alkaloid cluster gene ccsa of the ergot fungus …the ergot fungus claviceps purpurea is a...
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Mar. 2010, p. 1822–1830 Vol. 76, No. 60099-2240/10/$12.00 doi:10.1128/AEM.00737-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Alkaloid Cluster Gene ccsA of the Ergot Fungus Claviceps purpurea EncodesChanoclavine I Synthase, a Flavin Adenine Dinucleotide-Containing
Oxidoreductase Mediating the Transformation ofN-Methyl-Dimethylallyltryptophan
to Chanoclavine I�
Nicole Lorenz,1 Jana Olsovska,2 Miroslav Sulc,2 and Paul Tudzynski1*Institut fur Botanik, Westfalische Wilhelms-Universitat Munster, Schlossgarten 3, D-48149 Munster, Germany,1 and Institute of
Microbiology, Academy of Sciences of the Czech Republic, v.v.i., Vídenska 1083, 14220 Prague 4, Czech Republic2
Received 31 March 2009/Accepted 16 January 2010
Ergot alkaloids are indole-derived secondary metabolites synthesized by the phytopathogenic ascomyceteClaviceps purpurea. In wild-type strains, they are exclusively produced in the sclerotium, a hibernation struc-ture; for biotechnological applications, submerse production strains have been generated by mutagenesis. Itwas shown previously that the enzymes specific for alkaloid biosynthesis are encoded by a gene cluster of 68.5kb. This ergot alkaloid cluster consists of 14 genes coregulated and expressed under alkaloid-producingconditions. Although the role of some of the cluster genes in alkaloid biosynthesis could be confirmed by atargeted knockout approach, further functional analyses are needed, especially concerning the early pathway-specific steps up to the production of clavine alkaloids. Therefore, the gene ccsA, originally named easE andpreliminarily annotated as coding for a flavin adenine dinucleotide-containing oxidoreductase, was deleted inthe C. purpurea strain P1, which is able to synthesize ergot alkaloids in axenic culture. Five independentknockout mutants were analyzed with regard to alkaloid-producing capability. Thin-layer chromatography(TLC), ultrapressure liquid chromatography (UPLC), and mass spectrometry (MS) analyses revealed accu-mulation of N-methyl-dimethylallyltryptophan (Me-DMAT) and traces of dimethylallyltryptophan (DMAT),the first pathway-specific intermediate. Since other alkaloid intermediates could not be detected, we concludethat deletion of ccsA led to a block in alkaloid biosynthesis beyond Me-DMAT formation. Complementationwith a ccsA/gfp fusion construct restored alkaloid biosynthesis. These data indicate that ccsA encodes thechanoclavine I synthase or a component thereof catalyzing the conversion of N-methyl-dimethylallyltryptophanto chanoclavine I.
The ergot fungus Claviceps purpurea is a phytopathogenicascomycete which infects the ears of several grasses, replacingthe ovary and producing a hibernation structure, the so-calledsclerotium, in which the ergot alkaloids are formed. Thesesubstances show a high level of structural homology to someneurotransmitters like serotonin and dopamine and can there-fore bind to the same receptors in the central nervous system(CNS), which is the basis for the application of ergot alkaloidsin a variety of clinical conditions (15).
The biochemistry of ergot alkaloid biosynthesis was firstinvestigated by isolation of intermediates and postulation of ahypothetical pathway as well as enzymes needed for the suc-cessive biosynthetic steps of the production (Fig. 1). Most ofthe data were collected by pursuing the fate of radiolabeledprecursors in feeding experiments (4). The first enzyme whichcould be assigned to alkaloid production was dimethylallyltryp-tophan synthetase (DMATS), which is the key enzyme of thepathway and is encoded by the gene dmaW (18). These anal-yses were performed with a Claviceps fusiformis strain, but a
homolog of dmaW (AY259840) possessing a similar functioncould also be isolated in C. purpurea, as was confirmed by aknockout approach (N. Lorenz and P. Tudzynski, unpublisheddata). Using genome walking combined with cDNA screening,a 68.5-kb genomic region surrounding dmaW could be se-quenced and revealed 14 open reading frames (ORFs) (puta-tive genes) encoding, among others, nonribosomal peptidesynthetases (NRPSs), a putative catalase, a CYP450-1 mono-oxygenase, a putative methyltransferase, and several oxidoreduc-tases (6, 13, 19) (Fig. 2). Some of these genes were functionallyand biochemically analyzed by a gene replacement approachwhich revealed their function within the pathway (2, 5, 7). How-ever, there is still a deficit in functional analyses, especially withrespect to the early steps within this pathway. The conversionfrom N-methyl-dimethylallyltryptophan (Me-DMAT) to agrocla-vine via chanoclavine I and chanoclavine I aldehyde includessuccessive oxidation and reduction steps mediated by a specificclass of enzymes, the oxidoreductases (15) (Fig. 1).
These enzymes are involved in the biosynthesis of manyfungal secondary metabolites. A prominent example is thefamily of the cytochrome P450 monooxygenases (named afterthe characteristic peak of 450 nm when complexed with carbonmonoxide). Cytochrome P450 (CYP450) monooxygenases cat-alyze the transfer of one oxygen atom from molecular oxygen
* Corresponding author. Mailing address: Institut fur Botanik, West-falische Wilhelms-Universitat Munster, Schlossgarten 3, D-48149 Mun-ster, Germany. Phone: 49-251-8324998. Fax: 49-251-8321601. E-mail:[email protected].
� Published ahead of print on 29 January 2010.
1822
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

to various substrates, mostly accomplished by the involvementof NAD(P)H as an electron donor. The eas cluster of C. pur-purea also includes a gene encoding a CYP450 monooxygen-ase: cloA is involved in the oxidation of elymoclavine, leadingto the formation of paspalic acid (7).
No further monooxygenase-encoding genes seem to bepresent in the eas cluster, but several genes code for putativeoxidoreductases (easA, easD, easE, easG, and easH). Theseoxidoreductases are most likely involved in the early steps
within the pathway, but none of them has been functionallyanalyzed so far (15).
We initiated a functional analysis of the putative oxi-doreductase-encoding gene ccsA (formerly easE) (Fig. 2). Thecoding region of ccsA (AJ011965; 1,503 bp) is composed of twoexons interrupted by an intron of 52 bp, yielding a codingcapacity of 483 amino acids (aa). The gene product showshighest similarity to putative oxidoreductases of other ergotalkaloid-producing fungi: EasE of C. fusiformis (e�160;
FIG. 1. Biosynthetic pathway of the ergot alkaloid biosynthesis of C. purpurea. Genes analyzed so far have been assigned to the correspondingenzyme at the corresponding position within the pathway. DMAPP, dimethylallyldiphosphate; DMAT, dimethylallyltryptophan; Me-DMAT,N-methyl-DMAT. (Adapted from reference 7 with permission of Wiley-VCH Verlag GmbH & Co. KGaA.)
FIG. 2. Alkaloid biosynthesis gene cluster of C. purpurea. Highlighted in white is the gene of interest ccsA. (Adapted from reference 7 withpermission of Wiley-VCH Verlag GmbH & Co. KGaA.)
VOL. 76, 2010 CHANOCLAVINE SYNTHESIS GENE IN CLAVICEPS PURPUREA 1823
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

ABV57823), EasE of Neotyphodium lolii (e�118; ABM91450)and CpoX1 of Aspergillus fumigatus (e�96; XM_751049). Anal-yses of the protein sequence using the program PROSITErevealed a flavin adenine dinucleotide (FAD)-binding domain(pfam01565) spanning the region from amino acids 14 to 161and a berberine bridge enzyme domain (BBE domain;pfam08031) from amino acids 412 to 457. The role of CcsA inthe alkaloid biosynthesis pathway was investigated by knockoutof the corresponding gene, followed by functional and bio-chemical analyses of the deletion mutants.
MATERIALS AND METHODS
Strains and culture conditions. The ku70-deficient strain derived from Clavi-ceps purpurea strain P1 (ATCC 20102 [8, 19), which produces mainly ergotaminewith small amounts of ergocryptine, was described earlier (5), as were thestandard media and culture conditions (19). For alkaloid production, the funguswas cultivated in T25N medium with low (0.5 g/liter KH2PO4) and high (2.0g/liter KH2PO4) levels of phosphates.
Molecular biology techniques. Standard cloning and DNA analysis techniqueswere performed according to the methods of Sambrook et al. (14). The Esche-richia coli strains used for cloning by using plasmids pUC19 (Fermentas, St.Leon-Rot, Germany) and pCR2.1-TOPO (Invitrogen, Karlsruhe, Germany) andpropagation of clones was TOP10F� (Invitrogen, Karlsruhe, Germany). Extrac-tion of genomic DNA, Southern and Northern blot analyses, and DNA sequenc-ing were performed as described previously (11). For sequence comparisons andmultiple sequence alignments, DNA STAR (Madison, Wisconsin) was used. For
further analyses, the programs BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) and PROSITE (http://www.expasy.ch/prosite/) were utilized.
Design of a replacement vector and transformation of a ku70-deficient strainof C. purpurea. The replacement vector RV-ccsA is based on the vectorpAN8.1_UM, including two multiple cloning sites in front of the gpdA promoter(Aspergillus nidulans) and downstream of the gpdA terminator (Aspergillus nidu-lans) of the phleomycin resistance cassette, an ampicillin resistance gene forselection in bacteria, and an origin of replication (Fig. 3). (For primers used foramplification of the flanks, see below.)
Transformation of protoplasts of C. purpurea was performed as describedpreviously (2).
RNA isolation. Total RNA extraction was done as described earlier (17) from7-day-old mycelia using the RNAgents total RNA isolation system (Promega,Mannheim, Germany). Concentrations of purified RNA were determined usinga BioPhotometer (Eppendorf, Hamburg, Germany), and RNA integrity wasexamined by electrophoresis in 1% formaldehyde agarose gels.
PCR—primers and conditions. For PCR analysis, BioTherm (Genecraft, Lud-inghausen, Germany) polymerase was used according to the manufacturer’sinstructions.
For the construction of the replacement vector, the flanks were amplified bythe primer pairs ccsA_VF_v2 (5�-CCA TTC AGG TAC CCG TCC AG-3�) andccsA_VF_h (5�-AAG GAC AGA ATT CCT CCA CGC-3�) for the 5� flank andccsA_HF_v (5�-CGG AGC TAT CTA GAT AGC ATC-3�) and ccsA_HF_h(5�-TTG GAT GCC GCG GTG AGT GCA T-3�) for the 3� flank. To check thetransformants for homologous integration events, diagnostic PCR was per-formed using the primer pair d_ccsA_v (5�-GCA TCA AGA GCA CAA CAACAC GAG-3�) and PAN3 (5�-GGT CAC CAG TCG CTG GCT TCC CG-3�) forthe 5� flank and the primer pair PAN2 (5�-CCG TAA CAC CCA ATA CGCCGG-3�) and d_ccsA_h (5�-TGC AGC ATA GAC CCC AGA CAG AC-3�) for
FIG. 3. Construction of a replacement vector for knockout of the putative oxidoreductase gene ccsA. Top: genomic situation; orientation ofarrows indicates direction of transcription. Positions of primers utilized for amplification of the flanks are marked by black arrows. The 5� flankof 1,149 bp was amplified by the primer pair ccsA_VF_v2 and ccsA_VF_h and cloned in front of the gpdA promoter of the phleomycin resistancecassette using the restriction sites KpnI and EcoRI. The 3� flank of 1,030 bp was amplified by the primer pair ccsA_HF_v and ccsA_HF_h andcloned behind the gpdA terminator of the phleomycin resistance cassette using the restriction sites NotI and SacII. The complete vector RV-ccsA(8,935 bp) is based on vector pAN8.1_UM. phleo, phleomycin resistance gene; prom, gpdA promoter (Aspergillus nidulans); term, gpdA terminator(A. nidulans). Relative positions of introns are marked by light gray boxes.
1824 LORENZ ET AL. APPL. ENVIRON. MICROBIOL.
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

the 3� flank. Purification of the mutants was checked by the primer pair ccsA_easF_U (5�-ACC GTG GGT GCA GTA GGA GGC-3�) and ccsA_R (5�-GCTTCC GGC AAA TAC CTT CTG-3�).
For complementation of the deletion mutant, the gene ccsA was amplified bythe primer pair ccsA_GFP_v (5�-CAC CAA TTC TAG AAG CAC CG-3�) andccsA_GFP_h (5�-GGT ACC CAT TCC GAT AAG ACT GGA CGC-3�) andcloned into the pUC19 vector in front of the start codon of the gfp (greenfluorescent protein) gene from Aequorea jellyfish (16), leading to a fusion proteinwhere GFP is located at the C terminus of CcsA. The vector already containeda 1-kb fragment of the nitrate reductase gene (niaD) of C. purpurea (see Fig. 9).The niaD gene could be used for targeted integration of the complementationconstruct because of the increased rate of homologous integration of the ku70-deficient strain of C. purpurea. Additionally, a hygromycin resistance cassette(consisting of the oliC promoter, the hygromycin resistance gene, and the trpCterminator of Aspergillus nidulans) was cloned via the XbaI restriction site intothe vector, enabling direct selection of the transformants. Successful integrationwas tested by diagnostic PCR using the primer GFP_h (5�-TCG AAT TCT TACTTG TAC AGC TCG TCC-3�).
Extraction and analyses of ergot alkaloids. For alkaloid extraction and deter-mination, cultures were adjusted to pH 11 with concentrated aqueous ammo-nium hydroxide and extracted three times with chloroform, and after concentra-tion, the resulting liquid was applied onto thin-layer chromatography (TLC)plates (silica gel 60; Merck, Darmstadt, Germany). Alkaloids were identified bycomparison with the corresponding standards in chloroform-methanol-ammo-nium (80:20:0.2; vol/vol/vol). After separation, TLC plates were sprayed withEhrlich’s reagent for alkaloid visualization. Standards used were 1 mg/ml ergot-amine (Sigma, Taufkirchen, Germany), 1 mg/ml ergocryptine (Sigma,Taufkirchen, Germany), 1 mg/ml ergocristine (Sigma, Taufkirchen, Germany), 1mg/ml agroclavine (Sandoz AG, Basel, Switzerland), and 1 mg/ml D-lysergic acid(Sandoz AG, Basel, Switzerland).
High-pressure liquid chromatography (HPLC) separation was carried out on aLiChrospher 100 RP-18 (Merck/Hitachi, Darmstadt, Germany) column (250 � 4mm inner diameter; 5-�m particle size), tempered at 25°C, and operated at aflow rate of 1 ml/min. Compounds were isocratically eluted within 40 min usinga binary mobile phase containing 10 mM ammonium carbonate and acetonitrile(50:50, vol/vol). Ergot alkaloids (EA) were detected with an L-7400 UV detectoroperating at 230 nm. EA standards were used as described above.
UPLC method. An Acquity ultrapressure liquid chromatography (UPLC) sys-tem (Waters, Milford, Massachusetts), equipped with a 2996 polydiode array(PDA) detector operating at 225 nm and 310 nm, was used for EA analysis underUPLC conditions (12). Data were processed with Empower 2 software (Waters).Samples were analyzed on a Waters BEH C18 column (50 � 2.1 mm innerdiameter; 1.7-�m particle size) under the following conditions: column temper-ature, 35°C; data sample rate, 20 data points (pts)/s; filter constant, 0.5; injection
volume, 5 �l; analysis time, 12 min; flow rate, 0.4 ml/min. Mobile phases con-sisted of water (phase A) and acetonitrile (phase B), both containing 0.04%NH4OH. Gradient elution started at 5% acetonitrile (0 min), increasing linearlyto 61% acetonitrile within 12 min. Each analysis was followed with a column-washing step (95% acetonitrile, 1 min) and equilibration step (1 min).
Mass spectrometry analysis. Mass spectra were measured on a matrix-assistedlaser desorption–ionization reflectron time-of-flight (MALDI-TOF) mass spec-trometer (BIFLEX; Bruker-Franzen, Bremen, Germany) equipped with a nitro-gen laser (337 nm) and gridless delayed extraction ion source. The ion acceler-ation voltage was 19 kV, and the reflectron voltage was set to 20 kV. Spectra were
FIG. 4. Gene replacement strategy for ccsA (left) and diagnostic PCR of �ccsA (5) and �ccsA (6) deletion mutants compared to the wild type(right). Primers used for verification of homologous integration of the 5� flank were d_ccsA_v (1) and PAN3 (2), yielding a fragment of 1.8 kb.Integration of the 3� flank was checked using the primer pair PAN2 (3) and d_ccsA_h (4), resulting in a fragment of 1.3 kb. Purification of thetransformants was tested by the primer pair ccsA_easF_U (5) and ccsA_R (6), yielding a fragment of 1.1 kb, exclusively observed in the wild-typeP1. SacI restriction sites used for Southern analyses (Fig. 5) and the sizes of the fragments achieved were also indicated. phleo, phleomycinresistance gene; prom, gpdA promoter (Aspergillus nidulans); term, gpdA terminator (A. nidulans); WT, wild-type band.
FIG. 5. Southern analysis of three pure knockout transformants of�ccsA and the wild-type P1. Genomic DNA of the wild type and threedeletion mutants of ccsA were digested with the restriction enzymeSacI, separated in an agarose gel, blotted to a nylon membrane, andhybridized with the 5� flank of the replacement vector. In the wild type,a band of 4.8 kb could be observed, whereas in the deletion mutants atruncated fragment of 3.4 kb is detected, due to an additional SacIrestriction site in the replacement fragment (see also Fig. 4).
VOL. 76, 2010 CHANOCLAVINE SYNTHESIS GENE IN CLAVICEPS PURPUREA 1825
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

calibrated externally using the monoisotopic [M�H]� ion of matrix peaks 190.1m/z and 379.1 m/z and peptide MRFA (Met-Arg-Phe-Ala) 524.3 m/z. Solutions(10 mg/ml) of �-cyano-4-hydroxy-cinnamic acid or 2,5-dihydrobenzoic acid in50% acetonitrile with 0.3% acetic acid were used as MALDI matrices. Eachcollected UPLC fraction was dried on a vacuum concentrator, dissolved in water,and sonicated for 5 min prior to mass spectrometry analysis. A 1-�l sample of matrixsolution was mixed with 1 �l of the aqueous solution of the sample, and 1 �l of thispremix was loaded on the target and allowed to dry at ambient temperature. The
MALDI-TOF and postsource decay (PSD) spectra were collected in reflectronmode.
RESULTS
Generation of knockout mutants of ccsA in a ku70-deficientstrain of C. purpurea P1. For deletion of ccsA, a double-cross-over approach was used, resulting in replacement of the ccsAlocus by a phleomycin resistance cassette by homologous re-combination (Fig. 4). The replacement construct was based onthe phleomycin resistance cassette of vector pAN8.1_UMflanked by 1,149-bp and 1,030-bp 5� and 3� regions, respec-tively, of ccsA (Fig. 4).
As the recipient strain, a ku70-deficient derivative of theproducer strain P1 was used, characterized by an enhancedefficiency for homologous recombination (5).
Successful integration was checked by diagnostic PCR usingthe primer pair d_ccsA_v and PAN3 for homologous integra-tion of the 5� flank (yielding a fragment of 1.8 kb) and primerpair PAN2 and d_ccsA_h for verification of the 3� flank (yield-ing a band of 1.4 kb; Fig. 4). Of 22 transformants checked, 13showed homologous integration. Interestingly, 6 out of these13 also missed the wild-type band when the primer pairccsA_easF_U and ccsA_R was used, indicating that thesetransformants were already homokaryotic and contained onlytransformed nuclei.
Successful knockout was also confirmed by Southern analy-sis using SacI-digested DNA of wild-type strain P1 as well asfive homokaryotic transformants (Fig. 5). The gene ccsA lacksany SacI restriction site, so in the wild type only one band of 4.8kb could be observed. In contrast, the gene replacement con-
FIG. 6. Northern analysis of �ccsA (5) and �ccsA (6) mutants andwild-type P1. Total RNA of the wild-type P1 and of two deletionmutants of ccsA were isolated, separated in a formaldehyde agarosegel (1%), and transferred to a nylon membrane. Hybridization wascarried out using fragments of the genes ccsA, easD, and easF andribosomal DNA genes (rDNA) as a loading control.
FIG. 7. UPLC analyses of the transformant �ccsA (14). A peak at time point 4.32 min could be detected with a protonated molecule at m/z 287.2in MALDI-TOF analysis. The compound possessed a UV spectrum typical for ergot alkaloids showing maxima at 224 nm and 280 nm. The same UVspectrum could be observed for the compound detected at time point 4.58 min with a MALDI-TOF signal at m/z 272.2. AU, arbitrary units.
1826 LORENZ ET AL. APPL. ENVIRON. MICROBIOL.
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from
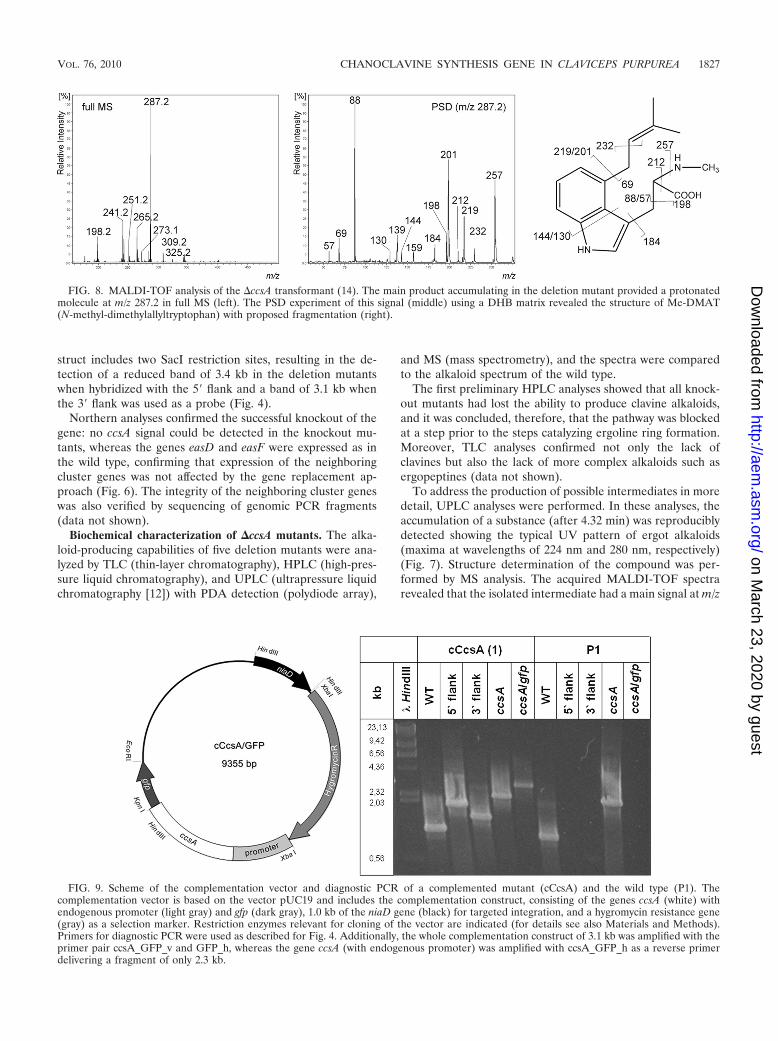
struct includes two SacI restriction sites, resulting in the de-tection of a reduced band of 3.4 kb in the deletion mutantswhen hybridized with the 5� flank and a band of 3.1 kb whenthe 3� flank was used as a probe (Fig. 4).
Northern analyses confirmed the successful knockout of thegene: no ccsA signal could be detected in the knockout mu-tants, whereas the genes easD and easF were expressed as inthe wild type, confirming that expression of the neighboringcluster genes was not affected by the gene replacement ap-proach (Fig. 6). The integrity of the neighboring cluster geneswas also verified by sequencing of genomic PCR fragments(data not shown).
Biochemical characterization of �ccsA mutants. The alka-loid-producing capabilities of five deletion mutants were ana-lyzed by TLC (thin-layer chromatography), HPLC (high-pres-sure liquid chromatography), and UPLC (ultrapressure liquidchromatography [12]) with PDA detection (polydiode array),
and MS (mass spectrometry), and the spectra were comparedto the alkaloid spectrum of the wild type.
The first preliminary HPLC analyses showed that all knock-out mutants had lost the ability to produce clavine alkaloids,and it was concluded, therefore, that the pathway was blockedat a step prior to the steps catalyzing ergoline ring formation.Moreover, TLC analyses confirmed not only the lack ofclavines but also the lack of more complex alkaloids such asergopeptines (data not shown).
To address the production of possible intermediates in moredetail, UPLC analyses were performed. In these analyses, theaccumulation of a substance (after 4.32 min) was reproduciblydetected showing the typical UV pattern of ergot alkaloids(maxima at wavelengths of 224 nm and 280 nm, respectively)(Fig. 7). Structure determination of the compound was per-formed by MS analysis. The acquired MALDI-TOF spectrarevealed that the isolated intermediate had a main signal at m/z
FIG. 8. MALDI-TOF analysis of the �ccsA transformant (14). The main product accumulating in the deletion mutant provided a protonatedmolecule at m/z 287.2 in full MS (left). The PSD experiment of this signal (middle) using a DHB matrix revealed the structure of Me-DMAT(N-methyl-dimethylallyltryptophan) with proposed fragmentation (right).
FIG. 9. Scheme of the complementation vector and diagnostic PCR of a complemented mutant (cCcsA) and the wild type (P1). Thecomplementation vector is based on the vector pUC19 and includes the complementation construct, consisting of the genes ccsA (white) withendogenous promoter (light gray) and gfp (dark gray), 1.0 kb of the niaD gene (black) for targeted integration, and a hygromycin resistance gene(gray) as a selection marker. Restriction enzymes relevant for cloning of the vector are indicated (for details see also Materials and Methods).Primers for diagnostic PCR were used as described for Fig. 4. Additionally, the whole complementation construct of 3.1 kb was amplified with theprimer pair ccsA_GFP_v and GFP_h, whereas the gene ccsA (with endogenous promoter) was amplified with ccsA_GFP_h as a reverse primerdelivering a fragment of only 2.3 kb.
VOL. 76, 2010 CHANOCLAVINE SYNTHESIS GENE IN CLAVICEPS PURPUREA 1827
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

287.2, and manual interpretation of its MS/MS fragmentationresulted in identification of N-methyl-dimethylallyltryptophan(Me-DMAT) as the main end product of the deletion mutants(Fig. 8). Dimethylallyltryptophan (DMAT) was also found inminor amounts in the UPLC fraction at the time point 4.58min, with a MALDI-TOF signal at m/z 272.2. The identifica-tion of Me-DMAT as the main product in contrast to DMATstrengthens the assumption that the pathway is blocked at thebiosynthetic step following DMAT methylation, which explainswhy Me-DMAT accumulates whereas DMAT is constantly
converted to Me-DMAT and thus present in only minoramounts. The next pathway-specific intermediate not detectedin the deletion mutants is chanoclavine I, supporting the pre-sumption that CcsA is essential for the conversion from Me-DMAT to chanoclavine I. Therefore, the gene—originallycalled easE (15)—was named ccsA (for chanoclavine synthase).
Complementation of the �ccsA mutants. To confirm that thefailure of the mutants to produce clavine and more-complexalkaloids was due to the targeted knockout, the transformantswere complemented with a gene construct designed to express
FIG. 10. HPLC analysis of the wild type (P1), the deletion mutant (�ccsA), and the complemented mutant (cCcsA:gfp). Peaks: 1, ergotamine;2, ergocryptine; 3, ergotaminine; 4, methyl-dimethylallyltryptophan.
1828 LORENZ ET AL. APPL. ENVIRON. MICROBIOL.
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

a CcsA::GFP fusion protein. This construct could allow notonly testing for restoration of alkaloid production but alsoanalysis of the cellular localization of alkaloid biosynthesis,which is described elsewhere as compartmentalized (15).
The complementation vector cCcsA/GFP contained the fullcoding region of ccsA with its endogenous promoter (to avoidoverexpression), the gfp gene fused in frame to the 3� end ofccsA, and part of the niaD (nitrate reductase) locus for tar-geted integration in this nonessential gene (for details, seeMaterials and Methods) (Fig. 9). Integration of the comple-mentation fragment in the niaD locus was confirmed by PCR(Fig. 9) as well as Southern analysis (data not shown), verifyingalso that—due to the integration outside the eas cluster—thegene replacement situation was still present.
The expression pattern, as well as the alkaloid spectrum ofthe complemented mutants, was analyzed by Northern blottingand HPLC. The expression of the ccsA/gfp fusion construct wasconfirmed by the presence of a larger ccsA homologous tran-script (data not shown), and functionality of the resulting fu-sion protein was shown by restoration of ergotamine produc-tion in the complemented mutant (Fig. 10). The GFP signalobtained, however, was rather diffuse, not supporting the as-sumption that the biosynthesis is located in specialized cellularcompartments (data not shown).
DISCUSSION
A knockout of the eas cluster gene ccsA resulted in accumu-lation of Me-DMAT and traces of DMAT, as indicated byUPLC and MS analyses, strongly suggesting that CcsA is in-volved in the “chanoclavine I synthase” activity convertingMe-DMAT to chanoclavine I (Fig. 1).
Two successive oxidation steps are required for chanoclavineI formation from Me-DMAT (Fig. 11). The reaction mostlikely starts with the generation of the diene by desaturation ofthe C8-C9 bond and loss of a proton at C17 (4). This is followedby rotation around the C8-C9 bond and oxidation of the C7-C8,proposed to give an epoxide intermediate (4). Decarboxylationof this intermediate by a (spontaneous) SN2� reaction could becoupled with bond formation between C5 and C10 and cleavageof the epoxide. In addition to binding the FAD cofactor, it isconceivable that the BBE domain is involved in the subsequentcyclization, either as a catalyst or by determining stereospeci-ficity, as has been suggested for the BBE (3, 9). Following this,ring opening of the epoxide occurs by proton attack with shiftof the double bond between C9 and C10 to the position be-tween C8 and C9.
We showed here that CcsA is essential for this biosyntheticstep, but it remains an open question whether the reaction iscarried out by CcsA alone or whether CcsA belongs to acomplex of enzymes catalyzing the two oxidation steps to formthe diene and then the epoxide, as was postulated by Schardl etal. (15). Experiments involving the knockout of other putativeoxidoreductase-encoding genes of the eas cluster, e.g., easA,easD, easG, or easH, could help in further elucidating thisprocess.
ACKNOWLEDGMENTS
We thank S. Pazoutova and M. Flieger, Prague, Czech Republic, fordiscussions and U. Keller, Berlin, Germany, for critical reading of themanuscript. The DFG (special focus program Evolution of MetabolicDiversity) provided financial support.
REFERENCES
1. Reference deleted.2. Correia, T., N. Grammel, I. Ortel, U. Keller, and P. Tudzynski. 2003. Mo-
lecular cloning and analysis of the ergopeptine assembly system in the ergotfungus Claviceps purpurea. Chem. Biol. 10:1281–1292.
3. Facchini, P. J., C. Penzes, A. G. Johnson, and D. Bull. 1996. Molecularcharacterization of berberine bridge enzyme genes from opium poppy. PlantPhysiol. 112:1669–1677.
4. Groger, D., and H. G. Floss. 1998. Biochemistry of ergot alkaloids—achieve-ments and challenges, p. 171. In G. A. Cordell (ed.), Alkaloids, vol. 50.Academic Press, New York, NY.
5. Haarmann, T., N. Lorenz, and P. Tudzynski. 2008. Use of a non-homologousend-joining deficient strain (�ku70) of the ergot fungus Claviceps purpureafor identification of a nonribosomal peptide synthetase gene involved inergotamine biosynthesis. Fungal Gen. Genet. 45:35–44.
6. Haarmann, T., C. Machado, Y. Lubbe, T. Correia, C. L. Schardl, D. G.Panaccione, and P. Tudzynski. 2005. The ergot alkaloid gene cluster inClaviceps purpurea: extension of the cluster sequence and intra species evo-lution. Phytochemistry 66:1312–1320.
7. Haarmann, T., I. Ortel, P. Tudzynski, and U. Keller. 2006. Identification ofthe cytochrome P450 monooxygenase that bridges the clavine and ergolinealkaloid pathways. Chembiochem 7:645–652.
8. Keller, U., M. Han, and M. Stoffler-Meilicke. 1988. D-lysergic acid activationand cell-free synthesis of D-lysergyl peptides in enzyme fractions from theergot fungus Claviceps purpurea. Biochemistry 27:6164–6170.
9. Kutchan, T. M., and H. Dittrich. 1995. Characterization and mechanism ofthe berberine bridge enzyme, a covalently flavinylated oxidase of benzo-phenanthridine alkaloid biosynthesis in plants. J. Biol. Chem. 270:24475–24481.
10. Reference deleted.11. Mey, G., K. Held, J. Scheffer, K. B. Tenberge, and P. Tudzynski. 2002.
CPMK2, a SLT2-homologous mitogen-activated protein (MAP) kinase, isessential for pathogenesis of Claviceps purpurea on rye: evidence for a secondconserved pathogenesis-related MAP kinase cascade in phytopathogenicfungi. Mol. Microbiol. 46:305–318.
12. Olsovska, J., M. Sulc, P. Novak, S. Pazoutova, and M. Flieger. 2008. Liquidchromatography-tandem mass spectrometry characterization of ergocristamdegradation products. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.873:165–172.
13. Rigbers, O., and S. M. Li. 2008. Ergot alkaloid biosynthesis in Aspergillusfumigatus. Overproduction and biochemical characterization of a 4-dimethyl-allyltryptophan N-methyltransferase. J. Biol. Chem. 283:26859–26868.
14. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a
FIG. 11. Early steps of the ergot alkaloid pathway, from N-methyl-dimethylallyltryptophan (Me-DMAT) to chanoclavine I. For details, seetext.
VOL. 76, 2010 CHANOCLAVINE SYNTHESIS GENE IN CLAVICEPS PURPUREA 1829
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from

laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, NY.
15. Schardl, C. L., D. G. Panaccione, and P. Tudzynski. 2006. Ergot alkaloids—biology and molecular biology. Alkaloids Chem. Biol. 63:45–86.
16. Shimomura, O., F. H. Johnson, and Y. Saiga. 1962. Extraction, purificationand properties of aequorin, a bioluminescent protein from the luminoushydromedusan, Aequorea. J. Cell. Comp. Physiol. 59:223–239.
17. Spiering, M. J., H. H. Wilkinson, J. D. Blankenship, and C. L. Schardl. 2002.Expressed sequence tags and genes associated with loline alkaloid expression
by the fungal endophyte Neotyphodium uncinatum. Fungal Genet. Biol. 36:242–254.
18. Tsai, H. F., H. Wang, J. C. Gebler, C. D. Poulter, and C. L. Schardl. 1995.The Claviceps purpurea gene encoding dimethylallyltryptophan synthase, thecommitted step for ergot alkaloid biosynthesis. Biochem. Biophys. Res.Commun. 216:119–125.
19. Tudzynski, P., K. Holter, T. Correia, C. Arntz, N. Grammel, and U. Keller.1999. Evidence for an ergot alkaloid gene cluster in Claviceps purpurea. Mol.Gen. Genet. 261:133–141.
1830 LORENZ ET AL. APPL. ENVIRON. MICROBIOL.
on March 23, 2020 by guest
http://aem.asm
.org/D
ownloaded from