analisi pixe e pige per la
TRANSCRIPT

Universita degli Studi di Firenze
Facolta di Scienze Matematiche Fisiche e Naturali
Tesi di Laurea in Fisica di I livello
ANALISI PIXE E PIGE PER LA
RIVELAZIONE DI ELEMENTI
LEGGERI: UN’APPLICAZIONE ALLA
DETERMINAZIONE DEL SODIO IN
VETRI ARCHEOLOGICI
Candidato: Matteo Viani
Relatore: Prof. Pier Andrea Mando
Anno Accademico 2004/05

Indice
1 PIXE 1
1.1 Aspetti generali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Analisi quantitativa . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.2.1 Target sottile . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.2 Target spesso . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2 PIGE 9
2.1 Aspetti generali . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.2 Analisi quantitativa . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
3 Analisi di alcuni vetri 14
3.1 Introduzione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
3.2 Analisi dati e commenti . . . . . . . . . . . . . . . . . . . . . . . . . 15
3.2.1 Analisi degli standard . . . . . . . . . . . . . . . . . . . . . . 15
3.2.2 Analisi delle tessere . . . . . . . . . . . . . . . . . . . . . . . . 18
Riferimenti bibliografici i

Il presente lavoro di tesi espone i risultati delle analisi fatte su vetri archeologici,
usando le tecniche PIXE e PIGE. L’esperimento e servito a fornire una caratter-
izzazione elementale dei reperti, con particolare attenzione al sodio, usato per dis-
criminare cronologicamente i vetri. Le due tecniche sono complementari nel rivelare
elementi medio-leggeri come il sodio, al limite della sensibilita di rivelazione per la
PIXE, ma ben rivelato dalla PIGE. L’esposizione dei risultati e preceduta da una
introduzione alle due tecniche di analisi. Tali risultati si basano sulle serie di misure
precedentemente effettuate presso l’acceleratore Tandem del LABEC di Firenze, ed
esposte in [2]
1 PIXE
1.1 Aspetti generali
La tecnica PIXE (Particle Induced X-ray Emission) si basa sull’interazione di un
fascio di particelle accelerate (tipicamente protoni a 2-3 MeV) con gli atomi di un
campione bersaglio. Questa interazione provoca l’emissione di raggi X con energie
caratteristiche di ogni elemento. Quando un protone incide su un atomo, infatti,
ha una certa probabilita di espellere un elettrone delle shell piu interne, ionizzando
l’atomo. La lacuna nell’orbitale interno viene prontamente riempita (entro un in-
tervallo di tempo dell’ordine di 10−15s), da un elettrone piu esterno, e si libera cosı
un’energia pari alla differenza ∆E = Eo − Ei delle energie di legame dell’elettrone
tra la shell esterna (di energia Eo) e quella interna (di energia Ei). Due sono i
processi con cui questa energia viene rilasciata.
Il primo e, appunto, la PIXE, cioe l’emissione di un fotone X di energia ∆E. Le
righe di emissione X, caratteristiche di ogni elemento, sono indicate con la notazione
K, L, M,. . . , a seconda che la shell che ha perso l’elettrone avesse numero quantico
principale n=1, n=2, n=3,. . . , rispettivamente. Compatibilmente con le regole di
selezione, ci sono piu transizioni possibili verso i livelli con n=1, n=2,. . . , e si parla
percio di serie K, serie L,. . . , all’interno di ciascuna delle quali si indicano le singole
righe mediante lettere greche accompagnate da numeri (ad esempio: Kα1, Kβ1, Lα1,
ecc).
L’altro processo e il cosiddetto effetto Auger, in cui l’energia e trasferita ad un
altro elettrone di una shell piu esterna, che viene rilasciato con una energia cinet-
ica pari a Ek = ∆E − Ej, essendo Ej l’energia di legame dell’elettrone a cui viene
trasferita l’energia ∆E.
1

I raggi X emessi sono raccolti da un opportuno rivelatore e ne viene fatta un’anal-
isi spettrale, cosı da mettere in evidenza, attraverso le energie caratteristiche della
radiazione, gli elementi che formano il campione. Tramite il conteggio dei raggi
X raccolti di una determinata energia, caratteristica di un determinato elemento,
si riesce a risalire alla quantita di quell’elemento all’interno del campione (l’analisi
quantitativa e trattata in maniera piu approfondita nel §1.2).
Si evidenzia cosı uno degli aspetti vantaggiosi della PIXE, quello di essere una anal-
isi multielementale, cioe che permette di rivelare contemporaneamente, in maniera
quantitativa, gli elementi che compongono il campione, consentendo anche di for-
mulare delle ipotesi stechiometriche. Un altro degli aspetti importanti della PIXE
e quello di poter essere una tecnica non invasiva, quando, come nel campo delle
applicazioni ai beni culturali, le correnti di fascio possono rimanere entro qualche
decina di pA; in queste condizioni non si produce alcuna alterazione visibile del mate-
riale analizzato. Inoltre e possibile eseguire PIXE tenendo il campione in atmosfera
(PIXE esterna), senza la necessita di mettere in vuoto l’oggetto da analizzare.
Questo comporta numerosi vantaggi: la presenza di aria, infatti, favorisce la disper-
sione di calore, limitando il riscaldamento del campione dovuto al fascio incidente
e riducendo i gia bassi rischi di danneggiamento; inoltre e possibile analizzare sen-
za grosse difficolta oggetti anche grandi (ad esempio dei dipinti), senza richiedere
il prelievo di campioni (non sempre possibile specialmente con oggetti di interesse
artistico come i dipinti); infine, in una misura con fascio esterno, non c’e alcun prob-
lema di disidratazione, come si avrebbe in vuoto per alcuni campioni, ad esempio
documenti cartacei.
L’energia minima rivelabile e dell’ordine del keV, che consente di rivelare le righe
K del sodio (1.04 keV); questo limite e dato principalmente dall’assorbimento nella
finestra d’ingresso del rivelatore (tipicamente un rivelatore a Si(Li)), oltre che dai
problemi di autoassorbimento da parte del campione (si veda anche §1.2 → effetti di
matrice). Cio significa che elementi piu leggeri (comprendendo quindi anche gli ele-
menti “organici” come l’ossigeno o il carbonio), che presentano differenze di energie
di legame inferiori al keV, non sono rivelabili. Di norma non sono rivelabili nem-
meno energie superiori a qualche decina di keV, la cui lunghezza di attenuazione nel
silicio diventa molto maggiore degli spessori dei rivelatori tipicamente usati. Tut-
tavia, come si vede anche dalla Fig.1, in corrispondenza di quegli elementi con righe
K troppo energetiche per essere rivelate con una buona efficienza, si hanno righe L
di energia minore e quindi rivelabili.
2

Figura 1: Energie X delle serie K e L in funzione del numero atomico. E stato messoin evidenza, mediante le due linee rosse, l’intervallo di energie che tipicamente e possibilerivelare con il rivelatore a Si(Li)
1.2 Analisi quantitativa
Come detto, una volta avvenuta la ionizzazione della shell, due sono i processi
con cui l’energia di diseccitazione puo venire rilasciata: tramite un fotone X o per
mezzo di un elettrone Auger. Per quantificare questo contributo si introduce la
efficienza di fluorescenza, definita come il rapporto tra il numero di emissioni di
raggi X e il numero totale di ionizzazioni prodotte in una data shell ; e usualmente
indicata con ω, specificando anche la shell a cui ci si riferisce (ωK , ωL,. . . ). Questo
parametro puo essere sia calcolato teoricamente sia misurato sperimentalmente e
risulta funzione del numero atomico Z. L’andamento di ω in funzione del numero
atomico e riportato in Fig.2. Un altro fattore da considerare per una corretta analisi
quantitativa di tipo PIXE e lo spessore del campione. Dovremo distinguere quindi
fra target sottile e target spesso. Per definire il significato di “sottile” e “spesso”
e necessario introdurre i cosiddetti effetti di matrice, cioe la perdita di energia
del fascio durante la penetrazione nel campione e l’autoassorbimento di raggi X
nel campione stesso. Un oggetto e considerato “sottile” se si possono trascurare
questi effetti, altrimenti si parla di target “spesso”. Trascurare gli effetti di matrice
significa sostanzialmente assumere che tutte le interazioni avvengano alla stessa
energia di fascio e che tutti i raggi X prodotti riescano ad uscire dal campione, con
3

Figura 2: Andamento dell’efficienza di fluorescenza delle shell K e L in funzione delnumero atomico Z
una probabilita trascurabile di essere riassorbiti dal campione stesso.
1.2.1 Target sottile
Il numero di ionizzazioni prodotte dal fascio di protoni in una data shell j (j =
K, L, M,. . . ) per una certa specie atomica Z e proporzionale alla concentrazione
di quella specie atomica nel campione, al numero di particelle Np che sono passate
attraverso il campione e allo spessore t del target :
Nion,j(Z) = σion,jNZtNp (1)
dove NZ e il numero di atomi con numero atomico Z per unita di volume. Il
termine σion,j e detto sezione d’urto di ionizzazione per una data shell j : questo
termine indica la probabilita che l’interazione dei protoni del fascio con gli atomi
del campione dia luogo ad una ionizzazione. Il prodotto
σXj= σion,jωj (2)
fra la sezione d’urto di ionizzazione σion,j e l’efficienza di fluorescenza ωj per una data
shell da la sezione d’urto totale per la produzione X della corrispondente serie; σXj
risulta ancora funzione del numero atomico Z e dell’energia del fascio. L’andamento
delle sezioni d’urto di produzione X per shell K e L e riportato in Fig. 3.
Dalla (1) e dalla (2) si puo ricavare il numero di raggi X prodotti:
NXj(Z) = ωjNion,j(Z) = σXj
NZtNp = σXj
NAρZ
AtQ
e(3)
4

Figura 3: Sezione d’urto di produzione X da parte di protoni per le shell K e L in funzionedelle energie dei protoni (fig. a) e del numero atomico (fig. b). (1 barn = 10−24 cm2)
5

dove ρZ e la densita dell’elemento Z, NA e il numero di Avogadro e A e la massa
atomica della specie, e dove Np e rappresentato dalla carica Q trasportata dal fascio
durante la misura divisa per la carica unitaria dell’elettrone e.
Tenendo conto del fattore geometrico Ω/4π dovuto all’angolo solido visto dal rivela-
tore, del fattore di trasmissione βZ , che tiene conto dell’assorbimento dei raggi X nel
percorso dal punto di emissione alla finestra d’ingresso del rivelatore, e dell’efficienza
intrinseca εZ del rivelatore stesso, dipendente dall’elemento tramite l’energia X che
lo identifica, dalla (3) si ottiene il numero di raggi X rivelati (yield), provenienti
dall’elemento Z:
Y0,Xj(Z) = σXj
(Z,E0)Ω
4πεZβZ
NAρZ
AtQ
e(4)
Questa puo anche essere riscritta in una forma piu compatta:
Y0,Xj(Z) = ηQρZt (5)
avendo definito il fattore di efficienza η per un dato raggio X, una data energia
di fascio, per un dato rivelatore, in una data geometria:
η = σXj(Z,E0)
Ω
4πεZβZ
NA
Ae(6)
Sebbene sia possibile conoscere η a priori, dalla conoscenza dei fattori geometrici
(misurabili) e di σXj, quello che si fa in pratica e di trovare il valore di η speri-
mentalmente, invertendo la (5) e misurando, nella stessa geometria di rivelazione,
la Y0,Xjper degli standard sottili con una densita areale ρZt nota.
1.2.2 Target spesso
Supponiamo ora che lo spessore del campione da analizzare sia tale da non poter piu
trascurare gli effetti di matrice. Per ottenere la yield di raggi X dovremo integrare
i contributi dei vari strati del target attraversati dal fascio con energie via via de-
crescenti, considerando per ciascuno di essi la corrispondente σXj(Ep) e la riduzione
dell’intensita di raggi X trasmessi al rivelatore, dovuta ad autoassorbimento. Con
riferimento alla Fig.4, si ha:
YXj(Z) =
Ω
4πεZβZ
Q
e
NA
AρZ
∫ T
0
σXj(Z,Ep(x)) e−
µxcos θ dx (7)
dove µ e il coefficiente di attenuazione dei raggi X nel materiale. Introducendo il
potere di frenamento (stopping power) S = dEp
ρ dx, funzione della composizione del
6

Figura 4: Diagramma di analisi su un target spesso
target e dell’energia del fascio Ep, dove ρ indica la densita complessiva del campione,
la (7) si puo riscrivere come:
YXj(Z) =
Ω
4πεZβZ
Q
e
NA
A
ρZ
ρ
∫ E0−∆E
E0
σXj(Z,Ep) e−
µx(Ep)
cos θdEp
S(Ep)(8)
dove E0 e l’energia iniziale del fascio (all’ingresso del target) e ∆E e la perdita totale
di energia attraverso lo spessore T , cioe
∆E =
∫ T
0
ρSdx (9)
Generalmente si introduce un fattore di correzione di matrice FXjper ogni energia
X, definito dal rapporto tra la yield ideale Y0, che si otterrebbe per quello stesso
spessore in assenza di effetti di matrice, e la yield con effetti di matrice:
FXj(Z) =
Y0,Xj(Z)
YXj(Z)
(10)
Con la (4) e la (7), la (10) si riscrive come
FXj(Z) =
TσXj(Z,E0)∫ T
0σXj
(Z,Ep(x)) e−µx
cos θ dx(11)
Dalla (5) e dalla (10) si ottiene una espressione per la densita areale:
(ρt)Z =YXj
(Z)
QηZ,E0
FXj(Z) (12)
7

dove η e il fattore di efficienza, risultante dalle misure su standard sottili. L’analisi
quantitativa, quindi, di target spessi si puo ottenere dalla YXj(Z) considerando il
target come sottile e applicando il fattore di correzione F appropriato per tenere
conto degli effetti di matrice.
Il problema della determinazione di FXjha una risoluzione piuttosto complessa.
Tipicamente, quando a priori non e nota la composizione del campione1, si ricorre
ad una procedura iterativa per calcolare FXj. L’approssimazione di ordine zero per
la composizione del target si ottiene trascurando tutti gli effetti di matrice, cioe
considerando il target sottile ed usando la (12) con FXj(Z) = 1 per tutte le Z. La
densita areale cosı ottenuta per gli elementi rivelati e usata per il calcolo del potere
di frenamento S e per il coefficiente di assorbimento µ da usare nella (11). Da
questo si ottiene un’approssimazione al prim’ordine della composizione del target
utilizzando nuovamente la (12), con cui ricavare un nuovo potere di frenamento e un
nuovo coefficiente di assorbimento da usare nella (11). La procedura si ripete quindi
iterativamente e, generalmente, converge rapidamente verso una buona stima della
composizione del campione (per una analisi piu in dettaglio si veda anche [1])
Per risolvere correttamente il problema della determinazione di FXjoccorre che
il campione si possa considerare di composizione omogenea alle diverse profon-
dita. In tal caso il problema e solo quello di conoscere lo spessore T . Se lo spes-
sore, per quanto sconosciuto, e maggiore del range dei protoni Rp (tipicamente
∼ 50− 100 µg cm−2, e si parla allora di target infinitamente spesso) si puo porre
T = Rp. Questa approssimazione e particolamente valida per elementi leggeri, in cui
l’effetto di matrice principale e l’autoassorbimento (in altri termini la distanza entro
cui vengono prodotti raggi X che riescono ad uscire dal campione e molto minore del-
la profondita a cui essi smettono di essere prodotti); tuttavia, la non omogeneita puo
influire molto sulla correttezza dei risultati. Una verifica sperimentale dell’ipotesi di
composizione omogenea del campione puo venire dalla PIXE differenziale: questa
variante della PIXE consiste nell’analizzare il campione variando l’energia del fascio
incidente, in modo da far variare la profondita di penetrazione del fascio stesso e
provocare emissione X da spessori differenti nel campione.
1In alcuni casi, si puo conoscere a priori la composizione della matrice dell’oggetto da analizzareed essere interessati a rilevare la presenza in traccia di alcuni elementi
8

2 PIGE
2.1 Aspetti generali
Simultaneamente alla PIXE e possibile usare anche un’altra tecnica, per certi aspetti
complementare alla PIXE: la PIGE.
L’analisi di tipo PIGE (Particle Induced Gamma-ray Emission) si basa sulla ec-
citazione dei nuclei del campione, da parte di particelle accelerate, mediante urti
anelastici: la diseccitazione dei nuclei avviene tramite emissione di radiazione γ di
energia pari alla differenza di energia dei livelli interessati; tali energie sono carat-
teristiche di ogni isotopo: questo permette di rivelare un elemento anche mediante
la presenza di isotopi, prendendoli come rappresentativi per quel nucleo.
Per avere urti di tipo anelastico, e necessario che le particelle incidenti abbiano
energia sufficiente per arrivare “sufficientemente vicino” al nucleo, essendo le forze
nucleari a corto range. Risulta quindi piuttosto evidente il fatto che la PIGE sia
piu efficace su elementi con Z basso, i cui nuclei producono una minore repulsione
coulombiana nei confronti delle particelle dei fasci, e sono quindi piu facilmente
avvicinabili (e quindi eccitabili) dalle particelle incidenti. Si puo avere un’idea di
questo considerando la distanza di minimo avvicinamento di una particella con car-
ica unitaria ad un bersaglio con carica Ze di uguale segno, in un’approssimazione
di particelle puntiformi e urto centrale. Uguagliando l’energia cinetica della parti-
cella a quella potenziale coulombiana, si ricava facilmente che la distanza di minimo
avvicinamento risulta:
d =2Ze2
mv2p
(13)
Dalla (13) si vede come, a parita di energia del fascio incidente, la distanza di mini-
mo avvicinamento diminuisca linearmente con Z, rendendo in parte ragione del fatto
che la PIGE sia piu adatta della PIXE nel rivelare elementi a Z basso2.
Si nota percio la complementarieta delle analisi PIXE e PIGE: la prima e piu efficace
su elementi medi o pesanti3, la seconda su quelli piu leggeri.
Le sezioni d’urto PIGE sono diversi ordini di grandezza piu piccole di quelle PIXE e
diminuiscono rapidamente al crescere di Z. Tuttavia, a differenza della PIXE, nella
2In realta il discorso risulta assai piu complesso; la sezione d’urto PIGE dipende anche dallastruttura specifica del nucleo interagente
3E pur vero, come si vede dalla Fig.3, che le sezioni d’urto di produzione X per le serie K vannorapidamente decrescendo all’aumentare di Z. Tuttavia, per quegli elementi con sezione d’urto per leserie K piuttosto bassa, si ha una sezione d’urto per le righe L sufficientemente alta da permettercidi rivelarli ugualmente bene
9

Reaction E γ (keV)23Na (p, p’, γ) 23Na 44025Mg (p, p’, γ) 25Mg 58524Mg (p, p’, γ) 24Mg 1369
27Al (p, p’, γ) 27Al 844, 101428Si (p, p’, γ) 28Si 177931P (p, p’, γ) 31P 1266
Tabella 1: Energie dei raggi γ di alcuni elementi rivelabili gia con fasci di protoni da 3MeV
PIGE non si presenta il problema dell’autoassorbimento, essendo le energie dei raggi
γ emessi molto piu elevate rispetto a quelle dei raggi X (mentre le energie dei raggi
X sono dell’ordine dei keV, le energie dei raggi γ sono dell’ordine delle centinaia o
migliaia di keV; si veda anche Tabella 1). Questo e l’altro fondamentale motivo per
cui la PIGE e piu adatta rispetto alla PIXE per quantificare elementi a Z basso.
Tuttavia, a causa dell’andamento della sezione d’urto con l’energia del fascio inci-
dente, che presenta spesso grosse variazioni anche per piccole differenze di energia,
occorre prestare molta attenzione alla perdita di energia del fascio all’interno del
campione. Per un target infinitamente spesso questo comunque non e un problema,
dal momento che la yield e integrata su tutte le energie delle particelle del fascio.
Il rivelatore utilizzato e un rivelatore a Ge iperpuro (HPGe).
2.2 Analisi quantitativa
Nell’approssimazione di bersaglio sottile (si veda anche §1.2), la yield γ si ottiene
da una espressione simile alla (4), cioe:
Yγ(Z) = σγ(Z,E0)Ω
4πεZ
NAρZ
AtQ
e(14)
dove σγ(Z,E0) e la sezione d’urto di produzione dei raggi γ da parte dell’elemento
con numero atomico Z, provocata da un fascio di ioni di energia E0, εZ e l’efficienza
intrinseca del rivelatore all’energia del γ considerato. A differenza della (4), qui si
e posto βZ = 1 poiche, essendo le energie dei γ dell’ordine delle centinaia o migliaia
di keV, l’assorbimento della radiazione dal punto di emissione al rivelatore si puo
considerare trascurabile.
Introducendo nuovamente il fattore di efficienza η definito nella (6), cioe
η = σγ(Z,E0)Ω
4πεZ
NA
Ae(15)
10

Figura 5: Andamento del potere di frenamento per alcuni elementi o composti in funzionedell’energia del fascio. I dati con cui e stato tracciato il grafico sono ricavati dai data sheetdel National Institute of Standards and Technology [7].
e possibile calcolare la densita areale per un dato elemento misurando la yield dei γ
e ricavando, mediante degli standard sottili, il fattore di efficienza η, analogamente
a quanto fatto per l’analisi PIXE. Si ottiene percio:
ρZt =Yγ(Z)
Qη(Z)(16)
Nel caso di target non sottili, ma di spessore finito, il problema si complica notevol-
mente. Riprendendo, cosı come fatto per il target sottile, le relazioni usate per
l’analisi PIXE, possiamo scrivere una formula analoga alla (7), riferendosi sempre
alla Fig. 4, ma trascurando il fattore esponenziale dell’autoassorbimento:
Yγ(Z) =Ω
4πεZ
Q
e
NA
AρZ
∫ T
0
σγ(Z,Ep) dx (17)
Questa, introducendo nuovamente il potere di frenamento definito nel §1.2.2, si puo
riscrivere nella forma:
Yγ(Z) =Ω
4πεZ
Q
e
NA
A
ρZ
ρ
∫ E0−∆E
E0
σγ(Z,Ep)dEp
S(Ep)(18)
11

SRM 93a SRM 620 SRM 621 SRM 1411 SRM 1412
SiO2 80.8 72.08 71.13 58.04 42.38
Li2O - - - - (4.50)
B2O3 12.56 - - 10.94 4.53
Na2O 3.98± 0.05 14.39± 0.06 12.74± 0.05 10.14± 0.23 4.69± 0.07
MgO - 3.69 - - (4.69)
Al2O3 2.28 1.80 2.76 5.68 7.52
K2O - - 2.01 2.97 4.14
CaO - 7.11 10.71 2.18 4.53
ZnO - - - 3.85 4.48
SrO - - - - 4.55
CdO - - - - 4.38
BaO - - - 5.00 4.67
PbO - - - - 4.40
Tabella 2: Gli standard usati per le misure. In tabella sono riportate le concentrazionicertificate (in % del peso) dei principali componenti degli standard stessi. I valori in par-entesi si riferiscono a quantita non certificate. Tali standard sono tutti dei vetri, ancorchecontenenti vari tipi di ossidi in percentuali differenti.
che per target infinitamente spessi diventa:
Yγ(Z) =Ω
4πεZ
Q
e
NA
A
ρZ
ρ
∫ 0
E0
σγ(Z,Ep)dEp
S(Ep)(19)
Come si puo notare anche dalla Fig. 5, l’andamento dei poteri di frenamento per
vari materiali e circa lo stesso su tutto il range di energia considerato. Cio comporta
che, dati due materiali con poteri di frenamento S1 e S2, si possa considerare valida
in buona approssimazione la relazione S1 = κS2, con κ costante al variare di Ep4.
Restringendo poi il confronto a materiali di natura simile, come per esempio gli
standard di vetro riportati in Tabella 2, si vede (Fig. 6) che ponendo κ = 1 si com-
mette un errore che, fatta eccezione per lo standard SRM 1412 (che contiene ossidi
di elementi a Z medio o alto come Sr, Cd e Pb in concentrazioni non trascurabili),
non supera il 2− 3% su tutto il range di energia considerato.
Misurando la Yγ(Z) di uno di questi standard, di natura simile al campione da anal-
4In realta κ non resta costante nel range di energia considerato, ma varia di qualche puntopercentuale. Tuttavia l’errore che si commette nel trascurare queste variazioni e accettabile nellamaggior parte delle misure composizionali in campo archeometrico
12
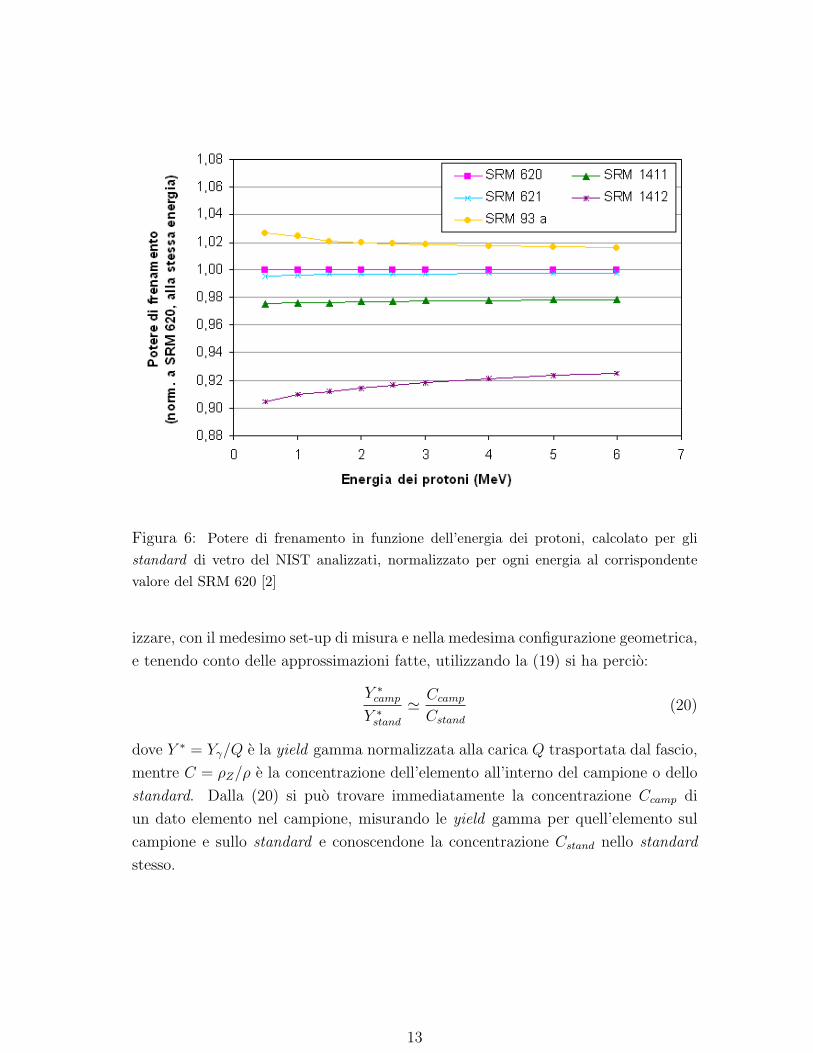
Figura 6: Potere di frenamento in funzione dell’energia dei protoni, calcolato per glistandard di vetro del NIST analizzati, normalizzato per ogni energia al corrispondentevalore del SRM 620 [2]
izzare, con il medesimo set-up di misura e nella medesima configurazione geometrica,
e tenendo conto delle approssimazioni fatte, utilizzando la (19) si ha percio:
Y ∗camp
Y ∗stand
' Ccamp
Cstand
(20)
dove Y ∗ = Yγ/Q e la yield gamma normalizzata alla carica Q trasportata dal fascio,
mentre C = ρZ/ρ e la concentrazione dell’elemento all’interno del campione o dello
standard. Dalla (20) si puo trovare immediatamente la concentrazione Ccamp di
un dato elemento nel campione, misurando le yield gamma per quell’elemento sul
campione e sullo standard e conoscendone la concentrazione Cstand nello standard
stesso.
13

Figura 7: Alcune delle tessere di vetro rinvenute durante gli scavi di Villa Adriana
(Tivoli)
3 Analisi di alcuni vetri
3.1 Introduzione
In collaborazione con la Sovrintendenza per i Beni Culturali del Lazio e l’Universita
di Roma - Tor Vergata, e stata condotta una campagna di analisi allo scopo di
studiare vari reperti archeologici rinvenuti durante gli scavi di Villa Adriana (Tivoli).
Durante tali scavi, nella costruzione cosiddetta “Edificio con tre esedre”, sono state
trovate, fra gli oggetti rinvenuti, delle tessere di vetro facenti parte di un mosaico
ornamentale (Fig. 7). Su tali tessere sono state eseguite misure PIXE e PIGE al fine
di stabilirne la composizione, dato utile agli archeologi per ottenere un discriminante
cronologico circa l’eta delle tessere, cosı da confermare, con un buon margine di
sicurezza, l’indicazione cronologica fornita dal luogo del ritrovamento. Nel periodo
romano e fino al VIII-IX sec., infatti, il composto alcalino aggiunto alla silice (SiO2)
per fare il vetro era una mistura di sali, contenenti principalmente soda (ossidi di
Na), chiamata natron; come stabilizzante era usata la calce (CaO). Successivamente,
a causa della difficolta di importare il natron dall’Egitto a seguito della caduta
dell’Impero Romano, si cominciarono ad usare altri tipi di sali alcalini, contenenti
K e Mg: percio, durante il medioevo, i sali di potassio sostituirono la soda e l’ossido
di magnesio fu usato al posto della calce5.
5Per un approfondimento in merito si rimanda a [2]
14

Per questi motivi, una concentrazione di Na nei vetri tra il 10% e il 20% ed una
bassa concentrazione di K e Mg (< 1%) indicherebbe l’appartenenza di tali vetri al
periodo romano; viceversa, una concentrazione alta di K e Mg con bassa presenza
di Na significherebbe l’appartenenza al periodo medioevale.
A causa dell’azione degli agenti atmosferici, si hanno varie reazioni di scambio ionico
che portano all’impoverimento degli strati superficiali dei vetri in ioni alcalini (come
Na+), sostituiti da H+. Al fine di una corretta valutazione della concentrazione bulk
di Na, percio, e necessario tenere in considerazione il fatto che i reperti presentano
uno strato piu esterno in cui tale concentrazione risulta profondamente alterata; a
seconda dell’estensione della corrosione, lo strato alterato puo variare da alcuni nm
a qualche centinaia di µm di spessore.
3.2 Analisi dati e commenti
Le misure sono state eseguite usando un fascio di protoni di energia Ep = 2.85 MeV
alla superficie del target, con una dimensione spaziale definita da un collimatore con
un diametro di 500 µm, posto in vuoto subito prima della finestra di uscita del fascio
in atmosfera (dove e collocato il bersaglio). Il rivelatore HPGe e stato posizionato
ad un angolo di circa 135, coprendo un angolo solido di circa 0.7 sr. E stata scelta
una bassa corrente di fascio (50 pA) al fine di evitare danni; il tempo di acquisizione
e stato di 3-4 minuti per punto misurato. Particolare attenzione e stata usata nello
schermare accuratamente il rivelatore con del piombo, al fine di evitare eventuali
radiazioni gamma spurie provenienti dal collimatore: per verificare l’effettiva effica-
cia della schermatura sono state eseguite misure di background senza bersaglio nelle
stesse condizioni delle misure sui campioni e sugli standard.
3.2.1 Analisi degli standard
Al fine di verificare sperimentalmente l’accuratezza dell’approssimazione introdotta
nel §2.2 per l’analisi quantitativa PIGE (cioe il considerare per tutti gli standard il
medesimo potere di frenamento, cosı da poterlo semplificare nel rapporto (20)), sono
state eseguite delle misure sui cinque standard, prodotti dal NIST (National Insti-
tute of Standards and Tecnology [7]), le cui composizioni sono riportate in Tabella
2. Tali misure costituiscono anche una “calibrazione” per il calcolo quantitativo di
Na nelle misure sui reperti di Villa Adriana.
Per ogni spettro relativo agli standard analizzati, e stata misurata la yield dei rag-
15

Figura 8: Yield dei raggi γ del sodio per degli standard in funzione della concentrazionedi Na2O negli stessi
gi γ del sodio, normalizzata alla carica trasportata dal fascio. Riportando le yield
normalizzate Y∗ in funzione della concentrazione di Na2O possiamo avere una in-
dicazione di quanto sia legittimo trascurare le differenze tra i poteri di frenamento
degli standard (Fig. 8). Minimizzando rispetto al parametro k il χ2(k), definito da
χ2(k) =1
n
5∑i=1
(yi − kxi)2
σ2yi
(21)
dove la somma e estesa ai cinque standard e dove yi rappresenta la Y∗, xi e la
concentrazione di Na nell’i-esimo standard, σyie l’incertezza sulla yield gamma6 e
n indica i gradi di liberta del problema (pari al numero di dati meno il numero di
parametri; nel nostro caso n = 4), si ricava il coefficiente lineare della miglior retta
per i cinque punti di Fig. 8. Il valore del χ2 minimo e pari a χ2min(k) = 2.84.
6La procedura seguita e leggermente piu complicata di quanto descritto, in quanto si e usata unaprocedura iterativa al fine di tenere conto anche degli errori sulle concentrazioni certificate. Infatti,siamo partiti da una approssimazione di ordine zero, in cui si sono trascurati gli errori sulle xi,trovando k1. Quindi si e usato tale coefficiente per correggere la σyi
tramite la relazione di somma
in quadratura σ′yi
=√
σ2yi
+ k21σ
2xi
in modo da tenere conto anche degli errori precedentementetrascurati. Tale incertezza e stata usata nella (21) per trovare un nuovo valore di k (k2). Laprocedura e stata arrestata alla prima iterazione, poiche la differenza relativa tra k2 e k1 e risultataessere dell’ordine di qualche punto per mille, ben al di sotto degli errori in gioco (dell’ordine del%)
16

Figura 9: Yield dei raggi γ del Na per gli standard, corretta con i poteri di frenamentocalcolati per un’energia iniziale di 2.85 MeV, in funzione della concentrazione di Na2Odegli standard stessi.
In molte applicazioni si puo ritenere sufficiente questo livello di incertezza e con-
siderare accettabile l’approssimazione fatta. Tuttavia, se fosse stato richiesto un
maggiore grado di precisione, si sarebbe potuto migliorare l’approssimazione andan-
do a calcolare i poteri di frenamento per i singoli standard, con cui correggere la Y∗
(si veda a tal proposito [2]): in Fig. 9 si sono corretti i valori di Y ∗ del grafico di
Fig. 8 moltiplicandoli per gli opportuni poteri di frenamento e, come si puo notare,
si ottiene un migliore grado di approssimazione con la retta. Questo lo si vede anche
analiticamente, ottenendo un minimo per il χ2(k) pari a χ2min(k) = 0.90.
A seconda, quindi, del grado di precisione richiesto dal particolare problema in
esame, in alcuni casi il semplice confronto fra le yield Y∗ (Fig. 8) puo essere suffi-
ciente a determinare la concentrazione di un dato elemento nel campione analizza-
to. Se questa “approssimazione di ordine zero” puo essere applicata, i calcoli sono
notevolmente semplificati, dal momento che non e necessario eseguire alcuna proce-
dura iterativa per calcolare il valore adeguato del potere di frenamento S(E0) del
campione incognito.
17

Figura 10: Spettri PIXE (a sinistra) e PIGE (a destra) delle misure di alcune tesserecolorate (in alto una verde, in basso una blu). Si noti come, mentre negli spettri PIXEil sodio sia vicino al limite di rivelabilita, negli spettri PIGE e sempre ben visibile ilcaratteristico picco a 440 keV.
3.2.2 Analisi delle tessere
L’alterazione degli strati superficiali dei reperti va ad incidere particolarmente sulle
misure PIXE, in cui l’autoassorbimento e piu significativo, specialmente per elemen-
ti a Z basso come il sodio7. Per quanto riguarda la PIGE, lo strato superficiale di
impoverimento in Na comporta il fatto che le reazioni che portano all’emissione dei
γ avvengano ad una energia inferiore a quella del fascio alla superficie del campione
(cio vuol dire che, nella (19), si deve integrare a partire da un’energia inferiore a E0).
Tuttavia, se la regione di svuotamento si estende in profondita al piu di qualche µm
(come nel caso discusso poco oltre), la yield γ ne risultera assai poco alterata8.
Come si vede anche in Fig. 10, nelle misure eseguite sulle tessere, a causa del-
lo strato superficiale di impoverimento, con la PIXE si riesce appena a rivelare in
maniera poco piu che qualitativa la presenza di Na, che invece risulta ben evidente
7La regione superficiale di impoverimento e le incrostazioni influiscono sulla misura perche,“spostando” in profondita la regione in cui si hanno il sodio e gli altri alcalini, fanno sı che i raggiX di questi elementi non riescano ad uscire dal campione.
8Un fascio di protoni a 3 MeV perde circa 25 keV/µm in una matrice di vetro
18

Figura 11: Confronto tra gli spettri PIXE (a sinistra) e PIGE (a destra) delle misureeseguite su una tessera turchese in un punto “fresco” (fig. in basso) e in un punto nonrotto (fig. in alto).
dagli spettri PIGE. Un’ulteriore conferma della presenza dello strato superficiale di
impoverimento in Na e venuta da una tessera che appariva rotta in tempi recenti (ad
un esame visivo accurato appariva di un colore piu vivo rispetto alle altre): infatti,
nella zona di rottura, non ancora alterata dagli agenti atmosferici, e stato possibile
rivelare la presenza di sodio anche con la PIXE. Il confronto degli spettri PIGE
relativi alla superficie “fresca” con quelli della zona alterata dello stesso campione
ci permette di dare una stima dell’effetto di “diminuzione” di yield gamma: tale
effetto e di circa il 10% (Fig. 11). Considerando l’andamento della sezione d’urto
σγ,Na in funzione dell’energia dei protoni Ep e conoscendo dEp/dx, si ricava che
tale diminuzione corrisponde ad uno spessore “svuotato” di qualche µm. Un tale
effetto di diminuzione puo essere ipotizzato anche per le altre tessere: questo lo si
puo dedurre dalla Fig. 10, in cui, negli spettri PIXE, il picco del sodio a 1.04 keV
e comunque visibile, seppur al limite di rivelabilita, e la stessa cosa puo dirsi dello
spettro relativo al punto nella zona non rotta di Fig. 11. Cio fa supporre che lo
spessore dello strato impoverito in Na sia dello stesso ordine di grandezza per tutti
i campioni e che quindi influisca per tutti circa allo stesso modo sulla yield γ. La
concentrazione di Na nelle tessere puo essere percio stimata dagli spettri PIGE per
19

Colore del vetro Concentrazione di Na2O
(%)
Verde 15− 20
Blu 14− 20
Turchese 15− 25
Rosso 7− 10
Giallo 7− 10
Tabella 3: Concentrazioni di Na2O misurate nelle tessere analizzate. I valori sono riportaticome range poiche sono state analizzate piu tessere per uno stesso colore: gli intervalliriportati tengono conto, oltre che degli errori sperimentali di misura, anche delle differentipercentuali di Na2O misurate in tessere diverse ma dello stesso colore.
confronto con gli standard di vetro del NIST, come spiegato in §2.2.
In Tabella 3 sono riportati i valori dedotti della concentrazione di Na2O nelle tessere,
raggruppandole per colore.
Data l’impossibilita, per quanto detto, di raggiungere un grado di precisione elevato
nelle misure, e tenendo conto anche della variabilita osservata misurando tessere
diverse dello stesso colore, i risultati sono forniti come range di concentrazioni di
Na2O: tuttavia, anche cosı e possibile trarre considerazioni ed informazioni utili per
gli archeologi. Dalle considerazioni fin qui esposte, si deduce che, nel complesso, la
PIGE puo essere adeguata per stimare la concentrazione di sodio in questo tipo di
campioni e darne una prima caratterizzazione della composizione. Infatti, possiamo
comunque differenziare le tipologie di vetro (romano e medioevale) nonostante le
incertezze nella quantificazione di Na2O siano dell’ordine del 10− 20%; il problema
infatti e di distinguere tra due concentrazioni largamente differenti tra loro, come
spiegato nel §3.1. Come si vede, le concentrazioni di Na2O stimate ci consentono di
dire che i reperti sono di eta romana.
La misura e stata utile, oltre che dal punto di vista archeologico (benche in questo ca-
so il contesto storico ed archeologico lasciasse pochi dubbi riguardo alla collocazione
temporale dei reperti), anche da un punto di vista metodologico, poiche mette in
luce la complementarieta delle tecniche PIXE e PIGE nella misura degli elementi:
infatti i risultati ottenuti possono essere successivamente utilizzati per una ulteriore
analisi della composizione dei reperti con la PIXE, avendo gia una indicazione sugli
elementi di matrice, come il sodio, che con tale tecnica non si riusciva a rivelare.
20

Riferimenti bibliografici
[1] P.A. Mando, PIXE (Particle induced X-ray Emission), in Encyclopedia of
Analytical Chemsestry, R.A. Meyers (Ed.), pp 12708 - 12740 (2000)
[2] N. Grassi, Recent developments and new perspectives of Ion Beam Analysis for
Cultural Heritage (PhD 24 gennaio 2006)
[3] S. Nava, Aerosol Characterisation by Ion Beam Analysis Tecnique (PhD 30 aprile
2003)
[4] F. Mazzei, Determinazione della concentrazione elementale dell’aerosol atmos-
ferico a Firenze tramite le tecniche PIXE e PIGE e nuovi sviluppi strumentali
del set-up di misura (Tesi di Laurea in Fisica - A.A. 2003/2004)
[5] A. Migliori, Nuclear non-invasive methods for the study of materials of
archaeometric interest (PhD A.A.2003/2004)
[6] C. Fiori, M. Vandini, V. Mazzotti, I colori del vetro antico, Ed. il prato, 2004
[7] http://physics.nist.gov/PhysRefData/Star/Text/PSTAR.html
i