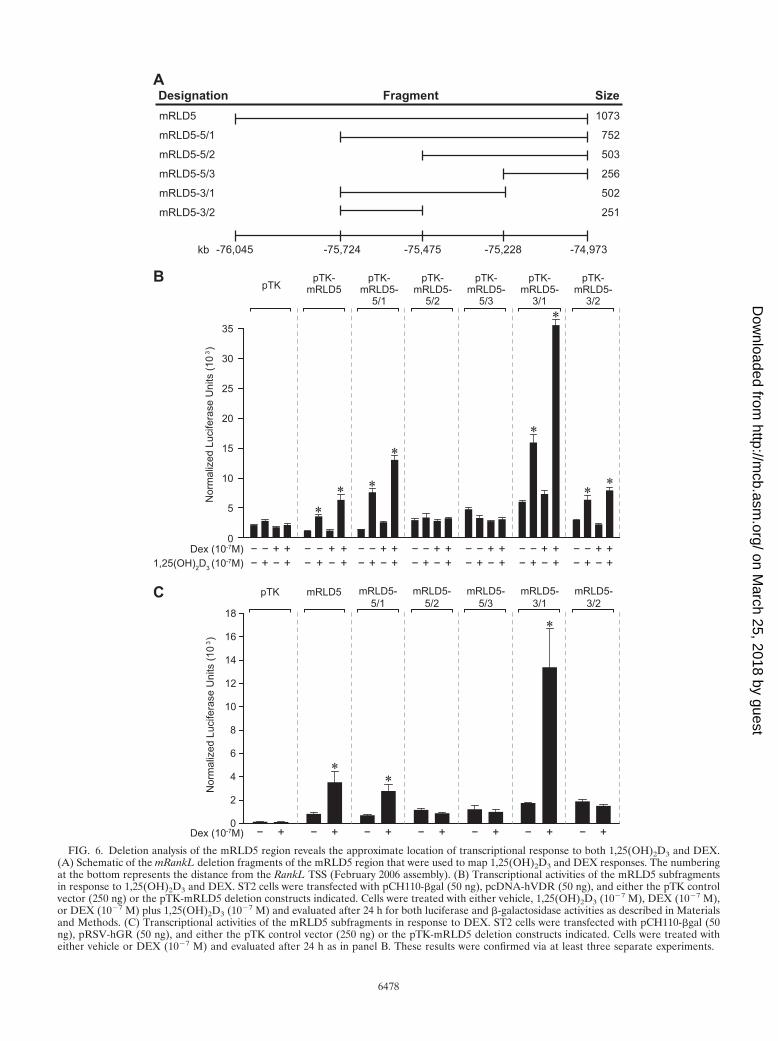
FIG. 6. Deletion analysis of the mRLD5 region reveals the approximate location of transcriptional response to both 1,25(OH)2D3 and DEX.(A) Schematic of the mRankL deletion fragments of the mRLD5 region that were used to map 1,25(OH)2D3 and DEX responses. The numberingat the bottom represents the distance from the RankL TSS (February 2006 assembly). (B) Transcriptional activities of the mRLD5 subfragmentsin response to 1,25(OH)2D3 and DEX. ST2 cells were transfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK controlvector (250 ng) or the pTK-mRLD5 deletion constructs indicated. Cells were treated with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M),or DEX (10�7 M) plus 1,25(OH)2D3 (10�7 M) and evaluated after 24 h for both luciferase and �-galactosidase activities as described in Materialsand Methods. (C) Transcriptional activities of the mRLD5 subfragments in response to DEX. ST2 cells were transfected with pCH110-�gal (50ng), pRSV-hGR (50 ng), and either the pTK control vector (250 ng) or the pTK-mRLD5 deletion constructs indicated. Cells were treated witheither vehicle or DEX (10�7 M) and evaluated after 24 h as in panel B. These results were confirmed via at least three separate experiments.
6478
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
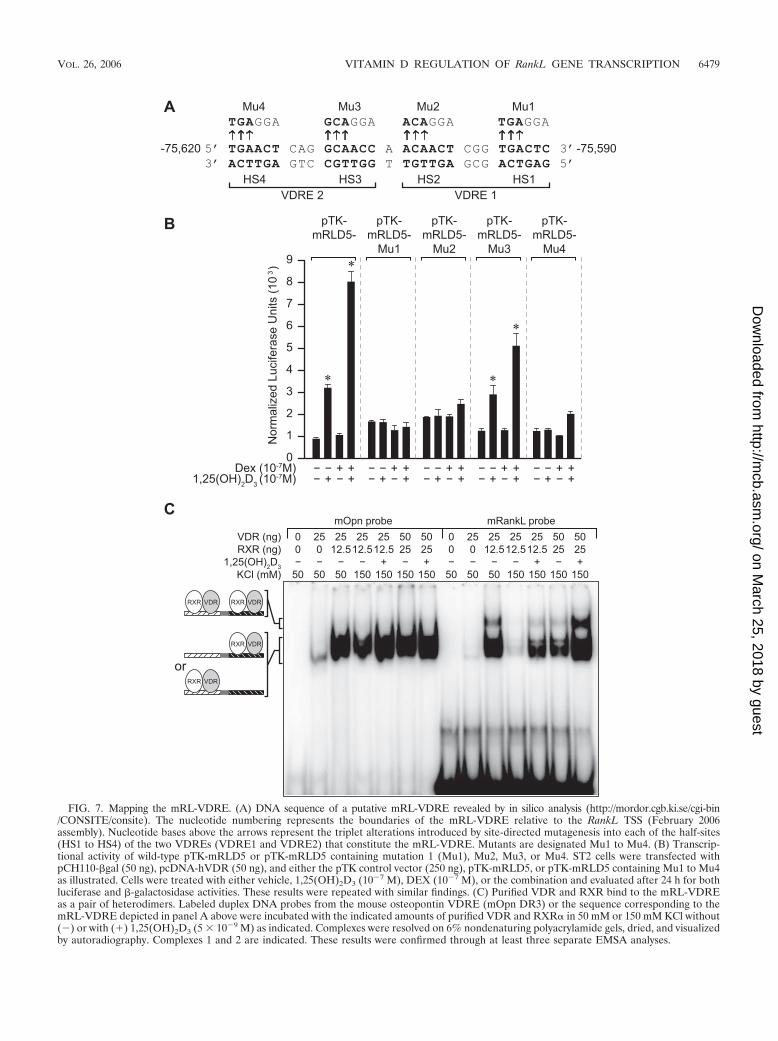
FIG. 7. Mapping the mRL-VDRE. (A) DNA sequence of a putative mRL-VDRE revealed by in silico analysis (http://mordor.cgb.ki.se/cgi-bin/CONSITE/consite). The nucleotide numbering represents the boundaries of the mRL-VDRE relative to the RankL TSS (February 2006assembly). Nucleotide bases above the arrows represent the triplet alterations introduced by site-directed mutagenesis into each of the half-sites(HS1 to HS4) of the two VDREs (VDRE1 and VDRE2) that constitute the mRL-VDRE. Mutants are designated Mu1 to Mu4. (B) Transcrip-tional activity of wild-type pTK-mRLD5 or pTK-mRLD5 containing mutation 1 (Mu1), Mu2, Mu3, or Mu4. ST2 cells were transfected withpCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK control vector (250 ng), pTK-mRLD5, or pTK-mRLD5 containing Mu1 to Mu4as illustrated. Cells were treated with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M), or the combination and evaluated after 24 h for bothluciferase and �-galactosidase activities. These results were repeated with similar findings. (C) Purified VDR and RXR bind to the mRL-VDREas a pair of heterodimers. Labeled duplex DNA probes from the mouse osteopontin VDRE (mOpn DR3) or the sequence corresponding to themRL-VDRE depicted in panel A above were incubated with the indicated amounts of purified VDR and RXR� in 50 mM or 150 mM KCl without(�) or with (�) 1,25(OH)2D3 (5 � 10�9 M) as indicated. Complexes were resolved on 6% nondenaturing polyacrylamide gels, dried, and visualizedby autoradiography. Complexes 1 and 2 are indicated. These results were confirmed through at least three separate EMSA analyses.
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6479
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
ase vector to prepare an authentic RankL promoter reporter,and then introduced the mRL regions mRLD1 to mRLD5 (aswell as a single copy of the mRL-VDRE) directly upstream ofthis TSS. The constructs were introduced into ST2 cells and
examined for responses to 1,25(OH)2D3, DEX, and the com-bination. As can be seen in Fig. S8 in the supplemental mate-rial, while neither pGL3 nor the modified pGL3 vector con-taining the minimal RankL promoter, mRL(100), manifested
FIG. 8. The mRL-VDRE confers significant VDR-dependent, 1,25(OH)2D3 response to the heterologous TK promoter. (A) Transcriptional activity of themRL-VDRE or its two-component VDREs in ST2 cells. ST2 cells were transfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTKcontrol vector (250 ng), pTK-mRL-VDRE, pTK-mRL-VDRE1, or pTK-mRL-VDRE2. Cells were treated with either vehicle or increasing concentrations of1,25(OH)2D3 (10�10 to 10�7 M) and evaluated after 24 h for both luciferase and �-galactosidase activities. (B) Effect of half-site mutations within themRL-VDRE on response to 1,25(OH)2D3. Duplex mRL-VDRE oligonucleotides containing either the wild-type sequence or individual triplet mutations asdepicted in Fig. 7A were cloned into the TK vector and transfected into ST2 cells, and their activity was evaluated in response to 1,25(OH)2D3 (10�7 M) after24 h. Due to baseline differences, induction (fold) is reported. (C) Ability of mVDR siRNA to block activation of mRL-VDRE-mediated transcription by1,25(OH)2D3 is rescued through the addition of a functional VDR expression vector. ST2 cells were cotransfected with 10 ng pCH110-�gal, pTK-mRL-VDRE,pcDNA-hVDR(wtp) (wild type), or pcDNA hVDR(m) (mutant) and 50 nM either nontargeted siRNA or mVDR siRNA as described in the legend to Fig. 5.Transfected cells were cultured for 48 h and then treated for an additional 24 h with either vehicle or 1,25(OH)2D3 (10�7 M) and processed as in panel B. Eachpoint represents the normalized relative light unit average � standard error of the mean for a triplicate set of transfections.
6480 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

any response, the profile of regulation of mRLD5 by both1,25(OH)2D3, DEX, and the combination was virtually identi-cal to that seen when mRLD5 when evaluated in the context ofthe heterologous TK promoter (Fig. 4). Unfortunately, thebasal and hormone-resistant activities of mRLD1 to mRLD4were also similar to that observed in the context of the TKpromoter, suggesting that the individual placement of each ofthese regions immediately upstream of its native proximal pro-moter was insufficient to manifest a transcriptional response to1,25(OH)2D3, DEX, or a combination of the two ligands (datanot shown). We therefore conclude that only mRLD5 mani-fests an autonomous response to the two ligands when exam-ined in the context of a reporter plasmid.
Activation of the five regulatory regions of the mouse RankLgene results in histone acetylation. While mRLD4 to mRLD1failed to exhibit inducible transcriptional activity in the contextof either the TK promoter or a homologous mRL promoter,the results of the ChIP/chip and direct ChIP assays using VDR,RXR, and RNA pol II strongly support a role for mRLD4 tomRLD1 in mediating the actions of 1,25(OH)2D3 on RankLgene expression. We therefore asked whether 1,25(OH)2D3-induced binding of VDR and RXR to the five RankL generegions was capable of inducing modifications to histoneswithin those regions of the gene. Previous studies have sug-gested that this modification is essential to the upregulationof Cyp24a1 (30). ST2 cells were therefore treated with1,25(OH)2D3, and the cells were subjected to ChIP analysis forincreasing times using antibodies to tetra-acetylated histone 4as well as VDR. 1,25(OH)2D3 induced both a time-dependentaccumulation of VDR at the mRLD1-D5 regions and an in-crease in histone 4 acetylation, as can be seen in Fig. 9 (and seeFig. S9 in the supplemental material). Surprisingly, however,increased H4 acetylation did not occur at mRLD1 to mRLD5in the RL gene locus (with the possible exception of mRLD3),but rather at sites located between the five enhancers and atthe RankL TSS. This finding suggests that while 1,25(OH)2D3
can induce changes in acetylation within the mRankL genelocus, these changes do not appear to correlate directly withregions of VDR/RXR heterodimer binding.
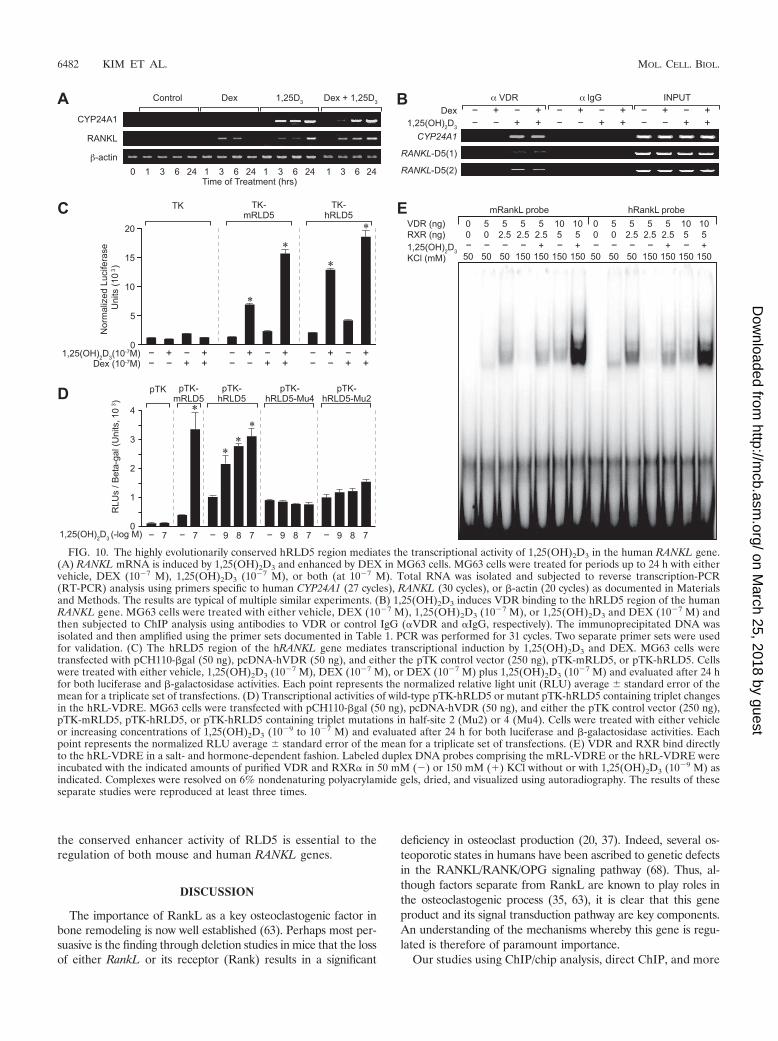
The highly evolutionarily conserved hRLD5 region locatedupstream of the human RANKL gene is controlled in a similarfashion by 1,25(OH)2D3 and DEX. The mRLD5 region of themouse RankL gene is highly conserved within vertebrate ge-nomes, including that of humans. In humans, the correspond-ing RANKL D5 region (hRLD5) is also similarly located up-stream of the TSS, although at a slightly greater distance ofapproximately �96 kb. To explore whether this region in thehuman RANKL gene might similarly mediate the actions of1,25(OH)2D3 and DEX on RANKL gene expression, we firstestablished a model for RANKL expression using the humanMG63 osteoblastic cell line. We treated MG63 cells with either1,25(OH)2D3, DEX, or both ligands and then evaluated theeffects of these ligands as a function of time on RANKL induc-tion. CYP24A1 was used as a positive control for 1,25(OH)2D3
induction. As can be seen in Fig. 10A, while both 1,25(OH)2D3
and DEX were able to modestly induce RANKL expression inthis cell line, the combination was slightly more effective. Next,we treated MG63 cells with either vehicle or 1,25(OH)2D3 for6 h and subjected the harvested cells to a ChIP analysis usingantibodies to either VDR or an IgG control. The precipitated
DNA was evaluated by PCR using primers to either CYP24A1or two separate primer sets to the hRLD5 region as indicatedin Materials and Methods and documented in Table 1. Asdepicted in Fig. 10B, 1,25(OH)2D3 induced VDR binding toboth control CYP24A1 and to the human hRLD5 region aswell. DEX did not appear to potentiate or influence the extentof VDR binding. Based upon these results, we cloned approx-imately 0.8 kb of the human hRLD5 region into the TK pro-moter, transfected this plasmid or the mouse mRLD5 TKplasmid into MG63 cells, and evaluated the transcriptionalresponse to 1,25(OH)2D3, DEX, or both ligands. The results inFig. 10C reveal that the hRLD5 region is comparable to themouse version in its capacity to mediate response to both1,25(OH)2D3 and DEX. DEX, however, appears able to exerta small but measurable effect directly on the hRLD5 regioneven in the absence of 1,25(OH)2D3. Importantly, mutagenesisof two of the putative human VDRE half-sites within thecontext of hRLD5 abrogated the response to 1,25(OH)2D3, asseen in Fig. 10D, thus confirming the role of these elements in1,25(OH)2D3-dependent induction. In a final set of experi-ments, we assessed the capacities of VDR and RXR to binddirectly to the hRL-VDRE using mRL-VDRE as a control. Ascan be seen in Fig. 10E, both VDR and RXR bind to each ofthe labeled RL-VDREs in a salt-sensitive, hormone-depen-dent fashion. The low input levels of VDR and RXR limit thebinding of two VDR/RXR heterodimers. These results clearlyestablish the ability of the hRLD5 region to mediate the ac-tions of 1,25(OH)2D3 and DEX on the human RANKL geneand provide additional supportive data for the hypothesis that
FIG. 9. 1,25(OH)2D3 induces histone acetylation in the upstreamregions of the mRankL gene. ST2 cells were treated with 1,25(OH)2D3(10�7 M) for periods of up to 6 h and then subjected to ChIP analysisusing antibodies to VDR, tetra-acetylated histone 4, or IgG (�VDR,�AcH4, and �IgG, respectively). Precipitated DNA was isolated andevaluated by PCR using Cyp24a1 primers (see Materials and Methods)or RankL primers whose locations are depicted in Fig. 3 (upper panel)and whose sequences are delineated in Table 1. Amplifications werecarried out as in Fig. 3. Similar results were obtained in at least fourseparate experiments.
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6481
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from
the conserved enhancer activity of RLD5 is essential to theregulation of both mouse and human RANKL genes.
DISCUSSION
The importance of RankL as a key osteoclastogenic factor inbone remodeling is now well established (63). Perhaps most per-suasive is the finding through deletion studies in mice that the lossof either RankL or its receptor (Rank) results in a significant
deficiency in osteoclast production (20, 37). Indeed, several os-teoporotic states in humans have been ascribed to genetic defectsin the RANKL/RANK/OPG signaling pathway (68). Thus, al-though factors separate from RankL are known to play roles inthe osteoclastogenic process (35, 63), it is clear that this geneproduct and its signal transduction pathway are key components.An understanding of the mechanisms whereby this gene is regu-lated is therefore of paramount importance.
Our studies using ChIP/chip analysis, direct ChIP, and more
FIG. 10. The highly evolutionarily conserved hRLD5 region mediates the transcriptional activity of 1,25(OH)2D3 in the human RANKL gene.(A) RANKL mRNA is induced by 1,25(OH)2D3 and enhanced by DEX in MG63 cells. MG63 cells were treated for periods up to 24 h with eithervehicle, DEX (10�7 M), 1,25(OH)2D3 (10�7 M), or both (at 10�7 M). Total RNA was isolated and subjected to reverse transcription-PCR(RT-PCR) analysis using primers specific to human CYP24A1 (27 cycles), RANKL (30 cycles), or �-actin (20 cycles) as documented in Materialsand Methods. The results are typical of multiple similar experiments. (B) 1,25(OH)2D3 induces VDR binding to the hRLD5 region of the humanRANKL gene. MG63 cells were treated with either vehicle, DEX (10�7 M), 1,25(OH)2D3 (10�7 M), or 1,25(OH)2D3 and DEX (10�7 M) andthen subjected to ChIP analysis using antibodies to VDR or control IgG (�VDR and �IgG, respectively). The immunoprecipitated DNA wasisolated and then amplified using the primer sets documented in Table 1. PCR was performed for 31 cycles. Two separate primer sets were usedfor validation. (C) The hRLD5 region of the hRANKL gene mediates transcriptional induction by 1,25(OH)2D3 and DEX. MG63 cells weretransfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK control vector (250 ng), pTK-mRLD5, or pTK-hRLD5. Cellswere treated with either vehicle, 1,25(OH)2D3 (10�7 M), DEX (10�7 M), or DEX (10�7 M) plus 1,25(OH)2D3 (10�7 M) and evaluated after 24 hfor both luciferase and �-galactosidase activities. Each point represents the normalized relative light unit (RLU) average � standard error of themean for a triplicate set of transfections. (D) Transcriptional activities of wild-type pTK-hRLD5 or mutant pTK-hRLD5 containing triplet changesin the hRL-VDRE. MG63 cells were transfected with pCH110-�gal (50 ng), pcDNA-hVDR (50 ng), and either the pTK control vector (250 ng),pTK-mRLD5, pTK-hRLD5, or pTK-hRLD5 containing triplet mutations in half-site 2 (Mu2) or 4 (Mu4). Cells were treated with either vehicleor increasing concentrations of 1,25(OH)2D3 (10�9 to 10�7 M) and evaluated after 24 h for both luciferase and �-galactosidase activities. Eachpoint represents the normalized RLU average � standard error of the mean for a triplicate set of transfections. (E) VDR and RXR bind directlyto the hRL-VDRE in a salt- and hormone-dependent fashion. Labeled duplex DNA probes comprising the mRL-VDRE or the hRL-VDRE wereincubated with the indicated amounts of purified VDR and RXR� in 50 mM (�) or 150 mM (�) KCl without or with 1,25(OH)2D3 (10�9 M) asindicated. Complexes were resolved on 6% nondenaturing polyacrylamide gels, dried, and visualized using autoradiography. The results of theseseparate studies were reproduced at least three times.
6482 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

traditional molecular approaches identified five distinct re-gions located at significant distances upstream of the TSS thatwere responsible for regulating the expression of the RankLgene. VDR and RXR localize to each of these regions inresponse to 1,25(OH)2D3 and appear to be accompanied byother factors as well, including GR. This supports the idea thateach of these regions may represent enhancer modules integralto RankL gene expression. A typical consequence of transac-tivation at enhancers is the rapid recruitment of coregulatoryenzymes and the subsequent modification of local chromatinstructure, in part through acetylation (5, 52, 55, 56). It is be-lieved that these epigenetic modifications function to promotethe chromatin decondensation that is often necessary for in-creased transcription. In our studies, while it is clear that1,25(OH)2D3 induced histone 4 acetylation at the RankL genelocus, these modifications did not occur at sites of VDR/RXRbinding, but rather at sites located in between. Thus, althoughdirect recruitment of histone-modifying enzymes by the VDR/RXR heterodimer may be involved, we believe it more likelythat the VDR precipitates events at these sites that lead in turnto the increased acetylation seen between the enhancer re-gions. The nature of these events remains to be determined.Interestingly, 1,25(OH)2D3 also induced the recruitment ofRNA pol II to the mRLD1-to-mRLD5 regions of the RankLgene. This finding suggests that the mRLD1-to-mRLD5 en-hancers may also act as recruitment centers for components ofthe transcriptional apparatus as well (59). Although specula-tive, it is possible that the recruitment of transcriptional com-ponents to the five enhancer regions and the consequences ofthis recruitment may represent the initiating event for thehistone modifications seen above. Regardless, our data suggestthat the ChIP/chip and ChIP approaches taken here werestrategic in identifying key regions within the RankL locus thatare responsible for transcriptional regulation of this gene by1,25(OH)2D3 and DEX.
While all of the sites appear to contribute to the responseinitiated by 1,25(OH)2D3, only the mRLD5 region was ca-pable of conferring sensitivity to 1,25(OH)2D3 in a transcrip-tional reporter plasmid assay. The respective responses to1,25(OH)2D3, DEX, and the combination are consistent withthat seen at the level of RankL mRNA, thereby validatingfurther the relevance of mRLD5 to RankL gene expression.The roles of VDR and RXR in this regulation were furtherestablished using both selective antagonists as well as siRNA,which reduced VDR mRNA levels and compromised the ca-pacity of 1,25(OH)2D3 to induce transcription via the mRLD5segment. Because ZK159222 and RU486 do not prevent re-ceptor DNA binding, but rather alter their ability to recruitcoregulators (16, 30), these results provide additional supportfor cofactor involvement in enhanced RankL gene expression.We defined the boundaries of mRLD5 based upon the VDRand RXR binding activities seen in the ChIP/chip analysis. Thefragment was comprised of approximately 1,100 bp of highlyconserved sequence present across multiple RankL genes. Anadditional highly conserved region of 800 bp immediately up-stream of the mRLD5 region was also noted. This segmentcorresponds directly to the region that mediates PTH re-sponse, as described in the accompanying article by Fu, Mano-lagas, and O’Brien (13). Our additional studies using ChIPanalysis, transient transcription assays, and mutagenesis fully
confirm these observations (S. Kim and J. W. Pike, unpub-lished observations). Thus, we propose that this entire con-served region be designated the RankL distal control region, orRL-DCR.
The transcriptional activity of the mRLD5 region allowed usboth to map the mRL-VDRE and to characterize a specific GCresponse. The mRL-VDRE retains a unique structure com-prised of two separate VDRE sequences linked via a singlebase pair. This element is clearly capable of the simultaneousbinding of two VDR/RXR heterodimers and is likewise re-sponsible for the 1,25(OH)2D3 sensitivity observed within themRLD5 region. Interestingly, the half-elements alone bindsingle VDR/RXR heterodimers, but do not elicit a significanttranscriptional response when cloned and analyzed indepen-dently. We did not map the existing GREs within this region ofthe RankL gene, however. The absence of activity in two con-tiguous but nonoverlapping fragments and the DEX sensitivityobserved in an mRLD5 fragment which spanned the boundarybetween these two fragments suggest the presence of severalGREs that may function synergistically to mediate DEX acti-vation. Further studies will be necessary to define the putativeGREs that we observed using in silico analysis (data notshown). Although DEX activity on mRLD5 appears to besynergistic, the addition of exogenous GR enabled us to detecta direct response to the GC. This effect was also noted whenthe human RLD5 region (hRLD5) was evaluated in the con-text of MG63 cells. Regardless of the nature of these overalleffects, it seems clear that 1,25(OH)2D3 action is mediated viaa single VDRE, whereas DEX activity is likely mediated byseveral independent GREs.
The appearance of VDR and RXR, histone acetylation, andRNA pol II recruitment all support the idea that mRLD1 tomRLD5 are active in the regulation of the RankL gene by1,25(OH)2D3. It is therefore curious as to why the mRLD1-to-mRLD4 upstream regions of the gene are incapable ofmediating a transcriptional response to 1,25(OH)2D3 in trans-fection studies. We believe that this resistance to hormonalinduction highlights the importance of context, wherein thepresence of both positive as well as negative cis elements andtheir respective transregulators exert significant influence ontranscriptional readouts obtained during transient transfectionassays. Interestingly, even the activity of a natural promoter ina plasmid context may be deceptive. The large distances thatexist between mRLD1 to mRLD5 and the location of the mostdistance element (�76 kb) made it difficult for us to assess thetransient activity of a “full-length” RankL promoter by thesemeans. Fu, Manolagas, and O’Brien overcame this problem,however, by using recombineering methods to produce large,bacterial artificial chromosome (BAC) clone-derived RankLconstructs which could be stably transfected into host cells(13). Interestingly, despite the differences in the two ap-proaches, both of our groups were able to identify the samehighly conserved distal region within the RankL gene thatdisplayed sensitivity to the two hormones 1,25(OH)2D3 andPTH. While the above results clearly demonstrate the overalldominance of the RL-DCR, the ability of 1,25(OH)2D3 toinduce a large RankL DNA fragment that no longer containedthe mRLD5 (13) provides further evidence that the mRLD1-to-mRLD4 regions are also important contributors to1,25(OH)2D3 response. Therefore, we believe that ChIP anal-
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6483
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

ysis may represent a more reliable method of assessing thepresence of functional enhancers than the alternative analysisusing plasmid transfection. Studies are ongoing in our labora-tory, however, to identify the precise locations of the VDREslodged within the mRLD1-to-mRLD4 regions that regulateRankL gene expression.
Regardless of the properties of mRLD1 to mRLD4, thestudies of both Fu, Manolagas, and O’Brien (13) as well as ourown highlight the important role of the mRL-DCR in mediat-ing both PTH and 1,25(OH)2D3 responses. It is thereforenoteworthy that in the accompanying article, Fu et al. (13)provide in vivo evidence that genetic deletion of the mRL-DCR in mice leads to a loss of PTH-mediated RankL induc-tion. We would predict that these mice will also display asimilar resistance to 1,25(OH)2D3, although whether this re-sistance will be only partial or complete remains to be deter-mined. Collaborative studies are currently under way to morefully investigate the properties of this altered RankL genelocus in vivo and, more importantly, perhaps, to determine thenature of the skeletal phenotype that results from this geneticchange.
The regulatory regions for RankL are widely dispersedacross a rather large segment of upstream DNA, the furthestsome 76 kb from the TSS. It is therefore clear that an under-standing of the function of each of these regions and theirindividual roles in modulating the expression of the RankLgene will require significant additional work. In that vein, thereis increasing evidence that many genes contain distant regula-tory elements such that this mechanism of regulation may bemore frequent than previously believed (2, 21, 39). The ex-tended pattern and distance of these elements within theRankL gene as well as in other genes almost certainly highlightthe crucial impact of chromatin structure and organization onthe expression of these genes such that the elements can di-rectly impact activity at the TSS. One might speculate thatthese regions converge directly on the proximal RankL pro-moter by virtue of extensive chromatin looping (6, 9, 49, 65).New technologies are currently available or in development totest distance relationships between regulatory regions andtheir functional promoters, thus making it possible to explorethis intriguing hypothesis (7).
A final question that emerges from these studies is why RNApol II might be recruited to each of the five enhancer domainswithin the RankL gene. One might imagine that the presenceof RNA pol II at these sites might also require the simulta-neous recruitment of basal transcription factors such as TF-IIA, TF-IIB and TAF-II250 as well (59). Indeed, we have seenin preliminary studies that 1,25(OH)2D3 can induce the re-cruitment of TAF-II250 to the mRLD5 region (S. Kim and J.Pike, unpublished data). One frequent consequence of theassembly of such factors at upstream regulatory regions, how-ever, is the production of nascent noncoding mRNAs (3, 4, 10,26, 27). The role of this transcription and these transcripts iscurrently unknown, although it has been suggested that theirproduction may be essential to the maintenance of an openchromatin state necessary for gene regulation (15). An alter-native proposal is that upstream enhancers may function ascenters for the recruitment of basal transcriptional machinery,thereby providing a source of such factors for authentic pro-moter-driven transcription (59). Now that some of the funda-
mentals of hormonal regulation have been established withinthe RankL gene locus, future studies will focus on these issuesinvolving RNA pol II recruitment and on understanding thespatial arrangement that likely exists between the five regula-tory regions and the TSS.
In summary, we have shown that 1,25(OH)2D3 and its re-ceptor induce the expression of RankL via five regulatory do-mains located at significant distances upstream of the TSS.This regulation is facilitated by DEX and the GR, the latterappearing to localize to several but not all of these regions.Transcription factor binding within these regions is associatedwith the recruitment of cofactors such as RNA pol II andadjacent histone acetylation. Mapping studies of mRLD5, per-haps the dominant control region for the RankL gene, led tothe delineation of an unusual VDRE. Support for mRLD5 asa primary enhancer of mouse RankL gene expression was in-creased by the discovery that an analogous component is lo-cated within the human RANKL gene.
ACKNOWLEDGMENTS
We thank members of the Pike laboratory for helpful discussion.We thank Adam Steinberg and Laura Vanderploeg for preparingthe figures for this article.
This work was supported by National Institutes of Health grantDK-74993 (to J.W.P.).
REFERENCES
1. Bettoun, D. J., T. P. Burris, K. A. Houck, D. W. Buck II, K. R. Stayrook, B.Khalifa, J. Lu, W. W. Chin, and S. Nagpal. 2003. Retinoid X receptor is anonsilent major contributor to vitamin D receptor-mediated transcriptionalactivation. Mol. Endocrinol. 17:2320–2328. [Epub ahead of print.]
2. Carroll, J. S., X. S. Liu, A. S. Brodsky, W. Li, C. A. Meyer, A. J. Szary, J.Eeckhoute, W. Shao, E. V. Hestermann, T. R. Geistlinger, E. A. Fox, P. A.Silver, and M. Brown. 2005. Chromosome-wide mapping of estrogen recep-tor binding reveals long-range regulation requiring the forkhead proteinFoxA1. Cell 122:33–43.
3. Cawley, S., S. Bekiranov, H. H. Ng, P. Kapranov, E. A. Sekinger, D. Kampa,A. Piccolboni, V. Sementchenko, J. Cheng, A. J. Williams, R. Wheeler, B.Wong, J. Drenkow, M. Yamanaka, S. Patel, S. Brubaker, H. Tammana, G.Helt, K. Struhl, and T. R. Gingeras. 2004. Unbiased mapping of transcrip-tion factor binding sites along human chromosomes 21 and 22 points towidespread regulation of noncoding RNAs. Cell 116:499–509.
4. Cheng, J., P. Kapranov, J. Drenkow, S. Dike, S. Brubaker, S. Patel, J. Long,D. Stern, H. Tammana, G. Helt, V. Sementchenko, A. Piccolboni, S.Bekiranov, D. K. Bailey, M. Ganesh, S. Ghosh, I. Bell, D. S. Gerhard, andT. R. Gingeras. 2005. Transcriptional maps of 10 human chromosomes at5-nucleotide resolution. Science 308:1149–1154. [Epub ahead of print.]
5. Cosgrove, M. S., J. D. Boeke, and C. Wolberger. 2004. Regulated nucleosomemobility and the histone code. Nat. Struct. Mol. Biol. 11:1037–1043.
6. de Bruin, D., Z. Zaman, R. A. Liberatore, and M. Ptashne. 2001. Telomerelooping permits gene activation by a downstream UAS in yeast. Nature409:109–113.
7. Dekker, J., K. Rippe, M. Dekker, and N. Kleckner. 2002. Capturing chro-mosome conformation. Science 295:1306–1311.
8. Dhawan, P., X. Peng, A. L. M. Sutton, P. N. MacDonald, C. M. Croniger, C.Trautwein, M. Centrella, T. L. McCarthy, and S. Christakos. 2005. Func-tional cooperation between CCAAT/enhancer-binding proteins and the vi-tamin D receptor in regulation of 25-hydroxyvitamin D3 24-hydroxylase.Mol. Cell. Biol. 25:472–487.
9. Drissen, R., R. J. Palstra, N. Gillemans, E. Splinter, F. Grosveld, S. Philipsen,and W. de Laat. 2004. The active spatial organization of the beta-globin locusrequires the transcription factor EKLF. Genes Dev. 18:2485–2490.
10. ENCODE Project Consortium. 2004. The ENCODE (ENCyclopedia OfDNA Elements) Project. Science 306:636–640.
11. Fan, X., E. M. Roy, T. C. Murphy, M. S. Nanes, S. Kim, J. W. Pike, and J.Rubin. 2004. Regulation of RANKL promoter activity is associated withhistone remodeling in murine bone stromal cells. J. Cell Biochem. 93:807–818.
12. Fu, Q., R. L. Jilka, S. C. Manolagas, and C. A. O’Brien. 2002. Parathyroidhormone stimulates receptor activator of NFkappa B ligand and inhibitsosteoprotegerin expression via protein kinase A activation of cAMP-re-sponse element-binding protein. J. Biol. Chem. 277:48868–48875. [Epubahead of print.]
6484 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

13. Fu, Q., S. C. Manolagas, and C. A. O’Brien. Parathyroid hormone controlsreceptor activator of NF-�B ligand gene expression via a distant transcrip-tional enhancer. Mol. Cell. Biol. 26:6453–6468.
14. Glynn, E. F., P. C. Megee, H. G. Yu, C. Mistrot, E. Unal, D. E. Koshland,J. L. Derisi, and J. L. Gerton. 2004. Genome-wide mapping of the cohesioncomplex in the yeast Saccharomyces cerevisiae. PLoS Biol. 2:E259. [Online.]
15. Gribnau, J., K. Diderich, S. Pruzina, R. Calzolari, and P. Fraser. 2000.Intergenic transcription and developmental remodeling of chromatin subdo-mains in the human beta-globin locus. Mol. Cell 5:377–386.
16. Groyer, A., G. Schweizer-Groyer, F. Cadepond, M. Mariller, and E. E.Baulieu. 1987. Antiglucocorticosteroid effects suggest why steroid hormoneis required for receptors to bind DNA in vivo but not in vitro. Nature328:624–626.
17. Harris, W. H., and R. P. Heaney. 1969. Skeletal renewal and metabolic bonedisease. N. Engl. J. Med. 280:193–202.
18. Hofbauer, L. C., F. Gori, L. Riggs, D. L. Lacey, C. R. Dunstan, T. C.Spelsberg, and S. Khosla. 1999. Stimulation of osteoprotegerin ligand andinhibition of osteoprotegerin production by glucocorticoids in human osteo-blastic lineage cells: potential paracrine mechanisms of glucocorticoid-in-duced osteoporosis. Endocrinology 140:4382–4389.
19. Hofbauer, L. C., S. Khosla, C. R. Dunstan, D. L. Lacey, W. J. Boyle, and B. L.Riggs. 2000. The roles of osteoprotegerin and osteoprotegerin ligand in theparacrine regulation of bone resorption. J. Bone Miner. Res. 15:2–12.
20. Hsu, H., D. L. Lacey, C. R. Dunstan, I. Solovyev, A. Colombero, E. Timms,H. L. Tan, G. Elliott, M. J. Kelley, I. Sarosi, L. Wang, X. Z. Xia, R. Elliott,L. Chiu, T. Black, S. Scully, C. Capparelli, S. Morony, G. Shimamoto, M. B.Bass, and W. J. Boyle. 1999. Tumor necrosis factor receptor family memberRANK mediates osteoclast differentiation and activation induced by osteo-protegerin ligand. Proc. Natl. Acad. Sci. USA 96:3540–3545.
21. Impey, S., S. R. McCorkle, H. Cha-Molstad, J. M. Dwyer, G. S. Yochum,J. M. Boss, S. McWeeney, J. J. Dunn, G. Mandel, and R. H. Goodman. 2004.Defining the CREB regulon: a genome-wide analysis of transcription factorregulatory regions. Cell 119:1041–1054.
22. Jimi, E., S. Akiyama, T. Tsurukai, N. Okahashi, K. Kobayashi, N. Udagawa,T. Nishihara, N. Takahashi, and T. Suda. 1999. Osteoclast differentiationfactor acts as a multifunctional regulator in murine osteoclast differentiationand function. J. Immunol. 163:434–442.
23. Jimi, E., I. Nakamura, L. T. Duong, T. Ikebe, N. Takahashi, G. A. Rodan,and T. Suda. 1999. Interleukin 1 induces multinucleation and bone-resorbingactivity of osteoclasts in the absence of osteoblasts/stromal cells. Exp. CellRes. 247:84–93.
24. Kabe, Y., J. Yamada, H. Uga, Y. Yamaguchi, T. Wada, and H. Handa. 2005.NF-Y is essential for the recruitment of RNA polymerase II and inducibletranscription of several CCAAT box-containing genes. Mol. Cell. Biol. 25:512–522.
25. Kaji, H., T. Sugimoto, M. Kanatani, M. Fukase, M. Kumegawa, and K.Chihara. 1996. Prostaglandin E2 stimulates osteoclast-like cell formationand bone resorbing activity via osteoblasts: role of cAMP-dependent proteinkinase. J. Bone Miner. Res. 11:62–71.
26. Kamp, D., J. Cheng, P. Kapranov, M. Yamanaka, S. Brubaker, S. Cawley, J.Drenkow, A. Piccolboni, S. Bekiranov, G. Helt, H. Tammana, and T. R.Gingeras. 2004. Novel RNAs identified from an in-depth analysis of thetranscriptome of human chromosomes 21 and 22. Genome Res. 14:331–342.
27. Kapranov, P., S. E. Cawley, J. Drenkow, S. Bekiranov, R. L. Strausberg, S. P.Fodor, and T. R. Gingeras. 2002. Large-scale transcriptional activity in chro-mosomes 21 and 22. Science 296:916–919.
28. Karst, M., G. Gorny, R. J. Galvin, and M. J. Oursler. 2004. Roles of stromalcell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulationof osteoclast differentiation. J. Cell Physiol. 200:99–106.
29. Kato, S., K. Sekine, T. Matsumoto, and T. Yoshizawa. 1998. Moleculargenetics of vitamin D receptor acting in bone. J. Bone Miner. Metab. 16:65–71.
30. Kim, S., N. K. Shevde, and J. W. Pike. 2005. 1,25-Dihydroxyvitamin D3stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J. BoneMiner. Res. 20:305–317. [Epub ahead of print.]
31. Kim, T. H., L. O. Barrera, M. Zheng, C. Qu, M. A. Singer, T. A. Richmond,Y. Wu, R. D. Green, and B. Ren. 2005. A high resolution map of activepromoters in the human genome. Nature 436:876–880.
32. Kim, T. H., L. O. Barrera, C. Qu, S. Van Calcar, N. D. Trinklein, S. J.Cooper, R. M. Luna, C. K. Glass, M. G. Rosenfeld, R. M. Myers, and B. Ren.2005. Direct isolation and identification of promoters in the human genome.Genome Res. 15:830–839.
33. Kirmizis, A., S. M. Bartley, A. Kuzmichev, R. Margueron, D. Reinberg, R.Green, and P. J. Farnham. 2004. Silencing of human polycomb target genesis associated with methylation of histone H3 lys 27. Genes Dev. 18:1592–1605.
34. Kitazawa, S., R. Kitazawa, and S. Maeda. 1999. Promoter structure of mouseRANKL/TRANCE/ODF gene. Biochim. Biophys. Acta 1445:134–141.
35. Koga, T., M. Inui, K. Inoue, S. Kim, A. Suematsu, E. Kobayashi, T. Iwata,H. Ohnishi, T. Matozaki, T. Kodama, T. Taniguchi, H. Takayanagi, and T.
Takai. 2004. Costimulatory signals mediated by the ITAM motif cooperatewith RANKL for bone homeostasis. Nature 428:758–763.
36. Kondo, H., J. Guo, and F. R. Bringhurst. 2002. Cyclic adenosine monophos-phate/protein kinase A mediates parathyroid hormone/parathyroid hor-mone-related protein receptor regulation of osteoclastogenesis and expres-sion of RANKL and osteoprotegerin mRNAs by marrow stromal cells.J. Bone Miner. Res. 17:1667–1679.
37. Kong, Y. Y., H. Yoshida, I. Sarosi, H. L. Tan, E. Timms, C. Capparelli, S.Morony, A. J. Oliveira-dos-Santos, G. Van, A. Itie, W. Khoo, A. Wakeham,C. R. Dunstan, D. L. Lacey, T. W. Mak, W. J. Boyle, and J. M. Penninger.1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte develop-ment and lymph-node organogenesis. Nature 397:315–323.
38. Lacey, D. L., E. Timm, H. L. Tan, M. J. Kelley, C. R. Dunstan, T. Burgess,R. Elliott, A. Colombero, G. Elliott, S. Scully, H. Hsu, J. Sullivan, N.Hawkins, E. Davy, C. Capparelli, A. Eli, Y. X. Qian, S. Kaufman, I. Sarosi,V. Shalhoub, G. Senaldi, J. Guo, J. Delaney, and W. J. Boyle. 1998. Osteo-protegerin ligand is a cytokine that regulates osteoclast differentiation andactivation. Cell 93:165–176.
39. Laganiere, J., G. Deblois, C. Lefebvre, A. R. Bataille, F. Robert, and V.Giguere. 2005. Location analysis of estrogen receptor alpha target promotersreveals that FOXA1 defines a domain of the estrogen response. Proc. Natl.Acad. Sci. USA 102:11651–11656. [Epub ahead of print.]
40. Lee, S. K., and J. A. Lorenzo. 1999. Parathyroid hormone stimulatesTRANCE and inhibits osteoprotegerin messenger ribonucleic acid expres-sion in murine bone marrow cultures: correlation with osteoclast-like cellformation. Endocrinology 140:3552–3561.
41. Liu, B. Y., J. Guo, B. Lanske, P. Divieti, H. M. Kronenberg, and F. R.Bringhurst. 1998. Conditionally immortalized murine bone marrow stromalcells mediate parathyroid hormone-dependent osteoclastogenesis in vitro.Endocrinology 139:1952–1964.
42. Manolagas, S. C. 2000. Birth and death of bone cells: basic regulatorymechanisms and implications for the pathogenesis and treatment of osteo-porosis. Endocr. Rev. 21:115–137.
43. Manolagas, S. C., R. S. Weinstein, R. L. Jilka, and A. M. Parfitt. 1998.Parathyroid hormone and corticosteroid-induced osteoporosis. Lancet 352:1940.
44. Matsuo, K., D. L. Galson, C. Zhao, L. Peng, C. Laplace, K. Z. Wang, M. A.Bachler, H. Amano, H. Aburatani, H. Ishikawa, and E. F. Wagner. 2004.Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis inprecursors lacking c-Fos. J. Biol. Chem. 279:26475–26480. [Epub ahead ofprint.]
45. Mundy, G. R. 2000. Pathogenesis of osteoporosis and challenges for drugdelivery. Adv. Drug Delivery Rev. 42:165–173.
46. Nagpal, S., S. Na, and R. Rathnachalam. 2005. Noncalcemic actions ofvitamin D receptor ligands. Endocr. Rev. 26:662–687. (First published 29March 2005; doi:10.1210/er.2004–0002.)
47. Oberley, M. J., J. Tsao, P. Yau, and P. J. Farnham. 2004. High throughputscreening of chromatin immunoprecipitates using CpG-island microarrays.Methods Enzymol. 376:315–334.
48. O’Brien, C. A., B. Kern, I. Gubrij, G. Karsenty, and S. C. Manolagas. 2002.Cbfa1 does not regulate RANKL gene activity in stromal/osteoblastic cells.Bone 30:453–462.
49. O’Sullivan, J. M., S. M. Tan-Wong, A. Morillon, B. Lee, J. Coles, J. Mellor,and N. J. Proudfoot. 2004. Gene loops juxtapose promoters and terminatorsin yeast. Nat. Genet. 36:1014–1018. [Epub ahead of print.]
50. Pike, J. W., and N. K. Shevde. 2005. The vitamin D receptor, p. 167–191. InD. Feldman, J. W. Pike, and F. H. Glorieux (ed.), Vitamin D, 2nd ed.Elsevier, San Diego, Calif.
51. Romas, E., N. Udagawa, H. Zhou, T. Tamura, M. Saito, T. Taga, D. J. Hilton,T. Suda, K. W. Ng, and T. J. Martin. 1996. The role of gp130-mediatedsignals in osteoclast development: regulation of interleukin 11 production byosteoblasts and distribution of its receptor in bone marrow cultures. J. Exp.Med. 183:2581–2591.
52. Schubeler, D., D. M. MacAlpine, D. Scalzo, C. Wirbelauer, C. Kooperberg,F. van Leeuwen, D. E. Gottschling, L. P. O’Neill, B. M. Turner, J. Delrow,S. P. Bell, and M. Groudine. 2004. The histone modification pattern of activegenes revealed through genome-wide chromatin analysis of a higher eu-karyote. Genes Dev. 18:1263–1271.
53. Shevde, N. K., L. A. Plum, M. Clagett-Dame, H. Yamamoto, J. W. Pike, andH. F. DeLuca. 2002. A potent analog of 1alpha,25-dihydroxyvitamin D3selectively induces bone formation. Proc. Natl. Acad. Sci. USA 99:13487–13491. [Epub ahead of print.]
54. Sivagurunathan, S., M. M. Muir, T. C. Brennan, J. P. Seale, and R. S.Mason. 2005. Influence of glucocorticoids on human osteoclast generationand activity. J. Bone Miner. Res. 20:390–398. [Epub ahead of print.]
55. Smith, C. L., and B. W. O’Malley. 2004. Coregulator function: a key tounderstanding tissue specificity of selective receptor modulators. Endocr.Rev. 25:45–71.
56. Spotswood, H. T., and B. M. Turner. 2002. An increasingly complex code.J. Clin. Investig. 110:577–582.
57. Suda, T., N. Takahashi, N. Udagawa, E. Jimi, M. T. Gillespie, and T. J.Martin. 1999. Modulation of osteoclast differentiation and function by the
VOL. 26, 2006 VITAMIN D REGULATION OF RankL GENE TRANSCRIPTION 6485
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from

new members of the tumor necrosis factor receptor and ligand families.Endocr. Rev. 20:345–357.
58. Sutton, A. L., and P. N. MacDonald. 2003. Vitamin D: more than a “bone-a-fide” hormone. Mol. Endocrinol. 17:777–791. [Epub ahead of print.]
59. Szutorisz, H., N. Dillon, and L. Tora. 2005. The role of enhancers as centresfor general transcription factor recruitment. Trends Biochem. Sci. 30:593–599. [Epub ahead of print.]
60. Takahashi, N., T. Akatsu, T. Sasaki, G. C. Nicholson, J. M. Moseley, T. J.Martin, and T. Suda. 1993. Induction of calcitonin receptors by 1,25-dihy-droxyvitamin D in osteoclast-like multinucleated cells formed from mousebone marrow cells. Endocrinology 123:1504–1510.
61. Takayanagi, H., S. Kim, T. Koga, H. Nishina, M. Isshiki, H. Yoshida, A.Saiura, M. Isobe, T. Yokochi, J. Inoue, E. F. Wagner, T. W. Mak, T. Kodama,and T. Taniguchi. 2002. Induction and activation of the transcription factorNFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation ofosteoclasts. Dev. Cell 3:889–901.
62. Tamura, T., N. Udagawa, N. Takahashi, C. Miyaura, S. Tanaka, Y. Yamada,Y. Koishihara, Y. Ohsugi, K. Kumaki, and T. Taga. 1993. Soluble interleu-kin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl.Acad. Sci. USA 90:11924–11928.
63. Teitelbaum, S. L. 2000. Bone resorption by osteoclasts. Science 289:1504–1508.
64. Teitelbaum, S. L., and F. P. Ross. 2003. Genetic regulation of osteoclastdevelopment and function. Nat. Rev. Genet. 4:638–649.
65. Tolhuis, B., R. J. Palstra, E. Splinter, F. Grosveld, and W. de Laat. 2002.
Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell 10:1453–1465.
66. Wani, M. R., K. Fuller, N. S. Kim, Y. Choi, and T. Chambers. 1999. Pros-taglandin E2 cooperates with TRANCE in osteoclast induction from hemo-poietic precursors: synergistic activation of differentiation, cell spreading,and fusion. Endocrinology 140:1927–1935.
67. Weinstein, R. S., R. L. Jilka, A. M. Parfitt, and S. C. Manolagas. 1998.Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblastsand osteocytes by glucocorticoids. Potential mechanisms of their deleteriouseffects on bone. J. Clin. Investig. 102:274–282.
68. Whyte, M. P., and S. Mumm. 2004. Heritable disorders of the RANKL/OPG/RANKL signaling pathway. Musculoskelet. Neuronal Interact. 4:254–267.
69. Yamada, K., D. T. Duong, D. K. Scott, J. C. Wang, and D. K. Granner. 1999.CCAAT/enhancer-binding protein beta is an accessory factor for the glu-cocorticoid response from the cAMP response element in the rat phos-phoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 274:5880–5888.
70. Yamamoto, H., N. K. Shevde, A. Warrier, L. A. Plum, H. F. DeLuca, andJ. W. Pike. 2003. 2-Methylene-19-nor-(20S)-1,25-dihydroxyvitamin D3 po-tently stimulates gene-specific DNA binding of the vitamin D receptor inosteoblasts. J. Biol. Chem. 278:31756–31765. [Epub ahead of print.]
71. Zella, L. A., S. Kim, N. K. Shevde, and J. W. Pike. 2006. Enhancers locatedwithin two introns of the vitamin D receptor gene mediate transcriptionalautoregulation by 1,25-dihydroxyvitamin D3. Mol. Endocrinol. 20:1231–1247. (First published 23 February 2006; 10.1210/me. 2000-0015.)
6486 KIM ET AL. MOL. CELL. BIOL.
on March 25, 2018 by guest
http://mcb.asm
.org/D
ownloaded from