aseptic processing
TRANSCRIPT

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
1
Aseptic Processing
Mrs Robyn Isaacson

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
2
Aseptic Processing - Overview
• Certain pharmaceutical products must be sterile– injections, ophthalmic preparations, irrigations
solutions, haemodialysis solutions
• Two categories of sterile products– those that can be sterilized in final container
(terminally sterilized)– those that cannot be terminally sterilized and
must be aseptically prepared

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
3
Aseptic Processing - Overview
Aseptic processing• Objective is to maintain the sterility of a product,
assembled from sterile components• Operating conditions so as to prevent microbial
contamination

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
4
Aseptic Processing - Overview
Objective• To review specific issues relating to the
manufacture of aseptically prepared products:– Manufacturing environment
• Clean areas• Personnel
– Preparation and filtration of solutions
– Pre-filtration bioburden
– Filter integrity/validation
– Equipment/container preparation and sterilization
– Filling Process
– Validation of aseptic processes
– Specific issues relating to Isolators, BFS and Bulk
Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
5
Manufacturing Environment
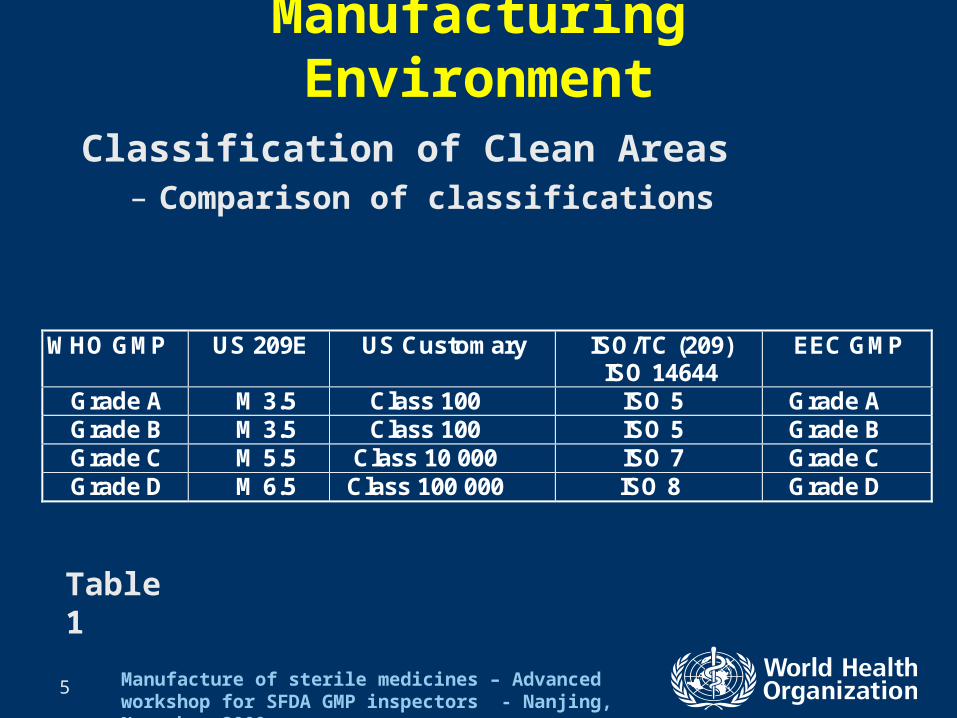
Classification of Clean Areas– Comparison of classifications
WHO GMP US 209E US Customary ISO/TC (209) ISO 14644
EEC GMP
Grade A M 3.5 Class 100 ISO 5 Grade A Grade B M 3.5 Class 100 ISO 5 Grade B Grade C M 5.5 Class 10 000 ISO 7 Grade C Grade D M 6.5 Class 100 000 ISO 8 Grade D
Table 1

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
6
Manufacturing Environment
Classification of Clean Areas– Classified in terms of airborne particles (Table 2)
Grade At rest In operation
maximum permitted number of particles/m3
0.5 - 5.0 µm > 5 µm 0.5 - 5.0 µm > 5 µ
A 3 500 0 3 500 0
B 3 500 0 350 000 2 000
C 350 000 2 000 3 500 000 20 000
D 3 500 000 20 000 not defined not defined
“At rest” - production equipment installed and operating
“In operation” - Installed equipment functioning in defined operating mode and specified number of personnel present

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
7
Manufacturing Environment
Four grades of clean areas:• Grade D (equivalent to Class 100,000, ISO 8):
– Clean area for carrying out less critical stages in manufacture of aseptically prepared products eg. handling of components after washing.
• Grade C (equivalent to Class 10,000, ISO 7):– Clean area for carrying out less critical stages in
manufacture of aseptically prepared products eg. preparation of solutions to be filtered.
• Grade B (equivalent to Class 100, ISO 5): – Background environment for Grade A zone, eg.
cleanroom in which laminar flow workstation is housed.

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
8
Manufacturing Environment
• Grade A (equivalent to Class 100 (US Federal Standard 209E), ISO 5 (ISO 14644-1):– Local zone for high risk operations eg. product filling,
stopper bowls, open vials, handling sterile materials, aseptic connections, transfer of partially stoppered containers to be lyophilized.
– Conditions usually provided by laminar air flow workstation.
• Each grade of cleanroom has specifications for viable and non-viable particles – Non-viable particles are defined by the air classification
(See Table 2)
Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
9
Manufacturing Environment
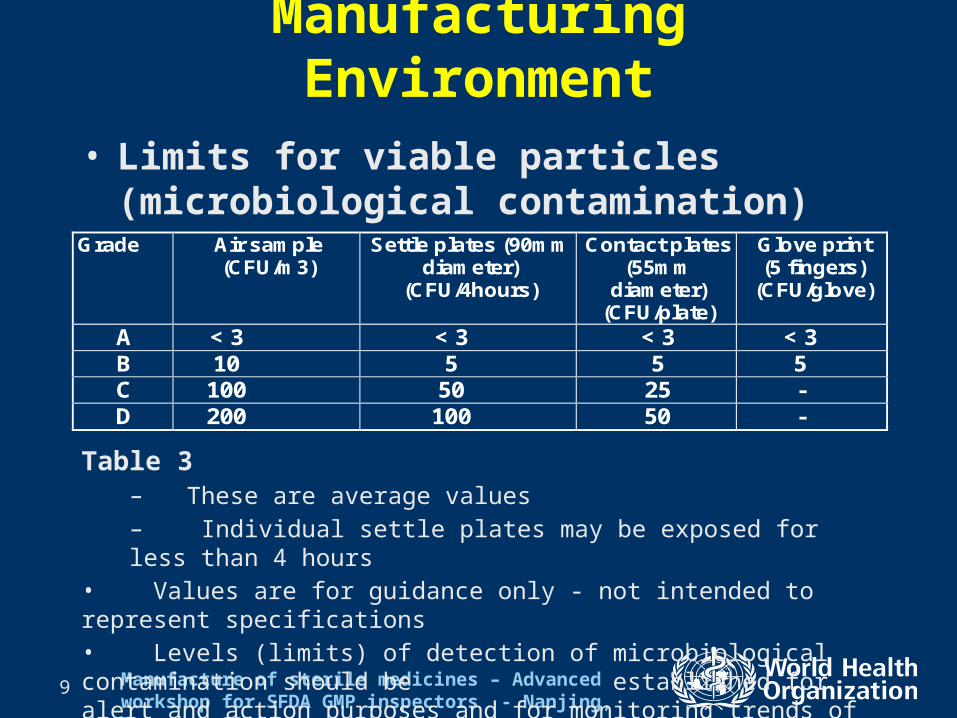
• Limits for viable particles (microbiological contamination)
Grade Air sample (CFU/m3)
Settle plates (90mm diameter)
(CFU/4hours)
Contact plates (55mm
diameter) (CFU/plate)
Glove print (5 fingers)
(CFU/glove)
A < 3 < 3 < 3 < 3 B 10 5 5 5 C 100 50 25 - D 200 100 50 -
Table 3– These are average values– Individual settle plates may be exposed for less than 4 hours
• Values are for guidance only - not intended to represent specifications• Levels (limits) of detection of microbiological contamination should be established for alert and action purposes and for monitoring trends of air quality in the facility

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
10
Manufacturing Environment
Environmental Monitoring• Physical
– Particulate matter
– Differential pressures
– Air changes, airflow patterns
– Clean up time/recovery
– Temperature and relative humidity
– Airflow velocity

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
11
Manufacturing Environment
Environmental Monitoring - Physical• Particulate matter
– Particles significant because they can contaminate and also carry organisms
– Critical environment should be measured not more than 30cm from worksite, within airflow and during filling/closing operations
– Preferably a remote probe that monitors continuously– Difficulties when process itself generates particles (e.g.
powder filling)– Appropriate alert and action limits should be set and
corrective actions defined if limits exceeded

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
12
Manufacturing Environment
Environmental Monitoring - Physical• Differential pressures
– Positive pressure differential of 10-15 Pascals should be maintained between adjacent rooms of different classification (with door closed)
– Most critical area should have the highest pressure– Pressures should be continuously monitored and
frequently recorded.
– Alarms should sound if pressures deviate
– Any deviations should be investigated and effect on environmental quality determined

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
13
Manufacturing Environment
Environmental Monitoring - Physical• Air Changes/Airflow patterns
– Air flow over critical areas should be uni-directional (laminar flow) at a velocity sufficient to sweep particles away from filling/closing area
– for B, C and D rooms at least 20 changes per hour are ususally required
• Clean up time/recovery– Particulate levels for the Grade A “at rest” state should
be achieved after a short “clean-up” period of 20 minutes after completion of operations (guidance value)
– Particle counts for Grade A “in operation” state should be maintained whenever product or open container is exposed

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
14
Manufacturing Environment
Environmental Monitoring - Physical• Temperature and Relative Humidity
– Ambient temperature and humidity should not be uncomfortably high (could cause operators to generate particles) (18°C)
• Airflow velocity– Laminar airflow workstation air speed of approx
0.45m/s ± 20% at working position (guidance value)

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
15
Manufacturing Environment
Personnel• Minimum number of personnel in clean areas
– especially during aseptic processing
• Inspections and controls from outside
• Training to all including cleaning and maintenance staff
– initial and regular
– manufacturing, hygiene, microbiology
– should be formally validated and authorized to enter aseptic area
• Special cases– supervision in case of outside staff
– decontamination procedures (e.g. staff who worked with animal tissue materials)

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
16
Manufacturing Environment
Personnel (2)• High standards of hygiene and cleanliness
– should not enter clean rooms if ill or with open wounds
• Periodic health checks
• No shedding of particles, movement slow and controlled
• No introduction of microbiological hazards
• No outdoor clothing brought into clean areas, should be clad in factory clothing
• Changing and washing procedure
• No watches, jewellery and cosmetics
• Eye checks if involved in visual inspection

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
17
Manufacturing Environment
Personnel (3)• Clothing of appropriate quality:
– Grade D• hair, beard, moustache covered• protective clothing and shoes
– Grade C• hair, beard, moustache covered• single or 2-piece suit (covering wrists, high neck),
shoes/overshoes• no fibres/particles to be shed
– Grade A and B• headgear, beard and moustache covered, masks,
gloves• not shedding fibres, and retain particles shed by
operators

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
18
Manufacturing Environment
Personnel (4)• Outdoor clothing not in change rooms leading to
Grade B and C rooms• Change at every working session, or once a day (if
supportive data)• Change gloves and masks at every working session• Frequent disinfection of gloves during operations• Washing of garments – separate laundry facility
– No damage, and according to validated procedures (washing and sterilization)
• Regular microbiological monitoring of operators

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
19
Aseptic Processing
• In aseptic processing, each component is individually sterilised, or several components are combined with the resulting mixture sterilized.– Most common is preparation of a solution which is
filtered through a sterilizing filter then filled into sterile containers (e.g active and excipients dissolved in Water for Injection)
– May involve aseptic compounding of previously sterilized components which is filled into sterile containers
– May involve filling of previously sterilized powder • sterilized by dry heat/irradiation
• produced from a sterile filtered solution which is then aseptically crystallized and precipitated
– requires more handling and manipulation with higher potential for contamination during processing

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
20
Aseptic Processing
Preparation and Filtration of Solutions• Solutions to be sterile filtered prepared in a Grade C
environment
• If not to be filtered, preparation should be prepared in a Grade A environment with Grade B background (e.g. ointments, creams, suspensions and emulsions)
• Prepared solutions filtered through a sterile 0.22μm (or less) membrane filter into a previously sterilized container– filters remove bacteria and moulds– do not remove all viruses or mycoplasmas
• filtration should be carried out under positive pressure

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
21
Aseptic Processing
Preparation and Filtration of Solutions (2)• consideration should be given to complementing
filtration process with some form of heat treatment
• Double filter or second filter at point of fill advisable
• Fitlers should not shed particles, asbestos containing filters should not be used
• Same filter should not be used for more than one day unless validated
• If bulk product is stored in sealed vessels, pressure release outlets should have hydrophobic microbial retentive air filters

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
22
Aseptic Processing
Preparation and Filtration of Solutions (3)• Time limits should be established for each phase of
processing, e.g.– maximum period between start of bulk product
compounding and sterilization (filtration)– maximum permitted holding time of bulk if held after
filtration prior to filling– product exposure on processing line– storage of sterilized containers/components– total time for product filtration to prevent organisms
from penetrating filter– maximum time for upstream filters used for clarification
or particle removal (can support microbial attachment)

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
23
Aseptic Processing
Preparation and Filtration of Solutions (4)• Filling of solution may be followed by lyophilization
(freeze drying)– stoppers partially seated, product transferred to
lyophilizer (Grade A/B conditions)– Release of air/nitrogen into lyophilizer chamber at
completion of process should be through sterilizing filter

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
24
Aseptic Processing
Prefiltration Bioburden (natural microbial load)• Limits should be stated and testing should be carried
out on each batch
• Frequency may be reduced after satisfactory history is established– and biobuden testing performed on components
• Should include action and alert limits (usually differ by a factor of 10) and action taken if limits are exceeded
• Limits should reasonably reflect bioburden routinely achieved

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
25
Aseptic Processing
Prefiltation Bioburden (2)• No defined “maximum” limit but the limit should not
exceed the validated retention capability of the filter
• Bioburden controls should also be included in “in-process” controls – particularly when product supports microbial growth
and/or manufacturing process involves use of culture media
• Excessive bioburden can have adverse effect on the quality of the product and cause excessive levels of endotoxins/pyrogens

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
26
Aseptic Processing
Filter integrity• Filters of 0.22μm or less should be used for filtration
of liquids and gasses (if applicable)– filters for gasses that may be used for purging or
overlaying of filled containers or to release vacuum in lyphilization chamber
• filter intergrity shoud be verified before filtration and confirmed after filtration– bubble point– pressure hold– forward flow
• methods are defined by filter manufacturers and limits determined during filter validation

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
27
Aseptic Processing
Filter Validaton• Filter must be validated to demonstrate ability to
remove bacteria– most common method is to show that filter can retain a
microbiological challenge of 107 CFU of Brevundimonas diminuta per cm2 of the filter surface
– a bioburden isolate may be more appropriate for filter retention studies than Brevundimonas diminuta
– Challenge concentration is intended to provide a margin of safety well beyond what would be expected in production
– preferably the microbial challenge is added to the fully formulated product which is then passed through the filter

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
28
Aseptic Processing
Filter validation (2)– if the product is bactericidal, product should be passed
through the filter first followed by modified product containing the microbial challenge (after removing any bactericidal activity remaining on the filter)
– filter validation should be carried out under worst case conditions e.g. maximum allowed filtration time and maximum pressure
– integrity testing specification for routine filtration should correlate with that identified during filter validation

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
29
Aseptic Processing
Equipment/container preparation and sterilization
• All equipment (including lyophilizers) and product containers/closures should be sterilized using validated cycles– same requirements apply for equipment sterilization that
apply to terminally sterilized product– particular attention to stoppers - should not be tightly
packed as may clump together and affect air removal during vacuum stage of sterilization process
– equipment wrapped and loaded to facilitate air removal– particular attention to filters, housings and tubing

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
30
Aseptic Processing
Equipment/container preparation and sterilization (2)
• CIP/SIP processes– particular attention to deadlegs - different orientation
requirements for CIP and SIP
• heat tunnels often used for sterilization/depyrogenation of glass vials/bottles– usually high temperature for short period of time– need to consider speed of conveyor– validation of depyrogenation (3 logs endotoxin units)
• worst case locations
– tunnel supplied with HEPA filtered air

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
31
Aseptic Processing
Equipment/container preparation and sterilization (2)
• equipment should be designed to be easily assembled and disassembled, cleaned, sanitised and/or sterilized– equipment should be appropriately cleaned - O-rings and
gaskets should be removed to prevent build up of dirt or residues
• rinse water should be WFI grade• equipment should be left dry unless sterilized immediately
after cleaning (to prevent build up of pyrogens) • washing of glass containers and rubber stoppers should be
validated for endotoxin removal• should be defined storage period between sterilization and
use (period should be justified)

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
32
Aseptic Processing
Process Validation• Not possible to define a sterility assurance level
for aseptic processing• Process is validated by simulating the
manufacturing process using microbiological growth medium (media fill)– Process simulation includes formulation
(compounding), filtration and filling with suitable media using the same processes involved in manufacture of the product
– modifications must be made for different dosage formats e.g. lyophilized products, ointments, sterile bulks, eye drops filled into semi-transparent/opaque containers, biological products

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
33
Aseptic Processing
Process Validation (2)• Media fill program should include worst case
activities– Factors associated with longest permitted run (e.g.
operator fatigue)
– Representative number, type, and complexity of normal interventions, non-routine interventions and events (e.g. maintenance, stoppages, etc)
– Lyophilisation
– Aseptic equipment assembly

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
34
Aseptic Processing
Process Validation (3)• Worst case activities (cont)
– No of personnel and their activities, shift changes, breaks, gown changes
– Representative number of aseptic additions (e.g. charging containers, closures, sterile ingredients) or transfers
– Aseptic equipment connections/disconnections
– Aseptic sample collections
– Line speed and configuration

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
35
Aseptic Processing
Process Validation (4)• Worst case activities (cont)
– Weight checks
– Container closure systems
– Specific provisions in processing instructions
• Written batch record documenting conditions and activities
• Should not be used to justify risky practices

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
36
Aseptic Processing
Process Validation (5)Duration
– Depends on type of operation
– BFS, Isolator processes - sufficient time to include manipulations and interventions
– For conventional operations should include the total filling time
Size– 5000 - 10000 generally acceptable or batch size if <5000
– For manually intensive processes larger numbers should be filled
– Lower numbers can be filled for isolators

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
37
Aseptic Processing
Process Validation (6)• Frequency and Number
– Three initial, consecutive per shift
– Subsequently semi-annual per shift and process
– All personnel should participate at least annually, consistent with routine duties
– Changes should be assessed and revalidation carried out as required
• Line Speed– Speed depends on type of process

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
38
Aseptic Processing
Process Validation (7)• Environmental conditions
– Representative of actual production conditions (no. of personnel, activity levels etc) - no special precautions (not including adjustment of HVAC)
– if nitrogen used for overlaying/purging need to substitute with air
• Media– Anaerobic media should be considered under certain
circumstances– Should be tested for growth promoting properties (including
factory isolates)

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
39
Aseptic Processing
Process Validation (8)• Incubation, Examination
– In the range 20-35ºC.
– If two temperatures are used, lower temperature first
– Inspection by qualified personnel.
– All integral units should be incubated. Should be justification for any units not incubated.
– Units removed (and not incubated) should be consistent with routine practices (although incubation would give information regarding risk of intervention)
– Batch reconciliation

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
40
Aseptic Processing
Process Validation (9)• Interpretation of Results
– When filling fewer than 5000 units: • no contaminated units should be detected
• One (1) contaminated unit is considered cause for revalidation, following an investigation
– When filling from 5000-10000 units• One (1) contaminated unit should result in an
investigation, including consideration of a repeat media fill
• Two (2) contaminated units are considered cause for revalidation, following investigation
– When filling more than 10000 units• One (1) contaminated unit should result in an investigation
• Two (2) contaminated units are considered cause for revalidation, following investigation

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
41
Aseptic Processing
Process Validation (10)• Interpretation of Results
– Media fills should be observed by QC and contaminated units reconcilable with time and activity being simulated (Video may help)
– Ideally - no contamination. Any contamination should be investigated.
– Any organisms isolated should be identified to species level (genotypic identification)
– Invalidation of a media fill run should be rare

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
42
Aseptic Processing
Process Validation (11)• Batch Record Review
– Process and environmental control activities should be included in batch records and reviewed as part of batch release
In-process and laboratory control results Environmental and personnel monitoring data Output from support systems(HEPA/HVAC, WFI, steam
generator) Equipment function (batch alarm reports, filter integrity) Interventions, Deviations, Stoppages - duration and
associated time Written instructions regarding need for line clearances Disruptions to power supply

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
43
Aseptic Processing
Additional issues specific to Isolator and BFS Technologies
• Isolators– Decontamination process requires a 4-6 log
reduction of appropriate Biological Indicator (BI)
– Minimum 6 log reduction of BI if surface is to be free of viable organisms
– Significant focus on glove integrity - daily checks, second pair of gloves inside isolator glove
– Traditional aseptic vigilance should be maintained

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
44
Aseptic Processing
• Blow-Fill-Seal (BFS)– Located in a Grade D environment
– Critial zone should meet Grade A (microbiological) requirements (particle count requirements may be difficult to meet in operation)
– Operators meet Grade C garment requirements
– Validation of extrusion process should demonstrate destruction of endotoxin and spore challenges in the polymeric material
– Final inspection should be capable of detecting leakers

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
45
Aseptic Processing
• Issues relating to Aseptic Bulk Processing• Applies to products which can not be filtered at point of
fill and require aseptic processing throughout entire manufacturing process.
• Entire aseptic process should be subject to process simulation studies under worst case conditions (maximum duration of "open" operations, maximum no of operators)
• Process simulations should incorporate storage and transport of bulk.
• Multiple uses of the same bulk with storage in between should also be included in process simulations
• Assurance of bulk vessel integrity for specified holding times.

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
46
Aseptic Processing
• Bulk Processing (2)• Process simulation for formulation stage should be
performed at least twice per year.
– Cellular therapies, cell derived products etc• products released before results of sterility tests
known (also TPNs, radioactive preps, cytotoxics)• should be manufactured in a closed system• Additional testing
– sterility testing of intermediates
– microscopic examination (e.g. gram stain)
– endotoxin testing

Manufacture of sterile medicines – Advanced workshop for SFDA GMP inspectors - Nanjing, November 2009
47
Useful Publications
• PIC/S Recommendation on the Validation of Aseptic Processes
• FDA Guidance for Industry- Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Process
• ISO 13408 Aseptic Processing of Health Care Products– Part 1: General Requirements
– Part 2: Filtration
– Part 3: Lyophilization
– Part 4: Clean-In-Place Technologies
– Part 5: Sterilization-In-Place
– Part 6: Isolator Systems