applied polymer science: 21st century || an overview of transition metal-mediated polymerizations
TRANSCRIPT

1021
AN OVERVIEW OF TRANSITION METAL-MEDIATED POLYMERIZATIONS: CATALYSTS FOR THE 21^^ CENTURY
LISAS.BOFFA
Exxon Research & Engineering Company Route 22 East Annandale, NJ 08801
Introduction Olefin Addition (Coordination-Insertion) Polymerization Olefin / Carbon Monoxide Copolymerization Olefin Metathesis Addition Polymerization of Polar Monomers Step-Growth Addition / Elimination (Coupling) Polymerization Beyond Addition and Condensation: Unique Organometallic Chain-Building Mechanisms
Introduction
The use of transition metal complexes in polymer synthesis is one of the most actively investigated areas in macromolecular science today. Over the last two decades, organometallic initiators have revolutionized strategies for controlled polymer syntheses, dramatically increased the available degree of control of polymer architectures and properties, and expanded the range of conditions under which commercial polymerizations can be carried out. In doing so, they have also greatly enhanced the technological applications of plastics and our fundamental chemical understanding of polymerization mechanisms.
Transition metal-mediated polymerizations may be defined as those having an organometallic active center covalently bound to the growing polymer chain. Depending on the nature of the organometallic fragment, the polymer-metal bond may be formally more covalent or ionic in character, but may be contrasted with anionic and cationic polymerizations in which the counterion is typically a dissociated entity. The useful feature of metal-mediated polymerization is the ability to influence the structure and / or kinetics of chain growth through the attached organometallic species. The metal's non-initiating or spectator ligands provide the steric and electronic means to do so. Organometallic polymerizations may be thought of as a logical refinement of anionic and cationic polymerizations in the sense that very active, "naked" sites have been replaced by more robust and selective propagating centers.

1022 LS.Boffa
Many monomers that are susceptible to unwanted side reactions wlien polymerized ionically may in fact be polymerized in a "living" manner through the use of a transition metal initiator.
Organometallic initiators are also a refinement of traditional heterogeneous catalysts, which tend to give polymers with broad property distributions due to the many types of active sites on their surfaces. Since it is impossible to know the exact roles of all sites, heterogeneous catalysts are very difficult to understand and modify in a rational manner. In contrast, discrete transition metal initiators have only a single site at which polymerization takes place. This not only gives a more uniform polymer product, but also allows the relationships between catalyst structure and polymer properties to be understood and manipulated for improvement.
Organometallic polymerizations have a number of advantages over traditional radical and ionic techniques but are not without challenges. Transition metal initiators can be quite complex, requiring several synthetic steps, and many catalysts need expensive cocatalysts or activators. As a result of their structural fine tuning, organometallic initiators that function well for certain monomers may be completely inactive for others. Additionally, a tradeoff often exists between catalyst selectivity and activity, in that making a catalyst more well-behaved may also mean making it slower (however, recent achievements in olefin polymerization have demonstrated that this obstacle is not insurmountable).
Transition metal complexes that carry out addition polymerization act as initiators, inserting a monomer unit into a metal-organic bond to begin propagation. However, most of them also activate the monomer unit first through precoordination, and in this sense also act as catalysts. Due to the fascinating variety of organometallic reaction mechanisms, transition metal catalysts can cany out a number of other chain-building mechanisms besides addition polymerization. Metal-mediated organic couplings based on two-electron addition-elimination steps provide an organometallic analogue to condensation polymerization, and there are also a number of recently discovered metal-mediated polymerizations that have no analogues in classical ionic-based polymerization. These systems perhaps best illustrate the possibilities that make organometallic polymerization initiators worthy of the phrase "catalysts for the 21st century."
Olefin Addition (Coordination-Insertion) Polvmerization
Metallocenes By far, the most significant example of transition metal-catalyzed polymerization is the use of metallocenes for polyolefin synthesis (1). Polyethylene and polypropylene are the world's largest volume thermoplastics, and taken together comprise an over 40 billion-dollar-a-year industry (2). At this time the fraction of polyolefins produced commercially by metallocenes is still very small (< 10%) conrlpared to Ziegler-Natta and radical-based processes (3); however, this number is certain to increase and greatly underrepresents the role metallocenes have played in advancing the understanding and development of olefin polymerization.
Metallocenes are soluble, single-site analogues of the traditional Ziegler-Natta initiators (TiCU / AIR3 / support) used to produce high-density and low-density polyethylene, isotactic polypropylene, and EPDM rubbers. The active polymerization
Over\/ie\N of Transition Metai-Mediated Polymerzations 1023
sites in Ziegler-Natta systems are heterogeneous, numerous in character, and generated In situ, and are thus very difficult to manipulate. Metallocene initiators arose from the early study of soluble Ziegler initiators comprising titanocene or zirconocene dichlorides (CpaTiCIa, or Cp2ZrCl2; Cp = C5H5) with a trialkylaluminum activator, which converts the Group IV complex to a highly active cation upon addition. It was later found that if methylaluminoxane ("MAO," a complex oligomer with the general structure [-AI(CH3)0-]) is used instead of a simple aluminum alkyl, the resultant polymerization systems exhibit amazingly high rates and activities (4). In fact, examples of metallocenes producing yields in excess of one ton polyethylene / g catalyst*hour and total efficiencies of 25 tons polyethylene / g catalyst have been noted (5). In the last decade, single-component metallocene initiators have been developed which do not require the use of MAO, which is structurally poorly understood. These systems comprise preformed Group IV metallocene cations combined with a discrete, weakly coordinating borane counterion (6). Although metallocenes, boranes, and MAO are all atmospherically sensitive, the enormous activities shown by these systems makes them industrially viable despite the need to exclude water and air from the polymerization process.
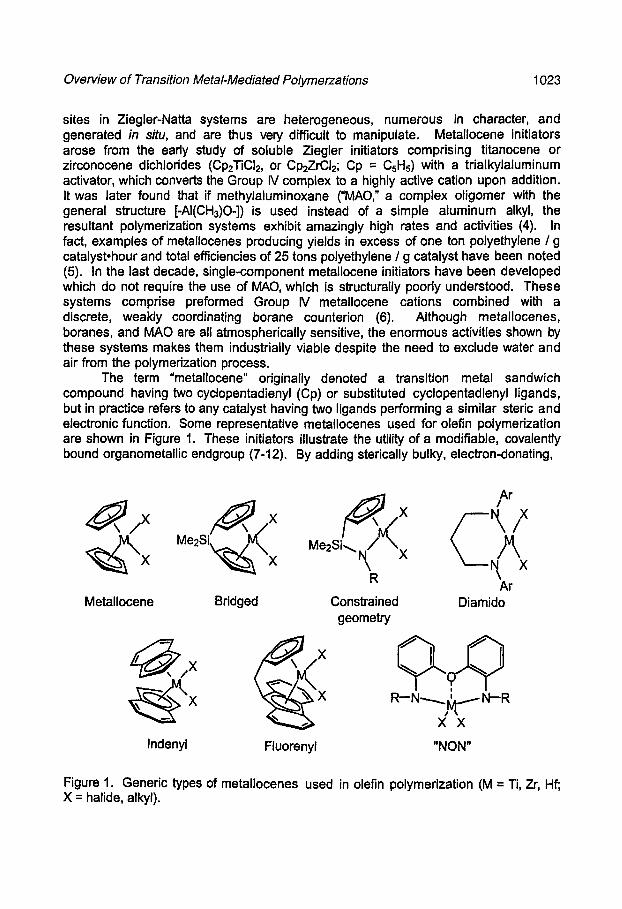
The term "metallocene" originally denoted a transition metal sandwich compound having two cyclopentadienyl (Op) or substituted cyclopentadienyl ligands, but in practice refers to any catalyst having two ligands performing a similar steric and electronic function. Some representative metallocenes used for olefin polymerization are shown in Figure 1. These initiators illustrate the utility of a modifiable, covalently bound organometallic endgroup (7-12). By adding sterically bulky, electron-donating,
\ /
Metallocene
Me2Si X
Indenyl
Bridged Constrained geometry
Fluorenyl
R-N
Ar Diamido
NHR
"NON"
Figure 1. Generic types of metallocenes used in olefin polymerization (M = Ti, Zr, Hf; X = halide, alkyl).

1024 LS.Boffa
or electron-withdrawing groups to the Cp groups (or Cp equivalents), rates of polymerization, catalyst activities, molecular weights, comonomer incorporation rates, and branching in the resultant polyolefins may be controlled to give monodisperse polymers or polymers with molecular weight distributions optimized for processing. Similarly, if the geometry of the catalyst is constrained by linking the Cp equivalents, catalysts with chiral or prochiral active sites for monomer insertion may be prepared that result in tactic polyolefins.
The active species in olefin polymerization is a cationic metallocene alkyl, generated by reaction of a neutral metallocene diaikyi with a borane Lewis acid, or similarly from a metallocene dichloride and a large excess of MAO, which also acts as an alkylating agent and scavenger for protic impurities (Eq. 1,2). As shown for
Zc y^sN\/\u^ 2r ^ Zr^CHs (1)
+ MAO-Me
V ^ " - B(CeFs)3, ^ \ ^ c H 3 (2)
CH3 ^ ^ < e + Me-B{C6F5)3
ethylene, initiation and propagation proceed through precoordination and insertion of the olefin into the alkyl group / polymer chain (Eq. 3). Chain transfer through p-hydride elimination produces metal hydrides and long-chain a-olefins (9). By modifying the
\® ru C2H4. <^®y^^^ C2H4 Zi—CH3 *- Zc V
^ ^ < / CH3
(3)
^ ^ HH

Overv\e\N of Transition Metai-Mediated Polymerzations 1025
structure of the catalyst, these a-olefin macromers can be excluded from the polymerization process (giving linear high-density polyethylene, HOPE) or reincorporated as comonomers to give long-chain-branched polyethylene. Alternately, a variety of comonomers including propylene, 1-hexene, 1-octene, and 1,5-hexadiene can be directly added to the ethylene feed to produce branched (linear-low density, LLDPE) or cyclic polyolefins with a wide range of properties. Homopolymerization of cycloolefins such as norbornene and cyclopentene with metallocenes is also possible and produces polymers with extremely high melting points (above 380 °C) (13). These polymers are not amenable to processing, although copolymers of cycloolefins with ethylene show improved properties such as transparency, chemical resistance, and high service temperatures, and are used in optoelectronic applications (1).
Commodity metallocene polyethylene is currently being produced in the U.S. for film applications, originally by Exxon in 1991 (Exact HOPE and Plastomer plasticized polyethylene, made with the Exxpol process) and subsequently by Dow (Affinity and Engage LLDPE, made through Insite), Phillips, Mobil, and a large number of licenses, alliances, and joint ventures (e.g., Exxon / Union Carbide Univation) (14). Exxon and Fina are also selling polypropylene for fiber applications. A number of specialty products, not available through traditional catalysts, are in development. These include Hoechst / Mitsui's Topas ethylene / norbornene copolymers (3), syndiotactic polystyrene from Dow / Idemitsu (15), and syndiotactic polypropylene from Fina (16).
Late Transition Metal Initiators A major drawback of Group IV metallocene catalysts is that, despite their extremely high activities for olefins, most are very air- and moisture-sensitive. They are also highly intolerant to heteroatom-containing monomers, whose lone pairs preferentially coordinate to the Lewis acidic metal center in place of the olefin monomer. Electron-rich late transition metal organometallics are more tolerant in these regards, but traditional thinking has held that these complexes are too limited in activity to compete with metallocenes. However, some breakthroughs in the last several years have shown that with proper spectator ligands to control sterics at the active site, middle- and late-transition metal complexes are quite useful for the polymerization of olefins and are promising catalysts for the potential copolymerization of olefins and polar monomers (10).
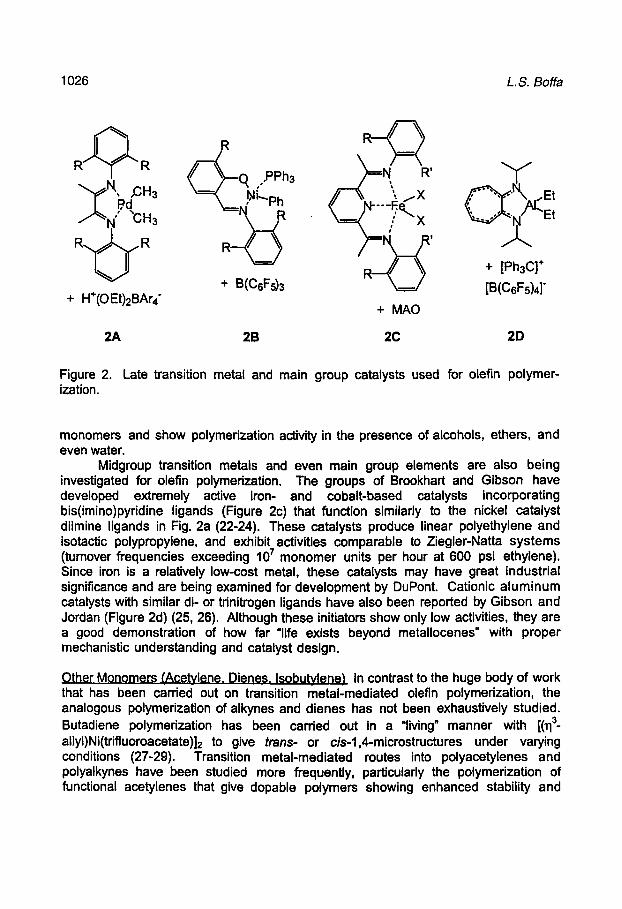
In 1995, Brookhart and coworkers developed a class of cationic Pd(ll) and Ni(ll) initiators incorporating a-diimine spectator ligands with N-aryl groups (Figure 2a) (17, 18). The 2,6-aryl substituents on the diimine ligands are crucial for controlling the steric hindrance above and below the active sites of the square planar complexes and allow for the precise tailoring of branching, activity, and molecular weight. The polyethylene produced by some types of these initiators is unique in that it is much more highly branched even than LDPE. These initiators are also unusual in that they are able to copolymerize small amounts (< 12 mol %) of acrylic monomers, which are selectively incorporated at the ends of the branches (19). Very recently, Grubbs and coworkers reported neutral salicylaldiminato nickel(ll) complexes that polymerize olefins (Figure 2b) (20, 21). These single-component initiators require only the presence of a phosphine sponge for activation, and as a consequence of their neutral and therefore more tolerant character, can incorporate functionalized norbornene
1026 LS. Boffa
+ H*{OEt)2BAr4-
2A
B(C6F5)3
2B
T A
+ [PhaC]*
[B(C6F5)4r
2D
Figure 2. Late transition metal and main group catalysts used for olefin polymerization.
monomers and show polymerization activity in the presence of alcohols, ethers, and even water.
Midgroup transition metals and even main group elements are also being investigated for olefin polymerization. The groups of Brookhart and Gibson have developed extremely active iron- and cobalt-based catalysts incorporating bis(imino)pyridine ligands (Figure 2c) that function similarly to the nickel catalyst diimine ligands in Fig. 2a (22-24). These catalysts produce linear polyethylene and isotactic polypropylene, and exhibit activities comparable to Ziegler-Natta systems (tumover frequencies exceeding 10^ monomer units per hour at 600 psi ethylene). Since iron is a relatively low-cost metal, these catalysts may have great industrial significance and are being examined for development by DuPont. Cationic aluminum catalysts with similar di- or trinitrogen ligands have also been reported by Gibson and Jordan (Figure 2d) (25, 26). Although these initiators show only low activities, they are a good demonstration of how far "life exists beyond metallocenes" with proper mechanistic understanding and catalyst design.
Other Monomers (Acetvlene. Dienes. Isobutylene) In contrast to the huge body of work that has been earned out on transition metal-mediated olefin polymerization, the analogous polymerization of alkynes and dienes has not been exhaustively studied. Butadiene polymerization has been carried out in a living" manner with [(TJ -allyl)Ni(trifluoroacetate)]2 to give trans- or c/s-1,4-microstructures under varying conditions (27-29). Transition metal-mediated routes into polyacetylenes and polyalkynes have been studied more frequently, particularly the polymerization of functional acetylenes that give dopable polymers showing enhanced stability and

Oyerv'\e\N of Transition l\/letai'l\/lediated Polymerzations 1027
solubility. Most of the organometallic initiators used are similar to the classical initiators used for ring-opening metathesis polymerization (ROMP) and comprise midgroup or late-transition metal chlorides, carbonyls, or alkoxides which form metal carbenes in situ when combined with an alkylating cocatalyst (30-34). These carbenes are the true active species of the polymerization; it is not clear whether polymerization takes place through an addition or metathesis mechanism.
Very recently, organometallics have been studied as initiators for the pseudocarbocationic polymerization of isobutylene. Cationic metallocenes ([Cp'zMMe]*; M = Zr, Hf) (35, 36) and aluminocenes ([Cp'2AI]*) (Cp' = C5H5, CsMes) (37, 38) have been used to prepare both high molecular weight homopoly(isobutylene) and isobutylene / isoprene copolymers. These systems utilize weakly coordinating borane counterions that play the same advantageous role as in metallocene polymerizations.
Olefin / Carbon Monoxide Copplymerization
The greater polar monomer tolerance of late transition metals makes it possible to copolymerize olefins with carbon monoxide, giving aliphatic polyketones. The copolymerization of ethylene and CO by radical means at elevated temperatures and pressures has been known for several decades, but the polymers formed have a random structure containing polyethylene sequences. However, perfectly alternating olefin / CO copolymers may be obtained through the use of organometallic nickel-and palladium-based catalysts (39-42). The alternating polyketone copolymers have a number of unusual properties due to their regular and functional structure, including very high crystallinities, melting points {Tu - 260 'C for ethylene-CO), high tensile strengths, photodegradabilty, and amenability to modification through the keto group. In addition, the low cost of ethylene and carbon monoxide makes these polymers commercially attractive, particularly since the palladium catalysts are not atmospherically sensitive.
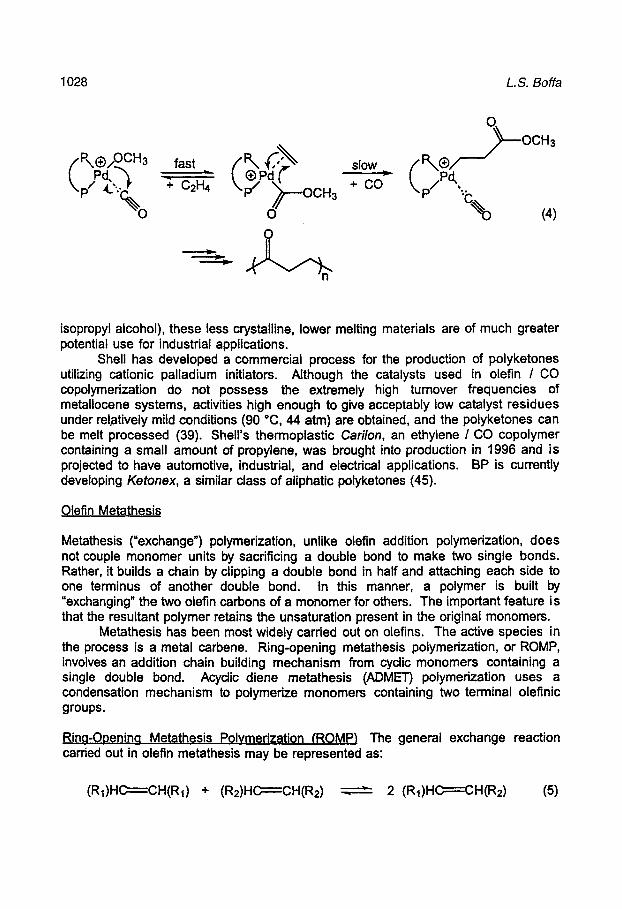
Alternating olefin / CO copolymerization is optimally carried out with dicationic palladium(ll) complexes incorporating a chelating dinitrogen or diphosphine ligand in the presence of methanol. These complexes can typically be prepared in situ from palladium(ll) acetate, free ligand, and the Bronsted acid of a weakly coordinating anion. Methanol plays a key role in the initiation step of polymerization, assisting in the formation of monocationic palladium hydride and alkoxide initiating species. Polymerization (Eq. 4) involves the rapid and reversible insertion of coordinated CO into a palladium alkyl (or initial palladium alkoxide) bond to give a palladium acyl; carbon monoxide is selected over the olefin in this step due to its higher coordinating ability. Ethylene is then irreversibly inserted into the acyl. A subsequent second insertion of CO into the growing polymer chain does not occur for thermodynamic reasons (39).
Substituted olefins can also be copolymerized with carbon monoxide to give alternating terpolymers. Propylene, styrene, allenes (43), functional olefins (44), and diolefins (giving cyclocopolymers) all may be incorporated with varying regio- and stereoregularity, depending on the nature of the ligands used (39-42). Since straight poly(ethylene / CO) is an extremely insoluble material (soluble only in hexafluoro-
1028 LS.Boffa
.p ^ - . ^ ^ C2H4 \ p / K 0 C H 3
^ o a ^ (4)
0CH3
isopropyl alcohol), these less crystalline, lower melting materials are of much greater potential use for industrial applications.
Shell has developed a commercial process for the production of polyketones utilizing cationic palladium initiators. Although the catalysts used in olefin / CO copolymerization do not possess the extremely high turnover frequencies of metallocene systems, activities high enough to give acceptably low catalyst residues under relatively mild conditions (90 **C, 44 atm) are obtained, and the polyketones can be melt processed (39). Shell's themrioplastic Carilon, an ethylene / CO copolymer containing a small amount of propylene, was brought into production in 1996 and is projected to have automotive, industrial, and electrical applications. BP is cun-ently developing Ketonex, a similar class of aliphatic polyketones (45).
Olefin Metathesis
Metathesis ("exchange") polymerization, unlike olefin addition polymerization, does not couple monomer units by sacrificing a double bond to make two single bonds. Rather, it builds a chain by clipping a double bond in half and attaching each side to one terminus of another double bond. In this manner, a polymer is built by "exchanging" the two olefin carbons of a monomer for others. The important feature is that the resultant polymer retains the unsaturation present in the original monomers.
Metathesis has been most widely carried out on olefins. The active species in the process is a metal carbene. Ring-opening metathesis polymerization, or ROMP, involves an addition chain building mechanism from cyclic monomers containing a single double bond. Acyclic diene metathesis (ADMET) polymerization uses a condensation mechanism to polymerize monomers containing two terminal olefinic groups.
Rina-Openino Metathesis Polvmerization (ROMP) The general exchange reaction earned out in olefin metathesis may be represented as:
(R^)HC=CH(Ri) + (R2)HC=CH(R2) ^ = ^ 2 (Ri)HC=CH(R2) (5)

Overview of Transition Metal-Mediated Polymerzatlons 1029
If a cyclic olefin is substituted for the generic olefin and each side of the double bond is thought of as an Ri or R2 moiety, it is apparent that a polymeric chain can be built (46, 47):
= ... (6) ..jDjD^a,._...j\. Ri R2 Ri R2 Ri R2 Ri R2 Ri R2 Ri R2
Since metathesis is an equilibrium process, the regiochemistry of the double bonds formed is ultimately subject to thermodynamic control. Polymers with varying amounts of cis and trans linkages are formed depending on catalyst and monomer structure. The driving force for polymerization is the release of monomer ring strain; therefore cycloalkenes such as norbornene and cyclobutene are better ROMP substrates than less-strained monomers such as cyclohexene.
ROMP was originally noted as a side reaction to olefin addition polymerization during the early years of Ziegler-Natta catalysis. Modern ROMP initiators are typically tungsten, molybdenum, or ruthenium carbenes bearing spectator ligands specifically tailored to provide "living" steric and electronic active site control. The most important of these are the Grubbs (48) and Schrock (49) initiators. Catalysts may also be prepared In situ by combining metal chlorides (particularly tungsten) with aluminum alkyls to give metal alkyls that undergo elimination to carbenes (50). Although ROMP polymerizations in aqueous media have been achieved (51-53), most catalysts and processes are highly air- and water-sensitive. Recently, a number of functional group-tolerant polymerizations have been reported (54, 55).
,Ph FfR3
RO tSu PR3
h
Ph
M = W, Mo "Grubbs" "Schrock"
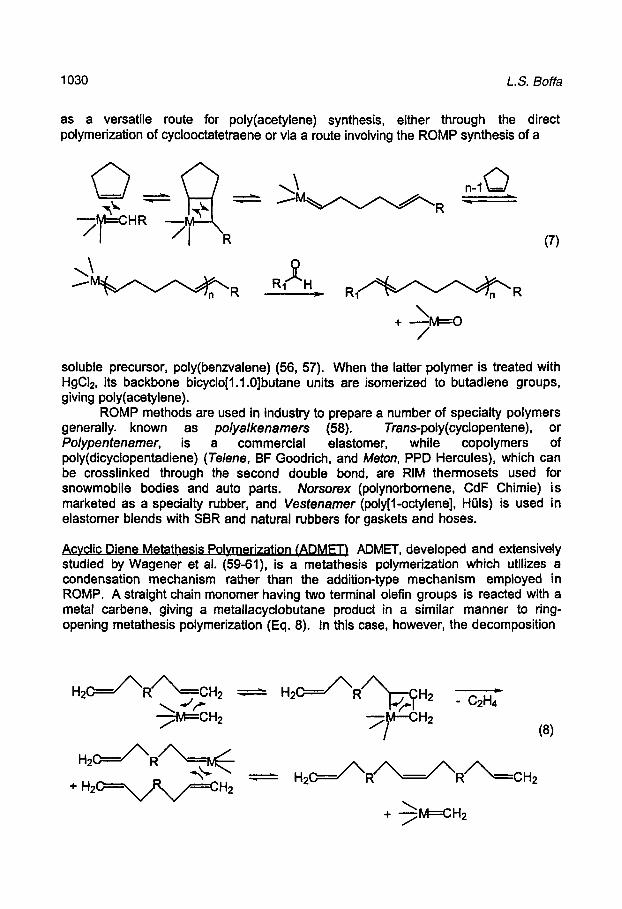
The ROMP mechanism involves metallacyclobutane formation via a [2+2] cycloaddition reaction. The metallacyclobutane then decomposes through a retro [2+2] mechanism where bond cleavage occurs in the opposite direction from formation. The polymerization may be terminated upon completion with an aldehyde to give a metal 0x0 complex (eq. 7).
Since unsaturation is preserved when olefins are polymerized through ROMP, it is a useful technique for producing conjugated polymers. ROMP particularly serves
1030 LS. Boffa
as a versatile route for poly(acetylene) synthesis, either through the direct polymerization of cyclooctatetraene or via a route involving the ROMP synthesis of a
O^O-^i --f^/l=CHR /T
.,o (7)
soluble precursor, poly(benzvalene) (56, 57). When the latter polymer is treated with HgCl2, its backbone bicyclo[1.1.0]butane units are isomerized to butadiene groups, giving poly(acetylene).
ROMP methods are used in industry to prepare a number of specialty polymers generally, known as polyalkenamers (58). Trans-poly(cydopentene), or Polypentenamer, is a commercial elastomer, while copolymers of poly(dicyclopentadiene) {Telene, BF Goodrich, and Meton, PPD Hercules), which can be crosslinked through the second double bond, are RIM thermosets used for snowmobile bodies and auto parts. Norsorex (polynorbomene, CdF Chimie) is marketed as a specialty rubber, and Vestenamer (poly[1-octylene], Huls) is used in elastomer blends with SBR and natural rubbers for gaskets and hoses.
Acvclic Diene Metathesis Polvmerization (AOMFH ADMET, developed and extensively studied by Wagener et al. (59-61), is a metathesis polymerization which utilizes a condensation mechanism rather than the addition-type mechanism employed in ROMP. A straight chain monomer having two terminal olefin groups is reacted with a metal carbene, giving a metallacyclobutane product in a similar manner to ring-opening metathesis polymerization (Eq. 8). In this case, however, the decomposition
H 2 0 = ^ R ' ^ = C H 2 =
^ M = C H 2
H 2 0 = / ^ R ^ \ = | ^
+ H 2 0 = \ J\ / = C H 2
—M—CH2
= ^ H 2 0 = = ^ F
C2H4
(8)
+ -3l^/t=CH2

Ovemew of Transition Metai-Mediated Polymerzations 1031
of the metallacycle produces ethylene in addition to a new carbene. A second monomer unit reacts with the product carbene and ethylene is again released to form a dimer. As the condensation process continues, removal of ethylene at reduced pressure pushes the ADMET system to high conversions and high polymer.
Equation 8 shows ADMET polymerization of a symmetrical monomer; however, asymmetrically substituted monomers can also be used. In this case, condensation may occur to give different regiochemistries depending on which "ends" of the monomers are joined together. As expected from an equilibrium polymerization, ADMET produces polymers with a high content of thermodynamically favored trans double bonds, typically 80:20 trans:cis (59).
ADMET polymerization may be carried out using the preformed Schrock and Grubbs catalysts, as well as classical tungsten-based, tin-activated initiators (62). With the proper catalyst choice it is tolerant to the presence of functionality at the monomer backbone center, as long as at least two CH2 units are present between the olefin and central functionality (63). Monomers incorporating ether, alcohol, sulfur, silane, and stannane groups have been polymerized (64-66). When the backbone double bonds of the resultant polymers are quantitatively hydrogenated, unusual functional polyethylenes are obtained. Similarly, ADMET has also been used to prepare a number of interesting segmented copolymers from a,Q)-olefin-substituted macromonomers (67, 68).
Another interesting use of ADMET stems from the ability to control the spacing between the incorporated R groups through the number of methylene units in the monomer. By choosing monomers with a variety of spacer lengths and a single methyl substituent on the central carbon, polymers with precisely spaced sidechains are formed (Eq. 9). These polymers can be hydrogenated to give polyethylenes with methyl branches on every nth carbon. Such "perfectly imperfect" polyethylenes serve as useful models for understanding the relationships between branching and thermal properties (69-72). Similar strategies using alcohol- and acetate-substituted monomers have provided property models for industrially important ethylene-vinyl alcohol and -vinyl acetate copolymers (69, 73).
"*=V-Hr^"^ ^ Ms. 4=.^/-^^^ n n
H2
Branch on every (2n+3) carbon
n ' 'n
Branch on every... TM 11th 11 "C 15th 39 "C 19th 57 'C 21st 62 "C Linear 134 "C
(9)

1032 LS. Boffa
Metathesis of Alkvnes Well-defined ROMP and ADMET have also been carried out on alkynes, although metal carbyne catalysts for well-controlled polymerizations are not as highly developed as those for alkene polymerization (74-76). Recently, ADIMET (acyclic diyne metathesis) has been used as an altemative to Pd-catalyzed couplings for the synthesis of high molecular weight poly(p-phenyleneethynylenes) (74, 77).
Addition Polvmerization of Polar Monomers
Group Transfer Polvmerization (GTP^ of (Meth^acrvlates Traditional group transfer polymerization (GTP) is a technique for the living" polymerization of (meth)acrylates involving a silyl ketene acetal initiator (producing a silyl enolate active species) and a carbonyl-activating catalyst (typically a Lewis acid) as an independent second component (78, 79). Until recently it was believed that GTP was similar to the other organometallic coordination-addition polymerizations discussed here, in that the silyl endgroup remained firmly bound to the growing polymer chain during propagation. However, recent work has shown that it may be better described as an anionic polymerization stabilized by a reversibly bound silyl group (80, 81). However, when early transition metal complexes are used as initiators, living" group transfer polymerizations of acrylic monomers are obtained which in fact do involve robust organometallic active sites, allowing for the control of polymer microstructure. Elegantly, the Lewis acid character of the organometallic initiators allows them to simultaneously serve as the second catalyst component of the GTP system.
Collins has developed a transition metal GTP technique for the polymerization of methyl methacrylate (MMA) which utilizes a neutral zirconocene enolate as an initiator and the conjugate zirconocene cation as a catalyst (82, 83). Each monomer addition step interconverts the two organometallic components (Eq. 10). The poly(methyl methacrylate) (PMMA) obtained is predominantly syndiotactic, although isotactic PMMA has been obtained by using chiral indenyl zirconocenes in combination with non-zirconocene Lewis acids (84).
OMe OMe 0
Cp2MeZr>v-^^x^^^ ^5^^;^;XV^.ZrCp2Me ^
(MMA)n.i ' ^ ' (10)
OMe
(MMA)'n-, ^Me Q
^^.ZrCpaMe
Oyerv\e\N of Transition Metal-Mediated Polymerzations 1033
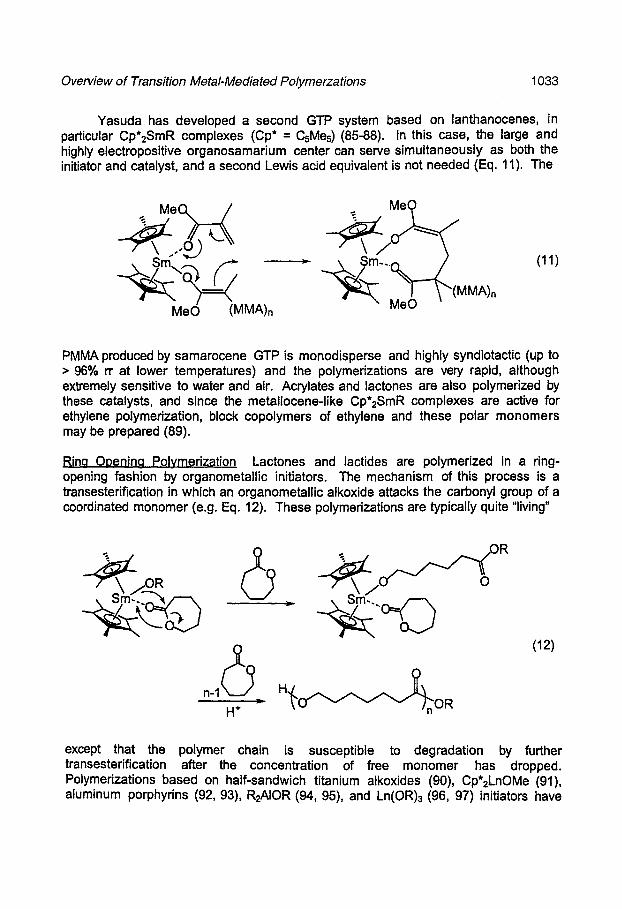
Yasuda has developed a second GTP system based on lanthanocenes, in particular Cp*2SmR complexes (Cp* = CsMes) (85-88). In this case, the large and highly electropositive organosamarium center can serve simultaneously as both the initiator and catalyst, and a second Lewis acid equivalent is not needed (Eq. 11). The
MeC
SnCX ^ \ Sm-n / (H)
{MMA)n
MeO (MMA)n ^^^
PMMA produced by samarocene GTP is monodisperse and highly syndiotactic (up to > 96% n at lower temperatures) and the polymerizations are very rapid, although extremely sensitive to water and air. Acrylates and lactones are also polymerized by these catalysts, and since the metallocene-like Cp*2SmR complexes are active for ethylene polymerization, block copolymers of ethylene and these polar monomers may be prepared (89).
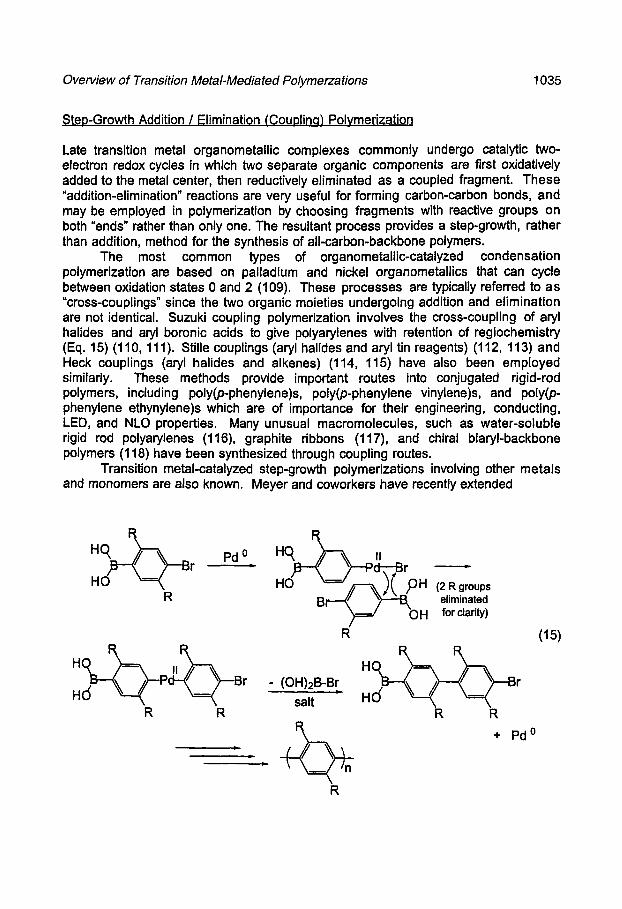
Ring Openino Polymerization Lactones and lactides are polymerized in a ring-opening fashion by organometallic initiators. The mechanism of this process is a transesterification in which an organometallic alkoxide attacks the carbonyl group of a coordinated monomer (e.g. Eq. 12). These polymerizations are typically quite "living"
(12)
except that the polymer chain is susceptible to degradation by further transesterification after the concentration of free monomer has dropped. Polymerizations based on half-sandwich titanium alkoxides (90), Cp*2LnOMe (91), aluminum porphyrins (92, 93), R2AIOR (94, 95), and Ln(0R)3 (96, 97) initiators have

1034 LS.Boffa
been reported (98). Cyclic carbonate homopolymerizations and copolymerizations are also possible (99).
Oxiranes are also polymerized in a ring-opening manner, most spectacularly by aluminum complexes with porphyrin or Schiff base ligands as initiators in conjunction with a second aluminum compound to act as a monomer-activating Lewis acid (92, 100). The steric protection provided by the bulky tetradentate ligands is so great that the polymerizations (termed "immortal" rather than "living") may be candied out in the presence of protic compounds such as water and even HCI. Aluminum porphyrin initiators also catalyze the alternating copolymerization of oxiranes with carbon dioxide (98,101,102).
Heterocumulenes Heterocumulenes and heterounsaturated monomers may be polymerized by mechanisms analogous to olefin addition polymerization. Novak and coworkers have developed a CpTiCl20R-based system for the "living" room temperature polymerization of isocyanates (Eq. 13) (103, 104) remarkable since well-
^ or O ° « . ^^Jf^ or ^^Jf^ (13)
N = C = N R R
controlled anionic polymerizations are possible only at -78 **C (105). This example illustrates the mitigative powers of organometallic polymerization catalysts nicely: in the anionic process, the extremely reactive "naked" amidinate chain end backbites into the polymer backbone at a carbonyl group to form cyclic trimers, while the less reactive titanium amidate present in the organometallic process is sterically and electronically prevented from doing so. Carbodiimides are also polymerized by CpTiCl20R-type initiators to produce polyguanidines (Eq. 13) (106); both types of poly(heterocumulenes) exhibit interesting helical chain conformations and rigid rod behavior.
Isocyanides, which cannot be polymerized anionically, may be polymerized in a "living" manner with allyl nickel carboxylate initiators using a system also developed by Novak et al. (Eq. 14) (29. 107). The nickel initiators are active for butadiene polymerization as well, allowing for the synthesis of polyisocyanide-polybutadiene elastomers. Polyisocyanides also exhibit helical behavior when bulky sidechains are incorporated (108).
R—NSC '^^"^NA II (14)
OvervleyN of Transition Metai-Mediated Polymerzations 1035
Step-Growth Addition / Elimination (Coupling) Polymerization
Late transition metal organometallic complexes commonly undergo catalytic two-electron redox cycles in which two separate organic components are first oxidatively added to the metal center, then reductively eliminated as a coupled fragment. These "addition-elimination" reactions are very useful for forming carbon-carbon bonds, and may be employed in polymerization by choosing fragments with reactive groups on both "ends" rather than only one. The resultant process provides a step-growth, rather than addition, method for the synthesis of all-carbon-backbone polymers.
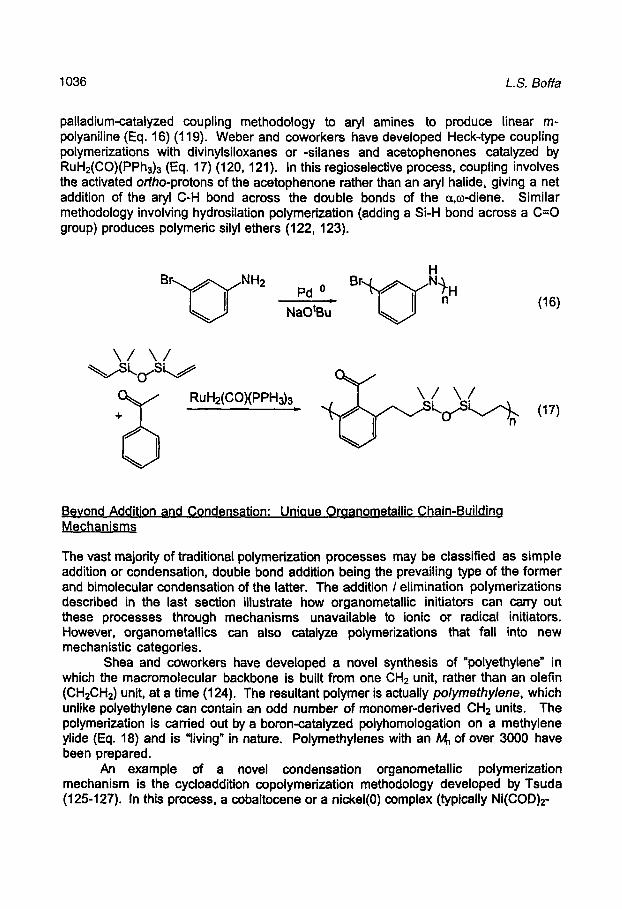
The most common types of organometallic-catalyzed condensation polymerization are based on palladium and nickel organometallics that can cycle between oxidation states 0 and 2 (109). These processes are typically referred to as "cross-couplings" since the two organic moieties undergoing addition and elimination are not identical. Suzuki coupling polymerization involves the cross-coupling of aryl halides and aryl boronic acids to give polyarylenes with retention of regiochemistry (Eq. 15) (110,111). Stille couplings (aryl halides and aryl tin reagents) (112, 113) and Heck couplings (aryl halides and alkenes) (114, 115) have also been employed similarly. These methods provide important routes into conjugated rigid-rod polymers, including poly(p-phenylene)s, poly(p-phenylene vinylene)s, and poly(p-phenylene ethynylene)s which are of importance for their engineering, conducting, LED, and NLO properties. Many unusual macromolecules, such as water-soluble rigid rod polyarylenes (116), graphite ribbons (117), and chiral biaryl-backbone polymers (118) have been synthesized through coupling routes.
Transition metal-catalyzed step-growth polymerizations involving other metals and monomers are also known. Meyer and coworkers have recently extended
HO \ = ( R
R R
HO ^ = / , ^ ^ J ^ pH (2R groups eliminated
O H for clarity)
- (0H)2B-Br
salt
1036 LS.Boffa
palladium-catalyzed coupling methodology to aryl amines to produce linear m-polyaniline (Eq. 16) (119). Weber and coworkers have developed Heck-type coupling polymerizations with divinylsiloxanes or -silanes and acetophenones catalyzed by RuH2(CO)(PPh3)3 (Eq. 17) (120,121). In this regioselective process, coupling involves the activated ortho-protons of the acetophenone rather than an aryl halide, giving a net addition of the aryl C-H bond across the double bonds of the a,(D-diene. Similar methodology involving hydrosilation polymerization (adding a Si-H bond across a C=0 group) produces polymeric silyl ethers (122, 123).
^ J l NaO Bu ^ J l
\ / \ /
RUH.(C0XPPH3), ., ] _ X ^ O i . (17,
(16)
Beyond Addition and Condensation: Unique Organometallic Chain-Buildino Mechanisms
The vast majority of traditional polymerization processes may be classified as simple addition or condensation, double bond addition being the prevailing type of the former and bimolecular condensation of the latter. The addition / elimination polymerizations described in the last section illustrate how organometallic initiators can cany out these processes through mechanisms unavailable to ionic or radical initiators. However, organometallics can also catalyze polymerizations that fall into new mechanistic categories.
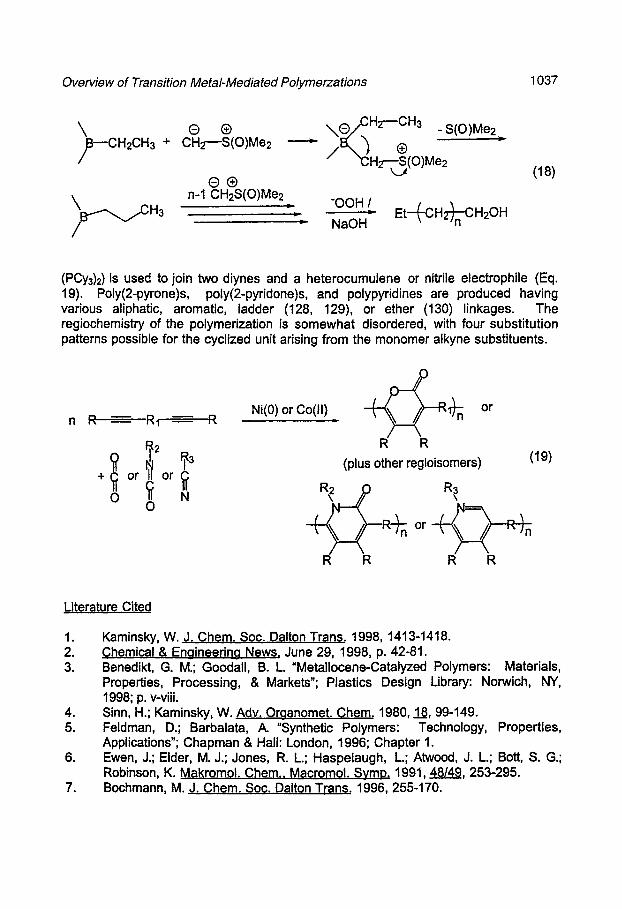
Shea and coworkers have developed a novel synthesis of "polyethylene" in which the macromolecular backbone is built from one CH2 unit, rather than an olefin (CH2CH2) unit, at a time (124). The resultant polymer is actually poly methylene, which unlike polyethylene can contain an odd number of monomer-derived CH2 units. The polymerization is carried out by a boron-catalyzed polyhomologation on a methylene ylide (Eq. 18) and is living" in nature. Polymethylenes with an H of over 3000 have been prepared.
An example of a novel condensation organometallic polymerization mechanism is the cycloaddition copolymerization methodology developed by Tsuda (125-127). In this process, a cobaltocene or a nickel(O) complex (typically Ni(C0D)2-
Overview of Transition Metal-Mediated Polymerzations 1037
\ . . . . _© © _ , . . \Q^^^^^^ -S(0)Me2^ ©
-S(0)Me2 KJ ' (18)
B—CH2CH3 + CH?—S{0)Me2 ^ \ \
© © n-1 CH2S(0)Me2
H3 = X . * Et-4cH2f-CH20H
(PCy3)2) is used to join two diynes and a lieterocumulene or nitrile electrophile (Eq. 19). Poly(2-pyrone)s, poly(2-pyridone)s, and polypyridines are produced having various alipliatic, aromatic, ladder (128, 129), or ether (130) linlcages. The regiochemistry of the polymerization is somewhat disordered, with four substitution patterns possible for the cyclized unit arising from the monomer alkyne substituents.
n R — ^ — R i =—R Ni(0) or Co(ll)
R2 R R ^ N f^ (plus other regioisomers) ^^'
. , j or I or J
0 I N
Literature Cited
1. Kaminsicy, W. J. Chem. Soc. Dalton Trans. 1998, 1413-1418. 2. Chemical & Engineering News. June 29, 1998, p. 42-81. 3. Benedil t, G. M.; Goodall, B. L. "Metallocene-Catalyzed Polymers: Materials,
Properties, Processing, & Marl<ets"; Plastics Design Library: NonA ich, NY, 1998;p. v-viii.
4. Sinn, H.; Kaminsky, W. Adv. Oraanomet. Chem. 1980, Ifi, 99-149. 5. Feldman, D.; Barbalata, A. "Synthetic Polymers: Technology, Properties,
Applications"; Chapman & Hall: London, 1996; Chapter 1. 6. Ewen, J.; Elder, M. J.; Jones, R. L; Haspelaugh, L; Atwood, J. L; Bott, S. G.;
Robinson. K. Mal<romol. Chem.. Macromoi. Symp. 1991,49/49, 253-295. 7. Bochmann, M. J. Chem. Soc. Dalton Trans. 1996, 255-170.

1038 LS.Boffa
8. Britzinger, H. H.; Fischer, D.; Mulhaupt, R.; Rieger, B.; Waymouth, R. M. Angew. Chem Int. Ed. Enal. 1995,34. 1143-1170.
9. Gupta, V. K.; Satish, S.; Bhardwaj, I. S. J. Macromol. Sci.. Rev. MacromoL Chem. Phys. 1994. C34. 439-514.
10. Britovsek, G. J. P.; Gibson, V. C; Wass. D. F. Anaew. Chem. Int. Ed. Enal. 1999, Sa. 429-447.
11. Soga, K.; Shiono, T. Proa. Polvm. Sci. 1997,22. 1503-1546. 12. Jordan, R. F. Adv. Oraanomet. Chem. 1991.32, 325-387. 13. Kaminsky, W.; Bark, A.; Amdt, M. Makromol. Chem.. Macromol. Svmo. 1991, 47,
83-93. 14. Chemical & Enaineerina News. July 6.1998, p. 11-16. 15. Ishihara, N.; Kuromoto, M.; Uoi, M. Macromolecules 1988,21, 3356-3360. 16. Ewen, J. J. Am. Chem. Soc. 1988, HO, 6255-6266. 17. Johnson, L K.; Killian, C. M.; Brookhart. M. J. Am. Chem. Soc. 1995, HZ, 6141-
6415. 18. Johnson, L. K. et al. (DuPont). World Patent Application 96/23010,1996. 19. Johnson, L K.; Mecking. S.; Brookhart, M. J. Am. Chem. Soc. 1996, 118, 267-
268. 20. Wang, C; Friedrich, S.; Younkin, T. R.; Li. R. T.; Grubbs, R. H.; Bansleben, D. A;
Day, M. W. Oraanometallics 1998,17, 3149-3151. 21. Bansleben, D. A.; Friedrich, S. K.; Younkin, T. R.; Grubbs, R. H.; Wang, C; Li, R.
T. Worid Patent Application 98/42665,1998. 22. Small, B. L; Brookhart, M.; Bennett, A. M. A. J. Am. Chem. Soc. 1998, 12Q, 4049-
4050. 23. Small, B. L.; Brookhart, M. Macromolecules 1999,32, 2120-2130. 24. Britovsek, G. J. P.; Gibson, V. C; Kimberiey. B. S.; Maddox, P. J.; McTavish, S. J.;
Solan, G. A.; White, A. J. P.; Williams, D. J. Chem. Commun. 1998, 849-850. 25. Ihara, E.; Young, V. G. J.; Jordan, R. F. J. Am. Chem. Soc. 1998, 12Q, 8277-
8278. 26. Bruce, M.; Gibson, V. C; Redshaw, C; Solan, G. A; White, A J. P.; Williams, D.
J. Chem. Commun. 1998, 2523-2524. 27. Hadjiandreou, P.; Julemont, M.; Teyssie, P. Macromolecules 1984, 17, 2455-
2456. 28. Fayt, R.; Hadjiandreou, P.; Teyssie, P. J. Polvm. Sci.. Polym. Chem. Ed. 1985,
22, 337-342. 29. Deming, T. J.; Novak, B. M.; Ziller, J. W. J. Am. Chem. Soc. 1994, 116, 2366-
2374 and references therein. 30. Tang, B.-Z.; Kotera, N. Macromolecules 1989. 1989. 4388-4390. 31. Nakayama, Y.; Mashima, K.; Nakamura, A Macromolecules 1993, 2fi, 6267-
6272. 32. Masuda, T.; Yoshimura, T.; Tamura, K.; Higashimura, T. Macromolecules 1987,
2Q. 1734-1739. 33. Yang, M.; Zheng, M.; Furiani, A; Russo, M. V. J. Polym. Sci.. Polym. Chem. Ed.
1994,32.2709-2713. 34. Xu, K.; Peng, H.; Tang. B. Z. Polvm. Mater. Sci. Enq. 1999, fiQ, 485-486.

Ovemew of Transition Metal-Mediated Polymerzations 1039
35. Carr, A. G.; Dawson, D. M.; Bochmann, M. Macromolecules 1998, 31. 2035-2040.
36. Shaffer, T. D.; Ashbaugh, J. R. J. Polym. Sci.. Polym. Chem. Ed. 1997, 35, 329-344.
37. Bochmann, M.; Dawson, D. M. Anqew. Chem. Int. Ed. Enal. 1996, 35, 2226-2228.
38. Bums, C. T.; Shapiro, C. J. Abstracts of Papers, 215th National Meeting of the American Chemical Society, Dallas, TX; American Chemical Society: Washington, DC, 1997; INOR 5.
39. Drent. E.: Budzelaar. H. M. Chem. Rev. 1996. 96. 663-681. 40. Sommazzi, A.; Garbassi, F. Proa. Polym. Sci. 1997,22, 1547-1605. 41. Sen, A. Ace. Chem. Res. 1993,2g. 303-310. 42. Amevor, E.; Bronco, S.; Consiglio, G.; DiBenedetto, S. Malcromol. Chem..
l\/lacromol. Symp. 1995, 89, 443-454. 43. Kacker, S.; Sen, A. Macromolecules 1997, US , 10028-10033. 44. Kacker, S.; Jiang, Z.; Sen, A. Macromolecules 1996,29, 5852-5858. 45. Bonner, J. G.; Powell, A. K. Polvm. Mater. Sci. Eno. 1997, 7g, 108-109. 46. Schrock, R. R. Ace. Chem. Re.c;. 1990,23. 158-165. 47. Amass, A. J. in "Comprehensive Polymer Science", Allen, G.; Bevington, J. C,
Eds.; Pergamon: Oxford, 1989; Vol. 4, p. 109-134. 48. Gilliom, L. R.; Grubbs, R. H. J. Am. Chem. Soc. 1986,108, 733-742. 49. Schrock, R. R.; Murdzek, J. S.; Bazan, G. C; Robbins, J.; DiMare, M.; O'Regan,
M. J. Am. Chem. Soc. 1990,112. 3875-3886. 50. Calderon, N.; Ofstead, E. A.; Judy, W. A J. Polvm. Sci.. Polvm. Chem. Ed. 1967,
5, 2209-2217. 51. Lynn, D. M.; Mohr, B.; Grubbs, R. H. J. Am. Chem. Soc. 1998,122. 1627-1628. 52. Lynn, D. M.; Kanaoka, S.; Grubbs, R. H. J. Am. Chem. Soc. 1996, H f i , 784-790. 53. Novak, B. M.; Grubbs. R. H. J. Am. Chem. Soc. 1988, Hf i , 7542-7543. 54. Pen-ott, M. G.; Novak, B. M. Macromolecules 1996, 22. 1817-1823 and
references therein. 55. Maughon, B. R.; Grubbs, R. H. Macromolecules 1997, 3fl. 3459-3469 and
references therein. 56. Klavetter, F. L; Grubbs, R. H. J. Am. Chem. Soc. 1988,110, 7807-7813. 57. Swager. T. M.; Dougherty, D. A; Grubbs, R. H. J. Am. Chem. Soc. 1988, 110.
2973-2974. 58. Ofetead, E. A. in "Encyclopedia of Polymer Science & Engineering", Mark, H. F.;
Kroschwitz, J. I., Eds.; Wiley-lnterscience: New York, 1988; Vol. 11, p. 287-214. 59. Wegener, K. B.; Boncella, J. M.; Nel, J. G. Macromolecules 1991, 24, 2649-
2657. 60. Tindall, D.; Pawlow, J. H.; Wagener, K. B. in "Trends in Organometallic
Chemistry: Alkene Metathesis in Organic Synthesis", Fuerstner, A, Ed Springer-Verlag: Berlin, 1998; p. 183-198.
61. Davidson, T. A.; Wagener, K. B. in "Synthesis of Polymers", Schluter. A.-D., Ed "Materials Science and Technology: A Comprehensive Treatmenf, Cahn, R W.; Haasen, P.; Kramer, E. J., Eds.; Wiley-VCH: New York, 1999; Vol. 19 Chapter 4.

1040 LS. Boffa
62. Gomez, F. J.; Wagener. K. B. Macromol. Chem. Phvs. 1998, 199, 1581-1587 and references therein.
63. Wagener, K. B.; Brzezinska, K.; Anderson, J. D.; Younkin, T. R.; Steppe, K.; DeBoer, W. Macromolecules 1997, 3Q, 7363-7369 and references therein.
64. Valenti, D. J.; Wagener, K. B. Polvm. Prepr.. Am. Chem. Soc. Div. Polym. Chem. 1996, 37 (2). 325-326 and references therein.
65. Wolfe, P. S.; Wagener, K. B. Macromol. Rapid Commun. 1998,19, 305-308. 66. Wolfe, P. S.; Gbmez, F. J.; Wagener, K. B. Macromolecules 1997, 30, 714-717. 67. Tindall, D.; Wagener. K. B. Polvm. Prepr.. Am. Chem. Soc. Div. Polvm. Chem.
1998. 39i2), 539. 68. G6mez, F. J.; Wagener, K. B. Polym. Prepr.. Am. Chem. Soc. Div. Polvm. Chem.
1998, 32i2). 540-541. 69. Wagener, K. B.; Valenti, D.; Watson, M. Polym. Preor.. Am. Chem. Soc. Div.
Polvm. Chem. 1998, 39 (1). 719-720. 70. Wagener, K. B.; Valenti, D. Macromolecules 1997,2Q, 6688-6690. 71. Wagener, K. B.; Valenti, D. J.; Hahn, S. F. Polym. Prepr.. Am. Chem. Soc. Div.
Polvm. Chem. 1997, 32^2), 394-395. 72. Wagener. K. B.. University of Florida, personal communication, 1999. 73. Valenti, D. J.; Wagener, K. B. Macromolecules 1998, 31. 2764-2773. 74. Weiss, K.; Michel, A. M.; Auth, E.-M.; Bunz. U. H. F.; Mangel, T.; Mullen, K. Anoew.
Chem. Int. Ed. Enol. 1997.36. 506-509. 75. Zhang. X.-L; Bazan, G. C. Macromolecules 1994,22, 4627-4628. 76. Krouse, S. A.; Schrock, R. R. Macromolecules 1989, 22. 2569-2576. 77. Kloppenburg, L; Song. D.; Bunz. U. H. F. J. Am. Chem. Soc. 1998,120, 7973-
7974. 78. Webster, O. W.; Hertler. W. R.; Sogah, D. Y.; Famham, W. B.; RajanBabu, T. V. J,
Am. Chem. Soc. 1983.125. 5706-5708. 79. Webster. O. W.; Sogah, D. Y. in "Comprehensive Polymer Science", Allen, G.;
Bevington, J. C, Eds.; Pergamon Press: Oxford, 1989; Vol. 4, p. 163-169 and references therein.
80. MiJIIer, A. H. E. Macromolecules 1994,27, 1685-1690. 81. Quirk. R. P.; Bidinger. G. P. Polvm. Bull. 1989.2Z. 63-70. 82. Collins, S.; Ward. D. G. J. Am..Chem. Soc. 1992, I H . 5461-5462. 83. Li, Y.; Ward, D. G.; Reddy. S. S.; Collins, S. Macromolecules 1997, 30, 1875-
1883. 84. Yano, T. et al. Polvm. Prepr. Jpn. 1994,43.141. 85. Yasuda. H.; Yamamoto, H.; Yamashita, M.; Yokota, K.; Nakamura, A.; Miyake, S.;
Kai. Y.; Kanehisa, N. Macromolecules 1993,26, 7134-7143. 86. Ihara, E.; Morimoto, M.; Yasuda, H. Macromolecules 1995.2S. 7886-7892. 87. Boffa, L. S.; Novak, B. M. Macromolecules 1997,3Q, 3494-3506. 88. Boffa, L S.; Novak, B. M. Tetrahedron 1997.53. 15367-15396. 89. Yasuda. H.; Furo. M.; Yamamoto, H.; Nakamura, A.; Miyake, S.; Kibino, N.
Macromolecules 1992. 25. 5115-5116. 90. Okuda. J.; Konig, P.; Rushkin, I. L; Kang, H.-C; Massa, W. J. Oroanomet.
Cl]finL 1995. 501.37-39.

Ovemew of Transition Metal-Mediated Polymerzations 1041
91. Yamashita, M.; Takemoto, Y.; Ihara, E.; Yasuda, H. Macromolecules 1996, 2 i , 1798-1806.
92. Aida. T.; Maekawa. Y; Asano, S.; Inoue, S. Macromolecules 1988, 21, 1195-1202.
93. Endo, M.; Alda, T.; Inoue. S. Macromolecules 1987,2Q, 2982-2988. 94. Dubois, P.; Ropson, N.; Jer6me, R.; Teyssie, P. Macromolecules 1996, 2S,
1965-1975. 95. Duda, A.; Florjanczyk, Z; Hofman, A.; Slomkowski, S.; Penczek, S.
Macromolecules 1990, 23, 1640-1646. 96. McLain, S. J.; Drysdale, N. E. Polvm. Prepr. Am. Chem. Soc. Div. Polym. Chem.
1992,3111). 174-175. 97. Stevels, W. M.; Ankone, M. J. K.; Dijkstra, P. J.; Feijen, J. Macromolecules 1996,
29, 3332-3333. 98. Kuran, W. Proo. Polvm. Sci. 1998.23. 919-992. 99. Evans, W. J.; Katsumata, H. Macromolecules 1994.2Z. 4011-4013. 100. Sugimoto, H.; Kawamura, C; Kurokl, M.; Aida, T.; Inoue, S. Macromolecules
1994. 2Z. 2013-2018. 101. Kuran, W.; Listos, T.; Abramczyk, M.; Dawidek, A. J. Macromol. Sci.. Chem.
1998, A3§. 427-437. 102. Aida, T.; Ishikawa, M.; Inoue. S. Macromolecules 1986.12, 8-13. 103. Patten, T. E.; Novak, B. M. J. Am. Chem. Soc. 1991.113. 5065-5066. 104. Patten, T. E.; Novak, B. M. J. Am. Chem. Soc. 1996.11S. 1906-1916. 105. Shashoua, V. E.; Sweeny, W.; Tietz, R. J. Am. Chem. Soc. 1960, SZ, 866-873. 106. Goodwin, A.; Novak, B. M. Macromolecules 1994.2Z. 5520-5522. 107. Deming, T. J.; Novak, B. M. Macromolecules 1991,24, 5478-5480. 108. Novak. B. M.; Goodwin. A A.; Schlitzer, D.; Patten, T. E.; Deming, T. J. Polym.
Preor.. Am. Chem. Soc. Div. Polvm. Chem. 1996.3112). 446-447. 109. Percec. V.; Hill, D. H. ACS Svmo. Ser. 1996, 4,2-56. 110. Rehahn, M.; Schiuter, A.-D.; Wegner, G.; Feast. W. J. Polymer 1989, 3fl. 1060-
1062. 111. Goodson, F. E.; Wallow, T. I.; Novak, B. M. Macromolecules 1998, 31, 2047-
2056. 112. Yu, L; Bao, Z.; Cai, R. Anaew. Chem. int. Ed. Enal. 1993, 32, 1345-1347. 113. Babudri, F.; Cicco, S. R.; Farinola, G. M.; Naso, F. Makromol. Rapid Commun.
1996.17.905-911. 114. Bao. Z.; Chen, Y.; Cai, R.; Yu, L. Macromolecules 1993,2S. 5281-5286. 115. Fu, D.-K.; Xu, B.; Swager. T. M. Tetrahedron 1997.53, 15487-15494. 116. Wallow, T. I.; Novak, B. M. J. Am. Chem. Soc. 1991,113. 7411-7412. 117. Goldfinger, M. B.; Swager, T. M. J. Am. Chem. Soc. 1994, US. 7895-7896. 118. Hu. Q.-S.; Vitharana. D.; Liu, G.; Jain, V.; Pu, L. Macromolecules 1996, 22. 5075-
5082. 119. Spetseris, N.; Ward, R. E.; Meyer, T. Y. Macromolecules 1998, M , 3158-3161. 120. Guo, H.; Weber. W. P. Polvm. Bull. 1994,1SS4, 525-528. 121. Guo, H.; Tapsak, M. A.; Weber, W. P. Macromolecules 1995,2S. 4714-4718. 122. Gupta, S.; Londergan, T. M.; Paulasaari, J. K.; Sargent. J. R.; Weber. W. P.
Polvm. Mater. Sci. Eng. 1999. fiQ. 445-446.

1042 LS.Boffa
123. Paulasaari, J. K.; Weber, W. P. Macromolecules 1998, M , 7105-7107. 124. Shea, K. J.; Walker, J. W.; Zhu, H.; Paz, M.; Greaves, J. J. Am. Chem. Soc. 1997,
119. 9049-9050. 125. Tsuda, T.; Maruta, K.; Kitaike, Y. J. Am. Chem. Soc. t992, I M , 1498-1499 and
references therein. 126. Tsuda, T.; Hokazono, H. Macromolecules 1993, 2S. 1796-1797 and references
therein. 127. Tsuda, T.; Maehara, H. Macromolecules 1996, 2S, 4544-4548 and references
therein. 128. Tsuda, T.; Yasukawa, H.; Hokazono, H.; Kitaike, Y. Macromolecules 1995, 2fi,
1312-1315. 129. Tsuda, T.; Hokazono, H. Macromolecules 1993,26, 5528-5529. 130. Tsuda, T.; Yasukawa, H.; Komori, K. Macromolecules 1995,28, 1356-1359.