−mediated enhanced catabolism of norepinephrine ... · where it principally catabolizes...
TRANSCRIPT

David A. Kass, Fabio Di Lisa and Nazareno PaolocciBedja, Kevin Chen, Kathleen L. Gabrielson, Randy D. Blakely, Jean C. Shih, Karel Pacak, Nina Kaludercic, Eiki Takimoto, Takahiro Nagayama, Ning Feng, Edwin W. Lai, Djahida
Adverse Remodeling and Pump Failure in Hearts With Pressure OverloadMediated Enhanced Catabolism of Norepinephrine Contributes to−Monoamine Oxidase A
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.109.1983662010;106:193-202; originally published online November 12, 2009;Circ Res.
http://circres.ahajournals.org/content/106/1/193World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2009/11/12/CIRCRESAHA.109.198366.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

Monoamine Oxidase A–Mediated Enhanced Catabolism ofNorepinephrine Contributes to Adverse Remodeling and
Pump Failure in Hearts With Pressure OverloadNina Kaludercic, Eiki Takimoto, Takahiro Nagayama, Ning Feng, Edwin W. Lai, Djahida Bedja,
Kevin Chen, Kathleen L. Gabrielson, Randy D. Blakely, Jean C. Shih, Karel Pacak, David A. Kass,Fabio Di Lisa, Nazareno Paolocci
Rationale: Monoamine oxidases (MAOs) are mitochondrial enzymes that catabolize prohypertrophic neurotrans-mitters, such as norepinephrine and serotonin, generating hydrogen peroxide. Because excess reactive oxygenspecies and catecholamines are major contributors to the pathophysiology of congestive heart failure, MAOscould play an important role in this process.
Objective: Here, we investigated the role of MAO-A in maladaptive hypertrophy and heart failure.Methods and Results: We report that MAO-A activity is triggered in isolated neonatal and adult myocytes on
stimulation with norepinephrine, followed by increase in cell size, reactive oxygen species production, and signsof maladaptive hypertrophy. All of these in vitro changes occur, in part, independently from �- and �-adrenergicreceptor–operated signaling and are inhibited by the specific MAO-A inhibitor clorgyline. In mice with leftventricular dilation and pump failure attributable to pressure overload, norepinephrine catabolism by MAO-Ais increased accompanied by exacerbated oxidative stress. MAO-A inhibition prevents these changes, and alsoreverses fetal gene reprogramming, metalloproteinase and caspase-3 activation, as well as myocardial apoptosis.The specific role of MAO-A was further tested in mice expressing a dominant-negative MAO-A (MAO-Aneo),which were more protected against pressure overload than their wild-type littermates.
Conclusions: In addition to adrenergic receptor– dependent mechanisms, enhanced MAO-A activity coupledwith increased intramyocardial norepinephrine availability results in augmented reactive oxygen speciesgeneration, contributing to maladaptive remodeling and left ventricular dysfunction in hearts subjected tochronic stress. (Circ Res. 2010;106:193-202.)
Key Words: monoamine oxidase A � congestive heart failure � oxidative stress � catecholamines� norepinephrine transporter
Hemodynamic overload leads to increased cardiac massand cardiomyocyte volume associated with characteris-
tic changes in gene and protein expression. This initialcompensatory hypertrophy is replaced by progressive struc-tural/functional cardiac remodeling involving alterations inextracellular matrix composition, myocardial energetics,Ca2� cycling, and myocyte viability.1–4 Reactive oxygenspecies (ROS) contribute to each of these abnormalities,5–10
but relevant sources of ROS remain to be fully defined. Thisis important, because recent studies suggest that specifictargeting of ROS sources provides a more effective therapyfor heart remodeling.11
Monoamine oxidases (MAOs) are mitochondrial flavoen-zymes which catalyze oxidative deamination of catechol-amines (CAs) and biogenic amines such as serotonin. Duringthis process, they generate hydrogen peroxide (H2O2) andthus can potentially be a source of oxidative stress in theheart, particularly under stress conditions. Yet, although therole of MAOs in terminating neurotransmitter signaling inthe brain is well established,12 little is known about itsmodulation of cardiac morphology and function. MAOs existin 2 isoforms, MAO-A and -B, with distinct substrate andinhibitor sensitivity.12 MAO-A, in particular, is present in themyocardium of several species from humans to rodents,13–15
Original received April 3, 2009; revision received October 28, 2009; accepted October 30, 2009.From the Division of Cardiology (N.K., E.T., T.N., N.F., D.A.K., N.P.) and Department of Comparative Pathobiology (D.B., K.L.G.), Johns Hopkins
Medical Institutions, Baltimore, Md; Department of Biomedical Sciences (N.K., F.D.L.), University of Padova, Italy; Section on MedicalNeuroendocrinology (E.W.L., K.P.), National Institute of Child Health and Development, NIH, Bethesda, Md; Department of Pharmacology andPharmaceutical Sciences (K.C., J.C.S.); and Department of Cell and Neurobiology (J.C.S.), Keck School of Medicine, University of Southern California,Los Angeles; Center for Molecular Neuroscience and Department of Pharmacology (R.D.B.), Vanderbilt University, Nashville, Tenn; and Departmentof Clinical Medicine (N.P.), University of Perugia, Italy.
This manuscript was sent to David Eisner, Consulting Editor, for review by expert referees, editorial decision, and final disposition.Correspondence to Nazareno Paolocci, MD, PhD, Division of Cardiology/Ross 858, Johns Hopkins School of Medicine, 720 Rutland Ave, Baltimore,
MD 21205. E-mail [email protected]© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.109.198366
193 at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

where it principally catabolizes serotonin, norepinephrine(NE) and epinephrine. All of these monoamine neurotrans-mitters have major functional implications in the heart,especially in the modulation of cardiac inotropy. A role forMAO-A and serotonin in ischemic/reperfused myocardiumhas been delineated showing that inhibiting MAO-A coun-termands oxidative stress, neutrophil accumulation andmitochondria-dependent cell death.16 Yet, the involvement ofMAO-A and its impact on neurotransmitter availability incongestive heart failure (CHF) remains poorly defined, al-though one could speculate that this is the type of settingwherein MAO-A might be chiefly at play. CHF is accompa-nied by enhanced sympathetic tone,17 excess circulating CAssuch as NE,18 and increased oxidative stress.19 In suchsituations, MAO-A may be upregulated, generating greateramounts of H2O2 thus exacerbating disease progression.
Here, we first determined whether cardiomyocyte (bothneonatal and adult) exposure to NE is coupled to augmentedMAO-A expression/activity, which in turn regulates a hyper-trophic response linked to the generation of intracellularROS. Adrenergic receptor–independent signaling was as-sessed using the MAO substrate tyramine. Second, we hy-pothesized that persistent hemodynamic stress imposed by invivo pressure overload leads to excess CAs (and/or serotonin)presence at the myocyte surface resulting in increased sarcolem-mal transport via extraneuronal monoamine transporter,20,21 thusenhancing substrate availability for intramyocyte MAO-Aactivity. To test this, we subjected mice to transverse aorticconstriction (TAC), in the absence and presence of the highlyselective MAO-A inhibitor clorgyline,22 and in mice express-ing a dominant negative MAO-A (MAO-Aneo). Here, weshow that NE triggers ROS and myocyte hypertrophy in partby a MAO-A dependent mechanism in vitro. In vivo, NEcatabolism and ROS production are markedly upregulated inpressure overloaded hearts and both effects are amelioratedby inhibiting MAO-A activity to suppress cardiac decompen-sation with pressure overload.
MethodsAnimalsFor pharmacological studies, male C57BL/6 mice (n�30, 9 to 11weeks old) were used and clorgyline was administered using salineas vehicle (1 mg/kg per day, IP). For experiments using geneticallymodified mice, MAO-Aneo mice and their wild-type (WT) litter-mates in 129/Sv background were used (n�30). The Johns HopkinsUniversity Institutional Animal Care and Use Committee approvedall animal experiments.
StatisticsAll values are expressed as means�SEM. Comparison betweengroups was performed by 1-way or 2-way ANOVA, followed by aTukey’s post hoc multiple comparison test. Comparisons betweentwo groups were performed using nonpaired 2-tailed Student t test. Avalue of P�0.05 was considered significant.
An expanded Methods section is available in the Online DataSupplement at http://circres.ahajournals.org.
ResultsNorepinephrine Triggers CardiomyocyteMaladaptive Hypertrophy in aMAO-A–Dependent MannerWe first tested whether externally applied NE (as whenreleased from varicosities into the neuroeffector junctionalareas) can enter myocytes, stimulating MAO-A activity andROS-dependent signaling. Cultured rat neonatal cardiomyo-cytes were incubated with NE (10 �mol/L, 24 hours), whichresulted in cell hypertrophy indexed by increased brainnatriuretic peptide (BNP) gene expression and 1.3-fold in-crease in myocyte area (Figure 1A and 1B). This effect wasaccompanied by a rise in MAO-A gene expression andassociated with a markedly increased production of mito-chondrial ROS, measured with Mitotracker Red (Figure 1Cand 1D). In these same experimental conditions, mitochon-drial membrane potential was unchanged (Online Figure IV).Coincubation with the selective MAO-A inhibitor clorgy-line23 (2 �mol/L) significantly blunted NE-induced ROSproduction and myocyte hypertrophy (Figure 1A, 1B, and1D). Similar inhibition was obtained with 5 �mol/L clorgy-line (40% to 50%, data not shown). To test the contributionof adrenergic receptor signaling, myocytes were incubatedwith tyramine, a MAO substrate that does not interact withadrenergic receptors yet is also transported into cells viathe extraneuronal monoamine transporter.24 Myocytestreated with tyramine for 24 hours displayed a markedincrease in mitochondrial oxidative stress (Figure 1D), andclorgyline prevented this change. Tyramine also increasedmyocyte area and BNP (Figure 1E and 1F), NFAT3(nuclear factor of activated T cells), and NFAT4 (Figure1G) gene expression; the latter are transcription factorsimplicated in maladaptive hypertrophy.25 Clorgyline inhib-ited tyramine-induced hypertrophy and suppressed in-creases in BNP and NFAT expression induced by eitherNE or tyramine.
We further tested these effects in adult cultured myocytesisolated from WT and MAO-Aneo mice, the latter expressinga dominant negative MAO-A, resulting in only 8% of residualMAO-A catalytic activity. WT myocytes challenged with NEdisplayed a marked arise in ROS production and thiswas significantly less in myocytes from MAO-Aneo mice
Non-standard Abbreviations and Acronyms
5-HIAA 5-hydroxyindoleacetic acid
5-HT 5-hydroxytryptamine
BNP brain natriuretic peptide
CA catecholamine
CHF congestive heart failure
DHPG dihydroxyphenylglycol
LV left ventricle
MAO monoamine oxidase
MHC myosin heavy chain
MMP matrix metalloproteinase
NE norepinephrine
NET norepinephrine transporter
PV pressure–volume
ROS reactive oxygen species
TAC transverse aortic constriction
WT wild type
194 Circulation Research January 8, 2010
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

(Figure 2A). Likewise, increased cell area from NE expo-sure in WT myocytes was reduced in those from MAO-Aneo hearts (Figure 2B). These data further support thehypothesis that, in addition to adrenergic receptors,MAO-A derived ROS also contribute to NE-induced hy-pertrophy. The fact that in 2 different systems (neonatal ratand adult mouse myocytes) clorgyline or the geneticsuppression of MAO-A activity produced a nearly equalreduction in ROS production and hypertrophy validatesclorgyline as a specific MAO-A inhibitor.
Norepinephrine Catabolism Is Increased in Failing(TAC) Hearts Because of EnhancedMAO-A ActivityWe next tested whether these findings pertained in vivo. Micewere subjected to TAC and MAO-A gene expression/activitywas examined. As expected,19 TAC increased left ventricle(LV) mass/body weight ratio (�330% versus sham,P�0.001), chamber dilation, and LV dysfunction, all featuresof adverse remodeling (Table 1). In these hearts, MAO-Agene expression was 3-fold higher (Figure 3A, top). To test
Figure 1. MAO-A triggers hypertrophy in neonatal rat cardiac myocytes. A, BNP gene expression levels in vehicle- or NE-treated myo-cytes (10 �mol/L, 24 hours) in the absence or presence of clorgyline (2 �mol/L). B, Myocyte area measured in control and NE-treatedcells (10 �mol/L NE, 24 hours) without or with clorgyline. C, MAO-A gene expression in vehicle and NE-treated cells. D, MitochondrialROS production determined by Mitotracker Red after 2 hours of incubation with 10 �mol/L NE (left) or 20 �mol/L tyramine (right), with-out or with clorgyline. E, Myocyte hypertrophy induced by tyramine determined by �-actinin staining and shown as increase in myocytearea (20 �mol/L, 24 hours). F, BNP gene expression levels in vehicle or tyramine-treated myocytes (20 �mol/L, 24 hours), without orwith clorgyline. G, NFAT3 and NFAT4 increase in gene expression. *P�0.05 vs respective control; **P�0.001 vs respective control;†P�0.05 treatment�clorgyline vs treatment alone.
Figure 2. NE induces ROS formationand hypertrophy in adult mouse ventric-ular myocytes. A, Mitochondrial ROSproduction determined by MitotrackerRed after 2 hours of incubation with10 �mol/L NE in WT and MAO-Aneo
myocytes. B, Myocyte area measured inWT and MAO-Aneo vehicle- andNE-treated myocytes (1 �mol/L NE, 24hours). *P�0.001 vs respective control,†P�0.005 vs NE treatment.
Kaludercic et al MAO-A and Congestive Heart Failure 195
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from
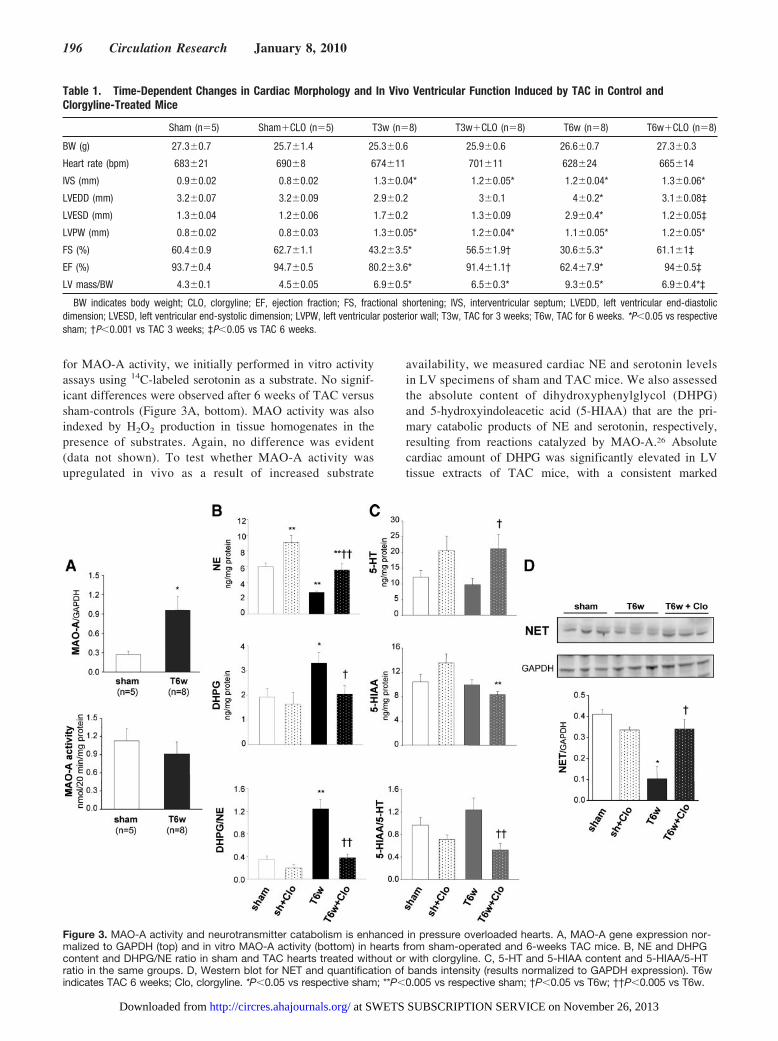
for MAO-A activity, we initially performed in vitro activityassays using 14C-labeled serotonin as a substrate. No signif-icant differences were observed after 6 weeks of TAC versussham-controls (Figure 3A, bottom). MAO activity was alsoindexed by H2O2 production in tissue homogenates in thepresence of substrates. Again, no difference was evident(data not shown). To test whether MAO-A activity wasupregulated in vivo as a result of increased substrate
availability, we measured cardiac NE and serotonin levelsin LV specimens of sham and TAC mice. We also assessedthe absolute content of dihydroxyphenylglycol (DHPG)and 5-hydroxyindoleacetic acid (5-HIAA) that are the pri-mary catabolic products of NE and serotonin, respectively,resulting from reactions catalyzed by MAO-A.26 Absolutecardiac amount of DHPG was significantly elevated in LVtissue extracts of TAC mice, with a consistent marked
Figure 3. MAO-A activity and neurotransmitter catabolism is enhanced in pressure overloaded hearts. A, MAO-A gene expression nor-malized to GAPDH (top) and in vitro MAO-A activity (bottom) in hearts from sham-operated and 6-weeks TAC mice. B, NE and DHPGcontent and DHPG/NE ratio in sham and TAC hearts treated without or with clorgyline. C, 5-HT and 5-HIAA content and 5-HIAA/5-HTratio in the same groups. D, Western blot for NET and quantification of bands intensity (results normalized to GAPDH expression). T6windicates TAC 6 weeks; Clo, clorgyline. *P�0.05 vs respective sham; **P�0.005 vs respective sham; †P�0.05 vs T6w; ††P�0.005 vs T6w.
Table 1. Time-Dependent Changes in Cardiac Morphology and In Vivo Ventricular Function Induced by TAC in Control andClorgyline-Treated Mice
Sham (n�5) Sham�CLO (n�5) T3w (n�8) T3w�CLO (n�8) T6w (n�8) T6w�CLO (n�8)
BW (g) 27.3�0.7 25.7�1.4 25.3�0.6 25.9�0.6 26.6�0.7 27.3�0.3
Heart rate (bpm) 683�21 690�8 674�11 701�11 628�24 665�14
IVS (mm) 0.9�0.02 0.8�0.02 1.3�0.04* 1.2�0.05* 1.2�0.04* 1.3�0.06*
LVEDD (mm) 3.2�0.07 3.2�0.09 2.9�0.2 3�0.1 4�0.2* 3.1�0.08‡
LVESD (mm) 1.3�0.04 1.2�0.06 1.7�0.2 1.3�0.09 2.9�0.4* 1.2�0.05‡
LVPW (mm) 0.8�0.02 0.8�0.03 1.3�0.05* 1.2�0.04* 1.1�0.05* 1.2�0.05*
FS (%) 60.4�0.9 62.7�1.1 43.2�3.5* 56.5�1.9† 30.6�5.3* 61.1�1‡
EF (%) 93.7�0.4 94.7�0.5 80.2�3.6* 91.4�1.1† 62.4�7.9* 94�0.5‡
LV mass/BW 4.3�0.1 4.5�0.05 6.9�0.5* 6.5�0.3* 9.3�0.5* 6.9�0.4*‡
BW indicates body weight; CLO, clorgyline; EF, ejection fraction; FS, fractional shortening; IVS, interventricular septum; LVEDD, left ventricular end-diastolicdimension; LVESD, left ventricular end-systolic dimension; LVPW, left ventricular posterior wall; T3w, TAC for 3 weeks; T6w, TAC for 6 weeks. *P�0.05 vs respectivesham; †P�0.001 vs TAC 3 weeks; ‡P�0.05 vs TAC 6 weeks.
196 Circulation Research January 8, 2010
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from
decrease in cardiac NE content (Figure 3B). When cardiacDHPG content was normalized to intracardiac NE levels, theDHPG/NE ratio was 4-fold higher in TAC hearts versusshams (Figure 3B). Thus, in TAC hearts NE catabolism isincreased. Evidence showing that MAO-A inhibition byclorgyline fully reverted the rise in DHPG and DHPG/NEratio while rescuing the intracardiac content of NE availablefor release (Figure 3B) supports the specific involvement ofMAO-A in this setting.
We also determined whether serotonin, another electiveMAO-A substrate, is equally important in this CHF model.Serotonin levels were unaltered by TAC, and rose only afterclorgyline treatment, consistent with MAO-A inhibition (Fig-ure 3C). In stark contrast to DHPG, serotonin catabolismmeasured by 5-HIAA and 5-HIAA/serotonin ratio was notaltered in TAC hearts compared to shams. However, clorgy-line significantly reduced 5-HIAA/serotonin ratio after TAC.Together, these data show NE to be the preferred substratefueling MAO-A activity in this model.
Intriguingly, the protein expression of the neuronal NEtransporter (NET) declined with TAC, likely reducing neu-ronal NE reuptake. Clorgyline treated TAC mice had normalNET expression (Figure 3D) and higher neuronal NE levels.
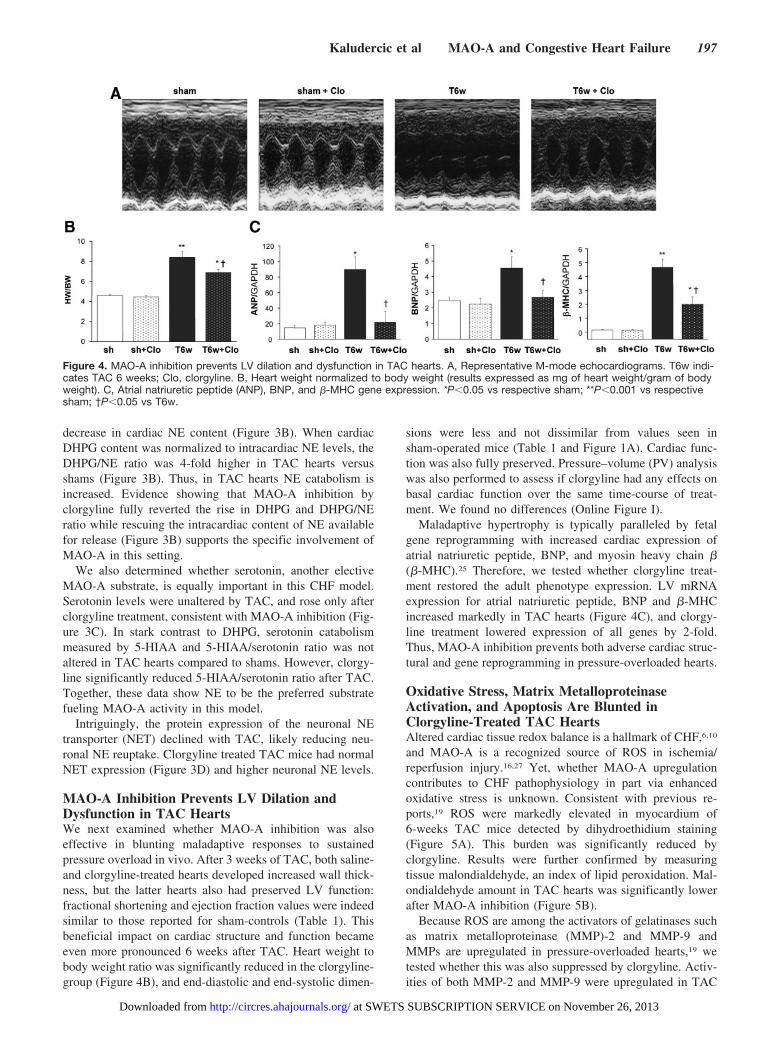
MAO-A Inhibition Prevents LV Dilation andDysfunction in TAC HeartsWe next examined whether MAO-A inhibition was alsoeffective in blunting maladaptive responses to sustainedpressure overload in vivo. After 3 weeks of TAC, both saline-and clorgyline-treated hearts developed increased wall thick-ness, but the latter hearts also had preserved LV function:fractional shortening and ejection fraction values were indeedsimilar to those reported for sham-controls (Table 1). Thisbeneficial impact on cardiac structure and function becameeven more pronounced 6 weeks after TAC. Heart weight tobody weight ratio was significantly reduced in the clorgyline-group (Figure 4B), and end-diastolic and end-systolic dimen-
sions were less and not dissimilar from values seen insham-operated mice (Table 1 and Figure 1A). Cardiac func-tion was also fully preserved. Pressure–volume (PV) analysiswas also performed to assess if clorgyline had any effects onbasal cardiac function over the same time-course of treat-ment. We found no differences (Online Figure I).
Maladaptive hypertrophy is typically paralleled by fetalgene reprogramming with increased cardiac expression ofatrial natriuretic peptide, BNP, and myosin heavy chain �(�-MHC).25 Therefore, we tested whether clorgyline treat-ment restored the adult phenotype expression. LV mRNAexpression for atrial natriuretic peptide, BNP and �-MHCincreased markedly in TAC hearts (Figure 4C), and clorgy-line treatment lowered expression of all genes by 2-fold.Thus, MAO-A inhibition prevents both adverse cardiac struc-tural and gene reprogramming in pressure-overloaded hearts.
Oxidative Stress, Matrix MetalloproteinaseActivation, and Apoptosis Are Blunted inClorgyline-Treated TAC HeartsAltered cardiac tissue redox balance is a hallmark of CHF,6,10
and MAO-A is a recognized source of ROS in ischemia/reperfusion injury.16,27 Yet, whether MAO-A upregulationcontributes to CHF pathophysiology in part via enhancedoxidative stress is unknown. Consistent with previous re-ports,19 ROS were markedly elevated in myocardium of6-weeks TAC mice detected by dihydroethidium staining(Figure 5A). This burden was significantly reduced byclorgyline. Results were further confirmed by measuringtissue malondialdehyde, an index of lipid peroxidation. Mal-ondialdehyde amount in TAC hearts was significantly lowerafter MAO-A inhibition (Figure 5B).
Because ROS are among the activators of gelatinases suchas matrix metalloproteinase (MMP)-2 and MMP-9 andMMPs are upregulated in pressure-overloaded hearts,19 wetested whether this was also suppressed by clorgyline. Activ-ities of both MMP-2 and MMP-9 were upregulated in TAC
Figure 4. MAO-A inhibition prevents LV dilation and dysfunction in TAC hearts. A, Representative M-mode echocardiograms. T6w indi-cates TAC 6 weeks; Clo, clorgyline. B, Heart weight normalized to body weight (results expressed as mg of heart weight/gram of bodyweight). C, Atrial natriuretic peptide (ANP), BNP, and �-MHC gene expression. *P�0.05 vs respective sham; **P�0.001 vs respectivesham; †P�0.05 vs T6w.
Kaludercic et al MAO-A and Congestive Heart Failure 197
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

hearts, and clorgyline reduced this (Figure 5C). We alsotested whether myocardial apoptosis was involved in TAC-induced CHF and blunted by clorgyline. Cleaved (activated)caspase-3 (Figure 5D) and the number of apoptotic cellsmeasured by TUNEL assay (Figure 5E) markedly rose after 6weeks of TAC. Clorgyline significantly blunted these adversephenomena.
MAO-Aneo Mice Display Improved LV Function,No Chamber Dilation, and Reduced Levels ofFibrosis After TACTo further establish the specific involvement of MAO-A inTAC-induced cardiac remodeling, we studied mice lackingenzyme activity because of expression of a dominant negativeMAO-A (MAO-Aneo). These mice display almost nullMAO-A activity (Figure 6A) but have preserved levels andactivity of MAO-B (Online Figure II). The cardiac phenotypeof these mice had yet to be fully characterized, thus load-independent LV function and hemodynamics were examinedusing in vivo PV relationships. WT and MAO-Aneo micewere somewhat different at baseline, with LV systolic pres-sure, dP/dtmax, and dP/dtmin all lower in MAO-Aneo comparedto WT littermates (Table 2). Contractile function assessed by
preload recruitable stroke work index was also lower inMAO-Aneo mice, so these differences were potentially relatedto loading changes. However, chamber volume and ejectionfraction were similar between the two strains. When WT andMAO-Aneo mice were subjected to 9 week of TAC, WThearts had a greater dilation, with a rightward shift in the PVloop (Figure 6A and 6B), whereas LV function becameimpaired. In contrast, MAO-Aneo mice showed a slightleftward shift of the PV relations, with preserved (basal)cardiac volumes, and maintained cardiac function. Consistentwith the data obtained in C57BL/6 mice, 129/Sv WT micealso displayed reduced NET protein abundance after CHF. Instark contrast, NET levels in MAO-Aneo mice were similar tothose reported for sham operated mice (Figure 6C). Com-pared to WT, MAO-Aneo mice subjected to TAC also had lessinterstitial fibrosis (Figure 6D). Thus, genetic inhibition ofMAO-A activity also helped ameliorate structural/functionalconsequences of chronic pressure overload.
DiscussionChanges in cardiac tissue redox balance participate in myo-cyte hypertrophy and failure, influencing extracellular matrixremodeling, Ca2� handling, and metabolic substrate.6,10,28
Figure 5. MAO-A inhibition prevents oxidative stress and apoptosis in pressure overloaded hearts. A, Examples of dihydroethidiumstaining performed on cryosections from sham-operated and 6-weeks TAC hearts without (T6w) or with clorgyline (T6w�Clo). Quantifi-cation of dihydroethidium fluorescence intensity is shown on the right. B, Malondialdehyde (MDA) quantification (results expressed asnanomoles of malondialdehyde per milligram of protein). C, Quantification results of gelatin zymography of myocardium in shams or6-weeks TAC hearts, without or with clorgyline. D, Western blot showing cleaved (activated) caspase-3 levels (densitometric results onthe right). E, Percentage of apoptotic nuclei determined by TUNEL staining vs total number of cells. *P�0.05 vs respective sham,†P�0.05 vs T6w.
198 Circulation Research January 8, 2010
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

Clarification of the important sources of ROS therefore haspathogenetic and therapeutic relevance. In the cytosol,NADPH oxidase, xanthine oxidase, and uncoupled nitricoxide synthase are recognized ROS sources.10,19,29 Mitochon-dria are another major source,30,31 largely from the respiratorychain and p66Shc,32 and are known to contribute to ischemia/reperfusion injury.33 Within mitochondria, the flavoenzymeMAO-A, located in the outer membrane of the organelle, is amajor ROS generator. MAO-A activity is implicated inserotonin-induced myocyte apoptosis16 and ischemia/reperfu-sion injury via a ROS-dependent process involving sphin-gosine kinase inhibition and accumulation of ceramide.27
MAO– derived ROS also appear relevant to serotonin-induced myocyte hypertrophy in vitro.34
In this study, we showed that in addition to serotonin, NEcatabolism by MAO-A plays a prominent role in hypertrophyin vitro and in its progression toward heart failure in vivo.Catecholamines, and NE in particular, are known to contrib-ute to cardiac disease and couple to ROS signaling.35 En-hanced CA synthesis and release may provide help foradaptation to increased workload; however, because NE is ahigh affinity substrate for MAO-A,36,37 this can also serve asa major factor for increased ROS, as supported by our in vitro
myocyte data. Clorgyline partly prevented this change,whereas the remaining prooxidant/hypertrophic effects werelikely attributable to adrenergic receptor–coupled mecha-nisms.35 Furthermore, NE metabolism by MAO-A increasedin TAC mice, which was also associated with exacerbatedoxidative stress, chamber dilation, and reduced systolic func-tion. Pharmacological inhibition of MAO-A suppressed thesechanges.
Impairment of NE neuronal reuptake and concomitantdownregulation of the �-adrenergic system are well docu-mented in human and experimental CHF,38 contributing to theloss of systolic performance in this syndrome. In normalhearts, 92% of the NE released by sympathetic nerves isrecaptured by NET, 4% is removed by extraneuronal uptake,and the remaining 4% enters the circulation.18 However, NETfunction declines in CHF,39,40 resulting in NE spillover andextraneuronal uptake which almost doubles in CHF pa-tients.18 Also, the balance between vesicular NE sequestration(which represents the “measurable” NE pool) and leakage inthe intracardiac sympathetic efferent fibers may be altered tofavor extraneuronal uptake,41 providing more substrate forMAO-A. The present results showing reduced NET expres-sion in TAC hearts support this hypothesis. Increased ROS
Figure 6. MAO-Aneo mice have preserved LV function and reduced chamber dimensions after pressure overload. A, MAO-A activity inWT and MAO-Aneo mice. B, Representative PV loops from WT and MAO-Aneo at baseline (dashed line) and 9 weeks after TAC (full line).C, Western blot for NET and quantification of bands intensity (results normalized to GAPDH expression). D, Masson’s trichrome stain-ing of paraffin sections from WT and MAO-Aneo mice showing the extent of interstitial fibrosis in shams and 9-weeks TAC hearts (T9w).The quantification of fibrotic areas is shown on the right and results are expressed as percent of myocardial area. *P�0.05 vs WTsham; †P�0.05 vs WT T9w; **P�0.05 vs MAO-Aneo sham.
Kaludercic et al MAO-A and Congestive Heart Failure 199
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

generation could stem from intramyocyte and/or intraneuro-nal MAO-A activity as clorgyline inhibits both. The former issupported by the present findings (eg, Figures 1 and 5A and5B), whereas proof of the latter would require studies inisolated sympathetic efferent fibers and/or nerve-musclepreparations. Our data also showed that clorgyline or geneticablation of MAO-A catalytic activity restored NET expres-sion back to control levels in TAC hearts, consistent withfindings that systemic administration of MAO inhibitorsincreases the number of NET recognition sites.42 This findinghints at the possibility that MAO-A inhibition may alsobenefit both NE reuptake and intraneuronal CAs recycle forrerelease, thereby reducing requirements for transmitterneosynthesis.20
Prior evidence for MAO-A involvement in cardiac remod-eling derives largely from microarray analyses, showingchanges in gene expression in models such as high-salt dietand myocardial infarction.43 More recently, the role ofMAO-A was explored in pressure overload-induced hyper-trophy.44 In that study, Lairez et al showed a role of enhancedserotonin signaling via the serotonin receptor 2A (5-hydroxy-tryptamine 2A [5-HT2A]) and found that although antagoniz-ing 5-HT2A was beneficial, genetic deletion of MAO-Aproved detrimental, exacerbating LV thickening and fibrosis.Although differences in the models could underlie the appar-ent contradiction with the present findings, other factorsmight also contribute, such as the severity of the pressureoverload. In the prior study, even the WT mice showed littledilation and preserved function. The authors also used WT asopposed to littermate controls (the latter employed in thepresent study). The C57BL/6 strain develops more severeresponses to TAC, and the benefits of clorgyline, whichavoids potential adaptive changes in the MAO-A gene dele-tion mouse models, supports the opposite response. Althoughour control mice for the MAO-Aneo studies (129/Sv back-ground) developed less hypertrophy (consistent with priorreports45), there was still substantial dilation that was ame-liorated in mice lacking active MAO-A. Other potentialcontributors to this discrepancy are difference in functionalassessment, with anesthesia-induced cardiodepression in theearlier study, versus conscious data in the present experi-ments. MAO-Aneo mice also had basal elevated intracardiacNE content. It remains to be determined whether myocyte-specific gene deletion would yield the same results. In theearlier study of Lairez et al,44 cardiac levels of CAs were notdetermined whereas serotonin levels were found to be in-creased at baseline in MAO-A�/� mice, consistent withpresent findings, and did not change when LV remodelingwas already established. The latter is also consistent with ourstudy in showing no alteration of serotonin content inTAC-induced CHF. As expected, in both studies MAO-Ainhibition increased serotonin levels in TAC hearts. Thus,considering that increased cardiac serotonin content is con-comitant with improved LV function and absence of remod-eling after TAC, an actual beneficial effect of serotonincannot be excluded. It is plausible that serotonin may sustaincardiac contractility46 during late stage CHF, particularly if adeficit in NE availability persists. In the end, this effect wouldprovide an additional explanation for the beneficial action ofMAO-A inhibition.
There are some limitations to this study. We did not test therole of MAO-B and catechol-O-methyl transferase (COMT),both additional monoamine catabolic enzymes. However,prior studies in mice and humans have not shown a major rolefor MAO-B in NE catabolism,36,37 and COMT gene expres-sion did not change in our TAC model (data not shown).Future studies with pharmacological and genetic ablation ofthese enzymes will be required to definitively examine theirrole. The mechanistic intricacies by which MAO-A inhibitionpreserves NET expression, and likely function (given clorgy-line effects on NE content in shams and TAC hearts),warrants further investigation, but a possible major involve-ment of oxidative/nitrosative stress seems plausible.47 Fi-
Table 2. Morphological and Functional Changes via PVRelationships in WT and MAO-Aneo Mice at Baseline and AfterNine Weeks of TAC
Parameter and Genotype Sham TAC 9 Weeks
Heart rate (bpm)
WT 537�10 503�23
MAO-Aneo 515�28 501�19
Peak LVPsys (mm Hg)
WT 119�6 162�14†
MAO-Aneo 97�6* 137�17†
LVPdia (mm Hg)
WT 1�0.3 2�1
MAO-Aneo 2�1 2�0.7
LVVes (�L)
WT 4.5�1 16.6�4.5‡
MAO-Aneo 5.7�2.4 5.5�1.4§
LVVed (�L)
WT 27.1�2.5 39.4�3.1†
MAO-Aneo 28.1�5 25.7�3.6§
EF (%)
WT 83.8�1.9 59.7�8.3‡
MAO-Aneo 80.6�5.3 79.3�2.6§
dP/dtmax (mm Hg/sec)
WT 16140�405 13796�226‡
MAO-Aneo 12681�1391* 12963�991
dP/dtmin (mm Hg/sec)
WT �11415�595 �10591�425
MAO-Aneo �8013�948* �9663�1192
� (ms)
WT 5.6�0.2 5.7�0.1
MAO-Aneo 5.9�0.1 5.8�0.5
PRSW (mm Hg)
WT 129.6�8.2 132.3�11
MAO-Aneo 95.3�11.1* 126.2�20.9
� indicates relaxation constant; EF, ejection fraction; LVPdia, left ventriculardiastolic pressure; LVPsys, left ventricular systolic pressure; LVVed, end-dia-stolic left ventricular volume; LVVes, end-systolic left ventricular volume;PRSW, preload recruitable stroke work. *P�0.05 vs WT sham; †P�0.05 TACvs sham; ‡P�0.01 vs WT sham; §P�0.05 vs WT TAC; comparison performedby t test (n�5 each group).
200 Circulation Research January 8, 2010
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

nally, any posttranscriptional and/or posttranslational regula-tion of MAO-A is currently under investigation.
In conclusion, the present data support MAO-A as animportant source of ROS that contributes to maladaptiveremodeling and myocardial dysfunction in hearts subjected tohemodynamic stress. The latter likely results in NE-mediatedMAO-A activation attributable to depressed neuronal uptake.Whether inhibited ROS production and improved NE cy-cling/availability are the only keys to interpret the beneficialimpact of MAO-A inhibition in pressure overloaded heartsneeds further investigation. However, present findings addMAO-dependent signaling as a cause of stress-induced mal-adaptive hypertrophy and pump failure. The data also suggestthat MAO-A inhibitors may prove useful in other models ofcardiac failure. In the past, nonselective MAO inhibition wasassociated with the so-called “cheese reaction,” consisting ofsevere hypertensive crises following the ingestion of foodrich in tyramine.37 However, the generation of new MAO-Ainhibitors lacking this limiting side effect makes even moreattractive the idea of advancing such therapy for clinical usein CHF patients.
AcknowledgmentsWe gratefully acknowledge Dr Graeme Eisenhofer for insightfulsuggestions and critical revision of the manuscript and RaymondJohnson at Vanderbilt Neuroscience Core for invaluable help withhigh-performance liquid chromatography assays. We also thank theRoss confocal facility (NIH/R24DK064388, The Hopkins BasicResearch Digestive Disease Development Core Center) for confocalmicroscope access and John Gibas for technical assistance.
Sources of FundingThis work was supported by the American Heart Association(Postdoctoral Grant to N.K. and Scientist Development Grant toN.P.); the Ministero dell’Universita e della Ricerca and ConsiglioNazionale delle Ricerche (to F.D.L.); the NIH (grants HL093432 toE.T., HL088649 to K.L.G., HL56693 to R.D.B., HL-089297 andHL-077180 to D.A.K., R01HL075265 and HL091923 to N.P.); and,in part, by the Intramural Research Program of the National Instituteof Child Health and Development, NIH; National Institute of MentalHealth grants R37 MH39085 (Merit Award) and R01 MH67968; anda Boyd and Elsie Welin Professorship Award (to J.C.S.).
DisclosuresNone.
References1. Spinale FG. Myocardial matrix remodeling and the matrix metallopro-
teinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342.
2. Taegtmeyer H, Wilson CR, Razeghi P, Sharma S. Metabolic energeticsand genetics in the heart. Ann N Y Acad Sci. 2005;1047:208–218.
3. Kranias EG, Bers DM. Calcium and cardiomyopathies. Subcell Biochem.2007;45:523–537.
4. Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overloadand apoptosis in heart failure: implications for therapy. J Card Fail.2000;6:1–7.
5. Gupte SA, Wolin MS. Oxidant and redox signaling in vascular oxygensensing: implications for systemic and pulmonary hypertension. AntioxidRedox Signal. 2008;10:1137–1152.
6. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophyand remodeling. Hypertension. 2007;49:241–248.
7. Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Roleof oxidative stress in myocardial hypertrophy and failure. J Mol CellCardiol. 2002;34:379–388.
8. Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, andNAD(P)H oxidases in the development and progression of heart failure.Congest Heart Fail. 2002;8:132–140.
9. Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition ofhypertrophy to heart failure. J Am Coll Cardiol. 1996;28:506–514.
10. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J ClinInvest. 2005;115:500–508.
11. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G,Ketner EA, Majmudar M, Gabrielson K, Halushka MK, Mitchell JB,Biswal S, Channon KM, Wolin MS, Alp NJ, Paolocci N, Champion HC,Kass DA. Reversal of cardiac hypertrophy and fibrosis from pressureoverload by tetrahydrobiopterin: efficacy of recoupling nitric oxidesynthase as a therapeutic strategy. Circulation. 2008;117:2626–2636.
12. Youdim MB, Edmondson D, Tipton KF. The therapeutic potential ofmonoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7:295–309.
13. Rodriguez MJ, Saura J, Billett EE, Finch CC, Mahy N. Cellular local-ization of monoamine oxidase A and B in human tissues outside of thecentral nervous system. Cell Tissue Res. 2001;304:215–220.
14. Saura J, Kettler R, Da PM, Richards JG. Quantitative enzyme radioau-tography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localizationand abundance of MAO-A and MAO-B in rat CNS, peripheral organs,and human brain. J Neurosci. 1992;12:1977–1999.
15. Lowe MC, Reichenbach DD, Horita A. Extraneuronal monoamineoxidase in rat heart: biochemical characterization and electron micro-scopic localization. J Pharmacol Exp Ther. 1975;194:522–536.
16. Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L,Seguelas MH, Nistri S, Colucci W, Leducq N, Parini A. Oxidative stressby monoamine oxidase mediates receptor-independent cardiomyocyteapoptosis by serotonin and postischemic myocardial injury. Circulation.2005;112:3297–3305.
17. Ferguson DW, Berg WJ, Sanders JS. Clinical and hemodynamic cor-relates of sympathetic nerve activity in normal humans and patients withheart failure: evidence from direct microneurographic recordings. J AmColl Cardiol. 1990;16:1125–1134.
18. Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, KayeDM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nervefunction in congestive heart failure. Circulation. 1996;93:1667–1676.
19. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B,Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidantstress from nitric oxide synthase-3 uncoupling stimulates cardiacpathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231.
20. Eisenhofer G. The role of neuronal and extraneuronal plasma membranetransporters in the inactivation of peripheral catecholamines. PharmacolTher. 2001;91:35–62.
21. Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S,O’Connell TD. Nuclear alpha1-adrenergic receptors signal activated ERKlocalization to caveolae in adult cardiac myocytes. Circ Res. 2008;103:992–1000.
22. Tipton KF, Boyce S, O’Sullivan J, Davey GP, Healy J. Monoamineoxidases: certainties and uncertainties. Curr Med Chem. 2004;11:1965–1982.
23. Youdim MB, Finberg JP. New directions in monoamine oxidase A and Bselective inhibitors and substrates. Biochem Pharmacol. 1991;41:155–162.
24. Coatrieux C, Sanson M, Negre-Salvayre A, Parini A, Hannun Y, ItoharaS, Salvayre R, Auge N. MAO-A-induced mitogenic signaling is mediatedby reactive oxygen species, MMP-2, and the sphingolipid pathway. FreeRadic Biol Med. 2007;43:80–89.
25. Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly.Annu Rev Physiol. 2003;65:45–79.
26. Fujii T, Yamazaki T, Akiyama T, Sano S, Mori H. Extraneuronalenzymatic degradation of myocardial interstitial norepinephrine in theischemic region. Cardiovasc Res. 2004;64:125–131.
27. Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, LeducqN, Seif I, Parini A, Cuvillier O. Oxidative stress-dependent sphingosinekinase-1 inhibition mediates monoamine oxidase A-associated cardiaccell apoptosis. Circ Res. 2007;100:41–49.
28. Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic andapoptotic signaling pathways by reactive oxygen species in cardiacmyocytes. Antioxid Redox Signal. 2003;5:731–740.
29. Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependentredox signalling in cardiac hypertrophy, remodelling and failure. Car-diovasc Res. 2006;71:208–215.
Kaludercic et al MAO-A and Congestive Heart Failure 201
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

30. Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M. Mitochondrialpathways for ROS formation and myocardial injury: the relevance ofp66(Shc) and monoamine oxidase. Basic Res Cardiol. 2009;104:131–139.
31. Orrenius S. Reactive oxygen species in mitochondria-mediated cell death.Drug Metab Rev. 2007;39:443–455.
32. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C,Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R,Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochromec and p66Shc generates reactive oxygen species that trigger mitochondrialapoptosis. Cell. 2005;122:221–233.
33. Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury ofthe heart: fixing a hole. Cardiovasc Res. 2006;70:191–199.
34. Bianchi P, Pimentel DR, Murphy MP, Colucci WS, Parini A. A newhypertrophic mechanism of serotonin in cardiac myocytes: receptor-independent ROS generation. FASEB J. 2005;19:641–643.
35. Amin JK, Xiao L, Pimental DR, Pagano PJ, Singh K, Sawyer DB,Colucci WS. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol CellCardiol. 2001;33:131–139.
36. Lenders JW, Eisenhofer G, Abeling NG, Berger W, Murphy DL, KoningsCH, Wagemakers LM, Kopin IJ, Karoum F, van Gennip AH, BrunnerHG. Specific genetic deficiencies of the A and B isoenzymes ofmonoamine oxidase are characterized by distinct neurochemical andclinical phenotypes. J Clin Invest. 1996;97:1010–1019.
37. Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: frompathophysiology to therapeutics. Adv Drug Deliv Rev. 2008;60:1527–1533.
38. Beau SL, Saffitz JE. Transmural heterogeneity of norepinephrine uptakein failing human hearts. J Am Coll Cardiol. 1994;23:579–585.
39. Backs J, Haunstetter A, Gerber SH, Metz J, Borst MM, Strasser RH,Kubler W, Haass M. The neuronal norepinephrine transporter in experi-
mental heart failure: evidence for a posttranscriptional downregulation.J Mol Cell Cardiol. 2001;33:461–472.
40. Liang CS, Fan TH, Sullebarger JT, Sakamoto S. Decreased adrenergicneuronal uptake activity in experimental right heart failure. A chamber-specific contributor to beta-adrenoceptor downregulation. J Clin Invest.1989;84:1267–1275.
41. Eisenhofer G, Kopin IJ, Goldstein DS. Leaky catecholamine stores:undue waste or a stress response coping mechanism? Ann N Y Acad Sci.2004;1018:224–230.
42. Lee CM, Javitch JA, Snyder SH. Recognition sites for norepinephrineuptake: regulation by neurotransmitter. Science. 1983;220:626–629.
43. Mialet-Perez J, Bianchi P, Kunduzova O, Parini A. New insights onreceptor-dependent and monoamine oxidase-dependent effects of seroto-nin in the heart. J Neural Transm. 2007;114:823–827.
44. Lairez O, Calise D, Bianchi P, Ordener C, Spreux-Varoquaux O,Guilbeau-Frugier C, Escourrou G, Seif I, Roncalli J, Pizzinat N, GalinierM, Parini A, Mialet-Perez J. Genetic deletion of MAO-A promotesserotonin-dependent ventricular hypertrophy by pressure overload. J MolCell Cardiol. 2009;46:587–595.
45. Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW. Cardiacresponse to pressure overload in 129S1/SvImJ and C57BL/6J mice:temporal- and background-dependent development of concentric left ven-tricular hypertrophy. Am J Physiol Heart Circ Physiol. 2007;292:H2119–H2130.
46. Kaumann AJ, Levy FO. 5-hydroxytryptamine receptors in the humancardiovascular system. Pharmacol Ther. 2006;111:674–706.
47. Mao W, Iwai C, Qin F, Liang CS. Norepinephrine induces endoplasmicreticulum stress and downregulation of norepinephrine transporter densityin PC12 cells via oxidative stress. Am J Physiol Heart Circ Physiol.2005;288:H2381–H2389.
202 Circulation Research January 8, 2010
at SWETS SUBSCRIPTION SERVICE on November 26, 2013http://circres.ahajournals.org/Downloaded from

Supplemental Material. Supplemental methods. Generation of MAO-Aneo mice. MAO-Aneo mouse was generated by alteration of a 9.0kb Bam HI fragment of MAO-A gene containing exon 10 to exon 13 of mouse MAO-A gene. A 0.28kb SmaI-EcoRI loxP fragment was blunt and ligated into a unique SphI site in intron 11 of MAO-A and a 1.6kb EcoRI-BglII floxed neomycin cassette was directionally cloned into in the same sites within the intron 12 of MAO-A gene (Online Figure V). MAO-Aneo mice do not have MAO-A activity. The full MAO-A activity was restored by crossing MAO-Aneo mice with CMV-cre mice and selectively deleting the 1.6 kb floxed neomycin insert in intron 12. Genotyping of MAO-Aneo mice. Primers flanking the loxP insert in intron 11 were designed for genotyping: forward primer (F): 5’-CCTCTCTTCCAAGTATTAGG-3’; reverse primer (R): 5’-GGAAAAGAGGGAGGAGTAAG-3’. Tail clipped genomic DNA was used as template, DNA fragment was amplified by a 4 min hot start at 94°C followed by 35 cycles of 30 sec at 94°C, 40 sec at the optimal annealing temperature (50°C), 45 sec at 72°C for elongation and a final 5 min extension at 72°C. The PCR product size is 300 bp and 500 bp for WT and MAO-Aneo mouse, respectively. TAC procedure. TAC was performed following a previously reported protocol1, 2. After induction of anesthesia and intubation, mice were placed on a volume ventilator (120 breaths/min, 1.2 ml/g/min) and anesthesia was maintained with 5% isofluorane. The aortic arch was isolated and tied against a 27-gauge needle, resulting in a 65-70% constriction after the removal of the needle. The chest and skin were closed and animals extubated and allowed to fully recover. Sham-operated mice underwent the same operation except that after the aortic arch was isolated, there was no ligature placed. Echocardiography. In vivo cardiac morphology and function were assessed by serial M-mode echocardiography (Acuson Sequioa C256, 13 MHz transducer, Siemens, PA) performed in conscious mice. LV end-systolic and end-diastolic dimensions were averaged from 3-5 beats. LV percent FS, EF and LV mass were calculated as described previously3. Thickness of posterior free wall and interventricular septum were averaged.
In Vivo Pressure-Volume Loop Studies. In vivo cardiac function was assessed by pressure-volume catheter in anesthetized mice employing a four-electrode pressure-volume catheter (model SPR-839, Millar Instruments, TX, USA) as previously described2, 4, 5. Anesthesia was maintained with i.p. etomidate (250 μg), urethane (30mg) and morphine (15 μg). Mouse was placed in supine position on a thermoregulated surgical table maintained at 37°C. Ventilation via endotracheal tube was maintained with 100% oxygen using a custom-designed constant flow ventilator delivering a tidal volume of 6.7 µl/g at 120 breaths/min. An internal jugular venous line was placed to provide a fluid and drug delivery port. The thorax was opened and a miniature PV catheter inserted into the left ventricle via the apex for continuous LV pressure-volume data. Calibration of the signal was performed using injection of hypertonic saline and direct measurement of cardiac output via an aortic flow probe (Transonic Instruments) placed around thoracic descending aorta or a pulse wave Doppler signal at the aortic outflow (Indus Instruments, Houston, TX). Pressure-volume loop analysis was made using a custom analysis program (WinPVAN 3.3). Neonatal rat cardiomyocytes studies. Myocytes were isolated from 1-2 days old Sprague Dawley rats, as previously described6. After 2 days of culture, myocytes were pretreated with
1

clorgyline for 1 hour and then subjected to stimulation with norepinephrine or tyramine (10 and 20 μmol/L respectively). After 24 hours, cells were harvested for RNA isolation using Trizol (Invitrogen). For cell area measurements, cells were fixed, incubated with α-actinin primary antibody (1:500 dilution, Sigma) and fluorescent secondary antibody (Molecular probes) and visualized by confocal microscopy (Zeiss, LSM 510 Meta). Approximately 150-200 cells were analyzed per condition in each experiment. All the experiments were repeated at least four times. For mitochondrial ROS production measurements, the reduced form of Mitotracker Red (Mitotracker Red CM-H2XRos, Molecular probes) was used. Cells were loaded with 100 nmol/L Mitotracker Red CM-H2XRos for 30 minutes at 37˚C, washed and then incubated with NE or vehicle for 2 hours. After this incubation time, cells were analyzed and images acquired by confocal microscopy. Images were analyzed and fluorescence intensities determined using computer-assisted image analysis systems (ImageJ, NIH). For the mitochondrial membrane potential measurements, cells were loaded with 20 nmol/L tetramethylrodamine (TMRM) for 30 minutes and then treated with NE for 2 hours. Images were acquired and analyzed using computer-assisted image analysis systems (ImageJ, NIH). Measurement of MAO-A activity. MAO-A activity in the heart (expressed as nmol per 20 min per milligram of protein) was determined by use of 14C labeled serotonin (5-HT) as a substrate7. Alternatively, MAO-A activity was determined by Amplex Red assay (Invitrogen), measuring hydrogen peroxide formation. Catecholamine measurement. In order to remove any contaminating blood from the LV specimens, after the excision the hearts were retrogradely perfused in a Langerdorff apparatus for 10 min using K-H solution. Then, LV specimens were dissected and homogenized in 0.4 mol/L perchloric acid containing 0.5 mmol/L EDTA, centrifuged at 3 000 rpm for 5 minutes and the supernatant stored for alumina extraction. CA and 5-HT were determined by HPLC as described previously8. CA/5-HT content is expressed as a function of protein content. Histology. Hearts were fixed in 10% formalin overnight, embedded in paraffin, sectioned at 5 μm thickness and stained using H&E and Masson’s trichrome6. Photomicrographs of the sections were evaluated for interstitial collagen fractions using computer-assisted image analysis systems (ImageJ, NIH). Apoptosis detection by TUNEL staining. The assay was performed using a commercially available kit according to manufacturer’s instructions (CardioTACs). TUNEL-positive nuclei were visualized using a light microscope equipped with a camera. The percentage of TUNEL-positive cardiomyocytes is expressed as a percentage of total nuclei. Measurement of oxidative stress. Oxidative stress was determined by MDA formation measurement and DHE staining. MDA, an end product of lipid peroxidation, was determined spectrophotometrically by measurement of 2-thiobarbituric acid reactive substances (TBARS), as described previously9. For DHE staining, 10 μm thick cryosections were incubated with 5 μmol/L DHE (Sigma) for 30 minutes at 37˚C, washed twice with PBS, mounted and visualized using a confocal microscope (Zeiss, LSM 510 Meta). MMP activity measurement. In vitro gelatin lysis by MMP-2 and MMP-9 was assessed by zymography as previously described10. Briefly, protein concentration was determined and equal amounts of lysed tissue samples were loaded on a 10% gelatin gels (Invitrogen Corp.). After electrophoresis, gels were washed twice with renaturing buffer at room temperature followed by developing buffer (Invitrogen Corp.) and then stained to visualize lytic bands (SimplyBlue;
2

Invitrogen Corp.). Lytic bands corresponding to active forms of MMP-2 and MMP-9 were quantified using the BioRad analysis software. Western Blot and real time PCR. Western blot was performed as previously described6. RNA was isolated, purified and reverse transcribed using commercially available kits (Qiagen, Invitrogen). cDNA was subjected to PCR amplification (Abi Prism 7000 Detection System, Applied Biosystems) using SYBR Green dye (Applied Biosystems). Primer sets for the specific target genes were designed to span one or more introns (IDT Technologies). Adult mouse ventricular myocytes (AMVM) isolation and culture. AMVMs were isolated from the hearts of adult (12 weeks) male MAO-Aneo mice and their WT littermates as described previously11. Cells were plated at a nonconfluent density of 25 000 rod shaped cells/ml on plastic culture dishes or glass coverslips precoated with laminin (20 µg/ml) and kept at 37°C in the culture medium (DMEM, Joklik modified MEM, NaHCO3 2 g/l, BSA 5 g/l, L-carnitine 1.5 mmol/L, creatine 5 mmol/L, taurine 7.5 mmol/L, ITS 1%, penicillin 100 IU/mL, streptomycin 10 µg/mL) for 1 hour before being used for the experiments. For mitochondrial ROS production measurements, cells were loaded with 100 nmol/L Mitotracker Red CM-H2XRos for 30 minutes at 37˚C, washed and then incubated with NE (10 μmol/L) or vehicle for 2 hours. After this incubation time, cells were analyzed and images acquired by confocal microscopy. Images were analyzed and fluorescence intensities determined using computer-assisted image analysis systems (ImageJ, NIH). For cell area measurements, cells were incubated with NE (1 μmol/L) for 24 hours and visualized by confocal microscopy (Zeiss, LSM 510 Meta). Approximately 150-200 cells were analyzed per condition in each experiment and all the experiments were repeated at least three times.
3

References
(1) Takimoto E, Yao A, Toko H, Takano H, Shimoyama M, Sonoda M, Wakimoto K, Takahashi T, Akazawa H, Mizukami M, Nagai T, Nagai R, Komuro I. Sodium calcium exchanger plays a key role in alteration of cardiac function in response to pressure overload. FASEB J. 2002;16:373-8.
(2) Isoda T, Paolocci N, Haghighi K, Wang C, Wang Y, Georgakopoulos D, Servillo G, Della Fazia MA, Kranias EG, Depaoli-Roach AA, Sassone-Corsi P, Kass DA. Novel regulation of cardiac force-frequency relation by CREM (cAMP response element modulator). FASEB J. 2003;17:144-51.
(3) Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221-31.
(4) Georgakopoulos D, Kass DA. Protocols for hemodynamic assessment of transgenic mice in vivo. Methods Mol Biol. 2003;219:233-43.
(5) Murphy AM, Kogler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marban E. Transgenic mouse model of stunned myocardium. Science. 2000;287:488-91.
(6) Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214-22.
(7) Wu HF, Chen K, Shih JC. Site-directed mutagenesis of monoamine oxidase A and B: role of cysteines. Mol Pharmacol. 1993;43:888-93.
(8) Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, Kopin IJ. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030-3.
(9) Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351-8.
(10) Paolocci N, Tavazzi B, Biondi R, Gluzband YA, Amorini AM, Tocchetti CG, Hejazi M, Caturegli PM, Kajstura J, Lazzarino G, Kass DA. Metalloproteinase inhibitor counters high-energy phosphate depletion and AMP deaminase activity enhancing ventricular diastolic compliance in subacute heart failure. J Pharmacol Exp Ther. 2006;317:506-13.
(11) Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, Di Benedetto G, O'Rourke B, Gao WD, Wink DA, Toscano JP, Zaccolo M, Bers DM, Valdivia HH, Cheng H, Kass DA, Paolocci N. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96-104.
4
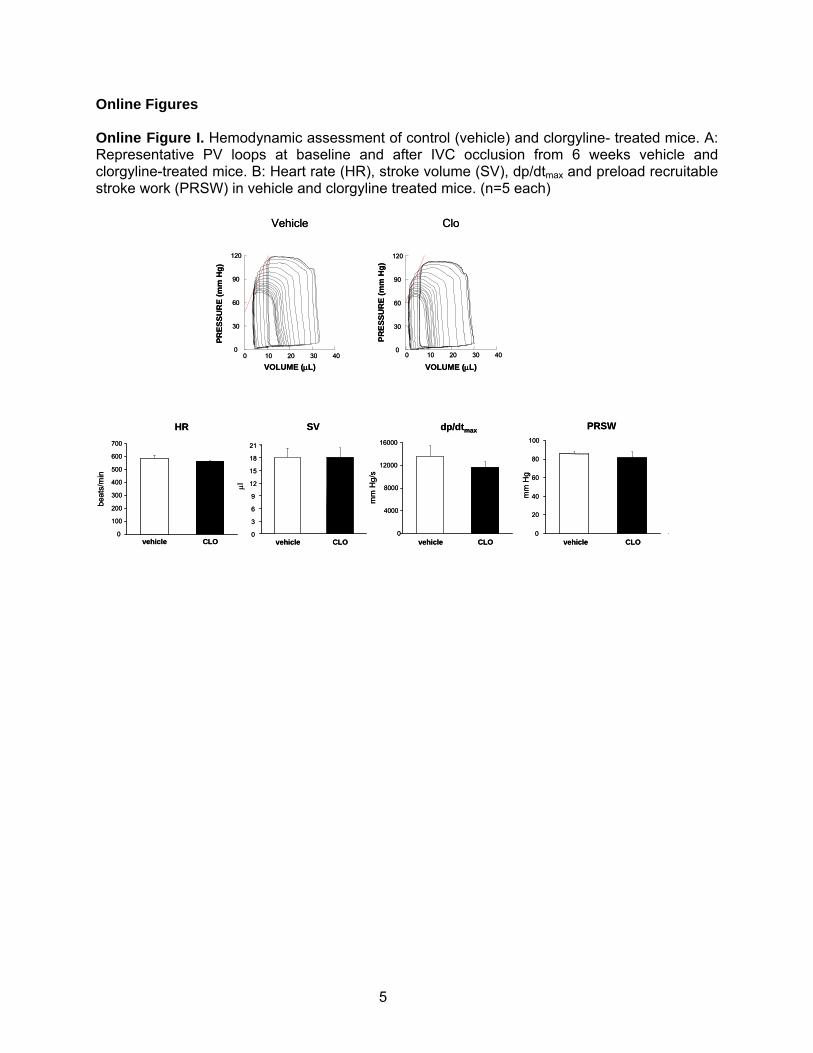
Online Figures Online Figure I. Hemodynamic assessment of control (vehicle) and clorgyline- treated mice. A: Representative PV loops at baseline and after IVC occlusion from 6 weeks vehicle and clorgyline-treated mice. B: Heart rate (HR), stroke volume (SV), dp/dtmax and preload recruitable stroke work (PRSW) in vehicle and clorgyline treated mice. (n=5 each)
PRES
SUR
E (m
m H
g)
VOLUME (μL)0 10 20 30 40
0
30
60
90
120
Vehicle
PRES
SUR
E (m
m H
g)VOLUME (μL)
0 10 20 30 400
30
60
90
120
Clo
vehicle CLO0
3
6
9
12
15
18
21
μl
SV
vehicle CLO0
4000
8000
12000
16000
mm
Hg/
s
dp/dtmax
vehicle CLO0
20
40
60
80
100
PRSWHR
vehicle CLO0
100
200
300
400
500
600
700
beat
s/m
in
mm
Hg
PRES
SUR
E (m
m H
g)
VOLUME (μL)0 10 20 30 40
0
30
60
90
120
Vehicle
PRES
SUR
E (m
m H
g)VOLUME (μL)
0 10 20 30 400
30
60
90
120
Clo
vehicle CLO0
3
6
9
12
15
18
21
μl
SV
vehicle CLO0
4000
8000
12000
16000
mm
Hg/
s
dp/dtmax
vehicle CLO0
20
40
60
80
100
PRSWHR
vehicle CLO0
100
200
300
400
500
600
700
beat
s/m
in
mm
Hg
5
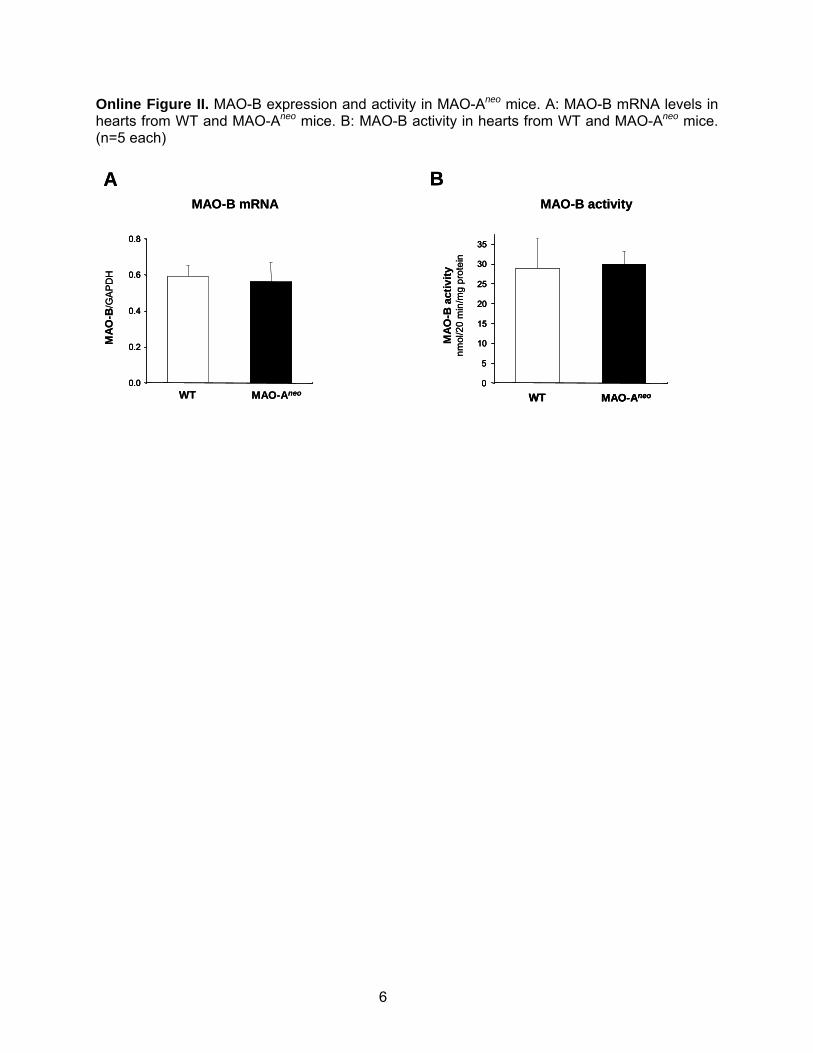
Online Figure II. MAO-B expression and activity in MAO-Aneo mice. A: MAO-B mRNA levels in hearts from WT and MAO-Aneo mice. B: MAO-B activity in hearts from WT and MAO-Aneo mice. (n=5 each)
WT MAO-Aneo0.0
0.2
0.4
0.6
0.8
MAO
-B/G
APD
H
MAO-B mRNA MAO-B activity
A B
0
5
10
15
20
25
30
35
WT MAO-Aneo
MAO
-B a
ctiv
itynm
ol/2
0 m
in/m
g pr
otei
n
WT MAO-Aneo0.0
0.2
0.4
0.6
0.8
0.0
0.2
0.4
0.6
0.8
MAO
-B/G
APD
H
MAO-B mRNA MAO-B activity
A B
0
5
10
15
20
25
30
35
0
5
10
15
20
25
30
35
WT MAO-AneoWT MAO-Aneo
MAO
-B a
ctiv
itynm
ol/2
0 m
in/m
g pr
otei
n
6

Online Figure III. NE (panel A) and serotonin (panel B) levels in hearts from WT and MAO-Aneo mice at baseline an after 9 weeks of TAC. §p<0.05 vs WT sham, #p<0.05 vs WT T9w. (n=5 each) A B
WT sh
MAO-Aneo sh
WT T9w0
2
4
6
8
10
NE,
ng/
mg
prot
MAO-Aneo T9w
§
0
4
8
12
16
20
24
5-H
T, n
g/m
g pr
ot
#
§
WT sh
MAO-Aneo sh
WT T9w
MAO-Aneo T9w
7

Online Figure IV. Mitochondrial membrane potential determined by TMRM after 2 hours of incubation with 10 μmol/L NE in the absence or in the presence of clorgyline (Clo).
CT NE NE+Clo0
20
40
60
80
100
120
TMR
M fl
uore
scen
ce, %
vs
cont
rol
CT NE NE+Clo0
20
40
60
80
100
120
TMR
M fl
uore
scen
ce, %
vs
cont
rol
8

Online Figure V. Schematic representation of MAO-Aneo mice generation. The insertion of 0.28kb loxP and 1.6 kb loxP-pgk1-Neo-loxP cassette into intron 11 and intron 12 of MAO A gene. A loxP (0.28kb SmaI-EcoRI) fragment was blunt end ligated into a unique Sp site in intron 11. Another 1.6kb E-Bg floxed neo cassette was directionally cloned into the E-Bg site in intron 12 of MAO-A gene. (Sp: SphI; E; EcoRI; Bg: Bgl II restriction enzyme).
PGK1
NeoR
E
loxPloxPloxP
Bg
11 12 13 B
E BgSp
10B
9.0 kb5` 3`
1.6 kb0.28 kb
ES
9

Advanced Search
Search My Library's Catalog: ISSN Search | Title Search
Search Workspace Ulrich's Update Admin
Enter a Title, ISSN, or search term to find journals or other periodicals:
Circulation Research
Log in to My Ulrich's
Macquarie University Library
Related Titles
Alternative MediaEdition (3)Supplement (2)
Lists
Marked Titles (0)
You have not performedany searches.
Save to List Email Download Print Corrections Expand All Collapse All
Title Circulation Research
ISSN 0009-7330
Publisher Lippincott Williams & Wilkins
Country United States
Status Active
Start Year 1953
Frequency Bi-weekly
Language of Text Text in: English
Refereed Yes
Abstracted / Indexed Yes
Serial Type Journal
Content Type Academic / Scholarly
Format Print
Website http://circres.ahajournals.org
Description Documents research advances in basic science, research, and experimentalmedicine.
Save to List Email Download Print Corrections Expand All Collapse All
Title Details Table of Contents
Contact Us | Privacy Policy | Terms and Conditions | Accessibility
Ulrichsweb.com™, Copyright © 2013 ProQuest LLC. All Rights Reserved
Basic Description
Subject Classifications
Additional Title Details
Publisher & Ordering Details
Price Data
Online Availability
Abstracting & Indexing
Other Availability
Demographics
ulrichsweb.com(TM) -- The Global Source for Periodicals http://ulrichsweb.serialssolutions.com/title/1385342146418/43821
1 of 1 25/11/2013 1:34 PM