amyloid transthyretin (attr) amyloidosis€¦ · ° ttr amyloid deposits have been detected in as...
TRANSCRIPT

Amyloid Transthyretin (ATTR) AmyloidosisAN OVERVIEW

AMYLOID TRANSTHYRETIN (ATTR) AMYLOIDOSIS
• ATTR amyloidosis is a rare, progressive, and fatal disease that manifests clinically with motor and sensory neuropathy, autonomic neuropathy, gastrointestinal disturbances, and cardiomyopathy1-3
• There are 2 main types of ATTR amyloidosis
° Hereditary ATTR amyloidosis—affects multiple organs and body systems (eg, heart, nervous system, gastrointestinal tract, and kidney)1,2,4
° Wild-type ATTR amyloidosis—primarily affects the heart1,2
PATHOPHYSIOLOGY
• ATTR amyloidosis is caused by the buildup of amyloid fibrils in organs and tissues in the body4
• The amyloidogenic precursor in ATTR amyloidosis is transthyretin (TTR), a transport protein synthesized primarily by the liver2,5,6
• Normally, TTR comprises 4 single-chain monomers (ie, tetramer)5
• TTR gene mutations are thought to induce changes that result in weaker interactions between monomer subunits5,7
° Weakened interactions render the normal tetrameric assembly prone to dissociation into monomers that misfold and have a greater propensity for aggregation
° Misfolded protein accumulates, forming fibrils that deposit in tissues and organs
MECHANISM OF AMYLOID FIBRIL FORMATION4,8,9
*Amyloid fibrils can be caused by a variety of toxic intermediates, including small oligomers and amorphous aggregates.
Modified with permission from www.thaos.net, which obtained image from J. Kelly, TSRI.

GENETICS
• TTR is encoded by a single-copy gene; however, more than 120 mutations have been identified10,11
• Hereditary ATTR amyloidosis is typically associated with a single amino acid substitution caused by a point mutation in the TTR gene2,10,12,13
° For example, valine to methionine substitution at position 30 (V30M)
• Most TTR mutations are amyloidogenic and promote instability of TTR tetramers2,11
• Hereditary ATTR amyloidosis is transmitted in an autosomal dominant manner, with variable penetrance1
GENOTYPE-PHENOTYPE CORRELATION
• Specific genotypes are associated with predominant polyneuropathy or cardiomyopathy features; however, most mutations affect multiple organs and there is considerable heterogeneity in disease manifestations1
• V30M is the most common mutation worldwide associated with hereditary ATTR amyloidosis with polyneuropathy1,4
° The highest occurrence of V30M is in northern Portugal (incidence, 1 in 538 individuals); however, this mutation is also common in other regions of world1,2,4
• Although V30M is commonly associated with polyneuropathy, many patients with V30M have cardiomyopathy1
NEUROLOGIC Cardiac
A36P
F33L
C10R
S50R
R34T
G47AA36P
T60A
L111M
I68L
V122I
V30M
Late onset
V30M
Early onset
F64L
T49A
E89Q
S23N
W41L H88R
Modified with permission from Rapezzi et al.14
• V122I is the most common mutation worldwide associated with hereditary ATTR amyloidosis with cardiomyopathy1,4
° In the United States, V122I is the most common mutation and primarily occurs in African American individuals4,15
° 3%-4% of African American individuals are V122I carriers compared with 0.44% of white individuals and 0% of Hispanic individuals2
• Although V122I is commonly associated with cardiomyopathy, many patients with V122I have polyneuropathy1
F33L
C10R
S50R
R34T
G47AA36P
T60A
L111M
I68L
V122I
V30M
Late onset
V30M
Early onset
F64L
T49A
E89Q
S23N
W41L H88R
Neurologic CARDIAC
Modified with permission from Rapezzi et al.14
CLINICAL PRESENTATION
• ATTR amyloidosis is characterized by substantial clinical heterogeneity
• Nonspecific symptoms and manifestations overlap with more common disorders, hindering recognition of amyloidosis
• Bilateral carpal tunnel syndrome (CTS) is a common presenting manifestation in patients with ATTR amyloidosis and can be diagnosed up to 10 years before confirmation of ATTR amyloidosis16-19
° Patients often have a history of CTS release surgery18; in a single report, 100% of patients with amyloidosis had bilateral CTS and 70% of patients had a history of CTS surgery19
• Spinal canal stenosis has been detected in patients with ATTR amyloidosis and is a common misdiagnosis in patients with hereditary ATTR amyloidosis16,20
° TTR amyloid deposits have been detected in as many as ~50% of surgical specimens from patients with lumbar spinal canal stenosis21
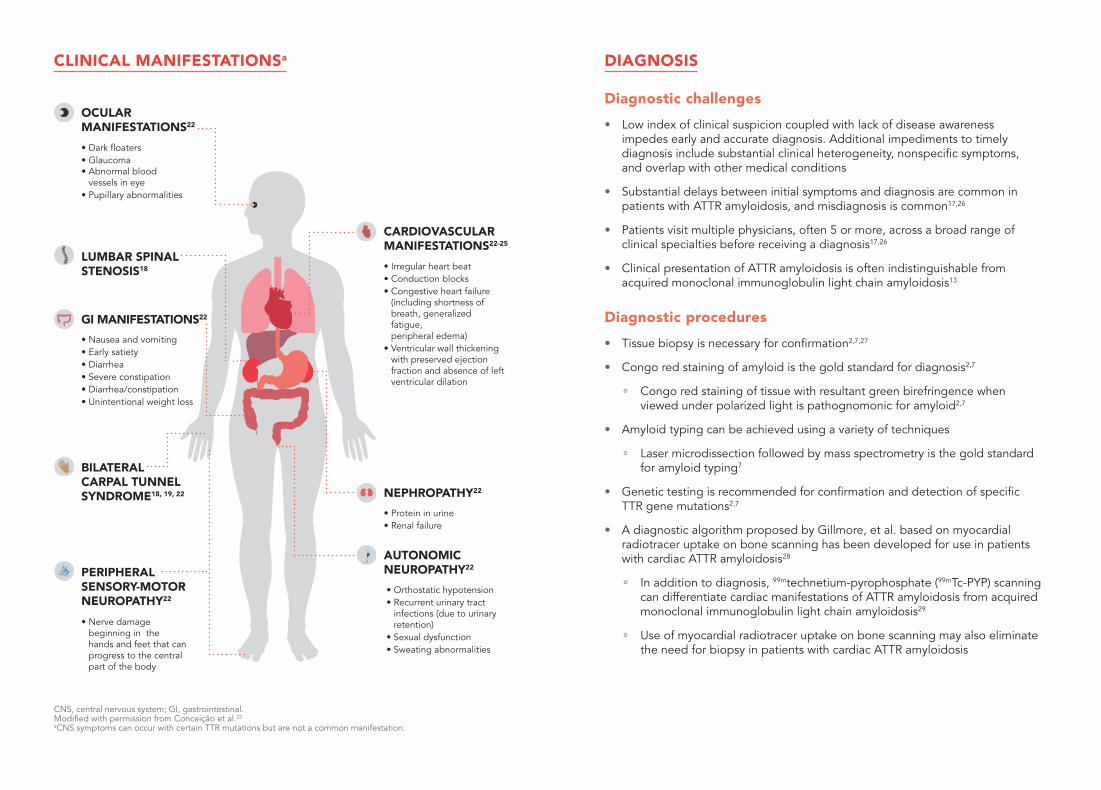
CLINICAL MANIFESTATIONSa
CNS, central nervous system; GI, gastrointestinal. Modified with permission from Conceição et al.22
aCNS symptoms can occur with certain TTR mutations but are not a common manifestation.
DIAGNOSIS
Diagnostic challenges
• Low index of clinical suspicion coupled with lack of disease awareness impedes early and accurate diagnosis. Additional impediments to timely diagnosis include substantial clinical heterogeneity, nonspecific symptoms, and overlap with other medical conditions
• Substantial delays between initial symptoms and diagnosis are common in patients with ATTR amyloidosis, and misdiagnosis is common17,26
• Patients visit multiple physicians, often 5 or more, across a broad range of clinical specialties before receiving a diagnosis17,26
• Clinical presentation of ATTR amyloidosis is often indistinguishable from acquired monoclonal immunoglobulin light chain amyloidosis13
Diagnostic procedures
• Tissue biopsy is necessary for confirmation2,7,27
• Congo red staining of amyloid is the gold standard for diagnosis2,7
° Congo red staining of tissue with resultant green birefringence when viewed under polarized light is pathognomonic for amyloid2,7
• Amyloid typing can be achieved using a variety of techniques
° Laser microdissection followed by mass spectrometry is the gold standard for amyloid typing7
• Genetic testing is recommended for confirmation and detection of specific TTR gene mutations2,7
• A diagnostic algorithm proposed by Gillmore, et al. based on myocardial radiotracer uptake on bone scanning has been developed for use in patients with cardiac ATTR amyloidosis28
° In addition to diagnosis, 99mtechnetium-pyrophosphate (99mTc-PYP) scanning can differentiate cardiac manifestations of ATTR amyloidosis from acquired monoclonal immunoglobulin light chain amyloidosis29
° Use of myocardial radiotracer uptake on bone scanning may also eliminate the need for biopsy in patients with cardiac ATTR amyloidosis
• Irregular heart beat • Conduction blocks• Congestive heart failure
(including shortness of breath, generalized fatigue, peripheral edema)
• Ventricular wall thickening with preserved ejection fraction and absence of left ventricular dilation
• Protein in urine• Renal failure
• Orthostatic hypotension• Recurrent urinary tract
infections (due to urinary retention)
• Sexual dysfunction• Sweating abnormalities
NEPHROPATHY22
AUTONOMIC NEUROPATHY22
CARDIOVASCULAR MANIFESTATIONS22-25
• Dark floaters• Glaucoma • Abnormal blood
vessels in eye• Pupillary abnormalities
BILATERAL CARPAL TUNNEL SYNDROME18, 19, 22
• Nerve damage beginning in the hands and feet that can progress to the central part of the body
OCULAR MANIFESTATIONS22
• Nausea and vomiting• Early satiety • Diarrhea• Severe constipation• Diarrhea/constipation• Unintentional weight loss
PERIPHERAL SENSORY-MOTOR NEUROPATHY22
LUMBAR SPINAL STENOSIS18
GI MANIFESTATIONS22

BURDEN OF DISEASE
• ATTR amyloidosis is a devastating, progressive disease that results in a rapid decline in quality of life
• Symptoms of hereditary ATTR amyloidosis impact multiple aspects of daily life, and disease burden increases with illness progression30-32
• Patients report difficulty with fine motor skills, bathroom/self-care, movement/mobility, activities of daily living, an inability to stand for hours, run, and work30-32
• Resource use is high among patients with hereditary ATTR amyloidosis, and hospitalization rates range from 14% to 30%, with a 14% to 24% rate for emergency visits32
• Functional health and well-being are severely compromised in patients with ATTR amyloidosis33
• More than half (55%) the patients with hereditary ATTR amyloidosis report that their mental health/outlook on life is impacted by amyloidosis and they have anxiety (71%), stress (62%), and depression (43%)31
CURRENT TREATMENT AND LIMITATIONS
• Treatment options are limited for patients with ATTR amyloidosis
° In the United States, no therapies for ATTR amyloidosis are approved by the US Food and Drug Administration (FDA)
° In Europe, tafamidis is approved for use in adults with stage 1 (stage 1 on a scale from 1 [lowest disease burden] to 3 [greatest disease burden]) symptomatic polyneuropathy to delay peripheral neurologic impairment34
• Current treatments, used off label, have limited effectiveness and are only used in specific patient populations2,4
• There is an urgent unmet need for treatments specifically designed for ATTR amyloidosis
UNMET NEEDS
DIAGNOSIS
• Increasing clinical suspicion and disease awareness are high unmet needs
• Increased efforts are needed to keep amyloidosis “front of mind” among clinicians
PATIENT ACCESS TO COORDINATED CARE
• Access to and coordination of care between amyloidosis centers of excellence and academic specialists is greatly needed
APPROVED THERAPIES TO TREAT ATTR AMYLOIDOSIS
• Safe and effective disease-modifying therapies are needed that optimize patient quality of life, which is severely compromised by the disease, and improve clinical outcomes
RESOURCES
Amyloidosis Support Groupwww.amyloidosissupport.org
Amyloidosis Foundationwww.amyloidosisfoundation.org
Amyloidosis Research Consortiumwww.arci.org
My Amyloidosis PathfinderDiscover treatment centers and clinical trials that match your amyloidosiswww.myamyloidosispathfinder.org

REFERENCES
1. Coelho T, et al. Curr Med Res Opin. 2013;29(1):63-76.
2. Ando Y, et al. Orphanet J Rare Dis. 2013;8:31.
3. Suhr O, et al. J Int Med. 1994;235(5):479-85.
4. Hawkins PN, et al. Ann Med. 2015;47(8):625-38.
5. Saraiva MJ. FEBS Lett. 2001;498:201-3.
6. Liz MA, et al. IUBMB Life. 2010;62(6):429-35.
7. Leung N, Nasr SH, Sethi S. Blood. 2012;120(16):3206-13.
8. Coelho T, et al. Neurology. 2012;79(8):785-92.
9. Disease Background - Transthyretin Amyloidosis. Transthyretin Amyloidosis Outcomes Survey (THAOS). http://www.thaos.net/Physicians/DiseaseBackground.cfm#01Transthyretin. Accessed January 3, 2018.
10. Rowczenio DM, et al. Hum Mutat. 2014;35(9):E2403-12.
11. Hou X, Aguilar MI, Small DH. FEBS J. 2007;274(7):1637-50.
12. Connors LH, et al. Amyloid. 2003;10(3):160-84.
13. Lachmann HJ, et al. N Engl J Med. 2002;346(23):1786-91.
14. Rapezzi C, et al. Eur Heart J. 2013;34(7):520-8.
15. Maurer MS, et al. J Am Coll Cardiol. 2016;68(2):161-72.
16. Nakagawa M, et al. Amyloid. 2016;23(1):58-63.
17. Lousada I, et al. Orphanet J Rare Dis. 2017;12(suppl 1):165.
18. Donnelly JP, Hanna M. Cleve Clin J Med. 2017;84(12 suppl 3):12-26.
19. Ikram A, et al. J Card Fail. 2017;23(8):S11-S12 (P021).
20. Cortese A, et al. J Neurol Neurosurg Psychiatry. 2017;88(5):457-8.
21. Yanagisawa A, et al. Mod Pathol. 2015;28(2):201-7.
22. Conceição I, et al. J Peripher Nerv Syst. 2016;21(1):5-9.
23. Coelho T, et al. A physician’s guide to transthyretin amyloidosis. Research Gate Amyloidosis Foundation, 2008. https://www.researchgate.net/publication/265490881_A_Physician’s_Guide_to_Transthyretin_Amyloidosis_Authored_by. Accessed January 3, 2018.
24. Gertz MA. Am J Manag Care. 2017;23(7 suppl):S107-12.
25. Galat A, et al. Eur Heart J. 2016;37(47):3525-31.
26. Lousada I, et al. Presented at: the First European Congress on Hereditary ATTR Amyloidosis. November 2015; Paris, France.
27. Castano A, et al. J Nucl Cardiol. 2016;23(6):1355-63.
28. Gillmore JD, et al. Circulation. 2016;133:2404-12.
29. Bokhari S, et al. Circ Cardiovasc Imaging. 2013;6(2):195-201.
30. Adams D, Amitay O, Coelho T. Orphanet J Rare Dis. 2015;10(suppl 1):P58
31. Amyloidosis Foundation. Understanding the patient voice in hereditary transthyretin-mediated amyloidosis (ATTR amyloidosis). http://www.amyloidosissupport.org/support_groups/fam_isabell_attr.pdf. Accessed January 3, 2018.
32. Stewart M, et al. Value Health. 2013;16:A386.
33. Lane T, et al. Orphanet J Rare Dis. 2015;10 (suppl 1):026.
34. Vyndaqel [summary of product characteristics]. Sandwich, Kent, UK: Pfizer Limited; July 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002294/WC500117862.pdf. Accessed January 3, 2018.
Cover image was created in PyMol using PDB ID 1DVQ obtained from the RCSB PDB (www.rcsb.org). V.B. Oza, P. Raman, J.W. Kelly, J.C. Sacchettini [2000] Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 7: 312-321).

© 2018 Akcea Therapeutics, Inc. All rights reserved. TTR-095-2 04/18