anti-pd-1 antibody therapy potently enhances the...
TRANSCRIPT

Cancer Therapy: PreclinicalSee related article by Morales-Kastresana, et al., p. 5546
Anti-PD-1 Antibody Therapy Potently Enhances theEradication of Established Tumors By Gene-ModifiedT Cells
Liza B. John1,3, Christel Devaud1,3, Connie P.M. Duong1,2,3, Carmen S. Yong1,3, Paul A. Beavis1,3, Nicole M.Haynes1,3, Melvyn T. Chow1,3,5,6, Mark J. Smyth1,2,3,4,5,6, Michael H. Kershaw1,2,3,4, and Phillip K. Darcy1,2,3,4
AbstractPurpose: To determine the antitumor efficacy and toxicity of a novel combination approach involving
adoptive T-cell immunotherapy using chimeric antigen receptor (CAR) T cells with an immunomodulatory
reagent for blocking immunosuppression.
ExperimentalDesign:Weexaminedwhether administration of a PD-1blocking antibody could increase
the therapeutic activity of CAR T cells against two different Her-2þ tumors. The use of a self-antigen mouse
model enabled investigation into the efficacy, mechanism, and toxicity of this combination approach.
Results: In this study, we first showed a significant increase in the level of PD-1 expressed on transduced
anti-Her-2 CD8þ T cells following antigen-specific stimulationwith PD-L1þ tumor cells and thatmarkers of
activation and proliferation were increased in anti-Her-2 T cells in the presence of anti-PD-1 antibody. In
adoptive transfer studies inHer-2 transgenic recipientmice,we showed a significant improvement in growth
inhibition of two different Her-2þ tumors treated with anti-Her-2 T cells in combination with anti-PD-1
antibody. The therapeutic effects observed correlated with increased function of anti-Her-2 T cells following
PD-1 blockade. Strikingly, a significant decrease in the percentage of Gr1þ CD11bþ myeloid-derived
suppressor cells (MDSC) was observed in the tumor microenvironment of mice treated with the combi-
nation therapy. Importantly, increased antitumor effects were not associated with any autoimmune
pathology in normal tissue expressing Her-2 antigen.
Conclusion: This study shows that specifically blocking PD-1 immunosuppression canpotently enhance
CAR T-cell therapy that has significant implications for potentially improving therapeutic outcomes of this
approach in patients with cancer. Clin Cancer Res; 19(20); 5636–46. �2013 AACR.
IntroductionAdoptive immunotherapy using gene-modified T cells
expressing antigen-specific chimeric antigen receptors(CAR) is a promising approach for the treatment of cancer(1–3). Recent clinical trials using CARs that target cellsurface tumor-associated antigens such as CD20 for thetreatment of non-Hodgkin lymphoma (4), GD2 in neuro-
blastoma (5), and CD19 in chronic lymphocytic leukemia(6, 7) have all displayed encouraging results to date result-ing in objective antitumor responses in a proportion ofpatients. These trials clearly illustrate the broad range oftumors that can be targeted by CAR T cells in a HLA-independent manner (8).
Despite encouraging results in preclinical models and inpatients, the existence of a number of different immuno-suppressive pathways can restrict the full potential of adop-tive T-cell therapy. This includes increased expression ofinhibitory immune receptors such as T-cell membraneprotein-3 (TIM-3), cytotoxic T lymphocyte-associated anti-gen 4 (CTLA-4), and/or programmed death-1 (PD-1) on Tcells following T-cell activation, which can limit the dura-tion and strength of the adaptive immune response (9).However, the recent development of checkpoint inhibitorssuch as ipilimumab targeting CTLA-4 (10), provide diverseopportunities to enhance antitumor immunity with thepotential to produce durable clinical responses (9).
The PD-1 pathway has emerged as another promisingtarget for cancer therapy. PD-1 binds to two known ligands;PD-L1, the predominant mediator of immunosuppressionwhich is upregulated on many different tumor types and
Authors' Affiliations: 1Cancer Immunology Program, Peter MacCallumCancer Centre, East Melbourne, Victoria; 2Department of Pathology, Uni-versity of Melbourne; 3Sir Peter MacCallum Department of Oncology,University of Melbourne, Parkville; 4Department of Immunology, MonashUniversity, Clayton; 5Immunology in Cancer and Infection Laboratory,Queensland Institute of Medical Research; and 6School of Medicine,University of Queensland, Herston, Queensland, Australia
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
M.H. Kershaw and P.K. Darcy contributed equally to this work.
Corresponding Author: Phillip K. Darcy, Cancer Immunology Program,Peter MacCallum Cancer Centre, Victoria, Australia. Phone 613-9656-1286; Fax: 613-9656-1411; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-13-0458
�2013 American Association for Cancer Research.
ClinicalCancer
Research
Clin Cancer Res; 19(20) October 15, 20135636
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

PD-L2 which has restricted expression onmacrophages anddendritic cells (11). Engagement of the PD-1/PD-L1 path-way results in the phosphorylation of tyrosine-basedmotifsin the cytoplasmic tail of the PD-1 inhibitory receptor,which promotes the recruitment of SHP2 phosphataseleading to dephosphorylation of PI3K. As a result, inhibi-tion of PI3K effects downstream activation of Akt kinasereducing T-cell activation, proliferation, and survival (12,13). Recent trials using a fully human IgG4 PD-1 mono-cloncal antibody (mAb; BMS-936558) have reported dura-ble clinical responses in patients with advancedmelanoma,non–small cell lung, renal cell, as well as hematologicmalignancies (11, 13–15). Furthermore, a significant cor-relation between level of PD-L1 expression on tumor cellsand objective responses was reported (14). Another clinicaltrial used a human IgG4 anti-PD-L1 mAb which showedpromising efficacy against a range of cancers with objectiveresponses reported in a proportion of patients (16). Giventhe early success of these checkpoint inhibitors in enhanc-ing T-cell immunity, it opens the possibility that thesereagents could be used to enhance adoptive T-cell strategies.Hence, in this study, we hypothesized that CAR T-cell
therapy in combination with PD-1 blockademay overcomePD-L1þ tumor immunosuppression, thereby leading toimproved therapeutic efficacy. Herein, we used self-antigenHer2 transgenic mice and used two different Her-2þ tumortargets to examineboth antitumor efficacy and safety ofCART cells and anti-PD-1 antibody in a combined immuno-therapeutic approach.
Materials and MethodsCell culture, antibodies, and miceThe C57BL/6 mouse 24JK sarcoma cell line was kindly
provided by Dr. Patrick Hwu (NIH, Bethesda, MD) and
maintained at 37�C in 5% CO2 in RPMI-1640 mediasupplemented with 10% heat-inactivated fetal calf serum(FCS), 2 mmol/L glutamine, 1 mmol/L sodium pyruvate,0.1 mmol/L nonessential amino acids, 100 U/mL penicil-lin, and 100 mg/mL streptomycin (Life technologies). TheB6 mouse e0771 (LMC variant) breast cancer line waskindly provided by Prof. Robin Anderson (Peter MacCal-lum Cancer Centre, Victoria, Australia) and maintained at37�C in 10% CO2 in Dulbecco’s modified Eagle medium(DMEM), supplemented as above with 5% FCS. These lineswere retrovirally transduced with the human Her-2 antigen(24JK-Her-2/e0771-Her-2) as described previously (17).The anti-PD-1 (clone RMP1-14, IgG, Protein G purified,endotoxin-free) and isotype (clone UB8-1B9) antibodieswere derived from Bio-XCell and in-house, respectively.C57BL/6 human Her-2 transgenic mice and congenicThy1.1þ Her-2 mice were bred at the Peter MacCallumCancer Centre and used for experimentation at 6 to 16weeks of age. All animal experimentation was approved inadvance by the Peter MacCallum Cancer Centre AnimalExperimentation Ethics Committee.
Generation of gene-modified T cellsThe GPþE86 packaging line expressing the CAR contain-
ing the extracellular scFv-anti-Her-2 human monoclonalantibody (mAb) region fused to the transmembrane andintracellular costimulatory domain CD28 and intracellularTCR-z domain was generated as previously described (18,19). For transduction, splenocytes from C57BL/6 Her-2 orThy1.1þ Her-2 mice transgenic mice were gene modified aspreviously described (20). Following transduction, anti-Her-2 T cells were cultured in RPMI-1640, supplementedwith 100 U/mL of IL-2 and 2 ng/mL of IL-7, and used for invitro and in vivo experiments.
Flow cytometryTransduction efficiency of T cells expressing the chimeric
anti-Her-2 receptor was determined by direct immunoflu-orescence with a myc-tag Alexa Fluor 488–conjugatedimmunoglobulin (Cell Signaling Technology). Backgroundimmunofluorescence was assessed using an IgG2a AlexaFluor 488–conjugated mouse isotype immunoglobulin(Invitrogen). Phenotypic characterization of transducedcells was determined using direct staining with myc-Tag(clone 9B11; Cell Signaling), TCRb (clone H57-597; JomarDiagnostics), CD4 (clone RM4-5; BD Pharmingen), CD8(clone 53-6.7; BD Pharmingen), and PD-1 (clone J43;Jomar Diagnostics) cell surface markers. Intracellular stain-ing for IFN-g (clone XMG1.2; Jomar Diagnostics) granzymeB (clone GB11; BD Bioscience) and Ki67 (clone B56; BDBioscience) was carried out using a Cytofix/Cytoperm,Fixation/Permeabilization Solution Kit in accordance withmanufacturers’ instructions (BD Bioscience). Tumors wereexamined for PD-L1 cell surface expression by doublestaining with biotinylated PD-L1 (cloneMIH5; JomarDiag-nostics) and Streptavidin-APC (Jomar Diagnostics). For T-cell function and localization studies, tumors and bloodwere removed from mice at day 2 and 8 posttreatment
Translational RelevanceIn this study, we show that blockade of the PD-1
immunosuppressive pathway using an anti-PD-1 anti-body can significantly enhance the antitumor efficacy ofgeneticallymodifiedT cells expressing a chimeric antigenreceptor (CAR). This combination therapy was shown tosignificantly inhibit tumor growth in two differentmouse models leading to eradication of disease in aproportion of mice. Both of these approaches have beenused singly in the clinic showing good safety profileswhere objective and complete responses have beenreported against various cancer types. However, manypatients do not respond to either treatment alone. Thecurrent study shows that combining these two modal-ities can dramatically increase antitumor effects againstestablished disease. Furthermore, we show that theincreased effects from combination therapy did notcause pathology in mice and that therapeutic responsesstrongly correlated with a decrease in MDSCs.
Anti-PD-1 Therapy Enhances Tumor Rejection By CAR T Cells
www.aacrjournals.org Clin Cancer Res; 19(20) October 15, 2013 5637
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

(n ¼ 6–8/group) and processed as previously described(21). Cells were stained with a cocktail of antibodies foridentifying presence of various immune cell populations(TCRb, Thy1.1, CD4, CD25, FR4, CD11b, Gr-1, F4/80) andintracellular IFN-g . Gr1þ cells were examined for PD-L1 andPD-1 cell surface expression by double staining with bio-tinylated PD-L1 (clone MIH5; Jomar Diagnostics) andStreptavidin-Pe-Cy7 (Jomar Diagnostics) or phycoerythrin(PE)-conjugated PD-1 (clone J43; JomarDiagnostics). Sam-ples were incubated for 25 minutes at 4�C and analyzed byflow cytometry (LSR-II, BD Biosciences, North Ryde, NewSouth Wales, Australia).
Adoptive cell therapy experimentsC57BL/6 human Her-2 transgenic mice (n ¼ 5–8) were
injected subcutaneously with 1 � 106 24JK-Her-2 cells ororthotopically with 5 � 105 e0771-Her-2 breast carcinomacells into the fourth mammary fat pad. Mice were thenpreconditioned (5 Gy) on Day 7 posttumor injection,before transfer of scFv-anti-Her-2–transduced T cells orcontrol LXSN–transduced T cells alone (1 � 107/dose ondays 7 and 8) or T cells in combination with either anti-PD-1 or isotype antibody (250 mg/injection on days 7, 11, and15). Control groups were left untreated. Tumor growth inmice was monitored by twice weekly measurements usingelectronic calipers (length � height) and the area of thetumor determined for each mouse. The mean tumor area�SEM was recorded for each treatment group. In someexperiments, recipient mice received transduced donor Tcells from congenic Thy1.1þ Her-2 mice to investigate thefunction and localization of adoptively transferred T cells invivo. Mice were also given twice daily intraperitoneal injec-tions of recombinant human IL-2 (Biological ResourceBranch, National Cancer Institute, Frederick, MD) involv-ing 9 doses of 50,000 IU/200 mL given subsequent to T-celltransfer.
HistologyMammary and brain tissue from nontreated and treated
Her-2 transgenic mice were removed, fixed with 10% neu-tral-buffered formalin, embedded in paraffin, and sec-tioned. Slides were stained with hematoxylin and eosin(H & E) to evaluate any potential signs of autoimmunitycaused by the therapy. Images were visualized with anOlympus BX51 microscope (Olympus Corporation),acquired using a RT SE Diagnostics Instruments SPOTcamera (Diagnostic Instruments) in conjunctionwith SPOTAdvanced Version 4.6 (Diagnostic Instruments), and com-piled with Adobe Photoshop and Illustrator CS5 (AdobeSystems).
Statistical analysisThe difference in tumor growth and survival distributions
between the treatment groups was analyzed by two-wayANOVAwith P < 0.05 considered significant. The differencein mean percentage� SEM of each immune cell type in theblood and tumors between treatment groups was analyzedby two-way ANOVA and Student t test.
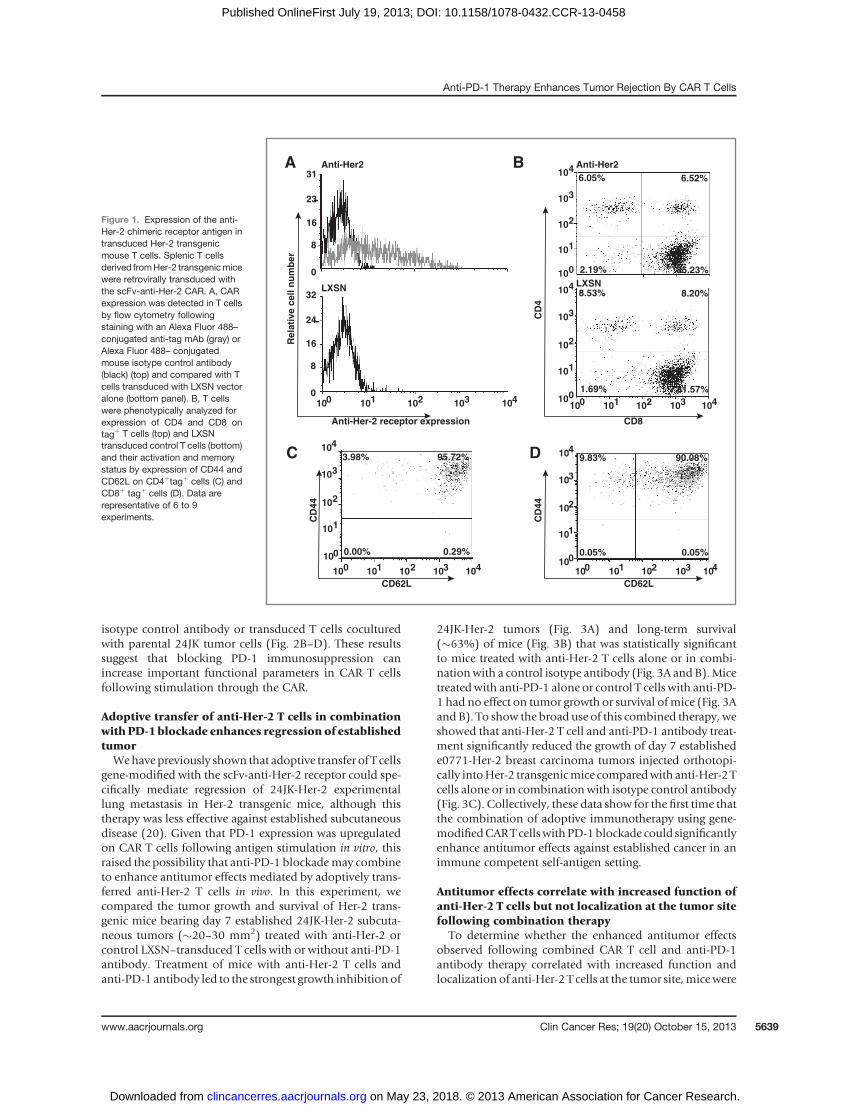
ResultsExpression of the anti-Her-2 chimeric receptor inHer-2transgenic mouse T cells
Splenic T cells fromHer-2 transgenic mice were activatedand retrovirally transduced with the anti-Her-2 CAR. Weobserved reproducible levels of anti-Her-2 receptor ex-pressed on transduced T cells compared with control LXSNvector-transduced T cells (Fig. 1A; 39.75� 8.23% SEM, n¼9). T cells transducedwith the anti-Her-2 receptor or controlvector alone were predominantly CD8þ (83.78 � 2.50%SEM, n ¼ 6), with only a small percentage of CD4þ T cellspresent in the culture (8.99� 14.72% SEM, n¼ 6; Fig. 1B).Further phenotypic analysis revealed that the majority ofCD4þ c-myc tagþ (Fig. 1C) and CD8þ c-myc tagþ (Fig. 1D)gated T cells displayed aCD62LhiCD44hi activated/memoryT-cell phenotype.
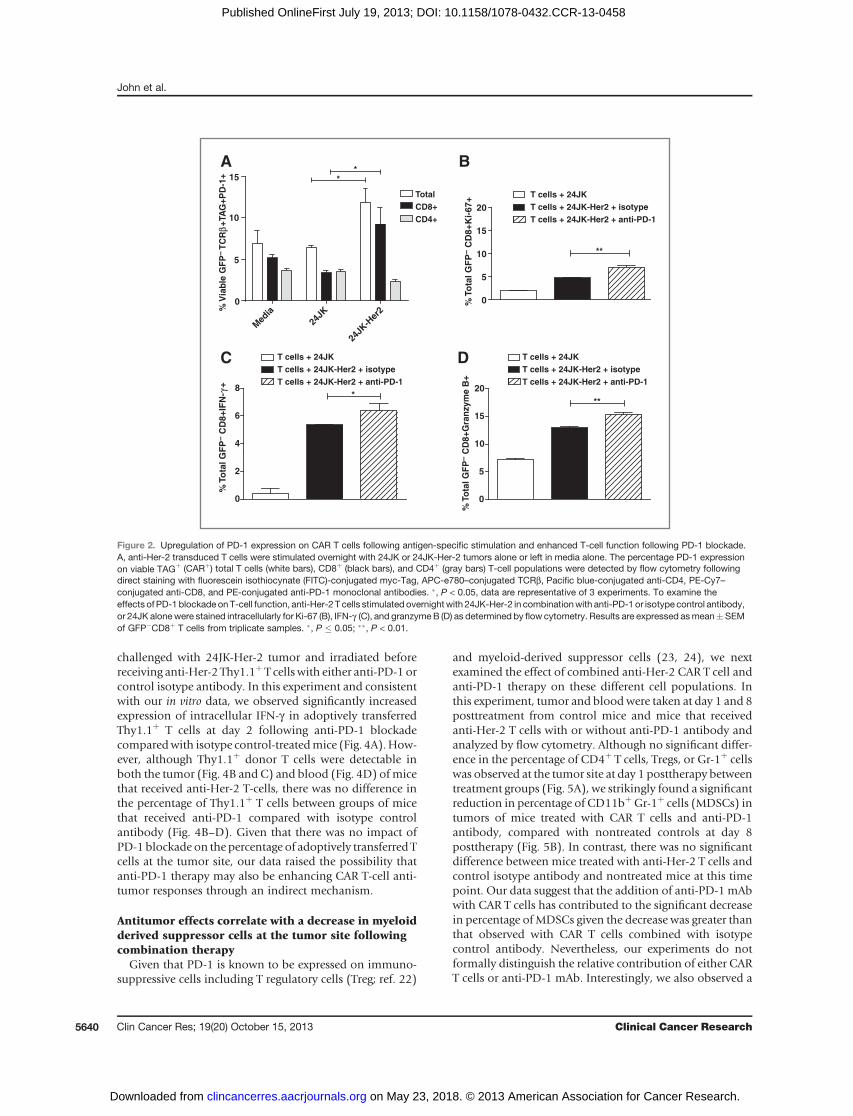
Increased PD-1 expression on anti-Her-2 T cellsfollowing antigen stimulation
Activation of T cells through TCR recognition of MHC/peptide leads to enhanced PD-1 expression which canreduce T-cell function and ultimately lead to T-cell exhaus-tion (13). To determine whether stimulation of T cellsthrough the chimeric antigen receptor similarly leads toincreased expression of PD-1, we first evaluated the expres-sion of the PD-1 ligand (PD-L1) on Her-2þ (24JK-Her-2,e0771-Her-2) and parental (24JK, e0771) tumor targets. Allof these tumor lines displayed high level expression of thePD-L1 surfacemarker (Supplementary Fig. 1A–D). To deter-mine whether CAR-stimulation upregulated PD-1 expres-sion, anti-Her-2 T cells were cocultured overnight with 24JKand 24JK-Her-2. Interestingly, we observed a significantlyincreased level of PD-1 receptor expression on the totaltransduced (Tagþ) T-cell population when cocultured withHer-2þ target cells (11.86 � 3.14% SEM) compared withstimulation with 24JK parental tumor cells (6.45 � 0.45%SEM; Fig. 2A). A similar findingwas also shownwhenT cellswere cocultured with e0771-Her2 tumor cells (Supplemen-tary Fig. S2). Further analysis revealed that increased PD-1expression was only observed on CD8þ T cells when cocul-tured with 24JK-Her-2 target cells (9.23 � 3.77% SEM),compared to stimulation with parental tumor cells (3.40�0.58% SEM) or in media alone (5.16 � 0.84% SEM; Fig.2A). In contrast, there was no significant increase in PD-1receptor expression on transduced CD4þ T cells after anti-gen stimulation (2.31 � 0.38% SEM; Fig. 2A).
PD-1 blockade enhances the proliferative andfunctional capacity of anti-Her-2 T cells in vitro
The functional capacity of anti-Her-2 T cells coculturedwith 24JK cells expressing humanHer-2 antigen (24JK-Her-2) in combination with anti-PD-1 or isotype antibody wasdetermined by staining for intracellular markers of activa-tion by flow cytometry. Following overnight stimulation,we found that blockade of PD-1 significantly enhancedthe intracellular expression of the proliferation markerKi-67, as well as IFN-g and granzyme B, compared with
John et al.
Clin Cancer Res; 19(20) October 15, 2013 Clinical Cancer Research5638
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458
isotype control antibody or transduced T cells coculturedwith parental 24JK tumor cells (Fig. 2B–D). These resultssuggest that blocking PD-1 immunosuppression canincrease important functional parameters in CAR T cellsfollowing stimulation through the CAR.
Adoptive transfer of anti-Her-2 T cells in combinationwith PD-1 blockade enhances regression of establishedtumorWehave previously shown that adoptive transfer of T cells
gene-modified with the scFv-anti-Her-2 receptor could spe-cifically mediate regression of 24JK-Her-2 experimentallung metastasis in Her-2 transgenic mice, although thistherapy was less effective against established subcutaneousdisease (20). Given that PD-1 expression was upregulatedon CAR T cells following antigen stimulation in vitro, thisraised the possibility that anti-PD-1 blockademay combineto enhance antitumor effects mediated by adoptively trans-ferred anti-Her-2 T cells in vivo. In this experiment, wecompared the tumor growth and survival of Her-2 trans-genic mice bearing day 7 established 24JK-Her-2 subcuta-neous tumors (�20–30 mm2) treated with anti-Her-2 orcontrol LXSN–transduced T cells with or without anti-PD-1antibody. Treatment of mice with anti-Her-2 T cells andanti-PD-1 antibody led to the strongest growth inhibition of
24JK-Her-2 tumors (Fig. 3A) and long-term survival(�63%) of mice (Fig. 3B) that was statistically significantto mice treated with anti-Her-2 T cells alone or in combi-nationwith a control isotype antibody (Fig. 3A and B).Micetreatedwith anti-PD-1 alone or control T cells with anti-PD-1 had no effect on tumor growth or survival ofmice (Fig. 3Aand B). To show the broad use of this combined therapy, weshowed that anti-Her-2 T cell and anti-PD-1 antibody treat-ment significantly reduced the growth of day 7 establishede0771-Her-2 breast carcinoma tumors injected orthotopi-cally intoHer-2 transgenicmice comparedwith anti-Her-2 Tcells alone or in combination with isotype control antibody(Fig. 3C). Collectively, these data show for the first time thatthe combination of adoptive immunotherapy using gene-modifiedCART cellswithPD-1blockade could significantlyenhance antitumor effects against established cancer in animmune competent self-antigen setting.
Antitumor effects correlate with increased function ofanti-Her-2 T cells but not localization at the tumor sitefollowing combination therapy
To determine whether the enhanced antitumor effectsobserved following combined CAR T cell and anti-PD-1antibody therapy correlated with increased function andlocalization of anti-Her-2 T cells at the tumor site,micewere
A
C
CD62L
CD
44
3.98% 95.72%
0.00% 0.29%
100 101 102 103 104
101
102
103
104
100
D
CD62L
CD
44
9.83% 90.08%
0.05% 0.05%
100 101 102 103 104
101
102
103
104
100
0
8
16
23
31
Anti-Her-2 receptor expression
Re
lati
ve
ce
ll n
um
be
r
100 101 102 103 1040
8
16
24
32
B
8.53% 8.20%
1.69% 81.57%
100 101 102 103 104
101
102
103
104
100
CD8
CD
4
101
102
103
104
100
6.05% 6.52%
2.19% 85.23%
Anti-Her2 Anti-Her2
LXSNLXSN
Figure 1. Expression of the anti-Her-2 chimeric receptor antigen intransduced Her-2 transgenicmouse T cells. Splenic T cellsderived fromHer-2 transgenicmicewere retrovirally transduced withthe scFv-anti-Her-2 CAR. A, CARexpression was detected in T cellsby flow cytometry followingstaining with an Alexa Fluor 488–conjugated anti-tag mAb (gray) orAlexa Fluor 488– conjugatedmouse isotype control antibody(black) (top) and compared with Tcells transduced with LXSN vectoralone (bottom panel). B, T cellswere phenotypically analyzed forexpression of CD4 and CD8 ontagþ T cells (top) and LXSNtransduced control T cells (bottom)and their activation and memorystatus by expression of CD44 andCD62L on CD4þtagþ cells (C) andCD8þ tagþ cells (D). Data arerepresentative of 6 to 9experiments.
Anti-PD-1 Therapy Enhances Tumor Rejection By CAR T Cells
www.aacrjournals.org Clin Cancer Res; 19(20) October 15, 2013 5639
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458
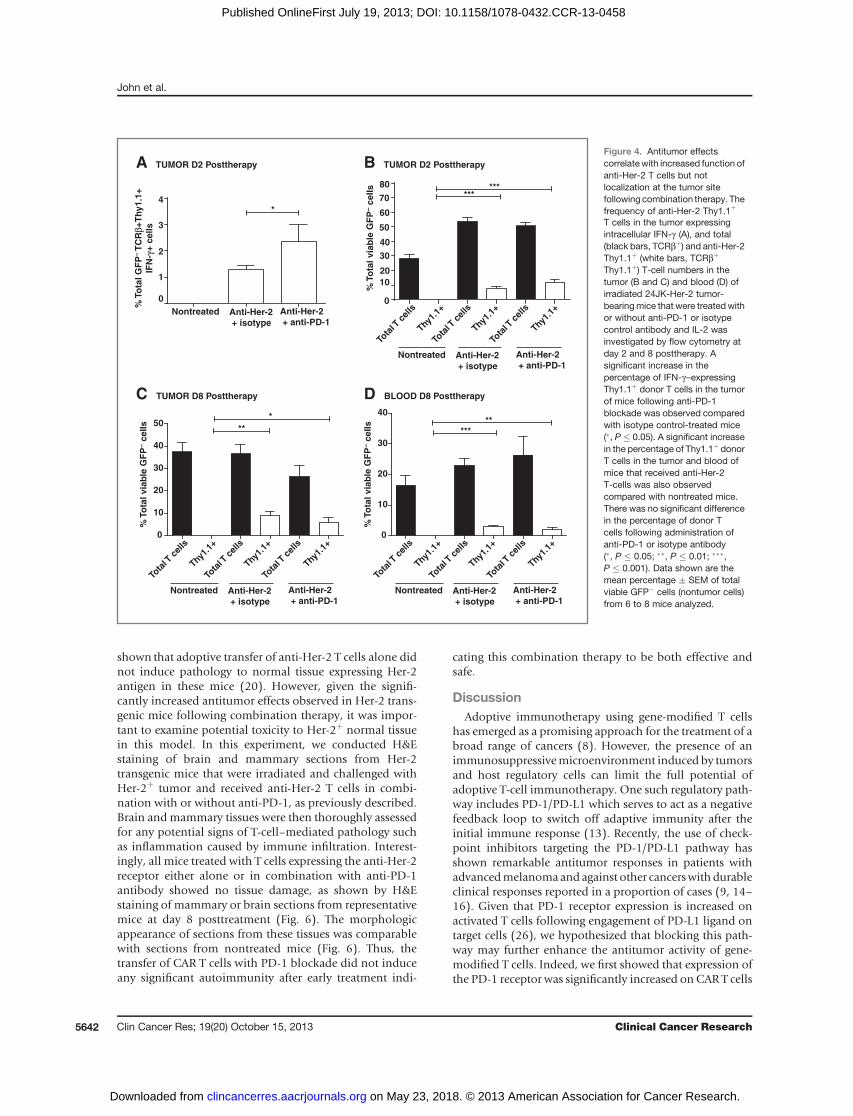
challenged with 24JK-Her-2 tumor and irradiated beforereceiving anti-Her-2 Thy1.1þ T cells with either anti-PD-1 orcontrol isotype antibody. In this experiment and consistentwith our in vitro data, we observed significantly increasedexpression of intracellular IFN-g in adoptively transferredThy1.1þ T cells at day 2 following anti-PD-1 blockadecomparedwith isotype control-treatedmice (Fig. 4A).How-ever, although Thy1.1þ donor T cells were detectable inboth the tumor (Fig. 4B and C) and blood (Fig. 4D) of micethat received anti-Her-2 T-cells, there was no difference inthe percentage of Thy1.1þ T cells between groups of micethat received anti-PD-1 compared with isotype controlantibody (Fig. 4B–D). Given that there was no impact ofPD-1 blockade on the percentage of adoptively transferred Tcells at the tumor site, our data raised the possibility thatanti-PD-1 therapy may also be enhancing CAR T-cell anti-tumor responses through an indirect mechanism.
Antitumor effects correlate with a decrease in myeloidderived suppressor cells at the tumor site followingcombination therapy
Given that PD-1 is known to be expressed on immuno-suppressive cells including T regulatory cells (Treg; ref. 22)
and myeloid-derived suppressor cells (23, 24), we nextexamined the effect of combined anti-Her-2 CAR T cell andanti-PD-1 therapy on these different cell populations. Inthis experiment, tumor and bloodwere taken at day 1 and 8posttreatment from control mice and mice that receivedanti-Her-2 T cells with or without anti-PD-1 antibody andanalyzed by flow cytometry. Although no significant differ-ence in the percentage of CD4þ T cells, Tregs, or Gr-1þ cellswas observed at the tumor site at day 1 posttherapy betweentreatment groups (Fig. 5A), we strikingly found a significantreduction in percentage of CD11bþGr-1þ cells (MDSCs) intumors of mice treated with CAR T cells and anti-PD-1antibody, compared with nontreated controls at day 8posttherapy (Fig. 5B). In contrast, there was no significantdifference between mice treated with anti-Her-2 T cells andcontrol isotype antibody and nontreated mice at this timepoint. Our data suggest that the addition of anti-PD-1 mAbwith CAR T cells has contributed to the significant decreasein percentage of MDSCs given the decrease was greater thanthat observed with CAR T cells combined with isotypecontrol antibody. Nevertheless, our experiments do notformally distinguish the relative contribution of either CART cells or anti-PD-1 mAb. Interestingly, we also observed a
0
5
10
15
20
**
% T
ota
l G
FP
– C
D8
+K
i-6
7+
T cells + 24JK-Her2 + anti-PD-1
T cells + 24JK
T cells + 24JK-Her2 + isotype
0
2
4
6
8*
% T
ota
l G
FP
– C
D8
+IF
N-γ
+
0
5
10
15
20
**
% T
ota
l G
FP
– C
D8+
Gra
nzy
me
B+
**
0
5
10
15
Med
ia
24JK
24JK
-Her
2
Total
CD8+
CD4+%
Via
ble
GF
P– T
CR
β+TA
G+
PD
-1+
T cells + 24JK-Her2 + anti-PD-1
T cells + 24JK
A B
C D
T cells + 24JK-Her2 + isotype
T cells + 24JK-Her2 + anti-PD-1
T cells + 24JK
T cells + 24JK-Her2 + isotype
Figure 2. Upregulation of PD-1 expression on CAR T cells following antigen-specific stimulation and enhanced T-cell function following PD-1 blockade.A, anti-Her-2 transduced T cells were stimulated overnight with 24JK or 24JK-Her-2 tumors alone or left in media alone. The percentage PD-1 expressionon viable TAGþ (CARþ) total T cells (white bars), CD8þ (black bars), and CD4þ (gray bars) T-cell populations were detected by flow cytometry followingdirect staining with fluorescein isothiocynate (FITC)-conjugated myc-Tag, APC-e780–conjugated TCRb, Pacific blue-conjugated anti-CD4, PE-Cy7–conjugated anti-CD8, and PE-conjugated anti-PD-1 monoclonal antibodies. �, P < 0.05, data are representative of 3 experiments. To examine theeffects of PD-1 blockade onT-cell function, anti-Her-2 T cells stimulated overnightwith 24JK-Her-2 in combinationwith anti-PD-1 or isotype control antibody,or 24JK alonewere stained intracellularly for Ki-67 (B), IFN-g (C), and granzymeB (D) as determined by flow cytometry. Results are expressed asmean�SEMof GFP�CD8þ T cells from triplicate samples. �, P � 0.05; ��, P < 0.01.
John et al.
Clin Cancer Res; 19(20) October 15, 2013 Clinical Cancer Research5640
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

significant decrease in percentage of CD11bþ Gr-1þ cells inthe blood of mice that received combination therapy com-pared with nontreated mice at day 1 posttherapy (Fig. 5C)and a moderate decrease in the blood at day 8 (Fig. 5D).Thesedata fitwellwith the fact that theseGr-1þ cells isolatedfrom 24JK-Her-2 tumors were found to express both PD-1(albeit at low levels) and PD-L1 (Supplementary Fig. S3). Incontrast with the effects onMDSCs, there was no additionalmodulation of Treg cells at the tumor site following com-bination therapy at day 8 (Fig. 5B). Notably though, thepercentage of total CD4 T cells and Treg cells in the tumor atday 8 posttherapy were similarly decreased in both anti-Her-2–treated mice with or without anti-PD-1 antibody
compared with untreated control mice (Fig. 5B). This cor-related with a concomitant increase in number of Thy1.1þ
donor T cells in the tumor of these mice (Fig. 4C). Collec-tively, our data showa strong correlation betweendecreasednumbers of MDSCs and increased therapeutic responsesmediated by combined CAR T cell and anti-PD-1 antibodytherapy.
Adoptive transfer of CAR T cells with PD-1 blockadedoes not cause autoimmunity in mice
ThehumanHer-2 antigen is reportedly expressed as a self-antigen in parts of the brain (cerebellum) and mammarytissue ofHer-2 transgenicmice (20, 25).Wehave previously
Nontreated
Anti-Her-2 T cells
Anti-Her-2 T cells + anti-PD-1
Anti-Her-2 T cells + isotype
Anti-PD-1
LXSN T cells + anti-PD-1
B
A
C
0 20 40 600
20
40
60
80
100
Days posttumor injection
e0771-Her2
24JK-Her2
24JK-Her2
Pe
rcen
t su
rviv
al
***
0 5 10 15 20 250
50
100
150
***
***
Days posttreatment
0 5 10 150
50
100
150
200
***
Days posttreatment
Tu
mo
r are
a (
mm
2)
Tu
mo
r are
a (
mm
2)
Figure 3. Adoptive transfer of anti-Her-2 T cells in combination withPD-1 blockade enhances growthinhibition of established tumor.Her-2 transgenic mice (n¼ 8 mice/group), were injectedsubcutaneouslywith 1� 106 24JK-Her-2 sarcoma or orthotopicallywith 5 � 105 e0771-Her-2 breastcarcinoma cells and grown to 20 to30 mm2 for 7 days. Mice were thenirradiated (5 Gy) before theadoptive transfer of anti-Her-2 orcontrol (LXSN) T cells alone(1 � 107/dose on days 0 and 1 oftreatment) or in combination withanti-PD-1 or control isotypeantibody at 250 mg/injection (ondays 0, 3, and 7) or left untreated.Mice were treated with IL-2 (9doses of 50,000 IU between days0 and 4). A, anti-Her-2 T cells withanti-PD-1 antibody significantlyenhanced growth inhibition of24JK-Her-2 subcutaneous tumorscomparedwith combinedanti-Her-2 T-cell and isotype controlantibody or anti-Her-2 T-celltreatment alone (���, P < 0.001). B,percentage survival ofmice treatedwith anti-Her-2 T cells and anti-PD-1 antibody was significantlyincreased compared with anti-Her-2 T cells alone or in combinationwith isotype antibody (���, P <0.001).C, anti-Her-2 T cell and anti-PD-1 antibody treatmentsignificantly enhanced growthinhibition of e0771-Her-2orthotopic tumors compared withanti-Her-2 T-cell treatment alone orin combination with isotype controlantibody (���, P < 0.001). Resultsare represented as the mean tumorsize (mm2) � SEM. Data from twocombined experiments are shown.
Anti-PD-1 Therapy Enhances Tumor Rejection By CAR T Cells
www.aacrjournals.org Clin Cancer Res; 19(20) October 15, 2013 5641
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458
shown that adoptive transfer of anti-Her-2 T cells alone didnot induce pathology to normal tissue expressing Her-2antigen in these mice (20). However, given the signifi-cantly increased antitumor effects observed in Her-2 trans-genic mice following combination therapy, it was impor-tant to examine potential toxicity to Her-2þ normal tissuein this model. In this experiment, we conducted H&Estaining of brain and mammary sections from Her-2transgenic mice that were irradiated and challenged withHer-2þ tumor and received anti-Her-2 T cells in combi-nation with or without anti-PD-1, as previously described.Brain andmammary tissues were then thoroughly assessedfor any potential signs of T-cell–mediated pathology suchas inflammation caused by immune infiltration. Interest-ingly, all mice treated with T cells expressing the anti-Her-2receptor either alone or in combination with anti-PD-1antibody showed no tissue damage, as shown by H&Estaining of mammary or brain sections from representativemice at day 8 posttreatment (Fig. 6). The morphologicappearance of sections from these tissues was comparablewith sections from nontreated mice (Fig. 6). Thus, thetransfer of CAR T cells with PD-1 blockade did not induceany significant autoimmunity after early treatment indi-
cating this combination therapy to be both effective andsafe.
DiscussionAdoptive immunotherapy using gene-modified T cells
has emerged as a promising approach for the treatment of abroad range of cancers (8). However, the presence of animmunosuppressivemicroenvironment induced by tumorsand host regulatory cells can limit the full potential ofadoptive T-cell immunotherapy. One such regulatory path-way includes PD-1/PD-L1 which serves to act as a negativefeedback loop to switch off adaptive immunity after theinitial immune response (13). Recently, the use of check-point inhibitors targeting the PD-1/PD-L1 pathway hasshown remarkable antitumor responses in patients withadvancedmelanomaand against other cancerswith durableclinical responses reported in a proportion of cases (9, 14–16). Given that PD-1 receptor expression is increased onactivated T cells following engagement of PD-L1 ligand ontarget cells (26), we hypothesized that blocking this path-way may further enhance the antitumor activity of gene-modified T cells. Indeed, we first showed that expression ofthe PD-1 receptor was significantly increased on CAR T cells
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
% T
ota
l via
ble
GF
P– c
ells
TUMOR D8 Posttherapy
0
10
20
30
40
50**
*
BLOOD D8 Posttherapy
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
0
10
20
30
40
*****
% T
ota
l via
ble
GF
P– c
ells
0
20
40
60
80
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Tota
l T c
ells
Thy1.1
+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
TUMOR D2 Posttherapy
******
% T
ota
l via
ble
GF
P– c
ells
% T
ota
l G
FP
– T
CR
β+T
hy1.1
+
IFN
-γ+
cells
10
30
50
70
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
0
1
2
3
4
TUMOR D2 PosttherapyA B
C D
*
Figure 4. Antitumor effectscorrelate with increased function ofanti-Her-2 T cells but notlocalization at the tumor sitefollowing combination therapy. Thefrequency of anti-Her-2 Thy1.1þ
T cells in the tumor expressingintracellular IFN-g (A), and total(black bars, TCRbþ) and anti-Her-2Thy1.1þ (white bars, TCRbþ
Thy1.1þ) T-cell numbers in thetumor (B and C) and blood (D) ofirradiated 24JK-Her-2 tumor-bearingmice that were treated withor without anti-PD-1 or isotypecontrol antibody and IL-2 wasinvestigated by flow cytometry atday 2 and 8 posttherapy. Asignificant increase in thepercentage of IFN-g–expressingThy1.1þ donor T cells in the tumorof mice following anti-PD-1blockade was observed comparedwith isotype control-treated mice(�, P � 0.05). A significant increasein the percentage of Thy1.1þ donorT cells in the tumor and blood ofmice that received anti-Her-2T-cells was also observedcompared with nontreated mice.There was no significant differencein the percentage of donor Tcells following administration ofanti-PD-1 or isotype antibody(�, P � 0.05; ��, P � 0.01; ���,P � 0.001). Data shown are themean percentage � SEM of totalviable GFP� cells (nontumor cells)from 6 to 8 mice analyzed.
John et al.
Clin Cancer Res; 19(20) October 15, 2013 Clinical Cancer Research5642
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

following coculture with PD-L1þ Her-2þ expressing tumortargets and that PD-1 blockade enhanced functional para-meters of CAR T cells in vitro and in vivo. In adoptive transferexperiments, we showed that combined CAR T-cell therapywith anti-PD-1 antibody significantly increased the growthinhibition of two different Her-2þ tumors and led toenhanced survival of transgenic Her-2 recipient mice. Inter-estingly, we found that increased antitumor responsesmediated by this combined therapy correlated well witha significant decrease in numbers of CD11bþGr-1þ mye-loid-derived suppressor cells (MDSCs) but not Treg cells atthe tumor site compared with nontreated control mice.Importantly CAR T-cell treatment combined with anti-PD-1 antibody administration was well tolerated causingno signs of autoimmunity in recipient mice. Overall, this isthe first study to show that blocking a major immunosup-pressive pathway such as PD-1 can significantly enhanceadoptive immunotherapy using gene-modified T cells.Although the question of whether PD-L1 ligand expressedon the tumor cells was critical for the therapeutic effectsobserved was not determined in this study, a significantcorrelation between the level of PD-L1 expression on tumorcells andobjective responses in patients has been previouslyreported (14).
We have previously shown that CAR T-cell therapy alonecould elicit significant antitumor effects on the growth ofestablished 24JK-Her-2þ lung metastases leading to long-term survival without causing any signs of pathology in vivo(20). However, in this current study, CAR T-cell therapyalone was not as effective against established subcutaneousdisease. Nevertheless, we were able to show in our in vivoexperiments striking growth inhibition of both 24JK-Her-2þ sarcoma cells and E0771-Her-2þ breast carcinoma cellswhen inoculated subcutaneously following CAR T-cell ther-apy in combination with PD-1 blockade compared withmice treated with CAR T cells and isotype control antibody.We found both in vitro and in vivo that blocking PD-1significantly enhanced the function of CAR T cells leadingto a more lethal hit against the tumor. Unlike other studiesthat usedMHCclass I restricted T cells (26, 27),we foundnoincrease in the percentage of adoptively transferred Thy1.1þ
T cells in the blood or tumor site following combinedtherapy compared with control treated mice. Future studiesusing other qualitative approaches such as luciferase trans-duced T cells would enable the kinetics of T-cell infiltrationto be better defined (27).
An interesting aspect of the current study was thatthe strong antitumor effects observed with CAR T cells
TUMOR D1 Posttherapy
D8 Posttherapy D8 Posttherapy
D1 PosttherapyBLOODCA
B DTUMOR BLOOD
0
5
10
15**
*
**
% T
ota
l v
iab
le G
FP
– c
ell
s
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
*ns
0
20
40
60
% T
ota
l v
iab
le G
FP
– c
ell
s
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
CD4 T c
ells
Treg
GR1+
Anti-Her-2
+ isotype
Nontreated Anti-Her-2
+ anti-PD-1
0
5
10
15
% T
ota
l v
iab
le G
FP
– c
ell
s0
20
40
60
ns
*
% T
ota
l v
iab
le G
FP
– c
ell
sFigure 5. Antitumor effectscorrelate with a decrease inMDSCs following combined CAR Tcell and anti-PD-1 therapy. Tumors(A and B) and blood (C and D) weretaken at day 1 and 8 from micetreated with anti-Her-2 T cells incombination with anti-PD-1 orisotype control antibody andnontreated mice, and examined byflow cytometry (n ¼ 6 mice/group).A–D, mean percentage of CD4þ Tcells (CD4þ TCRbþ), regulatory Tcells (CD4þ TCRbþ CD25þ FR4þ),and Gr-1þ myeloid cells (Gr-1þ
CD11bþ) at day 1 (A and C) and 8(B and D). Gr1þ cells weresignificantly reduced in tumor (day8) and blood (day 1) followingcombined CAR T cell and anti-PD-1 therapy compared withnontreated mice. Treg cells weresignificantly reduced in CART-cell–treated mice with orwithout anti-PD1 compared withnontreated control mice(�,P� 0.05, ��,P� 0.01 and ns; notsignificant between treatmentgroups). Data shown are the meanpercentage � SEM of viable GFP�
cells (nontumor cells) from 6 miceanalyzed.
Anti-PD-1 Therapy Enhances Tumor Rejection By CAR T Cells
www.aacrjournals.org Clin Cancer Res; 19(20) October 15, 2013 5643
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

combined with anti-PD-1 antibody correlated with a sig-nificant decrease in the percentage of Gr-1þ CD11bþ
MDSCs at the tumor site. Given that the percentage ofGr-1þ CD11bþ cells expressing PD-1 was found to be lowand that anti-PD-1 has been reported to be a blockingantibody rather than a depleting antibody (28, 29) webelieve that the increased antitumor effects observed usinganti-PD-1 in our tumor models were most likely due to anindirect effect on the MDSC population. Our results areconsistent with another study showing a reduction of mye-loid cells in tumors following anti-PD-1 therapy (30).Previous studies using anti-PD-1 alone or in combinationwith anti-CTLA-4have showna reduction in the suppressiveactivity of MDSCs through inhibition of arginase I (24).Other studies have shown that MDSCs can suppress anti-tumor responses through production of nitric oxidesynthase (31), reactive oxygen species (32), and cytokinessuch as TGF-b and IL-10 (33, 34). It remains to be deter-mined in future studies whether the increased antitumoreffects of CAR T-cell therapy in combination with PD-1blockade was through modulation of the immunosuppres-sive effects of MDSCs. In contrast with the effects on MDSCnumbers, we observed no additional effect on percentage of
Treg cells in the blood or tumor site following combinedCAR T-cell and anti-PD-1 treatment. The decreased percent-age of Treg cells and concomitant increase in Thy1.1þ T cellsin Her-2 recipient mice was most likely due to the effects ofirradiationwhich has been reported to decrease host immu-nosuppressive cell populations enabling expansion oftransferred T cells (35). Although PD-1 blockade alone haspreviously been reported to decrease Treg numbers (30),our findings are consistentwith another report showing thatanti-PD-1 antibody treatment did not modulate Treg cellnumbers following adoptive immunotherapy (27). It isimportant to note that these studies do not preclude aneffect of PD-1 blockade on the function of Treg cells withinthe tumor microenvironment.
Another important aspect of our work was that the use ofself-antigen Her-2 transgenic mice allowed us to assesswhetherCART-cell therapy combinedwith anti-PD-1 causedany pathology to normal tissue expressing theHer-2 antigen.Although we have previously reported no toxicity of anti-Her-2 CAR T cells alone in this model (20), it remainedpossible that significant increases in therapeutic activityusinganti-PD-1 couldhave concomitantly led toautoimmunity. Inthis current study, detailed immunohistochemical analysis
×100 ×200N
on
treate
dA
nti
-Her-
2 T
cells
An
ti-H
er-
2 T
cells
+ i
so
typ
e
+ a
nti
-PD
-1
Breast Brain
Figure 6. Adoptive transfer of anti-Her-2 T cells combined with PD-1blockade does not causeautoimmunity in Her-2 transgenicmice. Her-2 transgenic micebearing 24JK-Her-2 subcutaneoustumors were irradiated (5 Gy) andtreated with adoptively transferredanti-Her-2 T cells (1� 107/dose ondays 0 and 1 of treatment) or incombination with anti-PD-1 orisotype control antibody at 250 mg/injection or left untreated.Mammary (left) and brain (right)tissueswere harvested and stainedwith H&E at day 8 posttreatment.Analysis of tissue sectionsrevealed no structural damage orincreased immune cell infiltrationfor mice treated with anti-Her-2 Tcells and anti-PD-1 or isotypeantibody in mammary or braintissues compared with nontreatedmice. Mammary and brain sectionsare representative of 6 miceanalyzed per treatment group.
John et al.
Clin Cancer Res; 19(20) October 15, 2013 Clinical Cancer Research5644
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

revealed no damage to either Her-2þ mammary or braintissue following combined therapy. This may have been dueto the lower levels of Her-2 antigen expressed on normaltissue compared with 24JK-Her-2 tumor cells as we havereported previously (20) or due to insensitivity of the Her-2transgenic mouse model for detecting such autoimmuneevents. Nevertheless, our data are encouraging for movingtowards testing this combined approach in the clinic. Anumber of recent clinical trials involving adoptive transferof CAR T cells alone has shown promising results in patientswith chronic lymphoid leukaemia and neuroblastoma withreports of long-term remission in some cases (5, 7). Never-theless, other trials using CAR T cells have reported on targettoxicity following treatment (36–38). Similarly, recent clin-ical trials using antibodies targeting the PD-1 receptor and itsligand PD-L1 have reported durable responses against var-ious solid cancers (14–16). In these trials, treatment wasgenerally well tolerated although some drug-associatedadverse events and pneumonitis was reported in a smallpercentage of patients (14). Given these reported toxicities insomepatients, it will be important in future studies to furtheroptimize dose and timing regimens of CAR T cells with anti-PD-1 antibody in self-antigen mouse models before phase Itesting.In conclusion, our studies have shown for the first time
that administration of an anti-PD-1 antibody can signifi-cantly enhance the therapeutic efficacy of CAR T-cell ther-apy. The combination of these two modalities is likely tohave a significant impact on increasing the effectiveness ofimmunotherapy against a number of cancers that are cur-rently resistant to first-line treatment.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L.B. John, M.H. Kershaw, P.K. DarcyDevelopment of methodology: L.B. John, M.H. Kershaw, P.K. DarcyAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): L.B. John, C. Devaud, C.M. Duong, C. Yong,P.A. Beavis, N.M. Haynes, P.K. DarcyAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis): L.B. John, C. Devaud, M.H. Kershaw,P.K. DarcyWriting, review, and/or revision of the manuscript: L.B. John, C.M.Duong, C. Yong, P.A. Beavis, N.M. Haynes, M.T. Chow, M.J. Smyth,P.K. DarcyAdministrative, technical, or material support (i.e., reporting ororganizing data, constructing databases): L.B. John, M.T. Chow,P.K. DarcyStudy supervision: M.J. Smyth, M.H. Kershaw, P.K. Darcy
AcknowledgmentsThe authors thank the Peter MacCallum Cancer Centre Experimental
Animal Facility technicians for the assisting in animal care and the HistologyDepartment for processing of H&E sections of tissue following therapy.
Grant SupportThis work was funded by Project Grants from the National Health and
Medical Research Council (NHMRC), Cancer Council of Victoria, and theSusan Komen Breast Cancer Foundation. M.J. Smyth was supported by anNHMRC Australia Research Fellowship. M.H. Kershaw and P.K. Darcy weresupported by NHMRC Senior Research Fellowships.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received February 20, 2013; revised May 15, 2013; accepted July 4, 2013;published OnlineFirst July 19, 2013.
References1. PhamCD,Mitchell DA. Chasing cancer with chimeric antigen receptor
therapy. Immunotherapy 2012;4:365–7.2. Rosenberg SA. Raising the bar: the curative potential of human cancer
immunotherapy. Sci Transl Med 2012;4:127ps8.3. Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T
cell immunotherapy: current understanding and future directions.J Gene Med 2012;14:405–15.
4. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al.Adoptive immunotherapy for indolent non-Hodgkin lymphoma andmantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008;112:2261–71.
5. Louis C, Savoldo B, Dotti G, Pule M, Yvon E, Myers G, et al. Antitumoractivity and long-term fate of chimeric antigen receptor-positive T cellsin patients with neuroblastoma. Blood 2011;118:6050–6.
6. Brentjens R, Rivi�ere I, Park J, Davila M, Wang X, Stefanski J, et al.Safety and persistence of adoptively transferred autologous CD19-targetedTcells in patientswith relapsedor chemotherapy refractoryB-cell leukemias. Blood 2011;118:4817–28.
7. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigenreceptor–modified T cells in chronic lymphoid leukemia. N Engl J Med2011;365:725–33.
8. Chicaybam L, Sodr�e A, Bonamino M. Chimeric antigen receptors incancer immuno-gene therapy: current status and future directions. IntRev Immunol 2011;30:294–311.
9. Pardoll DM. The blockade of immune checkpoints in cancer immu-notherapy. Nat Rev Cancer 2012;12:252–64.
10. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, HaanenJB, et al. Improved survival with ipilimumab in patients with metastaticmelanoma. N Engl J Med 2010;363:711–23.
11. Ascierto PA, Simeone E, Sznol M, Fu Y-X, Melero I. Clinical experi-ences with anti-CD137 and anti-PD1 therapeutic antibodies. SeminOncol 2010;37:508–16.
12. BlankC,Gajewski T,MackensenA. Interaction of PD-L1 on tumor cellswith PD-1 on tumor-specific T cells as a mechanism of immuneevasion: implications for tumor immunotherapy. Cancer ImmunolImmunother 2005;54:307–14.
13. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands intolerance and immunity. Annu Rev Immunol 2008;26:677–704.
14. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDer-mott DF, et al. Safety, activity, and immune correlates of anti–PD-1antibody in cancer. N Engl J Med 2012;366:2443–54.
15. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M,et al. Phase I safety and pharmacokinetic study of CT-011, a human-ized antibody interacting with PD-1, in patients with advanced hema-tologic malignancies. Clin Cancer Res 2008;14:3044–51.
16. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P,et al. Safety and activity of anti–PD-L1 antibody in patients withadvanced cancer. N Engl J Med 2012;366:2455–65.
17. Kershaw MH, Jackson JT, Haynes NM, Teng MW, Moeller M, Haya-kawa Y, et al. Gene-engineered T cells as a superior adjuvant therapyfor metastatic cancer. J Immunol 2004;173:2143–50.
18. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ,et al. Redirecting mouse CTL against colon carcinoma: superiorsignaling efficacy of single-chain variable domain chimeras containingTCR-zeta vs Fc epsilon RI-gamma. J Immunol 2001;166:182–7.
19. DarcyPK,HaynesNM,SnookMB,Trapani JA,Cerruti L, JaneSM, et al.Redirected perforin-dependent lysis of colon carcinoma by ex vivogenetically engineered CTL. J Immunol 2000;164:3705–12.
Anti-PD-1 Therapy Enhances Tumor Rejection By CAR T Cells
www.aacrjournals.org Clin Cancer Res; 19(20) October 15, 2013 5645
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

20. Wang LXJ,Westwood JA,MoellerM, DuongCPM,WeiW-Z,MalaterreJ, et al. Tumor ablation by gene-modified T cells in the absence ofautoimmunity. Cancer Res 2010;70:9591–8.
21. John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, et al.Oncolytic virus and anti–4-1BB combination therapy elicits strongantitumor immunity against established cancer. Cancer Res 2012;72:1651–60.
22. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in toleranceand autoimmunity. Immunol Rev 2010;236:219–42.
23. Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R. B7-H1 on myeloid-derivedsuppressor cells in immune suppression by a mouse model of ovariancancer. Clin Immunol 2008;129:471–81.
24. Liu Y, Yu Y, Yang S, Zeng B, Zhang Z, Jiao G, et al. Regulation ofarginase I activity and expression by both PD-1 and CTLA-4 on themyeloid-derived suppressor cells. Cancer Immunol Immunother 2009;58:687–97.
25. Piechocki MP, Ho Y-S, Pilon S, Wei W-Z. Human ErbB-2 (Her-2)transgenic mice: a model system for testing Her-2 based vaccines.J Immunol 2003;171:5787–94.
26. Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H, et al. Interactionbetween B7-H1 and PD-1 determines initiation and reversal of T-cellanergy. Blood 2007;110:180–5.
27. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockadeenhances T-cell migration to tumors by elevating IFN-g induciblechemokines. Cancer Res 2012;72:5209–18.
28. Matsumoto K, Inoue H, Nakano T, Tsuda M, Yoshiura Y, Fukuyama S,et al. B7-DC regulates asthmatic response by an IFN-g-dependentmechanism. J Immunol 2004;172:2530–41.
29. Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K.Blockade of B7-H1 on macrophages suppresses CD4þ T cell prolif-eration by augmenting IFN-g-induced nitric oxide production. J Immu-nol 2005;175:1586–92.
30. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4combination blockade expands infiltrating T cells and reduces regu-latory T and myeloid cells within B16 melanoma tumors. Proc NatlAcad Sci U S A 2010;107:4275–80.
31. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible dis-ruption of the Jak3/STAT5 signaling pathway. J Immunol 1998;160:5729–34.
32. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specificinhibition of CD8þ T cell response by immature myeloid cells incancer is mediated by reactive oxygen species. J Immunol 2004;172:989–99.
33. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as reg-ulators of the immune system. Nat Rev Immunol 2009;9:162–74.
34. Stewart T, Smyth M. Improving cancer immunotherapy by targetingtumor-induced immune suppression. Cancer Metastasis Rev 2011;30:125–40.
35. Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immu-notherapy for cancer: building on success. Nat Rev Immunol 2006;6:383–93.
36. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al.Treatment of metastatic renal cell carcinoma with autologous T-lym-phocytes genetically retargeted against carbonic anhydrase IX: firstclinical experience. J Clin Oncol 2006;24:e20–2.
37. MorganRA,Yang JC,KitanoM,DudleyME, LaurencotCM,RosenbergSA.Case report of a serious adverse event following the administrationof T cells transduced with a chimeric antigen receptor recognizingERBB2. Mol Ther 2010;18:843–51.
38. Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment ofchronic lymphocytic leukemia with genetically targeted autologous Tcells: case report of an unforeseen adverse event in a Phase I clinicaltrial. Mol Ther 2010;18:666–8.
John et al.
Clin Cancer Res; 19(20) October 15, 2013 Clinical Cancer Research5646
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458

2013;19:5636-5646. Published OnlineFirst July 19, 2013.Clin Cancer Res Liza B. John, Christel Devaud, Connie P.M. Duong, et al. Established Tumors By Gene-Modified T CellsAnti-PD-1 Antibody Therapy Potently Enhances the Eradication of
Updated version
10.1158/1078-0432.CCR-13-0458doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2013/07/22/1078-0432.CCR-13-0458.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/19/20/5636.full#ref-list-1
This article cites 38 articles, 19 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/19/20/5636.full#related-urls
This article has been cited by 32 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/19/20/5636To request permission to re-use all or part of this article, use this link
on May 23, 2018. © 2013 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst July 19, 2013; DOI: 10.1158/1078-0432.CCR-13-0458