applicazioni dei metodi computazionali in chimica organica · •la chimica computazionale permette...
TRANSCRIPT

Opportunità in chimica computazionale
Applicazioni dei metodi computazionali in chimica organica
Alessandro BagnoDip. Scienze Chimiche, Università di Padova
http://www.chimica.unipd.it/alessandro.bagno

La chimica è la finestra che l'uomo utilizza per sondare la propria essenza materiale e per venire a patti con i propri limiti
S. Shaik (2003)
Chimica e materia

Chimica e proprietà
Chimica Energia in funzione delle coordinate dei nuclei
Come varia l'energia in risposta ad una perturbazioneProprietà

La chimica computazionale consiste nel trattare tutti gli aspetti della chimica tramite il calcolo piuttosto che l'esperimento
Paul v. R. Schleyer (1987)
Cos'è la chimica computazionale?

"Tanto maggiore è il progresso delle scienze fisiche, tanto più esse
tendono ad entrare nel dominio della matematica, il centro a cui
tutte convergono. Si potrebbe persino valutare il grado di
perfezione a cui una scienza ègiunta dalla facilità con cui essa può essere soggetta al calcolo"
A. Quetelet, 1828
Chimica e calcoli: un rapporto difficile?
"Ogni tentativo di utilizzare metodi matematici nello studio dei problemi chimici deve essere considerato profondamente irrazionale e contrario allo spirito della chimica. Se l'analisi matematica occupasse un posto di rilievo nella chimica – un'aberrazione per fortuna quasi impossibile – ne provocherebbe la sua rapida e completa degenerazione"A. Compte, 1830

La chimica computazionale non è una scatola nera!
Per valutare la qualità dei calcoli è necessario•Comprendere i concetti teorici sottostanti•Guardare con occhio critico sia all'esperimento che alla teoria
Perché fare chimica (una scienza “sperimentale”) sui computer?
•Migliore comprensione della chimica (e della fisica)•Veloce circolazione delle idee•Costi ridotti•Sicurezza•Migliore accuratezza (per sistemi molto piccoli)

Metodi della chimica computazionale
Classici
Quanto-meccanici
Meccanica molecolare
Funzione d'onda(ab initio)
Densità (DFT)
Hartree-Fock
Semiempirici
Post-HF

•Facile da capire e da programmare•Estremamente veloce•Non ci sono elettroni: interpretazione limitata
Modello: oscillatore armonico classicoCiascun legame caratterizzato da una costante di forza per stiramento, piegamento, etc.
Basati sul “tipo di atomo”
Meccanica molecolare (force field)AMBER, Sybyl, Merck, etc.
Accuratezza tipica:Lunghezze di legame: 0.01 ÅAngoli di legame: 1°Angoli diedri: qualche gradoEnergie conformazionali: > 1 kcal/molFrequenze vibrazionali: > 20-30 cm–1
Tipicamente impiegati per “grandi” sistemi (proteine, etc.)
Strutture inconsuete, stati di transizione etc. non trattabili!

Metodi semiempirici (AM1, PM3…)
•Trattati solo gli elettroni di valenza•Base minima, alcune grandezze derivate da parametri sperimentali
Alcune deficienze:Le molecole stericamente impedite sono troppo instabiliGli anelli a 4 membri sono troppo stabiliLe barriere rotazionali sono spesso sottostimate (es. etano)I legami a idrogeno sono troppo deboli e lunghi (es. dimero dell'acqua)Barriere di attivazione troppo alte
Accuratezza tipicaLunghezze di legame: 0.014 ÅAngoli di legame: 2.8°Potenziali di ionizzazione: 0.48 eVCalori di formazione: 6.3 kcal/mol
Molecole organiche semplici, fino a 103 atomi

•Completamente quantistici•Solo costanti fisiche fondamentali•Grande accuratezza•Sono comprese tutte le interazioni•Molto costosi•Possibile miglioramento sistematico
Metodi ab initio
Metodo Hartree-FockGli elettroni interagiscono uno con l'altro solo attraverso il campo medio degli altri elettroni
Molecole organiche fino a 102 atomiStati di transizione
N2-3

Repulsione istantanea energia di correlazioneL'equazione di Schrödinger non è più risolvibile in forma analitica
•Teorie perturbative (MPn, tipicamente MP2)•Interazione delle configurazioni (CI)•Coupled Cluster (CC)
Metodi post-HF
Molecole organiche fino a 101 atomiStati di transizioneStati eccitati
N4-N7

L'energia e tutte le proprietà dello stato fondamentale sono determinate unicamente dalla densità di carica
Il teorema non specifica la forma funzionale di questa relazioneLe loro prestazioni debbono essere verificate empiricamente
Teoria del funzionale di densità(DFT)
•Completamente quantistici•In linea di principio, “esatti”•Molto più veloci degli ab initio•Accuratezza variabile•Miglioramento sistematico impossibile
Molecole organiche fino a 102 atomiStati di transizione
N2

•Molte specie sono instabili (intermedi reattivi)•Molte specie sono stabili ma non isolabili•Stati di transizione
Struttura molecolare
Spesso la struttura è nota (ad es. raggi X) proprietà
La struttura non è nota o non èdeterminabile!
Chimica Energia in funzione delle coordinate dei nuclei

Coordinate atomiche iniziali nuove cooordinate tali da minimizzare l'energia e annullare le forze agenti sugli atomi
Minimo (locale o globale)?Stato di transizione?
Fase gas vs. soluzioneGeneralmente i calcoli si riferiscono alle specie nel vuoto a 0 K Modelli del solvente
Ottimizzazione della geometria
•Ottimizzazione ad un minimo Struttura, stabilità e proprietà molecolari
•Ottimizzazione ad un TS Struttura, reattività, meccanismo di reazione

Analisi di popolazioneStruttura (distribuzione) elettronica molecolare intuizione chimica!Cariche atomiche, densità di spin, momento di dipolo, polarizzabilità
Proprietà molecolari e spettroscopiche, stabilità, reattività
Spettri elettroniciSpettro UV-VIS, dicroismo circolare
Spettri vibrazionaliSpettro IR e Raman, dicroismo circolare vibrazionaleInformazioni sulla natura della struttura ottenuta
Spettroscopie magneticheSpettro EPR (frequenze, costanti di accoppiamento iperfine, fattore g)Spettro NMR (chemical shift, costanti di accoppiamento)
Energie dei minimi e degli stati di transizioneStabilità relativa di specie isomeriche
Barriera di attivazione per una reazione
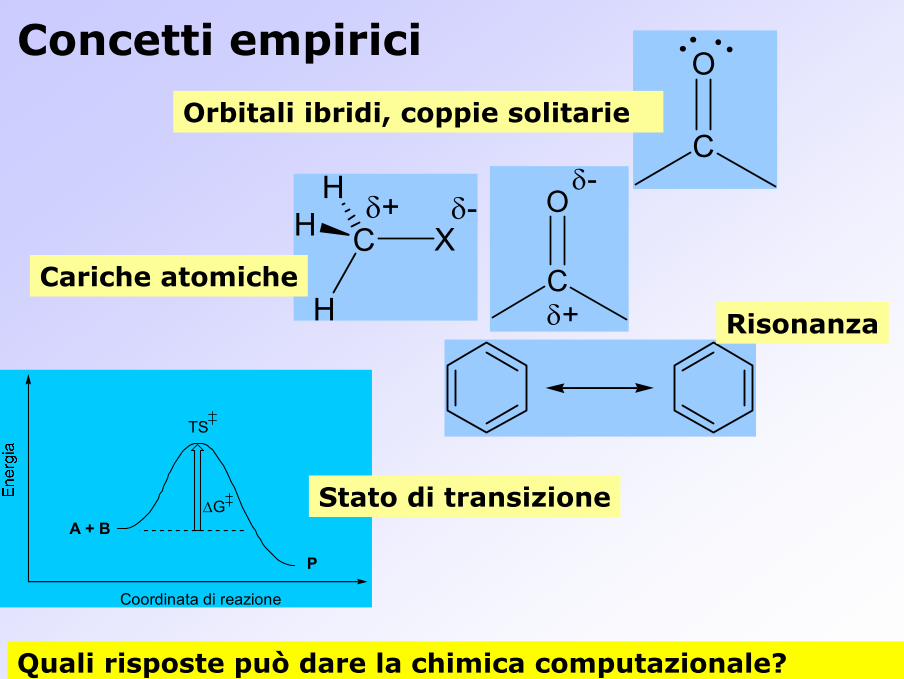
Concetti empirici
C
H
HH X
δ+ δ-
C
O
δ+
δ-
Cariche atomiche
Quali risposte può dare la chimica computazionale?
Risonanza
C
OOrbitali ibridi, coppie solitarie
A + B
Coordinata di reazione
P
TS
∆G Stato di transizione

Orbitali ibridi vs. MO: CH4
MO #5C(2pz),H(1s)
MO #2C(2s),H(1s)
MO #3C(2px),H(1s)
MO #4C(2py),H(1s)
CHH
H
H
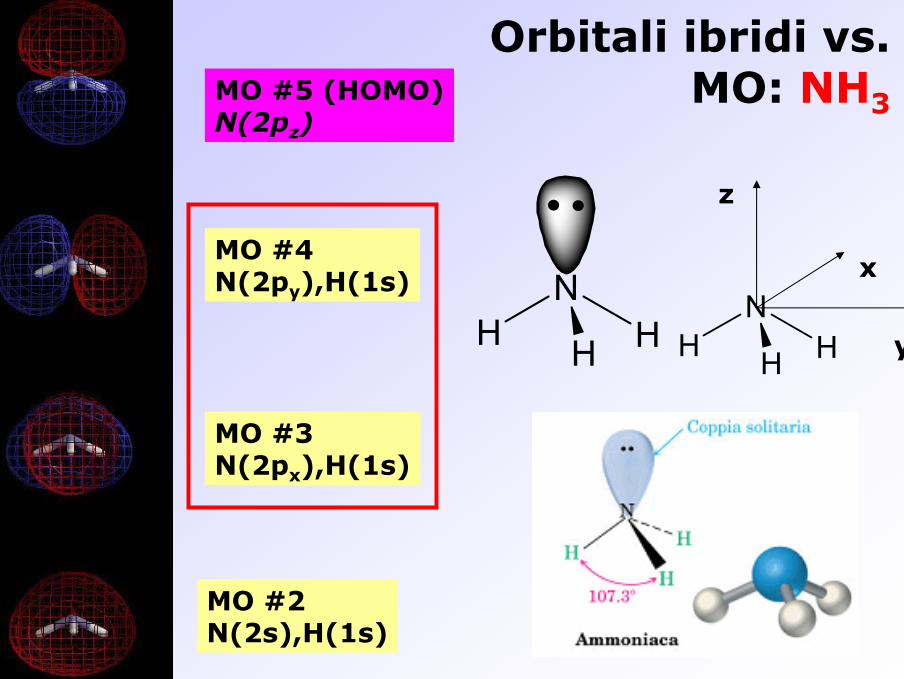
Orbitali ibridi vs. MO: NH3MO #5 (HOMO)
N(2pz)
MO #2N(2s),H(1s)
MO #3N(2px),H(1s)
MO #4N(2py),H(1s) N
H HHN
H HH y
z
x

La funzione d'onda ψ non ha un significato fisico immediatoLa densità elettronica ρ ha un significato:
2|)(|)( rr Ψ=ρ
Interpretazione della funzione d'onda
La presentazione e l'interpretazione sono difficili

Analisi di popolazione
Le cariche atomiche q:•Non sono osservabili in senso quantomeccanico•Non esiste un metodo univoco per calcolarle
Calcolo delle “cariche atomiche” q
Li+ F–
Natural Population Analysis
q(Li) = +0.815
q(Li) = +0.977
Parzialmente covalente
Prevalentemente ionico
Li–H

BHH
H H
-0.47 -0.13
AlHH
H H
0.78 -0.44
Distribuzione di carica in BH4- e AlH4
-
Superfici di potenziale elettrostatico (ESP)

•Carica negativa localizzata su B e H terminali•H a ponte carichi positivamente
BHH
H HB
H
H
+0.08-0.04
-0.02
Distribuzione di carica in B2H6
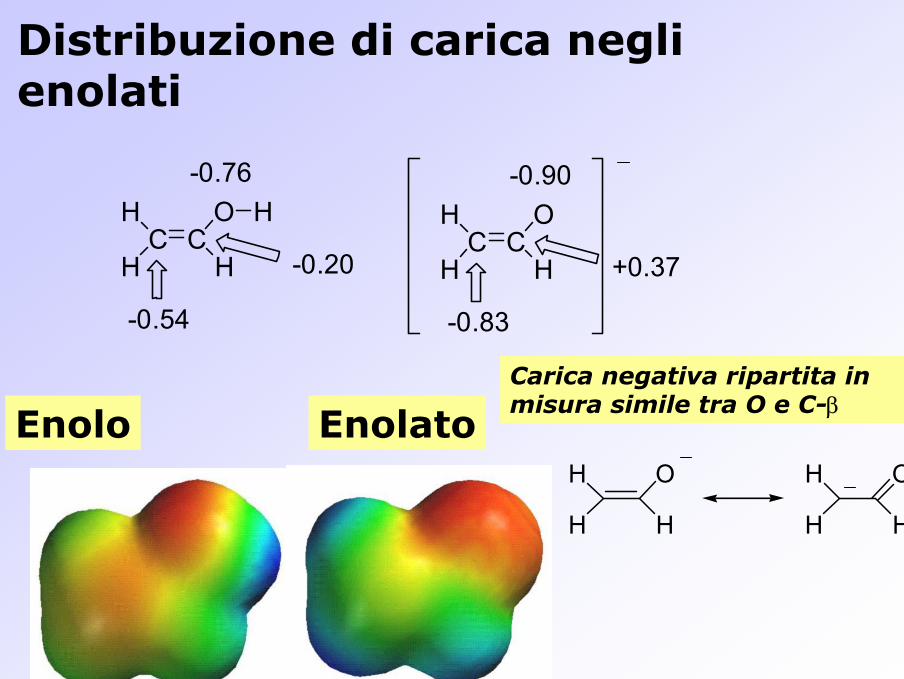
Carica negativa ripartita in misura simile tra O e C-β
C CO
HH
H H
-0.54
-0.20
-0.76
C CO
HH
H
-0.83
+0.37
-0.90
O
HH
H O
HH
H
Distribuzione di carica negli enolati
Enolo Enolato

Distribuzione di carica in intermedi di Wheland
Cl H
+0.06
-0.21+0.16
Carica positiva distribuita su C-2, C-4, C-6
Addizione di Cl+ al benzene

Densità di spin nel radicale PhO.
O O O O
Elettrone spaiato su C-2, C-4, C-6, e in parte su O
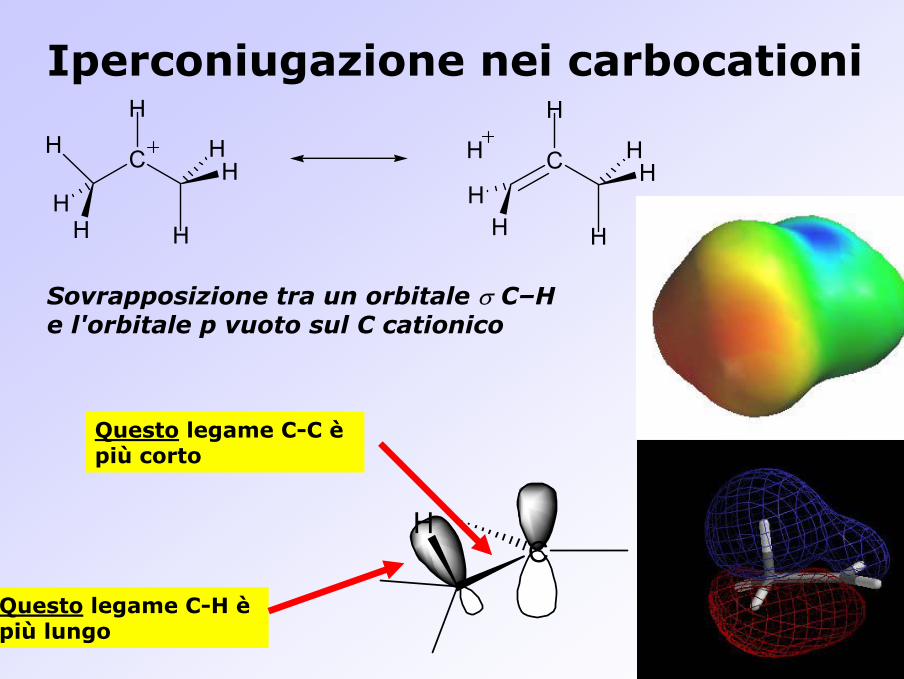
Iperconiugazione nei carbocationi
Sovrapposizione tra un orbitale σ C–H e l'orbitale p vuoto sul C cationico
CH
Questo legame C-C èpiù corto
Questo legame C-H èpiù lungo
C
H
HH
HH H
H
C
H
HH
HH
HH

Proprietà spettroscopiche
Spettri elettroniciSpettro UV-VIS, dicroismo circolare
Spettri vibrazionaliSpettro IR e Raman, dicroismo circolare vibrazionaleInformazioni sulla natura della struttura ottenuta
Spettroscopie magneticheSpettro EPR (frequenze, costanti di accoppiamento iperfine, fattore g)Spettro NMR (chemical shift, costanti di accoppiamento)
Come varia l'energia in risposta ad una perturbazioneProprietà

Spettri vibrazionali
Stretching asimmetrico Bending
•Calcolare lo spettro IR•Assegnare le bande ai modi di vibrazione•Caratterizzare una struttura (minimo, TS…)
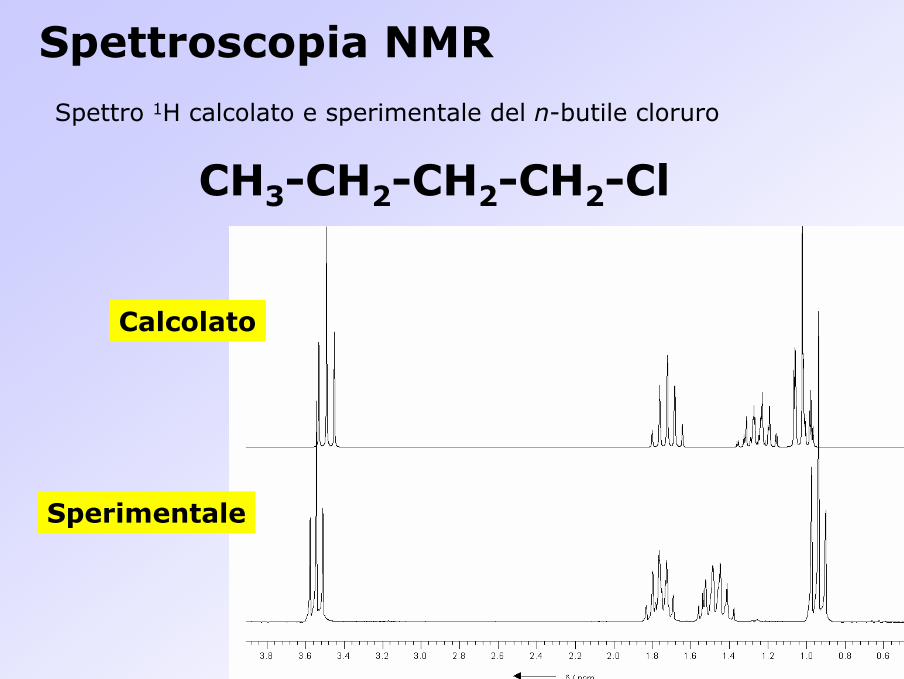
Spettro 1H calcolato e sperimentale del n-butile cloruro
Spettroscopia NMR
CH3-CH2-CH2-CH2-Cl
Calcolato
Sperimentale

N
N
OO
1
2
3
4
17a17b
18a 18b
20a
20b
23a
22
23b
14
15a13
1211b11a
15b
8
16
13C
Calc.
Sper.
303540455055606570758085 ppm
303540455055606570758085 ppm
12 23 16 8
20
7
18
1317 11 14
15
12 2316 8 7 20 1813
1711 14 15
Previsione di spettri NMR: stricnina

Meccanismi di reazioni organiche: la trasposizione di Fries
Estere arilico p- e o-idrossichetone
O
O
R OH
O
R
OH
O
RR' R' R'
BCl3
Lewis acid
Nota da > 100 anniScarse informazioni sul meccanismo!Intramolecolare o intermolecolare?Orto o para?

O
O
HCH3O O
O
HCH3O+ BCl3
BCl3
O
O
HCH3O
BCl3
OBCl2
O
HCH3O
+Cl
OBCl2
O
HCH3O
+Cl
OBCl2CH3O
CHO+ HCl
1 1a
1a 2
2 3
Passaggi principali della trasposizione di Fries sugli aril formiati
1
2
3

O H
O
CH3O O H
O
CH3O
Escludiamo una acilazione FC intramolecolare diretta
Cerchiamo un cammino con scissione del legame ArO-CHO e formazione di formile cloruro, con successiva acilazione FC
Studio meccanicistico tramite calcoli DFT

Passaggio 2: scissione del legame ArO-CHO
O
O
HCH3O
BCl3
OCH3O OBCl2H Cl
OCH3O O
H Cl
Cl2B
+ BCl3
OBCl2CH3OHCOCl BCl3
1a4
OCH3O O
H Cl
BCl2BCl
ClCl
2
TS42a TS42b
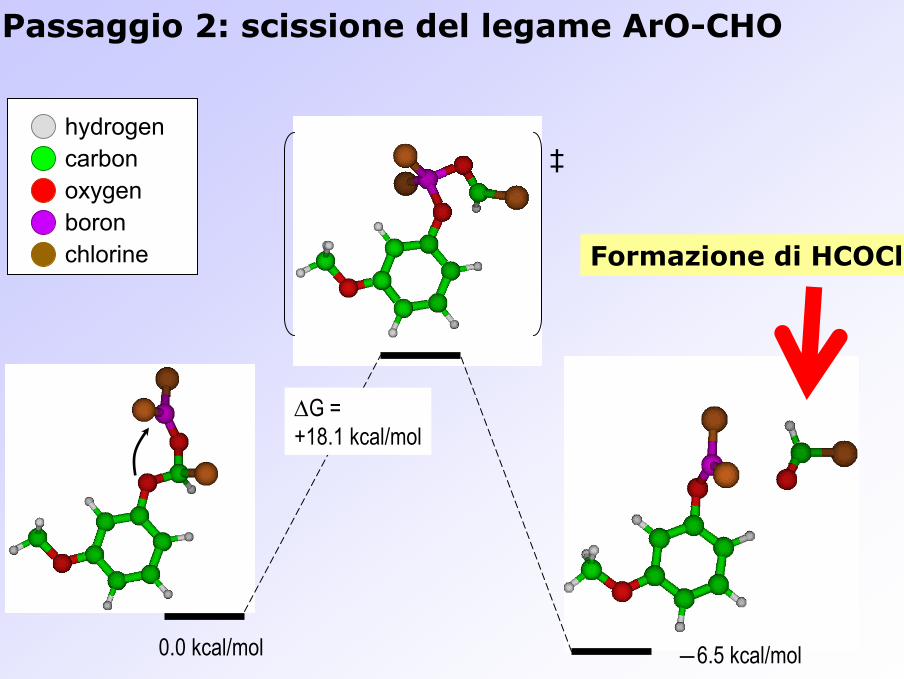
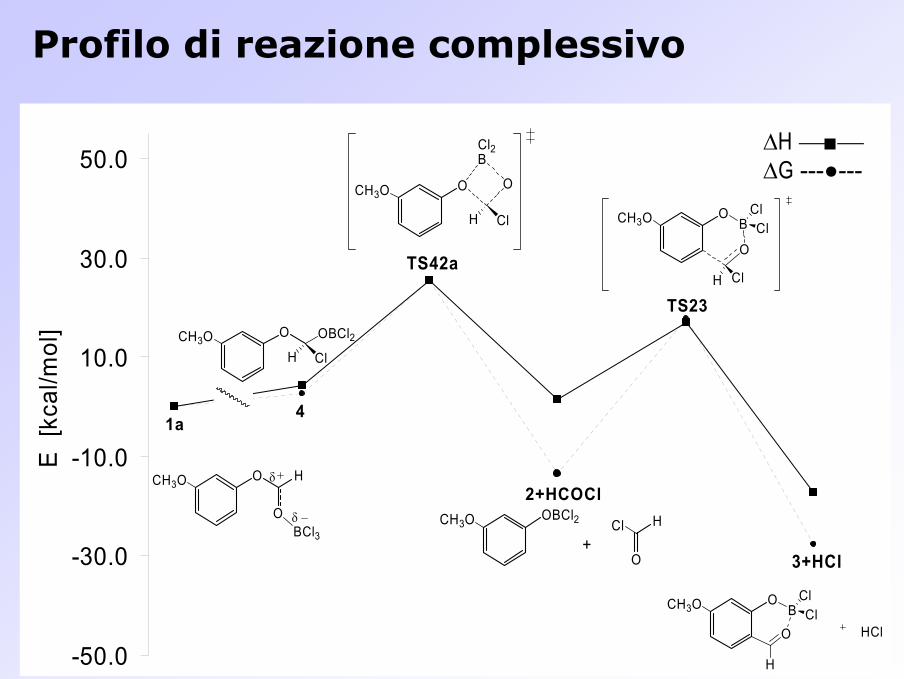
0.0 kcal/mol ―6.5 kcal/mol
∆G = +18.1 kcal/mol
‡hydrogencarbonoxygenboronchlorine
Passaggio 2: scissione del legame ArO-CHO
Formazione di HCOCl
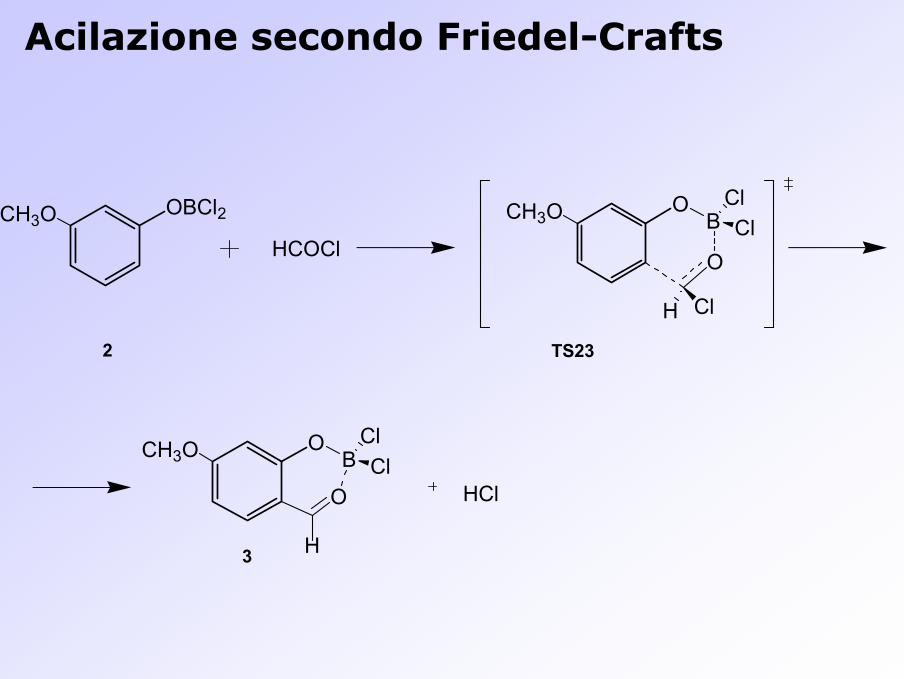
OBCl2CH3OHCOCl
OCH3O B
O
ClCl
H Cl
OCH3O BClCl
HClO
H
2
3
TS23
Acilazione secondo Friedel-Crafts
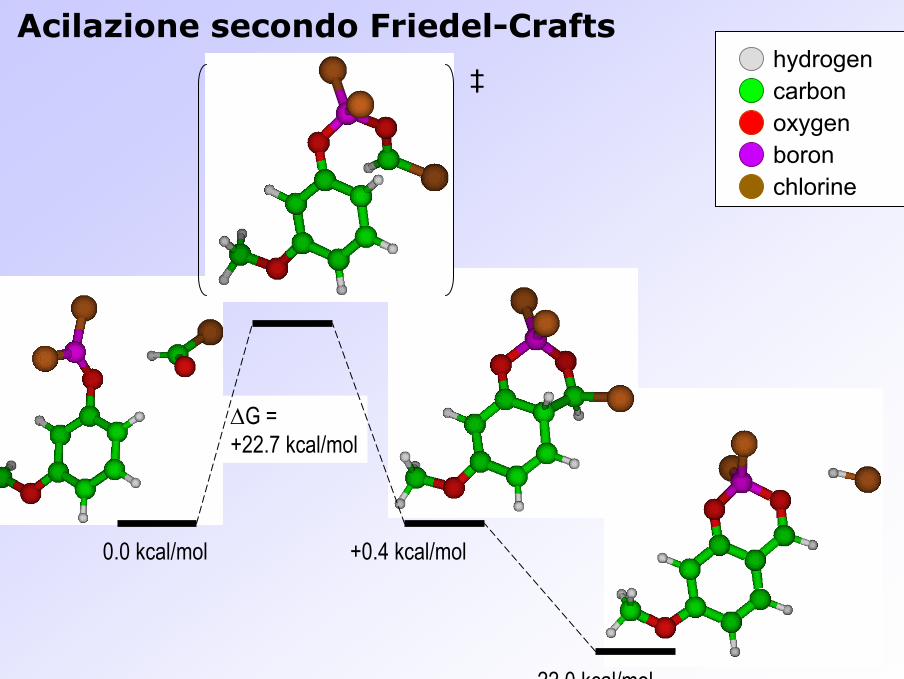
∆G =+22.7 kcal/mol
0.0 kcal/mol +0.4 kcal/mol
―22.0 kcal/mol
‡hydrogencarbonoxygenboronchlorine
Acilazione secondo Friedel-Crafts
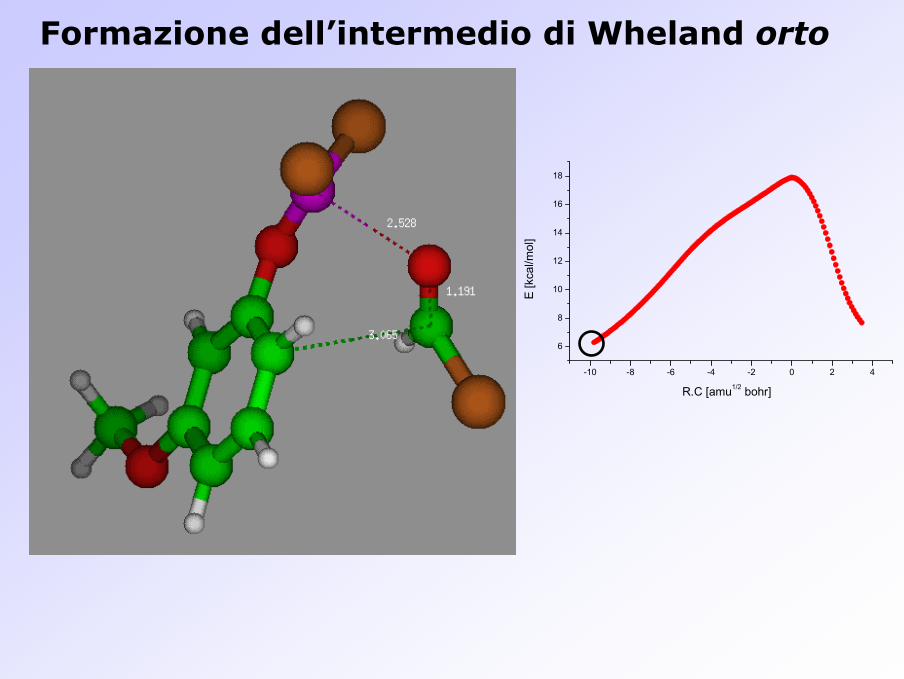
Formazione dell’intermedio di Wheland orto
-10 -8 -6 -4 -2 0 2 4
6
8
10
12
14
16
18
E [k
cal/m
ol]
R.C [amu1/2 bohr]

-10 -8 -6 -4 -2 0 2 4
6
8
10
12
14
16
18
E [k
cal/m
ol]
R.C [amu1/2 bohr]
Formazione dell’intermedio di Wheland orto

-10 -8 -6 -4 -2 0 2 4
6
8
10
12
14
16
18
E [k
cal/m
ol]
R.C [amu1/2 bohr]
Formazione dell’intermedio di Wheland orto

-10 -8 -6 -4 -2 0 2 4
6
8
10
12
14
16
18
E [k
cal/m
ol]
R.C [amu1/2 bohr]
Formazione dell’intermedio di Wheland orto
-50.0
-30.0
-10.0
10.0
30.0
50.0
E [
kcal
/mol
]
O
O
HCH3O
BCl3
δ
δ
1a4
TS42a
2+HCOCl
TS23
3+HCl
OCH3O OBCl2H Cl
OCH3O O
H Cl
Cl2B
OBCl2
O
HCH3O
+Cl
OCH3O B
O
ClCl
H Cl
OCH3O BClCl
HClO
H
∆H ■∆G ---●---
Profilo di reazione complessivo

Sommario
•La chimica computazionale permette di accedere ad un punto di vista “indipendente” su tutti gli aspetti della chimica
•La potenza dei comuni PC è già sufficiente a consentire lo studio di sistemi di reale interesse
•La chimica computazionale è uno strumento ormai “indispensabile”