an exploration of the structural and
TRANSCRIPT

An Exploration of the Structural,
Electronic, and Anion Binding Properties
of 2-Indolylphosphines
by
Joanne Yu
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Graduate Department of Chemistry
University of Toronto
© Copyright by Joanne Yu 2008

An Exploration of the Structural, Electronic, and Anion
Binding Properties of 2-Indolylphosphines
Joanne Yu Doctor of Philosophy Department of Chemistry University of Toronto 2008
Abstract
2-Indolylphosphines are unique ligands which have the capability for further
phosphine modification by introducing substituents on an indolyl nitrogen centre.
Substituents can vary in electronics, sterics, chirality, and can contain amino or
phosphino groups which result in a multidentate (P,N)- or (P,P)-2-indolylphosphine.
X-ray crystallography was used predominantly to examine and analyze the
structural features of 2-indolyphosphines and their metal complexes. While the cone
angles could not be determined crystallographically, the sum of the <CPC bond angles
provided some information on the steric crowding around a phosphorus atom in selected
2-indolylphosphines.
The symmetric tris-2-(3-methylindolyl)phosphine demonstrated anion binding
ability through its three indolyl NH sites. Titrations to a series of selected anions were
carried out; it was determined that tris-2-(3-methylindolyl)phosphine binds to these
selected anions in a 1 : 1 receptor to anion binding ratio. Crystal structures of the fluoride
and acetate complexes confirm the binding stoichiometry, and demonstrate the
cooperative interaction of all three indolyl NH sites with the anion guest. Synthetic
routes to new anion receptors with three or two indolyl NH donors were explored. The
second type yielded a molecular cleft that was used in anion binding studies.
The net basicity of a 2-indolylphosphine was determined through formation of a
Ni(CO)3L complex. Net basicity can be tuned by changing the substituents on
ii

phosphorus or on an indolyl nitrogen centre. The [Cu(tris-2-(3-
methylindolyl)phosphine)(phenanthroline)]BF4 complex is a discrete ion pair complex,
exhibiting coordination chemistry at the phosphorus centre of the phosphine, while
simultaneously hydrogen bonding through the indolyl NH sites to the BF4- anion.
Complexes of the type [Pd(L)Cl(μ-Cl)]2 were analyzed by crystallography and the effect
of net basicity on Pd-P bond length examined.
The solid-state structures of (P,N)- and (P,P)-2-indolylphosphines were evaluated.
In general, the sum of the <CPC bond angles increased from the parent unfunctionalized
2-indolylphosphine. The metal complexes of (P,N)- and (P,P)-2-indolylphosphines were
assessed by crystallography to find possible trends of trans-influence.
Lastly, a tetradentate tripodal ligand was synthesized by furnishing
diphenylphosphino substituents on the indolyl nitrogen centres of tris-2-(3-
methylindolyl)phosphine. The coordination of the tetradentate tripodal ligand to Pt(II) or
Rh(I) resulted in five-coordinate trigonal bipyramidal complexes.
iii

Acknowledgments
I would like to thank my advisor Prof. David H. Farrar, first and foremost, for
presenting me an opportunity to learn in a relatively un-structured, and therefore,
independent environment. Thank you for making the time to provide guidance and
criticism, yet allowing me the freedom to find my own way. Dave your dedication to life
outside the walls of academia and gift of positivity are truly inspiring. I would also like
to thank my co-advisor Dr. C. Scott Browning; I would not have been able to string the
story of my research together so eloquently without his valuable input. Scott you are a
talented instructor and your enthusiasm for teaching has been instrumental for me – not
only observing as a student, but observing as an instructor myself.
A warm thank you to my PhD committee: Professors Anthony J. Poë and Robert
H. Morris; and Professors Stephen J. Loeb and Douglas W. Stephan for serving as my
external examiners. I am grateful to you all for investing the time into assessing my work,
and providing helpful feedback.
I owe a great deal of gratitude to Dr. Alan J. Lough for teaching me the
techniques of X-ray crystallography. Alan your wealth of knowledge and expertise in
this field is above and beyond admirable. I appreciate all the times you would let me cut
the queue to run my own samples and answer all of my questions, no matter how simple
they were. I hope I can take the skills that you have taught me and be successful on my
own. I’d also like to thank Dr. Timothy Burrow for all of his NMR advice.
I would especially like to thank the past members of the Farrar group, first for
being superb friends, and second for providing crystalline compounds for my analysis.
The particulars: Dr. Edmond Lam for taking the time to read my thesis and providing
non-stop encouragement; Trisha Ang for being an excellent labmate and conference
buddy; Mengxin Zhao for her infectious happiness and knowledge of phosphine
synthesis; Amina Mulani and Megan Oh for being great labmates; and Dr. Claudia Babij
Krywiak for her wise words. And to the honorary members of the Farrar group: Brian
Mariampillai for his unwavering friendship and support, and for lending me chemicals;
Dr. Alan Hadzovic for his astounding wealth of knowledge and always providing a
shoulder.
iv

Without my family and friends I would not be where I am today. I am indebted to
Mom, Andy, and Allen for their unconditional love, support, and encouragement. I
would not have the courage to see this through without Hiro, you are my constant.
For Shing Chung Yu
v

Wabi-sabi – imperfect and incomplete.
vi

Table of Contents
Abstract ii
Acknowledgments iv
Table of Contents vii
List of Tables xi
List of Figures xiv
List of Schemes xix
List of Compounds xxii
List of Abbreviations xxxii
Chapter 1 Introduction 1
1.1. Tertiary Phosphine Ligands 1
1.1.1 Determining Steric Profiles of Phosphine Molecules 2
1.1.2. Determining Phosphine Ligand Basicity 3
1.1.3. The Utility of Phosphine Ligands 4
1.2. N-Heterocyclic Phosphine Ligands 6
1.3. Reactivity at Indole Nitrogen 8
1.4. 2-Indolylphosphine Ligands 10
1.5. Synthetic Anion Receptors 12
1.6. Scope of the Thesis 17
1.7 References 19
Chapter 2 Monodentate 2-Indolylphosphines: A Study in Structure by X-ray
Crystallography
24
2.1 Introduction 24
2.1.1. Crystallographic Analysis of Common Phosphines in the CSD 25
2.1.2. Crystallographic Analysis of Indole-Containing Structures in the CSD 29
vii

2.2 Results and Discussion 30
2.2.1. Synthesis of Unsubstituted 2-Indolylphosphines 30
2.2.2. X-ray Crystallographic Analysis of Unsubstituted 2-Indolylphosphines 31
2.2.3. Synthesis of Monodentate N-Alkylated 2-Indolylphosphines 40
2.2.4. Crystallographic Analysis of N-Alkylated 2-Indolylphosphines 41
2.3. Examining the Σ{<CPC} of 2-Indolylphosphines 45
2.3.1. Examining Indolyl Aromaticity in 2-Indolylphosphines 48
2.4. Conclusions 50
2.5. Experimental 51
2.6. References 54
Chapter 3 Tris-2-(3-methylindolyl)phosphine: Synthesis, Reactivity and Anion
Binding 55
3.1. Introduction 55
3.1.1. Hydrogen Bonding in 2-Indolylphosphines 56
3.2 Results and Discussion 57
3.2.1. Synthesis of Tris-2-(3-methylindolyl)phosphine 57
3.2.2. Anion Binding Properties of Tris-2-(3-methylindolyl)phosphine 59
3.3. Design of a New C3-Symmetric Anion Receptor 73
3.3.1. Synthesis of New C3-Symmetric Anion Receptor 75
3.3.2. Design of a New Diindolyl-Based Anion Receptor 81
3.3.3. Synthesis of New Diindolyl-Based Anion Receptor 82
3.3.4. Anion Binding Studies of Molecular Cleft 18 85
3.4. Conclusions 88
3.5. Experimental 89
3.6. References 101
viii

Chapter 4 Coordination Chemistry of Monodentate 2-Indolylphosphines 104
4.1. Introduction 104
4.2. Results and Discussion 105
4.2.1. An Evaluation of 2-Indolylphosphine Net-Basicity: Coordination to
Ni(CO)3
105
4.2.2. [5·X]- Complexes: Simultaneous Coordination at Phosphorus with
Ni(II)
108
4.2.3. [5·Anion]- Complexes: Simultaneous Coordination at Phosphorus
with Cu(I)
110
4.3. Pd(II) and Pt(II) Complexes of Monodentate 2-Indolylphosphines 116
4.3.1. X-ray Crystallographic Analysis of Pd(II) and Pt(II) Complexes of
Monodentate 2-Indolylphosphines
118
4.3.2. Examining the Effect of Phosphine Net-Basicity on Pd(II)-P Bond
Lengths
128
4.3.3. The Effect of Metal Coordination on the Σ{<CPC} of Monodentate 2-
Indolylphosphines
129
4.3.4. The Effect of Metal Coordination on Indolyl Aromaticity 131
4.4. Conclusions 132
4.5. Experimental 134
4.6. References 137
Chapter 5 Multidentate N-Functionalized 2-Indolylphosphines and their Metal
Complexes
138
5.1. Introduction 138
5.1.1. Bidentate (P,N)- and (P,P)-Ligands 138
5.1.2. Symmetric Tetradentate Ligand PP3 and Its Reactivity 139
5.2. Results and Discussion 140
5.2.1. Synthesis of Multidentate 2-Indolylphosphines 140
ix

5.2.2. X-ray Crystallographic Analysis of (P,N)- and (P,P)-2-
Indolylphosphines
142
5.2.3. The Effect of N-Functionalization on the Σ{<CPC} of (P,N)-and
(P,P)-2-Indolylphosphines
150
5.2.4. The Effect of N-Functionalization on Indolyl Aromaticity of (P,N)-
and (P,P)-2-Indolylphosphines
153
5.3. Pd(II) Complexes of (P,N)-2-Indolylphosphines 153
5.3.1. X-ray Crystallographic Analysis of Pd(II) and Pt(II) Complexes of
(P,N)-2-Indolylphosphines
155
5.3.2. X-ray Crystallographic Analysis of Pd(II) Complexes of (P,P)-2-
Indolylphosphines
162
5.4. Examining Trans-Influence Properties of (P,N)- and (P,P)-2-Indolylphosphines 168
5.5. Synthesis and Characterization of Multidentate N-Functionalized Ligands based
on Phosphine 5
171
5.5.1. X-ray Crystallographic Analysis of Metal Complexes of (N-PPh2)3-5 175
5.5.2. The Reactivity of [PtCl(N-PPh2)3-5]BF4 and [RhCl(N-PPh2)3-5] 180
5.6. Metal Coordination on the Σ{<CPC} of (P,N)- and (P,P)-2-Indolylphosphines 182
5.6.1. The Effect of Metal Coordination on Indolyl Aromaticity of
Multidentate 2-Indolylphosphines
184
5.7. Conclusions 185
5.8. Experimental 186
5.9. References 191
Chapter 6 - Conclusion 193
x

List of Tables
Chapter 1 Introduction Table 1.1. The variation in geometry of some common anions. 12 Chapter 2 Monodentate 2-Indolylphosphines: A Study in Structure by X-ray
Crystallography Table 2.1. Summary of computed average cone angles of selected common
phosphines. 27
Table 2.2. X-ray diffraction data from the CSD for PPh3, PCy3, and PtBu3. 27 Table 2.3. Selected averaged bond distances (Å) and bond angles (o) of indole-
based structures from the CSD. 29
Table 2.4. X-ray crystallographic experimental data of unsubstituted 2-
indolylphosphines. 32
Table 2.5. Selected bond lengths (Å) and bond angles (o) for unsubstituted 2-
indolylphosphines. 35
Table 2.6. X-ray crystallographic experimental data of N-alkylated monodentate 2-
indolylphosphines. 41
Table 2.7. Selected bond lengths (Å) and bond angles (o) for N-alkylated 2-
indolylphosphines. 42
Table 2.8. Sum of <CPC bond angles (o) and 31P (ppm) resonances for
unfunctionalized and alkylated 2-indolylphosphines 46
Chapter 3 Tris-2-(3-methylindolyl)phosphine: Synthesis, Reactivity and Anion
Binding Table 3.1. Stability constants Ka (M-1) of 5 with a selection of anion guests as
determined by 1H NMR titration techniques performed at 298 K following NH resonances in 5.
62
Table 3.2. X-ray crystallographic experimental data of [5·X]- complexes. 67 Table 3.3. Selected bond lengths (Å) and bond angles (o) for [5.X]- complexes. 71
xi

Table 3.4. Stability constants Ka (M-1) of Pfeffer’s indole-based short receptor and indole-based long receptor with chloride and acetate guests as determined by 1H NMR titration techniques performed at 298 K.
74
Table 3.5. Stability constants Ka (M-1) of Chang’s indole-based macrocycle and
indole-based molecular cleft with a selection of anion guests as determined by spectroscopic titration techniques performed at 298 K.
82
Table 3.6. X-ray crystallographic experimental data of molecular cleft 18. 84 Table 3.7. Selected bond lengths (Å) and angles (o) for molecular cleft 18. 85 Table 3.8. Stability constants Ka (M-1) of 18 and 19 with a selection of anion guests
as determined by 1H NMR titration techniques performed at 298 K , following the indolyl NH resonances.
86
Table 3.9. Stability constants Ka (M-1) of Sessler’s diindolylquinoxaline-based
anion receptors with a selection of anion guests as determined by UV-visible spectroscopic titration techniques performed at 298 K in CH2Cl2.
88
Chapter 4 Coordination Chemistry of Monodentate 2-Indolylphosphines Table 4.1. Infrared CO stretching frequencies of Ni(CO)3L in heptanes. 106 Table 4.2. Infrared CO stretching frequencies of [Ni(CO)3(5·X)]- in heptanes. 109 Table 4.3. X-ray crystallographic experimental data for complexes: 23, 24, and 25. 112 Table 4.4. Selected bond lengths (Å) and bond angles (o) for complex 23. 114 Table 4.5. X-ray crystallographic experimental data for complexes: 26, 27, and 28. 118 Table 4.6. Selected bond lengths (Å) and bond angles (o) for complexes 24 and 25. 120 Table 4.7. Selected bond lengths (Å) and bond angles (o) for complexes 26, 27, and
28. 123
Table 4.8. Sum of <CPC bond angles (o) and 31P (ppm) resonances for
monodentate 2-indolylphosphine metal complexes. 130
xii

Chapter 5 Multidentate N-Functionalized 2-Indolylphosphines and their Metal Complexes
Table 5.1. X-ray crystallographic experimental data for (P,N)- and (P,P)-2-
indolylphosphines. 143
Table 5.2. Selected bond lengths (Å) and bond angles (o) for (P,N)- and (P,P)-2-
indolylphosphines. 144
Table 5.3. Sum of <CPC bond angles (o) and 31P (ppm) resonances for
uncomplexed multidentate 2-indolylphosphines. 150
Table 5.4. X-ray crystallographic experimental data for Pd(II) and Pt(II) complexes
of (P,N)-2-indolylphosphines. 155
Table 5.5. Selected bond lengths (Å) and bond angles (o) for Pd(II) and Pt(II)
complexes of (P,N)-2-indolylphosphines. 157
Table 5.6. X-ray crystallographic experimental data for Pd(II) complexes of (P,P)-
2-indolylphosphines. 162
Table 5.7. Selected bond lengths (Å) and bond angles (o) for Pd(II) complexes of
(P,P)-2-indolylphosphines. 163
Table 5.8. Increasing order of Pd(II)-Cl bond length that is trans to the phosphine
in Pd(II) complexes of (P,N)-2-indolylphosphines. 168
Table 5.9. Increasing order of Pd(II)-Cl bond length that is trans to the phosphine
in Pd(II) complexes of (P,P)-2-indolylphosphines. 170
Table 5.10. 31P NMR resonances of phosphine 5 and (N-PPh2)3-5 recorded in
CDCl3. 172
Table 5.11. 31P NMR resonances of metal complexes of (N-PPh2)3-5 recorded in
DMSO-d6. 174
Table 5.12. X-ray crystallographic experimental data for complexes 37 and 38. 176 Table 5.13. Selected bond lengths (Å) and bond angles (o) for complexes 37 and 38. 177 Table 5.14. Sum of <CPC bond angles (o) and 31P (ppm) resonances for metal
complexes of multidentate 2-indolylphosphines. 183
xiii

List of Figures
Chapter 1 Introduction Figure 1.1. A basic schematic illustrating the orbitals involved in PH3 bonding to a
generic transition metal, M. 1
Figure 1.2. Method of measuring cone angles for unsymmetrical ligands. 2 Figure 1.3. Early examples of chelating bisphosphines. (a) DIOP (b) DIPAMP, (c)
and BINAP ligands. 5
Figure 1.4. Chiral bidentate (P,N)-ligands: (a) (S)-PHOX ligand, and (b) (R,R)-
nBu-QUINAPHOS. 5
Figure 1.5. N-Heterocyclic phosphine ligands (a) diphenyl(2-pyridyl)phosphine
and (b) diphenyl(2-pyrrolyl)phosphine. 6
Figure 1.6. The numbering scheme of indole. 7 Figure 1.7. N-Phosphination of indole examples: (a) tri(N-3-
methylindolyl)phosphine, and (b) and phosphatri(3-methylindolyl)methane.
9
Figure 1.8. N-metallation of indole examples: (a) (η5-C5H5)Re(NO)(PPh3)(η1-
C8H6N), and (b) (κ1-N-C10H10)2Sm(THF)4. 10
Figure 1.9. Positively charged anion receptors (a) polyammonium tren-based
receptor, and (b) tetrapyridinium macrocycle. 13
Figure 1.10. Metal complex-based anion receptor that functions with both hydrogen
bond donors on the ligand and electrostatic interactions on the metal. 14
Figure 1.11. Examples of neutral anion receptors: (a) amide-based, (b) pyrrole-
based and (c) urea-based. 15
Figure 1.12. Example of an indole-based anion receptor with additional hydrogen
bond donors at the 2- and 7-positions on indole. 15
Figure 1.13. An example of metal coordinated phosphine-based anion receptor. 16
xiv

Chapter 2 Monodentate 2-Indolylphosphines: A Study in Structure by X-ray Crystallography
Figure 2.1. Description of the van der Waals surface of a phosphine ligand to
determine the Tolman cone angle from crystallographic data. 26
Figure 2.2. Selected monodentate unsubstituted 2-indolylphosphines. 31 Figure 2.3. Unit cell diagram of the four molecules of 1. 33 Figure 2.4. Numbering scheme of 1a in unit cell. 34 Figure 2.5. Numbering scheme of 2 in unit cell. 36 Figure 2.6. Numbering scheme of 3. 38 Figure 2.7. Numbering scheme of 5. 39 Figure 2.8. Selected N-alkylated 2-indolylphosphines for crystallographic
analysis: (N-Bn)-1 and (N-F5Bn)2-4. 40
Figure 2.9. Numbering scheme of (N-Bn)-1. 43 Figure 2.10. Numbering scheme of (N-F5Bn)2-4. 44 Chapter 3 Tris-2-(3-methylindolyl)phosphine: Synthesis, Reactivity and Anion
Binding Figure 3.1. Hydrogen bonding in the [Pd(1)Cl(μ-Cl)]2 and [Pd(4)Cl(μ-Cl)]2
complexes. 56
Figure 3.2. Bowl shaped hydrogen bonding cavity of 5. 59 Figure 3.3. A representative example of partial titration stack plots for the
addition of Cl- anion to 5. 61
Figure 3.4. Fit plot for titration experiment of 5 with tetrabutylammonium
chloride in CD2Cl2. 62
Figure 3.5. Sessler and co-workers’ diindolylquinoxaline-based anion receptors. 63 Figure 3.6. ORTEP diagram of one of the two independent sets of molecules in
the asymmetric unit of complex 6 with concomitant uncomplexed 5. 64
Figure 3.7. ORTEP diagram illustrating the hydrogen bonding in complex 6. 65
xv

Figure 3.8. ORTEP diagram of 7. 68 Figure 3.9. ORTEP diagram of symmetric dimer of 8 in the crystallographic ab-
plane. 72
Figure 3.10. Flexible indole-based anion receptors by Pfeffer and co-workers. 73 Figure 3.11. (a) New C3-symmetric 5-based anion receptor target, P(C9H8N)3, 9.
(b) Model phosphine P(C9H8N)(C6H5)2, 10. 75
Figure 3.12. Indole-based anion receptors by Chang and co-workers. 81 Figure 3.13. ORTEP diagram of 18. 83 Figure 3.14. A representative of partial stack plots of 18 titrations with Cl- in
CD2Cl2 at 298 K. 86
Figure 3.15. Fit plot for titration experiment of 18 with tetrabutylammonium
chloride in CD2Cl2. 87
Figure 3.16. Fit plot for titration experiment of 5 with tetrabutylammonium
bromide in CD2Cl2. 95
Figure 3.17. Fit plot for titration experiment of 5 with tetrabutylammonium iodide
in CD2Cl2. 96
Figure 3.18. Fit plot for titration experiment of 5 with tetrabutylammonium acetate
in CD2Cl2. 96
Figure 3.19. Fit plot for titration experiment of 5 with tetrabutylammonium
hydrogensulfate in CD2Cl2. 97
Figure 3.20. Fit plot for titration experiment of 5 with tetrabutylammonium nitrate
in CD2Cl2. 97
Figure 3.21. Fit plot for titration experiment of 5 with tetrabutylammonium
tetrafluoroborate in CD2Cl2. 98
Figure 3.22. Fit plot for titration experiment of 18 with tetrabutylammonium
bromide in CD2Cl2. 98
Figure 3.23. Fit plot for titration experiment of 18 with tetrabutylammonium
acetate in CD2Cl2. 99
Figure 3.24. Fit plot for titration experiment of 18 with tetrabutylammonium
hydrogensulfate in CD2Cl2. 99
xvi

Figure 3.25. Fit plot for titration experiment of 18 with tetrabutylammonium nitrate in CD2Cl2.
100
Figure 3.26. Fit plot for titration experiment of 18 with tetrabutylammonium
tetrafluoroborate in CD2Cl2. 100
Chapter 4 Coordination Chemistry of Monodentate 2-Indolylphosphines Figure 4.1. The ORTEP diagram and numbering scheme of [Cu(5)(phen)]BF4, 23. 113 Figure 4.2. The unit cell diagram of [Cu(5)(phen)]+, viewing along the
crystallographic c-face. 115
Figure 4.3. Metal complexes of monodentate 2-indolylphosphines 117 Figure 4.4. The ORTEP diagram and numbering scheme of 24. 119 Figure 4.5. The ORTEP diagram and numbering scheme of 25. 121 Figure 4.6. The ORTEP diagram and numbering scheme of 26. 122 Figure 4.7. The ORTEP diagram and numbering scheme of 27. 124 Figure 4.8. Extended hydrogen bonding network of 27 along the crystallographic
a-axis. 125
Figure 4.9. The ORTEP diagram and numbering scheme of 28. 126 Figure 4.10. The varied intra- and intermolecular hydrogen bonding of 28. 127 Chapter 5 Multidentate N-Functionalized 2-Indolylphosphines and their Metal
Complexes Figure 5.1. (a) Tris(2-(diphenylphosphino)ethyl)phosphine, PP3. (b) A generic
metal complex of PP3 exhibiting trigonal bipyramidal coordination geometry enforced by the PP3 ligand.
139
Figure 5.2. The multidentate 2-indolylphosphines that will be structurally
characterized with X-ray crystallography: (a) (N-CH2NMe2)-20 (b) (N-CH2NMe2)2-4 (c) (N-PCy2)-1 (d) 29.
142
Figure 5.3. ORTEP diagram and numbering scheme of (N-CH2NMe2)2-20. 145 Figure 5.4. ORTEP diagram and numbering scheme of (N-CH2NMe2)2-4. 146
xvii

Figure 5.5. ORTEP diagram and numbering scheme of (N-PCy2)-1. 147 Figure 5.6. ORTEP diagram and numbering scheme of 29. 149 Figure 5.7. The selected metal complexes of (P,N)-2-indolylphosphines to be
assessed by X-ray crystallography: (a) 30 (b) 31 (c) 32 (d) 33. The selected Pd(II) complexes of (P,P)-2-indolylphosphines to be analyzed by X-ray crystallography: (e) 34 (f) 35 (g) 36.
154
Figure 5.8. ORTEP diagram and numbering scheme of 30. 156 Figure 5.9. ORTEP diagram and numbering scheme of 31. 158 Figure 5.10. ORTEP diagram and numbering scheme of 32. 159 Figure 5.11. ORTEP diagram and numbering scheme of 33. 161 Figure 5.12. Brown and co-workers demonstrate absence of axial chirality in 1-
methyl-2-diphenylphosphino-3-(1’-isoquinolyl)indole upon coordination to a chiral Pd(II) complex.
161
Figure 5.13. ORTEP diagram and numbering scheme of 34. 164 Figure 5.14. ORTEP diagram and numbering schemes of the two independent
molecules of 35 in the asymmetric unit. 166
Figure 5.15. ORTEP diagram and numbering scheme of complex 36. 167 Figure 5.16. Ciclosi and co-workers’ example of an indolyl-based C3-symmetric
ligand, CP3, that binds to Pd(II). 172
Figure 5.17. ORTEP diagram and numbering scheme of complex 37. 178 Figure 5.18. ORTEP diagram and numbering scheme of complex 38. 179
xviii

List of Schemes
Chapter 1 Introduction Scheme 1.1. Examples of reactivity at the nitrogen centre of indole: (a) N-
alkylation, (b) N-arylation, (c) N-phosphination, and (d) N-metallation.
8
Scheme 1.2. Reactivity at the 1- and 3-positions of indole to form tris(3-methyl-
1H-indol-1-yl)phosphine. 9
Scheme 1.3. Synthesis of 2,2’-bis-diphenylphosphino[3,3’]biindolyl. 11 Scheme 1.4. General reaction scheme for the synthesis of 2-indolylphosphine
ligands 11
Chapter 2 Monodentate 2-Indolylphosphines: A Study in Structure by X-ray
Crystallography Scheme 2.1. A general scheme demonstrating the aufbau assembly of N-
substituted 2-indolylphosphines. 24
Scheme 2.2. The synthesis of phosphine 1 from the commercially available 3-
methylindole. 30
Scheme 2.3. The synthesis of (N-Bn)-1. 40 Chapter 3 Tris-2-(3-methylindolyl)phosphine: Synthesis, Reactivity and Anion
Binding Scheme 3.1. The synthesis of 5 from the aminal-protected 3-methylindole. 58 Scheme 3.2. The synthesis of 5 from 3-methylindole with CO2 as a protecting
group for the indole nitrogen. 59
Scheme 3.3. General titration scheme of 5 with anions as their
tetrabutylammonium salts in CD2Cl2. 60
Scheme 3.4. Reaction scheme to form 8 from 5 and CH3I. 69 Scheme 3.5. The two unsuccessful methods used to install the N,N’-
dimethylaminomethylene protecting group on the nitrogen centre of 11.
76
xix

Scheme 3.6. Reaction scheme of t-Boc-protected indole-2-carboxylic acid ethyl ester, 12, to the t-Boc-protected indole-2-methyl alcohol, 13.
77
Scheme 3.7. Reaction scheme of Cbz-protected indole-2-carboxylic acid ethyl
ester, 14, to the Cbz-protected indole-2-methyl bromide, 16. 78
Scheme 3.8. Attempted synthesis of Cbz-protected 10 from 16 using n-BuLi and
PPh2Cl. 79
Scheme 3.9. Attempted synthesis of Cbz-protected 10 from 16 through Grignard
reagent and PPh2Cl. 79
Scheme 3.10. Attempted synthesis of Cbz-protected 10 from 16 using a
Ni(dppe)Cl2 catalyst in the presence of Zn and PPh2Cl. 80
Scheme 3.11. Synthesis of 4-tert-butyl-7-(4-(4-tert-butyl-1H-indol-7-yl)phenyl)-
1H-indole, 18, from 1-bromo-4-tert-butylbenzene. 83
Scheme 3.12. General titration scheme of 18 and selected anions as their
tetrabutylammonium salts in CD2Cl2. 85
Scheme 3.13. Proposed mode of acetate interaction with molecular cleft 18. 87 Chapter 4 Coordination Chemistry of Monodentate 2-Indolylphosphines Scheme 4.1. General scheme for the formation of a monodentate
Ni(CO)3(phosphine) complex. 105
Scheme 4.2. General scheme for the formation of a monodentate
[NEt4][Ni(CO)3(5·X)] complex. X = F-, Cl-, Br-. 108
Scheme 4.3. Anticipated four-coordinate tetrahedral Cu(I) complex in the
reaction of 5 (2 eq), phen , and [Cu(MeCN)4]BF4. 110
Scheme 4.4. Three coordinate trigonal planar Cu(I) complex, 23, yielded from
reaction of 5 (2 eq), phen, and [Cu(MeCN)4]BF4. 111
Scheme 4.5. Reaction scheme for the formation of [Pd(1)Cl(μ-Cl)]2, 24. 116 Scheme 4.6. Reaction scheme for the formation of cis-Pt(3)2Cl2, 28. 117
xx

Chapter 5 Multidentate N-Functionalized 2-Indolylphosphines and their Metal Complexes
Scheme 5.1. General reaction sequence to generate CH2NMe2-functionalized 2-
indolylphosphines. 140
Scheme 5.2. Reaction scheme for the synthesis of pyridyl-functionalized (N-
py)-1. 140
Scheme 5.3. Reaction scheme for the synthesis of isoquinoline-functionalized
(N-isoquin)-1. 141
Scheme 5.4. General reaction scheme to generate phosphorus-functionalized 2-
indolylphosphines. 141
Scheme 5.5. Reaction scheme for the synthesis of diphosphine, 29. 142 Scheme 5.6. The reaction sequence for the synthesis of complex 30. 154 Scheme 5.7. General reaction scheme for the synthesis of multidentate
phosphine ligand (N-PPh2)3-5. 171
Scheme 5.8. Reaction schemes for the synthesis of metal coordinated (N-
PPh2)3-5 complexes. (a) [PtCl(N-PPh2)3-5]BPh4, 37. (b) [RhCl(N-PPh2)3-5], 38.
173
Scheme 5.9. The attempted synthesis of [PtH(N-PPh2)3-5]BPh4 by reaction of
complex 37 and NaBH4. 180
Scheme 5.10. The attempted SnCl2 insertion into the Pt(II)-Cl bond of complex
37. 181
Scheme 5.11. The attempted metathesis of the chloro ligand in complex 38 for
either a methyl or phenyl ligand. 181
xxi

List of Compounds
Compound Number
HN
P
1 P(C9H8N)(C6H5)2
*
HN
P
2 P(C9H8N)(C6H11)2
*
NH
PHN
3 P(C9H8N)2(C6H5)*
HN
PNH
4 P(C17H12N2)(C6H5)*
HN
PNH
HN
5 P(C9H8N)3
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxii

NP
(N-Bn)-1 P(C9H8NC7H7)(C6H5)2
*
NP
N
F
FF
FF
F F
F
FF
(N-F5Bn)2-4 P(C17H12N2C14H4F10)(C6H5) *
P
HN
3
O
NEt4
O
6 [NEt4][P(C9H8N)3
.(CH3COO)]
P
HN
3
F
NEt4
7 [NEt4][P(C9H8N)3
.F]
P
HN
3
I
NEt4
8 [CH3P(C9H8N)3]I
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxiii

HN
HN
NHP
9 P(C9H8N)3
HN
P
10 P(C9H8N)(C6H5)2
HN
OEt
O
11 1H-indole-2-carboxylic acid ethyl ester
N
OEt
O
O O
12 tert-butyl ethyl 1H-indole-1,2-dicarboxylate
N
OH
O O
13 tert-butyl 2-(hydroxymethyl)-1H-indole-1-carboxylate
N
OEt
O
OO
14 benzyl ethyl 1H-indole-1,2-dicarboxylate
xxiv

N
OH
OO
15 benzyl 2-(hydroxymethyl)-1H-indole-1-carboxylate
N
Br
OO
16 benzyl 2-(bromomethyl)-1H-indole-1-carboxylate
HN
Br
17 4-tert-butyl-7-bromoindole
NH HN
18 4-tert-butyl-7-(4-(4-tert-butyl-1H-indol-7-yl)phenyl)-1H-indole
NH HN
19 7-(4-(1H-indol-7-yl)phenyl)-1H-indole **
HN
P
20 P(C9H8N)(C4H9)2
*
NP
(N-Me)-1 P(C10H10N)(C6H5)2
*
** Refer to M. Trisha C. Ang’s MSc thesis (University of Toronto, 2007) for synthetic protocol. * Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxv

P
HN
3
Cl
NEt4
21 [NEt4][P(C9H8N)3
.Cl]
P
HN
3
Br
NEt4
22 [NEt4][P(C9H8N)3
.Br]
CuN
N
P
HNHN
HN BF4
23 {Cu[P(C9H8N)3](phen)}BF4
NH
PPd
Cl
ClCl
HN
PPd
Cl
24 {Pd[P(C9H8N)(C6H5)2]Cl(μ-Cl)}2
*
NH
PPd
Cl
ClCl
HN
PPd
Cl
25 {Pd[P(C9H8N)(C6H11)2]Cl(μ-Cl)}2
*
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxvi

NH
PPd
Cl
ClCl
HN
PPd
ClHN N
H
26 {Pd[P(C9H8N)2(C6H5)]Cl(μ-Cl)}2
*
NHP
PdCl
ClClPd
ClNH
HNP H
N
27 {Pd[P(C17H12N2)(C6H5)]Cl(μ-Cl)}2
*
HN
PPt
ClCl
HN
NH
P HN
28 cis-Pt[P(C9H8N)2(C6H5)]Cl2
NP
N
(N-CH2NMe2)-20 P(C12H15N2)(C4H9)2
*
NP
N
N N
(N-CH2NMe2)2-4 P(C23H26N4)(C6H5)*
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxvii

NP
N
(N-py)-1 P(C14H11N2)(C6H5)2
*
NP
N
(N-isoquin)-1 P(C18H13N2)(C6H5)2
*
NP
P
(N-PPh2)-1 P(C9H7NP(C6H5)2)(C6H5)2
*
NP
P
(N-PCy2)-1 P(C9H7NP(C6H11)2)(C6H5)2
*
NP
PO O
(N-(R)-BINO)-1 P(C9H7NP(O2C20H12))(C6H5)2
*
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxviii
HN
P
P
HN
NH
NH
29 [P(C18H16N2)(CH2)]2
NP
NPd
Cl
Cl
30 [PdCl2P(C12H15N2)(C6H5)2
]*
NP
NPt
Cl
Cl
31 [PtCl2P(C12H15N2)(C6H5)2]
NP
NPd
Cl
Cl
32 [PdCl2P(C14H11N2)(C6H5)2] *
NP
NPd
Cl
Cl
33 [PdCl2P(C18H13N2)(C6H5)2]*
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxix

NP
PPd Cl
Cl
34 [PdCl2P(C9H7NP(C6H5)2)(C6H5)2]*
NP
PPd Cl
Cl
35 [PdCl2P(C9H7NP(C6H11)2)(C6H5)2]*
NP
Pd Cl
ClOO P
36 [PdCl2P(C9H7NP(O2C20H12))(C6H5)2]*
NP
N
N
PPh2
PPh2
PPh2
(N-PPh2)3-5 P[C9H7NP(C6H5)2]3
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
37 {PtClP[C9H7NP(C6H5)2]3}BF4
* Refer to Dr. Edmond Lam’s PhD thesis (University of Toronto, 2007) for synthetic protocol.
xxx

Ph2P RhI
PPh2
PPh2
P
Cl
N NN
38 {RhClP[C9H7NP(C6H5)2]3}
xxxi

List of Abbreviations
Å angstroms
BF4- tetrafluoroborate
BINAP 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
BnBr benzyl bromide
Boc2O di-tert-butyl dicarbonate
t-Boc tert-butoxycarbonyl
BPh4- tetraphenylborate
br broad tBu tert-butyl
Bu n-butyl
bpy 2,2’-bipyridine
Br- bromide 13C NMR carbon-13 nuclear magnetic resonance spectrometry
Cbz carbobenzyloxy
CbzCl carbobenzyloxy chloride
CDCl3 deuterated chloroform
CD2Cl2 deuterated dichloromethane
CHCl3 chloroform
CH2Cl2 dichloromethane
CH3COO- acetate
Cl- chloride
COD 1,5-cyclooctadiene
CP3 tris(3-methyl-1-(diphenylphosphino)-1H-indol-2-yl)methane
CSD Cambridge Structural Database
Cy cyclohexyl oC degrees Centigrade
δ chemical shift
Δ reflux
d doublet
xxxii

dd doublet of doublets
DIBAL-H diisobutylaluminum hydride
DMAP 4-dimethylaminopyridine
DMSO dimethylsulfoxide
DMSO-d6 deuterated dimethylsulfoxide
dppe 1,2-bis(diphenylphosphino)ethane
e- electron
ee enantiomeric excess
EI-MS electron impact mass spectrometry
Et ethyl
Et2O diethyl ether
EtOAc ethyl acetate
EtOH ethanol
F- fluoride
g gram
h hour 1H NMR proton nuclear magnetic resonance spectrometry
H3PO4 phosphoric acid
HCl hydrochloric acid
HNO3 nitric acid
HRMS high resolution mass spectrometry
HSO3- hydrogen sulfate
Hz hertz
I- iodide
IR infrared spectroscopy
L litre
M molar
m multiplet
m.p. melting point
Me methyl
MeCN acetonitrile
xxxiii

MeI methyl iodide
MeOH methanol
MgSO4 magnesium sulfate
MHz megahertz
μmol micromole
μL microlitre
mL mililitre
mmol milimole
mol mole
NaBH4 sodium borohydride
Na2SO4 sodium sulfate
NaH sodium hydride
NaHCO3 sodium bicarbonate
NBu4 tetrabutylammonium
NEt4 tetraethylammonium
NH4Cl ammonium chloride
NMR nuclear magnetic resonance
NO3- nitrate
O=PCl3 phosphorus oxychloride
O=PCy3 tricyclohexylphosphine oxide
O=PPh3 triphenylphosphine oxide
OAc- acetate
ORTEP Oak Ridge Thermal Ellipsoids Plot 31P NMR phosphorus nuclear magnetic resonance spectrometry
P(tBu)3 tri-tert-butylphosphine
PCl3 trichlorophosphine
PCy3 tricyclohexylphosphine
Ph phenyl
P(o-tol)3 tri-ortho-tolylphosphine
PP3 tris(2-(diphenylphosphino)ethyl)phosphine
PPh2Cl chlorodiphenylphosphine
xxxiv

xxxv
PPh3 triphenylphosphine
ppm parts per million
phen 1,10-phenanthroline
q quartet
QUINAP 1-(2-diphenylphosphino-1-naphthyl)isoquinoline
rms root mean squared
rt room temperature
s singlet
satd. saturated
SiMe4 tetramethylsilane
t triplet
TBSCl tert-butyldimethylsilyl chloride
THF tetrahydrofuran
TLC thin layer chromatography
TMS tetramethylsilane
v/v volume / volume
X- halide

Chapter 1
Introduction
1.1. Tertiary Phosphine Ligands
Tertiary phosphines, PR3, are recognized as a class of important ligands for
coordination chemistry as they are excellent 2-electron neutral donor ligands that have
the potential for steric and electronic adjustments. Phosphine ligands primarily function
as σ-donors via the phosphorus lone pair, but can also behave as π-acceptors through the
P-R σ*-antibonding orbitals.1-3 Because of this dual property, phosphine ligands can
effectively stabilize transition metals of low oxidation states. A fundamental scheme of
the orbitals involved in the bonding of PH3 with a generic metal centre is illustrated in
Figure 1.1.
HH
P
H
HH
P
HM
(a) (b)
M
Figure 1.1. A basic schematic illustrating the orbitals involved in PH3 bonding to a generic transition
metal, M. (a) σ-Donation from the phosphine to an empty d-orbital of a metal. (b) π-Acceptance into the
σ*-antibonding orbitals of the phosphine from the appropriate occupied d-orbitals of the metal.
Changing the R-substituents on PR3, can lead to several results: an
increase / decrease in the σ-donicity or π-acidity of the phosphine molecule, and / or
large variations in the steric profile of the phosphine ligand. Their utility in catalysis has
led to the development of numerous examples of phosphine molecules that have
properties ranging in sterics and basicities.4,5 There is continuing interest to improve
1

2
phosphine-mediated catalysis, which also requires the modification or tailoring of
phosphine ligands to incorporate additional functional groups. The steric bulk and
electron-donating ability of a phosphine ligand are difficult properties to quantify
exclusively since both properties are closely related.6,7
1.1.1. Determining Steric Profiles of Phosphine Molecules
Tolman suggested that a geometrical cone angle could be utilized as a description
of a ligand’s steric size.6 This parameter is estimated from idealized space-filling CPK
models and requires the van der Waals surface of the phosphine to be known, particularly
the surface generated by the tangential hydrogen atoms. The cone angle for PR3 is
defined as the vertex of a cylindrical cone, situated 2.28 Å from the centre of the
phosphorus atom, which diverges outwards toward the R groups and borders the van der
Waals radii of the peripheral atoms (Figure 1.2). This model can be used to estimate
cone angles for unsymmetrical phosphines PR1R2R3 by minimizing the sum of the half-
angles θi of each R substituent (Equation 1.1).
Figure 1.2. Method of measuring cone angles for unsymmetrical ligands.6

3
Θ = 2/3 Σ θi/23
i=1 (1.1)
While Tolman’s method appears to be quite simple and crude, the values obtained by this
technique have been useful in correlating the chemical reactivity of a variety of
coordination compounds on the dependence of ligand steric properties. Attempts to
improve the Tolman cone angle model are also based on the geometrical parameters of a
phosphine compound, either acquired from crystallographic data8 or from quantum
mechanically calculated structures of the ligand-metal complex.9,10
1.1.2. Determining Phosphine Ligand Basicity
Strohmeier first proposed that the CO stretching frequencies of monosubstituted
transition-metal carbonyls could be used to evaluate the electron-donor ability of
phosphine ligands, and Tolman expanded on this concept with Ni(CO)3L complexes,
where L is the monodentate phosphine ligand.7,11 The Ni(CO)3L complexes are formed
quickly upon mixing the phosphine molecule and Ni(CO)4 in a 1 : 1 ratio at room
temperature, even if the ligands are large in size. Ligands with strong electron-donating
ability contribute to a larger degree of back-bonding from the metal centre into the C≡O
π*-antibonding orbitals, which therefore result in decreased C≡O bond order and leads to
lower C≡O stretching frequencies. On the other hand, weakly donating ligands induce
less M-C≡O back-bonding, which leads to higher C≡O stretching frequencies.
As a standard measure, the A1 carbonyl mode of Ni(CO)3L complexes is
referenced against the analogous stretching frequency of Ni(CO)3(PtBu3) to give the
electronic parameter χ (Equation 1.2).
χ χ1χ2χ3 = νCO(A1) - 2056.1 cm-1 = Σ3
i=1χi
P (1.2)

4
Tolman noted a linear additive electronic effect of each substituent on phosphorus
(χi in Equation 1.2) to estimate the net-basicity of phosphines that have not been
quantified.6 Bartik and co-workers have clarified that the additivity rule is not wholly
linear, especially concerning aromatic substituents, and the deviations from Tolman’s χ-
values are dependent on the combined donor and acceptor character of the atoms in the
α-position to the phosphorus.12
The basicity of phosphines towards protons has also been measured by several
groups, where the pKa values obtained give a quantitative measurement of the σ-donicity
of a ligand.13-16 The method of quantitative analysis of ligand effects,17,18 QALE, is a
more generalized approach to measure the combined effects of electron-donor ability and
steric size. This approach considers other electronic parameters such as the so-called
‘aryl effect’ and a size parameter called ‘steric threshold’.
1.1.3. The Utility of Phosphine Ligands
Phosphine ligands are often used in transition metal-mediated catalysis because of
the potential to tune the steric and electronic properties by simply changing the
substituents on phosphorus. Minor adjustments in the electronic and / or steric size of a
phosphine can alter the reactivity of a catalyst by increasing its stability, promoting
reactions, and influencing enantioselectivity in organic transformation reactions.19
The utility of PPh3, PCy3, and PtBu3 as ligands in transition metal catalysis is well
documented in the literature.4,20-23 These common monodentate phosphines are found to
be active species in C-C and C-heteroatom bond formation reactions such as Negishi
coupling,21 Suzuki coupling,23 and Kumada coupling.20 While monodentate phosphines
continue to flourish as co-catalysts, the development of chelating bidentate bisphosphines
has led to superior enantioselectivity compared to monodentate ligands.24 Early
examples such as DIOP,25-27 DIPAMP,28,29 and BINAP30-32 (Figure 1.3) have led to the
optimization of transition metal-catalyzed asymmetric hydrogenation reactions and
inspired the emergence of new chiral bisphosphine ligands. The common thread among
DIOP, DIPAMP, and BINAP is that they are chelating bisphosphines. These ligands

5
differ in the basis of their chirality: DIOP contains a rigid chiral backbone; the
phosphorus atoms of DIPAMP are chiral centres, having different substituents; the C2-
symmetric BINAP is atropisomeric by hindered rotation about the C-C single bond.
PPh2PPh2
P
POO
PPh2
PPh2O
O
(a) (c)(b) Figure 1.3. Early examples of chelating bisphosphines. (a) DIOP25-27 (b) DIPAMP,28,29 (c) and BINAP30-32
ligands.
The combination of (P,N)-donors as chiral bidentate ligands has proven to be
more effective than (P,P)-donors at directing enantioselectivity of a reaction. This is in
part due to the distinct differences in the σ-donor abilities of the soft P-ligand with π-
acceptor properties compared to the hard N-ligand functioning primarily as a σ-donor.
For example the mixed (P,N)-ligands of the PHOX19 series are particularly well-suited
for Ir-catalyzed asymmetric hydrogenation of simple olefins, and (R,R)-n-Bu-
QUINAPHOS33 is a highly efficient ligand in Rh-catalyzed asymmetric hydrogenation of
itaconic acid (Figure 1.4).
P N
O
tBu(o-Tol)2 O
OP N
Ph2P
Bun
(a) (b) Figure 1.4. Chiral bidentate (P,N)-ligands: (a) (S)-PHOX ligand,19 and (b) (R,R)-nBu-QUINAPHOS.33

6
1.2. N-Heterocyclic Phosphine Ligands
Phosphines with aromatic heterocyclic substituents are interesting ligands in
coordination chemistry because they offer several potential modes of coordination to
transition metals. The most frequently studied of these polydentate ligands are the
pyridylphosphines34 in which the pyridyl substituents are bound to phosphorus at the 2-
position (Figure 1.5a).
N
P P
NH
(a) (b) Figure 1.5. N-Heterocyclic phosphine ligands (a) diphenyl(2-pyridyl)phosphine and (b) diphenyl(2-
pyrrolyl)phosphine.
Mono-, bis-, and tris(2-pyridyl)phosphines have been prepared and their coordination
chemistry remains an area of considerable activity in part because of the difference in
character between the two centres of Lewis basicity. As a softer σ-donor and stronger π-
acceptor than the nitrogen atom, the phosphorus centre of diphenylpyridylphosphine
preferentially coordinates to transition metals to obtain mononuclear complexes.34
In principle, diphenyl(2-pyrrolyl)phosphine (Figure 1.5b) should provide a
similarly rich and varied chemistry in reaction with transition metals upon deprotonation
of its pyrrolyl nitrogen centre. However, its low yielding synthesis, coupled with its
propensity for complicated and unpredictable behaviour in metal coordination have
rendered diphenyl(2-pyrrolyl)phosphine a rarely used ligand in reaction with transition
metals.35,36
Electron-rich heteroaromatic indole, like pyrrole, also offers further modes of
coordination through its nitrogen atom in a potentially polydentate ligand (Figure 1.6).

7
NH
12
345
67
Figure 1.6. The numbering scheme of indole.
The required deprotonation of the indolyl nitrogen centre provides the opportunity to
control secondary N-coordination. It is reasoned that relative to pyrrole, indole has lesser
reactivity due to its fused benzene moiety, which permits an easier synthesis of mono-
and di- substituted indolylphenylphosphines and that these ligands provide a rich and
interesting coordination chemistry that is based upon their expected polydenticity rather
than the propensity for P-C bond cleavage and other complications observed upon
coordination of diphenyl(2-pyrrolyl)phosphine.35
Traditionally, the functionalization of a phosphine ligand after P-C bond
formation is an arduous process that can require several time- and atom-consuming steps.
Therefore in many cases, the tailoring of a phosphine ligand requires a “ground-up”
strategy where the substituents must be modified prior to the formation of a phosphine
molecule. Changes to the substituents on a preformed phosphine ligand without extra
protection and deprotection steps would be a valuable commodity with which a large
variety of phosphine ligands having varied electronic and steric properties can be
generated easily and efficiently. An indolyl group would be an ideal candidate as a
phosphine substituent as it will provide the necessary site of reactivity for future
functionalization, but it can also be capable of coordinating to transition metal centres.
An additional attractive feature of indoles is the availability of substituted indoles, either
commercially or via well-documented indole syntheses37 in the literature.

8
1.3. Reactivity at Indole Nitrogen
One of the most reactive sites on indole is at the nitrogen centre; generally
deprotonation of the acidic NH proton occurs prior to the formation of any new bond at
this site. Some common reactions that involve an indole nitrogen centre are shown in
Scheme 1.1. One of the most common reactions at an indolyl nitrogen centre is N-
alkylation (Scheme 1.1a).37 Addition of the appropriate alkyl halide after deprotonation
of the NH proton yields the desired N-alkylated indole. Several bases can be utilized to
perform the same deprotonation and they include: NEt3, n-BuLi, NaOH and NaH. Aryl
groups can be installed on the nitrogen centre of indole with the aid of a Cu(I) catalyst
(Scheme 1.1b). This reaction has shown to be useful for furnishing N-heterocycles on the
indole nitrogen centre.38
HN
NR
NAr
NPR2
NM
(a) (b)
(d)(c)
baseR-X Ar-X
base basePR2Cl
Cu(I)
M
Scheme 1.1. Examples of reactivity at the nitrogen centre of indole: (a) N-alkylation, (b) N-arylation, (c) N-
phosphination, and (d) N-metallation.
N-Phosphination of indole (Scheme 1.1c) is well documented in the literature.
Direct reaction of indole with dichlorophenylphosphine, in the presence of NEt3, forms
phosphine ligands that have a combination of N-bound and C3-bound indolyl substituents
on phosphorus (Scheme 1.2).39

9
HN
HN
P NNEt3
PPhCl2
Scheme 1.2. Reactivity at the 1- and 3-positions of indole to form tris(3-methyl-1H-indol-1-yl)phosphine.39
Similarly, the indolyl substituents are all N-bound to the phosphorus atom in tri(N-3-
methylindolyl)phosphine (Figure 1.7a) and phosphatri(3-methylindolyl)methane (Figure
1.7b).40
NP
NN
N NPN
(a) (b) Figure 1.7. N-Phosphination of indole examples: (a) tri(N-3-methylindolyl)phosphine, and (b) and
phosphatri(3-methylindolyl)methane.40
This reactivity at indole nitrogen with chlorophosphines poses a synthetic challenge in
forming 2-indolylphosphines. Thus an appropriate protecting group for the indole
nitrogen centre, which does not hinder the reactivity at the C2-position in reaction, will
be necessary.
The N-metallation of indole (Scheme 1.1d) involves coordinating deprotonated
indole to a transition metal. The indole ligand acts as an anionic 2-electron N-donor in
(η5-C5H5)Re(NO)(PPh3)(η1-C8H6N)41 and (κ1-N-C10H10)2Sm(THF)442
(Figure 1.8).

10
N
Re PPh3ONSmTHF
THF THFTHF
N
N
(a) (b) Figure 1.8. N-metallation of indole examples: (a) (η5-C5H5)Re(NO)(PPh3)(η1-C8H6N),41 and (b) (κ1-N-
C10H10)2Sm(THF)4.42
1.4. 2-Indolylphosphine Ligands
One of the goals in our research is to be able to modify a phosphine ligand post P-
C bond formation. Indolyl is a suitable substituent on phosphorus for this objective only
if the nitrogen centre is free to react further; this can be accomplished through the
synthesis of 2-indolylphosphine ligands, in which the phosphorus centre is bonded to
indole at its C2-position. This also allows the phosphorus centre to be proximal to the
reactive nitrogen centre, which would allow for communication between the two centres.
Brown and co-workers43 reported the synthesis of 2,2’-bis-
diphenylphosphino[3,3’]biindolyl, the first example of a phosphine ligand with the
phosphorus atom bonded to the 2-position on indole (Scheme 1.3). The indolyl nitrogen
centres in Brown and co-workers’ system is protected with an N,N’-
dimethylaminomethylene protecting group, which has been shown by Katrizky and co-
workers44 to be effective at directing lithiation to the C2-position on indole.

11
N
N
N
N
N
N
N
N
PPh2
PPh2
1. nBuLi2. Ph2PCl
Scheme 1.3. Synthesis of 2,2’-bis-diphenylphosphino[3,3’]biindolyl.43
We required explicit reactivity at the C2-position on indole, however the C3-
position is also susceptible to attack by chlorophosphines45 due to contributing resonance
structures in which there is extra electron density at this position. Therefore, we chose 3-
methylindole as a starting material; by blocking the C3-position on indole with a methyl
group we have removed the reactivity at this site. A general reaction scheme for the
synthesis of 2-indolylphosphine ligands is shown in Scheme 1.4. Modifying Katrizky
and co-workers protocol,44 lithiation of indole is achieved at the C2-position, and the
subsequent addition the appropriate chlorophosphine reagent leads to the desired aminal-
protected phosphine. The N,N’-dimethylaminomethylene aminal protecting group can be
straightforwardly removed upon reaction of the protected phosphine with NaBH4.
N
N
N
N
PR2
HN
PR2
1. n-BuLi2. PR2Cl
THF, -78oC
NaBH4EtOH/THF
Δ
Scheme 1.4. General reaction scheme for the synthesis of 2-indolylphosphine ligands.

12
1.5. Synthetic Anion Receptors
Anions are pervasive in the natural world. They are essential to a broad range of
biological and chemical processes. For example, research is being conducted on using
anion receptors as membrane transport agents for chloride in biological systems.46 The
interest in anion recognition has been quickly gaining momentum, as seen in the large
number of reviews describing the chemistry involved in making and evaluating anion
receptors.47-58
The basic concepts which are considered when designing anion receptors are:
hydrogen bonding and / or electrostatic interactions; metal-anion complex formation; and
size and shape complementarity. In contrast to isoelectronic cations, anions are larger in
size, thus having lower charge-to-radius ratio and weaker electrostatic interactions.
Anions may be susceptible to protonation and only exist above a certain pH level.
Anions come in a wider range of geometries compared to common cations (Table 1.1),
necessitating a higher degree of design and complementarity to generate receptors that
are selective for one anion guest over another.58 Anion binding strength and selectivity
are dependent upon the type of solvent in which the interaction occurs. This is especially
true for neutral anion receptors that function primarily by hydrogen bonding; these
receptors will have difficulty competing with polar protic solvents that form strong
hydrogen bonds with anions.
Table 1.1. The variation in geometry of some common anions.
Geometrical Shape Anion
Spherical F-, Cl-, Br-, I-
Linear N3-, CN-, SCN-, OH-
Trigonal Planar CO32-, NO3
-
Tetrahedral PO43-, SO4
2-, BF4-
Octahedral Fe(CN)64-, Co(CN)6
3-
There are several methods used to determine the strength of interaction between
an anion receptor and an anion guest.58 Generally these techniques require titration of
anion guest into a host solution. The titration may be monitored by NMR, UV-visible or

13
fluorescence-emission spectroscopy, or isothermal titration calorimetry. 1H NMR
spectroscopy can be useful for following the NH proton chemical shifts of receptors
containing these functionalities, and can offer insight into the immediate interaction of an
anion guest with a receptor’s hydrogen bond donors. Both UV-visible and fluorescence-
emission spectroscopy exhibit the changes in the optical properties of the light
absorbing / emitting segments of an anion receptor upon binding. Isothermal calorimetry
affords information about a receptor’s energy changes upon anion interaction. These
titration techniques operate at different sensitivity limits: ~10-3 M for NMR; ~10-4 M for
isothermal calorimetry; ~10-5 M (or lower) for UV-visible and fluorescence-emission
spectroscopy. Titrations are often performed in several different solvents, as solvent
polarity has a direct impact on the strength of anion-to-receptor interaction. Anion
binding strengths tend to be higher in less polar organic solvents (such as CH2Cl2), and
often tetrabutylammonium anion salts are used as they are soluble in these less
competitive solvents.
Anion recognition hosts fall into two categories: positively charged receptors and
neutral receptors. Positively charged anion receptors often contain groups such as
ammonium55,59,60 or pyridinium61 that can interact with an anion guest through a
combination of electrostatic and hydrogen bonds. For example, Bowman-James and co-
workers60 examined the anion binding ability of a polyammonium tren-based tripodal
species (Figure 1.9a) and determined that the receptor has high affinity for dihydrogen
phosphate and hydrogen sulfate (log Ka = 3.25 and 3.20 in CDCl3, respectively). A
tetrapyridinium macrocycle (Figure 1.9b) from Shinoda and co-workers61 exhibits
binding to tricarboxylate anions with high affinities (log Ka = 5.1 in D2O at pH 7-8).
N
N
N
N
(a) (b)
N N N
NH
HH
H H
H
Figure 1.9. Positively charged anion receptors (a) polyammonium tren-based receptor,60 and (b)
tetrapyridinium macrocycle.61

14
Transition metal centres can provide a positive charge to the receptor to increase
the electrostatic interaction between the host and anionic guest, and impart geometrical
constraints by coordination geometry to organize the hydrogen-bonding groups for
optimal guest interaction.48,62-68 For example, Loeb and co-workers63 have demonstrated
the use of Pt(II) to arrange four isoquinolyl ligands with pendant urea functionalities into
close proximity (Figure 1.10). The receptor was found to have high binding affinity for
sulfate and dihydrogen phosphate in 1 : 1 receptor to anion ratio (Ka values in excess of
105 M-1) through a combination of hydrogen bonding and electrostatic interactions
between the anion and the Pt(II).
N
NH
NHO
Bu
Pt2+
4 Figure 1.10. Metal complex-based anion receptor that functions with both hydrogen bond donors on the
ligand and electrostatic interactions on the metal.63
Commonly, electroneutral anion recognition hosts have suitable NH donors and
are frequently based on small molecules with organic structures that can contain
combinations of functional groups such as amides,69-74 pyrroles,46,75-81 and ureas.82-87 For
example, Gale and co-workers58 have demonstrated that two amide pendant arms on a
receptor (Figure 1.11a) has improved anion binding affinity and increased selectivity
towards dihydrogen phosphate anion (Ka = 2.6 x 104 M-1 in MeCN containing 0.5%
DMSO) relative to a receptor only containing one amide pendant arm. In addition,
Gale’s group76 has constructed an anion receptor which incorporates a pyrrolyl moiety
between two amide functionalities (Figure 1.11b). The crystal structure of this pyrrolyl-
based anion receptor with benzoate anion demonstrates the interaction between the anion
and the receptor, and more importantly that all three of the hydrogen bond donors are
engaged in the interaction. Fabbrizzi and co-workers82 reported a urea-based anion

15
receptor that contains two electron withdrawing nitro substituents (Figure 1.11c) which
act to increase the acidity of the hydrogen bond donors. This receptor shows strong
binding to fluoride, acetate and benzoate in a 1 : 1 receptor to anion stoichiometry (log Ka
= 7.38, 6.61, and 6.42, respectively in CH3CN).
O
NH
O
HN
NH HN OO NH
NH
OHN
O
HN
O
NHBu Bu
(a) (b) (c)
NO2O2N
Figure 1.11. Examples of neutral anion receptors: (a) amide-based,58 (b) pyrrole-based76 and (c) urea-
based82.
The limited number of reports of indole-based anion receptors is somewhat
remarkable, considering for example, the indole-containing amino acid tryptophan is a
component of a sulfate binding protein that helps to stabilize sulfate binding in biological
systems.88 Only recently attention has been given to indole-based hosts and they have
demonstrated anion binding capability and high selectivity for various anions.88-104 For
instance, Gale and co-workers90 have synthesized a series of 2,7-functionalized indoles
that have additional amide and / or urea or thiourea moieties. Figure 1.12 shows one of
the indole-based receptors in the series that has urea functionality at the 7-position and an
amide functionality at the 2-position. This particular receptor shows very strong binding
to acetate anions with a stability constant of Ka = 10000 M-1 in DMSO-d6 / 0.5% water.
HN
O
HNNH
NH
O
Figure 1.12. Example of an indole-based anion receptor with additional hydrogen bond donors at the 2-
and 7-positions on indole.90

16
A survey of the literature for phosphine ligand-based anion receptors yields a
small number of reports. Often phosphine ligands are designed such that they contain a
hydrogen bond donor displaced at some distance from the phosphorus centre, and the
phosphorus atom itself is used for metal coordination.84-87 For example, Knight and co-
workers86 have reported a Pd(II) with two urea-functionalized phosphine ligands that
exhibit intramolecular hydrogen bonding (Figure 1.13a). In the presence of a chloride
guest, the self-association is disrupted and the urea functionalities concurrently hydrogen
bond to the anion (Figure 1.13b). The association constant for this Pd(II) system was
found to be 1000 M-1 in CDCl3, and is considered a relatively strong interaction in this
solvent.
PPh2
Pd
P
MeCl
Ph2
NH
NH
HN
HN
O
O
EtOOC
EtOOC
Cl
-
PPh2
Pd
P
MeCl
Ph2
NH
NH
NH
O
EtOOC
NH
EtOOC
OCl-
CDCl3
(a) (b) Figure 1.13. An example of metal coordinated phosphine-based anion receptor. (a) The urea functionalities
are engaged in intramolecular hydrogen bonding in the absence of an anion guest. (b) Upon the addition of
chloride, the intramolecular hydrogen bonding is disrupted to allow the urea functionalities to act as an
anion receptor.86
One of the goals of this thesis is to evaluate 2-indolylphosphine ligands as anion
receptors and to assess how hydrogen bonding can alter their properties. Furthermore,
since indole-based receptors have shown to be as promising as their pyrrole-based
counterparts, and there is a general absence of phosphine ligands acting as anion
receptors, it is thought that 2-indolylphosphines as anion receptors would be a timely
addition to this burgeoning field.

17
1.6. Scope of the Thesis
A series of unfunctionalized and N-functionalized 2-indolylphosphine ligands
have been synthesized in our group, and further coordinated to Pd(II). The main goal of
this research is to structurally analyze these new 2-indolylphosphines and their
subsequent metal complexes, as well as examine the anion binding properties of tris-2-(3-
methylindolyl)phosphine. This thesis is divided into four parts:
Chapter 2 describes the solid-state structures of monodentate 2-indolylphosphine
ligands and their N-alkylated derivatives. A detailed investigation of the structural
features, namely implications to the steric bulk around a phosphorus atom, and indole
aromaticity upon N-functionalization will be addressed.
The anion binding properties of tris-2-(3-methylindolyl)phosphine will be
examined in Chapter 3. Various anions have been examined to determine the utility of
tris-2-(3-methylindolyl)phosphine as an anion receptor. In addition, attempted syntheses
for new indole-based anion receptors will be presented.
In Chapter 4, the metal complexes of monodentate 2-indolylphosphines will be
examined. The net-basicities of these ligands will be determined through coordination to
Ni(CO)3, it will also be demonstrated how N-alkylation affects the net-basicity of the
parent phosphine molecule. Complexation of tris-2-(3-methylindolyl)phosphine to
[Cu(MeCN)4]BF4 generates a discrete ion-pair complex in which the BF4- anion interacts
with the phosphine ligand through hydrogen bonds. The coordination chemistry of
monodentate 2-indolylphosphines will be investigated by X-ray crystallography.
Chapter 5 will present multidentate 2-indolylphosphine ligands, where the indole
nitrogen centres are functionalized with substituents that have the ability to further
coordinate to metal complexes. The structural analyses of these new bidentate (P,N)- and
(P,P)-2-indolylphosphine ligands and their metal complexes will be presented.
Furthermore, a new tetradentate 2-indolylphosphine ligand will be examined along with
its coordination chemistry to both Pt(II) and Rh(I).
All the crystallographic and synthetic work presented in this thesis was performed
by Joanne Yu, with the exception of the syntheses of the unfunctionalized and N-

18
functionalized 2-indolylphosphines and their Pd(II) complexes, which were conducted by
Dr. Edmond Lam.105

19
1.7. References
(1) Marynick, D. S. J. Am. Chem. Soc. 1984, 106, 4064-4065.
(2) Orpen, G. A.; Connelly, N. G. J. Chem. Soc., Chem. Commun. 1985, 1310-1311.
(3) Xiao, S.-X.; Trogler, W. C.; Ellis, D. E.; Berkovitch-Yellin, Z. J. Am. Chem. Soc.
1983, 105.
(4) Bedford, R. B.; Cazin, C. S. J.; Holder, D. Coord. Chem. Rev. 2004, 248, 2283-
2321.
(5) Chinchilla, R.; Najera, C. Chem. Rev. 2007, 107, 874-922.
(6) Tolman, C. A. J. Am. Chem. Soc. 1970, 90, 2953 - 2956.
(7) Tolman, C. A. Chem. Rev. 1977, 77, 313 - 348.
(8) Muller, T. E.; Mingos, D. M. P. Transition Met. Chem. 1995, 20, 533 - 539.
(9) Mathew, J.; Thomas, T.; Suresh, C. H. Inorg. Chem. 2007, 46, 10800 - 10809.
(10) Suresh, C. H. Inorg. Chem. 2006, 45, 4982 - 4986.
(11) Strohmeier, W. Chem. Ber. 1964, 97, 1877 - 1885.
(12) Bartik, T.; Himmler, H.-G.; Seevogel, K. J. Organomet. Chem. 1984, 272, 29 - 41.
(13) Henderson, W. A.; Streuli, C. A. J. Am. Chem. Soc. 1960, 82, 5791-5794.
(14) Streuli, C. A. Anal. Chem. 1960, 32, 985-987.
(15) Abdur-Rashid, K.; Fong, T. P.; Greaves, B.; Gusev, D. G.; Hinman, J. G.; Landau,
S. E.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2000, 122, 9155-9171.
(16) Allman, T.; Goel, R. G. Can. J. Chem. 1982, 60, 716 - 722.
(17) Woska, D.; Prock, A.; Giering, W. P. Organometallics 2000, 19, 4629-4638.
(18) Wilson, M. R.; Woska, D. C.; Prock, A.; Giering, W. P. Organometallics 1993,
12, 1742-1752.
(19) Pfaltz, A.; Drury, W. J., III Proc. Nat. Acad. Sci. 2004, 101, 5723-5726.
(20) Frisch, A. C.; Shaikh, N.; Zapf, A.; Beller, M. Angew. Chem. Int. Ed. 2002, 41,
4056-4059.
(21) Huo, S. Org. Lett. 2003, 5, 423-425.
(22) McLaughlin, M. Org. Lett. 2005, 7, 4875-4878.
(23) Shimizu, M.; Nakamaki, C.; Shimono, K.; Schelper, M.; Kurahashi, T.; Hiyama,
T. J. Am. Chem. Soc. 2005, 127, 12506-12507.
19

20
(24) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029-3070.
(25) Dang, T. P.; Kagan, H. B. Chem. Commun. 1971, 481.
(26) Kagan, H. B.; Dang, T. P. J. Am. Chem. Soc. 1972, 94, 6429-6433.
(27) Kagan, H. B.; Langlois, N.; Dang, T. P. J. Organomet. Chem. 1975, 90, 353-365.
(28) Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D.
J. J. Am. Chem. Soc. 1977, 99.
(29) Knowles, W. S. Acc. Chem. Res. 1983, 16, 106-112.
(30) Noyori, R.; Takaya, H. Acc. Chem. Res. 1990, 23, 345-350.
(31) Noyori, R. Chem. Soc. Rev. 1989, 18, 187-208.
(32) Noyori, R. Science 1990, 248, 1194-1199.
(33) Giancarlo Franciò, F. F. W. L. Angew. Chem. Int. Ed. 2000, 39, 1428-1430.
(34) Newkome, G. R. Chem. Rev. 1993, 93, 2067-2089.
(35) Arce, A. J.; Deeming, A. J.; De Sanctis, Y.; Johal, S. K.; Martin, C. M.; Shinhmar,
M.; Speel, D. M.; Vassos, A. Chem. Commun. 1998, 233-234.
(36) Deeming, A. J.; Shinhmar, M. K. J. Organomet. Chem. 1999, 592, 235-239.
(37) Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105.
(38) Antilla, J. C.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 11684-
11688.
(39) Gurevich, P. A.; Razumov, A. I.; Komina, T. V.; Klimentova, G. Y.; Zykora, T. V.
J. Gen. Chem. USSR (Engl. Trans.) 1985, 55, 1121.
(40) Barnard, T. S.; Mason, M. R. Organometallics 2001, 20, 206-214.
(41) Johnson, T. J.; Arif, A. M.; Gladysz, J. A. Organometallics 1994, 13, 3182-3193.
(42) Evans, W. J.; Brady, J. C.; Ziller, J. W. Inorg. Chem. 2002, 41, 3340-3346.
(43) Berens, U.; Brown, J. M.; Long, J.; Selke, R. Tetrahedron: Asymmetry 1996, 7,
285-292.
(44) Katritzky, A. R.; Lue, P.; Chen, Y.-X. J. Org. Chem. 1990, 55, 3688-3691.
(45) Tolmachev, A. A.; Chaikovskaja, A. A.; Terikovskaja, T. E.; Ivonin, S. P.;
Pinchuk, A. M. Heteroatom Chemistry 1996, 7, 525-531.
(46) Gale, P. A. Chem. Commun. 2005, 3761-3772.
(47) Amendola, V.; Bonizzoni, M.; Esteban-Gómez, D.; Fabbrizzi, L.; Licchelli, M.;
Sancenón, F.; Taglietti, A. Coord. Chem. Rev. 2006, 250, 1451-1470.

21
(48) Beer, P. D.; Hayes, E. J. Coord. Chem. Rev. 2003, 240, 167-189.
(49) Best, M. D.; Tobey, S. L.; Anslyn, E. V. Coord. Chem. Rev. 2003, 240, 3-15.
(50) Bondy, C. R.; Loeb, S. J. Coord. Chem. Rev. 2003, 240, 77-99.
(51) Choi, K.; Hamilton, A. D. Coord. Chem. Rev. 2003, 240, 101-110.
(52) Gale, P. A. Coord. Chem. Rev. 2003, 240, 191-221.
(53) Gale, P. A.; García-Garrido, S. E.; Garric, J. Chem. Soc. Rev. 2008, 37, 151-190.
(54) García-España, E.; Díaz, P.; Llinares, J. M.; Bianchi, A. Coord. Chem. Rev. 2006,
250, 2952-2986.
(55) Llinares, J. M.; Powell, D.; Bowman-James, K. Coord. Chem. Rev. 2003, 240, 57-
75.
(56) Martínez-Máñez, R.; Sancenón, F. Chem. Rev. 2003, 103, 4419-4476.
(57) Sessler, J. L.; Camiolo, S.; Gale, P. A. Coord. Chem. Rev. 2003, 240, 17-55.
(58) Sessler, J. L.; Gale, P. A.; Cho, W.-S. Anion Receptor Chemistry; Royal Society
of Chemistry: Cambridge, UK, 2006.
(59) Bowman-James, K. Acc. Chem. Res. 2005, 38, 671-678.
(60) Hossain, M. A.; Liljegren, J. A.; Powell, D.; Bowman-James, K. Inorg. Chem.
2004, 43, 3751-3755.
(61) Shinoda, S.; Tadokoro, M.; Tsukube, H.; Arakawa, R. Chem. Commun. 1998,
181-182.
(62) Amendola, V.; Boiocchi, M.; Colasson, B.; Fabbrizzi, L. Inorg. Chem. 2006, 45,
6138-6147.
(63) Bondy, C. R.; Gale, P. A.; Loeb, S. J. J. Am. Chem. Soc. 2004, 126, 5030-5031.
(64) Bondy, C. R.; Loeb, S. J.; Gale, P. A. Chem. Commun. 2001, 729-730.
(65) Oton, F.; Tarraga, A.; Espinosa, A.; Velasco, M. D.; Bautista, D.; Molina, P. J.
Org. Chem. 2005, 70, 6603-6608.
(66) Sun, S.-S.; Lees, A. J. Chem. Commun. 2000, 1687-1688.
(67) Amendola, V.; Bastianello, E.; Fabbrizzi, L.; Mangano, C.; Pallavicini, P.; Perotti,
A.; Lanfredi, A. M.; Ugozzoli, F. Angew. Chem. Int. Ed. 2000, 39, 2917-2920.
(68) Vega, I. E. D.; Gale, P. A.; Light, M. E.; Loeb, S. J. Chem. Commun. 2005, 4913-
4915.

22
(69) Brooks, S. J.; Evans, L. S.; Gale, P. A.; Hursthouse, M. B.; Light, M. E. Chem.
Commun. 2005, 734-736.
(70) Coles, S. J.; Frey, J. G.; Gale, P. A.; Hursthouse, M. B.; Light, M. E.; Navakhun,
K.; Thomas, G. L. Chem. Commun. 2003, 568-569.
(71) Kingston, E. J.; Ashford, L.; Beer, P. D.; Drew, M. G. B. Dalton Trans. 1999,
251-258.
(72) Kavallieratos, K.; Bertao, C. M.; Crabtree, R. H. J. Org. Chem. 1999, 64, 1675-
1683.
(73) Kondo, S.-i.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005,
1720-1722.
(74) Lakshminarayanan, P. S.; Ravikumar, I.; Suresh, E.; Ghosh, P. Inorg. Chem. 2007,
46, 4769-4771.
(75) Evans, L. S.; Gale, P. A.; Light, M. E.; Quesada, R. Chem. Commun. 2006, 965-
967.
(76) Gale, P. A.; Camiolo, S.; Chapman, C. P.; Light, M. E.; Hursthouse, M. B.
Tetrahedron Lett. 2001, 5095 - 5097.
(77) Gale, P. A.; Camiolo, S.; Tizzard, G. J.; Chapman, C. P.; Light, M. E.; Coles, S.
J.; Hursthouse, M. B. J. Org. Chem. 2001, 66, 7849-7853.
(78) Gale, P. A.; Sessler, J. L.; Kral, V.; Lynch, V. J. Am. Chem. Soc. 1996, 118, 5140-
5141.
(79) Maeda, H.; Ito, Y. Inorg. Chem. 2006, 45, 8205-8210.
(80) Navakhun, K.; Gale, P. A.; Camiolo, S.; Light, M. E.; Hursthouse, M. B. Chem.
Commun. 2002, 2084-2085.
(81) Sessler, J. L.; Gross, D. E.; Cho, W. S.; Lynch, V. M.; Schmidtchen, F. P.; Bates,
G. W.; Light, M. E.; Gale, P. A. J. Am. Chem. Soc. 2006, 128, 12281-12288.
(82) Boiocchi, M.; DelBoca, L.; Gomez, D. E.; Fabbrizzi, L.; Licchelli, M.; Monzani,
E. J. Am. Chem. Soc. 2004, 126, 16507-16514.
(83) Brooks, S. J.; Gale, P. A.; Light, M. E. Chem. Commun. 2006, 4344-4346.
(84) Duckmanton, P. A.; Blake, A. J.; Love, J. B. Inorg. Chem. 2005, 44, 7708-7710.
(85) Eisler, D. J.; Puddephatt, R. J. Inorg. Chem. 2003, 42, 8192-8202.

23
(86) Knight, L. K.; Freixa, Z.; vanLeeuwen, P. W. N. M.; Reek, J. N. H.
Organometallics 2006, 25, 954-960.
(87) Tovilla, J. A.; Vilar, R.; White, A. J. P. Chem. Commun. 2005, 4839 - 4841.
(88) He, J. J.; Quiocho, F. A. Science 1991, 251, 1479-1481.
(89) Bates, G. W.; Gale, P. A.; Light, M. E. Chem. Commun. 2007, 2121-2123.
(90) Bates, G. W.; Triyanti; Light, M. E.; Albrecht, M.; Gale, P. A. J. Org. Chem.
2007, 72, 8921-8927.
(91) Black, D. S.; Craig, D. C.; Kumar, N.; McConnell, D. B. Tetrahedron Lett. 1996,
37, 241-244.
(92) Chae, M. K.; Lee, J. I.; Kim, N. K.; Jeong, K. S. Tetrahedron Lett. 2007, 48,
6624-6627.
(93) Chang, K. J.; Chae, M. K.; Lee, C.; Lee, J. Y.; Jeong, K. S. Tetrahedron Lett.
2006, 47, 6385-6388.
(94) Chang, K. J.; Kang, B. N.; Lee, M. H.; Jeong, K. S. J. Am. Chem. Soc. 2005, 127,
12214-12215.
(95) Curiel, D.; Cowley, A.; Beer, P. D. Chem. Commun. 2005, 236-238.
(96) He, X.; Hu, S.; Liu, K.; Guo, Y.; Xu, J.; Shao, S. Org. Lett. 2006, 8, 333-336.
(97) Kwon, T. H.; Jeong, K. S. Tetrahedron Lett. 2006, 47, 8539-8541.
(98) Chang, D. -J.; Moon, D.; Lah, M. S.; Jeong, K. -S. Angew. Chem. Int. Ed. 2005,
44, 7926-7929.
(99) Lee, J.-Y.; Lee, M.-H.; Jeong, K.-S. Supramol. Chem. 2007, 19, 257 - 263.
(100) Papageorgiou, G.; Corrie, J. E. T. Tetrahedron 2007, 63, 9668-9676.
(101) Pfeffer, F. M.; Lim, K. F.; Sedgwick, K. J. Org. & Biomol. Chem. 2007, 5, 1795-
1799.
(102) Sessler, J. L.; Cho, D.-G.; Lynch, V. J. Am. Chem. Soc. 2006, 128, 16518-16519.
(103) Wang, Y.; Lin, H.; Shao, J.; Cai, Z. S.; Lin, H. K. Talanta 2008, 74, 1122-1125.
(104) Zielinski, T.; Dydio, P.; Jurczak, J. Tetrahedron 2008, 64, 568-574.
(105) Lam, E., PhD Thesis, University of Toronto, 2007.

Chapter 2
Monodentate 2-Indolylphosphines: A Study in
Structure by X-ray Crystallography*
2.1. Introduction
2-Indolylphosphines are ideal ligands due to their ability to coordinate Lewis
acids via the phosphorus centre, and the indolyl NH moiety can serve as a secondary site
of reactivity. The indolyl substituent provides a basis for aufbau or stepwise assembly of
potentially more complex phosphines. Functionality can be incorporated on the
phosphine at the nitrogen centre upon deprotonation with a variety of substituents that
can possess properties which alter the overall bulkiness, electronics, or chirality of the
phosphine (Scheme 2.1). This modular approach to creating new phosphines via N-
modification following P-substituent bond formation, lies in contrast to the more
customary methods of phosphine synthesis. Therein, the challenging and often
substituent-specific intricacies and sometimes unpredictability of P-C bond formation
must be overcome each time that a different structural element of interest is incorporated
into the phosphine.
HN
HN
PR1
R1
NP
R1
R1
R2
(a) (b) (c) Scheme 2.1. A general scheme demonstrating the Aufbau assembly of N-substituted 2-indolylphosphines.
(a) 3-Methylindole is the starting point (b) A generic unsubstituted 2-indolylphosphine (c) An N-
functionalized 2-indolylphosphine. * Reproduced in part with permission from: J. O. Yu, E. Lam, J. L. Sereda, N. C. Rampersad, A. J. Lough, C. S. Browning, and D. H. Farrar. Organometallics 2005, 24, 37-47. Copyright 2006 American Chemical Society.
24

25
A variety of methods can be utilized to characterize the 2-indolylphosphines that
we have generated, most commonly are spectroscopic methods for routine analysis of
these compounds. Single crystal X-ray diffraction has proven to be a useful method to
analyze the bond lengths and angles of phosphines to provide insight into the steric bulk
associated with the ligands, which is an important aspect in phosphine design particularly
for transition metal catalysis.1,2 Additionally, X-ray crystallography can be used to
examine the effect of substituents on the indolyl moiety to determine how they impact the
aromaticity of the pyrrole ring.
In this Chapter, X-ray crystallography will be used to examine the structural
features of unfunctionalized and N-alkylated 2-indolylphosphines. A correlation between
steric crowding around a phosphorus atom and a phosphine ligand’s size will also be
discussed.
2.1.1. Crystallographic Analysis of Common Phosphines
in the CSD
While the Tolman cone angle is a good estimate of a phosphine’s steric bulk, the
Tolman cone angle does not consider the variation in ligand conformation or the impact
of these conformations on a ligand’s cone angle.1 Mingos and co-workers devised a
method to determine a phosphine’s cone angle based on crystallographic data, and used
statistical information from the Cambridge Crystallographic Structural Database (CSD)
to analyze the method.3 The crystallographic method used the geometric relationship
originally proposed by Tolman where the metal-phosphorus bond in phosphine
complexes was adjusted to 2.28 Å and a van der Waals radius of 1.00 Å was used for
hydrogen atoms (Figure 2.1).

26
Figure 2.1. Description of the van der Waals surface of a phosphine ligand to determine the Tolman cone
angle from crystallographic data.3
Mingos et al. gathered statistical data on the phosphines’ cone angles from the CSD and
determined that the statistics of these data could be characterized well with a normal
distribution.3 Based on the normal distributions, they were able to determine the mean
and median parameters which allowed them to calculate the average values of cone
angles for several common phosphines (Table 2.1). The average calculated cone angles
for the common phosphines correlated reasonably well with Tolman’s cone angles based
on space-filling models, however the statistical spread of crystallographic cone angles
due to varied conformation in the phosphine’s substituents was greater than had been
originally expected. The advantage of this approach is that a ligand’s cone angle can be
easily determined from its crystal structure; the disadvantage is that a crystal structure of
the phosphine must be available. The crystal structure must have the ligand coordinated
to a metal atom and free of secondary interactions, such as hydrogen bonding or ionic
interactions that may affect its cone angle.
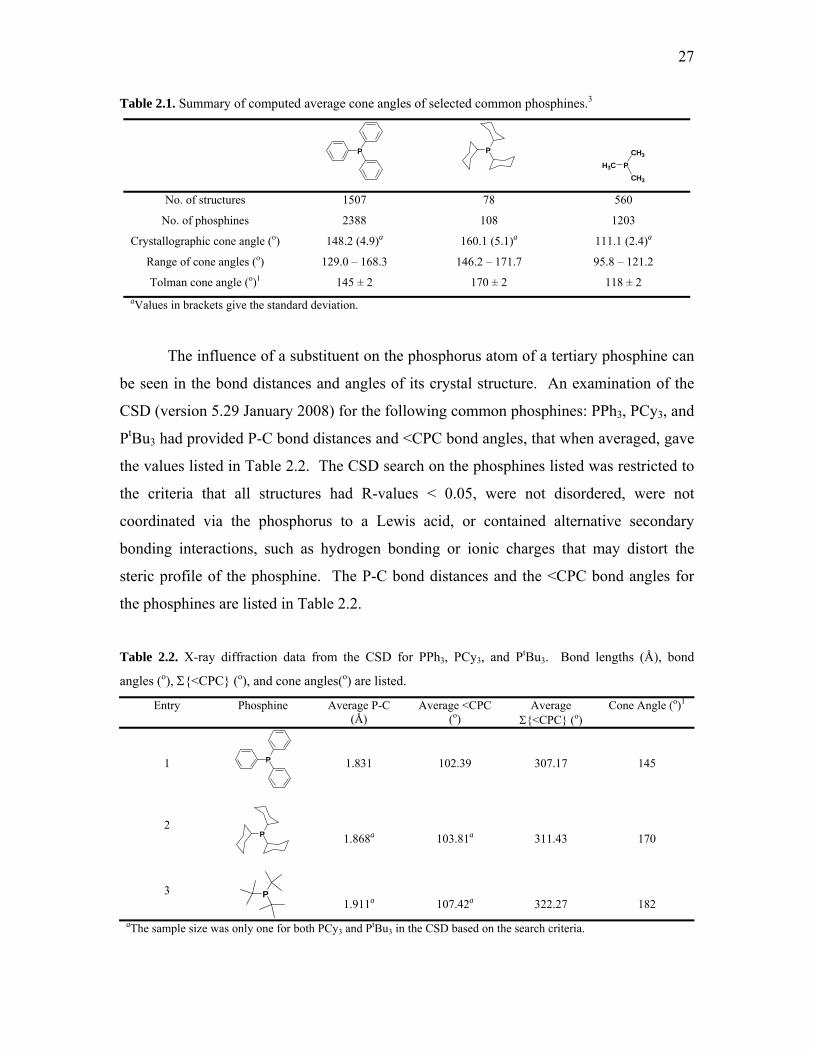
27
Table 2.1. Summary of computed average cone angles of selected common phosphines.3
P
P
CH3
PH3CCH3
No. of structures 1507 78 560
No. of phosphines 2388 108 1203
Crystallographic cone angle (o) 148.2 (4.9)a 160.1 (5.1)a 111.1 (2.4)a
Range of cone angles (o) 129.0 – 168.3 146.2 – 171.7 95.8 – 121.2
Tolman cone angle (o)1 145 ± 2 170 ± 2 118 ± 2 aValues in brackets give the standard deviation.
The influence of a substituent on the phosphorus atom of a tertiary phosphine can
be seen in the bond distances and angles of its crystal structure. An examination of the
CSD (version 5.29 January 2008) for the following common phosphines: PPh3, PCy3, and
PtBu3 had provided P-C bond distances and <CPC bond angles, that when averaged, gave
the values listed in Table 2.2. The CSD search on the phosphines listed was restricted to
the criteria that all structures had R-values < 0.05, were not disordered, were not
coordinated via the phosphorus to a Lewis acid, or contained alternative secondary
bonding interactions, such as hydrogen bonding or ionic charges that may distort the
steric profile of the phosphine. The P-C bond distances and the <CPC bond angles for
the phosphines are listed in Table 2.2.
Table 2.2. X-ray diffraction data from the CSD for PPh3, PCy3, and PtBu3. Bond lengths (Å), bond
angles (o), Σ{<CPC} (o), and cone angles(o) are listed.
Entry Phosphine Average P-C (Å)
Average <CPC (o)
Average Σ{<CPC} (o)
Cone Angle (o)1
1 P
1.831
102.39
307.17
145
2
P
1.868a
103.81a
311.43
170
3 P
1.911a
107.42a
322.27
182 aThe sample size was only one for both PCy3 and PtBu3 in the CSD based on the search criteria.

28
The P-C bond distances can provide a measure of carbon atom hybridization of
the substituent on phosphorus, for example an sp3-hybridized carbon will have a longer
P-C bond length compared to a carbon with sp2-hybridization. Additionally, the
phosphines in Table 2.2 can be categorized by their basicity4,5 and the order of least basic
to most basic is PPh3 < PCy3 < PtBu3. The basicity of the phosphine is directly related to
the nature of substituents on the phosphorus atom, and generally the more electron-
withdrawing a substituent is, the less basic the phosphine will be. This example of
basicity approximation is also reflected in the P-C bond lengths of the phosphines listed
in Table 2.2, with PPh3 being the least basic it has the shortest P-C bond lengths, whereas
the PtBu3 being the most basic has the longest P-C bond lengths.
While the traditional gauge of phosphine steric bulk is measured using Tolman’s
cone angle method,1 an alternative simple method to assess steric bulk at the phosphorus
centre can be achieved by comparing the sums of the angles around the phosphorus,
Σ{<CPC}.6 For a three-coordinate molecule possessing absolute trigonal planar
geometry, the Σ{<CPC} would be 360o, whereas a perfect trigonal pyramidal structure
having ideal tetrahedral bond angles will have a Σ{<CPC} of 328o. The average
Σ{<CPC} are calculated for PPh3, PCy3, and PtBu3 and are listed in Table 2.2. The
average Σ{<CPC} for the phosphines in Table 2.2 are consistent with their cone angle
values determined using Tolman’s method: PPh3 has the smallest Σ{<CPC} and PtBu3
has the largest Σ{<CPC}. While bond angles can be affected immensely by
crystallographic packing forces, in silico studies comparing Σ{<CPC} of
triarylphosphines to crystallographically obtained Σ{<CPC} values indicate that the bond
angles are only slightly affected by crystallographic packing forces, and the experimental
and calculated Σ{<CPC} values are comparable.6 Therefore, in the event that suitable
crystal structures of a phosphine-metal complex are not available, and thus Tolman’s
method to determine the phosphine’s cone angle is impossible by analysis of
crystallographic data, then an approximation of the phosphine’s steric bulk can be
estimated from its Σ{<CPC}.

29
2.1.2. Crystallographic Analysis of Indole-Containing
Structures in the CSD
Changes in the aromaticity of the pyrrolyl ring on indole may be seen
crystallographically in the bond distances and bond angles of its crystal structures. In
order to determine the standard of aromaticity in the pyrrole ring of indole, a CSD search
was conducted to find aromatic indole-based structures and examine their bond distances
and angles. The following search restrictions were applied to maintain consistency on the
structures that were returned: R-values < 0.05, no disorder was present, the absence of
coordination to either the nitrogen centre or either of the rings (in η-fashion), alternative
secondary bonding interactions were omitted, such as hydrogen bonding or ionic charges
that may distort the aromaticity of the pyrrolyl moiety, and finally carbazole-based
structures were not permitted. The search criteria produced a sample size of 11
structures.7 Selected bond distances and angles are listed in Table 2.3 along with the
numbering scheme.
Table 2.3. Selected averaged bond distances (Å) and bond angles (o) of indole-based structures from the
CSD.
C4
C9
C3
C2
N1
R3
R2
R1
N1-C2 1.389 C2-C3 1.364 C3-C4 1.439 C4-C9 1.413 C9-N1 1.382
C9-N1-C2 108.251 N1-C2-C3 109.789 C2-C3-C4 107.313 C3-C4-C9 106.44 C4-C9-N1 108.156

30
The average pyrrolyl-moiety bond distances were found to be midway between
true N-C or C-C single bonds and N=C or C=C double bonds, consistent with the
delocalized characteristic of a pyrrole ring, with the exception of the C2-C3 bond
distance which is closer to a true C=C double bond.(note C-C, C-N, C=C, and C=N) The
bond angles were all averaged to be about 108o, the only angle being smaller than the
ideal 108o was the <C3-C4-C9 (106.44o) poses no significant influence on the structural
features of the pyrrolyl moiety. Thus, these bond distances and angles may be used as a
benchmark for aromaticity when analyzing our 2-indolylphosphines.
2.2. Results and Discussion
2.2.1. Synthesis of Unsubstituted 2-Indolylphosphines
Our group has established methods for the preparation of a variety of 2-
indolylphosphines.8,9 A general reaction sequence is shown in Scheme 2.2, the details of
the synthesis will be discussed in Chapter 3.
HN
P
HN
N
N
NP
N1. n-BuLi
2. PCl3-xRxTHF
1. NaBH4EtOH/THF
reflux2. HCl workup
Me2NHH2O
reflux
H H
O
quantitative 89% overall 85% Scheme 2.2. The synthesis of phosphine 1 from the commercially available 3-methylindole. This is a
representative example of the facile method for 2-indolylphosphine synthesis.
These phosphines can be varied in several ways just by adding the desired
chlorophosphine. For example, the number of indolyl substituents can be modified from
one indolyl substituent in 1, to three indolyl substituents in the C3-symmetric 5.
Alternatively, the two other substituents on the phosphorus can also be altered from
phenyl in 1 to cyclohexyl in 2. A library of 2-indolylphosphines has been generated by
this simple method8 and are shown in Figure 2.2.

31
HN
P
NH
P HN
NH
P
HN
HNH
NP
(a) (b) (c) (d)
1 2 3 5
Figure 2.2. Selected monodentate unsubstituted 2-indolylphosphines. (a) P(C9H8N)(C6H5)2, 1
(b) P(C9H8N)(C6H11)2, 2 (c) P(C9H8N)2(C6H5), 3 (d) P(C9H8N)3, 5.
2.2.2. X-ray Crystallographic Analysis of Unsubstituted 2-
Indolylphosphines
Discussion of the crystallographic analysis of the 2-indolylphosphines will be
confined to the structures shown in Figure 2.2. X-ray diffraction data will be used to
interpret how exchanging the substituents can affect the steric crowding of the
phosphorus centre by inspecting the bond lengths and angles associated with the
phosphorus centre (Section 2.3), and the influence of N- or C2-substitution on the
aromaticity of the pyrrole moiety of the indolyl substituent (Section 2.3.1). The X-ray
diffraction experimental data are shown in Table 2.4.

32
Table 2.4. X-ray crystallographic experimental data of unsubstituted 2-indolylphosphines.
1
HN
P
2
HN
P
3
NH
PHN
5
NH
P
HN
HN
Formula C21H18NP C21H30NP C24H21N2P C27H24N3P Formula Weight 315.33 327.43 368.40 421.46 Crystal colour, shape colourless, block colourless, block colourless, block colourless, plate Crystal size, mm 0.35 x 0.30 x 0.30 0.26 x 0.08 x 0.06 0.20 x 0.18 x 0.18 0.16 x 0.12 x 0.08 Crystal system Triclinic Monoclinic Monoclinic Rhombohedral Space group Pī P21/c P21/c R3 a, Å 9.9678(2) 11.1906(9) 10.3563(3) 14.415(3) b, Å 10.3453(2) 14.3772(12) 10.3027(6) 14.415(3) c, Å 17.4704(4) 11.7375(5) 19.0016(8) 9.4877(16) α deg 79.973(8) 90 90 90 β, deg 75.992(7) 98.016(5) 104.383(3) 90 γ deg 74.133(9) 90 90 120 V, Å3 1670.09(6) 1870.0(2) 1963.9(2) 1707.3(6) Z 4 4 4 3 Dcalcd, g cm-3 1.254 1.163 1.246 1.230 F(000) 664 712 776 666 μ, mm-1 0.163 0.148 0.150 0.140 λ (Mo Kα), Å 0.71073 0.71073 0.71073 0.71073 Limiting indices 0<=h<=12 -14<=h<=14 -13<=h<=13 -18<=h<=15 -12<=k<=13 -16<=k<=18 -13<=k<=13 -16<=k<=18 -21<=l<=22 -15<=l<=15 -24<=l<=24 -11<=l<=12 2θ range, deg 2.55 to 27.48 1.84 to 27.55 2.27 to 27.50 2.70 to 27.43 Max. and min. transmission 0.9526 and 0.9450 0.993 and 0.834 0.993 and 0.602 1.003 and 0.937 No. of reflections collected 22759 14623 16135 2314 No. of independent reflections/Rint 7596 / 0.046 4469 / 0.081 4736 / 0.0904 1510 / 0.0571 Absolute structure parameter - - - 0.2(2) Extinction coefficient 0.0014(16) 0.0027(12) 0.0111(16) 0.0033(7) No. of refined parameters 426 210 255 96 Final R1, wR2 0.0449, 0.1140 0.0513, 0.1093 0.0546, 0.1221 0.0548, 0.1002 Final R1, wR2 (all data) 0.0767, 0.1320 0.1023, 0.1268 0.1017, 0.1442 0.1142, 0.1208 Goodness of fit 1.029 1.027 1.020 0.950 Δρmin, Δρmax, eÅ-3 -0.258, 0.257 0.212 and -0.280 -0.318, 0.359 0.193 and -0.187
Colourless block-shaped single crystals of phosphine 1 were grown by slow
evaporation from toluene. Phosphine 1 crystallizes in the triclinic space group Pī, with
two crystallographically independent molecules in the asymmetric unit (Figure 2.3). The
molecular structures and numbering scheme of the independent molecules are shown in
Figure 2.4, and Table 2.5 lists selected bond distances and angles. The independent
molecules are adequately similar in structure such that discussion is confined to one of
them (the most significant difference between the two molecules is the <C2-P1-C17 bond
angle of 99.94(8)o and 101.94(9)o). The geometry about the phosphorus atoms is best

33
described as trigonal pyramidal with the aromatic substituents adopting a twisted,
propeller blade-like orientation presumably to minimize the steric repulsion associated
with each ring. All of the hydrogen atoms on carbon were placed in calculated positions,
while the indolyl NH protons could have been located in the electron density map, they
were included in the refinement in the riding model on nitrogen. The solid-state structure
of 1 does not appear to possess secondary bonding interactions, as the closest non-
bonding distance is 2.70 Å between N1A-H1A and the phenyl ring of a symmetry
equivalent molecule.
Figure 2.3. Unit cell diagram of the four molecules of 1. Crystallographically independent molecules are
labeled. Hydrogen atoms are removed for clarity.

34
Figure 2.4. Numbering scheme of 1a in unit cell. Non-acidic protons removed for clarity. Thermal
ellipsoids are drawn at 35% probability.
It can be seen clearly in Figure 2.3 that both independent molecules of 1 have
their indolyl rings oriented in a fashion where the indolyl NH’s are pointing away from
the lone pair of the phosphorus atoms. This indolyl ring orientation may be a result of the
presence of the methyl group at the C3-position and the lack of substitution on the
nitrogen centre. The smaller proton substituent on nitrogen would assist in the indolyl
ring to be oriented in the lowest energy conformation prior to crystallization. While the
methyl group on the C3-position is directed towards the phosphorus lone pair, it is distant
enough not to induce any chemical changes to the phosphorus centre (average of
3.336(3) Å).
The P1-C2indolyl bond distance of 1.813(2) Å is significantly shorter than the bond
distances from P1-C11phenyl and P1-C21phenyl (average = 1.838(2) Å). No chemical
significance is associated with the bond angle between the indolyl and one of the phenyl
substituents, <C2A-P1A-C17A (99.9(8)°), being significantly smaller than the other bond
angles of <C2A-P1A-C11A (101.6(8)°) and <C11A-P1A-C17A (103.0(8)°).

35
Table 2.5. Selected bond lengths (Å) and bond angles (o) for unsubstituted 2-indolylphosphines.
1a in asymmetric unit
HN
P
1b in asymmetric unit
HN
P
2
HN
P
3
NH
PHN
5
NH
P
HN
HN
P1-C2 1.813(2) 1.811(2) 1.818(2) 1.812(2) 1.812(3) P1-C12 1.837(2) 1.833(2) 1.858(2) 1.809(2) 1.812(3) P1-C22 1.837(2) 1.834(2) 1.858(2) 1.841(2) 1.812(3)
N1-C2 1.399(2) 1.396(2) 1.395(2) 1.398(3) 1.390(5) C2-C3 1.374(3) 1.372(3) 1.375(3) 1.372(3) 1.378(5) C3-C4 1.438(3) 1.438(3) 1.438(3) 1.436(3) 1.427(5) C4-C9 1.409(3) 1.411(3) 1.407(3) 1.410(3) 1.427(5) C9-N1 1.378(2) 1.378(2) 1.373(2) 1.373(3) 1.389(4)
C9-N1-C2 109.2(2) 109.1(2) 110.1(2) 109.2(2) 109.5(3) N1-C2-C3 109.0(2) 109.0(2) 108.1(2) 109.1(2) 108.4(3) C2-C3-C4 106.8(2) 107.0(2) 107.3(2) 106.8(2) 107.6(3) C3-C4-C9 107.8(2) 107.4(2) 107.4(2) 107.6(2) 107.6(3) C4-C9-N1 107.3(2) 107.4(2) 107.1(2) 107.4(2) 106.9(3)
N11-C12 - - - 1.397(3) - C12-C13 - - - 1.379(3) - C13-C14 - - - 1.439(3) - C14-C19 - - - 1.407(3) - C19-N11 - - - 1.381(3) -
C19-N11-C12 - - - 109.2(2) - N11-C12-C13 - - - 108.8(2) - C12-C13-C14 - - - 106.8(2) - C13-C14-C19 - - - 107.6(2) - C14-C19-N11 - - - 107.4(2) -
It is evident from the crystal structure of 1 that the C2-bound methylindolyl group
constitutes a markedly larger phosphorus substituent than the phenyl ring. An average
separation of 2.40(6) Å between the two phenyl carbon atoms positioned ortho to
phosphorus on each ring (C12..C16 and C18..C22) serves as an effective measure of the
width of the phosphine’s two phenyl groups. In comparison, measured from its nitrogen
atom to the methyl carbon atom C10A, the equivalent width of 3.713(4) Å for the
methylindolyl group of 1 suggests that the 2-indolylphosphines possess cone angles
larger than that of PPh3,1 perhaps bearing a closer similarity in size to P(o-tolyl)3.1
Colourless block-shaped single crystals of 2 were obtained from hexane vapour
diffusion into a solution of the phosphine in CH2Cl2. The numbering scheme of 2 is
shown in Figure 2.5 and selected bond distances and angles listed in Table 2.5. The

36
phosphine crystallized in the monoclinic space group P21/c with one molecule in the
asymmetric unit. The hydrogen atoms were placed in calculated positions and included
in the refinement as riding atoms. There are no secondary bonding interactions in the
solid-state structure of 2 as the closest N1-H1A distance is 3.86 Å to the pyrrolyl ring of a
symmetry equivalent molecule. Similar to 1, the geometry about the phosphorus atom of
2 can best be described as trigonal pyramidal.
The P1-C2indolyl distance of 1.818(2) Å is similar to the analogous P-Cindolyl bond
distance of 1. Additionally, the coordination of a phosphorus atom at the C2-position on
indole does not result in any significant changes to its structural features. The P-Ccyclohexyl
bonds (1.858(2) Å) are significantly longer than the analogous P-Cphenyl bonds found in 1
(average 1.835(4) Å) as anticipated for the sp3-hybridized carbons of the cyclohexyl rings
compared to the sp2-hybridized carbons of the phenyl rings.
Figure 2.5. Numbering scheme of 2 in unit cell. Non-acidic protons removed for clarity. Thermal
ellipsoids are drawn at 35% probability.
The indolyl NH moiety of 2 points away from the phosphorus lone pair, which is
similar to the solid-state structure of 1. In this case, the orientation of the indolyl ring is
likely to alleviate any steric crowding that may occur between the methyl group at the
C3-position and the two bulky cyclohexyl substituents on phosphorus. The methyl group
is sufficiently distant from the phosphorus atom (3.390(2) Å) that it should not impart
any significant chemical changes to its lone pair.
A measure of the width of the phosphine’s two cyclohexyl groups was conducted
by determining the average separation between the two cyclohexyl carbon atoms
positioned ortho to phosphorus on each ring (C12..C16 and C18..C22) gave a value of

37
2.506(3) Å. In comparison to the analogously measured average separation for the
phenyl groups on 1 (2.40(6) Å), as it would be expected the cyclohexyl substituents are
slightly larger. However, performing the same measurement with the indolyl substituent
on 2 (from N1...C10) gave a distance of 3.705(3) Å, suggesting that a 3-methylindolyl is a
wider substituent than cyclohexyl.
Phosphine 3 crystals were grown as colourless blocks from a saturated solution in
CH2Cl2 via slow evaporation. Figure 2.6 shows the molecular structure and numbering
scheme of the ligand and Table 2.5 lists selected bond distances and angles. Phosphine 3
crystallizes in the space group P21/c with one molecule in the asymmetric unit. All of the
hydrogen atoms were placed in calculated positions, even though the indolyl protons
could be located in the electron density map, and included in the refinement as riding
atoms. There are no secondary bonding interactions in the solid-state structure of 3, since
the closest N11-H11A distance is to an indolyl moiety of a symmetry equivalent
molecule at 2.64 Å.
The phosphorus atom adopts a trigonal pyramidal geometry with the aromatic
substituents situated in a twisted, propeller blade-like fashion, similar to the solid-state
structures of 1 and 2. Likewise, the indolyl substituents are oriented such that the methyl
moieties at the C3 and C13 positions point away from the pyramid formed by the
substituents around P1. In addition, the P1-C2indolyl and P1-C12indolyl bond distances of
1.812(2) Å and 1.809(2) Å, respectively, are shorter than the P1-C21phenyl bond distance
of 1.841(2) Å.

38
Figure 2.6. Numbering scheme of 3. Non-acidic protons removed for clarity. Thermal ellipsoids are
drawn at 35% probability.
Single crystals of 5 were acquired by slow evaporation of a saturated solution of
the phosphine in CH2Cl2. The numbering scheme of 5 is shown in Figure 2.7 and
selected bond distances and angles listed in Table 2.5. C3-Symmetric 5 crystallized in the
chiral rhombohedral space group R3 indicating resolution of phosphine 5 in the solid-
state. One-third of a molecule exists in the asymmetric unit with a crystallographic C3-
axis that is parallel to the molecular C3-axis running through the phosphorus atom to
generate two symmetry equivalent indolyl rings. The indolyl substituents are oriented in
a propeller-like fashion, not unlike the previously discussed 2-indolylphosphines.

39
Figure 2.7. Numbering scheme of 5. Non-acidic protons removed for clarity. Thermal ellipsoids are
drawn at 35% probability.
As was determined for phosphines 1, 2, and 3, the indolyl NH functionalities of 5
point inwards toward the pyramid created by its three indolyl substituents and the
phosphorus atom as the apex. The importance of the indolyl rings orientation of
phosphine 5 will become apparent in Chapter 3 when the anion binding ability of this
molecule will be discussed in detail. While the indolyl NH proton could have been
located in the electron density map, it was refined as a riding model on nitrogen, as were
the hydrogen atoms on carbon that were placed in calculated positions. The solid-state
structure of 5 does not appear to possess secondary bonding interactions, since the closest
N1-H1A distance is to a phenyl moiety of a symmetry equivalent molecule at 2.71 Å.
The P1-C2indolyl bond distances for 5 were determined to be 1.812(3) Å, which correlates
well to analogous P-Cindolyl bonding distances of the previously described 2-
indolylphosphines.

40
2.2.3. Synthesis of Monodentate N-Alkylated 2-
Indolylphosphines
More complex 2-indolylphosphines can be synthesized easily by a stepwise
approach that involves making the phosphine scaffold first and introducing modifying
groups on the indolyl nitrogen centre second. Just by adding the desired alkyl group, the
overall physical and chemical characteristics of the phosphine can be transformed to be
electron withdrawing, electron donating, or even have additional stereocentres. In the
presence of NaH, the nitrogen centre is deprotonated, and the addition of the desired alkyl
halide will produce a newly functionalized 2-indolylphosphine (Scheme 2.3), without the
extra steps of protection and deprotection of the phosphorus centre. Selected examples of
N-functionalization post P-C bond formation are shown in Figure 2.8.
HN
P1. NaH2. BnBr
THFN
P
52% Scheme 2.3. The synthesis of (N-Bn)-1. This is a representative scheme for N-substitution of 2-
indolylphosphines.
NP
N
F
FF
FF
F F
F
FFNP
(a) (b)
N-Bn-1 (N-CH2C6F5)2-4
Figure 2.8. Selected N-alkylated 2-indolylphosphines. (a) P(C9H8NC7H7)(C6H5)2, (N-Bn)-1
(b) P(C17H12N2C14H4F10)(C6H5), (N-F5Bn)2-4.

41
2.2.4. Crystallographic Analysis of N-Alkylated 2-
Indolylphosphines
Discussion of the crystallographic analysis of the N-alkylated 2-indolylphosphines
will be confined to the structures shown in Figure 2.8. The X-ray diffraction
experimental data for the N-alkylated 2-indolylphosphines are shown in Table 2.6.
Selected bond distances and angles for the crystal structures of the N-alkylated 2-
indolylphosphines are listed in Table 2.7.
Table 2.6. X-ray crystallographic experimental data of N-alkylated monodentate 2-indolylphosphines.
(N-Bn)-1
NP
(N-F5Bn)2-4
NP
N
F
FF
FF
F F
F
FF
Formula C28H24NP C37H19F10N2P.CHCl3 Formula Weight 405.45 831.88 Crystal colour, shape colourless, block colourless, block Crystal size, mm 0.46 x 0.30 x 0.22 0.32 x 0.30 x 0.22 Crystal system Monoclinic Monoclinic Space group P21/c P21/n a, Å 13.7780(5) 16.5993(5) b, Å 8.7875(4) 12.0034(3) c, Å 18.6350(7) 17.1393(4) α deg 90 90 β, deg 105.301(2) 98.8110(17) γ deg 90 90 V, Å3 2176.24(15) 3374.67(15) Z 4 4 Dcalcd, g cm-3 1.237 1.637 F(000) 856 1672 μ, mm-1 0.141 0.409 λ (Mo Kα), Å 0.71073 0.71073 Limiting indices -17<=h<=17 -21<=h<=21 -10<=k<=11 -15<=k<=15 -23<=l<=24 -22<=l<=22 2θ range, deg 2.38 to 27.50 1.59 to 27.53 Max. and min. transmission 0.971 and 0.909 0.964 and 0.852 No. of reflections collected 19191 32929 No. of independent reflections/Rint 5299 / 0.0466 8134 / 0.0528 Extinction coefficient 0.0073(17) 0.0013(4) No. of refined parameters 273 488 Final R1, wR2 0.0520, 0.1298 0.0438, 0.1071 Final R1, wR2 (all data) 0.0866, 0.1529 0.0721, 0.1221 Goodness of fit 1.029 1.027 Δρmin, Δρmax, eÅ-3 0.346 and -0.391 0.367 and -0.494

42
Table 2.7. Selected bond lengths (Å) and bond angles (o) for N-alkylated 2-indolylphosphines.
(N-Bn)-1
NP
(N-F5Bn)2-4
NP
N
F
FF
FF
F F
F
FF
P1-C2 1.819(2) 1.802(2) P1-Cb 1.831(2) 1.813(2) P1-Cc 1.836(2) 1.844(2)
N1-C2 1.400(3) 1.383(3) C2-C3 1.385(3) 1.378(3) C3-C4 1.425(3) 1.436(3) C4-C9 1.401(4) 1.410(3) C9-N1 1.377(3) 1.399(3)
C9-N1-C2 108.8(2) 108.5(2) N1-C2-C3 108.4(2) 109.3(2) C2-C3-C4 107.2(2) 106.9(2) C3-C4-C9 107.6(2) 107.3(2) C4-C9-N1 108.1(2) 108.0(2)
N11-C12 - 1.396(3) C12-C13 - 1.370(3) C13-C14 - 1.442(3) C14-C19 - 1.412(3) C19-N11 - 1.378(3)
C19-N11-C12 - 108.9(2) N11-C12-C13 - 109.4(2) C12-C13-C14 - 106.9(2) C13-C14-C19 - 107.0(2) C14-C19-N11 - 107.8(2)
Single crystals of (N-Bn)-1 were obtained as colourless blocks from slow
evaporation of a saturated solution of the phosphine in CH2Cl2. Figure 2.9 shows the
molecular structure and numbering scheme of the ligand, and Table 2.7 lists selected
bond distances and angles. The ligand (N-Bn)-1 crystallizes in the monoclinic space
group P21/c with one molecule in the asymmetric unit. The geometry about P1 is trigonal
pyramidal and the aromatic substituents are oriented in a similar propeller-fashion as the
unsubstituted 2-indolylphosphines. There appear to be no secondary bonding interactions
in the crystal structure of (N-Bn)-1.

43
Figure 2.9. Numbering scheme of (N-Bn)-1. Hydrogen atoms removed for clarity. Thermal ellipsoids are
drawn at 35% probability.
In contrast to the unsubstituted 2-indolylphosphines, the methyl group at the C3-
position of the indolyl substituent of (N-Bn)-1 points away from the phosphorus lone
pair. The much larger benzyl moiety on the nitrogen centre results in the indolyl ring
being oriented such that the bulkier group is not pointing inwards towards the pyramid
created by the three P-substituents. The distance between the methylene C23 and P1 was
found to be 3.138(3) Å, thus the benzyl moiety should not cause significant chemical
changes to the phosphorus lone pair.
The benzyl group adopts an orientation in which it forms a slipped pseudo-
eclipsed π-stacking interaction with one of the phenyl moieties on P1 with a ring-to-ring
distance of 4.413 Å. The benzylated-methylindolyl substituent is a significantly larger
phosphorus substituent than the phenyl moieties. The analogous width of the benzylated-
methylindolyl, measured from indolyl’s methyl carbon atom C10 to the N-substituted
methylene carbon C23 was determined to be 5.061(4) Å; suggesting that the introduction
of any group on the indolyl nitrogen renders that phosphine substituent a cone angle
markedly greater than that of a phenyl group.

44
The P1-C2indolyl bonding distance of 1.819(2) Å is consistent with the
unsubstituted 2-indolylphosphines, as are the P1-C11phenyl and P1-C17phenyl bond
distances comparable to the analogous bond distances found within 1.
The formation of the phosphacycle was evident in the structure determination of
(N-F5Bn)2-4, single crystals of which were obtained by slow solvent evaporation from a
CDCl3 solution of the phosphine. (N-F5Bn)2-4 crystallizes in the monoclinic space group
P21/n with one deuterochloroform solvent molecule present. There was an absence of
intermolecular hydrogen bonding even though there are many hydrogen bond acceptors
located on the pentafluoro moieties. Figure 2.10 shows the molecular structure and
numbering scheme of (N-F5Bn)2-4 and Table 2.7 lists selected bond distances and angles.
Figure 2.10. Numbering scheme of (N-F5Bn)2-4. Hydrogen atoms and solvent molecule removed for
clarity. Thermal ellipsoids are drawn at 35% probability.
The P1-C2indolyl and P1-C12indolyl bond lengths of 1.802(2) Å and 1.813(2) Å
respectively in (N-F5Bn)2-4 are not significantly different from the P1-C2indolyl separation
of 1.813(2) Å observed in 1. The structural data for (N-F5Bn)2-4 suggests that its six-
membered phosphacycle does not experience substantial ring strain. The <C3-C10-C13
bond angle of 112.9(2)° about the methylene carbon of the phosphacycles of (N-F5Bn)2-4
is only slightly smaller than the corresponding bond angle of 115.4(2)° measured in 3,3’-
diindolylmethane.10

45
Interestingly, coupling of the indolyl substituents does produce a compression of
almost 6° upon the <C2-P1-C12 bond angle of 94.36(9)° in (N-F5Bn)2-4 compared with
that of 100.3(1)° measured between the uncoupled indolyl substituents of 3. This is not
considered to result in significant ring strain to the phosphacycle because of the facility
with which phosphorus subtly changes hybridization in order to accommodate
substituents of differing electronic or steric requirements. These changes in <CPC bond
angles are undeniably reflected in the Σ{<CPC} value for (N-F5Bn)2-4, which will be
discussed in greater detail in Section 2.3.
In the solid state, the pentafluorobenzyl groups of (N-F5Bn)2-4 adopt
conformations in which they are oriented in the same direction as the phenyl substituent
on the phosphorus atom. The fluorinated benzyl groups additionally exhibit a slipped
pseudo-eclipsed π-stacking conformation with the lone phenyl substituent of P1.
Distances between ring centroids of 3.547Å and 3.805Å, and an angle of 146° described
by them suggest the near-planar π-stacking of the three rings.
2.3. Examining the Σ{<CPC} of 2-Indolylphosphines
The sums of the <CPC bond angles from crystallographic data will be used to
asses the steric crowding around the phosphorus atoms of each of the 2-
indolylphosphines, and how the alkylation of an indolyl nitrogen centre will affect the
Σ{<CPC} values. A possible correlation with Σ{<CPC} and the 31P signal of a
phosphine will also be investigated. The Σ{<CPC} values are listed in Table 2.8.

46
Table 2.8. Sum of <CPC bond angles (o) and 31P resonances (ppm) for unfunctionalized and alkylated 2-indolylphosphines.
Entry
Complex
<C2-P1-C12
<C12-P1-C22
<C22-P1-C2
avg. Σ{<CPC}
31P δ
1
1
HN
P
99.94(8)b
101.94(9)c
102.97(8)b
101.60(9)c
101.64(9)b
100.43(9)c
304.6(1)b
304.0(2)c
-33.1
2
2 HN
P
103.17(9)
104.13(8)
101.12(9)
308.4(2)
-26.9
3
3
NH
PHN
100.3(1)
101.3(1)
102.2(1)
303.8(2)
-59.3
4
5
NH
P
HN
HN
102.3(1)
102.3(1)
102.3(1)
306.9(2)
-81.7
5
(N-Bn)-1
NP
101.0(1)
103.60(9)
103.24(9)
307.8(2)
-32.0
6
(N-F5Bn)2-4
NP
N
F
FF
FF
F F
F
FF
94.36(9)
101.63(9)
103.20(9)
299.2(2)
-30.8
aSpectra collected in CDCl3. b<C-P-C of 1a in asymmetric unit. c<C-P-C of 1b in asymmetric unit.
The average Σ{<CPC} for 1 was calculated to be 304.3(2)o from both
independent molecules in the asymmetric unit (the contrast between their individual
Σ{<CPC} was less than 3σ indicating no crystallographic distinction in their bond angles
around the phosphorus), which would suggest that the steric crowding around the
phosphorus is less than that of PPh3 (307.17o) and implies that the basicity of the lone
pair on phosphorus in 1 is correspondingly less donating than that of PPh3.

47
The average Σ{<CPC} for 2 was determined to be 308.4(2)o implying less steric
bulk around the phosphorus atom than PCy3 (311.43o), but slightly more steric crowding
than PPh3 (307.17o) and 1 (304.3(2)o). The increase in steric crowding of 2 in
comparison to 1 is most likely due to the change in substituents on phosphorus. The
bulkier cyclohexyl moieties would in principle cause the <CPC in 2 to “spread out” to
relieve steric crowding in contrast to the planar phenyl rings of 1. The basicity of the
phosphorus lone pair should change accordingly to the change in P-substituents, it is
reasonable to expect the donating ability of 2 to be greater than 1.
The average Σ{<CPC} for 3 was determined to be 303.8(2)o and while it appears
to be slightly less than the corresponding Σ{<CPC} for 1 (304.3(2)o), they are
crystallographically the same with a difference less than 2σ. The parity of the Σ{<CPC}
values of 1 and 3 is somewhat surprising since the exchange of one phenyl substituent for
an indolyl substituent appeared to have little to no effect on the sum of the <CPC. In the
case of 3, the lower than expected Σ{<CPC} may be due to the orientation of the three
aryl rings in the solid-state structure and perhaps to a lesser extent, crystallographic
packing forces. The two methyl groups at the C3 and C13 positions on the indolyl rings
are almost perpendicular in space with respect to each other, and this rotation of the
indolyl rings is possibly to avoid steric interactions between the two indolyl rings. Due to
the comparability of the Σ{<CPC} values for 1 and 3, one might predict the donating
ability of the phosphorus lone pair to be alike.
The Σ{<CPC} for 5 was calculated to be 306.87(24)o, a value that is larger than
the Σ{<CPC} of 1 (304.3(2)o) and 3 (303.8(2)o) but slightly less than PPh3 (307.17o).
The increased value of Σ{<CPC} of 5 is plausible due to the replacement of all of the
phenyl rings for indolyl rings and the orientation of the indolyl rings in the solid-state can
cause a “spreading out” of the phosphorus substituents. What is interesting to note, is
that the Σ{<CPC} of 5 is approaching the value of Σ{<CPC} of PPh3, which suggests
that the steric crowding around the phosphorus atoms in both phosphines is relatively
similar, and as such the basicity of the phosphorus lone pair should also reflect this
likeness.

48
The Σ{<CPC} value for (N-Bn)-1 was determined to be 307.8(2)o, and is larger
than the Σ{<CPC} value for 1 presumably due to the introduction of the benzyl group at
the nitrogen centre. The greater steric crowding seen in (N-Bn)-1 from the Σ{<CPC}
determination is complementary to the description of the size of the benzylated-
methylindolyl substituent discussed in Section 2.2.4.
Analysis of the Σ{<CPC} in (N-F5Bn)2-4 shows that the coupled indolyl
substituent causes a consequential decrease of this value to 299.2(2)o. Interestingly the
average Σ{<HPH} for PH3 is 280.8o, the geometry about the phosphorus atom is
generally regarded as highly pyramidal and the molecule as having almost perfect
tetrahedral bond angles.11 The Σ{<CPC} for (N-F5Bn)2-4 might suggest that the
coupling of the indolyl moieties to create one bidentate substituent on phosphorus has
caused the phosphorus atom in the N-alkylated derivative of 4 to become more
pyramidal.
Upon closer inspection, it seems there is little correlation with the Σ{<CPC} and
the 31P chemical shifts of the 2-indolylphosphines. While the Σ{<CPC} values do show
some relationship with phosphine basicity (as will be discussed in Chapter 4), the 31P
chemical shifts appear to be more sensitive to the type of substituent on the phosphorus
atom.
2.3.1. Examining Indolyl Aromaticity in 2-
Indolylphosphines
Two methods were employed to determine if the introduction of a phosphino
substituent to the C2-position on indole would affect the aromaticity of the indole,
particularly the five-membered pyrrolyl moiety. The first method examined the bond
distances that make up the pyrrolyl moiety of the indolyl ring of 2-indolylphosphines and
compared to those listed in Table 2.3. The second method to examine pyrrolyl
aromaticity was to define a least-squares mean plane by the five atoms making up the
pyrrolyl ring: N1-C2-C3-C4-C9, then compute the root mean squared (rms) deviation of

49
the five atoms in that plane. If any of the atoms of the pyrrolyl ring diverged appreciably
from the plane of the pyrrolyl ring, this deviation would suggest a loss of aromaticity in
the five-membered ring.
It was found that the distances in 1 were comparable to the analogous bond
distances in indoles from the CSD search. The likeness in bond distances would suggest
no loss of aromaticity in the pyrrolyl ring of indole upon phosphorus bonding at the C2-
position. The pyrrolyl ring in one of the crystallographically independent molecules of 1
has an rms deviation of 0.0044 Å, while the other independent molecule has an analogous
rms deviation of 0.0092 Å. The minor rms deviations of each of the pyrrolyl rings imply
planarity of the five atoms that make up the rings, and as such aromaticity is retained.
Comparing the bond distances of the pyrrolyl ring in 2 and the analogous bond
distances listed in Table 2.3 shows that the five-membered ring appears to be structurally
unaffected by the incorporation of a phosphorus at the C2-position. The rms deviation of
the five atoms in the pyrrolyl ring was calculated based on the plane described by the
same ring and found to be 0.0035 Å, suggestive of ideal planarity.
As with 1, the bonding of the phosphorus atom at the C2-positions on indolyl of 3
do not impose chemically significant changes in the structural features of the pyrrolyl
rings in its 3-methylindolyl substituents. The rms deviation of the five atoms in each of
pyrrolyl rings were found to be 0.0047 Å and 0.0108 Å with respect to the planes
described by the pyrrolyl rings, however the difference in the rms deviation values bear
no chemical significance, and the indolyl substituents of 3 are entirely aromatic.
The bond distances of the pyrrolyl moiety in the indolyl substituent of 5 were
comparable to the corresponding distances listed in Table 2.3, also an rms deviation of
0.0037 Å was calculated for the five atoms in the pyrrolyl ring with regard to the plane
described by the same ring. These properties are consistent with the aforementioned 2-
indolylphosphines as aromatic substituents. As demonstrated in these other 2-
indolylphosphines, the installation of a phosphorus atom at the C2-position on indole
does not perturb the aromaticity of the pyrrole moiety of the indolyl substituents.
For phosphine (N-Bn)-1, neither the bonding of the phosphorus atom at the C2-
position of the indolyl substituent, nor the installation of an alkyl group on the indolyl
nitrogen, display any structural changes that would suggest a departure from aromaticity.

50
Furthermore, the rms deviation of the atoms in the pyrrolyl ring with respect to the plane
described by the pyrrolyl ring was computed to be 0.0043 Å, consistent with the indolyl
substituent remaining aromatic with substituents at N1- and C2-positions.
The solid-state structure of (N-F5Bn)2-4 reveals that the introduction of the
electron withdrawing pentafluorobenzyl substituents imparts little structural change upon
the rest of the molecule. For example, the bond distances of the pyrrolyl moieties in the
coupled indolyl substituent are comparable to the bond distances listed in Table 2.3.
Additionally, the rms deviation of the atoms in the pyrrolyl rings with respect to the plane
described by the pyrrolyl rings were determined to be 0.0035 Å and 0.0046 Å, consistent
with the indolyl substituent remaining aromatic with substituents at N1- and C2-positions
as seen in phosphine (N-Bn)-1.
2.4. Conclusions
In the multiple derivatives of 2-indolylphosphines, the presence of the phosphorus
and the introduction of an alkyl group on the indolyl nitrogen do not seem to impart any
structural changes in pyrrolyl ring that would alter the aromaticity of this ring. The rings
remain planar and the five bond distances that make up the pyrrolyl moiety are consistent
with the CSD search on aromatic indolyl compounds. For the unsubstituted 2-
indolylphosphines, it was found that the methyl group on the indolyl substituents
preferentially crystallized pointing towards the phosphorus lone pair and away from the
pyramid created by the three substituents on phosphorus. In contrast, the methyl group
would point towards the pyramid in the crystal structures of the N-alkylated 2-
indolylphosphines, possibly to reduce the strain within the pyramid created by the
presence of the alkyl group on the indolyl nitrogen.
While the cone angles of these 2-indolylphosphines could not be determined
through the crystal structure data available, the steric crowding of the phosphorus atom
and therefore the relative size of the phosphine may be discussed using Σ{<CPC} values.
The Σ{<CPC} values for the unsubstituted 2-indolylphosphines exhibited a small
relationship where replacing the number of phenyl substituents with indolyl ones resulted

51
in a slight increase in Σ{<CPC}. The Σ{<CPC} did not change appreciably from DPIP
to PDIP, and it may have been due to crystal packing effects. However, as expected, the
exchange of phenyl substituents for bulkier cyclohexyl ones did increase the Σ{<CPC}
drastically from 1 to 2. The Σ{<CPC} calculated for (N-Bn)-1 was considerably larger
than 1, which follows that the introduction of a substituent on the indolyl nitrogen results
in greater steric crowding of the phosphorus lone pair. Contrary to (N-F5Bn)2-4, the
coupled indolyl rings are effectively one substituent on phosphorus that occupies two
coordination sites, and as such the Σ{<CPC} will be smaller for the coupled indolyl
substituent than the analogous uncoupled 2.
In (N-F5Bn)2-4, Additionally, the atoms of the pyrrolyl rings were found to have
rms deviations from the plane described by the pyrrolyl rings of 0.0035 Å and 0.0046 Å,
in agreement with pyrrolyl aromaticity.
2.5. Experimental
Unless otherwise stated, the synthetic protocol for 2-indolylphosphines are reported in
Dr. Edmond Lam’s thesis.8
General Considerations. All reactions and manipulations were carried out under an
atmosphere of dinitrogen using standard Schlenk techniques unless otherwise stated.
PCl3, 1.6 M n-BuLi, NaBH4 were purchased from Aldrich and used as received. 1-[(N,N-
Dimethylamino)methyl]-3-methylindole12 was synthesized according to literature
procedures. THF was distilled from sodium benzophenone ketyl under a dinitrogen
atmosphere. 1H, 13C, and 31P NMR spectra were recorded on Varian 400 MHz or 300
MHz NMR systems, and referenced to SiMe4 (TMS) and 85% H3PO4, respectively.
Splitting patterns are indicated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet;
br, broad peak.

52
PNH NH
HN
Tris-2-(3-methylindolyl)phosphine, P(C9H8N)3, 5. 1-[(N,N-Dimethylamino)methyl]-3-
methylindole (4.25 g, 22.6 mmol) was dissolved in THF (60 mL) and cooled to -78oC. n-
BuLi (17.0 mL, 27.2 mmol) was added dropwise over 10 minutes. The reaction was
stirred at -78oC for 10 minutes, warmed to ambient temperature and stirred for 3 hours.
The resulting orange mixture was cooled to -78oC, and PCl3 (0.66 mL, 7.56 mmol) was
added dropwise over 5 minutes. The mixture was allowed to warm to ambient
temperature over 5 hours and then quenched with MeOH. Solvents were removed in
vacuo to yield a yellow solid. Water (100 mL) was added to the solid, and the suspension
extracted with DCM (3 x 100 mL). The organic layers were combined, dried over
anhydrous MgSO4, filtered, and concentrated in vacuo to afford dark orange oil. MeOH
was added to triturate pure aminal-protected product as a white solid, which was isolated
by filtration and dried in vacuo (2.09 g, 47%). Mp: 122oC; 31P NMR (CD2Cl2,
121 MHz): δ -71.3 (s); 1H NMR (CD2Cl2, 300 MHz): δ 7.48 (d, J = 8.1 Hz, 6H, Ar-H),
7.20 (t, J = 7.4 Hz, 3H, Ar-H), 7.07 (t, J = 7.4 Hz, 3H, Ar-H), 4.78 (d, J = 12 Hz, 3H,
amine NCH2), 4.62 (d, J = 12 Hz, 3H, anime NCH2), 2.15 (s, 18H, amine N(CH3)2), 1.84
(s, 9H, indole CH3); 13C NMR (CD2Cl2, 75 MHz):.139.54, 130.14, 126.86, 123.08,
121.39, 119.64, 119.08, 111.06, 67.92, 42.86, 9.11; HRMS EI for C36H45N6P: calcd m/z
592.344334, found m/z 592.344809; Microanalysis for C36H45N6P: calcd (%) C = 72.94,
H = 7.65, N = 14.18, found (%) C = 72.56, H = 7.57, N = 13.78. A solution of
THF/EtOH (120 mL, 1:1 v/v) was added to a solid mixture of the aminal-protected
product (3.75 g, 6.33 mmol) and NaBH4 (1.50 g, 39.7 mmol). The mixture was refluxed
for 5 hours. The solution was reduced to dryness in vacuo and acidified water was added
to the resultant white solid, which was extracted with DCM (3 x 100 mL). The organic
phases were combined and dried over anhydrous MgSO4, filtered and taken to dryness to
afford a dark yellow coloured oil. MeOH was added to triturate pure product as a white
solid, which was isolated by filtration and dried in vacuo (1.84 g, 69 %). Mp: 166oC; 31P

53
NMR (CD2Cl2, 121 MHz): δ -81.7 (s); 1H NMR (CD2Cl2, 300 MHz): δ 7.90 (br s, 3H,
indole NH), 7.60 (d, J = 7.2 Hz, 3H, Ar-H), 7.27 (d, J = 7.5 Hz, 3H, Ar-H), 7.21 – 7.12
(m, 6H, Ar-H), 2.41 (s, 9H, indole CH3); 13C NMR (CD2Cl2, 75 MHz): 138.94, 130.29,
125.53, 123.69, 121.34, 120.17, 119.54, 111.80, 9.90; HRMS EI for C27H24N3P: calcd
m/z 421.170357, found m/z 421.170786; Microanalysis for C27H24N3P: calcd (%)
C = 76.94, H = 5.74, N = 9.97, found (%)C = 76.70, H = 5.78, N = 9.79.
X-ray Crystallography. X-ray data were collected on a Nonius Kappa CCD
diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). A
combination of 1º φ and ω (with κ offsets) scans were used to collect sufficient data. The
data frames were integrated and scaled using the Denzo-SMN package.13 The structures
were solved and refined with the SHELXTL-PC v6.12 software package.14 Refinement
was by full-matrix least squares on F2 using data (including negative intensities) with
hydrogen atoms bonded to carbon and nitrogen atoms included in calculated positions
and treated as riding atoms. Absorption corrections were made for every structure. All
the heavy atoms were refined anisotropically.

54
2.6. References
(1) Tolman, C. A. Chem. Rev. 1977, 77, 313 - 348.
(2) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029-3070.
(3) Muller, T. E.; Mingos, D. M. P. Transition Met. Chem. 1995, 20, 533 - 539.
(4) Bartik, T.; Himmler, H.-G.; Seevogel, K. J. Organomet. Chem. 1984, 272, 29 -
41.
(5) Tolman, C. A. J. Am. Chem. Soc. 1970, 90, 2953 - 2956.
(6) Boere, R. T.; Zhang, Y. J. Organomet. Chem. 2005, 690, 2651 - 2657.
(7) CSD Database (version 5.29, January 2008).
(8) Lam, E., PhD Thesis, University of Toronto, 2007.
(9) Kondo, S.-i.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005,
1720-1722.
(10) Rampersad, N. C., MSc Thesis, University of Toronto, 2001.
(11) Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements; 2nd ed.;
Butterworth-Heinemann: Oxford, 1997.
(12) Katritzky, A. R.; Lue, P.; Chen, Y.-X. J. Org. Chem. 1990, 55, 3688-3691.
(13) Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307-326.
(14) Sheldrick, G. M. In SHELXTL-Windows NT. V6.12, Bruker Analytical X-Ray
Systems Inc. Madison, WI, 2001.
54

Chapter 3
Tris-2-(3-methylindolyl)phosphine:
Synthesis, Reactivity and Anion Binding
3.1. Introduction
Electroneutral anion recognition hosts have suitable NH donors to bind anion
guests, and are commonly based on small molecules with organic scaffolds that usually
incorporate a combination of amides,1-4 pyrroles,5-8 and / or ureas.9-12 The acidity of the
hydrogen bond donors can be increased in order to improve anion binding, often through
the incorporation of electron-withdrawing substituents on the receptor.13,14 Another
method to increase the acidity of a hydrogen bond donor is to fuse a pyrrole ring to a
benzene ring, which creates an indole moiety; this changes the pKa of the NH from 23.0
in pyrrole to 20.9 in indole (in DMSO).15 Interestingly, indole-based hosts have only
recently been investigated, and they have demonstrated anion binding capability and high
selectivity for various anions (refer to Chapter 1 for a detailed review).
Only several reports of phosphines acting as anion receptors have been reported,
where the phosphorus centres are situated more than two bonds away from the hydrogen
bond donor sites.10-12,16 In these examples the phosphorus centre appears to provide a
metal coordination site, and provide precedence for simultaneous coordination at the
phosphorus site while the receptor is engaged in hydrogen bonding to anion guests.
55

56
3.1.1. Hydrogen Bonding in 2-Indolylphosphines
The palladium chloride dimer complexes of the mono- and di-indolylphosphines
have demonstrated the propensity to form intramolecular (Figure 3.1a) as well as
intermolecular hydrogen bonds (Figure 3.1b) in their solid-state structures, which will be
discussed in greater detail in Chapter 4.3 The C3-symmetric phosphine 5, was
synthesized to investigate whether the three indolyl NH sites are suitably positioned to act
collectively to form strong hydrogen bonding interactions with anions. Its phosphorus
centre serves as an additional site of reactivity through which the receptor may, in
principle, simultaneously coordinate to transition metals or other Lewis acids. The close
spatial proximity of the phosphorus centre to the NH moieties in 5 lies in contrast to
phosphines which have previously been reported to act as anion receptors in which the
phosphorus centres are well-displaced from the sites of hydrogen-bonding.10-12,16
(a) (b)
Figure 3.1. (a) Intramolecular hydrogen bonding in the [Pd(1)Cl(μ-Cl)]2 between N1 and Cl2 [3.109(3) Å].
(b) Hydrogen bonding in the [Pd(4)Cl(μ-Cl)]2·H2O complex: intramolecular hydrogen bonding between
N11 and Cl2 [3.243(3) Å], and intermolecular hydrogen bonding between N1 and O1 [2.941(4) Å].

57
In this Chapter, symmetric phosphine 5 will be examined for anion binding
capability through a series of anion titrations. Based on the utility of 5 to bind anions, a
new indolylphosphine-based anion receptor is designed, and the attempted synthesis of
this new phosphine molecule will be presented. Furthermore, a molecular cleft based on
two indole rings separated by a rigid phenyl linker, will be investigated for its anion
binding ability.
3.2. Results and Discussion
3.2.1. Synthesis of Tris-2-(3-methylindolyl)phosphine
Lithiation of indole preferentially occurs at the nitrogen or at the C3-position; by
controlling the reactivity at these more vulnerable centres with protecting groups the
desired lithiation at the C2-position is achieved. The C3-position is easily blocked from
reactivity by the introduction of a methyl group, and this convenient 3-methylindole
starting material is commercially available. Several protecting groups commonly used
for amine protection have been investigated and it was determined that the N,N’-
dimethylaminomethylene protecting group is the most suitable for our purpose as it
directs lithiation to the C2-position on indole. Following the work of Katrizky and co-
workers, the N,N’-dimethylaminomethylene moiety can be introduced easily by refluxing
3-methylindole with formaldehyde and dimethylamine in water to afford the protected 3-
methylindole in quantitative yield following aqueous work-up.17 Lithiation of indole is
achieved at the C2-position with n-BuLi at -78oC, and the subsequent addition of
trichlorophosphine leads to chloride substitution by the indolyl groups to provide the
desired aminal-protected phosphine in 47% yield after reacting for 24h. The 31P NMR
spectrum of aminal-protected 5 exhibited one signal indicating a single phosphorus
environment at δ = -71.3 ppm in CD2Cl2, and in the 1H NMR the presence of the NMe2
resonances for the protecting group attached to the indole nitrogen.

58
The N,N’-dimethylaminomethylene aminal protecting group can be
straightforwardly removed upon reaction of the protected phosphine with NaBH4 in a
THF / EtOH reflux. After successive aqueous work-up, phosphine 5 is afforded in 32%
yield from trituration with MeOH (Scheme 3.1). The 31P NMR spectrum of 5 exhibited
only one phosphorus environment at δ = -81.7 ppm in CD2Cl2; the 1H NMR spectrum
showed the absence of the aminal protecting group and the emergence of an indole NH
proton resonance at δ = 7.87 ppm in CD2Cl2.
N
N
N
P
N
1. n-BuLi2. PCl3
THF-78oC
1. NaBH4EtOH/THF
reflux2. HCl workup
47% overall 32%
N NN
N
HN
P
HN NH
Scheme 3.1. The synthesis of 5 from the aminal-protected 3-methylindole.
An alternative one-pot synthesis of ligand 5 can be carried out by using CO2 as a
protecting agent for the indolyl nitrogen18 that is also capable of directing lithiation to the
C2-position on indole. The indolyl NH proton of 3-methylindole is deprotonated with n-
BuLi; subsequent bubbling of the reaction with a stream of CO2 gas furnishes the
protecting group onto the nitrogen centre. The C2-position on the CO2-protected indole
is deprotonated with t-BuLi, and addition of trichlorophosphine generates the CO2-
protected derivative of phosphine 5. The CO2 protecting group is conveniently cleaved
upon aqueous workup of 5 (Scheme 3.2). Phosphine 5 is easily recovered by trituration
from MeOH in yields that are comparable to the aforementioned synthetic procedure.

59
1. n-BuLi2. CO2
THF, -78oC
HN
N
OOLi 1. t-BuLi
2. PCl3THF, -78oC3. Aqueous
workup
NH
P
HN
HN
39% Scheme 3.2. The synthesis of 5 from 3-methylindole with CO2 as a protecting group for the indole
nitrogen.
3.2.2. Anion Binding Properties of Tris-2-(3-
methylindolyl)phosphine
It is evident from the crystal structure of 5 that the indolyl substituents are
arranged such that the indolyl NH functionalities are in proximity to form the necessary
hydrogen bond donor cavity (Figure 3.2). Refer to Chapter 2 for a detailed discussion on
the solid-state structure of 5. 1H NMR titration techniques with anions will provide
information on the extent to which 5 can function as an anion receptor.
Figure 3.2. Bowl shaped hydrogen bonding cavity of 5. Selected bond angle: <C2-P1-C2 102.3(1)o,
Σ{<CPC} = 306.9(2)o.
1H NMR titration techniques in CD2Cl2 were employed to assess the anion binding
capability of 5 with selected anions as their tetrabutylammonium salts (Scheme 3.3).

60
PNH NH
HNCD2Cl2
+ NBu4X P
HN
3
X
NBu4
Scheme 3.3. General titration scheme of 5 with anions as their tetrabutylammonium salts in CD2Cl2.
Selected anions: X- = Cl-, Br-, CH3COO-, HSO4-, NO3
-, and BF4-.
The interaction of 5 with anions was monitored by the increasing downfield shift of the
indolyl NH resonances (δ = 7.87 ppm for the uncomplexed 5) with increasing anion
concentration until saturation was obtained. Figure 3.3 shows a general compilation of 1H NMR spectra from a typical titration experiment using the Cl- anion. The ambient
temperature 1H NMR titrations reveal that the hydrogen bonding interactions between 5
and the anion are fast on the NMR time scale, suggested by only one resonance being
observed for the indolyl NH sites. In general, the larger the downfield chemical shift of
the protons being monitored at any point during the titration, the stronger the binding to
that particular anion.

61
NH
0 eq
0.5 eq
1 eq
2 eq
4 eq
ppm (t1)7.08.09.010.011.012.013.0
6 eq
Figure 3.3. A representative example of partial titration stack plots for the addition of Cl- anion to 5 in
CD2Cl2 at 298K.
In all cases the data could be fitted to a 1 : 1 receptor : anion binding model by means
of the EQNMR software19 to obtain the stability constants listed in Table 3.1.
Figure 3.4 is a fit plot illustration for the titration of 5 with Cl-, it is a representative
example of the saturation curve for the titration experiment. A minor amount of 2 : 1
receptor : anion binding was observed at low concentrations of strongly coordinating
anions (Cl-, Br-, CH3COO-). The fit plots for the remainder of the titration
experiments are deposited in the Experimental section of this chapter. The errors
associated with the stability constants were determined by the fit of the experimental
curve to the calculated curve, and the overall errors computed as the quotient between
the error in Ka and the value of Ka. The fit of the acetate anion titration curve
(Figure 3.18) is imperfect even though the error on the stability constant is
approxiamtely 4%. The poorer fit is most likely due to the interaction of additional
hydrogen atoms on the indolyl moiety with the acetate anion (Figure 3.7). The errors

62
associated with the iodide, hydrogensulfate, and tetrafluoroborate are expected to be
higher since these titration experiments do not appear to reach a saturation point.
Thus, the stability constants for these experiments may not be entirely accurate.
Figure 3.4. Fit plot for titration experiment of 5 with tetrabutylammonium chloride in CD2Cl2.
Table 3.1. Stability constants Ka (M-1) of 5 with a selection of anion guests as determined by 1H NMR
titration techniques performed at 298 K following NH resonances in 5.a
Anion Ka Error in Ka Overall error (%)
Cl- 3920 +/- 86 2.2
Br- 320 +/- 0.5 0.2
I- 20 +/- 0.3 1.5
CH3COO- 2730 +/- 120 4.4
HSO4- 35 +/- 0.6 1.7
NO3- 100 +/- 0.4 0.4
BF4- 150 +/- 14 9.3
The symmetry and position of the NH groups of the receptor provide a well-
defined hydrogen bonding cavity of geometry and size suitable for binding well to
spherical anions such as halides, in particular to Cl- as it has the largest stability constant.
The high charge densities on the oxygen atoms of CH3COO- are likely responsible for the
strong interaction that it exhibits with 5. While the NO3-, HSO4
-, and BF4- anions provide
an improved complement in symmetry to the hydrogen bond cavity of receptor 5, the

63
reduced ability of 5 to bind these anions may be due to their diffuse singly negative
charges being delocalized over several terminal atoms.
The stability constants of 5 are relatively modest (considering the anion
binding studies are carried out in dichloromethane), in comparison to most indole-based
receptors reported in the literature, which are predominantly studied in more polar
solvents such as DMSO or acetonitrile.20-27 Phosphine 5 does show better binding to
chloride anion (Ka = 3920 M-1) than the two diindolylquinoxaline-based anion receptors
reported by Sessler and co-workers (Ka = 170 M-1 for the unfunctionalized receptor, and
250 M-1 for the nitro functionalized receptor) (Figure 3.5 and Table 3.9).28 It is suspected
that the better binding to chloride seen in 5 is most likely due to the additional hydrogen
bond donor that is not present in Sessler’s two diindolylquinoxaline-based receptors.
N N
NH
NH
NO2
N N
NH
NH
(a) (b) Figure 3.5. Sessler and co-workers’ diindolylquinoxaline-based anion receptors. (a) Unfunctionalized
bisindole. (b) Nitro functionalized bisindole.28
Single crystals of the [NEt4][5·CH3COO] complex, 6, were grown from slow
evaporation from a MeOH/CH2Cl2 solution of the complex, and X-ray diffraction
experimental data are listed in Table 3.2. Complex 6 crystallizes in the monoclinic space
group P21/n, with two independent sets of molecules in the asymmetric unit (Figure 3.6).
Selected bond distances and angles are listed in Table 3.3. One molecule of
uncomplexed 5 concomitantly crystallizes with one complex of 6; the NEt4+ cation is
disordered over two sites with the major component modeled at 60% occupancy and the
minor component at 40% occupancy; two methanol solvent molecules are hydrogen
bonded to the acetate anion.

64
Figure 3.6. ORTEP diagram of one of the two independent sets of molecules in the asymmetric unit of
complex 6 with concomitant uncomplexed 5. Non-acidic protons removed for clarity. Disordered NEt4+
cation removed for clarity. Thermal ellipsoids drawn at 35%.
The crystal structure of 6 confirms the 1 : 1 receptor to anion binding ratio
determined through 1H NMR titration techniques. The acetate anion hydrogen bonds to 5
through one of its oxygen atoms, O1A, with typical hydrogen bonding distances in the
range N…O- of 2.817(2) Å to 2.927(2) Å; coupled with the hydrogen bond angles
<NH…O- (range from 155.8o to 172.7o) these can be considered of moderate interaction
strength.29 An additional interaction from C18B to O2A can be seen in Figure 3.7, and is
thought to give rise to the poorer fitting of the titration plot of acetate with phosphine 5
(Figure 3.18). While the interaction between C18B and O2A is not considered a
hydrogen bond (the 3.488(3) Å distance is longer than accepted hydrogen bond lengths),
presumably it contributes to the binding strength of phosphine 5 to the acetate anion.

65
Figure 3.7. ORTEP diagram illustrating the hydrogen bonding in complex 6. Only selected protons are
shown for clarity. Disordered NEt4+ cation, and concomitantly crystallized phosphine 5 removed for
clarity. Thermal ellipsoids drawn at 35%.
Two methanol solvates are seen to hydrogen bond to the other oxygen atom, O2A,
of the acetate anion. The C-O bond lengths of the acetate anion are indistinguishable
from each other (C1AA-O1A 1.264(3) Å and C1AA-O2A 1.257(3) Å), indicating
delocalization of the C-O double bond even in the presence of hydrogen bonding to 5. It
is interesting to note that while the possibility exists for the acetate anion to hydrogen
bond to 5 in two modes (such as both oxygen atoms hydrogen bonding to the cavity), the
acetate anion preferentially coordinates to 5 via one oxygen atom; a result of all three
indolyl NH sites collectively engaged in hydrogen bonding and possibly a small
hydrogen bond donor cavity size.
While the acetate anion does not appear to impart important structural changes in
the bond lengths and angles of 5, the Σ{<CPC} value for 6 was determined to be
313.2(2)o, and is larger than the corresponding Σ{<CPC} value for 5. The concomitantly
crystallized uncomplexed 5 moiety has a Σ{<CPC} value of 305.0(2)o, and is comparable
to the crystal structure of 5 (306.9(2)o) as discussed in Chapter 2. The increase in the
Σ{<CPC} value in 6 may be due to the necessary expansion of the <CPC bond angles to
accommodate a hydrogen bonded guest; it suggests that hydrogen bonding to acetate does
not pull the indolyl substituents together but rather forces them apart. The uncomplexed
5 in the 6 structure additionally hydrogen bonds to the two methanol solvent molecules

66
that are hydrogen bonding to an acetate anion of an adjacent asymmetric unit with
ordinary hydrogen bond donor-to-acceptor distances (2.856(3) Å to 3.106(2) Å), and
hydrogen bond angles <NH…O- (141o to 145o). While there is considerable hydrogen
bonding among the components of the unit cell, it does not extend beyond the unit cell.
While the absence of a clean fit to a 1 : 1 receptor : anion binding model
precluded an accurate determination of its stability constant, the 1H NMR of 5 in the
presence of F- anion exhibited the largest downfield change in chemical shift of the
indolyl NH signals (ca. 6.7 ppm), suggesting a very strong interaction with them.30 This
is consistent with the well-known propensity of F- to serve as a strong hydrogen bond
acceptor. To confirm the interaction between F- and 5, single crystals of the [5·F]-
complex, as its tetraethylammonium salt, 7, were obtained from a solution of the complex
in a MeCN / MeOH mixture layered with Et2O. The X-ray diffraction experimental data
are shown in Table 3.2, whereas Table 3.3 lists selected bond distances and angles. The
numbering scheme of complex 7 is shown in Figure 3.8.

67
Table 3.2. X-ray crystallographic experimental data of [5·X]- complexes.
6
P
HN
3
O
NEt4
O
7
P
HN
3
F
NEt4
8
P
HN
3
I
CH3+
Formula C66H79N7O4P2 C73H100F2N8O3P2 C32H37IN3OP Formula Weight 548.15 1237.55 637.52 Crystal colour, shape colourless, block colourless, block colourless, plates Crystal size, mm 0.38 x 0.14 x 0.10 0.58 x 0.30 x 0.20 0.24 x 0.14 x 0.08 Crystal system Monoclinic Orthorhombic Monoclinic Space group P21/n P212121 P21/c a, Å 15.2163(2) 11.3758(1) 10.4385(5) b, Å 20.6034(3) 24.2008(2) 22.9652(14) c, Å 20.4701(3) 24.9237(2) 12.8667(7) α deg 90 90 90 β, deg 111.1640(10) 90 91.542(4) γ deg 90 90 90 V, Å3 5984.67(15) 6861.6(1) 3083.3(3) Z 4 4 4 Dcalcd, g cm-3 1.217 1.198 1.373 F(000) 2344 2664 1304 μ, mm-1 0.127 0.121 1.119 λ (Mo Kα), Å 0.71073 0.71073 0.71073 Limiting indices -19<=h<=19 -11<=h<=14 -13<=h<=13 -24<=k<=26 -31<=k<=31 -29<=k<=29 -26<=l<=26 -31<=l<=32 -13<=l<=16 2θ range, deg 2.85 to 27.48 2.57 to 27.55 1.77 to 27.50 Max. and min. transmission 1.008 and 0.479 0.998 and 0.591 0.899 and 0.490 No. of reflections collected 55668 46395 21801 No. of independent reflections/Rint 13676 /0.0971 15463 / 0.0807 7160 / 0.1133 Absolute structure parameter - 0.01(7) - Extinction coefficient 0.0028(3) 0.0014(5) 0.0029(5) No. of refined parameters 763 811 350 Final R1, wR2 0.0558, 0.1231 0.0532, 0.1278 0.0713, 0.1497 Final R1, wR2 (all data) 0.1072, 0.1470 0.0707, 0.1389 0.1775, 0.2038 Goodness of fit 1.022 1.056 1.057 Δρmin, Δρmax, eÅ-3 -0.411, 0.372 -0.345, 0.572 1.490, -0.921

68
Complex 7 crystallizes in the orthorhombic space group P212121 with two
independent complexes, 7a and 7b, and three methanol solvate molecules in the
asymmetric unit (Figure 3.8). The solid-state structure confirms the interaction between
fluoride and 5 to be a 1 : 1 anion to receptor stoichiometry. Phosphine 5 exhibits
hydrogen bonding to the fluoride with all three of the indolyl NH sites in both complexes,
with typical hydrogen bond donor-to-acceptor distances that range from N…F- 2.632(3) Å
to 2.790(2) Å;31 along with the hydrogen bond angles <NH…F- (159.1o to 176.2o) these
interactions can be considered hydrogen bonds of moderate strength.29 The fluoride
anion does not appear to impart significant structural changes to the phosphine, and bond
lengths and angles are comparable to the uncomplexed 5. Correspondingly, the
aromaticity of the pyrrolyl ring is not affected by the hydrogen bonded fluoride anion.
Figure 3.8. ORTEP diagram of 7. Non-acidic protons removed for clarity. NEt4
+ cations removed for
clarity. Thermal ellipsoids drawn at 35% probability.
The fluoride anion of complex 7a hydrogen bonds to one MeOH solvate, and has a
distorted four-coordinate tetrahedral coordination geometry, with hydrogen bond donor-
to-acceptor distance and angle of 2.597(3) Å and 171.4o, respectively. The fluoride anion
of 7b hydrogen bonds to two MeOH solvent molecules, and has a distorted five-
coordinate geometry, bearing average hydrogen bond donor-to-acceptor distance and
angle of 2.669(4) Å and 178.3o, respectively. Despite the fact that there is extensive
hydrogen bonding between the components in the unit cell, the hydrogen bonding is
considered zero-dimensional and does not form an extended array.

69
Even though F- and CH3COO- strongly bind to 5, the hydrogen bonding distance
of the CH3COO- is slightly longer than the analogous interaction of 7 (N…F- 2.722(1) Å),
presumably due to the more compatible fit of the spherical halide to that of the bulkier
acetate. Compared to uncomplexed 5 (Σ{<CPC) = 306.9(2)o), the hydrogen bonding of
the fluoride anion results in an increase in the C-P-C bond angles of the phosphine as
seen in the Σ{<CPC} bond angles in 7, which are determined to be 311.6(2)o and
312.2(2)o for 7a and 7b, respectively. This suggests the necessity for 5’s <CPC bond
angles to “open up” in order to accommodate hydrogen bonding to a guest. Presumably,
the larger the guest, the greater is the increase in the Σ{<CPC} value. Therefore 6 with
its larger acetate guest has a larger Σ{<CPC} value than 7.
The titration of NBu4I with 5 indicated a 1 : 1 receptor : anion interaction, albeit a
weak one with a stability constant of 20 M-1. However, the reaction with
tetraethylammonium iodide with 5 did not yield crystals as the analogous reactions
between 5 and F- or CH3COO- did. However it was thought that the formation of a
phosphonium cation would increase indolyl NH acidity. The generation of the
phosphonium cation of 5 was carried out by reaction with a stoichiometric amount of
methyl iodide in THF (Scheme 3.4).
P
NH
HN
NH + CH3I THF
P
HN
CH3
3
I
Scheme 3.4. Reaction scheme to form [CH3-5]I, 8, from 5 and CH3I.
The 31P NMR spectrum of the [CH3-5]I species, 8, contains the presence of one signal
occurring at -12.1 ppm, indicating a single phosphorus environment that is shifted
downfield from 5 (δ -81.7 ppm) and is consistent with methylation at the phosphorus
centre.32 Similarly, the downfield chemical shift of the indolyl NH resonances in the 1H
NMR spectrum is also consistent with some degree of NH-iodide interaction. Single
crystals of 8 were obtained from Et2O vapour diffusion into a solution of CH2Cl2

70
containing the complex; the X-ray diffraction experimental data are listed in Table 3.2,
while selected bond angles and distances are listed in Table 3.3. Complex 8 crystallizes
in the monoclinic space group P21/c, with one [CH3-5]+ complex, one iodide anion, and a
hydrogen bonded Et2O solvate in the asymmetric unit. The complex forms a hydrogen
bonded dimer in the solid state, one half of which is related to the other by a
crystallographic 21 screw axis along the b-axis. Two iodide anions interact with two
[CH3-5]+ cations via symmetry equivalent indolyl NH sites (Figure 3.9).

71
Table 3.3. Selected bond lengths (Å) and bond angles (o) for [5.X]- complexes. 6
P
HN
3
O
NEt4
O
5 in crystal of 6
PNH NH
HN
7a
P
HN
3
F
NEt4
7b
P
HN
3
F
NEt4
8
P
HN
3
I
CH3+
P1-C2 1.817(2) 1.815(2) 1.814(3) 1.817(2) 1.767(7)
P1-C12 1.823(2) 1.805(2) 1.812(3) 1.819(2) 1.773(6) P1-C22 1.805(2) 1.821(2) 1.813(2) 1.808(2) 1.780(7)
<C2-P1-C12 103.2(1) 103.2(1) 101.9(1) 102.7(1) 110.2(3) <C12-P1-C22 105.6(1) 102.1(1) 106.2(1) 105.5(1) 108.2(3) <C22-P1-C2 104.5(1) 99.7(1) 103.5(1) 104.0(1) 111.1(3)
avg. Σ{<CPC} 313.2(2) 305.0(2) 311.6(2) 312.2(2) 329.5(5)
N1-C2 1.393(3) 1.387(3) 1.391(3) 1.389(3) 1.394(8) C2-C3 1.382(3) 1.378(3) 1.388(4) 1.382(3) 1.362(9) C3-C4 1.431(3) 1.435(3) 1.431(4) 1.437(4) 1.426(10) C4-C9 1.406(3) 1.412(3) 1.415(4) 1.416(4) 1.421(9) C9-N1 1.371(3) 1.375(3) 1.367(3) 1.368(3) 1.373(8)
<C9-N1-C2 109.3(2) 109.5(2) 109.6(2) 109.6(2) 108.6(5) <N1-C2-C3 108.4(2) 109.1(2) 108.5(2) 108.9(2) 109.6(6) <C2-C3-C4 107.2(2) 106.7(2) 106.9(2) 106.8(2) 107.3(6) <C3-C4-C9 107.2(2) 107.5(2) 107.2(2) 107.1(2) 107.0(6) <C4-C9-N1 107.9(2) 107.3(2) 107.8(2) 107.7(2) 107.5(6)
N11-C12 1.389(3) 1.392(3) 1.392(3) 1.400(3) 1.385(8) C12-C13 1.372(3) 1.382(3) 1.380(4) 1.376(4) 1.385(9) C13-C14 1.437(3) 1.436(3) 1.438(4) 1.431(4) 1.429(9) C14-C19 1.410(3) 1.410(3) 1.419(4) 1.407(4) 1.407(9) C19-N11 1.370(3) 1.368(3) 1.371(3) 1.367(3) 1.363(8)
<C19-N11-C12 109.3(2) 109.3(2) 109.2(2) 109.0(2) 108.8(5) <N11-C12-C13 108.8 (2) 109.0(2) 109.1(2) 108.7(2) 109.8(6) <C12-C13-C14 107.3(2) 106.5(2) 107.2(2) 107.1(2) 105.7(6) <C13-C14-C19 106.7(2) 107.4(2) 106.6(2) 107.2(2) 107.8(6) <C14-C19-N11 107.9(2) 107.8(2) 108.0(2) 108.1(2) 108.0(6)
N21-C22 1.394(3) 1.392(3) 1.404(3) 1.401(3) 1.388(9) C22-C23 1.382(3) 1.376(3) 1.373(3) 1.374(3) 1.370(9) C23-C24 1.435(3) 1.435(3) 1.442(3) 1.432(3) 1.432(9) C24-C29 1.410(3) 1.409(3) 1.407(3) 1.414(3) 1.396(9) C29-N21 1.369(3) 1.377(3) 1.362(3) 1.369(3) 1.381(8)
<C29-N21-C22 109.5(2) 109.3(2) 108.3(2) 108.8(2) 108.0(5) <N21-C22-C23 108.3(2) 108.9(2) 109.1(2) 108.9(2) 110.4(6) <C22-C23-C24 107.4(2) 107.0(2) 107.1(2) 107.3(2) 105.6(6) <C23-C24-C29 106.8(2) 107.3(2) 106.4(2) 106.9(2) 108.3(6) <C24-C29-N21 108.0(2) 107.5(2) 109.1(2) 108.1(2) 107.6(6)
N1 .. X 2.870(2) - 2.727(3) 2.726(3) - N11 .. X 2.817(2) - 2.716(3) 2.790(2) 3.427(5) N21 .. X 2.927(2) - 2.632(3) 2.711(2) 3.490(5)
<N1-H1-X 155.8 - 172.5 175.7 -
<N11-H11-X 163.8 - 159.1 168.6 167.6 <N21-H21-X 172.7 - 175.1 176.2 156.0

72
Figure 3.9. ORTEP diagram of symmetric dimer of 8 in the crystallographic ab-plane. Non-acidic protons
removed for clarity. Thermal ellipsoids are drawn at 35% probability.
The unsymmetrical intermolecular iodide to indolyl NH interaction distances
(3.427(5) Å and 3.490(5) Å) are in agreement with previously reported hydrogen bonds
to iodide.31 The indolyl NH interaction with iodide has <NH…I- bond angles (156.0o and
167.6o) suggesting near linear interactions. The Et2O solvent molecule hydrogen bonds
to one of the indolyl NH sites not already interacting with the iodide anion, with a typical
hydrogen bond distance of 2.950(7) Å. Three C-H…I- interactions contribute to the three-
dimensional arrangement of the molecules in the unit cell. The C…I- interaction distances
[3.978(7),33 3.970(7), and 3.794(8) Å34] are comparable to those found in the solid-state
structure of (benzoylmethyl)triphenylphosphonium iodide (C…I- range 3.696(2) to
3.983(2) Å).35 The Σ{<CPC} bond angle was found to be 329.5(5)o and the increase in
this value from 5 is most likely due to the methylation of the phosphorus centre which
has altered its hybridization,35 as opposed to the indolyl NH interaction with iodide.

73
3.3. Design of a New Symmetric Anion Receptor
While compound 5 has shown capacity for binding anions, greater anion
selectivity cannot be achieved in phosphine 5 without further structural modifications to
it. One design strategy is to build an anion receptor that has greater flexibility in its
structure that may allow for discriminatory binding to larger and more complex anions.
Pfeffer and co-workers have shown how changing the size of an anion receptor,
through the addition of an elongated flexible linker, results in its subsequent selectivity
for acetate over other anions.36 The authors present two indole-based receptors that are
structurally similar in that both receptors contain indolyl and amido NH functionalities;
the short receptor contains a urea functionality which is separated from the indolyl and
amido hydrogen bond donors through an ethyl linker; the long receptor instead contains a
nitro-functionalized thiourea substituent, and is separated from the indolyl and amido
groups by a hexyl linker (Figure 3.10). Although the presence of the thiourea and the
electron withdrawing nitro substituent in the longer receptor should increase the acidity
of the hydrogen bond donors, thus leading to stronger anion interaction, the authors’ main
concern was whether the flexibility and the length of the linker would have an influence
on the anion binding ability of the receptors.36 The stability constants from titrations of
selected anions with these varied length receptors are listed in Table 3.4.
NH
N
O
H NH
O
NH
NH
N
O
H NH
S
NH NO2
(b)(a) Figure 3.10. Flexible indole-based anion receptors by Pfeffer and co-workers:36 (a) short indole-based
anion receptor, (b) long indole-based anion receptor.
In the case of the short receptor, the lack of chloride binding suggested that the
ethyl linker was not long enough to allow for cooperative interaction of all four of the
hydrogen bond donors. Similarly, the binding to acetate was predominantly through the
urea NH donor site. The long receptor has preference for binding acetate, and it has been

74
suggested that the length of the linker is responsible for allowing the four hydrogen bond
donors to function collectively, which consequently leads to a strong interaction with
acetate.
Table 3.4. Stability constants Ka (M-1) of indole-based short receptor and indole-based long receptor with
chloride and acetate guests as determined by 1H NMR titration techniques performed at 298 K.36
Anion
Ka
NH
N
O
H NH
O
NH
Ka
NH
N
O
H NH
S
NH NO2
Cl- 125 no binding detected
CH3COO- 1260 7940
A new anion receptor based on C3-symmetric 5 would incorporate an additional
methylene-bridge between the indolyl substituent and the phosphorus atom. The added
methylene functionality should provide an extra degree of freedom that may allow for
greater anion selectivity, and greater conformational flexibility to accommodate a larger
number of anions.

75
3.3.1. Synthesis of New C3-Symmetric Anion Receptor
A retrosynthetic methodology was used in determining the best route to take to
generate the target C3-symmetric 5-based anion receptor, (CH2)3-5, 9 (Figure 3.11a).
However, prior to embarking on the synthesis of the target anion receptor, a model
phosphine, (CH2indolyl)PPh2, 10 (Figure 3.11b), would be synthesized first to establish
the viability of the reaction routes. The commercially available 1H-indole-2-carboxylic
acid ethyl ester, 11, was chosen as a suitable starting material, as there has been literature
precedence for the formation of the desired methylene-bridged indolyl substituent using
this material.37
HN
HN
NHP
HN
P
9 1
(a) (b)
0
Figure 3.11. (a) New C3-symmetric 5-based anion receptor target, P(C9H8N)3, 9. (b) Model phosphine
P(C9H8N)(C6H5)2, 10.
Initially the familiar N,N’-dimethylaminomethylene protecting group, normally
utilized for the protection of 3-methylindole to generate the aminal-protected starting
material to 2-indolylphosphines, could not be installed on the nitrogen centre. Two
known methods (reacting 11 with NEt3 followed by addition of N,N’-
dimethylmethyleneammonium chloride,3 or alternatively reacting 11 with formaldehyde
and dimethylamine17) were unsuccessful in the protection of the indole nitrogen
(Scheme 3.5).

76
HN O
OEt
N O
OEt
N
Me2NHH2O
reflux
H H
O
NEt3THFr.t.
no reactionMe2N CH2Cl11
Scheme 3.5. The two unsuccessful methods used to install the N,N’-dimethylaminomethylene protecting
group on the nitrogen centre of 11. (a) Reaction conditions: formaldehyde, dimethylamine, and 11 refluxed
in water. (b) Reaction conditions: Dimethylmethyleneammonium chloride, triethylamine, and 11 stirred in
THF at room temperature.
The t-Boc-protecting group is a versatile protecting group that can be used to
protect a variety of nitrogen centres and is resistant to diverse reaction conditions.38 The
t-Boc group is easily furnished on the indole nitrogen in near quantitative yield by
reaction of 11 with di-tert-butyl dicarbonate in the presence of DMAP at ambient
temperature. The reaction was completed in 1.5 h as monitored by TLC. Aqueous
workup, extraction, and subsequent removal of volatile solvents gave t-Boc-protected
indole-2-carboxylic acid ethyl ester, 12 (Scheme 3.6). The 1H NMR spectrum of 12
lacked the indole NH proton resonance, and exhibited a large singlet at δ = 1.63 ppm (in
CDCl3) that is associated with the t-Boc protecting group.
Compound 12 was reduced to the alcohol with DIBAL-H in toluene at -40oC
according to literature procedures.37 The reaction was allowed to stir at -40oC for 50
minutes, allowed to warm to -10oC and then quenched with MeOH and water. Following
an extraction and removal of volatile solvents, a dark-orange oil was recovered. Column
chromatography was required to purify the t-Boc-protected indole-2-methyl alcohol, 13,
which was recovered in 78 % yield. The characterization of 13 is identical to the one
reported for the same compound by Belanger and co-workers.37 The 1H NMR of 13
clearly exhibits the appearance of the methylene functionality at the C2-position on
indole as a singlet at δ = 1.68 ppm (in CDCl3), and the EI-HRMS is consistent with the
formation of the alcohol.

77
Alcohol 13 can be transformed into the useful bromide reagent through several
means: the classic method of alcohol conversion to bromide uses PBr3,39 or by reaction of
the alcohol with PPh3Br2, a milder reaction that proceeds at room temperature under 30
minutes.40,41 A solution of 13 in CH2Cl2 is added dropwise to a suspension of PPh3Br2 in
CH2Cl2. After stirring for 10 minutes the reaction is quenched with water. Upon workup
and subsequent chromatography, it was discovered that the acid sensitive t-Boc-
protecting group was cleaved from the indole due to minute, but sufficient amounts of
HBr generated as a side product in the reaction. Thus re-evaluation of the protecting
group was necessary in order to make the useful indole-2-methyl bromide.
HN
OEt
ON
OEt
O
O ON
OH
O ON
Br
O ODMAPBoc2O
THFr.t.
DIBAL-HCH2Cl2-40oC
PPh3Br2CH2Cl2
r.t.
quantitative 78% no reaction
12 1311 Scheme 3.6. Reaction scheme of t-Boc-protected indole-2-carboxylic acid ethyl ester, 12, to the t-Boc-
protected indole-2-methyl alcohol, 13.
The Cbz-group can be utilized to protect amines and has been found to resist
cleavage in a variety of reaction conditions.38 The installation of the Cbz-protecting
group on the indole nitrogen is a facile process (Scheme 3.7); deprotonation with NaH,
and subsequent addition of carbobenzyloxy chloride affords the Cbz-protected indole-2-
carboxylic acid ethyl ester, 14, in 96 % yield. The 1H NMR spectrum of 14 exhibited the
loss of the indole NH proton and the presence of the benzyl moiety of the Cbz-protecting
group.
Similar reaction conditions for 12 were carried out for 14. The Cbz-protected
indole-2-methyl alcohol, 15, was acquired in 65 % yield after reaction of 14 with
DIBAL-H in CH2Cl2 at -40oC. The characterization of 15 by 1H NMR was consistent
with the reduction of the carboxylate moiety of 14 to form the methyl alcohol moiety of
15; the appearance of the methylene protons at δ = 4.81 ppm (CDCl3) as a doublet from

78
coupling to the alcohol proton, and the alcohol proton resonates at δ = 2.73 ppm (CDCl3)
as a broad singlet.
A solution of alcohol 15 in CH2Cl2 was added dropwise to a suspension of
PPh3Br2 in CH2Cl2. After 10 minutes of stirring, the reaction was quenched with water
and extracted. Removal of volatile solvents, and subsequent column chromatography
revealed that Cbz-protected indole-2-methyl bromide, 16, was successfully synthesized
and purified in 40 % yield. The 1H NMR spectrum of 16 shows the methylene protons as
a singlet at δ = 4.94 ppm (CDCl3), and the EI-HRMS confirms the presence of the bromo
moiety.
N
OEt
O
OO
N
OH
OO
N
Br
OO
HN
OEt
O NaHCbzClTHFr.t.
DIBAL-HCH2Cl2-40oC
PPh3Br2CH2Cl2
r.t.
96% 65% 40%
11 14 15 16 Scheme 3.7. Reaction scheme of Cbz-protected indole-2-carboxylic acid ethyl ester, 14, to the Cbz-
protected indole-2-methyl bromide, 16.
Several methods are proposed to synthesize the model phosphine, 10, from Cbz-
protected indole-2-methyl bromide 16. The first method is to follow the protocol for 2-
indolylphosphines synthesis,3 through the generation of a lithiated indole moiety (Scheme
3.8). A solution of bromide 16 in THF was cooled to -78oC, n-BuLi was added dropwise.
The reaction was allowed to stir at -78oC for several hours prior to the addition of PPh2Cl.
Aqueous workup followed after 12 h of reaction with PPh2Cl. The 31P NMR of the crude
reaction indicated mostly hydrolyzed PPh2Cl, suggesting the phosphine chloride did not
react with the proposed lithiated 16. Further inspection of the 1H NMR spectrum of the
crude material showed that the Cbz-group had been cleaved off since an indole NH peak
was present. Generally hydrogenolysis is required to remove the Cbz-protecting group,38
however Shieh and co-workers42 have reported that selective deprotection of Cbz-
protected heterocycles can be achieved with bases such as sodium methoxide or DBU

79
(diaza(1,3)bicyclo[5.4.0]undecane) in methanol, while aliphatic amines remained
unaffected.
N
P
OO
N
Br
OO 1. n-BuLi
2. PPh2ClTHF, -78oC3. Aqueous
workup
no reaction
16
Scheme 3.8. Attempted synthesis of Cbz-protected 10 from 16 using n-BuLi and PPh2Cl.
The second method proposed to synthesize target phosphine 10 is based on the
reactivity of a Grignard reagent generated in situ through the treatment of 16 with
activated Mg dust in THF.43 Addition of PPh2Cl followed after the Mg was consumed,
and the resultant reaction mixture was refluxed overnight. Following an aqueous
workup, the 31P NMR spectrum of the crude reaction mixture was disappointingly mostly
hydrolyzed PPh2Cl. Interestingly, the major product formed in this reaction is Cbz-
protected 2-ethylindole, which suggests the formation of the Grignard reagent. While
Landaeta and co-workers43 were successful in the formation of tribenzylphosphine using
this protocol, the synthesis of the Cbz-protected model phosphine 10 could not be
realized by this method.
N
P
OO
N
Br
OO 1. Mg(s), r.t. to reflux, THF
2. PPh2Cl, -10oC to reflux3. Aqueous workup
no reaction
16
Scheme 3.9. Attempted synthesis of Cbz-protected 10 from 16 through Grignard reagent and PPh2Cl.

80
The third method proposed to generate the target model phosphine 10 involves a
nickel catalyst in the presence of zinc for P-C bond formation, and has shown to be
widely applicable for the synthesis of tertiary phosphines.44 The reaction conditions
involve the addition of Ni(PPh3)2Cl2 to 16 in DMF at 0oC, followed by the addition of
activated Zn dust and PPh2Cl at 10oC (Scheme 3.10). The Zn dust serves two roles: to
reduce the Ni(II) catalyst to the reactive Ni(0) species, and to generate the Ph2PZnCl for
transmetallation. The resultant mixture is heated to 100oC and maintained at this
temperature for 18 hours. The mixture is then cooled to 80oC, filtered, and rinsed with a
minimal amount of DMF. The combined filtrate and rinses were further cooled to 5oC, at
which point the pure product should crystallize from the solution. When no solids were
present, the mixture was concentrated in vacuo to afford a dark-orange coloured oil that
appeared to be a mixture containing three different species by TLC. The oil was
subjected to column chromatography and the three compounds separated. Surprisingly,
the major product was found to be Cbz-protected 2-ethylindole, and the other two
compounds did not have any 31P NMR resonances.
N
P
OO
N
Br
OO 1. Ni(dppe)Cl2
PPh2Cl, ZnDMF
15oC to 85oC2. Aqueous
workup
no reaction
16
Scheme 3.10. Attempted synthesis of Cbz-protected 10 from 16 using a Ni(dppe)Cl2 catalyst in the
presence of Zn and PPh2Cl.

81
3.3.2. Design of a New Diindolyl-Based Anion Receptor
While the synthesis of a new C3-symmetric 5-based anion receptor was
unsuccessful, alternative designs for indole-based anion receptors are limitless. It has
been demonstrated that anion receptors with deliberately placed hydrogen bond donors
can have greater selectivity for anion guests. For example, indole-based anion receptors
reported by Chang and co-workers are methodically designed to consist of indolyl NH
donors confined in macrocyles24 or molecular clefts22 (Figure 3.12).
HN
HN
NH
NH
NN
N
O
N
O
H HH H
H H
(a) (b) Figure 3.12. Indole-based anion receptors by Chang and co-workers. (a) Indole-based macrocycle.24
(b) Indole-based molecular cleft.22
The anion binding capabilities of the two indole-based receptors were determined using
spectroscopic titration techniques and the stability constants determined are listed in
Table 3.5. The macrocycle clearly binds anions with higher anion binding affinities,
presumably due to the rigidity of the hydrogen bond donors. However, the molecular
cleft demonstrates better anion selectivity toward the acetate anion over others. This
selectivity is most likely due to the flexibility of the molecular cleft by free rotation to
arrange its amide donors around the acetate anion to provide a better binding pocket.

82
Table 3.5. Stability constants Ka (M-1) of indole-based macrocyclea and indole-based molecular cleftb with
a selection of anion guests as determined by spectroscopic titration techniques performed at 298 K.22,24
Anion Ka
HN
HN
NH
NH
Ka
NN
N
O
N
O
H HH H
H H
Cl- 1.5 x 106 5.1 x 103
Br- K1 = 1.9 x 103, K2 = 10 2.1 x 102
CH3COO- 5.9 x 106 1.4 x 105
HSO4- 6.5 x 105 77
aTitrations performed in CD3CN and monitored with 1H NMR. bTitrations performed in CH3CN and monitored with
UV-visible spectroscopy.
3.3.3. Synthesis of New Diindolyl-Based Anion Receptor
Based on the work of Chang and co-workers, the design of a new molecular cleft-
based anion receptor bearing two indolyl moieties separated by a rigid phenyl linker
should allow for more selective binding of anions compared to 5. The molecular cleft, 4-
tert-butyl-7-(4-(4-tert-butyl-1H-indol-7-yl)phenyl)-1H-indole, 18, is based on a similar
one previously synthesized and investigated for its anion binding properties;45 the
difference in the new molecular cleft features two tert-butyl substituents at the C4-
position on indole to increase solubility of the receptor in less polar organic solvents.
The synthesis of 17 is shown in Scheme 3.11 and began with the nitration ortho to
the bromo substituent of 1-bromo-4-tert-butylbenzene with nitric acid and sulfuric acid at
ambient temperature. The nitrated species was then treated with vinyl magnesium
bromide in a Bartoli indole synthesis to form 4-tert-butyl-7-bromoindole, 17. Subsequent
Suzuki coupling with benzene-1,4-diboronic acid in the presence of Pd(PPh3)4 catalyst
gave the desired molecular cleft, 18, in moderate yields.

83
HN
BrBr Br
HN
NH
HNO3H2SO4
3oC to r.t.
H2C CHMgBrTHF
-40oC
Pd(PPh3)41,4-(BOH)2benzene
toluene / EtOHNa2CO3, H2O
reflux
NO2
89% 40%
34%17
18 Scheme 3.11. Synthesis of 4-tert-butyl-7-(4-(4-tert-butyl-1H-indol-7-yl)phenyl)-1H-indole, 18, from 1-
bromo-4-tert-butylbenzene.
Single crystals of the molecular cleft were acquired from slow evaporation of a
concentrated solution of 18 in CH2Cl2; the experimental X-ray diffraction data are listed
in Table 3.6. The numbering scheme for the molecular cleft are shown in Figure 3.13,
while Table 3.7 lists selected bond distances and angles.
Figure 3.13. ORTEP diagram of 18. Non-acidic hydrogen atoms removed for clarity. Thermal ellipsoids
are drawn at 35% probability.
The molecular cleft crystallizes in the monoclinic space group P21/c, where half of the
molecule lies on a crystallographic inversion centre that generates the other half through
symmetry. The phenyl linker is nearly perpendicular from the plane of the indoles, with a
dihedral angle <C16-C14-C8-C9 of -94.5(3)o that suggests this orientation to minimize
repulsion from the protons on the phenyl linker and the indolyl NH sites. Additionally,
the indolyl NH functionalities are oriented in opposite directions with respect to each

84
other, with a distance of 6.96 Å separating the two indolyl NH across the phenyl linker.
There are no significant structural changes with the addition of either the tert-butyl or the
phenyl moieties on the aromaticity of the indole, as the bond distances and angles are
comparable to indole (refer to Chapter 2).
Table 3.6. X-ray crystallographic experimental data of molecular cleft 18.
NH HN
Formula C30H32N2 Formula Weight 420.58 Crystal colour, shape colourless, plate Crystal size, mm 0.30 x 0.06 x 0.04 Crystal system Monoclinic Space group P21/c a, Å 11.9000(15) b, Å 6.6563(5) c, Å 16.244(2) α deg 90 β, deg 110.922(4) γ deg 90 V, Å3 1201.9(2) Z 2 Dcalcd, g cm-3 1.162 F(000) 452 μ, mm-1 0.067 λ (Mo Kα), Å 0.71073 Limiting indices -15<=h<=15 -8<=k<=8 -20<=l<=21 2θ range, deg 2.66 to 27.63 Max. and min. transmission 1.054 and 0.834 No. of reflections collected 9294 No. of independent reflections/Rint 2966 / 0.0947 Extinction coefficient 0.004(2) No. of refined parameters 149 Final R1, wR2 0.0575, 0.1162 Final R1, wR2 (all data) 0.1670, 0.1509 Goodness of fit 0.958 Δρmin, Δρmax, eÅ-3 -0.181, 0.176

85
Table 3.7. Selected bond lengths (Å) and angles (o) for molecular cleft 18.
NH HN
N1-C2 1.370(3) C2-C3 1.357(3) C3-C4 1.439(3) C4-C9 1.416(3) C9-N1 1.376(3)
<C9-N1-C2 108.7(2) <N1-C2-C3 109.8(2) <C2-C3-C4 107.7(2) <C3-C4-C9 105.5(2) <C4-C9-N1 108.2(2)
3.3.4. Anion Binding Studies of Molecular Cleft 18
Similar to 5, the anion binding capability of 18 was examined by 1H NMR
titration techniques in CD2Cl2 with selected anions (Scheme 3.12); the chemical shifts of
the indolyl NH protons were monitored via 1H NMR with increasing anion concentration
(Figure 3.14); the stability constants were determined by investigation of the titration data
by non-linear regression analysis and which were best fit a 1 : 1 receptor to anion binding
model (Figure 3.15 and Table 3.8). The stability constants of the unsubstituted
derivative, 7-(4-(1H-indol-7-yl)phenyl)-1H-indole, 19, are included in Table 3.8 for
comparison.45
HN
NH
+ NBu4XCD2Cl2
HN
NH
X- NBu4+
18 Scheme 3.12. General titration scheme of 18 and selected anions as their tetrabutylammonium salts in
CD2Cl2. Selected anions: X- = Cl-, Br-, CH3COO-, HSO4-, NO3
-, and BF4-.
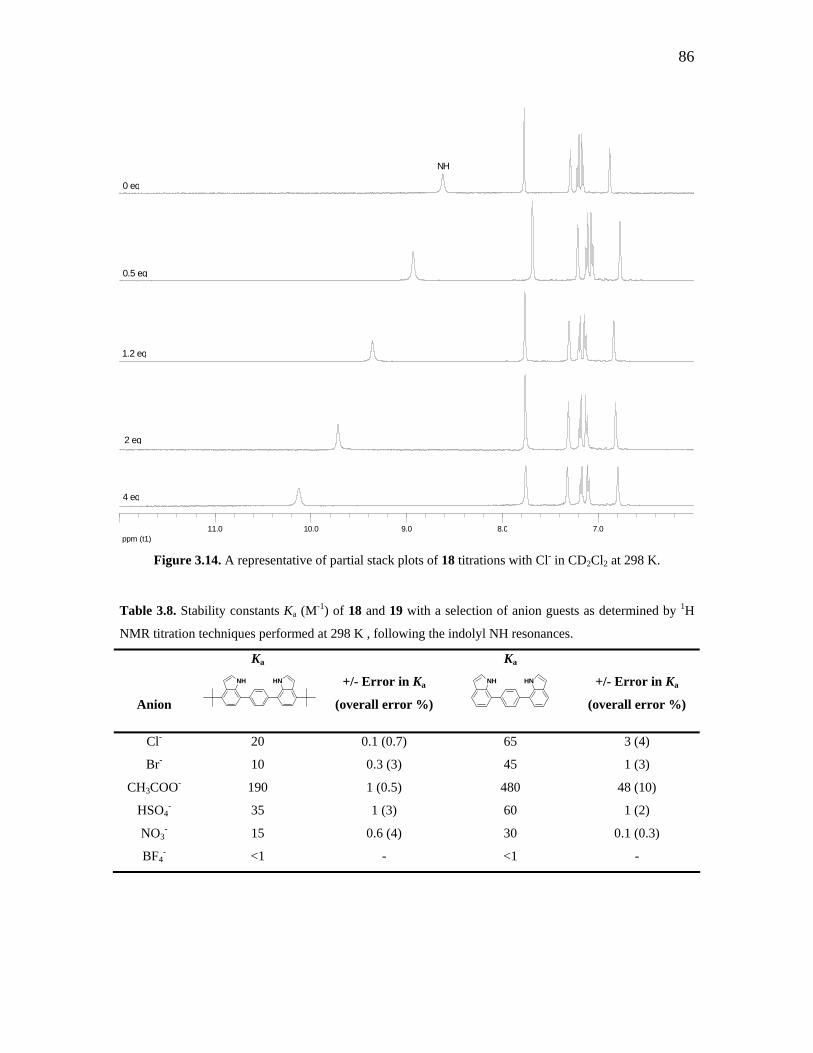
86
0 eq
NH
0.5 eq
1.2 eq
2 eq
ppm (t1)7.08.09.010.011.0
4 eq
Figure 3.14. A representative of partial stack plots of 18 titrations with Cl- in CD2Cl2 at 298 K.
Table 3.8. Stability constants Ka (M-1) of 18 and 19 with a selection of anion guests as determined by 1H
NMR titration techniques performed at 298 K , following the indolyl NH resonances.
Anion
Ka
NH HN
+/- Error in Ka
(overall error %)
Ka
NH HN
+/- Error in Ka
(overall error %)
Cl- 20 0.1 (0.7) 65 3 (4)
Br- 10 0.3 (3) 45 1 (3)
CH3COO- 190 1 (0.5) 480 48 (10)
HSO4- 35 1 (3) 60 1 (2)
NO3- 15 0.6 (4) 30 0.1 (0.3)
BF4- <1 - <1 -

87
Figure 3.15. . Fit plot for titration experiment of 18 with tetrabutylammonium chloride in CD2Cl2.
The stability constants indicate a strong selectivity for acetate anion over the other
anions titrated for both molecular clefts 18 and 19. This affinity for acetate by both
molecular clefts suggests a better complementary fit of the anion in the hydrogen bonding
pocket of the receptor because the indole NH hydrogen bond donors are spatially
predisposed to bind acetate. In contrast to the [5·CH3COO]- complex, it is speculated
that both oxygen atoms of acetate are binding to both indolyl NH donors in molecular
clefts 18 and 19 (Scheme 3.13). However, the stability constant determined for complex
6 (Ka = 2730 M-1) is greater than that for both molecular clefts, which is most likely due
to the additional hydrogen bond donor in receptor 5.
N NH HO O
CH3
Scheme 3.13. Proposed mode of acetate interaction with molecular cleft 18.
As can be seen from the stability constants listed in Table 3.8, the tert-butyl
functionalized molecular cleft does not have as strong anion binding affinities as that of
the unfunctionalized molecular cleft. The decrease in anion binding is most likely due to
the electron donating power of the tert-butyl substituents at the C4- and C4’-positions on

88
indole. While the tert-butyl moieties are situated four bonds away from the indolyl NH
centres, they do appear to have an effect on the acidity of these protons. Similarly,
Sessler and co-workers have demonstrated that the installation of an electron withdrawing
nitro substituent on their indole-based anion receptor has profoundly increased the acidity
of indolyl NH sites that are reflected in the stability constants (Table 3.9).28 Even though
the nitro group is seven bonds away from the hydrogen bond donors, it not only increases
the anion binding affinities for all anions, but it also enhances selectivity for phosphate
anion.
Table 3.9. Stability constants Ka (M-1) of diindolylquinoxaline-based anion receptors with a selection of
anion guests as determined by UV-visible spectroscopic titration techniques performed at 298 K in
CH2Cl2.28
Anion
Ka
N N
NH
NH Ka
N N
NH
NH
NO2
Cl- 170 470
HSO4- 80 250
C6H5COO- 600 2700
H2PO4- 6800 20 000
3.4. Conclusions
It was found that the C3-symmetric 2-indolylphosphine, 5, has demonstrated anion
binding ability. The solid-state structure of 5 exhibits how the three indolyl NH hydrogen
bond donors are oriented in the same direction, thus are available to capture anion guests.
Through 1H NMR titration techniques in CD2Cl2, it was determined that 5 has the
strongest affinity for chloride and acetate in a 1 : 1 receptor : anion ratio, and that the
interactions are confirmed by the solid-state structure involving the acetate anion.
Although an accurate stability constant could not be determined for the fluoride titration

89
with 5, its titration series did have the largest overall shift in the indolyl NH resonances,
implying a strong interaction. This interaction between 5 and F- was demonstrated in the
solid-state structure of its [NEt4][5·F] complex, 7, as a 1 : 1 receptor to anion binding
stoichiometry.
The solid-state structure of the phosphonium complex, 8, shows iodide
interactions with indolyl NH functionalities. It was thought that the methylation of the
phosphorus centre would encourage iodide interaction, which is seen with only one of the
indolyl NH sites. The remaining two indolyl NH sites of 5 are either hydrogen bonding
to Et2O solvate, or are not engaged in secondary bonding interactions.
Attempts to design and synthesize a new C3-symmetric anion receptor based on 5
were unsuccessful; complications arose from the reactivity of the nitrogen protecting
groups, and P-C bond formation procedures that were unpredictable. However, a
diindolyl-based molecular cleft, 18, was synthesized and found to have anion binding
capability.45 While the unfunctionalized molecular cleft is capable of anion binding, its
relative insolubility in most organic solvents makes it difficult to examine. The
installation of tert-butyl substituents on the molecular cleft has improved its solubility,
but has decreased the acidity of the indolyl NH donors, thus resulting in lower stability
constants compared to the unfunctionalized analogue.
3.5. Experimental
General Considerations. All reactions and manipulations were carried out under an
atmosphere of dinitrogen using standard Schlenk techniques unless otherwise stated.
PCl3, 1.6 M n-BuLi, NaBH4, MeI, 1H-indole-2-carboxylic acid ethyl ester, DMAP,
Boc2O, CbzCl, 1.0 M DIBAL-H, NaH, PPh2Cl, benzene 1,4-diboronic acid, 1-bromo-4-
tert-butylbenzene, tetraethylammonium and tetrabutylammonium salts (fluoride, chloride,
bromide, iodide, acetate, sulfate, nitrate, tetrafluoroborate) were purchased from Aldrich
and used as received. Pd(PPh3)4 was purchased from Strem and used without further

90
purification. 1-[(N,N-Dimethylamino)methyl]-3-methylindole,17 and 1-bromo-4-tert-
butyl-2-nitrobenzene46 were synthesized according to literature procedures. THF was
distilled from sodium benzophenone ketyl under a dinitrogen atmosphere. CH2Cl2 was
distilled from calcium hydride under a dinitrogen atmosphere. 1H, 13C, and 31P NMR
spectra were recorded on Varian 400 MHz or 300 MHz NMR systems, and referenced to
SiMe4 (TMS) and 85% H3PO4, respectively. Splitting patterns are indicated as s, singlet;
d, doublet; t, triplet; q, quartet; m, multiplet; br, broad peak. High resolution mass spectra
were obtained from a Micromass 70S-250 spectrometer (EI).
P
HN
CH3
3
I
Methyltris-2-(3-methylindolyl)phosphonium iodide, 8. To a clear colourless solution
of tris-2-(3-methylindolyl)phosphine (0.307 g, 0.73 mmol) in THF (15 mL) was added
dropwise methyl iodide (0.25 mL, 4.0 mmol). The resultant reaction was left to stir at
ambient temperature for 18 h during which a white precipitate formed. The product was
isolated by filtration, washed with Et2O (2 x 5 mL), and air dried (0.254 g, 62 %). 31P
NMR (CDCl3, 161 MHz): δ -12.7 (s); 1H NMR (CDCl3, 400 MHz): δ 10.29 (br s, 3H,
indole NH), 7.72 (d, J = 8.3 Hz, 3H, Ar-H), 7.58 (d, J = 8.1 Hz, 3H, Ar-H), 7.30 (t,
J = 7.2, 15.2 Hz, 3H, Ar-H) 7.18 (t, J = 7.9, 15.8 Hz, 3H, Ar-H), 3.39 (d, J = 13.8 Hz, 3H,
P+-CH3), 2.01 (d, J = 1.8 Hz, 9H, indole CH3); 13C NMR (CDCl3, 75 MHz): 140.0, 128.5,
126.8, 121.1, 120.0, 113.2, 107.9, 106.2, 15.9, 9.1.
N
OEt
O
O O
Indole-1,2-dicarboxylic acid 1-tert-butyl ester 2-ethyl ester, 12. THF (50 mL) was
added to 1H-Indole-2-carboxylic acid ethyl ester (2.45 g, 13.0 mmol), DMAP (1.74 g,
14.3 mmol), and Boc2O (3.57 g, 16.4 mmol) to afford a clear pale yellow-coloured
solution. The reaction was stirred at rt for 1.5 h and monitored by TLC. Solvents were

91
removed in vacuo to yield a pale orange-yellow oil. Added CH2Cl2 (50 mL) and
extracted with 2N HCl (2 x 50 mL). Dried organic layer over anhydrous MgSO4, filtered,
and concentrated in vacuo to afford the pure product as an orange-coloured oil (3.73 g,
99%). 1H NMR (CDCl3, 300 MHz): δ 8.08 (d, J = 8.4 Hz, 1H, Ar-H), 7.60 (d, J =
7.8 Hz, 1H, Ar-H), 7.41 (t, J = 7.8 Hz, 1H, Ar-H), 7.26 (t, J = 7.5 Hz, 1H, Ar-H), 7.10 (s,
1H, Ar-H), 4.38 (q, J = 7.2, 14.1 Hz, 2H, CH2CH3), 1.63 (s, 9H, C(CH3)3), 1.40 (t,
J = 6.8 Hz, 3H, CH2CH3); 13C NMR (CDCl3, 75 MHz): 162.1, 152.4, 149.5, 138.0, 131.0,
127.8, 126.9, 123.5, 122.3, 115.0, 84.8, 61.6, 28.1, 14.5; HRMS EI for C16H19NO4Na:
calcd m/z 312.1206, found m/z 312.1215.
N
OH
O O
2-Hydroxymethyl-indole-1-carboxylic acid tert-butyl ester, 13. Added toluene
(14 mL) to indole-1,2-dicarboxylic acid 1-tert-butyl ester 2-ethyl ester (1.99 g,
6.88 mmol) and cooled to -40oC. Added DIBAL-H (1 M in CH2Cl2, 16.5 mL,
16.5 mmol) dropwise over 10 minutes. The resultant mixture was allowed to stir for 50
minutes at -40oC. The reaction vessel was vented and MeOH (8 mL) was added
dropwise, followed by the dropwise addition of water (4 mL), which formed a precipitate.
The reaction was warmed to rt and filtered. The filtrate was extracted with CH2Cl2 (3 x
50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and
reduced to afford a dark orange coloured oil. The crude material was purified by column
chromatography (silica gel, 4 : 1 hexanes-EtOAc) to yield the pure product (1.39 g, 78 %)
as a colourless oil. 1H NMR (CDCl3, 400 MHz): δ 8.14 (d, J = 8.4 Hz, 1H, Ar-H), 7.51
(d, J = 8 Hz, 1H, Ar-H), 7.29 (t, J = 7.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.6 Hz, 1H, Ar-H),
6.66 (s, 1H, Ar-H), 5.43 (s, 2H, CH2OH), 1.68 (s, 9H, C(CH3)3); 13C NMR (CDCl3,
75 MHz): 153.5, 140.0, 137.1, 134.9, 128.9, 124.7, 123.1, 120.8, 115.8, 110.0, 84.6, 63.5,
28.3; HRMS EI for C14H17NO3: calcd m/z 247.120844, found m/z 247.121422.

92
N
OEt
O
OO
Indole-1,2-dicarboxylic acid 1-benzyl ester 2-ethyl ester, 14. THF (50 mL) was added
slowly to 1H-indole-2-carboxylic acid ethyl ester (7.09 g, 37.5 mmol) and NaH (60 %wt,
2.53 g, 63.3 mmol) to afford a bright pink solution. Allowed the reaction mixture to stir
for 3 h before adding benzyl chloroformate (12 mL, 85.3 mmol) dropwise, and allowed
the reaction to stir at rt overnight. The orange-coloured reaction mixture was poured into
satd. NH4Cl and extracted with EtOAc (2 x 200 mL). The combined organic layers were
washed with brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo to
give a golden-orange-coloured oil. The crude material was purified by flash
chromatography (silica gel, 10 : 5 : 1 hexanes-CH2Cl2-Et2O) to afford the pure product
(11.5 g, 96 %) as a golden-orange oil. 1H NMR (CD2Cl2, 400 MHz): δ 8.13 (d, J =
8.4 Hz, 1H, Ar-H), 7.66 (d, J = 7.6 Hz, 1H, Ar-H), 7.51 – 7.39 (m, 6H, Ar-H), 7.32 (t,
J = 7.5 Hz, 1H, Ar-H), 7.17 (s, 1H, Ar-H), 5.46 (s, 2H, CH2Ph), 4.27 (q, J = 7.2, 14 Hz,
2H, CH2CH3), 1.32 (t, J = 7.2 Hz, 3H, CH2CH3); 13C NMR (CD2Cl2, 75 MHz): 162.1,
156.2, 151.3, 138.2, 136.2, 135.3, 131.5, 129.2, 128.3, 127.5, 124.1, 122.8, 115.6, 115.4,
70.2, 62.1, 14.5; HRMS EI for C19H17NO4: calcd m/z 323.115758, found m/z 323.115143.
N
OH
OO
2-Hydroxymethyl-indole-1-carboxylic acid benzyl ester, 15. CH2Cl2 (50 mL) was
added to indole-1,2-dicarboxylic acid 1-benzyl ester 2-ethyl ester (6.41 g, 19.8 mmol)
and cooled to -40oC. DIBAL-H (47 mL, 47 mmol, 1.0 M in CH2Cl2) was added dropwise
to afford an orange-coloured solution and the mixture allowed to stir at -40oC overnight.
The reaction was warmed to 0oC and MeOH (50 mL) was added dropwise to give a
bright yellow mixture, followed by the dropwise addition of H2O (40 mL), which formed

93
a white precipitate. The bright yellow suspension was warmed to rt over 3 h. The
mixture was filtered, the filtrate was extracted with CH2Cl2 (3 x 100 mL), the organic
layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give a
cream-coloured oil. The crude material was purified by flash chromatography (silica gel,
4 : 1 hexanes-EtOAc) to afford the pure product as a white solid (3.6 g, 65 %). 1H NMR
(CD2Cl2, 400 MHz): δ 8.05 (d, J = 8 Hz, 1H, Ar-H), 7.56 - 7.24 (m, 8H, Ar-H), 6.63 (s,
1H, Ar-H), 5.51 (s, 2H, CH2Ph), 4.81 (d, J = 7 Hz, 2H, CH2OH), 2.73 (bs, 1H, OH); 13C
NMR (CD2Cl2, 75 MHz): 152.9, 142.0, 141.2, 135.4, 129.8, 129.3, 127.9, 127.4, 125.1,
123.9, 121.4, 116.3, 110.2, 69.6, 59.4; HRMS EI for C17H15NO3: calcd m/z 281.105194,
found m/z 281.105106.
N
Br
OO
2-Bromomethyl-indole-1-carboxylic acid benzyl ester, 16. To a suspension of PPh3Br2
(0.65 g, 1.54 mmol) in CH2Cl2 (0.8 mL), was added dropwise a solution of 2-
hydroxymethyl-indole-1-carboxylic acid benzyl ester (1.0 g, 3.6 mmol) in CH2Cl2
(1.5 mL). The mixture becomes clear and dark pink coloured after 5 minutes of stirring.
The reaction was quenched with water (1.5 mL), and the layers separated. The organic
layer was washed with water (3 x 10 mL), dried over anhydrous MgSO4, filtered and
concentrated in vacuo. The residue was fused to silica and purified by flash
chromatography (silica gel, 9 : 1 hexanes-EtOAc) to afford the pure product as a white
solid (2.2 g, 40 %). 1H NMR (CD2Cl2, 400 MHz): δ 8.13 (d, J = 8.4 Hz, 1H, Ar-H), 7.56
(dd, J = 1.6, 8 Hz, 1H, Ar-H), 7.53 (d, J = 7.6 Hz, 2H, Ar-H), 7.44 - 7.38 (m, 3H, Ar-H),
7.32 (td, J = 1.2, 7.8 Hz, 1H, Ar-H), 7.24 (td, J = 0.8, 7.6 Hz, 1H, Ar-H) 6.77 (s, 1H, Ar-
H), 5.52 (s, 2H, CH2Ph), 4.94 (s, 2H, CH2Br); 13C NMR (CD2Cl2, 75 MHz): 151.7, 137.8,
136.9, 135.5, 129.4, 129.0, 126.0, 124.0, 121.5, 116.4, 112.9, 69.8, 27.7; HRMS EI for
C17H14NO2Br: calcd m/z 343.020790, found m/z 343.020130.

94
HN
Br
7-Bromo-4-tert-butylindole, 17. A solution of 1-bromo-4-tert-butyl-2-nitrobenzene
(2.82 g, 10.9 mmol) in THF (20 mL) was cooled to -40oC. Vinylmagnesium bromide
(33 mL, 1.0 M in THF, 33 mmol) was added dropwise via an addition funnel over 10
minutes. The resultant dark brown solution was allowed to warm to ambient temperature
overnight. The reaction was quenched with the addition of saturated aqueous NH4Cl, the
organic layer was separated, and the aqueous layer was extracted with CH2Cl2
(2 x 50 mL). The organic layers were combined, dried over anhydrous MgSO4, filtered,
and concentrated in vacuo to afford a dark brown oil. The product was obtained pure
after flash chromatography (silica gel, 5% EtOAc in hexanes) as a pale brown coloured
oil (0.9 g, 30%). 1H NMR (CDCl3, 400 MHz): δ 8.52 (s, 1H, indole NH), 7.33 (d, J =
8 Hz, 1H, Ar-H), 7.24 (t, J = 3.2, 5.8 Hz, 1H, Ar-H), 6.99 (d, J = 8 Hz, 1H, Ar-H), 6.87
(dd, J = 2.2, 3.3 Hz, 1H, Ar-H), 1.56 (s, 9H, C(CH3)3); 13C NMR (CDCl3, 100 MHz):
143.0, 135.1, 126.7, 124.1, 123.5, 117.4, 105.3, 103.0, 35.8, 30.8; HRMS EI for
C12H14NBr: calcd m/z 251.030961, found m/z 251.031127.
NH HN
4-tert-butyl-7-(4-(4-tert-butyl-1H-indol-7-yl)phenyl)-1H-indole, 18. A mixture of
toluene / EtOH (65 mL, 1 : 1, v / v) was added to a solid mixture of 7-bromo-4-tert-
butylindole (2.054 g, 8.2 mmol), benzene 1,4-diboronic acid (0.682 g, 4.1 mmol), and
Pd(PPh3)4 (0.475 g, 0.4 mmol) to give a dark yellow coloured suspension. The mixture
was stirred at r.t. for 15 minutes until all of the boronic acid was in solution. A clear
colourless solution of Na2CO3 (2.52 g, 20.4 mmol) in H2O (19.5 mL) was added quickly
via syringe to the reaction mixture. The biphasic mixture was refluxed for 1 h. A
suspension of Pd(PPh3)4 (0.50 g, 0.4 mmol) in toluene / EtOH (33 mL, 1 : 1, v / v) was
added via addition funnel. The reaction continued to reflux overnight. Subsequent
cooling to r.t., followed by removal of solvent in vacuo afforded a dark brown residue,

95
which was extracted with CH2Cl2 (2 x 100 mL). The organic layers were combined,
washed with brine, dried over anhydrous MgSO4, filtered and reduced in vacuo. The
residue was fused to silica gel and purified by flash chromatography (silica gel, 10%
EtOAc in hexanes) to produce the pure product as a pale yellow solid (0.58 g, 34%). 1H
NMR (CDCl3, 400 MHz): δ 8.51 (s, 2H, indole NH), 7.75 (s, 4H, Ph-H), 7.22 (m, 2H, Ar-
H), 7.20 (q, J = 7.6 Hz, 4H, Ar-H), 6.88 (dd, J = 2.1, 3.3 Hz, 2H, Ar-H), 1.56 (s, 18H,
C(CH3)3); 13C NMR (CDCl3, 100 MHz): 143.1, 138.5, 134.6, 129.2, 125.9, 123.7, 123.2,
122.0, 116.8, 104.7, 36.0, 31.0; HRMS EI for C30H32N2: calcd m/z 420.256549, found m/z
420.256767.
Titration Methods. 1H NMR titration experiments were carried out by dissolving 5 or
18 (15.4 μmol) in CD2Cl2 (0.65 mL) and adding sequential 10 μL aliquots of a solution
of the selected anions as their tetrabutylammonium salts (154 μmol) in CD2Cl2 (1 mL)
(which corresponds to 0.1eq of anion) to 5 or 18 until saturation. Association constants,
Ka, were determined by non-linear least-squares fit of the data using the program
EQNMR.19
Figure 3.16. Fit plot for titration experiment of 5 with tetrabutylammonium bromide in CD2Cl2.

96
Figure 3.17. Fit plot for titration experiment of 5 with tetrabutylammonium iodide in CD2Cl2.
Figure 3.18. Fit plot for titration experiment of 5 with tetrabutylammonium acetate in CD2Cl2.

97
Figure 3.19. Fit plot for titration experiment of 5 with tetrabutylammonium hydrogensulfate in CD2Cl2.
Figure 3.20. Fit plot for titration experiment of 5 with tetrabutylammonium nitrate in CD2Cl2.

98
Figure 3.21. Fit plot for titration experiment of 5 with tetrabutylammonium tetrafluoroborate in CD2Cl2.
Figure 3.22. Fit plot for titration experiment of 18 with tetrabutylammonium bromide in CD2Cl2.

99
Figure 3.23. Fit plot for titration experiment of 18 with tetrabutylammonium acetate in CD2Cl2.
Figure 3.24. Fit plot for titration experiment of 18 with tetrabutylammonium hydrogensulfate in CD2Cl2.

100
Figure 3.25. Fit plot for titration experiment of 18 with tetrabutylammonium nitrate in CD2Cl2.
Figure 3.26. Fit plot for titration experiment of 18 with tetrabutylammonium tetrafluoroborate in CD2Cl2.
X-ray Crystallography. X-ray data were collected on a Nonius Kappa CCD
diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). A
combination of 1º φ and ω (with κ offsets) scans were used to collect sufficient data. The
data frames were integrated and scaled using the Denzo-SMN package.47 The structures
were solved and refined with the SHELXTL-PC v6.12 software package.48 Refinement
was by full-matrix least squares on F2 using data (including negative intensities) with
hydrogen atoms bonded to carbon and nitrogen atoms included in calculated positions
and treated as riding atoms. Absorption corrections were made for every structure. All
the heavy atoms were refined anisotropically.

101
3.6. References
(1) Brooks, S. J.; Evans, L. S.; Gale, P. A.; Hursthouse, M. B.; Light, M. E. Chem.
Commun. 2005, 734-736.
(2) Coles, S. J.; Frey, J. G.; Gale, P. A.; Hursthouse, M. B.; Light, M. E.; Navakhun,
K.; Thomas, G. L. Chem. Commun. 2003, 568-569.
(3) Kondo, S.-i.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005,
1720-1722.
(4) Lakshminarayanan, P. S.; Ravikumar, I.; Suresh, E.; Ghosh, P. Inorg. Chem.
2007, 46, 4769-4771.
(5) Evans, L. S.; Gale, P. A.; Light, M. E.; Quesada, R. Chem. Commun. 2006, 965-
967.
(6) Gale, P. A. Chem. Commun. 2005, 3761-3772.
(7) Gale, P. A.; Camiolo, S.; Chapman, C. P.; Light, M. E.; Hursthouse, M. B.
Tetrahedron Lett. 2001, 5095 - 5097.
(8) Gale, P. A.; Camiolo, S.; Tizzard, G. J.; Chapman, C. P.; Light, M. E.; Coles, S.
J.; Hursthouse, M. B. J. Org. Chem. 2001, 66, 7849-7853.
(9) Brooks, S. J.; Gale, P. A.; Light, M. E. Chem. Commun. 2006, 4344-4346.
(10) Duckmanton, P. A.; Blake, A. J.; Love, J. B. Inorg. Chem. 2005, 44, 7708-7710.
(11) Knight, L. K.; Freixa, Z.; vanLeeuwen, P. W. N. M.; Reek, J. N. H.
Organometallics 2006, 25, 954-960.
(12) Tovilla, J. A.; Vilar, R.; White, A. J. P. Chem. Commun. 2005, 4839 - 4841.
(13) Ayling, A. J.; Perez-Payan, M. N.; Davis, A. P. J. Am. Chem. Soc. 2001, 123,
12716-12717.
(14) Kwon, J. Y.; Jang, Y. J.; Kim, S. K.; Lee, K. H.; Kim, J. S.; Yoon, J. J. Org.
Chem. 2004, 69, 5155-5157.
(15) Bordwell, F. G.; Zhang, X.; Cheng, J.-P. J. Org. Chem. 1991, 56, 3216-3219.
(16) Eisler, D. J.; Puddephatt, R. J. Inorg. Chem. 2003, 42, 8192-8202.
(17) Katritzky, A. R.; Lue, P.; Chen, Y.-X. J. Org. Chem. 1990, 55, 3688-3691.
(18) Katritzky, A. R.; Akutagawa, K. Tetrahedron Lett. 1985, 26, 5935-5938.
(19) Hynes, M. J. J. Chem. Soc. Dalton Trans. 1993, 311-312.
101

102
(20) Bates, G. W.; Gale, P. A.; Light, M. E. Chem. Commun. 2007, 2121-2123.
(21) Bates, G. W.; Triyanti; Light, M. E.; Albrecht, M.; Gale, P. A. J. Org. Chem.
2007, 72, 8921-8927.
(22) Chang, K. J.; Chae, M. K.; Lee, C.; Lee, J. Y.; Jeong, K. S. Tetrahedron Lett.
2006, 47, 6385-6388.
(23) Kwon, T. H.; Jeong, K. S. Tetrahedron Lett. 2006, 47, 8539-8541.
(24) Chang, D. -J.; Moon, D.; Lah, M. S.; Jeong, K. -S. Angew. Chem. Int. Ed. 2005,
44, 7926-7929.
(25) Lee, J.-Y.; Lee, M.-H.; Jeong, K.-S. Supramol. Chem. 2007, 19, 257 - 263.
(26) Wang, Y.; Lin, H.; Shao, J.; Cai, Z. S.; Lin, H. K. Talanta 2008, 74, 1122-1125.
(27) Zielinski, T.; Dydio, P.; Jurczak, J. Tetrahedron 2008, 64, 568-574.
(28) Sessler, J. L.; Cho, D.-G.; Lynch, V. J. Am. Chem. Soc. 2006, 128, 16518-16519.
(29) Steed, J. W.; Atwood, J. L. Supramolecular Chemistry; John Wiley & Sons, Ltd.:
New York, NY, 2001.
(30) Deprotonation of the indolyl NH sites was not observed, even though it is a well
documented phenomenon: Boiocchi, M.; DelBoca, L.; Gomez, D. E.; Fabbrizz, L.;
Licchelli, M.; Monzani, E. J. Am. Chem. Soc. 2004, 126, 16507-16514.
(31) Steiner, T. Acta Cryst. 1998, B54, 456-463.
(32) Bugner, D. E. J. Org. Chem. 1989, 54, 2580-2586.
(33) Symmetry code: -x,-y+1,-z+1
(34) Symmetry code: x,-y+1/2,z-1/2
(35) Baby Mariyatra, M.; Kayanasundari, B.; Panchanatheswaran, K.; Goeta, A. E.
Acta Cryst. 2003, E59, o255-o257.
(36) Pfeffer, F. M.; Lim, K. F.; Sedgwick, K. J. Org. & Biomol. Chem. 2007, 5, 1795-
1799.
(37) Belanger, G.; Larouche-Gauthier, R.; Menard, F.; Nantel, M.; Barabe, F. J. Org.
Chem. 2006, 71, 704-712.
(38) Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; Third
Edition ed.; John Wiley & Sons, Ltd.: New York, NY, 1999.
(39) March, J. Advanced Organic Chemistry; 4th ed.; John Wiley & Sons, Ltd.: New
York, NY, 1992.

103
(40) Bressy, C.; Alberico, D.; Lautens, M. J. Am. Chem. Soc. 2005, 127, 13148-13149.
(41) Vizitiu, D.; Walkinshaw, C. S.; Gorin, B. I.; Thatcher, G. R. J. J. Org. Chem.
1997, 62, 8760-8766.
(42) Shieh, W. C.; Xue, S.; McKenna, J.; Prasad, K.; Repic, O.; Blacklock, T.
Tetrahedron Lett. 2006, 47, 5645-5648.
(43) Landaeta, V. R.; Peruzzini, M.; Herrera, V.; Bianchini, C.; Sanchez-Delgado, R.
A.; Goeta, A. E.; Zanobini, F. J. Organomet. Chem. 2006, 691, 1039-1050.
(44) Ager, D. J.; East, M. B.; Eisenstadt, A.; Laneman, S. A. Chem. Commun. 1997,
2359-2360.
(45) Ang, M. T. C., MSc Thesis, University of Toronto, 2007.
(46) Wegner, H. A.; Reisch, H.; Rauch, K.; Demeter, A.; Zachariasse, K. A.; de
Meijere, A.; Scott, L. T. J. Org. Chem. 2006, 71, 9080-9087.
(47) Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307-326.
(48) Sheldrick, G. M. In SHELXTL-Windows NT. V6.12, Bruker Analytical X-Ray
Systems Inc. Madison, WI, 2001.

Chapter 4
Coordination Chemistry of Monodentate
2-Indolylphosphines*
4.1. Introduction
One method to study a phosphine molecule is to examine its coordination
chemistry to transition metal centres. It is well documented that the coordination of a
phosphine ligand to Ni(CO)3 can provide information on the net-basicity of the ligand
through the complex’s infrared CO stretches.1,2 Likewise, studies have shown that the
trans-influence of ligands can be seen in the bond lengths of their metal complexes, and
can be correlated to the basicity of that ligand.3 The trans-influence of a ligand
influences the ground state properties of transition metal coordination complexes, such as
metal-ligand bond distances and NMR chemical shifts. Often the experimental measure
of a ligand’s trans-influence is to examine the M-X bond trans to the ligand in square
planar Pd(II) or Pt(II) complexes.4
In this Chapter, the coordination of monodentate 2-indolylphosphines to Ni(CO)3
will be examined, and the net-basicities of several 2-indolylphosphines deduced by this
technique. The reaction of phosphine 5 to Cu(I) in the presence of phenanthroline yields
a complex that exhibits simultaneous coordination at the phosphorus centre and anion
binding through the indolyl NH hydrogen bond donors 5. The solid-state structures of
Pd(II) and Pt(II) 2-indolylphosphine complexes will be discussed, with focus on the
trans-influence of different 2-indolylphosphine ligands.
* Reproduced in part with permission from: J. O. Yu, E. Lam, J. L. Sereda, N. C. Rampersad, A. J. Lough, C. S. Browning, and D. H. Farrar. Organometallics 2005, 24, 37-47. Copyright 2006 American Chemical Society.
104

105
4.2. Results and Discussion
4.2.1. An Evaluation of 2-Indolylphosphine Net-Basicity:
Coordination to Ni(CO)3
The net-basicities of several 2-indolylphosphines were determined by reacting
them with Ni(CO)4 in THF as shown in Scheme 4.1. The reaction proceeds under inert
dinitrogen atmosphere where Ni(CO)4 is added dropwise from its lecture bottle via static
vacuum into a Schlenk flask containing a THF solution of the phosphine. After several
hours of stirring the unreacted Ni(CO)4, CO, and THF are removed in vacuo into a
secondary trap containing bleach, which renders the Ni(CO)4 unreactive. The remaining
solid is the desired Ni(CO)3L complex, which has an unambiguous IR pattern consistent
with monosubstitution of Ni(CO)4.
R3R1
R2P + Ni(CO)4
THF R2R1
R3P Ni(CO)3
Scheme 4.1. General scheme for the formation of a monodentate Ni(CO)3(phosphine) complex.
In accordance with Tolman’s method of determining phosphine basicity, the A1
carbonyl stretching frequency of each of the monosubstituted Ni(CO)3L complexes
(where L = 2-indolylphosphine) were measured by IR spectroscopy in heptanes.
Hydrocarbon solvents tend to produce sharper peaks for the Ni(CO)3L complexes,
whereas halogenated solvents result in broadening of the CO stretching bands.5 These A1
CO stretching frequencies are subsequently compared to the analogous carbonyl stretch
of Ni(CO)3(PtBu3) (νCO = 2056.1 cm-1), the most basic phosphine in Tolman’s series, to
give the net-basicity value denoted χ.2 The χ-values determined for the selected 2-
indolylphosphines are listed in Table 4.1 along with some common phosphines from the
literature to provide frames of references for comparison.

106
Table 4.1. Infrared CO stretching frequencies of Ni(CO)3L in heptanes.
Entry
L = Phosphine
νCO (A1) (cm-1)
χ (cm-1)
1
P(tBu)3
2056.1a
0
2 PMe3 2064.1 a 8.0
3
HN
P (20)
2065.7
9.6
4
P(o-tolyl)3
2066.6 a
10.5
5 PPh3 2068.9 a 12.8
6
NP (N-Me)-1
2070.9
14.8
7
HN
PNH (4)
2071.7
15.6
8
HN
P (1)
2072.8
16.7
9
NH
PHN (3)
2074.9
18.8
10
NP
N
F
FF
FF
F F
F
FF (N-F5Bn)2-4
2076.2
20.1
11
NH
P
HN
HN
(5)
2076.5
20.4
12 P(2-CH2CH2CN)3 2077.9 a 21.8 aValues determined in CH2Cl2.2

107
Ligand 20 is found to be the most basic of the 2-indolylphosphines, with a χ-
value of 9.6 cm-1, and is most likely due to the electron donating power of the tert-butyl
substituents on phosphorus. The χ-value of 20 makes it less basic than PMe3, but more
basic than P(o-tolyl)3. By exchanging the tert-butyl substituents for less electron rich
phenyl moieties, the net-basicity of 1 has decreased substantially (χ-value = 16.7 cm-1)
and renders it less basic than PPh3. As the phenyl substituents are replaced with
increasing number of 3-methylindolyl groups, the χ-values increase as well, suggesting a
weaker donating ability for 3-methylindolyl compared to phenyl. The exception to this is
the coupled indolyl phosphine, 4, which has a χ-value greater than 1. A possible reason
for the increase in net-basicity of 4 may be a change in the hybridization of the
phosphorus from the coupling of the indolyl substituents, resulting in greater p-character
of the lone pair.
As it was discussed in Section 2.3.1, 2-indolylphosphines can be modified at the
nitrogen centre to produce functionalized ligands. The utility of the indolyl NH site is
demonstrated with the N-alkylated 2-indolylphosphines, whereby the net-basicity of the
parent phosphine can be altered depending on the substituent on nitrogen. For example,
the electron donating methyl group of (N-Me)-1 (χ = 14.8 cm-1) increases the net-
basicity of 1 (χ = 16.7 cm-1); similarly as expected, the electron withdrawing
pentafluorobenzyl moieties of (N-F5Bn)2-4 appear to decrease the net-basicity of 4 from
χ = 15.6 cm-1 to χ = 20.1 cm-1. Consequently, the net-basicity of 2-indolylphosphines
can be tuned easily by two methods: changing the substituents on phosphorus, or
furnishing different substituents on nitrogen. The symmetric phosphine 5 possesses the
largest χ-value (20.4 cm-1) of the 2-indolylphosphines investigated, with a net-basicity
that approaches P(2-CH2CH2CN)3 (χ = 21.8 cm-1). The three indolyl NH sites of 5 have
demonstrated collective hydrogen bonding to bind anion guests, as discussed in
Section 3.2.2. Alternatively, the nitrogen centres can be modified with a variety of
different substituents, analogous to (N-Me)-1 and (N-F5Bn)2-4, which would increase the
range of net-basicities that can be achieved with 2-indolylphosphines.

108
4.2.2. [5·X]- Complexes: Simultaneous Coordination at
Phosphorus with Ni(II)
As it was detailed in Section 3.2.2, phosphine 5 has the ability to act as an anion
receptor through its three indolyl NH functionalities. It was anticipated that simultaneous
coordination at the phosphorus centre may be feasible, while the NH sites were engaged
in hydrogen bonding. To explore the possibility of concurrent P-coordination and
hydrogen bonding, three [5·X]- complexes were isolated by mixing 5 with F-, Cl-, or Br-
(as their tetraethylammonium salts), which were subsequently reacted with Ni(CO)4 in
THF (Scheme 4.2).
P
HN
3
X
NEt4THF+ Ni(CO)4 P
HN
3
X
NEt4 Ni(CO)3
Scheme 4.2. General scheme for the formation of a monodentate [NEt4][Ni(CO)3(5·X)] complex. X = F-,
Cl-, Br-.
The reaction produced a product which exhibited an unambiguous IR spectrum consistent
with a monosubstituted Ni(CO)3L complex. The χ-values for the three [5·X]- complexes
were determined, and are listed in Table 4.2.

109
Table 4.2. Infrared CO stretching frequencies of [Ni(CO)3(5·X)]- in heptanes.
Entry
[5.X]-
νCO (A1) (cm-1)
χ (cm-1)
1
P
HN
3
F
(7)
2069.6
13.5
2
P
HN
3
Cl
(21)
2074.2
18.1
3
P
HN
3
Br
(22)
2074.2
18.1
The χ-values determined for the [5·X]- complexes show an increase in the net-
basicity compared to uncomplexed 5 (χ = 20.4 cm-1). This increase in χ-value suggests
that the presence of the hydrogen bonded anion not only influences the net-basicity of the
phosphine, but provides evidence that simultaneous coordination chemistry can occur at
the phosphorus centre while the phosphine is acting as an anion receptor. Although the
Cl- and Br- complexes of 5 do not appear to have a significant impact on its net-basicity,
complex [5·F]- has a χ-value (13.5 cm-1) that approaches PPh3 (χ = 12.8cm-1). The
hydrogen bonds are strong enough (as seen in the hydrogen bond distances and stability
constants, Section 3.2.2) that they can survive solvation and simultaneous coordination at
the phosphorus centre. Thus the anion binding ability of 5 provides a supplementary
means to alter the net-basicities of 2-indolylphosphines.

110
It is unlikely that the χ-values determined for the [5·X]- complexes are due to the
coordination of the halide to Ni(CO)3. According to Cassar and co-workers,6 the
treatment of a [Ni(CO)3X]- complex with PPh3 irreversibly yields Ni(CO)3PPh3 and free
halide. Additionally, the A1 carbonyl stretching frequencies for the [Ni(CO)3X]-
complexes are substantially lower than those of the [Ni(CO)3(5·X)]- complexes: Br-
νCO = 2051.0 cm-1 and Cl- νCO = 2049.0 cm-1.
4.2.3. [5·Anion]- Complexes: Simultaneous Coordination
at Phosphorus with Cu(I)
In continuing to explore the simultaneous coordination at phosphorus while
hydrogen bonding at nitrogen, the initial intent was to develop a four-coordinate
tetrahedral [Cu(5)2(phen)]+ complex as a building block for supramolecular assemblies
(Scheme 4.3). One can envisage the addition of an appropriately shaped dianion that can
result in the self-assembly of higher architectures through hydrogen bonding to the two
hydrogen bond donating cavities of the [Cu(5)2(phen)]+ complex.
N
NHN
P
HN NH
N
N Cu PP
HNHN
3
3
BF4
NCMe
CuMeCN NCMe
NCMe
BF4
MeCN
2 +
no reaction
Scheme 4.3. Anticipated four-coordinate tetrahedral Cu(I) complex in the reaction of 5 (2 eq), phen , and
[Cu(MeCN)4]BF4.

111
The reaction of 5 and phen with [Cu(MeCN)4]BF4 in CH2Cl2 in a 2 : 1 : 1 stoichiometry
resulted in the formation of a [Cu(5)(phen)]BF4 complex (Scheme 4.4).
N
NHN
P
HN NH
NCMe
CuMeCN NCMe
NCMe
BF4
MeCN
2 +
34%
CuN
N
P
HNHN
HN BF4
23 Scheme 4.4. Three coordinate trigonal planar Cu(I) complex, 23, yielded from reaction of 5 (2 eq), phen,
and [Cu(MeCN)4]BF4.
The stoichiometry of the [Cu(5)(phen)]BF4 complex, 23, was confirmed by single crystal
X-ray diffraction, and experimental details are listed in Table 4.3. Crystals of 23 were
grown from Et2O vapour diffusion into a CH2Cl2 solution of the complex. Tables 4.4 and
4.8 lists selected bond angles and distances and the numbering scheme is shown in Figure
4.1.
Complex 23 crystallizes in the space group Pī with two molecules in the unit cell.
The Cu(I) adopts a three-coordinate essentially trigonal planar geometry where P1 of 5
deviates from the plane defined by the chelated phen ligand and Cu(I) by 0.654(3) Å.
The Cu(I)-P1 distance is 2.1729(8) Å and is short for Cu(I)-P bond lengths. A survey of
the literature does not produce three-coordinate copper(I)-phosphine complexes; however,
several examples of four-coordinate copper(I)-phosphine complexes were found to have
Cu(I)-P bond lengths that ranged from 2.24 to 2.37 Å.7-9 The shorter Cu(I)-P bond length
of 23 is most likely due to the smaller coordination number around the Cu(I) centre,
which eliminates some of the steric crowding from the ligands observed in the four-
coordinate copper(I) complexes.

112
Table 4.3. X-ray crystallographic experimental data for complexes: 23, 24, and 25.
23
CuN
N
P
HNHN
HN BF4
24
NH
PPd
Cl
ClCl
HN
PPd
Cl
25
NH
PPd
Cl
ClCl
HN
PPd
Cl
Formula C39H32CuN5PBF4 C42H36Cl4N2P2Pd2 C44H64Cl8N2P2Pd2 Formula Weight 752.02 985.27 1179.31 Crystal colour, shape yellow, cubes dark orange, plates orange, block Crystal size, mm 0.26 x 0.20 x 0.10 0.56 x 0.30 x 0.02 0.14 x 0.12 x 0.06 Crystal system Triclinic Monoclinic Rhombohedral Space group P -1 P21/c R -3 a, Å 12.1112(4) 15.6092(5) 26.5804(11 b, Å 12.1741(4) 8.0167(2) 26.5804(11) c, Å 13.4482(6) 15.9823(4) 21.1789(7) α deg 68.866(2) 90 90 β, deg 70.427(2) 96.823(1) 90 γ deg 72.803(2) 90 120 V, Å3 1707.76(11) 1985.77(9) 12958.6(9) Z 2 2 9 Dcalcd, g cm-3 1.462 1.648 1.360 F(000) 772 984 5400 μ, mm-1 0.746 1.289 1.080 λ (Mo Kα), Å 0.71073 0.71073 0.71073 Limiting indices -15<=h<=15 -20<=h<=20 -34<=h<=26 -15<=k<=15 -8<=k<=10 -30<=k<=34 -15<=l<=17 -19<=l<=20 -27<=l<=27 2θ range, deg 2.78 to 27.52 2.85 to 27.54 2.61 to 27.50 Max. and min. transmission 0.950 and 0.564 0.978 and 0.763 0.9380 and 0.8635 No. of reflections collected 18062 14479 15629 No. of independent reflections/Rint 7758 / 0.1151 4839 / 0.0501 6527 / 0.0579 Extinction coefficient - 0.0021(5) 0.00006(5) No. of refined parameters 463 237 285 Final R1, wR2 0.0549, 0.1299 0.0321, /0.0827 0.0627, 0.1593 Final R1, wR2 (all data) 0.1176, 0.1534 0.0408, 0.0880 0.1206, 0.1852 Goodness of fit 1.042 1.032 0.988 Δρmin, Δρmax, eÅ-3 -0.731, 0.575 -1.065, 0.675 -0.841, 1.795

113
Figure 4.1. The ORTEP diagram and numbering scheme of [Cu(5)(phen)]BF4, 23. The broken lines show
the hydrogen bonding between the indolyl NH sites and the BF4- anion. Selected hydrogen atoms shown,
others removed for clarity. Thermal ellipsoids shown at 35% probability.
It appears from Figure 4.1 that the methyl groups of 5 are in relatively close
proximity to the Cu(I) centre in complex 23, suggesting a possible Cu(I)…H-C agostic
interaction. Agostic interactions are typically characterized by interaction distances and
angles ranging from M…H ≈ 1.8 – 2.3 Å, and M…H-C ≈ 90 – 140o, respectively.10
Additionally, an agostic interaction would be indicated by proton signals in the hydride
region in its 1H NMR spectrum. The Cu(I)…H non-bonding distances range from 2.477
to 3.016 Å, and the <Cu(I)…H-C angles range from 134.2 to 146.8o. Even the shortest
Cu(I)…H10A interaction (2.477 Å) with a Cu(I)…H10A-C10 interaction angle at 134.2o
would be considered too large to be an agostic interaction. Furthermore, all the methyl
resonances of 23 appear as a singlet at 2.49 ppm. Thus, the Cu(I)…H interactions of 23
may be classified as electrostatic interactions: the methyl resonances of 23 appear
downfield compared to the uncomplexed ligand; the Cu(I)…H distances and the Cu(I)…H-
C angles are larger than typical agostic interactions.

114
Table 4.4. Selected bond lengths (Å) and bond angles (o) for complex 23.
CuN
N
P
HNHN
HN BF4
(23) Cu1-P1 2.1729(8) N1-C2 1.392(4)
Cu1-N31 2.021(3) C2-C3 1.378(4) Cu1-N34 2.063(3) C3-C4 1.427(5)
C4-C9 1.408(4) N(31)-Cu(1)-P(1) 140.38(8) C9-N1 1.376(4) N(34)-Cu(1)-P(1) 135.01(8)
N(31)-Cu(1)-N(34) 82.24(10) <C9-N1-C2 108.8(2) <N1-C2-C3 109.2(3)
P1-C2 1.806(3) <C2-C3-C4 106.6(3) P1-C12 1.801(3) <C3-C4-C9 107.7(3) P1-C22 1.808(3) <C4-C9-N1 107.6(3)
N21-C22 1.389(4) N11-C12 1.394(3) C22-C23 1.381(4) C12-C13 1.363(4) C23-C24 1.428(4) C13-C14 1.435(4) C24-C29 1.420(4) C14-C19 1.406(4) C29-N21 1.371(4) C19-N11 1.366(4)
<C29-N21-C22 109.4(3) <C19-N11-C12 109.3(2) <N21-C22-C23 109.1(3) <N11-C12-C13 109.3(3) <C22-C23-C24 106.8(3) <C12-C13-C14 106.5(3) <C23-C24-C29 107.5(3) <C13-C14-C19 107.7(3) <C24-C29-N21 107.3(3) <C14-C19-N11 107.2(3)
N1…F4 2.879(3) <N1-H1…F4 151.8
N11…F3 2.994(3) <N11-H11…F3 140.4 N21...F1 2.983(3) <N21-H21…F1 129.9
In contrast to the weak BF4- binding ability of 5 in solution (Ka = 150 M-1) the
solid-state structure of 23 clearly shows the indolylphosphine ligand binding the
tetrafluoroborate counteranion. The anion does not interact with 5 through one of its
fluoride atoms only, as might be anticipated from the crystal structure of [NEt4][5·F].
Rather, the hydrogen bonding between 5 and BF4- demonstrates the C3-symmetric
complementarity between the tetrahedral geometry of the anion and the three hydrogen
bond donors of 5. It appears that the BF4- anion is too large in size to fit well inside the
receptor cavity of 5 as the hydrogen bonding distances are longer than typical N...F-
distances11 and the NH…F- bond angles (130 to 152o) are much less than ideally linear
hydrogen bonding interactions.

115
The complex stacks in alternating head-to-head and tail-to-tail fashion (Figure
4.2). The head-to-head stacking appears to consist of intermolecular π-cation interactions
at a distance of 3.84 Å between the central fused benzene ring of the phen ligand of one
complex and the Cu(I) ion of another complex.
Figure 4.2. The unit cell diagram of [Cu(5)(phen)]+, viewing along the crystallographic c-face. Cation-π
interactions are likely between symmetry equivalent complexes about the crystallographic inversion centre.
Hydrogen atoms have been removed for clarity.
Three-coordinate trigonal planar Cu(I) complexes can result from the dissociation
of ligands as a result of steric interactions between bulky groups, as is seen in
[Cu(dmp)(PPh3)2]+, which has unfavourable contacts between the phenyl substituents of
PPh3 and the methyl groups of 2,9-dimethyl-1,10-phenanthroline (dmp).12 It is likely that
the bulkiness of 5, by virtue of the three methyl groups on the indolyl substituents,
prevented the coordination of a second phosphine to Cu(I) and resulted in the metal
centre to be three-coordinate. Alternatively, the larger steric crowding of the three
methyl moieties of 5 may have caused the dissociation of a second phosphine on the
Cu(I) centre. The ligand stoichiometry of [Cu(5)(phen)]+ suggests that 5 is larger than
PPh3 (cone angle 145o)13 which can readily form four-coordinate tetrahedral
[Cu(PPh3)2(phen)]+ complexes.12 It had been previously suggested that phosphines 1 and
3 are likely to possess cone angles larger than PPh3, and may be similar in size to P(o-

116
tolyl)3.13 Therefore, it is in agreement that 5 should also be similar in size to P(o-tolyl)3,
as is suggested by the ligand stoichiometry of [Cu(5)(phen)]+.
4.3. Pd(II) and Pt(II) Complexes of Monodentate 2-
Indolylphosphines
2-Indolylphosphines can serve as neutral 2e- monodentate phosphorus ligands,
and their coordination chemistry with Pd(II) has been well documented elsewhere.14 A
general reaction scheme for the synthesis of dimeric [Pd(2-indolylphosphine)Cl(μ-Cl)]2
complexes is illustrated in Scheme 4.5. Generally, the reaction involves adding a
stoichiometric amount of phosphine to a stirring solution of Pd(COD)Cl2 in MeCN, the
complex is precipitated from the reaction solution with Et2O. With the ligand to Pd(II)
ratio restricted to 1 : 1, the dimeric species is always formed.
NH
PPd
Cl
ClCl
HN
PPd
ClHN
PPd(COD)Cl2MeCN, r.t.
15 min
24 Scheme 4.5. Reaction scheme for the formation of [Pd(1)Cl(μ-Cl)]2, 24. This is a representative example
of the general reaction sequence for the synthesis of [Pd(2-indolylphosphine)Cl(μ-Cl)]2 complexes.
The addition of 2 : 1 ligand to metal stoichiometry results in the formation of
monomeric species. The cis-Pt(3)2Cl2 complex, 28, was formed by reaction of Zeise’s
salt and two equivalents of phosphine 3 in acetone (Scheme 4.6). Complex 28
precipitated from the reaction solution and was acquired in moderate yield. The 31P
NMR spectrum of 28 exhibited one phosphorus environment at δ = -17.5 ppm (JPt-P =

117
3579 Hz), which is consistent with the 31P chemical shift of cis-Pt(PPh3)2Cl2 (δ =
11.7 ppm, JPt-P = 3679 Hz).15 Attempts to isomerize complex 28 into the trans-isomer by
refluxing 28 in CH2Cl2 led to intractable species. It is thought that the complicated
reactivity of complex 28 may be a result of the indolyl NH hydrogen bond donors
interacting with the chloro ligands on Pt(II), either inter- or intramolecularly.
HN
PPt
ClCl
HN
NH
P HNNH
P HN
K[PtCl3(C2H4)]acetone, r.t.
15 min
66%
28 Scheme 4.6. Reaction scheme for the formation of cis-Pt(3)2Cl2, 28.
Crystallographic analyses of the metal complexes of various monodentate 2-
indolylphosphines are shown in Figure 4.3.
NH
PPd
Cl
ClCl
HN
PPd
Cl
NH
PPd
Cl
ClCl
HN
PPd
ClHN N
H
NHP
PdCl
ClClPd
ClNH
HNP H
N
NH
PPd
Cl
ClCl
HN
PPd
Cl
HN
PPt
ClCl
HN
NH
P HN
(a) (b)
(c) (d) (e)
24 25
26 27 28
Figure 4.3. Metal complexes of monodentate 2-indolylphosphines. (a) [Pd(1)Cl(μ-Cl)]2 (24).
(b) [Pd(2)Cl(μ-Cl)]2 (25). (c) [Pd(3)Cl(μ-Cl)]2 (26). (d) [Pd(4)Cl(μ-Cl)]2 (27). (e) cis-Pt(3)2Cl2 (28).

118
4.3.1. X-ray Crystallographic Analysis of Pd(II) and Pt(II)
Complexes of Monodentate 2-Indolylphosphines
The X-ray crystallographic experimental data for [Pd(1)Cl(μ-Cl)]2, 24, and
[Pd(3)Cl(μ-Cl)]2, 25, are listed in Table 4.3, while the remaining metal complexes are
listed in Table 4.5.
Table 4.5. X-ray crystallographic experimental data for complexes: 26, 27, and 28.
26
NH
PPd
Cl
ClCl
HN
PPd
ClHN N
H
27
NHP
PdCl
ClClPd
ClNH
HNP H
N
28
HN
PPt
ClCl
HN
NH
P HN
Formula C48H42Cl4N4P2Pd2 C48H42Cl8N4O2P2Pd2 C50H48Cl2N4OP2Pt Formula Weight 1091.40 1265.20 1048.85 Crystal colour, shape dark orange, needles dark orange, block colourless, block Crystal size, mm 0.08 x 0.05 x 0.04 0.32 x 0.18 x 0.02 0.40 x 0.36 x 0.26 Crystal system Monoclinic Triclinic Monoclinic Space group P21/c P -1 P21/c a, Å 8.2002(4) 8.7188(2) 11.0184(3) b, Å 19.3095(10) 11.8646(4) 34.3286(5) c, Å 14.4641(9) 12.2122(3) 11.8324(3) α deg 90 79.429(14) 90 β, deg 100.594(2) 79.387(13) 92.221(9) γ deg 90 87.593(13) 90 V, Å3 2251.2(2) 1220.56(6) 4472.2(2) Z 2 1 4 Dcalcd, g cm-3 1.610 1.721 1.558 F(000) 1096 632 2104 μ, mm-1 1.147 1.285 3.371 λ (Mo Kα), Å 0.71073 0.71073 0.71073 Limiting indices -10<=h<=10 -11<=h<=11 -14<=h<=14 -25<=k<=25 -15<=k<=15 -44<=k<=44 -19<=l<=18 -15<=l<=15 -14<=l<=15 2θ range, deg 1.78 to 27.59 1.72 to 27.58 1.19 to 27.50 Max. and min. transmission 0.9556 and 0.9138 0.981 and 0.814 0.430 and 0.336 No. of reflections collected 18674 19119 31469 No. of independent reflections/Rint 5154 / 0.1572 5618 / 0.0635 10223 / 0.0544 Extinction coefficient 0.0138(9) 0.0042(9) 0.00035(10) No. of refined parameters 274 307 547 Final R1, wR2 0.0625, 0.1260 0.0387, 0.0943 0.0421, 0.0925 Final R1, wR2 (all data) 0.1546, 0.1721 0.0510, 0.1018 0.0590, 0.0995 Goodness of fit 0.985 1.050 1.053 Δρmin, Δρmax, eÅ-3 -1.056, 1.167 -1.367, 0.887 -1.455, 1.836

119
Single crystals of 24 were grown from solvent vapour diffusion of hexanes into a
solution of the complex in CH2Cl2. The numbering scheme of the complex is shown in
Figure 4.4 and Tables 4.6 and 4.8 lists selected bond angles and distances. The solid-
state structure of 24 consists of discrete molecules of the dimer sitting about
crystallographic inversion centres in the monoclinic space group P21/c, with one
molecule of the dimer in the asymmetric unit. The Pd(II) exhibits the expected square
planar coordination geometry and share a number of structural similarities with
previously reported [Pd(L)Cl(μ-Cl)]2 dimers. Most notably, the Pd-P bond lengths of
2.2331(7) Å is consistent with Pd-P bond lengths reported for other palladium-phosphine
complexes.16,17 Intramolecular hydrogen-bonding contacts are evident between N1-H1A
and the terminally bound Cl2 at a distance of 3.109(3) Å and a bond angle of 134.0o.
There appear to be no intermolecular hydrogen bonding interactions in the extended
solid-state.
Compared to the crystal structure of 1, the most significant differences in 24 can
be found in the P-C bond lengths. The P-C bond distances in 1 ranged from 1.811(2) to
1.837(4) Å, whereas the corresponding bond distances in 24 are shorter and range from
1.798(3) to 1.810(3) Å. The shortening of P-C bond lengths are consistent with
phosphine coordination to Pd(II).
Figure 4.4. The ORTEP diagram and numbering scheme of 24. The broken lines show the intramolecular
hydrogen bonding between the indolyl NH and the terminal chloro ligand. Non-acidic protons removed for
clarity. Thermal ellipsoids are drawn at 35% probability.

120
Table 4.6. Selected bond lengths (Å) and bond angles (o) for complexes 24 and 25.
24
NH
PPd
Cl
ClCl
HN
PPd
Cl
25
NH
PPd
Cl
ClCl
HN
PPd
Cl
Pd1-P1 2.2331(7) 2.260(2) Pd1-Cl1 2.3208(6) 2.324(2) Pd1-Cl2 2.2794(7) 2.276(2)
Pd1-Cl1#1 2.4312(6) 2.446(1)
<P1-Pd1-Cl2 89.45(3) 90.24(5) <P1-Pd1-Cl1 94.51(2) 95.60(5)
<Cl2-Pd1-Cl1#1 91.27(2) 89.53(5) <Cl1-Pd1-Cl1#1 84.66(2) 84.68(5)
P1-C2 1.798(3) 1.803(6)
P1-C12 1.810(3) 1.825(6) P1-C22 1.808(3) 1.841(6)
N1-C2 1.380(4) 1.399(7) C2-C3 1.388(4) 1.350(8) C3-C4 1.424(4) 1.433(7) C4-C9 1.408(4) 1.395(8) C9-N1 1.366(4) 1.361(7)
<C9-N1-C2 108.9(2) 108.6(5) <N1-C2-C3 109.6(3) 108.8(5) <C2-C3-C4 105.9(3) 107.6(5) <C3-C4-C9 107.8(3) 106.7(5) <C4-C9-N1 107.8(3) 108.3(5)
N1...Cl2 3.109(3) 3.284(5)a
<N1-H1…Cl2 134.0 163.4a
a -x+y+4/3, -x+2/3, z-1/3, intermolecular hydrogen bonding.
The solid-state structure of 25 was acquired by vapour diffusion of hexanes into a
solution of the complex CH2Cl2. A molecular diagram and numbering scheme for 25 is
illustrated in Figure 4.5, selected bond distances and angles are listed in Tables 4.6 and
4.8. Complex 25 crystallizes in the rhombohedral space group R-3. Within the
asymmetric unit are contained: one dimer molecule; two CH2Cl2 solvate molecules, one
molecule which is disordered about a crystallographic 3-fold rotational axis and modeled
at 33% occupancy, while the other molecule is modeled at 66% occupancy.

121
Figure 4.5. The ORTEP diagram and numbering scheme of 25 Non-acidic protons and CH2Cl2 solvent
molecules removed for clarity. Thermal ellipsoids are drawn at 35% probability.
Similar to the solid-state structure of 24, the Pd(II) centre in complex 25 has
expected square planar coordination geometry. The Pd(II)-P1 bond length of 25
(2.260(2) Å) is comparable to that belonging to 24. While there are no intramolecular
hydrogen bond interactions there is evidence for intermolecular interactions. The
secondary interactions occur between symmetry related dimmers from the terminal Cl2 to
an indolyl NH with a hydrogen bond donor-to-acceptor distance of 3.284(5) Å and an
angle of 163.4o. In comparing the phosphine of complex 25 to the solid-state structure of
the free phosphine, 2, the largest differentiation is the shortening of the P-C bond
distances upon coordination of Pd(II). The P-C bond lengths range from 1.818(2) to
1.858(2) Å in 2, whereas the same bond lengths in 25 ranges from 1.803(6) to 1.841(6) Å.
Single crystals of 26 were obtained from solvent vapour diffusion of Et2O into a
solution of the complex in CH2Cl2. Figure 4.6 shows the molecular structure and
numbering scheme for the complex, while Tables 4.7 and 4.8 lists selected bond distances
and angles. Complex 26 is isostructural to the solid-state structure of 24, also
crystallizing in the monoclinic space group P21/c with one dimer molecule in the
asymmetric unit that sits about a crystallographic inversion centre. The Pd(II) centre is
square planar with a Pd(II)-P1 bond length of 2.220(2) Å, and is comparable to the
analogous bond length found in 24. Similar intramolecular hydrogen-bonding

122
interactions exist between the terminal Cl2 in 26 and the N11-H11A group of the suitably
orientated indolyl substituent. However, intermolecular hydrogen bonding is not seen in
the extended structure between dimers. Consistent with the P-C bond lengths shortening
upon coordination seen in complexes 24 and 25, the phosphine in 26 also exhibits shorter
P-C bond lengths (1.790(8) to 1.805(7) Å) in contrast to the free ligand 3 (1.809(2) to
1.841(2) Å).
Figure 4.6. The ORTEP diagram and numbering scheme of 26. The broken lines show the intramolecular
hydrogen bonding of an indolyl NH and the terminal chloro ligand. Non-acidic protons removed for clarity.
Thermal ellipsoids are drawn at 35% probability.

123
Table 4.7. Selected bond lengths (Å) and bond angles (o) for complexes 26, 27, and 28.
26
NH
PPd
Cl
ClCl
HN
PPd
ClHN N
H
27
NHP
PdCl
ClClPd
ClNH
HNP H
N
28
HN
PPt
ClCl
HN
NH
P HN
28
HN
PPt
ClCl
HN
NH
P HN
Pd1-P1 2.220(2) 2.2162(8) 2.246(1)c 2.246(1)c Pd1-Cl1 2.319(2) 2.3163(7) 2.334(1)c 2.334(1)c Pd1-Cl2 2.269(2) 2.2901(8) 2.356(1)c 2.356(1)c
Pd1-Cl1#1 2.421(2) 2.4385(8) 2.267(1)d 2.267(1)d
<P1-Pd1-Cl2 86.90(8) 87.42(3) 96.31(4)e 96.31(4)e <P1-Pd1-Cl1 95.43(7) 94.68(3) 90.85(4)f 90.85(4)f
<Cl2-Pd1-Cl1#1 91.72(7) 91.39(3) 86.93(4)f 86.93(4)f <Cl1-Pd1-Cl1#1 86.04(7) 86.75(3) 85.89(4)g 85.89(4)g
P1-C2 1.795(8) 1.772(3) 1.803(5) 1.814(5)i P1-C12 1.790(8) 1.778(3) 1.797(5) 1.809(5)i P1-C22 1.805(7) 1.807(3) 1.820(5) 1.816(5)i
N1-C2 1.388(9) 1.385(4) 1.388(6) 1.395(6)i C2-C3 1.385(10) 1.377(4) 1.379(6) 1.388(7)i C3-C4 1.416(10) 1.426(4) 1.443(7) 1.436(6)i C4-C9 1.438(10) 1.422(4) 1.403(7) 1.394(7)i C9-N1 1.370(10) 1.366(4) 1.385(6) 1.368(6)i
<C9-N1-C2 108.8(6) 108.5(2) 108.0(4) 109.0(4)i <N1-C2-C3 109.7(6) 109.9(3) 110.5(4) 109.1(4)i <C2-C3-C4 106.8(6) 106.6(3) 105.6(4) 105.7(4)i <C3-C4-C9 107.1(7) 106.9(3) 107.9(4) 108.4(4)i <C4-C9-N1 107.5(6) 108.1(3) 108.0(4) 107.9(4)i
N11-C12 1.403(9) 1.390(4) 1.402(6) 1.402(6)i C12-C13 1.365(10) 1.375(4) 1.381(6) 1.384(6)i C13-C14 1.426(10) 1.429(4) 1.427(7) 1.432(7)i C14-C19 1.409(11) 1.408(4) 1.407(7) 1.416(6)i C19-N11 1.380(9) 1.378(4) 1.374(6) 1.376(6)i
<C19-N11-C12 107.2(6) 108.0(2) 109.0(4) 109.5(4)i <N11-C12-C13 110.0(6) 110.2(2) 108.6(4) 108.9(4)i <C12-C13-C14 107.2(6) 106.1(3) 106.9(4) 106.3(4)i <C13-C14-C19 106.9(6) 107.7(2) 107.8(4) 108.4(4)i <C14-C19-N11 108.7(6) 107.9(3) 107.6(4) 106.8(4)i
N1...Cl 3.096(7) 3.243(3)a - 2.994(6)j
N11...Cl - 2.941(4)b 3.195(5)h 3.414(4)k N21...Cl - - - 2.982(6)m
<N1-H1…Cl 130.6 129.2 - 138.4j
<N11-H11…Cl - 160.3 155.6h 123.6k <N21-H21…Cl - - - 152.8m
a-x+2,-y+2,-z+1, intramolecular hydrogen bonding. b-x+1,-y+2,-z+1, intermolecular hydrogen bonding. cPt1-atom bond distance. dPt1-P2 bond distance. e<P1-Pt1-P2 bond angle. f<atom-Pt1-atom bond angle. g<P2-Pt1-Cl2 bond angle. hN11…N41 intramolecular hydrogen bonding. iAngles associated with indolyl substituents on P2. jN31…N11 intramolecular hydrogen bonding. kN41…O1S intermolecular hydrogen bonding. mN41…O1S intermolecular bonding.

124
Single crystals of 27 were obtained from solvent vapour diffusion of hexanes into
a solution of the complex in CH2Cl2. Figure 4.7 shows the molecular structure and
numbering scheme of the complex and Tables 4.7 and 4.8 lists selected bond distances
and angles. The dimeric complex crystallizes in Pī with each half of the dimer sitting
about a crystallographic inversion centre. Per asymmetric unit, there is one molecule of
water and one molecule of CH2Cl2 also present in the lattice.
Figure 4.7. The ORTEP diagram and numbering scheme of 27. The broken lines show the intramolecular
hydrogen bonding from one of the indolyl NH to the terminal chloro ligand. Non-acidic protons, CH2Cl2,
and water solvate removed for clarity. Thermal ellipsoids are drawn at 35% probability.
As exhibited in complexes 24-26, the Pd(II) centre in 27 adopts the expected
square-planar coordination geometry and monodentate P-coordination of the ligand is
obtained. Structural consequences derived from coordination of 4 in 27 can be acquired
from a comparison of its solid-state structure with those of the uncomplexed N-
substituted derivatives of 4. This reveals that the <C3-C10-C13 bond angle of 113.9(2)°
in the phosphacycle of 27 remains essentially unchanged from the values observed in (N-
CH2NMe2)2-4 and (N-F5Bz)2-4 (which are 113.0(2) and 112.9(2)o, respectively).
Nevertheless, the 4-5° increase in bond angle between indolyl substituents observed upon
P-coordination to Pd(II) in 26 is still seen in 27 and implies that coupling of the indolyl
substituents does not inhibit their “opening up” upon P-coordination (Table 4.8). A
deviation from coplanarity of 5.6(1)° between the two indolyl groups indicates that the
coupled indole remains effectively planar upon P-coordination.

125
Symmetry-related dimers of 27 pack in a laddered head-to-tail fashion that
extends up the a-axis of the unit cell. Both crystallographically independent indolyl NH
groups of each dimer are involved in hydrogen-bonding either within or between the
dimeric units of the ladder as shown in Figure 4.8. One NH group intramolecularly
hydrogen-bonds to its neighbouring terminal chloride as observed in 24 and 26. The
other NH group hydrogen-bonds to the oxygen atom of solvate water which completes a
cyclic network by hydrogen-bonding in turn to the terminal chloride of a symmetry-
related dimer. The CH2Cl2 solvate has no significant intermolecular contacts.
Figure 4.8. Extended hydrogen bonding network of 27 along the crystallographic a-axis. The broken lines
show the intramolecular hydrogen bonding from one of the indolyl NH to the terminal chloro ligand, and
intermolecular hydrogen bonding from the opposite indolyl NH to adventitious water in the lattice. Non-
acidic protons and CH2Cl2 solvent molecules have been omitted for clarity. Thermal ellipsoids are drawn
at 35% probability.

126
Complex 28 has a 31P NMR signal (δ -17.5 ppm, JPt-P = 3579 Hz) consistent with
cis-coordination of the ligands about a Pt(II) centre. The solid-state structure of 28
confirms the isomeric arrangement. Selected bond distances and angles are listed in
Tables 4.7 and 4.8, the molecular structure and numbering scheme is shown in Figure 4.9.
Figure 4.9. The ORTEP diagram and numbering scheme of 28. Non-acidic protons and EtOH solvent
molecule removed for clarity. Thermal ellipsoids are drawn at 35% probability.
Complex 28 crystallizes in the monoclinic space group P21/c with one molecule
of the complex and one additional molecule of EtOH solvate in the asymmetric unit. The
Pt(II) has the expected square planar coordination geometry and the Pt(II)-P1 bond
distances are ordinary (average Pt(II)-P bond length 2.257(2) Å) compared to an
assessment of similar cis-Pt(II)(phosphine)2Cl2 structures in the CSD (average Pt(II)-P
bond length 2.246(3) Å).18 In accord with the Pd(II) complexes, the P-C bond lengths of
ligand 3 shorten upon coordination of the phosphine to Pt(II) (from 1.809(2) -1.841(2) Å
for the free ligand to 1.797(5)-1.820(5) Å for the complexed ligand).
Secondary hydrogen bonding interactions are evident in the solid-state structure
of 28 (Figure 4.10). Interestingly, more intramolecular hydrogen bonding interactions
occur between adjacent NH sites, rather than the conventional intramolecular NH…Cl-
interactions. The close proximity of the indolyl NH sites probably result in the formation
of secondary interactions seen between N11-H11a…N41 (3.195(5) Å, 155.6o), and N31-
H31A…N11 (2.994(6) Å, and 138.4o). Conventional intramolecular hydrogen bonding

127
between N41-H41A…Cl2 is also found in 28 (H41A…Cl2 2.85 Å); however, the
interaction is weaker than the alternate NH…N contacts, as it is approaching the limit of
the sum of van der Waals radii. Intermolecular hydrogen bonding is exhibited between
the OH moiety of the EtOH solvate and one of the indolyl NH sites, N41-H41. An
interaction is formed from N41-H41A to O1S, and the proton of O1S forms an
interaction with N41 in a cyclic fashion. Despite the fact that widespread hydrogen
bonding is apparent in the solid-state structure of 28, these secondary interactions are
restricted to the discrete complex and do not continue into the extended lattice.
Figure 4.10. The varied intra- and intermolecular hydrogen bonding of 28. Broken lines indicate
intramolecular hydrogen bonds from N11…N41, N31…N11, and N41…Cl2. Cyclic intermolecular hydrogen
bonding is exhibited between N41…O1S. Non-acidic hydrogen atoms removed for clarity. Thermal
ellipsoids are drawn at 35% probability.

128
4.3.2. Examining the Effect of Phosphine Net-Basicity on
Pd(II)-P Bond Lengths
The correlation between phosphine net-basicity and Pd(II)-P bond lengths in
complexes 24-27 are examined. While the net-basicity of ligand 2 was not measured, its
net-basicity can be approximated using Tolman’s substituent additivity rule.2 The
individual electronic parameter, χi for a cyclohexyl substituent is 0.1 cm-1 and the
individual electronic parameter of the indolyl substituent of C3-symmetric phosphine 5 is
6.8 cm-1, as determined by Equation 4.1.
χ χ1χ2χ3 = νCO(A1) - 2056.1 cm-1 = Σ3
i=1χi
P (4.1)
By Tolman’s additivity rule, the net-basicity of 2 is estimated to be 2063.1 cm-1, which
makes it the more net-basic than ligands 1, 3, and 4. Therefore, the order of increasing
net-basicity of the 2-indolylphosphines for which there are dimeric [Pd(2-
indolylphosphine)Cl(μ-Cl)]2 complexes is: the diindolylphenylphosphine 3 is the least
net-basic, followed by the indolyldiphenylphosphine 1, then the coupled
indolylphenylphosphine 4, and finally the most net-basic dicyclohexylindolylphosphine 2.
One would expect the Pd(II)-P bond lengths to decrease with increasing
phosphine basicity. On inspection of the Pd(II)-P bond lengths of complexes 24, 25, 26,
and 27, the shortest Pd(II)-P bond length occurs in complex 25 and does indeed belong to
the most net-basic phosphine 2. The trend is consistent with complexes 24 and 26 which
have increasing Pd(II)-P bond lengths, suggesting a decrease in phosphine basicity.
However, the Pd(II)-P bond distance of complex 27 is inconsistent with this argument, it
is the shortest of the four complexes, and can be a result of the difference in hybridization
of the phosphorus atom of 4.
It is anticipated that the more basic the phosphine is, the stronger its trans
influence, which can be seen in a lengthening of the Pd(II)-Cl bond situated trans to the
phosphine. Thus the trans Pd(II)-Cl1#1 bond distances were examined. The order of

129
increasing Pd(II)-Cl1#1 bond lengths is consistent with increasing net-basicity of the 2-
indolylphosphines. The shortest Pd(II)-Cl1#1 bond length is found in complex 26, where
ligand 3 is the least net-basic. The longest Pd(II)-Cl1#1 bond length belongs to complex
25, with ligand 2 being the most net-basic and suggests it has the strongest trans influence.
4.3.3. The Effect of Metal Coordination on the Σ{<CPC}
of Monodentate 2-Indolylphosphines
In general, the average Σ{<CPC} for all of the monodentate 2-indolylphosphines
have shown an increase from the uncoordinated free ligands, and is consistent with the
subtle change of the coordination geometry of the phosphorus atom upon metal
complexation (Table 4.8). This trend however, appears to be irregular and does not seem
to be due to systematic changes in the ligands. The coordination of ligands 1 and 3 with
Pd(II) show an increase in the average Σ{<CPC} to the same value (320.0(2) and
320.1(6)o, respectively) which is expected since both phosphines have similar average
Σ{<CPC} (304.3(2) and 303.8(2)o, respectively). Complex 28 is structurally different
from complex 26, however they both share the same phosphine ligand. Assessment of
the average Σ{<CPC} for 28 reveals that these values (315.6(4) and 312.0(4)o) are
considerably smaller than 26; this appears to be attributable to the cis-conformation of the
two ligands which experience increased steric crowding from the neighbouring ligand’s
substituents, and are thus forced to have a decreased Σ{<CPC}. In addition, the
increased intramolecular hydrogen bonding seen in complex 28 may be a contributing
factor for the smaller Σ{<CPC}, as these secondary interactions function to hold the
substituents together.

130
Table 4.8. Sum of <CPC bond angles (o) and 31P resonances for monodentate 2-indolylphosphine metal
complexes.
Entry
Complex
<C2-P1-C12
<C12-P1-C22
<C22-P1-C2
avg. Σ{<CPC}
31P δ (ppm)a
1
23 Cu
N
N
P
HNHN
HN BF4
102.2(1)
103.7(1)
102.5(1)
308.3(2)
-63.2b
2
24 NH
PPd
Cl
ClCl
HN
PPd
Cl
102.9(1)
107.2(1)
109.9(1)
320.0(2)
14.6
3
25 NH
PPd
Cl
ClCl
HN
PPd
Cl
105.5(3)
106.2(3)
105.4(3)
317.1(5)
20.5
4
26 NH
PPd
Cl
ClCl
HN
PPd
ClHN N
H
104.6(3)
106.2(3)
109.3(4)
320.1(6)
-5.7
5
27 NH
PPd
Cl
ClClPd
ClNH
HNP H
N
99.1(1)
110.4(1)
107.9(1)
317.4(2)
-14.9
6
28 HN
PPt
ClCl
HN
NH
P HN
107.6(2)
101.8(2)b
102.6(2)
107.5(2)b
105.4(2)
102.7(2)b
315.6(4)
312.0(4)b
-17.5
aSpectra collected in CDCl3. bSpectra collected in CD2Cl2. bAngles associated with indolyl substituents on P2.
It was determined that ligand 2 has greater steric crowding in comparison to
ligand 1 (Section 2.2.2), and it was anticipated that the Σ{<CPC} would increase more
for the complexation of a bulkier phosphine, yet this is not the case for 25. The smaller
Σ{<CPC} exhibited in 25 may be a result of the lack of intramolecular hydrogen bonding
which is demonstrated in complexes 24 and 26. Presumably, intramolecular hydrogen
bonding between an indolyl NH and a terminal chloro ligand of the dimeric [Pd(L)Cl(μ-
Cl)]2 complex will encourage the <CPC bond angles to open up to allow for these
secondary interactions.

131
Although a solid-state structure of ligand 4 was not obtained, the average
Σ{<CPC} from the crystal structure of (N-F5Bn)2-4 can be extrapolated to estimate an
average Σ{<CPC} for unsubstituted 4. The average Σ{<CPC} for 4 can be approximated
by the differences in (N-Bn)-1 and ligand 1. The average Σ{<CPC} for 1 is 304.3(2)o
and (N-Bn)-1 is 307.8(2)o. By this extension, the average Σ{<CPC} for (N-F5Bn)-4
(299.2(2)o) should be slightly larger than unfunctionalized 4. Using the approximated
average Σ{<CPC}, the coordination of 4 to Pd(II) has resulted in the largest increase in
the average Σ{<CPC} (317.4(2)o) for all the monodentate 2-indolylphosphines. It is
speculated that the considerable increase in <CPC bond angles may be due to a change in
the hybridization of the phosphorus atom when it coordinates to Pd(II).
Notably, the average Σ{<CPC} for complex 23 did not vary significantly from the
free phosphine, 5. Although the Σ{<CPC} did increase from 306.9(2)o to 308.3(2)o, the
increase is not very substantial in comparison to the other monodentate 2-
indolylphosphines. One explanation may be the collective hydrogen bonding of the
indolyl NH sites to the BF4- anion, which does not allow the <CPC bond angles to open
up as demonstrated with the other ligands.
As it was observed in Section 2.3.1., there is little correlation between the
Σ{<CPC} and the 31P chemical shift of a particular 2-indolylphosphine. Especially when
a 2-indolylphosphine is acting as a monodentate ligand through phosphorus coordination,
the 31P resonance is more greatly influenced by the substituents bonded to it, as well as
the type of transition metal it is coordinating to, rather than the Σ{<CPC} bond angles.
4.3.4. The Effect of Metal Coordination on Indolyl
Aromaticity
The five bond angles within the pyrrolyl ring of the indolyl substituents of each
metal complex were compared to the equivalent bond angles of the free ligands. It was
found that coordination to a metal centre does little to the aromaticity of the indolyl
substituent. Perhaps because the nitrogen centre is not participating in metal coordination,

132
the bond angles of the indolyl ring remain virtually the same as the uncomplexed ligand.
Predictably, the most significant changes to the ligands were to the P-C bond distances
and the <CPC bond angles.
4.4. Conclusions
The coordination of 2-indolylphosphines as monodentate ligands has been
realized with complexes consisting of Ni(0), Cu(I), Pd(II), and Pt(II) species. The
Ni(CO)3(2-indolylphosphine) complexes have allowed for the determination of
phosphine net-basicity based on Tolman’s method. The various 2-indolylphosphines
exhibit a diverse range of net-basicity values: from the most basic ligand 20 with two
electron-donating tert-butyl substituents on phosphorus, to the least basic ligand 5, which
is C3-symmetric with three indolyl substituents on phosphorus. Additionally, the
nitrogen centre can be functionalized with groups of altering electronics that influence the
net-basicity of the parent 2-indolylphosphine.
It was initially introduced in Chapter 3 the ability of phosphine 5 to behave as an
anion receptor through its three indolyl NH functionalities. The coordination of [5·X]-
(where X- = F-, Cl-, Br-) to Ni(CO)3 demonstrates the facility for simultaneous
coordination chemistry at the phosphorus atom while the anion remained hydrogen
bonded to 5. Infrared measurements of the CO stretching frequencies of each
[Ni(CO)3(5·X)]- show that the presence of the hydrogen bonded anions have an impact
on the net-basicity of parent phosphine 5 by increasing it. The fluoride anion imparts the
most significant change, where the [Ni(CO)3(5·X)]- complex has a net-basicity that
approaches PPh3. Consequently, the net-basicity of 2-indolylphosphines can be tuned
relatively easily: by altering the substituents on phosphorus; by adding substituents on
nitrogen; by introducing anions for hydrogen bonding to 5.
Complex 23 also substantiates the ability for simultaneous coordination at
phosphorus while hydrogen bonding to anions at nitrogen for phosphine 5. Most notably,
the three indolyl NH sites collectively hydrogen bond to the BF4- anion in a C3-symmetric
fashion to create a discrete ion pair complex. The stoichiometry of 23 suggests

133
phosphine 5 is quite bulky, which resulted in the formation of the monophosphinated
complex, as opposed to the diphosphinated as originally proposed with the reaction
conditions.
The reaction of one equivalence of 2-indolylphosphine to one equivalence of
Pd(II) produces dimeric [Pd(2-indolylphosphine)Cl(μ-Cl)]2 complexes. Whereas the
addition of an extra equivalent of phosphine ligand, yields a monomeric species, such as
complex 28. It was observed that in all the crystal structures of the monodentate 2-
indolylphosphines that P-C bond distances decreased upon metal coordination from the
uncomplexed ligands.
The net-basicity of a phosphine can be correlated to its trans-influence strength.
Generally, the more basic a phosphine, the shorter the metal-phosphorus bond, and the
longer the metal-atom bond trans to the phosphine. The Pd(II)-P bond distances in
complexes 24-27 demonstrate this trend, with the exception of 26 which can be a result of
the difference in hybridization of the phosphorus atom of ligand 4. Alternatively, the
trans Pd(II)-Cl1#1 bond length is consistent with the expected observation that the more
basic phosphine results in a longer trans Pd(II)-Cl1#1 bonding distance as seen in
complex 25.
Additionally, the Σ{<CPC} of the metal complexes increased from the free
ligands. These changes are most likely from the subtle alteration in the hybridization of
the phosphorus atom when coordinating a metal centre. Since the 2-indolylphosphines
were acting as monodentate ligands through the phosphorus centre, the aromaticity of the
indolyl ring is not affected. Therefore, in the metal complexes, the bond angles within
the pyrrolyl moiety of the indolyl substituents are comparable to the analogous bond
angles of the free ligands.

134
4.5. Experimental
Unless otherwise stated, the synthetic protocol for 2-indolylphosphines and their metal
complexes are reported in Dr. Edmond Lam’s thesis.14
General Considerations. All reactions and manipulations were carried out under an
atmosphere of dinitrogen using standard Schlenk techniques unless otherwise stated.
Ni(CO)4 was purchased from Strem Chemicals and used as received. [Cu(MeCN)4]BF419
and K[PtCl3(C2H4)]20 were synthesized according to literature procedures.
Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl still under a
dinitrogen atmosphere. 1H, 13C, and 31P NMR spectra were recorded on Varian 400 MHz
or 300 MHz NMR systems, and referenced to SiMe4 (TMS) and 85% H3PO4, respectively.
Splitting patterns are indicated as s, singlet; d, doublet; t, triplet; q, quartet;, m, multiplet;
br, broad peak. IR spectra were obtained using a Perkin-Elmer 1000FT-IR spectrometer
as solutions between NaCl plates. High resolution mass spectra were obtained on an
ABI/Sciex Qstar mass spectrometer (ESI).
General preparation of Ni(CO)3(2-indolylphosphines). To a solution of a 2-
indolylphosphine ligand (~0.12 mmol) in THF (5 mL) is added Ni(CO)4 (~1 mL,
7.6 mmol) dropwise. The resultant mixture is left to stir at room temperature for 30
minutes. Unreacted Ni(CO)4, CO and THF are removed into a secondary trap containing
bleach under vacuum. The remaining residues are washed with minimal hexanes.
Ni(CO)3(1) IR (heptane) ν 2072.8 cm-1 (νCO A1), 2006 (νCO A1). Ni(CO)3(3) IR (heptane)
ν 2074.9 cm-1 (νCO A1), 2005 (νCO A1). Ni(CO)3(4) IR (heptane) ν 2071.7 cm-1 (νCO A1),
2008 (νCO A1). Ni(CO)3(5) IR (heptane) ν 2076.5 cm-1 (νCO A1), 2008 (νCO A1).
Ni(CO)3(20) IR (heptane) ν 2065.9 cm-1 (νCO A1), 1996 (νCO A1). Ni(CO)3(N-Me)-1 IR
(heptane) ν 2076.2 cm-1 (νCO A1), 2004 (νCO A1). Ni(CO)3(N-F5Bn)2-4 IR (heptane) ν
2072.8 cm-1 (νCO A1), 2006 (νCO A1).

135
General preparation of NEt4[Ni(CO)3(5·X)]. To a solution of complex NEt4[5·X]
(~0.12 mmol) in THF (5 mL) is added Ni(CO)4 (~1 mL, 7.6 mmol) dropwise. The
resultant mixture is left to stir at room temperature for 30 minutes. Unreacted Ni(CO)4,
CO and THF are removed into a secondary trap containing bleach under vacuum. The
remaining residues are washed with minimal hexanes. NEt4[Ni(CO)3(5·F)] IR (heptane)
ν 2069.6 cm-1 (νCO A1), 1999 (νCO A1). NEt4[Ni(CO)3(5·Cl)] IR (heptane) ν 2074.2 cm-1
(νCO A1), 1997 (νCO A1). NEt4[Ni(CO)3(5·Br)] IR (heptane) ν 2074.1 cm-1 (νCO A1), 1997
(νCO A1).
CuN
N
P
HNHN
HN BF4
[Cu(5)(1,10-phenanthroline)]BF4, 23. Dissolved [Cu(MeCN)4]BF4 (0.168 g,
0.242 mmol), 1 (0.229 g, 0.543 mmol) and 1,10-phenanthroline (0.049 g, 0.272 mmol) in
DCM (20 mL) to give a clear yellow solution. The resultant yellow solution was placed
into a vial containing Et2O. X-ray quality single crystals were formed within one week
(0.061 g, 34 %). 31P NMR (CD2Cl2, 161 MHz): δ -63.2 (br s); 1H NMR (CD2Cl2,
400 MHz): δ 8.92 (br s, JNHF = 3.6 Hz, 3H, indole NH), 8.65 (d, J = 8.4 Hz, 2H, Ar-H),
8.57 (s, 2H, Ar-H), 8.10 (s, 2H, Ar-H), 7.93 (dd, J = 4.4, 8.0 Hz, 2H, Ar-H), 7.64 (d,
J = 8.0 Hz, 3H, Ar-H), 7.44 (d, J = 8.0 Hz, 3H, Ar-H), 7.29 (t, J = 7.6 Hz, 3H, Ar-H),
7.18 (t, J = 7.6 Hz, 3H, Ar-H), 2.49 (s, 9H, indole CH3); 13C NMR (CD2Cl2, 100 MHz):
150.9, 143.9, 139.6, 138.8, 129.6, 127.5, 126.0, 125.6, 124.5, 121.6, 120.3, 119.4, 112.3,
10.4; HRMS ESI+ for [M+] ion C39N32N5PCu+: calcd m/z 664.1657, found m/z 664.1685.
HN
PPt
ClCl
HN
NH
P HN

136
cis-Pt(3)2Cl2, 28. A solution of ligand 3 (0.88 g, 2.4 mmol) in acetone (5 mL) was added
dropwise to a suspension of K[PtCl3(C2H4)] (0.44 g, 1.2 g) in acetone (5 mL) with
stirring. A pale yellow solid precipitated out of solution within 10 minutes. The solid
was isolated by filtration, washed with minimal acetone, and dried in air. The solid was
determined to be the cis-isomer of the product (0.88 g, 66%). cis-28: 31P NMR (CDCl3,
161 MHz): δ -17.5 (JPt-P = 3579 Hz); 1H NMR (CDCl3, 300 MHz): δ 8.50 (br s, 4H,
indole NH), 7.66 – 7.10 (m, 22H, Ar-H), 6.52 (d, J = 7.5 Hz, 4H, Ar-H), 2.17 (s, 18H,
indole CH3); 13C NMR (CDCl3, 100 MHz): 138.8, 135.3, 132.1, 129.6, 129.1, 128.9,
126.2, 123.3, 122.0, 119.7, 119.5, 113.2, 111.8, 9.9.
X-ray Crystallography. X-ray data were collected on a Nonius Kappa CCD
diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). A
combination of 1º φ and ω (with κ offsets) scans were used to collect sufficient data. The
data frames were integrated and scaled using the Denzo-SMN package.21 The structures
were solved and refined with the SHELXTL-PC v6.12 software package.22 Refinement
was by full-matrix least squares on F2 using data (including negative intensities) with
hydrogen atoms bonded to carbon and nitrogen atoms included in calculated positions
and treated as riding atoms. Absorption corrections were made for every structure. All
the heavy atoms were refined anisotropically.

137
4.6. References
(1) Bartik, T.; Himmler, H.-G.; Seevogel, K. J. Organomet. Chem. 1984, 272, 29 - 41.
(2) Tolman, C. A. J. Am. Chem. Soc. 1970, 90, 2953 - 2956.
(3) Kapoor, P. N.; Kakkar, R. Theochem 2004, 679, 149-156.
(4) Oksarsson, Å. Acta Cryst. 1990, B46, 748-752.
(5) Bor, G. Spectrochim. Acta 1962, 18, 817-822.
(6) Cassar, L.; Foá, M. Inorg. Nucl. Chem. Letters 1970, 6, 291-294.
(7) Dori, Z.; Ziolo, R. F.; Gaughan, A. P.; Pierpont, C. G.; Eisenberg, R. Inorg. Chem.
1971, 10, 1289-1296.
(8) Darensbourg, D. J.; Longridge, E. M.; Atnip, E. V.; Reibenspies, J. H. Inorg.
Chem. 1991, 30, 357-358.
(9) Saturnino, D. J.; Arif, A. M. Inorg. Chem. 1993, 32, 4157-4160.
(10) Brookhart, M.; Green, M. L. H.; Parkin, G. Proc. Nat. Acad. Sci. 2007, 104, 6908-
6914.
(11) Steiner, T. Acta. Cryst. 1998, B54, 456-463.
(12) Palmer, C. E. A.; McMillin, D. R. Inorg. Chem. 1987, 26, 3837-3840.
(13) Tolman, C. A. Chem. Rev. 1977, 77, 313 - 348.
(14) Lam, E., PhD Thesis, University of Toronto, 2007.
(15) de Jong, F.; Bour, J. J.; Schlebos, P. P. J. Inorg. Chim. Acta 1988, 154, 89-93.
(16) Coles, S. J.; Faulds, P.; Hursthouse, M. B.; Kelly, D. G.; Ranger, D. C.; Toner, A.
J.; Walker, N. M. J. Organomet. Chem. 1999, 586, 234.
(17) Slawin, A. M. Z.; Woollins, J. D.; Zhang, Q. Inorg. Chem. Commun. 1999, 2, 386.
(18) CSD Database (version 5.29, January 2008).
(19) Kubas, G. J. Inorg. Synth. 1979, 19, 90.
(20) Chock, P. B.; Halpern, J.; Paulik, F. E. Inorg. Synth. 1973, 14, 90-92.
(21) Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307-326.
(22) Sheldrick, G. M. In SHELXTL-Windows NT. V6.12, Bruker Analytical X-Ray
Systems Inc. Madison, WI, 2001.
137

Chapter 5
Multidentate N-Functionalized 2-Indolylphosphines
and their Metal Complexes
5.1. Introduction
5.1.1. Bidentate (P,N)- and (P,P)-Ligands
Bidentate phosphines with hetero-donors such as QUINAP,1 and diphosphines,
such as BINAP,2 are common ligands used in catalysis. Their catalytic success is in part
due to the ability to tune the steric and electronic properties at the various donor sites. It
was revealed in Chapter 2 the ease with which alkyl groups can be furnished at the
indolyl nitrogen; furthermore in Chapter 4 the Ni(CO)3 complexes of the N-alkylated 2-
indolylphosphines demonstrated diverse net-basicities. By this extension, it is possible to
functionalize the nitrogen centre of 2-indolylphopshines to form unsymmetrical bidentate
phosphines with phosphino or amino substituents.
Parent phosphine 1 will serve as the platform for new (P,N)- and (P,P)-2-
indolylphosphines. These new bidentate ligands are coordinated to Pd(II) and Pt(II)
species and their structural features examined by X-ray crystallography. Generally,
phosphines are stronger trans-influence ligands due to their stronger σ-donicity than
amines.3 In this Chapter, the metal complexes of chelating (P,N)- and (P,P)-2-
indolylphosphines will be examined via crystallography to assess the trans-influence of
amino donors versus phosphino donors.
138

139
5.1.2. Symmetric Tetradentate Ligand PP3 and Its
Reactivity
There is continuing interest in the symmetric tripodal tris(2-
diphenylphosphino)ethyl)phosphine, PP3 (Figure 5.1a).4-7 The PP3 ligand has shown to
induce five-coordinate trigonal bipyramidal geometry when it binds to metal centres
(Figure 5.1b). Generally the PP3 ligand binds in a tetradentate fashion to the metal centre
resulting in a trigonal bipyramidal coordination geometry; the remaining fifth
coordination site on the metal is occupied by either an anionic ligand (such as Cl-, Br-, or
I-) or a neutral ligand such as a monophosphine.5,8
P
P
P
P
PP3
(a)
Ph2P MPPh2
PPh2
P
L
ML(PP3)
(b) Figure 5.1. (a) Tris(2-(diphenylphosphino)ethyl)phosphine, PP3. (b) A generic metal complex of PP3
exhibiting trigonal bipyramidal coordination geometry enforced by the PP3 ligand. M denotes a metal
cation, and L is a coordinating ligand.
Ligand 5 has the capacity to be converted to a potential tetradentate phosphine by
functionalizing the nitrogen centres with diphenylphosphino substituents. The utility of
the diphenylphosphino-functionalized 5, (N-PPh2)3-5, will be discussed in this Chapter.
Its coordination chemistry to Pt(II) and Rh(I) and subsequent reactivity will be presented.

140
5.2. Results and Discussion
5.2.1. Synthesis of Multidentate 2-Indolylphosphines
It was introduced in Section 2.2.3. the ease with which a nitrogen centre of 2-
indolylphosphines could be functionalized. Multidentate ligands can be generated by
installing groups that have the capacity to coordinate to metal centres, such groups will
contain for example nitrogen or phosphorus donors. One simple method of obtaining an
N-functionalized 2-indolylphosphine is during its general synthesis (Scheme 5.1); the
aminal-protecting group remains on the nitrogen centre following phosphine molecule
formation, which will produce a (P,N)-ligand where N refers to the donor of the newly
added functional group, and P refers to the phosphorus centre of the 2-indolylphosphine.
The variety in substituents on phosphorus will increase the diversity in (P,N)-ligands.
N
N
NP
R
R
N1. n-BuLi
2. PCl3-xRxTHF
Scheme 5.1. General reaction sequence to generate CH2NMe2-functionalized 2-indolylphosphines.
It has been demonstrated that rigid heteroaromatic rings, such as 2-pyridyl and 2-
isoquinolyl, can be furnished on the indolyl nitrogen centre with a variety of methods.
The pyridyl derivative, (N-py)-1, is synthesized using a method similar for the generation
of N-alkylated 2-indolylphosphines, with the exception of the base used to deprotonate
the nitrogen centre.9
NN
NP
N1. n-BuLi, THF, -78oC
2. Ph2PCl, -78oC3. Workup
(N-py)-1 Scheme 5.2. Reaction scheme for the synthesis of pyridyl-functionalized (N-py)-1.

141
The isoquinolyl derivative, (N-isoquin)-1, is synthesized by initially installing an
isoquinolyl substituent on the indolyl nitrogen by Cu(I) catalyzed cross-coupling between
3-methylindole and 2-bromoisoquinoline.9 Subsequent reaction of the N-functionalized
3-methylindole with n-BuLi, and chlorodiphenylphosphine oxide generates the oxide of
the desired ligand. Reduction with trichlorosilane affords (N-isoquin)-1 in moderate
yields.
NN
NN
PPh2
O NN
PPh2
1. n-BuLiTHF, -78oC
2. Ph2P(O)Cl-78oC
3. Workup
HSiCl3, NEt3dioxane, Δ
(N-isoquin)-1 Scheme 5.3. Reaction scheme for the synthesis of isoquinoline-functionalized (N-isoquin)-1.
Similarly, new (P,P)-ligands can be formed by reacting a 2-indolylphosphine with
a base to deprotonate the nitrogen centre and the ensuing reaction with the desired
phosphino substituent.9 Scheme 5.4 outlines several phosphine functionalized derivatives
of ligand 1. The additional phosphine moieties can range in bulkiness, such as (N-PCy2)-
1, or introduce a second centre of chirality, as in the (N-(R)-BINO)-1 ligand.
HN
PN
P
PR2
1. NaH, THF2. PR2Cl
R = Ph, (N-PPh2)-1R = Cy, (N-PCy2)-1
R = BINAP, (N-(R)-BINO)-1 Scheme 5.4. General reaction scheme to generate phosphorus-functionalized 2-indolylphosphines.
An indolyl-based diphosphine that is related to 2-bis(diphenylphosphino)ethane,
dppe, can be created in the same fashion as other 2-indolylphosphines. Simply reacting
two equivalents of the lithiated aminal-protected 3-methylindole with one equivalent of

142
1,2-bis(dichlorophosphino)ethane, followed by reaction with NaBH4 to cleave the
protecting group affords diphosphine 29 in good yields (Scheme 5.5).
NH
PPHN
HN
NH
N
PPN
N
NN
N
N
NNN 1. n-BuLi, -78oC
2. Cl2P(CH2)2PCl2THF
1. NaBH4EtOH/THF
reflux2. HCl workup
29 Scheme 5.5. Reaction scheme for the synthesis of diphosphine, 29.
Figure 5.2 shows the multidentate 2-indolylphosphines which will be analyzed by X-ray
crystallography.
HN
P
P
HN
NH
NH
NP
P
NP
N
N NN N
P
(N-CH2NMe2)-20
(a)
(N-CH2NMe2)2-4
(b)
(N-PCy2)-1
(c)
29
(d) Figure 5.2. The multidentate 2-indolylphosphines that will be structurally characterized with X-ray
crystallography: (a) P(C12H15N2)(C4H9)2, (N-CH2NMe2)-20 (b) P(C23H26N4)(C6H5), (N-CH2NMe2)2-4
(c) P(C9H7NP(C6H11)2)(C6H5)2, (N-PCy2)-1 (d) [P(C18H16N2)(CH2)]2, 29.
5.2.2. X-ray Crystallographic Analysis of (P,N)- and (P,P)-
2-Indolylphosphines
As it was discussed in Sections 2.2.2. and 2.3., X-ray crystallography will be
employed to determine the impact of altering the substituents on phosphorus and the
introduction of non-alkyl groups on nitrogen on the Σ{<CPC} of 2-indolylphosphines. It

143
was demonstrated in Section 2.3.1. that exchanging substituents on phosphorus or the
incorporation of alkyl groups on nitrogen had very little effect on indolyl aromaticity.
The (P,N)- and (P,P)-ligands will be crystallographically examined to determine if the
presence of non-alkyl moieties on nitrogen will affect indolyl aromaticity. The X-ray
diffraction experimental data for the phosphines in question are listed in Table 5.1.
Table 5.1. X-ray crystallographic experimental data for (P,N)- and (P,P)-2-indolylphosphines.
(N-CH2NMe2)-20
NP
N
(N-CH2NMe2)2-4
NP
N
N
N
(N-PCy2)-1
NP
P
29
HN
P
P
HN
NH
NH
Formula C20H33N2P C29H31N4P C33H39NP2 C38H36N4P2 Formula Weight 332.45 466.55 511.59 610.65 Crystal colour, shape colourless, block colourless, plate colourless, plate colourless, plate Crystal size, mm 0.70 x 0.40 x 0.36 0.30 x 0.25 x 0.12 0.70 x 0.66 x 0.32 0.28 x 0.24 x 0.20 Crystal system Triclinic Monoclinic Monoclinic Monoclinic Space group Pī P21/c P21/c C2/c a, Å 8.2258(2) 12.8780(3) 19.8367(5) 22.6480(7) b, Å 8.8026(2) 18.6590(6) 8.9714(3) 10.8290(2) c, Å 14.4148(4) 11.6680(3) 17.3784(6) 25.8980(8) α deg 83.331(2) 90 90 90 β, deg 74.118(2) 116.2791(18) 115.480(2) 91.9580(12) γ deg 87.283(2) 90 90 90 V, Å3 996.98(4) 2513.94(12) 2791.90(15) 6347.9(3) Z 2 4 4 8 Dcalcd, g cm-3 1.107 1.233 1.217 1.278 F(000) 364 992 1096 2576 μ, mm-1 0.140 0.134 0.178 0.171 λ (Mo Kα), Å 0.71073 0.71073 0.71073 0.71073 Limiting indices -10<=h<=10 -15<=h<=13 -21<=h<=25 -28<=h<=29 -11<=k<=11 -22<=k<=22 -11<=k<=10 -12<=k<=14 -16<=l<=18 -13<=l<=13 -22<=l<=21 -25<=l<=33 2θ range, deg 1.48 to 27.52 2.93 to 25.04 1.14 to 27.49 2.59 to 27.51 Max. and min. transmission 0.959 and 0.450 0.9841 and 0.9609 0.951 and 0.495 0.9666 and 0.9536 No. of reflections collected 9193 17602 17475 21969 No. of independent reflections/Rint 4489 / 0.0778 4431 / 0.065 6795 / 0.0480 7268 / 0.0491 Extinction coefficient 0.38(3) 0.0064(12) 0.0078(17) 0.00044(16) No. of refined parameters 218 312 327 398 Final R1, wR2 0.0626, 0.1586 0.0473, 0.1095 0.0450, 0.1126 0.0522, 0.1208 Final R1, wR2 (all data) 0.0811, 0.1898 0.0725, 0.1237 0.0621, 0.1217 0.0845, 0.1362 Goodness of fit 1.120 1.019 1.022 1.039 Δρmin, Δρmax, eÅ-3 0.516 and -0.973 0.580, -0.229 -0.481, 0.377 -0.329, 0.662

144
Colourless block-shaped crystals of phosphine (N-CH2NMe2)-20 were harvested
from MeOH at -30oC. The molecular structure and numbering scheme of (N-
CH2NMe2)-20 is shown in Figure 5.3, and selected bond distances and angles are listed
in Table 5.2. The solid-state structure of phosphine (N-CH2NMe2)-20 is ordinary; it
crystallizes in the triclinic space group Pī, with one molecule in the asymmetric unit.
There do not appear to be any secondary bonding interactions within the solid-state
structure of the phosphine.
Table 5.2. Selected bond lengths (Å) and bond angles (o) for (P,N)- and (P,P)-2-indolylphosphines.
(N-CH2NMe2)-20
NP
N
(N-CH2NMe2)2-4
NP
N
N
N
(N-PCy2)-1
NP
P
29a
HN
P
P
HN
NH
NH
29b
HN
P
P
HN
NH
NH
P1-C2 1.834(2) 1.811(2) 1.823(2) 1.817(2) 1.807(2)
P1-C12 1.884(2) 1.814(2) 1.830(2) 1.804(2) 1.816(2) P1-C22 1.898(2) 1.845(2) 1.845(2) 1.856(2) 1.851(2)
P2-N1 - - 1.757(1) - - P2-C11 - - 1.858(2) - - P2-C17 - - 1.853(2) - -
N1-C2 1.420(2) 1.404(3) 1.429(2) 1.395(3) 1.400(3) C2-C3 1.385(2) 1.373(3) 1.376(2) 1.376(3) 1.378(3) C3-C4 1.436(2) 1.434(3) 1.432(2) 1.446(4) 1.434(3) C4-C9 1.407(2) 1.408(3) 1.407(2) 1.404(4) 1.406(3) C9-N1 1.375(2) 1.383(3) 1.394(2) 1.363(3) 1.382(3)
<C9-N1-C2 109.2(1) 108.0(2) 107.1(1) 109.3(2) 109.1(2) <N1-C2-C3 107.6(1) 109.4(2) 109.0(1) 109.2(2) 108.6(2) <C2-C3-C4 107.7(2) 107.1(2) 107.8(1) 106.1(2) 107.2(2) <C3-C4-C9 107.4(2) 107.2(2) 107.2(1) 107.4(2) 107.6(2) <C4-C9-N1 108.1(2) 108.3(2) 108.9(1) 107.9(2) 107.5(2)
N11-C12 - 1.397(3) - 1.396(3) 1.386(3) C12-C13 - 1.368(3) - 1.377(3) 1.381(3) C13-C14 - 1.432(3) - 1.430(3) 1.436(3) C14-C19 - 1.410(3) - 1.407(3) 1.408(3) C19-N11 - 1.379(3) - 1.382(3) 1.382(3)
<C19-N11-C12 - 108.6(2) - 109.4(2) 109.4(2) <N11-C12-C13 - 109.2(2) - 108.4(2) 108.7(2) <C12-C13-C14 - 107.3(2) - 107.4(2) 107.2(2) <C13-C14-C19 - 107.1(2) - 107.6(2) 107.1(2) <C14-C19-N11 - 107.8(2) - 107.2(2) 107.6(2)
aIndolyl rings on P1. bIndolyl rings on P2.

145
The P1-C2indolyl bond distance of 1.834(2) Å in (N-CH2NMe2)-20 is considerably
longer than the analogous bond length found in both the unfunctionalized and N-alkylated
2-indolylphosphines described in Chapter 2 (which averages to 1.813(5) Å for the
unfunctionalized phosphines, and 1.811(5) Å for the N-alkylated derivatives). Several
reasons may be considered for the lengthening of the P1-C2indolyl bond but the most
evident is the steric bulk associated with the tert-butyl groups that cause a lengthening of
all the P-C bonds. In order to relieve the steric crowding around the phosphorus atom, all
of the P-C bonds lengthen and the Σ{<CPC} increases. These effects are observed in the
increase of the Σ{<CPC} to 322.9(1)o for (N-CH2NMe2)-20 compared to the less bulky
dicyclohexyl derivative, 2, which has a Σ{<CPC} of 308.4o.
Figure 5.3. ORTEP diagram and numbering scheme of (N-CH2NMe2)2-20. Hydrogen atoms removed for
clarity. Thermal ellipsoids are drawn at 35% probability.
As exhibited in the crystal structure of (N-Bn)-1, the methyl group on C3 in (N-
CH2NMe2)-20 is oriented away from the phosphorus lone pair and points towards the
pyramid created by the three substituents of phosphorus. The argument for this methyl
group orientation is likely to be similar to the one described for (N-Bn)-1, a relief of
steric crowding near the lone pair results in the methyl group be oriented into the pyramid.
The width of the indolyl substituent determined by measuring the distance from the
methyl carbon, C10, to the methylene carbon on nitrogen, C11, indicates a length of
5.113(3) Å, which is in agreement with the analogous width measurement in (N-Bn)-1
(5.061(4) Å). The average width of the tert-butyl substituents was found to be

146
2.492(7) Å, determined by calculating the distance from each methyl group to its adjacent
methyl group on the same tert-butyl moiety. As expected, the (N-CH2NMe2)-3-
methylindolyl substituent is larger than the tert-butyl moieties, and a larger cone angle
would be expected for this phosphine in comparison with an unsubstituted derivative.
Crystals of aminal-protected 4, (N-CH2NMe2)2-4, were grown as colourless
plates by solvent vapour diffusion of pentane into a solution of the phosphine in CH2Cl2
Phosphine (N-CH2NMe2)2-4 crystallizes in the monoclinic space group P21/c with one
molecule in the asymmetric unit. Figure 5.4 shows the molecular structure and
numbering scheme of the phosphine, and Table 5.2 lists selected bond distances and
angles. The X-ray structure determination of (N-CH2NMe2)2-4 confirmed the
incorporation of the phosphorus atom P1 into the six-membered ring through bonding to
the coupled indolyl substituent at its two C2-positions: C2 and C12. Secondary bonding
interactions were absent in the solid-state structure of (N-CH2NMe2)2-4.
Figure 5.4. ORTEP diagram and numbering scheme of (N-CH2NMe2)2-4. Hydrogen atoms removed for
clarity. Thermal ellipsoids are drawn at 35% probability.
A comparison of the solid-state structures of (N-CH2NMe2)2-4 and (N-F5Bn)2-4
reveals that the replacement of the electron-rich aminal-protecting groups with the
electron withdrawing pentafluorobenzyl substituents imparts little structural change upon
the rest of the molecule. For example, the P1-C2indolyl and P1-C12indolyl bond lengths of
1.802(2) Å and 1.813(2) Å respectively in (N-F5Bn)2-4 are effectively the same as the

147
analogous bond lengths of 1.811(2) Å and 1.814(2) Å in (N-CH2NMe2)2-4 and are not
significantly different from the P1-C2indolyl separation of 1.813(2) Å observed in 1.
The structural data for both N-alkylated derivatives of 4 suggest that their six-
membered rings do not experience substantial ring strain. The respective <C3-C10-C13
bond angles of 113.0(2)° and 112.9(2)° about the methylene carbon of the phosphacycles
of (N-CH2NMe2)2-4 and (N-F5Bn)2-4, respectively, are only slightly smaller than the
corresponding bond angle of 115.4(2)° measured in 3,3’-diindolylmethane.10
Interestingly, coupling of the indolyl substituents does produce a compression of
almost 6° upon the <C2-P1-C12 bond angle from comparison of the values of 94.7 (1)°
and 94.36(9)° in (N-CH2NMe2)2-4 and (N-F5Bn)2-4 respectively, with that of 100.3(1)°
measured between the uncoupled indolyl substituents of 3. This is not considered to
result in significant ring strain to the phosphacycle because of the facility with which
phosphorus subtly changes hybridization in order to accommodate substituents of
differing electronic or steric requirements.
Single crystals of (N-PCy2)-1 were acquired by slow evaporation of the
phosphine in a 1 : 1 mixture of hexane-CH2Cl2 as colourless plates. An ORTEP
representation and numbering scheme of (N-PCy2)-1 is illustrated in Figure 5.5, selected
bond lengths and angles are listed in Table 5.2. (N-PCy2)-1 crystallizes in the
monoclinic space group P21/c with one molecule in the asymmetric unit. The solid-state
structure of (N-PCy2)-1 is ordinary and there are no significant secondary interactions.
Figure 5.5. ORTEP diagram and numbering scheme of (N-PCy2)-1. Hydrogen atoms removed for clarity.
Thermal ellipsoids are drawn at 35% probability.

148
Both phosphorus atoms, P1 and P2, adopt trigonal pyramidal geometry. The P-C
bond lengths differ according to the type of substituent on phosphorus. The P1-Cphenyl
bond distances are significantly shorter (average 1.838(3) Å) than the P2-Ccyclohexyl bond
lengths (average 1.856(3) Å). The disparity is most likely due to the bulkiness of the
cyclohexyl groups that require longer P-C bond lengths to be accommodated around the
phosphorus centre. The P1-Cindolyl bond length of 1.823(2) Å is appreciably longer than
the P2-N1 bond distance (1.757(1) Å). The lone pair of electrons on P1 is approximately
orthogonal with respect to the lone pair on P2, this causes the phenyl groups on P1 to be
oriented away from the cyclohexyl moieties on P2. Similar to the solid-state structure of
(N-CH2NMe2)-20, the methyl group on C3 of (N-PCy2)-1 is directed into the pyramid
formed by the three substituents on P1.
The P-Cphenyl bond distances are in (N-PCy2)-1 are equivalent to the same bonds
in unfunctionalized 1 (average 1.812(2) Å); while the P-Cindolyl bond length is longer in
(N-PCy2)-1 considering the dicyclohexylphosphino substituent on nitrogen likely makes
the indolyl moiety larger than the unfunctionalized derivative. The width of the
dicyclohexylphosphino substituted 3-methylindole was determined by measuring the
distance from C10 of the methyl substituent to P2 bonded to nitrogen, and a value of
5.441(2) Å was obtained. In comparison to unfunctionalized 1, which has an analogous
width of 3.713(4) Å, the dicyclohexylphosphino substituted 3-methylindole is a much
larger substituent, thus resulting in the longer P-Cindolyl bond lengths observed in (N-
PCy2)-1.
Crystals of diphosphine 29 were acquired as colourless plates from hexane vapour
diffusion into a solution of 29 in CH2Cl2. Figure 5.6 shows the molecular structure and
the numbering scheme of 29, while Table 5.2 lists selected bond distances and angles.
Diphosphine 29 crystallizes in the monoclinic space group C2/c with one molecule in the
asymmetric unit. The diphosphine packs in a herringbone fashion along the
crystallographic c-axis. There are no apparent secondary interactions in the solid-state
structure of 29.
The P1-Cindolyl bond distances are statistically different (~4σ) from the P2-Cindolyl
bond distances, however, the discrepancies are chemically insignificant. The indolyl
rings on each phosphorus atoms are located near perpendicular to each other. The P1-

149
Csp3 (1.856(2) Å) and P2-Csp3 (1.851(2) Å) distances are equivalent, although they are
longer than the analogous P-Csp3 bond lengths in the solid-state structure of dppe
(average 1.829(3) Å). The ethyl bridge between the two phosphorus atoms has a bond
length of 1.526(3) Å, which is consistent with the Csp3-Csp3 bond distance of 1.521(7) Å
in dppe. The P-C-C-P backbone is constrained to the trans-configuration with a dihedral
angle of 159.1(1)o. In contrast to the N-functionalized 2-indolylphosphine derivatives,
the methyl groups of 29 point away from the pyramid created by the three substituents on
phosphorus.
Figure 5.6. ORTEP diagram and numbering scheme of 29. Hydrogen atoms removed for clarity. Thermal
ellipsoids are drawn at 35% probability.

150
5.2.3. The Effect of N-Functionalization on the Σ{<CPC}
of (P,N)- and (P,P)-2-Indolylphosphines
The influence on the sum of the <CPC bond angles with changing phosphine
substituents and N-alkylation was introduced in Section 2.3. The effect of N-
modification with additional amino or phosphino functionalities on the Σ{<CPC} will be
examined for the (P,N)- and (P,P)-2-indolylphosphines. The Σ{<CPC} values are listed
in Table 5.3.
Table 5.3. Sum of <CPC bond angles (o) and 31P (ppm) chemical shifts for uncomplexed multidentate 2-
indolylphosphines.
Entry
Complex
<C2-P1-C12
<C12-P1-C22
<C22-P1-C2
avg. Σ{<CPC}
31P δ (ppm)i
1
(N-CH2NMe2)-1
NP
N
102.84(8)
111.61(9)
108.46(8)
322.9(1)
11.2
2
(N-CH2NMe2)-4
NP
N
N
N
94.7(1)
101.7(1)
101.3(1)
297.7(2)
-61.9
3
(N-PCy2)-1
NP
P
102.94(8)
100.47(7)a
102.70(8)
104.34(7)b
104.69(8)
104.76(7)c
310.3(1)
309.5(1)d
-28.5
53.9
4
29
HN
P
P
HN
NH
NH
100.5(1)e
103.2(1)f
102.3(1)e
98.1(1)f
97.9(1)e
102.1(1)f
300.7(2)g
303.4(2)h
-47.7j
-47.7j
a<N1-P2-C11. b<C11-P2-C17. c<C17-P2-N1. dΣ{<C-P2-C}. e<C-P1-C. f<C-P2-C. gΣ{<C-P1-C}. hΣ{<C-P2-C}. iSpectra collected in CDCl3. jSpectra collected in CD2Cl2.

151
It was observed in the solid-state structure of (N-CH2NMe2)-20 that the P1-
C2indolyl bond distances are longer than the corresponding distances in other 2-
indolylphosphines; the difference is attributed to the steric bulk associated with the tert-
butyl groups that cause a lengthening of all the P-C bonds, and simultaneous increase in
the Σ{<CPC} to 322.9(2)o. Phosphine (N-CH2NMe2)-20 appears to have the largest
Σ{<CPC} value for all 2-indolylphosphines regardless of functionalization at nitrogen,
which is expected for the large bulky tert-butyl groups on phosphorus and the presence of
an aminal-protecting group on the nitrogen centre.
The value of Σ{<CPC} of (N-CH2NMe2)-20 is essentially the same as that of
PtBu3 at 322.27o.11 While this likeness in the sum of the <CPC bond angles suggests that
(N-CH2NMe2)-20 will have a basicity that correlates with PtBu3, the large Σ{<CPC} for
(N-CH2NMe2)-20 is likely due to the orientation of the indolyl substituent around
phosphorus in the solid-state as opposed to a likeness to a tert-butyl group in electron
donating power.
Analysis of the Σ{<CPC} for (N-CH2NMe2)2-4 show that the coupled indolyl
substituent causes a consequential decrease of this value to 297.7(2)o from the uncoupled
diindolyl phosphine 3 (303.8(2)o). Not surprisingly, the Σ{<CPC} for (N-CH2NMe2)2-4
is slightly smaller than that of (N-F5Bn)2-4 since the pentafluorophenyl groups are quite
wide. However, the significance in the difference of these two Σ{<CPC} values for the
N-functionalized derivatives of 4 is debatable. Interestingly the average Σ{<HPH} for
PH3 is 280.8o, the geometry about the phosphorus atom is generally regarded as highly
pyramidal and the molecule as having almost perfect tetrahedral bond angles.12 The
Σ{<CPC} for (N-CH2NMe2)2-4 and (N-F5Bn)2-4 might suggest that the coupling of the
indolyl moieties to create one bidentate substituent on phosphorus has caused the
phosphorus atom in the N-functionalized derivatives of 4 to become more pyramidal.
Phosphine (N-PCy2)-1 contains two Σ{<CPC} values, one for P1 (310.3(1)o) and
the other for P2 (309.5(1)o). In essence, the Σ{<CPC} values are statistically equivalent
even though the substituents on phosphorus are quite different. The increase in Σ{<CPC}
of P1 from 304.3(2)o in the parent phosphine 1 is most likely due to the presence of the
dicyclohexylphosphino substituent on nitrogen, as seen with the N-functionalized

152
derivatives of 1. While the substituents on P2 are identical to the ones on the phosphorus
atom of ligand 2, with the only difference being the site of connectivity of the indolyl
moiety, the Σ{<CPC} of P2 has not increased considerably from that of phosphine 2
(Σ{<CPC} = 308.4(2)o). It would be expected that since the Σ{<CPC} increases for N-
functionalized 1 compared to parent phosphine 1, the Σ{<CPC} of P2 should also
increase correspondingly as there is additional substitution at C2 with the
diphenylphosphino group.
It was anticipated that the averaged Σ{<CPC} for phosphine 29 (302.1(3)o) would
be less than that of diindolylphenylphosphine 3 (303.8(2)o) as the methylene group is a
smaller substituent than the phenyl moiety of 3. Interestingly, the average Σ{<CPC} for
dppe is 304.2(7)o. It was determined in Section 2.2.2. that the 3-methylindolyl
substituent was wider than a phenyl substituent, thus one would expect phosphine 29 to
have a larger average Σ{<CPC} value. The smaller Σ{<CPC} in 29 may be due to
packing forces in the solid-state, and not a direct consequence of the size of the indolyl
substituents.
While there may be a Σ{<CPC} contribution to the 31P chemical shift of a 2-
indolylphosphine, the 31P resonance appears to be dominated by the type of substituents
that are on the phosphorus centre. The prime example would be phosphine (N-PCy2)-1
in which the Σ{<CPC} values for P1 (310.3(1)o) and P2 (309.5(1)o) are nearly equivalent,
yet are quite chemically different. The chemical environment of P1 causes it to resonate
at -28.5 ppm, while P2 resonates at 53.9 ppm. Clearly the substituents around each
phosphorus atom have a greater impact on their respective chemical shifts as opposed to
their Σ{<CPC} values.

153
5.2.4. The Effect of N-Functionalization on Indolyl
Aromaticity of (P,N)- and (P,P)-2-Indolylphosphines
The bond distances that make up the pyrrolyl moiety of the indolyl ring of (P,N)-
and (P,P)-2-indolylphosphines were examined to determine if the installation of an amino
or phosphino substituent would affect the indolyl aromaticity. It was found that the bond
distances of amino-functionalized phosphines: (N-CH2NMe2)-20, (N-CH2NMe2)2-4, and
(N-PCy2)-1 are consistent with those found in the solid-state structure of phosphine 1.
The largest difference from 1 is the N1-C2 bond distance of phosphine (N-PCy2)-1
(1.429(2) Å), however the nitrogen atom appears to stay sp2-hybridized as the remaining
bond lengths in the pyrrolyl ring are consistent with those in 1. Additionally,
diphosphine 29 has pyrrolyl bond distances that are statistically equivalent to those of 1,
as it would be expected for an unfunctionalized indolyl substituent. The uniformity of
the bond distances of unfunctionalized and functionalized indolyl rings do not seem to
change, thus suggesting that the aromaticity of the pyrrolyl ring remains intact.
5.3. Pd(II) Complexes of (P,N)-2-Indolylphosphines
Bidentate (P,N)- and the (P,P)-ligands tend to bind metal centres in a chelating
fashion to form 1 : 1 ligand-to-metal complexes. A general reaction sequence for the
formation of (P,N)- or (P,P)-chelated metal complexes is shown in Scheme 5.6. Usually
an (P,N)- or (P,P)-2-indolylphosphine is reacted in a 1 : 1 stoichiometry with a metal
species such as Pd(COD)Cl2 in acetonitrile and precipitated from solution with Et2O.

154
NP
Pd(COD)Cl2MeCN, r.t.
15 minN
P
NPd
Cl
ClN
30 Scheme 5.6. The reaction sequence for the synthesis of complex 30. This is a representative example for
the synthesis of metal coordinated (P,N)-2-indolylphosphines.
While the synthetic and spectroscopic characterization of selected metal complexes of
(P,N)- or (P,P)-2-indolylphosphines are discussed elsewhere,9 their structural
characterization by X-ray crystallography will be detailed in the following section. The
selected metal complexes for crystallographic analyses are shown in Figure 5.7.
NP
NPd
Cl
Cl
NP
NPd
Cl
Cl
NP
NPt
Cl
Cl
NP
NPd
Cl
Cl
NP
PPd Cl
Cl
NP
PPd Cl
Cl
NP
Pd Cl
ClOO P
30
(a)
31
(b)
32
(c)
33
(d)
34
(e)
35
(f)
36
(g) Figure 5.7. The selected metal complexes of (P,N)-2-indolylphosphines to be assessed by X-ray
crystallography: (a) [PdCl2(N-CH2NMe2)-1], 30 (b) [PtCl2(N-CH2NMe2)-1], 31 (c) [PdCl2(N-py)-1], 32
(d) [PdCl2(N-isoquin)-1], 33. The selected Pd(II) complexes of (P,P)-2-indolylphosphines to be analyzed
by X-ray crystallography: (e) [PdCl2(N-PPh2)-1], 34 (f) [PdCl2(N-PCy2)-1], 35 (g) [PdCl2(N-BINO)-1], 36.

155
5.3.1. X-ray Crystallographic Analysis of Pd(II) and Pt(II)
Complexes of (P,N)-2-Indolylphosphines
The experimental X-ray crystallographic data for the Pd(II) and Pt(II) complexes
of selected (P,N)-2-indolylphosphines are listed in Table 5.4.
Table 5.4. X-ray crystallographic experimental data for Pd(II) and Pt(II) complexes of (P,N)-2-
indolylphosphines.
30
NP
NPd
Cl
Cl
31
NP
NPt
Cl
Cl
32
NP
NPd
Cl
Cl
33
NP
NPd
Cl
Cl
Formula C24H25Cl2N2PPd C24H25Cl2N2PPt C27H23Cl4N2PPd C30H23Cl2N2PPd Formula Weight 549.73 638.42 654.64 619.77 Crystal colour, shape orange, plate yellow, plate dark orange, plate orange, block Crystal size, mm 0.64 x 0.34 x 0.16 0.40 x 0.32 x 0.12 0.50 x 0.28 x 0.06 0.20 x 0.08 x 0.04 Crystal system Monoclinic Monoclinic Monoclinic Monoclinic Space group P21/c P21/c P21/c P21/n a, Å 7.38770(10) 7.4109(1) 13.416(3) 9.0904(3) b, Å 17.2844(4) 17.3060(6) 15.114(3) 20.658(1) c, Å 18.4832(4) 18.4665(5) 14.307(3) 14.0685(7) α deg 90 90 90 90 β, deg 97.9730(12) 98.225(2) 109.21(3) 100.073(3) γ deg 90 90 90 90 V, Å3 2337.34(8) 2344.02(11) 2739.4(10) 2601.2(2) Z 4 4 4 4 Dcalcd, g cm-3 1.562 1.809 1.587 1.583 F(000) 1112 1240 1312 1248 μ, mm-1 1.105 6.296 1.146 1.003 λ (Mo Kα), Å 0.71073 0.71073 0.71073 0.71073 Limiting indices -9<=h<=9 -9<=h<=9 -17<=h<=16 -11<=h<=11 -22<=k<=22 -22<=k<=22 -19<=k<=19 -24<=k<=26 -20<=l<=23 -23<=l<=23 -15<=l<=18 -17<=l<=18 2θ range, deg 1.62 to 27.54 2.35 to 27.59 1.61 to 27.50 1.77 to 27.44 Max. and min. transmission 0.840 and 0.516 0.535 and 0.190 0.951 and 0.743 0.972 and 0.732 No. of reflections collected 18928 18829 20981 26117 No. of independent reflections/Rint 5530 / 0.0577 5569 / 0.0710 6499 / 0.0494 5930 / 0.1272 Extinction coefficient 0.0086(7) 0.0029(5) 0.0068(6) 0.0042(5) No. of refined parameters 275 275 318 327 Final R1, wR2 0.0354, 0.0967 0.0548, 0.1450 0.0500, 0.1303 0.0520, 0.1075 Final R1, wR2 (all data) 0.0423, 0.1059 0.0619, 0.1514 0.0689, 0.1458 0.1296, 0.1435 Goodness of fit 1.065 1.039 1.091 0.999 Δρmin, Δρmax, eÅ-3 -1.082, 1.330 -3.344, 4.282 -1.206, 1.498 -0.906, 1.388

156
Single crystals of complex 30 were acquired as orange coloured plates from Et2O
vapour diffusion into a CH2Cl2 solution containing the complex. The molecular structure
and numbering scheme for 30 is shown in Figure 5.8 and Table 5.5 lists selected bond
lengths and angles. Complex 30 crystallizes in the monoclinic space group P21/c with
one molecule in the asymmetric unit. The Pd(II) centre adopts square planar coordination
geometry with the (N-CH2NMe2)-1 ligand cis-chelated. The Pd(II)-P bond length of
2.2373(6) Å is consistent with those determined for the [Pd(2-indolylphosphine)Cl(μ-
Cl)]2 dimers (which range from 2.2162(8) to 2.260(2) Å) discussed in Chapter 2. The
six-membered metallocycle is non-planar due to the methylene carbon linking the two
nitrogen centres of the aminal protecting group; as such, the metallocycle takes on a boat
conformation. All three of the aromatic rings are virtually perpendicular to one another.
The P-Cindolyl and P-Cphenyl bond lengths in 30 are consistent with those found in the
dimeric [Pd(1)Cl(μ-Cl)]2 complex, 24. However the average <CPC bond angles differ by
an overall 3.8(3)o between complex 30 and complex 24; complex 30 possesses the
smaller Σ{<CPC} angles probably as a result of the added strain from the formation of
the metallocycle. Secondary interactions are absent in the solid-state structure of 30.
Figure 5.8. ORTEP diagram and numbering scheme of 30. Hydrogen atoms removed for clarity. Thermal
ellipsoids are drawn at 35% probability.

157
Table 5.5. Selected bond lengths (Å) and bond angles (o) for Pd(II) and Pt(II) complexes of (P,N)-2-
indolylphosphines.
30
NP
NPd
Cl
Cl
31
NP
NPt
Cl
Cl
32
NP
NPd
Cl
Cl
33
NP
NPd
Cl
Cl
Pd1-P1 2.2373(6) 2.222(2)a 2.218(1) 2.228(1) Pd1-N2 2.125(2) 2.120(6)a 2.063(3) 2.084(4) Pd1-Cl1 2.3791(6) 2.294(2)a 2.274(1) 2.374(1) Pd1-Cl2 2.2860(7) 2.372(2)a 2.370(1) 2.276(1)
<N2-Pd1-P1 94.04(6) 94.3(2) 88.9(1) 88.8(1) <P1-Pd1-Cl2 86.73(2) 88.30(6)b 90.05(4)d 87.73(5) <N2-Pd1-Cl1 90.06(6) 89.36(16)c 91.9(1)e 90.9(1) <Cl2-Pd1-Cl1 90.77(2) 89.12(6) 89.77(4) 93.20(5)
P1-C2 1.800(3) 1.795(6) 1.796(4) 1.807(5) P1-C12 1.812(3) 1.825(6) 1.807(4) 1.809(5) P1-C22 1.809(3) 1.810(6) 1.815(4) 1.798(5)
N1-C2 1.399(3) 1.409(7) 1.426(5) 1.416(6) C2-C3 1.380(3) 1.373(8) 1.363(6) 1.369(7) C3-C4 1.436(4) 1.446(10) 1.449(6) 1.440(7) C4-C9 1.405(4) 1.402(10) 1.396(5) 1.405(8) C9-N1 1.382(3) 1.368(8) 1.410(5) 1.401(6)
<C9-N1-C2 108.6(2) 108.7(5) 107.5(3) 107.7(4) <N1-C2-C3 109.6(2) 109.1(5) 109.4(3) 109.0(4) <C2-C3-C4 105.9(2) 106.3(5) 107.2(4) 107.8(5) <C3-C4-C9 108.4(2) 107.6(5) 108.2(3) 107.1(5) <C4-C9-N1 107.3(2) 108.1(6) 107.6(3) 108.3(5)
aBond distances to Pt1. b<P1-Pt1-Cl1. c<N2-Pt1-Cl2. d<P1-Pd1-Cl1. e<N2-Pd1-Cl2.
Yellow plate-like crystals of the Pt(II) complex of (N-CH2NMe2)-1 were formed
by Et2O vapour diffusion into a CH2Cl2 solution containing the complex. The molecular
structure and numbering scheme are shown in Figure 5.9, while selected bond angles and
distances are listed in Table 5.5. Complex 31 is isostructural to complex 30, so it
crystallizes in the monoclinic space group P21/c with one molecule in the asymmetric
unit. The Pt(II) exhibits square planar coordination geometry, the ligand is coordinated
in a cis-conformation, and the six-membered metallocycle adopts a boat conformation in
the same manner as complex 30. The Pt(II)-P1 distance is 2.222(2) Å is shorter than the
Pd(II)-P1 distance in 30, and the Pt(II)-N1 distance is 2.120(6) Å which is consistent with
the Pd(II)-N1 distance in 30. The P-C bond distances in 31 are crystallographically

158
equivalent to the ones in 30. The three aromatic rings are near orthogonal with respect to
each other, and no secondary interactions are present in the solid-state.
Figure 5.9. ORTEP diagram and numbering scheme of 31. Hydrogen atoms removed for clarity. Thermal
ellipsoids are drawn at 35% probability.
X-ray quality single crystals of complex 32 were obtained as dark-orange plates
from Et2O vapour diffusion into a solution of CH2Cl2 containing the complex. Figure
5.10 shows the molecular structure and the numbering scheme of the complex, and Table
5.5 lists selected bond angles and distances. Complex 32 crystallizes in the monoclinic
space group P21/c with one molecule and one CH2Cl2 solvate in the asymmetric unit.
The Pd(II) centre has square planar coordination geometry, with a Pd(II)-P bond length of
2.218(1) Å and a Pd(II)-N1 bond distance of 2.063(3) Å. The Pd(II)-P1 bond distance is
equivalent to the one found in complex 30. While the Pd(II)-N1 bond length in 32 is
consistent with those reported in the literature,13,14 it is considerably shorter than the
Pd(II)-N bond distance found in 30; this may be a result of the inherent rigidity of the
pyridyl ring to coordinate to Pd(II), whereas the methylene carbon has a degree of
freedom to conformationally adjust when the amino group coordinates to Pd(II).

159
Figure 5.10. ORTEP diagram and numbering scheme of 32. Hydrogen atoms and CH2Cl2 solvent
molecule removed for clarity. Thermal ellipsoids are drawn at 35% probability.
The P-C bond lengths in 32 are also crystallographically equivalent to those in
complexes 30 and 31. The indolyl and the two phenyl rings are oriented near orthogonal
to each other. The indolyl and pyridyl rings deviate from co-planarity by 43.0(1)o. The
biaryl twist angle between the pyridyl and indolyl rings is 50.0(5)o as determined by the
dihedral angle ascribed by the atoms N2-C11-N1-C2. This twist angle is smaller than the
corresponding twist angle found for QUINAP (60o)14 which is necessarily larger to
accommodate steric repulsion between the two biaryl rings. The pyridyl ring is situated
approximately parallel to one of the phenyl rings (ascribed by atoms C16 through C21)
(Figure 5.10); the distance between the two rings was determined to be 5.281 Å (by two
centrally placed points within the pyridyl ring and the phenyl ring in question), much
further than expected solid-state π-π interactions.15 Similar to complexes 30 and 31, the
six-membered metallocycle of 32 adopts a boat conformation.
Multiple weak intermolecular hydrogen bonding interactions are exhibited in the
solid-state structure of 32 between the pyridyl and indolyl rings to the chloro ligands of
symmetry equivalent complexes; the weak C…Cl bond distances range from 3.465(4) to
3.698(4) Å and are within the sum of the van der Waals’ radii. Additional weak
intermolecular hydrogen bonds occur between the CH2Cl2 molecule to the chloro ligands
of symmetry equivalent complexes.

160
Orange coloured block-like crystals of complex 33 were obtained from Et2O
vapour diffusion into a CH2Cl2 solution containing the complex. The molecular structure
and numbering scheme is shown in Figure 5.11, selected bond distances and angles are
listed in Table 5.5. Complex 33 crystallizes in the monoclinic space group P21/n with
one molecule in the asymmetric unit. The solid-state structure of 33 shares very similar
features with complex 32: the Pd(II) takes on a square planar coordination geometry with
the ligand binding in a cis-chelating fashion; the metallocycle adopts a boat
conformation; the Pd(II)-P and Pd(II)-N bond distances, 2.228(1) Å and 2.084(2) Å,
respectively, are crystallographically identical to the ones found in complex 32;
additionally, the P-C bonds lengths of 33 are the same as those in 32. Multiple weak
intermolecular hydrogen bonding interactions are demonstrated in the crystal structure of
33: atoms C16 on the isoquinolyl ring and C28 on a phenyl ring interact with chloro
ligands of symmetry equivalent molecules; weak intramolecular hydrogen bonding
between C19 and a chloro ligand is also evident.
The indolyl and phenyl substituents on phosphorus are nearly orthogonal with one
another. However, the isoquinolyl and indolyl rings deviate from co-planarity by
56.6(8)o, which is larger than the deviation of the pyridyl and indolyl rings in 32. The
dihedral angle ascribed by atoms N2-C11-N1-C2 was determined to be 61.3(6)o. The
deviation from co-planarity coupled with the larger dihedral angle of 33 confirms that the
isoquinolyl functionality on nitrogen is much larger than the pyridyl moiety of 32, the
larger angles also suggest steric repulsion exists between the hydrogen atoms on C8 and
C13 of the indolyl ring and isoquinolyl rings, respectively.

161
Figure 5.11. ORTEP diagram and numbering scheme of 33. Hydrogen atoms removed for clarity.
Thermal ellipsoids are drawn at 35% probability.
The (N-isoquin)-1 ligand of 33 may appear to exhibit axial chirality through
hindered rotation about the N1-C11 bond. However, coordination to a chiral metal
species would be necessary to assess whether (N-isoquin)-1 does indeed possess an axis
of chirality. A structurally related ligand by Brown and co-workers13 (Figure 5.12)
demonstrates racemisation of either the ligand or its chiral Pd(II) complex, or both, to
form one enantiopure product upon coordination.
NP
N NPd
Cl
2Pd
PPh2
Me2N
N
N+ KPF6
MeOH
PF6
Figure 5.12. Brown and co-workers demonstrate absence of axial chirality in 1-methyl-2-
diphenylphosphino-3-(1’-isoquinolyl)indole upon coordination to a chiral Pd(II) complex.13
Additionally, the solid-state structure of the chiral Pd(II) complex possessed a biaryl twist
of 46.6o, which was smaller than the QUINAP derivative. Based on the observations by
Brown and co-workers, it is suspected that (N-isoquin)-1 would not possess an axis of
chirality either.

162
5.3.2. X-ray Crystallographic Analysis of Pd(II)
Complexes of (P,P)-2-Indolylphosphines
The experimental X-ray crystallographic data for the Pd(II) complexes of selected
(P,P)-2-indolylphosphines are listed in Table 5.6. Selected bond distances and angles for
complexes 34 – 36 are listed in Table 5.7.
Table 5.6. X-ray crystallographic experimental data for Pd(II) complexes of (P,P)-2-indolylphosphines.
34
NP
PPd Cl
Cl
35
NP
PPd Cl
Cl
36
NP
Pd Cl
ClOO P
Formula C33H27Cl2NP2Pd C33H39Cl2NP2Pd C41H29Cl2 2P2Pd NOFormula Weight
ape ss, needle edle ss, needle 6
4(5) 58(4) 038(11)
76(3) .6730(12)
4.8(3) 1.3(2)
cd, g cm-3 1.521 79 88
0.941 ), Å 3 3 3
s 13 14 26
θ range, deg transmission 5 2 2
s/Rint .0765 0.1059 .1002
0081(14) 0011(3) rs
4, 0.0863 3, 0.1339 3, 0.1425 all data)
-3 0.543 0.880 1.241
676.80 688.89 806.89 Crystal colour, sh colourle yellow, ne colourleCrystal size, mm 0.30 x 0.08 x 0.07 0.24 x 0.10 x 0.0 0.57 x 0.12 x 0.06 Crystal system Monoclinic Triclinic Monoclinic Space group P21/n P-1 C2 a, Å 10.288 11.46 25.3b, Å 14.6103(8) 14.2463(4) 9.8766(2) c, Å 19.9589(9) 19.9934(6) 18.4572(8)α deg 90 97.138(2) 90 β, deg 99.9 91.627(2) 128γ deg 90 106.855(2) 90 V, Å3 295 3094.16(17) 360Z 4 4 4 Dcal 1.4 1.4F(000) 1368 1416 1632 μ, mm-1 0.900 0.790 λ (Mo Kα 0.7107 0.7107 0.7107Limiting indice -13<=h<= -14<=h<= -32<=h<= -18<=k<=18 -18<=k<=18 -12<=k<=12 -22<=l<=25 -24<=l<=25 -21<=l<=23 2 2.79 to 27.49 2.68 to 27.54 1.41 to 27.52 Max. and min. 0.939 and 0.85 0.961 and 0.47 0.963 and 0.45No. of reflections collected 18123 40112 14561 No. of independent reflection 6730 / 0 14125 / 4368 / 0Absolute structure parameter - - -0.01(4) Extinction coefficient 0.0 0. 0.0038(4)No. of refined paramete 386 752 444 Final R1, wR2 0.050 0.055 0.060Final R1, wR2 ( 0.1370, 0.1097 0.1031, 0.1621 0.0861, 0.1638 Goodness of fit 0.962 1.041 1.066 Δρmin, Δρmax, eÅ -0.889, -1.224, -1.827,

163
Table 5.7. Selected bond lengths (Å) and bond angles (o) for Pd(II) complexes of (P,P)-2-
indolylphosphines.
NP
PPd Cl
Cl
34
Major component
(70%)
N P
PPd Cl
Cl
34
Minor component
(30%)
NP
PPd Cl
Cl
35 Independent molecule A
NP
PPd Cl
Cl
35 Independent molecule B
NP
Pd Cl
ClOO P
36
Pd1-P1 2.219(1) 2.219(1) 2.216(1) 2.225(1) 2.235(2) Pd1-P2 2.219(1) 2.219(2) 2.244(1) 2.239(1) 2.189(2) Pd1-Cl1 2.352(1) 2.352(2) 2.341(1) 2.354(1) 2.326(2) Pd1-Cl2 2.358(1) 2.358(2) 2.340(1) 2.347(1) 2.329(2)
<P2-Pd1-P1 88.27(4) 88.27(4) 89.08(4) 88.70(4) 85.22(6) <P1-Pd1-Cl1 88.71(4) 88.71(4) 86.52(4) 89.56(4) 90.33(6) <P2-Pd1-Cl2 87.48(4) 87.48(4) 90.95(4) 88.69(4) 90.87(7) <Cl2-Pd1-Cl1 95.68(4) 95.68(4) 93.46(5) 93.03(4) 93.80(7
P1-C2 1.77(1) 1.69(2) 1.789(5) 1.796(4) 1.795(7)
P1-C11 1.810(5) 1.803(5) 1.826(5) 1.808(5) 1.818(7) P1-C17 1.811(5) 1.800(5) 1.812(5) 1.822(5) 1.795(7)
P2-N1 1.69(2)a 1.69(2) 1.739(4) 1.731(4) 1.679(5) P2-C23 1.803(5) 1.803(5) 1.836(5) 1.827(4) 1.598(5)d P2-C29 1.800(5) 1.800(5) 1.844(5) 1.825(4) 1.589(5)e
N1-C2 1.408(7) 1.408(8) 1.417(5) 1.422(6) 1.428(8) C2-C3 1.36(1) 1.36(1) 1.367(6) 1.363(6) 1.369(9) C3-C4 1.44(1) 1.44(1) 1.422(7) 1.429(7) 1.44(1) C4-C9 1.41(1) 1.41(1) 1.411(6) 1.407(7) 1.402(9) C9-N1 1.41(1) 1.41(1) 1.403(6) 1.410(5) 1.410(8)
<C9-N1-C2 108.0(8) 108.0(8) 107.6(3) 107.0(4) 106.6(5) <N1-C2-C3 109.6(8) 109.6(9) 109.8(4) 110.0(4) 109.8(6) <C2-C3-C4 107.4(7) 107.4(8) 106.9(4) 107.2(4) 107.3(6) <C3-C4-C9 107.9(4) 107.9(4) 108.9(4) 108.5(4) 107.9(6) <C4-C9-N1 107.0(7) 107.0(8) 106.9(4) 107.4(4) 108.4(5)
aP2-C2A bond distance. b<C2a-P2-C23. c<C29-P2-C2a. dP2-O1 bond distance. eP2-O2 bond distance.
Colourless needle-shaped crystals of complex 34 were produced by Et2O vapour
diffusion into a CH2Cl2 solution containing the complex. Figure 5.13 shows the
molecular structure and the numbering scheme of 34, which crystallizes in the
monoclinic space group P21/c with one molecule in the asymmetric unit. The indolyl
ring in the complex is disordered over the same site. The major component of the indolyl
disorder is modeled at 70% (Figure 5.13a) and the minor component modeled at 30%
(Figure 5.13b). One can envisage that in the absence of the methyl group on the indolyl

164
ring, complex 34 can have pseudo C2-symmetry. The rotational axis would bisect the
Pd(II) and the indolyl ring with the chloro ligands and the diphenylphosphino substituents
on either side of it. Only N1 and C2 would prevent the complex from having true C2-
symmetry. By rotating the complex about this pseudo-C2-rotational axis, N1 and C2 can
switch positions while the remainder of the atoms stay in the same positions. Thus, in the
solid-state structure of 34, the complex has crystallized about 30% of the time with the
indolyl ring flipped “upside down” in other unit cells to give rise to its positional disorder.
(a) (b)
Figure 5.13. ORTEP diagram and numbering scheme of 34. Hydrogen atoms removed for clarity.
Thermal ellipsoids are drawn at 35% probability. (a) Major component of indolyl disorder modeled at 70%
occupancy. (b) Minor component of indolyl disorder modeled at 30% occupancy.
The Pd(II) adopts square planar coordination geometry where the ligand binds in
a cis-chelating conformation. The Pd(II)-P1 and Pd(II)-P2 bond lengths in 34 are
identical at 2.219(1) Å, most likely due to the disorder of the indolyl ring in the solid-
state. These Pd(II)-P bond distances are consistent with those of the Pd(II) complexes of
(P,N)-2-indolylphosphine ligands. The P-Cindolyl bond length of 1.77(1) Å is statistically
equivalent to the analogous bond length found in complexes 30 – 32, as are the P-Cphenyl
bond lengths (average 1.811(7) Å). Weak intermolecular hydrogen bonding between C5
and Cl2 of a symmetry related complex is exhibited at a donor-to-acceptor distance of

165
3.483(6) Å. Alternate classic hydrogen bonding interactions are not demonstrated in the
crystal structure of 34.
Each set of phenyl substituents are near orthogonal to the indolyl ring. The five-
membered metallocycle is approaching planarity with a rms deviation of 0.0697 Å for the
metallocycle ascribed by atoms: Pd1-P1-C2-N1-P2, and a rms deviation of 0.0791 Å for
the minor disordered component ascribed by atoms: Pd1-P1-N1A-C2A-P2. The dihedral
angle of the chelating (N-PPh2)-1 ligand, created by atoms P2-N1-C2-P1 is -20(2)o,
while the minor disordered component has a dihedral angle of -22(7)o. It is evident that
the dihedral angle of 34 is much smaller than those of the (P,N)-2-indolylphosphine
complexes because the coordinating moiety is bonded directly to nitrogen, as opposed to
the (P,N)-ligands with have a carbon spacer between the nitrogen and the coordinating
centre.
Single crystals of complex 35 were produced by Et2O vapour diffusion into a
CH2Cl2 solution containing the complex as yellow needles. Figure 5.14 shows the
molecular structure and the numbering scheme of 35, which crystallizes in the triclinic
space group Pī with two independent molecules in the asymmetric unit. Independent
molecule A possesses a cyclohexyl ring disordered over two positions (Figure 5.14a), the
major component is modeled at 64% occupancy and the minor component is modeled at
36% occupancy.
The (N-PCy2)-1 ligand coordinates to Pd(II) in a cis-chelating manner. The
Pd(II)-P1 and Pd(II)-P2 bond distances in both independent molecules are statistically
different by approximately 9σ and 5σ, respectively. However, the P-C bond distances
are statistically equivalent between the two independent molecules. Thus, the deviation
in Pd(II)-P bond lengths may be a result of crystal packing forces. In comparison the
solid-state structure of the free ligand (N-PCy2)-1, all the P-C and P-N bond distances
shorten upon coordination to Pd(II). In evaluation against the structure of 35, the Pd(II)-
PPh2 bond lengths are equivalent; however, as anticipated the Pd(II)-PCy2 bond lengths
are considerably longer (average 2.242(1) Å) as a result of the bulkier cyclohexyl
substituents on P2 that would prohibit closer Pd(II)-P contacts.

166
The phenyl rings are almost perpendicular to the indolyl ring in both independent
molecules. The five-membered metallocycles are near planar with rms deviations of
0.0387 Å for independent molecule A, and 0.0548 Å for independent molecule B. The
dihedral angle P1-C2-N1-P2 found in independent molecule A is -15.5(5)o, whereas in
molecule B it is 8.1(5)o. While these two dihedral angles are statistically dissimilar, the
disparity may be an artifact of crystal packing, and it is unlikely that the differences
contribute to any chemical distinction between the two molecules.
(a) (b)
Figure 5.14. ORTEP diagram and numbering schemes of the two independent molecules of 35 in the
asymmetric unit. Hydrogen atoms removed for clarity. Thermal ellipsoids are drawn at 35% probability.
(a) Independent molecule A: the major component of the disordered cyclohexyl is modeled at 64%
occupancy, and the minor component (shown with transparent bonds) modeled at 36% occupancy.
(b) Independent molecule B.
Colourless needle-shaped crystals of complex 36 were produced by Et2O vapour
diffusion into a CH2Cl2 solution containing the complex. Figure 5.15 shows the
molecular structure and the numbering scheme of 36, which crystallizes in the chiral
monoclinic space group C2 with one molecule in the asymmetric unit. The chiral Flack
parameter was found to be -0.01(4) which indicates the correct stereochemistry for
complex 36. Additionally, the (N-(R)-BINO)-1 ligand was synthesized using (R)-
BINOL, the solid-state structure of 36 confirms that the chirality of the ligand remained
intact upon chelating to Pd(II). The Pd(II) centre has square planar coordination
geometry with a Pd(II)-P1 bond distance of 2.235(2) Å, which is longer than the

167
analogous distances in the (P,N)-2-indolylphosphine complexes, as well as complexes 34
and 35. The Pd(II)-P2 bond distance is considerably shorter than the corresponding bond
lengths in complexes 34 and 35, this is owing to the two oxygen moieties on P2 which
alter the donating ability of the lone pair on phosphorus. While the phosphine P1-C bond
distances in 36 remain equivalent to those of 34 and 35, the phosphite P2-N1 bond
distance is appreciably shorter at 1.679(5) Å.
Figure 5.15. ORTEP diagram and numbering scheme of complex 36. Hydrogen atoms omitted for clarity.
Thermal ellipsoids are drawn at 35% probability.
The phenyl and indolyl rings are near perpendicular with each other. The five-
membered palladacycle is near planarity with a rms deviation of 0.1617 Å. A dihedral
angle ascribed by the atoms P1-C2-N1-P2 was determined to be -4.1(7)o, and is
significantly smaller than the same dihedral angle in complexes 34 and 35. The dihedral
angle of the phosphite’s atoms C33-C34-C24-C23 was found to be -53.1(9)o, and is
consistent with other complexes featuring BINAP.16
Reek and co-workers17 recently introduced a phosphine-phosphoramidite that is
the opposite enantiomer of (N-(R)-BINO)-1 and have evaluated its catalytic performance
in Rh-catalyzed hydrogenation and hydroformylation reactions. Their ligand was found
to have good activity, with moderate enantioselectivity. They had found that by
introducing sterically bulky substituents to the BINOL at the positions ortho to the
oxygen atoms that catalytic activity was greatly enhanced with higher ee’s.

168
5.4. Examining Trans-Influence Properties of (P,N)- and
(P,P)-2-Indolylphosphines
The trans-influence of the phosphino and amino ligands on PdCl2 or PtCl2 are
examined for complexes 30 – 33, while the variation in phosphine and phosphite
substituents are investigated for their trans-influence in complexes 34 – 36. In general,
the stronger the ligand’s σ-donicity, the more trans-influence that particular ligand has.
This correlates into a decreased bond strength of the bond trans to that particular ligand.
In other words, the bond trans to the ligand exerting the trans-influence, experiences
bond lengthening. Relative ligand trans-influence can be assessed by examining the
bond lengths of the ligand trans to the phosphine.12 Table 5.8 lists, in increasing order,
the Pd(II)-Cl bond distances that are trans to the diphenylphosphino substituent on indole
for the Pd(II) complexes of (P,N)-2-indolylphosphines.
Table 5.8. Increasing order of Pd(II)-Cl bond length that is trans to the phosphine in Pd(II) complexes of
(P,N)-2-indolylphosphines.
Complex Pd-Cltrans to phosphine (Å)
32
NP
NPd
Cl
Cl
2.370(1)
33
NP
NPd
Cl
Cl
2.374(1)
30
NP
NPd
Cl
Cl
2.3791(6)
In all cases, the Pd(II)-Cl bond trans to the amino group is shorter than the Pd(II)-
Cl bonds trans to the diphenylphosphino moiety. It is reasonable to say that the amino
group (regardless of whether the nitrogen is sp3- or sp2-hybridized) is a weaker trans-
influence ligand than the diphenylphosphino moiety. In examining the Pd(II)-Cl bond
lengths trans to the diphenylphosphino ligand, the Pd(II) complexes have comparable

169
Pd(II)-Cl bond distances, however they differ by more than 4σ. This suggests that the
nature of the amino substituent on the indolyl nitrogen does have a slight impact on the
σ-donicity of the diphenylphosphino group. According to Table 5.8, the order of
increasing Pd(II)-Cl bond distances, and therefore increasing trans-influence, is 32 < 33
< 30. The shortest Pd(II)-Cl bond distance in complex 32 suggests that the pyridyl
moiety on the indolyl nitrogen does not impart very significant σ-donating power to the
diphenylphosphino substituent. The isoquinolyl group improves the σ-donicity of the
diphenylphosphino substituent in 33. By changing the amino donor from sp2-
hybridization in complexes 32 and 33, to an sp3-donor in complex 30, the σ-donicity of
the amino substituent is enhanced to the diphenylphosphino group, and thus the trans
Pd(II)-Cl bond is the longest of the three Pd(II) complexes.
Interestingly, the trans Pt(II)-Cl bond of complex 31 (2.372(2) Å) would place it
between complexes 32 and 33 on the scale of increasing Pd(II)-Cl bond lengths. This is
in contrast to the analogous Pd(II) complex with the longest Pd(II)-Cl bond. The only
apparent modification between 30 and 31 are the metal centres involved; thus the
variance in trans M-Cl bond distances can be attributed to the subtleties of the chloro
ligand binding to either Pt(II) or Pd(II) as opposed to the trans-influence of the phosphine
ligand.
The order of increasing Pd(II)-Cl bond lengths for the Pd(II) complexes of (P,P)-
2-indolylphosphines are listed in Table 5.9. Since the indolyl is functionalized with
phosphino or phosphito substituents, the trans-influence of each P-donor, as well as the
overall (P,P)-2-indolylphosphine ligand can be assessed.

170
Table 5.9. Increasing order of Pd(II)-Cl bond length that is trans to the phosphine in Pd(II) complexes of
(P,P)-2-indolylphosphines.
Complex
Pd-Cltrans to P1 (Å) Pd-Cltrans to P2 (Å)
36 N
P
Pd Cl
ClOO P
2.329(2)
2.326(2)
35 N
P
PPd Cl
Cl
2.344(1)a
2.348(1)a
34 N
P
PPd Cl
Cl
2.358(1)
2.352(2)
aThe Pd(II)-Cl bond lengths are averaged between independent molecules A and B.
Complexes 34 – 36 can be categorized according to increasing Pd(II)-Cl bond
lengths that are trans to P1 and P2: 36 < 35 < 34. It is interesting to note that the order of
increasing Pd(II)-Cl bond length, which implies increase in trans-influence of the P-
donor, is the same for P1 and P2. This observation suggests that the phosphito or
phosphino substituent on the indolyl nitrogen affects the σ-donicity of the
diphenylphosphino substituent at the C2 position on indole. Not surprisingly, complex
36 has the shortest Pd(II)-Cl bond lengths as it contains a phosphite moiety which is a
better π-acid ligand than a σ-donor ligand.18 Surprisingly, the dicyclohexylphosphine
group does not impart a greater affect on the σ-donicity of the overall (P,P)-ligand. This
may be in part due to the significant deviations in bond lengths among independent
molecules A and B in the solid-state structure of 35, as the Pd(II)-Cl bond distances
shown in Table 5.9 are the averaged values of these independent molecules. Complex 34
has the longest Pd(II)-Cl bonds, suggesting (N-PPh2)-1 has the strongest trans-influence
of the three (P,P)-2-indolylphosphines investigated.

171
In comparing the trans-influence of the (P,N)- and (P,P)-2-indolylphosphines, the
phosphito and phosphino functionalities are weaker at inducing an increase in σ-donating
power of the diphenylphosphino substituent at C2. The amino functionalities increase the
trans-influence of the diphenylphosphino moiety more effectively. The order of
increasing Pd(II)-Cl bond distances, and thus increase in ligand trans-influence: 36 < 35
< 34 <32 < 33 < 30.
5.5. Synthesis and Characterization of Multidentate N-
Functionalized Ligands based on Phosphine 5
Phosphine 5 can be easily transformed into a multidentate ligand by furnishing
diphenylphosphino groups on the nitrogen centre. The multidentate trisubstituted ligand,
(N-PPh2)3-5, is formed by the reaction of 5 with NaH in THF, followed by the addition
of three equivalents of PPh2Cl (Scheme 5.7). The product is isolated by column
chromatography in 35% yield as a white microcrystalline solid.
1. 10 eq NaH2. 3eq PPh2Cl
THF24h
NP
N
N
PPh2
PPh2
PPh2
(N-PPh2)3-5
35%
HN
P
NH
NH
Scheme 5.7. General reaction scheme for the synthesis of multidentate phosphine ligand (N-PPh2)3-5.
The 31P NMR spectrum of (N-PPh2)3-5 is consistent with an AX3 spin system that
confirms trisubstitution of all three indolyl nitrogen centres in phosphine ligand 5 (Table
5.10). The signal pertaining to the bridgehead phosphorus atom is split into a quartet by
coupling to the three equivalent diphenylphosphino substituents, while the resonances of

172
the diphenylphosphino substituents are split into a doublet through coupling to the
bridgehead phosphorus atom.
Table 5.10. 31P NMR resonances of phosphine 5 and (N-PPh2)3-5 recorded in CDCl3.
Phosphine 31P NMR Resonances (ppm) JP-P (Hz)
5
-82.3 (s)
-
(N-PPh2)3-5
36.2 (d) -76.2 (q)
159 159
(s) = singlet, (d) = doublet, (q) = quartet
Recently, Ciclosi and co-workers19 have introduced an indolyl-based C3-
symmetric tripodal ligand, CP3 (Figure 5.16), that is reminiscent of (N-PPh2)3-5. The
bridgehead atom is an sp3-hybridized carbon instead of a phosphorus as in the case of (N-
PPh2)3-5. Ciclosi’s CP3 ligand is capable of binding Pd(II) to induce five-coordinate
trigonal bipyramidal coordination geometry about the metal centre. They have examined
the utility of this [PdCl(CP3)] complex in various Suzuki cross-coupling reactions and
found that it is efficient and reusable for the formation of a variety of biaryls. It was
found that the inherent axial chirality of the complex in solution is retained when the
chloro ligand is replaced with a chiral (2S,5S)-2,5-dimethyl-1-phenylphospholane. The
efficacy of an enantiopure complex was tested in the asymmetric cross-coupling of 1-
iodo-2-methoxynaphthalene and 1-naphthylboronic acid, which led to the formation of
(S)-1-(2-methoxynaphthalen-1-yl)naphthalene, but only in 7% ee. Nevertheless,
Ciclosi’s intrinsically chiral complex can be further explored to increase its potential as a
chiral catalyst.
NCH
N
N
PPh2
PPh2
PPh2
Ph2P PdPPh2
PPh2
Cl
N NN
PdCl2, THFAgBF4, r.t.
Figure 5.16. Ciclosi and co-workers’ example of an indolyl-based C3-symmetric ligand, CP3, that binds to
Pd(II).19

173
The coordination chemistry of trisubstituted 5, (N-PPh2)3-5, is demonstrated with
Pt(II) and Rh(I) (Scheme 5.8). The synthesis of the Pt(II) complex, 37, involves mixing
equimolar (N-PPh2)3-5 with Pt(COD)Cl2 and NaBPh4 in acetone. The orange complex
precipitates from solution in 51% yield. The analogous dark red Rh(I) complex, 38, is
generated in 94% yield by reacting half an equivalence of [Rh(COD)Cl] with (N-PPh2)3-
5 in THF.
NP
N
N
PPh2
PPh2
PPh2
(a) 1eq Pt(COD)Cl21eq NaBPh4
acetone
(b) 0.5eq [Rh(COD)Cl]2THF
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
51%
37
94%
38 Scheme 5.8. Reaction schemes for the synthesis of metal coordinated (N-PPh2)3-5 complexes. (a) [PtCl(N-
PPh2)3-5]BPh4, 37. (b) [RhCl(N-PPh2)3-5], 38.
The 31P NMR spectra of both complexes are simple and confirm coordination of all four
phosphorus donors of (N-PPh2)3-5 in a trigonal bipyramidal geometry to a metal centre
by their downfield chemical shifts and splitting patterns, respectively (Table 5.11).
Complex 37 should be expected to exhibit the anticipated splitting pattern for an AMX3
spin system which consists of two resonances where the axial phosphorus signal is split
into a quartet by coupling to the three equivalent equatorial phosphorus donors, and the
equatorial phosphorus signal is split into a doublet from coupling to the axial phosphorus.
However, both the axial and equatorial phosphorus signals appears as singlets with small
splitting associated with Pax-Peq coupling (JP-P = 10 Hz). Each of the 31P resonances are
flanked by 195Pt satellites (natural abundance 33.8%). The 195Pt-Peq coupling constant of

174
2826 Hz is larger compared to the analogous coupling constant of the [PtCl(PP3)]+
complex (JPt-Peq = 2516 Hz); whereas the 195Pt-Pax coupling constant in 37 is smaller than
the one found in [PtCl(PP3)]+ complex (JPt-Pax = 2585 Hz).20 Interestingly, the δPeq is
shifted more downfield than δPax, which is inconsistent with all of the metal complexes
of PP3; further examination of complex 37 is necessary to reveal the reasons for this
unpredictability.
Table 5.11. 31P NMR resonances of metal complexes of (N-PPh2)3-5 recorded in DMSO-d6.
Complex
31P NMR Resonances (ppm) and J (Hz)
Coordinated Chemical Shifts Δ (ppm)
δPax δPeq ΔPax ΔPeq
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
37
3.3 (d) JPt-Pax = 2225 JPax-Peq = 10
49.4 (d) JPt-Peq = 2826
79.5
13.2
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
38
42.4 (dq) JRh-Pax = 84 JPax-Peq = 24
72.8 (dd) JRh-Peq = 159
118.6
36.6
(s) = singlet, (d) = doublet, (dd) = doublet of doublets, (dq) = doublet of quartets; Pax = axial or bridgehead phosphorus atom, Peq = equatorial phosphorus atom.
The 31P NMR spectrum of complex 38 is consistent with an AMX3 spin system, with
additional coupling to 103Rh (natural abundance 100%); the signal of the axial phosphorus
donor is split into a quartet from coupling to the three equivalent equatorial
diphenylphosphino groups, which is then further split into a doublet by coupling to the 103Rh. Similarly, the signal of the equatorial phosphorus donors is split into a doublet
from coupling to the axial phosphorus atom, and split further into another doublet from
coupling to 103Rh. The Pax-Peq coupling constant is observed in the 31P NMR spectrum of
complex 38 at 24 Hz, and is slightly larger than the analogous coupling observed for
[Rh(PP3)Cl] (JPax-Peq = 17 Hz).21 Similar to complex 37, the JRh-Peq coupling constant is
larger in 38 (JRh-Peq = 159 Hz) than the analogous [Rh(PP3)Cl] complex (JRh-Peq = 126 Hz),
while the JRh-Pax coupling is smaller in 38 (JRh-Pax = 84 Hz) than that observed in the

175
[Rh(PP3)Cl] example (JRh-Pax = 148 Hz).21 Comparable to 37, the δPeq is shifted more
downfield than δPax, which is contradictory to other metal complexes of PP3.
In general, the 31P NMR resonance of a phosphine ligand shifts downfield upon
metal coordination in comparison to the free ligand. The downfield shift is treated
quantitatively as the coordination chemical shift denoted Δ, and is defined as the
difference between the δ(Pcoordinated) and δ(Pfree ligand).7 Additionally, if a phosphorus atom
is incorporated into a 5-membered ring, the δ(Pcoordinated) is shifted even more downfield
due to an additional deshielding contribution denoted ΔR. It has been observed with PP3
metal complexes that if the phosphorus atom is located at the bridgehead of two or three
five-membered chelate rings, then the Δ values for each chelate ring are nearly additive.
This also appears to be the case for complex 38, where the ΔPax is three times larger than
ΔPeq; for complex 37, the difference between ΔPax and ΔPeq is almost six times larger. It
has been observed that the chemical shifts of Pax are sensitive to the variation of the fifth
ligand in the trigonal bipyramidal metal complex.5,7,8,21 Since both complexes 37 and 38
have Cl- as the fifth ligand, the deviation in δPax cannot be commented on.
5.5.1. X-ray Crystallographic Analysis of Metal
Complexes of (N-PPh2)3-5
Single crystals of complex 37 were acquired from Et2O vapour diffusion into a
solution of CH2Cl2 containing the complex. The experimental X-ray data for 37 is listed
in Table 5.12, selected bond distances and angles are listed in Tables 5.13 and 5.14. The
molecular structure and numbering scheme of complex 37 is shown in Figure 5.17.
Complex 37 crystallizes in the triclinic space group Pī with one molecule, consisting of
[PtCl(N-PPh2)3-5]BPh4, and one disordered CH2Cl2 solvate in the asymmetric unit.
Repeated attempts to model the disordered CH2Cl2 were unsuccessful; the SQUEEZE
option in PLATON22 was used and indicated a solvent accessible void of volume 265 Å3
containing approximately 86 e-. In the final cycles of refinement, this contribution to the
electron density was removed from the observed data. The density, the F(000) value, the

176
molecular weight and the formula are given without taking into account the results
obtained with the SQUEEZE option in PLATON.
Table 5.12. X-ray crystallographic experimental data for complexes 37 and 38.
37
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
38
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
Formula C87H71BClN3P4Pt C63H51ClN3P4Rh Formula Weight 1523.70 1112.31 Crystal colour, shape dark orange, block dark red, block Crystal size, mm 0.16 x 0.10 x 0.08 0.34 x 0.24 x 0.20 Crystal system Triclinic Triclinic Space group Pī Pī a, Å 13.481(3) 11.7691(3) b, Å 16.857(3) 15.0323(3) c, Å 17.476(4) 16.9382(5) α deg 67.71(3)°. 83.9670(14) β, deg 85.77(3)°. 86.4380(11) γ deg 87.25(3)°. 69.9640(13) V, Å3 3663.7(13) 2798.60(12) Z 2 2 Dcalcd, g cm-3 1.381 1.320 F(000) 1548 1144 μ, mm-1 2.087 0.509 λ (Mo Kα), Å 0.71073 0.71073 Limiting indices -17<=h<=17 -15<=h<=15 -20<=k<=21 -19<=k<=19 -21<=l<=22 -19<=l<=22 2θ range, deg 1.26 to 27.48 1.21 to 27.53 Max. and min. transmission 0.866 and 0.528 0.910 and 0.776 No. of reflections collected 41593 27186 No. of independent reflections/Rint 16610 / 0.1019 12670 / 0.0508 Extinction coefficient 0.00032(13) 0.0030(5) No. of refined parameters 878 653 Final R1, wR2 0.0543, 0.1010 0.0436, 0.1123 Final R1, wR2 (all data) 0.0924, 0.1107 0.0568, 0.1187 Goodness of fit 0.965 1.057 Δρmin, Δρmax, eÅ-3 -1.786, 1.947 -1.054, 0.707

177
Table 5.13. Selected bond lengths (Å) and bond angles (o) for complexes 37 and 38. 37
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
38
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
M-P1 2.206(2)a 2.1614(7) M-P2 2.348(1)a 2.3001(7) M-P3 2.368(1)a 2.2996(7) M-P4 2.335(1)a 2.3007(7) M-Cl1 2.400(2)a 2.4428(7)
P4-M-P2 123.57(5)a 121.64(2) P4-M-P3 117.76(5)a 116.89(2) P2-M-P3 117.56(5)a 119.87(3)
P1-C2 1.784(5) 1.803(3) P1-C12 1.788(5) 1.806(3) P1-C22 1.796(4) 1.803(3) P2-N1 1.731(4) 1.743(2) P3-N11 1.729(4) 1.745(2) P4-N21 1.716(4) 1.749(2)
N1-C2 1.423(6) 1.417(3) C2-C3 1.372(6) 1.368(4) C3-C4 1.437(7) 1.443(4) C4-C9 1.392(6) 1.394(4) C9-N1 1.407(6) 1.405(3)
<C9-N1-C2 106.9(4) 106.8(2) <N1-C2-C3 110.0(4) 110.5(2) <C2-C3-C4 106.3(4) 106.2(2) <C3-C4-C9 108.9(4) 108.4(2) <C4-C9-N1 107.8(4) 108.1(2)
N11-C12 1.448(6) 1.424(3) C12-C13 1.351(6) 1.371(4) C13-C14 1.439(7) 1.440(4) C14-C19 1.405(7) 1.403(4) C19-N11 1.404(6) 1.399(3)
<C19-N11-C12 105.8(4) 107.0(2) <N11-C12-C13 110.5(4) 110.2(2) <C12-C13-C14 107.0(4) 106.2(3) <C13-C14-C19 108.2(4) 108.6(2) <C14-C19-N11 108.5(4) 108.0(2)
N21-C22 1.432(6) 1.415(3) C22-C23 1.363(7) 1.370(4) C23-C24 1.443(7) 1.431(4) C24-C29 1.389(7) 1.405(4) C29-N21 1.415(6) 1.395(3)
<C29-N21-C22 106.0(4) 107.1(2) <N21-C22-C23 110.6(4) 110.2(2) <C22-C23-C24 106.3(4) 106.4(2) <C23-C24-C29 108.8(4) 108.3(2) <C24-C29-N21 108.3(4) 107.9(2)
aM = Pt1. bM = Rh1.

178
(a) (b)
Figure 5.17. ORTEP diagram and numbering scheme of complex 37. Hydrogen atoms and BPh4+
countercation removed for clarity, thermal ellipsoids drawn at 35% probability. (a) ORTEP diagram
showing coordination sphere about the Pt(II) centre. (b) View along the virtual threefold axis with the
phenyl substituents removed for clarity.
The solid-state structure of 37 confirms the trigonal bipyramidal coordination
geometry of the Pt(II) centre. Although the ligand is C3-symmetric, the three equivalent
phosphorus atoms of the diphenylphosphino groups are crystallographically inequivalent.
The Pt(II) centre deviates from the plane defined by the three equatorial phosphorus
donors by 0.1440(7) Å towards the Cl- ligand. The Pt-Pax bond length is significantly
shorter at 2.206(2) Å than the Pt-Peq bond distances (range from 2.335(1) to 2.368(1) Å),
which is consistent with what is observed in the solid-state structures of [M(PP3)X]+ and
Ciclosi’s [PdCl(CP3)].19 The disparity in the Pt-Pax and Pt-Peq bond lengths may be
attributed to the strong σ-interaction of the axial bond in the trigonal bipyramidal
geometry leading to a shorter Pt-Pax bond, while the participation of the three equatorial
phosphorus donors in three five-membered chelate rings to the Pt(II) facilitate the
lengthening the Pt-Peq interaction. The BPh4- counteranion is ordinary and the [PtCl(N-
PPh2)3-5]+ cation does not possess additional secondary interactions.
Similar to the solid-state structure of 37, complex 38 also crystallizes in the
triclinic space group Pī with one molecule of [RhCl(N-PPh2)3-5], and one disordered
CH2Cl2 solvate in the asymmetric unit. Table 5.12 lists the experimental X-ray
diffraction data, whereas selected bond distances and angles are listed in Tables 5.13 and
5.14. The molecular structure and numbering scheme for 38 is shown in Figure 5.18.

179
The disordered CH2Cl2 could not be successfully modeled, thus the SQUEEZE option in
PLATON22 was used to remove the disorder. A solvent accessible cavity of volume
322 Å3 containing approximately 50 e- was suggested. The density, the F(000) value, the
molecular weight and the formula are given without taking into account the results
obtained with the SQUEEZE option in PLATON.
(a) (b)
Figure 5.18. ORTEP diagram and numbering scheme of complex 38. Hydrogen atoms removed for clarity,
thermal ellipsoids drawn at 35% probability. (a) ORTEP diagram showing coordination sphere about the
Rh(I) centre. (b) View along the virtual threefold axis with the phenyl substituents removed for clarity.
The solid-state structure of 38 confirms the trigonal bipyramidal coordination
geometry of the Rh(I) centre. There are many common similarities between the solid-
state structures of 37 and 38: the three equivalent phosphorus atoms of the
diphenylphosphino groups are crystallographically inequivalent in 38; the Rh(I) centre
deviates from the plane defined by the three equatorial phosphorus donors by
0.1686(4) Å towards the Cl- ligand; the Rh-Pax bond length is significantly shorter at
2.1614(7) Å than the Rh-Peq bond distances (range from 2.2996(7) to 2.3007(7) Å).
Complex 38 does not possess additional secondary interactions.

180
5.5.2. The Reactivity of [PtCl(N-PPh2)3-5]BF4 and
[RhCl(N-PPh2)3-5]
The reactivity of [M(PP3)X]0/+ has been well documented in the literature.4-8,21 In
particular, the metathesis of the fifth ligand appears to be quite facile, and the stability of
these complexes have been investigated thoroughly. In most cases, the ligand remains
coordinated to the metal, which retains trigonal bipyramidal coordination geometry. It
has been noted that small ligands, such as halides and small phosphine ligands (for
instance PMe3 or P(OMe)3) can be easily accommodated in the fifth position of the
metal’s coordination sphere.
Following the work by Brüggeller and co-workers,6 the metathesis of the chloro
ligand in [PtCl(PP3)]Cl was achieved on reaction with NaBH4 at room temperature for
30 minutes to give [PtH(PP3)]BPh4. Repeated attempts to generate the analogous
[PtH(N-PPh2)3-5]BPh4 from complex 37 with Brüggeller’s reaction conditions (Scheme
5.9) were unsuccessful. Complex 37 remained intact despite varying the time and the
temperature of the reaction. Refluxing conditions however, resulted in the cleavage of
the diphenylphosphino substituents from the indolyl nitrogen centre as seen in the 31P
NMR with the absence of the AMX3 spin system. In hindsight, the reaction of 37 with
NaBH4 would not have been successful in the first place, since the deprotection of
aminal-protected 2-indolylphosphines is accomplished by reacting it with NaBH4.
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
37
NaBH4, EtOHr.t., 12 hours Ph2P PtII
PPh2
PPh2
P
H
N NN
BPh4
not formed Scheme 5.9. The attempted synthesis of [PtH(N-PPh2)3-5]BPh4 by reaction of complex 37 and NaBH4.
Fernández and co-workers20 have demonstrated the insertion of SnCl2 into the Pt-
Cl bond of [PtCl(PP3)]+, and shown that the Pt-SnCl3 systems have catalytic activity

181
towards hydroformylation. The clean reaction of [PtCl(PP3)]Cl in CHCl3 with solid
SnCl2 in the presence of PPh3 to form [Pt(SnCl3)(PP3)]SnCl3 requires stirring at room
temperature for 12 hours. Similar reaction conditions were investigated on complex 37
(Scheme 5.10), and the reaction was monitored using 31P NMR. The reaction was
disappointingly unclean compared to the formation of [Pt(SnCl3)(PP3)]SnCl3, it was
found that multiple species were formed and that P-N cleavage was rampant leading to a
loss of the trigonal bipyramidal coordination geometry of the Pt(II) centre.
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
37
SnCl2, PPh3 CH2Cl2
r.t., 24 hours Ph2P PtII
PPh2
PPh2
P
SnCl3
N NN
SnCl3
not formed Scheme 5.10. The attempted SnCl2 insertion into the Pt(II)-Cl bond of complex 37.
The capacity for complex 38 to undergo metathesis of the fifth ligand was
examined by adhering to the reaction conditions set forth by Bianchini and co-workers
for [RhCl(PP3)].5 Bianchini and co-workers were able to replace the chloro ligand in
[RhCl(PP3)] for either methyl or phenyl groups by simply reacting the complex with the
appropriate alkyllithium reagent in THF. Repeated attempts of reacting complex 38 with
either methyllithium or phenyllithium resulted in no reaction (Scheme 5.11). The starting
complex was recovered almost quantitatively.
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
38
RLi, THF-78oC to r.t., 24 hours Ph2P RhI
PPh2
PPh2
P
R
N NN
R = Me or Ph
no reaction Scheme 5.11. The attempted metathesis of the chloro ligand in complex 38 for either a methyl or phenyl
ligand.

182
The lack of reactivity of complex 38 to alkyllithium reagents for the metathesis of
the chloro ligand may be due to the congested environment created by the three
equatorial diphenylphosphino groups on the nitrogen. The inability for even the small
methyllithium reagent to approach the small cavity remaining for the fifth ligand suggests
that the steric crowding of the three equatorial diphenylphosphino groups is greater in
comparison to the analogous [RhCl(PP3)] complex. Presumably, the bite angles <Pax-C-
N-Peq in 38 increase the proximity of the diphenylphosphino substituents; the five-
membered pyrrolyl ring establishes the maximum limit that the diphenylphosphino
groups can be oriented to avoid steric congestion with each other and with the phenyl
backbone of the indolyl moiety. Therefore the closeness of the diphenylphosphino
groups create a smaller cavity for the fifth ligand, and prevents reactivity of incoming
metathesis reagents, and rather reactivity occurs at the more accessible P-N bond sites.
5.6. Metal Coordination on the Σ{<CPC} of (P,N)- and (P,P)-
2-Indolylphosphines
The average Σ{<CPC} angles for metal complexes of (P,N)- and (P,P)-2-
indolylphosphines are listed in Table 5.14. The largest average Σ{<CPC} value for P1
is exhibited in complex 36 at 324.0(5)o. This Σ{<CPC} suggests much steric crowding
around P1, which is not particularly apparent from the solid-state structure (Figure 5.15).
It is speculated that the (R)-BINO substituent on nitrogen plays a dual role in affecting
the average Σ{<CPC} values, firstly it is inherently large, and like complexes 34 and 35,
the phosphite is also coordinating to Pd(II), so it enforces a rigid ligand conformation
about the metal centre which necessitates the opening up of the <C-P1-C angles.
Secondly, the (R)-BINO substituent is a phosphite, the oxygen atoms that link the
biphenyl to the phosphorus donor are not rigid, and therefore can be arranged such that
the <O-P2-O bond angles are much more compact than those around P1. The oxygen
atoms about P2 result in a modest average Σ{<CPC} of 311.5(5)o for the phosphite
moiety.

183
Table 5.14. Sum of <CPC bond angles (o) and 31P (ppm) resonances for metal complexes of multidentate 2-
indolylphosphines.
Entry
Complex
<C2-P1-C12
<C12-P1-C22
<C22-P1-C2
avg. Σ{<CPC}
31P δ (ppm)
1
30 N
P
NPd
Cl
Cl
105.5(1)
105.3(1)
105.4(1)
316.2(2)
4.9v
2
31 N
P
NPt
Cl
Cl
105.6(3)
104.8(3)
105.1(3)
315.5(5)
-2.1v
3
32
NP
NPd
Cl
Cl
109.4(2)
106.3(2)
105.0(2)
320.5(3)
12.5w
4
33
NP
NPd
Cl
Cl
109.7(2)
107.8(2)
105.0(2)
322.5(3)
13.9w
5
34
NP
PPd Cl
Cl
104.5(7)a
104(3)b
108.9(2)a
109.9(2)b
106.8(7)a
102(3)b
320.2(11)a
316(4)b
28.1w
85.7w
6
35
NP
PPd Cl
Cl
108.8(2)c 107.0(2)d 107.7(2)e 103.4(2)f
107.0(2)c 105.6(2)d 106.2(2)e 110.2(2)f
104.1(2)c 105.3(2)d 106.1(2)e 109.4(2)f
319.9(4)c 317.9(4)d 320.0(4)e 323.0(4)f
27.5w 127.4w 27.5w 127.4w
7
36
NP
Pd Cl
ClOO P
106.9(3)g
99.0(3)h
110.2(3)g
103.6(2)i
106.9(3)g
108.9(3)j
324.0(5)g
311.5(5)
24.6w
122.6w
8
37
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
109.3(2) 101.6(2)k 105.2(2)p 105.1(2)s
113.8(2)
105.5(2)m
100.4(2)q 102.9(2)t
109.5(2) 106.4(2)n 104.4(2)r
106.1(2)u
332.6(3) 313.5(3) 310.0(3) 314.1(3)
3.3v
49.4v 49.4v 49.4v
9
38
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
109.2(1) 104.2(1)k 104.5(1)p 103.1(1)s
109.4(1) 99.9(1)m 100.0(1)q 101.5(1)t
107.9(1) 101.9(1)n 102.4(1)r 102.4(1)u
326.6(2) 306.0(2) 306.9(2) 307.0(2)
42.4v 72.8v 72.8v 72.8v
a<C-P1-C of major component (70%). b<C-P2-C of major component (70%). c<C-P1-C of molecule A. d<N-P1-C of molecule A. e<C-P2-C of molecule B. f<N1-P2-C of molecule B. g<C-P1-C. h< N1-P2-O1. i<O1-P2-O2. j<O2-P2-N1. k< N1-P2-C31. m<N1-P2-C37. n<C31-P2-C37. p< N11-P3-C43. q<N11-P3-C49. r<C43-P3-C49. s<N21-P4-C55. t<N21-P4-C61. u<C55-P4-C61. vSpectra collected in DMSO-d6. wSpectra collected in CDCl3.

184
The functionalization of phosphine 5 with diphenylphosphino substituents on
nitrogen and subsequent coordination of the (N-PPh2)3-5 ligand to Pt(II) and Rh(I) have
undoubtedly increased the average Σ{<CPC} of P1 from 306.9(2)o in the free ligand to
332.6(3)o and 326.6(2)o, respectively. The discrepancy between the average Σ{<CPC}
values for complexes 37 and 38 is a result of the larger van der Waals radius of Pt(II)
(1.72 Å) compared to Rh(I) (1.63 Å). The larger Pt(II) causes greater spreading out of
the <C-Pax-C bond angles. Likewise, the equatorial diphenylphosphino substituents on
nitrogen experience greater spreading out of the <C-Peq-C bond angles due to the larger
Pt(II) centre.
The multidentate (P,N)- and (P,P)-2-indolylphosphines in general undergo an
increase in average Σ{<CPC} values upon metal coordination. The increase of Σ{<CPC}
may not necessarily reflect an increase in steric crowding, and care must be taken when
using this estimation to determine a ligand’s steric bulk.
As it was observed in Section 5.2.3., there is little correlation between the
Σ{<CPC} and the 31P chemical shift of a particular 2-indolylphosphine. Especially when
a 2-indolylphosphine is acting as a monodentate ligand through phosphorus coordination,
the 31P resonance is more greatly influenced by the substituents bonded to it, as well as
the type of transition metal it is coordinating to, rather than the Σ{<CPC} bond angles.
5.6.1. The Effect of Metal Coordination on Indolyl
Aromaticity of Multidentate 2-Indolylphosphines
It was realized in Section 5.2.4. the introduction of amino or phosphino
substituents on the indolyl nitrogen does not affect the aromaticity of the pyrrolyl moiety
to an appreciable amount that can be determined by X-ray crystallography. The pyrrolyl
bond distances in the metal complexes of (P,N)- and (P,P)-2-indolylphosphines were
examined (Table 5.5). The overall trend with the metal complexes shows the N1-C2
bonds lengthening, especially in the cases with the phosphino substituents on nitrogen.
Also, the C2-C3 bonds tend to shorten to lengths that range from 1.36(1) Å to 1.380(3) Å

185
from the analogous bonds in 1 (average 1.373(4) Å). Generally, the pyrrolyl bond
lengths do not vary significantly from those found in 1; therefore, even when the (P,N)-
and (P,P)-2-indolylphosphines are acting as cis-chelating ligands, the pyrrolyl ring
remains aromatic.
The pyrrolyl bond distances in complexes 37 and 38 exhibit similar trends seen in
complexes 30 – 36. The N1-C2 and N1-C9 bond lengths have a propensity to lengthen,
while the C2-C3 bond distances shorten, upon coordination to either Pt(II) or Rh(I).
However, the pyrrolyl bond lengths are consistent with aromaticity seen in phosphine 5.
Therefore, functionalization of the indolyl nitrogens with diphenylphosphino substituents
and their subsequent coordination to metal complexes does not impart changes in
aromaticity of the indolyl ring.
5.7. Conclusions
New multidentate (P,N)- and (P,P)-2-indolylphosphine ligands have been
synthesized and subsequently coordinated to Pd(II) or Pt(II). Reacting the multidentate
ligands in a 1 : 1 ligand to metal complex ratio produces cis-chelated complexes. Using
X-ray crystallography, the structural details of the (P,N)- and (P,P)-2-indolylphosphines
show that the aromaticity of the pyrrolyl rings do not change upon N-functionalization or
metal binding.
The Pd(II)-Cl bonds trans to the phosphino substituents of the cis-chelated Pd(II)
complexes, 30 and 32 - 36, were examined for possible trans-influence trends. It was
found that the (P,P)-2-indolylphosphines have the weakest trans-influence, while the
amino functionalized (P,N)-2-indolylphosphines have stronger trans-influence on the
Pd(II)-Cl bond. The weakest trans-influence ligand was determined to be (N-(R)-
BINO)-1, whereas the strongest trans-influence ligand was (N-CH2NMe2)-1.
The average Σ{<CPC} angles of the free (P,N)- and (P,P)-2-indolylphosphines
were assessed. In general the average Σ{<CPC} values increased for the parent
phosphine 1 when substituents were furnished on the nitrogen centre. As anticipated,
variation of the substituents on phosphorus is a has greater influence on the Σ{<CPC}

186
than the addition of functionality at the indolyl nitrogen. The average Σ{<CPC} angles
typically increase from 1 by 15o - 20o.
Installing diphenylphosphino substituents on all three of the indolyl nitrogen
centres of C3-symmetric phosphine 5 resulted in the formation of a tetradentate ligand,
(N-PPh2)3-5. Coordination of the (N-PPh2)3-5 ligand to Pt(II) or Rh(I) enforces five-
coordinate trigonal bipyramidal geometry of the metal centres. Crystallographic analyses
shows the pyrrolyl rings remain aromatic in the indolyl substituents upon coordination.
Furthermore, the average Σ{<CPC} angles increase with N-functionalization and metal
binding. Complexes 37 and 38 were subjected to a variety of reaction conditions that
included attempted metathesis of the fifth ligand. However, these reactions were
unsuccessful, largely resulting in P-N bond cleavage. The reactivity at the P-N bonds
could be a result of the steric crowding of the fifth ligand by the three equatorial
diphenylphosphino substituents.
5.8. Experimental
Unless otherwise stated, the synthetic protocol for the functionalized 2-indolylphosphines
and their metal complexes are reported in Dr. Edmond Lam’s thesis.9
General Considerations. All reactions and manipulations were carried out under an
atmosphere of dinitrogen using standard Schlenk techniques unless otherwise stated. 1,2-
Bis(dichlorophosphino)ethane (Cl2P(CH2)2PCl2), 1.6 M n-BuLi, NaBH4, were purchased
from Aldrich and used as received. 1-[(N,N-Dimethylamino)methyl]-3-methylindole23
and Pt(COD)Cl224 were synthesized according to literature procedures. Tetrahydrofuran
(THF) was distilled from sodium benzophenone ketyl still under a dinitrogen atmosphere. 1H, 13C, and 31P NMR spectra were recorded on Varian 400 MHz or 300 MHz NMR
systems, and referenced to SiMe4 (TMS) and 85% H3PO4, respectively. Splitting patterns
are indicated as s, singlet; d, doublet; t, triplet; q, quartet;, m, multiplet; br, broad peak.

187
HN
P
P
HN
NH
NH
1,2-Bis(diindolylphosphino)ethane, (C9H8N)2P(CH2)2P(C9H8N)2, 29. 1-[(N,N-
Dimethylamino)methyl]-3-methylindole (5.0 g, 26.6 mmol) was dissolved in THF
(50 mL) and cooled to -78oC. n-BuLi (20.0 mL, 32.0 mmol) was added dropwise over 10
minutes. The reaction was stirred at -78oC for 10 minutes, warmed to ambient
temperature and stirred for 3 hours. The resulting orange mixture was cooled to -78oC,
and Cl2P(CH2)2Cl2 (0.65 mL, 4.3 mmol) was added dropwise over 5 minutes. The
mixture was allowed to warm to ambient temperature overnight and then quenched with
MeOH. Solvents were removed in vacuo to yield a dark yellow residue. Water (100 mL)
was added to the residue, and the suspension extracted with DCM (3 x 100 mL). The
organic layers were combined, washed with brine, dried over anhydrous MgSO4, filtered,
and concentrated in vacuo to afford dark orange oil. MeOH was added to triturate pure
aminal-protected product as a white solid, which was isolated by filtration and dried in
vacuo (0.84 g, 23%). 31P NMR (CDCl3, 161 MHz): δ -47.7 (s); 1H NMR (CDCl3,
400 MHz): δ 7.47 (d, J = 8 Hz, 4H, Ar-H), 7.30 (d, J = 8.4 Hz, 4H, Ar-H), 7.14 (t,
J = 7.6 Hz, 4H, Ar-H), 7.05 (t, J = 7.6 Hz, 4H, Ar-H), 4.40 (d, J = 12 Hz, 4H, amine
NCH2), 4.19 (d, J = 12 Hz, 4H, anime NCH2), 2.54 (t, J = 4.8 Hz, 10 Hz, 4H, CH2CH2),
2.29 (s, 12H, indole CH3), 1.84 (s, 24H, amine N(CH3)2); 13C NMR (CDCl3,
101 MHz):.139.5, 129.7, 129.3, 122.7, 119.8, 119.4, 118.7, 110.6, 67.0, 42.4, 21.7, 10.7;
HRMS EI for C50H64N8P2: calcd m/z 838.4729, found m/z 838.4719. A solution of
THF/EtOH (13 mL, 1:1 v/v) was added to a solid mixture of the aminal-protected product
(0.812 g, 0.97 mmol) and NaBH4 (0.18 g, 4.8 mmol). The mixture was refluxed for 5
hours. The solution was reduced to dryness in vacuo and acidified water was added to
the resultant white solid, which was extracted with DCM (3 x 50 mL). The organic
phases were combined and dried over anhydrous MgSO4, filtered and taken to dryness to
afford a dark yellow coloured oil. MeOH was added to triturate pure product as a white

188
solid, which was isolated by filtration and dried in vacuo (0.48 g, 81 %). 31P NMR
(CDCl3, 161 MHz): δ -66.9 (s); 1H NMR (CD2Cl2, 300 MHz): δ 7.90 (br s, 3H, indole
NH), 7.60 (d, J = 7.2 Hz, 3H, Ar-H), 7.27 (d, J = 7.5 Hz, 3H, Ar-H), 7.21 – 7.12 (m, 6H,
Ar-H), 2.41 (s, 9H, indole CH3); 13C NMR (CD2Cl2, 75 MHz): 138.9, 130.3, 125.5, 123.7,
121.3, 120.2, 119.5, 111.8, 9.9; HRMS EI for C27H24N3P: calcd m/z 421.170357, found
m/z 421.170786; Microanalysis for C27H24N3P: calcd (%) C = 76.94, H = 5.74, N = 9.97,
found (%)C = 76.70, H = 5.78, N = 9.79.
NP
N
N
PPh2
PPh2
PPh2
2-(Bis(3-methyl-1-(diphenylphosphino)-1H-indol-2-yl)phosphino)-3-methyl-1-
(diphenylphosphino)-1H-indole, P[C9H7NP(C6H5)2]3, (N-PPh2)3-5. Phosphine 5
(0.2211 g, 0.70 mmol) was dissolved in THF (10 mL) to yield a clear colourless solution.
NaH (0.179 g, 4.5 mmol) was added to the solution, immediate effervescence was
observed. The mixture gradually changed colour from yellow to orange to bright orange
over 24 h. The resultant solution was cooled to -78oC, and PPh2Cl (0.38 mL, 2.1 mmol)
was added dropwise via syringe over 10 minutes. The temperature was maintained at -
78oC for 30 minutes while the reaction stirred. The reaction was allowed to warm to
ambient temperature overnight. Volatile solvents were removed in vacuo to give an oily
residue which was fused to silica gel and subjected to column chromatography (silica gel,
2 : 1 hexanes-CH2Cl2). The product (N-PPh2)3-5 is a microcrystalline white solid (0.18 g,
35%). 31P NMR (CD2Cl2, 161 MHz): δ 36.2 (d, J = 159 Hz, 3P, Peq), -76.2 (q, J = 159,
318 Hz, 1P, Pax); 1H NMR (CD2Cl2, 300 MHz): δ 7.44 (d, 3H, J = 7.8 Hz, Ar-H), 7.32-
7.12 (m, 30H, Ar-H), 7.01 (t, J = 7.9, 14.8 Hz, 3H, Ar-H), 6.81 (t, J = 8.2, 15.3 Hz, 3H,
Ar-H), 6.71 (d, J = 8.3 Hz, 3H, Ar-H), 2.07 (s, 9H, indole CH3); 13C NMR (CD2Cl2,
75 MHz): 140.8, 136.2, 136.0, 135.9, 129.2, 128.6, 128.3, 122.7, 120.4, 119.2, 114.6,

189
66.0. Microanalysis for C63H51N3P4: calcd (%) C = 77.69, H = 5.28, N = 4.31, found (%)
C = 76.88, H = 5.40, N = 4.41.
Ph2P PtII
PPh2
PPh2
P
Cl
N NN
BPh4
[PtCl(N-PPh2)3-5]BPh4, 37. Phosphine ligand (N-PPh2)3-5 (0.061 g, 0.063 mmol) was
dissolved in CH2Cl2 (2 mL) and added dropwise to a stirring solution of Pt(COD)Cl2
(0.024 g, 0.063 g) and NaBPh4 (0.022 g, 0.063 mmol) in acetone (2 mL). Gradually the
solution turns bright orange in colour. The solution was left to stir overnight upon which
an orange solid precipitated from solution. The solid was isolated by filtration and
washed with minimal acetone and dried in air (0.049 g, 51%). 31P NMR (DMSO-d6,
161 MHz): δ 49.4 (s, JPt-Peq = 2826 Hz, 3P, Peq), 3.3 (s, JPt-Pax = 2225 Hz, JPax-Peq = 10 Hz,
1P, Pax); 1H NMR (DMSO-d6, 300 MHz): δ 7.85 (d, 3H, J = 8.2 Hz, Ar-H), 7.38-7.05 (m,
30H, Ar-H), 6.92 (t, J = 7.3, 14.8 Hz, 3H, Ar-H), 6.81 – 6.76 (m, 3H, Ar-H), 6.38 (d, J =
8.4 Hz, 3H, Ar-H), 3.34 (s, 9H, indole CH3); 13C NMR (DMSO-d6, 75 MHz): 142.5,
137.2, 136.3, 135.1, 130.2, 128.6, 127.3, 122.2, 121.4, 118.8, 113.2, 68.5.
Ph2P RhI
PPh2
PPh2
P
Cl
N NN
[RhCl(N-PPh2)3-5], 38. Phosphine ligand (N-PPh2)3-5 (0.22 g, 0.23 mmol) was
dissolved in THF (5 mL) and added dropwise to a stirring solution of [Rh(COD)Cl]2
(0.056 g, 0.11 g) in THF (3 mL). Immediately the solution turned dark red in colour, and
allowed to stir for 30 minutes. The solution was concentrated to dryness to afford a dark
red solid. The solid was re-dissolved in minimal CH2Cl2, and added to a copious amount
of Et2O which precipitated out a fine dark red coloured solid. The solid was isolated by
filtration and washed with minimal Et2O and dried in air (0.24 g, 94%). 31P NMR

190
(DMSO-d6, 161 MHz): δ 72.8 (dd, JPt-Peq = 159 Hz, 3P, Peq), 42.4 (dq, JRh-Pax = 84 Hz,
JPax-Peq = 24 Hz, 1P, Pax); 1H NMR (DMSO-d6, 300 MHz): δ 7.88 (d, 3H, J = 7.8 Hz, Ar-
H), 7.36-7.04 (m, 30H, Ar-H), 6.95 (t, J = 7.9, 14.8 Hz, 3H, Ar-H), 6.81 (t, J = 7.9,
14.8 Hz, 3H, Ar-H), 6.36 (d, J = 8.3 Hz, 3H, Ar-H), 3.34 (s, 9H, indole CH3); 13C NMR
(DMSO-d6, 75 MHz): 141.2, 137.8, 136.1, 135.2, 133.2, 128.9, 126.0, 123.1, 120.9,
119.2, 111.6, 68.6.
X-ray Crystallography. X-ray data were collected on a Nonius Kappa CCD
diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). A
combination of 1º φ and ω (with κ offsets) scans were used to collect sufficient data. The
data frames were integrated and scaled using the Denzo-SMN package.25 The structures
were solved and refined with the SHELXTL-PC v6.12 software package.26 Refinement
was by full-matrix least squares on F2 using data (including negative intensities) with
hydrogen atoms bonded to carbon and nitrogen atoms included in calculated positions
and treated as riding atoms. Absorption corrections were made for every structure. All
the heavy atoms were refined anisotropically.

191
5.9. References
(1) Alcock, N. W.; Brown, J. M.; Hulmes, D. I. Tetrahedron: Asymmetry 1993, 4,
743-756.
(2) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029-3070.
(3) Bridgeman, A. J.; Gerloch, M. J. Chem. Soc. Dalton Trans. 1995, 197-204.
(4) Aizawa, S.-I.; Funahashi, S. Anal. Sci. 1996, 12, 27-30.
(5) Bianchini, C.; Masi, D.; Meli, A.; Peruzzini, M.; Zanobini, F. J. Am. Chem. Soc.
1988, 110, 6411-6423.
(6) Brüggeller, P. Inorg. Chem. 1987, 26, 4125-4127.
(7) Hohman, W. H.; Kountz, D. J.; Meek, D. W. Inorg. Chem. 1986, 25, 612-623.
(8) Fernández, D.; García-Seijo, M. I.; Kégl, R.; Peťócz, G.; László, K.; García-
Fernádez, M. E. Inorg. Chem. 2002, 41, 4435-4443.
(9) Lam, E., PhD Thesis, University of Toronto, 2007.
(10) Rampersad, N. C., MSc Thesis, University of Toronto, 2001.
(11) CSD Database (version 5.29, January 2008).
(12) Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements; 2nd ed.;
Butterworth-Heinemann: Oxford, 1997.
(13) Claridge, T. D. W.; Long, J. M.; Brown, J. M.; Hibbs, D.; Hursthouse, M. B.
Tetrahedron 1997, 53, 4035-4050.
(14) Alcock, N. W.; Hulmes, D. I.; Brown, J. M. J. Chem. Soc., Chem. Commun. 1995,
395-397.
(15) Steed, J. W.; Atwood, J. L. Supramolecular Chemistry; John Wiley & Sons, Ltd.:
New York, NY, 2001.
(16) Filipuzzi, S.; Pregosin, P. S.; Albinati, A.; Rizzato, S. Organometallics 2006, 25,
5955-5964.
(17) Wassenaar, J.; Reek, J. N. H. Dalton Trans. 2007, 3750-3753.
(18) Tolman, C. A. J. Am. Chem. Soc. 1970, 90, 2953 - 2956.
(19) Ciclosi, M.; Lloret, J.; Estevan, F.; Lahuerta, P.; Sanaú; Pérez-Prieto, J. Angew.
Chem. Int. Ed. 2006, 45, 6741-6744.
191

192
(20) Wilson, M. R.; Prock, A.; Giering, W. P.; Fernandez, A. L.; Haar, C. M.; Nolan, S.
P.; Foxman, B. M. Organometallics 2002, 21, 2758-2763.
(21) Gambaro, J. J.; Hohman, W. H.; Meek, D. W. Inorg. Chem. 1989, 28.
(22) Spek, A. L. J. Appl. Cryst. 2003, 36, 7-13.
(23) Katritzky, A. R.; Lue, P.; Chen, Y.-X. J. Org. Chem. 1990, 55, 3688-3691.
(24) Drew, D.; Doyle, A. R. Inorg. Synth. 1972, 13, 47-55.
(25) Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307-326.
(26) Sheldrick, G. M. In SHELXTL-Windows NT. V6.12, Bruker Analytical X-Ray
Systems Inc. Madison, WI, 2001.

Chapter 6
Conclusion
2-Indolylphosphines represent an interesting class of compounds that can be
systematically modified at an indolyl nitrogen centre with relative ease. It has been
shown that the a variety of functional groups can be incorporated on an indolyl nitrogen
centre that can alter the overall bulkiness, electronics, or chirality of the resultant
phosphine molecule.
The solid-state structures of selected 2-indolylphosphines were analyzed using X-
ray crystallography. Several general trends emerged from this analysis: the orientation of
the methyl substituent on indole in the solid-state appears to have a degree of correlation
with substitution at the nitrogen centre; the aromaticity of the pyrrole ring of each indolyl
substituent is unaffected by the introduction of a phosphorus atom at the C2-position or in
the presence of a substituent on the indolyl nitrogen. A value called the sum of the <CPC
bond angles, Σ{<CPC}, can provide information on the steric crowding around a
phosphorus atom based on the crystallographic bond angles of a phosphine ligand. This
technique is a good estimate for the steric bulk of a phosphine ligand in the absence of
cone angle data. It was determined that the Σ{<CPC} values for N-functionalized 2-
indolylphosphines are in general larger than the unfunctionalized compounds.
The crystal structures of PdCl2 complexes of 2-indolylphosphines exhibit both
inter- and intramolecular hydrogen bonding from an indolyl NH hydrogen bond donor to
a chloro ligand. Thus the utility of the inherent hydrogen bonding ability of tris-2-(3-
methylindolyl)phosphine was examined through a series of anion titrations. The titration
data indicated strong binding of fluoride, chloride, and acetate to tris-2-(3-
methylindolyl)phosphine in a 1 : 1 anion to phosphine receptor ratio. Crystal structures
of the fluoride and acetate complexes to tris-2-(3-methylindolyl)phosphine confirm the
binding stoichiometry. Based on these results, a new indolylphosphine-based anion
receptor was designed to increase anion selectivity. A series of attempts to generate the
193

194
new anion receptor were met with synthetic challenges that prevented the formation of
the desired phosphine compound. However, a molecular cleft that is based on two
indolyl rings linked via a phenyl moiety was synthesized and shown to have anion
binding capability. The molecular cleft exhibited high selectivity for acetate over other
anions.
The coordination of 2-indolylphosphine ligands to transition metal complexes
provides additional ways to investigate their properties. Reaction of a phosphine ligand
with Ni(CO)4, and subsequent examination of the resultant complex’s CO stretching
frequencies gives information about the phosphine’s net-basicity. It was found that 2-
indolylphosphines can have a range of net-basicities, and by introducing substituents at
nitrogen, more subtle changes to the net-basicity of the parent ligand can be made. The
[Cu(tris-2-(3-methylindolyl)phosphine)(phenanthroline)]BF4 complex exhibits
simultaneous P-coordination while the indolyl NH groups are engaging in intermolecular
hydrogen bonding to the BF4- to create a discrete ion pair complex. The trans-influence
of selected 2-indolylphosphines was speculated by examining the Pd-Cl bond lengths
trans to the 2-indolylphosphine in [Pd(L)Cl(μ-Cl)]2 complexes.
Amino and phosphino substituents can be used to functionalize an indolyl
nitrogen centre of 2-indolylphosphines to give (P,N)- and (P,P)-ligands. These
substituents are particularly useful as they provide an additional site for metal
coordination. Metal complexes of (P,N)- and (P,P)-2-indolylphosphines can be used to
examine the trans-influence of amino versus phosphino donors, and correlate σ-donating
strength to trans-influence.
Finally, a tetradentate tripodal ligand can be generated from tris-2-(3-
methylindolyl)phosphine by introducing diphenylphosphino substituents on each indolyl
nitrogen centre. This trisubstituted tripod induces five-coordinate trigonal bipyramidal
coordination geometry on Pt(II) and Rh(I). A variety of attempts were made to exchange
the fifth ligand in both the Pt(II) and Rh(I) complexes, but were unsuccessful due to
complex decomposition.