ampk activation and metabolic reprogramming by tamoxifen...
TRANSCRIPT

Therapeutics, Targets, and Chemical Biology
AMPK Activation and Metabolic Reprogrammingby Tamoxifen through Estrogen Receptor–Independent Mechanisms Suggests New Uses forThis Therapeutic Modality in Cancer TreatmentNatalie A. Daurio1, Stephen W. Tuttle1, Andrew J.Worth2, Ethan Y. Song1,Julianne M. Davis3, Nathaniel W. Snyder4, Ian A. Blair2, and Constantinos Koumenis1
Abstract
Tamoxifen is themost widely used adjuvant chemotherapeuticfor the treatment of estrogen receptor (ER)–positive breast cancer,yet a large body of clinical and preclinical data indicates thattamoxifen canmodulatemultiple cellular processes independent-ly of ER status. Here, we describe the ER-independent effects oftamoxifen on tumor metabolism. Using combined pharmaco-logic and genetic knockout approaches, we demonstrate thattamoxifen inhibits oxygen consumption via inhibition of mito-chondrial complex I, resulting in an increase in the AMP/ATP ratioand activation of the AMP-activated protein kinase (AMPK)signaling pathway in vitro and in vivo. AMPK in turn promotes
glycolysis and alters fatty acid metabolism. We also show thattamoxifen-induced cytotoxicity is modulated by isoform-specificeffects of AMPK signaling, in which AMPKa1 promotes cell deaththrough inhibition of the mTOR pathway and translation. Byusing agents that concurrently target distinct adaptive responses totamoxifen-mediated metabolic reprogramming, we demonstrateincreased cytotoxicity through synergistic therapeutic approaches.Our results demonstrate novelmetabolic perturbations by tamox-ifen in tumor cells, which can be exploited to expand the ther-apeutic potential of tamoxifen treatment beyond ERþ breastcancer. Cancer Res; 76(11); 1–12. �2016 AACR.
IntroductionTamoxifen is the most widely used nonsteroidal selective
estrogen receptor modulator (SERM) for adjuvant therapy ofestrogen receptor–positive (ERþ) breast cancer. Patients on stan-dard treatment receive tamoxifen at 20mg/day for 5 years during,or shortly after surgical removal of the primary tumor andtreatment with ionizing radiation or cytotoxic chemotherapy.Tamoxifen therapy has reduced disease recurrence by half andoverall mortality by one third in ERþ breast cancer patients.However, the development of tamoxifen resistance remains asignificant clinical problem (1, 2).
The antitumor effects of tamoxifen are attributed primarily toits antiproliferative activity via competitive inhibition of the ER inbreast tissue (2). Several studies have previously described non-ER–dependent effects of tamoxifen, such as the inhibition ofprotein kinase C (PKC; ref. 3) and the sensitization of ER-negative
(ER�) tumors to chemotherapeutic agents (4). Furthermore,during the past two decades, more than 25 clinical trials havebeen published reporting the effects of "high dose" tamoxifen(i.e., doses above those needed to inhibit ER activity) rangingfrom 80 to 720 mg/day (5) to treat non-breast cancers, includingglioma (6),melanoma (7), andothers (8, 9), as a single agent or incombinationwith chemotherapeutics. Although, of these, phase Iand II trials showed variable clinical benefit, they clearly demon-strated the safety of high-dose tamoxifen in diverse patient popu-lations. Pharmacodynamic analyses indicated that tamoxifencould reach plasma concentrations as high as 8 mmol/L (8), andadditional preclinical studies indicated that tamoxifen accumu-lates in tumor tissue at 60 to 70 times the plasma concentration(10). Therefore, further studies on "high-dose" tamoxifen areclinically relevant and may result in expanded indications forthis affordable and well-tolerated chemotherapeutic.
The chemical properties of tamoxifen as a lipophilic weakbase contribute to its high partition into lipid bilayers, and tam-oxifen has been previously described to have inhibitory effectson mitochondrial respiratory rate (11) and membrane potential(12). Cells in which oxidative phosphorylation (OXPHOS)is inhibited by pharmacologically targeting members of themitochondrial respiratory chain, or due to lack of oxygen (hyp-oxia), exhibit a compensatory increase in glycolysis. For example,metformin, a diabetes drug showing promise as a cancer thera-peutic, was recently demonstrated to inhibit mitochondrialcomplex I and to upregulate glycolysis (13). A defining charac-teristic of many cancers is the Warburg effect, which denotes anelevated aerobic glycolytic rate, irrespective of any hypoxic bur-den, as well as an increased dependence on ATP and macro-molecule intermediates to support rapid cell growth and division
1Department of Radiation Oncology, University of Pennsylvania, Phi-ladelphia, Pennsylvania. 2Department of Pharmacology, University ofPennsylvania, Philadelphia, Pennsylvania. 3SUPERS Program, Univer-sity of Pennsylvania, Philadelphia, Pennsylvania. 4A.J. Drexel AutismInstitute, Drexel University, Philadelphia, Pennsylvania.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Constantinos Koumenis, University of PennsylvaniaSchool ofMedicine, TRCBldg Room8-087, 3400Civic Center Blvd, Philadelphia,PA 19104-5156. Phone: 215-898-0076; Fax: 215-898-0090; E-mail:[email protected].
doi: 10.1158/0008-5472.CAN-15-2197
�2016 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org OF1
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

(14). Tumor promotion by certain oncogenes can alter the met-abolic characteristics of cells as a requirement for transformation(15). As a result, strategies that target these vulnerable metabolicpathways have shown significant therapeutic promise. For exam-ple, both BRAF and KRASmutations promote glycolysis (16, 17).Resistance to targeted therapies limits the therapeutic options incancers such asmelanomaandpancreatic cancer that are drivenbythese pathways. The drug-resistant populations are thus depen-dent on elevated OXPHOS, and drugs that target mitochondrialmetabolism, including metformin, have potential utility in tar-geting such resistant populations (16, 18–20).
One of the consequences of OXPHOS inhibition, especiallyin metabolically active cancer cells, is the activation of the AMP-activated protein kinase (AMPK), the major regulator of cellularenergy homeostasis. AMPK is activated when the AMP:ATPratio increases, signaling a loss in cellular energy charge. AMPbinds to the g subunit of AMPK, inducing a conformationalchange that prevents the dephosphorylation of the catalytic asubunit of AMPK by phosphatases. This enhances phosphor-ylation of AMPK by upstream kinases, including LKB1 andCaMKK2. Once activated, AMPK restores cellular energy levelsby promoting catabolic and inhibiting anabolic processes (21).The role of AMPK in cancer is quite complex, as it has beenreported to function contextually as both a tumor suppressorand promoter (22).
On the basis of these studies, we investigated whethertamoxifen had an impact on cellular metabolism and meta-bolic signaling. Here, we report the ability of tamoxifen toinhibit mitochondrial oxygen consumption, increase glycoly-sis, and alter fatty acid metabolism. We also observe rapid (<10minutes) and acute activation of AMPK in vitro and in vivo in anER-independent manner. We demonstrate that activation of theAMPK pathway by tamoxifen promotes cell death in triple-negative breast cancer cells in an AMPK isoform–specific man-ner. Moreover, we demonstrate that cells treated with lowmicromolar doses of tamoxifen exhibit increased dependenceon glucose metabolism and that pharmacologic inhibition ofthese processes is synergistically lethal with tamoxifen. Overall,these data suggest that tamoxifen functions as a potent regu-lator of tumor metabolism and AMPK activity and that thesemetabolic effects can be exploited with combination therapiesto increase tumor cytotoxicity.
Materials and MethodsChemicals and antibodies
Tamoxifen, fulvestrant, methyl succinate, and spautin-1 werepurchased from Sigma Aldrich. The following rabbit polyclonalantibodies were obtained from Cell Signaling Technology:AMPKa, AMPKa1, phos-AMPKa, phos-ACC, acetyl-CoA carbox-ylase (ACC), phos-p70s6K, phos-s6, phos-4EBP1, cleaved PARP,cleaved caspase-3, and LC3B. ERa antibody was purchased fromGeneTex, AMPKa2 antibody was purchased from Millipore, andsiRNA against PRKAA2 was purchased from Dharmacon.
Cell cultureCell lineswere cultured inDMEMsupplementedwith10%FBS,
penicillin/streptomycin, and HEPES at 37�C humidified 5% CO2
atmosphere. MCF7, MDA-MB-231, and 4175 cell lines were a giftfrom Andy Minn (University of Pennsylvania, Philadelphia, PA),LCC1 cellswere a gift fromRobertClarke (GeorgetownUniversity,
Washington DC), and WM9 and 1205Lu cells were a gift fromMeenhard Herlyn (The Wistar Institute, Philadelphia, PA). Celllines were authenticated through genetic testing by ATCC.
Oxygen consumption measurementsOxygen consumption was measured using a YSI 5300a Bio-
logical Oxygen Monitor. Electrodes were equilibrated in mediafor 10 minutes. Cells were trypsinized and suspended at 2 � 107
cells/mL and injected into the chamber at a 1:10 dilution. Afterthe oxygen consumption rate was recorded, drug was injectedinto the chambers at 10-mmol/L increments, allowing the con-sumption rate to be determined after each injection. Total proteinper chamber was quantified by Lowry assay.
Isolation of liver mitochondriaLiver was removed from a sacrificed mouse and homogenized
in 8 volumes of sucrose buffer [0.25 mol/L sucrose, 0.02 mol/LKCl, 0.005 mol/L MgCl2, and 0.01 mol/L KH2PO4 (pH ¼ 7.4)].Homogenate was spun at 600 � g for 5 minutes at 4�C. Thesupernatant was then transferred to a new tube and centrifugationwas repeated. Cleared supernatant was spun at 9,000 � g for 10minutes at 0�C, and the pellet was resuspended in sucrose buffer.The following conditions were used for respiration measure-ments: complex 1 respiration: sucrose bufferþ 0.1 mmol/L ADP,5 mmol/L glutamate, and 5 mmol/L malate; complex 2 respira-tion: sucrose buffer þ 0.1 mmol/L ADP and 5 mmol/L sodiumsuccinate.
Cell survival assaysFor more details, see Supplementary Methods.
Immunoblot analysisFor more details, see Supplementary Methods.
Generation of CRISPR-Cas9 knockout cell linesKnockout cell lines were generated using the CRISPR-Cas9
system as described previously (23). sgRNAs were designed totarget the first exon of the genes of interest using the CRISPRdesign tool at http://crispr.mit.edu/. Two sgRNA sequences werechosen for each geneof interest (Supplementary Table S1). Briefly,sgRNAs were cloned into CRISPR-Cas9 plasmid. pSpCas9(BB)-2A-Puro (PX459) was a gift from Feng Zhang (Addgene plasmid# 48139). Cells were transfected using lipofectamine 2000 andcarrier DNA. Media were changed 24 hours after transfection,and cells were selected with puromycin for 48 hours. Remainingcells were grown up, and clones were selected. Clones werescreened for gene excision ormutation byDNAgel and expressionof the protein of interest by Western blot analysis. Knockout wassuccessful using both PRKAA1 sgRNAs 1 and 2. Knockout of ESR1expression required combined transfection of sgRNA 1 and 2.
HPLC quantification of AMP and ATPAMP and ATP were quantified using a previously published
method (24). Briefly, 2–4� 106 cells were plated in 10-cm2 dishesand treated with tamoxifen. Cells were then pelleted and nucleo-tides extracted with perchloric acid. Extracts were analyzed by ionpair reverse-phase high-performance liquid chromatography(HPLC). Analysis was carried out on Jasco HPLC. Separation wasperformed on SUPELCOSIL C18 Column (15 cm � 4.6 mm,3-mm particle size) in combination with a Supelguard AscentisC18 guard column (2 cm � 4.0 mm) from Sigma.
Daurio et al.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF2
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

LC/MS-MS metabolite analysisLiquid chromatography–selected reaction monitoring
(LC-SRM)/MS analysis of acyl-CoA thioesters was performedas described previously (25). CoAs were separated using aReversed Phase HPLC Phenomenex Luna C18 Column(2.0 mm � 150 mm; particle size, 3.5 mm) with 5 mmol/Lammonium acetate in water as solvent A, 5 mmol/L ammo-nium acetate in acetonitrile (ACN)/water (95:5; v/v) as solventB, and ACN/water/formic acid (80:20:0.1; v/v/v) as solvent C.Samples were analyzed using an API 4000 triple-quadrupolemass spectrometer (Applied Biosystems) in the positive elec-trospray mode. Samples (10 mL) were injected using a LeapCTC Autosampler (CTC Analytics), where they were main-tained at 4�C.
Short-chain and long-chain acyl-CoA extractionExtractions were performed as described in detail previously
(25, 26). For more details, see Supplementary Methods.
Lactate extraction and analysis by LC/MS-MSMedia were aspirated and cells were quenched by the direct
addition of 1-mL �80�C 4:1 methanol:water (v/v) to the cellculture dish. Plates were placed at�80�C for 20minutes, scraped,and transferred into Eppendorf tubes. Samples were pulse son-icated on ice for 30 seconds at a rate of 1 pulse/sec prior tocentrifugation at 16,000 � g at 4�C for 10 minutes. The super-natants were then transferred to clean glass tubes and evaporatedto dryness under nitrogen. Dried residues were resuspended in100mL ofmobile phase A for LC/MS analysis. For labeling studies,cells were grown in media omitting glucose supplemented with1 mg/mL [13C6]-glucose. Details of the LC/MS-MS analysis havebeen described in detail previously (27).
Quantification of tamoxifen and metabolites in tumor tissue. Fro-zen tissue was weighed and homogenized by electric homoge-nizer on ice in 4:1 methanol:water (v/v). Samples were thencentrifuged at 16,000 � g at 4�C for 10 minutes. Ten microlitersof the resulting supernatant was analyzed by reverse-phaseLC/MS-MS, utilizing teniposide as the internal standard forabsolute quantification.
siRNA transfectionFor more details, see Supplementary Methods.
BODIPY 493/503 stainingBODIPY lipid probes were purchased from Molecular
Probes. Staining solution was made up at 2 mg/mL in PBS.After drug treatments, cells were fixed using 4% paraformalde-hyde and stored at 4�C. Neutral lipid staining was performed bypelleting the cells and resuspending in staining solution andincubated for 10 minutes at room temperature. Cells werewashed twice with PBS, suspended in 500 mL PBS, and strained.Cells were analyzed for mean BODIPY stain intensity by flowcytometry (FL1-H).
[35S] labelingCells were plated in 60-mm dish and treated for experi-
mental conditions. Cells were incubated with methionine/cysteine–free media for 30 minutes. Hot labeling mediamade up at 0.075 mCi/mL [35S]-methionine/cysteine. Cellswere incubated with labeling media for 30 minutes. Cells were
washed with cold PBS and harvested for protein. Proteinassay preformed on cold samples and equal amounts of[35S]-labeled protein were resolved on SDS-PAGE. Gel waswashed with fixing solution (water with 20% methanol, 10%acetic acid) for 30 minutes, washed with deionized water, andthen washed with enlightening solution (PerkinElmer) for 30minutes. Gel was dried overnight using a Bio-Rad gel dryingapparatus. Gel was exposed to autoradiography film at �80�Cand film was developed.
Orthotopic xenograft tumorsFour-week-old female nu/nu mice were purchased from
Charles River Laboratories and housed in the University of Penn-sylvania animal facility maintained on a 12-hour light/darkcycle and given free access to water and food ad libitum. 4175cells (2� 106) suspended in 50 mL of a 50/50 mixture of PBS andMatrigel Matrix Basement Membrane (Corning) were injectedinto the mammary fat pad.
In vivo tamoxifen treatmentTamoxifen (20 mg) was dissolved in 200-mL ethanol and
800-mL peanut oil. Ethanol was evaporated off overnight. Micewere dosed via intraperitoneal injection once per day for 3 daysat 100 mg/kg.
Protein analysis from tumorsFor more details, see Supplementary Methods.
Statistical analysisAll statistical analyses were performed using a two-tailed Stu-
dent t test assuming homoscedasticity. P < 0.05 was consideredstatistically significant.
ResultsTamoxifen inhibits OXPHOS and cellular oxygen consumptionindependently of ER inhibition
Although tamoxifen has been reported to affect mitochon-drial electron transport and membrane potential, its effects onoxygen consumption have not been adequately described. AClark-type electrode was used to measure oxygen consumptionof cells. Tamoxifen, at low micromolar doses, inhibited oxygenconsumption in a dose-dependent manner in both ERþ MCF7cells and triple-negative MDA-MB-231 cells (Fig. 1A). Tamox-ifen also inhibited oxygen consumption in a panel of 6 non-breast cancer cell lines (Supplementary Table S2). The activemetabolite of tamoxifen, 4-hydroxy-tamoxifen (4OH-Tam)also inhibited oxygen consumption, whereas the ER antagonistfulvestrant, which inhibits ER activity by blocking ER dimer-ization and promoting degradation of the ER (28), did not havea significant effect (Supplementary Fig. S1A). This suggests thatinhibition of ER signaling is not responsible for the rapidinhibition of oxygen consumption in response to tamoxifentreatment. To further rule out the involvement of ER signalingin tamoxifen-mediated inhibition of oxygen consumption, weused the CRISPR-Cas9 system to knock out ERa from LCC1cells, a variant of the MCF7 line, which expresses functional ERbut is not dependent on its signaling for proliferation (29). Theability of tamoxifen to inhibit oxygen consumption in theLCC1-crspER cells was no different from the parental LCC1
AMPK Activation and Metabolic Reprogramming by Tamoxifen
www.aacrjournals.org Cancer Res; 76(11) June 1, 2016 OF3
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
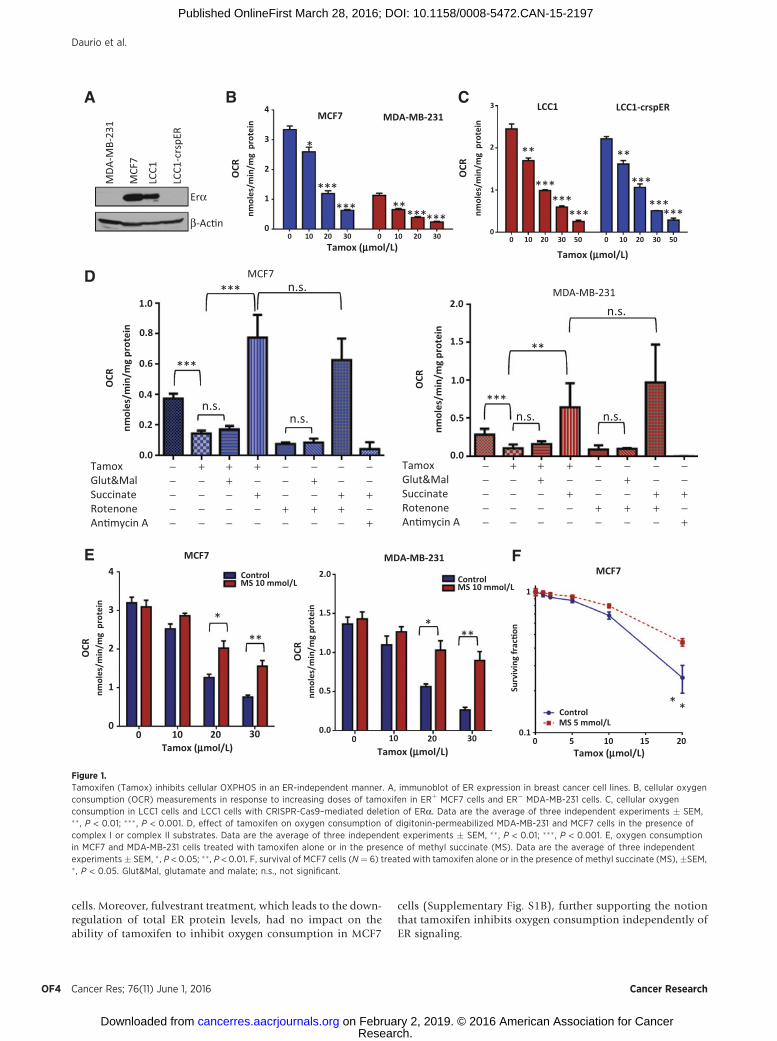
cells. Moreover, fulvestrant treatment, which leads to the down-regulation of total ER protein levels, had no impact on theability of tamoxifen to inhibit oxygen consumption in MCF7
cells (Supplementary Fig. S1B), further supporting the notionthat tamoxifen inhibits oxygen consumption independently ofER signaling.
E
****
**
F
*
MCF7
TamoxGlut&MalSuccinateRotenoneAn�mycin A
*
****** ********
CA B
****
******
***
***
******
***
n.s.
D
n.s.
MDA-MB-231*** n.s.
TamoxGlut&MalSuccinateRotenoneAn�mycin A
***n.s. n.s.
n.s.
**
*
−−
−−
−−
−
−−−
−−
−−−−−
−−−
−−−−−
+
+++
−
−+−++
+
++++ −
−
−−
−−
−
−−−
−−
−−−−−
−−−
−−−−−
+
+++
−
−+−++
+
++++
4 2.0
1
0.1
1.5
1.0
0.5
0.00 0 5 10
ControlMS 5 mmol/L
15 2010 20 30
ControlMS 10 mmol/L Control
MS 10 mmol/L
MCF7MCF7
MDA-MB-231
3
2
1
00
OCR
nmol
es/m
in/m
g p
rote
in
1.0
0.8
0.6
0.4
0.2
0.0
2.0
1.5
1.0
0.5
0.0
OCR
nmol
es/m
in/m
g pr
otei
n4
3
MCF7M
DA-M
B-23
1
MCF
7
LCC1
Erα
β-Ac�n
LCC1
-crs
pER
MDA-MB-231LCC1 LCC1-crspER
2
1
00 010 1020 20 0
0
1
2
3
010 1020 2030 3050 5030Tamox (mmol/L)
Tamox (mmol/L)
30O
CRnm
oles
/min
/mg
pro
tein
OCR
nmol
es/m
in/m
g p
rote
in
OCR
nmol
es/m
in/m
g pr
otei
n
OCR
nmol
es/m
in/m
g pr
otei
n
Surv
ivin
g fr
ac�o
n
10 20 30Tamox (mmol/L) Tamox (mmol/L) Tamox (mmol/L)
Figure 1.Tamoxifen (Tamox) inhibits cellular OXPHOS in an ER-independent manner. A, immunoblot of ER expression in breast cancer cell lines. B, cellular oxygenconsumption (OCR) measurements in response to increasing doses of tamoxifen in ERþ MCF7 cells and ER� MDA-MB-231 cells. C, cellular oxygenconsumption in LCC1 cells and LCC1 cells with CRISPR-Cas9–mediated deletion of ERa. Data are the average of three independent experiments � SEM,�� , P < 0.01; ��� , P < 0.001. D, effect of tamoxifen on oxygen consumption of digitonin-permeabilized MDA-MB-231 and MCF7 cells in the presence ofcomplex I or complex II substrates. Data are the average of three independent experiments � SEM, �� , P < 0.01; ��� , P < 0.001. E, oxygen consumptionin MCF7 and MDA-MB-231 cells treated with tamoxifen alone or in the presence of methyl succinate (MS). Data are the average of three independentexperiments � SEM, � , P < 0.05; ��, P < 0.01. F, survival of MCF7 cells (N ¼ 6) treated with tamoxifen alone or in the presence of methyl succinate (MS), �SEM,� , P < 0.05. Glut&Mal, glutamate and malate; n.s., not significant.
Daurio et al.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF4
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
Tamoxifen inhibits oxygen consumption by blocking complex Iof the mitochondrial respiratory chain
To determine whether the effect of tamoxifen on oxygenconsumption was due to a direct interaction with mitochondria,we permeabilized MCF7 and MDA-231 cells using digitonin.Tamoxifen inhibited oxygen consumption of mitochondria byapproximately 60% in both permeabilized cell lines. Respirationcould be rescued by supplementation with electron transportchain complex II substrate succinate but not with complex Isubstrates, glutamate and malate (Fig. 1D). These effects oftamoxifen on mitochondria respiration are similar to what isseen with rotenone, a known complex I inhibitor (Fig. 1D). Inmitochondria isolated frommouse liver, tamoxifen also inhibitedoxygen consumption in the presence of glutamate andmalate butnot in the presence of succinate (Supplementary Fig. S1C). Thisindicates that the tamoxifen-mediated inhibition of oxygen con-sumption occurs through a direct effect on mitochondria,upstream of complex II. The addition of methyl succinate, acell-permeable analogue of succinate, to media, reduced the
ability of tamoxifen to inhibit oxygen consumption in intactbreast cancer cells (Fig. 1E) and significantly increased cell survivalof MCF7 cells treated with tamoxifen (Fig. 1F). These resultssuggest that inhibition of mitochondrial function upstream ofcomplex IImediates the effects of tamoxifen on oxygen consump-tion and downstream effects.
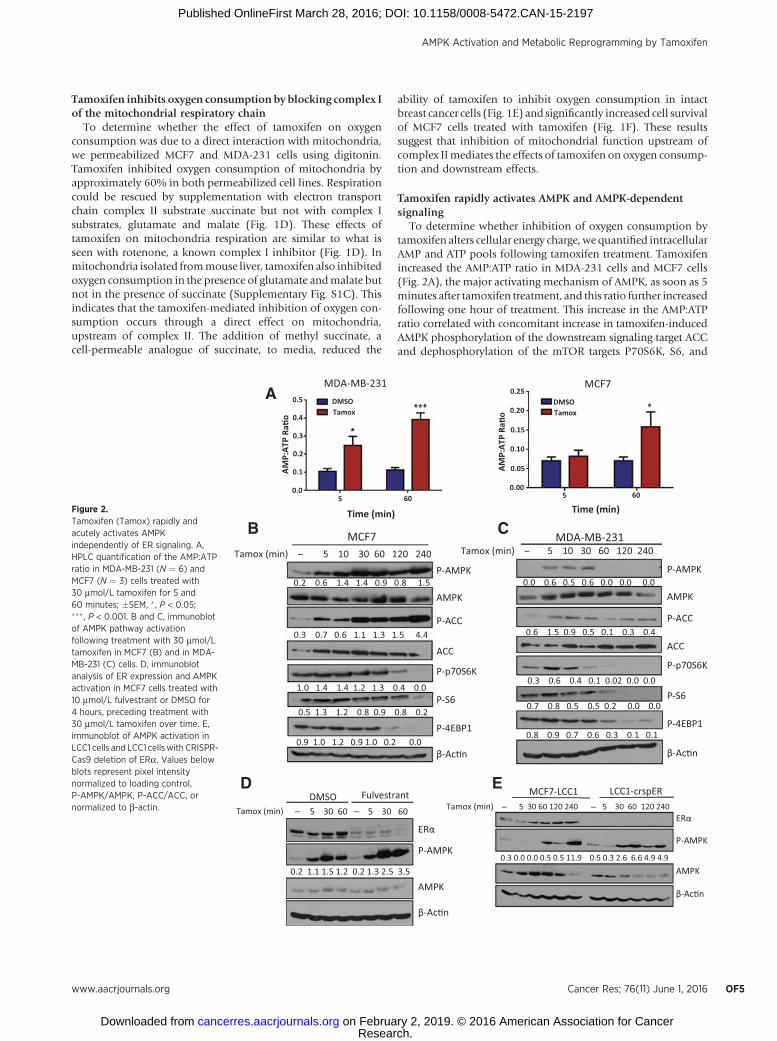
Tamoxifen rapidly activates AMPK and AMPK-dependentsignaling
To determine whether inhibition of oxygen consumption bytamoxifen alters cellular energy charge, we quantified intracellularAMP and ATP pools following tamoxifen treatment. Tamoxifenincreased the AMP:ATP ratio in MDA-231 cells and MCF7 cells(Fig. 2A), the major activating mechanism of AMPK, as soon as 5minutes after tamoxifen treatment, and this ratio further increasedfollowing one hour of treatment. This increase in the AMP:ATPratio correlated with concomitant increase in tamoxifen-inducedAMPK phosphorylation of the downstream signaling target ACCand dephosphorylation of the mTOR targets P70S6K, S6, and
A
B
P-AMPK
AMPK
ERα
β-Ac�n
5 30 60 – – –5 30 60DMSO Fulvestrant
P-ACC
P-AMPK
AMPK
ACC
P-p70S6K
P-S6
β-Ac�n
5 10 30 60 120 240MDA-MB-231
P-4EBP1
ERα
P-AMPK
AMPK
β-Ac�n
MCF7-LCC1 LCC1-crspERTamox (min) –
–
Tamox (min) 5 30 60 120 240 5 30 60 120 240
Tamox (min) –Tamox (min)5 10 30 60 120 240
0.0
0.1
0.2
0.3
0.4
0.50.25
0.20
0.15
0.10
0.05
0.005 605 60
Time (min)Time (min)
DMSOTamox
DMSOTamox
AMP:
ATP
Ra�o
AMP:
ATP
Ra�o
MCF7
P-ACC
P-AMPK
AMPK
ACC
P-p70S6K
P-S6
β-Ac�n
P-4EBP1
D E
C
0.2 0.6 1.4 1.4 0.9 0.8 1.5
0.3 0.7 0.6 1.1 1.3 1.5 4.4
1.0 1.4 1.4 1.2 1.3 0.4 0.0
0.5 1.3 1.2 0.8 0.9 0.8 0.2
0.9 1.0 1.2 0.9 1.0 0.2 0.0
0.0 0.6 0.5 0.6 0.0 0.0 0.0
0.6 1.5 0.9 0.5 0.1 0.3 0.4
0.3 0.6 0.4 0.1 0.02 0.0 0.0
0.7 0.8 0.5 0.5 0.2 0.0 0.0
0.8 0.9 0.7 0.6 0.3 0.1 0.1
0.3 0.0 0.0 0.5 0.5 11.9 0.5 0.3 2.6 6.6 4.9 4.9
0.2 1.1 1.5 1.2 0.2 1.3 2.5 3.5
*
MDA-MB-231 MCF7
Figure 2.Tamoxifen (Tamox) rapidly andacutely activates AMPKindependently of ER signaling. A,HPLC quantification of the AMP:ATPratio in MDA-MB-231 (N ¼ 6) andMCF7 (N ¼ 3) cells treated with30 mmol/L tamoxifen for 5 and60 minutes; �SEM, � , P < 0.05;��� , P < 0.001. B and C, immunoblotof AMPK pathway activationfollowing treatment with 30 mmol/Ltamoxifen in MCF7 (B) and in MDA-MB-231 (C) cells. D, immunoblotanalysis of ER expression and AMPKactivation in MCF7 cells treated with10 mmol/L fulvestrant or DMSO for4 hours, preceding treatment with30 mmol/L tamoxifen over time. E,immunoblot of AMPK activation inLCC1 cells and LCC1 cellswith CRISPR-Cas9 deletion of ERa. Values belowblots represent pixel intensitynormalized to loading control,P-AMPK/AMPK, P-ACC/ACC, ornormalized to b-actin.
AMPK Activation and Metabolic Reprogramming by Tamoxifen
www.aacrjournals.org Cancer Res; 76(11) June 1, 2016 OF5
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
4EBP1 in both MCF7 (Fig. 2B) and MDA-MB-231 cells (Fig. 2C).To investigate whether ER signaling is necessary for tamoxifenactivation of the AMPK pathway, we used pharmacologic andgenetic methods to block ER signaling. Pretreatment with fulves-trant did not inhibit tamoxifen activation of AMPK (Fig. 2D).Furthermore, activation of AMPK in LCC1-crspER was compara-ble with that in LCC1 parental cells (Fig. 2E), indicating that theeffects of tamoxifen on AMPK signaling are also ER independent.
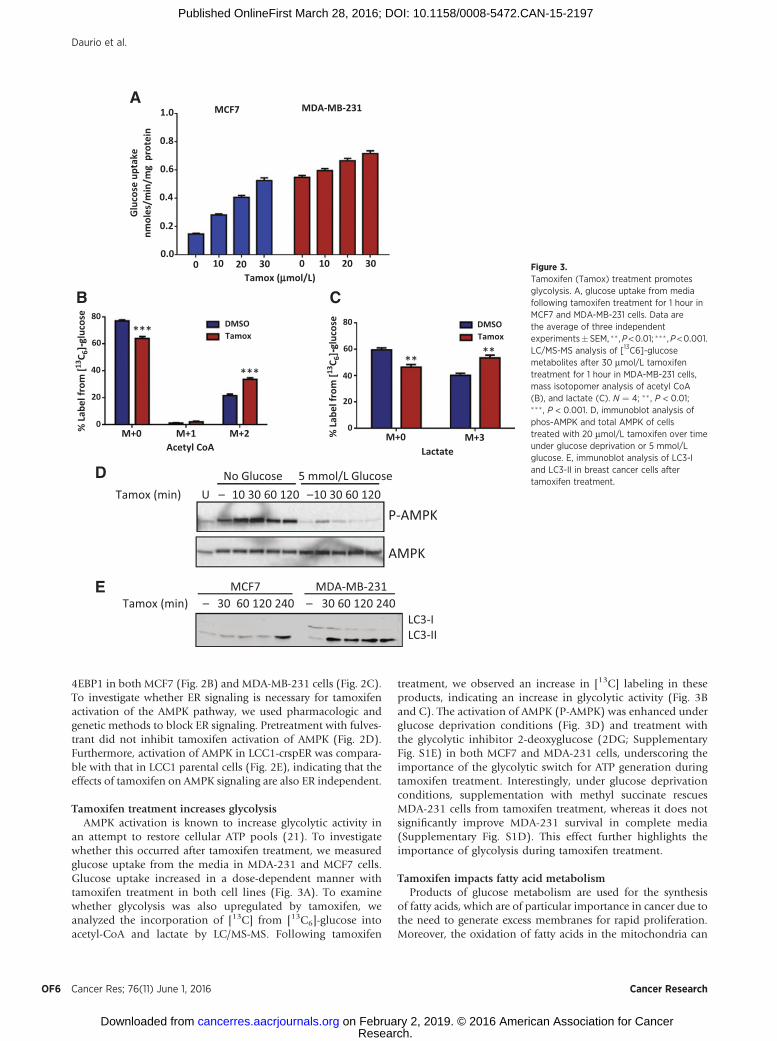
Tamoxifen treatment increases glycolysisAMPK activation is known to increase glycolytic activity in
an attempt to restore cellular ATP pools (21). To investigatewhether this occurred after tamoxifen treatment, we measuredglucose uptake from the media in MDA-231 and MCF7 cells.Glucose uptake increased in a dose-dependent manner withtamoxifen treatment in both cell lines (Fig. 3A). To examinewhether glycolysis was also upregulated by tamoxifen, weanalyzed the incorporation of [13C] from [13C6]-glucose intoacetyl-CoA and lactate by LC/MS-MS. Following tamoxifen
treatment, we observed an increase in [13C] labeling in theseproducts, indicating an increase in glycolytic activity (Fig. 3Band C). The activation of AMPK (P-AMPK) was enhanced underglucose deprivation conditions (Fig. 3D) and treatment withthe glycolytic inhibitor 2-deoxyglucose (2DG; SupplementaryFig. S1E) in both MCF7 and MDA-231 cells, underscoring theimportance of the glycolytic switch for ATP generation duringtamoxifen treatment. Interestingly, under glucose deprivationconditions, supplementation with methyl succinate rescuesMDA-231 cells from tamoxifen treatment, whereas it does notsignificantly improve MDA-231 survival in complete media(Supplementary Fig. S1D). This effect further highlights theimportance of glycolysis during tamoxifen treatment.
Tamoxifen impacts fatty acid metabolismProducts of glucose metabolism are used for the synthesis
of fatty acids, which are of particular importance in cancer due tothe need to generate excess membranes for rapid proliferation.Moreover, the oxidation of fatty acids in the mitochondria can
No Glucose
1.0 MCF7 MDA-MB-231
0.8
0.6
Glu
cose
upt
ake
nmol
es/m
in/m
g p
rote
in
% L
abel
from
[13C 6
]-glu
cose
0.4
0.2
80DMSOTamox
DMSOTamox
60
40
20
M+0 M+1Acetyl CoA Lactate
M+2 M+0 M+30
% L
abel
from
[13C 6
]-glu
cose 80
60
40
20
0
0.00 10 20 30
Tamox (mmol/L)0 10 20 30
5 mmol/L GlucoseTamox (min) U 10 30 60 120 10 30 60 120
D
C
P-AMPK
AMPK
B
Tamox (min) – 30 60 120 240 – 30 60 120 240MCF7 MDA-MB-231
LC3-ILC3-II
E
***
*****
**
A
– –
Figure 3.Tamoxifen (Tamox) treatment promotesglycolysis. A, glucose uptake from mediafollowing tamoxifen treatment for 1 hour inMCF7 and MDA-MB-231 cells. Data arethe average of three independentexperiments�SEM, �� ,P<0.01; ��� ,P<0.001.LC/MS-MS analysis of [13C6]-glucosemetabolites after 30 mmol/L tamoxifentreatment for 1 hour in MDA-MB-231 cells,mass isotopomer analysis of acetyl CoA(B), and lactate (C). N ¼ 4; �� , P < 0.01;��� , P < 0.001. D, immunoblot analysis ofphos-AMPK and total AMPK of cellstreated with 20 mmol/L tamoxifen over timeunder glucose deprivation or 5 mmol/Lglucose. E, immunoblot analysis of LC3-Iand LC3-II in breast cancer cells aftertamoxifen treatment.
Daurio et al.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF6
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
replenish cellular ATP levels during times of metabolic stress(30). To examine whether tamoxifen also impacted fatty acidmetabolism, we preformed metabolite analysis of fatty acyl-CoAspecies by LC/MS-MS. This analysis showed an accumulation ofacetyl-CoA and malonyl-CoA following tamoxifen treatment(Supplementary Fig. S2A). We also observed a reduction insuccinyl-CoA and other medium chain acyl-CoA species (C3:0,C6:0), consistent with reports of the effects of other complex Iinhibitors (31) and an accumulation of longer chain (C16-C20)acyl-CoA species (Supplementary Fig. S2A). The addition of[13C]-glucose or [13C]-glutamine resulted in no labeling of thelong-chain CoAmetabolites, indicating that these fatty acyl-CoAsare not synthesized from glucose or glutamine de novo in responseto tamoxifen treatment. Activation of the AMPK pathway is alsoknown to promote b-oxidation of fatty acids as a strategy toincrease ATP pools. The degradation of lipid droplets by autop-
hagy, termed lipophagy, increases the supply of free fatty acids,which are then transported into the mitochondria for oxidation(30). We also observed a rapid activation of autophagy upontamoxifen treatment (Fig. 3E). BODIPY stain, which measurestotal neutral lipid content in the cell, decreased modestly after 4hours of tamoxifen treatment, by 14%, in cells treated withtamoxifen (Supplementary Fig. S2B and S2C). These data suggestthat the accumulation of the long chain acyl-CoA species resultsfrom breakdown of lipid droplets.
AMPK isoforms differentially regulate survival in response totamoxifen treatment
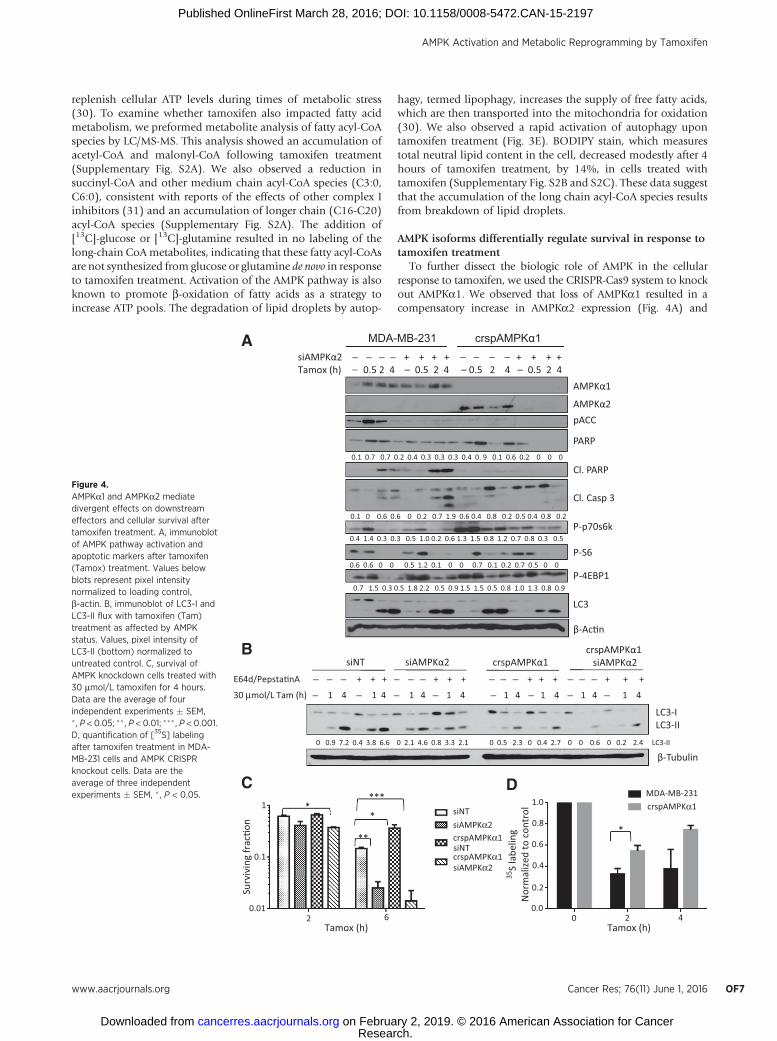
To further dissect the biologic role of AMPK in the cellularresponse to tamoxifen, we used the CRISPR-Cas9 system to knockout AMPKa1. We observed that loss of AMPKa1 resulted in acompensatory increase in AMPKa2 expression (Fig. 4A) and
P-S6
AMPKα1
Cl. Casp 3
P-p70s6k
P-4EBP1
AMPKα2pACC
Cl. PARP
siAMPKα2 + + + + + + + +Tamox (h) 0.5 2 4 – 0.5 2 4 – 0.5 2 4 – 0.5 2 4
crspAMPKα1MDA-MB-231
LC3
A
C*
*
**
***
β-Ac�n
PARP
LC3-ILC3-II
β-Tubulin
30 μmol/L Tam (h) 1 4
1 1.0MDA-MB-231crspAMPKα1
crspAMPKα1
crspAMPKα1siAMPKα2
siAMPKα2
siNT
siNT0.8
0.6
0.4
0.2
0.00 2 4
Tamox (h)
0.1
0.012 6
Tamox (h)
Surv
ivin
g fr
ac�o
n
35S
labe
ling
Nor
mal
ized
to c
ontr
ol
1 4 1 4 1 4 1 4 1 4 1 4 1 4
E64d/Pepsta�nA + + + + + + + + + + + +
crspAMPKα1siAMPKα2crspAMPKα1siAMPKα2siNT
0 0.9 7.2 0.4 3.8 6.6 0 2.1 4.6 0.8 3.3 2.1 0 0.5 2.3 0 0.4 2.7 0 0 0.6 0 0.2 2.4 LC3-II
B
D
*
0.7 1.5 0.3 0.5 1.8 2.2 0.5 0.9 1.5 1.5 0.5 0.8 1.0 1.3 0.8 0.9
0.6 0.6 0 0 0.5 1.2 0.1 0 0 0.7 0.1 0.2 0.7 0.5 0 0
0.4 1.4 0.3 0.3 0.5 1.0 0.2 0.6 1.3 1.5 0.8 1.2 0.7 0.8 0.3 0.5
0.1 0 0.6 0.6 0 0.2 0.7 1.9 0.6 0.4 0.8 0.2 0.5 0.4 0.8 0.2
0.1 0.7 0.7 0.2 0.4 0.3 0.3 0.3 0.4 0. 9 0.1 0.6 0.2 0 0 0
−
−−
− − − − − − −
− −− −
− − −− −
− − −− −
− − −−−
Figure 4.AMPKa1 and AMPKa2 mediatedivergent effects on downstreameffectors and cellular survival aftertamoxifen treatment. A, immunoblotof AMPK pathway activation andapoptotic markers after tamoxifen(Tamox) treatment. Values belowblots represent pixel intensitynormalized to loading control,b-actin. B, immunoblot of LC3-I andLC3-II flux with tamoxifen (Tam)treatment as affected by AMPKstatus. Values, pixel intensity ofLC3-II (bottom) normalized tountreated control. C, survival ofAMPK knockdown cells treated with30 mmol/L tamoxifen for 4 hours.Data are the average of fourindependent experiments � SEM,� , P < 0.05; �� , P < 0.01; ��� , P < 0.001.D, quantification of [35S] labelingafter tamoxifen treatment in MDA-MB-231 cells and AMPK CRISPRknockout cells. Data are theaverage of three independentexperiments � SEM, � , P < 0.05.
AMPK Activation and Metabolic Reprogramming by Tamoxifen
www.aacrjournals.org Cancer Res; 76(11) June 1, 2016 OF7
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
had increased survival in response to tamoxifen treatment(Fig. 4C). This effect was recapitulated in MCF7 cells, whereAMPKa1 inhibition by shRNA resulted in reduced levels ofcleaved PARP and increased survival upon tamoxifen treat-ment compared with shNT-transfected cells (SupplementaryFig. S3A and S3B). To evaluate the impact of completeinhibition of AMPK catalytic activity on cellular response totamoxifen, we used siRNA to knock down AMPKa2 in bothwild-type MDA-231 cells and in crspAMPKa1 cells. Loss ofAMPKa2 in MDA-231 cells resulted in increased cell deathupon tamoxifen treatment (Fig. 4C), whereas knockdown ofAMPKa2 in the AMPKa1 CRISPR knockout cells resulted infurther increase in tamoxifen-induced cytotoxicity.
Interestingly, knockdown of either isoform of AMPKaresulted in loss of phospho-ACC upon tamoxifen treatment,whereas loss of AMPKa1 alone ameliorated the inhibition ofthe mTOR pathway following tamoxifen treatment. Inductionof autophagy remained unchanged upon AMPKa1 inhibition;however, knockdown of both isoforms substantially reducedautophagy levels as seen by a reduction in LC3 processing (Fig.4A). To further investigate the role of AMPK isoforms in theautophagic response of breast cancer cells to tamoxifen treat-ment, we pretreated cells with E64d and pepstatin A to preventLC3-II recycling and monitored autophagic flux by immuno-blot. Although we saw reduced levels of LC3 in both siAMPKa2and crspAMPKa1 conditions, MDA-231 cells require bothAMPKa1 and AMPKa2 to be knocked down to significantlyreduce autophagy induction with tamoxifen treatment (Fig.4B). These data suggest that AMPK activation promotes autop-hagy in response to tamoxifen treatment. To determine wheth-er the AMPK isoform–specific effects on the mTOR pathwayimpacted upon cellular translation, we pulse labeled MDA-231cells and crspAMPKa1 cells with [35S]-cysteine and methio-nine. Translation was inhibited in both cell lines, albeit to asubstantially lesser extent in the crspAMPKa1 cells upontamoxifen treatment (Fig. 4D and Supplementary Fig. S3E).
Notably, although the dual AMPKa1/2 knockdown showedthe highest levels of tamoxifen toxicity as determined by mod-ified MTT assay, the levels of apoptosis markers cleaved PARPand cleaved caspase-3 were quite low. This could be due to morerapid processing of these markers under these conditions asindicated by complete loss of total PARP in these cells. Caspase-3/7 activity assay suggests a decrease in apoptosis upon AMPKa1knockout, whereas Annexin V and propidium iodide stainingsupport an increase in apoptosis with AMPKa2 knockdown(Supplementary Fig. S3C). However, the data from these apo-ptosis assays were not statistically significant and showed only asmall trend toward different levels of apoptosis dependent onAMPK status. Crystal violet staining of cells 7 days followingtamoxifen treatment also shows that AMPKa1 knockout rescuescells from tamoxifen treatment, whereas AMPKa2 knockdownpromotes cell death (Supplementary Fig. S3D). Therefore,although increased apoptosis may contribute to reduced viabil-ity of AMPKa2 knockout cells following tamoxifen treatment,additional mechanisms leading to reduced overall survival arelikely involved.
Tamoxifen activates the AMPK pathway in an orthotropicmodel of breast cancer
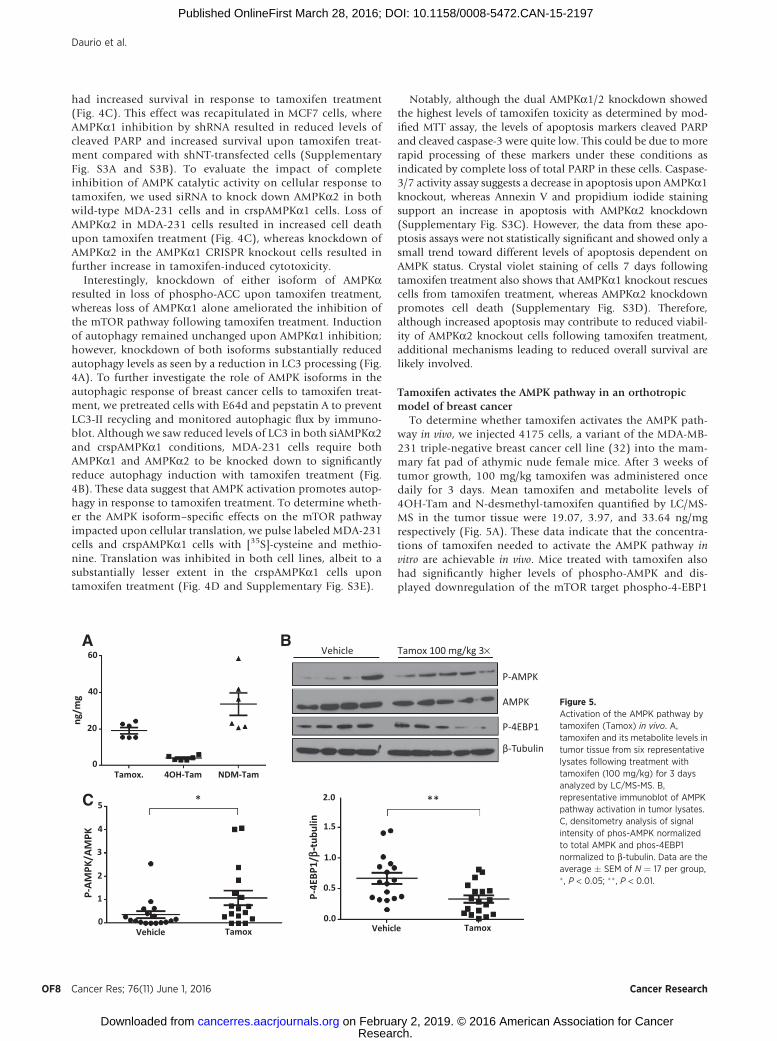
To determine whether tamoxifen activates the AMPK path-way in vivo, we injected 4175 cells, a variant of the MDA-MB-231 triple-negative breast cancer cell line (32) into the mam-mary fat pad of athymic nude female mice. After 3 weeks oftumor growth, 100 mg/kg tamoxifen was administered oncedaily for 3 days. Mean tamoxifen and metabolite levels of4OH-Tam and N-desmethyl-tamoxifen quantified by LC/MS-MS in the tumor tissue were 19.07, 3.97, and 33.64 ng/mgrespectively (Fig. 5A). These data indicate that the concentra-tions of tamoxifen needed to activate the AMPK pathway invitro are achievable in vivo. Mice treated with tamoxifen alsohad significantly higher levels of phospho-AMPK and dis-played downregulation of the mTOR target phospho-4-EBP1
A B
P-AMPK
60
40
20
0Tamox. 4OH-Tam NDM-Tam
Vehicle
52.0
1.5
1.0
0.5
0.0
4
3
2
1
0
P-AM
PK/A
MPK
P-4E
BP1/
b-tu
bulin
Tamox Vehicle Tamox
ng/m
g AMPK
Vehicle Tamox 100 mg/kg 3×
β-Tubulin
P-4EBP1
***C
Figure 5.Activation of the AMPK pathway bytamoxifen (Tamox) in vivo. A,tamoxifen and its metabolite levels intumor tissue from six representativelysates following treatment withtamoxifen (100 mg/kg) for 3 daysanalyzed by LC/MS-MS. B,representative immunoblot of AMPKpathway activation in tumor lysates.C, densitometry analysis of signalintensity of phos-AMPK normalizedto total AMPK and phos-4EBP1normalized to b-tubulin. Data are theaverage � SEM of N ¼ 17 per group,� , P < 0.05; �� , P < 0.01.
Daurio et al.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF8
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197
(Fig. 5B and C). These results are in agreement with AMPKpathway activation seen in vitro.
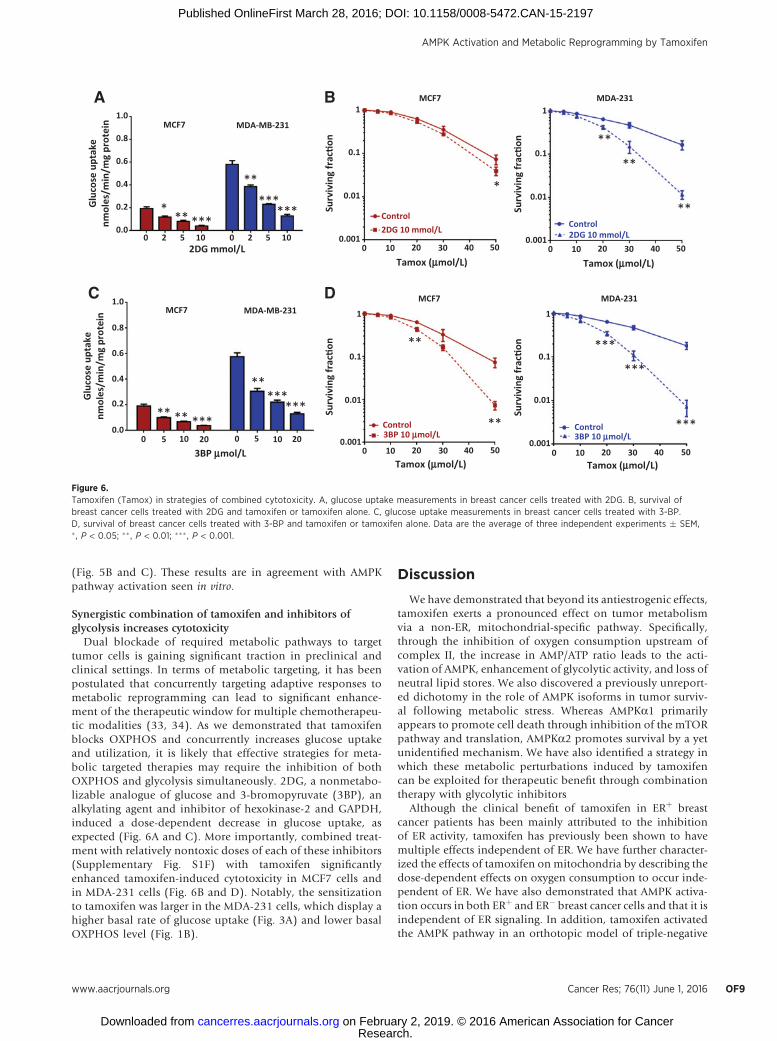
Synergistic combination of tamoxifen and inhibitors ofglycolysis increases cytotoxicity
Dual blockade of required metabolic pathways to targettumor cells is gaining significant traction in preclinical andclinical settings. In terms of metabolic targeting, it has beenpostulated that concurrently targeting adaptive responses tometabolic reprogramming can lead to significant enhance-ment of the therapeutic window for multiple chemotherapeu-tic modalities (33, 34). As we demonstrated that tamoxifenblocks OXPHOS and concurrently increases glucose uptakeand utilization, it is likely that effective strategies for meta-bolic targeted therapies may require the inhibition of bothOXPHOS and glycolysis simultaneously. 2DG, a nonmetabo-lizable analogue of glucose and 3-bromopyruvate (3BP), analkylating agent and inhibitor of hexokinase-2 and GAPDH,induced a dose-dependent decrease in glucose uptake, asexpected (Fig. 6A and C). More importantly, combined treat-ment with relatively nontoxic doses of each of these inhibitors(Supplementary Fig. S1F) with tamoxifen significantlyenhanced tamoxifen-induced cytotoxicity in MCF7 cells andin MDA-231 cells (Fig. 6B and D). Notably, the sensitizationto tamoxifen was larger in the MDA-231 cells, which display ahigher basal rate of glucose uptake (Fig. 3A) and lower basalOXPHOS level (Fig. 1B).
Discussion
We have demonstrated that beyond its antiestrogenic effects,tamoxifen exerts a pronounced effect on tumor metabolismvia a non-ER, mitochondrial-specific pathway. Specifically,through the inhibition of oxygen consumption upstream ofcomplex II, the increase in AMP/ATP ratio leads to the acti-vation of AMPK, enhancement of glycolytic activity, and loss ofneutral lipid stores. We also discovered a previously unreport-ed dichotomy in the role of AMPK isoforms in tumor surviv-al following metabolic stress. Whereas AMPKa1 primarilyappears to promote cell death through inhibition of the mTORpathway and translation, AMPKa2 promotes survival by a yetunidentified mechanism. We have also identified a strategy inwhich these metabolic perturbations induced by tamoxifencan be exploited for therapeutic benefit through combinationtherapy with glycolytic inhibitors
Although the clinical benefit of tamoxifen in ERþ breastcancer patients has been mainly attributed to the inhibitionof ER activity, tamoxifen has previously been shown to havemultiple effects independent of ER. We have further character-ized the effects of tamoxifen on mitochondria by describing thedose-dependent effects on oxygen consumption to occur inde-pendent of ER. We have also demonstrated that AMPK activa-tion occurs in both ERþ and ER� breast cancer cells and that it isindependent of ER signaling. In addition, tamoxifen activatedthe AMPK pathway in an orthotopic model of triple-negative
1.01
0.1
Surv
ivin
g fr
ac�o
nSu
rviv
ing
frac
�on
Surv
ivin
g fr
ac�o
nSu
rviv
ing
frac
�on
0.01
Control2DG 10 mmol/L
Control3BP 10 mmol/L
Control2DG 10 mmol/L
Control3BP 10 mmol/L
0.001
1
0.1
0.01
0.001
1
0.1
0.01
0.001
1
0.1
0.01
0.0010 10 20
Tamox (mmol/L) Tamox (mmol/L)
Tamox (mmol/L) Tamox (mmol/L)
30 40 50
0 10 20 30 40 50 0 10 20 30 40 50
0 10 20 30 40 50
MCF7
MCF7
MDA-MB-231
MDA-231
MCF7MCF7
MDA-MB-231MDA-231
0.8
0.6
0.4
0.2
0.0
1.0
0.8
0.6
0.4
0.2
0.0
0 2 5 102DG mmol/L
0 2 5 10
0 5 10
3BP mmol/L020 5 10 20
Glu
cose
upt
ake
nmol
es/m
in/m
g pr
otei
nG
luco
se u
ptak
enm
oles
/min
/mg
prot
ein
**
**
**
**
**
***
***
***
A
*
C
B
D
* ** ***
**
******
** ** ***
**
******
Figure 6.Tamoxifen (Tamox) in strategies of combined cytotoxicity. A, glucose uptake measurements in breast cancer cells treated with 2DG. B, survival ofbreast cancer cells treated with 2DG and tamoxifen or tamoxifen alone. C, glucose uptake measurements in breast cancer cells treated with 3-BP.D, survival of breast cancer cells treated with 3-BP and tamoxifen or tamoxifen alone. Data are the average of three independent experiments � SEM,� , P < 0.05; ��, P < 0.01; ��� , P < 0.001.
AMPK Activation and Metabolic Reprogramming by Tamoxifen
www.aacrjournals.org Cancer Res; 76(11) June 1, 2016 OF9
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

breast cancer. Although we treated these animals for a shorttime at a high dose, we would anticipate similar effects at alower dose over a longer period of time, as tamoxifen has beenshown to accumulate in tumor tissues to levels up to 60 to 70times the plasma concentration (10).
The non-ER–dependent effects of tamoxifen described herehave the potential to expand the therapeutic window for tamox-ifen into ER� BC and non-breast cancers. Furthermore, most ERþ
BC related deaths have been attributed to metastatic disease,which is often tamoxifen resistant (2). Although the mechanismby which tamoxifen resistance occurs remains unclear, it isthought to involve alterations in ER itself and alterations in thelevels of coregulators or downstream effectors in the signalingpathway (2). Our results suggest that it would be valuable tofurther explore whether combination therapies that take advan-tage of themetabolic effects of tamoxifen could restore tamoxifensensitivity or prevent resistance.
The precise target for tamoxifen in the mitochondrial respi-ration chain is not currently clear. The evidence presented inthis article supports the hypothesis that tamoxifen inhibitscomplex I in the electron transport chain (see Fig. 7). Theability of complex II substrate succinate to restore oxygenconsumption in tamoxifen-treated cells points to the inhibitionof mitochondrial respiration upstream of complex II. More-over, the inability of complex I substrates glutamate and malateto restore any oxygen consumption in permeabilized breastcancer cells indicates that complex I may be the site of tamox-ifen action (Fig. 7). However, we cannot completely rule outpossible effects of tamoxifen on upstream processes, such as theTCA cycle, that contribute electron donors to the respiratorychain. Another implication of our findings with translationalpotential is the fact that inhibition of oxygen consumption is avalidated strategy to increase tumor pO2 and decrease hypoxia,a major cause of cancer resistance to genotoxic therapy, includ-ing radiation (35). Therefore, tamoxifen has the potential tofunction as a radiation and chemotherapeutic sensitizer. In fact,other complex I inhibitors, such as metformin, have shownanticancer effects through both their metabolic effects andactivation of AMPK. Although our results are reminiscent ofthe metabolic effects of metformin treatment, both the inhi-bition of oxygen consumption and activation of AMPK occur ata far more rapid rate (5–10 minutes) than that seen withmetformin (12–24 hours). Also important is the distinctionthat although in vitro metformin exerts its OXPHOS-inhibitoryeffects at doses ranging from 1 to 50 mmol/L, tamoxifen doesso at low micromolar concentrations.
We also show that like metformin, tamoxifen inhibitedOXPHOS and increased dependence on glycolysis (Fig. 7).Intriguingly, soon after breast cancer patients begin treatmentwith tamoxifen, a "metabolic flare" is often seen in their FDG-PETscans (36). Although the cause for this radiologic abnormalityhas not been clearly delineated, it is reasonable to hypothesizethat this could be due to increased uptake of 2[18F]fluoro-2-deoxy-D-glucose (FDG) in response tometabolic reprogrammingand AMPK activation, as GLUT 1 transporters, the major trans-porter system of glucose and FDG uptake, are AMPK targets (37).
Our results are also in agreement with other metabolic effectsof tamoxifen in patient populations. Women on tamoxifentherapy have an increased risk for developing nonalcoholicfatty liver disease (38). However, the effects of tamoxifen ontumor fatty acid metabolism have not been investigated in
detail. Accumulation of malonyl-CoA following tamoxifentreatment has been reported previously (39), and we alsoobserved this in breast cancer cells. The synthesis of malo-nyl-CoA is the rate-limiting step in fatty acid synthesis; thus, itsaccumulation leads us to suspect that there is a decrease in theactivity of FASN, which is consistent with AMPK activation. It isalso possible that increased upstream flux through glycolysiscontributes to the accumulation of malonyl-CoA with tamox-ifen treatment. It is worth noting that AMPK is also known toinhibit fatty acid synthesis and promote b-oxidation, primarilythrough inhibiting ACC (21).
The antitumorigenic effects of metformin in breast cancerhave been shown to be dependent on its activation of AMPK(40). We asked whether AMPK had a similar role in beast cancersurvival of tamoxifen treatment. We used the CRISPR-Cas9system to delete AMPKa1 from MDA-MB-231 cells andobserved a compensatory increase in AMPKa2 expression. Thiswould lead us to hypothesize that the isoforms have redundanteffects, yet we observed opposing effects on cell survival. Otherwork examining the isoforms of AMPKa has identified somedifferences, including the suppression of a2 expression inprimary breast cancer (41). Knockout of AMPKa1 alone res-cued cells from tamoxifen treatment, whereas knockdown of
Figure 7.Model of the effects of tamoxifen (Tamox) on cellular metabolism andAMPK signaling. Tamoxifen directly inhibits the mitochondrial electrontransport chain at complex I. This results in a decrease in consumption ofO2 by complex IV and an increase in the AMP, ATP ratio, activating AMPK,and promoting glycolysis and autophagy. AMPK signaling inhibits themTOR pathway and translation, resulting in cell death following tamoxifentreatment. Modulation of fatty acid metabolism may involve directinhibitory action of tamoxifen on FASN activity.
Daurio et al.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF10
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

AMPKa2 increased cell death. Immunoblot analysis and [35S]labeling of proteins indicated that the a1 subunit is responsiblefor downregulation of the MTOR pathway and translation.However, both isoforms are needed to inhibit ACC, and eitherisoform can activate autophagy. We suspect that it is the loss ofrobust autophagy induction in the dual AMPKa1a2 knock-down that results in increased tamoxifen cytotoxicity underthose conditions. The ability of tamoxifen to activate autop-hagy is well established. In fact, the autophagic response totamoxifen has been implicated in breast cancer resistance totamoxifen treatment (42, 43). In addition, the ability of AMPKto activate autophagy via ULK1 is also well described (44). Thedata reported in this manuscript are the first to directly identifyAMPK activation as a mechanism of autophagy induction intamoxifen treatment. In summary, we have identified AMPK asa mediator of tamoxifen toxicity through isoform-specificeffects.
We have also demonstrated the potential for increased ther-apeutic benefit through drug combinations with tamoxifen thathave a synergistic effect via blockade of upregulated metabolicpathways. Because of the fact that inhibition of oxygen con-sumption by tamoxifen results in upregulated glycolysis, effec-tive strategies for metabolic interventions may require theinhibition of both OXPHOS and glycolysis simultaneously.Glycolytic inhibitors have long been attractive clinical agentsdue to the Warburg effect, yet systemic toxicity has limited theirsuccess in the clinic. 2DG has been tested in clinical trials, and3BP has shown significant preclinical potential (45). Our datasuggest that clinical use of these inhibitors in combination withtamoxifen may result in increased efficacy and use of reduceddoses that may limit toxicity.
In conclusion, we have characterized the ER-independenteffects of tamoxifen on cancer metabolism and described how
these effects can be exploited for therapeutic benefit throughnovel drug combinations with the potential to expand the clinicaluse of tamoxifen.
Disclosure of Potential Conflicts of InterestC. Koumenis is a consultant/advisory board member for Ruga Pharma-
ceuticals. No potential conflicts of interest were disclosed by the otherauthors.
Authors' ContributionsConception and design: N.A. Daurio, S.W. Tuttle, E.Y. Song, N.W. Snyder,I.A. Blair, C. KoumenisDevelopment of methodology: N.A. Daurio, S.W. Tuttle, A.J. Worth,N.W. Snyder, I.A. BlairAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): N.A. Daurio, S.W. Tuttle, A.J. Worth, E.Y. Song,J.M. Davis, N.W. SnyderAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): N.A. Daurio, S.W. Tuttle, A.J. Worth, E.Y. Song,N.W. Snyder, I.A. Blair, C. KoumenisWriting, review, and/or revision of the manuscript: N.A. Daurio, S.W. Tuttle,A.J. Worth, J.M. Davis, N.W. Snyder, I.A. Blair, C. KoumenisAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): C. KoumenisStudy supervision: I.A. Blair, C. Koumenis
Grant SupportThis studywas supported byNIH grants 2R25CA140116-06, P30CA016520,
P30ES013508, T32ES019851, and R01CA182747.The costs of publication of this article were defrayed in part by the
payment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received August 13, 2015; revised February 26, 2016; accepted March 14,2016; published OnlineFirst March 28, 2016.
References1. EBCTCG. Effects of chemotherapy and hormonal therapy for early breast
cancer on recurrence and 15-year survival: an overview of the randomisedtrials. Lancet 2005;365:1687–717.
2. Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med1998;339:1609–18.
3. O'Brian CA, Liskamp RM, Solomon DH, Weinstein IB. Inhibition ofprotein kinase C by tamoxifen. Cancer Res 1985;45:2462–5.
4. McClay EF, Albright KD, Jones Ja, Christen RD, Howell SB. Tamoxifenmodulation of cisplatin sensitivity in human malignant melanoma cells.Cancer Res 1993;53:1571–6.
5. Stuart NS, Philip P, Harris a L, Tonkin K, Houlbrook S, Kirk J, et al. High-dose tamoxifen as an enhancer of etoposide cytotoxicity. Clinical effectsand in vitro assessment in p-glycoprotein expressing cell lines. Br J Cancer1992;66:833–9.
6. Tang PA, RoldanG, Brasher PMA, FultonD, RoaW,Murtha A, et al. A phaseII study of carboplatin and chronic high-dose tamoxifen in patients withrecurrent malignant glioma. J Neurooncol 2006;78:311–6.
7. McClay EF, McClay ME, Jones JA, Winski PJ, Christen RD, Howell SB,et al. A phase I and pharmacokinetic study of high dose tamoxifen andweekly cisplatin in patients with metastatic melanoma. Cancer 1997;79:1037–43.
8. Perez EA, Gandara DR, Edelman MJ, O'Donnell R, Lauder IJ, DeGregorioM. Phase I trial of high-dose tamoxifen in combination with cisplatin inpatients with lung cancer and other advanced malignancies. Cancer Invest2003;21:1–6.
9. Bergan RC, Reed E,Myers CE, Headlee D, BrawleyO, ChoHK, et al. A PhaseII study of high-dose tamoxifen in patients with hormone-refractoryprostate cancer. Clin cancer Res 1999;5:2366–73.
10. Lien EA, Solheim E, Ueland PM. Distribution of tamoxifen and its meta-bolites in rat and human tissues during steady-state treatment. Cancer Res1991;51:4837–44.
11. Moreira PI, Cust�odio J, Moreno A, Oliveira CR, Santos MS. Tamox-ifen and estradiol interact with the flavin mononucleotide site ofcomplex I leading to mitochondrial failure. J Biol Chem 2006;281:10143–52.
12. Cardoso CMP, Moreno AJM, Almeida LM, Cust�odio JBa. 4-Hydroxy-tamoxifen induces slight uncoupling of mitochondrial oxidative phos-phorylation system in relation to the deleterious effects of tamoxifen.Toxicology 2002;179:221–32.
13. WheatonWW,Weinberg SE,Hamanaka RB, Soberanes S, Sullivan LB, AnsoE, et al. Metformin inhibits mitochondrial complex I of cancer cells toreduce tumorigenesis. Elife 2014;2014:1–18.
14. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the War-burg effect: the metabolic requirements of cell proliferation. Science2009;324:1029–33.
15. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cellsurvival and growth. Nat Cell Biol 2015;17:351–9.
16. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sanani-kone E, et al. Oncogenic kras maintains pancreatic tumors through regu-lation of anabolic glucose metabolism. Cell 2012;149:656–70.
17. Haq R, Fisher DE, Widlund HR. Molecular pathways: BRAF inducesbioenergetic adaptation by attenuating oxidative phosphorylation. ClinCancer Res 2014;20:2257–63.
18. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al.Oncogenic BRAF regulates oxidative metabolism via PGC1a and MITF.Cancer Cell 2013;23:302–15.
AMPK Activation and Metabolic Reprogramming by Tamoxifen
www.aacrjournals.org Cancer Res; 76(11) June 1, 2016 OF11
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

19. Yuan P, Ito K, Perez-lorenzo R, Del Guzzo C, Lee JH, Shen C-H, et al.Phenformin enhances the therapeutic benefit of BRAF V600E inhibition inmelanoma. Proc Natl Acad Sci U S A 2013;110:18226–31.
20. Viale A, Pettazzoni P, Lyssiotis Ca., Ying H, S�anchez N, Marchesini M, et al.Oncogene ablation-resistant pancreatic cancer cells depend on mitochon-drial function. Nature 2014;514:628–32.
21. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cellgrowth, autophagy and metabolism. Nat Cell Biol 2011;13:1016–23.
22. Liang J, Mills GB. AMPK: A contextual oncogene or tumor suppressor?Cancer Res 2013;73:2929–35.
23. Ran F, Hsu P,Wright J, Agarwala V. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–308.
24. Kochanowski N, Blanchard F, Cacan R, Chirat F, Guedon E, Marc a, et al.Intracellular nucleotide and nucleotide sugar contents of cultured CHOcells determined by a fast, sensitive, and high-resolution ion-pair RP-HPLC. Anal Biochem 2006;348:243–51.
25. Basu SS, Blair IA. SILEC: a protocol for generating and using isotopi-cally labeled coenzyme A mass spectrometry standards. Nat Protoc2012;7:1–12.
26. Snyder NW, Basu SS, Zhou Z, Worth AJ, Blair IA. Stable isotope dilutionliquid chromatography/mass spectrometry analysis of cellular and tissuemedium- and long-chain acyl-coenzyme A thioesters. Rapid CommunMass Spectrom 2014;28:1840–8.
27. Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, et al. ATMcouples replication stress and metabolic reprogramming during cellularsenescence. Cell Rep 2015;11:893–901.
28. Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogenreceptor antagonist with a novel mechanism of action. Br J Cancer2004;90:S2–6.
29. Br€unner N, Boulay V, Fojo A, Freter CE, LippmanME, Clarke R. Acquisitionof hormone-independent growth in MCF-7 cells is accompanied byincreased expression of estrogen-regulated genes but without detectableDNA amplifications. Cancer Res 1993;53:283–90.
30. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking instarved cells: Regulation by lipid droplet lipolysis, autophagy, and mito-chondrial fusion dynamics. Dev Cell . 2015;32:1–15.
31. Worth AJ, Basu SS, Snyder NW, Mesaros C, Blair Ia. Inhibition ofneuronal cell mitochondrial complex I with rotenone increases lipid-oxidation supporting acetyl-coenzyme A levels. J Biol Chem 2014;289:26895–903.
32. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes thatmediate breast cancer metastasis to lung. Nature 2005;436:518–24.
33. D€orr JR, Yu Y, Milanovic M, Beuster G, Zasada C, D€abritz JHM, et al.Synthetic lethal metabolic targeting of cellular senescence in cancer ther-apy. Nature 2013;501:421–5.
34. Sonveaux P, V�egran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al.Targeting lactate-fueled respiration selectively kills hypoxic tumor cells inmice. J Clin Invest 2008;118:3930–42.
35. Lin A, Maity A. Molecular pathways: a novel approach to targeting hypoxiaand improving radiotherapy efficacy via reduction in oxygen demand.Clin cancer Res 2015;21:1995–2000.
36. Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ,Siegel BA. Positron emission tomographic assessment of "metabolic flare "to predict response of metastatic breast cancer to antiestrogen therapy.Eur J Nucl Med 1999;26:51–6.
37. Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhancedglucose uptake via GLUT1. Mol Cell 2013;49:1167–75.
38. Saphner T, Triest-Robertson S, Li H, Holzman P. The association ofnonalcoholic steatohepatitis and tamoxifen in patients with breast cancer.Cancer 2009;115:3189–95.
39. L�opez M, Lelliott CJ, Tovar S, Kimber W, Gallego R, Virtue S, et al.Tamoxifen-induced anorexia is associated with fatty acid synthase inhibi-tion in the ventromedial nucleus of the hypothalamus and accumulationof malonyl-CoA. Diabetes 2006;55:1327–36.
40. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metforminis an AMP kinase-dependent growth inhibitor for breast cancer cells.Cancer Res 2006;66:10269–73.
41. Fox MM, Phoenix KN, Kopsiaftis SG, Claffey KP. AMP-activated proteinkinase a 2 isoform suppression in primary breast cancer alters AMPKgrowth control and apoptotic signaling. Genes Cancer 2013;4:3–14.
42. Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ,et al. A role formacroautophagy inprotection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance.Mol Cancer Ther 2008;7:2977–87.
43. Cook KL, Shajahan AN, W€arri A, Jin L, Hilakivi-Clarke LA, Clarke R.Glucose-regulated protein 78 controls cross-talk between apoptosis andautophagy to determine antiestrogen responsiveness. Cancer Res 2012;72:3337–49.
44. Kim J, KunduM, Viollet B, GuanK-L. AMPK andmTOR regulate autophagythrough direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132–41.
45. Ko YH, Smith BL, Wang Y, Pomper MG, Rini Da, Torbenson MS, et al.Advanced cancers: Eradication in all cases using 3-bromopyruvate therapyto deplete ATP. Biochem Biophys Res Commun 2004;324:269–75.
Cancer Res; 76(11) June 1, 2016 Cancer ResearchOF12
Daurio et al.
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197

Published OnlineFirst March 28, 2016.Cancer Res Natalie A. Daurio, Stephen W. Tuttle, Andrew J. Worth, et al. Modality in Cancer TreatmentMechanisms Suggests New Uses for This Therapeutic
Independent−Tamoxifen through Estrogen Receptor AMPK Activation and Metabolic Reprogramming by
Updated version
10.1158/0008-5472.CAN-15-2197doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2016/03/26/0008-5472.CAN-15-2197.DC1
Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/early/2016/05/20/0008-5472.CAN-15-2197To request permission to re-use all or part of this article, use this link
Research. on February 2, 2019. © 2016 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 28, 2016; DOI: 10.1158/0008-5472.CAN-15-2197