analysis of genome-wide association studies lecture 1: introduction
TRANSCRIPT

Analysis of genome-wide association studies
Lecture 1: Introduction

Linkage studies
• Traditional approach to identifying genes for human traits and diseases was through linkage.
• For Mendelian diseases (e.g. Huntington’s disease) there is a clear co-segregation of genetic markers with disease within pedigrees.
• For complex traits (e.g. type 2 diabetes), linkage analysis has been less successful because the relationship between phenotype and genotype is less clear.• Non-genetic risk factors influence the outcome;• Many genes have an impact on the trait, each having only a
small effect on the outcome.

Association studies
• Common disease, common variant hypothesis: complex traits will be determined by variants that occur frequently in the population, but each have only a small impact.
• Ascertain sample of affected cases and unaffected controls (or a random sample for a quantitative trait) from the population.
• Compare allele frequencies in cases and controls (or mean trait values between alleles for a quantitative traits).
• With sufficient sample size, powerful approach to identify loci contributing to complex traits.

Common variation in the genome
• Impractical to genotype all common variants in the genome in large samples.
• International HapMap project genotyped more than three million SNPs in samples from multiple ancestry groups.
• Common variation is arranged on relatively few haplotypes that occur within blocks of strong linkage disequilibrium between recombination hotspots...

• SNPs located within the same block will be in strong LD with each other. However, a pair of SNPs located in adjacent blocks will be uncorrelated due to the high levels of recombination at the flanking hotspot.
• The strong LD between SNPs in the same block means that we tend to observe fewer haplotypes than we might expect by chance. In fact, much of the diversity is accounted for by a small number of common haplotypes.
BLOCK 1
HOTSPOT
BLOCK 2
HOTSPOT
BLOCK 3
SNPs in strong LD
SNPs in strong LD SNPs in strong LD
Low diversity of haplotypes
Low diversity of haplotypes
Low diversity of haplotypes
Common variation in the genome

3-
• Jeffreys et al. (2001) looked for evidence of recombination in the sperm of six men across ~200kb region of the MHC.• Crossovers cluster in six hotspots: recombination extremely rare elsewhere in the region.• High-resolution LD analysis in sample of 50 unrelated individuals identified blocks with boundaries corresponding to these recombination hotspots.
(Nature Genetics 29: 217-222)
Common variation in the genome

(Thanks to Lon Cardon)
• Dawson et al. (2002) genotyped 90 unrelated UK individuals at over 1500 SNPs across chromosome 22.• Defined haplotype blocks as regions of limited haplotype diversity.
Common variation in the genome

Common variation in the genome
Genotyping this subset of SNPs accounts for all common genetic variation in the block

Measures of LD• Linkage disequilibrium between two marker allele M and
disease variant X quantified by measure:
• Adjustment for allele frequencies:
• May not have typed disease variant: but understanding patterns of LD between markers will help interpretation of results of association studies.
XPMPMXPD
MAXD
DD xPXPmPMP
Dr
22

Genome-wide association arrays
• Possible to “cover” much of the common genetic variation in the human genome by genotyping only a subset of SNPs.
• Most efficient approach is to select “tags” that cover common SNPs (at some r2 threshold).

11

• Samples genotyped with Affymetrix GeneChip 500K Mapping Array Set.
• Identified novel loci and replicated association signals for five diseases.
• Established standard protocols for quality control and analysis of GWAS.
• Publicly available control cohort.

Published Genome-Wide Associations through 07/2012Published GWA at p≤5X10-8 for 18 trait categories
NHGRI GWA Catalogwww.genome.gov/GWAStudieswww.ebi.ac.uk/fgpt/gwas/

Design issues
• Is the trait heritable?!• Phenotype definition, case-control selection,
and availability of non-genetic risk factors.• Choice of genotyping product.• Sample size requirements.• The GENETIC POWER CALCULATOR
(http://statgen.iop.kcl.ac.uk/gpc) can be used to calculate power of case-control studies.

QUALITY CONTROL

Introduction
• Poor study design and errors in genotype calling can introduce systematic bias in association studies.• Increase in false positive error rate and decrease
in power.• Assess data quality to remove sub-standard
genotypes, samples and SNPs from subsequent association analysis.

Genotype calling
• For large-scale GWA studies, automated genotype calling algorithms have been developed, e.g. GENCALL and GENOSNP.• Estimate probability that any specific genotype is AA, AB or
BB.• Apply threshold to probabilities in order to call genotype,
otherwise treated as missing.• Choice of calling threshold will impact results:
• Too low: include poor quality genotypes.• Too high: unnecessarily remove high quality genotypes, or
may introduce bias by preferentially calling specific genotypes (e.g. rare homozygotes).

Genotype calling

Genotype calling
• For large-scale GWA studies, automated genotype calling algorithms have been developed, e.g. GENCALL and GENOSNP.• Estimate probability that any specific genotype is AA, AB or
BB.• Apply threshold to probabilities in order to call genotype,
otherwise treated as missing.• Choice of calling threshold will impact results:
• Too low: include poor quality genotypes.• Too high: unnecessarily remove high quality genotypes, or
may introduce bias by preferentially calling specific genotypes (e.g. rare homozygotes).

Sample quality control
• Remove samples on the basis of:• Low call rate (poor DNA quality).• Outlying heterozygosity across autosomes (DNA
sample contamination or inbreeding).• Duplication or relatedness based on identity-by-
state (samples should be independent).• Mismatches with external information (sample
mix-up).• Outlying population ancestry (confounding due to
population structure).

Call rate and heterozygosity
3% missing
23-30% heterozygosity

Identity-by-state (IBS)• Over M markers, the IBS between the ith and jth individuals is
given by
where Gik denotes the number of minor alleles (0, 1 or 2) carried by the ith individual at SNP k.
• Identical samples will share IBS near to 100% (allowing for genotyping errors).
• Related individuals will share higher IBS than unrelated individuals.
• Common to plot histogram of IBS of each individual with “nearest neighbour”.
k
jkikij GGM
IBS2
11

IBS distribution
Duplicates
Relateds
Remove one sample from each duplicate or related pair (usually one with lowest call rate).

X chromosome
• Distribution of heterozygosity different in males and females.• Should be no heterozygosity in males, but expect
some genotyping error.• Discrepancies with external gender
information may reflect:• Errors in external data;• Sample mix-up;• Gender inconsistent with sex chromosomes.

X chromosome
Each individual plotted twice according to reported gender: females in red and males in blue.
Should these samples be removed from the study or the sex corrected based on heterozygosity? May impact on results if sex is adjusted for in the analysis or if sex specific analyses are to be undertaken.

SNP quality control
• Remove SNPs on the basis of:• Low call rate, variable by MAF (poor quality SNP).• Extreme deviation from Hardy-Weinberg equilibrium in
cases, controls or both for autosomes (genotyping error).• Extreme differential call rates between cases and controls
(calling bias).• Study specific SNP QC filters (such as differences in allele
frequencies between multiple control cohorts).• Low frequency SNPs (more prone to bias due to
genotyping error and low power to detect association).• Visual inspection of cluster plots.

Effect of differential call rate
Individuals called as missing?Fewer heterozygotes among cases.

Cluster plot inspection: good SNP

Cluster plot inspection: bad SNP

Summary
• QC criteria are subjective and vary from one study to another.
• Sample QC filters should not be so stringent as to remove the majority of the analysis cohort!
• SNP QC filters should eliminate the worst quality markers without “throwing the baby out with the bathwater”.
• All SNPs demonstrating evidence for association should be followed up with visual inspection of cluster plots.

BASIC ANALYSIS OF GENOME-WIDE ASSOCIATION STUDIES

Introduction
• Association analyses focus on the identification of SNPs that differ in allele (genotype) frequency between cases and controls.
• Basic analyses utilise standard epidemiological tools, rather than specialised methods that have been developed for analysing more traditional pedigree and family studies:• contingency table analysis; • logistic regression modelling.

• Assuming the sample to be typed at a SNP marker of interest, we can represent genotype data in a 2 x 3 contingency table.
• The usual test for independence of rows and columns in contingency tables can be applied to test the null hypothesis of no disease-marker association
where
• X2 has distribution with 2 degrees of freedom under null hypothesis.
Cases Controls Total
MM n2A n2U n2·
Mm n1A n1U n1·
mm n0A n0U n0·
Total n·A n·U n··
210
22
,, ,i UAj ij
ijij
nE
nEnX
n
nnnE jiij
• Odds ratio for genotype MM relative to mm
• Affected individual times more likely to have marker genotype MM than mm.
UAUAmmMM nnnn 2002|
2
2mmMM|
Genotype-based test

• Assume a multiplicative model of disease risks:
• The Cochran-Armitage trend test of association between disease and the marker SNP is given by
where
• X2 has distribution with 1 degree of freedom under null hypothesis.
Cases Controls Total
MM n2A n2U n2·
Mm n1A n1U n1·
mm n0A n0U n0·
Total n·A n·U n··
2
21212
2
12122
21
41111
21
21
..........
nnnnnnnn
pppp
X
UA
UUAA
j
ijij n
np
• Odds ratio for allele M relative to allele m
• Affected individual times more likely to have marker genotype MM than mm, and times more likely to have genotype Mm than mm.
......
......|
20
21
0022
21
21
10
10
20
02
21
12
10
01
4
4
nnnnnn
nnnn
nnnn
nnnn
nnnn
nnnn
UAUAUAUA
UAUAUA
mM
2
2mM|
mM|
Cochran-Armitage trend test
2mmMmmmMM ||
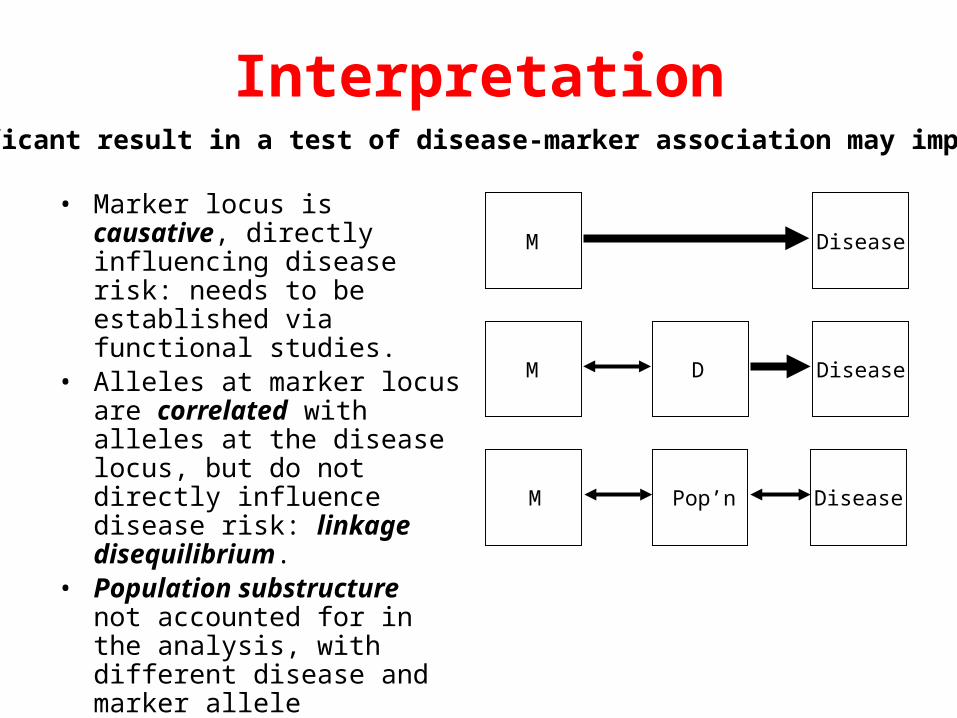
Interpretation
• Marker locus is causative, directly influencing disease risk: needs to be established via functional studies.
• Alleles at marker locus are correlated with alleles at the disease locus, but do not directly influence disease risk: linkage disequilibrium.
• Population substructure not accounted for in the analysis, with different disease and marker allele frequencies in each subpopulation.
• False positive signal of association.
M Disease
M D Disease
M DiseasePop’n
A significant result in a test of disease-marker association may imply:

Genome-wide significance
• A type I error occurs when we reject the null hypothesis of no association, when in fact the null hypothesis is true.
• Specify type I error rate – or significance level – at the design stage of the analysis.• Lower type I error rate reduces the probability of detecting a
false positive association, but with the penalty of reducing the power to detect association when it truly exists.
• It is important to correct for multiple testing to maintain the type I error rate for the experiment overall (i.e. all the SNPs tested in the association study).
• Genome-wide significance threshold: p<5x10-8.• Replication is necessary to confirm association.

• We model the log odds of disease for the ith individual as
where pi is the probability that the ith individual is affected by disease, xi is a vector of measures of the risk factors, and β is a vector of the corresponding risk effects.
• Over all individuals, we can obtain estimates of the risk effects by maximising the log-likelihood
where yi denotes the phenotype of the ith individual (0 for control and 1
for case).• Extremely flexible modelling framework: can test for joint effects of risk
factors (multi-locus analysis) and allow for covariates (adjustment for non-genetic effects).
iT
i
i
p
pxβ
1
log
i iiii pypyl 1log1log,βxy
Logistic regression modelling

• Assuming multiplicative model of disease risk (additive on the log scale), we can model the log-odds of disease for the ith individual by
where βM denotes the log-odds ratio of allele M relative to allele m, and xMi is an indicator variable that counts the number of M alleles (0, 1 or 2) carried by the ith individual.
• Test for association by maximising the likelihood of the model, and comparing with the maximised likelihood under the null hypothesis of no association (i.e. βM = 0):
• X2 has χ2 distribution with one degree of freedom under the null hypothesis.
• Asymptotically equivalent to Cochran-Armitage trend test.
MiMi
i xp
p
01
log
0,,,,2 002 MMMM llX xyxy
Additive test of association

• To allow for deviations from the multiplicative disease model , we can model the log-odds of disease for the ith individual by
where βMm and βMM denote the log-odds ratios of genotypes Mm and MM relative to genotype mm, and xMmi and xMMi are variables indicating that the ith individual carries genotype Mm and MM, respectively.
• Test for association by maximising the likelihood of the model, and comparing with the maximised likelihood under the null hypothesis of no association (i.e. βMm = βMM = 0).
• X2 has χ2 distribution with two degrees of freedom under the null hypothesis.
MMiMMMmiMmi
i xxp
p
01
log
0,,,,,,,2 002 MMMmMMMmMMMmMMMm xlxlX xyxy
Genotypic test of association

• The same modelling framework can be used to test for association under alternative disease models, such as heterozygote advantage, recessive and dominant.
• Testing many different models of association requires correction for multiple testing.
• We can compare the likelihoods of the genotypic and trend models of association to test for a deviation from the multiplicative model of disease risks.
• Trend test is generally most powerful unless there is extreme deviation from a multiplicative disease model.
• Testing for association at a marker SNP in LD with the causal SNP weakens the effect of the underlying disease model.
Genotype M recessive M dominant Heterozygote advantage
MM 1 1 0
Mm 0 1 1
mm 0 0 0
General disease models

• For complex diseases, we may wish to take account of non-genetic risk factors, such as exposure to specific environments, or the effects of established genetic loci.
• Assuming multiplicative model of disease risk, we can model the log-odds of disease for the ith individual by
where βC denotes the effect of a covariate xC, and xCi denotes the value of the covariate for the ith individual.
• Obtain χ2 test of association with one degree of freedom by maximising the likelihood of the model, and comparing with the maximised likelihood under the null hypothesis of no association (i.e. βM = 0).
• Estimated log-odds ratio of allele M adjusted for the effect of the covariate.• Can be generalised to allow for any number of covariates and general models
of disease risk.
MiMCiCi
i xxp
p
01
log
Allowing for covariates

• The methodology described here generalises to quantitative (continuous) traits. It is straightforward to compare the mean response for each marker genotype by analysis of variance, assuming a normally distributed trait, within the standard linear regression framework.
• A powerful strategy is to ascertain individuals from the extremes of the quantitative trait distribution: cases and hyper controls.• We can analyse trait values by linear regression, although this leads to
biased estimates of mean trait values for marker genotypes.• We can ignore the trait values, and analyse as a standard case-control
sample. • Are hyper controls representative, or are there polygenic effects
involved?• This strategy may not be cost effective if phenotyping is expensive
relative to genotyping.
Quantitative traits

• Contingency table analysis and generalised linear modelling can be performed using standard statistical software. • Define indicator variables for specific genetic models from the
observed SNP genotype data.• Some statistical software packages include specific libraries of
routines to perform genetic analyses (R, STATA)• Specialised genetic analysis software:
• PLINK. Whole genome association analysis toolset designed to perform a range of basic, large-scale analyses. Allows for data management and basic QC analyses. Performs simple case-control tests of association.
• SNPTEST. Designed for analysis of whole genome association studies. Allows for flexible single-locus analysis of genotype data allowing for covariates.
Software

Replication• To confirm positive association signals from an initial
study, it is essential to replicate the result in independent samples from the same and/or different populations.
• Replication of positive association signals has not proved to be easy: will depend on power of both initial and replication studies.
• Multi-stage designs: genotype a proportion of samples with GWAS array and follow-up the strongest signals of association in the remaining samples through de novo genotyping.
• Collaboration between international groups studying the same trait allows for in silico replication.

Meta-analysis• We can increase power to detect rarer variants of
more modest effect by collecting larger and larger samples.
• Alternatively, we can combine the results of GWA studies of the same trait through meta-analysis, without direct exchange of genotype data.
• Exchange summary statistics for each SNP including “risk” allele, p-value, odds ratio (effect) and 95% confidence interval (standard error).
• GWA studies can be combined through meta-analysis even if genotyped directly for different sets of SNPs through imputation.
• Software: GWAMA and METAL.

Summary
• Standard statistical procedures available for the analysis of genotype data from genetic association studies.
• Logistic regression provides a flexible framework for modelling SNP association with disease.
• Signals of association should be validated through replication and/or meta-analysis.