an fgfr3 autocrine loop sustains acquired...
TRANSCRIPT

Cancer Therapy: Preclinical
An FGFR3 Autocrine Loop Sustains AcquiredResistance to Trastuzumab in Gastric CancerPatientsGeny Piro1,2, Carmine Carbone1, Ivana Cataldo3, Federica Di Nicolantonio4,5,Simone Giacopuzzi6, Giuseppe Aprile7, Francesca Simionato8, Federico Boschi9,Marco Zanotto1, Maria Mihaela Mina2, Raffaela Santoro1, Valeria Merz8,Andrea Sbarbati10, Giovanni de Manzoni6, Aldo Scarpa3, Giampaolo Tortora2,8,and Davide Melisi1,8
Abstract
Purpose: The majority of gastric cancer patients who achievean initial response to trastuzumab-based regimens developresistance within 1 year of treatment. This study was aimedat identifying the molecular mechanisms responsible forresistance.
Experimental Design: A HER2þ-trastuzumab sensitive NCI-N87 gastric cancer orthotopic nude mouse model was treatedwith trastuzumab until resistance emerged. Differentiallyexpressed transcripts between trastuzumab-resistant and sen-sitive gastric cancer cell lines were annotated for functionalinterrelatedness by Ingenuity Pathway Analysis software.Immunohistochemical analyses were performed in pretreat-ment versus posttreatment biopsies from gastric cancerpatients receiving trastuzumab-based treatments. All statisticaltests were two-sided.
Results: Four NCI-N87 trastuzumab-resistant (N87-TR) celllines were established. Microarray analysis showed HER2 down-
regulation, induction of epithelial-to-mesenchymal transition,and indicated fibroblast growth factor receptor 3 (FGFR3) as oneof the top upregulated genes in N87-TR cell lines. In vitro, N87-TRcell lines demonstrated a higher sensitivity than did trastuzumab-sensitive parental cells to the FGFR3 inhibitor dovitinib, whichreduced expression of pAKT, ZEB1, and cell migration. Oraldovitinib significantly (P ¼ 0.0006) reduced tumor burden andprolonged mice survival duration in N87-TR mouse models. Ahigher expression of FGFR3, phosphorylated AKT, and ZEB1wereobserved in biopsies from patients progressing under trastuzu-mab-based therapies if compared with matched pretreatmentbiopsies.
Conclusions: This study identified the FGFR3/AKT axis as anescape pathway responsible for trastuzumab resistance in gastriccancer, thus indicating the inhibition of FGFR3 as a potentialstrategy to modulate this resistance. Clin Cancer Res; 1–12. �2016AACR.
IntroductionGastric cancer is the fourth most commonly diagnosed cancer
and the third leading cause of cancer-related death worldwide(1, 2). Trastuzumab, a recombinant humanized mAb directedagainst the human epidermal growth factor receptor 2 (HER2),is the only targeted agent to be approved for the first-linetreatment of patients with HER2-overexpressing metastatic gas-tric or gastroesophageal junction adenocarcinoma (3). Howev-er, after an initial period of clinical benefit, patients almostinevitably progress, as the tumors become refractory to trastu-zumab (4).
Several mechanisms involved in acquired resistance to tras-tuzumab have been described in breast cancer (5), includingoverexpression of Cyclin E (6), cross-talk between HER2 andother tyrosine kinase receptors (7, 8), or activation of down-stream intracellular signal transducers such as SRC (9).
In contrast, themolecularmechanisms involved in resistance totrastuzumab in gastric cancer remain largely uncharacterized.Recently, integrated SNP array–based copy number and whole-exome sequencing analyses of data from HER2-amplified gastro-esophageal adenocarcinoma revealed that more than half of thecases had additional oncogenic alterations at diagnosis that could
1Digestive Molecular Clinical Oncology Research Unit, Departmentof Medicine, Universit�a degli studi di Verona, Verona, Italy. 2Labo-ratory of Oncology and Molecular Therapy, Department of Medi-cine, Universit�a degli studi di Verona, Verona, Italy. 3ARC-NetResearch Centre and Department of Pathology and Diagnostics,Universit�a degli studi di Verona, Verona, Italy. 4Department ofOncology, University of Turin, Candiolo, Italy. 5Candiolo CancerInstitute–FPO, IRCCS, Candiolo, Torino, Italy. 6Esophageal and Gas-tric Surgery Unit, Department of Surgery, Azienda OspedalieraUniversitaria Integrata,Verona, Italy. 7Department of Medical Oncol-ogy, Azienda Ospedaliero-Universitaria, Udine, Italy. 8MedicalOncology Unit, Azienda Ospedaliera Universitaria Integrata, Ver-ona, Italy. 9Department of Computer Science, Universit�a degli studidi Verona, Verona, Italy. 10Section of Anatomy and Histology,Department of Neurological, Neuropsychological, Morphologicaland Movement Sciences, Universit�a degli studi di Verona, Verona,Italy.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
G. Piro and C. Carbone contributed equally to this article.
Corresponding Author: Davide Melisi, University of Verona, AOUI Policlinico,Piazzale L.A. Scuro, 10, Verona 37134, Italy. Phone: 3904-5812-8148; Fax: 3904-5802-7410; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-16-0178
�2016 American Association for Cancer Research.
ClinicalCancerResearch
www.aacrjournals.org OF1
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

potentially hamper the antitumor effect of anti-HER2 agents,including amplifications of cell cycle–related genes CCNE1 andCDK6, PI3K pathway activation by PI3KCA mutations, and geneamplification of other tyrosine kinase receptors such as EGFRand MET (10).
Solid cancers are molecularly heterogeneous, but among thelarge number of genetic alterations present at diagnosis onlyfew of them represent relevant driver gene mutations thatdirectly or indirectly confer a selective growth advantage(11). Although we recognize the potential contributions ofadditional oncogenic alterations present at diagnosis to tumorprogression during therapy with trastuzumab, a number ofgenes that contain few or no mutations could be insteadoverexpressed, downregulated, or epigenetically altered, thusplaying an equally important role in the development of drugresistance (12).
Therefore, in this study we aimed at directly identifying tran-scriptional mechanisms responsible for the resistance of gastriccancer to trastuzumab, which may represent new therapeutictargets in the search for ways to reverse the invariable escape ofthis disease from anti-HER2 therapies.
Materials and MethodsCell lines, reagents, and in vitro studies
Human gastric cancer cell line NCI-N87 and human breastcancer cell lines MCF-7 and BT474 were obtained from ATCC.Human gastric cancer cell line YCC-2 was obtained from KCLB.Cell lines were authenticated by standard short tandem repeat(STR) DNA typing methodology before being purchased fromcell bank. Cells were daily checked by morphology and rou-tinely tested to be mycoplasma free by PCR assay. Generationof GFPþ/luciferaseþ NCI-N87 cell line (13), in vitro cell pro-liferation assay, wound-healing assay (14), IHC, protein extrac-tion, and Western blotting (15) were performed as previouslydescribed, and for further details see Supplementary Materialsand Methods.
Gene expression microarray and pathway analysisRNA isolation and quantitative RT-PCR assay were performed
as detailed in Supplementary Materials andMethods. Differencesin gene expression between parental and resistant cells wereexamined using Illumina Human 48k-gene-chips (Illumina) asindicated in SupplementaryMaterials andMethods. Gene expres-sion microarray data have been deposited in the GEO database(accession no. GSE77346). Differentially expressed transcriptswere tested for network and functional interrelatedness using theIPA software program (Ingenuity Systems).
Establishment of gastric cancer cell lines in vivo resistant totrastuzumab and in vivo studies
Six- to 8-week-old female BALB/c athymic (nu/nu) mice werepurchased from Harlan Laboratories. All mice were housed andtreated in accordance with the guidelines of the Italian Ministryof Health Animal Care and Use Committee, and maintained inspecific pathogen-free conditions at CIRSAL Animal Care Facil-ity. The orthotopic implantation of gastric cancer cells wasperformed in six mice as described previously (16). Briefly,GFPþ/luciferaseþ NCI-N87 cell line was harvested from sub-confluent culture by exposure to trypsin. Trypsinization wasstopped with medium containing 10% FBS, and the cells werewashed once with PBS. To produce orthotopic gastric tumors,1 � 106 cells resuspended in 50 mL of PBS/Matrigel solution(1:1) were injected into the gastric wall of nude mice anesthe-tized with a 1.5% isoflurane–air mixture. To prevent suchleakage, a cotton swab was held over the injection site for 1minute. One layer of the abdominal wound was closed withwound clips (Auto-clip; Clay Adams). The mice tolerated thesurgical procedure well, and no anesthesia-related deathsoccurred. Tumor growth was monitored by bioluminescentimaging performed using a cryogenically cooled IVIS 100imaging system coupled with a data-acquisition computerrunning the Living Image software program (Xenogen). Whenthe resulting tumors became detectable, the mice were given 20mg/kg of trastuzumab i.p. twice a week until the tumorssuddenly recurred during continuous therapy. Treatment resis-tance developed in 4 of 6 mice. At evidence of advanced bulkydisease, mice were euthanized using carbon dioxide inhalation.Four trastuzumab-resistant cell lines were established fromexcised tumors via repeated green fluorescent protein flowcytometric sorting with FACSAria II sorter (Becton Dickinson).
The subcutaneous heterotopic implantation of gastric cancercells was performed as described previously (17). Tumor bearingmicewere randomly assigned (n¼10per group) to receive 20mg/kg of trastuzumab i.p. twice a week for 4 weeks, or 40 mg/kg ofdovitinib oral gavage daily for 4 weeks, or respective vehicles as acontrol. Tumor size was measured with a caliper by the modifiedellipsoid formula (p/6) x AB2, where A is the longest and B is theshortest perpendicular axis of an assumed ellipsoid correspond-ing to tumor mass, as reported previously (18). All mice wereweighed weekly and observed for tumor growth. When at least 6of the 10mice in a treatment group presented with bulky disease,the median survival duration for that group was considered tohave been reached. At themedian survival duration of the controlgroup, the tumor growth in mice in all groups was evaluated. Themice were euthanized using carbon dioxide inhalation whenevidence of advanced bulky disease developed or at cut-offvolume of 2 cm3, which was considered the day of death for thepurpose of survival evaluation.
Translational Relevance
Themajority of gastric cancer patients who achieve an initialresponse to trastuzumab-based regimens develop resistancewithin 1 year of treatment. The molecular mechanismsinvolved in trastuzumab resistance of gastric cancer remainuncharacterized. In this study, we propose an in vivomodel inwhich trastuzumab therapy induces the selection of gastriccancers overexpressing FGFR3, which activates the PI3K/AKT/mTOR signaling pathway, sustaining, in turn, tumor growthand a more aggressive EMT phenotype. We confirmed ourfinding by demonstrating for the first time the overexpressionof FGFR3 in paired pretreatment and postprogression biopticsamples from patients affected by advanced gastric cancer thatrelapsed upon trastuzumab therapy. Of translational rele-vance, we showed the therapeutic efficacy in vivo of the FGFR3inhibitor dovitinib in models of trastuzumab-resistant gastriccancer. Our study provides the preclinical rationale to inves-tigate the inhibition of FGFR3 as second-line treatment strat-egy in gastric cancer patients refractory to first-line trastuzu-mab-containing therapies.
Piro et al.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF2
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

PatientsThree patients were considered for analyses. For each patient,
pre- and postresistance tumor samples were compared by immu-nohistochemical analyses. Informed consent was obtained fromall patients. See SupplementaryMaterials andMethods for clinicalhistory and details.
Statistical analysisThe results of in vitro proliferation were analyzed for statis-
tical significance of differences by nonlinear regression analysisand are expressed as means and 95% confidence intervals (CI)for at least three independent experiments performed in qua-druplicate. Statistical significance of differences in tumorgrowth was determined by the Mann–Whitney test; differencesin survival duration were determined using a log-rank test. Allstatistical tests were two-sided, and a P value less than 0.05indicated statistical significance. All statistical analyses wereperformed using GraphPad Prism software version 4.0c forMacintosh (GraphPad Software).
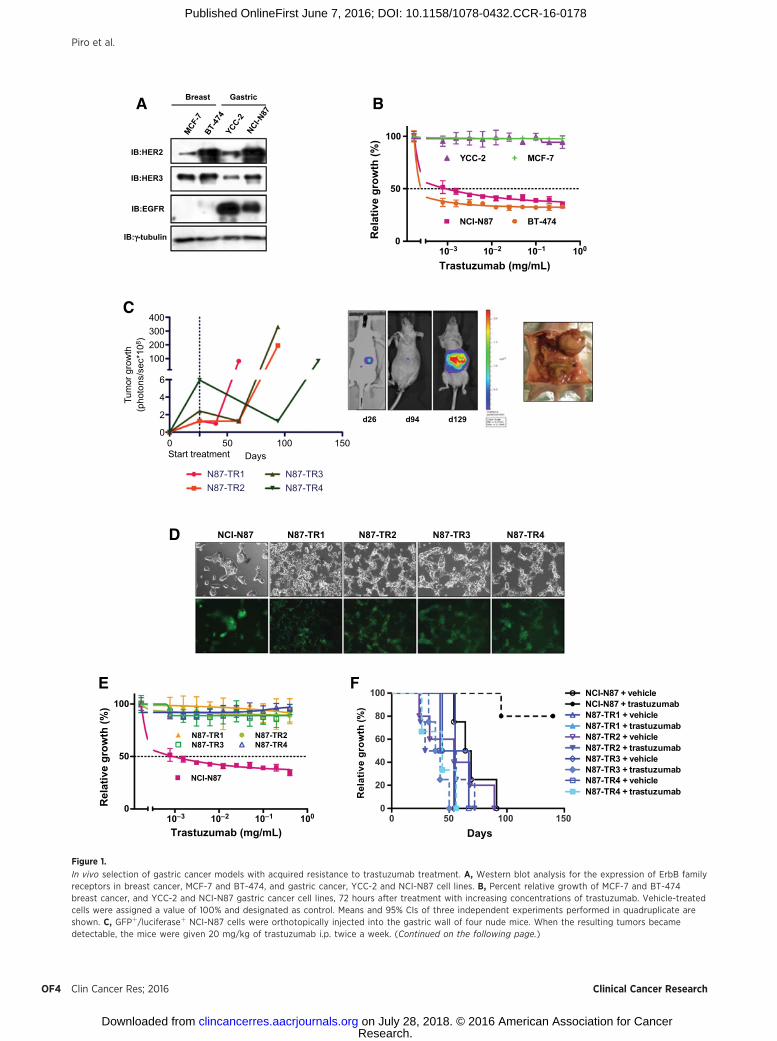
ResultsIn vivo selection of four gastric cancer models with acquiredresistance to trastuzumab
We initially measured the expression levels of several membersof the EGFR protein family in YCC-2 and NCI-N87 gastric cancer,and in the BT-474 andMCF-7 breast cancer cell lines (19), used asHER2-positive and HER2-negative controls, respectively. NCI-N87 gastric cancer cells showed a high basal expression of HER2comparable with that of the HER2-positive BT-474 breast cancercells (Fig. 1A). Consistently, with the expression of HER2, theNCI-N87 and BT-474 cells demonstrated in vitro a significantlyhigher sensitivity to trastuzumab (NCI-N87 IC50 ¼ 4.74 mg/mL;BT-474 IC50¼ 1.05mg/mL) than did YCC-2 andMCF-7 cells (IC50
> 400 mg/mL; Fig. 1B).To study the molecular mechanisms of acquired resistance to
HER2-targeted agents in gastric cancer, we established andvalidated four novel trastuzumab-resistant cell lines, N87-TR1,N87-TR2, N87-TR3, and N87-TR4 (Fig. 1C). Trastuzumab-resistant cell lines had a more spindle-shaped morphology(Fig. 1D) and exhibited significantly higher migration ratescompared with trastuzumab-sensitive NCI-N87 (P < 0.001;Supplementary Fig. S1).
N87-TR1, N87-TR2, N87-TR3, and N87-TR4 cells demonstrat-ed a significantly higher resistance in vitro to trastuzumab than didthe parental NCI-N87 cells (NCI-N87 IC50¼ 4.74 mg/mL vs. N87-TR1, N87-TR2, N87-TR3, or N87-TR4 IC50 > 400 mg/mL; Fig. 1E).As expected, NCI-N87 tumors were sensitive to drug treatment,whereas mice bearing trastuzumab-resistant tumors had survivalrates comparable with untreated control mice (Fig. 1F).
Identification of relevant biologic processes and genes by usingglobal transcript profiling
To gain insight into the molecular mechanisms underlyingtrastuzumab-resistant phenotype in gastric cancer cells, wecompared gene expression profiles in sensitive and resistantcells by microarray analysis to identify groups of genes asso-ciated with a specific signaling pathway or biological process.Differentially modulated transcripts in trastuzumab-resistantcells were enriched for genes implicated in mTOR signalingpathway and regulation of the eukaryotic initiation factors
(eIF) 2 and 4 (Fig. 2A). Top differentially regulated transcriptsin trastuzumab-resistant gastric cancer cell lines compared withNCI-N87 control cell line are summarized in SupplementaryTable S1.
Downregulation of HER2, EGFR, and HER3 expression intrastuzumab-resistant lines was evident, which was confirmedat the protein level by Western blot analysis (Fig. 2B). Con-sistent with this finding, although trastuzumab blocked theactivation of AKT in sensitive NCI-N87 cells, it was complete-ly ineffective in trastuzumab-resistant lines (SupplementaryFig. S2).
However, transcriptome analysis revealed that the expressionof ZEB1, a key member of the transcriptional complexes essen-tial for the development of the epithelial-to-mesenchymaltransition (EMT) genetic program, and that of the mesenchy-mal marker vimentin (VIM) were strongly upregulated, andthat the expression of the epithelial marker gene E-cadherin(CDH1) was strongly downregulated in trastuzumab-resistantgastric cancer cell lines compared with sensitive control cell line(Supplementary Table S1; Fig. 2C). Consistently, the N87-TR1,N87-TR2, N87-TR3, and N87-TR4 cell lines had considerablylower levels of E-cadherin and higher levels of ZEB1 andvimentin protein expression than did their trastuzumab-sensi-tive cell lines when cultured in vitro (Fig. 2D). These resultsindicate EMT as a plausible underlying molecular mechanismresponsible for the phenotypic changes observed in trastuzu-mab-resistant cells.
Among the top differentially regulated genes, we also found aconsistent overexpression of the membrane receptor geneFGFR3 and of the gene coding for its ligand FGF9 in theN87-TR1, N87-TR2, N87-TR3, and N87-TR4 trastuzumab-resis-tant gastric cancer cell lines compared with NCI-N87–sensitivecontrol cell lines (Fig. 2E). We validated that resistant cell lineshad significantly higher levels of FGFR3, and of total andphosphorylated AKT than did their trastuzumab-sensitiveparental counterparts. However, in all trastuzumab-resistantlines, we found a profound suppression of basal ERK1/2phosphorylation, the other main intracellular signaling path-way activated by tyrosine kinase receptors (Fig. 2F).
We corroborated the most relevant changes in protein levelsin vivo. All trastuzumab-resistant tumors had undetectable orlow levels of HER2 expression. NCI-N87 tumors had lowexpression levels of FGFR3 and of phosphorylated AKT, highexpression of E-cadherin, and no expression of ZEB1 andvimentin. In contrast, all trastuzumab-resistant tumors exhib-ited a significantly stronger expression of FGFR3 and phos-phorylated AKT, no expression of E-cadherin, but high expres-sion levels of ZEB1 and vimentin (Fig. 3).
On the basis of the overall results, we propose a mechanisticmodel for the resistance to trastuzumab in gastric cancer in whichan autocrine loop established through the overexpression ofFGFR3 and of its ligand FGF9 could be responsible for theactivation of the PI3K/AKT/mTOR signaling pathway, sustaining,in turn, tumor growth and EMT irrespectively of the anti-HER2treatment (Fig. 2G).
Effects of FGFR inhibition in trastuzumab-resistant gastriccancer models
To validate our proposed model, we tested the activityof two different FGFR3 inhibitors, dovitinib and AZD4547,in vitro. All four trastuzumab-resistant cell lines were
FGFR3 in Trastuzumab-Resistant Gastric Cancer
www.aacrjournals.org Clin Cancer Res; 2016 OF3
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178
d26 d94 d129
IB:HER3
IB:HER2
IB:EGFR
IB:g-tubulin
GastricBreast
NCI-N87 N87-TR1 N87-TR2 N87-TR3 N87-TR4D
E F
A
C
B
0 50 100 1500
20
40
60
80
100 NCI-N87 + vehicleNCI-N87 + trastuzumabN87-TR1 + vehicleN87-TR1 + trastuzumabN87-TR2 + vehicleN87-TR2 + trastuzumabN87-TR3 + vehicleN87-TR3 + trastuzumabN87-TR4 + vehicleN87-TR4 + trastuzumab
Days
10–3 10–2 10–1 1000
50
100
NCI-N87 BT-474
YCC-2 MCF-7
Trastuzumab (mg/mL)
Rel
ativ
e gr
owth
(%)
10–3 10–2 10–1 1000
50
100
NCI-N87
N87-TR1 N87-TR2N87-TR3 N87-TR4
Trastuzumab (mg/mL)
400300200100
6
4
2
00 50 100 150Start treatment Days
Tum
or g
row
th(p
hoto
ns/s
ec*1
08)
N87-TR1 N87-TR3N87-TR2 N87-TR4
Rel
ativ
e gr
owth
(%)
Rel
ativ
e gr
owth
(%)
Figure 1.
In vivo selection of gastric cancer models with acquired resistance to trastuzumab treatment. A, Western blot analysis for the expression of ErbB familyreceptors in breast cancer, MCF-7 and BT-474, and gastric cancer, YCC-2 and NCI-N87 cell lines. B, Percent relative growth of MCF-7 and BT-474breast cancer, and YCC-2 and NCI-N87 gastric cancer cell lines, 72 hours after treatment with increasing concentrations of trastuzumab. Vehicle-treatedcells were assigned a value of 100% and designated as control. Means and 95% CIs of three independent experiments performed in quadruplicate areshown. C, GFPþ/luciferaseþ NCI-N87 cells were orthotopically injected into the gastric wall of four nude mice. When the resulting tumors becamedetectable, the mice were given 20 mg/kg of trastuzumab i.p. twice a week. (Continued on the following page.)
Piro et al.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF4
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

significantly more sensitive than NCI-N87 parental cells(dashed line) to the same extent to the inhibition of FGFR3(All P < 0.0001; Fig. 4A). Furthermore, we found that in aclinically relevant concentration range, dovitinib was able toinduce a measurable reduction of the phosphorylation of AKT(Fig. 4B), suppressed the expression of ZEB1 (Fig. 4C), andsignificantly (P < 0.01) inhibited migration in all trastuzumab-resistant cell lines (Fig. 4D and E).
To demonstrate that FGFR3 is a druggable target to overcomeacquired resistance to trastuzumab in gastric cancer, 40 micewere injected with trastuzumab-sensitive NCI-N87 or trastuzu-mab-resistant N87-TR4 gastric cancer cells and randomlyassigned to receive oral dovitinib or its vehicle as a control.Dovitinib was completely inactive in NCI-N87 tumor bearingmice (Fig. 5A). Conversely, N87-TR4 gastric cancer bearingmice treated with dovitinib experienced a significant reductionin tumor burden. Accordingly, only the N87-TR4 tumor bearingmice treated with dovitinib demonstrated a significantly longermedian survival duration (P ¼ 0.0006; Fig. 5B). In this regard,control N87-TR4 tumors from mice treated with oral vehicleshowed a moderate to strong expression of phosphorylatedAKT. Conversely, N87-TR4 tumors from mice treated withdovitinib demonstrated only very weak or no expression ofphosphorylated AKT (Fig. 5C).
Validation of the FGFR3/AKT axis as an escape pathway fortrastuzumab resistance in gastric cancer patients
To corroborate the clinical relevance of our findings, weidentified three patients affected by advanced gastric cancerwho received trastuzumab-containing treatments and for whichthere were available both pretreatment and postprogressionbioptic samples. The clinical history for each patient is depictedin Fig. 6. Immunohistochemical analyses were performed onboth pretreatment and postprogression samples to determinethe expression of FGFR3, activated AKT, and of the EMT markerZEB1. We demonstrated that in all three patients, samples takenat trastuzumab progression exhibited significantly higherFGFR3 expression than their matched pretreatment samples.Consistently, we observed higher expression levels of activatedAKT and ZEB1 in postprogression bioptic samples when com-pared with their respective pretreatment samples (Fig. 6).
DiscussionIn this study, we sought to identify the molecular mechanisms
responsible for the resistance of gastric cancer to anti-HER2treatment trastuzumab. We demonstrated that trastuzumabinduces the selection of gastric cancer cells overexpressing FGFR3and its specific ligand FGF9. This autocrine loop activates thePI3K/AKT/mTOR signaling pathway, sustaining, in turn, tumorgrowth and amore aggressive EMTphenotype. To our knowledge,this is the first study to provide evidences that targeting the kinaseactivity of FGFR3 could be a valid approach tomodulate acquiredresistance to trastuzumab in gastric cancer.
FGFR3 is a member of a family of four tyrosine kinasereceptors that contribute to carcinogenesis by stimulatingtumor proliferation, survival, and neoangiogenesis (20). Recentstudies provide evidence for an increasing role or FGFR signal-ing as a key mediator of resistance to several anticancer ther-apies (21, 22), including the dual HER2 and EGFR inhibitorlapatinib (23). In our study, we demonstrated that FGFR3 isoverexpressed in gastric cancer tumor models selected forresistance to trastuzumab. Most importantly, we confirmed ourfinding by demonstrating for the first time the overexpressionof FGFR3 in paired pretreatment and postprogression biopticsamples from patients affected by advanced gastric cancer thatrelapsed upon therapies with trastuzumab.
FGF9 is oneof themembers of the heparin binding polypeptideligands of the FGF family, which presents a unique affinity (Kd:0.25 nmol/L) for FGFR3 (24). FGF9 was initially identified as animportant secreted mediator for the epithelial-to-mesenchymalcell signaling during embryonic development (25). In this study,we measured a significantly higher expression of FGF9 in trastu-zumab-resistant gastric cancer cell lines than in the sensitiveparental line, thus supporting the existence of an autocrine loopbetween FGF9 and overexpressed FGFR3.
Either the interaction of FGFR3 with the specific ligand FGF9or the ligand-independent dimerization induced by the over-expression of FGF receptors may lead to the transphosphoryla-tion of their tyrosine kinase domains, activating, in turn, theMAPK and the PI3K/AKT/mTOR signaling pathways (20). Thecontinued activation of the PI3K/AKT/mTOR signaling path-way is the most common alteration associated with the resis-tance to anti-HER2 therapies in breast cancer (26, 27). More
(Continued.) As expected, the tumors responded dramatically to the treatment. The mice received treatment until the tumors suddenly recurredduring continuous therapy with trastuzumab. At evidence of advanced bulky disease, mice were euthanized. Four novel trastuzumab-resistant cell lines, N87-TR1, N87-TR2, N87-TR3, and N87-TR4, were established from excised tumors via repeated GFP flow cytometric sorting. The tumor growth wasquantified weekly on the basis of bioluminescence emitted by the tumor cells as the sum of all detected photons within the region of the tumor persecond using a cryogenically cooled IVIS 100 imaging system coupled with a data-acquisition computer running the Living Image software program(Xenogen). A digital grayscale image was acquired, followed by the acquisition and overlay of a pseudocolor image representing the spatial distribution ofdetected photons emerging from the active luciferase within the mouse (representative image). D, Light- and fluorescent microscopic phenotype of thetrastuzumab-sensitive NCI-N87 cells and trastuzumab-resistant N87-TR1, N87-TR2, N87-TR3, and N87-TR4 gastric cancer cells. E, Percent relative growth oftrastuzumab-sensitive NCI-N87 cells and trastuzumab-resistant N87-TR1, N87-TR2, N87-TR3, and N87-TR4 gastric cancer cells, 72 hours after treatmentwith increasing concentrations of trastuzumab. Vehicle-treated cells were assigned a value of 100% and designated as control. Means and 95% CIs ofthree independent experiments performed in quadruplicate are shown. F, Fifty athymic nude mice bearing heterotopic NCI-N87, N87-TR1, N87-TR2, N87-TR3,and N87-TR4 gastric tumors were randomly assigned to 10 groups (n ¼ 5 per group) to receive 20 mg/kg of either trastuzumab or saline (control) i.p.twice a week. Mice were sacrificed by carbon dioxide inhalation when evidence of advanced bulky disease developed. The day of sacrifice wasconsidered the day of death from disease for the purpose of survival evaluation. Differences among survival duration of mice in each group were determinedby log-rank test. NCI-N87, control versus trastuzumab, median survival ¼ 66.5 days vs. undefined days, P¼ 0.0027, HR ¼ 0.03652, 95% CI¼ 0.004217–0.3163;N87-TR1, control versus trastuzumab, median survival ¼ 55 days vs. 49 days, HR ¼ 0.7026, 95% CI ¼ 0.04055–12.17, P ¼ 0.8; N87-TR2, control versustrastuzumab, median survival ¼ 54 days versus 42.5 days, HR ¼ 1.338, 95% CI ¼ 0.3258–5.491, P ¼ 0.686; N87-TR3, control versus trastuzumab,median survival ¼ 49 days versus 40 days, HR ¼ 3.621, 95% CI ¼ 0.5385–19.75, P ¼ 0.1983; N87-TR4, control versus trastuzumab, median survival ¼ 54.5 daysvs. 44 days, HR ¼ 2.143, 95% CI ¼ 0.2905–15.8, P ¼ 0.4547.
FGFR3 in Trastuzumab-Resistant Gastric Cancer
www.aacrjournals.org Clin Cancer Res; 2016 OF5
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

IB:HER3
IB:HER2
IB:EGFR
IB:g-tubulin
B
IB:p-ERK1/2
IB:ERK 1/2
IB:g-tubulin
IB:FGFR3
IB:AKT
IB:p-AKT
F
IB:Vimentin
IB:E-cadherin
IB:g-tubulin
IB:ZEB1
D
NCI-N87
N87-TR1
N87-TR2
N87-TR3
N87-TR4
0
50
100
150 FGF9
NCI-N87
N87-TR1
N87-TR2
N87-TR3
N87-TR4
0
1,000
2,000
3,000 FGFR3
NCI-N87
N87-TR1
N87-TR2
N87-TR3
N87-TR4
0
20
40
60 ZEB1
Fold
cha
nge
(2–Δ
Δ Ct)
Fold
cha
nge
(2–Δ
Δ Ct)
Fold
cha
nge
(2–Δ
Δ Ct)
Fold
cha
nge
(2–Δ
Δ Ct)
Fold
cha
nge
(2–Δ
Δ Ct)
C
NCI-N87
N87-TR1
N87-TR2
N87-TR3
N87-TR4
0.0
0.5
1.0
1.5 CDH1
NCI-N87
N87-TR1
N87-TR2
N87-TR3
N87-TR4
0
10,000
20,000
30,000
40,000
50,000 VIM
E
Thre
shol
d
A0 5 10 15 20 25 30 35
–log(P value)
EIF2 Signaling
Regulation of eIF4 and p70S6K signaling
Protein ubiquitination pathway
Mitochondrial dysfunction
mTOR Signaling
G
Ratio
Piro et al.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF6
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

recent clinical evidences suggested that activating mutations inthe PI3KCA gene encoding the p110a catalytic subunit of thePI3K enzyme (28, 29), rather than the loss or inactivatingmutations of phosphatase and tensin homolog on chromo-some 10 (PTEN; ref. 30), might be important biomarkers toidentify breast cancer patients resistant to anti-HER2 therapies.However, less is known about the role of FGFR pathway in anti-HER2 resistance in breast cancer. It has been demonstrated thatFGFR2 is a pivotal molecule for the survival of lapatinib-
resistant cells, suggesting that a switch of addiction from theHER2 to the FGFR2 pathway enable cancer cells to becomeresistant to HER2-targeted therapy (23). In our study, wedemonstrated that the AKT signaling pathway is significantlyactivated in trastuzumab-resistant gastric cancer models, ifcompared with sensitive control. More significantly, we dem-onstrated for the first time the overactivation of the AKTsignaling pathway in postprogression bioptic samples fromgastric cancer patients receiving therapies with trastuzumab, if
E-cadherin
ZEB1
FGFR3
HER2
N87-TR4N87-TR3N87-TR2N87-TR1NCI-N87
Vimentin
p-AKT
Figure 3.
Immunohistochemical analysis oftrastuzumab-resistant versustrastuzumab-sensitive gastricxenograft tumors. Serial paraffin-embedded tumor sections werestained with antibodies against HER2,FGFR3, ZEB1, pAKT, E-cadherin, andvimentin proteins (scale bar: 200 mm;10� magnification).
Figure 2.Identification of relevant biological processes and genes by using global transcript profiling. A, Signaling pathways enriched among genes differentiallyexpressed in trastuzumab-resistant gastric cancer cells versus their sensitive control cell line. The X-axis represents the �log(10) P value for enrichment,with the threshold drawn at P ¼ 0.05. B, Western blot analysis for the expression of ErbB family receptors in N87-TR gastric cancer cells comparedto trastuzumab-sensitive NCI-N87 gastric cancer and BT-474 breast cancer cells, and trastuzumab-resistant YCC2 gastric cancer and MCF-7 breast cancercells. C, Validation of ZEB-1, E-cadherin (CDH1), and vimentin (VIM) genes differentially expressed in trastuzumab-resistant cells versus parentalsensitive cells. qRT-PCR data are expressed as the fold change in RNA expression between the gene of interest and b-actin. Mean and 95% CIs are shown.D, Western blot analysis for the expression of EMT markers. E, Validation of fibroblast growth factor (FGF)9 and FGF receptor 3 (FGFR3) genesdifferentially expressed in trastuzumab-resistant cells versus parental sensitive cells. qRT-PCR data are expressed as the fold change in RNA expressionbetween the gene of interest and b-actin. Mean and 95% CIs are shown. F, Western blot analysis for the expression of FGFR3, and the activation of AKT andERK1/2, in N87-TR cells and their control cell line. G, Interaction network derived from genes upregulated in trastuzumab-resistant gastric cancer cell linesversus the respective sensitive control cell line. Each interaction is supported by at least one literature reference identified in the Ingenuity PathwayKnowledge Base, with solid lines representing direct interactions and dashed lines representing indirect interactions. FGFR3, fibroblast growth factor receptor3; FGF9, fibroblast growth factor 9; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; AKT, V-Akt murinethymoma viral oncogene homolog; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PDK1, pyruvate dehydrogenase kinase isozyme 1; CDH1, cadherin 1;TSC1, tuberous sclerosis 1; TSC2, tuberous sclerosis 2; Rheb, RAS-homolog enriched in brain; mTORC1, mechanistic target of rapamycin complex 1; mTORC2,mechanistic target of rapamycin complex 2; Rho, rhodopsin; Rock, Rho-associated coiled-coil containing protein kinase; VIM, vimentin; Raptor, regulatoryassociated protein of MTOR complex 1; GBL, G protein beta subunit-like; PRAS40, proline-rich Akt substrate of 40 kDa; mTOR, mammalian target ofrapamycin; Rictor, RPTOR independent companion of MTOR complex 2; Proctor, protein observed with rictor-1; SIN1, SAPK-interacting protein.
FGFR3 in Trastuzumab-Resistant Gastric Cancer
www.aacrjournals.org Clin Cancer Res; 2016 OF7
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

compared with their respective pretreatment samples. Mostimportantly, we demonstrated that the overactivation of theAKT signaling pathway in trastuzumab-resistant gastric cancermodels is dependent on the overexpression of FGFR3 and canbe modulated by the inhibition of this receptor.
EMT is a transdifferentiation process that converts tumorcells with an epithelial phenotype into highly motile mesen-chymal cells (31). The PI3K/AKT/mTOR signaling pathwaydemonstrated an essential role in mediating EMT, thought theactivation of both mTOR complex 1 (mTORC1; ref. 32), and
A
B
D
C
E
100500
N87-TR4
N87-TR3
N87-TR2
N87-TR1
NCI-N87ctrDovitinib 5 mmol/L**
***
***
***
***
Migration (%)
NCI-N87 N87-TR2N87-TR1 N87-TR3 N87-TR4
CTR
Dovitinib 5 mmol/L
36 h
IB:g-tubulin
IB:ZEB1
Dovitinib 5 mmol/L +−+ −+ −+ −−0 2 5 0 2 55 0 2 0 0 2 5 Dovitinib mmol/L
IB:p-AKT
IB:g-tubulin
IB:AKT
10–7
10–6
.5
10–6
10–5
.50
50
100 NCI-N87
N87-TR1N87-TR2N87-TR3N87-TR4
Dovitinib (mol/L)
Rel
ativ
e gr
owth
(%)
10–5
.9
10–5
.5
10–5
.1
10–4
.70
50
100 NCI-N87
N87-TR1N87-TR2N87-TR3N87-TR4
AZD4547 (mol/L)
Rel
ativ
e gr
owth
(%)
Figure 4.
In vitro antitumor activity of fibroblast growth factor receptor 3 inhibitor dovitinib in trastuzumab-resistant N87-TR cell lines. A, Percent relative growthof N87-TR cells and their control cell line, 72 hours after treatment with increasing concentrations of dovitinib or AZD4547. Vehicle-treated cells wereassigned a value of 100% and designated as control. Means and 95% CIs of three independent experiments performed in quadruplicate are shown.Dovitinib: NCI-N87 IC50 ¼ 2.189E�006 M versus N87-TR1 IC50 ¼ 4.122E�007 M, or versus N87-TR2 IC50 ¼ 5.297E�007 M, N87-TR3 IC50 ¼ 6.127E�007 M, N87-TR4 IC50 ¼ 5.764E�007 M, all P < 0.0001; AZD4547: NCI-N87 IC50 ¼ 1.697E�005 M versus N87-TR1 IC50 ¼ 1.038E�005 M, or versus N87-TR2 IC50 ¼9.465E�006 M, N87-TR3 IC50 ¼ 9.161E�006 M, N87-TR4 IC50¼ 6.875E�006 M, all P < 0.0001. B,Western blot analysis for the activation of AKT after 24 hourstreatment with dovitinib. C, Western blot analysis for the expression of ZEB1 after 72 hours treatment with dovitinib. D, Photographs of the wound area weretaken by using phase-contrast microscopy immediately and 36 hours after the incision in untreated and treated cells. E, Levels of cancer cell migration of N87-TR cells and their control cell line after dovitinib treatment. Results are presented as percentages of the total distances between the wound edges enclosedby cancer cells. The mean values and 95% CIs from three independent experiments done in quadruplicate are shown. Relative migration: NCI-N87, control,mean ¼ 15.91, 95% CI ¼ 12.75–19.06, versus dovitinib, mean ¼ 8.39, 95% CI ¼ 5.24–11.55, P < 0.01; N87-TR1, control, mean ¼ 93.85, 95% CI ¼ 86.05–101.65,versus dovitinib, mean ¼ 3.51, 95% CI ¼ 1.84–5.17, P < 0.001; N87-TR2, control, mean ¼ 100, 95% CI ¼ 99.12–100.88, versus dovitinib, mean ¼ 4.41, 95% CI ¼3.53–5.28, P < 0.001; N87-TR3, control, mean ¼ 93.84, 95% CI ¼ 85.42–102.25, versus dovitinib, mean ¼ 4.06, 95% CI ¼ 2.14–5.99, P < 0.001; N87-TR4, control,mean ¼ 100, 95% CI ¼ 99.12–100.88, versus dovitinib, mean ¼ 6.33, 95% CI ¼ 5.11–7.55, P < 0.001. �� , P < 0.01; ��� , P < 0.001 (unpaired Student t test).
Piro et al.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF8
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

mTORC2 (33, 34). In our study, we found an EMT phenotypeand increased cell motility in trastuzumab-resistant gastriccancer models. We demonstrated that the induction of thisphenotype parallels the overactivation of the AKT signalingpathway, and is sustained by the autocrine loop betweenoverexpressed FGF9 and FGFR3, as it can be modulated by theFGFR3 inhibitor dovitinib. Of clinical relevance, we showed forthe first time the overexpression of the EMT marker ZEB1 inpostprogression bioptic samples from gastric cancer patientsreceiving therapies with trastuzumab, if compared with theirrespective pretreatment samples.
Dovitinib (TKI258, Novartis) is a multitargeted receptortyrosine kinase inhibitor, which strongly binds to FGFR3 andinhibits its phosphorylation (NCI Drug Dictionary). This agentis currently explored as a monotherapy in the second- and third-line setting [NCT01719549], and in combination with docetaxelin the second-line setting (NCT01921673) for patients withgastric cancer. In our study, we showed that dovitinib signifi-cantly reduced the activation of AKT and the expression of ZEB1,inhibiting, in turn, the growth and the migratory properties of
trastuzumab-resistant cells. Of translational relevance, weshowed the therapeutic efficacy of treatment with dovitinib inan in vivomodel of trastuzumab-resistant N87-TR4 gastric cancerxenografts.
This study, however, had some limitations. The orthotopicxenograft tumor models used are limited by the artificial micro-environment in an immunocompromised host and could notfaithfully recapitulate the histopathologic features of the humandisease, thus, impairing the representativeness of the resultsobserved to human patients. Moreover, the four different TR celllines developed and analyzed in this study derive from NCI-N87cells, thus, we could not exclude that different mechanisms ofresistance could have been emerged by starting the selectionprocess from different HER2-positive gastric cancer cell models.In this regard, we corroborated the clinical relevance of ourfindings for human gastric cancer patients by detecting for thefirst time a significantly higher expression of FGFR3, activatedAKT, andof the EMTmarker ZEB1 in posttreatment biopsieswhencompared with their respective pretreatment biopsies from threegastric cancer patients receiving trastuzumab-based treatments.
A
B
NCI-N87
C
N87-TR4
NCI-N87
N87-TR4
N87-TR4
DovitinibVehicle0.0
0.5
1.0
1.5
2.0
2.5
**
Tum
or v
olum
e (c
m3 )
DovitinibVehicle0.0
0.5
1.0
1.5
2.0
2.5
Tum
or v
olum
e (c
m3 )
8060402000
20
40
60
80
100
VehicleDovitinib
Days after injection
Surv
ival
(%)
1008060402000
20
40
60
80
100 VehicleDovitinib
Days after injection
Surv
ival
(%)
ctr
Dovitinib
p-A
KT
Figure 5.
Antitumor activity of oral fibroblast growthfactor receptor 3 inhibitor dovitinib in vivoin N87-TR4 and NCI-N87 gastric tumorxenografts. A and B, Left, 40 athymic nudemice bearing subcutaneous heterotopicNCI-N87 or N87-TR4 gastric tumors wererandomly assigned to four groups (n ¼ 10per group) to receive daily oral dovitinib(40 mg/kg) or vehicle for 4 weeks. Micewere sacrificed by carbon dioxideinhalation when reached cut-off volume of2 cm3. The day of sacrifice was consideredthe day of death from disease for thepurpose of survival evaluation. Differencesamong survival duration of mice in eachgroup were determined by log-rank test.Right, tumor growth at the median survivalduration of the control group (65 and 40days for NCI-N87 and N87-TR4,respectively). Error bars are 95% CIs. (NCI-N87 tumor volume: control, mean ¼ 1,921mm3, 95% CI ¼ 1,836–2,007; dovitinib,mean ¼ 1,813, 95% CI ¼ 1,574–2,051;survival: control versus dovitinib, mediansurvival ¼ 65 days versus 62 days, HR ¼0.7750, 95% CI ¼ 0.2664–2.255, P ¼ 0.64;N87-TR4 tumor volume: control, mean¼ 1,910, 95% CI ¼ 1,459–2,362; dovitinib,mean ¼ 1,086, 95% CI ¼ 803–1,369;survival: control versus dovitinib, mediansurvival ¼ 40 versus 63 days, HR ¼ 0.1413,95% CI ¼ 0.04611–0.4331, P ¼ 0.0006)�� , P < 0.01. C, Six mice bearing N87-TR4gastric tumors were randomly allocated (n¼ 3 per group) to receive 40 mg/kg dailyoral dovitinib or vehicle as control.Treatments were continued for 4 weeks.Tumors were excised 1 day after theend of treatments. Paraffin-embeddedgastric tumor sections stainedimmunohistochemically with antibodiesagainst pAKT.
FGFR3 in Trastuzumab-Resistant Gastric Cancer
www.aacrjournals.org Clin Cancer Res; 2016 OF9
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

2013 2014
DiagnosisGastrectomy +liver resection
Taxotere + 5-FU + oxaliplatinCapecitabine + oxaliplatinTrastuzumab
Hemicolectomy
RT
Disease relapse
FGFR3
p-AKT
ZEB1
FGFR3
p-AKT
ZEB1
2012 2013
DiagnosisEsophagectomy
Taxotere + cisplatin + 5FUTrastuzumab
Liver metastasis resection
2014
Lung metastasis resection
Disease relapseDisease relapse
RTCisplatin + 5FU
2011
FGFR3
p-AKT
ZEB1
FGFR3
p-AKT
ZEB1
Diagnosis
Cisplatin + 5FUTrastuzumab
Pretreatment sample Postprogression sample
2015
Liver metastasis biopsy
Disease relapse
2012
Patient 1
Patient 2
Patient 3
FGFR3
p-AKT
ZEB1ZEB1
p-AKT
FGFR3
2015
Pretreatment sample Postprogression sample
Pretreatment sample Postprogression sample
Figure 6.
Analysis of pretreatment and postprogression biopsies from gastric cancer patients and clinical history. Paraffin-embedded sections from patients treated withtrastuzumabwho underwent relapsewere immunohistochemically stained with antibodies against FGFR3, p-AKT, and ZEB1 (scale bar, 200 mm; 10�magnification).
Piro et al.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF10
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

The limited number of patients' samples available and the poten-tial heterogeneity in the primary tumor and among the differentsites of metastasis analyzed could represent additional potentiallimitations of this study.
Two prior studies reported the development of trastuzumab-resistant NCI-N87 cells (35, 36). Eto and colleagues establisheda resistant cell line by culturing NCI-N87 cells in vitro in thepresence of increasing concentration of trastuzumab from 5 to100 mg/mL along 6 months (35). Similarly, Yang and collea-gues selected in vitro another resistant cell line by growing NCI-N87 over a 1-year period in increasing concentration of tras-tuzumab up to 10 mg/mL. During the development of theresistance, NCI-N87 cells underwent EMT, as shown by theincrease of ZEB1 expression and of their migratory properties.However, trastuzumab was still able to suppress the phosphor-ylation of AKT in this cell model, and the activation of an IL6/STAT3/Jagged-1/Notch positive feedback signaling loop wasindicated as associated with the acquisition of trastuzumabresistance (36). In our study, we developed four differenttrastuzumab-resistant models through, not an in vitro, but anin vivo process of selection by using a clinically relevant modelof orthotopic growth of tumors in the mouse stomach. Weverified the resistance of these models to trastuzumab in vitro,up to 400 mg/mL, as well as in vivo. Although we confirmed theacquisition of an EMT phenotype and an overexpression ofZEB1 in the trastuzumab-resistant models when compared withsensitive cells, we did not measure any significant overexpres-sion of IL6 or Jagged-1 in our models (data not shown).
In conclusion, we propose a model in which trastuzumabinduces the selection of gastric cancer cells overexpressingFGFR3 and its ligand FGF9. This autocrine signaling loopsustains the activation of the PI3K/AKT/mTOR signaling path-way, and in turn, tumor growth and a more aggressive EMTphenotype. Most importantly, our study provides the preclin-ical rationale to investigate the inihibition of FGFR3 as second-line treatment strategy in gastric cancer patients refractory tofirst-line trastuzumab-containing therapies.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: G. Piro, C. Carbone, S. Giacopuzzi, D. MelisiDevelopment of methodology: G. Piro, I. Cataldo, A. Sbarbati, D. MelisiAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.):G. Piro, C. Carbone, F.D.Nicolantonio, S. Giacopuzzi,G. Aprile, F. Simionato, F. Boschi, M. Zanotto, R. Santoro, G. de Manzoni,A. Scarpa, D. MelisiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): G. Piro, C. Carbone, I. Cataldo, F.D. Nicolantonio,G. Aprile, V. Merz, A. Scarpa, D. MelisiWriting, review, and/or revisionof themanuscript:G. Piro, F.D.Nicolantonio,G. Aprile, V. Merz, A. Scarpa, G. Tortora, D. MelisiAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): F. Simionato, M. Mihaela Mina, G. deManzoni,D. MelisiStudy supervision: G. de Manzoni, G. Tortora, D. Melisi
AcknowledgmentsWe thank Licia Montagna for the technical execution of immunohis-
tochemistry. Part of the work was performed at the Laboratorio Universitariodi Ricerca Medica (LURM) Research Center, University of Verona.
Grant SupportThisworkwas supported inpart by theAssociazione Italiana per la Ricerca sul
Cancro (AIRC) Start-Upno. 10129, and5permille no. 10016 (toD.Melisi), andby the AIRC grants IG 11930, 5 per mille 12182, 12214, and PRIN no.2009�23L78_005 (to G. Tortora). Partial support was also provided by grantFarmacogenomica' 5 per mille 2009 MIUR from Fondazione Piemontese per laRicerca sul Cancro—ONLUS (to F.D. Nicolantonio) and Fondo per la RicercaLocale (ex 60%), Universit�a di Torino, 2014 (to F.D. Nicolantonio).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received January 29, 2016; revised May 26, 2016; accepted June 1, 2016;published OnlineFirst June 7, 2016.
References1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin
2015;65:5–29.2. Lordick F, Allum W, Carneiro F, Mitry E, Tabernero J, Tan P, et al. Unmet
needs and challenges in gastric cancer: the way forward. Cancer Treat Rev2014;40:692–700.
3. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al.Trastuzumab in combination with chemotherapy versus chemotherapyalone for treatment of HER2-positive advanced gastric or gastro-oesopha-geal junction cancer (ToGA): a phase 3, open-label, randomised controlledtrial. Lancet 2010;376:687–97.
4. Aprile G, Giampieri R, Bonotto M, Bittoni A, Ongaro E, Cardellino GG,et al. The challenge of targeted therapies for gastric cancer patients: thebeginning of a long journey. Expert Opin Investig Drugs 2014;23:925–42.
5. Tortora G. Mechanisms of resistance to HER2 target therapy. J Natl CancerInst Monogr 2011;2011:95–8.
6. ScaltritiM, Eichhorn PJ, Cortes J, Prudkin L, AuraC, Jimenez J, et al. Cyclin Eamplification/overexpression is a mechanism of trastuzumab resistance inHER2þ breast cancer patients. Proc Natl Acad Sci U S A 2011;108:3761–6.
7. Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al.Heterotrimerization of the growth factor receptors erbB2, erbB3, andinsulin-like growth factor-i receptor in breast cancer cells resistant toherceptin. Cancer Res 2010;70:1204–14.
8. Ritter CA, Perez-TorresM, Rinehart C,GuixM,Dugger T, Engelman JA, et al.Human breast cancer cells selected for resistance to trastuzumab in vivo
overexpress epidermal growth factor receptor and ErbB ligands andremain dependent on the ErbB receptor network. Clin Cancer Res 2007;13:4909–19.
9. Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combatingtrastuzumab resistance by targeting SRC, a common node downstream ofmultiple resistance pathways. Nat Med 2011;17:461–9.
10. Kim J, Fox C, Peng S, Pusung M, Pectasides E, Matthee E, et al.Preexisting oncogenic events impact trastuzumab sensitivity inERBB2-amplified gastroesophageal adenocarcinoma. J Clin Invest2014;124:5145–58.
11. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., KinzlerKW. Cancer genome landscapes. Science 2013;339:1546–58.
12. Melisi D, Piro G, Tamburrino A, Carbone C, Tortora G. Rationale andclinical use of multitargeting anticancer agents. Curr Opin Pharmacol2013;13:536–42.
13. Carbone C, Tamburrino A, Piro G, Boschi F, Cataldo I, Zanotto M, et al.Combined inhibition of IL1, CXCR1/2, and TGFbeta signaling pathwaysmodulates in vivo resistance to anti-VEGF treatment. Anticancer Drugs2016;27:29–40.
14. CarboneC, PiroG, FassanM,TamburrinoA,MinaMM,ZanottoM, et al. Anangiopoietin-like protein 2 autocrine signaling promotes EMT duringpancreatic ductal carcinogenesis. Oncotarget 2015;6:13822–34.
15. Piro G, Giacopuzzi S, Bencivenga M, Carbone C, Verlato G, Frizziero M,et al. TAK1-regulated expression of BIRC3 predicts resistance to
FGFR3 in Trastuzumab-Resistant Gastric Cancer
www.aacrjournals.org Clin Cancer Res; 2016 OF11
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

preoperative chemoradiotherapy in oesophageal adenocarcinoma patients.Br J Cancer 2015 Sep 15;113:878–85.
16. Sumida T, Kitadai Y, Shinagawa K, TanakaM, KodamaM,Ohnishi M, et al.Anti-stromal therapy with imatinib inhibits growth and metastasis ofgastric carcinoma in an orthotopic nude mouse model. Int J Cancer 2011;128:2050–62.
17. Dalla Pozza E, Dando I, Biondani G, Brandi J, Costanzo C, Zoratti E, et al.Pancreatic ductal adenocarcinoma cell lines display a plastic ability tobi-directionally convert into cancer stem cells. Int J Oncol 2015;46:1099–108.
18. Zhuang Z, Ju HQ, Aguilar M, Gocho T, Li H, Iida T, et al. IL1 ReceptorAntagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-kB Acti-vation. Clin Cancer Res 2016;22:1432–44.
19. Isola J, Chu L, DeVries S, Matsumura K, Chew K, Ljung BM, et al. Geneticalterations in ERBB2-amplified breast carcinomas. Clin Cancer Res 1999;5:4140–5.
20. Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFRsignaling in cancer. Clin Cancer Res 2015;21:2684–94.
21. Oliveras-FerrarosC,Cufi S,Queralt B, Vazquez-MartinA,Martin-Castillo B,de Llorens R, et al. Cross-suppression of EGFR ligands amphiregulin andepiregulin and de-repression of FGFR3 signalling contribute to cetuximabresistance in wild-type KRAS tumour cells. Br J Cancer 2012;106:1406–14.
22. Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation ofmitogen-activated protein kinase (MAPK) pathway by FGF receptor 3(FGFR3)/Ras mediates resistance to vemurafenib in human B-RAFV600E mutant melanoma. J Biol Chem 2012;287:28087–98.
23. Azuma K, Tsurutani J, Sakai K, Kaneda H, Fujisaka Y, Takeda M, et al.Switching addictions between HER2 and FGFR2 in HER2-positive breasttumor cells: FGFR2 as a potential target for salvage after lapatinib failure.Biochem Biophys Res Commun 2011;407:219–24.
24. Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A. Identification offibroblast growth factor 9 (FGF9) as a high affinity, heparin dependentligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. GrowthFactors 1995;12:223–33.
25. Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonataldeath in Fgf9-null mice identify this gene as an essential regulator of lungmesenchyme. Development 2001;128:2095–106.
26. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K,et al. A functional genetic approach identifies the PI3K pathway as a major
determinant of trastuzumab resistance in breast cancer. Cancer Cell2007;12:395–402.
27. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTENactivation contributes to tumor inhibition by trastuzumab, and loss ofPTEN predicts trastuzumab resistance in patients. Cancer Cell 2004;6:117–27.
28. Majewski IJ, Nuciforo P,Mittempergher L, Bosma AJ, EidtmannH,HolmesE, et al. PIK3CA mutations are associated with decreased benefit toneoadjuvant human epidermal growth factor receptor 2-targeted therapiesin breast cancer. J Clin Oncol 2015;33:1334–9.
29. Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M,et al. PIK3CA mutations are associated with lower rates of pathologiccomplete response to anti-human epidermal growth factor receptor 2(her2) therapy in primaryHER2-overexpressing breast cancer. J ClinOncol2014;32:3212–20.
30. Nuciforo PG, Aura C, Holmes E, Prudkin L, Jimenez J, Martinez P, et al.Benefit to neoadjuvant anti-human epidermal growth factor receptor 2(HER2)-targeted therapies in HER2-positive primary breast cancer is inde-pendent of phosphatase and tensin homolog deleted from chromosome10 (PTEN) status. Ann Oncol 2015;26:1494–500.
31. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mes-enchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96.
32. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-inducedepithelial to mesenchymal transition is regulated by activation of themTOR pathway. J Cell Biol 2007;178:437–51.
33. Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, et al.mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colo-rectal cancer via RhoA and Rac1 signaling pathways. Cancer Res 2011;71:3246–56.
34. Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymaltransition and cell invasion. J Cell Sci 2012;125:1259–73.
35. Eto K, Iwatsuki M, Watanabe M, Ishimoto T, Ida S, Imamura Y, et al. Thesensitivity of gastric cancer to trastuzumab is regulated by the miR-223/FBXW7 pathway. Int J Cancer 2015;136:1537–45.
36. Yang Z, Guo L, Liu D, Sun L, Chen H, Deng Q, et al. Acquisitionof resistance to trastuzumab in gastric cancer cells is associatedwith activation of IL-6/STAT3/Jagged-1/Notch positive feedback loop.Oncotarget 2015;6:5072–87.
Clin Cancer Res; 2016 Clinical Cancer ResearchOF12
Piro et al.
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178

Published OnlineFirst June 7, 2016.Clin Cancer Res Geny Piro, Carmine Carbone, Ivana Cataldo, et al. Trastuzumab in Gastric Cancer PatientsAn FGFR3 Autocrine Loop Sustains Acquired Resistance to
Updated version
10.1158/1078-0432.CCR-16-0178doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2016/06/07/1078-0432.CCR-16-0178.DC1Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/early/2016/11/09/1078-0432.CCR-16-0178To request permission to re-use all or part of this article, use this link
Research. on July 28, 2018. © 2016 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst June 7, 2016; DOI: 10.1158/1078-0432.CCR-16-0178