associative properties in water of copolymers of sodium 2-(acrylamido)-2-methylpropanesulfonate and...
TRANSCRIPT

Associative Properties in Water of Copolymers of Sodium2-(Acrylamido)-2-methylpropanesulfonate and MethacrylamidesSubstituted with Alkyl Groups of Varying Lengths
Hiroshi Yamamoto, Itsuro Tomatsu, Akihito Hashidzume, and Yotaro Morishima*
Department of Macromolecular Science, Graduate School of Science, Osaka University,Toyonaka, Osaka 560-0043, Japan
Received December 7, 1999; Revised Manuscript Received August 7, 2000
ABSTRACT: Associative properties of random copolymers of sodium 2-(acrylamido)-2-methylpropane-sulfonate (AMPS) with N-hexylmethacrylamide (C6MAm) and with N-octadecylmethacrylamide (C18MAm)in aqueous solutions were investigated comparing with those of AMPS copolymers with N-dodecyl-methacrylamide (C12MAm) reported previously. For characterization by fluorescence, all the polymerswere labeled with 1 mol % of naphthalene by terpolymerization using N-(1-naphthylmethyl)methacryl-amide. The polymers possessing C6 or C12 alkyl chains exhibit a strong tendency for intramolecularhydrophobic association to form unimolecular micelles when the CnMAm (n ) 6, 12) contents in thepolymers are in the ranges of 30-70 and 10-50 mol % for the C6 and C12 chains, respectively. A similartendency was observed for the polymers possessing C18 chains when the C18MAm contents are in therange of 5-20 mol %, but polymer micelles formed were not completely unimolecular. Both the lowerand upper limits of the CnMAm contents for all the three polymers decrease significantly with increasingthe length of the alkyl chain. When the CnMAm contents are either lower or higher than these limits,the polymer-bound alkyl chains undergo interpolymer associations. Fluorescence depolarization and 1HNMR relaxation times indicate that local motions of naphthalene labels and alkyl chains are morepronouncedly restricted in hydrophobic domains formed by longer alkyl chains. An inclination to theself-association of these polymers was discussed in terms of the ratio of the total number of carbon atomsin hydrophobes to the number of SO3
- in a polymer chain. A conclusion is that with C6 chains one needsto incorporate more carbon atoms into a polymer than with C12 and C18 chains to attain the same extentof hydrophobe associations. In other words, the CH2 and CH3 residues in the C6 chain are much lesseffective than those in the C12 and C18 chains for the self-association when compared at the samehydrophobe/charge ratio in a polymer chain, whereas there is no significant difference between those inthe C12 and C18 chains. Thus, the ability of the C6, C12, and C18 chains per carbon atom to associate is inthe order C18 g C12 . C6.
IntroductionHydrophobically modified water-soluble polymers have
been a target of extensive studies in the past decadesbecause of their potential in industrial applications suchas water-borne paints, coating fluids, cosmetics, food-stuff, drug delivery systems, oil recovery, and watertreatment and also because of their relevance to biologi-cal macromolecular systems.1-6
In amphiphilic polyelectrolytes, hydrophobic interac-tions among hydrophobic residues compete with repul-sive electrostatic interactions among charged segmentsin aqueous solution, resulting in the formation ofvarious types of micellelike nanostructures. The self-association behavior of amphiphilic random copolymerscan be controlled by molecular architectures whichinclude the sequence distribution of hydrophilic andhydrophobic monomer units,7-12 the spacer bond be-tween hydrophobes and the polymer backbone,13-16 andthe type of hydrophobes and their content in thepolymer.13,17-21 The type of micellelike nanostructuresis greatly different depending on whether hydrophobeassociations occur in an intra- or interpolymer fashionin aqueous solution. Highly preferential intrapolymerassociations lead to the formation of single-macromo-lecular or unimolecular micelles, whereas interpolymerassociations yield network structures, giving rise to“thickening” phenomena. For example, random copoly-mers of sodium 2-(acrylamido)-2-methylpropanesulfonate(AMPS) and methacrylamides substituted with bulky
hydrophobes at the N-position, in which hydrophobesare linked to the polymer backbone via an amide spacerbond, show a strong preference for intrapolymer as-sociations in water.17,22,23 As a result, they form compactunimolecular micelles even at high polymer concentra-tions. In contrast, random copolymers of AMPS and ahydrophobic methacrylate, in which hydrophobes arelinked via an ester spacer bond, show a tendency forinterpolymer associations even in dilute polymer solu-tions, resulting in the formation of multipolymer mi-cellelike aggregates.15,16
Previously, we reported on the self-association be-havior of copolymers of AMPS and methacrylamideN-substituted with a C12 alkyl chain (C12MAm) char-acterized by various techniques including fluorescence(lifetime, depolarization, quenching, and nonradiativeenergy transfer) using polymers labeled with pyreneand/or naphthalene, viscosity, gel permeation chroma-tography (GPC), static light scattering (SLS), quasielas-tic light scattering (QELS), and 1H NMR relaxationtimes.22-24 The self-associative properties of these co-polymers were found to be a strong function of C12MAmcontent (fC12) in the copolymer. When fC12 was lowerthan ca. 10 mol %, the polymer adopted a relatively openchain conformation, but there was a weak tendency forinterpolymer association, giving rise to hydrophobicallycross-linked chains depending on the polymer concen-tration.25 In the range 10 < fC12 < 50 mol %, thecopolymers showed a strong tendency for intrapolymer
7852 Macromolecules 2000, 33, 7852-7861
10.1021/ma9920487 CCC: $19.00 © 2000 American Chemical SocietyPublished on Web 09/23/2000

hydrophobe associations, leading to the formation ofunimolecular micelles. However, when fC12 exceeded 50mol %, multipolymer aggregates were formed, and allpolymer chains were found in the aggregate with nounimolecular micelles coexisting.23
The association properties of the polymer shoulddepend on the length of the alkyl chain, i.e., hydropho-bicity, and also on the hydrophobe/charge ratio in apolymer chain, i.e., the ratio of the total number ofcarbon atoms (methylene and methyl groups) in alkylsubstituents and AMPS charges in a polymer chain.Thus, it is important to elucidate how the “effectiveness”of the hydrophobe to cause the polymer to micellizedepends on the length of the alkyl chain when thehydrophobe/charge ratios are the same.
In this work, we synthesized copolymers of AMPSwith N-hexylmethacrylamide (C6MAm) with the con-tents of C6MAm (fC6) ranging 0-80 mol % and alsocopolymers of AMPS with N-octadecylmethacrylamide(C18MAm) with C18MAm contents (fC18) ranging 0-40mol %. These copolymers were labeled with naphthalene(1 mol %) for fluorescence studies (Chart 1). We mainlyfocused on the association behavior of the AMPS-C6MAm copolymers as characterized using fluorescencedecay and depolarization, FT-IR, 1H NMR relaxation,and QELS techniques. To discuss the effects of thelength of the alkyl chain among C6, C12, and C18 chainsin a systematic manner, characterization data for theAMPS-C6MAm copolymers were compared with thosefor the AMPS-C18MAm copolymers and also with thosefor the AMPS-C12MAm copolymers obtained in ourearlier studies.22-24
Experimental Section
Monomers. N-Hexylmethacrylamide (C6MAm) was syn-thesized from hexylamine (Tokyo Chemical Industry Co.) andmethacryloyl chloride (Tokyo Chemical Industry Co.) in amanner analogous to the synthesis of N-dodecylmethacryl-amide (C12MAm) as reported previously.26 C6MAm was puri-fied by distillation under reduced pressure: yield 73.2%; bp77-78 °C (at 0.2 mmHg). 1H NMR (270 MHz, CDCl3): δ 0.9(t, 3H), 1.3-1.5 (m, 8H), 2.0 (m, 3H), 3.3 (q, 2H), 5.3 (m, 1H),5.6 (s, 1H), and 5.9 (s, 1H). Anal. Calcd for C10H19NO: C, 70.96;H, 11.31; N, 8.27. Found: C, 70.44; H, 11.23; N, 8.16.
N-Octadecylmethacrylamide (C18MAm) was synthesizedfrom octadecylamine (Sigma-Aldrich Co.) and methacryloylchloride in a manner analogous to the synthesis of C12MAm.The crude product was recrystallized twice from ethanol anddried in vacuo at room temperature overnight: yield 65.8%;mp 65-66 °C. 1H NMR (270 MHz, CDCl3): δ 0.9 (t, 3H), 1.3-1.5 (m, 32H), 2.0 (m, 3H), 3.3 (q, 2H), 5.3 (m, 1H), 5.6 (s, 1H),and 5.9 (s, 1H). Anal. Calcd for C22H43NO: C, 78.27; H, 12.84;N, 4.15. Found: C, 78.08; H, 12.79; N, 4.22.
N-(1-Naphthylmethyl)methacrylamide (1NpMAm) was syn-thesized as reported previously.27 2-(Acrylamido)-2-methyl-propanesulfonic acid (AMPS) was purchased from Wako PureChemicals and used without further purification. 2,2′-Azobis-(isobutyronitrile) (AIBN) (Wako Pure Chemicals) was recrys-tallized twice from ethanol prior to use.
Terpolymerization. A general procedure for the prepara-tion of naphthalene-labeled polymers is as follows: A mixtureof AMPS, an alkylmethacrylamide, 1 mol % of 1NpMAm, and0.1 mol % (based on the total monomers) of AIBN weredissolved in distilled N,N-dimethylformamide (DMF) in a glassampule. The mole percents of the alkylmethacrylamidesemployed were 0-80 mol % for C6MAm and 0-40 mol % forC18MAm. The ampule was outgassed by six freeze-pump-thaw cycles on a vacuum line before sealing. The sealedampule was immersed in a temperature-controlled water bathat 60 °C for 12 h. The reaction mixture was poured into excessether to precipitate polymers. The polymer was purified byreprecipitation from a methanol solution into excess etherthree times and then dissolved in a dilute aqueous NaOHsolution. The aqueous solution was dialyzed against purewater for a week and finally lyophilized. The compositions ofthe terpolymers were determined by elemental analysis (N/Cratios) and UV absorption spectroscopy.
Other Materials. KBr (IR spectroscopy grade), NaCl, andLiClO4 were purchased from Wako Pure Chemicals and usedwithout further purification. Water purified with a MilliporeMilli-Q system was used for all measurements.
Measurements. a. GPC. Measurements were performedat 40 °C with a JASCO GPC-900 equipped with ShodexAsahipak GF-7M HQ columns in combination with a JASCOUV-975 detector. A methanol solution containing 0.2 M LiClO4
was used as eluent at a flow rate of 1 mL/min. Molecularweights of the polymer samples were calibrated with poly-(ethylene oxide) standard samples purchased from ScientificPolymer Products.
b. Absorption and Fluorescence Spectra. Absorptionspectra were recorded with a JASCO V-550 spectrophotometer.Steady-state fluorescence spectra were measured on a HitachiF-4500 fluorescence spectrophotometer using a 1 cm pathlength quartz cuvette with excitation at 290 nm.
c. FT-IR. IR spectra were taken with KBr pellets on aJASCO FT/IR-410 spectrophotometer. The KBr pellets wereprepared with polymer samples recovered from their aqueoussolutions by a freeze-drying technique.
d. Fluorescence Lifetime. Fluorescence decays weremeasured by a time-correlated single-photon counting tech-nique using a Horiba NAES-550 system equipped with a flashlamp filled with H2. Sample solutions were excited at 290 nmand monitored with a cutoff filter (HOYA UV32) placedbetween the sample and detector. Both decay and responsefunctions were measured simultaneously. Decay data wereanalyzed by a conventional deconvolution technique.
e. Fluorescence Depolarization. Fluorescence anisotropy(r) was measured on a Hitachi F-4500 fluorescence spectro-photometer equipped with a polarizer (excitation side) and ananalyzer (detection side).23 Fluorescence spectra for the naph-thalene-labeled polymers were obtained at 25 °C by excitationat 290 nm and monitored at 325 nm. Both the excitation andemission slit widths were maintained at 10.0 nm. The r valuewas calculated from
where Ivv and Ivh are the fluorescence intensities measuredwith parallel and perpendicular orientations of the excitationand emission polarizers, respectively, and g is the factor forinstrumental correction (i.e., g ) Ihv/Ihh). The rotational cor-relation time (τφ) was calculated from28
where r0 is the limiting value of r for steady-state fluorescencein rigid media where no rotation occurs and τ is the fluores-
Chart 1
r ) (Ivv - gIvh)/(Ivv + 2gIvh) (1)
r0/r ) 1 + 3τ/τφ (2)
Macromolecules, Vol. 33, No. 21, 2000 Association of Polyelectrolyte-Bound Alkyl Chains 7853

cence lifetime. The value of r for 1NpMAm in glycerolmeasured at 265 K (i.e., r ) 0.18) was used as a value for r0.This value was in agreement with that obtained by time-resolved fluorescence depolarization measurement.23
f. 1H NMR Relaxation Times. 1H NMR spectra wererecorded with a JEOL EX-270 spectrometer using a deuteriumlock at a constant temperature of 30 °C during the whole run.Sample solutions of 1.0 mg/mL polymer in D2O containing 0.05M NaCl were deaerated in NMR tubes by purging with Ar gasfor 30 min prior to measurement. Proton spin-lattice relax-ation times (T1) were determined using a conventional inver-sion-recovery technique with a 180°-τ-90° pulse sequence.29-31
Proton spin-spin relaxation times (T2) were determined bythe Carr-Purcell-Meiboom-Gill (CPMG) method using a{90°xτ(180°y2τ)n} pulse sequence.32
g. Quasielastic Light Scattering (QELS). QELS datawere obtained at 25 ( 0.1 °C using an Otsuka ElectronicsPhotal DLS-7000 light scattering spectrometer equipped withan Ar laser lamp (60 mW at 488 nm) and detector optics. AnALV-5000E digital multiple-τ correlator (Langen-GmbH) wasemployed for data collection. The autocorrelation function wasmeasured at different angles (50°-130°). Sample solutionswere filtered with a 0.2 µm pore size membrane filter prior tomeasurement.
The observed intensity autocorrelation function, g(2)(t), wasmeasured experimentally, which is related to normalizedautocorrelation function, g(1)(t), by the Siegert relation
where â is a constant parameter for an optical system and Bis a baseline term. To obtain the relaxation time distribution,τA(τ), the inverse Laplace transform (ILT) analysis for g(2)(t)was performed by conforming the REPES algorithm,33 accord-ing to the equation
where τ is the relaxation time. The relaxation time distribu-tions are given as a τA(τ) vs log τ profile with an equal area.
The translational diffusion coefficient, D, are calculated fromILT moments as
where Γ is the relaxation rate and q represents the magnitudeof scattering vector expressed as
where θ is the scattering angle and n is the refractive indexof the solution. The hydrodynamic radius, Rh, is given by theEinstein-Stokes equation
where kB is the Boltzmann constant, T is the absolutetemperature, and η is the solvent viscosity. The details ofQELS instrumentation and theory are described in the litera-ture.34,35
h. Static Light Scattering (SLS). SLS data were obtainedat 25 °C with an Otsuka Electronics Photal DLS-7000 lightscattering spectrometer equipped with an Ar laser (50 mW at488 nm). Sample solutions of polymers in 0.2 M NaCl werefiltered with a 0.2 µm pore size membrane filter prior tomeasurement. The details of SLS measurements and dataanalysis are described elsewhere.24
i. Preparation of Sample Solutions. For all measure-ments sample solutions were prepared as follows: Solidpolymer samples (recovered by a freeze-drying technique) weredissolved into pure water or D2O (for NMR measurements) at90 °C with vigorous stirring for 15 min. After cooling the
solutions to room temperature, a predetermined amount ofNaCl was added to the polymer solutions to adjust ionicstrength.
ResultsCharacteristics of the Polymers. We reported
previously that the copolymerization of AMPS andC12MAm36 and also the copolymerization of AMPS and1-NpMAm27 were characterized as “ideal copolymeri-zation” systems, resulting in copolymer compositionsequal to monomer feed compositions and completelyrandom distributions of the two monomer units alongthe polymer chain. In the case of the terpolymerizationof AMPS, C6MAm or C18MAm, and 1-NpMAm, weconfirmed that the composition in the terpolymer isvirtually the same as that in the monomer feed as inthe case of the terpolymer of AMPS, C12MAm, and1-NpMAm synthesized previously.22 Therefore, the dis-tributions of the monomer units along the polymer chainfor the terpolymers are essentially random.
In Table 1 are listed weight-average molecular weights(Mw) estimated by GPC in methanol containing 0.2 MLiClO4 for the three terpolymers with varying hydro-phobe contents. In our earlier work on AMPS-C12MAmcopolymers, we confirmed that Mw values estimated byGPC in methanol containing 0.2 M LiClO4 using poly-(ethylene oxide) standards for calibration are a roughmeasure of “real” Mw determined by SLS.24 Thus, weperformed GPC measurements to roughly assess Mw.As can be seen from Table 1, Mw values for all thepolymers are of the same order of magnitude, (0.8-2.2)× 104, (1.1-3.1) × 104, and (0.8-1.5) × 104 for theC6MAm, C12MAm, and C18MAm containing terpoly-mers, respectively. Dry samples of these terpolymers aresoluble in water, yielding optically clear solutions, upto the alkylmethacrylamide contents of 80, 60, and 40mol % for the C6MAm, C12MAm, and C18MAm con-taining terpolymers, respectively. When the alkyl-methacrylamide contents in the respective polymers
g(2)(t) ) B[1 + â|g(1)(t)|2] (3)
g(1)(t) ) ∫τA(τ) exp(-t/τ) d ln τ (4)
D ) (Γ/q2)qf0 (5)
q ) 4πnλ
sin(θ/2) (6)
Rh )kBT
6πηD(7)
Table 1. Molecular Weights and Hydrodynamic Radii ofthe C6-, C12-, and C18-Carrying Polymers
hydrophobiccomonomer
hydrophobecontent (mol %)
Mwa
(×104) Mw/Mna
Rhb
(nm)
C6MAm 0 1.1 2.1 7.35 0.9 2.5 5.7
10 0.8 2.6 4.4, 5420 1.8 3.3 1230 2.1 4.2 7.340 2.2 4.0 5.950 2.2 3.7 4.860 2.2 3.1 4.070 1.8 2.3 3.880 2.0 2.2 8.1
C12MAm 10 2.8 2.2 7.120 3.1 2.2 4.930 2.6 2.4 3.940 3.0 2.2 4.050 1.5 1.9 4.355 1.5 2.1 5.760 1.9 1.8 8.1
C18MAm 5 1.5 2.9 9.810 1.3 3.3 7.120 0.8 2.1 6.725 0.9 2.5 1630 1.5 2.2 6-10, 4835 1.0 2.2 1540 0.9 1.5 31
a Determined by GPC using methanol containing 0.2 M LiClO4as an eluent at 40 °C. b Determined by QELS in 0.05 M NaClaqueous solutions at 25 °C.
7854 Yamamoto et al. Macromolecules, Vol. 33, No. 21, 2000

exceed these levels, polymers become insoluble in water.Therefore, in this study, we employed polymers withhydrophobe contents lower than these solubility limits.
Fluorescence Lifetime. The polarity of microenvi-ronments around naphthalene labels is known to bereflected in their fluorescence lifetimes; the lifetime islonger in less polar media.37 When the hydrophobecontent is sufficiently high, polymer-bound hydrophobesaggregate into a microdomain in which naphthalenelabels may be entrapped. On the other hand, if hydro-phobe contents are low, naphthalene labels would beexposed to the aqueous phase.
Fluorescence lifetime measurements for the naph-thalene-labeled polymers were performed in 0.05 MNaCl aqueous solution at 25 °C. All the fluorescencedecay data for polymers possessing C6, C12, and C18 alkylchains (Chart 1) were best-fitted to a single-exponentialfunction with ø2 ranging from 1.0 to 1.3 independent ofthe hydrophobe content. Fluorescence lifetimes areplotted in Figure 1 against the hydrophobe content forthe three polymers with different alkyl chain lengths.When the hydrophobe content is zero (i.e., naphthalene-labeled polyAMPS), the fluorescence lifetime is 36 ns.However, the lifetime increases up to ca. 53 ns whenthe hydrophobe contents are sufficiently high for all thepolymers. These observations indicate that, with in-creasing hydrophobe content, the formation of hydro-phobic domains proceeds, and naphthalene labels areincorporated in hydrophobic domains. Hence, the fluo-rescence lifetime provides information about the forma-tion of hydrophobic domains. The longest lifetimeobserved (ca. 53 ns) thus reflects a situation where thelabels are completely incorporated in hydrophobic do-mains formed by the polymer-bound alkyl chains.
As can be seen from Figure 1, the lifetime increasessignificantly in a relatively narrow range of the hydro-phobe contents in the polymer depending on the lengthof the alkyl chain. In the case of the C6 alkyl chain, thefluorescence lifetimes are practically the same as thatof polyAMPS-bound naphthalene up to a C6MAm con-tent (fC6) of ca. 20 mol %, suggesting all naphthalenelabels are exposed to the aqueous phase. The lifetimestarts to increase significantly near fC6 ≈ 30 mol % andcontinues to increase with increasing fC6 up to 80 mol% although the increase slows down at fC6 > 60 mol %,exhibiting a tendency for saturation near a lifetime ofca. 53 ns. On the other hand, in the cases of the C12
and C18 chains, the lifetime increases to a saturatedlevel at much lower hydrophobe contents than in thecase of the C6 chain. In our earlier work on AMPS-C12MAm copolymers, we confirmed that the formationof hydrophobic domains could be monitored through themicropolarity reported by fluorescence labels.22 Thus,we can interpret the results in Figure 1 as indicatingthat the self-association of polymer-bound C6 chainsstarts to occur at a much higher hydrophobe contentthan in the cases of the C12 and C18 chains. It should benoted that the saturated value of the lifetime is constantat ca. 53 ns regardless of the length of the alkyl chain.
The sigmoidal curve observed for the C6 polymerssuggests that the hydrophobic association occurs in acooperative manner. Namely, once hydrophobic associa-tion starts to occur at a certain hydrophobe content,polymer-bound hydrophobes associate more favorably,leading to a large increase in the formation of hydro-phobic domains in a relatively narrow range of hydro-phobe contents.
Associations of polyelectrolyte-bound hydrophobesoccur competing with electrostatic repulsion. Therefore,added salt enhances the hydrophobic association byshielding electrostatic repulsive interactions. Fluores-cence lifetimes for the C6 polymers at varying concen-trations of added salt are presented in Figure 2. Withincreasing ionic strength, the hydrophobe content atwhich the lifetime starts to increase shifts toward lowerfC6. At fC6 < 20 mol %, the effect of added salt on thefluorescence lifetime is small because the numbers ofthe C6 chains per polymer are not enough to formhydrophobic domains even though electrostatic repul-sions are shielded by added salt. On the other hand,when fC6 is sufficiently high (i.e., fC6 > 70 mol %), theeffect of added salt on the fluorescence lifetime is smallbecause the hydrophobic association prevails over elec-trostatic repulsion even without salt added due to alarge enough number of hydrophobes relative to thenumber of charge in the polymer. A most pronouncedsalt effect was observed in the regime 40 < fC6 < 50mol %. For example, the lifetime for fC6 ) 40 mol % isca. 36 ns in pure water, but it increases to ca. 48 ns ina 0.5 M NaCl aqueous solution. These observationssuggest that the AMPS-C6MAm copolymers may findan application as a salt-responsive water-soluble poly-mer because they adopt an open chain conformation inpure water but collapse into a compact conformation
Figure 1. Fluorescence lifetimes for naphthalene labels inthe C6-, C12-, and C18-carrying polymers plotted as a functionof the hydrophobe content: Polymer concentration: 1 g/L in a0.05 M NaCl aqueous solution.
Figure 2. Plots of the fluorescence lifetimes for naphthalenelabels in the C6-carrying polymers as a function of fC6 atvarying concentrations of added NaCl. Polymer concentra-tion: 1 g/L.
Macromolecules, Vol. 33, No. 21, 2000 Association of Polyelectrolyte-Bound Alkyl Chains 7855

(see later subsection) accompanied by the formation ofhydrophobic domains in saltwater. It is to be noted herethat the copolymers with fC6 ) 70 and 80 mol % werefound to be insoluble in 0.5 M NaCl aqueous solutionsbecause of a salting-out effect.
FT-IR. Figure 3 shows an example of FT-IR spectrafor the C6 polymers with fC6 ) 60 and 80 mol %compared in the range 1500-1700 cm-1. An IR spec-trum for naphthalene-labeled polyAMPS (i.e., fC6 ) 0mol %) is also presented as a reference in Figure 3. ForFT-IR measurements, we used solid polymer samplesrecovered from ca. 1 g/L aqueous solutions of thepolymers by a freeze-drying technique. All the polymersshow two characteristic IR absorption bands in thiswavenumber region due to the amide bond, the peaksat higher and lower wavenumbers attributable to amideCdO stretching vibration (amide I band) and amideN-H deformation (amide II band), respectively. Anobservation to be noted is that the amide II band forthe C6 polymers shifts toward lower wavenumberscomparing to that for the reference polymer. This lower-wavenumber shift is ascribable to the hydrogen bondingof amide spacer bonds.
As we discussed previously,17 micellar structuresformed from associations of polyelectrolyte-bound hy-drophobes in aqueous solutions are retained, if notperfectly, in freeze-dried solid samples. Hydrophobicdomains in the micellar structures are microscopicallyphase-separated from the water phase and sustainedin water by hydrated polyelectrolyte segments aroundthe hydrophobic domains. This microphase-separatedstructure should remain intact when aqueous solutionsare frozen. When the frozen solution is subjected tofreeze-drying, water molecules in the bulk phase andin the hydration layer are removed, leaving a polymersolid phase behind with the microphase structuresremaining as such. Therefore, it is reasonable to con-sider that the hydrogen bonds detected by FT-IR (Fig-ure 3) are those existing in association structures inaqueous solutions.
We measured IR spectra on the C6 polymers ofvarying fC6, and the wavenumbers of the amide II band
are plotted in Figure 4 as a function of fC6. Thewavenumbers are practically constant at fC6 < 40 mol%. However, the wavenumber commences to decreasenear fC6 ≈ 50 mol % with increasing fC6 and eventuallysaturates in the range 70 < fC6 < 80 mol %, exhibitinga wavenumber shift of ca. 20 cm-1 compared to theamide II band of the reference polymer of fC6 ) 0 mol%. This tendency fairly agrees with the tendency of thefluorescence lifetime shown in Figure 1, thus suggestingthat hydrogen bonding is more favorably formed be-tween the spacer amide bonds when the C6 chains formhydrophobic domains. Hence, we can monitor the for-mation of hydrophobic domains by IR spectroscopyfocusing on the amide II band.
Fluorescence Depolarization. Fluorescence depo-larization of naphthalene labels covalently linked to apolymer chain is a useful tool for studies of dynamicproperties of the polymer chain.38,39 Previously, wedemonstrated that fluorescence depolarization of naph-thalene labels on hydrophobically modified polyelectro-lytes provided information on the formation of hydro-phobic domains due to the fact that local motions ofnaphthalene labels are restricted when the labels areincorporated in the hydrophobic domains.23
In Figure 5, rotational correlation times (τφ) fornaphthalene labels in the C6, C12, and C18 polymers,calculated from anisotropy (r) values using eq 2, are
Figure 3. FT-IR spectra for the C6-carrying polymers withfC6 ) 0, 60, and 80 mol % measured as KBr pellets. Arrowsindicate the amide II band.
Figure 4. Plot of the wavenumber of the amide II band forthe C6 polymers as a function of fC6.
Figure 5. Rotational correlation times for the C6-, C12-, andC18-carrying polymers determined by fluorescence depolariza-tion plotted as a function of the hydrophobe content. Thedepolarization experiments were performed with 1 g/L polymersolutions containing 0.05 M NaCl at 25 °C. The data for theC12 copolymers are cited from ref 23.
7856 Yamamoto et al. Macromolecules, Vol. 33, No. 21, 2000
plotted as a function of the hydrophobe content. Ingeneral, not only local motions of a fluorescence labelwithin a polymer environment but also motions of thewhole polymer molecule would contribute to the fluo-rescence depolarization. The rotational correlation timefor a rigid sphere (τs) of the radius of R is given by
In the case of the C6 polymer, the hydrodynamic radius(Rh) for fC6 ) 80 mol % was observed to be 8.1 nm byQELS (see later subsection). Assuming this polymer asa rigid sphere with a radius of 8 nm, the rotationalcorrelation time for the polymer molecule may beroughly estimated from eq 8 to be 1.4 µs, which is muchlonger than a value of τφ (360 ns) observed from thefluorescence depolarization of naphthalene labels on theC6 polymer (Figure 5). From similar estimations of therotational correlation times for the other polymers ascompared to the observed τφ values, we concluded thatthe τφ values observed for naphthalene labels reflectessentially the rotational motions of the label itself inpolymer environments.
In the case of the C6 polymer, τφ values are practicallyconstant at fC6 < 50 mol %, and the value starts toincrease near fC6 ≈ 60 mol % with increasing fC6,followed by a sharp increase between fC6 ) 70 and 80mol %. This tendency is in contrast to the tendencyobserved for the fluorescence lifetime (Figure 1). In theregion 60 < fC6 < 80 mol %, the τφ value increasessignificantly, whereas the fluorescence lifetime exhibitsa saturation tendency, that is, when fC6 is increasedfrom 60 to 80 mol %, the fluorescence lifetime increasesonly by ca. 3 ns, whereas the τφ value greatly increasesfrom ca. 40 to 360 ns. This indicates that at fC6 ≈ 60mol % the labels are incorporated into hydrophobicdomains formed by C6 chains and experience a hydro-phobic microenvironment, but the labels can rotate quitefreely within the hydrophobic domain. With an increasein fC6 beyond 60 mol %, motions of the labels becomemore restricted although the micropolarity around thelabels has already reached a saturated level.
In the cases of the C12 and C18 polymers, the τφ valuesincrease gradually first as the hydrophobe contents areincreased and then start to increase sharply at fC12 ≈55 and fC18 ≈ 35 mol %, respectively. The lifetimes ofnaphthalene fluorescence for these two polymers reacha saturated level of ca. 53 ns at hydrophobe contentsnear 30 mol % (Figure 1), but there is a large differencein the τφ values between the C12 and C18 polymers athigher hydrophobe contents. In the case of the C18polymer, the τφ value jumps up to ca. 950 ns at fC18 )40 mol %, while the τφ value for the C12 polymer at thesame hydrophobe content is only ca. 100 ns.
Results in Figure 5, taken together with results inFigure 1, led us to conclude as follows: The C6, C12, andC18 alkyl chains undergo self-association to form hydro-phobic domains when their contents in the polymersexceed a certain level that is dependent on the lengthof the alkyl chain. The size of the hydrophobic domaingrows with increasing hydrophobe content to an extentwhere naphthalene labels are completely entrapped.Even after the labels are completely incorporated inhydrophobic domains, thus reporting a saturated mi-cropolarity in their fluorescence lifetimes, the rigidityof the hydrophobic domain greatly increases with fur-ther increasing hydrophobe content, hence the labels
reporting a large increase in their motional restriction.The rigidity or tightness of the hydrophobic domaindepends strongly on the length of the alkyl chain. Asthe number of carbon atoms in an alkyl chain isincreased, the tightness of hydrophobic domains in-creases greatly. Thus, the largest extent of the motionalrestriction of the naphthalene label is observed for theC18 chains whereas the smallest extent was observedfor the C6 chains. These results suggest that the rigidityof hydrophobic domain is related to the dynamic motionof each alkyl chain within the domain; i.e., shorterchains can move more freely in the domain.
1H NMR Relaxation Times. 1H NMR relaxationtechniques provide a useful tool to study local motionsof polymer chains.29-31 We performed 1H NMR relax-ation time measurements on the C6, C12, and C18polymers in D2O containing 0.05 M NaCl to obtaininformation about changes in the dynamic motion of thealkyl chains upon hydrophobic association. For thecalculation of the spin-lattice relaxation time (T1) andthe spin-spin relaxation time (T2), we chose the methylproton peak in the C6, C12, and C18 alkyl chains becausethe methyl protons in these alkyl chains give an isolatedpeak at ca. 0.9 ppm, whereas the methylene proton peakin the alkyl chains (at ca. 1.3 ppm) overlaps with peaksdue to the methyl protons in the AMPS unit and themethyl and methylene protons in the main chain.23 InFigure 6, T1 and T2 values are plotted against thehydrophobe content. (Resonance peaks due to the meth-yl protons for 10 mol % hydrophobe content were sosmall that we were unable to determine T1 and T2values.) The spin-lattice relaxation occurs most ef-
τs ) 4πR3η/kBT (8)
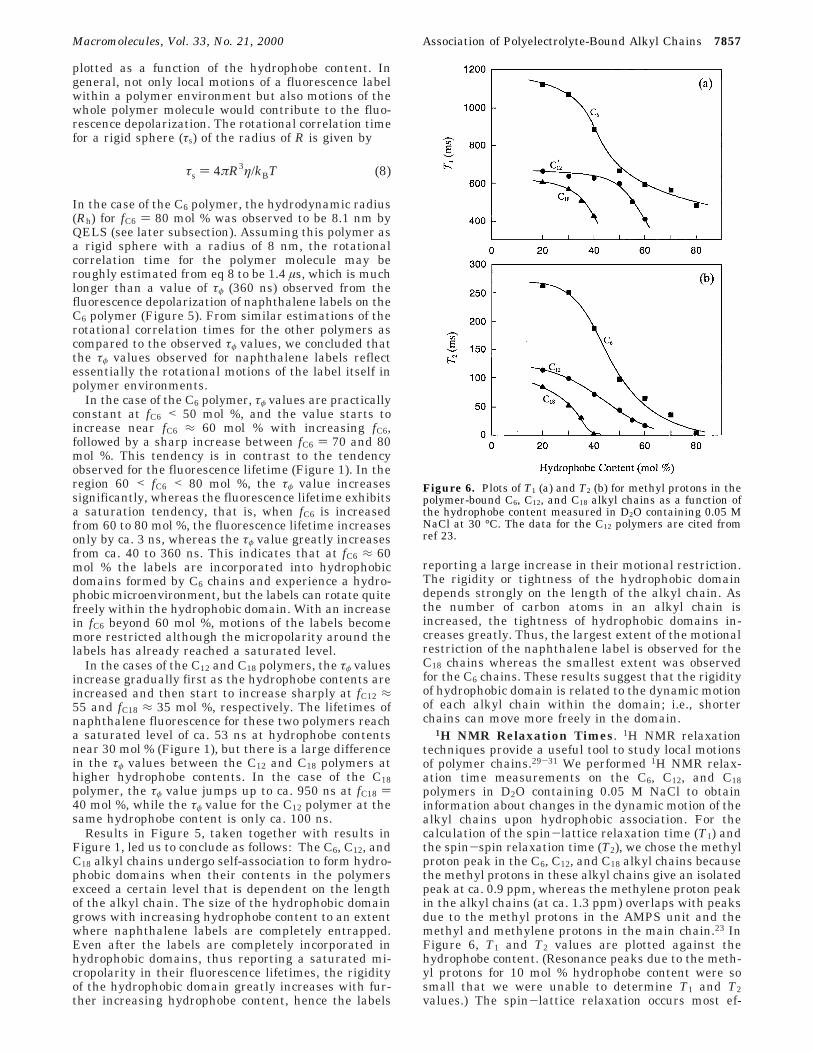
Figure 6. Plots of T1 (a) and T2 (b) for methyl protons in thepolymer-bound C6, C12, and C18 alkyl chains as a function ofthe hydrophobe content measured in D2O containing 0.05 MNaCl at 30 °C. The data for the C12 polymers are cited fromref 23.
Macromolecules, Vol. 33, No. 21, 2000 Association of Polyelectrolyte-Bound Alkyl Chains 7857

ficiently through molecular motions whose frequency iscomparable to the NMR frequency.40 Therefore, T1decreases concurrently with T2 as the molecular motiondecreases. In the case of the C6 chain, both T1 and T2values decrease significantly in the range 30 < fC6 < 60mol %. This tendency is similar to that observed for thefluorescence lifetime of naphthalene labels (Figure 1)but different from the tendency observed for the fluo-rescence depolarization (Figure 5). In the cases of theC12 and C18 chains, both T1 and T2 values are alreadydecreased greatly at their content of 20 mol %, indicat-ing that hydrophobic domains are formed at lowerhydrophobe contents than in the case of the C6 chain,consistent with the results in Figure 1. The T1 valuesfor the C12 and C18 chains show a tendency to decreasemore significantly with increasing their contents near50 and 30 mol %, respectively. This tendency is some-what similar to the tendency observed for the fluores-cence depolarization (Figure 5). These observationsindicate that the motional freedom of the terminalmethyl group in alkyl chains depends strongly on thelength of the alkyl chain when they form hydrophobicdomains.
QELS. Distributions of the relaxation times (τ) inQELS for the C6 polymers with varying fC6 in 0.05 MNaCl aqueous solutions observed at a scattering angleof 90° are presented in Figure 7. The relaxation timedistributions were also measured at varying scatteringangles (50°-130°), and the relaxation rate (Γ) (i.e., thereciprocal of the relaxation time) at each peak top wasplotted against the square of scattering vector (q2),yielding a straight line passing through the origin (datanot shown). From the translational diffusion coefficient(D) determined from the slope of the Γ-q2 plot, Rh wascalculated using eq 7. Values of Rh thus estimated are
listed in Table 1. The distribution is unimodal for fC6 )5 mol %, but it becomes bimodal when fC6 is increasedto 10 mol %. In our earlier work on AMPS-C12MAmcopolymers,25 we observed that at fC12 < 10 mol % QELSrelaxation time distributions were bimodal with a fastrelaxation mode attributable to unimers (a single mo-lecular state) and a slow mode to multipolymer ag-gregates. The slow mode peak in the bimodal distribu-tion observed for the C6 polymer with fC6 ) 10 mol %(Figure 7) is an indication of the interpolymer associa-tion of C6 chains yielding multipolymer aggregates withRh ) 54 nm. The fast mode peak corresponding to Rh )4.4 nm is presumably due to unimers. When fC6 isincreased to 20 mol %, the distribution becomes uni-modal, but Rh is significantly larger than those for fC6) 0 and 5 mol % (Table 1), suggesting that the polymerundergoes interpolymer association as in the case of theAMPS-C12MAm copolymers of fC12 < 10 mol %.25 Withincreasing fC6 in the range 20 e fC6 e 70 mol %, Rhdecreases from 12 to 3.8 nm. This decrease in Rh in thisfC6 range indicates that hydrophobic associations of C6chains occur progressively in a intrapolymer fashion,leading to the formation of unimolecular micelles as inthe case of the AMPS-C12MAm copolymers with hydro-phobe contents in the range 20 < fC12 < 50 mol %.23
However, when fC6 is further increased to 80 mol %, Rhincreases rather abruptly to 8.1 nm. A similar tendencywas also observed for the AMPS-C12MAm copolymers.23
When fC12 in the AMPS-C12MAm copolymer was in-creased to 55 mol % or higher, Rh increased abruptly,indicating the formation of multipolymer aggregates.23
These multipolymer aggregates formed at fC12 g 55 mol% are markedly different from those observed at fC12 <10 mol % in that the relaxation distributions for fC12 g55 mol % are unimodal with a narrow width, whereasthe distributions for fC12 < 10 mol % are bimodal.23 Thesame unimodal distribution with a narrow width ob-served for the C6 polymer with fC6 ) 80 mol % (Figure7) corresponds to the same type of multipolymer ag-gregates observed for the AMPS-C12MAm copolymerswith fC12 g 55 mol %.
Relaxation time distributions for the C18 polymerswith varying fC18 in 0.05 M NaCl aqueous solutionsobserved at a scattering angle of 90° are presented inFigure 8. Values of Rh estimated from Γ-q2 plots arelisted in Table 1. In contrast to the case of the C6polymer, Rh increases from 7.3 to 9.8 nm when fC18 isincreased from 0 to 5 mol % and then decreases to 6.7nm as fC18 is further increased to 20 mol %. The largerRh observed for fC18 ) 5 mol % may be due to the sametype of the intermolecularly associated polymer chainsobserved for the C6 and C12 polymers when hydrophobecontents are low. In the case of the C18 polymers, aprogressive decrease in Rh is observed in the range 5 <fC18 < 20 mol %, followed by an increase in Rh as fC18 isfurther increased. This suggests that an increase in fC18in this fC18 region leads to an increase in the tendencyfor intrapolymer association of the C18 chains whereasa further increase in fC18 leads to an inclination forinterpolymer associations to form multipolymer ag-gregates. At fC18 ) 25 mol %, the distribution appearsto be bimodal with a major peak apparently due tomultipolymer aggregates with Rh ) 16 nm and a smallshoulder on the longer relaxation time side correspond-ing to larger aggregates. When fC18 is increased to 30mol %, the distribution remains bimodal but with alarge peak of a much longer relaxation time correspond-
Figure 7. Relaxation time distributions in QELS observedat θ ) 90° for the C6 polymers at 25 °C. Polymer concentra-tion: 1 g/L in a 0.05 M NaCl aqueous solution.
7858 Yamamoto et al. Macromolecules, Vol. 33, No. 21, 2000

ing to multipolymer aggregates with Rh ) 48 nm and asmall shoulder on the side of shorter relaxation timesapparently due to unimers or small interpolymer ag-gregates. At present, it is not well understood why therelaxation time distributions at fC18 ) 25 and 30 mol %are so different and the polymer with fC18 ) 30 mol %shows the presence of considerably larger aggregatesas a major component. The structure of the largeraggregates remains an open question. This type ofassociation behavior is unique to the C18 alkyl chainbecause no such larger aggregates were observed for theC6 and C12 polymers. As fC18 is further increased to 35mol % or more, the distributions become unimodal andnarrower, suggesting the formation of the same type ofmultipolymer aggregates as that for the AMPS-C12MAmcopolymers with fC12 g 55 mol %.23
Discussion
The polymers with C6, C12, and C18 alkyl chainsexhibit the smallest Rh values of 3.8, 3.9, and 6.7 nm athydrophobe contents of 70, 30, and 20 mol %, respec-tively (Table 1). At these hydrophobe contents, polymeraggregates formed are likely to be unimolecular micellesor multipolymer micelles formed from a small numberof polymer chains. We attempted to estimate thecompactness of these micelles by comparing apparentMw and Rh values for the micelles. We determined Mwby SLS for the C6, C12, and C18 polymers with thesmallest Rh to be 6.5 × 104, 5.3 × 104, and 11.5 × 104,respectively. Since we found it difficult to measure SLSfor polymer solutions in 0.05 M NaCl, we chose to usepolymer solutions in 0.2 M NaCl. In a separate QELSexperiment, we confirmed that in 0.2 M NaCl thepolymers form practically the same type of aggregatesas that formed in 0.05 M NaCl. From these Mw and Rhvalues, apparent densities (i.e., mass/hydrodynamicvolume) of the polymer aggregates are calculated to be0.47, 0.36, and 0.15 g/cm3 for the polymers of fC6 ) 70mol %, fC12 ) 30 mol %, and fC18 ) 20 mol %, respec-tively. These apparently low densities are presumably
due to the fact that the hydrodynamic volume of thepolymer aggregate is mainly determined by the size ofextended hydrophilic loops of AMPS segments sur-rounding hydrophobic microdomains. Because the sizeof the hydrophilic loop increases with the AMPS content,the apparent density decreases in the order of (fC6 ) 70mol %) > (fC12 ) 30 mol %) > (fC18 ) 20 mol %).
Association structures depend on protocol for thepreparation of aqueous solutions of the polymers.24 Inthe present work, polymers were purified by reprecipi-tation from methanol solutions into excess diethyl ether,followed by dialysis of aqueous polymer solutions againstpure water. Solid polymer samples were recovered fromthe dialyzed aqueous solutions by freeze-drying. Ourprevious study revealed that there existed multipolymermicelles in the freeze-dried samples that had beenformed by hydrophobic associations of entangled poly-mer chains during the purification procedure.24 Whenthe solid polymer samples are added to pure water atroom temperature, the already existing multipolymermicelles are simply redissolved in water as such.However, if the aqueous solution was heated to 90 °C,followed by the addition of a predetermined amount ofNaCl to adjust ionic strength of the solution, the chainentanglement could be eliminated, forming unimolecu-lar micelles if the polymers has a strong tendency, asits intrinsic nature, for preferential intrapolymer as-sociation.24 In the present study, we employed theseprocedures to prepare polymer solutions in 0.05 or 0.2M NaCl. Thus, it is expected that virtually unimolecularmicelles were obtained for polymers having a strongtendency for intrapolymer association.24
In our earlier work on the C12 polymers, we observedthat Mw values for unimers determined by SLS in 0.2M NaCl were 2-4 times larger than those estimatedby GPC in methanol calibrated with poly(ethyleneoxide).24 Thus, the Mw values presently determined bySLS for the 70 mol % C6 and 30 mol % C12 polymers(ca. 4 and 2 times larger than those estimated by GPC,respectively) suggest that the polymer aggregates aremostly unimolecular micelles. However, micelles formedfrom the C6 and C12 polymers with higher hydrophobecontents (i.e., fC6 > 70 mol % and fC12 > 50 mol %) aremultipolymer micelles probably because chain entangle-ments could not be eliminated completely during theprocess of the preparation of polymer solutions. In thecase of the 20 mol % C18 polymer, however, the Mw valueby SLS is ca. 14 times larger than that estimated byGPC, suggesting that the polymer aggregates are notunimolecular but multipolymer micelles. These observa-tions indicate that the tendency for intrapolymer hy-drophobic association is much smaller for the C18 alkylchain than the C6 and C12 chains. At present, it is notunderstood whether this is due to an intrinsic natureof the longer alkyl chain or an increased difficulty ofchain disentanglement during the process of polymersolution preparation.
Results of the present study, together with results ofour earlier work on AMPS-C12MAm copolymers, indi-cate that intrapolymer associations occur dominantlyfor the C6 and C12 polymers in the range 30 < fC6 < 70mol % and 10 < fC12 < 50 mol %. A similar tendencywas observed for the C18 polymers in the range 5 < fC18< 20 mol % although polymer aggregates formed in thisrange were not purely unimolecular. When hydrophobecontents are lower than these ranges, a significantextent of interpolymer associations occur concurrently,
Figure 8. Relaxation time distributions in QELS observedat θ ) 90° for the C18 polymers at 25 °C. Polymer concentra-tion: 1 g/L in a 0.05 M NaCl aqueous solution.
Macromolecules, Vol. 33, No. 21, 2000 Association of Polyelectrolyte-Bound Alkyl Chains 7859

yielding larger aggregates, and QELS relaxation timesexhibit bimodal distributions attributed to multipolymeraggregates and coexisting unimers. On the other hand,when hydrophobe contents are higher than these ranges,multipolymer aggregates are formed predominantly,and QELS relaxation times exhibit unimodal distribu-tions with a narrow width. Lower and upper limits forthe preferential intrapolymer association to occur shifttoward lower hydrophobe contents, and the widthbetween the lower and upper limits becomes narroweras the length of the alkyl chain increases.
To compare the “effectiveness” of the C6, C12, and C18alkyl chains per carbon atom for hydrophobic self-association, we define a parameter Q which representsa hydrophobe/charge ratio in a polymer chain as
where N is the number of carbon atoms in an alkylchain, and fCN is the mole percent content of a meth-acrylamide substituted with an alkyl chain with Ncarbon atoms. The solubility limits of 80, 60, and 40 mol% alkylmethacrylamide contents for the respective C6,C12, and C18 polymers correspond to Q ≈ 25, 18, and12, respectively. Thus, the maximum hydrophobe/chargeratio of the polymer soluble in water decreases signifi-cantly with increasing length of the alkyl chain. In otherwords, with a shorter alkyl chain as a hydrophobe, onecan incorporate more carbon atoms into a polymerkeeping the polymer water-soluble.
In Figure 9, the fluorescence lifetimes and rotationalcorrelation times for fluorescence depolarization are
plotted as a function of Q. As can be seen from Figure9a, the lifetime plots for the C12 and C18 polymers followsimilar lines, indicating that there is no significantdifference in the effectiveness of the C12 and C18 alkylchains per carbon atom for the formation of hydrophobicdomains. The C12 and C18 chains can form hydrophobicdomains even at a low level of Q (i.e., Q ≈ 1), and thedomain formation further proceeds with increasing Q,reaching a saturated level near Q ≈ 10. In the case ofthe C6 chains, by contrast, the domain formation is notsignificant at Q < 3, but it proceeds with increasing Qover the range of 3 < Q < 15. The formation ofhydrophobic domains for the C6 chains is more gradualthan in the cases of the C12 and C18 chains. Thesefindings indicate that the C6 alkyl chains are lesseffective for the self-association than the C12 and C18chains per carbon atom. In other words, with C6 chains,one needs more carbon atoms to incorporate into apolymer than with C12 and C18 chains to attain the sameextent of the hydrophobic domain formation.
Although the effectiveness of the C12 and C18 chainsin hydrophobic associations looks similar when moni-tored by the fluorescence lifetime of naphthalene labels,the extent of the motional restriction imposed on thelabels in the hydrophobic domains monitored by fluo-rescence depolarization is quite different at Q > 10(Figure 9b). At Q < 8, hydrophobic domains formed bythe C12 and C18 chains are similar in the extent of themotional restriction of the labels, but at Q > 10, theC18 domains impose much more restriction on the labelsthan do the C12 domains. In the case of the C6 chains,the extent of the motional restriction increases gradu-ally with increasing Q over the whole range of Q shownin Figure 9b and is much less than the cases of the C12and C18 chains. A similar conclusion can be drawn fromthe dependence of 1H NMR relaxation times on Q (plotsnot shown).
Conclusions
The self-association properties in water of AMPScopolymers with methacrylamides N-substituted witha C6 or C18 alkyl chain were investigated by fluorescencedecay and depolarization, FT-IR, 1H NMR relaxation,and QELS techniques, focusing on the effect of thelength of the alkyl chain on the hydrophobic self-association behavior. Experimental results were com-pared with those of the same type of AMPS copolymerscarrying C12 alkyl chains in our earlier work. Thepolymer-linked C6 chains commence to undergo self-association at a much higher hydrophobe content thanin the cases of the C12 and C18 chains. The polymer-bound C6 and C12 alkyl chains undergo predominantlyintrapolymer self-associations when the hydrophobecontents are in the range of 30-70 and 10-50 mol %,respectively. In the case of the polymer-bound C18chains, there is a similar tendency for intrapolymerassociation when the hydrophobe contents are in therange of 5-20 mol %, but polymer aggregates formedare not completely unimolecular. If the hydrophobecontents are either lower or higher than these limits,intermolecularly associated structures with larger hy-drodynamic volumes are formed. It was found that thedynamic properties of hydrophobic domains formed byalkyl chains with different lengths are considerablydifferent. Although micropolarity experienced by naph-thalene labels incorporated in the hydrophobic domainis the same, motional restrictions imposed on the labels
Figure 9. Plots of the fluorescence lifetime (a) and rotationalcorrelation time (b) for naphthalene labels in the C6-, C12-, andC18-carrying polymers plotted as a function of the hydrophobe/charge ratio Q.
Q )NfCN
99 - fCN(9)
7860 Yamamoto et al. Macromolecules, Vol. 33, No. 21, 2000

are more pronounced when the domains are formedfrom longer alkyl chains. The same tendency of re-stricted motions of the alkyl chains was indicated by1H NMR relaxation times. The effectiveness of themethylene and methyl residues in the polymer-boundC6, C12, and C18 chains to self-associate was comparedin terms of a parameter representing the hydrophobe/charge ratio. The CH2 and CH3 residues in the C6 chainare much less effective than those in the C12 and C18chains in hydrophobic associations when compared atthe same hydrophobe/charge ratio in a polymer chain,whereas there is no significant difference between thosein the C12 and C18 chains.
Acknowledgment. This work was supported in partby a Grant-in-Aid for Scientific Research No. 10450354from the Ministry of Education, Science, Sports andCulture, Japan, and in part by Shorai Foundation forScience and Technology.
References and Notes
(1) Principles of Polymer Science and Technology in Cosmeticsand Personal Care; Goddard, E. D., Gruber, J. V., Eds.;Marcel Dekker: New York, 1999.
(2) McCormick, C. L.; Bock, J.; Schulz, D. N. Encyclopedia ofPolymer Science and Engineering; John Wiley: New York,1989; Vol. 17, p 730.
(3) Bock, J.; Varadaraj, R.; Schulz, D. N.; Maurer, J. J. InMacromolecular Complexes in Chemistry and Biology; Dubin,P., Bock, J., Davies, R. M., Schulz, D. N., Thies, C., Eds.;Springer-Verlag: Berlin, 1994; p 33.
(4) Polymers as Rheology Modifiers; Schulz, D. N., Glass, J. E.,Eds.; Advances in Chemistry Series 462; American ChemicalSociety: Washington, DC, 1991.
(5) Hydrophilic Polymer, Performance with Environmental Ac-ceptability; Glass, J. E., Ed.; Advances in Chemistry Series248; American Chemical Society: Washington, DC, 1996.
(6) Valint, P. L., Jr.; Bock, J.; Schulz, D. N. In Polymers inAqueous Media: Performance through Association; Glass, J.E., Ed.; Advances in Chemistry Series 223; American Chemi-cal Society: Washington, DC, 1989; p 399.
(7) Kamioka, K.; Webber, S. E.; Morishima, Y. Macromolecules1988, 21, 972.
(8) Morishima, Y.; Lim, H. S.; Nozakura, S.; Strutevant, J. L.Macromolecules 1989, 22, 1148.
(9) McCormick, C. L.; Chang, Y. Macromolecules 1994, 27, 2151.(10) Prochazka, K.; Kiserow, D.; Ramireddy, C.; Tuzar, Z.; Munk,
P.; Webber, S. E. Macromolecules 1992, 25, 454.
(11) McCormick, C. L.; Salazar, L. C. Polymer 1992, 33, 4617.(12) Chang, Y.; McCormick, C. L. Macromolecules 1993, 26, 6121.(13) Morishima, Y. Trends Polym. Sci. 1994, 2, 31.(14) Morishima, Y. In Solvents and Self-Organization of Polymers;
Webber, S. E., Tuzar, D., Munk, P., Eds.; Kluwer AcademicPublishers: Dordrecht, The Netherlands, 1996; p 331.
(15) Yusa, S.; Kamachi, M.; Morishima, Y. Langmuir 1998, 14,6059.
(16) Noda, T.; Morishima, Y. Macromolecules 1999, 32, 4631.(17) Morishima, Y.; Nomura, S.; Ikeda, T.; Seki, M.; Kamachi, M.
Macromolecules 1995, 28, 2874.(18) Morishima, Y. Bio-Industry 1995, 12, 20.(19) Morishima, Y. In Multidimensional Spectroscopy of Poly-
mers: Vibrational, NMR, and Fluorescence Techniques;Urban, M. W., Provder, T., Eds.; ACS Symposium Series 598;American Chemical Society: Washington, DC, 1995; p 490.
(20) Seki, M.; Morishima, Y.; Kamachi, M. Macromolecules 1992,25, 6540.
(21) Morishima, Y.; Tsuji, M.; Seki, M.; Kamachi, M. Macromol-ecules 1993, 26, 3299.
(22) Yamamoto, H.; Mizusaki, M.; Yoda, K.; Morishima, Y.Macromolecules 1998, 31, 3588.
(23) Yamamoto, H.; Morishima, Y. Macromolecules 1999, 32, 7469.(24) Yamamoto, H.; Hashidzume, A.; Morishima. Y. Polym. J., in
press.(25) Hashidzume, A.; Yamamoto, H.; Mizusaki, M.; Morishima,
Y. Polym. J. 1999, 31, 1009.(26) Morishima, Y.; Kobayashi, T.; Nozakura, S. Polym. J. 1989,
21, 267.(27) Morishima, Y.; Tominaga, Y.; Nomura, S.; Kamachi, M.
Macromolecules 1992, 25, 861.(28) Perrin, F. J. Phys. Radium 1926, 7, 39.(29) Erdmann, K.; Gutsze, A. Colloid Polym. Sci. 1987, 256, 667.(30) Rady, P.; Budd, P. M.; Heatiey, F.; Price, C. J. Polym. Sci.,
Polym. Phys. Ed. 1991, 29, 451.(31) Brereton, M. G.; Ward, I. M.; Boden, N.; Wright, P. Macro-
molecules 1991, 24, 2068.(32) Meiboom, S.; Gill, D. Rev. Sci. Instrum. 1958, 29, 688.(33) Jakes, J. Czech. J. Phys. 1988, B38, 1305.(34) Phillies, G. D. J. Anal. Chem. 1990, 62, 1049A.(35) Phillies, G. D. J. J. Chem. Phys. 1988, 89, 91.(36) Morishima, Y.; Tominaga, Y.; Kamachi, M.; Okada, T.; Hirata,
Y.; Mataga, N. J. Phys. Chem. 1991, 95, 6027.(37) Kalyanasundaram, K.; Thomas, J. K. J. Am. Chem. Soc. 1977,
99, 2039.(38) Kiserow, D.; Chan, J.; Ramireddy, C.; Munk, P.; Webber, S.
E. Macromolecules 1992, 25, 5338.(39) Chan, J.; Fox, S.; Kiserow, D.; Ramireddy, C.; Munk, P.;
Webber, S. E. Macromolecules 1993, 26, 7016.(40) Pake, G. E. In Solid State Physics; Seitz, F., Turnbull, D.,
Eds.; Academic Press: New York, 1965; Vol. 2, p 1.
MA9920487
Macromolecules, Vol. 33, No. 21, 2000 Association of Polyelectrolyte-Bound Alkyl Chains 7861