assessment tool based on the model quality … · working document qas/13.555 august 2013...
TRANSCRIPT

Working document QAS/13.555
August 2013
RESTRICTED
1 2
ASSESSMENT TOOL 3
BASED ON THE MODEL QUALITY ASSURANCE SYSTEM 4
FOR PROCUREMENT AGENCIES 5
AIDE-MEMOIRE FOR INSPECTION 6
7
8
DRAFT FOR COMMENT 9
10
11
12
13
© World Health Organization 2013 14
All rights reserved. 15
This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft 16 may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in 17 any form or by any means outside these individuals and organizations (including the organizations' concerned staff and 18 member organizations) without the permission of the World Health Organization. The draft should not be displayed on any 19 web site. 20
Please send any request for permission to: 21
Dr Sabine Kopp, Manager, Medicines Quality Assurance Programme, Quality Assurance & Safety: Medicines, Department 22 of Essential Medicines and Health Products, World Health Organization, CH-1211 Geneva 27, Switzerland; e-mail: 23 [email protected]. 24
The designations employed and the presentation of the material in this draft do not imply the expression of any opinion 25 whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or 26 of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate 27 border lines for which there may not yet be full agreement. 28
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or 29 recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors 30 and omissions excepted, the names of proprietary products are distinguished by initial capital letters. 31
All reasonable precautions have been taken by the World Health Organization to verify the information contained in this 32 draft. However, the printed material is being distributed without warranty of any kind, either expressed or implied. The 33 responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health 34 Organization be liable for damages arising from its use. 35
This draft does not necessarily represent the decisions or the stated policy of the World Health Organization. 36 37
Should you have any comments on the attached text, please send these to
Dr Sabine Kopp, Manager, Medicines Quality Assurance Programme, Quality
Assurance and Safety: Medicines, World Health Organization, 1211 Geneva 27,
Switzerland; e-mail: [email protected]; fax: (+41 22) 791 4730 ([email protected]) and to
Ms Marie Gaspard ([email protected]), by 23 September 2013.
Working documents are sent out electronically and they will also be placed on the
Medicines web site for comment. If you do not already receive directly our draft
guidelines please let us have your e-mail address (to [email protected]) and we will
add it to our electronic mailing list.

Working document QAS/13.555
page 2
38
SCHEDULE FOR THE PROPOSED ADOPTION PROCESS OF DOCUMENT QAS/13.555: 39
ASSESSMENT TOOL 40
BASED ON THE MODEL QUALITY ASSURANCE SYSTEM FOR PROCUREMENT 41
AGENCIES 42
AIDE-MEMOIRE FOR INSPECTION 43
44
45
46
47
48
Need for a Model Quality Assurance System Assessment
Tool identified in a WHO-Global Fund to Fight Aids,
Tuberculosis and Malaria (GFATM) joint stakeholders
meeting
August 2011
Working group, consisting of representatives from:
CHMP, Crown Agents, GDF, GFATM, ICRC, IDA,
MSF, MSH, PFSC, QUAMED, UNICEF, UNION,
UNOPS, USAID and WHO, created to develop a
harmonized assessment tool
Several meetings were held, along
with communications via email
Outcome: Model Quality Assurance System Assessment
Tool – inspection of a procurement agency circulated for
comments
August 2013
Compilation of comments
September 2013
Presentation to 48th meeting of the WHO Expert
Committee on Specifications for Pharmaceutical
Preparations
14-18 October 2013
Any further action as required, as recommended by the
WHO Expert Committee on Specifications for
Pharmaceutical Preparations

Working document QAS/13.555
page 3
ASSESSMENT TOOL 49
BASED ON THE MODEL QUALITY ASSURANCE SYSTEM FOR PROCUREMENT 50
AGENCIES 51
AIDE-MEMOIRE FOR INSPECTION 52
53
54
55
CONTENTS 56
page 57
58
1. Introduction ………………………………………………………………………... 59
2. Purpose …………………………………………………………………………….. 60
3. Scope ………………………………………………………………………………. 61
4. Assessment tool ……………………………………………………………………. 62
5. Revision history ……………………………………………………………………. 63 64
65
1. INTRODUCTION 66
67
The Expert Committee on Specifications for Pharmaceutical Preparations of the World 68
Health Organization (WHO) adopted a Model Quality Assurance System for Procurement 69
Agencies (MQAS) during a meeting in Geneva, Switzerland in 2005. This was subsequently 70
published as Annex 6 in the Technical Report Series, No. 937 in 2006. 71
72
The Global Fund to Fight Aids, Tuberculosis and Malaria (GFATM) Secretariat coordinated 73
this project with the aim to prepare a harmonized assessment tool based on the WHO 74
document: Model quality assurance system for procurement agencies (MQAS); WHO 75
guidelines on good storage practices (GSP) and WHO Guidelines on good distribution 76
practices (GDP). (For current versions, see www.who.int/medicines.) 77
78
This harmonized tool was developed by a working group consisting of representatives from 79
the following organizations: CHMP, Crown Agents, GDF, ICRC, IDA, MSF, MSH, PFSCM, 80
QUAMED, UNICEF, UNION, UNOPS and USAID. 81
82
2. PURPOSE 83
84
This harmonized tool was developed by the working group with the objective that it could be 85
utilized with the aim of better use of resources by coordinating procurement agency (PA) 86
assessments; and working towards mutual recognition of PA assessment findings. 87

Working document QAS/13.555
page 4
88
3. SCOPE 89
90
The assessment tool is based on the six modules in the MQAS: 91
92
Module I General requirements for procurement agencies 93
Module II Prequalification 94
Module III Purchasing 95
Module IV Receiving and storage 96
Module V Distribution 97
Module VI Reassessment 98
99
The tool covers these topics in different modules below. The logical flow considered is the 100
quality system and infrastructure of the PA under assessment, how the PA performed 101
prequalification, then purchasing of the products followed by the receiving and storage 102
thereof. The last two modules then focus on the receiving of orders and dispatch of products 103
followed by the reevaluation concept. 104
105
4. ASSESSMENT TOOL 106
107
The tool should be used by qualified, experienced persons when assessing PA (including 108
wholesalers, distributors) for compliance with recommended international standards. It can 109
also be useful for a PA when doing a self-assessment. 110
111
The tool is not checklist, but serves as a document to help and remind inspectors as to what 112
should be assessed during inspections of PAs. 113
114
Module I: General requirements for procurement agencies 115
116
This Module covers general requirements for procurement agencies including premises, 117
equipment, transport and documentation (such as SOPs, confidentiality, code of conduct and 118
complaint handling). It should be used in all cases of assessment of a procurement agency. 119
(Modules 2 to 6 may be used depending on the activities performed by the procurement 120
agency.) 121

Working document QAS/13.555
page 5
122
Area of
operation
Note Critical aspects
Premises,
Equipment,
Furniture,
Transport
General
• Licensed to operate
• Sufficient space (offices for
personnel, products, documents,
samples, etc.)
• Suitable conditions
• Necessary furniture
• Working office equipment
• Stationery and consumables
• Telephone and email access
• Appropriate transport available
Compliance with legislation
(licence)
There must be a sufficient and
functional infrastructure to enable
the PA to perform its activities
Human
resources
Personnel
• Compliance with national
legislation (e.g. responsible
person)
• Sufficient number of people
• Key personnel – quality
assurance, prequalification,
purchasing, storage and
distribution
• Quality assurance/
prequalification and purchasing
independent of one another
• Support staff
• Contracted personnel and
agreements
Training, education and
experience
Compliance with legislation
Quality assurance/prequalification
and purchasing independent of one
another (personnel – and reporting
structure)

Working document QAS/13.555
page 6
Organization Organization chart
• Authorized and current
• In line with the job descriptions
Job descriptions
• Written job descriptions
• Signed and dated
Ethical
considerations
Conflict of interest
• Policy on conflict of interest is
observed
• Signed declaration of interest.
No vested interests
Code of conduct
• Written, authorized and
implemented
• Covers conduct of personnel
• All personnel to comply with a
code of conduct
Confidentiality
• Relevant product information
kept confidential
• Confidentiality agreements exist
Declaration and management of
conflict of interest
Computers Appropriate hardware and software
• Sufficient capacity and memory
• Access control
• Data transfer procedures
• Reliable and accurate quality
and management of data and
information
• Data storage (e.g. hard copies)
If used, reliable data management
(including access control)

Working document QAS/13.555
page 7
• Back-up at defined intervals,
storage, access, readable
• Virus protection programme
and firewall
• Technical support
• Maintenance
• Trained personnel
Financial
systems
• Adequate banking facilities
• Signatories of bank accounts
appointed
• Accounting system in place
• National and international
financial transactions
• Financial transactions
performed without delay
• Funds available
• Regular financial audits are
performed.
Documentation Comprehensive documented system
• Covers policies, guidelines,
norms, standards, manuals,
procedures, records and related
documents
• SOPs for activities
Quality manual (QM)
• Contains a quality policy
• Evidence of QM
implementation, QM
maintained, reviewed and
amended as necessary
Activities and responsibilities
described in standard operating
procedures (SOPs) which are
implemented and followed;
Records reflecting activities

Working document QAS/13.555
page 8
Standard operating procedures
• SOP for writing an SOP
followed
• Written, clear, detailed SOPs for
activities
• Controlled, distributed and
retrieved when required
• Available for use
• SOPs are reviewed periodically
• Quality risk management
(QRM) principles applied
Style and layout
• SOPs in defined format
• Signed and dated
Activities to be
covered by
SOPs
All activities should be covered by
SOPs and include:
• prequalification
• purchasing
• receiving and storage
• distribution
• training
• handling of complaints
• handling of recalls
• document/record control
including distribution and
retrieval of SOPs
• self-inspection
• monitoring of environmental
conditions (e.g. temperature)
• monitoring supplier
performance
Written SOPs followed for
prequalification, purchasing,
storage, distribution, complaints,
recalls, identifying and reporting
substandard/spurious/falsely-
labelled/falsified/counterfeit
(SSFFC) medical products;
Change control
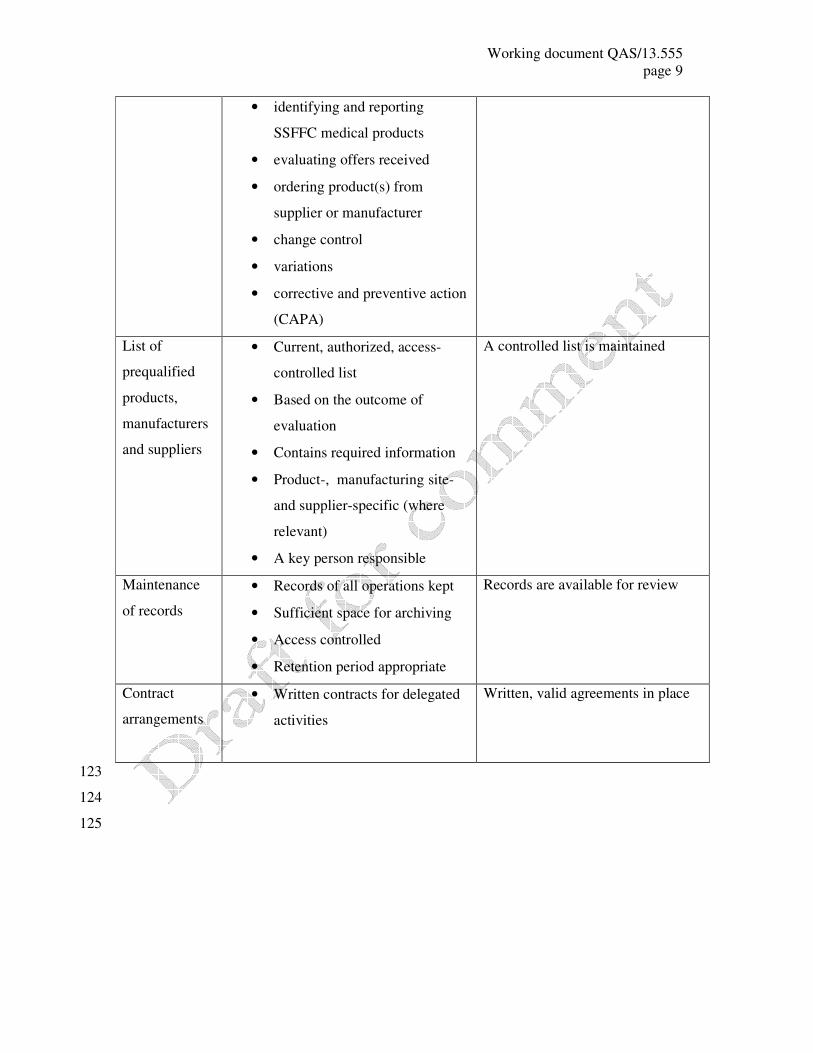
Working document QAS/13.555
page 9
• identifying and reporting
SSFFC medical products
• evaluating offers received
• ordering product(s) from
supplier or manufacturer
• change control
• variations
• corrective and preventive action
(CAPA)
List of
prequalified
products,
manufacturers
and suppliers
• Current, authorized, access-
controlled list
• Based on the outcome of
evaluation
• Contains required information
• Product-, manufacturing site-
and supplier-specific (where
relevant)
• A key person responsible
A controlled list is maintained
Maintenance
of records
• Records of all operations kept
• Sufficient space for archiving
• Access controlled
• Retention period appropriate
Records are available for review
Contract
arrangements
• Written contracts for delegated
activities
Written, valid agreements in place
123
124
125
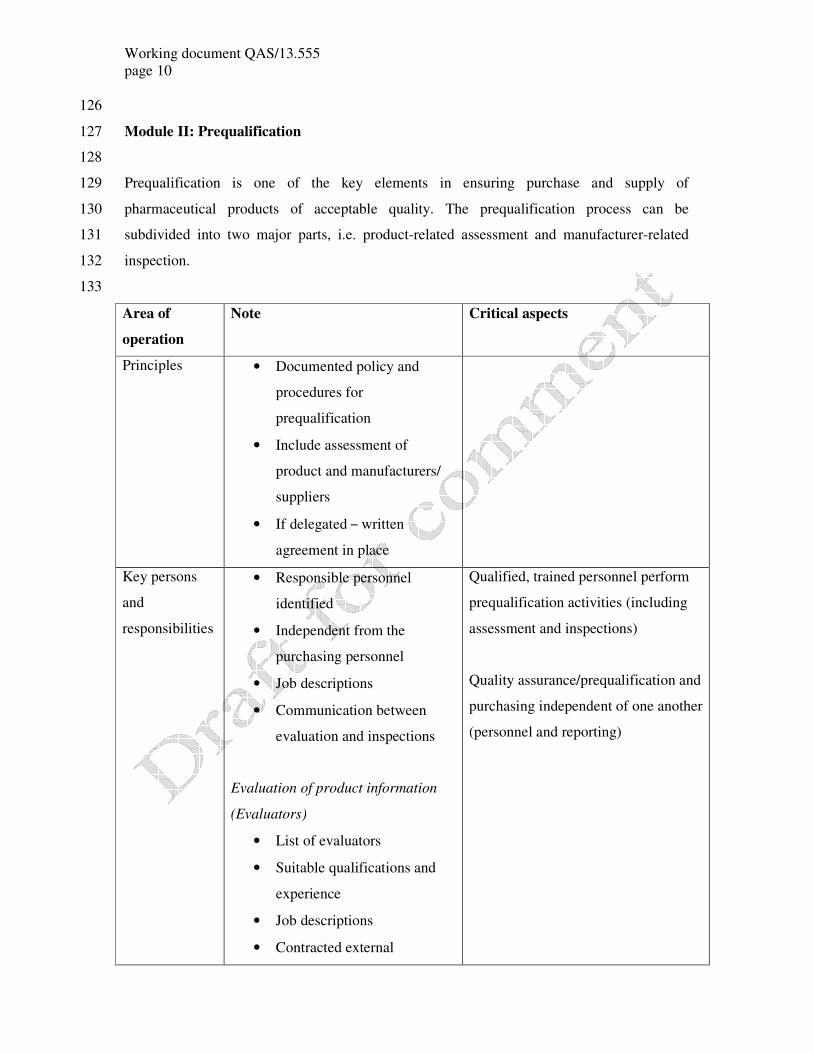
Working document QAS/13.555
page 10
126
Module II: Prequalification 127
128
Prequalification is one of the key elements in ensuring purchase and supply of 129
pharmaceutical products of acceptable quality. The prequalification process can be 130
subdivided into two major parts, i.e. product-related assessment and manufacturer-related 131
inspection. 132
133
Area of
operation
Note Critical aspects
Principles • Documented policy and
procedures for
prequalification
• Include assessment of
product and manufacturers/
suppliers
• If delegated – written
agreement in place
Key persons
and
responsibilities
• Responsible personnel
identified
• Independent from the
purchasing personnel
• Job descriptions
• Communication between
evaluation and inspections
Evaluation of product information
(Evaluators)
• List of evaluators
• Suitable qualifications and
experience
• Job descriptions
• Contracted external
Qualified, trained personnel perform
prequalification activities (including
assessment and inspections)
Quality assurance/prequalification and
purchasing independent of one another
(personnel and reporting)
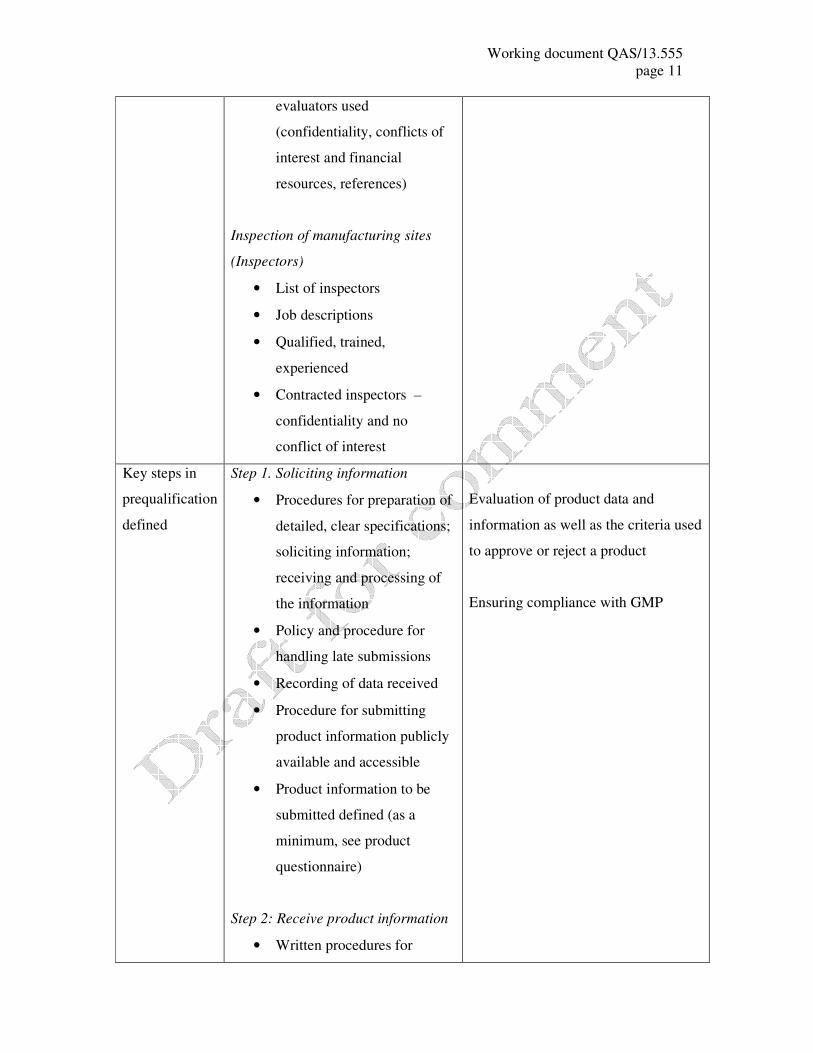
Working document QAS/13.555
page 11
evaluators used
(confidentiality, conflicts of
interest and financial
resources, references)
Inspection of manufacturing sites
(Inspectors)
• List of inspectors
• Job descriptions
• Qualified, trained,
experienced
• Contracted inspectors –
confidentiality and no
conflict of interest
Key steps in
prequalification
defined
Step 1. Soliciting information
• Procedures for preparation of
detailed, clear specifications;
soliciting information;
receiving and processing of
the information
• Policy and procedure for
handling late submissions
• Recording of data received
• Procedure for submitting
product information publicly
available and accessible
• Product information to be
submitted defined (as a
minimum, see product
questionnaire)
Step 2: Receive product information
• Written procedures for
Evaluation of product data and
information as well as the criteria used
to approve or reject a product
Ensuring compliance with GMP

Working document QAS/13.555
page 12
receiving, identification,
marking files, containers and
samples, and sufficient space
for unpacking and storage
• Procedure to ensure
traceability of the product
information
• Personnel available
Step 3: Screen product information
• SOP: screen for completeness
• A screening form used
• Record of screening kept
• Outcome communicated to
manufacturer/supplier
Step 4: Evaluate product information
• Follow SOP to evaluate
against requirement
• Time frames
• Evaluation report for each
product exists
• Outcome communicated to
the manufacturer/supplier
• Response invited where
needed
• Outcome accepted or rejected
• Evaluation report kept as
record
• Samples analysed if needed
(see also monitoring below)

Working document QAS/13.555
page 13
Step 5: Plan, prepare and perform
inspections
General points
• Evidence of GMP
compliance
• Site of manufacture known
• Site inspection policy
• Contract manufacturing sites
known
Control over active
pharmaceutical ingredients
(APIs) (inspection risk based)
Plan
• SOP and recording system
for inspection planning
• Procedure and data reviewed
as part of preparation for
inspection (e.g. site master
file)
Conduct
• SOP: how to perform an
inspection
• Scope: data and information
verified and WHO GMP
compliance assessed
• If not done – conditions for
waiving on-site inspections
Inspection report
• Inspection report for each site
inspected
• Outcome communicated
• CAPA requested, received

Working document QAS/13.555
page 14
and reviewed
• Conclusion or outcome
• Copy of report kept
Step 6: Finalize assessment process
• Written procedure followed
• Covers product evaluation
plus laboratory results and
inspection outcome
• Responsible persons
(decision-taking) and reasons
for decision
• Outcome communicated
• List of prequalified products,
manufacturers and suppliers
• Agreement between PA and
supplier/manufacturer
• List reviewed and updated at
regular intervals
Cost recovery
• If used, transparent procedure
• Fee-for-services structure
134
Module III: Purchasing 135
136
Procurement should be done with the aim of purchasing effective, quality assured products, 137
and not be focused on price alone. The term “procurement” in this Module relates specifically 138
to the purchase of health sector goods from manufacturers or suppliers. The module goes on 139
to describe the key activities in purchasing pharmaceutical products, as well as the 140
recommended organizational structure of the procurement agencies which carry out these key 141
activities. 142
143
144

Working document QAS/13.555
page 15
Area of operation Note Critical aspects
Procurement strategies • Policy: suppliers are selected
and monitored through a
process that takes into account
product quality, service
reliability and performance,
delivery time, ethics, legal
status, financial viability and
minimum order quantities;
Purchase prequalified products (from
manufacturers/suppliers)
• Efficient and transparent
management
• Financial management
procedures
• Competitive procurement
methods
• Procedure to calculate lowest
possible total cost
• Procurement and purchasing
procedures are transparent
• Independent contract review
• Purchasing and tender
documents list all
pharmaceutical products by
their international
nonproprietary name (INN)
or national generic names
• Intellectual property rights are
respected in accordance with
best practice and national law
Purchasing prequalified
products
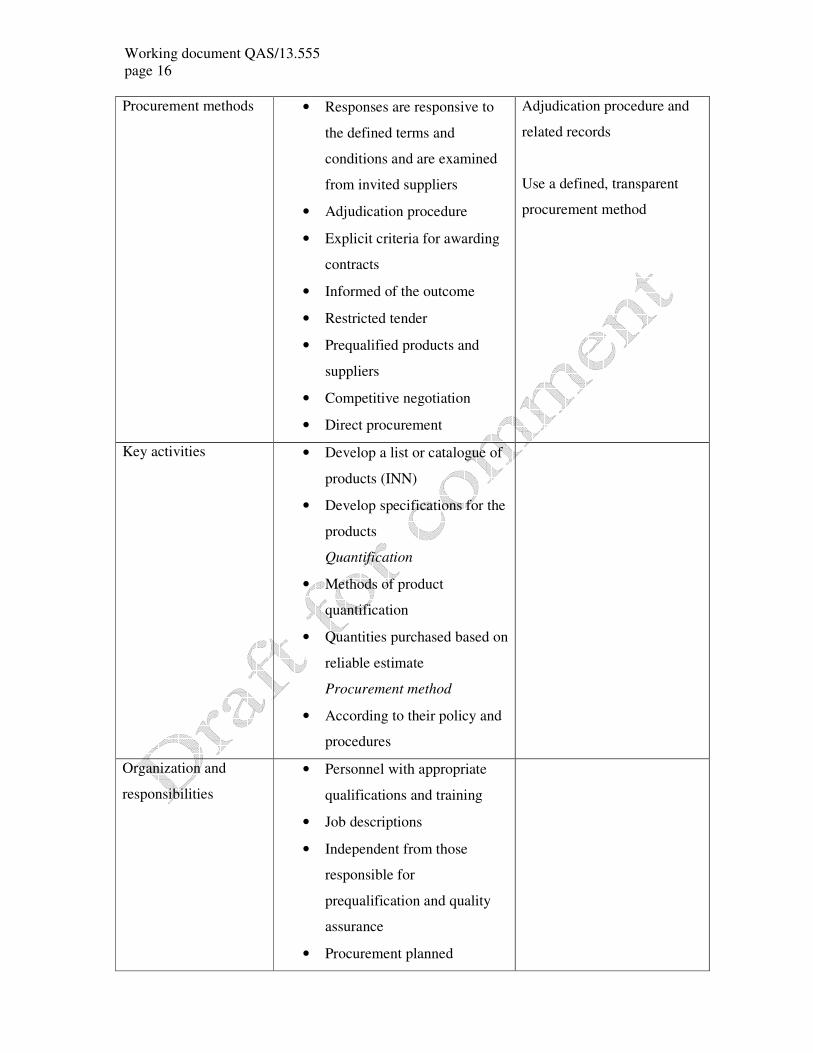
Working document QAS/13.555
page 16
Procurement methods • Responses are responsive to
the defined terms and
conditions and are examined
from invited suppliers
• Adjudication procedure
• Explicit criteria for awarding
contracts
• Informed of the outcome
• Restricted tender
• Prequalified products and
suppliers
• Competitive negotiation
• Direct procurement
Adjudication procedure and
related records
Use a defined, transparent
procurement method
Key activities • Develop a list or catalogue of
products (INN)
• Develop specifications for the
products
Quantification
• Methods of product
quantification
• Quantities purchased based on
reliable estimate
Procurement method
• According to their policy and
procedures
Organization and
responsibilities
• Personnel with appropriate
qualifications and training
• Job descriptions
• Independent from those
responsible for
prequalification and quality
assurance
• Procurement planned
Working document QAS/13.555
page 17
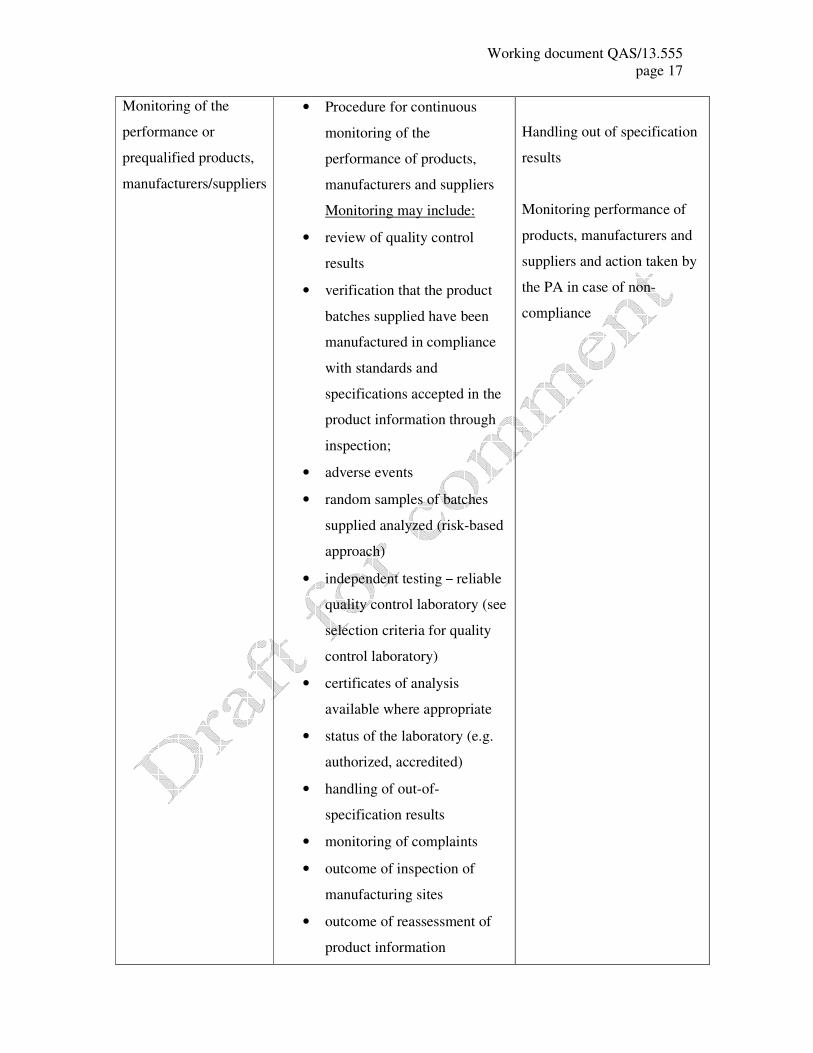
Monitoring of the
performance or
prequalified products,
manufacturers/suppliers
• Procedure for continuous
monitoring of the
performance of products,
manufacturers and suppliers
Monitoring may include:
• review of quality control
results
• verification that the product
batches supplied have been
manufactured in compliance
with standards and
specifications accepted in the
product information through
inspection;
• adverse events
• random samples of batches
supplied analyzed (risk-based
approach)
• independent testing – reliable
quality control laboratory (see
selection criteria for quality
control laboratory)
• certificates of analysis
available where appropriate
• status of the laboratory (e.g.
authorized, accredited)
• handling of out-of-
specification results
• monitoring of complaints
• outcome of inspection of
manufacturing sites
• outcome of reassessment of
product information
Handling out of specification
results
Monitoring performance of
products, manufacturers and
suppliers and action taken by
the PA in case of non-
compliance

Working document QAS/13.555
page 18
• monitoring of direct and
indirect product costs
• monitoring of adherence to
delivery schedules
• contract terms and conditions
• tracking system (values of
contracts awarded, total
purchases, performance)
Donations • Written procedure
145
Module IV: Receiving and storage 146
147
The procurement agency should ensure that the pharmaceutical products purchased are 148
received and stored correctly and in compliance with applicable legislation and regulations. 149
Products should be received and stored in such a way that their quality and integrity is 150
preserved, batch traceability is maintained and stock can be rotated. 151
152
Area of
operation
Note Critical aspects
General
arrangements
• Received and stored correctly
• Quality and integrity is
maintained
• Batch traceability
• Stock rotation
• Unidirectional flow
• Security of materials and
products
• Subcontracting
Procedures followed for receiving
and storage
Batch traceability
Pre-shipment
quality control
• Batches released by the
manufacturer (certificate of
analysis (CoA))
• Batches additionally tested
(risk-based approach) prior to
Batch release with CoA (meeting
specifications)

Working document QAS/13.555
page 19
shipment to PA
• Selection criteria for quality
control laboratory
Receiving of
stock
• Receiving and dispatch bays
• Incoming containers cleaned,
quarantined
• Review of CoAs
• Released for use or
distribution (responsible
person involved)
Checks on receipt:
• order, delivery note, labels and
transport conditions, integrity
of packages and seals and for
uniformity of the containers
Visual inspection for:
• contamination; tampering and
damage; expiry date,
compliance with labelling,
packaging instructions
• suspect containers and
damaged containers – recorded
and investigated
Goods received and checked
according to an appropriate SOP –
supported by records
Products released by responsible
person
Post-
procurement
control
• Random sampling for
independent laboratory
analysis
• Selection criteria for quality
control laboratory
• SOP and national legislation
• Representative samples –
sampling plans and
instructions (risk assessment)
Action taken in case of non-
conforming product

Working document QAS/13.555
page 20
• Appropriately trained and
qualified personnel
Rejected
materials
• SOP for rejected products
• Separate storage or validated
computerized system
• Action approved by authorized
personnel and recorded
Rejected materials kept separately,
access controlled and handled
appropriately
Storage of
materials/
products
Personnel
• Trained
• Personal hygiene and
sanitation
• Appropriate garments
Storage areas
• No unauthorized access
• Sufficient space
• Adequate ventilation,
temperature and relative
humidity
• Conditions checked,
monitored and recorded
• Segregation of rejected,
expired, recalled or returned
stock
• Toilet and washing facilities
separated from storage areas
• Narcotics/psychotropic
medicines as per national
legislation
• SOP for fire control
• No smoking and eating
• SOP and records for cleaning
• Waste management
• Pest-control
Access controlled and sufficient
space
Appropriate conditions for storage

Working document QAS/13.555
page 21
• SOP for handling spillages
Storage conditions
• As established by the
manufacturer
• Orderly, batch segregation,
stock rotation, first expired-
first out (FEFO)
• Stored off the floor
• Space to permit cleaning and
inspection
• Pallets in a good state of
cleanliness and repair
• Stacking of products without
damage
• Freeze-sensitive products –
use monitoring devices
• Cold rooms (qualification,
temperature mapping, alarm,
monitoring, records, back-up
system in case of failure)
Monitoring of storage conditions
• Temperature mapping protocol
and report
• Calibrated sensors/devices
• Ongoing monitoring with
records
• Out-of-limit and out-of-trend
results investigated, action
taken
Miscellaneous and hazardous
materials
• Rodenticides, insecticides,
fumigating agents and

Working document QAS/13.555
page 22
sanitizing materials
• Toxic substances and
flammable materials
Re-packaging
and re-labelling
• If performed – in compliance
with national legislation and
WHO GMP
Compliance with national legislation
and WHO GMP
Stock control
• Validated stock control system
• Batch number control and
expiry dating
• Periodic stock reconciliation
• Significant stock discrepancies
investigated
• Records maintained
• Damaged containers handled
Control of obsolete and outdated
materials and products
• SOP
• Regular checks
Recalled materials and products
• SOP
• Written records of actions with
signatures
• Products identified, recorded,
reconciled and stored
separately
• Decision by appropriately
qualified and experienced
member of staff
Returned goods
• SOP
• Quarantined and assessed
• Resale conditions
• Destruction in compliance
Stock control in place (e.g.
reconciliation, obsolete materials,
recalled products, returned goods,
FEFO and waste)

Working document QAS/13.555
page 23
with national requirements
• Records
Waste materials
• SOP
• Safe storage while awaiting
disposal
• Toxic substances and
flammable materials
• No accumulation
• Safe disposal, national
regulations
Documentation:
written
instructions and
records
• SOPs for activities
• Handling of expired stock
• Ensuring batch traceability
• Records for storage
conditions, precautions
• National regulations
concerning labels and
containers
• Comprehensive records of all
activities
• Retention of records
Record keeping ensuring traceability
(e.g. receiving, issuing, expired
goods)
153
Module V: Distribution 154
155
The PA (or contracted party) should have a well-managed distribution system meeting the 156
objectives of ensuring constant supply of quality medicines. Distribution should be done in 157
accordance with general principles of GMP. 158
159
Working document QAS/13.555
page 24
160
Area of
operation
Note Critical aspects
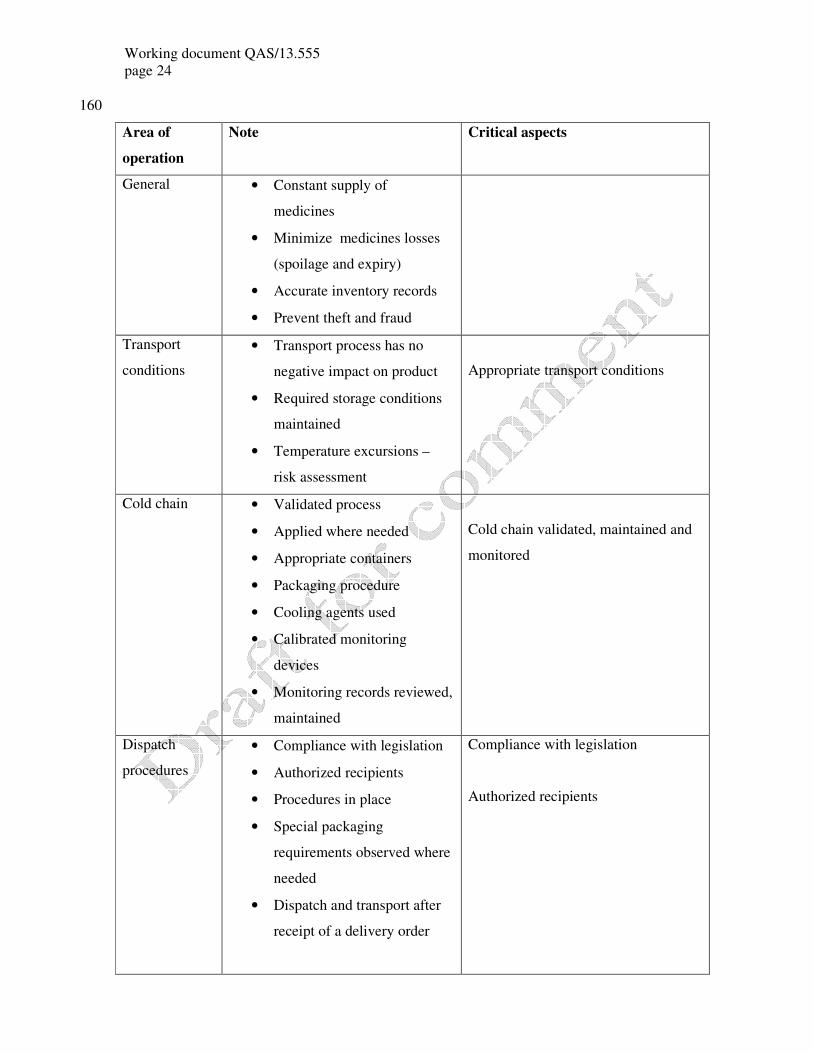
General • Constant supply of
medicines
• Minimize medicines losses
(spoilage and expiry)
• Accurate inventory records
• Prevent theft and fraud
Transport
conditions
• Transport process has no
negative impact on product
• Required storage conditions
maintained
• Temperature excursions –
risk assessment
Appropriate transport conditions
Cold chain • Validated process
• Applied where needed
• Appropriate containers
• Packaging procedure
• Cooling agents used
• Calibrated monitoring
devices
• Monitoring records reviewed,
maintained
Cold chain validated, maintained and
monitored
Dispatch
procedures
• Compliance with legislation
• Authorized recipients
• Procedures in place
• Special packaging
requirements observed where
needed
• Dispatch and transport after
receipt of a delivery order
Compliance with legislation
Authorized recipients

Working document QAS/13.555
page 25
Dispatch
containers
• Provide protection
• Appropriately labelled
• Prevent theft (e.g.
locked/wrapped)
Dispatch
records
• Detailed records kept (e.g.
date, customer name and
address; product name and
batch number and quantity)
• Products and batches
traceable
• Discrepancies investigated
Records ensure traceability of goods
Port of entry • Storage conditions met
• Temperature-sensitive
products handled
appropriately
• Security measures in place
(e.g. prevent theft, fraud and
bribery)
161
Module VI: Reassessment 162
163
Quality of products and services should be continuously monitored. This process includes 164
reassessment. 165
166
Area of
operation
Note Critical aspects
Reevaluation
of
manufacturers
• Reinspection frequency based
on risk assessment
• Within five-year cycle
• Change control
• Mechanism for suspension
and withdrawal
Reinspection policy and procedure
followed

Working document QAS/13.555
page 26
Reevaluation
of products
• Reevaluation procedure
• Within five-ear cycle
• Variations procedure
Reevaluation of product policy and
procedure followed
Monitoring
performance of
contractors
• Written procedure
• Covers continuous
monitoring, periodic review
and renewal of contracts
• System for documenting
service problems
Procedure followed for monitoring
performance
167
5. REVISION HISTORY 168
169
Date Version number Reasons for revision
February 2013 1.2013 Original
June 2013 2.2013 Editing in line with revisions proposed in
MQAS
170
171
172

Working document QAS/13.555
page 27
ANNEX 1 173
INTER-AGENCY FINISHED PHARMACEUTICAL PRODUCT QUESTIONNAIRE V2 174
175
176
Please fill out one form separately for each pharmaceutical product 177
Section 1: Administrative section 178
179
1.1 Product Identification 180
181 1.1.1 Active pharmaceutical ingredient(s) (use INN if any):_____________________ 182
183
1.1.2 Generic name of the product:______________________________________ 184
185
1.1.3 Trade (proprietary) name (if any):___________________________________ 186
187
1.1.4 Dosage form: 188
� Tablets � Capsules � Injectable � Syrups/oral liquids 189
190
� Other: (Please specify) 191
192
1.1.5 Strength per dosage unit: ______________________________________ 193
194
1.1.6 Route of administration: 195
�Oral � I.M. � I.V. � S.C. � Other (Please specify) 196
197
1.1.7 Please provide the formulation of the product (complete qualitative and quantitative 198
composition including active ingredient(s), overages if any and excipients). Mention 199
specifically if the product is a fixed-dose combination (FDC) or Copackaged 200
(Annex A) 201
_________________________________________________________ 202
203
_________________________________________________________ 204
205
_________________________________________________________ 206
207
1.2 Packaging 208 209
1.2.1 Description and materials used for primary packaging1 and pack size (quantity of 210
dosage-form units per pack): Annex B 211
212
1.2.2 Pack size and Description of secondary packaging materials: Annex C 213
214
1 For example, HDPE bottle, Alu-Alu strip, neutral glass vial.

Working document QAS/13.555
page 28
Contact details 215
216
1.3 Manufacturer identification 217 218
Name, address and activities of the manufacturer and manufacturing site(s) (or contract 219
manufacturer(s): 220
221
Name of
manufacturer,
contract
manufacturer if
any
Reference of
manufacturing
licence, date and
expiry date, if
any
Physical address.
Please specify
units, block if
existing
Telephone
number,
facsimile
number and
email contact
details
Activity (e.g.
packaging)
222
1.4 Supplier identification 223
(to be filled in if not identical to that indicated in 1.3) 224
225
Name of company:_________________________________________ 226
227
Physical address (complete details required):_____________________________ 228
_____________________________________________________________ 229
230
Telephone number:__________________ 231
232
Fax:_____________________________ 233
234
Website:___________________________________________________________ 235
236
Email:___________________________________ 237
238
Link with the product 239 240
Marketing licence holder Manufacturer 241
Distributor/wholesaler Other 242
243
1.5 Note for the applicant 244 245
Please note that the information in this questionnaire can be shared confidentially among 246
ICRC, MSF, WHO and UNICEF for procurement purposes. If you have any objection, please 247
indicate to the relevant agency that you are dealing with. 248
249
Has the dossier been submitted to any of the following agencies (ERP, ICRC, MSF, The 250
Union, WHO procurement centre, UNICEF)? 251

Working document QAS/13.555
page 29
252
Please precise the date of the submission: ______________________________________ 253
254
1.6 Regulatory (Licencing) status 255
256
1.6.1 In the country of manufacture 257
258
� Product registered and currently marketed – Licence no.:________________________ 259
Provide a copy in Annex D 260
261
� Product registered for marketing in the country of manufacturing but not currently 262
marketed – Licence no.:_________________________________________________ 263
264
265
� Product registered for export only – Licence no.:_____________________________ 266
267
� Product not registered (please clarify):_____________________________________ 268
269
270
� Please attach a certificate of pharmaceutical product (CPP) according to the WHO 271
Certification Scheme (WHO Technical Report Series, No. 863 in Annex E. An earlier 272
version is not acceptable). 273
274
� If a CPP cannot be obtained from the national drug regulatory authority (NDRA), 275
please state the reason and send equivalent document if any. 276
277
� Submit recent as well as historical deficiency/acceptance letters issued by the WHO 278
Prequalification Programme (PQP)/SRA in relation to the specific product dossier in 279
Annex F. 280
281
1.6.2 In other countries 282
283 List other countries where the product is registered and is currently marketed (please provide 284
registration number) 285
________________________________________________________________ 286
287
________________________________________________________________ 288
289
1.6.3 WHO prequalification status, if applicable 290
291 This product is prequalified by WHO/PQP.
2 292
293
� Yes � No 294
295
If yes, please attach a copy of the relevant WHO/PQP approval letter signed by your 296
company (Annex G) 297
298
299
2 WHO Prequalification website link: http://apps.who.int/prequal/.

Working document QAS/13.555
page 30
1.6.4 Submitted for prequalification: indicate date of submission, WHO acceptance letter 300
for product dossier review mentioning the WHO reference number assigned by 301
WHO for this specific product (Annex H) 302
_____________________________________________________________ 303
304
1.7 Samples for technical evaluation 305
306 1.7.1 Samples of finished product and insert information 307
308
You are required to please provide a sample of the finished product(s) offered, and relevant 309
inserts/leaflets. (If you cannot submit any of the above with the questionnaire, please state 310
why not and when you will do so.) (Annex I) 311
312
313
1.7.2 Label language (attach a copy): primary packaging 314
315
� Bilingual English/French � English � French � Other(specify) 316
317
1.7.3 Label language (attach a copy): secondary packaging 318
319
� Bilingual English/French � English � French � Other (specify) 320
321
For oral powder for suspension and powder for injection, in-use periods and storage 322
conditions after reconstitution should be stated on the product label 323
324
1.7.4 Patient information leaflet (Annex J) 325
326
� Yes (attach a copy) � No 327
Section 2. Active pharmaceutical ingredients 328
329
(In case of more than one active ingredient or more than one manufacturer is used, please 330
replicate this section) 331
2.1. Details of API used (INN if any): 332
333
……………………………………………………………………………………………… 334
335
2.1.1 Manufacturer 336
337 Manufacturer (name, physical address + country)/manufacturing site (please list all 338
alternative sources): 339
___________________________________________________________ 340
__________________________________________________________________ 341
__________________________________________________________________ 342
343
GMP certificate from the country of origin: attach a copy of the GMP certificate if available 344
in Annex K 345

Working document QAS/13.555
page 31
Last inspection of API manufacturing sites performed when available 346
(please attached GMP certificate or relevant letter) by: 347
348
� Finished product manufacturer 349
� WHO Prequalification Programme, Geneva 350
� EDQM 351
� US FDA 352
� PIC/S members 353
� Others (specify) 354
� None of above 355
Outcomes and date: ___________________________________________ 356
357
____________________________________________________________ 358 359
Is the API(s) used to manufacture this product is/are WHO-prequalified? 360
361
Yes No 362
363
2.1.2 API specifications 364
365
Specifications and standard test methods exist for this API: 366
367
� Yes � No 368
� Attach a copy of the internal API(s ) specifications in Annex L 369
370
API specifications: 371
372
� BP (Edition/Year): 373
� USP (Edition/Year): 374
� The International Pharmacopoeia (Ph.Int.) (Edition/Year) 375
� Others (specify): 376
� Specifications additional to those in the pharmacopoeia referred to above if available. 377
378
379
� If specifications are in-house specifications, different from BP, USP and Ph.Int., 380
attach also validated analytical methods in Annex M 381
382
2.1.3 Certificate of analysis 383
384
Please provide a copy of the certificate of analysis of the API from the API manufacturer as 385
well as from the finished pharmaceutical product (FPP) manufacturer in Annex N 386
387
388
389

Working document QAS/13.555
page 32
2.1.4 Suitability of monograph for API 390
391
Are you in a possession of the following information for APIs? 392
393
Certificate of suitability to the European Pharmacopoeia (CEP): please attach a copy of the 394
CEP and its annexes (Annex O) 395
396
397
Certificate No.: ______________________________________________________ 398
399
Open part of drug master file (DMF) registered in (country): 400
401
____________________________________________________________________ 402
403
Technical file (please attach): 404
405
� Yes � No 406
407
Section 3: Finished product 408
3.1 Manufacturing site GMP status 409
410
GMP inspections carried out by a NDRA 411
412
NRA of country of origin
Any other inspection of PIC/s member
GMP certificate no.
Valid until
Country
413
Please attach the recent/valid GMP certificates/letter (Annex P) 414
415
Other GMP inspections carried out by (tick all that apply): 416
417
Agency Date of audit Outcome
WHO Prequalification Programme
UNICEF Supply Division
MSF International
ICRC
Other (specify)

Working document QAS/13.555
page 33
3.2 Finished product specification 418
419 3.2.1 Non-sterile products or sterile products 420
421
422
Standard Edition Year published
BP
USP
Ph.Int.
In-house Year documented
Specifications additional to those in the
pharmacopoeia referred to above (e.g. dissolution,
syringeability): explain
Other ( specify)
423
� If specifications are in house specifications, different from BP, USP and Ph.Int., attach 424
copy of the in-house finished product specifications and also validated analytical methods 425
in Annex Q 426
427
� Please attach a copy of the certificate of analysis for the three last batches released in 428
Annex R 429
430
� Have the manufacturing methods for each standard batch size been validated? 431
432
� Yes � No 433
434
If no, please clarify:____________________________________________ 435
436
___________________________________________________________ 437
438
If yes, please provide details of validation status in the table below: 439
440
The batch size of the validated batches
The batch numbers of the validated batches
Manufacturing dates of the validated batches
Reference number for the process validation report
If processes are yet to be validated then reference number
for the process validation protocol should be indicated.
441
Provide batch formulae for all proposed batch sizes: 442
_______________________________________________________________ 443
444

Working document QAS/13.555
page 34
________________________________________________________________ 445
446
3.2.2 Additional information for sterile products 447
448
Provide the data on validation of the sterile aspects of the product including recent media fill 449
validation data as applicable in Annex S 450
451
Describe the method of sterilization used when applicable 452
________________________________________________________________ 453
454
455
3.2.3 Inactive ingredients (excipients) of medical/pharmaceutical relevance, amount in 456
dosage form or per dosage unit (e.g. contains alcohol 10%, paraben) 457
458
Specifications:_________________________________________________________ 459
460
Justification of specifications:______________________________________________ 461
___________________________________________________________________ 462
___________________________________________________________________ 463
464
3.3 Stability of finished product 465
466
3.3.1 Is stability testing data available? 467
468
� Yes � No 469
470
Please provide the protocol and the report for accelerated and real-time stability testing, 471
including: type and material of container; conditions (temperature/relative humidity/duration 472
of stability study); number of batches involved in the study (minimum three); batch sizes for 473
each lot tested; date of beginning of the study; and study conclusions. (These can be provided 474
in Annex T.) 475
476
3.3.2 Was the stability testing done on a product of the same formula, same API source, 477
manufactured on the same site and packed in the same packaging material as the 478
product that will be supplied? 479
480
� Yes � No 481
482
483
If no, describe the differences:_______________________________________________ 484
485
_________________________________________________________________ 486
487
488
489

Working document QAS/13.555
page 35
3.3.3 Please specify whether stability studies have been done or are on-going with all 490
declared API sources 491
492
� Yes � No 493
494
Submit a declaration in Annex U that stability studies have been done or are being done with 495
all declared API sources 496
497
If no, explain why: ________________________________________________________ 498
499
__________________________________________________________________ 500
501
502
3.3.4 Do you have on going stability data for this product? 503
504
� Yes � No 505
506
Attach status report of any ongoing stability studies in Annex V 507
508
3.3.5 Shelf-life as it appears on packaging 509
510
� 2 years � 3 years � 4 years � 5 years 511
512
Other: (specify) 513
514
515
3.3.6 Specific storage conditions for this product as they appear on the packaging and based 516
on stability studies (e.g. «Do not store above 30 °C – Protect from light»): 517
518
Temperature
Light
Humidity
Other (specify)
519
3.3.7 Product suitable for use in: 520
521
Zone I 522
Zone II 523
Zone III 524
Zone IVa 525
Zone IVb 526
Other (specify): 527
3.3.8 For oral powder for suspension and powder for injection, provide in-use 528
stability data and storage condition after reconstitution in Annex W 529

Working document QAS/13.555
page 36
530
Indicate the period until which the product is stable after reconstitution based on the 531
available in-use stability data: 532
533
Section 4: Safety/efficacy and or therapeutic equivalence (WHO Technical Report Series, 534
No. 902 Annex 11) 535
4.1 For innovator products 536
537
Please attach a summary of pharmacology, toxicology and efficacy of the product 538
539
4.2 For generic products: THERAPEUTIC EQUIVALENCE 540
541
� Demonstrated 542
543
� Not demonstrated 544
545
Not relevant, please explain why:_____________________________ 546
547
_______________________________________________________ 548 549
550
If demonstrated, 551 552
4.2.1 By in vivo bioequivalence studies 553
554
Indicate Biopharmaceutics Classification System (BCS) classification: 555
_________________________________________________________________ 556
557
_________________________________________________________________ 558
559
Justify above classification:______________________________________________ 560
561
____________________________________________________________ 562
563
Study period (dd/mm/yyyy): from to 564
565
Reference product 566
Generic name:
Dosage form:
Strength:
Brand/trade name:
Manufacturer:
Manufacture site:
Batch number:
Expiry date:
567

Working document QAS/13.555
page 37
Study protocol 568
Contract research organization
(CRO) name:
Country of study:
Number of volunteers:
Study design (describe in detail):
569
Bio batch size:
Bio batch number:
Bio batch API(s) source(s):
Study conclusion:
570
Attach graphic/pictorial representation of summary study results 571
572
Study results 573 F2 (similarity factor) value: (Standard 50–100%) 574
575
___________________________________________________________ 576
F1 (difference factor) value: 577
___________________________________________________________ 578
579
Study conclusion: 580 ____________________________________________________________ 581
582
____________________________________________________________ 583
584
Attach graphic/pictorial representation of summary study results 585
586
� Provide a copy of the report of the proof of therapeutic equivalence (BE study) 587
comparative dissolution profile, dissolution tests, others if any in Annex X 588
� For bioequivalence studies indicate the stringent regulatory authority 589
(SRA)/WHO/PIC(S) inspection status of the CRO (if the CRO has ever received 590
inspections in relation to the current or other studies). 591
592
� Attach schematic representation of study design 593
594
� Attach study protocol summary 595
596
4.2.2 By comparative in vitro dissolution tests according to conditions described in WHO 597
BCS classification document (WHO Technical Report Series, No. 937, or later) 598
599 Yes 600
601
No (explain):________________________________________________ 602
603
___________________________________________________________ 604
605
606
607

Working document QAS/13.555
page 38
Reference product 608
Generic name
Dosage form
Strength
Brand/trade name
Manufacturer
Manufacture site
Batch number
Expiry date
609 610
Name and contact details of laboratory performing tests: 611
______________________________________________________________ 612
613
614
4.2.3 By another method (please describe study conclusion briefly): 615
616
________________________________________________________ 617
618 Attach graphic/pictorial representation of summary study results in Annex Y 619
620
4.3 The product used in the therapeutic equivalence study is essentially the same as 621
the one that will be supplied (same materials from the same suppliers, same 622
formula and same manufacturing method) 623 624
Yes 625
626
No (explain what the differences are and justify that the differences do not have any 627
impact on the bioavailability):__________________________________________ 628
629
______________________________________________________________________ 630
631
� Please provide in Annex Z a flow diagram describing the manufacturing and control 632
process of this product with relevant parameters. 633
Section 5: Commitment and authorization 634
635
5.1 Commitment 636 637
I, the undersigned, 638
………………………………….………………………………………………… ................... …., 639
(position in the company, e.g. General Manager, Authorized Person, Responsible 640
Pharmacist), acting as responsible for the company……………………………………(name 641
of the company), certify that the information provided (above) is correct and true, 642
643
(if the product is marketed in the country of origin, select the appropriate box below) 644
645

Working document QAS/13.555
page 39
� and I certify that the product offered is identical in all aspects of manufacturing and 646
quality to that marketed in ……………………………………..(country of 647
origin),including formulation, method and site of manufacture, sources of active and 648
excipient starting materials, quality control of the product and starting material, 649
packaging, shelf-life and product information. 650
651
� and I certify that the product offered is identical to that marketed in 652
………………………………………………………………….(name of country),except: 653
……………………………………………………………………………… .................. …….. 654
…………………………………………………………………………………… . …………… 655
(e.g. formulation, method and site of manufacture, sources of active and excipient starting 656
materials, quality control of the finished product and starting material, packaging, shelf-657
life, indications, product information) 658
If any changes occur to the information after the submission of this product questionnaire, the 659
manufacturer/supplier undertakes to provide the relevant update as soon as possible. 660
661
662
Date: …………..…………………………… Signature: 663
664
……………………………………………… 665
666
5.2 Power of attorney 667
668
The manufacturer authorizes a distributor to submit the questionnaire 669
670 [SIGNATURE] 671
672 Distributor (Signed by Distributor for Manufacturer under power of attorney ) 673
674
Please provide a copy of the power of attorney (Annex AA) 675
676
677
5.3 Authorization for sharing information with other agency 678
679 I, the undersigned confirm that the company has no objection to the information contained 680
herein being shared with the agencies listed on page 2 (1.4) except: 681
682
___________________________________________________________________ 683
684
___________________________________________________________________ 685
686
687
688
689

Working document QAS/13.555
page 40
I, the undersigned, certify that the information provided above is accurate, correct, complete, 690
up-to-date and true at the time of submission 691
692
Full name:__________________________________________________________ 693
___________________________________________________________________ 694
695
Full title/position in company:____________________________________________ 696
697
____________________________________________________________________ 698
699
Company name:_______________________________________________________ 700
701
____________________________________________________________________ 702
703
704
705
Signature Date
706
707
Company seal/stamp: 708
709
710
711

Working document QAS/13.555
page 41
712
713
714
Section 6: Attachments/annexes 715
716
Attachments or Annexes to the questionnaire should be in PDF format and should be 717
well indexed to facilitate review 718 719
Please ensure that all documents necessary to enable objective evaluation of your product are 720
attached. This checklist may not be exhaustive. 721
722
A. Formulation of the product (complete qualitative and quantitative composition 723
including active ingredient(s) and excipients (1.1.7) 724
725
B. Description and composition of primary packing materials (1.2.1) 726
727
C. Description and composition of secondary packaging materials (1.2.2) 728
729
D. Copy of product registered and currently marketed – Licence no. (1.6.1) 730
731
E. Certificate of pharmaceutical product (CPP) according to the WHO Certification 732
Scheme (WHO Technical Report Series, No. 863 in the Annex. Earlier version is not 733
acceptable) (1.6.1) 734
735
F. Submit recent as well as historical deficiency/acceptance letters issued by PQP/SRA 736
in relation to the specific product dossier.(1.6.1) 737
738
G. Copy of the relevant WHO Prequalification approval letter signed by your company 739
(1.6.3) 740
741
H. WHO acceptance letter for product dossier review mentioning the WHO reference 742
number assigned by WHO for this specific product (1.6.4) 743
744
I. Package insert/leaflet (1.7.1) 745
746
J. Patient Information Leaflet (1.7.4) 747
748
K. GMP certificate from the country of origin (2.1.1) 749
750
L. Attach a copy of the internal API(s) specifications (2.1.2) 751
752
M. Validated analytical methods if specifications for finished product are in-house 753
specifications, different from BP, USP and Ph.Int. (2.1.2) 754
755
N. Copy of the certificate(s) of analysis of the API from the API manufacturer as well as 756
from the FP manufacturer (2.1.3) 757
758
O. Copy of the certificate of suitability to the European Pharmacopoeia CEP and its 759
annexes (2.1.4) 760

Working document QAS/13.555
page 42
761
P. Recent/valid GMP certificates/letter (3.1) 762
763
Q. If specifications are in house specifications, different from BP, USP and Ph.Int., 764
attach copy of the in-house finished product specifications and also validated 765
analytical methods (3.2.1) 766
767
R. Copy of the certificate of analysis for the three last batches released (3.2.1) 768
769
S. Data on validation of the sterile aspects of the product including recent media fill 770
validation data as applicable (3.2.2) 771
772
T. Protocol and report for accelerated and real time stability testing (3.3.1) 773
774
U. Submit a declaration that stability studies have been done or being done with all 775
declared API source (3.3.3) 776
777
V. Attach status report of any ongoing stability studies (3.3.4) 778
779
W. For oral powder for suspension and powder for injection, provide in-use stability data 780
and storage condition after reconstitution (3.3.8) 781
782
X. Provide a copy of the report of the proof of therapeutic equivalence (BE study) 783
comparative dissolution profile, dissolution tests, others if any (4.2.1) 784
785
Y. Attach graphic/pictorial representation of summary study results (4.2.3) 786
787
Z. Flow diagram describing the manufacturing and control process of this product with 788
relevant parameters (4.3) 789
790
AA. Copy of the power of attorney (5.2) 791
792

Working document QAS/13.555
page 43
793
ANNEX 2 794
INSPECTION REPORT: PROCUREMENT AGENCY 795
796
Section 1. General information 797
798
Name of organization:
Website reference:
Physical address:
Postal address:
Tel.:
Fax:
Contact person:
Email address:
Activities: Prequalification
Purchasing
Receiving and storage
Distribution
Reassessment
Date of assessment/inspection:
Products and/or product category
(e.g. pharmaceuticals, diagnostics,
medical devices)
Inspector:
799

Working document QAS/13.555
page 44
800
Section 2. Summary 801
General information about the procurement agent and site
___________________________________________________________________________
___________________________________________________________________________
History of inspections
___________________________________________________________________________
___________________________________________________________________________
Focus of the inspection and inspected areas
___________________________________________________________________________
___________________________________________________________________________
Summary of findings
General activities:
___________________________________________________________________________

Working document QAS/13.555
page 45
___________________________________________________________________________
Prequalification:
___________________________________________________________________________
___________________________________________________________________________
Purchasing:
___________________________________________________________________________
___________________________________________________________________________
Receiving and storage:
___________________________________________________________________________
___________________________________________________________________________
Distribution (including the ability to supply the needed products in quantities required):
___________________________________________________________________________
___________________________________________________________________________
Reassessment:
___________________________________________________________________________

Working document QAS/13.555
page 46
___________________________________________________________________________
802
Section 3. Observations and deficiencies/non-compliance 803
(Note: Module I should be used in all cases of assessment of a procurement agency. Modules 804
2 to 6 may be used depending on the activities performed by the procurement agency) 805
806
Module I: General requirements Classification
(C, M, O)*
1.
2.
3.
4.
5.
Module II: Prequalification
6.
7.
8.
9.
10.
Module III: Purchasing
11.
12.
13.
14.
Module IV: Receiving and Storage
15.

Working document QAS/13.555
page 47
16.
17.
18.
Module V: Distribution
19.
20.
21.
22.
Module VI: Reassessment
23.
24.
25.
26.
807
* 808
Critical Observation (C) 809
An observation relating to any activity, action or omission thereof, by the procurement 810
agency, in relation to product(s), that may result in a significant risk to the user. 811
812 Major Observation (M) 813
A non-critical observation that: 814
• may have a negative impact on a product in relation to prequalification, purchasing, 815
storage, distribution or requalification; and/or 816
• indicates a major deviation from the MQAS; and/or 817
• consists of several other deficiencies, none of which on its own may be major, but 818
which may together represent a major deficiency and should be explained and 819
reported as such. 820
821 Other Observation (O) 822
An observation that cannot be classified as either critical or major, but indicates a departure 823
from the recommendations in the MQAS (including GSP and GDP). 824
825
826

Working document QAS/13.555
page 48
Section 4. Outcome of the inspection (select one of the following options) 827
Based on the areas inspected, the personnel met and the documents reviewed, and considering
the findings of the inspection, including the observations listed above – the agency was
considered to be operating in compliance with the MQAS for the following activities (select
the appropriate one(s)) prequalification, purchasing, storage, distribution, requalification).
Or
Based on the areas inspected, the personnel met and the documents reviewed, and considering
the findings of the inspection, including the observations listed above – the agency was
considered not yet to be operating at an acceptable level of compliance with the MQAS for
the following activities (select the appropriate one(s)) prequalification, purchasing, storage,
distribution, requalification). The CAPAs will be reviewed after which a conclusion will be
made whether the procurement agency is operating in compliance with the MQAS. (A
reinspection may be considered before the conclusion is reached.)
Or
Based on the areas inspected, the personnel met and the documents reviewed, and considering
the findings of the inspection, including the observations listed above – the agency was
considered to be operating at an unacceptable level of compliance with the MQAS for the
following activities (select the appropriate one(s)): prequalification, purchasing, storage,
distribution, requalification).
828
829
830
_____________________________ _________________________ 831
Signature: Date: 832
833

Working document QAS/13.555
page 49
(Name):______________________ 834
(Print) 835
836
History of changes 837
Version 1. Initial format
Version 2. Additions Add under section 3
(Note: Module I should be used in all cases
of assessment of a procurement agency.
Modules 2 to 6 may be used depending on the
activities performed by the procurement
agency)
Add classification of observations as critical,
major and minor and definitions
Edit wording in section 4 for the outcome
838
839
*** 840