acute myeloid leukemia - hematology ash education...
TRANSCRIPT

Hematology 2002 73
Acute Myeloid Leukemia
Francis J. Giles, Armand Keating, Anthony H. Goldstone, Irit Avivi,Cheryl L. Willman, and Hagop M. Kantarjian
In this chapter, Drs. Keating and Willman reviewrecent advances in our understanding of thepathophysiology of acute myeloid leukemia (AML)and allied conditions, including the advancedmyelodysplastic syndromes (MDS), while Drs.Goldstone, Avivi, Giles, and Kantarjian focus ontherapeutic data with an emphasis on currentpatient care and future research studies.
In Section I, Dr. Armand Keating reviews therole of the hematopoietic microenvironment in theinitiation and progression of leukemia. He alsodiscusses recent data on the stromal, ornonhematopoietic, marrow mesenchymal cellpopulation and its possible role in AML.
In Section II, Drs. Anthony Goldstone and IritAvivi review the current role of stem cell transplan-
tation as therapy for AML and MDS. They focus ondata generated on recent Medical Research Councilstudies and promising investigation approaches.
In Section III, Dr. Cheryl Willman reviews thecurrent role of molecular genetics and gene ex-pression analysis as tools to assist in AML diseaseclassification systems, modeling of gene expres-sion profiles associated with response or resis-tance to various interventions, and identifyingnovel therapeutic targets.
In Section IV, Drs. Hagop Kantarjian andFrancis Giles review some promising agents andstrategies under investigation in the therapy ofAML and MDS with an emphasis on novel deliverysystems for cytotoxic therapy and on targetedbiologic agents.
I. BIOLOGY OF ACUTE MYELOID LEUKEMIA:THE ROLE OF STROMA
Armand Keating, MD*
Over the past decade, rapid advances have been made inelucidating some of the key molecular lesions that lead toacute myeloid leukemia (AML).1 The therapeutic impli-cations of a detailed knowledge of aberrant signal trans-duction in malignant cells are evident, as the success ofimatinib mesylate in the treatment of chronic myeloid leu-kemia dramatically attests. It is perhaps not surprising, then,that considerably less attention has been directed towardexamining the role of the hematopoietic microenvironment(HM) in the initiation and progression of leukemia. Thedefinition of the HM as an entity that regulates hemato-poiesis through interactions with progenitor cells, hemato-poietic cytokines, and the biosynthetic products of stromaland other cells suggests, however, that much may be learnedabout the leukemic state by a better understanding of this
area. The recent resurgence of interest in the stromal ornonhematopoietic marrow mesenchymal cell populationmay serve as a springboard for further studies of howstroma influences, or is influenced by, leukemia. A briefoverview of the normal HM will serve as a prelude to areview of stromal cells in AML.
The Normal Hematopoietic MicroenvironmentThe HM in the bone marrow consists of a heterogeneouspopulation of hematopoietic and nonhematopoietic stro-mal cells, their extracellular biosynthetic products, andhematopoietic cytokines (reviewed in Clark andKeating2). The cells include myofibroblasts, other fibro-blastoid cells, endothelial cells, osteogenic precursors,adipocytes, and macrophages. These cells produce acomplex array of extracellular matrix (ECM) moleculesconsisting of proteoglycans and their constituent sulfatedglycosaminoglycans, chondroitin, heparan, and dermatanspecies as well as hyaluronic acid.2 In addition, they makea variety of interstitial (fibril-forming) and basal laminacollagens, including collagen types I, III, IV, V, and VI.Stromal cells also synthesize other matrix molecules,such as fibronectin, thrombospondin, hemonectin,sialoadhesin, laminin, and the tenascin glycoproteins (re-viewed in Klein3 and Verfaillie et al4) (Table 1). Cells
* Medical Oncology & Hematology, Princess MargaretHospital, 610 University Avenue, Suite 5-211, Toronto, ONM5G 2M9, Canada

74 American Society of Hematology
comprising the HM also provide a source of many he-matopoietic cytokines, either secreted or membranebound, including GM-CSF, G-CSF, and stem cell factor(kit ligand) (Table 2).
The growth, differentiation, and survival of hemato-poietic stem/progenitor cells is regulated in the HM byat least three different mechanisms that involve the fol-lowing:
1. Interactions of hematopoietic progenitor cells withhematopoietic cytokines, present in the HM, in partin association with ECM components such as gly-cosaminoglycans
2.Interactions between hematopoietic and stromal cellsby means of cell adhesion molecules
3. Interactions of adhesion molecules on hematopoi-etic cells with appropriate ligands on ECM compo-nents
Evidence is emerging that, in addition to hemato-poietic cytokines, adhesion molecules are involved inmediating signal transduction in hematopoietic precur-sors and hence play an important role in cell prolifera-tion that extends beyond brokering hematopoietic cellcontact and adhesion to stromal cells and ECM elements(reviewed in Levesque and Simmons5).
Interaction of Adhesion Molecules onHematopoietic Precursors and Marrow Stroma
Hematopoietic progenitors express cell adhesion mol-ecules (CAMs) that can be classified into six structur-ally distinct superfamilies: integrins; selectins; sialo-mucins, including CD34; immunoglobulins; CD44 cellsurface proteoglycans; and cadherins.5,6
IntegrinsThe integrins are particularly important because they areinvolved in stromal cell–stem cell, as well as stem cell–
ECM, adhesion. They are heterodimeric proteins con-sisting of noncovalently linked α and β chains thatuniquely pair to form at least 20 different integrin re-ceptors. The receptors are transmembrane structures inwhich the cytoplasmic domain initiates intracellular sig-naling involving the phosphorylation of cytoplasmic pro-teins and modulation of cell proliferation by activationof the ras and other pathways.5 The cytoplasmic portionof the integrin interacts with cytoskeletal elements (talin)that in turn influence the development of focal adhesioncontacts between the cell and the ECM.
There are two main β integrin families involvedin hematopoiesis, the β1 and β2 integrins. Bindingspecificity of the integrins is largely conferred by theparticular α chain.
βββββ1 IntegrinsThe β1 common chain (CD29) combines with differenta chains to form a variety of VLA (very late antigen)molecules that mediate the adhesion of hematopoieticcells to ECM components and ligands on stromal andendothelial cells. CD34(+) cells express the integrin re-ceptor VLA-4 (α4β1) (CD49d), whose ligand is VCAM-1 (vascular adhesion molecule-1) on marrow stromalcells and fibronectin in the ECM. VCAM-1 is variablyexpressed on marrow stromal and endothelial cells andcan be upregulated by several cytokines, including
Table 1. Extracellular matrix constituents.
Proteoglycans and constituent glycosaminoglycans
Heparan sulfate
Chondroitin sulfate
Dermatan sulfate
Hyaluronic acid
Collagen:types I, III, IV, V, VI
Fibronectin
Thrombospondin
Sialoadhesin
Laminin
Tenascin
Table 2. Factors constitutively or inducibly expressed bymarrow stromal cells.
G-CSF IL-1
GM-CSF IL-1β
M-CSF IL-6
Flt-3 ligand IL-7
SCF IL-8
LIF IL-12
Thrombopoietin IL-14
TNF-α IL-15
TNF-β IL-16
TGF-β IL-17
HGF IL-18
NGF
BDNF
SDF-1
Abbreviations: G-CSF, granulocyte colony-stimulating factor; IL,interleukin; GM-CSF, granulocyte-macrophage colony-stimulatingfactor; M-CSF, macrophage colony-stimulating factor; SCF, stemcell factor; LIF, leukemia inhibitory factor; TNF, tumor necrosisfactor; TGF, transforming growth factor; HGF, hepatocyte growthfactor; NGF, nerve growth factor; BDNF, brain-derived neurotropicfactor; SDF-1, stromal-derived factor-1.

Hematology 2002 75
interleukin-1 (IL-1). Because VLA-4 and its ligands arewidely distributed, specificity is most likely conferredby the coexpression of other adhesion molecules andcan be modulated by hematopoietic cytokines. The VLA-4/VCAM-1 interaction is a critical component of thecomplex process of stem cell homing.7 The chemokineand chemoattractant stromal-derived factor 1 (SDF-1),another important element in the homing of hematopoi-etic stem cells to the bone marrow, is secreted by stro-mal cells and strongly upregulates the VLA-4-mediatedadhesion of CD34(+) cells to stroma and ECM fibro-nectin.8 Early precursors also express VLA-5 (α
5β
1)
(CD49e), which can bind to ECM fibronectin.
βββββ2 IntegrinsOf the three β2 integrins (CD18), the best characterizedis LFA-1 (lymphocyte function antigen-1) (CD11a),which is associated with the adhesion of more matureleukocytes to the ligand ICAM1 on endothelium but isalso found on early hematopoietic precursors. Mac-1(CD11b), another β2 integrin, is found on mature mono-cytic and granulocytic cells but not on early hematopoi-etic precursors.
SelectinsThe selectins, a family of three glycoproteins, are alsoinvolved in adhesion and signaling. L-selectin is ex-pressed not only on mature leukocytes but also on earlyhematopoietic precursors. Its role in adhesion is bestdocumented in leukocyte attachment to endothelium. Incontrast, P- and E-selectin are found on endothelial cells,while their ligands are found on early hematopoieticcells.
SialomucinsOther adhesion molecules include the sialomucins, gly-coproteins carrying O-linked sugars: CD34, CD45RA,leukosialin (CD43), and the more recently identifiedCD164 molecule (reviewed in Simmons et al9). CD164(MGC-24) is expressed on both marrow stromal andCD34(+) cells and mediates adhesion between the twocell populations. Recent studies show that in addition,this molecule is involved in modulating the prolifera-tion of early precursors.10 Other sialomucins, includingleukosialin, also appear to act as negative regulators ofhematopoiesis.11
Immunoglobulin superfamilyThese cell adhesion molecules (CAMs) share a degreeof sequence homology with immunoglobulins and areinvolved in cell-cell interactions. There are three mainCAMs of relevance to hematopoiesis: VCAM-1, theICAMs, and NCAM. VCAM-1 is expressed on marrow
stromal cells and endothelial cells and interacts with theβ1 integrin VLA-4 (α
4β
1) on early hematopoietic pro-
genitors. Expression of VCAM-1 can be upregulated bya variety of cytokines, notably IL-1β and tumor necro-sis factor-α (TNF-α). Stromal cells also express ICAM1,which can interact with the β2 integrins such as LFA-1and Mac-1. NCAM-1 (CD56), which is a marker of natu-ral killer (NK) cells and is found on neuronal tissue, isalso expressed on marrow stromal cells and is involvedin supporting lymphopoiesis.
CD44 proteoglycansThe CD44 family, highly expressed on stromal cells,binds the nonsulfated glycosaminoglycan hyaluronicacid, a major component of the ECM present in long-term marrow culture adherent layers. Although CD44 isalso expressed on hematopoietic precursors, only a smallproportion (presumably high-affinity receptors) bindshyaluronate.12 CD44 can also bind to other ECM com-ponents, including fibronectin.13 Because CD44 is widelyexpressed, specificity of interaction is conferred by nu-merous isoforms generated by alternative splicing. Anti-CD44 antibody-blocking studies suggest that CD44 in-teractions are important in maintaining hematopoiesisin long-term marrow cultures.14
CadherinsThe cadherins (E-, N-, and P-cadherin) are transmem-brane glycoproteins that mediate calcium-dependent celladhesion in embryonic development and in the mainte-nance of tissue architecture. Their role in hematopoiesisis unclear, although recent studies indicate that both E-and N-cadherin are expressed on stromal cells and a sub-set of CD34(+) cells and erythroid progenitors.15-17 Theirrole in affecting leukemic cell development is unknown,but E-cadherin expression can be downregulated in AMLblasts by hypermethylation mechanisms.
The Hematopoietic Microenvironment in AMLGiven the multitude of interactions possible betweenhematopoietic progenitor cells and the HM, the acquisi-tion of a leukemic clone may have numerous effects onthis relationship and influence the clinical characteris-tics of the leukemia. At least three possible consequencesto changes in leukemia cell–HM interactions have beenproposed,11 as shown in Table 3.
Adhesion Molecule Expression on Leukemic CellsAML blasts express many of the adhesion moleculesidentified on normal hematopoietic precursors. Althoughdifferential expression has been documented, results havebeen variable, perhaps reflecting the heterogeneity ofAML as defined by morphology. For example, while

76 American Society of Hematology
AML blasts from one subset of patients express theintegrins VLA-1, -2, -3, and -6, not usually found onnormal CD34(+) cells,18 results of another study showreduced VLA-2, -3, and L-selectin levels and increasedVLA-5 expression19 or, indeed, the absence of transcriptsfor the α2, α3, or α6 chains of the β1 integrins in theblasts of yet other AML patients.20 Perhaps the most in-teresting observation is from Lyon, where a correlationwas shown between the expression of VLA-4 on leuke-mic blasts and a high initial white count as well as ex-tensive marrow involvement.21
Studies by the Westmead group in Sydney confirmthat adhesion of AML blasts, at least in part, is mediatedby the interaction of VLA-5 with ECM fibronectin aswell as via both β1 (VLA-4) and β2 (LFA-1) integrininteractions with stromal cells.22-24 Bendall and col-leagues have also shown that the adhesion of AML blaststo marrow fibroblasts can be modulated by a variety ofmechanisms, including the upregulation of stromalVCAM-1 by TNF-α and interferon γ.25
The adhesion of AML cells to ECM elements mayexplain the tenacity with which residual leukemic blastsmay persist in the marrow. The blasts from all patientsin a small cohort with AML expressed the sialylatedLewis x antigen, a ligand for E-selectin, on endothelialcells, suggesting a mechanism for migration across thevascular wall and into extravascular tissue.26
Further studies are required to determine whetherdifferences in the expression of adhesion molecules onleukemic blasts influence cell trafficking and the clini-cal phenotype in AML, as appears to be the case forchronic myeloid leukemia.27
Stromal Interaction with Leukemic CellsLeukemic cells, like their normal hematopoietic counter-parts, are subject to the influence of the HM. Encountersbetween the HM and leukemic cells can affect the
apoptosis, differentiation, and proliferation of AML blasts.
Cell-Cell InteractionsDirect contact of leukemic cells with stromal layersstrongly inhibits the apoptosis of the leukemic cells.28
The reduction in apoptosis correlates with enhancedgrowth of clonogenic leukemic cells. The growth fac-tors, stem cell factor (SCF), GM-CSF, and TNF-α in se-rum-free medium could achieve the same degree of in-hibition of apoptosis in only half the cases studied. Theco-culture of primary untreated AML cells with a stro-mal cell line in the presence of chemotherapy agentsinhibits drug-induced apoptosis and increases the viabil-ity of leukemic clonogenic cells.29 The interaction ofacute lymphoblastic leukemia (ALL) cells with stromalcells in the presence of chemotherapy drugs reduces thelevel of caspase 3 in leukemic cells and may account forthe reduced apoptosis.30 These studies indicate that di-rect contact between leukemic and stromal cells enhancesthe survival of clonogenic leukemic cells and may ex-plain how the small numbers of malignant cells remain-ing after chemotherapy are protected in vivo.
These observations contrast with the demonstrationof the relative inhospitability of long-term marrow cul-tures to CML and AML progenitors, which are prefer-entially lost, allowing normal precursors to be re-expressed,31,32 and formed the basis for the clinical purg-ing of grafts for patients undergoing intensive therapyand autotransplant.33,34 It is possible that, over the ex-tended period of culture, the system does not providecritical survival factors for neoplastic clones. Loss ofmalignant progenitors is much less likely in the culturesof patients with advanced disease,35 suggesting that suchlimitations to growth in vitro are overcome and that nor-mal hematopoiesis remains severely suppressed, if itexists at all.
CytokinesThe growth-promoting effects on leukemic blasts ofcytokines such as G-CSF, SCF, GM-CSF, macrophagecolony-stimulating factor (M-CSF), and IL-6 secretedby stromal cells have been documented.11 There is evi-dence that the secretion of IL-1β by leukemic cells canstimulate the release of G-CSF and GM-CSF from en-dothelial cells, which, in turn, may affect the prolifera-tion of leukemic blasts.36 Hepatocyte growth factor(HGF) or scatter factor, a pleiotropic cytokine involvedin hepatocyte morphogenesis, is secreted by marrow stro-mal cells and, in conjunction with other growth factors(GM-CSF, IL-3), can augment the growth of committedprogenitors through interaction with its receptor, c-met,found on CD34(+) cells.37,38 Alone, HGF appears to se-lectively stimulate AML blast colony growth and pro-
Table 3. Possible interactions between leukemic and stromalcells.
• Promotion of leukemic cell growth
Inhibition of apoptosis
Blockade of differentiation
Stimulation of proliferation
Growth stimulation by hepatocyte growth factors (HGFs)
• Inhibition of leukemic cell growth
Induction of differentiation
Inhibition of proliferation
• Inhibition of stromal cell growth
Adapted from Duhrsen and Hossfeld.11

Hematology 2002 77
motes migration of leukemic cells.39
In addition to cytokines, stromal cells release otherfactors that may influence the behavior of leukemic cells.For example, SDF-1, a chemokine and chemoattractantinvolved in the homing of stem cells, may affect leuke-mic cell trafficking.40 SDF-1 appears to selectively at-tract FAB M4/5 AML blasts that express the chemokinereceptor CXCR4, a mechanism that may, in part, explainthe marrow and tissue infiltration in this AML subtype.41
Changes in Cellular Composition of the HMStudies comparing bone marrow biopsies obtained fromAML patients with those from normal donors revealnormal numbers of macrophages but increased numbersof alkaline phosphatase–positive stromal cells and en-dothelial cells.42 Increased microvessel density in un-treated AML correlates with vascular endothelial growthfactor (VEGF) and VEGF receptor levels on AML blasts.The frequency of endothelial cells normalizes when re-mission is achieved,43,44 suggesting that AML blasts maystimulate endothelial cell growth in a paracrine fashionand change the HM in AML. AML blasts also secreteplatelet-derived growth factor (PDGF),45 a potent mito-gen for marrow stromal cells,46 possibly accounting forthe increased frequency of stromal cells in active AML.An alternative explanation is that marrow adipocytestransdifferentiate to fibroblastic cells in response to othersignals.11,47
A more detailed understanding of the compositionand characteristics of the AML stromal population hasbeen provided by studying the long-term marrow cul-ture adherent layer, arguably the best in vitro model ofthe HM, than has been possible by conducting investi-gations in vivo. Stromal layers generated from patientswith AML frequently appear abnormal because the ad-herent population either is sparse or absent, or appearsmorphologically disorganized. Several studies show thatfibroblast progenitors (CFU-F) are reduced in frequencyin the majority of patients with AML48 but can be re-stored to normal levels during remission.49 The reduc-tion in CFU-F levels is probably not due to dilution byhigh numbers of leukemic blasts. Macrophages andadipocytes can also be reduced in AML adherent lay-ers.50 Although the elaboration of inhibitors of stromalprogenitors by AML blasts has been proposed as an ex-planation,51 mechanisms are needed to reconcile the ob-servation of increased stromal cells in biopsy specimensand the report that CFU-F levels can be increased dur-ing leukemia relapse.52 At least one consequence of re-duced cellularity in the stromal population has been docu-mented. Mayani and colleagues showed that for some pa-tients with AML, the reduced frequency of CFU-F corre-lated with reduced M-CSF levels in the adherent layer con-
ditioned medium.50 Of interest, AML layers with normalcellularity had normal levels of CFU-F and M-CSF.
A Functional Defect in AML Stromal LayersThe inability of AML stromal layers, including thosewith normal cellularity, to adequately support normalhematopoiesis is well documented.50 However, fibroblas-tic cells derived from AML cultures free of macrophageshave a normal capacity to support committed progenitorgrowth.53 Suppression of hematopoiesis is attributed, inpart, to the production of TNF-α and possibly, prostag-landin E by macrophages.54 More recent work shows bycytogenetic analysis that the macrophages in AML long-term culture adherent layers are part of the leukemicclone in some cases but that defective support of normalhematopoiesis is also observed, with layers lacking leu-kemic adherent cells.55 There is defective differentiationof normal CD34(+)CD38(–) cells but not the more com-mitted CD34(+)CD38(+) cells,56 suggesting selective in-hibition of primitive hematopoietic precursors.
Functional defects in stromal layers may also be dueto abnormalities in cytokine production, in addition tothe reduced M-CSF and increased TNF-α levels de-scribed previously. Constitutive expression of IL-1β, IL-6, and G-CSF transcripts was observed in AML but notin normal adherent layers.57 Leukemia inhibitory factor(LIF) protein levels were significantly raised in AMLstromal layer supernatants compared with normal con-trols.58 The significance of raised LIF levels in this con-text is unclear, but in conjunction with marrow stromalcells, the factor is known to enhance the proliferation ofCD34(+)CD90(+) cells.59
It is evident that the interactions of hematopoieticand stromal growth factors and inhibitors on normal andleukemic hematopoiesis in in vitro cultures are highlycomplex. Results from different studies may be difficultto compare because culture conditions and cellular com-position of the stromal layers can vary significantly, es-pecially for levels of stromal macrophages. The relativeproportions of cellular components of primary stromallayers are rarely reported but may have a profound ef-fect on cytokine profiles, making comparisons of stud-ies with cloned stromal lines even more problematic.Caution should also be exercised in evaluating studiesthat report cytokine messenger RNA versus protein lev-els because of the uncertain significance of subliminallevels of the former.
Interaction of Leukemic Cells withStromal Extracellular Matrix
AML cells can adhere to a wide variety of ECM compo-nents.23 As discussed above, AML blasts adhere to ECMfibronectin and laminin through the β1 (VLA-4, VLA-

78 American Society of Hematology
5, VLA-6) and β2 (LFA-1) integrins found on the leu-kemic cells.22,24 Antibody-blocking studies show that thisonly partially accounts for the adhesion24 and that othermechanisms, including CD44 binding, have been in-voked.23 Many more CD44 variants are expressed on leu-kemic than on normal hematopoietic cells, and they mayaffect the interaction of AML cells with stroma.60 A 67-kd receptor present on CD14(+)CD11a(+) AML cellswith monocytic morphology but absent on normal bonemarrow cells mediates specific adhesion to laminin andrepresents a novel mechanism for the interaction of leu-kemic cells with the HM.61
The reduced apoptosis of AML cells documentedin several in vitro models is not restricted to stromal cellcontact-mediated mechanisms. The adhesion of c-kit(+)AML blasts to fibronectin is enhanced by SCF, whichaugments the fibronectin/VLA-5-mediated inhibition ofapoptosis and increases leukemic cell proliferation.62
Evidence for Malignant Stromal CellsAlthough stromal macrophages have been shown to beof leukemic origin,55 definitive studies demonstrating thatnonhematopoietic stromal cells are part of the neoplas-tic clone are lacking. Moreover, marrow stromal cellsfrom patients with the best-characterized multipotentstem cell disorder, Ph(+) chronic myeloid leukemia, arePh negative.63 However, in a study of patients with my-eloproliferative disorders heterozygous for glucose-6-
Figure 1. Interactions of normal and leukemic progenitors with the hematopoietic microenvironment (HM).
Abbreviations: SDF, stromal-derived factor; VLA, very late antigen; LFA, lymphocyte function antigen; CAM, cell adhesion molecule; VCAM,vascular adhesion molecule; SCF, stem cell factor.
phosphate dehydrogenase, the nonhematopoietic stro-mal cells from adherent layers of long-term marrow cul-tures were derived from the same clonal progenitors in-volved in the multipotent stem cell disorder.64 In con-trast, the stromal cells were nonclonal in a patient with arestricted-type AML (clonal granulopoiesis and non-clonal erythropoiesis). Further investigation is neededwith better clonal markers and methodologies that com-pletely eliminate contaminating stromal macrophages totest this intriguing possibility, especially in light of re-cent work demonstrating stem cell plasticity. Nonethe-less, the paucity of positive data makes the possibilitythat there are malignant stromal cells less likely; or atleast these cells are at best probably uncommon.ConclusionIt is evident that leukemic cells interact with the HM atmany levels and mimic the action of normal early pre-cursors to a variable extent, as summarized in Figure 1and Table 4. Like normal cells, AML blasts adhere tostromal cells and ECM components, but in contrast, theymay receive additional protection from endogenousapoptotic mechanisms or apoptosis-mediated chemo-therapy. In situ, leukemic cells may proliferate in re-sponse to any, or all, of the adhesive interactions withstromal cells, ECM components such as fibronectin andlaminin, or local gradients of cytokines in the HM thatare secreted by stromal cells, are generated by autocrinemechanisms, or are found in association with glycosami-

Hematology 2002 79
noglycans (for example, heparan sulfate). Aberrant ex-pression of CAMs on leukemic blasts may account fordifferent patterns of trafficking and possibly the clinicalpresentation of AML subtypes. In these many steps thereare opportunities for therapeutic intervention. One ap-proach that merits further investigation is to develop waysto interfere with the suppression of the apoptosis of AMLcells that is mediated in particular by ECM fibronectin.
II. STEM CELL TRANSPLANTATION IN ACUTE MYELOID
LEUKEMIA IN THE YOUNGER ADULT
Anthony H. Goldstone, MD,* and Irit Avivi, MD
Despite the fact that 70-80% of patients with AMLachieve complete remission (CR) most of them eventu-ally relapse and die of the disease.1,2 Once remission hasbeen achieved, further intensive therapy is needed toprevent relapse. Patients under the age of 60 have threemain options after going into remission: intensive che-motherapy (IC), autologous stem cell transplantation(ASCT), or allogeneic stem cell transplantation (alloSCT) of some kind.3-8 Patients with standard-risk diseasehave traditionally been referred for a matched sibling
allograft if a donor is availableand the patient’s performancestatus is adequate. In recentyears, chemotherapy post-remission has improved patients’outcome, with a 50% 5-yearoverall survival (OS) and 40%disease-free survival (DFS) simi-lar to that achieved with alloSCT, narrowing the differencesbetween chemotherapy and suchtransplants8 (MRC AML 12,Burnett et al, unpublished data,2002) (Figure 2). The high mor-tality of allograft (20%-25%),even today, is therefore makingit less attractive in comparisonwith chemotherapy, and selec-tion of patients to be given al-lografts in first remission (CR1)should be done very carefully.
Risk Group DesignationAlthough all patients included in
these studies were diagnosed with AML, there is a con-siderable variation in their risk of relapse. Cytogeneticsis now considered the most powerful single prognosticfactor.9,10 Whilst patients with t(15;17) or chromosomalabnormalities involving the core binding factor [t(8;21)and inversion 16] are classified as having a favorableprognosis, with approximately 30% risk for relapse, pa-tients with abnormalities in chromosome 5/7/3(q–) ormultiple chromosomal abnormalities have an approxi-mately 75% chance of relapse (Table 5).9,11 However,most patients do not belong to these two categories andare classified as having standard-risk disease (5-year OS= 43%).9,11
Recent studies have indicated that an internal tan-dem duplication (ITD) in the FLT3 gene may adverselyaffect clinical outcome in AML patients.12
Prevention of Relapse in Young PatientsA significant reduction in relapse rate (RR) has beenobserved since the introduction of intensified post-remission therapy.1,3 The Cancer and Leukemia GroupB (CALGB) randomly assigned 596 patients in CR1 toreceive 4 courses of cytarabine at 1 of 3 doses.1 High-dose cytarabine (18 g/m2/course) was demonstrated tobe superior to 2 g/m2/course, with a DFS of 44% versus29% (P = 0.003) and an OS of 52% versus 40% (P = 0.02).1
A different consolidation regimen, containing nomore than 1 g/m2 cytarabine rather than high dose, hasbeen successfully used by the Medical Research Coun-
* University College Hospital, Grafton Way, London WC1E6AU, United Kingdom
Dr. Goldstone is a consultant to Roche and Novartis.
Table 4. Adhesion molecule interactions.
Adhesion Molecule Location Ligand Location
β1 integrinsα4β1 (VLA-4) HP Fibronectin (CS1 domain) ECM
AML cells VCAM-1 MSC
α5β1 (VLA-5) HP Fibronectin (RGD sequence) ECM
β2 integrins
LFA-1 HP, AML cells ICAM-1 MSC, EC
Mac-1 DHC, AML cells
Selectins
E-selectin EC Sialylated sugar moieties; Myeloid cells,Lewis x; CD44 T cells
P-selectin EC, platelets P selectin, Myeloid cells,glycoprotein ligand 1 (CD162) AML blasts
L-selectin HP Glycoprotein ligand HP, AML blasts
CD44 HP Hyaluronic acid ECM
AML blasts Fibronectin
Abbreviations: VLA, very late antigen; HP, hematopoietic progenitors; AML, acute myeloidleukemia; VCAM, vascular adhesion molecule; ECM, extracellular matrix; MSC, marrow stromalcells; LFA, lymphocyte function antigen; DHC, differentiated hematopoietic cells; ICAM,intercellular adhesion molecule; EC, endothelial cells.

80 American Society of Hematology
cil (MRC) AML 10 trial (DFS and OS were 43% and40%, respectively).2,3
The number of postremission chemotherapy coursesrequired is unresolved. Recent data of the MRC AML12 trial seem to show no advantage for 4 consolidationscompared with 3.13
Main prospective trialsSeveral prospective trials ofpostremission therapies have been de-signed to evaluate the efficacy of thethree treatment options: chemotherapy,HSCT, and allo SCT.3-8,13
EORTC/GIMEMA AML 8 trial(1986-1991)The European Organization for Re-search and Treatment of Cancer(EORTC) and the Gruppo ItalianoMalattie Ematologiche Malignedell’Adulto (GIMEMA) LeukemiaCooperative Groups5 carried out thefirst published prospective randomizedstudy designed to review the best con-solidation therapy for AML patients(Table 6).5
AML patients 10 to 45 years old who entered CRafter 1 or 2 courses of induction were then treated withintermediate cytarabine dose (6 g/m2/course + amsacrine,–1 course), followed by allograft if matched related do-nor was available, or randomized to ASCT versus che-motherapy (cytarabine 16 g/m²/course + daunorubicin)if no donor was available. There was no significant sur-
Table 5. Risk group definition used by transplant trial groups.
MRC MRC EORTC/GIMEMA EORTC/GIMEMAAML 10 AML 12 Intergroup AML 8 AML10 GOELAM
Good M3, t(15;17) Good karyotype, t(15;17) with CR first course t(15;17) M2, M3 + WBC < 30†
t(8;21) irrespective of other: inv(16) + FAB M2/M3/M4e inv 16inv(16) % blasts post-first t(8;21) without t(8;21)
course del(9q) or complex M1/M4 + WBC < 25†
Standard Not good or Not good or +8, –y, +6, CR first course with Normal or -y M0, 1, 2, 4, 5, 6, 7poor poor del (12p) unfavorable FAB or and WBC < 30†
normal WBC > 25†
CR > first course withfavorable FAB +WBC < 25
Poor –5/del(5q) Poor karyotype, –5/del(5q), CR > first course Not good or M0, 1, 2, 4, 5, 6, 7,del(7q) 3q–, or >15% blasts –7/del(7q), FAB 5, 6, 7 standard and WBC > 30†
complex ≥ 5 post-first course inv(3q), 11q, M1, 2, 3, 4abnormalities 20q, 21q, 17p, + WBC > 25†
del(9q), t(6.9),t(9;22)complex ≥ 3abnormalities
Abbreviations: MRC, Medical Research Council; AML, acute myeloid leukemia; EORTC/GIMEMA, European Organization for Research andTreatment of Cancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; GOELAM, Groupe Ouest Est Leucemies AiguesMyeloblastiques; inv, inversion; del, deletion; CR, complete remission; FAB,French-American-British classification of acute leukemias; M,myeloid leukemia subtype; WBC, white blood cells.†Number of WBC × 109/L.
Figure 2. MRC AML 12: Overall survival on a donor versus no donor basis.
Figure presented with permission of Prof. Burnett, chairman of the MRC AML trials.

Hematology 2002 81
Tab
le 6
. Maj
or r
and
om
ized
pro
spec
tive
tria
ls.
Po
stre
mis
sio
n T
her
apy
Inte
nt t
o T
reat
/Act
ual
ly T
reat
ed +
Reg
imen
Stu
dy
Gro
up
,N
o. o
f Pat
ien
tsB
efo
re R
ecei
vin
g th
eR
and
om
-T
ime
of
Year
s,A
ttai
ned
CR
/A
ssig
ned
Th
erap
yiz
ed,
Ran
do
m-
Ag
e ra
ng
eTo
tal P
atie
nts
Ind
uct
ion
(IC
/AS
CT
/Allo
SC
T)
%iz
atio
nA
llo S
CT
Ass
ign
ed IC
AS
CT
EO
RT
C/G
IME
MA
623/
941
Dau
noru
bici
nC
ytar
abin
e 6
g/m
2 /co
urse
63A
fter 2
(or 3
)14
4/16
810
4/12
6 (8
3%)
95/1
28
AM
L 8
(66%
)45
mg/
m2
+ a
msa
crin
eco
urse
s†(8
6%)
cyta
rabi
ne 1
6 g/
m2 /
cour
se(7
4%)
1986
–199
1+
cyt
arab
ine
+ da
unor
ubic
in10
-59
y20
0 m
g/m
2
1-2
cour
ses†
EO
RT
C/G
IME
MA
1445
/203
8A
nthr
acyc
line
Ant
hrac
yclin
e +
cyt
arab
ine
67A
BM
T v
s19
8/29
2N
o ar
m fo
r onl
y IC
87/1
46-B
M
AM
L 10
(71%
)+
cyta
rabi
neP
BS
CT
(68%
)99
/146
-PB
11/1
993–
12/1
999
+ et
opos
ide
16-6
0 y
GO
ELA
M36
7/51
7C
ytar
abin
eC
ytar
abin
e 50
0 m
g/m
2 /co
urse
61A
fter 2
(or 3
)73
/88
71/7
8 (9
1%)
75/8
611
/199
3–12
/199
9(7
1%)
200
mg/
m2
+ a
msa
crin
e fo
r pt a
ssig
ned
allo
SC
Tco
urse
s†(8
3%)
amsa
crin
e +
eto
posi
de(8
7%)
15-5
0 y
+ an
thra
cycl
ine
or(1
-2 c
ours
es)†
cyta
rabi
ne 2
4 g/
m2 /
cour
se+
ant
hrac
yclin
e fo
r pt a
ssig
ned
AS
CT
/IC
MR
C A
ML
1016
09/1
966‡
DAT
or A
DE
All
patie
nts
rece
ived
3 a
dditi
onal
34A
fter 3
rd25
7/41
9N
o ad
ditio
nal
126/
190
1988
–196
6(8
3%)
(1-2
cou
rses
)†co
urse
s of
DAT
/AD
E (1
), M
AC
E (1
),co
urse
(61%
)(4
cou
rses
pre
viou
sly)
(66%
)<
55
yM
IDA
C (1
) if t
hey
had
not
rece
ived
2 c
ours
es a
lread
y
MR
C A
ML1
2n
= 3
459
2 co
urse
s of
MA
CE
(1)
Afte
r 3rd
Stil
l un-
Ran
dom
izat
ion
betw
een
1 an
dS
till u
n-19
95–2
002
(85%
CR
)A
DE
vs
MA
Eco
urse
publ
ishe
d2
addi
tiona
l cou
rses
:pu
blis
hed
< 6
0 y
repl
aced
by
ICE
vs
S-D
AT v
s H
-DAT
ICE
follo
wed
by
MID
AC
Inte
rgro
up s
tudy
518/
740
Cyt
arab
ine
All
patie
nts
who
ach
ieve
d C
R60
Afte
r 2 (o
r 3)
92/1
1310
6/11
763
/116
1990
–199
5(7
0%)
100
mg/
m2
rece
ived
1 c
ours
e of
cour
ses†
(81%
)(9
1%)
(54%
)16
-55
y+
idar
ubic
incy
tara
bine
500
mg/
m2 /
cour
secy
tara
bine
+ id
arub
icin
36 g
/m2 /
cour
se
Abb
revi
atio
ns: C
R, c
ompl
ete
rem
issi
on; I
C, i
nten
sive
che
mot
hera
py; A
SC
T, a
utol
ogou
s st
em c
ell t
rans
plan
tatio
n; a
llo S
CT,
allo
gene
ic s
tem
cel
l tra
nspl
anta
tion;
EO
RT
C/G
IME
MA
, Eur
opea
nO
rgan
izat
ion
for R
esea
rch
and
Trea
tmen
t of C
ance
r/G
rupp
o Ita
liano
Mal
attie
Em
atol
ogic
he M
alig
ne d
ell’A
dulto
; AM
L, a
cute
mye
loid
leuk
emia
; AB
MT,
aut
olog
ous
bone
mar
row
tran
spla
ntat
ion;
PB
SC
T, p
erip
hera
l blo
od s
tem
cel
l tra
nspl
anta
tion;
BM
, bon
e m
arro
w; P
B, p
erip
hera
l blo
od; G
OE
LAM
, Gro
upe
Oue
st E
st L
euce
mie
s A
igue
s M
yelo
blas
tique
s; M
RC
, Med
ical
Res
earc
h C
ounc
il;D
AT, d
auno
rubi
cin,
cyt
arab
ine,
thio
guan
ine;
AD
E, c
ytar
abin
e, d
auno
rubi
cin,
eto
posi
de; M
AC
E, a
msa
crin
e, c
ytar
abin
e, e
topo
side
; MID
AC
, mito
xant
rone
, cyt
arab
ine;
CT
X, c
yclo
phos
pham
ide;
TB
I,to
tal b
ody
irrad
iatio
n; M
AE
, mito
xant
rone
, cyt
arab
ine,
eto
posi
de; S
-DAT
, dau
noru
bici
n, c
ytar
abin
e 10
0/m
2 /d,
thio
guan
ine;
H-D
AT, d
auno
rubi
cin,
cyt
arab
ine
200/
m2 /
d, th
iogu
anin
e; B
u, b
usul
fan.
† S
econ
d co
urse
has
bee
n gi
ven
to p
atie
nts
who
faile
d to
ach
ieve
CR
with
firs
t ind
uctio
n.
‡196
6 pa
tient
s en
tere
d th
e st
udy,
but
som
e w
ere
take
n ou
t bec
ause
of i
ncor
rect
dia
gnos
is; 8
3% o
f the
pat
ient
s w
ith c
onfir
med
AM
L at
tain
ed C
R.

82 American Society of Hematology
vival advantage for allo SCT compared with chemo-therapy or ASCT (4-year OS for allo SCT = 59%, ASCT= 56%, IC = 46%). However, both allo SCT and ASCTseemed to have a better antileukemic effect comparedwith chemotherapy, reflected by reduced RR and im-proved DFS (Tables 7 and 8). It seems that these resultsreflect the relatively poor outcome observed with che-motherapy, rather than being a true “superior effect” ofthe transplant (alloSCT/ASCT) itself.
EORTC/GIMEMA AML 10 trial (1993-1999)Building on their previous study, the EORTC/GIMEMAconducted another study, aiming to clarify the role ofASCT versus allo SCT. Patients who achieved CR re-ceived IC followed by allo SCT if they had a matchedrelated donor, or ASCT if no matched related donor wasavailable.7
Nearly half of the patients without a donor hadASCT. A donor versus no donor analysis revealed a re-duced RR with increased DFS in the donor group (RR:31.2% vs 52.9%, P = 0.0001; DFS = 51.4% vs 41.2%, P= 0.046). A survival advantage was most noticeable inpoor-risk patients who had a donor (4-year DFS = 43.8%vs 19%; OS = 50.4% vs 27.7%).
GOELAM trialIn the Groupe Ouest Est Leucemies Aigues Myelo-blastiques (GOELAM) trial, patients with de novo AMLwho entered CR were assigned to allograft if they had amatched related donor and were less than 40 years old.6
All other patients were consolidated with 1 chemotherapycourse of high-dose cytarabine and anthracycline. At thisstage, patients were randomized to receive ASCT, or asecond course of IC, containing amsacrine and etoposide
(Table 6).Despite the relatively low mortality rate
(22%) reported in those assigned to allograft,there was no increase in OS compared with pa-tients without a donor, partially because of thehigh RR (37%) observed in the donor group.There were no significant differences in DFSand OS between patients assigned to ASCT ver-sus IC (Table 8). However, ASCT was associ-ated with high myelotoxicity, especially pro-longed thrombocytopenia (109.5 days withASCT vs 18.5 days with IC).
MRC AML 10Between 1988 and 1996, 1966 patients with denovo or secondary leukemia were recruited tothe MRC AML 10 trial. All patients were treatedwith 4 chemotherapy courses and were then as-signed to allo SCT if they had a matched re-lated donor (n = 419) or randomized betweenASCT (n = 190) and no further treatment (n =
191) if no donor was available(Table 6).3,8 Analysis of donor ver-sus no donor groups revealed a sig-nificantly higher transplant-relatedmortality (TRM) rate in the donorgroup: 19% versus 9% (P < 0.001).Twenty-four percent (62/255) of thepatients who received allo SCT diedin remission, compared with 17 of156 (11%) who had a donor avail-able but did not receive an al-lograft.8 Relapse risk was signifi-cantly lower and DFS was signifi-cantly higher in the donor groupcompared with the “no available
Table 7. Recent trials evaluating allogeneic transplant on a donor versusno donor basis.¶
Disease-Free OverallSurvival* (%) Survival* (%)
Trial Donor No Donor Donor No Donor
EORTC/GIMEMA AML 8 46 33† 48 40
GOELAM 44 38 53 53
MRC AML 10 50 42 55 50
Intergroup 43 35 46 52
EORTC/GIMEMA AML 10 51.4 41.2‡ 58 49.4
Abbreviations: EORTC/GIMEMA, European Organization for Research andTreatment of Cancer/Gruppo Italiano Malattie Ematologiche Malignedell’Adulto; AML, acute myeloid leukemia; GOELAM, Groupe Ouest EstLeucemies Aigues Myeloblastiques; MRC, Medical Research Council.¶ All patients having a donor available were regarded as transplant recipients.
* % at 4 years or beyond† P = .01‡ P = .046
Table 8. Prospective trials of autologous transplantation in adults.
Disease-Free OverallRelapse* (%) Survival* (%) Survival* (%)
Trial Autograft Chemo Autograft Chemo Autograft Chemo
EORTC/GIMEMA AML8 40 57 48 30 56 46
GOELAM NA NA 44 40 50 55
MRC AML 10 37 58 53 40 57 45P = 0.0007 P = 0.04
Abbreviations: EORTC/GIMEMA, European Organization for Research and Treatment ofCancer/Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto; AML, acute myeloidleukemia; GOELAM, Groupe Ouest Est Leucemies Aigues Myeloblastiques; NA, notapplicable; MRC, Medical Research Council.
* % at 4 years or beyond
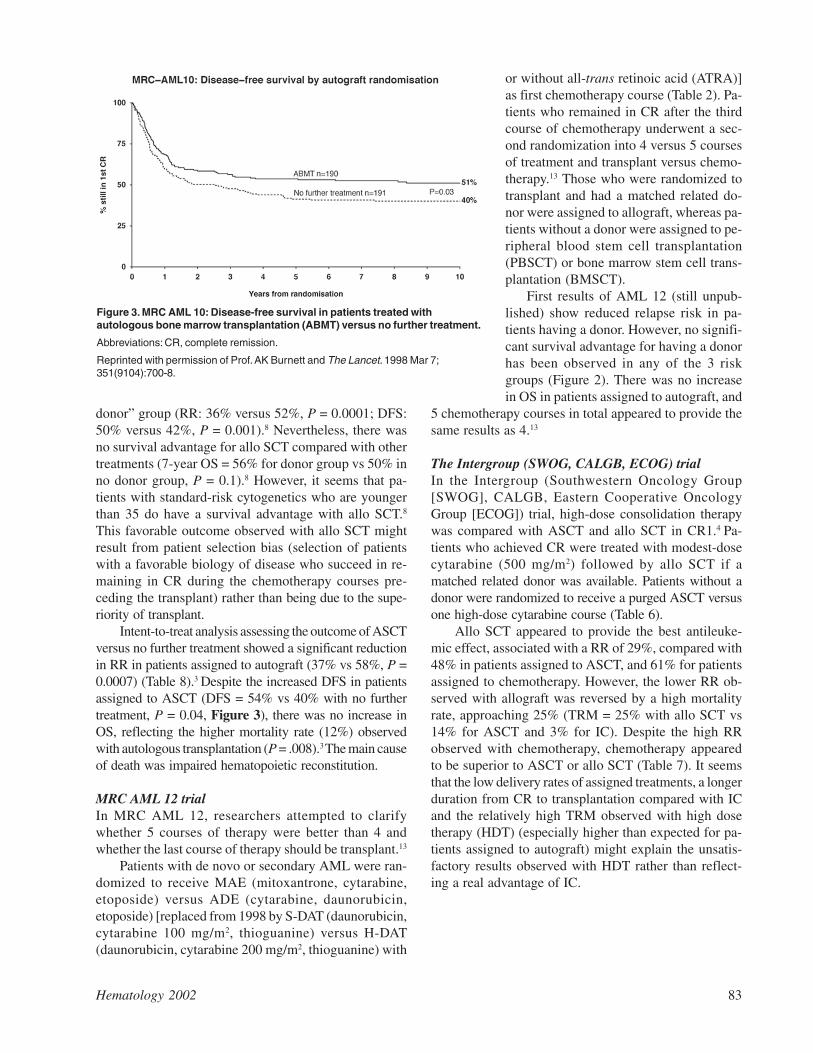
Hematology 2002 83
donor” group (RR: 36% versus 52%, P = 0.0001; DFS:50% versus 42%, P = 0.001).8 Nevertheless, there wasno survival advantage for allo SCT compared with othertreatments (7-year OS = 56% for donor group vs 50% inno donor group, P = 0.1).8 However, it seems that pa-tients with standard-risk cytogenetics who are youngerthan 35 do have a survival advantage with allo SCT.8
This favorable outcome observed with allo SCT mightresult from patient selection bias (selection of patientswith a favorable biology of disease who succeed in re-maining in CR during the chemotherapy courses pre-ceding the transplant) rather than being due to the supe-riority of transplant.
Intent-to-treat analysis assessing the outcome of ASCTversus no further treatment showed a significant reductionin RR in patients assigned to autograft (37% vs 58%, P =0.0007) (Table 8).3 Despite the increased DFS in patientsassigned to ASCT (DFS = 54% vs 40% with no furthertreatment, P = 0.04, Figure 3), there was no increase inOS, reflecting the higher mortality rate (12%) observedwith autologous transplantation (P = .008).3 The main causeof death was impaired hematopoietic reconstitution.
MRC AML 12 trialIn MRC AML 12, researchers attempted to clarifywhether 5 courses of therapy were better than 4 andwhether the last course of therapy should be transplant.13
Patients with de novo or secondary AML were ran-domized to receive MAE (mitoxantrone, cytarabine,etoposide) versus ADE (cytarabine, daunorubicin,etoposide) [replaced from 1998 by S-DAT (daunorubicin,cytarabine 100 mg/m2, thioguanine) versus H-DAT(daunorubicin, cytarabine 200 mg/m2, thioguanine) with
or without all-trans retinoic acid (ATRA)]as first chemotherapy course (Table 2). Pa-tients who remained in CR after the thirdcourse of chemotherapy underwent a sec-ond randomization into 4 versus 5 coursesof treatment and transplant versus chemo-therapy.13 Those who were randomized totransplant and had a matched related do-nor were assigned to allograft, whereas pa-tients without a donor were assigned to pe-ripheral blood stem cell transplantation(PBSCT) or bone marrow stem cell trans-plantation (BMSCT).
First results of AML 12 (still unpub-lished) show reduced relapse risk in pa-tients having a donor. However, no signifi-cant survival advantage for having a donorhas been observed in any of the 3 riskgroups (Figure 2). There was no increasein OS in patients assigned to autograft, and
5 chemotherapy courses in total appeared to provide thesame results as 4.13
The Intergroup (SWOG, CALGB, ECOG) trialIn the Intergroup (Southwestern Oncology Group[SWOG], CALGB, Eastern Cooperative OncologyGroup [ECOG]) trial, high-dose consolidation therapywas compared with ASCT and allo SCT in CR1.4 Pa-tients who achieved CR were treated with modest-dosecytarabine (500 mg/m2) followed by allo SCT if amatched related donor was available. Patients without adonor were randomized to receive a purged ASCT versusone high-dose cytarabine course (Table 6).
Allo SCT appeared to provide the best antileuke-mic effect, associated with a RR of 29%, compared with48% in patients assigned to ASCT, and 61% for patientsassigned to chemotherapy. However, the lower RR ob-served with allograft was reversed by a high mortalityrate, approaching 25% (TRM = 25% with allo SCT vs14% for ASCT and 3% for IC). Despite the high RRobserved with chemotherapy, chemotherapy appearedto be superior to ASCT or allo SCT (Table 7). It seemsthat the low delivery rates of assigned treatments, a longerduration from CR to transplantation compared with ICand the relatively high TRM observed with high dosetherapy (HDT) (especially higher than expected for pa-tients assigned to autograft) might explain the unsatis-factory results observed with HDT rather than reflect-ing a real advantage of IC.
Figure 3. MRC AML 10: Disease-free survival in patients treated withautologous bone marrow transplantation (ABMT) versus no further treatment.
Abbreviations: CR, complete remission.
Reprinted with permission of Prof. AK Burnett and The Lancet. 1998 Mar 7;351(9104):700-8.

84 American Society of Hematology
Main Problems Interpreting Resultsof Prospective Trials
There remain some problems with interpreting the re-sults of major prospective AML trials.14 Patients whoreceive allo SCT are clearly selected, because a propor-tion of patients with a matched sibling donor do not re-ceive a transplant. Patients may be excluded from trans-plant because of early relapse/death, previous compli-cations with chemotherapy, or other medical problems.It is not possible to predict the direction of such biases14
and indeed they are at the core of problems with inter-preting registry data.
• On intent to treat basis, “crossover” between armsand/or a failure to receive intended treatment, mayin some circumstances radically underestimate dif-ferences between arms, if happening to a signifi-cant degree.
• Both compliance and randomization were quite poorin most prospective trials (Table 6). As a result, bothbeneficial and harmful effects might be underesti-mated.
• Allo SCT is usually delayed, which might lead togreater selection of patients with favorable diseasewho remain in CR until transplant.
• A large number of patients are needed for a differ-ence in the efficacy between postremission therapyoptions to be detected. For example, to detect a 10%difference in survival from 40% to 50% (P = 0.05with 90 power), 1000 patients are needed.14
Allo SCT in AMLWith chemotherapy producing a less than 20% chanceof survival before the early 1980s, durable survivals upto 50%, with a low relapse risk of 15-25%, were reportedin patients receiving allografts in CR1.15,16 Today, theDFS and OS in nontransplanted patients begin to achievethis rate of durable survival and make such treatmentcomparable to that achieved with allograft3,8 (MRC AML12, unpublished data).
Allo SCT in the main prospective trialsAll trials confirmed allo SCT to be the best antileuke-mic treatment, associated with a relapse risk of 24%-36%compared with 46%-61% observed with ASCT/IC.3-8,17
However, almost all prospective studies failed to showan improved OS in patients assigned to allo SCT (seeTable 7).
The MRC AML 10 trial observed a survival advan-tage for patients treated with allograft compared withpatients treated with IC who had no available donor. 8
However, the survival of patients who had a donor but
were eventually treated with only IC was inferior to thesurvival of not only allografted patients but also the “nodonor” group. These data indicate that patients who even-tually received the transplant were biologically selectedto have a favorable prognosis, as patients with poorerprognosis did not get the transplant. However, theEORTC/GIMEMA AML 10 trial has recently reporteda higher survival rate in poor-risk patients assigned toallograft (OS = 50.4% versus 27.7%).7
Summarizing the data regarding allograft in CR1 inAML patients remains difficult:
• None of the trials is truly prospective with full bio-logical assignment based on donor availability assurrogate for intent-to-treat analysis.
• Pretransplant chemotherapy varies in its number ofcourses in some trials (Table 6). This variability inthe number of courses might affect transplant out-come in relation to both toxicity and time to treat-ment bias.
• All studies had problems in delivering the assignedtreatment (Table 6). This might underestimate bothits efficacy and toxicity.
• The superiority of allo SCT depends upon compari-son with the best available IC, but the best availableIC was not always used in every trial (Table 6).
• Upper age limit for transplant will affect outcome,as toxicity increases with age, probably more thanwith chemotherapy treatment alone (Table 6).
• Most of the major studies were initiated more than10 years ago. The currently improved HLA-match-ing stem cell transplant technology and supportivecare may now make many of the toxicity figuresmeaningless. PBSCT may also reduce relapserisk.18,19 The problem remains, however, that bigstudies with a large number of patients take a num-ber of years to conduct, and during that period vari-ous aspects of treatment can change radically.
• Varying RRs of different risk groups mean thattherapy should be tailored according to each indi-vidual patient’s risk.
BMSC or PBSC for Allo SCT?The main reason for the increasing use of PBSC relieson the rapid hematopoietic recovery observed with PBSCcompared with BMSC.18,19 A few studies have also re-ported improved immune reconstitution using PBSC.18,20
However, PBSCT might be associated with increasedincidence/severity of acute graft-versus-host disease(AGVHD) and chronic graft-versus-host disease(CGVHD).21 Champlin et al, on behalf of the EuropeanGroup for Blood and Marrow Transplantation (EBMT)

Hematology 2002 85
and the International Bone Marrow Transplant Registry(IBMTR) working committee, have retrospectively com-pared the outcome of patients treated with PBSCT (n =288) and BMSCT (n = 536).21 Incidence of CGVHD,but not AGVHD, appeared to be higher in patients whoreceived PBSCT. Patients with advanced disease at trans-plantation (AML patients in second remission [CR2] andchronic myelogenous leukemia patients in acceleratedphase) seemed to have reduced TRM and increased leu-kemia-free survival (LFS) when receiving PBSC com-pared with BMSC.21 However, these advantages werenot observed in AML patients receiving PBSCT in CR1.21
Conversely, Russell et al have reported an improved DFSin patients who received PBSCT in CR1.22
Randomized trials confirmed an improved engraft-ment with PBSCT compared with BMSCT.19 The SeattleGroup randomized 172 patients with hematologicalmalignancies (including 21% AML patients) to receivePBSCT versus BMSCT.19 Despite the improved DFSobserved with PBSCT compared to that with BMSCT(2-year RR: 14% vs 25%, P = 0.04, DFS: 65% versus45%, P = 0.03), there was no significant increase in OS(OS = 66% versus 54%, P = 0.06). TRM, though lowerin patients treated with PBSC, was still high (21% ver-sus 30%, hazard ratio 0.7; 95% confidence interval 0.38-1.28; P = 0.24). In contrast to the higher incidence ofCGVHD reported in some retrospective studies,21 therewas no significant difference in the incidence ofAGVHD/CGVHD in this prospective trial. Further stud-ies comparing PBSCT and BMSCT and allowing longerfollow-up are needed before any one stem cell sourcecan be deemed superior.
T-cell depletionGVHD is a major cause of mortality and morbidity afterallo SCT, and removing the T lymphocytes from the do-nor bone marrow can decrease the incidence of both.However, unselected T-cell depletion may theoreticallyincrease RR (preventing the graft-versus-leukemia ef-fect) and enhance treatment-related infections due todelayed immunological recovery.
Nevertheless, it seems that T-cell depletion may notnecessarily increase relapse risk in AML allografted pa-tients, though decreasing GVHD.23,24
However, it is still unclear whether T-cell depletioncan provide any benefit beyond reducing GVHD andimproving the quality of life.23,24
Low-intensity stem cell transplantionLow-intensity stem cell transplantation (LI SCT) is be-ing increasingly used, aiming to exploit the curative po-tential of allo SCT by inducing graft-versus-tumor ef-fect without the morbidity and mortality associated with
conventional transplantation. Low-intensity SCT is lesstoxic and therefore may be considered for some patientswho are otherwise not eligible for conventional alloge-neic transplant. However, its efficacy in AML patientshas still not proven to be as good as that of high-inten-sity allo BMT. The EBMT reported on 69 patients (me-dian age 51) treated with LI BMT for AML ormyelodysplastic syndrome (MDS).25 More than half ofthe patients had refractory/relapsed disease at transplan-tation. Graft failure was more frequent than observedwith conventional allo SCT, though 77% achieved > 95%donor chimerism. Patients’ outcome was highly depen-dent on disease status at transplantation: 1-year TRM,RR, and OS were 47%, 30%, and 41% for the wholegroup, compared with 17%, 21%, and 67% in patientstransplanted in CR (CR1 or later). However, follow-up wasstill short at time of publication.25
Peggs et al have recently reported 44% progression-free survival (PFS) and 53% OS (median follow-up, 18months) in 24 patients aged 18-60 years (median 47)treated with low-intensity matched related/unrelated SCT(total body irradiation, melphalan, fludarabine, Campath-1H) for MDS/AML (n = 17).26 DFS and OS were higherin AML patients transplanted in CR1 (n = 15), approach-ing 57% and 62%, respectively.26 Longer follow-up andprospective comparison with IC are needed in order todefine the role of LI SCT in selected AML patients. TheMRC AML 15 trial intends to allow the possibility of LISCT in patients aged 35-45 years who have a matchedrelated donor, while suggesting conventional transplantfor the younger recipients.
Autologous Stem Cell Transplantation
ASCT in CR1On the assumption that HDT has value in AML, severalgroups conducted nonrandomized trials using ASCT asconsolidation therapy in CR1.27-29 Encouraging results,observing a reduced RR with improved DFS comparedwith chemotherapy, were reported.27-29 Patient selection,however, might have played a role.
Does stem cell purging improve patient’s outcome?Despite the superior DFS observed with an ASCT com-pared with chemotherapy,27-29 RR remained higher thanobserved with allo SCT and the main cause of death wasrecurrence of disease. Some investigators have tried topurge stem cells pretransplantation, aiming to reduce RRand improve OS.28,30,31However, results were vari-able.4,30,31 Furthermore, pretransplant purging may carrya risk of loss of accessory and progenitor cells, resulting indelayed hematopoietic and immunological reconstitution.

86 American Society of Hematology
ASCT in prospective randomized trialsOne of the main unsolved questions is whether there isany place for ASCT in CR1 and which patients (if any)should be transplanted. All three prospective trials de-signed to compare IC with ASCT 4-7 observed a reducedRR in patients assigned for autograft, reflecting its su-perior antileukemic effect compared with chemotherapy(Table 8).4-7 The MRC AML 10 trial, comparing ASCTwith no further treatment, reported a significantly in-creased DFS in patients assigned to ASCT (Figure 3).3
Failure to deliver assigned treatment, observed with alltrials (Table 6), might underestimate the real efficacy ofASCT. All prospective trials failed to show a survivaladvantage in patients assigned to autograft comparedwith chemotherapy. The high mortality rate reported withASCT (12%) offsets the antileukemic advantage pro-vided with autograft. In addition, some patients who re-lapsed postchemotherapy were salvaged with HDT.3,5
However, the TRM associated with ASCT is no longertoday significantly higher than that observed with IC. Areduced TRM might therefore be translated into in-creased OS.
ASCT for patients in second or later CRASCT might have a place in rescuing patients who haverelapsed postchemotherapy, with up to 20-50% relapse-free survival in selected patients.3,5,32-34
Linker et al34 have recently reported a 5-year DFSof 54% in patients with advanced leukemia (patients withprimary induction failure who remitted with salvagetherapy/patients in CR2 or later CR) who were treatedwith high dose cytarabine/etoposide consolidation, fol-lowed by an ASCT.34
It is still unclear if patients in CR2 or later without adonor should be referred to matched unrelated donor(MUD) transplantation instead of ASCT. A prospectivecomparison of MUD versus ASCT in this situation isvery difficult because time to treatment might be delayedand some MUD patients may not be in true remissionwhile others, proposed autograft, might fail to obtain anadequate stem cell harvest.
BMSCT or PBSCT in ASCT?Retrospective studies observed a faster hematopoieticrecovery, associated with a lower morbidity and mortal-ity (TRM = 5%) in patients treated with PBSCT com-pared with BMSCT.28,29
Most studies did not show any differences in RR orDFS between stem cell sources.35-37 The EORTC/GIMEMA AML 10 trial randomized patients with noavailable donor to BMT (n = 146) or PBSCT (n = 146).37
There were no significant differences in DFS and OSbetween the two groups (4-year DFS = 49.8% with
BMSCT vs 42.6% with PBSCT, P = 0.33; OS = 55% vs55.3%, respectively).37 It is still unclear whether PBSCTprovides any other advantage compared with BMT, ex-cept of facilitating stem cell engraftment.
Outcome of Allo SCT in Different Risk Groups
Good-risk patientsAll prospective studies3,5-8 except the Intergroup trial4,10
failed to show a survival advantage with allo SCT inCR1 in patients with favorable cytogenetics. It appearsthat the Intergroup result might reflect a random resultrather than a genuine tendency, because of the smallnumber of patients included.
Acute promyelocytic leukemia (APL) patients, but notpatients with t(8:21) or inversion 16, were reported to havea significantly lower RR with allograft in CR1 (donor vsno donor analysis: 22% vs 43%, P = 0.02).8 However, itseems that since the introduction of ATRA, nontransplanttherapy can provide the same good result. The only studythat observed a survival advantage in good-risk patientstreated with autograft was the Intergroup study.4,10 How-ever, this “superior” outcome might actually reflect thepoor results achieved with chemotherapy.4 It now seemsthat there is no place for allo SCT or ASCT in CR1 inpatients with favorable-risk cytogenetics.
Standard-risk patientsBoth the MRC AML 10 trial3,8 and the EORTC/GIMEMA AML 10 trial7 reported reduced RR and im-proved DFS in patients with standard-risk disease as-signed to allo SCT. However, it is still unclear whetherallo SCT can improve patients’ OS (MRC AML 12, un-published data).7
There is still significant heterogeneity in this groupof patients and RR is influenced (independent of cyto-genetics) by response to first induction and the presenceof the FLT3 mutation. It is possible that some of thesestandard-risk patients (e.g., those who express the FLT3mutation or failed to remit with first course of induc-tion) will do better with allo SCT, while others will gainno advantage from being transplanted.
Poor-risk patientsResearchers from the EORTC/GIMEMA AML 10 trialhave recently reported their current results.7 When pa-tients are divided into risk subgroups based on their cy-togenetics, it seems that patients with unfavorable cyto-genetics get the maximal benefit from having allo SCTcompared with ASCT or IC.7 Of interest, the EORTC/GIMEMA AML 10 study considered all patients with-out a favorable/normal karyotype as having poor-riskdisease (Table 5). However, the MRC AML 10,8 which

Hematology 2002 87
failed to show an advantage for allograft in poor-riskpatients but showed one for the standard-risk group, in-cluded approximately half of these patients with unfa-vorable cytogenetics in the standard-risk group ratherthan in the unfavorable-risk one.
Other Options for Transplantation for PatientsWithout a Matched Related Donor
Matched unrelated donor transplantation (MUD)MUD in CR1. Despite the controversy about the advan-tage of matched related donor allograft in CR1 in poor-risk patients,7,8,10 the very grim prognosis observed withchemotherapy (less than 30% DFS) might justify usingMUD in CR1 in this group of patients. However, thereis currently little evidence that any kind of allo SCT cancure large numbers of these poor-risk patients.
MUD in CR2. Patients with unfavorable cytogeneticswho achieve a second CR and have no matched relateddonor are often referred to MUD SCT. However, MUD inCR2 in patients with favorable-/standard-risk cytogenet-ics remains controversial and its superiority compared withASCT is still questionable.38-40 Lazarus et al, on behalf ofthe IBMTR, have retrospectively compared the outcomeof AML patients (CR1/CR2) treated with MUD versusASCT between 1989-1996.39 Three-year LFS was 33%in MUD patients versus 40% with an autograft. How-ever, long-term side effects were significantly higher inMUD patients and selection bias is unknown.
On the basis of these incomplete retrospective data,it may be reasonable to consider MUD in young patientswith adverse cytogenetics and short CR1s who have amatched unrelated donor (at least 10 antigen matching).40
Conversely, patients older than 40 with a long CR1 maydo better with ASCT, if their disease is in genuine re-mission and enough cells can be harvested.40
T-replete versus T-depleted MUD. The reducedGVHD-related mortality achieved with T-cell depletionis often balanced by increased RR and overwhelminginfections.
In contrast to patients with some other malignan-cies, AML patients may achieve a survival advantagewith T-depleted marrows compared with T-replete ones,40
justifying T cell depleted MUD in selected AML pa-tients. Nevertheless, data justifying this are still scantyand further studies are needed to support this strategy.
Haploidentical BMTHaploidentical BMT is an option for patients who donot have a matched related donor (approximately 70%of patients). The historical data concerning haploidenticalBMT in AML patients were disappointing. These dis-appointing results might be attributed to patient selec-
tion for transplant (advanced disease) as well as to ahigh rate of transplant-related complications, mainlyGVHD, which in turn was replaced by a high incidenceof graft failure as T-cell depletion has been introducedto prevent GVHD. T-cell depletion by itself was associ-ated with delayed immune recovery, resulting in highincidence of severe infections. However, it seems thatrecent modifications have succeeded in making someprogress. Stem cell megadose (106 CD34 cells/kg) isessential to overcome the HLA barrier in full haplotype-mismatched transplants.41 Further reduction of T-celldose infused reduces the frequency and the severity ofGVHD significantly. Posttransplant granulocyte colony-stimulating factor (G-CSF) appears to interfere with natu-ral killer (NK) cell recovery and has therefore been ex-cluded.42 Donor’s NK cell alloreactivity, a unique phe-nomenon of mismatched transplants, appears to play anessential role in preventing relapse and supporting stemcell engraftment.43 A recent update from Perugia sug-gests that the current morbidity and mortality in AMLpatients having haploidentical BMT is not higher thanreported with matched allogeneic BMT.44 Event-freesurvival in high-risk patients transplanted in CR1/CR2approached 45%, with an RR of less than 15%.44 A do-nor versus recipient NK cell alloreactivity is essentialfor achieving graft-versus-tumor effect and may there-fore become a major criterion for donor selection inmismatched SCT.
Acute Promyelocytic LeukemiaNone of the large randomized trials showed a survivaladvantage with ASCT/allo SCT in CR1 compared withthe outcome achieved with ATRA-containing regimens.However, for patients in second CR, there might be anadvantage with both kinds of transplants.45 An achieve-ment of molecular remission pre-autograft is essentialand is associated with a high cure rate.45,46 Conversely,patients transplanted with evidence of minimal residualdisease (MRD) (morphological remission without mo-lecular remission) have high risk for relapse46,47 andshould therefore be considered for an alternative therapysuch as allo SCT or arsenic trioxide.45,47 Postautograftmaintenance therapy with ATRA might further reduceRR, although this has to be confirmed.
Options for Treatment in Elderly PatientsThe age-related differences in biology of disease partlyexplain the poor outcome observed in patients older than60 than in younger patients. However, undertreatmentmight also contribute to these inferior results.
Postremission therapy with high-dose cytarabinefailed to improve the outcome of patients older than 60and has been associated with high toxicity.1

88 American Society of Hematology
The advantage of ASCT in CR1 is not proven ei-ther, though some investigators have reported an im-proved survival with ASCT;48,49 however, a selection biasmight be involved. Allo SCT, being associated withhigher TRM in elderly patients (especially GVHD re-lated), is even less feasible. Deeg et al50 have recentlyreported a nonrelapse mortality of 39% in 50 MDS pa-tients (16 with transformation to leukemia) aged 55-66years (median 58.8) treated with conventional allo SCT.However, encouraging results with low-intensity SCTin elderly AML patients have recently been reported.26
It appears that stem cell transplantation (ASCT or alloSCT), being often associated with high toxicity, will notbe the routine treatment in elderly patients. Overcomingthe drug resistance that frequently exists in elderly pa-tients with AML has remained one of the main chal-lenges to treating them.
Minimal Residual DiseaseDetection of MRD posttherapy, aiming to identify pa-tients who are at higher risk for relapse, remains a majorchallenge.51,52 Molecular methods (polymerase chainreaction [PCR]),51,52 as well as immunological methods(multi-parametric flow cytometry analysis)52,53 have beenemployed. Whereas immunophenotype analysis can beinformative in 80%-85% of AML patients, molecularanalysis can be useful in less than 30% of patients, asonly the minority of AML patients express a traceblemolecular marker (e.g., AML1/ETO, PML/RARα, andpossibly the FLT-3 mutation).51,52 There are some patientswhose leukemic cells present chromosomal abnormali-ties that can be monitored with fluorescence in situ hy-bridization (FISH).51 However, FISH is usually less sen-sitive than molecular monitoring, and the sensitivity ofit may be inadequate.51,52 Furthermore, 30%-50% ofAML patients have normal karyotype.
The significance of a positive MRD finding is notalways clear and a different level of residual diseaseseems to be significant for different types of AML.54,55
Tobal et al reported a tenfold difference in MRD levelsbetween patients in long-term remission from APL54
compared with patients with t(8;21).55 Molecular remis-sion in APL patients seems to be associated with long-term DFS, whereas failure to achieve a molecular re-mission does not.45-47,56,57 In contrast, a positive molecu-lar result in patients with t(8;21) who obtained clinicalremission is not necessarily associated with impendingrelapse.51,58 APL patients who achieved a molecular re-mission have an excellent prognosis having IC only.45-47
However, data are still too scanty to apply this strategyto other AML subtypes, and further studies includingquantitative monitoring should be investigated.
Conclusions and Future DirectionsDespite the contributions of large prospective trials com-paring the efficacy of allo SCT with that of IC or ASCT,it is still unclear what the best postremission therapy isfor AML patients.
ASCT in CR1It appears that patients in the favorable-risk group donot require HDT in CR1, as their outcome with chemo-therapy is excellent.8 There is no clear evidence that stan-dard-risk patients are doing better with allo SCT thanwith IC either (MRC AML 12 unpublished data, Figure2). With the lack of major benefit for allo SCT, the poorerquality of life often observed post allograft should beconsidered.59 Patients will need to decide whether in-creasing their chance of survival by a few percentagepoints is worth the potential long-term morbidity. Also,issues of infertility will be perceived differently by thepatients in different age groups and those with differentfamily circumstances.60
The role of allo SCT in CR1 in poor-risk patientsremains unclear,8 although some studies have showedsome advantage compared with chemotherapy.4,7,10
However, all these large prospective studies haveused BMSC, whereas recent studies have shown a pos-sible reduced RR with PBSCT compared withBMSCT.18,19 Prospective studies comparing PBSCT withBMSCT are needed to clarify these issues.
Better understanding of prognostic factors (e.g.,FLT3 status, MRD status) might help in selection of thepatients who will benefit from allo SCT. The number ofchemotherapy courses needed pretransplantation is un-clear61 and might be influenced by patients’ risk group(including evaluation for MRD) and planned HDT.
New kinds of allo SCT based on reduced chemo-therapy doses (LI SCT) or T-cell depletion allograft maysucceed in reducing TRM observed with conventionalallo SCT and allow transplantation to be considered inpatients who are not eligible for conventional allograft,including elderly patients.
ASCT/IC in CR1The role of ASCT in CR1 is not clear. All prospectivetrials showed reduced RR in autografted patients. How-ever, survival was not higher than observed with che-motherapy, mainly because of the high TRM reportedwith ASCT. A current prospective study, including a largenumber of patients, comparing IC with PBSCT andBMSCT is needed. Improving the outcome achieved withchemotherapy remains one of the main challenges to beaddressed by current studies. Attempts to overcome che-motherapy resistance, which is most noticeable in eld-erly patients, need to be ongoing.

Hematology 2002 89
Figure 4. Eastern Cooperative Oncology Group (ECOG) protocol trial overview.
Abbreviations: Ara-C, cytarabine; CR, complete remission; HDAC, high-dose Ara-C; PBSC, peripheral blood stem cell.
Figure 5. AML 15 protocol flowchart.
Abbreviations: AML, acute myeloid leukemia; DAT, daunorubicin, cytarabine, thioguanine; CR, complete remission; Mab,monoclonal antibody;FLAG, fludarabine, cytarabine; Ida, Idarubicin; allo SCT, allogenic stem cell transplantation; ARA-C, cytarabine.

90 American Society of Hematology
Future studiesIn the absence of any clear evidence for the role of bothallo SCT and ASCT in CR1, the main trial groups havedesigned different directions for future research. ECOGis soon starting a new treatment protocol retaining theoption for autotransplant compared with allo SCT andchemotherapy. Chemotherapy intensification will be fur-ther investigated, including an increased dose of dauno-rubicin and combining anti-CD33 with chemotherapy(Figure 4).
In contrast, the MRC AML15 trial has decided toomit ASCT, having seen no evidence yet of a survivaladvantage with ASCT. Patients with standard- or poor-risk disease who are younger than 35 and have a matchedrelated donor will receive a PBSC/BMSC allograft. Theiroutcome will be compared with that of patients with noavailable donor, treated with further chemotherapycourses (Figure 5). Patients 35-45 years old who have amatched related donor can receive a conventional al-lograft, or an LI BMT, depending on investigator or pa-tient choice (but not randomized).
Hopefully, new ongoing prospective trials will givea better idea about the current value of allo SCT com-pared with ASCT and IC in adult AML patients. How-ever, if nontransplant options continue to improve at agreater rate than transplant options, then selection of ap-propriate patients for transplant will become more andmore difficult.
III. BIOLOGIC AND GENETIC RISK ASSESSMENT OF
AML IN THE GENOMIC ERA
Cheryl L. Willman, MD*
In the past two decades, important scientific advanceshave yielded new insights into the epidemiologic, ge-netic, and biologic features of the AMLs.1-6 Yet despitethese scientific advances, the majority of patients affectedby AML still die of their disease.6 With the exception ofacute promyelocytic leukemia (APL), we have not yetsucceeded in translating our scientific discoveries intomore effective treatments for the majority of AML pa-tients. While therapeutic intensification and improvedsupportive care have led to gradual improvements inoutcome in children and younger adults with AML (par-ticularly those with more favorable cytogenetic abnor-malities), overall survival in this age group still ap-proaches only 50%.6 In older individuals (> 55-60 years)and in secondary AML patients, in whom resistance tocurrent therapies and an increase in unfavorable cytoge-netics characterizes the disease, the outlook is even moredismal, with overall survivals of 10% to 15%.7-14 As the
majority of individuals affected by AML are in this olderage group,15 we thus lack effective therapeutic ap-proaches for the majority of patients with this disease.New laboratory and clinical approaches that can be suc-cessfully translated into more effective diagnostic andtherapeutic strategies are desperately needed for AML.We must define the most important questions to advanceour knowledge and focus on the discovery of new thera-peutic strategies for AML patients with high-risk as wellas low-risk disease. Molecular genetics and gene expres-sion profiling hold promise to improve AML disease clas-sification systems, to model gene expression profilesassociated with chemoresistance or response to variousagents, and to identify novel therapeutic targets.16-23 Thissection will briefly review preliminary accomplishmentsand ongoing studies in this field of investigation.
AML Prognostic Factors 2002:Age, Antecedent Disease, Cytogenetics
From the past two decades to the present day, the mostimportant prognostic factors in AML have remained: (1)patient age; (2) presenting white blood cell count; (3)whether the AML presents clinically as de novo diseaseor secondary to antecedent myelodysplasia (MDS) orleukemogenic therapies (therapy-related AML, t-AML);and (4) the presence of specific cytogenetic abnormali-ties, usually clustered as “favorable,” “intermediate,” or“unfavorable” (Table 9).1,3,6 While AML accounts foronly 13% to 14% of leukemia cases in the first 10 yearsof age (the majority being acute lymphoblastic leuke-mia or ALL), AML constitutes nearly 36% of the leuke-mia cases in older children.15 In adults, AML rates be-gin to rise exponentially after 50 years of age; age-spe-cific incidence rates are 3.5 per 100,000 in adults 50years of age, increasing significantly to 15 at age 70 and
* UNM Cancer Research and Treatment Center, 2325 Caminode Salud, N.E., Albuquerque, NM 87131-5636
Acknowledgement of Research Support: DHHS NIHU01CA32012, U01CA60433, U01CA30969, R01CA186026and the W.M. Keck Foundation, Los Angeles, Calif.
Acknowledgments: I would like to thank my colleaguesassociated with the Southwest Oncology Group and thePediatric Oncology Group/Children’s Oncology Group, whowith many colleagues throughout the world have madesignificant contributions to both scientific and clinicaladvances in leukemia. I would also like to thank my colleaguesand research team at the University of New Mexico (UNM)Cancer Research and Treatment Center, the UNM HighPerformance Computing Education and Research Center, andSandia National Laboratories, whose collaborations have beenessential and critical for our genomic studies of acute leukemia.

Hematology 2002 91
35 at age 90. The mean age of AML in the United Statesis 63 years. Currently accounting for 10% to 20% of allAML cases, the incidence of secondary AML appearsto be increasing due to the use of DNA damaging agentsin cancer therapy at higher dose intensities and an in-creased length of survival for many cancer patients.24
Each of the broadly defined groups of AML (AMLin infants and children, AML in younger adults, AMLin older individuals, and secondary AML) is character-ized by distinct but overlapping cytogenetic and molecu-lar genetic abnormalities (Table 9). Although “favorable”cytogenetic abnormalities [such as t(8;21), inv(16), or
t(15;17)] are more frequently seen in children andyounger adults less than 55 years of age, it should benoted that these “good prognosis” cases still constitute aminority (< 35%) of the AML cases seen in this agegroup. In contrast to AML in younger patients, AML inolder individuals is more frequently associated with unfa-vorable cytogenetic abnormalities (such as –7, 7q–, –5,5q–, or complex karyotypes) and other poor prognosis bio-logic features (trilineage dysplasia suggestive of ante-cedent myelodysplasia or marrow injury and an increasedfrequency of intrinsic drug resistance).7-14,25 The signifi-cantly poorer outcome seen in older AML patients is, in
Table 9. Cytogenetic classification for risk grouping in acute myeloid leukemia (AML).*†
Cytogenetic Frequen cy Frequency Genetic and BiologicAbnormality in Children in Adults Fusion Genes Modifiers of Outcome
“Favorable” Cytogenetics
t(8;21)(q22;q22) 12% 5-8% (< 55 yrs), Rare (> 55 yrs) AML1/ETO FLT3 ITD (9%) + del(9q),+ Complex
Inv(16)(p13q22) 12% 10% (< 45 yrs), Rare (> 45 yrs) CBFβ/MYH11 FLT3 ITD (7%)t(16;16)(p13;q22)
t(15;17)(q21;q11) 7% 15% (< 45 years), Rare (> 45 years) PML-RARα FLT3 ITD (37%)Variants:t(11;17)(q23;q11) PLZF-RARαt(5;17)(q32;q11) NPM-RARαt(11;17)(q13;q11) NuMA-RARα
“Intermediate” Cytogenetics
+8 Rare 10% FLT3 ITD (28%)
Normal karyotype 10-15% 15-20% FLT3 ITD (34%)MLL ITD (10%)
Others: –Y, +6,All other karyotypes not FLT3 ITD (20-30%)considered favorable or unfavorable
“Unfavorable” Cytogenetics
Abn 11q23 > 50% of 5-7% MLL FLT3 ITD (0%)Common Variants: infant AML casest(4;11)(q21;q23) 7% t(9;11) MLL/AF4t(9;11)(p22;q23) MLL/AF9t(11;19)(q23;p13.1) MLL/ELLt(11;19)(q23;p13.3) MLL/ENL
t(6;9)(p23;q34) Rare <1% DEK/CAN Inv(3)(q21q26),
t(3;3)(q21;q26) 3% 3-5% Ribophorin/EVI1 FLT3 ITD (17%)
–5/del(5q) Rare <10% (< 45 yrs) >10% (> 45 yrs) MDR drug resistanceFLT3 ITD (0%)
–7/del(7q) 10% <10% (< 45 yrs) >10% (> 45 yrs) MDR drug resistanceFLT3 ITD (7%)
Others: 20q, 21q, del (9q), t(9;22), abn 17p, complex karyotypes (≥ 3)
*Adapted from References 1, 2, 11, 24.†It should be noted that this classification scheme is that used by the US Cooperative Groups.24 The MRC11 uses an altered classificationscheme in which abn11q23, del(9), del(7q) without other abnormalities and complex abn ≥ 3 but ≤ 5 are considered “intermediate” risk, not“unfavorable” as shown in this table. Awareness of these differences in cytogenetic risk grouping is critical for comparison of outcome onclinical trials conducted by these groups.

92 American Society of Hematology
part, explained by a higher frequency of these unfavor-able cytogenetic abnormalities and biologic features.7-14
While younger and older AML patients define the ex-tremes of response, the majority of patients affected byAML in all age groups fall into the “intermediate” cyto-genetics category, in which risk classification and strati-fication are currently ill defined. While large clinical tri-als have demonstrated that AML-associated cytogeneticabnormalities confer important prognostic information,striking differences in therapeutic response and outcomemay still be observed in leukemias with the same cyto-genetic profile, implying that other more subtle geneticabnormalities and functional activation or inactivationof critical cellular pathways (signal transduction abnor-malities, apoptosis, drug resistance, angiogenesis, cellcycle regulation, DNA repair) also impact disease biol-ogy and therapeutic response. Although all of these criti-cal cellular pathways provide potentially important thera-peutic targets for AML, we must determine how to besttarget genetically and biologically heterogeneous AMLpatients to each specific therapy. While karyotype pro-vides critical prognostic information for AML patientsin the context of current therapeutic approaches, it re-mains to be determined whether morphologic and cyto-genetic classification schemes are the best approach tothe classification of AML, particularly when consider-ing targeted therapy. This is a question that can be di-rectly tested using gene expression profiling.
Gene Expression Profiling in AMLMany investigators throughout the world are engaged inthe use of microarray technology to derive gene expres-sion profiles in the acute leukemias and other diseases.The goals of many of these studies are to understand theintrinsic biology of these heterogeneous diseases, to de-termine whether expression profiling can improve diag-nosis and risk classification of the leukemias, to deter-mine if expression profiles can be derived that predictfor clinical outcome and response or resistance to cur-rent therapies, and to use expression profiles to identifynew therapeutic targets. Such goals will require obtain-ing gene expression profiles on large, statistically de-signed cohorts of well-characterized leukemic patientsin whom detailed cytogenetics, biologic covariables, andclinical outcome parameters are known and available.As described in several reviews and original reports,16-23
gene expression profiles may be derived using both oli-gonucleotide or cDNA microarray technology. Methodshave been derived that allow for the amplification andhybridization of small amounts of RNA,26 allowing geneexpression profiles in AML and normal hematopoieticstem and progenitor cells to be compared. Although geneexpression profiling studies in leukemia and MDS are
still in their infancy, several preliminary studies havebeen reported. Miyazato and colleagues19 isolated AC133surface marker-positive hematopoietic stem cells from5 AML, 2 secondary AML, and 3 MDS patients; devel-oped cDNA libraries; and compared gene expressionprofiles on custom oligonucleotide arrays containingapproximately 1100 gene sequences. Although theseauthors attempted to identify genes more strongly asso-ciated with MDS (Dlk, Tec, inositol 1,4,5-triphosphatereceptor 1) and AML (solute carrier family members,opioid receptor delta 1, and the leptin receptor), the ex-pression of each of these genes was very heterogeneousand did not appear to precisely distinguish these relateddiseases. Focusing on AML, Guzman and colleagues20
compared the expression of 1400 genes in CD34+/CD38–
cells isolated from 7 AML patients and from 3 normalbone marrow controls. Two tumor suppressor genes,IRF1 and death-associated protein kinase (DAPK), andseveral other genes (AML1, Af-4, EWS, Ikaros, andSTAT6) were shown to be overexpressed in all AMLprogenitor samples relative to controls. Whether the ex-pression of these genes can be used as markers of AMLvs normal progenitor cells remains to be determined inlarger confirmatory studies.
Not surprisingly, the results and conclusions fromeach microarray study for gene expression profiling de-pend on the characteristics of the patient sample set, howthe questions to be addressed using microarrays wereposed, the available clinical and laboratory data, and themethods employed for computational analysis of geneexpression data.23 To date, the computational analysis ofgene expression has centered on two different ap-proaches. Unsupervised learning approaches, oftentermed “class discovery” or “clustering,” are used touncover inherent “clusters” or biologic cohorts within adata set. Such “agglomerative” algorithms may be usedto cluster a group of patients based on the similarity oftheir aggregate expression profiles or to alternativelycluster genes that are related to a specific biologic pa-rameter. A number of methodologies have been em-ployed for clustering, including hierarchical clusteringas applied originally to microarray data sets by Eisenand colleagues,23 self-organizing maps (SOM) first ap-plied to the distinction of AML and ALL by Golub etal,16 k-means, and principal component analysis (PCA).Unsupervised learning approaches have the advantageof being unbiased and allow for the identification ofstructure in large and complex data sets. However, be-cause many different relationships or clusters may bepossible in complex data sets, there may not be one“right” answer, a fact that is rarely appreciated by cur-rent readers and reviewers of the literature. Addition-ally, the clusters derived may not reflect important bio-

Hematology 2002 93
logical or clinical parameters.In contrast to unsupervised learning tools, supervised
learning methods, also termed “class predictors,” usevarious computational algorithms to model or “fit” a dataset in order to be able to predict a specific label (such asAML vs ALL, remission vs failure, the presence of aspecific cytogenetic abnormality vs not). In supervisedlearning, a data set is usually divided into a “training”set in which the parameter to be predicted is known tothe analyst who then focuses on defining genes and pro-files that can be used to predict for this parameter. Heldin reserve, and blinded to the analyst, is a set of “test”cases in which the predictive profiles developed fromthe training set can be tested. One of the problems withsupervised learning is that sample labels must be accu-rate and precise, which can be difficult in heterogeneousdiseases. Another frequent problem with supervisedlearning approaches is that it is very easy to “over-fit”the data on a training set, resulting in an inability to makeaccurate predictions on the test set. Many supervisedlearning methods have been employed for the analysisof gene expression profiles, including Bayesian networks(P. Helman, R.Veroff, S. Atlas, C. L. Willman, unpub-lished data) and Support Vector Machines with Recur-sive Feature Elimination (SVM-RFE).27 These compu-tational approaches require significant computationalresources and parallel computing, particularly if predic-tive genes are to be rigorously validated. Many of thesetechniques, particularly when “leave one out” cross-vali-dation (LOCV) is performed on large data sets, requireextensive parametric studies or the solution of largematrix problems that can only be done using massivelyparallel computers. Ultimately, hypotheses generatedfrom the computational analysis of an initial microarraydata set must ideally be tested and validated on an inde-pendent sample set.
Discovery of Novel AML Classesby Using Gene Expression Profiling
Two recent studies have unexpectedly demonstrated thatgene expression profiling can identify novel intrinsicbiologic groups of AML patients that cannot be preciselydefined by traditional morphologic, immunophenotypic,and cytogenetic classification schemes. While not sur-prising in the context of solid tumors, these results wereindeed surprising in acute leukemia, in which we holddetailed knowledge of the cytogenetic and moleculargenetic features of the disease. If validated, these stud-ies hold promise to enhance and significantly alter ourdiagnostic and risk classification schemes for acute leu-kemia and to reveal unique clusters of cases that maybenefit from specific therapeutic approaches.
Therapy-related AMLGene expression profiles were recently obtained fromenriched CD34+ AML progenitor cells from 14 t-AMLpatients using oligonucleotide arrays by Le Beau andcolleagues.28,29 Unexpectedly, using hierarchical cluster-ing, these investigators found 2 intrinsic groups of t-AMLthat would not have been predicted by clinical course,morphology, or cytogenetic abnormalities. The firstgroup, containing t-AML patients with complex rear-rangements involving chromosome 5q, had relative in-creases in expression of genes involved in cell cycle orcheckpoint control (CCNA2, CCNE2, CDC2, andBUB1), cell growth (MYC), and loss of expression ofthe gene encoding interferon consensus sequence bind-ing protein (ICSBP). The second group with a distinctgene expression profile clustered several t-AML patientswith heterogeneous karyotypes including those with lossof chromosome 7, normal karyotypes, and t(3;3) ort(3;21). Despite cytogenetic heterogeneity, this group ofpatients had a similar gene expression profile with a rela-tive decrease in expression of key transcriptional regu-lators (TAL1, GATA1, and EKLF) and overexpressionof FLT3 and BCL2. These preliminary studies suggestthat expression profiling may define novel biologicgroups of t-AML not precisely related to karyotype thatmay benefit from a common therapeutic approach.
Infant AML/ALLOur studies in infant leukemia have also yielded inter-esting and unexpected results. With its distinguishinggenetic, biologic, and epidemiologic features, infantacute leukemia affords a unique investigational modelfor the study of leukemogenesis. Epidemiologic andmolecular genetic studies have confirmed that most, ifnot all, infant leukemias arise in utero,30 suggesting aprenatal exposure or initiating event. AML and ALL alsooccur with relatively equal frequency in this age group.Although the mechanism is not well understood, struc-tural rearrangements of the MLL/ALL1/HRX gene onchromosome 11q23 (hereafter referred to as MLL) ap-pear to occur more frequently during fetal development,accounting for the relatively high incidence of abnor-malities involving this gene in infant leukemia. Indeed,nearly 60% of infant leukemia cases of both ALL andAML morphologic subtypes have balanced, reciprocaltranslocations involving MLL fused to a number of dif-ferent partner genes. With an overall survival of only25%, there is an urgent need to better understand theetiology and pathogenesis of this disease in order to de-velop more effective therapies.
To improve diagnosis and risk classification and toidentify new therapeutic targets in infant leukemia, weperformed gene expression profiling using oligonucle-

94 American Society of Hematology
otide arrays (Affymetrix U-95A.v2) in a retrospectivecohort of 126 infant leukemias (78 ALL, 48 AML, 57/126 with MLL rearrangements) registered to NCI-spon-sored clinical trials. To determine whether there wereinherent biologic classes of infant leukemia, gene ex-pression profiles were correlated with a large number ofbiologic and clinical covariables using several unsuper-vised clustering methods and novel data visualizationtools (VxInsight) (Figure 6, see Color Figures, page513). For “class prediction” (AML vs ALL, MLL vs not,remission vs failure, the presence of specific cytogeneticabnormalities, and novel diagnostic and therapeutic tar-gets), several supervised learning methods were em-ployed (Bayesian networks, SVM-RFE, and Neuro-Fuzzy Logic).
VxInsight,31 a very powerful tool for class discov-ery and data visualization, was developed by our col-laborator George Davidson and colleagues at SandiaNational Laboratories in Albuquerque, NM (Figure 6,page 513).28,32 VxInsight has the capacity to cluster pa-tients or genes, using all of the gene expression datawithout having to select smaller subsets of genes foractual clustering, in a novel and intuitive way. Similargenes are clustered together spatially and represented ina 3-D terrain map, where large mountains represent largeclusters of similar genes, and smaller hills represent clus-ters with fewer genes (Figure 6, page 513). Clusters thatare the most similar (genes or patients) are also sitednearer to each other and farther away from less similarclusters. The analogy to real terrain allows memoriza-tion of the landscape, which makes subsequent explora-tions very easy. The level of detail can be changed to“fly” over the terrain and view smaller clusters withinclusters. The results of database queries can then beshown within the context of the clusters by highlightingthose genes matching the queries. VxInsight also pro-vides a simple way to select genes and display Web-based information for those genes. In addition, clinicaland laboratory covariables can be loaded into the pro-gram, and the visual display and clusters can be queriedin real time and reconfigured to show the interactionbetween the patients or genes in a cluster and these clini-cal and laboratory covariables.
When VxInsight31 was applied to our infant leuke-mia data, we discovered 3 discrete, statistically robustbiologic clusters of infant leukemia. These intrinsic bio-logic groups could not be identified using traditional di-agnostic parameters (morphology: ALL vs AML,immunophenotyping or cytogenetics: MLL rearrange-ment vs not) (Figure 7, see Color Figures, page 513).We had anticipated that we might see 2 clusters (ALLvs AML or MLL vs not) or 3 clusters (ALL vs AML vsMLL leukemias) based on the data by Armstrong et al17
suggesting that MLL is a homogeneous and distinct formof leukemia. In each cluster, an individual patient is rep-resented by a pyramid (highly similar patients in eachcluster will be overlapping in this “high level” view)(Figure 7, page 513). By querying VxInsight in “real-time,” cases previously labeled ALL vs AML were iden-tified (ALL cases are colored white and AML coloredgreen), and using analysis of variance (ANOVA), themost statistically significant genes that were unique toeach cluster were identified (Figure 7, page 513). Thesegenes serve as potentially new diagnostic and therapeu-tic targets. One cluster (n = 52) (left, VX-GB, Figure 7,page 513), predominantly lymphoid (51 ALL, 1 AML;28 MLL), was uniquely characterized by expression oflymphoid, viral, and viral-induced genes. A second het-erogeneous cluster (n = 54, 12 ALL, 42 AML; 16 MLL)(bottom right, VX-GC, Figure 7, page 513) was associ-ated with activation of DNA repair, metabolic, and RASfamily genes, suggesting environmental initiating events.The third cluster (n = 21, 16 ALL, 5 AML; 10 MLL)(top, VX-GA, Figure 7, page 513), with the highest rateof treatment failure, had a highly unique expression pro-file with activation of novel embryonal, neuronal, tumorsuppressor, vascular, and adhesion genes, as well asgenes associated with the earliest phases of hematopoie-sis. We speculate that these “leukemic” cases, never be-fore recognized or identified, represent transformedmultipotential human stem cells. Each of the clusterswe have identified may represent distinct etiologic path-ways in infant leukemia in which MLL rearrangementsact as a secondary, not a primary, transforming event.We hypothesize that identification of these novel groupsmay be important for future risk classification and thera-peutic targeting in infants. Ongoing expression profil-ing studies of large retrospective cohorts of adult de novoand secondary AML cases are currently under way inour group and others.
Discovery of Gene Expression Profiles Associatedwith AML-Associated Cytogenetic Abnormalities:New Modes of Classification andTherapeutic Targets
Trisomy 8Focusing on obtaining gene expression profiles associ-ated with trisomy 8 and with normal cytogenetics inAML, Virtaneva et al21 used oligonucleotide arrays con-taining over 6000 genes to compare expression profilesin 10 AML samples with normal cytogenetics, 10 AMLsamples with trisomy 8 (+8), and bone marrow controls.While all AML samples had different gene expressionprofiles than normal CD34+ cells, it was difficult to dis-cern differences in profiles between AML cases with

Hematology 2002 95
Table 10. Genes shared and distinctive between MLLtranslocation variants (MLL/ALL1/HRX gene on chromosome11q23) in acute myeloid leukemia (AML)/acute lymphoblasticleukemia (ALL).
Genes Sharedin AllMLL Variants t(4;11) t(11;19) t(9;11) t(10;11)
Lyn Bmi-1 HRAFY TRADD RUNX3
NFATC3 DOK1 ERH TCFL4 HMGCR
RAD9 WAS IL2Rγ COX7 HGF
CD44 PPKAR IRAK1 HSF1 CDKN1CD
AC133 YES IGBP1 AHR RARαBAG1 TIMP1 TACT PLCγ2 RARES2
CRADD ELF4 LPXN Rab33A TNFSF9
Blk JUND Serpine RASA1 GIT2
FLT3† FLT3† MAGED2 DOC1R SH3BP1
† FLT3 expression is more significantly associated with t(4;11) thanwith all MLL variants.
+8 and those with normal cytogenetics. Cases with +8clearly had increased expression of chromosome 8 associ-ated genes, reflecting increased gene dosage and down-regulation of genes involved in the control of cell death.
Favorable cytogenetics: t(8;21), inv(16), t(15;17)Schoch et al22 have used oligonucleotide arrays in 37AML patients characterized by favorable cytogeneticabnormalities. A limited number of genes (n = 36) accu-rately separated AML patients with these different cy-togenetic abnormalities. It remains to be determinedwhether an expansion of these results will lead to re-finements in cytogenetic classification schemes. Withthe limited number of patients reported to date, the ex-tent of heterogeneity in gene expression profiles in eachcytogenetic abnormality is also unknown. Whether anyof the genes identified are useful as new therapeutic tar-gets remains to be determined.
MLL rearrangementsAs discussed above, translocations involving the MLLgene at chromosome band 11q23 are the most commongenetic alteration in both ALL and AML in infants andoccur frequently in adults with both de novo and sec-ondary AML (Table 9). While the t(4;11) is most com-mon (occurring in 20%-30% of infant cases, primarilyALL), the t(9;11), t(1;11), t(11;19) in 2 variants, t(10;11),and many other translocation fusion partners may be seen(Table 9). While there is evidence that most of these chro-mosomal rearrangements carry a poor prognosis, thepathogenesis and unique gene expression profiles foreach MLL translocation variant remain undefined. Us-ing hierarchical clustering and PCA, Armstrong andcolleagues17 concluded that leukemias with MLL rear-rangements were a homogeneous and unique form ofacute leukemia, distinct from AML and ALL. Severalgenes, including FLT3, were identified as being uniquelyexpressed in MLL-associated cases.17 However, theseconclusions must be carefully considered in light of thecohort of MLL patients examined and their comparisonAML and ALL groups. Of the 37 cases tested, 17 hadMLL rearrangements, all but one of which was a t(4;11).The ALL comparison group contained a large numberof cases with t(12;21). Thus, in this context, it is per-haps not surprising that MLL cases were deemed homo-geneous and distinct from ALL cases with t(12;21) andother ALL and AML cases.
We recently used oligonucleotide arrays and highperformance computing methods for both unsupervisedand supervised data analysis to study MLL-associatedgene expression profiles from the infant leukemia co-hort, 54/126 of whom had heterogeneous MLL rear-rangements: 30 t(4;11), 10 t(11;19), 6 t(10;11), 4 t(9;11),
4 t(1;11). As mentioned earlier, in the infant cohort, thepresence or absence of MLL rearrangements did notdefine a distinct class during unsupervised learning ap-proaches (Figure 7, page 513). Using supervised learn-ing algorithms for class prediction (Bayesian networks,SVM-RFE, and Discriminant Analysis), we found thatMLL-associated gene expression profiles were quite het-erogeneous among different MLL translocation variants.In addition, in the context of an appropriate control groupof other infant cases, we found that the MLL genotypealone did not define a distinct group of leukemia cases,distinct from other ALL and AML cases arising in in-fants. Using these methods, we ordered the most statis-tically significant genes associated with those infant casescontaining vs lacking any MLL rearrangement, whichare therefore common to all MLL variants, as well asthose genes that were unique to each translocation vari-ant (Table 10). We believe that these genes representnovel diagnostic and therapeutic targets for MLL-asso-ciated disease. Although we do not agree with Armstronget al17 that MLL cases are homogeneous, it is interestingthat the lists of statistically significant genes that areshared by all MLL cases, particularly t(4;11), are virtu-ally identical in our different laboratories. This resultadds further support to the validity and reproducibilityof this approach.
Discovery of Gene Expression Profiles thatPredict for Treatment Failure in AML
Since Golub et al16 first identified genes, such as HoxA9, potentially associated with poorer outcome in AML,several groups have embarked on obtaining gene expres-

96 American Society of Hematology
sion profiles that might predict for remission or failurein acute leukemia. Modeling of these complex clinicalparameters is far more difficult than modeling other moreuniform biologic and cytogenetic features. A second goalof our studies was to identify sets of genes that could beused to predict outcome as well as therapeutic resistanceand treatment failure in AML and ALL. This is a morecomplex task, as the biologic or clinical labels for “con-tinuous complete remission (CCR)” and “failure/relapse”are not pure or precisely defined and are dependent ontime and numerous host and tumor factors. In addition,there is a tendency to “over-fit” algorithms and compu-tational approaches to make these predictions, particu-larly in leukemia data sets where the failure rate to cur-rent therapies exceeds 50%. Nonetheless, identificationof genes and pathways associated with treatment failureis essential to understanding therapeutic resistance andthe development of appropriate therapies that overcomeresistance.
In our studies, Bayesian nets and SVM-RFE haveshown classification rates in excess of 90% in discern-ing between the type classes ALL vs AML (Table 11).However, only SVM-RFE was able to identify remis-sion vs failure across the unconditioned data set with atotal error rate differing from random prediction at a sig-nificance level of P < .10. Interestingly, far greater suc-cess is obtained when remission vs failure is conditionedon the VxInsight clusters (Figure 7, page 513), ratherthan on AML vs ALL or in modeling the entire group asone biologic cohort. These results speak to the furtherbiologic validity of the novel clusters that we have iden-tified. In particular, significance levels of better than P= .01 were achieved within cluster VX-GC by the Bayes-ian nets (better than P = .05 for SVM), better than P =.10 within cluster VX-GA by both methods, and betterthan P = .175 within VX-GB by Bayesian nets. It is rather
unlikely that random chance alone would produce suchhigh accuracy levels. The results are taken as strong evi-dence that the VxInsight clusters reflect biologically realand clinically exploitable groupings of the patients. Incontrast, comparable accuracy was not achieved condi-tioning on either of the traditional criteria of ALL ver-sus AML or MLL versus not MLL.
Striking in VxInsight Group C (VX-GC, bottomright, Figure 7, page 513), containing the majority ofAML as well as a significant number of ALL cases, wasthe identification of genes and pathways associated withtreatment failure. Interestingly, nearly 100 genes weredetermined to be significantly associated with failure,and there was very significant overlap in the list of genesderived from SVM-RFE and Bayesian approaches. Themajority of the 20 most significant genes associated withtreatment failure were those involved in specific signaltransduction pathways (Table 11). We were struck bythe appearance of a series of genes that encode signal-ing proteins (Table 11), all of which could potentiallybe placed in signaling cascades leading to RAS by bothconventional and novel pathways. Other critical path-ways involved DNA repair, apoptosis, and methylation.
Kinnunen et al33 have identified a statistically sig-nificant (P < .01) associated with relapse in pediatricAML. These include genes involved in proliferation(TTK S/T kinase, CRIP1, FTH1, BUB3, S100, and CA9),transcription (PRO2000, ZNF268, JUNB, MBLR, andEGR1), cellular trafficking (TRAM, RABEX5, MYO1B,and RANBP2L1), and oncogenesis (TTK, FOS, andCD68). The ability of these genes to predict for relapseand a poorer outcome are being tested in an indepen-dent set of cases.
Therapeutic Targets and Novel ApproachesDerived from Gene Expression Profiling
Signal transduction targetsFLT3. One of the most interesting genes to emerge inthe past 2 years as a diagnostic and therapeutic target inAML is FLT3. FLT3 is a transmembrane tyrosine ki-nase growth factor receptor (a member of the PDGF,FMS, and KIT receptor family) that is selectively ex-pressed on hematopoietic cells, where it mediates stemcell proliferation and differentiation. Activation of theFLT3 receptor in AML precursor cells appears to stimu-late proliferation and inhibit apoptosis. Several recentstudies34-39 have shown that an FLT3 mutation in AML(an in-frame internal tandem duplication of exon 11 and12, termed the FLT3 ITD), results in auto-activation ofthe receptor in the absence of ligand, leading to inde-pendent activation of cell growth mediated by the RASand STAT5 signaling pathways. Based on data recently
Table 11. Genes and pathways associated with treatmentfailure in infant leukemia
Genes and Pathways Individual Genes
DNA repair pathways Helicase RecQ1RAD1
Chromatin methylation Methyltransferases
Signal transduction TGFβ signaling pathways
Ras signaling pathways Farnesyl transferasePI3 KinasePLCεRANTESRASDYRK3
TGFβ signaling pathways BMP 1, 2, 4TGFβ binding proteins
Interferon-stimulated genes

Hematology 2002 97
published by several groups, FLT3 mutations are nowconsidered one of the most common mutations in AML,increasing in frequency from 17% in pediatric AML35 to36% in older AML patients.39 Although initially associ-ated with a high WBC and normal karyotypes in AML,the FLT3 ITD has now been reported in many cytoge-netic categories in both favorable and intermediate riskgroups (Table 9).36 Although the prognostic significanceof FLT3 ITD may be blunted by more intensive treat-ment regimens,37 multivariate analysis has shown that theFLT3 ITD is a striking predictor of poor outcome in pe-diatric and adult AML.35,36 In adults, FLT3 ITD was themost significant prognostic factor in predicting relapserisk and disease-free survival, as well as overall sur-vival.36 The impact of FLT3 ITD may also be influencedby loss of the wild-type FLT3 allele; AML patients withFLT3 ITD and loss of the wild-type allele had the worstoutcomes overall.38 With the development of selectivetyrosine kinase inhibitors for FLT3-mediated signalingentering early phase clinical trials,40 there is a lot of ex-citement. Ultimately, careful consideration about howsuch selective inhibitors might be targeted with otheragents will likely be essential for increased therapeuticoutcomes in AML.
Many gene expression profiling studies have foundrelatively high levels of expression of presumably wildtype FLT3 in AML, including AML and ALL cases as-sociated with MLL rearrangements, cases of t-AML as-sociated with abnormalities of chromosomes 3 and 7,and AML cases with t(15;17).17,22 However, as the statusof the FLT3 gene was not determined in these samples,it remains possible that arrays could be detecting ex-pression of both wild-type and mutant alleles. These datalead to speculation that FLT3 might be an attractive thera-peutic target in these diseases, as well as in AML caseswith FLT3 ITD. Very preliminary data (D. Small and J.Griffin, personal communications) have revealed thatFLT3 inhibitors can inhibit signaling from overexpressedwild-type receptors as well as from mutant FLT3 recep-tors. If confirmed, the identification of high FLT3 ex-pression in certain AML cases using arrays would bethe first proof of principle that arrays can indeed iden-tify novel therapeutic targets.
RAS. RAS mutations and RAS activation occur in asignificant number of AML and MDS cases.6,41-43 Newertherapies using farnesyl transferase inhibitors (FTI) (suchas R115777; which target RAS to the plasma membrane,essential for RAS activation) or monoterpenes holdpromise for therapeutic efficacy in AML.6,43 Interestingly,no correlation was observed between RAS mutations andresponse in early phase clinical trials testing these in-hibitors,43 which suggests that RAS activation may bepresent in many patients regardless of the presence or
absence of RAS mutations (as suggested in our arraystudies). Alternatively, FTIs may impact other key sig-naling elements in the RAS signaling pathways, such asRho proteins or PI3 Kinase/AKT, which promotes cellsurvival. Our recent studies in pediatric leukemia sug-gest that perturbations in RAS signaling pathways arecommon in AML as well as in related cases of ALL(Figure 7, page 513). In many AML cases, RAS may beactivated by perturbations in upstream signaling path-ways (such as FLT3, KIT, or PDGF).
Intrinsic drug resistance, apoptosis, angiogenesis,and adhesion. Each of these biologic features of leuke-mic cells may mediate drug resistance and promote cellsurvival. Each pathway also represents an important po-tential avenue for new drug development and therapeu-tic targeting.12,13,44-46 It is hoped that as more gene ex-pression profiling data are compiled, we can determinehow these pathways are affected in each type of AMLin relation to existing cytogenetic and other prognosticabnormalities. This knowledge may allow us to appro-priately target groups of patients to more effective ex-perimental treatment approaches.
Summary and Future Scientific QuestionsExciting preliminary gene expression profiling studiesare providing new insights into the etiology and patho-genesis of AML. These studies hold promise to impactdisease classification, risk assessment, and therapeutictargeting and will likely lead to the identification of noveltherapeutic targets. To make continued progress in thesestudies, it will be essential to obtain gene expression pro-files on large, statistically designed cohorts of well-char-acterized leukemic patients for whom detailed cytoge-netics, biologic covariables, and clinical outcome param-eters are known and available. Particularly fruitful areasfor future investigation include the application of ge-nomic and proteomic technologies to understand alter-ations in the global patterns of gene expression and pro-tein function in AML, the development of computationalmethods to place genes into pathways in order to deter-mine the impact of aggregate expression profiles on cellbehavior, the development of computational methods toprioritize target selection for drug development, and thedevelopment of common database architectures so thatdata may be exchanged and shared to facilitate progressin this field.

98 American Society of Hematology
Table 13. Outcome in acute myeloid leukemia (AML) witht(8;21) by number of high-dose ara-C consolidations.*
Number of High-Dose Ara-C CoursesParameter 1 ≥≥≥≥≥ 3 P value
Relapse, % 62 19 0.004
Median CR duration (mos) 10.5 > 35
5-year DFS, % 38 61 0.03
Median survival (mos) 24 > 43
5-year survival, % 44 76 0.04
*Data from Byrd et al.26
Abbreviations: CR, complete remission; DFS, disease-free survival.
IV. NEW AGENTS AND STRATEGIES IN
ACUTE MYELOID LEUKEMIA AND
HIGH-RISK MYELODYSPLASTIC SYNDROMES
Francis J. Giles, MD,*and Hagop M. Kantarjian, MD*
With current standard induction regimens for adult pa-tients with AML, the CR rates range from 40% to 90%,and the cure rates from less than 10% to 70%, depend-ing on several characteristics, including leukemia karyo-type, patient age and performance status, and organ func-tion (Table 12).1-4 Patients with high-risk myelodysplasticsyndrome (MDS), defined here as MDS with 10% ormore marrow blasts, have a prognosis similar to that ofpatients with AML.5-7 High-risk MDS and AML havedifferent pathophysiologies (e.g., rates of apoptosis, me-thylation profiles, incidence of FLT-3 internal tandem du-plications [ITD] or mutations, aberrant angiogenesis, an-giogenic factor profiles).8-18 Some anti-AML programshave been applied successfully to high-risk MDS,1,6,19-21
while other approaches are being designed to specifi-cally target the pathophysiologic events in MDS.22-25
Herein we will review some of the novel agents underinvestigation by our group in adult patients with AMLor high-risk MDS.
Special Therapeutic Considerations in AML withFavorable Karyotypes
These include t(8;21), inversion 16, and APL, i.e.,t(15;17) and variants. In AML with t(8;21), multiplehigh-dose ara-C regimens have improved outcome. In areview of CALGB studies, 29 patients with t(8;21) re-ceived 3 or more cycles of high-dose ara-C and 21 pa-tients received only 1 cycle. The results, shown in Table13, indicated significantly lower relapse rates, better 5-year survival, and better overall disease-free survival(DFS) rates with more high-dose ara-C consolidations.26
Inversion 16 AML also benefits from high-dose ara-Cin relation to decreased incidence of central nervoussystem disease (from 30% to less than 5%). However,the importance of multiple high-dose ara-C courses, al-though possibly as significant as with t(8;21), has notbeen analyzed.
The introduction of ATRA to APL therapy has im-proved prognosis. ATRA plus chemotherapy has im-proved the cure rate from less than 40% to over 70%.27-31
Anthracyclines are very important for improved out-come,29,32 while ara-C plays little if any role.30,33 Poly-
merase chain reaction (PCR) monitoring for promyelo-cytic leukemia gene-retinoic acid receptor alpha (PML-RARα) gene is part of the management of patients ontherapy in first CR, as molecular relapses can still becured in 70% of cases.34-36 Arsenic trioxide (As
2O
3) is
important for salvage therapy and is probably superiorto ATRA.36 The updated results of the pivotal trial ofAs
2O
3 in APL salvage included 52 patients, 45 of whom
achieved CR (87%). Molecular CR was noted in 51% ofpatients at CR and in 78% after 1 consolidation course.Persistent DFS was observed in 1 of 7 patients (14%) whoreceived only 1 course of As
2O
3, in 8 of 20 patients (40%)
who received 4 or more courses of AS2O
3 with or without
other therapies, and in 11 of 12 patients (92%) who under-went allogeneic stem cell transplant (SCT) in CR.
Studies of As2O
3 plus ATRA in frontline therapy are
ongoing and may obviate the need for any chemotherapy.Gemtuzumab ozogamicin (GO; Mylotarg) is also effec-tive against APL (strong CD33 expression) and is cur-rently being investigated in combination with ATRA infrontline and salvage APL.37 In a recent M.D. AndersonCancer Center (MDACC) frontline study of ATRA + GOin APL, patients received induction with ATRA 45 mg/
Table 12. Prognosis in acute myeloid leukemia (AML) byleukemia karyotype.
Karyotype % % CR % EFS
Favorablet(8;21) 5-10 90 50-70inversion 16 5-10 90 50-70t(15;17) 5-10 80-90 70
Intermediatediploid, –Y 40-50 70-80 20-40
Unfavorable–5/–7 20-30 40 5-10+8 10 60 10-2011q23, 20q–, other 10-120 60 10
Abbreviations: EFS, event-free survival.
* M.D. Anderson Cancer Center, Department of Leukemia,1400 Holcombe Blvd., Box 428, Houston, TX 77030

Hematology 2002 99
m2 daily (D) until CR, then for 2 weeks every 4 weeksfor 9 months, and GO 9 mg/m2 intravenously (IV) onday 1 for induction, then every month. Idarubicin 12 mg/m2/D × 3 was given during induction for high-risk pa-tients (peripheral blasts more than 10 × 109/L) or if PCRpersisted as positive or reverted to positive after 3 monthsinto CR. Among 20 patients treated, 16 achieved CRincluding 14 of 17 low-risk and 2 of 3 high-risk patients.After a median follow-up of 9 months, all 16 patientsremain in CR; only 1 became transiently positive. Thisexperience suggests that future regimens in APL neednot incorporate ara-C, and, in most patients, may notrequire anthracyclines, and that long-term event-free sur-vival (EFS) would be obtained with nonchemotherapyregimens (e.g., ATRA plus As
2O
3) or without anthracy-
clines (e.g., ATRA plus GO). Novel approaches that arebeing investigated as induction and/or maintenance/con-solidation approaches include liposomal ATRA, Am80,GO, HuM195, and tetra-arsenic tetra-sulfide.37-42 The roleof SCT in patients with APL is unclear, with most re-ports based on registry data or small patient series.43 Thesevere, often fatal, coagulopathy that is often present inhigh-risk APL patients remains a serious challenge.44
Strategies to Improve Prognosis ofAML and High-Risk MDS
Current promising investigational avenues in AML-MDSinclude both traditional cytotoxic approaches and novel“targeted” or immunomodulatory therapies.
• Intensification strategies, e.g., dexamethasone,cytarabine, thioguanine, etoposide, and daunorubicin(DCTER) regimen as used in pediatric AML.45,46
• Investigating new formulations of established drugs(e.g., liposomal formulations of daunorubicin,47,48 as-paraginase,49 pegylated interferon,49 topoisomeraseI inhibitors,50 deposomal ara-C for intrathecaltherapy51).
• Investigating drugs with mechanisms of actions notfully exploited in current AML-MDS therapies. Ex-amples include:
o Topoisomerase I inhibitors (e.g., topotecan,52 9nitrocamptothecin, OSI 211,50 DE-31053)
o Nucleoside analogues54 (e.g., clofarabine,55
troxacitabine,56 decitabine57)
o Multi-drug resistance (MDR)-reversal agents(e.g., cyclosporine A,58 PSC 83359)
• Strategies directed at relatively leukemia-specificmechanisms or antigens, including:
o Targeting surface antigens (e.g., with mono-clonal antibodies such as GO)60
o Signal transduction targeting (e.g., FLT-3,61
farnesylation,62 methylation,63 angiogenesis64)
Exploring improved formulations of establishedagents that offer pharmacologic or safety advantagesA recent example of such strategies is the developmentof liposomal daunorubicin.47,48 We initially studied lipo-somal daunorubicin in a Phase I study as a single agentat doses of 75, 100, 150, and 200 mg/m2/D × 3 (225 to600 mg/m2 per course) in patients with refractory leuke-mia.48 Among 24 patients studied, the maximum toler-ated dose (MTD) was defined at 150 mg/m2/D × 3, andthe dose limiting toxicity (DLT) was mucositis. No car-diac events were noted; 2 patients achieved CR. Thesedata led to the next study, a Phase I-II study of liposo-mal daunorubicin 75 to 150 mg/m2/D × 3 plus ara-C 1 g/m2 by continuous infusion daily × 4 in 62 patients.47 Over-all 18 patients (29%) achieved CR and 7 marrow CRwith low platelets (CRp, 11%), for an overall responserate of 40%. The observed:expected CR ratio was 1.7, sug-gesting a beneficial effect. The median CR duration was14 months, and median survival 6 months. Mucositis wasDLT occurring in 4 of 9 patients at 150 mg/m2/D × 3 but inonly 2 of 32 (6%) at 125 mg/m2 and in 1 of 13 (8%) at 135mg/m2. Severe cardiotoxicity was noted in 4 patients(6%). Liposomal daunorubicin combination regimens arebeing investigated as frontline therapy in AML and ALL,at doses of 125 mg/m2/D × 3, by several groups.
Investigating agents withdifferent mechanisms of action
Combinations of topotecan + ara-C and fludarabine +ara-C. These regimens have been extensively used insalvage and frontline therapy of AML, MDS, andALL.1,6,21,65 Topotecan + ara-C has high activity and rela-tively low toxicity in advanced MDS.66 Among 59 pa-tients with MDS and 27 with chronic myelomonocyticleukemia (CMML) treated with topotecan 1.25 mg/m2
continuous infusion (CI)/D × 5 and ara-C 1 g/m2 over 2h/D x 5, the CR rate was 56% and the induction deathrate 7%. These regimens are probably equivalent toidarubicin + high-dose ara-C (IA) in AML and high-risk MDS and could be used as alternative or comple-mentary strategies (Table 14).1,6 New topoisomerase Iinhibitors under study include liposomal lurtotecan (OSI211),50 DX-8951f,67,68 and pegylated preparations(GL147211C).69
Nucleoside analogues. Clofarabine is an adenosinenucleoside analog that was designed to combine the bet-ter properties of fludarabine and chlorodeoxyadenoside(Figure 8).54,55 Phase I studies of clofarabine 2-55 mg/m2/D × 5 established the phase II dose at 40 mg/m2/D ×

100 American Society of Hematology
5; the DLTs were liver dysfunction and skin rashes. Re-sponses were noted in 6 of 35 patients (17%). In theongoing Phase II study, 39 patients have been treated.Responses in the 36 evaluable patients with refractory-relapse leukemias were encouraging (Table 15).Clofarabine is currently undergoing multi-institutionalstudies in pediatric and adult leukemias. Combinationstudies will include clofarabine + ara-C ± idarubicin.
Troxacitabine is an L isomer cytosine analogue thathas significant activity in AML and CML blastic phase(Figure 8).56,70 It has a number of metabolic features that
may allow activity in ara-C resistant AML.71-73 Deoxy-cytidine kinase (dCK), which lacks chiral specificity,catalyses the monophosphorylation of both ara-C andtroxacitabine. Deoxycytidine deaminase (dCD) is morechiral specific and cannot inactivate troxacitabine.Troxacitabine has a unique pattern of cellular uptake andmetabolism, which may also allow it to circumvent an-other mechanism of resistance to cytotoxic nucleosideanalogs. Nucleoside-specific membrane transporters(NSMT) mediate plasma membrane permeation of manynucleoside analogs including ara-C and fludarabine. Insome preclinical models, troxacitabine is transportedrapidly into cells by both equilibrative sensitive and in-sensitive nucleoside transport systems with subsequentaccumulation of troxacitabine monophosphate, diphos-phate, and triphosphate in a time- and concentration-dependent manner. As troxacitabine is not dependent onNSMT to achieve a lethal intracellular concentration, itmay thus not be susceptible to NSMT-mediated mecha-nisms of resistance to ara-C. Unlike ara-C, which lacksproportionality between ara-C TP formation and extra-cellular ara-C concentration, the formation of troxa-citabine diphosphate increases linearly with increasingextracellular drug concentration. Troxacitabine does notinhibit ribonucleotide reductase and may thus be mecha-nistically complementary to several nucleosides that arecytotoxic in part via ribonucleotide reductase inhibitione.g., Gemcitabine, FMdC, fludarabine. The pharmaco-kinetic behavior of troxacitabine is substantially differ-ent from that of other nucleoside analogs possessing aD configuration, which are characterized by rapid dis-appearance from plasma due to deamination. In contrast,troxacitabine has a long terminal half-life (82 hours) anda systemic clearance comparable to the glomerular fil-tration rate (137 mL/min).56
In a Phase I study, doses ranging from 0.72 to 10mg/m2 IV over 1 hour daily × 5 were investigated inpatients with refractory leukemia.56 Response was asfollows: AML, 3 CR + 1 PR in 31 patients (13%); MDS,
Table 14. Topotecan + ara-C and fludarabine + ara-C regimensin acute myeloid leukemia (AML).
A. Regimen Schedule
1. IA IDA 12 mg/m2/D × 3ara-C 1.5 g/m2/CI/D × 3-4
2. FAIA fludarabine 30 mg/m2/D × 5ara-C 2 g/m2 over 4 h/D × 5
3. TA Topotecan 1.25 mg/m2 CI/D × 5ara-C 1-2 g/m2 over 2-4 h/D × 5
B. Results
MedianRegimen No. % CR EFS Survival (wks)
IA 322 77 63 77
FA 600 55 40 30
TA 357 59 36 41
Abbreviations: IDA indicates idarubicin; CI, confidence interval; CR,complete remission; EFS, event-free survival.
Table 15. Preliminary results of the Phase II study ofclofarabine in refractory relapse leukemia.
No. No.Disease Evaluable CR + CRp (%)
AML 20 11 + 1 (60)
ALL 6 1 + 1 (33)
CML-blastic phase 6 3 + 1 (67)
MDS/CML 4 1 + 2 (75)
Abbreviations: AML, acute myeloid leukemia; ALL, acute lympho-cytic leukemia; CML, chronic myeloid leukemia; MDS,myelodysplastic syndrome; CR, complete remission; CRp, CR withlow platelets.
Figure 8. Nucleoside analogues.
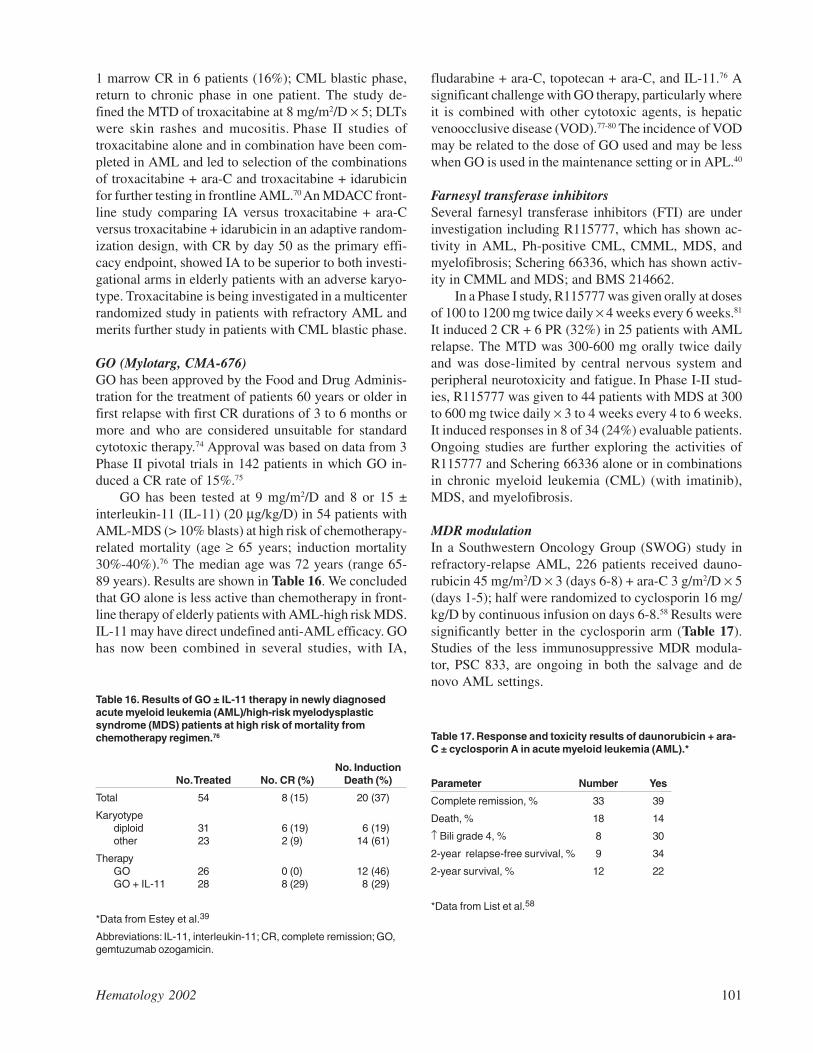
Hematology 2002 101
1 marrow CR in 6 patients (16%); CML blastic phase,return to chronic phase in one patient. The study de-fined the MTD of troxacitabine at 8 mg/m2/D × 5; DLTswere skin rashes and mucositis. Phase II studies oftroxacitabine alone and in combination have been com-pleted in AML and led to selection of the combinationsof troxacitabine + ara-C and troxacitabine + idarubicinfor further testing in frontline AML.70 An MDACC front-line study comparing IA versus troxacitabine + ara-Cversus troxacitabine + idarubicin in an adaptive random-ization design, with CR by day 50 as the primary effi-cacy endpoint, showed IA to be superior to both investi-gational arms in elderly patients with an adverse karyo-type. Troxacitabine is being investigated in a multicenterrandomized study in patients with refractory AML andmerits further study in patients with CML blastic phase.
GO (Mylotarg, CMA-676)GO has been approved by the Food and Drug Adminis-tration for the treatment of patients 60 years or older infirst relapse with first CR durations of 3 to 6 months ormore and who are considered unsuitable for standardcytotoxic therapy.74 Approval was based on data from 3Phase II pivotal trials in 142 patients in which GO in-duced a CR rate of 15%.75
GO has been tested at 9 mg/m2/D and 8 or 15 ±interleukin-11 (IL-11) (20 µg/kg/D) in 54 patients withAML-MDS (> 10% blasts) at high risk of chemotherapy-related mortality (age ≥ 65 years; induction mortality30%-40%).76 The median age was 72 years (range 65-89 years). Results are shown in Table 16. We concludedthat GO alone is less active than chemotherapy in front-line therapy of elderly patients with AML-high risk MDS.IL-11 may have direct undefined anti-AML efficacy. GOhas now been combined in several studies, with IA,
fludarabine + ara-C, topotecan + ara-C, and IL-11.76 Asignificant challenge with GO therapy, particularly whereit is combined with other cytotoxic agents, is hepaticvenoocclusive disease (VOD).77-80 The incidence of VODmay be related to the dose of GO used and may be lesswhen GO is used in the maintenance setting or in APL.40
Farnesyl transferase inhibitorsSeveral farnesyl transferase inhibitors (FTI) are underinvestigation including R115777, which has shown ac-tivity in AML, Ph-positive CML, CMML, MDS, andmyelofibrosis; Schering 66336, which has shown activ-ity in CMML and MDS; and BMS 214662.
In a Phase I study, R115777 was given orally at dosesof 100 to 1200 mg twice daily × 4 weeks every 6 weeks.81
It induced 2 CR + 6 PR (32%) in 25 patients with AMLrelapse. The MTD was 300-600 mg orally twice dailyand was dose-limited by central nervous system andperipheral neurotoxicity and fatigue. In Phase I-II stud-ies, R115777 was given to 44 patients with MDS at 300to 600 mg twice daily × 3 to 4 weeks every 4 to 6 weeks.It induced responses in 8 of 34 (24%) evaluable patients.Ongoing studies are further exploring the activities ofR115777 and Schering 66336 alone or in combinationsin chronic myeloid leukemia (CML) (with imatinib),MDS, and myelofibrosis.
MDR modulationIn a Southwestern Oncology Group (SWOG) study inrefractory-relapse AML, 226 patients received dauno-rubicin 45 mg/m2/D × 3 (days 6-8) + ara-C 3 g/m2/D × 5(days 1-5); half were randomized to cyclosporin 16 mg/kg/D by continuous infusion on days 6-8.58 Results weresignificantly better in the cyclosporin arm (Table 17).Studies of the less immunosuppressive MDR modula-tor, PSC 833, are ongoing in both the salvage and denovo AML settings.
Table 16. Results of GO ± IL-11 therapy in newly diagnosedacute myeloid leukemia (AML)/high-risk myelodysplasticsyndrome (MDS) patients at high risk of mortality fromchemotherapy regimen.76
No. InductionNo. Treated No. CR (%) Death (%)
Total 54 8 (15) 20 (37)
Karyotypediploid 31 6 (19) 6 (19)other 23 2 (9) 14 (61)
TherapyGO 26 0 (0) 12 (46)GO + IL-11 28 8 (29) 8 (29)
*Data from Estey et al.39
Abbreviations: IL-11, interleukin-11; CR, complete remission; GO,gemtuzumab ozogamicin.
Table 17. Response and toxicity results of daunorubicin + ara-C ± cyclosporin A in acute myeloid leukemia (AML).*
Parameter Number Yes
Complete remission, % 33 39
Death, % 18 14
↑ Bili grade 4, % 8 30
2-year relapse-free survival, % 9 34
2-year survival, % 12 22
*Data from List et al.58

102 American Society of Hematology
Targeting angiogenesisIncreased marrow vascularity and increased levels ofplasma-cellular vascular endothelial growth factor(VEGF) and other angiogenesis modulators have beendemonstrated in AML, MDS, CML, and other leuke-mias and have been associated with poor prognosis inAML and CML.82 Thus, targeting angiogenesis may im-prove outcome. Thalidomide, a putative angiogenesisinhibitor, has shown activity in MDS, particularly inRA.23,83,84 A study at MDACC of liposomal daunorubicin+ high-dose ara-C ± thalidomide did not show an addedbenefit with thalidomide. Other angiogenesis inhibitors(SU5416, SU11248, PTK787, GFKI-258, CPE-7055,VEGF MoAb) are undergoing investigation.11,64,82,85,86
Both in vitro direct inhibition of AML blast growth inresponse to cKit inhibition by SU5416, and clinical re-sponses on a Phase II study have been reported in pa-tients with AML.87,88 The assessment of these agents inPhase I studies (in which there may not be a traditionalDLT) or in Phase II studies with statistics designedaround expectations of achievement of objective re-sponses, which in turn are based on our experience withstandard cytotoxic agents, may not be optimal. Cyto-static agents may well be of most benefit in AML as amaintenance agent or in relatively indolent phases ofMDS. Predictive methods to identify potential respond-ers to angiogenic modulators are required.
Targeting methylationDNA methylation is associated with suppression of regu-latory genes and with disease progression.63 Azacytidine(AZA) and decitabine are active hypomethylating agentsin MDS. Decitabine is also active in AML and CML.55,89,90
A randomized study of AZA versus supportive careshowed a benefit from AZA, as measured by responserate, time to transformation, and quality of life (Table18).91,92 Decitabine given at 45 mg/m2/D × 3 to 121 pa-tients with MDS induced CR in 20%, PR in 10%, andimprovement in 19%. Induction death was 8%. Responserates by IPSS risk were 58% in high-risk, 45% in inter-
mediate-2, and 39% in intermediate-1. A Phase I low-dose longer exposure biologic study of decitabine de-fined the Phase II dose at 10 mg/m2/D × 10. Among 42evaluable patients, 7 (17%) achieved CR, 10 (24%) hadPR, and 7 (17%) improved. Combinations of decitabine(with ara-C in AML, with imatinib in CML, withtopotecan or ara-C in MDS) are under consideration.
Other targetsa. Mutations and ITD of FLT-3 in AML are common
(30%-40%) and associated with poor prognosis.4,14-16,61
Several studies are now investigating FLT-3 inhibi-tors in Phase I-II studies of AML with FLT-3 muta-tion/ITD.
b. Overexpression of bcl-2, an anti-apoptosis signal,may be involved in AML resistance to therapy.Genasense (bcl-2 antisense) is undergoing evalua-tion in AML.55
c. A novel diphtheria toxin—granulocyte-macrophagecolony-stimulating factor, fusion protein (DT388GMCSF)—has completed Phase I evaluation in re-fractory-relapse AML.93 The drug was given as ashort infusion daily × 5. The MTD was 4 µg/kg/Dand DLT was liver toxicity. Among 31 patientstreated, 1 had CR and 2 had PR (10%).
ConclusionsThe plethora of available novel agents engenders hopethat real progress will soon be made in achieving cure inall patients with AML or MDS. Above we have focusedon some aspects of our investigational programs—spacedoes not allow a discussion of other very important ap-proaches including immunomodulation, vaccination, andSCT. A challenge with the increasing number of avail-able agents and their diverse mechanisms of anti-leuke-mic activity is the rational design and conduct of stud-ies—recent advances in biostatistics should aid clinicalresearch.94-96 The top priority is to ensure access for pa-tients to adequately designed clinical studies.
REFERENCES
I. Biology of Acute Myeloid Leukemia:The Role of Stroma
1. Appelbaum FR, Rowe JM, Radich J, Dick JE. Acute myeloidleukemia. In: Schechter GP, Broudy VC, Williams ME, eds.Hematology 2001. Washington, DC: American Society ofHematology; 2001:62-86.
2. Clark BR, Keating A. Biology of bone marrow stroma. Ann NY Acad Sci. 1995;770:70-78.
3. Klein G. The extracellular matrix of the hematopoieticmicroenvironment. Experientia. 1995;51:914-926.
4. Verfaillie C, Hurley R, Bhatia R, McCarthy J. Role of bonemarrow matrix in normal and abnormal hematopoiesis. Crit Rev
Table 18. Results of azacytidine (AZA) versus supportive carein myelodysplastic syndrome (MDS).*
SupportiveAZA Care P Value
Number treated 99 92
Complete remission, % 6 0
Partial remission/improved, % 10/47 0/7 <0.001
Median time to transformation (mos) 22 12 0.003
Median survival (mos) 18 14 0.1
*Data from Silverman et al.92

Hematology 2002 103
Oncol Hematol. 1994;16:201-224.5. Levesque J-P, Simmons PJ. Cytoskeleton and integrin-mediated
adhesion signaling in human CD34+ hemopoietic progenitorcells. Exp Hematol. 1999;27:579-586.
6. Liesveld JL, Dipersio JF, Abboud CN. Integrins and adhesivereceptors in normal and leukemic CD34+ progenitor cells:potential regulatory checkpoints for cellular traffic. LeukLymphoma. 1994;14:19-28.
7. Whetton AD, Graham GJ. Homing and mobilization in the stemcell niche. Trends Cell Biol. 199;9(6):233-238.
8. Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL, et al.Chemokine stromal cell-derived factor-1a modulates VLA-4integrin-dependent adhesion to fibronectin and VCAM-1 onbone marrow hematopoietic progenitor cells. Exp Hematol.2001;29:345-355.
9. Simmons PJ, Levesque J-P, Zannettino ACW. Adhesionmolecules in haemopoiesis. Baillieres Clin Haematol.1997;10(3):485-505.
10. Zannettino AC, Buhring HJ, Niutta S, Watt SM, Benton MA,Simmons JP. The sialomucin CD164 (MGC-24v) is an adhesiveglycoprotein expressed by human hematopoietic progenitorsand bone marrow stromal cells that serves as a potent negativeregulator of hematopoiesis. Blood. 1998;92(8):2613-2628.
11. Duhrsen U, Hossfeld DK. Stromal abnormalities in neoplasticbone marrow diseases. Ann Hematol. 1996;73(2):53-70.
12. Smadja-Joffe F, Legras S, Girard N, et al. CD44 and hyaluronanbinding by human myeloid cells. Leuk Lymphoma.1996;21:407-420.
13. Jalkanen S, Jalkanen M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J Cell Biol.1992;116(3):817-825.
14. Miyake K, Medina KL, Hayashi S, Ono S, Hamaoka T, KincadePW. Monoclonal antibodies to Pgp-1/CD44 block lympho-hemopoiesis in long-term bone marrow cultures. J Exp Med.1990;171(2):477-488.
15. Turel KR, Rao SG. Expression of the cell adhesion molecule E-cadherin by the human bone marrow stromal cells and itsprobable role in CD34(+) stem cell adhesion. Cell Biol Int.1998;22(9-10):641-648.
16. Puch S, Armeanu S, Kibler C, et al. N-cadherin is developmen-tally regulated and functionally involved in early hematopoieticcell differentiation. J Cell Sci. 2001;114(pt 8):1567-1577.
17. Corn PG, Smith BD, Ruckdeschel ES, Douglas D, Baylin SB,Herman JG. E-cadherin expression is silenced by 5’ CpG islandmethylation in acute leukemia. Clin Cancer Res.2000;6(11):4243-4248.
18. Reuss-Borst MA, Klein G, Waller HD, Muller CA. Differentialexpression of adhesion molecules in acute leukemia. Leukemia.1995;9(5):869-874.
19. De Waele M, Renmans W, Jochmans K, et al. Differentexpression of adhesion molecules on CD34+ cells in AML andB-lineage ALL and their normal bone marrow counterparts. EurJ Haematol. 1999;63(3):192-201.
20. Janiak M, Sawicki G, Janowska-Wieczorek A. Expression ofbeta 1 integrin mRNAs in human leukemic blasts. Leukemia.1994;8(9):1544-1550.
21. Vila L, Thomas X, Campos L, Sabido O, Archimbaud E.Expression of VLA molecules on acute leukemia cells:relationship with disease characteristics. Exp Hematol.1995;23(6):514-518.
22. Kortlepel K, Bendall LJ, Gottlieb DJ. Human acute myeloidleukemia cells express adhesion proteins and bind to bonemarrow fibroblast monolayers and extracellular matrix proteins.Leukemia. 1993;7(8):1174-1179.
23. Bradstock KF, Gottlieb DJ. Interaction of acute leukemia cells
with the bone marrow microenvironment: implications forcontrol of minimal residual disease. Leuk Lymphoma.1995;18:1-16.
24. Bendall LJ, Kortlepel K, Gottlieb DJ. Human acute myeloidleukemia cells bind to bone marrow stroma via a combinationof beta-1 and beta-2 integrin mechanisms. Blood.1993;82(10):3125-3132.
25. Bendall LJ, Kortlepel K, Gottlieb DJ. Bone marrow fibroblastexposure to the inflammatory cytokines tumor necrosis factor-alpha and interferon-gamma increases adhesion of acutemyeloid leukemia cells and alters the adhesive mechanism. ExpHematol. 1997;25(2):132-139.
26. Cavenagh JD, Gordon-Smith EC, Gibson FM, Gordon MY.Acute myeloid leukemia blast cells bind to human endotheliumin vitro utilizing E-selectin and vascular cell adhesionmolecule-1 (VCAM-1). Br J Haematol. 1993;85(2):285-291.
27. Verfaillie CM, McCarthy JB, McGlave PB. Mechanismsunderlying abnormal trafficking of malignant progenitors inchronic myelogenous leukemia: decreased adhesion to stromaand fibronectin but increased adhesion to the basementmembrane components laminin and collagen type IV. J ClinInvest. 1992;90(4):1232-1241.
28. Bendall LJ, Daniel A, Kortlepel K, Gottlieb DJ. Bone marrowadherent layers inhibit apoptosis of acute myeloid leukemiacells. Exp Hematol. 1994;22(13):1252-1260.
29. Garrido SM, Appelbaum FR, Willman CL, Banker DE. Acutemyeloid leukemia cells are protected from spontaneous anddrug-induced apoptosis by direct contact with a human bonemarrow stromal cell line (HS-5). Exp Hematol. 2001;29:448-457.
30. Fortney JE, Zhao W, Wenger SL, Gibson LF. Bone marrowstromal cells regulate caspase 3 in leukemic cells duringchemotherapy. Leuk Res. 2001;25:901-907.
31. Coulombel L, Kalousek DK, Eaves CJ, Gupta CM, Eaves AC.Long-term marrow culture reveals chromosomally normalhematopoietic progenitor cells in patients with Philadelphiachromosome-positive chronic myelogenous leukemia. N Engl JMed. 1983;308:1493-1498.
32. Coulombel L, Eaves CJ, Kalousek DK, Gupta C, Eaves AC.Long-term marrow cultures of cells from patients with acutemyelogenous leukemia (AML): selection in favor of normalphenotypes in some but not all cases. J Clin Invest.1985;75:961-969.
33. Barnett MJ, Eaves CJ, Phillips GL, et al. Successfulautografting in chronic myeloid leukemia after maintenance ofmarrow in culture. Bone Marrow Transplant. 1989;4:345-351.
34. Chang J, Coutinho L, Morgenstern G, et al. Reconstitution ofhaematopoietic system with autologous marrow taken duringrelapse of acute myeloblastic leukaemia and grown in long-term culture. Lancet. 1986;I:294-295.
35. Brandwein JM, Dube ID, Laraya P, Keating A. Maintenance ofPhiladelphia-chromosome-positive progenitors in long-termmarrow cultures from patients with advanced chronic myeloidleukemia. Leukemia. 1992;6(6):556-561.
36. Griffin JD, Rambaldi A, Vellenga E, Young DC, Ostapovicz D,Cannistra SA. Secretion of interleukin-1 by acute myeloblasticleukemia cells in vitro induces endothelial cells to secretecolony stimulating factors. Blood. 1987;70(4):1218-1221.
37. Takai K, Hara J, Matsumoto K, et al. Hepatocyte growth factoris constitutively produced by human bone marrow stromal cellsand indirectly promotes hematopoiesis. Blood.1997;89(5):1560-1565.
38. Weimar IS, Miranda N, Muller EJ, et al. Hepatocyte growthfactor/scatter factor (HGF/SF) is produced by human bonemarrow stromal cells and promotes proliferation, adhesion and

104 American Society of Hematology
survival of human hematopoietic progenitor cells (CD34+). ExpHematol. 1998;29(6):885-894.
39. Weimar IS, Voermans C, Hourhis J-H, et al. Hepatocyte growthfactor/scatter factor (HGF/SF) affects proliferation andmigration of myeloid leukemic cells. Leukemia. 1998;12:1195-1203.
40. Mohle R, Bautz F, Fafii S, Moore MAS, Brugger W, Kanz L.The chemokine receptor CXCR-4 is expressed on CD34+hematopoietic progenitors and leukemic cells and mediatestransendothelial migration induced by stromal cell-derivedfactor-1. Blood. 1998;91:4523-4530.
41. Mohle R, Schittenhelm M, Failenschmid C, et al. Functionalresponse of leukaemic blasts to stromal cell-derived factor-1correlates with preferential expression of the chemokinereceptor CXCR4 in acute myelomonocytic and lymphoblasticleukaemia. Br J Haematol. 2000;110:563-572.
42. Dilly SA, Jagger CJ. Bone marrow stromal cell changes inhaematological malignancies. J Clin Pathol. 1990;43:942-946.
43. Hussong JW, Rodgers GM, Shami PJ. Evidence of increasedangiogenesis in patients with acute myeloid leukemia. Blood.2000;95(1):309-313.
44. De Bont ES, Rosati S, Jacobs S, Kamps WA, Vellenga E.Increased bone marrow vascularization in patients with acutemyeloid leukaemia: a possible role for vascular endothelialgrowth factor. Br J Haematol. 2001;113(2):296-304.
45. Foss B, Ulvestad E, Bruserud O. Platelet-derived growth factor(PDGF) in human acute myelogenous leukemia: PDGF receptorexpression, endogenous PDGF release and responsiveness toexogenous PDF isoforms by in vitro cultured acute myelog-enous leukemia blasts. Eur J Haematol. 2001;67:267-278.
46. Rosenfeld M, Keating A, Bowen-Pope D, Singer JW, Ross R.Responsiveness of the in vitro hematopoietic microenvironmentto platelet-derived growth factor. Leuk Res. 1985;9(4):427-434.
47. Bianco P, Costantini M, Dearden LC, Bonucci E. Alkalinephosphatase-positive precursors of adipocytes in the humanbone marrow. Br J Haematol. 1988;68:401-403.
48. Mayani H. Composition and function of the hematopoieticmicroenvironment in human myeloid leukemia. Leukemia.1996;10:1041-1047.
49. Nagao T, Yamauchi K, Komatsuda M. Serial in vitro bonemarrow fibroblast culture in human leukemia. Blood.1983;61:589-593.
50. Mayani H, Guilbert LK, Clark SC, Belch AR, Janowksa-Wieczorek A. Composition and functional integrity of the invitro hemopoietic microenvironment in acute myelogenousleukemia: effect of macrophage colony-stimulating factor. ExpHematol. 1992;20(9):1077-1084.
51. Nagao T, Yamauchi K, Kometsuda M, et al. Inhibition of humanbone marrow fibroblast colony formation by leukemic cells.Blood. 1983;62:1261-1265.
52. Hirata J, Katsuno M, Kaneko S, et al. Clinical significance ofhuman bone marrow stromal cell colonies in acute leukemias.Leuk Res. 1986;10:1441-1445.
53. Mayani H, Guilbert LK, Janowska-Wieczorek A. Functionalcharacterization of fibroblastic cells in long-term marrowcultures from patients with acute myelogenous leukemia.Leukemia. 1993;7(10):1564-1569.
54. Mayani H, Guilbert LJ, Sych I, Janowska-Wieczorek A.Production of tumor necrosis factor-alpha in human long-termmarrow cultures from normal subjects and patients with acutemyelogenous leukemia: effect of recombinant macrophagecolony-stimulating factor. Leukemia. 1992;6(11):1148-1154.
55. Zhang W, Knieling G, Vohwinkel G, et al. Origin of stromacells in long-term bone marrow cultures from patients withacute myeloid leukemia. Ann Hematol. 1999;78:305-314.
56. Sparrow RL, O’Flaherty E, Blanksby TM, Szer J, Van DerWeyden MB. Perturbation in the ability of bone marrow stromafrom patients with acute myeloid leukemia but not chronicmyeloid leukemia to support normal early hematopoieticprogenitor cells. Leuk Res. 1997;21(1):29-36.
57. Wetzler M, Kurzrock R, Estrov Z, Estey E, Talpaz M. Cytokineexpression in adherent layers from patients withmyelodysplastic syndrome and acute myelogenous leukemia.Leuk Res. 1995;19(1):23-34.
58. Wetzler M, Estrov Z, Talpaz M, et al. Leukemia inhibitoryfactor in long-term adherent layer cultures: increased levels ofbioactive protein in leukemia and modulation by IL-4, IL-1beta, and TNF-alpha. Cancer Res. 1994;54(7):1837-1842.
59. Shih CC, Hu MC, Hu, J, Medeiros J, Forman SJ. Long-term exvivo maintenance and expansion of transplantable humanhematopoietic stem cells. Blood. 1999;94(5):1623-1636.
60. Bendall LJ, Bradstock KF, Gottlieb DJ. Expression of CD44variant exons in acute myeloid leukemia is more common andmore complex than that observed in normal blood, bonemarrow or CD34+ cells. Leukemia. 2000;14(7):1239-1246.
61. Montuori N, Selleri C, Risitano AN, et al. Expression of the 67-kDa laminin receptor in acute myeloid leukemia cells mediatesadhesion to laminin and is frequently associated with mono-cytic differentiation. Clin Cancer Res. 1999;5:1465-1472.
62. Bendall LJ, Makrynikola V, Hutchinson A, Bianchi AC,Bradstock KF, Gottlieb DJ. Stem cell factor enhances theadhesion of AML cells to fibronectin and augments fibronectin-mediated and anti-apoptotic and proliferative signals. Leuke-mia. 1998;12(9):1375-1382.
63. Keating A, Gown A, Klein M, et al. Passaged CML marrowstromal cells exhibit a normal phenotype. Blood. 1986;68(suppl1):1449.
64. Singer JW, Keating A, Cuttner J, et al. Evidence for a stem cellcommon to hematopoiesis and its in vitro microenvironment:studies of patients with clonal hematopoietic neoplasia.Leukemia. 1984;8(4):535-545.
II. Stem Cell Transplantation in Acute MyeloidLeukemia in the Younger Adult
1. Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremissionchemotherapy in adults with acute myeloid leukemia. Cancerand Leukemia Group B. N Engl J Med. 1994;331: 896-903.
2. Hann IM, Stevens RF, Goldstone AH, et al. Randomizedcomparison of DAT versus ADE as induction chemotherapy inchildren and younger adults with acute myeloid leukemia:results of the Medical Research Council’s 10th AML trial(MRC AML 10). Adult and Childhood Leukaemia WorkingParties of the Medical Research Council. Blood. 1997;89:2311-2318.
3. Burnett AK, Goldstone AH, Stevens RM, et al. Randomisedcomparison of addition of autologous bone-marrow transplanta-tion to intensive chemotherapy for acute myeloid leukaemia infirst remission: results of MRC AML 10 trial. UK MedicalResearch Council Adult and Children’s Leukaemia WorkingParties. Lancet. 1998;351:700-708.
4. Cassileth PA, Harrington DP, Appelbaum FR, et al. Chemo-therapy compared with autologous or allogeneic bone marrowtransplantation in the management of acute myeloid leukemiain first remission. N Engl J Med. 1998;339:1649-1656.
5. Zittoun RA, Mandelli F, Willemze R, et al. Autologous orallogeneic bone marrow transplantation compared withintensive chemotherapy in acute myelogenous leukemia.European Organization for Research and Treatment of Cancer(EORTC) and the Gruppo Italiano Malattie EmatologicheMaligne dell’Adulto (GIMEMA) Leukemia Cooperative

Hematology 2002 105
Groups. N Engl J Med. 1995;332:217-223.6. Harousseau JL, Cahn JY, Pignon B, et al. Comparison of
autologous bone marrow transplantation and intensivechemotherapy as postremission therapy in adult acute myeloidleukemia. The Groupe Ouest Est Leucemies AiguesMyeloblastiques (GOELAM). Blood. 1997;90:2978-2986.
7. Suciu S, Zittoun R, Mandelli F, et al. Allogeneic vs autologousstem cell transplantation according to cytogenetic features inAML patients (pts) 45 years old in first CR: results of theEORTC-GIMEMA AML-10 Trial. [abstract] Blood.2001;98:481a.
8. Burnett AK, Wheatley K, Goldstone AH, Stevens RF, Hann IM,Rees JH, Harrison G. The value of allogeneic bone marrowtransplant in patients with acute myeloid leukaemia at differingrisk of relapse: results of the UK MRC AML 10 trial. Br JHaematol. 2002;118:385-400.
9. Grimwade D, Walker H, Oliver F, et al. The importance ofdiagnostic cytogenetics on outcome in AML: analysis of 1,612patients entered into the MRC AML 10 trial. The MedicalResearch Council Adult and Children’s Leukaemia WorkingParties. Blood. 1998;92:2322-2333.
10. Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypicanalysis predicts outcome of preremission and postremissiontherapy in adult acute myeloid leukemia: a Southwest OncologyGroup/Eastern Cooperative Oncology Group study. Blood.2000;96:4075-4083.
11. Wheatley K, Burnett AK, Goldstone AH, et al. A simple,robust, validated and highly predictive index for the determina-tion of risk-directed therapy in acute myeloid leukaemiaderived from the MRC AML 10 trial. United Kingdom MedicalResearch Council’s Adult and Childhood Leukaemia WorkingParties. Br J Haematol. 1999;107:69-79.
12. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3internal tandem duplication in patients with acute myeloidleukemia (AML) adds important prognostic information tocytogenetic risk group and response to the first cycle ofchemotherapy: analysis of 854 patients from the UnitedKingdom Medical Research Council AML 10 and 12 trials.Blood. 2001;98:1752-1759.
13. Wheatley K, Burnett AK, Gibson B, et al, on behalf of the MRCLeukaemia Working Parties. Optimizing consolidation therapy:four versus five courses of SCT versus chemotherapy—preliminary results of MRC AML 12. Haematology J.2002;3(suppl 1):159.
14. Wheatley K. Current controversies: which patients with acutemyeloid leukaemia should receive a bone marrow transplanta-tion? A statistician’s view. Br J Haematol. 2002;118:351-6.
15. Reiffers J, Gaspard MH, Maraninchi D, et al. Comparison ofallogeneic or autologous bone marrow transplantation andchemotherapy in patients with acute myeloid leukaemia in firstremission: a prospective controlled trial. Br J Haematol.1989;72:57-63.
16. Gratwohl A, Ljungman P, de Witte T, et al. Bone marrowtransplantation for acute myeloid leukemia: the EBMTexperience: a prospective analysis from HLA-typing. TheEMBT Leukemia Working Party. Leukemia. 1992;6(suppl2):110-113.
17. Reiffers J, Stoppa AM, Attal M, et al. Allogeneic vs autologousstem cell transplantation vs emotherapy in patients with acutemyeloid leukemia in first remission: the BGMT 87 study.Leukemia. 1996;10:1874-1882.
18. Powles R, Mehta J, Kulkarni S, et al. Allogeneic blood andbone-marrow stem-cell transplantation in haematologicalmalignant diseases: a randomised trial. Lancet. 2000;355:1231-1237.
19. Bensinger WI, Martin PJ, Storer B, et al. Transplantation ofbone marrow as compared with peripheral-blood cells fromHLA-identical relatives in patients with hematologic cancers. NEngl J Med. 2001;344:175-181.
20. Ottinger HD, Beelen DW, Scheulen B, Schaefer UW, Grosse-Wilde H. Improved immune reconstitution after allotransplanta-tion of peripheral blood stem cells instead of bone marrow.Blood. 1996;88:2775-2779.
21. Champlin RE, Schmitz N, Horowitz MM, et al. Blood stem cellscompared with bone marrow as a source of hematopoietic cellsfor allogeneic transplantation. IBMTR Histocompatibility andStem Cell Sources Working Committee and the EuropeanGroup for Blood and Marrow Transplantation (EBMT). Blood.2000;95:3702-3709.
22. Russell JA, Larratt L, Brown C, et al. Allogeneic blood stemcell and bone marrow transplantation for acute myelogenousleukemia and myelodysplasia: influence of stem cell source onoutcome. Bone Marrow Transplant. 1999;24(11):1177-1183.
23. Kroger N, Zabelina T, Kruger W, et al. In vivo T cell depletionwith pretransplant anti-thymocyte globulin reduces graft-versus-host disease without increasing relapse in good riskmyeloid leukemia patients after stem cell transplantation frommatched related donors. Bone Marrow Transplant.2002;29:683-9.
24. Papadopoulos EB, Carabasi MH, Castro-Malaspina H, et al. T-cell-depleted allogeneic bone marrow transplantation aspostremission therapy for acute myelogenous leukemia:freedom from relapse in the absence of graft-versus-hostdisease. Blood. 1998;91:1083-1090.
25. Rezavani K, Lalancette M, Szydlo R. Non myeloablative stemcell transplantation (NMSCT) in AML, ALL and MDS:disappointing outcome of patients with advanced phase disease.Bone Marrow Transplant. 2001;27(suppl 1):S279.
26. Peggs K, Craddock C, Milligan D, et al. Non-myeloablativeallogeneic transplantation for high-risk acute leukemia andmyelodysplasia [abstract]. Blood. 2001:98:410a.
27. Sanz MA, de la Rubia J, Sanz GF, et al. Busulfan plus cyclo-phosphamide followed by autologous blood stem-cell trans-plantation for patients with acute myeloblastic leukemia in firstcomplete remission: a report from a single institution. J ClinOncol. 1993;11:1661-1667.
28. Korbling M, Fliedner TM, Holle R, et al. Autologous bloodstem cell (ABSCT) versus purged bone marrow transplantation(pABMT) in standard risk AML: influence of source and cellcomposition of the autograft on hemopoietic reconstitution anddisease-free survival. Bone Marrow Transplant. 1991;7:343-349.
29. Martin C, Torres A, Leon A, et al. Autologous peripheral bloodstem cell transplantation (PBSCT) mobilized with G-CSF inAML in first complete remission: role of intensification therapyin outcome. Bone Marrow Transplant. 1998;21:375-382.
30. Yeager AM, Kaizer H, Santos GW, et al. Autologous bonemarrow transplantation in patients with acute nonlymphocyticleukemia, using ex vivo marrow treatment with 4-hydroperoxycyclophosphamide. N Engl J Med. 1986;315:141-147.
31. Gorin NC, Aegerter P, Auvert B, et al. Autologous bone marrowtransplantation for acute myelocytic leukemia in first remission:a European survey of the role of marrow purging. Blood.1990;75:1606-1614.
32. Meloni G, Vignetti M, Avvisati G, et al. BAVC regimen andautograft for acute myelogenous leukemia in second completeremission. Bone Marrow Transplant. 1996;18:693-698.
33. Tomas F, Gomez-Garcia de Soria V, Lopez-Lorenzo JL, et al.Autologous or allogeneic bone marrow transplantation for acute

106 American Society of Hematology
myeloblastic leukemia in second complete remission: impor-tance of duration of first complete remission in final outcome.Bone Marrow Transplant. 1996;17:979-984.
34. Linker CA, Damon LE, Ries CA, Navarro WA, Case D, WolfJL. Autologous stem cell transplantation for advanced acutemyeloid leukemia. Bone Marrow Transplant. 2002;29:297-301.
35. Reiffers J, Labopin M, Sanz MA, et al. The source of stem cellsdose not affect the outcome of patients undergoing autologousstem cell transplantation for acute myeloid leukemia in firstremission. [abstract] Blood. 1996;88(suppl 1):684a.
36. Reiffers J, Stoppa AM, Attal M, Michallet M. Is there a placefor blood stem-cell transplantation for the younger adult patientwith acute myelogenous leukemia? BGMT Group. J Clin Oncol.1994;12:1100-1102.
37. de Witte T, Keating S, Suciu S, et al. A randomized comparisonof the value of autologous BMT versus autologous PSCT forpatients with AML in first CR in the AML 10 Trial of theEORTC LCG and GIMEMA. [abstract] Blood. 2001;98:859a.
38. Busca A, Anasetti C, Anderson G, et al. Unrelated donor orautologous marrow transplantation for treatment of acuteleukemia. Blood. 1994;83:3077-3084.
39. Lazarus HM, Perez WS, Weisdorf D, et al. Autologous versusunrelated donor transplantation for acute myeloid leukemia infirst or second remission. Blood. [abstract] 2000;96;414a.
40. Mark DI. Allogeneic transplantation for leukemia usingunrelated donors. Baillieres Best Pract Res Clin Haematol.2001;14:793-805.
41. Reisner Y, Martelli MF. Transplantation tolerance induced by“mega dose” CD34+ cell transplants [review]. Exp Hematol.2000;28:119-127.
42. Volpi I, Perruccio K, Tosti A, et al. Postgrafting administrationof granulocyte colony-stimulating factor impairs functionalimmune recovery in recipients of human leukocyte antigenhaplotype-mismatched hematopoietic transplants. Blood.2001;97:2514-2521.
43. Ruggeri L, Capanni M, Casucci M, et al. Role of natural killercell alloreactivity in HLA-mismatched hematopoietic stem celltransplantation. Blood. 1999;94:333-339.
44. Aversa F, Tabilio A, Velardi A, et al. Full haplotype mismatchedtransplant in high risk acute leukemia patients. [abstract] Blood.2001;98:669a.
45. Nabhan C, Mehta J, Tallman MS. The role of bone marrowtransplantation in acute promyelocytic leukemia. [review]Bone Marrow Transplant. 2001;28:219-26.
46. Meloni G, Diverio D, Vignetti M, et al. Autologous bonemarrow transplantation for acute promyelocytic leukemia insecond remission: prognostic relevance of pretransplantminimal residual disease assessment by reverse-transcriptionpolymerase chain reaction of the PML/RAR alpha fusion gene.Blood. 1997;90:1321-1325.
47. Tallman MS, Nabhan C, Feusner JH, Rowe JM. Acutepromyelocytic leukemia: evolving therapeutic strategies[review]. Blood. 2002;99:759-767.
48. Gorin NC, Labopin M, Pichard P, et al. Feasibility and recentimprovement of autologous stem cell transplantation for acutemyelocytic leukaemia in patients over 60 years of age:importance of the source of stem cells. Br J Haematol.2000;110:887-893.
49. Cahn JY, Labopin M, Mandelli F, et al. Autologous bonemarrow transplantation for first remission acute myeloblasticleukemia in patients older than 50 years: a retrospectiveanalysis of the European Bone Marrow Transplant Group.Blood. 1995;85:575-579.
50. Deeg HJ, Shulman HM, Anderson JE, et al. Allogeneic andsyngeneic marrow transplantation for myelodysplastic
syndrome in patients 55 to 66 years of age. Blood.2000;95:1188-1194.
51. Paietta E. Assessing minimal residual disease (MRD) inleukemia: a changing definition and concept? [review]BoneMarrow Transplant. 2002 ;29:459-65.
52. Liu Yin JA, Grimwade D. Minimal residual disease evaluationin acute meyloid leukemia. Lancet. 2002;360:160-162.
53. San Miguel JF, Vidriales MB, Lopez-Berges C,et al. Earlyimmunophenotypical evaluation of minimal residual disease inacute myeloid leukemia identifies different patient risk groupsand may contribute to postinduction treatment stratification.Blood. 2001;98:1746-51
54. Tobal K, Moore H, Macheta M, Yin JA. Monitoring minimalresidual disease and predicting relapse in APL by quantitatingPML-RARalpha transcripts with a sensitive competitive RT-PCR method. Leukemia. 2001;15:1060-5.
55. Tobal K, Newton J, Macheta M, et al.. Molecular quantitationof minimal residual disease in acute myeloid leukemia witht(8;21) can identify patients in durable remission and predictclinical relapse. Blood. 2000 ;95:815-9.
56. Lo Coco F, Diverio D, Pandolfi PP, et al. Molecular evaluationof residual disease as a predictor of relapse in acutepromyelocytic leukaemia. Lancet. 1992;340:1437-1438.
57. Burnett AK, Grimwade D, Solomon E, Wheatley K, GoldstoneAH. Presenting white blood cell count and kinetics of molecu-lar remission predict prognosis in acute promyelocyticleukemia treated with all-trans retinoic acid: result of theRandomized MRC Trial. Blood. 1999;93:4131-4143.
58. Nucifora G, Larson RA, Rowley JD. Persistence of the 8;21translocation in patients with acute myeloid leukemia type M2in long-term remission. Blood. 1993;82:712.
59. Zittoun R, Suciu S, Watson M, et al. Quality of life in patientswith acute myelogenous leukemia in prolonged first completeremission after bone marrow transplantation (allogeneic orautologous) or chemotherapy: a cross-sectional study of theEORTC-GIMEMA AML 8A trial. Bone Marrow Transplant.1997;20:307-15.
60. Leung W, Hudson MM, Strickland DK, et al. Late effects oftreatment in survivors of childhood acute myeloid leukemia.JClin Oncol. 2000;18:3273-9.
61. Tallman MS, Rowlings PA, Milone G, et al. Effect ofpostremission chemotherapy before human leukocyte antigen-identical sibling transplantation for acute myelogenousleukemia in first complete remission. Blood. 2000;96:1254-8.
III. Biologic and Genetic Risk Assessment of AMLin the Genomic Era
1. Bloomfield CD, Herzig GP, Caligiuri MD. Introduction: Acuteleukemia: recent advances. Semin Oncol. 1997;24(1):1-2.
2. Willman CL. Acute leukemias: A paradigm for the integrationof new technologies in diagnosis and classification. ModernPathol. 1999;12:218-228.
3. Willman CL. Molecular evaluation of acute myeloid leukemias.Semin Hematol. 1999;13(4):390-400.
4. Look AT. Oncogenic transcription factors in the human acuteleukemias. Science. 1997;278:1059-1064.
5. Tenen DG, Hromas R, Licht JD, Zhang DE. Transcriptionfactors, normal myeloid development, and leukemia. Blood.1997;90:489-519.
6. Appelbaum FR, Rowe JM, Radich JR, Dick JE. Acute myeloidleukemia. In: Schechter GP, Broudy VC, Williams ME, eds.Hematology 2001. American Society of Hematology: Washing-ton, DC; 2001:62-86.
7. Rowley JD, Alimena G, Garson MO, et al. A collaborativestudy of the relationship of the morphologic type of acute non-

Hematology 2002 107
lymphocytic leukemia with patient age and karyotype. Blood.1982;59:1013.
8. Dastugue N, Payen C, Lafage-Pochitaloff MM, et al. Prognosticsignificance of karyotype in adult de novo acute myeloidleukemia. Leukemia. 1995;9:1291.
9. Leith CP, Kopecky KJ, Godwin J, et al. Acute myeloid leukemiain the elderly: Assessment of multidrug resistance (MDR1) andcytogenetics distinguishes biologic subgroups with remarkablydistinct responses to standard chemotherapy: A SouthwestOncology Group Study. Blood. 1997;89:3323.
10. Lancet JE, Willman CL, Bennett JM. Acute myelogenousleukemia in the elderly. Hematol/Oncol Clin North Am.2000;14(10):251-267.
11. Grimwade D, Walker H, Harrison G, et al. The predictive valueof hierarchical cytogenetic classification in older adults withacute myeloid leukemia (AML): analysis of 1065 patientsentered into the United Kingdom Medical Research CouncilAML11 trial. Blood. 2001;98:1312-1320.
12. Willman CL. The prognostic significance of the expression andfunction of multidrug resistance transporter proteins in acutemyeloid leukemia: Studies of the Southwest Oncology GroupLeukemia Research Program. Semin Hematol. 1997;34(suppl5):25-33.
13. List AF, Kopecky KJ, Willman CL, et al. Benefit ofcyclosporine modulation of drug resistance in patients withpoor risk acute myeloid leukemia: a Southwest OncologyGroup study. Blood. 2001;98:3212-3220.
14. Head DR. Revised classification of acute myeloid leukemia.Leukemia. 1996;10:1826.
15. Available at: http://www.seer.cancer.gov16. Golub TR, Slonim DK, Tamayo P, et al. Molecular classification
of cancer: class discovery and class prediction by geneexpression monitoring. Science. 1999;286:531-537.
17. Armstrong SA, Staunton JE, Silverman LB, et al. MLLtranslocations specify a distinct gene expression profile thatdistinguishes a unique leukemia. Nat Genetics. 2002;30:41-47.
18. Staunton JE, Slonim DK, Coller HA, et al. Chemosensitivityprediction by transcriptional profiling. Proc Natl Acad Sci U SA. 2001;98:10787-10792.
19. Miyazato A, Ueno S, Ohmine K, et al. Identification ofmyelodysplastic syndrome-specific genes by DNA microarrayanalysis with purified hematopoietic stem cell fraction. Blood.2001;98:422-427.
20. Guzman ML, Upchurch D, Grimes B, et al. Expression of tumorsuppressor genes interferon regulatory factor 1 and death-associated protein kinase in primitive acute myelogenousleukemia cells. Blood. 2001;97:2177-2179.
21. Virtaneva K, Wright FA, Tanner SM, et al. Expression profilingreveals fundamental biologic differences in acute myeloidleukemia with isolated trisomy 8 and normal cytogenetics. ProcNatl Acad Sci U S A. 2001;98:11124-11129.
22. Schoch C, Kohlmann A, Schnittger S, et al. Acute myeloidleukemias with reciprocal rearrangements can be distinguishedby specific gene expression profiles. Blood. 2002;99:10008-10013.
23. Ramaswamy S, Golub TR. DNA microarrays in clinicaloncology. J Clin Oncol. 2002;20(7):1932-1941.
24. Smith MA, McCaffrey RP, Karp JE. The secondary leukemias:challenges and research directions. J Natl Cancer Inst.1996;88(7):407-418.
25. Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypicanalysis predicts outcome of pre- and post-remission therapy inadult acute myeloid leukemia (AML): A SWOG (S9034)/ECOG(E3489) intergroup study. Blood. 2000;96:4075-4083.
26. Available at: http://dc.nci.nih.gov.
27. Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection forcancer classification using support vector machines. MachineLearning. In press.
28. Davidson GS, Hendrickson B, Johnson DK, et al. KnowledgeMining with VxInsight: Discovery Through Exploration. NewYork, NY: Kluwer Academic Publishers; 2001:1-20.
29. Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM.Expression Profiling of CD34+ Hematopoietic Stem/ProgenitorCells Reveals Distinct Subtypes of Therapy-Related AML, ProcNatl Acad U S A. In press.
30. Biondi A, Cimino G, Pieters R, Pui CH. Biologic and therapeu-tic aspects of infant leukemia. Blood. 2000;96:24-33.
31. Available at: http://www.cs.sandia.gov/projects/VxInsight.html.32. Kim SK, Lund J, Kiraly M, et al. A gene expression map for
Caenorhabditis elegans. Science. 2001;293:2087-2092.33. Lacayo N, Kinnunen P, Meshinchi S, et al. Gene expression
profiling of pediatric acute myeloid leukemia (AML) in de novoand relapsed patients reveals an expression signature thatcorrelates with FLT3-internal tandem duplications (ITD), FLT3point mutations, and KIT point mutations. Blood, AbstractSubmitted to ASH 2002.
34. Yokota S, Kiyoi H, Nakao M, et al. Internal tandem duplicationof the FLT3 gene is preferentially seen in acute myeloidleukemia and myelodysplastic syndrome among varioushematologic malignancies. A study of a large series of patientsand cell lines. Leukemia. 1997;11:1605-1609.
35. Meschinchi S, Woods WG, Stirewalt DL, et al. Prevalence andprognostic significance of Flt3 internal tandem duplication inpediatric acute myeloid leukemia. Blood. 2001;97:89-94.
36. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3internal tandem duplication in patients with acute myeloidleukemia (AML) adds important prognostic information tocytogenetic risk group and response to the first cycle ofchemotherapy: analysis of 854 patients from the UnitedKingdom MRC AML 10 and 12 Trials. Blood. 2001;98:1752-1759.
37. Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3length mutations in 1003 patients with acute myeloid leukemia:correlation to cytogenetics, FAB subtype, and prognosis in theAMLCG study and usefulness as a marker of minimal residualdisease. Blood. 2002;100:59-66.
38. Whitman DP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acutemyeloid leukemia with normal cytogenetics and the internaltandem duplication of FLT3: a Cancer and Leukemia Group BStudy. Cancer Res. 2001;61:7233-7239.
39. Stirewalt DL, Kopecky KJ, Meshinchi S, et al. FLT3, RAS, andTP53 mutations in elderly patients with acute myeloidleukemia. Blood. 2001;97:3589-3595.
40. Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosinekinase inhibitor is cytotoxic to leukemia cells in vitro and invivo. Blood. 2002;99:3885-3891.
41. Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results inactivation of the RAS pathway and leads to aberrant growth inhematopoietic cells. Nat Genetics. 1996;12:144.
42. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implications ofFLT3 and N-RAS mutations in acute myeloid leukemia. Blood.1999;93:3074-3080.
43. Karp JE, Lancet JE, Kaufmann SH, et al. Clinical and biologicactivity of the farnesyltransferase inhibitor R115777 in adultswith refractory and relapsed acute leukemias: a phase 1clinical-laboratory correlative trial. Blood. 2001;97:3361-3369.
44. Albitar M. Angiogenesis in acute myeloid leukemia andmyelodysplastic syndrome. Acta Haematol. 2001;106(4):170-176.

108 American Society of Hematology
45. Bellamy WT, Ritcher L, Frutiger Y, Grogan TM. Expression ofvascular endothelial derived growth factor and its receptors inhematopoietic malignancies. Cancer Res. 1999;59:728-733.
46. Hussong JW, Rodgers GM, Shami PJ. Evidence of increasedangiogenesis in patients with acute myeloid leukemia. Blood.2000;95:309-313.
IV. New Agents and Strategies in Acute MyeloidLeukemia and High-Risk MyelodysplasticSyndromes
1. Estey EH, Thall PF, Cortes JE, et al. Comparison of idarubicin +ara-C-, fludarabine + ara-C-, and topotecan + ara-C-basedregimens in treatment of newly diagnosed acute myeloidleukemia, refractory anemia with excess blasts in transforma-tion, or refractory anemia with excess blasts. Blood.2001;98:3575-3583.
2. Grimwade D, Walker H, Oliver F, et al. The importance ofdiagnostic cytogenetics on outcome in AML: analysis of 1,612patients entered into the MRC AML 10 trial. The MedicalResearch Council Adult and Children’s Leukaemia WorkingParties. Blood. 1998;92:2322-2333.
3. Grimwade D, Walker H, Harrison G, et al. The predictive valueof hierarchical cytogenetic classification in older adults withacute myeloid leukemia (AML): analysis of 1065 patientsentered into the United Kingdom Medical Research CouncilAML11 trial. Blood. 2001;98:1312-1320.
4. Burnett AK. Acute myeloid leukemia: treatment of adults under60 years. Rev Clin Exp Hematol. 2002;6:26-45; discussion 86-27.
5. Estey E, Thall P, Beran M, et al. Effect of diagnosis (refractoryanemia with excess blasts, refractory anemia with excess blastsin transformation, or acute myeloid leukemia ) on outcome ofAML-type chemotherapy. Blood. 1997;90:2969-2977.
6. Beran M, Shen Y, Kantarjian H, et al. High-dose chemotherapyin high-risk myelodysplastic syndrome: covariate-adjustedcomparison of five regimens. Cancer. 2001;92:1999-2015.
7. Estey EH, Keating MJ, Dixon DO, et al. Karyotype isprognostically more important than the FAB system’s distinc-tion between myelodysplastic syndrome and acute myelog-enous leukemia. Hematol Pathol. 1987;1:203-208.
8. Albitar M, Manshouri T, Shen Y, et al. Myelodysplasticsyndrome is not merely “preleukemia”. Blood. 2002;100:791-798.
9. Lin CW, Manshouri T, Jilani I, et al. Proliferation and apoptosisin acute and chronic leukemias and myelodysplastic syndrome.Leuk Res. 2002;26:551-559.
10. Lai R, Estey E, Shen Y, et al. Clinical significance of plasmaendostatin in acute myeloid leukemia/myelodysplasticsyndrome. Cancer. 2002;94:14-17.
11. Thomas DA, Giles FJ, Cortes J, et al. Antiangiogenic therapy inleukemia. Acta Haematol. 2001;106:190-207.
12. Albitar M, Beran M, O’Brien S, et al. Differences betweenrefractory anemia with excess blasts in transformation andacute myeloid leukemia. Blood. 2000;96:372-373.
13. Aguayo A, Kantarjian H, Manshouri T, et al. Angiogenesis inacute and chronic leukemias and myelodysplastic syndromes.Blood. 2000;96:2240-2245.
14. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3internal tandem duplication in patients with acute myeloidleukemia (AML) adds important prognostic information tocytogenetic risk group and response to the first cycle ofchemotherapy: analysis of 854 patients from the UnitedKingdom Medical Research Council AML 10 and 12 trials.Blood. 2001;98:1752-1759.
15. Kiyoi H, Ohno R, Ueda R, et al. Mechanism of constitutiveactivation of FLT3 with internal tandem duplication in thejuxtamembrane domain. Oncogene. 2002;21:2555-2563.
16. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication ofFLT3 and N-RAS gene mutations in acute myeloid leukemia.Blood. 1999;93:3074-3080.
17. Parker JE, Mufti GJ, Rasool F, et al. The role of apoptosis,proliferation, and the Bcl-2-related proteins in themyelodysplastic syndromes and acute myeloid leukemiasecondary to MDS. Blood. 2000;96:3932-3938.
18. Raza A, Gezer S, Mundle S, et al. Apoptosis in bone marrowbiopsy samples involving stromal and hematopoietic cells in 50patients with myelodysplastic syndromes. Blood. 1995;86:268-276.
19. Beran M. Intensive chemotherapy for patients with high-riskmyelodysplastic syndrome. Int J Hematol. 2000;72:139-150.
20. Estey E, Thall P, Andreeff M, et al. Use of granulocyte colony-stimulating factor before, during, and after fludarabine pluscytarabine induction therapy of newly diagnosed acutemyelogenous leukemia or myelodysplastic syndromes:comparison with fludarabine plus cytarabine without granulo-cyte colony-stimulating factor [see comments]. J Clin Oncol.1994;12:671-678.
21. Estey EH, Thall PF, Pierce S, et al. Randomized phase II studyof fludarabine + cytosine arabinoside + idarubicin +/- all-transretinoic acid +/- granulocyte colony- stimulating factor in poorprognosis newly diagnosed acute myeloid leukemia andmyelodysplastic syndrome. Blood. 1999;93:2478-2484.
22. List AF. New approaches to the treatment of myelodysplasia.Oncologist. 2002;7:39-49.
23. Zorat F, Shetty V, Dutt D, et al. The clinical and biologicaleffects of thalidomide in patients with myelodysplasticsyndromes. Br J Haematol. 2001;115:881-894.
24. Rosenfeld C, List A. A hypothesis for the pathogenesis ofmyelodysplastic syndromes: implications for new therapies.Leukemia. 2000;14:2-8.
25. List AF. Use of amifostine in hematologic malignancies,myelodysplastic syndrome, and acute leukemia. Semin Oncol.1999;26:61-65.
26. Byrd JC, Dodge RK, Carroll A, et al. Patients witht(8;21)(q22;q22) and acute myeloid leukemia have superiorfailure-free and overall survival when repetitive cycles of high-dose cytarabine are administered. J Clin Oncol. 1999;17:3767-3775.
27. Tallman MS, Nabhan C, Feusner JH, et al. Acute promyelocyticleukemia: evolving therapeutic strategies. Blood. 2002;99:759-767.
28. Fenaux P, Chastang C, Chevret S, et al. A randomizedcomparison of all transretinoic acid (ATRA) followed bychemotherapy and ATRA plus chemotherapy and the role ofmaintenance therapy in newly diagnosed acute promyelocyticleukemia. The European APL Group. Blood. 1999;94:1192-1200.
29. Sanz MA, Martin G, Rayon C, et al. A modified AIDA protocolwith anthracycline-based consolidation results in highantileukemic efficacy and reduced toxicity in newly diagnosedPML/RARalpha-positive acute promyelocytic leukemia.PETHEMA group. Blood. 1999;94:3015-3021.
30. Sanz MA, Lo Coco F, Martin G, et al. Definition of relapse riskand role of nonanthracycline drugs for consolidation in patientswith acute promyelocytic leukemia: a joint study of thePETHEMA and GIMEMA cooperative groups. Blood.2000;96:1247-1253.
31. Mandelli F, Diverio D, Avvisati G, et al. Molecular remission inPML/RAR alpha-positive acute promyelocytic leukemia by

Hematology 2002 109
combined all-trans retinoic acid and idarubicin (AIDA) therapy.Gruppo Italiano-Malattie Ematologiche Maligne dell’Adultoand Associazione Italiana di Ematologia ed OncologiaPediatrica Cooperative Groups. Blood. 1997;90:1014-1021.
32. Head D, Kopecky KJ, Weick J, et al. Effect of aggressivedaunomycin therapy on survival in acute promyelocyticleukemia. Blood. 1995;86:1717-1728.
33. Estey E, Thall PF, Pierce S, et al. Treatment of newly diagnosedacute promyelocytic leukemia without cytarabine. J Clin Oncol.1997;15:483-490.
34. Diverio D, Rossi V, Avvisati G, et al. Early detection of relapseby prospective reverse transcriptase- polymerase chain reactionanalysis of the PML/RARalpha fusion gene in patients withacute promyelocytic leukemia enrolled in the GIMEMA-AIEOPmulticenter “AIDA” trial. GIMEMA-AIEOP Multicenter“AIDA” Trial. Blood. 1998;92:784-789.
35. Lo Coco F, Diverio D, Avvisati G, et al. Therapy of molecularrelapse in acute promyelocytic leukemia. Blood. 1999;94:2225-2229.
36. Soignet SL, Frankel SR, Douer D, et al. United Statesmulticenter study of arsenic trioxide in relapsed acutepromyelocytic leukemia. J Clin Oncol. 2001;19:3852-3860.
37. Estey EH, Giles FJ, Kantarjian H, et al. Molecular remissionsinduced by liposomal-encapsulated all-trans retinoic acid innewly diagnosed acute promyelocytic leukemia. Blood.1999;94:2230-2235.
38. Lu DP, Qiu JY, Jiang B, et al. Tetra-arsenic tetra-sulfide for thetreatment of acute promyelocytic leukemia: a pilot report.Blood. 2002;99:3136-3143.
39. Estey E, Koller C, Cortes J, et al. Treatment of newly-diagnosedacute promyelocytic leukemia with liposomal all-trans retinoicacid. Leuk Lymphoma. 2001;42:309-316.
40. Estey EH, Giles FJ, Beran M, et al. Experience withgemtuzumab ozogamycin (“mylotarg”) and all-trans retinoicacid in untreated acute promyelocytic leukemia. Blood.2002;99:4222-4224.
41. Takeuchi M, Yano T, Omoto E, et al. Relapsed acutepromyelocytic leukemia previously treated with all-transretinoic acid: clinical experience with a new synthetic retinoid,Am-80. Leuk Lymphoma. 1998;31:441-451.
42. Jurcic JG, DeBlasio T, Dumont L, et al. Molecular remissioninduction with retinoic acid and anti-CD33 monoclonalantibody HuM195 in acute promyelocytic leukemia. ClinCancer Res. 2000;6:372-380.
43. Nabhan C, Mehta J, Tallman MS. The role of bone marrowtransplantation in acute promyelocytic leukemia. Bone MarrowTransplant. 2001;28:219-226.
44. Visani G, Gugliotta L, Tosi P, et al. All-trans retinoic acidsignificantly reduces the incidence of early hemorrhagic deathduring induction therapy of acute promyelocytic leukemia. EurJ Haematol. 2000;64:139-144.
45. Woods WG, Kobrinsky N, Buckley JD, et al. Timed-sequentialinduction therapy improves postremission outcome in acutemyeloid leukemia: a report from the Children’s Cancer Group.Blood. 1996;87:4979-4989.
46. Woods WG, Kobrinsky N, Buckley J, et al. Intensively timedinduction therapy followed by autologous or allogeneic bonemarrow transplantation for children with acute myeloidleukemia or myelodysplastic syndrome: a Childrens CancerGroup pilot study. J Clin Oncol. 1993;11:1448-1457.
47. Cortes J, Estey E, O’Brien S, et al. High-dose liposomaldaunorubicin and high-dose cytarabine combination in patientswith refractory or relapsed acute myelogenous leukemia.Cancer. 2001:92:7-14.
48. Cortes J, O’Brien S, Estey E, et al. Phase I study of liposomal
daunorubicin in patients with acute leukemia. Invest NewDrugs. 1999;17:81-87.
49. Gaspar MM, Perez-Soler R, Cruz ME. Biological characteriza-tion of L-asparaginase liposomal formulations. CancerChemother Pharmacol. 1996;38:373-377.
50. Emerson DL, Bendele R, Brown E, et al. Antitumor efficacy,pharmacokinetics, and biodistribution of NX 211: a low-clearance liposomal formulation of lurtotecan. Clin Cancer Res.2000;6:2903-2912.
51. Glantz MJ, LaFollette S, Jaeckle KA, et al. Randomized trial ofa slow-release versus a standard formulation of cytarabine forthe intrathecal treatment of lymphomatous meningitis. J ClinOncol. 1999;17:3110-3116.
52. Kantarjian H. New developments in the treatment of acutemyeloid leukemia: focus on topotecan. Semin Hematol.1999;36:16-25.
53. Inoue K. [Drug delivery systems for cancer chemotherapy].Nippon Geka Gakkai Zasshi. 2002;103:224-228.
54. Johnson SA. Nucleoside analogues in the treatment ofhaematological malignancies. Expert Opin Pharmacother.2001;2:929-943.
55. Giles FJ. New drugs in acute myeloid leukemia. Curr OncolRep. 2002;4:369-374.
56. Giles FJ, Cortes JE, Baker SD, et al. Troxacitabine, a noveldioxolane nucleoside analog, has activity in patients withadvanced leukemia. J Clin Oncol. 2001;19:762-771.
57. Kurzrock R. Myelodysplastic syndrome overview. SeminHematol. 2002;39:18-25.
58. List AF, Kopecky KJ, Willman CL, et al. Benefit ofcyclosporine modulation of drug resistance in patients withpoor-risk acute myeloid leukemia: a Southwest OncologyGroup study. Blood. 2001;98:3212-3220.
59. Matsui H, Takeshita A, Naito K, et al. Reduced effect ofgemtuzumab ozogamicin (CMA-676) on P-glycoprotein and/orCD34-positive leukemia cells and its restoration by multidrugresistance modifiers. Leukemia. 2002;16:813-819.
60. Bernstein ID. CD33 as a target for selective ablation of acutemyeloid leukemia. Clin Lymphoma. 2002;2(Suppl 1):S9-S11.
61. Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosinekinase inhibitor is cytotoxic to leukemia cells in vitro and invivo. Blood. 2002;99:3885-3891.
62. Karp JE. Farnesyl protein transferase inhibitors as targetedtherapies for hematologic malignancies. Semin Hematol.2001;38:16-23.
63. Willman CL. Targeted AML therapy: new biologic paradigmsand therapeutic opportunities. Leukemia. 2001;15:690-694.
64. Giles FJ. The vascular endothelial growth factor (VEGF)signaling pathway: a therapeutic target in patients withhematologic malignancies. Oncologist. 2001;6:32-39.
65. Seiter K, Feldman EJ, Halicka HD, et al. Phase I clinical andlaboratory evaluation of topotecan and cytarabine in patientswith acute leukemia. J Clin Oncol. 1997;15:44-51.
66. Beran M, Estey E, O’Brien S, et al. Topotecan and cytarabine isan active combination regimen in myelodysplastic syndromesand chronic myelomonocytic leukemia. J Clin Oncol.1999;17:2819-2830.
67. Giles FJ, Cortes JE, Thomas DA, et al. Phase I and Pharmacoki-netic Study of DX-8951f (Exatecan Mesylate), a HexacyclicCamptothecin, on a Daily-Times-Five Schedule in Patients withAdvanced Leukemia. Clin Cancer Res. 2002;8:2134-2141.
68. Vey N, Giles FJ, Kantarjian H, et al. The topoisomerase Iinhibitor DX-8951f is active in a severe combined immunodefi-cient mouse model of human acute myelogenous leukemia. ClinCancer Res. 2000;6:731-736.
69. Gerrits CJ, Schellens JH, Creemers GJ, et al. The bioavailability

110 American Society of Hematology
of oral GI147211 (GG211), a new topoisomerase I inhibitor. BrJ Cancer. 1997;76:946-951.
70. Giles FJ, Garcia-Manero G, Cortes JE, et al. Phase II Study oftroxacitabine, a novel dioxolane nucleoside analog, in patientswith refractory leukemia. J Clin Oncol. 2002;20:656-664.
71. Gourdeau H, Bibeau L, Ouellet F, et al. Comparative study of anovel nucleoside analogue (Troxatyl, troxacitabine, BCH-4556)and AraC against leukemic human tumor xenografts expressinghigh or low cytidine deaminase activity. Cancer ChemotherPharmacol. 2001;47:236-240.
72. Gourdeau H, Clarke ML, Ouellet F, et al. Mechanisms of uptakeand resistance to troxacitabine, a novel deoxycytidinenucleoside analogue, in human leukemic and solid tumor celllines. Cancer Res. 2001;61:7217-7224.
73. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues:mechanisms of drug resistance and reversal strategies.Leukemia. 2001;15:875-890.
74. Bross PF, Beitz J, Chen G, et al. Approval summary:gemtuzumab ozogamicin in relapsed acute myeloid leukemia.Clin Cancer Res. 2001;7:1490-1496.
75. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy andsafety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol.2001;19:3244-3254.
76. Estey EH, Thall PF, Giles FJ, et al. Gemtuzumab ozogamicinwith or without interleukin 11 in patients 65 years of age orolder with untreated acute myeloid leukemia and high- riskmyelodysplastic syndrome: comparison with idarubicin pluscontinuous-infusion, high-dose cytosine arabinoside. Blood.2002;99:4343-4349.
77. Giles FJ, Kantarjian HM, Kornblau SM, et al.: Mylotarg(gemtuzumab ozogamicin) therapy is associated with hepaticvenoocclusive disease in patients who have not received stemcell transplantation. Cancer. 2001;92:406-413.
78. Cohen AD, Luger SM, Sickles C, et al. Gemtuzumabozogamicin (Mylotarg) monotherapy for relapsed AML afterhematopoietic stem cell transplant: efficacy and incidence ofhepatic veno-occlusive disease. Bone Marrow Transplant.2002;30:23-28.
79. Rajvanshi P, Shulman HM, Sievers EL, et al. Hepatic sinusoidalobstruction after gemtuzumab ozogamicin (Mylotarg) therapy.Blood. 2002;99:2310-2314.
80. McDonald GB. Management of hepatic sinusoidal obstructionsyndrome following treatment with gemtuzumab ozogamicin(mylotarg(r)). Clin Lymphoma. 2002;2(Suppl 1):S35-39.
81. Karp JE, Lancet JE, Kaufmann SH, et al. Clinical and biologicactivity of the farnesyltransferase inhibitor R115777 in adultswith refractory and relapsed acute leukemias: a phase 1clinical-laboratory correlative trial. Blood. 2001;97:3361-3369.
82. Giles FJ. The emerging role of angiogenesis inhibitors inhematologic malignancies. Oncology (Huntingt). 2002;16:23-29.
83. Raza A, Meyer P, Dutt D, et al. Thalidomide producestransfusion independence in long-standing refractory anemiasof patients with myelodysplastic syndromes. Blood.2001;98:958-965.
84. Strupp C, Germing U, Aivado M, et al. Thalidomide for thetreatment of patients with myelodysplastic syndromes.Leukemia. 2002;16:1-6.
85. Lin B, Podar K, Gupta D, et al. The vascular endothelial growthfactor receptor tyrosine kinase inhibitor PTK787/ZK222584inhibits growth and migration of multiple myeloma cells in thebone marrow microenvironment. Cancer Res. 2002;62:5019-5026.
86. Gordon MS, Margolin K, Talpaz M, et al. Phase I safety andpharmacokinetic study of recombinant human anti- vascularendothelial growth factor in patients with advanced cancer. JClin Oncol. 2001;19:843-850.
87. Smolich BD, Yuen HA, West KA, et al. The antiangiogenicprotein kinase inhibitors SU5416 and SU6668 inhibit the SCFreceptor (c-kit) in a human myeloid leukemia cell line and inacute myeloid leukemia blasts. Blood. 2001;97:1413-1421.
88. Mesters RM, Padro T, Bieker R, et al. Stable remission afteradministration of the receptor tyrosine kinase inhibitor SU5416in a patient with refractory acute myeloid leukemia. Blood.2001;98:241-243.
89. Cheson BD, Zwiebel JA, Dancey J, et al. Novel therapeuticagents for the treatment of myelodysplastic syndromes. SeminOncol. 2000;27:560-577.
90. Kantarjian HM, O’Brien SM, Keating M, et al. Results ofdecitabine therapy in the accelerated and blastic phases ofchronic myelogenous leukemia. Leukemia. 1997;11:1617-1620.
91. Kornblith AB, Herndon JE, 2nd, Silverman LR, et al. Impact ofazacytidine on the quality of life of patients withmyelodysplastic syndrome treated in a randomized phase IIItrial: a Cancer and Leukemia Group B study. J Clin Oncol.2002;20:2441-2452.
92. Silverman LR, Demakos EP, Peterson BL, et al. Randomizedcontrolled trial of azacitidine in patients with themyelodysplastic syndrome: a study of the Cancer and LeukemiaGroup B. J Clin Oncol. 2002;20:2429-2440.
93. Frankel AE, Powell BL, Hall PD, et al. Phase I trial of a noveldiphtheria toxin/granulocyte macrophage colony- stimulatingfactor fusion protein (DT388GMCSF) for refractory or relapsedacute myeloid leukemia. Clin Cancer Res. 2002;8:1004-1013.
94. Stallard N, Thall PF. Decision-theoretic designs for pre-phase IIscreening trials in oncology. Biometrics. 2001;57:1089-1095.
95. Thall PF, Lee JJ, Tseng CH, et al. Accrual strategies for phase Itrials with delayed patient outcome. Stat Med. 1999;18:1155-1169.
96. Estey E, Thall P, David C. Design and analysis of trials ofsalvage therapy in acute myelogenous leukemia. CancerChemother Pharmacol. 1997;40:S9-12.