aulas práticas de orgânica experimental
TRANSCRIPT

Universidade Federal do Espírito Santo
Centro de ciências Agrárias de Alegre
Aulas Práticas de Química
Orgânica Experimental
CURSO DE LICENCIATURA EM QUÍMICA
Prof. Daniel Rinaldo
Alegre ‐ 2012

ÍNDICE
1. INTRODUÇÃO ................................................................................................................................ 1 2. INSTRUMENTOS UTILIZADOS EM LABORATÓRIO .................................................................... 2 3. RECOMENDAÇÕES AOS ALUNOS ............................................................................................ 10
3.1. Cuidados Pessoais ................................................................................................................ 10 3.2. Trabalho com Vidro ................................................................................................................ 11 3.3. Incêndios ................................................................................................................................ 12 3.4. Explosões ............................................................................................................................... 12 3.5. Substâncias tóxicas ............................................................................................................... 12 3.6. Produtos químicos ................................................................................................................. 13 3.7. Capelas .................................................................................................................................. 14 3.8. Limpeza .................................................................................................................................. 14 3.9. Balança .................................................................................................................................. 14 3.10. Banho-Maria ......................................................................................................................... 14 3.11. Manta Elétrica ...................................................................................................................... 14 3.12. Trompa D´Água.................................................................................................................... 15 3.13. Estufa ................................................................................................................................... 15 3.14. Termômetro .......................................................................................................................... 15 3.15. Funil de vidro sinterizado ..................................................................................................... 15 3.16. Livros .................................................................................................................................... 15 3.17. Pias e Canaletas .................................................................................................................. 16
4. CONSTANTES FÍSICAS DE COMPOSTOS ORGÂNICOS ......................................................... 16
4.1. Temperatura de fusão ............................................................................................................ 16 4.2. Temperatura de ebulição ....................................................................................................... 19 4.3. Densidade .............................................................................................................................. 21
5. AULAS PRÁTICAS ........................................................................................................................ 23
Aula prática 01: Extração Líquido-Líquido .................................................................................. 23 Aula prática 02: Extração Líquido-Líquido com solventes quimicamente ativos ........................ 34 Aula prática 03: Destilação simples, fracionada e a vácuo......................................................... 40 Aula prática 04: Síntese e Recristalização da acetanilida .......................................................... 52 Aula prática 05: Extração de óleos essenciais através de destilação por arraste de vapor e sua análise por Cromatografia em Camada Delgada (CCD) .............................................................. 62 Aula prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre ....................................................................................................................................... 80 Aula prática 07: Síntese de biodiesel da soja ............................................................................. 91

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
1
1. INTRODUÇÃO
Nesta disciplina serão executadas experiências que permitirão ao estudante conhecer os
princípios e as técnicas básicas necessárias para o trabalho no laboratório de química, bem como
reforçar os aspectos teóricos de cada assunto.
As práticas selecionadas serão o caminho para estudar:
os materiais e equipamentos básicos do laboratório;
cálculos e técnicas utilizadas para preparo de soluções.
Para um bom aproveitamento do curso é necessário uma preparação prévia de cada
experiência, seguindo por exemplo, o esquema apresentado abaixo:
consultar a bibliografia e estudar o procedimento experimental a ser realizado;
organizar um roteiro de todas as operações a serem executadas;
desenhar o(s) esquema(s) dos sistemas a serem montados;
escrever todas as equações das reações a serem realizadas e suas relações
estequiométricas;
anotar as constantes físicas dos reagentes e dos solventes;
anotar os cuidados a serem tomados na realização da experiência (substâncias tóxicas,
inflamáveis, corrosivas etc);
verificar a compreensão da sequência completa das etapas experimentais e dos princípios
envolvidos, como por exemplo:
a) experiência preparativa, na qual um composto é sintetizado a partir de outros
reagentes: cálculo estequiométrico, reagente limitante, rendimento, mecanismo da
reação, outros métodos de preparação, esquema da separação e purificação do
produto.
b) encarar o procedimento experimental como uma sugestão a ser interpretada e
não como uma receita a ser executada.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
2
2. INSTRUMENTOS UTILIZADOS EM LABORATÓRIO
Quem entrar em um laboratório de pesquisas ou de ensino em Química se deparará
com uma grande quantidade de peças que denominamos de aparelhagem de laboratório. Cada
uma destas peças tem um uso específico e é confeccionada de um determinado material. Uma
grande quantidade delas é confeccionada em vidro, normalmente vidro pirex ou vidro de
borossilicato, metal ou plástico. Estas aparelhagens de laboratório não fazem parte do nosso dia a
dia, mas vários têm formato muito semelhantes instrumentos que não fazem parte do dia a dia.
Muitos apresentam formato ou funções semelhante aos equipamentos que possuímos nas
cozinhas de nossas residências, enquanto que outros tem formatos e aplicações totalmente e as
vezes até não imagináveis para alguém que ainda não estudou um pouco de química.
Além destes equipamentos confeccionados em vidro ou plástico, em laboratório há
também muitos outros equipamentos como microscópios, aquecedores elétricos, aparelhos de
refrigeração entre outros, que necessitam de energia elétrica para o seu funcionamento.
Todas estas aparelhagens ou equipamentos são fruto de séculos de desenvolvimento
da ciência, em particular da química, da biologia e da física, sendo portanto, resultado de uma
evolução lenta e gradativa.
Os primeiros laboratórios que realizavam transformações químicas (ainda que de
forma primitiva) remontam à época dos alquimistas. Num misto de magia, superstição e ciência
primitiva, seus adeptos realizavam diversas sínteses, como do ácido acético e sulfúrico (conhecido
como óleo de vitríolo). Entre os mais famosos podemos citar Hermes Trimegisto, Geber e
Paracelso, famoso por utilizar alguns conceitos alquímicos na cura de doenças
A química, no entanto, somente ganharia características de ciência formal com os
primeiros trabalhos do físico Robert Boyle (1627-1691). De formação bastante ampla, Boyle foi
físico, químico e filósofo, sendo o primeiro a apresentar a noção de “elemento químico”. Os
trabalhos de Boyle serviram de base para o surgimento da Química Experimental que, embora de
forma primitiva, alcançaria sua maturidade alguns anos depois.
Coube a Antoine Laurent de Lavoisier o papel de estabelecer em definitivo a Química
entre as ciências já fundamentadas. De origem nobre, Lavoisier se ocupou com grande paixão da
pesquisa química, a tal ponto que seu laboratório caseiro era um dos mais bem montados de sua
época. Na impossibilidade de dispor de fornecedores de equipamentos, ele mesmo desenhava e
encomendava aos vidreiros e artesãos a aparelhagem de que necessitaria. Suas intensas
pesquisas o levaram, entre outras coisas, à descoberta da conservação das massas durante uma
reação química.
Apesar de todo o desenvolvimento técnico e científico, qualquer laboratório químico,
por mais sofisticado que seja, ainda utiliza um conjunto muito simples de equipamentos que têm
suas origens ligadas ao desenvolvimento da química, como os béqueres, tubos de ensaio,
erlenmeyers, buretas, etc.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
3
As atividades de laboratório exigem parte do aluno não só um conhecimento das
peças e aparelhos utilizados, como também o emprego correto de cada um deles. Portanto, antes
de tudo, é necessário que observem bem cada uma das peças, memorizem a sua forma e
conheçam a utilidade de cada uma.
Estante para tubos de ensaio e tubos de ensaio: Os tubos de ensaio são empregados para fazer reações em pequena escala, principalmente na realização de testes de reação. Eles podem ser aquecidos com cuidado sobre a chama do bico de gás ou bico de Bunsen, desde que segurados por pinça de madeira. A estante para tubos de ensaio são feitas de madeira ou metal e servem como suporte para manter os tubos de ensaio em posição vertical. Os tubos de ensaio podem ter de 5 a 20 cm de altura e de podem ter diferentes diâmetros.
Béquer: Serve para reações entre soluções, dissolver substâncias, efetuar reações de precipitação e aquecer líquidos. Apresentam escala para medir volumes aproximados, portanto, constituem vidraria graduada. Eles possuem um bico para facilitar a transferência de líquidos Pode ser aquecido sobre a tela de amianto. Os béqueres são vidrarias graduadas e que apresentam capacidade variando de 5 mL até 2000 mL.
Erlenmeyer: Utilizado para titulações, aquecimento de líquidos, dissolução de substâncias e reações entre soluções. Para seu aquecimento, usa-se o tripé com tela de amianto. Os erlenmeyers também são utilizados em titulação para conter a solução a ser titulada e sobre a qual será adicionada a solução tittulante. São graduados e, assim como os béqueres, os valores de volume são aproximados devido ao seu grande diâmetro. Normalmente são utilizados erlenmeyers com capacidade de 125 ml, 250 mL ou 500 mL.
Funil: Usado na filtração, para retenção de partículas sólidas em misturas sólido-líquido. No funil é adaptado o papel de filtro que retém o sólido e permite a passagem do material líquido. Podem ter a haste inferior curta ou longa.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
4
Balão de fundo chato: Empregado para aquecer líquidos ou soluções ou ainda fazer reações com desprendimento de gases. Pode ser aquecido sobre tripé com tela de amianto. Os balões de fundo chato podem ter apenas uma saída como podem ter duas ou três saídas laterais.
Condensadores: Utilizados na destilação, têm por finalidade condensar os vapores do líquido. Condensar significa transformar os vapores em líquidos por resfriamento. A entrada e saída lateral dos condensadores servem para manter um fluxo constante de água ou de outro líquido refrigerante e com isto manter uma temperatura baixa no interior do condensador para permitir o resfriamento do vapor e consequentemente sua condensação.
Bastão de vidro ou bagueta: Corresponde a um bastão maciço de vidro. Serve para agitar e facilitar as dissoluções, manter massas líquidas em constante movimento, ou ainda, na transferência de líquidos de um recipiente a outro.
Proveta ou cilindro graduado: Serve para medidas aproximadas de volumes de líquidos. Não pode ser aquecida por ser considerada uma vidraria de maior precisão que os béqueres ou erlenmeyers. As provetas apresentam capacidade de 10 mL até 2000 mL de solução.
Pipetas: Usadas para medir e transferir pequenos volumes de líquidos. Não pode ser aquecida por ser vidraria de grande precisão de volume. A capacidade das pipetas pode variar de 0,5 mL até 200 mL. São classificadas em graduadas e volumétricas. As pipetas volumétricas são altamente precisas e são utilizadas para tomar um único e fixo volume de solução. As pipetas graduadas apresentam escala e podem tomar diferentes volumes de líquido em função da capacidade máxima da pipeta.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
5
Bico de Bunsen ou bico de gás: O bico de gás é a fonte de aquecimento mais usada em laboratório. Consiste de um sistema de metal que apresenta uma entrada de gás na parte inferior e uma parte superior na qual é produzida a chama que servirá de aquecimento. Os bicos de gás também apresentam um anel na parte inferior que permite regular a entrada de oxigênio e, com isso, controlar a temperatura da chama.
Suporte Universal: Utilizado em várias operações como filtração, suporte de condensador, sustentação de peças, etc. São confeccionados em metal e a base permite sustentação da haste na qual serão presas as peças e vidrarias.
Anel ou argola para funil: Empregado como suporte do funil na filtração, ou para sustentação do funil de decantação. São confeccionadas em metal e apresentam diferentes diâmetros. Apresenta um sistema de rosca (mufa) que permite prendê-la ao suporte universal.
Garra com mufa: Presa ao suporte serve para segurar várias outras peças como buretas, condensadores, colunas de refluxo, balão de destilação. Apresentam diferentes formatos e tamanhos. Uma das extremidades (mufa) é presa ao suporte universal e a outra prende a peça que deseja manter fixa ao suporte universal como bureta, condensador, erlenmeyer, balão, dentre outras.
Tripé de ferro: Sustentáculo na qual se coloca a tela de amianto e sobre a qual se coloca o recipiente que contém o líquido a ser aquecido. É usado com tela de amianto. É colocado sobre o bico de Bunsen.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
6
Tela de amianto: Suporte para as peças a serem aquecidas. A função do amianto é distribuir uniformemente o calor recebido pelo bico de Bunsen e distribuí-lo uniformetemente para o recipiente que contém o líquido ou solução que está sendo aquecido.
Pinça de madeira: Usada para segurar tubos de ensaio durante o aquecimento e para transportar tubos de ensaio aquecidos. Pinça de Hoffmann e pinça de Mohr: Usadas para reduzir ou impedir a passagem de gases ou líquidos através de tubos flexíveis.
Cápsula de porcelana: Peça de porcelana usada para evaporar líquidos das soluções.
Vidro de relógio: Peça de vidro de forma côncava. O vidro de relógio é usado para cobrir béqueres em evaporações, para pesagens e diversos fins como tampar frascos para impedir que caia poeira ou qualquer outro contaminante.
Bureta: Usada para medidas precisas de líquidos. Usada em análises volumétricas para determinar o volume de solução titulante que reage com uma determinada quantidade de solução a ser titulada. Podem ser utilizadas também para a transferência de volumes precisos de líquidos. Na parte inferior das buretas há uma torneira por onde escoa o liquido a ser transferido. É sempre utilizada presa ao suporte universal por garras próprias para isto.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
7
Almofariz e pistilo: Confeccionados em ágata ou porcelana, são utilizados na trituração e pulverização de sólidos.
Balão volumétrico: Usado para preparar e diluir soluções. Apresenta fundo chato, um gargalo, junta esmirilhada e um tampa que se ajusta perfeitamente na junta esmirilhada. No gargalho há uma marca que indica a capacidade exata do balão. Existem balões com capacidade variando de 5 mL até 5000 mL.
Funil de decantação, funil de separação ou funil de bromo: Usado para separação de líquidos imiscíveis. Na parte inferior dos funis há uma torneira que permite escoar o líquido de maior densidade e na parte superior há uma entrada com junta esmirilhada que possui tampa que se ajusta perfeitamente à junta esmirilhada. São afixados ao suporte universal utilizando argolas.
Espátulas: Usadas para transferência de substâncias sólidas do frasco que a contém para outro frasco ou para o recipiente que está sobre a balança para o material sólido ser pesado. Também podem ser utilizadas para quaisquer outras transferências de materiais sólidos. São confeccionadas em metal ou plástico e apresentam diferentes formatos e tamanhos.
Funil de Büchner: Funil de porcelana utilizado para realizar filtração rápida de sistemas heterogêneos sólido-líquidoNa parte interna apresenta uma superfície com furos na qual se fixa o papel de filtro. Kitassato: Usados em conjunto para filtrações a vácuo. O kitassato é o recipiente na qual ficará o líquido da mistura sólido-líquido. O kitassato tem formato de erlenmeyer, entretanto, as paredes são mais grossas para evitar que se quebre devido á diminuição da pressão e apresenta uma saída lateral por onde é retirado o ar. Trompa de vácuo: Usada em conjunto com o kitassato e o funil de Büchner, é responsável

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
8
para remoção do ar dentro do sistema para acelerar a filtração. Para retirar o ar a trompa de vácuo é fixada a uma torneira e o fluxo de água que passa pela trompa é responsável pela remoção do ar diminuindo a pressão interna do sistema.
Pissetas: Usadas para lavagem de materiais ou recipientes através de jatos de água, álcool ou outros solventes ou para adicionar líquidos em outros recipientes e até para adicionar líquidos a sólidos para realizar a dissolução dos sólidos. Normalmente são de polietileno e apresentam volume de 250 mL ou de 500 mL.
Dessecador: Usado para armazenar substâncias em atmosfera contendo baixo índice de umidade. Na parte inferior coloca-se uma substância capaz de absorver água (higroscópica). Os dessecadores são de vidro e apresenta paredes extremamente grossas para suportar baixa pressão interna por o armazenamento da substância pode ser feito em baixa pressão.
Placa de Petri: Recipiente de vidro utilizada para armazenar materiais sólidos que poderão ser armazenados no dessecador ou em estufa para secagem. Podem ser utilizadas também para cobrir reagente impedindo assim sua contaminação.
Balão de destilação: É utilizado em processos de destilação. O tubo lateral permite a saída de vapores obtidos a partir do aquecimento de líquidos ou soluções contidos no balão. Destilação consiste no processo de separação sólido-líquido ou líquido-líquido por aquecimento da solução seguida da evaporação de um dos líquidos.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
9
Triângulo de porcelana: O triângulo de porcelana é utilizado para sustentar cadinhos de porcelana em operações de aquecimentos na qual o cadinho é aquecido diretamente no bico de Bunsen durante uma calcinação. O triângulo de porcelana é adaptado sobre o tripé ou sobre a argola.
Cadinho: O cadinho de porcelana é utilizado para aquecimento a seco com o objetivo de remover totalmente o solvente que umidece o material uma vez que ele resiste ao aquecimento diretamente no bico de gás e não trinca quando o material estiver totalmente seco. Também é utilizado para operações de calcinação na qual o material sólido é convertido a outro quando aquecido, pode ser utilizado para a eliminação de substâncias orgânicas, secagem e fusões, no bico de Bunsen ou mufla pois pode ser aquecido a temperaturas superiores a 1000º C.
2.1. Fontes de aquecimento e seu uso
BICO DE GÁS: incompatível com substâncias inflamáveis. Entre o bico e o frasco, interpõe-se uma
tela de amianto, para que o aquecimento seja praticamente uniforme. O contato direto da chama
pode superaquecer alguma parte do frasco o que levaria a trincá-lo ou a decompor a mistura em
reação.
BANHOS DE AQUECIMENTO:
Banho- maria: utilizado para líquidos de baixo ponto de ebulição. O banho de água pode ser
aquecido com bico de gás ou com aquecedor elétrico, neste último caso o banho pode ser usado,
para refluxar líquidos inflamáveis;
Banho de óleo: utilizado quando é necessário aquecer acima de 95oC. Utiliza-se óleos de baixa
pressão de vapor como óleos vegetais hidrogenados que não inflamam até 300oC. Parafina e
outros derivados de petróleo podem ser usados, porém inflamam à temperatura bem inferior a
aquela.
MANTAS ELÉTRICAS: o aquecimento é obtido por uma resistência elétrica a qual se encontra
envolvida por lã de vidro.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
10
3. RECOMENDAÇÕES AOS ALUNOS
O uso da apostila é imprescindível a partir da primeira aula.
O aluno deverá tomar conhecimento, a partir da primeira aula, das instalações do
laboratório, bem como de suas normas de funcionamento.
É obrigatório, por razões de segurança, o uso de avental e dos óculos de segurança
durante as aulas.
O material do laboratório deve ser usado sempre de maneira adequada e somente aqueles
reagentes e soluções especificadas.
Não é permitido fumar, comer ou beber nos laboratórios.
Todo o material usado deve ser lavado ao final de cada aula e organizado no local
apropriado (mesas, bancadas ou armários).
Devem ser evitadas conversas em voz alta, e sobre assuntos alheios à aula.
3.1. Cuidados Pessoais
Não trabalhe sozinho no laboratório. Um companheiro, ao menos, sempre será uma ajuda
em caso de acidente.
Saiba onde se encontra o material de emergência para primeiros socorros.
Tome conhecimento dos cuidados descritos na "Tabela de Primeiros Socorros para
Laboratório".
Em caso de acidente, procure imediatamente o professor, mesmo que não haja danos
pessoais ou materiais.
Encare todos os produtos químicos como venenos em potencial enquanto não verificar sua
inocuidade, consultando a literatura especializada (por exemplo o Merck Index).
Caindo produto químico nos olhos, boca ou pele, o primeiro cuidado é lavar
abundantemente com água imediatamente. A seguir, procure o tratamento específico para
cada caso. Se os olhos forem atingidos, esta medida é particularmente importante,
devendo lavá-los e procurar o médico.
Use óculos ao trabalhar com sódio metálico, com maçarico, ao preparar soluções ácidas
ou básicas e ao transferi-las de um frasco para outro.
Substituir, sempre que possível, operações de pipetar com a boca por outro método (usar
pêra de borracha, por ex.). Em particular, não aspire líquidos corrosivos ou venenosos com
a boca.
Execute as experiências em pequena escala, sempre que possível.
Não deixe fios elétricos descobertos ligados para evitar curto-circuitos.
Se algum ácido ou produto químico for derramado, lave o local imediatamente.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
11
Todo aparelho em funcionamento deve ficar sob vigilância constante.
Faça o possível para não contaminar a atmosfera do laboratório. Para isso, não deixe
líquidos em recipientes de grande superfície, (como um bequer), não deixe frascos
abertos, recolha o destilado diretamente do bico do condensador em um recipiente
apropriado, deixando apenas um pequena abertura para equilíbrio de pressão.- Informe
seus colegas sobre o andamento de qualquer experiência que possa oferecer perigo.
Não deixe torneiras de gás abertas. Se notar algum vazamento de gás, avise ao
laboratorista.
Nunca aqueça um tubo de ensaio, apontando sua extremidade aberta para um colega.
Consulte o professor antes de fazer qualquer modificação no andamento da experiência e
na quantidade ou espécie de reagentes a serem usados.
Evite a inalação de vapores. Nunca cheire diretamente o conteúdo de algum recipiente.
Ao preparar soluções aquosas diluídas de um ácido, coloque o ácido concentrado na água,
nunca o contrário.
3.2. Trabalho com Vidro
O vidro é uma causa muito comum de acidentes e deve-se proceder sempre com muito
cuidado quando se trabalha com objetos de vidro. A sua quebra forma extremidades pontiagudas e
cortantes de extrema periculosidade.
Se for necessário introduzir uma peça de vidro em uma rolha (tubo, termômetro,
alongamento, etc.) proceda com cuidado, envolvendo o pedaço de vidro com uma toalha e
umedecendo o tubo e a rolha antes e no decorrer da operação. Mantenha as mãos
próximas e gire a rolha até introduzir o tubo.
Deixe qualquer peça de vidro quente esfriar bastante tempo antes de manuseá-la. Repare
bem onde colocá-la, pois o vidro quente tem a mesma aparência de vidro frio.
Polir no fogo todas as bordas pontiagudas de vidro quebrado (inclusive bastão de vidro,
evitando assim danificar o fundo de béquer, erlenmeyer, etc). Esta operação custa alguns
segundos e evita acidentes, como cortes.
Se uma rolha de vidro aderir a um frasco, bater levemente na rolha com um bastão de
madeira até conseguir soltá-la. Caso isto não ocorra, chame o professor.
Nunca use material de vidro trincado ou quebrado, que podem arruinar uma experiência ou
causar um acidente. material danificado deve ser substituído imediatamente.
Para remover tubos de vidro de rolhas de cortiça ou borracha, lubrifique inicialmente,
gotejando água. Gire então a rolha para ambos os lados até retirar o tubo. Se não
conseguir, não force. O vidro pode quebrar-se. O melhor é cortar a rolha com uma gilete.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
12
3.3. Incêndios
Além de materiais usualmente inflamáveis (madeira, cortiça, gás, o próprio vestuário,
cabelos) todo laboratório contém solventes altamente inflamáveis (éter, acetona, álcool, benzeno e
outros). Além disso, durante o trabalho experimental podem ser formadas substâncias inflamáveis.
Para evitar acidentes:
Use a chama do bico de bunsen apenas quando necessário, apagando-a imediatamente
após terminada a operação.
Nunca acenda um bico de bunsen perto de material inflamável.
Não aqueça líquidos inflamáveis em chama direta.
Não deixe chamas acesas ao sair do laboratório.
Já na la. vez, que entrar no laboratório, trate de familiarizar-se com a localização do
extintores de incêndio, toalhas, chuveiros, cobertores, etc.
Em caso de incêndio:
Se for um acidente de pequenas proporções, abafe imediatamente com uma toalha.
Feche os bicos de gás e desligue aparelhos elétricos das proximidades.
Apague o fogo com extintor de incêndio.
Coloque-se em segurança.
3.4. Explosões
Podem ocorrer especialmente por causa do vazamento de gás ou ignição espontânea de
materiais finamente divididos (carvão ativo, pó de alumínio), de vapores de solventes inflamáveis
ou então por aquecimento de substâncias oxidantes (ácido nítrico, ácido perclórico, cloratos, nitrato
de amônio e outros) à temperatura acima do necessário ou em presença de substâncias orgânicas.
3.5. Substâncias tóxicas
Grande número de substâncias empregadas no laboratório são tóxicas em maior ou menor
escala.
Notoriamente tóxicos são os cianetos, arsênio, gás sulfídrico, fósforo branco, compostos de
mercúrio, entre outros, mas de um modo geral evite o contacto de qualquer droga com a pele
(H2SO4 concentrado, HNO3, etc).
Tome especial cuidado com os olhos

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
13
Não leve à boca nenhuma substância desconhecida.
Não aspire profundamente nenhuma substância desconhecida.
Para sentir o odor de uma substância não coloque diretamente o nariz sobre o recipiente,
mas com a mão traga, um pouco do vapor até ele.
Produtos voláteis, tóxicos ou corrosivos devem ser abertos e usados na capela. Ex.: ácido
nítrico, ácido clorídrico, hidróxido de amônio, bissulfeto de carbono, piridina, cloreto de
alumínio, haletos de acila, ácido acético, anidrido acético, entre outros.
3.6. Produtos químicos
Antes de usar qualquer reagente, leia cuidadosamente o rótulo do frasco para ter certeza
de que é aquele o do reagente desejado.
Antes de abrir um frasco novo de uma substância, verifique se há algum outro já aberto.
Consulte os funcionários e o professor.
Abra frascos o mais longe possível do rosto e evite aspirar ar naquele exato momento.
Nunca torne a colocar no frasco uma droga retirada em excesso e não usada. Ela pode ter
sido contaminada.
Não coloque objeto algum nos frascos de reagentes, exceto o conta-gotas próprio de que
alguns são providos.
Imediatamente após o uso, feche perfeitamente o frasco com a sua rolha ou tampa própria.
Tome cuidado para não trocar as rolhas quando estiver usando vários reagentes. O melhor
é abrir um frasco e colocar a rolha sobre um papel de filtro limpo ou segurá-la na mão,
retirar a quantidade necessária de reagente, fechar o frasco e a seguir realizar estas
mesmas operações com os demais reagentes, um de cada vez.
Lave os resíduos que tenham ficado nas paredes externas do frasco antes de colocá-lo
sobre a mesa.
Ao esvaziar-se um frasco, limpe-o imediatamente e guarde-o num local adequado.
Ao usar um frasco observe se:
a) a tampa usada é conveniente ao conteúdo;
b) o rótulo e o número de classificação estão bem legíveis;
c) Se preciso, lembre ao funcionário do laboratório as correções.
Soluções alcalinas devem ser colocadas em frascos de polietileno, nunca em vidro. Ex.:
hidróxido de sódio, de potássio e de amônio, carbonatos de sódio e potássio.
Não use espátulas de metal com cloreto de alumínio e zinco.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
14
3.7. Capelas
Use a capela para experiências em que haja desenvolvimento de gases tóxicos ou
corrosivos ou quando receber instruções para isso:
a) Abaixe as janelas da parte em uso até a última borboleta e a daquela diretamente usada
até o máximo possível, se não for possível atingir aquela borboleta;
b) Desligue o motor tão logo termine o trabalho e os gases tenham sido eliminados;
c) Retire seu material e limpe o local.
3.8. Limpeza
Conserve limpos seu equipamento e seu balcão de trabalho.
Evite derramar líquidos, mas, se o fizer, lave imediatamente o local.
Jogue todos os sólidos e pedaços de papel usados numa cesta de lixo. Nunca jogue nas
pias fósforos, papel de filtro ou qualquer sólido, ainda que ligeiramente solúvel.
Ao terminar o trabalho num local (capela, mesa, balança, furador de rolhas, mesa de
reagentes, etc.), deixe-o perfeitamente limpo.
O material usado principalmente vidraria, deve ser lavado logo após o uso.
Ao término do período de laboratório, guarde seu próprio equipamento no lugar apropriado
e leve qualquer aparelho especial para local designado.
3.9. Balança
Conserve perfeitamente limpos as balanças e o balcão em que estão colocados.
Se, por descuido, deixar cair algum sólido nos pratos ou no interior da balança, limpe com
o pincel apropriado.
Não se encoste nos balcões das balanças.
3.10. Banho-Maria
Faça circular água e regule seu nível. Só então ligue a resistência à corrente elétrica.
Somente banho-maria com resistência de imersão pode ser usado próximo de líquido
inflamável.
Isole sempre a mesa da fonte de calor com tijolos perfurados.
3.11. Manta Elétrica

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
15
Deve ser usada exclusivamente para aquecer balões, preferivelmente de fundo redondo.
A adição de materiais ao balão deve ser feita estando este fora da manta.
Limpe e seque a parte externa do balão antes de colocá-lo na manta.
3.12. Trompa D´Água
Deve ser presa à torneira com arame e dela não deve ser retirada sem forte motivo.
Entre a trompa e o frasco onde se vai reduzir a pressão, deve-se colocar um frasco
intermediário com torneira, ficando este firmemente ligado ao balcão (frasco de
segurança).
3.13. Estufa
Antes de colocar algum material na estufa, consulte a literatura para ver se o material pode
se decompor, e seu ponto de fusão.
A temperatura da estufa só pode ser alterada depois de consultas a todos os usuários e
comunicado ao funcionário.
O que nela for colocado deve ser retirado no mesmo dia.
3.14. Termômetro
Sempre que fora de uso deve ficar na caixa.
Não deve ser usado para agitar. Para isto existe a bagueta.
Não deve ser colocado em ambiente já muito quente, nem esfriado rapidamente (sob a
água, por exemplo).
3.15. Funil de vidro sinterizado
Não passe água de torneira através da placa porosa.
Use "POLICEMAN" de borracha látex ou outro material macio para retirar o sólido, e não
espátula de metal.
3.16. Livros
Não os deixe próximo à mesa de trabalho.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
16
3.17. Pias e Canaletas
O escoamento deve ser mantido livre, principalmente nas canaletas.
Soluções ácidas devem ser neutralizadas antes de vertidas na pia.
4. CONSTANTES FÍSICAS DE COMPOSTOS ORGÂNICOS
(texto elaborado pelo Prof. Robson R. Teixeira da Universidade Federal de Viçosa)
As substâncias químicas apresentam propriedades físicas que são utilizadas para sua
caracterização ou mesmo para determinação do seu grau de pureza.
Em geral, as constantes físicas estão associadas às forças intermoleculares (forças
eletrostáticas, ligações de hidrogênio, interações dipolo-dipolo, etc) a que cada substância está
sujeita.
Propriedades físicas clássicas incluem cor, temperatura de fusão, temperatura de ebulição,
densidade, índice de refração, massa molecular e rotação específica. Fórmulas de compostos
orgânicos podem ser confirmadas por análises elementares de carbono, nitrogênio, hidrogênio e
enxofre em equipamentos apropriados. Além disso, para a caracterização de substâncias podem
ser utilizados métodos espectroscópicos modernos como espectroscopias no infravermelho, no
ultravioleta-visível e de ressonância magnética nuclear, e espectrometria de massas.
4.1. Temperatura de fusão
A temperatura de fusão de uma amostra em análise é determinada a partir do momento em
que se observa a formação da primeira gota de fase líquida (Figura 1, tubo 3) até a temperatura
em que o último cristal desaparece, situação entre aquelas representadas pelos tubos 4 e 5 na
Figura 1. Portanto, o que normalmente se obtém é uma faixa de fusão. Durante a análise da
amostra, com o aquecimento, podem ocorrer mudanças estruturais nos cristais sem que surja uma
fase líquida, como exemplifica o tubo 2 na Figura 1. Essas alterações visuais da amostra não
constituem ainda a sua fusão.
Em geral, a faixa de temperatura de fusão não excede 2 ºC para uma substância pura.
Uma pequena quantidade de impurezas na amostra é suficiente para alargar consideravelmente
essa faixa de temperatura, além de abaixar a temperatura inicial de fusão. Assim, essas medidas
podem ser utilizadas para avaliar a pureza de uma dada amostra.
Deve-se considerar também que algumas substâncias sólidas podem apresentar diferentes
arranjos cristalinos, que possuem cada um a sua temperatura de fusão. Os valores de temperatura

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
17
de fusão de substâncias conhecidas estão descritos na literatura, sendo dados de referência úteis
na caracterização de amostras químicas.
Figura 1. Transformações ocorridas no intervalo de fusão.
É comum existirem compostos diferentes com temperaturas de fusão iguais. Assim, deve-
se recorrer a outras propriedades físicas para se conhecer inequivocamente a identidade de um
composto sólido. Um procedimento muito utilizado para verificar a identidade de dois sólidos que
apresentam mesma temperatura de fusão consiste em misturar pequenas quantidades do sólido
desconhecido com o conhecido e determinar a temperatura de fusão da mistura. Se houver
variação no valor da temperatura de fusão após a mistura, conclui-se que os sólidos são
substâncias diferentes.
A temperatura de fusão pode ser determinada empregando-se o tubo de Thiele. Ele
consiste em um tubo de vidro desenhado para conter um óleo de aquecimento e um termômetro ao
qual se liga um tubo capilar que contém a amostra (Figura 2). Ele é usualmente aquecido com um
bico de Bunsen, sendo que a velocidade de aumento da temperatura deve ser controlada.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
18
Figura 2. Tubo de Thiele.
Embora existam diversos instrumentos comerciais para determinação da temperatura de
fusão (Melt-Temp, Fisher-Johns, Thomas-Hoover e Koffler Micro Melting, etc, Figura 3) todos
envolvem basicamente a mesma técnica e utilizam sistema elétrico de aquecimento, que permite o
aumento gradual e preciso da temperatura, um termômetro ou termopar e um artefato ótico (jogo
de lentes e/ou objetivas) para observação da amostra. Duas temperaturas são obtidas: a primeira é
o ponto em que a primeira gota de líquido se forma entre os cristais e a segunda, aquele em que
toda a amostra transforma-se em um líquido límpido (Figura 1).
Figura 3. Aparelhos de ponto de fusão.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
19
Quando a temperatura de fusão da substância é desconhecida, o procedimento mais
comum envolve inicialmente a determinação de uma temperatura de fusão aproximada, através do
aquecimento da amostra a uma taxa de 10 ºC/min, e, então, a determinação da temperatura de
fusão com maior exatidão, utilizando-se uma velocidade de aquecimento mais lenta, a uma taxa de
1-2 ºC/min.
4.2. Temperatura de ebulição
Uma das propriedades físicas características, de fácil determinação e que contribuem para
a identificação de líquidos orgânicos bem como para verificação do seu grau de pureza é a
temperatura de ebulição (te). Isso acontece quando a pressão do vapor do líquido se iguala à
pressão aplicada sobre ele (usualmente a pressão atmosférica). Em tal temperatura, considera-se
que o líquido ferve. A temperatura de ebulição normal é essa temperatura medida sob pressão de
760 mmHg (ou 1 atm). Quando a pressão aplicada sobre o líquido, ou também chamada de
pressão externa, é inferior à pressão atmosférica, a pressão de vapor necessária para que o
líquido entre em ebulição também diminui e o líquido ferve em uma temperatura mais baixa.
Portanto, a temperatura de ebulição de um líquido está intimamente relacionada com a pressão
externa. O monógrafo da Figura 4 é utilizado para correlacionar a temperatura de ebulição de um
líquido com a pressão externa, permitindo uma previsão da temperatura de ebulição de um líquido
puro em variadas situações de pressão.
Para entender como se usa o monógrafo, imagine que o ponto de ebulição dado é 100 ºC
(coluna A) a 1 mmHg (coluna C). Para determinar o ponto de ebulição em 18 mmHg, ligue por uma
reta 100 ºC (coluna A) a 1 mmHg (coluna C) e observe onde essa reta intercepta a coluna B (cerca
de 280 ºC). Este valor corresponde ao ponto de ebulição normal. A seguir, ligue 280 ºC (coluna B)
com 18 mmHg (coluna C) e observe onde a linha intercepta a coluna A (151 ºC). O ponto de ebulição
aproximado será 151 ºC em 18 mmHg.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
20
Figura 4. Monógrafo de alinhamento de pressão e temperatura.
Analogamente à temperatura de fusão, a existência de impurezas no líquido também causa
um desvio na temperatura de ebulição. Existem dois métodos principais para determinação de
temperaturas de ebulição, dependendo da quantidade de amostra disponível. A destilação, cujos
princípios serão discutidos na Experiência 5, é utilizada quando se tem uma grande quantidade de
amostra. Nesse caso, a determinação da temperatura de ebulição é feita através do registro da
temperatura do vapor observada no termômetro durante a destilação. Por esse método, as
medidas com uma substância pura apresentarão intervalo menor que 0,5 ºC durante a ebulição,
considerando-se a temperatura lida quando da destilação da primeira gota e a temperatura do
vapor no decorrer da destilação.
Para a determinação da temperatura de ebulição de pequenas quantidades de amostra
(0,25 a 0,5 mL), utiliza-se, por exemplo, o sistema ilustrado na Figura 5 utilizando um tubo de
Thiele ou outro frasco contendo óleo sob agitação e uma fonte de calor. Nesse processo,
conhecido como Método de Siwoloboff, utiliza-se um microtubo de ensaio contendo a substância
que para a qual se deseja determinar a temperatura de ebulição e um capilar com a extremidade
superior fechada. Esse microtubo está preso ao termômetro (Figura 5). À medida que o líquido é
aquecido, bolhas do ar contido no capilar começam a ser liberadas. Em temperatura próxima à da
ebulição, observa-se um fluxo intenso de bolhas a partir da saída do capilar (um “colar” de bolhas).
Para determinar a temperatura de ebulição com maior precisão, nesse momento remove-se a fonte
de calor. Quando a pressão de vapor do líquido se igualar à pressão atmosférica, o líquido
começará a entrar no capilar. Nesse instante deve-se ler a temperatura no termômetro novamente.
Essa será a temperatura de ebulição. As duas leituras de temperatura (quando se observa o “colar”
de bolhas e quando o líquido começa a entrar no capilar) não devem diferir em mais de 1 oC. Caso
contrário, o experimento deve ser repetido com um aquecimento mais brando para se confirmar a
pureza da amostra e a temperatura de ebulição.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
21
Figura 5. Sistema para determinação do ponto de ebulição.
4.3. Densidade
A densidade é uma propriedade física que independe da quantidade de matéria. Ela é
definida como a razão entre a massa e volume, em determinada temperatura. Pelo Sistema
Internacional de Unidades, ela é expressa em kg.m-3. Entretanto, ela é geralmente expressa em
g.cm-3.
volume
massadensidade ou
V
md
A densidade é uma propriedade física específica, ou seja, cada substância pura tem uma
densidade própria, que pode identificá-la e diferenciá-la de outras substâncias. Por isso, essa
densidade é conhecida como densidade absoluta. Já a densidade relativa de uma substância é a
relação entre a sua densidade absoluta e a densidade absoluta de uma substância estabelecida
como padrão. O padrão usualmente escolhido é a água, cuja densidade absoluta é de 1,000 g.cm-3
a 4 ºC.
A maioria dos líquidos e sólidos expande-se ligeiramente sob calor. O volume de uma
amostra de água, por exemplo, aumenta cerca de 4% quando aquecida de 4 ºC para 100 ºC.
Entretanto, a água apresenta comportamento anômalo quando aquecida de 0 a 4 ºC, pois sua
densidade aumenta ligeiramente durante esse aquecimento. A partir desse ponto, o aumento de
temperatura ocasiona a diminuição da densidade da água, analogamente ao que ocorre com os
demais líquidos. Por causa da variação da densidade com a temperatura, é necessário especificar
a temperatura quando se determina a densidade de um sólido ou líquido.
A densidade de um sólido, além de ser função da temperatura, depende da natureza da
sua rede cristalina, pois diferentes polimorfos de uma substância exibem diferentes densidades.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
22
Na química orgânica, o valor da densidade é muito utilizado para converter o volume de um
líquido em massa e vice-versa, com a finalidade de se medir uma quantidade de amostra com a
instrumentação disponível.
Existem equipamentos comerciais para a determinação da densidade de líquidos,
conhecidos como densímetros. Em laboratório, a densidade também pode ser determinada com o
auxílio de uma vidraria denominada picnômetro (Figura 6) e de uma balança semi-analítica ou
analítica ou mesmo através do uso de vidrarias volumétricas e da balança.
Figura 6. Picnômetro.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 23
5. AULAS PRÁTICAS (Aulas práticas elaboradas pelo Prof. Robson R. Teixeira da Universidade Federal de Viçosa)
Aula prática 01: Extração Líquido-Líquido
INTRODUÇÃO
As substâncias orgânicas obtidas de fonte natural ou mesmo através de uma reação
química raramente estão puras. No caso de uma reação química que gerou uma mistura de
substâncias, o produto principal pode ser removido por filtração, caso seja um sólido insolúvel no
meio reacional, ou pode ser destilado, se for um líquido de baixa temperatura de ebulição. No
entanto, esses casos são minoria e o que se observa é a necessidade de realizar extrações
seletivas ou lavagens do meio reacional para remoção das impurezas.
Na maioria das vezes, as impurezas e reagentes inorgânicos solúveis em água podem ser
removidos com facilidade pela lavagem do composto orgânico ou de sua solução em um solvente
orgânico volátil insolúvel em água, que em uma segunda etapa pode ser eliminado por
evaporação.
Alguns compostos orgânicos, apesar da baixa solubilidade em água, podem facilmente ser
transformados em derivados solúveis em meio aquoso. Compostos contendo grupos ácidos ou
básicos, após tratamento com bases ou ácidos inorgânicos, respectivamente, originam sais
solúveis.
Ambos os processos de separação e purificação descritos anteriormente envolvem a
técnica denominada de extração, ou mais precisamente, extração líquido-líquido.
Teoria da extração líquido-líquido
A extração envolve a transferência de um soluto de um solvente para outro. Essa
transferência ocorre porque o soluto é mais solúvel no segundo solvente do que no primeiro. Os
dois solventes devem ser imiscíveis para que ocorra a formação de um sistema bifásico. Em geral,
utiliza-se um funil de separação (Figura 1) para se realizar a extração líquido-líquido em escala
laboratorial.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 24
Figura 1. Esquema de montagem para extração líquido-líquido.
A Figura 2 ilustra de maneira geral o processo de extração. No início, as substâncias
hipotéticas A e B estão misturadas na fase aquosa. Após a adição de um solvente orgânico
imiscível com a água, serão formadas duas fases e haverá uma migração diferencial da substância
A para a fase orgânica, até o estabelecimento de um estado de equilíbrio. Com a separação das
duas fases, há a consequente separação das substâncias A e B.
Figura 2. Esquema do processo de extração líquido-líquido.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 25
Portanto, o processo de extração envolve uma distribuição de um dado soluto entre duas
fases líquidas imiscíveis (fase orgânica e fase aquosa). Quando as fases se separam em duas
camadas, estabelece-se um equilíbrio tal que a razão das concentrações do soluto em cada fase é
uma constante, chamada de coeficiente de distribuição (ou coeficiente de partição). O coeficiente
de distribuição K é definido como
1
2
C
CK
onde
C1 = concentração (em g.L-1, mg.mL-1 ou outra unidade) de equilíbrio do soluto na fase aquosa;
C2 = concentração (em g.L-1, mg.mL-1 ou outra unidade) de equilíbrio do soluto na fase orgânica.
O valor do coeficiente de distribuição é constante para cada soluto e independe das
quantidades dos dois solventes em contato. Entretanto, o coeficiente de distribuição depende da
natureza dos solventes utilizados e da temperatura. Assim, o valor do coeficiente de distribuição é
fixo para um determinado par de solventes a uma dada temperatura.
Como a concentração em cada fase pode ser expressa pela relação entre a massa do
soluto e o volume do solvente, é possível expressar diretamente a massa do soluto extraído e a
massa restante através do coeficiente de partição (K). Pela equação apresentada a seguir, verifica-
se que quanto maior o volume do solvente extrator, maior será a quantidade extraída.
2
2
1
1
2
1
V
mV
m
C
CK
2
121 .
V
VKmm
O processo de extração não é 100 % eficiente, o que significa dizer que nem todo soluto
será transferido totalmente de uma fase para outra em uma única etapa de extração, a não ser que
o valor de K seja muito elevado. Geralmente, é necessário que várias extrações sejam realizadas
para remover todo o soluto de uma das fases. De fato, o processo de extração é muito mais
eficiente quando efetuado várias vezes com um pequeno volume de solvente do que quando
efetuado uma única vez com um grande volume de solvente. O exemplo seguinte procura ilustrar
esse aspecto.
Considere que são empregados dois solventes A e B em igual volume, 100 mL, onde B
contém inicialmente um soluto X em concentração de 40 % (m/v). O coeficiente de partição para
este sistema é de 0,8. Observa-se que nas condições descritas, o solvente extrator A, que
inicialmente não continha nenhuma quantidade de soluto dissolvida, contém agora 17,8 g de X,
enquanto no solvente B permaneceram ainda 22,2 g, após o estabelecimento do estado de
equilíbrio. A distribuição do soluto X entre os solventes A e B é mostrada na Figura 3.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 26
Figura 3. Distribuição do soluto X em uma extração líquido-líquido em uma única etapa.
Pode-se comparar a eficiência do processo extração em uma única etapa com a extração
múltipla empregando-se um mesmo volume total. Por exemplo, caso se faça a extração descrita
anteriormente com duas porções de 50 mL serão removidos pelo solvente A na primeira extração
cerca de 11,4 g e na segunda 8,20 g, totalizando 19,6 g contra 17,8 g com uma única extração,
conforme demonstrado na Figura 4.
Portanto, as Figuras 3 e 4 demonstram que o processo de extração em várias etapas é
mais eficiente do que o processo de extração realizado em uma única etapa.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 27
Figura 4. Distribuição do soluto X em uma extração líquido-líquido múltipla.
Escolha do solvente extrator
Conforme discutido anteriormente, a extração líquido-líquido envolve a distribuição
diferencial de um soluto entre dois líquidos imiscíveis. Em geral, uma das fases é constituída pela
água e, portanto, denominada fase aquosa e a outra fase é constituída por um solvente que seja
imiscível na água, que é conhecido como fase orgânica ou solvente extrator.
Na escolha do solvente adequado para cada extração devem ser considerados alguns
critérios:
a) O solvente deve apresentar baixa solubilidade na água, possibilitando a formação de duas
fases;

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 28
b) O solvente deve solubilizar consideravelmente a substância que se deseja extrair. Neste
ponto, deve ser levado em conta que “semelhante dissolve semelhante”, ou seja, para
extrair substâncias pouco polares, por exemplo, deve ser utilizado um solvente de baixa
polaridade;
c) O solvente deve ser quimicamente inerte, não reagindo com as substâncias a serem
extraídas;
d) O solvente deve apresentar volatilidade razoável (baixa temperatura de ebulição) para que
possa ser removido facilmente, permitindo o rápido isolamento do produto desejado;
e) É desejável ainda que o solvente seja de baixo custo e de baixa toxicidade.
Na Tabela 1 estão listados os solventes mais utilizados para extrações líquido-líquido. Observe
que dentre os solventes listados, apenas aqueles que possuem cloro em sua composição são mais
densos que a água. Portanto, apenas quando for utilizado um solvente clorado para extração, que
a fase orgânica será a inferior.
Tabela 1. Propriedades físicas dos solventes mais utilizados para extração.
Solvente Massa Molar
(g mol-1)
Ponto de Ebulição
(ºC)
Densidade a 20 ºC
(g cm-3)
Éter dietílico 74 35 0,714
Pentano 72 36 0,626
Diclorometano 85 41 1,335
Clorofórmio 119 61 1,492
Hexano 86 66 0,659
Tetracloreto de carbono 154 77 1,594
Benzeno 78 80 0,879
Tolueno 92 111 0,867
Aspectos práticos da extração líquido-líquido
A seguir são descritos aspectos práticos relevantes quando se realiza uma extração
líquido-líquido em escala laboratorial.
Funil de separação
Em escala laboratorial, o processo de extração líquido-líquido é geralmente realizado em
um funil de separação (Figura 1). Deve-se ter o cuidado para que o volume de líquido no funil de
separação nunca ultrapasse ¾ de sua capacidade nominal. Deve-se sempre checar se a torneira
está fechada e manter um erlenmeyer embaixo do funil de separação.
O processo de extração inicia-se com a adição da solução a ser extraída e do solvente
extrator ao funil de separação. Tampa-se o funil e realiza-se a agitação, com movimentos

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 29
circulares, mantendo o funil em um ângulo de 45º em relação à vertical. Com uma das mãos,
segura-se firmemente a tampa e com a outra a torneira (Figura 5).
Figura 5. Forma correta de agitar e liberar a pressão de um funil de separação.
Ao agitar a mistura, devido à alta volatilidade do solvente, poderá haver aumento da
pressão interna. Por isso, após o procedimento de agitação, deve-se realizar o “alívio” de pressão,
inclinando-se a parte inferior do funil para cima e abrindo lentamente a torneira. Ao abri-la, deve-se
tomar o máximo de cuidado para não dirigir a saída dos vapores para si ou para alguém próximo.
O processo de agitação e alívio de pressão é usualmente realizado três vezes e, então,
deve-se colocar o funil de separação em suporte apropriado, retirar a tampa e esperar a formação
das duas fases, para realizar a separação.
Solventes utilizados
Normalmente os solventes utilizados na extração possuem baixa temperatura de ebulição
(Tabela 1) ou mesmo pode-se realizar uma extração com solvente quimicamente ativo, como
solução de bicarbonato de sódio, que leva à liberação de gases quando esta solução entra em
contato com um ácido. Por isso, é necessário que se faça o “alívio” de pressão. Entretanto, isto
deve ser feito lentamente, porque os vapores podem se expandir de forma violenta, projetando o
líquido para fora do funil.
Emulsão
Em alguns casos, após o processo de extração, não se observa separação completa das
fases orgânica e aquosa, ficando gotículas de uma fase dispersas na outra. Isto leva à formação de
uma interface não definida, chamada de emulsão, que compromete a eficiência da separação.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 30
Uma das alternativas para “quebrar” a emulsão (completa separação das fases) consiste
em deixar o funil de separação em repouso por alguns minutos. Entretanto, se isso não for
suficiente, outros métodos podem ser empregados. Por exemplo, a simples adição de mais
solvente orgânico ou água pode ser suficiente. Se a emulsão persistir, a adição de sais inorgânicos
(NaCl, Na2SO4, etc) pode ser uma alternativa eficaz para desfazer a emulsão. Nos casos mais
persistentes, a centrifugação pode ser usada.
Descarte de fases
Um dos enganos mais comuns durante um processo de extração líquido-líquido é o
descarte da fase errada. Como discutido anteriormente, somente quando se utilizam solventes
orgânicos clorados, que possuem densidades maiores que da água (Tabela 1), a fase orgânica
será a inferior.
Cuidado redobrado deve ser tomado quando se realiza a extração com solventes
quimicamente ativos, pois, muitas vezes, a substância de interesse está na fase aquosa na forma
de um sal.
Considerando os fatos acima, é aconselhável que ambas as fases sejam guardas até que
se tenha isolado o produto desejado.
Agentes de secagem
Após o processo de extração e de separação das fases, a fase orgânica, que usualmente
contém o composto de interesse, deve ser seca através da utilização de agentes de secagem (ou
agentes secantes). Isto é necessário, pois qualquer solvente orgânico, mesmo com baixa afinidade
pela água, absorverá água quando agitado com uma solução aquosa. É claro que a quantidade de
água dissolvida irá variar de um solvente para outro.
Os agentes secantes são sais inorgânicos anidros capazes de adicionar água de
hidratação quando expostos à umidade do ar ou às soluções úmidas. Os mais comumente usados
são: sulfato de sódio, sulfato de magnésio, cloreto de cálcio, sulfato de cálcio e carbonato de
potássio. Estes sais variam em propriedades e aplicações. Assim, eles não absorvem a mesma
quantidade de água em uma dada massa de solvente. A Tabela 2 compara os agentes secantes
mais comuns.
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 31
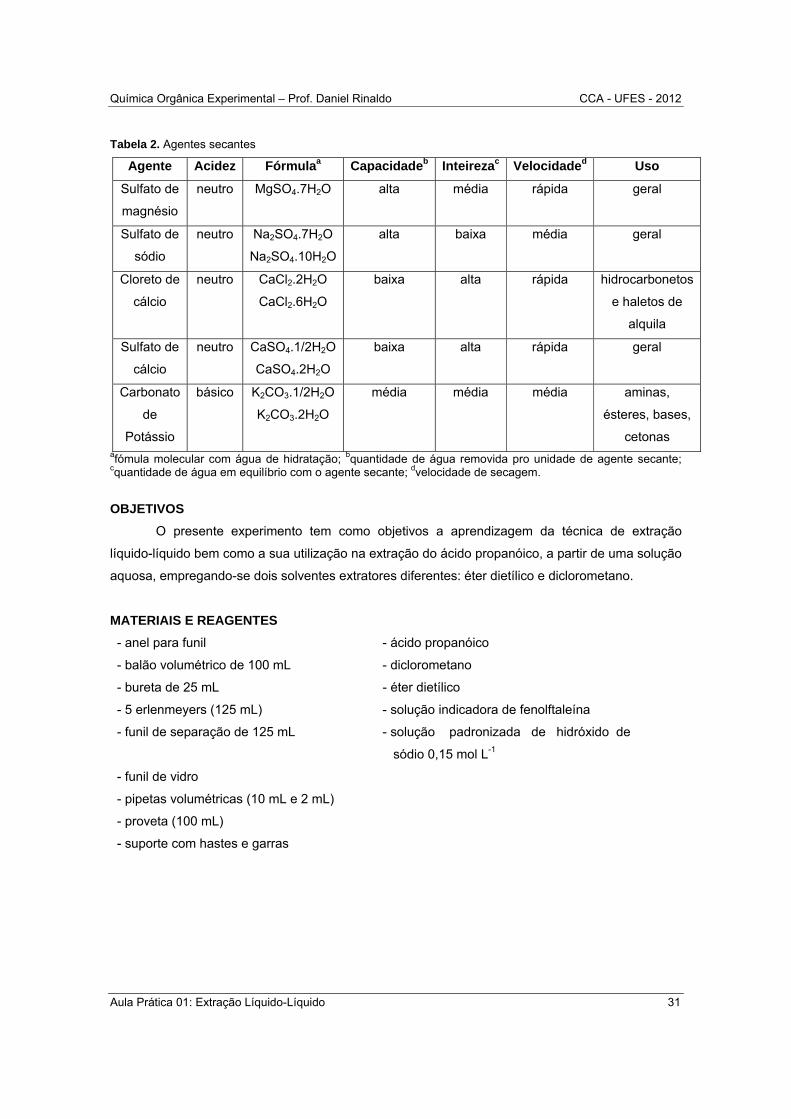
Tabela 2. Agentes secantes
Agente Acidez Fórmulaa Capacidadeb Inteirezac Velocidaded Uso
Sulfato de
magnésio
neutro MgSO4.7H2O alta média rápida geral
Sulfato de
sódio
neutro Na2SO4.7H2O
Na2SO4.10H2O
alta baixa média geral
Cloreto de
cálcio
neutro CaCl2.2H2O
CaCl2.6H2O
baixa alta rápida hidrocarbonetos
e haletos de
alquila
Sulfato de
cálcio
neutro CaSO4.1/2H2O
CaSO4.2H2O
baixa alta rápida geral
Carbonato
de
Potássio
básico K2CO3.1/2H2O
K2CO3.2H2O
média média média aminas,
ésteres, bases,
cetonas afómula molecular com água de hidratação; bquantidade de água removida pro unidade de agente secante; cquantidade de água em equilíbrio com o agente secante; dvelocidade de secagem.
OBJETIVOS
O presente experimento tem como objetivos a aprendizagem da técnica de extração
líquido-líquido bem como a sua utilização na extração do ácido propanóico, a partir de uma solução
aquosa, empregando-se dois solventes extratores diferentes: éter dietílico e diclorometano.
MATERIAIS E REAGENTES
- anel para funil - ácido propanóico
- balão volumétrico de 100 mL - diclorometano
- bureta de 25 mL - éter dietílico
- 5 erlenmeyers (125 mL) - solução indicadora de fenolftaleína
- funil de separação de 125 mL - solução padronizada de hidróxido de
sódio 0,15 mol L-1
- funil de vidro
- pipetas volumétricas (10 mL e 2 mL)
- proveta (100 mL)
- suporte com hastes e garras

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 32
PROCEDIMENTO EXPERIMENTAL
1) Preparo de uma solução aquosa de ácido propanóico
a) A um balão volumétrico de 100 mL, adicione 2 mL de ácido propanóico e complete com água
destilada. Agite até a homogeneização da solução resultante (solução A).
b) Pipete uma alíquota de 10 mL da solução A e transfira para um erlenmeyer de 125 mL.
Adicione 3 gotas de solução indicadora de fenolftaleína.
c) Encha a bureta com solução padronizada de NaOH (0,15 mol L-1) e titule a solução. O ponto
final da titulação é alcançado quando surge e permanece a cor rósea. Anote o volume consumido
de solução de NaOH. Complete o volume da bureta e titule uma nova amostra de solução de ácido
propanóico (10 mL). Anote o volume consumido da solução de NaOH. A massa de ácido
propanóico presente na solução aquosa será calculada utilizando-se a média das duas medidas
obtidas na titulação.
2) Extração simples empregando-se como solvente extrator éter etílico
Pipete 10 mL da solução ácida (solução A) e transfira para um funil de separação.
Adicione 30 mL de éter dietílico. Agite a mistura, conforme ilustrado na Figura 5, tomando o
cuidado para aliviar a pressão interna no funil. Esta operação deve ser realizada no interior de uma
capela de exaustão, uma vez que o éter dietílico é muito volátil. Deixe o sistema em repouso até a
separação completa das fases. Recolha a camada aquosa (fase inferior no funil de separação) em
um erlenmeyer de 125 mL e adicione 3 gotas de solução indicadora de fenolftaleína. Complete o
volume da bureta com solução padronizada de NaOH (0,15 mol L-1) e titule a solução do ácido até
que surja e permaneça a cor rósea. Anote o volume consumido de solução de NaOH.
3) Extração múltipla empregando-se como solvente extrator éter etílico
Pipete 10 mL da solução aquosa de ácido propanóico anteriormente preparada (solução
A); transfira para um funil de separação e faça a extração com 15 mL de éter etílico, conforme
descrito no item 2. Separe a fase aquosa da fase orgânica e retorne-a para o funil de separação.
Extraia novamente a fase aquosa com mais 15 mL de éter dietílico. Recolha a fase aquosa em um
erlenmeyer de 125 mL e adicione 3 gotas de fenolftaleína. Complete o volume da bureta com
solução padronizada de NaOH (0,15 mol L-1) e titule a fase aquosa, conforme descrito
anteriormente. Anote o volume consumido de solução de NaOH.
4) Extração simples empregando-se como solvente extrator diclorometano
Repita o procedimento descrito no item 2, porém utilize como solvente extrator
diclorometano. O processo de extração também deverá ser realizado em capela de exaustão.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 01: Extração Líquido-Líquido 33
QUESTÕES
1) Calcule a massa (em gramas) de ácido propanóico presente na solução aquosa, que foi titulada
no item 1.
2) Calcule a massa (em gramas) de ácido propanóico restante nas soluções aquosas, após as
extrações realizadas nos itens 2 e 3.
3) Calcule a porcentagem do ácido propanóico que foi extraída nos itens 2 e 3. Com base nas
porcentagens de ácido propanóico calculadas, o que é possível concluir sobre a eficiência dos dois
tipos de extração?
4) Calcule a massa (em gramas) de ácido propanóico restante nas soluções aquosas, após as
extrações realizadas no item 4. Considerando os solventes éter dietílico e diclorometano, qual
possui maior eficiência para a extração do ácido propanóico?
5) Calcule o coeficiente de partição para o ácido propanóico utilizando os resultados obtidos nas
extrações simples com éter dietílico (procedimento 2) e com diclorometano (procedimento 4).
6) O que se deve fazer quando há dúvida sobre qual das fases é a orgânica em um procedimento
de extração?
7) A solubilidade do ácido subérico é 0,14 g/100 mL de água e 0,56 g/100 mL de éter dietílico.
Calcule o volume de éter dietílico necessário para remover 90 % do ácido subérico a partir de 100
mL de uma solução aquosa saturada, em uma única extração.
8) Mostre que para um composto hipotético A, cujo coeficiente de distribuição em água e éter
dietílico é 5, a realização de duas extrações com 50 mL de éter dietílico é mais eficiente que uma
extração com 100 mL de éter dietílico. Realize os cálculos das quantidades extraídas para
comprovar a maior eficiência do processo de extração múltipla.
BIBLIOGRAFIA
DIAS, A.G.; DA COSTA, M.A.; GUIMARÃES, P.I.C. Guia Prático de Química Orgânica. Volume I
– Técnicas e Procedimentos: Aprendendo a Fazer. Editora Interciência, Rio de Janeiro, 2004.
MARQUES, J.A.; BORGES, C.P.F. Práticas de Química Orgânica. Editora Átomo, Campinas,
2007.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 34
Aula prática 02: Extração Líquido-Líquido com solventes quimicamente ativos
INTRODUÇÃO
A extração líquido-líquido convencional permite principalmente a separação de substâncias
orgânicas neutras. No caso da substância orgânica apresentar característica básica ou ácida, é
possível aumentar a eficiência da extração aproveitando-se dessa característica e desenvolvendo
um método especial de extração. Esse método, denominado extração por solventes quimicamente
ativos, baseia-se na facilidade com que certas substâncias podem ser transformadas em derivados
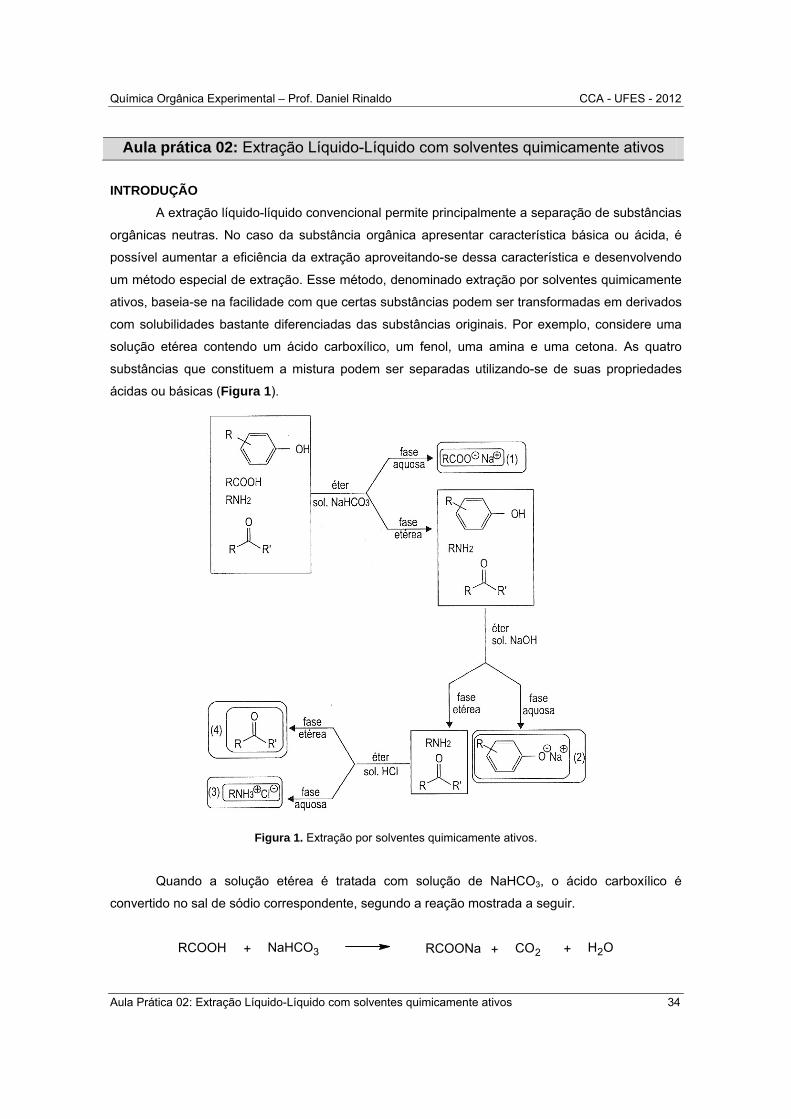
com solubilidades bastante diferenciadas das substâncias originais. Por exemplo, considere uma
solução etérea contendo um ácido carboxílico, um fenol, uma amina e uma cetona. As quatro
substâncias que constituem a mistura podem ser separadas utilizando-se de suas propriedades
ácidas ou básicas (Figura 1).
Figura 1. Extração por solventes quimicamente ativos.
Quando a solução etérea é tratada com solução de NaHCO3, o ácido carboxílico é
convertido no sal de sódio correspondente, segundo a reação mostrada a seguir.
RCOOH + NaHCO3 RCOONa + CO2 + H2O

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 35
O sal do ácido carboxílico é mais solúvel em água, o que provoca a sua migração para a
fase aquosa. Para obtenção do ácido carboxílico, deve-se acidificar a fase aquosa com HCl,
ocasionando a seguinte reação:
RCOONa + HCl RCOOH + NaCl
Nesse caso, o ácido carboxílico deve ser extraído da fase aquosa através da adição de
solvente orgânico
Na fase orgânica original restaram o fenol, a amina e a cetona. A essa fase pode-se
adicionar uma solução de NaOH, que irá reagir com o fenol (uma substância cujo caráter ácido é
menor que o do ácido carboxílico) resultando na formação de um fenóxido de sódio.
Analogamente ao que foi descrito para o sal do ácido carboxílico, o fenóxido de sódio pode ser
separado e, posteriormente, reconvertido ao fenol através da reação com HCl, segundo as
reações:
OH
+ NaOH
ONa
+ H2O
ONa
+ HCl
OH
+ NaCl
Assim, na fase orgânica ainda ficaram a amina e a cetona. Com a adição de uma solução
aquosa de HCl à fase orgânica, ocorrerá a reação de conversão da amina no cloridrato
correspondente, que por ser um sal é mais solúvel em água, e migrará para a fase aquosa.
RNH2 + HCl RNH3Cl
Após a separação das fases, a cetona estará presente na fase orgânica e o cloridrato na
fase aquosa. A posterior reação da fase aquosa com NaHCO3 e a extração com éter etílico
fornecerá a amina, segundo a reação mostrada a seguir.
RNH3Cl + NaHCO3 RNH2 + NaCl + CO2 + H2O

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 36
Para a separação final dos componentes nas respectivas fases etéreas pode-se proceder a
destilações ou, se conveniente, à evaporação do éter.
OBJETIVOS
O presente experimento tem como objetivos utilizar a técnica de extração líquido-líquido
com solventes ativos para separação dos componentes de uma mistura constituída por m-
nitroanilina, ácido benzóico e naftaleno.
MATERIAL E REAGENTES
- béquer de 50 mL - espátula
- erlenmeyer de 125 mL - banho de gelo
- funil de separação de 125 mL - ácido benzóico
- funil de vidro - ácido clorídrico (concentrado e 11 %, v/v)
- proveta de 10 mL - éter dietílico
- proveta de 25 mL - hidróxido de sódio (10 %, m/v)
- papel filtro - m-Nitroanilina
- suporte para funil - naftaleno
- papel indicador universal - sulfato de magnésio anidro
PROCEDIMENTO EXPERIMENTAL
a) Antes de iniciar os procedimentos experimentais, verifique possíveis vazamentos na torneira e
na tampa do funil de separação, utilizando-se água destilada. Em um béquer de 50 mL, dissolva
1,50 g da mistura1 contendo m-nitroanilina, ácido benzóico e naftaleno em 15 mL de éter dietílico.
Transfira essa solução para um funil de separação de 125 mL.
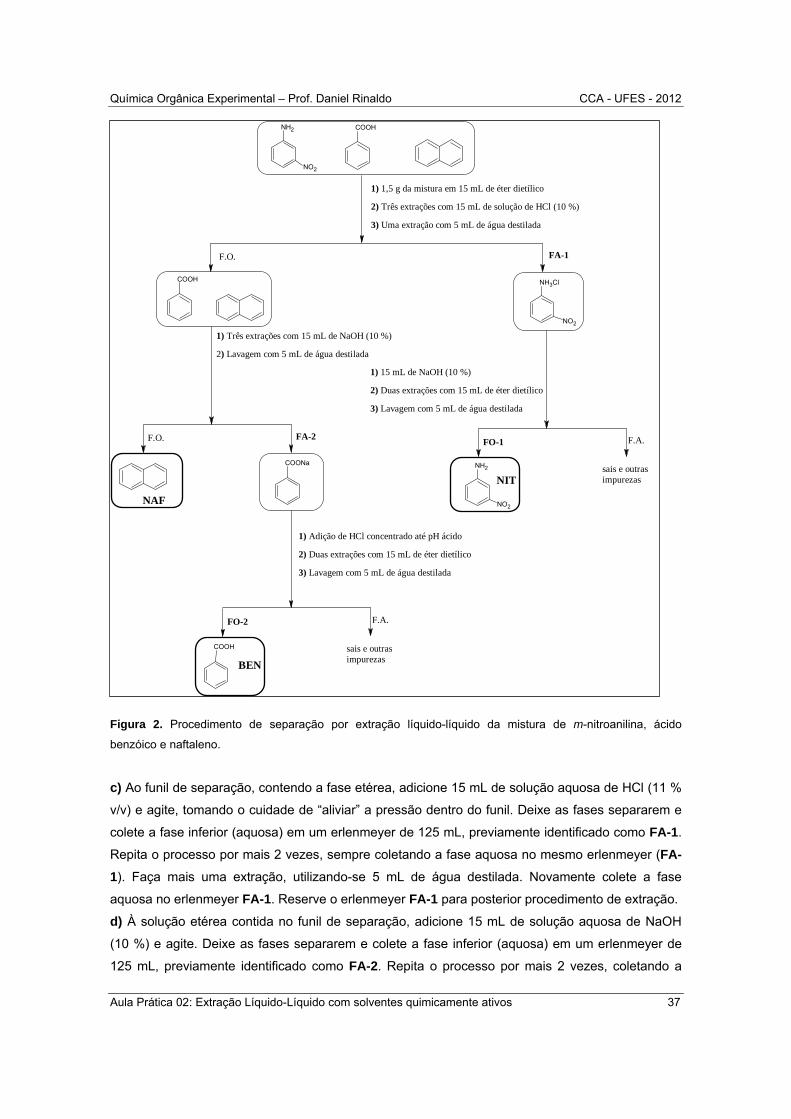
b) O procedimento da separação da mistura está esquematizado na Figura 2 e está descrito em
detalhes a seguir.
1 A mistura contém 0,50 g de cada componente.
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 37
Figura 2. Procedimento de separação por extração líquido-líquido da mistura de m-nitroanilina, ácido
benzóico e naftaleno.
c) Ao funil de separação, contendo a fase etérea, adicione 15 mL de solução aquosa de HCl (11 %
v/v) e agite, tomando o cuidade de “aliviar” a pressão dentro do funil. Deixe as fases separarem e
colete a fase inferior (aquosa) em um erlenmeyer de 125 mL, previamente identificado como FA-1.
Repita o processo por mais 2 vezes, sempre coletando a fase aquosa no mesmo erlenmeyer (FA-
1). Faça mais uma extração, utilizando-se 5 mL de água destilada. Novamente colete a fase
aquosa no erlenmeyer FA-1. Reserve o erlenmeyer FA-1 para posterior procedimento de extração.
d) À solução etérea contida no funil de separação, adicione 15 mL de solução aquosa de NaOH
(10 %) e agite. Deixe as fases separarem e colete a fase inferior (aquosa) em um erlenmeyer de
125 mL, previamente identificado como FA-2. Repita o processo por mais 2 vezes, coletando a
NH2
NO2
COOH
1) 1,5 g da mistura em 15 mL de éter dietílico
2) Três extrações com 15 mL de solução de HCl (10 %)
3) Uma extração com 5 mL de água destilada
F.O. FA-1
NH3Cl
NO2
COOH
1) 15 mL de NaOH (10 %)
2) Duas extrações com 15 mL de éter dietílico
3) Lavagem com 5 mL de água destilada
F.A.FO-1
NH2
NO2
sais e outras impurezas
FA-2F.O.
NIT
NAF
COONa
1) Adição de HCl concentrado até pH ácido
2) Duas extrações com 15 mL de éter dietílico
3) Lavagem com 5 mL de água destilada
F.A.FO-2
sais e outras impurezas
COOH
BEN
1) Três extrações com 15 mL de NaOH (10 %)
2) Lavagem com 5 mL de água destilada

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 38
fase aquosa sempre no mesmo erlenmeyer (FA-2). Faça mais uma extração, utilizando-se 5 mL de
água destilada. Novamente colete a fase aquosa no erlenmeyer FA-2. Reserve o erlenmeyer FA-2
para posterior procedimento de extração.
e) A solução etérea que ficou no funil de separação, deve ser transferida para um erlenmeyer de
50 mL. Em seguida, adicione sulfato de magnésio anidro em quantidade suficiente para que a fase
etérea fique seca (observa-se que o sulfato de magnésio fica solto quando isso acontece). Filtre a
fase orgânica seca por gravidade, coletando a solução em um béquer de 50 mL previamente
pesado e identificado como NAF. Coloque esse béquer em capela de exaustão para a evaporação
do solvente e, então, pese o béquer novamente.
f) Adicione a fase aquosa FA-1 ao funil de separação de 125 mL, juntamente com 15 mL de
solução aquosa de NaOH (10 %) e 15 mL de éter dietílico. Agite e deixe as fases separarem,
colete a fase inferior (aquosa) em um erlenmeyer de 125 mL e a fase superior (orgânica) em outro
erlenmeyer de 125 mL previamente identificado como FO-1. Retorne a fase aquosa ao funil de
separação e adicione mais 15 mL de éter dietílico e agite. Deixe as fases separarem e colete a
fase aquosa no mesmo erlenmeyer anteriormente utilizado para a fase aquosa e a fase orgânica
no erlenmeyer FO-1. Voltar a fase orgânica FO-1 ao funil de separação e adicionar 5 mL de água
destilada. Agite, deixe as fases separarem e colete a fase aquosa em um erlenmeyer e a fase
orgânica no erlenmeyer FO-1. Adicione sulfato de magnésio anidro em quantidade suficiente para
secar a fase etérea contida no erlenmeyer FO-1. Filtre a fase orgânica seca por gravidade,
coletando a solução em um béquer de 50 mL previamente pesado e identificado como NIT.
Coloque esse béquer em capela de exaustão para a evaporação do solvente e, então, pese o
béquer novamente.
g) Adicione lentamente HCl concentrado à fase aquosa FA-2, até obter pH ácido (pH
aproximadamente 1). Para verificar o pH, utilize o papel indicador universal. Durante a adição do
ácido, mantenha o erlenmeyer FA-2 em banho de gelo. Transfira a fase aquosa para o funil de
separação de 125 mL, juntamente com 15 mL de éter dietílico. Agite, deixe as fases separarem e
colete a fase inferior (aquosa) em um erlenmeyer de 125 mL e a fase superior (orgânica) em outro
erlenmeyer de 125 mL previamente identificado como FO-2. Retorne a fase aquosa ao funil de
separação e adicione mais 15 mL de éter dietílico. Agite, deixe as fases separarem e colete a fase
aquosa no mesmo erlenmeyer anteriormente utilizado para a fase aquosa, e a fase orgânica no
erlenmeyer FO-2. Retorne a fase orgânica FO-2 ao funil de separação e adicione 5 mL de água
destilada. Agite, deixe as fases separarem e colete a fase aquosa em um erlenmeyer e a fase
orgânica no erlenmeyer FO-2. Adicione sulfato de magnésio anidro em quantidade suficiente para
secar a fase etérea contida no erlenmeyer FO-2. Filtre a fase orgânica seca por gravidade,
coletando a solução em um béquer de 50 mL previamente pesado e identificado como BEN.
Coloque esse béquer em capela de exaustão para a evaporação do solvente e, então, pese o
béquer novamente.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 02: Extração Líquido-Líquido com solventes quimicamente ativos 39
QUESTÕES
1) Discuta sobre a eficiência do processo de extração líquido-líquido para separação dos
componentes da mistura constituída por m-nitroanilina, ácido benzóico e naftaleno. Baseie suas
discussões na quantidade obtida de cada substância.
2) Quais as vantagens e desvantagens de se usar éter dietílico como solvente extrator?
3) Escreva todas as reações envolvidas no experimento.
4) Proponha um procedimento para separação dos seguintes compostos:
OH
Br
Br
N
CH3
CH3
CH3
COOH
5) A Panacetina® é um medicamento que contém ácido acetilsalicílico, sacarose e acetanilida ou
fenacetina. Estes compostos têm as seguintes características de solubilidade:
a) A sacarose é solúvel em água e insolúvel em diclorometano (CH2Cl2);
b) O ácido acetilsalicílico é solúvel em diclorometano e relativamente insolúvel em água. O
hidróxido de sódio converte o ácido no correspondente sal, que é solúvel em água;
c) A acetanilida e a fenacetina são solúveis em diclorometano e insolúveis em água.
Com base nessas informações, descreva um procedimento experimental para separação dos
constituintes da Panacetina através da extração com solventes quimicamente ativos.
BIBLIOGRAFIA
DIAS, A.G.; DA COSTA, M.A.; GUIMARÃES, P.I.C. “Guia Prático de Química Orgânica. Volume I – Técnicas e Procedimentos: Aprendendo a Fazer”. Editora Interciência, Rio de Janeiro, 2004. MARQUES, J.A.; BORGES, C.P.F. “Práticas de Química Orgânica”. Editora Átomo, Campinas, 2007. PAVIA, D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental – técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009. HART, H.; CRAINE, L.E.; HART, D.J. “Organic Chemisty Laboratory Manual – A short Course”. Houghton Mifflin Company, Tenth Edition, Boston, 1999.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 40
Aula prática 03: Destilação simples, fracionada e a vácuo
INTRODUÇÃO
A destilação é um processo que consiste na vaporização do líquido, ou seja, na passagem
de um líquido para o estado gasoso com auxílio de calor e/ou por redução da pressão, seguido da
condensação desse vapor (liquefação) e coleta do condensado em outro recipiente. Um líquido só
entrará em ebulição quando sua pressão de vapor se igualar à pressão exercida sobre ele, ou seja,
à pressão atmosférica ou à pressão do sistema. O líquido obtido por esse processo na maioria das
vezes apresenta um elevado grau de pureza.
A destilação é um dos processos mais baratos e eficientes para purificação e/ou separação
de líquidos, sendo amplamente empregada em várias indústrias, especialmente para o refino de
petróleo e de outros produtos.
As técnicas existentes para se destilar um líquido variam de acordo com o grau de pureza
desejado e com o tipo de líquido que se deseja purificar ou separar. A dificuldade da separação
depende da volatilidade relativa dos componentes da mistura, ou seja, da diferença entre suas
temperaturas de ebulição. Existem quatro métodos principais de destilação: simples, fracionada, a
vácuo (ou à pressão reduzida) e por arraste de vapor.
Destilação simples
A destilação simples é empregada quando se deseja separar dois ou mais líquidos que
apresentem uma diferença entre suas temperaturas de ebulição superior a 80 ºC, ou separar um
componente mais volátil de uma solução onde os solutos são líquidos com elevadas temperaturas
de ebulição e estão presentes em baixa concentração (menor que 10 %). É utilizada ainda para
separar um líquido volátil que contém sólidos não voláteis dissolvidos.
A Figura 1 mostra uma aparelhagem típica para realização de uma destilação simples, que
consiste basicamente de: fonte de aquecimento, balão de destilação, cabeça de destilação,
termômetro, condensador (de Liebig), adaptador de vácuo (alonga) e frasco de coleta. O líquido a
ser destilado é colocado no balão de destilação e aquecido. O líquido aquecido se vaporiza e sobe,
passando pelo termômetro e entrando no condensador. O vapor é resfriado e se condensa,
escorrendo pela alonga até o frasco coletor.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 41
Figura 1. Sistema utilizado em uma destilação simples.
A temperatura observada durante a destilação de uma substância pura permanece
constante durante o processo, enquanto vapor e líquido estiverem presentes no sistema em torno
do bulbo do termômetro. Quando uma mistura líquida é destilada, a temperatura não permanece
constante. Ela aumenta durante a destilação porque a composição do vapor varia durante a
destilação, sendo a composição do primeiro destilado mais rica no componente mais volátil.
Um exemplo da aplicação da destilação simples seria a separação de uma mistura
contendo o componente desejado A (te 140 ºC), contaminado com uma impureza B (te 250 ºC),
ambos dissolvidos em éter dietílico (te 36 ºC). O éter dietílico é removido facilmente em baixa
temperatura; o componente A é destilado em temperatura mais alta e coletado em um recipiente
diferente; o componente B pode ser destilado ou ser tratado como resíduo.
A) Aspectos práticos da destilação simples
Fonte de calor
As fontes de calor mais empregadas em um processo de destilação são: bico de Bunsen,
manta e banho de aquecimento. A escolha da fonte depende da disponibilidade e, principalmente,
do tipo de líquido ou mistura líquida que se deseja destilar. O bico de Bunsen pode ser empregado
prioritariamente para destilação de líquidos de temperaturas de ebulição mais elevadas (> 180 ºC).
Para líquidos voláteis apresentando temperaturas de ebulição inferiores a 60 ºC utiliza-se manta de

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 42
aquecimento ou banho-maria. Já para líquidos com temperaturas de ebulição entre 60 e 180 ºC
recomenda-se o emprego de banho de óleo (mineral ou de silicone) ou de glicerina. O aquecimento
deve ser controlado, permitindo uma liberação constante de gotas do destilado, a uma velocidade
de destilação de uma a duas gotas por segundo.
Balão de destilação
O balão de destilação é um balão de fundo redondo, que é desenhado para resistir ao calor
e ao processo de destilação. O balão utilizado não deve ter mais do que 2/3 e não menos do que a
metade de sua capacidade nominal preenchida com a mistura líquida a ser destilada. Assim, o
volume do balão dependerá da quantidade de líquido a ser destilada. Deve-se ter cuidado para
que, ao final da destilação, o balão não seja levado à secura, de forma a evitar um eventual
acidente, caso haja resíduos ou mistura de resíduos sólidos no balão.
“Pedras” de ebulição
Antes de se iniciar uma destilação devem ser introduzidas no balão de destilação algumas
pedras de porcelana porosa, pérolas de vidro ou pedra-pomes, a fim de se evitar o fenômeno de
superaquecimento, também conhecido como ebulição tumultuosa ou em saltos. Uma alternativa ao
uso das “pedras” de ebulição seria utilizar um sistema de agitação magnética.
O uso das pedras de ebulição garante a ebulição suave do líquido, sem a ocorrência de
projeções. As bolhas de ar contidas nas pedras porosas são eliminadas pelo aquecimento,
auxiliando as microbolhas, que são formadas no interior do líquido, a vencer a pressão da coluna
do próprio líquido. Assim, a formação de bolhas de grande volume é minimizada.
As pedras nunca devem ser adicionadas ao líquido já aquecido, pois grandes quantidades
de vapor serão liberadas, levando à projeção do líquido para fora do balão. Além disso, caso a
destilação seja interrompida, novas pedras devem ser utilizadas.
Cabeça de destilação
A cabeça de destilação dirige os vapores da destilação para o condensador e permite a
conexão do termômetro ao sistema. O termômetro deve ser colocado na cabeça de destilação de
forma que seu bulbo esteja completamente imerso no vapor (vide destaque da Figura 1). Isto
significa que o bulbo deve estar situado imediatamente abaixo da saída lateral da cabeça de
destilação, para que a leitura da temperatura seja acurada.
Condensador
O condensador é um sistema de refrigeração no qual o vapor é liquefeito. Na destilação,
emprega-se o condensador de tubo reto ou condensador de Liebig. Ele deve ser montado
inclinado, para facilitar o escoamento do líquido condensado.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 43
O resfriamento é feito em contra-fluxo com o vapor, ou seja, a água entra pela parte de
baixo do condensador e sai por cima da camisa de refrigeração (Figura 1). O fluxo de água no
condensador deve ser moderado, para evitar vazamentos.
Adaptador
Uma destilação simples não pode ser efetuada em sistemas fechados, pois o aumento da
pressão do sistema pode levar à explosão da aparelhagem. Para evitar esse tipo de problema,
entre o condensador de Liebig e o frasco coletor se utiliza um adaptador, também conhecido com
adaptador de vácuo, unha ou alonga. Esse adaptador possui uma saída lateral, que permite ao
sistema permanecer aberto, além, é claro, de evitar a projeção do destilado para fora do frasco
coletor. Ele também pode proteger o destilado da umidade do ar, pois é possível conectar um tubo
com agente dessecante (como CaCl2 e Na2SO4 anidro) em sua saída lateral.
Frasco coletor
O frasco coletor, que fica após o adaptador, pode ser um balão de fundo redondo,
erlenmeyer ou qualquer outro recipiente. Com frequência, o destilado é coletado em porções ou
frações, substituindo-se o frasco de coleta por outro em intervalos regulares.
Se o líquido destilado for muito volátil e existir possibilidade de perdas por evaporação, é
aconselhável resfriar o balão de coleta em banho de água e gelo.
Todo o sistema (balão de destilação, cabeça de destilação, condensador, adaptador e
frasco coletor) deve ser bem preso por garras e mufas a suportes universais, de forma que as
garras estejam apertadas com firmeza, mas não causem tensão na vidraria.
As conexões entre as diversas peças de uma aparelhagem de destilação são feitas por
juntas de vidro esmerilhado. Nesse caso, é necessário lubrificar as juntas esmerilhadas com graxa
de silicone ou fitas de Teflon. Entretanto, deve-se tomar o máximo de cuidado para evitar a
contaminação do produto, quando da utilização de graxa de silicone.
Destilação fracionada
A destilação fracionada é empregada quando se deseja separar dois ou mais líquidos
miscíveis, cuja diferença entre suas temperaturas de ebulição seja inferior a 80 ºC. O sistema utilizado
para a realização da destilação fracionada (Figura 2) é bastante similar ao da destilação simples
(Figura 1), com exceção da presença de uma coluna de fracionamento entre o balão de destilação
e a cabeça de destilação. Uma coluna de fracionamento é basicamente um tubo de vidro recheado
ou com saliências internas.
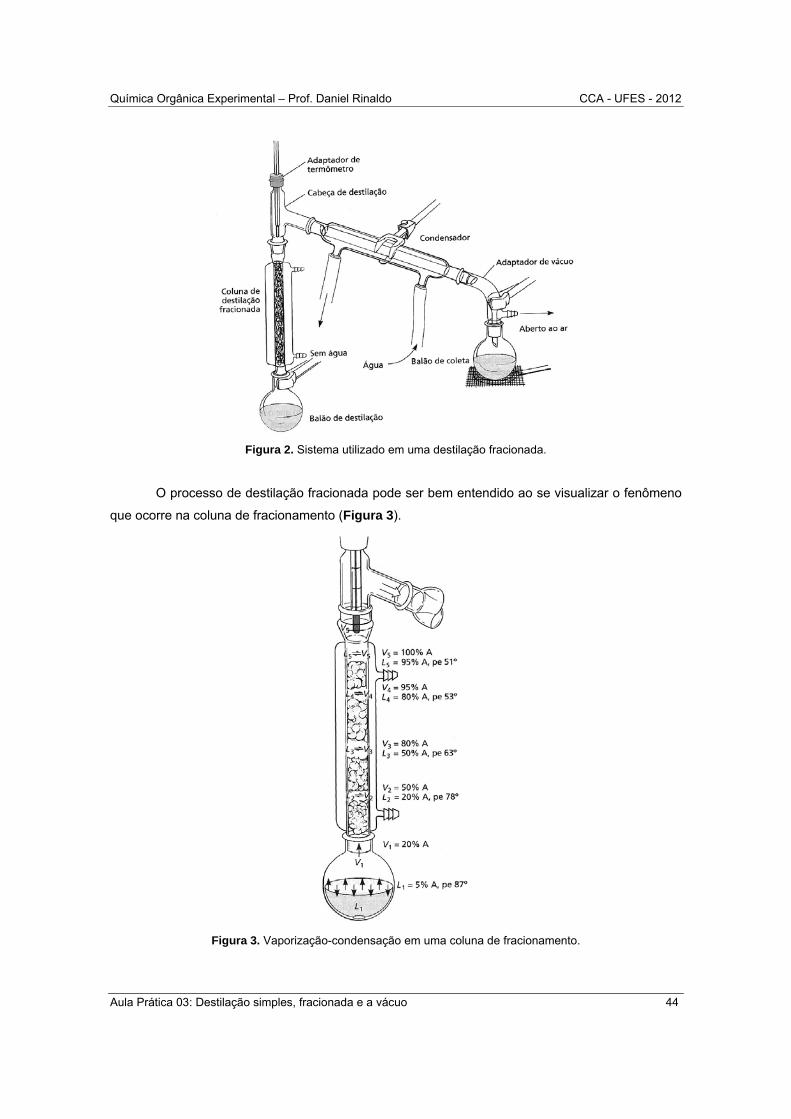
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 44
Figura 2. Sistema utilizado em uma destilação fracionada.
O processo de destilação fracionada pode ser bem entendido ao se visualizar o fenômeno
que ocorre na coluna de fracionamento (Figura 3).
Figura 3. Vaporização-condensação em uma coluna de fracionamento.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 45
A Figura 3 mostra um exemplo de destilação fracionada de uma mistura de dois líquidos A
(te 50 ºC) e B (te 90 ºC), que contém 5 % de A e 95 % de B. Essa solução é aquecida até a
ebulição (que se inicia a 87 ºC) e o primeiro vapor resultante (V1) contém 20 % de A e 80 % de B.
Portanto, o vapor ficou mais rico no componente mais volátil, mas A não está puro. Esse vapor se
condensa na coluna de fracionamento para dar o líquido L2 (20 % de A e 80 % de B), que evapora
parcialmente ainda dentro da coluna (te 78 ºC) para dar o vapor V2, que contém agora 50 % de A e
50 % de B. Portanto, a cada processo de vaporização-condensação, há o enriquecimento do vapor
no componente mais volátil A, de forma que o processo continua até o vapor V5, que contém 100
% de A. A condensação desse vapor e a coleta do destilado permitem a obtenção do líquido A
puro. Com a continuidade do processo, todo o líquido A é removido do balão de destilação,
deixando B quase puro. Se a temperatura aumentar, o líquido B pode destilar como uma fração
pura. Quanto maior for diferença entre as pressões de vapor (ou entre as temperaturas de
ebulição) de A e B, maior a concentração do componente mais volátil na fase vapor e mais fácil é a
separação de A e B por destilação fracionada. Portanto, em uma coluna de fracionamento ocorrem
sucessivas vaporizações e condensações.
Vários tipos de colunas de fracionamento podem ser utilizados. Os mais comuns são a
coluna de Vigreux, Hempel e Yung (Figura 4). A coluna de Vigreux é formada por um tubo de vidro
com protuberâncias internas que quase se tocam; a de Hempel é um tipo de coluna empacotada
com anéis ou pérolas de vidro; a de Yung é formada por uma espiral metálica ou de vidro em torno
de uma haste.
Figura 4. Tipos mais comuns de colunas de fracionamento.
A escolha da coluna de fracionamento é determinada pela dificuldade de se conseguir a
separação (ou seja, pela diferença entre as temperaturas de ebulição dos líquidos da mistura), pela
pressão em que será realizada a destilação e pela quantidade de líquido a destilar.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 46
A eficiência de uma coluna de fracionamento está relacionada ao número de pratos
teóricos da coluna. Prato teórico é uma grandeza que se refere ao número de ciclos de
vaporização-condensação que ocorrem quando uma mistura líquida percorre a coluna, ou seja,
cada prato teórico corresponde a um ciclo de vaporização-condensação ou a uma destilação
simples que ocorre na coluna. Portanto, quanto maior o número de pratos teóricos, maior a
eficiência da coluna e, consequentemente, quanto maior a coluna, maior a sua eficiência. No
exemplo da Figura 3, seriam necessários cinco pratos teóricos para separar a mistura que
começou com a composição L1. O prato teórico também pode ser definido como um comprimento
X da coluna onde ocorre uma destilação simples. Esse comprimento X é chamado de altura
equivalente a um prato teórico (AEPT), sendo inversamente proporcional à eficiência da coluna.
Quanto menor for a diferença entre as temperaturas de ebulição dos líquidos, maior deve
ser o número de pratos teóricos e, portanto, mais eficiente deve ser a coluna de fracionamento. Em
geral, a coluna mais eficiente é a de Hempel, mas existem colunas como a de spinning-band que
são tão eficientes que permitem a separação de líquidos com uma diferença de temperatura de
ebulição de 0,5 e 1 ºC.
Cabe ressaltar que a eficiência da coluna também é influenciada pela temperatura, de
forma que o isolamento térmico adequado da coluna acarreta um aumento significativo de sua
eficiência. Tal isolamento pode ser feito com: amianto, amianto e papel laminado, lã de vidro e
papel laminado, algodão e papel laminado, etc.
Destilação a vácuo
A destilação a vácuo (ou destilação a pressão reduzida) é usada quando os compostos
têm elevadas temperaturas de ebulição (acima de 200 ºC), pois eles frequentemente se
decompõem nas temperaturas necessárias para a destilação em pressão atmosférica. Como
discutido anteriormente (Seção 4.3), a temperatura de ebulição de uma substância se reduz
substancialmente quando a pressão aplicada diminui. A destilação a vácuo também é usada no
caso de compostos que podem reagir com o oxigênio do ar, quando aquecidos, e, ainda, quando é
mais conveniente realizar a destilação em temperaturas mais baixas por causa das limitações
experimentais.
A destilação a vácuo pode ser realizada empregando-se tanto uma aparelhagem para
destilação simples (Figura 1) quanto fracionada (Figura 2). O vácuo é aplicado no sistema através
da saída lateral do adaptador de vácuo. Um esquema de destilação simples a vácuo é ilustrado na
Figura 5.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 47
Figura 5. Sistema utilizado em uma destilação simples a vácuo.
Geralmente, a redução de pressão necessária para a destilação a vácuo é conseguida por
meio de uma bomba de vácuo. Existem dois tipos de bombas de vácuo mais usadas em
laboratório: a bomba d’água e a bomba de óleo. A primeira pode reduzir a pressão em até 30
mmHg, enquanto que a segunda chega a atingir uma pressão de 0,01 mmHg, sendo, portanto,
mais eficiente. Em ambos os casos, é recomendado que entre a aparelhagem de destilação e a
bomba seja colocado um frasco de segurança (trap) para se evitar que, ao se utilizar uma bomba
d’água haja a contaminação do destilado por refluxo de água para o sistema, quando há uma
queda de pressão ou, a contaminação da bomba a óleo por vapores provenientes da destilação.
Vários cuidados devem ser tomados ao se realizar uma destilação a pressão reduzida.
Antes de iniciar a destilação à vácuo, é aconselhável verificar se a vidraria que constitui o sistema
não apresenta nenhuma trinca e se está devidamente conectada, sem nenhum vazamento. O
sistema só pode ser fechado lentamente após se ligar a bomba e, então, ser aquecido. A abertura
do sistema é realizada após o resfriamento do balão de destilação, sendo a pressão normalizada
lentamente e, só, então a bomba de vácuo deve ser desligada.
A) Evaporador rotatório (Rotaevaporador)
O evaporador rotatório (ou rotaevaporador) é um equipamento utilizado para evaporar o
solvente de uma mistura sob pressão reduzida. Ele é constituído por um motor para rotação do
balão de destilação, um banho de aquecimento e um sistema de destilação simples a vácuo
(Figura 6).

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 48
Figura 6. Evaporador rotatório.
Aplica-se vácuo ao sistema e o motor gira o balão de destilação. A rotação espalha um
filme fino de líquido pela superfície do vidro, aumentando a área superficial do líquido e,
consequentemente, acelerando a evaporação. Além disso, a rotação evita o problema de ebulição
tumultuosa. Tanto a rotação quanto o aquecimento do balão de destilação podem ser controlados,
de forma a atingir uma velocidade desejada de evaporação. Quando o solvente evapora, os
vapores são resfriados pelo condensador e recolhidos no outro balão (coletor de solvente). O
produto desejado (soluto não volátil ou pouco volátil) permanece no balão de destilação.
Portanto, o evaporador rotatório permite a evaporação rápida da maior parte dos solventes
(te ≤ 120ºC) devido à grande área superficial de líquido formada com a rotação do balão e à
pressão reduzida do sistema.
OBJETIVOS
A presente experiência tem os seguintes objetivos:
- aprendizagem das técnicas de destilação simples, destilação fracionada e destilação a
vácuo (especificamente do uso de evaporador rotatório);
- purificação de água por destilação simples a partir de uma solução aquosa de sulfato de
cobre;
- separação de misturas acetona:água (2:5) e diclorometano:etanol (2:5) por destilação
fracionada;
- evaporação de acetona em evaporador rotativo.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 49
MATERIAL E REAGENTES
- adaptador de vácuo - tubos de ensaio
- algodão - glicerina
- balão de fundo redondo de 125 e 250 mL - silicone
- cabeça de destilação - acetona
- cápsula de porcelana - diclorometano
- chapa de aquecimento - etanol
- coluna de Vigreux - solução de 2,4-dinitrofenilidrazina*
- condensador de Liebig - sulfato de cobre
- funil de vidro
- papel de alumínio * Preparação da solução de 2,4-
dinitrofenilidrazina
Dissolver 3 g de 2,4-
dinitrofenilidrazina em 15 mL de ácido
sulfúrico concentrado. Adicionar a
solução, com agitação, a 20 mL de água
e 70 mL de etanol 95%. Misturar bem e
filtrar.
- pedras de porcelana
- presilhas de segurança
- suporte para tubo de ensaio
- termômetro (0-200 ºC),
com e sem junta esmerilhada
- mangueiras de látex ou silicone
- suporte universal, garras e mufas
PROCEDIMENTO EXPERIMENTAL
1) Destilação simples da solução aquosa de sulfato de cobre
a) Coloque em um balão de fundo redondo de 250 mL, 125 mL da solução aquosa de sulfato de
cobre, juntamente com algumas pedras de porcelana.
b) Faça a montagem da aparelhagem necessária para a realização de uma destilação simples
(Figura 1). A fonte de aquecimento será um banho de glicerina contido em uma cápsula de
porcelana, que ficará sobre a chapa de aquecimento. Lubrifique as conexões com graxa de
silicone, verificando se estão bem adaptadas. Abra a torneira e regule a saída de água do
condensador até que se estabeleça um fluxo contínuo de água. A temperatura do banho de
aquecimento deve ser controlada por um termômetro mergulhado na glicerina.
c) Inicie a destilação, aquecendo lentamente o balão de destilação. Observe o comportamento dos
vapores ao longo da montagem da destilação. Recolha o destilado (água) em um balão de fundo
redondo de 125 mL, registrando a temperatura de ebulição. Colete aproximadamente 70 mL de
água destilada.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 50
2) destilação fracionada das misturas acetona/água e diclorometano/etanol
a) Coloque em um balão de fundo redondo de 250 mL, 70 mL da mistura acetona/água (2:5) ou da
mistura diclorometano:etanol (2/5), juntamente com algumas pedras de porcelana. A mistura a ser
destilada será indicada pelo seu professor.
b) Faça a montagem da aparelhagem necessária para a realização de uma destilação fracionada
(Figura 2). A fonte de aquecimento será um banho de glicerina contido em uma cápsula de
porcelana, que ficará sobre a chapa de aquecimento. Lubrifique as conexões com graxa de
silicone, verificando se estão bem adaptadas. Abra a torneira e regule a saída de água do
condensador até que se estabeleça um fluxo contínuo de água. A temperatura do banho de
aquecimento deve ser controlada por um termômetro mergulhado na glicerina. Cubra a coluna de
Vigreux com algodão e papel alumínio.
c) Inicie a destilação, aquecendo lentamente o balão de destilação. Observe o comportamento dos
vapores ao longo da montagem da destilação.
d) Recolha frações de 5 mL do destilado em tubos de ensaio, registrando sempre a temperatura de
cada fração coletada.
e) No caso da destilação da mistura de acetona/água, faça o teste das frações com a solução de
2,4-dinitrofenilidrazina. Para isso, adicione em um tubo de ensaio, 3 mL dessa solução e 2 mL de
uma das frações coletadas. Observe a ocorrência de um precipitado alaranjado, que é indicativo da
presença de carbonila de aldeído ou cetona na amostra. Para comparação, realize este teste com
a acetona pura.
f) Após coletar 8 frações, desligue o aquecimento.
3) Evaporação da acetona em evaporador rotatório
O uso do evaporador rotatório será demonstrado pelo seu professor.
QUESTÕES
1. Por que a destilação simples não deve ser empregada na separação de líquidos com
temperaturas de ebulição próximas?
2. Quais dos seguintes pares de solventes podem ser separados por destilação simples?
a) Acetona (56 ºC) e anilina (184 ºC)
b) Acetato de butila (126 ºC) e Butanol (117 ºC)
c) Cicloexano (81 ºC) e Cicloexanol (161 ºC)
d) Hexano (69 ºC) e Tolueno (111 ºC)
3. O petróleo é constituído principalmente por uma mistura bastante complexa de hidrocarbonetos,
com diferentes temperaturas de ebulição. Qual a técnica de destilação mais adequada para o
refino do petróleo? Justifique. Como a eficiência da separação dos constituintes do petróleo pode
ser aumentada?

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 03: Destilação simples, fracionada e a vácuo 51
4. Na purificação de cicloexanona (te=156 ºC) seria conveniente a utilização da destilação a vácuo,
sob uma pressão de 2.0 mmHg? Use o monógrafo dado na Experiência 1 para auxiliar na sua
resposta. Que cuidados especiais deveriam ser tomados nessas circunstâncias?
BIBLIOGRAFIA
DEMUNER, A.J.; MALTHA, C.R.A.; BARBOSA, L.C.A.; PERES, V. “Experimentos de Química
Orgânica”. Editora UFV, 2ª ed, Viçosa, 2004.
DIAS, A.G.; DA COSTA, M.A.; GUIMARÃES, P.I.C. “Guia Prático de Química Orgânica. Volume I –
Técnicas e Procedimentos: Aprendendo a Fazer”. Editora Interciência, Rio de Janeiro, 2004.
MARQUES, J.A.; BORGES, C.P.F. “Práticas de Química Orgânica”. Editora Átomo, Campinas,
2007.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 52
Aula prática 04: Síntese e Recristalização da acetanilida
INTRODUÇÃO
Muitas reações químicas produzem substâncias orgânicas sólidas que, geralmente, estão
contaminadas com subprodutos ou resíduos de reagentes. A purificação desses sólidos orgânicos
pode ser realizada utilizando-se o processo da recristalização, sendo esse um método amplamente
empregado seja em escala laboratorial seja em escala industrial. O processo de recristalização
fundamenta-se na diferença de solubilidade do composto de interesse e das impurezas em um
solvente adequado ou em mistura de solventes a uma dada temperatura.
Etapas envolvidas no processo de recristalização
A técnica geral envolve a dissolução do material em um solvente (ou em uma mistura de
solventes) sob aquecimento e o resfriamento lento da solução resultante.
As etapas envolvidas no processo de recristalização podem ser assim descritas:
1) Escolher o solvente.
2) Dissolver a amostra no solvente a quente ou em ebulição.
3) Filtrar a mistura, a quente, para remover impurezas insolúveis.
4) Deixar o filtrado resfriar lentamente, até temperatura ambiente, para ocorrer a cristalização.
5) Separar os cristais da solução por meio de filtração a vácuo. O filtrado nesse caso é chamado
de “água-mãe”.
6) Lavar os cristais com solvente adequado para remover a “água-mãe” residual que fica aderida à
superfície dos cristais.
7) Secagem dos cristais para remover o solvente residual.
8) Verificar a pureza da substância.
Cabe destacar que as impurezas que forem insolúveis no solvente a quente serão
removidas durante a primeira filtração. Entretanto, as impurezas solúveis nesse solvente a quente
deverão também ser solúveis a frio, pois serão separadas dos cristais durante a segunda filtração.
A escolha do solvente
A escolha do solvente é um dos aspectos mais importantes quando se deseja realizar uma
recristalização. As características ideais que um solvente deve possuir para ser utilizado numa
recristalização são:
a) Ser quimicamente inerte, isto é, ele não deve reagir com nenhum dos componentes da
mistura a ser purificada;
b) Solubilizar muito bem a substância a ser purificada à temperatura próxima a sua
temperatura de ebulição e não solubilizar a substância à temperatura ambiente;
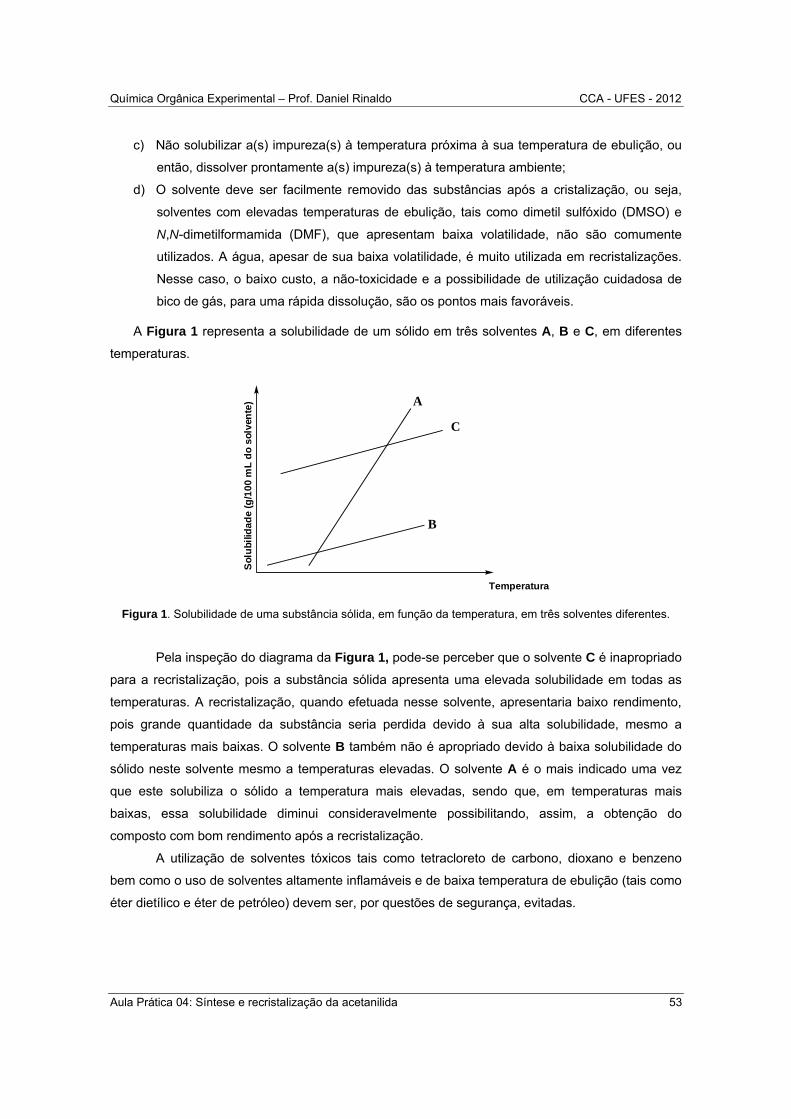
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 53
c) Não solubilizar a(s) impureza(s) à temperatura próxima à sua temperatura de ebulição, ou
então, dissolver prontamente a(s) impureza(s) à temperatura ambiente;
d) O solvente deve ser facilmente removido das substâncias após a cristalização, ou seja,
solventes com elevadas temperaturas de ebulição, tais como dimetil sulfóxido (DMSO) e
N,N-dimetilformamida (DMF), que apresentam baixa volatilidade, não são comumente
utilizados. A água, apesar de sua baixa volatilidade, é muito utilizada em recristalizações.
Nesse caso, o baixo custo, a não-toxicidade e a possibilidade de utilização cuidadosa de
bico de gás, para uma rápida dissolução, são os pontos mais favoráveis.
A Figura 1 representa a solubilidade de um sólido em três solventes A, B e C, em diferentes
temperaturas.
So
lub
ilid
ad
e(g
/10
0m
Ld
os
olv
ente
)
C
B
A
Temperatura
Figura 1. Solubilidade de uma substância sólida, em função da temperatura, em três solventes diferentes.
Pela inspeção do diagrama da Figura 1, pode-se perceber que o solvente C é inapropriado
para a recristalização, pois a substância sólida apresenta uma elevada solubilidade em todas as
temperaturas. A recristalização, quando efetuada nesse solvente, apresentaria baixo rendimento,
pois grande quantidade da substância seria perdida devido à sua alta solubilidade, mesmo a
temperaturas mais baixas. O solvente B também não é apropriado devido à baixa solubilidade do
sólido neste solvente mesmo a temperaturas elevadas. O solvente A é o mais indicado uma vez
que este solubiliza o sólido a temperatura mais elevadas, sendo que, em temperaturas mais
baixas, essa solubilidade diminui consideravelmente possibilitando, assim, a obtenção do
composto com bom rendimento após a recristalização.
A utilização de solventes tóxicos tais como tetracloreto de carbono, dioxano e benzeno
bem como o uso de solventes altamente inflamáveis e de baixa temperatura de ebulição (tais como
éter dietílico e éter de petróleo) devem ser, por questões de segurança, evitadas.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 54
Aspectos práticos do processo de recristalização
Durante o processo de recristalização, é desejável que o solvente a quente (ou em
ebulição) seja saturado de modo a minimizar as perdas de material na “água-mãe”. Para tal efeito,
é importante que o solvente seja aquecido até a temperatura de ebulição e a amostra, contendo a
substância a ser recristalizada seja dissolvida na menor quantidade possível de solvente. Neste
contexto, um procedimento útil é manter um recipiente com solvente em ebulição (em uma placa
de aquecimento). Transfere-se uma pequena quantidade de solvente deste recipiente para o
Erlenmeyer que contém o sólido a ser recristalizado. A mistura é aquecida com agitação eventual
(por movimentos circulares) até que volte a ferver.
Caso não haja dissolução do sólido após a adição da primeira porção de solvente a quente
(ou em ebulição), uma nova porção de solvente deve ser adicionada ao frasco contendo o sólido. O
sistema deve ser aquecido e agitado novamente até que volte a ferver. Essas etapas descritas
devem ser repetidas até que ocorra a dissolução completa do sólido. É importante enfatizar que as
porções adicionadas de solvente devem ser pequenas de modo a garantir que o menor volume
possível de solvente seja utilizado no processo de recristalização.
Se após o processo de dissolução do sólido restarem impurezas insolúveis, deve ser
realizada a filtração a quente e empregando-se uma montagem como a mostrada na Figura 2.
Figura 2. Montagem para filtração a quente.
Nesse caso, deve-se aquecer o funil e o papel de filtro pregueado. Para isso, coloca-se o
filtro pregueado no funil e este último é posicionado sobre o topo do Erlenmeyer que irá recolher o
filtrado. (Figura 2). O Erlemeyer com o funil e o filtro deve, então, ser colocado sobre uma placa de
aquecimento. Em seguida, molha-se o papel de filtro e o funil com o solvente quente usado no
processo de dissolução da amostra sólida a ser recristalizada. A mistura a ser filtrada deve ser,
então, levada à ebulição e passada pelo filtro em pequenas porções. É necessário manter as
soluções de ambos os frascos na temperatura de ebulição para evitar a cristalização prematura. O

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 55
refluxo do filtrado mantém o funil aquecido e reduz a possibilidade de entupimento do filtro pelos
cristais que eventualmente se formem durante a filtração. Com solventes mais voláteis haverá
perdas significativas durante a execução das operações supracitadas. Esse problema deve ser
contornado pela adição de mais solvente. Caso haja formação de cristais no papel de filtro, é
aconselhável adicionar um pouco de solvente em ebulição para removê-los e permitir que a
solução passe pelo papel. O solvente de lavagem é combinado, posteriormente, com o filtrado
original.
Uso do carvão ativo (carvão ativado)
É bastante frequente que a solução do composto que se deseja purificar por recristalização
esteja contaminada com substâncias coloridas ou resinosas. Essas impurezas devem ser
removidas da solução, pois, caso contrário, serão obtidos cristais impuros. O emprego de carvão
ativo (ou ativado) é indicado para esse propósito. O carvão ativado é um sólido de grande área
superficial (1 grama de carvão ativado possui cerca de 500 m2 de área superficial), onde as
impurezas são facilmente adsorvidas. A quantidade de carvão ativado não deve ser superior a 2-
3% da massa da substância a ser purificada; caso contrário haverá perda significativa por adsorção
da substância, comprometendo o rendimento do processo de recristalização. Recomenda-se que a
adição de carvão ativado seja feita antes da etapa de filtração descrita no item 1.3.
Cristalização
Após a etapa de filtração descrita no item 1.3, a solução resultante deve ser deixada
esfriar lentamente. Esse resfriamento lento favorece a formação de cristais mais perfeitos e de
maior grau de pureza, enquanto que, o resfriamento brusco da solução, favorece a formação de
cristais menores.
Terminada a cristalização, os cristais são coletados por filtração a vácuo em um funil de
Büchner. Os cristais devem ser lavados com uma pequena quantidade de solvente gelado (para
remover a água-mãe que adere à superfície dos cristais) e secos, resultando na obtenção da
substância pura desejada. A determinação da temperatura de fusão ou a cromatografia em
camada delgada são métodos simples que podem ser utilizados para verificação da pureza dos
cristais obtidos.
Comentários adicionais sobre o processo de recristalização
a) O processo de recristalização deve ser conduzido em frasco de Ernlenmeyer e não em um
béquer. Não deve ser utilizado o béquer porque a abertura larga faz com que o solvente evapore
muito rapidamente. Além disso, o uso do Erlenmeyer evita a contaminação do sistema com
partículas de poeira.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 56
b) É de fundamental importância que o sólido a ser recristalizado não seja aquecido antes da
adição de uma porção de solvente a quente. Algumas substâncias são termicamente instáveis e
podem se decompor com o aquecimento excessivo.
c) Pode ocorrer o não aparecimento de cristais após o resfriamento da solução à temperatura
ambiente. Nesses casos, o procedimento correto seria inocular a solução com um pequeno cristal
da substância que se deseja cristalizar. Esse cristal servirá como germe de cristalização induzindo
a cristalização. Caso não se disponha desse cristal no laboratório, pode-se, alternativamente,
utilizar um bastão de vidro para atritar cuidadosamente as paredes laterais do frasco contendo a
solução. Esse atrito causará pequenas ranhuras nas paredes do frasco que servirão como sítios,
por onde se iniciará a cristalização. Caso ainda não se obtenha o resultado esperado, o
resfriamento da solução abaixo da temperatura ambiente poderá funcionar.
d) Após a cristalização, é desejável esfriar o frasco contendo os cristais em um banho de água e
gelo. Uma vez que o soluto é menos solúvel em temperaturas mais baixas, este procedimento faz
aumentar o rendimento dos cristais. Cabe salientar que este procedimento não resultará na
cristalização completa da substância a partir da “água-mãe”. Em outras palavras, haverá sempre
parte do composto de interesse dissolvida na “água-mãe”.
Síntese e recristalização da acetanilida
A acetanilida pertence a um grupo de fármacos usados para aliviar dores de cabeça.
Acetanilida, fenacetina e acetaminofeno (Figura 3) são analgésicos (aliviam a dor) leves e
antipiréticos (reduzem a febre) e são importantes, juntamente com a aspirina, em muitos
medicamentos de uso livre2.
N N NH C H C H C
O O O
CH3 CH3 CH3
Acetanilida
OCH2CH3 OH
Fenacetina Acetaminofeno
Figura 3. Estruturas da acetanilida, fenacetina e acetaminofeno.
Aminas primárias e secundárias são normalmente acetiladas com anidrido acético. A
acetilação com cloreto de acetila nem sempre é satisfatória, pois uma quantidade do sal de amônio
é simultaneamente produzida. No entanto, fornece bons resultados na presença de uma base
(podendo ser outro equivalente da amina) para neutralizar o ácido formado.
2 Medicamentos de uso livre são aqueles que podem ser comprados sem receita médica.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 57
2 RNH2 + CH3COCl CH3CONHR + RNH3+Cl-
Aminas primárias reagem sob aquecimento com anidrido acético produzindo o derivado
monoacetilado. Se o aquecimento é prolongado na presença de excesso de anidrido acético, o
produto diacetilado é formado juntamente com o produto monoacetilado. Em geral, os derivados
diacetilados são hidrolisados ao produto monoacetilado na presença de água.
Nesse experimento a acetanilida será preparada a partir da anilina, segundo a reação de
acetilação mostrada no Esquema 1.
NH2
Anilina
NH C
O
CH3
H3C O CH3
O O
+
Anidrido acéticoAcetanilida
+ CH3COOH
Ácido acético
Esquema 1. Síntese da acetanilida a partir do anidrido acético.
A acetanilida bruta contém impurezas coloridas3 que serão removidas pelo carvão ativado. Além
disso, a acetanilida será purificada por recristalização em água quente. A Tabela 1 contém
informações sobre as substâncias envolvidas nesse experimento.
Tabela 1. Alguns dados sobre os principais compostos envolvidos nesse experimento
Substância Aparência Propriedades físicas
Solubilidade Toxicidade Aplicações
Anilina Líquido incolor
Te= 185 oC ; Tf = -6 oC; d = 1,02 g cm-
3
1g/ 28,6 mL de água; 1g/15,7 mL de água fervente.
Tóxica por inalação, ingestão, absorção cutânea. LD50-ratos
= 0,440 g/kg
Manufatura de corantes, remédios, resinas, vernizes, perfumes, borracha vulcanizada.
Acetanilida Sólido branco
Tf = 113-115 oC
1g/185 mL água; 1g/3,4 mL etanol; 1g/20 mL água fervente .
LD50-ratos = 0,800 g/kg
Antipirético; analgésico; corantes; estabilizante para H2O2; componente de vernizes.
3 As impurezas coloridas são oriundas da anilina impura utilizada neste experimento.
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 58
Tabela 1. cont.
Anidrido Acético
Líquido incolor
Te = 139 oC; d = 1,08 g cm-
3
Reage com água e álcool; solúvel em éter
Irritante para a pele e olhos. LD50-ratos
= 1,78 g/kg
Manufatura de compostos acetilados; em sínteses orgânicas fornece grupos protetores; agente desidratante (remoção de água)
Ácido acético
Líquido incolor
Te = 118 oC Solúvel em água, álcool e éter.
Irritante para olhos e pele. Por ingestão causa corrosão da boca e trato intestinal. LD50-ratos
= 3,53 g/kg
Um dos componentes do vinagre; solvente; manufatura de plásticos e borrachas; conservante de alimentos; acidificante.
Tf: temperatura de fusão Te: temperatura de ebulição LD: Dose letal mediana (dose letal para 50% da população em teste) d: densidade
OBJETIVOS
A presente experiência tem os seguintes objetivos:
- sintetizar a acetanilida por meio da acetilação da anilina com anidrido acético;
- purificar a acetanilida sintetizada empregando-se a técnica de recristalização.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 59
MATERIAL E REAGENTES
- balão de fundo redondo de 50 mL - cápsula de porcelana
- condensador de Liebig - espátula metálica
- chapa de aquecimento - anidrido acético
- barra de agitação magnética - anilina
- béquer de 100 mL - gelo
- funil de Büchner - carvão ativado
- bomba de vácuo - aparato para determinação de temperatura de
fusão
- 3 Erlenmeyers de 125 mL - vidro de relógio
- balança - etanol
- funil de vidro - estufa
- Kitasato - bastão de vidro
- papel de filtro qualitativo - proveta de 100 mL
- suporte universal com haste e anel
para filtração
- vidro de relógio
- etanol
PROCEDIMENTO EXPERIMENTAL
1) Síntese da acetanilida
Em um balão de fundo redondo (50 mL), coloque 5 mL de anilina e adapte o condensador
de Liebig. Inicie a mistura sob agitação magnética. Adicione pelo topo do condensador 15 mL de
anidrido acético e agite por 30 minutos. Após este período, verta a mistura reacional sobre água
destilada e gelo triturado em um béquer (100 mL). Agite com um bastão de vidro e filtre sob vácuo.
Lave os cristais com água destilada gelada e transfira o sólido para um vidro de relógio
previamente pesado. Deixe secar em estufa a 50 oC por 30 minutos ou até massa constante e
determine o rendimento bruto da reação.
2) Recristalização da acetanilida
a) Em um Erlenmeyer de 125 mL, contendo 50 mL de água destilada e 10 mL de etanol, adicione 2
g de acetanilida recém preparada, agitando com bastão de vidro para tentar dissolvê-la.
b) Aqueça a mistura até a ebulição e continue agitando até que todo o sólido tenha se dissolvido.
Se a solução estiver colorida, retire o Erlenmeyer da placa e adicione uma ponta de espátula de
carvão ativo (lembre-se que normalmente a quantidade de carvão ativo utilizada é de

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 60
aproximadamente 2 a 3% da massa da amostra a ser recristalizada). Filtre a mistura a quente,
conforme mostrado na Figura 2, recolhendo o filtrado em outro Erlenmeyer
c) Deixe o filtrado resfriar até a temperatura ambiente. Coloque o Erlemeyer em banho de gelo e
deixe em repouso até que a acetanilida se recristalize completamente. Quando isso acontecer,
filtre a mistura a vácuo, em funil de Büchner, lavando os cristais com água gelada.
d) Coloque a acetanilida recristalizada em um vidro de relógio e deixe secando em estufa a 50 oC.
Após secagem completa dos cristais, determine a massa de acetanilida obtida bem como sua
temperatura de fusão.
QUESTÕES
1. Complete a tabela abaixo com os resultados experimentais encontrados durante a síntese e
purificação da acetanilida.
Temperatura de fusão determinada para a acetanilida sintetizada
Rendimento da acetanilida bruta
Rendimento do processo de recristalização
Deixe indicados os cálculos de modo a explicitar o seu raciocínio. 2. Escreva o mecanismo da reação que você realizou, supondo a formação do produto
monoacetilado.
3. Qual composto deve apresentar o menor pKb, a anilina ou a acetanilida? Qual seria a relação
entre esse fato e a obtenção do produto monoacetilado ser favorecida em relação à obtenção do
diacetilado?
4. Escreva o mecanismo da hidrólise de um possível subproduto diacetilado dessa reação
[C6H5N(COCH3)2], na presença de água.
5. Para dissolver 1 g de acetanilida são necessários 185 mL de água. Para dissolver 1 g de anilina,
são necessários 28,6 mL de água (temperatura ambiente). Explique a diferença.
6. Explique como a amostra de acetanilida pode se tornar mais pura através do processo de
recristalização.
7. Como se deve escolher um solvente (ou mistura de solventes) para recristalização?
8. Por que foi usado carvão ativo na purificação da acetanilida?
9. Por que os cristais, após a filtração, foram lavados com água destilada fria?
10. Ao purificar um composto por recristalização, é aconselhável esfriar a solução lenta ou
rapidamente? Explique.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 04: Síntese e recristalização da acetanilida 61
BIBLIOGRAFIA
DEMUNER, A.J.; MALTHA, C.R.A.; BARBOSA, L.C.A.; PERES, V. “Experimentos de Química
Orgânica”. Editora UFV, 2ª ed, Viçosa, 2004.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.
MARQUES, J.A.; BORGES, C.P.F. “Práticas de Química Orgânica”. Editora Átomo, Campinas,
2007.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 62
Aula prática 05: Extração de óleos essenciais através de destilação por arraste de vapor e sua análise por Cromatografia em Camada Delgada (CCD)
INTRODUÇÃO
Os óleos essenciais são misturas complexas de substâncias voláteis, lipofílicas, odoríferas
e líquidas. A denominação “óleo essencial” está relacionada à aparência oleosa (à temperatura
ambiente) e ao aroma agradável e intenso (essências) dessas misturas.
Além das funções biológicas (inibidores de germinação, proteção contra predadores,
atração de polinizadores, proteção contra a perda de água e aumento de temperatura), os óleos
essenciais têm importância econômica e medicinal. Atualmente, existem pelo menos 200 óleos
essenciais de importância econômica.
Os óleos essenciais são encontrados, freqüentemente, nas glândulas ou nos espaços
intercelulares dos tecidos de plantas. Eles podem ocorrem em todos os órgãos das plantas (flores,
folhas, cascas dos caules, raízes, frutos, sementes, etc), mas são encontrados com maior
freqüência nas folhas, sementes e flores. Sua composição química, características físico-químicas
e odores podem variar com a localização na planta.
Os óleos essenciais são misturas que podem conter mais de 300 substâncias dentre elas
hidrocarbonetos, álcoois, compostos carbonilados e ésteres. Os componentes dos óleos essenciais
usualmente pertencem a dois grupos de produtos naturais: terpenos (ou terpenóides) e
fenilpropanóides.
Os produtos naturais são classificados em função da rota metabólica pela qual são obtidos
e não por seus grupos funcionais. Os terpenos são biossintetizados a partir do
isopentenilpirofosfato (IPP) e do ácido mevalônico. O IPP é isomerizado a dimetilalilpirofosfafo
(DMAPP) (Figura 1).
Figura 1. Biossíntese de terpenos.
Várias unidades de IPP e DMAP combinam-se dando origem a compostos que possuem
número de átomos de carbonos múltiplo de cinco. As unidades se juntam formando estruturas
abertas ou cíclicas. Desta forma, os terpenos podem ser classificados em monoterpenos (C10),
sesquiterpenos (C15), diterpenos (C20), sesterterpenos (C25), triterpenos (C30) e tetraterpenos (C40).
Um aspecto histórico relativo aos terpenos merece destaque nesse ponto. Antes do
conhecimento dos detalhes da rota metabólica envolvida na biossíntese dos terpenos, acreditava-
HOOCOH
OH
Ácido mevalônico
OPP OPP
Terpenos
IPP DMAPP
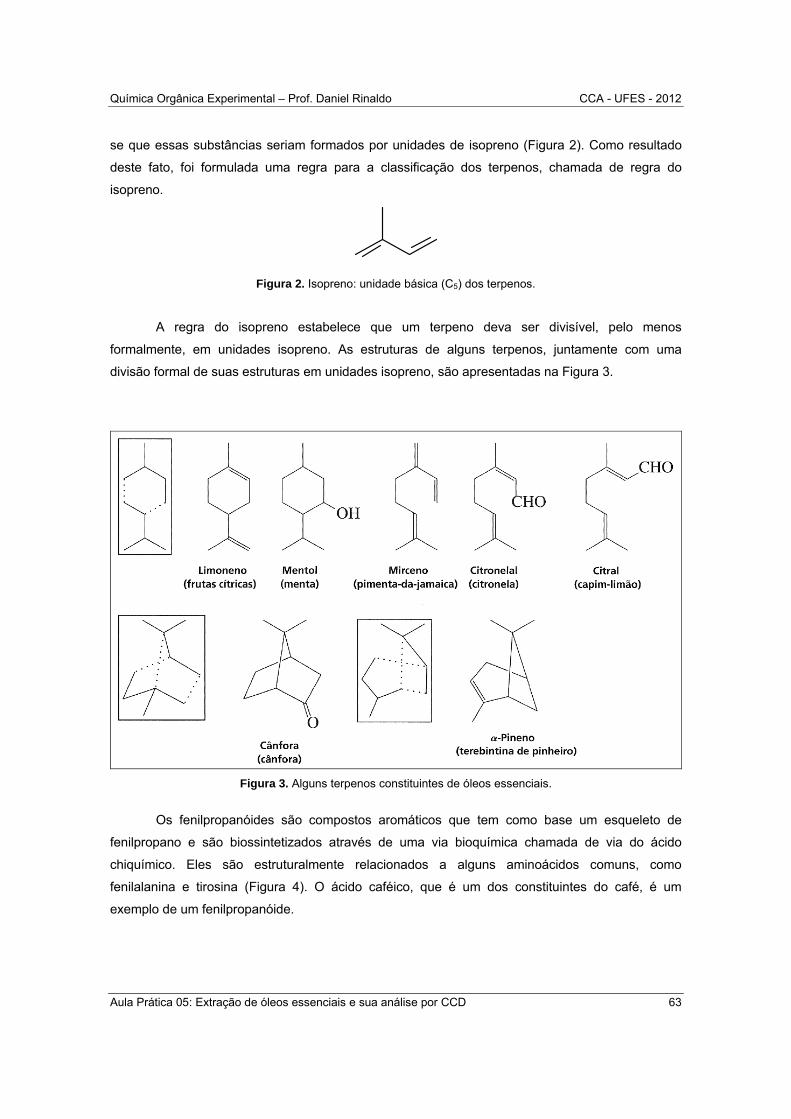
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 63
se que essas substâncias seriam formados por unidades de isopreno (Figura 2). Como resultado
deste fato, foi formulada uma regra para a classificação dos terpenos, chamada de regra do
isopreno.
Figura 2. Isopreno: unidade básica (C5) dos terpenos.
A regra do isopreno estabelece que um terpeno deva ser divisível, pelo menos
formalmente, em unidades isopreno. As estruturas de alguns terpenos, juntamente com uma
divisão formal de suas estruturas em unidades isopreno, são apresentadas na Figura 3.
Figura 3. Alguns terpenos constituintes de óleos essenciais.
Os fenilpropanóides são compostos aromáticos que tem como base um esqueleto de
fenilpropano e são biossintetizados através de uma via bioquímica chamada de via do ácido
chiquímico. Eles são estruturalmente relacionados a alguns aminoácidos comuns, como
fenilalanina e tirosina (Figura 4). O ácido caféico, que é um dos constituintes do café, é um
exemplo de um fenilpropanóide.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 64
Figura 4. Fenilpropanóides e aminoácidos estruturalmente relacionados.
Óleo essencial de cravo
O cravo é uma planta usada como tempero desde a antiguidade. Ela também é utilizada,
há mais de 2.000 anos, para fins medicinais pois apresenta propriedades antiséptica, bactericida e
anestésica.
Os principais consumidores mundiais de cravo são os habitantes da Indonésia,
responsável pelo consumo de mais de 50 % da produção mundial. Neste país, o principal uso do
óleo essencial de cravo é na aromatização de cigarros.
O conteúdo total de óleo em cravos pode chegar a 15 %. O óleo é constituído,
basicamente, por eugenol (70 a 80%), acetato de eugenol (15%) e -cariofileno (5 a 12%) (Figura
5). Os dois primeiros são fenilpropanóides e o último é um sesquiterpeno.
Figura 5. Constituintes majoritários do óleo essencial de cravo.
Óleo essencial de eucalipto
Entre as aproximadamente 600 espécies de eucaliptos descritas, pouco mais de 200 foram
examinadas com relação à produção e ao teor de óleo essencial e menos de 20 têm sido usadas
na exploração comercial. No Brasil, as principais espécies cultivadas para a produção de óleo são
Eucalyptus globulus, E. citriodora e E. staigeriana. E. globulus é a principal espécie utilizada para
COOH
NH2
R OHOH
CH=CHCOOH
fenilpropano R = H, fenilalanina
R = OH, tirosina
ácido caféico
H3CO
RO
R = H, eugenol
R = Ac, acetato de eugenol
-cariofileno

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 65
obtenção de óleo essencial com fins medicinais. Já a espécie E. citriodora é usada para se obter
óleo para indústria de perfumes.
Com relação ao óleo essencial de E. citriodora, os rendimentos variam de 1-1,6 %, ou seja,
para cada tonelada de folhas, 10-16 kg de óleo essencial são obtidos. A concentração do seu
componente principal, o citronelal (Figura 6), varia entre 65-85 %.
H
O
Figura 6. Fórmula estrutural do citronelal, o componente principal do óleo essencial de Eucalyptus
citirodora.
Óleo essencial de limão
O óleo essencial obtido de cascas de limão é composto basicamente por monoterpenos.
Em pesquisas realizadas com folhas e cascas de limão das espécies Citrus limon (limão siciliano) e
C. aurantifolia (limão taiti e galego), determinou-se que nos óleos essenciais das cascas dessas
espécies a quantidade de substâncias olefínicas é maior que a de substâncias oxigenadas. O
limoneno é o principal constituinte de todos esses óleos, cuja concentração varia de 39,9 a 94,4 %.
Dentre os terpenos oxigenados, -terpineol, linalol, acetato de linalila, neral, geranial, acetato de
nerila e acetato de geranila estão presentes em praticamente todas as amostras. A Figura 7
apresenta alguns terpenos representativos dos constituintes do óleo de limão.
Figura 7. Estruturas químicas de alguns terpenos constituintes do óleo essencial de limão.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 66
Métodos de extração de óleos essenciais
Os métodos de extração de óleos essenciais variam conforme a localização do óleo volátil
na planta e com a proposta de utilização do mesmo.
Existem vários métodos de extração de óleos essenciais. Dependendo do método
empregado para extração de um óleo essencial, suas características químicas poderão ser
totalmente alteradas. O calor e a pressão usados no ato da extração podem, por exemplo, interferir
na qualidade final do óleo essencial, pois no momento da extração as moléculas sensíveis de um
princípio ativo podem ser quebradas e oxidadas em produtos de menor eficácia, ou às vezes, até
tóxicos. Um exemplo é o óleo de bergamota que perde bergapteno (furanocumarina que causa
manchas de pele) se destilado e não extraído por prensagem a frio das cascas. Métodos mais
rápidos de extração podem reduzir o custo de um produto. No entanto, conforme o óleo a ser
extraído, isso poderá alterar drasticamente suas qualidades terapêuticas para um tratamento.
As técnicas mais comuns de extração são: enfloração (enflurage), destilação por arraste de
vapor d’água; prensagem; extração com solventes orgânicos (de forma contínua ou descontínua) e
extração por dióxido de carbono (CO2) supercrítico. Hoje com a tecnologia disponível, os óleos
essenciais podem ser extraídos com alta pureza e concentração. É o caso da extração por CO2
que permite a obtenção de um produto final de extrema pureza e qualidade.
A) Destilação por arraste de vapor
A destilação por arraste de vapor é o método mais comum de extração de óleos
essenciais. Normalmente é empregado para obtenção de óleos essenciais a partir de folhas e
ervas, mas nem sempre é indicado para extrair-se o óleo essencial de sementes, raízes, madeiras
e algumas flores.
As partes frescas ou secas da planta são colocadas em contato com a água ou vapor
d’água (Figuras 8 e 9). O vapor d’água força a quebra das bolsas intercelulares ou a abertura das
paredes celulares, fazendo liberar os óleos essenciais presentes na planta. Os óleos voláteis
apresentam pressão de vapor mais elevadas que a da água, sendo, por isso, arrastadas pelo vapor
d'água. Então, a água e óleo são condensados. Nesse produto de saída pode se ver a diferença de
duas fases, óleo na parte superior e a água na fase inferior. As fases são separadas por um
processo de decantação ou por extração líquido-líquido (conceitos apresentados no Experimento
3). A água que sobra deste processo recebe o nome de água floral, destilado, hidrosol ou hidrolato.
O óleo essencial extraído deve ser seco com um agente secante.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 67
Figura 8. Destilação por arraste de vapor pelo método direto.
Figura 9. Destilação por arraste de vapor pelo método indireto.
De modo geral, a destilação por arraste de vapor é utilizada para: separar líquidos
imiscíveis com o solvente de arraste; isolar e purificar sólidos que sejam insolúveis a frio e solúveis
a quente no solvente de arraste; separar ou purificar líquidos que se decompõem na temperatura
de ebulição a pressão atmosférica; e, para extrair óleos essenciais.
A destilação por arraste de vapor se baseia no fato de que quando dois ou mais
componentes imiscíveis ou levemente miscíveis formam uma mistura, a pressão de vapor da

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 68
mistura será a soma das pressões de vapor que esses componentes exerceriam se estivessem
sozinhos.
Quando uma mistura de líquidos imiscíveis é destilada, ela entra em ebulição no momento
em que a pressão de vapor da mistura (ou a soma das pressões de vapor das substâncias) se
iguala à pressão externa (geralmente, a pressão atmosférica). Portanto, a temperatura na qual uma
mistura destila é menor do que o ponto de ebulição de qualquer componente puro. Quando se usa
água como um dos componentes na destilação por arraste de vapor, a destilação dos compostos
orgânicos ocorre a uma temperatura inferior a 100 C.
A composição do destilado, ou seja, a relação dos dois líquidos será diretamente
proporcional às suas pressões de vapor. Além disso, quanto maior a pressão de vapor de uma
dada substância, menor a quantidade de vapor de água necessária para destilar certa massa
dessa substância. Por isso a destilação por arraste de vapor é um método eficiente para obtenção
dos constituintes voláteis que compõem os óleos essenciais com alto rendimento.
As únicas restrições para que a destilação por arraste de vapor possa ser usada são que:
as substâncias a serem “arrastadas” devem ser imiscíveis ou apenas levemente solúveis em água;
essas substâncias não podem reagir com a água; e, elas devem exercer alguma pressão de vapor,
mesmo que pequena, a 100 C.
As Figuras 8 e 9 mostram duas montagens utilizadas na destilação por arraste de vapor. A
principal diferença entre elas é na geração do vapor. No método direto (Figura 8), não há um balão
gerador de vapor. O vapor é gerado in situ no próprio balão de destilação. Nesse caso, pode-se
adicionar a quantidade total de água que será usada na destilação por arraste de vapor ou
adicioná-la por um funil de adição, que tem a vantagem de evitar a interrupção da destilação para
reposição do líquido de arraste. No método indireto (Figura 9), o vapor é gerado em outro balão,
que funciona como uma caldeira. Esse método é mais usado quando o material é sensível ao
calor.
Quando se realiza a destilação por arraste de vapor para extração de óleos essenciais a
partir de material vegetal, recomenda-se que o material sólido seja macerado ou cortado em
pequenas partes logo antes da destilação. Esse procedimento aumentará a superfície de contato
do material e assim diminuirá o tempo necessário para a obtenção do óleo essencial.
Cromatografia em Camada Delgada (CCD)
A cromatografia é definida como um método físico-químico de separação dos componentes
de uma mistura, que é realizada através da distribuição de tais componentes entre duas fases,
uma estacionária e outra móvel, que estão em contato íntimo. A fase estacionária é fixa e possui
grande área superficial, enquanto a fase móvel é um fluido que percola através da fase
estacionária. O termo cromatografia origina-se das palavras gregas chrom (cor) e graphe
(escrever), pois era inicialmente uma técnica para separação de misturas de substâncias coloridas.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 69
Existem vários critérios para a classificação das técnicas cromatográficas, dentre eles:
mecanismo de separação, técnica empregada, tipo de fase móvel. O critério mais importante para
classificação das cromatografias baseia-se no mecanismo de separação.
Todos os métodos cromatográficos dependem basicamente das solubilidades ou das
adsortividades diferenciais das substâncias, que constituem a mistura, em relação às fases
estacionária e móvel.
Quando a fase estacionária é líquida, o processo ocorre por absorção ou partição e,
portanto, está baseado na solubilidade dos componentes de uma dada mistura na fase
estacionária e na fase móvel. Por exemplo, quando se tem uma fase móvel líquida na qual está
dissolvido um analito, movendo-se através da fase estacionária, o analito se moverá mais ou
menos rápido, dependendo da relação de solubilidade na fase móvel e na fase estacionária. Este
fenômeno é conhecido como partição. Portanto, este tipo de cromatografia envolve a partição da
amostra entre duas fases líquidas imiscíveis, devido à diferença de solubilidade dos componentes
da amostra entre as fases. Neste tipo de cromatografia geralmente a fase estacionária é a água.
Cromatografia com fase estacionária líquida normalmente requer um suporte, que pode
ser, por exemplo, sílica gel, celite (terra diatomácea) ou celulose. O tipo mais simples dessa
cromatografia é a cromatografia em papel, na qual a água (fase estacionária) fica presa sobre os
polímeros de celulose. O papel pode conter de 5 – 20 % de água. As técnicas mais sofisticadas
desse tipo envolvem a cromatografia gás-líquido.
Quando a fase estacionária é sólida, o principal mecanismo de separação está baseado no
fenômeno de adsorção. O sólido utilizado como fase estacionária pode ser qualquer material que
não se dissolva na fase líquida. As fases estacionárias mais comuns são sílica gel (SiO2.xH2O) e
alumina Al2O3.xH2O, que são utilizados na forma pulverizada.
A adsorção da amostra ocorre na interface entre as fases móvel e estacionária, devido à
presença de grupos ativos na superfície da fase sólida. Por exemplo, se alumina finamente dividida
é utilizada como fase estacionária (Figura 10), as substâncias orgânicas irão adsorver (aderir) nas
partículas de sólido. Forças intermoleculares de intensidades diferentes estão envolvidas no
processo de adsorção: atração eletrostática (íon-íon), forças de van der Waals, interações dipolo-
dipolo, ligações de hidrogênio, etc. A Figura 10 ilustra estes tipos de interação. Por conveniência, a
Figura 10 mostra somente uma parte da estrutura da alumina. Interações semelhantes ocorrem
com sílica gel.
A força de interação varia, portanto, com os tipos de compostos. Quanto mais polar o
composto, maior a sua interação com a alumina e com a sílica gel.
O equilíbrio de distribuição das moléculas sobre a superfície da fase estacionária sólida é
dinâmico, com moléculas sendo constantemente adsorvidas e dessorvidas. Esse equilíbrio e as
diferenças de adsorção entre os compostos de uma mistura são a base do processo de separação
cromatográfica.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 70
Figura 10. Principais tipos de interações entre compostos orgânicos e alumina.
Uma classificação bastante comum dos métodos cromatográficos baseia-se na geometria
da superfície na qual a separação ocorre: se dentro de um tubo (de vidro ou metal), a
cromatografia é denominada em coluna; se em uma superfície plana (placa de vidro ou metal
impregnada com a fase estacionária ou então uma folha de papel de filtro embebida com solvente),
a cromatografia é planar.
A cromatografia em camada delgada (CCD) é um caso particular de cromatografia planar e
de adsorção. Na química orgânica, a CCD é utilizada, principalmente como uma ferramenta eficaz
de análise qualitativa da pureza de uma amostra, avaliação do número de componentes de uma
mistura, determinação da identidade de uma amostra por comparação com um padrão,
identificação de uma ou mais substâncias presentes em uma mistura por comparação com
padrões, monitoramento do progresso de uma reação química, escolha de uma fase móvel
apropriada para uma separação cromatográfica em coluna e monitoramento de uma separação por
cromatografia em coluna.
A CCD consiste em uma fase estacionária sobre uma placa (de vidro, alumínio ou material
plástico), na qual se aplica a amostra (Figura 11). Realiza-se a eluição da placa de forma
ascendente em uma câmara fechada (Figura 12), chamada de cuba cromatográfica, contendo a
fase móvel apropriada.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 71
Figura 11. Aplicação de uma amostra em placa cromatográfica.
Figura 12. Cuba cromatográfica para eluição da placa de CCD.
A) Aspectos práticos da CCD
Placas de CCD
As principais fases estacionárias utilizadas na CCD são sílica gel, alumina, sílica gel de
fase reversa, celulose e poliamida. Essas fases são impregnadas sobre placas de vidro, plástico ou
alumínio.
As placas de CCD podem ser adquiridas comercialmente ou ser preparadas em
laboratório. Nesse último caso, elas devem ser secas ao ar e, então, “ativadas” em estufa a 110-
120 ºC, durante 1 hora. As placas devem ser guardadas em lugares onde a atmosfera seja a mais
seca possível.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 72
Aplicação das amostras
As amostras a serem analisadas por CCD devem ser previamente dissolvidas em solvente
orgânico volátil. A aplicação dessa solução sobre a placa cromatográfica deve ser efetuada a
aproximadamente 1 cm de sua base inferior (Figura 11).
Nessa aplicação, pode-se utilizar um tubo capilar, cuja extremidade inferior esteja
uniformemente seccionada. O capilar não pode danificar a fina camada de adsorvente, pois os
resultados não serão reprodutíveis.
Quando mais de uma amostra for aplicada sobre uma mesma placa, os pontos de
aplicação devem ser separados por, no mínimo, 1 cm.
Desenvolvimento do cromatograma (Eluição)
Após a aplicação da amostra sobre a placa, ela deve ser introduzida em uma cuba
cromatográfica contendo a fase móvel apropriada.
A altura da fase móvel na cuba não pode ultrapassar o ponto de aplicação da amostra na
placa. Para que bons resultados sejam obtidos na CCD, é necessário que a cuba fique saturada
com vapores da fase móvel. Para isso, as paredes laterais internas da cuba devem ser recobertas
com papel filtro (Figura 12).
Uma vez introduzida a placa na cuba cromatográfica, o solvente ascenderá, por
capilaridade, até a extremidade superior. Ao ascender, o solvente arrastará mais os compostos
menos adsorvidos na fase estacionária, separando-os dos compostos mais adsorvidos. A placa
deve ser retirada da cuba um pouco antes da frente do solvente alcançar a extremidade superior
da placa.
Após a eluição da placa, seca-se a placa por simples exposição ao ar ou com um secador
de ar quente. Como a maioria dos compostos orgânicos é incolor, deve-se realizar a revelação da
placa cromatográfica.
Um exemplo de cromatograma final é mostrado na Figura 13. Nessa figura, pode-se
observar que a amostra continha dois componentes: o 1, que ficou mais adsorvido na fase
estacionária, e o 2, que ficou menos adsorvido na fase estacionária, tendo uma maior afinidade
pela fase móvel. Se, para esse exemplo, a fase estacionária fosse sílica, a conclusão que se
poderia chegar é que a substância 1 era mais polar que a substância 2.
Na Figura 13, são apresentados os valores de Rf (fator de retenção) para os componentes
1 e 2. O Rf é definido como a razão entre a distância X percorrida por um dado componente da
amostra (desde o ponto de aplicação até o centro da mancha) e a distância Y percorrida pelo
solvente (desde o ponto de aplicação até a linha final de avanço do solvente).
Y
XRf

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 73
Figura 13. Exemplo de cromatograma e de cálculo de Rf.
O valor de Rf é um número adimensional, que pode variar entre 0 e 1. Dentro dessa faixa,
substâncias com baixos valores de Rf possuem maior afinidade pela fase estacionária, e
substâncias com valores de Rf próximos a 1 possuem maior afinidade pela fase móvel do que pela
fase estacionária.
Embora seja característico de cada substância, o valor de Rf pode sofrer alterações devido
a condições experimentais (variação de fase estacionária, eluente, temperatura, etc). Portanto,
para fins comparativos, é essencial realizar a análise sob as mesmas condições.
Escolha da fase móvel
A escolha da fase móvel adequada para uma determinada separação por CCD é uma
tarefa trabalhosa, para qual não existem regras fixas. Entretanto, considerando-se os conceitos
básicos de polaridade de moléculas orgânicas e, através da experiência adquirida, a escolha da
fase móvel se torna mais fácil.
Para se obter um bom cromatograma por CCD, o composto deve alcançar uma altura entre
a metade e 2/3 da placa. Na Figura 14(a), a fase móvel (também chamada de eluente) é tão pouco
polar, que os dois componentes da amostra têm muita afinidade pela fase estacionária e nenhuma
pela fase móvel. Na Figura 14(c), a situação é totalmente oposta, ou seja, como a polaridade do
eluente é muito alta, os dois componentes têm igual afinidade pela fase móvel, percorrendo a placa
cromatográfica juntamente com a linha de frente do solvente. Assim, não se observa separação
alguma.
Na Figura 14(b), têm-se o eluente ideal, ou seja, com polaridade intermediária, que permite
a interação das substâncias tanto com a fase móvel quanto com a fase estacionária. Isto permite a

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 74
diferenciação entre as interações dos dois componentes pelo eluente e fase estacionária,
causando a separação.
Figura 14. Escolha da fase móvel.
Nos casos em que a utilização de solventes puros não é suficientemente eficaz
para se obter a separação desejada, pode-se usar mistura de solventes. A Tabela 1 mostra alguns
solventes em ordem crescente de polaridade.
Tabela 1. Poder de eluição de alguns solventes
Reveladores
Como a maioria das substâncias orgânicas é incolor, após a eluição e secagem da placa
cromatográfica, ela deve ser revelada. Os reveladores mais comuns são: luz UV, iodo, e reagentes
químicos (ácido fosfomolibídico, solução de permanganato de potássio, etc).
Para a placa ser revelada em câmara UV, a fase estacionária deve conter substâncias
fluorescentes, na concentração de aproximadamente 1%. A substância fluorescente absorverá luz
na região do ultravioleta ( = 254 nm) e emitirá luz em outra região do espectro. Essa emissão tem
uma coloração característica esverdeada e brilhante. Assim, os locais onde houver substâncias

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 75
orgânicas, principalmente aquelas contendo duplas ligações conjugadas ou sistemas aromáticos,
impedirão a emissão de luz, aparecendo como manchas escuras.
Outra forma de revelação é introduzir a placa cromatográfica em uma câmara contendo
cristais de iodo. Este, na forma de vapor, interage com os compostos orgânicos insaturados da
placa, resultando em manchas de coloração marrom. Normalmente a interação com o iodo é
reversível e a placa revelada dessa maneira, deixada em repouso por algum tempo, após o
desaparecimento das manchas pode ser revelada por outros métodos químicos. Somente
compostos orgânicos que interagem com o iodo podem ser revelados dessa forma. Compostos,
saturados, por exemplo, não se revelam e devem ser visualizados utilizando outros reagentes
químicos, que constituem métodos destrutivos de revelação.
Os métodos destrutivos de revelação consistem usualmente na oxidação dos compostos
orgânicos que estão sobre a superfície da placa, utilizando-se oxidantes fortes e às vezes
temperaturas elevadas. Exemplos desses reagentes são o ácido fosfomolíbdico e o permanganato
de potássio. Esses reveladores são preparados na forma de solução, que é aspergida sobre a
placa. A placa deve ser então aquecida em estufa, chapa de aquecimento ou com soprador
térmico. As substâncias orgânicas serão oxidadas e reveladas na forma de pontos escuros.
Além dos reveladores de aplicação geral, existem reveladores que detectam apenas
alguns compostos contendo certos grupos funcionais. Solução de 2,4-dinitrofenilidrazina, por
exemplo, produz manchas amarelo-avermelhadas quando o composto possui função aldeído e/ou
cetona, e o reagente de Dragendorff origina manchas alaranjadas para alcalóides. A Tabela 2
descreve alguns reveladores gerais e específicos.
Tabela 2. Reveladores para CCD
Reagente Revelador
Vanilina/H2SO4 Universal
Ácido fosfomolibídico Universal
Ninidrina Específico: aminas
Anisaldeído Específico: compostos carbonílicos
Ftalato de anilínio Específico: açúcares redutores
OBJETIVOS
A presente experiência tem os seguintes objetivos:
- aprendizagem da técnica de destilação por arraste de vapor e da cromatografia em
camada delgada (CCD);
- extração de óleos essenciais (de cravo, eucalipto e limão) por destilação por arraste de
vapor;
- caracterização dos óleos essenciais por CCD e por testes químicos.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 76
MATERIAL E REAGENTES
- adaptador de vácuo - água de bromo (1 gota de Br2 para 4 mL de
água destilada)
- balão de fundo redondo de 100 e 250 mL - botões de cravo
- béquer de 50 mL - diclorometano
- cabeça de destilação - cascas de limão
- capilares para cromatografia - éter dietílico
- cápsula de porcelana - folhas de eucalipto
- chapa de aquecimento - glicerina
- condensador de Liebig - hexano
- cuba cromatográfica - solução de ácido fosfomobídico (1 g ácido em
25 mL de Etanol)
- erlenmeyer de 125 mL - solução de 2,4-dinitrofenilidrazina*
- funil de vidro - solução de permanganato de potássio 1%
- funil de separação de 250 mL - sulfato de magnésio anidro
- lâmpada para revelação no UV
- mangueiras de látex * Preparação da solução de 2,4-
difenilidrazina
Dissolver 3 g de 2,4-dinitrofenilidrazina
em 15 mL de ácido sulfúrico concentrado.
Adicionar a solução, com agitação, a 20 mL de
água e 70 mL de etanol 95%. Misturar bem e
filtrar.
- placas cromatográficas de sílica
- pipeta Pasteur
- proveta de 10 mL
- termômetro (0-200 ºC), com e sem junta
esmerilhada
- tubos de ensaio

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 77
PROCEDIMENTO EXPERIMENTAL
1) Extração de óleos essenciais
a) Faça a montagem necessária para uma destilação por arraste de vapor pelo método direto
(Figura 8) ou para uma destilação simples (Aula Prática 3, Figura 1). Abra a torneira e regule a
saída de água até que se estabeleça um fluxo contínuo de água pelo condensador.
b) Coloque 10 g do material vegetal (botões de cravo da índia ou folhas de eucalipto) no balão de
fundo redondo de 250 mL e adicione água destilada até completar, no máximo, 2/3 do balão. No
caso dos limões, utilizam-se as cascas obtidas de dois limões. Descasque os limões de modo a
não permitir a presença de bagaço e suco.
c) Inicie a destilação, aquecendo a mistura em banho de glicerina. Observe as mudanças ocorridas
na mistura.
d) Recolha 50 mL de destilado em um balão de fundo redondo de 125 mL. Separe 10 mL desse
destilado em um béquer, para a realização de testes químicos (seção 4.3), e transfira o restante do
destilado para um funil de separação.
e) Ao funil de separação, adicione 10 mL de diclorometano e agite suavemente, tomando o cuidado
em aliviar a pressão interna, como descrito na Aula Prática 1. Essa operação deve ser feita em
capela de exaustão. Deixe o sistema em repouso até que ocorra a separação completa das fases.
Recolha a fase orgânica em um erlenmeyer de 125 mL previamente identificado como FO. Lembre-
se que, nesse caso, a FO será a inferior.
f) Adicione mais 10 mL de diclorometano à fase aquosa contida no funil de separação e repita o
procedimento de extração líquido-líquido. Recolha a fase orgânica no mesmo erlenmeyer
identificado como FO.
g) Adicione sulfato de sódio anidro à solução do erlenmeyer FO em quantidade suficiente para que
a fase orgânica fique seca. Filtre, então, a fase orgânica seca por gravidade.
2) Análise por CCD
a) Aplique em três placas cromatográficas distintas, a amostra de destilado obtida no item 4.1. A
aplicação deve ser realizada com o auxílio de capilar e da forma descrita na seção 1.5.1 e ilustrada
na Figura 11.
b) Prepare a fase móvel, misturando hexano e éter dietílico na proporção 2:1 (v/v). Elua,
separadamente, as placas em câmara cromatográfica, conforme descrito na seção 1.5.1 e ilustrado
na Figura 12.
c) Após o término da eluição, retire a placa da cuba cromatografia e marque com um lápis a
distância percorrido pelo solvente. Deixe o solvente evaporar da placa e observe-a placa em
câmara de luz ultravioleta. Marque com um lápis as manchas observadas na placa.
d) Separadamente, revele as placas cromatográficas com solução de ácido fosfomolíbdico, solução
de permanganato de potássio e solução de 2,4-dinitrofenilidrazina.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 78
e) Faça o desenho de cada uma das placas reveladas e marque a distância percorrida pela
substância. Compare os resultados obtidos com o uso de cada revelador.
3) Testes químicos
A) Ensaio de Bayer
Em um tubo de ensaio, coloque 3 mL de solução aquosa de permanganato de potássio
(1%) e adicione 2 mL do destilado, que foi reservado para testes químicos. Observe as mudanças
ocorridas na mistura.
B) Ensaio com a água de bromo
Em um tubo de ensaio, coloque 3 mL de solução aquosa de bromo (água de bromo) e
adicione 2 mL do destilado, que foi reservado para testes químicos. Observe as mudanças
ocorridas na mistura.
C) Ensaio com solução de 2,4-dinitrofenilidrazina
Em um tubo de ensaio, coloque 3 mL de solução de 2,4-dinitrofenilidrazina e adicione 2 mL
do destilado, que foi reservado para testes químicos. Observe as mudanças ocorridas na mistura.
QUESTÕES
a) Faça um desenho das placas cromatográficas revelada em câmara de UV. Faça o mesmo após
a revelação das placas com os diferentes agentes reveladores. Determine os valores de Rf para as
substâncias reveladas nas placas cromatográficas. Qual a utilidade em se revelar as placas com
diferentes reagentes?
b) Com base nas informações sobre a constituição química de cada óleo essencial, indique quais
das substâncias apresentadas nas Figuras 5, 6 e 7 são quirais.
c) Considere uma mistura constituída por bifenila, ácido benzóico e álcool benzílico. A mistura é
submetida à análise por CCD. Coloque as substâncias em ordem crescente de Rf na CCD.
d) Uma mistura contendo um ácido dicarboxílico e um ácido tricarboxílico foi analisada por CCD,
utilizando como eluente a acetona. Entretanto, verificou-se que, após a eluição, a mancha da
mistura continuava no ponto de aplicação. O que se pode fazer nesse caso, para se conseguir uma
análise cromatográfica que forneça a separação dos componentes da mistura?
e) Quais são os grupos funcionais que podem ser identificados pelos ensaios com água de bromo,
solução de Baeyer e solução de 2,4-dinitrofenilidrazina? Escreva as equações químicas das
reações envolvidas quando esses reagentes são utilizados.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 05: Extração de óleos essenciais e sua análise por CCD 79
BIBLIOGRAFIA
BARBOSA, L.C.A. “Introdução à Química Orgânica”. Editora Pearson, 2a ed., São Paulo, 2004.
COLLINS, C.H.; BRAGA, G.L.; BONATO, P.S. “Fundamentos de Cromatografia”, Editora
UNICAMP, Campinas, 2006.
DEMUNER, A.J.; MALTHA, C.R.A.; BARBOSA, L.C.A.; PERES, V. “Experimentos de Química
Orgânica”. Editora UFV, 2ª ed, Viçosa, 2004.
DIAS, A.G.; DA COSTA, M.A.; GUIMARÃES, P.I.C. “Guia Prático de Química Orgânica. Volume I –
Técnicas e Procedimentos: Aprendendo a Fazer”. Editora Interciência, Rio de Janeiro, 2004.
LANÇAS, F.M. “Cromatografia Líquida Moderna”. Editora Átomo, Campinas, 2009.
MARQUES, J.A.; BORGES, C.P.F. “Práticas de Química Orgânica”. Editora Átomo, Campinas,
2007.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.
SILVA, R.S.; RIBEIRO, C.M.R.; BORGES, M.N.; BLOIS, G.S.O. “Óleo essencial de limão no ensino
de cromatografia em camada delgada”. Quim. Nova, 32(8): 2234-2237, 2009.
SIMÕES, C.M.O.; SCHENKEL, E.P.; GOSMANN, G.; MELLO, J.C.P.D.; MENTZ, L.A.;
PETROVICK, P.R. “Farmacognosia – da planta ao medicamento”. Ed. UFRGS, Porto Alegre, 5a
ed, 2003.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 80
Aula prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre
INTRODUÇÃO
A cromatografia é um dos métodos mais modernos e eficazes para separação,
identificação e quantificação de compostos orgânicos. Ela se baseia na distribuição diferencial dos
componentes de uma dada mistura entre duas fases (estacionária e móvel). Conforme discutido no
Experimento 6, vários são os critérios para classificação das técnicas cromatográficas. Dentre
esses, o mecanismo envolvido na separação é um dos mais importantes. Também a geometria da
superfície na qual a separação ocorre é outra maneira de se classificar a cromatografia. Nesse
sentido, no Experimento 6 foi apresentada a cromatografia em camada delgada (CCD), que é uma
cromatografia planar e por adsorção, muito útil na análise qualitativa da pureza de uma amostra,
avaliação do número de componentes de uma mistura, determinação da identidade de uma
amostra, monitoramento do progresso de uma reação química, etc.
Se a fase estacionária sólida não estiver sobre a superfície de um suporte, mas contida em
um tubo cilíndrico, normalmente de vidro, a cromatografia será classificada como cromatografia em
coluna. Analogamente à CCD, a cromatografia em coluna também é uma cromatografia de
adsorção. Ela é utilizada para separação e purificação de substâncias e os princípios que regem
esse tipo de cromatografia são similares aos discutidos para CCD no Experimento 6.
A técnica da cromatografia em coluna consiste em adicionar um sólido adsorvente
apropriado (fase estacionária) a um tubo de vidro na posição vertical, acrescentando-se, no topo da
coluna, a mistura que se deseja fracionar (Figura 1). Um eluente líquido (fase móvel), que pode ser
constituído por um ou mais solventes miscíveis entre si, é adicionado ao sistema e, ao percorrer a
coluna, “arrasta” as substâncias adsorvidas na fase estacionária. Os componentes da mistura que
interagirem mais fortemente com a fase estacionária apresentarão um deslocamento mais lento
pela coluna. Os componentes que apresentarem interações mais fracas com a fase estacionária e
mais fortes com a fase móvel se deslocarão mais rápido pela coluna. A diferença na velocidade de
eluição dos componentes da mistura, devido às suas diferentes interações com a fase móvel e
estacionária, farão com que eles se separem e formem bandas na coluna, que podem, então, ser
coletadas. Assim, se alumina for utilizada como adsorvente, um composto de menor polaridade
formará uma banda que será eluída mais rapidamente do que a banda de um composto mais polar
(Figura 2).
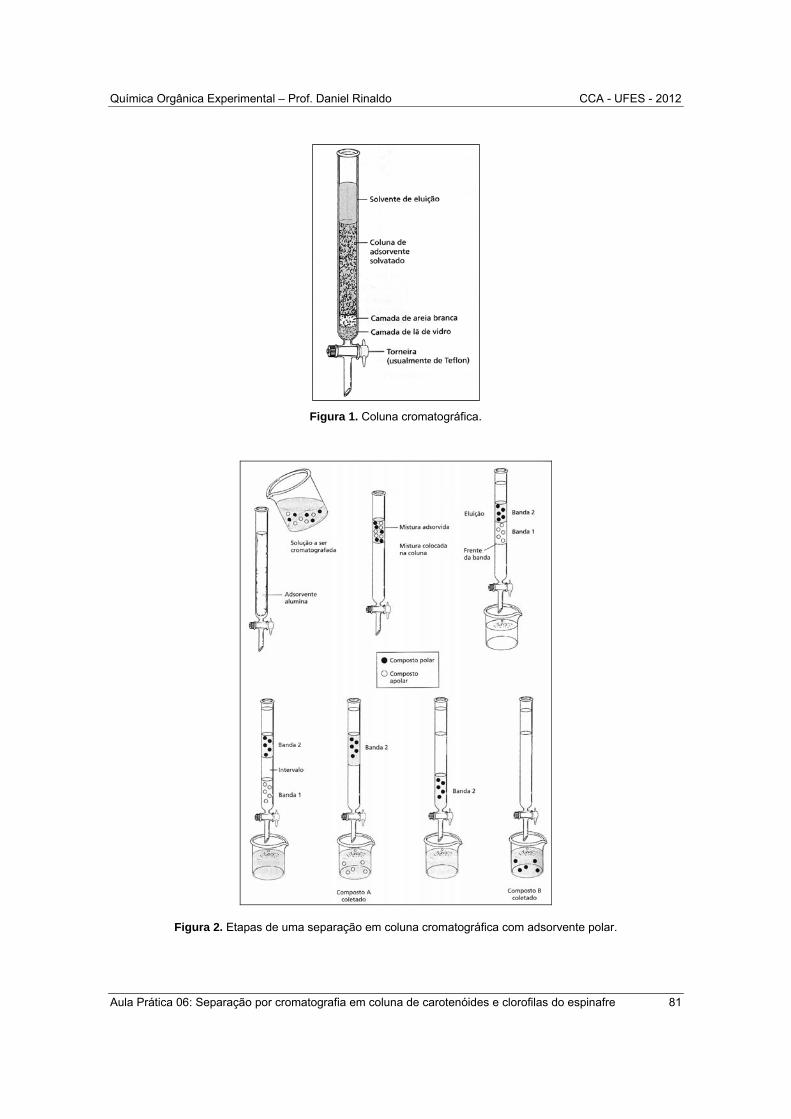
Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 81
Figura 1. Coluna cromatográfica.
Figura 2. Etapas de uma separação em coluna cromatográfica com adsorvente polar.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 82
Aspectos práticos da cromatografia em coluna
Apesar da cromatografia em coluna ser uma técnica versátil, sua eficiência depende do
ajuste de alguns fatores, tais como: escolha do adsorvente, polaridade da fase móvel, tamanho
(comprimento e diâmetro) da coluna em relação ao material a ser cromatografado e velocidade de
eluição (fluxo). Além disso, uma separação cromatográfica eficiente depende da realização correta
dos procedimentos envolvidos na cromatografia em coluna (empacotamento, aplicação da
amostra, etc).
Escolha do adsorvente
Vários tipos de adsorventes podem ser usados em cromatografia em coluna (sílica gel,
alumina, celulose, florisil, etc). A escolha do adsorvente depende dos tipos de compostos a serem
separados. Celulose, amido e açúcares são usados para separar compostos polifuncionais de
origem natural, que são muito sensíveis a interações ácido-base. Sílica gel e florisil são usados
para separar muitos tipos de compostos (hidrocarbonetos, álcoois, cetonas, ésteres,
azocompostos, aminas, etc). Alumina também é amplamente empregada para separações por
cromatografia em coluna.
Escolha da fase móvel
Os mesmos solventes empregados na CCD (Experimento 6, Tabela 1) podem ser
utilizados como fase móvel na cromatografia em coluna. A CCD pode ser, inclusive, empregada
para a escolha da fase móvel que será utilizada na cromatografia em coluna. Às vezes, um único
solvente é capaz de separar todos os componentes de uma mistura. Outras vezes, deve-se utilizar
uma mistura de solventes. Entretanto, uma prática muito comum é começar a eluição com um
solvente apolar e aumentar gradualmente a polaridade da fase móvel. Isto é chamado de eluição
gradiente. A eluição é dita isocrática quando se mantém a mesma polaridade da fase móvel
durante todo o processo de separação.
Em geral, os compostos apolares são eluídos mais rapidamente que os compostos
polares, quando se utiliza um adsorvente polar como sílica ou alumina.
Escolha do tamanho da coluna e da quantidade de adsorvente
O tamanho da coluna e quantidade de adsorvente são parâmetros que também devem ser
corretamente selecionados para que se tenha uma separação eficiente por cromatografia em
coluna.
Como regra geral, a quantidade recomendada de adsorvente é de 25 a 30 vezes (em
massa) da quantidade de material a ser separado e a coluna deve ter uma razão de 8:1 entre o
comprimento e o diâmetro. Entretanto, a escolha do tamanho da coluna e da quantidade de

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 83
adsorvente depende significativamente da separação entre os componentes da mistura.
Compostos que se separam com dificuldade podem exigir colunas mais longas e mais adsorvente
do que o especificado pela regra geral. Contrariamente, se os compostos se separam facilmente,
uma coluna mais curta e menos quantidade de adsorvente podem ser utilizados.
Fluxo da fase móvel
A velocidade com que a fase móvel flui pela coluna também é um importante fator na
eficiência da separação. Em geral, o tempo que a mistura a ser separada fica na coluna é
diretamente proporcional ao grau atingido pelo equilíbrio entre as fases móvel e estacionária.
Assim, compostos semelhantes podem ser separados, se ficarem na coluna tempo suficiente.
Entretanto, se o fluxo é muito lento, as substâncias dissolvidas da mistura podem difundir mais
rapidamente do que serem eluídas. Neste caso, as bandas ficam mais largas e a separação piora.
Portanto, um fluxo adequado de fase móvel deve ser utilizado para permitir a separação eficiente
dos constituintes da mistura. Como regra geral, fluxo de 5-50 gotas por minuto costuma fornecer
separações satisfatórias.
Empacotamento da coluna
O empacotamento da coluna deve ser “homogêneo”, ou seja, livre de irregularidades,
bolhas de ar ou falhas. Além disso, a face superior do adsorvente empacotado deve estar na
horizontal assim como a coluna deve estar perfeitamente na vertical. A falha desses fatores pode
ocasionar bandas não-horizontais (Figura 3), que prejudicam a separação.
Figura 3. Comparação das bandas obtidas com e sem nivelamento.
Outro fenômeno, que pode prejudicar a separação, é a ondulação ou canalização (Figura
4). Isto ocorre quando a superfície do adsorvente tem falhas ou irregularidades ou se existirem

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 84
bolhas de ar. Nesse caso, a parte da frente da banda avança mais rapidamente do que a maior
parte da banda, pois se move pelo canal.
Figura 4. Problemas de canalização na coluna cromatográfica.
Antes do empacotamento, coloca-se uma pequena quantidade de algodão na extremidade
inferior da coluna, de forma a evitar o escoamento da fase estacionária. Pode-se adicionar uma
camada de areia sobre o algodão e depois sobre a fase estacionária empacotada (Figura 1). As
camadas de areia têm a finalidade de manter a coluna de fase estacionária nivelada.
A introdução da fase estacionária na coluna pode ser feita na forma de uma suspensão na
fase móvel. Essa suspensão deve ser adicionada lentamente à coluna, batendo-se continuamente
ao longo da mesma, para que todo ar seja expulso e uma compactação uniforme seja obtida.
Nunca o nível do solvente pode ficar abaixo do nível da fase estacionária, pois isso poderia causar
a entrada de ar e a conseqüente formação de rachaduras na fase estacionária, que prejudicariam a
separação.
Aplicação da amostra
Se a amostra a ser purificada for líquida (e de baixa viscosidade), ela pode ser diretamente
aplicada na coluna. Entretanto, se a amostra for um líquido viscoso ou um sólido, a amostra pode
ser introduzida na coluna na forma de uma solução, que contém a amostra dissolvida em uma
pequena quantidade de solvente (em geral, 2-3 mL). Essa pequena quantidade de solvente é
necessária para que uma banda estreita da amostra seja obtida, que é ideal para a melhor
separação dos constituintes da amostra.
Em casos em que a amostra é muito viscosa ou de solubilidade muito baixa na fase móvel,
incorpora-se essa amostra em pequena quantidade da fase estacionária e, então, distribui-se
uniformemente a amostra na forma de pó no topo da coluna.
Eluição

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 85
Uma vez introduzida a amostra, deve-se iniciar a eluição, utilizando a fase móvel
adequadamente escolhida. Ao contrário da CCD, na qual se utiliza eluição isocrática, na
cromatografia em coluna é mais comum utilizar-se a eluição gradiente, que permite alterar
gradativamente a composição da fase móvel. Assim, a eluição da amostra começa com um
solvente apolar e, ao longo da análise, aumenta-se gradativamente a polaridade da fase móvel,
através da adição de maior porcentagem de solventes mais polares à fase móvel. Geralmente, a
polaridade da fase móvel não pode ser drasticamente mudada. Se os calores de solvatação do
adsorvente nos dois solventes forem muito diferentes, pode-se gerar aumento de temperatura na
coluna, provocando “rachaduras” no seu interior, que prejudicam a eficiência da separação. A
coluna pode rachar também se o nível da fase móvel ficar abaixo do nível da fase estacionária, ou
seja, se a coluna secar. O uso de colunas com reservatório de solvente ou mesmo de funil de
adição é conveniente para evitar a adição contínua de pequenas quantidades de solvente,
aumentando a chance da ocorrência da secagem da coluna.
Monitoramento das frações coletadas
Quando os compostos a serem separados são coloridos, pode-se monitorar visualmente a
separação das bandas e coletá-las separadamente. Entretanto, como a maior parte dos compostos
orgânicos não é colorida, outros métodos devem ser utilizados para a análise das frações
coletadas. O mais comum deles, é a CCD.
Então, o que se faz é coletar frações de volume fixo. O volume coletado depende da
quantidade de amostra e da facilidade de separação. Em separações mais difíceis, devem ser
recolhidas frações com volumes menores. Após a coleta, as frações são analisadas por CCD
(Figura 5). De acordo com os resultados obtidos nessa análise, algumas frações podem ser
reunidas e o processo de cromatografia em coluna encerrado.
Figura 5. Monitoramento por CCD das frações coletadas em uma separação em coluna.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 86
Pigmentos vegetais
Os vegetais produzem uma infinidade de substâncias química, conhecidas como
metabólitos. Estes metabólitos podem ser classificados em dois grandes grupos: metabólitos
primários, que estão envolvidos no metabolismo básico da planta, e metabólitos secundários, que
possuem diversas outras funções (defesa contra predadores, atração de polinizadores, etc).
Os carotenóides constituem um grupo de metabólitos secundários encontrado em muitas
plantas, que trazem benefícios para a saúde por sua atividade antioxidante e anticancerígena.
Alguns desses compostos apresentam também atividade pró-vitamínica A.
As substâncias pertencentes a esse grupo são caracterizadas pela presença de um grande
número de ligações duplas conjugadas, fazendo com que absorvam luz na região do visível e,
portanto, contribuindo para a coloração das plantas. A coloração dos carotenóides varia do
amarelo, passando pelo laranja, até o vermelho. O -caroteno (Figura 6) é o pigmento amarelo-
alaranjado da cenoura e o licopeno é a substância avermelhada do tomate.
Os carotenóides podem ser classificados de duas maneiras. A primeira considera a
existência de duas grandes famílias: os carotenos (carotenóides hidrocarbonetos) e as xantofilas
(carotenóides oxigenados). O segundo sistema divide os carotenóides em três grupos: acíclicos
(por exemplo, o licopeno), monocíclicos (por exemplo, o -caroteno) e bicíclicos (por exemplo, o -
caroteno e o -caroteno).
Outros metabólitos associados à coloração das plantas são as clorofilas, que são os
pigmentos verdes das plantas que funcionam com fotorreceptores. As estruturas das clorofilas a e
b diferem apenas quanto a um dos grupos substituintes, como mostra a (Figura 7).
Figura 6. Fórmula estrutural do -caroteno.
Figura 7. Fórmula estrutural das clorofilas a e b.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 87
OBJETIVOS
A presente experiência tem os seguintes objetivos:
- aprendizagem da técnica de cromatografia em coluna;
- separação de pigmentos do espinafre por cromatografia em coluna;
- monitoramento da cromatografia em coluna por CCD.
MATERIAL E REAGENTES
- algodão - Acetona
- almofariz e pistilo - Espinafre
- capilar para CCD - Etanol
- coluna cromatográfica (bureta de 25 mL) - Hexano
- cuba cromatográfica - Sílica Gel
- erlenmeyer de 125 mL
- funil de vidro
- papel de filtro
- pipeta de Pasteur
- placa cromatográfica
- provetas de 50 e 100 mL
- suporte universal com garras
- suporte para tubos de ensaio
- 15 tubos de ensaio
PROCEDIMENTO EXPERIMENTAL
1) Extração dos pigmentos do espinafre
A) Preparo da mistura de solventes para extração das folhas
Em capela de exaustão, adicione 40 mL de hexano e 10 mL de acetona a um Erlenmeyer de 125 mL, tampe e reserve.
B) Preparo da amostra
Essa parte do procedimento deverá ser realizada em conjunto com outro grupo.
Pese cerca de 25 g de folhas de espinafre. Corte ou rasgue as folhas em pequenas partes
e coloque-as em um almofariz, juntamente com 50 mL de uma mistura de hexano:acetona ( 4:1
v/v). Amasse as folhas até que a solução adquira uma coloração esverdeada. Com uma pipeta,
transfira a solução para um erlenmeyer de 125 mL.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 88
2) Separação dos pigmentos em coluna cromatográfica
A) Empacotamento da coluna
a) Com o auxílio de uma vareta, coloque um pequeno pedaço de algodão na ponta da coluna.
Ajuste a coluna na posição vertical.
b) A um erlenmeyer de 125 mL, adicione cerca de 9 g de sílica gel e hexano até que se obtenha
uma suspensão de sílica no solvente. Este procedimento deve ser realizado em capela de
exaustão.
c) Agite e transfira a suspensão para a coluna com o auxílio de um funil de boca larga. Utilize
pequenas porções de hexano para auxiliar na transferência quantitativa da sílica do erlenmeyer
para a coluna. À medida que a sílica decanta, bata suavemente ao longo do tubo de vidro ou
exerça leve pressão no topo da coluna com uma bomba manual (tipo pêra de borracha) ou
compressor de ar. Isto fornecerá um empacotamento mais perfeito da fase estacionária, livre de
bolhas de ar.
d) Feche a torneira quando o eluente estiver aproximadamente 0,2 cm acima da sílica. O eluente
coletado pode ser reutilizado no fracionamento da amostra.
B) Aplicação da amostra
a) Com uma pipeta de Pasteur, adicione 4 mL do extrato bruto (obtido na seção 4.1.2) ao topo da
coluna. Faça a aplicação cuidadosa da amostra de forma a não alterar a superfície da sílica.
b) Abra a torneira e deixe a amostra penetrar na sílica, coletando o eluente em um erlenmeyer, até
que reste cerca de 0,1 cm de líquido acima da coluna de sílica.
c) Adicione, cuidadosamente, cerca de 0,5 mL de hexano à coluna, fazendo com que escorra pelas
paredes internas da coluna. Abra a torneira e deixe escoar o eluente até que reste uma coluna de
0,1 cm de solvente acima da sílica.
Repita esta operação (c) duas ou três vezes, até que a amostra já tenha “penetrado”
totalmente na coluna de sílica, ou seja, até que a coluna de líquido acima da sílica esteja incolor. O
eluente coletado até esse ponto pode ser reutilizado no fracionamento da amostra.
C) Fracionamento da amostra
a) Encha a coluna com hexano. Colete o eluente em erlenmeyer até que a banda amarela, relativa
ao -caroteno, esteja próxima à saída. A partir daí, colete frações de 5 mL em tubos de ensaio.
b) Após a separação da banda amarela, deixe o nível do hexano ficar até 0,1 cm acima da sílica e
encha a coluna com álcool etílico. Colete o eluente em erlenmeyer até que a banda verde, relativa
às clorofilas, esteja próxima à saída. A partir daí, colete frações de 5 mL em tubos de ensaio até
que toda a banda de coloração verde tenha sido eluída da coluna.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 89
3) Análise das frações por CCD
Antes da realização dessa etapa, reveja os procedimentos da CCD discutidos na
Aula Prática 5.
a) Em uma placa cromatográfica, aplique as frações coloridas coletadas durante o procedimento
descrito na seção 4.2.3.
b) Prepare a fase móvel, misturando hexano e acetona na proporção 4:1 (v/v) e elua a placa em
câmara cromatográfica.
c) Após o término da eluição, retire a placa da cuba cromatográfica e marque com um lápis a
distância percorrida pelo solvente. Deixe o solvente evaporar da placa e observe as manchas
coloridas na placa. Faça o desenho da placa e marque a distância percorrida por cada substância.
QUESTÕES
1. Com base nas estruturas moleculares apresentadas nas Figuras 6 e 7, explique por que o -
caroteno foi eluído da coluna cromatográfica antes das clorofilas a e b.
2. Calcule os Rfs das manchas obtidas na análise cromatográfica da seção 4.3. Com base nesses
valores, discuta por que a separação dos pigmentos do espinafre foi possível. Se as substâncias
separadas não tivessem cor, que método de revelação poderia ser utilizado? Explique sua
resposta.
3. A páprica é um condimento de cor vermelha-intensa preparado a partir do pimentão vermelho
(Capsicum annuum), seco e moído, sendo utilizada tanto na culinária como na agroindústria. Os
principais pigmentos encontrados nesse condimento são o β-caroteno (Figura 6) e a capsatina.
A cromatografia em coluna é uma técnica que pode ser utilizada para a separação dos pigmentos
da páprica. Nesse caso, qual das substâncias seria eluída primeiro, se fosse utilizada sílica como
fase estacionária?
4. Uma amostra foi submetida à cromatografia em coluna. A fase estacionária utilizada foi sílica gel
e a fase móvel foi diclorometano. Todos os componentes da amostra foram eluídos, mas não se
obteve separação. Por que isso aconteceu e o que poderia ser feito de forma a se conseguir a
separação?

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 06: Separação por cromatografia em coluna de carotenóides e clorofilas do espinafre 90
BIBLIOGRAFIA
COLLINS, C.H.; BRAGA, G.L.; BONATO, P.S. “Fundamentos de Cromatografia”, Editora
UNICAMP, Campinas, 2006.
DIAS, A.G.; DA COSTA, M.A.; GUIMARÃES, P.I.C. “Guia Prático de Química Orgânica. Volume I –
Técnicas e Procedimentos: Aprendendo a Fazer”. Editora Interciência, Rio de Janeiro, 2004.
LANÇAS, F.M. “Cromatografia Líquida Moderna”. Editora Átomo, Campinas, 2009.
MARQUES, J.A.; BORGES, C.P.F. “Práticas de Química Orgânica”. Editora Átomo, Campinas,
2007.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 91
Aula prática 07: Síntese de biodiesel da soja
INTRODUÇÃO
A história da aplicação de óleos vegetais como combustível começou em 1898 na Feira
Mundial de Paris, onde Rudolf Diesel apresentou um motor abastecido com óleo de amendoim
mais eficiente que os motores a vapor usados na época. Entretanto, desde o início do século XX, o
óleo mineral tornou-se o combustível para esse tipo de motor, devido ao seu menor custo e a
melhores propriedades físico-químicas. O óleo mineral é comumente chamado óleo diesel em
reconhecimento a Rudolf Diesel.
Atualmente, as mudanças climáticas associadas à liberação de gases da queima de
combustíveis minerais, o alto preço internacional do petróleo e a preocupação com o
desenvolvimento sustentável começam a retomar a intenção original de Diesel de empregar óleos
vegetais nos motores ciclo diesel.
Os óleos vegetais apresentam várias vantagens para uso como combustível, como elevado
poder calorífico, ausência de enxofre em suas composições e são de origem renovável. Contudo, o
uso direto de óleos vegetais nos motores ciclo diesel é problemático devido a sua alta viscosidade,
maior densidade e baixa volatilidade.
Essas características geram vários problemas como combustão incompleta, formação de
depósitos de carbono nos sistemas de injeção, diminuição da eficiência de lubrificação, obstrução
nos filtros de óleo e sistemas de injeção de combustível das máquinas, comprometimento da
durabilidade do motor e emissão de acroleína (substância altamente tóxica e cancerígena). Várias
abordagens têm sido consideradas para contornar esses problemas, sendo que a transformação
de óleos vegetais e gorduras animais em ésteres de ácidos graxos tem importância estratégica
para o setor energético, pois possibilita a obtenção do chamado biodiesel, com características
físico-químicas semelhantes ao óleo diesel. Além disso, este processo relativamente simples reduz
a massa molecular para um terço em relação aos triacilglicerídeos, reduzindo a viscosidade e
aumentando a volatilidade.
O biodiesel é um combustível renovável, biodegradável, que apresenta menor emissão de
poluentes, maior ponto de fulgor e maior lubricidade quando comparado ao diesel mineral. Ele é
perfeitamente miscível com o diesel mineral, podendo ser utilizado puro ou em mistura, sem que
qualquer adaptação nos motores seja necessária. As misturas binárias de biodiesel e diesel
mineral são designadas pela abreviação BX, onde X é a porcentagem de biodiesel adicionada à
mistura. Desde 1º de janeiro de 2010, a lei brasileira estabelece a obrigatoriedade da adição de 5
% de biodiesel ao diesel mineral (B5). Só o Brasil produziu 1,6 bilhões de litros de biodiesel em
2010 e essa quantidade pode levá-lo ao terceiro lugar no ranking mundial de produção de
biodiesel.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 92
Reação de transesterificação de óleos e gorduras
Os óleos vegetais e as gorduras animais são constituídos predominantemente de
substâncias conhecidas como triglicerídeos (também chamadas de triacilgliceróis ou
triacilglicerídeos), que são ésteres formados a partir de ácidos carboxílicos de cadeia longa (ácidos
graxos) e glicerol. Além dos triglicerídeos, os óleos vegetais apresentam em sua composição
quantidades apreciáveis de ácidos graxos livres, fosfolipídeos, esteróis e tocoferóis.
Assim, os óleos e gorduras são majoritariamente constituídos por triglicerídeos, que
diferem apenas no tipo de ácido graxo que esterifica a unidade glicerol. Os ácidos graxos variam
na extensão da cadeia carbônica, no número, orientação e posição das ligações duplas. Cerca de
20 a 30 ácidos graxos podem ser encontrados nas gorduras e óleos e é bastante comum eles
serem compostos por 10 a 12 ácidos graxos diferentes. Os ácidos graxos de ocorrência mais
ampla possuem 12, 14, 16 ou 18 carbonos. A Tabela 1 apresenta os ácidos graxos mais comuns e
suas estruturas.
Tabela 1. Ácidos graxos de ampla ocorrência na natureza
Nome Número de carbonos
Estrutura
Ácido láurico 12 CH3(CH2)10COOH
Ácido mirístico 14 CH3(CH2)12COOH
Ácido palmítico 16 CH3(CH2)14COOH
Ácido palmitoléico 16 CH3(CH2)5CH=CH(CH2)7COOH
Esteárico 18 CH3(CH2)16COOH
Oléico 18 CH3(CH2)7CH=CH(CH2)7COOH
Linoléico 18 CH3(CH2)4(CH=CHCH2)2(CH2)6COOH
Linolênico 18 CH3CH2(CH=CHCH2)3(CH2)6COOH
A composição média do óleo de soja, por exemplo, é: 50-59 % de ácido linoléico, 21-29 %
de ácido oléico, 6-10 % de ácido palmítico, 4-8 % de ácido linolênico, 2-6 % de ácido esteárico e 0-
1 % de ácido mirístico.
Os ácidos graxos saturados tendem a ser sólidos e os insaturados líquidos. Desta forma, o
tipo de ácido graxo que esterifica a unidade glicerol influencia nas propriedades físicas e químicas
do óleo e da gordura. De maneira geral, as gorduras tendem a ser sólidas a temperatura ambiente,
devido à presença majoritária de ácidos graxos saturados, enquanto os óleos tendem a ser
líquidos, pois possuem uma quantidade maior de ácidos graxos insaturados.
O biodiesel é obtido através da transesterificação dos triglicerídeos de óleos e gorduras de
origem vegetal ou animal com um monoálcool de cadeia curta, tipicamente metanol ou etanol, na
presença de um catalisador, produzindo uma mistura de ésteres alquílicos de ácidos graxos e

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 93
glicerol (Figura 1). O processo mais comum de transesterificação para obtenção do biodiesel
utiliza metanol e um catalisador básico, tal como hidróxido de sódio ou potássio.
O glicerol é o principal co-produto da transesterificação, mas ácidos graxos livres, mono-,
di-, triglicerídeos, água, álcool, catalisador residual e outras impurezas podem também constituir o
produto final.
Figura 1. Reação de transesterificação de um triglicerídeo.
Transesterificação é um termo geral usado para descrever uma importante classe de
reações orgânicas onde um éster é transformado em outro através da troca do resíduo alcoxila.
Quando o éster original reage com um álcool, o processo de transesterificação é denominado
alcoólise. Esta reação é reversível e prossegue essencialmente misturando os reagentes. Contudo,
a presença de um catalisador (ácido ou base) acelera consideravelmente esta conversão e
contribui para aumentar o rendimento da mesma.
O processo geral é uma sequência de três reações consecutivas, na qual mono- e
diglicerídeos são formados como intermediários (Figura 2). Para uma transesterificação
estequiometricamente completa, uma proporção molar 3:1 de álcool por triglicerídeo é necessária.
Entretanto, devido ao caráter reversível da reação, o agente transesterificante (álcool) geralmente
é adicionado em excesso contribuindo, assim, para aumentar o rendimento do éster, bem como
permitir a sua separação do glicerol formado.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 94
Figura 2. Etapas da transesterificação de um triglicerídeo.
As características físico-químicas de cada biodiesel dependerão do tipo de fonte utilizada
para sua obtenção. O Brasil é um país que contém grandes plantações de oleaginosas e,
consequentemente, usufrui de uma diversidade de opções para produção de biodiesel a partir de
plantas como palma, babaçu, soja, girassol, amendoim, mamona e dendê. Óleos vegetais usados
também são considerados como uma fonte promissora para obtenção do biocombustível, em
função do baixo custo e por envolver reciclagem de resíduos. O produto obtido é comparável com
o biodiesel obtido a partir do óleo refinado. Em 2009, mais de 80 % do biodiesel comercializado no
Brasil eram oriundos de soja.
Com relação ao agente transesterificante, o processo reacional ocorre preferencialmente
com álcoois de baixa massa molecular (metanol, etanol, propanol, butanol e álcool amílico), mas
metanol e etanol são os mais frequentemente empregados. Metanol é o mais utilizado devido ao
seu baixo custo na maioria dos países e às suas vantagens físicas e químicas (polaridade, reação
rápida com triglicerídeo e fácil dissolução do catalisador básico). Além disso, permite a separação
simultânea do glicerol. A mesma reação usando etanol é mais complicada, pois requer um álcool
anidro e um óleo com baixo teor de água.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 95
OBJETIVOS
A presente experiência tem como principal objetivo a síntese de biodiesel de soja a partir
da reação de transesterificação de óleo de soja refinado com metanol, sob catálise básica.
MATERIAL E REAGENTES
- balão de fundo redondo de 250 mL - Ácido acético
- cápsula de porcelana - Hidróxido de potássio
- chapa de aquecimento - Iodo (cristais para revelação)
- cuba cromatográfica - Metanol
- erlenmeyer de 125 mL e 250 mL - Óleo de soja refinado
- funil de separação de 250 mL - solução de ácido clorídrico (0,5 %)
- funil de vidro - solução saturada de NaCl
- papel de filtro - sulfato de sódio anidro
- papel indicador de pH - acetona
- picnômetro de 5 mL - éter dietílico
- placa cromatográfica - hexano
- proveta de 50 mL, 100 mL
- termômetro (0-100 ºC)
PROCEDIMENTO EXPERIMENTAL
1) Preparação do metóxido de potássio
Em um erlenmeyer de 125 mL, dissolva 1,5 g de hidróxido de potássio (KOH) em 35 mL de
metanol. A dissolução deve ser feita com o auxílio de agitação magnética e sob aquecimento em
banho-maria (45 ºC). Este procedimento deve ser realizado em capela.
2) Reação de transesterificação do óleo de soja
a) Em um balão de fundo redondo de 250 mL, adicione 100 mL do óleo de soja e aqueça até 45 ºC
em banho-maria e sob agitação magnética.
b) Adicione toda a solução de metóxido de potássio recentemente preparada segundo
procedimento descrito na seção 4.1.
c) Mantenha a mistura reacional sob agitação durante 10 minutos a 45 ºC.
3) Etapas de elaboração do biodiesel
Antes de realizar os procedimentos desta seção, reveja os conceitos apresentados
na Aula Prática 1.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 96
a) Transfira a mistura reacional (obtida através dos procedimentos descritos na seção 4.2.) para
um funil de separação de 250 mL. Deixe as fases separarem, o que deve ocorrer no período de 15
minutos. Para os casos onde houver formação de emulsão, as mesmas deverão ser desfeitas com
o auxílio de um bastão de vidro, agitando-se lentamente a camada emulsificada. Colete a fase
inferior (glicerol e impurezas) em um erlenmeyer de 125 mL.
b) A fase superior (biodiesel), que ficou no funil de separação, deve ser lavada com 50 mL de
solução aquosa de ácido clorídrico (0,5%, v/v). Realize o processo de extração líquido-líquido,
agitando a mistura e fazendo o alívio de pressão por 3 vezes. Deixe as fases separarem e colete a
fase inferior (aquosa) no mesmo erlenmeyer de 125 mL.
c) A fase superior (biodiesel), que ficou no funil de separação, deve ser então lavada com 50 mL
de solução saturada de NaCl. Realize o processo de extração da mesma forma que descrito
anteriormente e colete a fase inferior (aquosa) em um erlenmeyer de 125 mL.
d) Finalmente, a fase superior (biodiesel), que ficou no funil de separação, deve ser lavada com 50
mL de água destilada. Realize o processo de extração da mesma forma que descrito anteriormente
e colete a fase inferior (aquosa) em um erlenmeyer de 125 mL. Deve-se medir o pH da fase
aquosa com papel indicador de pH, sendo que este deve estar aproximadamente neutro. Caso
necessário, repita o processo de lavagem com água destilada.
e) Colete a fase superior em um erlenmeyer de 250 mL e adicione sulfato de sódio anidro, até que
a fase orgânica esteja seca. Filtre então a mistura por gravidade, empregando papel de filtro
pregueado, coletando o biodiesel em erlenmeyer de 250 mL. O biodiesel obtido deve ser um
líquido límpido de coloração amarela.
4) Caracterização do biodiesel
A) Análise por CCD
Antes de realizar os procedimentos desta seção, reveja os conceitos apresentados
na Aula Prática 5.
Dissolva uma alíquota do biodiesel de soja e outra de óleo de soja em hexano. Aplique
ambas em uma placa cromatográfica de sílica. Faça a eluição da placa em uma câmara
cromatográfica contendo a fase móvel, constituída por uma mistura de hexano:éter etílico (8:1).
Após a eluição, a cromatoplaca deve ser revelada com vapores de iodo.
B) Determinação da densidade Antes de realizar os procedimentos desta seção, reveja os conceitos apresentados
na Seção 4.3.
a) Determine a massa do picnômetro seco e vazio.

Química Orgânica Experimental – Prof. Daniel Rinaldo CCA - UFES - 2012
Aula Prática 07: Síntese de Biodiesel da soja 97
b) Coloque o biodiesel no picnômetro, completando seu volume, de modo a não deixar espaço
vazio.
c) Tampe o picnômetro e limpe-o com papel absorvente. Determine a massa do picnômetro.
d) Esvazie o picnômetro, lave-o com hexano, com acetona e, em seguida, com água destilada.
e) Complete o volume do picnômetro com água destilada, enxugando o excesso com papel
absorvente. Determine a massa do picnômetro com água.
QUESTÕES
1. Dê o mecanismo da reação de transesterificação do óleo de soja para obtenção do biodiesel de
soja.
2. Quando se utiliza catálise básica no processo de transesterificação, por que o óleo vegetal deve
apresentar baixo conteúdo de ácidos graxos livres? Explique com base nas reações laterais que
podem acontecer.
3. Calcule os Rfs do biodiesel e do óleo de soja. Faça um esquema da placa cromatográfica
observada após revelação com iodo.
4. Calcule, a partir dos dados experimentais obtidos, a densidade do biodiesel e compare com o
valor da literatura.4
5. Sabendo-se que o óleo de dendê é composto majoritariamente por triglicerídeos com cadeias
graxas saturadas, o que você poderia dizer sobre a facilidade de solidificação desse biodiesel em
comparação com o biodiesel de soja? O biodiesel de dendê seria mais ou menos suscetível a
reações de degradação oxidativa? Explique.
BIBLIOGRAFIA
GERIS, R.; DOS SANTOS, N.A.C.; AMARAL, B.A.; MAIA, I.S.; CASTRO, V.D.; CARVALHO, J.R.M.
“Biodiesel de soja – reação de transesterificação para aulas práticas de química orgânica”. Quim.
Nova, 30(5): 1369-1373, 2007.
PAVIA D.L.; LAMPMAN, G.M.; KRIZ, G.S.; ENGEL, R.G. “Química Orgânica Experimental –
técnicas de escala pequena”. Editora Bookman, 2ª ed, São Paulo, 2009.
Revista Biodieselbr.
RINALDI, R.; GARCIA, C.; MARCINIUK, L.L.; ROSSI, A.V.; SCHUCHARDT, U. “Síntese de
biodiesel: uma proposta contextualizada de experimento para laboratório de química geral”. Quim.
Nova, 30(5): 1374-1380, 2007. 4 Nas condições utilizadas no experimento proposto, os biodieseis obtidos a partir de óleo de soja in natura possuem densidade de 0,877 g/mL a 25 oC.