australian public assessment report for sapropterin
TRANSCRIPT

Australian Public Assessment Report for Sapropterin dihydrochloride
Proprietary Product Name: Kuvan
Sponsor: Merck Serono Australia Pty Ltd
January 2011

Therapeutic Goods Administration
Copyright © Commonwealth of Australia 2011 This work is copyright. Apart from any use as permitted under the Copyright Act 1968, no part may be reproduced by any process without prior written permission from the Commonwealth. Requests and inquiries concerning reproduction and rights should be addressed to the Commonwealth Copyright Administration, Attorney General’s Department, National Circuit, Barton ACT 2600 or posted at http://www.ag.gov.au/cca
About the Therapeutic Goods Administration (TGA) · The TGA is a division of the Australian Government Department of Health and Ageing, and is
responsible for regulating medicines and medical devices.
· TGA administers the Therapeutic Goods Act 1989 (the Act), applying a risk management approach designed to ensure therapeutic goods supplied in Australia meet acceptable standards of quality, safety and efficacy (performance), when necessary.
· The work of the TGA is based on applying scientific and clinical expertise to decision-making, to ensure that the benefits to consumers outweigh any risks associated with the use of medicines and medical devices.
· The TGA relies on the public, healthcare professionals and industry to report problems with medicines or medical devices. TGA investigates reports received by it to determine any necessary regulatory action.
· To report a problem with a medicine or medical device, please see the information on the TGA website.
About AusPARs · An Australian Public Assessment Record (AusPAR) provides information about the evaluation of a
prescription medicine and the considerations that led the TGA to approve or not approve a prescription medicine submission.
· AusPARs are prepared and published by the TGA.
· An AusPAR is prepared for submissions that relate to new chemical entities, generic medicines, major variations, and extensions of indications.
· An AusPAR is a static document, in that it will provide information that relates to a submission at a particular point in time.
· A new AusPAR will be developed to reflect changes to indications and/or major variations to a prescription medicine subject to evaluation by the TGA.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 2 of 123

Therapeutic Goods Administration
Contents
I. Introduction to Product Submission .............................................................. 4 Submission Details........................................................................................................ 4 Product Background ..................................................................................................... 4 Regulatory Status .......................................................................................................... 6 Product Information ...................................................................................................... 6
II. Quality Findings .................................................................................................. 6 Drug Substance (active ingredient)............................................................................ 6 Drug Product .................................................................................................................. 7 Bioavailability ................................................................................................................. 7 Consideration by PSC ................................................................................................... 9 Quality Summary and Conclusions............................................................................ 9
III. Nonclinical Findings .......................................................................................... 9 Introduction..................................................................................................................... 9 Pharmacology .............................................................................................................. 10 Pharmacokinetics ........................................................................................................ 14 Toxicology ..................................................................................................................... 16 Nonclinical Summary and Conclusions .................................................................. 21
IV. Clinical Findings ............................................................................................... 23 Introduction................................................................................................................... 23 Pharmacology .............................................................................................................. 25 Pharmacokinetics ........................................................................................................ 27 Pharmacodynamics ..................................................................................................... 33 Efficacy .......................................................................................................................... 35 Safety ............................................................................................................................. 68 Clinical Summary and Conclusions ......................................................................... 79
V. Pharmacovigilance Findings .......................................................................... 83 Risk Management Plan ............................................................................................... 83
VI. Overall Conclusion and Risk/Benefit Assessment .................................... 87 Quality ............................................................................................................................ 87 Nonclinical .................................................................................................................... 88 Clinical ........................................................................................................................... 89 Risk Management Plan ............................................................................................... 98 Risk-Benefit Analysis ................................................................................................ 100 Outcome ...................................................................................................................... 106
Attachment 1. Product Information .................................................................... 107
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 3 of 123

Therapeutic Goods Administration
I. Introduction to Product Submission Submission Details Type of Submission New Chemical Entity
Decision: Approved Date of Decision: 21 October 2010
Active ingredient(s): Sapropterin dihydrochloride
Product Name(s): Kuvan Sponsor’s Name and Address:
Merck Serono Australia Pty Ltd Units 3-4, 25 Frenchs Forest Road East Frenchs Forest NSW 2086
Dose form(s): Soluble tablets Strength(s): 100 mg
Container(s): HDPE Bottle, LDPE Child-Resistant Closure Pack size(s): 30, 120, 240 tablets
Approved Therapeutic use: Kuvan is indicated for the treatment of hyperphenylalaninaemia (HPA) in sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency (see Dosage and Administration for definition of sapropterin responsiveness).
Route(s) of administration: Oral Dosage: The dosage instructions are complex – see Dosage and
Administration section of the Product Information (PI). ARTG Number: 165738
Product Background This is an application to register a new chemical entity, sapropterin dihydrochloride (Kuvan) 100 mg soluble tablets. Sapropterin is a synthetic form of the naturally occurring R-diastereoisomer of tetrahydrobiopterin (BH4). Hyperphenylalaninaemia (HPA) is defined as a chronic abnormal elevation in blood phenylalanine levels. Phenylalanine (Phe) is one of 8 essential amino acids that cannot be synthesised de novo in the human body. Physiological requirements for Phe are met exclusively by dietary protein intake. In addition, Phe is a necessary precursor for synthesis of tyrosine, which is considered a conditionally essential amino acid due to its dependence on Phe metabolism. Tyrosine, in turn, serves as precursor for neurotransmitter and thyroid hormone syntheses. Usual dietary intake of protein provides excess amounts of Phe and thus blood Phe levels are maintained within non-toxic levels via utilisation, metabolism and excretion.
Each of the hyperphenylalaninaemias results from the reduced activity of phenylalanine hydroxylase (PAH). In humans the complete enzyme system is expressed only in the liver. Phenylalanine and molecular oxygen are substrates and a reduced pteridine,
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 4 of 123

Therapeutic Goods Administration
tetrahydrobiopterin, is a cofactor. Tyrosine and dihydrobiopterin are the products of this catalytic system, with the dihydrobiopterin being reconverted to BH4 by a second enzyme, dihydropteridine reductase (DHPR). Since the conversion of Phe to tyrosine by PAH normally accounts for approximately 75% of dietary Phe disposal, disorders of Phe metabolism (for example, HPA) can cause abnormal elevations in blood Phe levels. Although such conditions are rare, severe HPA has serious clinical consequences for affected individuals, including severe neurocognitive delay and mental retardation, neuromotor disability and adverse pregnancy outcomes for affected women. Two genetic conditions, phenylketonuria (PKU) and BH4 deficiency, account for the majority of cases of clinically significant HPA. Both of these conditions are detectable in newborn screening programs. HPA in both conditions can be controlled by dietary restriction of whole protein, with concomitant administration of commercial Phe-free protein supplements to provide adequate nutritional intake of protein. Although this dietary therapy has proven to be beneficial in lowering Phe levels and preventing the severe neurological consequences of HPA, most affected individuals are unable to maintain adequate control of blood Phe levels with diet therapy since the severe restriction of dietary protein is impractical in daily life. In addition, the Phe-free protein supplements, which are necessary to maintain protein nutrition, are widely acknowledged to be unpalatable. PKU, a rare disorder in the general population, is one of the most common clinically significant inborn errors of metabolism with an estimated worldwide live birth prevalence (population incidence) of approximately 1 in 10,000. The 2006 Orphan Drug designation application for sapropterin estimated the overall population prevalence for HPA in Australia, including PKU, to be 1,610 cases or approximately 0.8 in 10,000. PKU is an autosomal recessive condition caused by deficient activity of PAH, the enzyme which metabolises Phe to tyrosine. Blood levels in non-PKU populations have an approximate range between 25 and 100 µmol/L, with variability related to age, Phe intake and assay methodology. In individuals with PKU, blood Phe levels can range above 2000 µmol/L, depending on the degree of PAH deficiency and dietary Phe intake. BH4 deficiency, a very rare inborn error of metabolism, is estimated to account for 1-2% of cases of HPA. Like PKU, BH4 deficiency is an autosomal recessive genetic condition. However, unlike PKU in which a single enzyme (that is, PAH) is involved, BH4 deficiency can result from mutations or deletions in any of the genes controlling the five different enzymes involved in BH4 synthesis and regeneration.1
1 Examples include dihydropteridine reductase (DHPR) deficiency, 6-pyruvoyltetrahydrobiopterin synthase
(PTPS) deficiency and guanosine triphosphate cyclohydrolase (GCHI) deficiency, all autosomal recessive conditions.
Usual dietary intake of BH4 is physiologically insignificant. Instead daily requirements for BH4 are met either via BH4 synthesis or regeneration. As mentioned earlier, BH4 is a necessary co-factor for PAH. Therefore, BH4 deficiency impairs PAH activity leading to a biochemical situation similar to PKU, with HPA resulting from deficient conversion of Phe to tyrosine. In addition, BH4 is also a necessary co-factor for both the enzymes tyrosine hydroxylase and tryptophan hydroxylase and so a deficiency in BH4 will lead to deficiencies downstream in the neurotransmitter products of both tyrosine and tryptophan including catecholamines like L-dopa and noradrenaline from tyrosine and serotonin from tryptophan. Based on prevalence data from the Orphan Drug designation application, 16 to 32 patients are estimated to suffer from this disorder in Australia.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 5 of 123

Therapeutic Goods Administration
The indication proposed in the application was: Kuvan is indicated for the treatment of Hyperphenylalaninaemia (HPA) in sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency. Regulatory Status Sapropterin received TGA designation as an Orphan Drug for the treatment of patients with Hyperphenylalaninaemia (HPA) in August 2006.
Kuvan was approved in the USA on 13 December 2007. The approved indication is as follows:
Kuvan is indicated to reduce blood phenylalanine (Phe) levels in patients with hyperphenylalaninemia (HPA) due to tetrahydrobiopterin – (BH4-) responsive phenylketonuria (PKU). Kuvan is to be used in conjunction with a Phe-restricted diet. Kuvan was approved in the European Union (EU) on 2 December 2008. The approved indication is as follows: Kuvan is indicated for the treatment of hyperphenylalaninemia (HPA) in adult and paediatric patients of 4 years of age and over with phenylketonuria (PKU) who have been shown to be responsive to such treatment.
Kuvan is also indicated for the treatment of hyperphenylalaninemia (HPA) in adult and paediatric patients with tetrahydrobiopterin (BH4) deficiency, who have been shown to be responsive to such treatment. Product Information The approved product information (PI) current at the time this AusPAR was prepared can be found as Attachment 1.
II. Quality Findings
Drug Substance (active ingredient) This submission is to register Kuvan soluble tablets which contain 100 mg of sapropterin dihydrochloride. This drug substance is a new chemical entity that is chemically synthesised. However the free base occurs naturally where it is called R-tetrahydrobiopterin. R-tetrahydrobiopterin is a cofactor of the hydroxylases for phenylalanine, tyrosine and tryptophan. There are no compendial monographs for drug substance or finished products containing this drug substance. The drug substance has three chiral centres but is presented as a single enantiomer. It can exist in two conformations.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 6 of 123

Therapeutic Goods Administration
(6R)-2-amino-6[(3R,2S)-dihydroxypropyl]-4-oxo-5,6,7,8-tetrahydropteridine dihydrochloride C9H15N5O3.2HCl (C9H15N5O3 for free base) Molecular mass = 314.17 (241.21 for free base) CAS # = [69056-38-8] ([62989-33-7] for free base) pKa = 1.2 (NH at position 1), 5.0 (NH at position 5) and 10.3 (carbonyl at position 4) aqueous solubility = 2 mg/mL (0.2 %w/v, slightly soluble) to give pH 2.3 Biopharmaceutical Classification System (BCS) Class 3 The synthesis starts from the sugar L-rhamnose with known stereochemistry.
The drug substance is prone to oxidation and hydrolysis and, once manufactured, it is stored in double low density polyethylene (LDPE) bags under nitrogen (with a desiccant between the layers and an external foil pouch).
Thirteen polymorphic forms, hydrates and solvates have been identified, but anhydrate Form B is thermodynamically stable and is the form obtained. As the drug substance is dissolved prior to ingestion neither polymorphic form nor particle size distribution are critical parameters. The specifications of sapropterin dihydrochloride (referred to as sapropterin in the remainder of this AusPAR) drug substance include satisfactory limits for assay, most related substances and the residual solvents and the platinum catalyst were limited to at or below the limits allowed by the International Council of Harmonisation (ICH) guidance.
However, the limit for the synthetic impurity S-tetrahydrobiopterin (S-BH4) is above the ICH qualification threshold of 0.15% and was referred to the Delegate.2
Drug Product
The soluble tablets are to be manufactured and packaged by Lyne Laboratories in Massachusetts USA. The process involves simply dry granulation and compression. The bulk soluble tablets can be stored for 12 months prior to bottling and data to support this storage was provided. The tablets were found to undergo a humidity and temperature dependent oxidation on storage. In order to minimise this oxidation, the product is to be supplied in bottles containing a desiccant and the storage conditions ‘Keep the bottle tightly closed in order to protect from moisture’ and ‘Product should be used within 2 months of first opening the bottle’. The specifications have acceptable expiry limits and release limits that allow for the change over the shelf life.
Stability data was provided to support an unopened shelf life of 2 years when stored below 25ºC in the bottles containing desiccant and an in-use shelf life of 2 months.
Bioavailability Clinical Background
Some of the Phase III clinical efficacy studies were performed with the formulation soluble tablet proposed for registration but most were performed with a slightly different formulation in that the amount of mannitol was very slightly different and the amount of sodium stearyl fumarate was about two thirds. The amounts of the other excipients were the same in both tablets.
2 Qualification is the process of acquiring and evaluating data that establishes the biological safety of an
individual impurity or a given impurity profile at the level(s) specified.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 7 of 123

Therapeutic Goods Administration
As the product is fully dissolved before ingestion, it was accepted that these differences in formulations would not affect bioavailability.
The evaluator believed that dosing during the Phase III clinical efficacy studies used the tablets dissolved in water given with food. Studies submitted
The submission included one bioavailability study to compare the bioavailability with and without food and with and without orange juice. Study PKU-005 was a four-way cross-over study in 28 subjects (27 completed) with the following treatments.
(a) 100 mg tablet dissolved in water/fasted (b) 100 mg tablet dissolved in water/fed (c) 100 mg tablet dissolved in orange juice/fasted (d) 100 mg tablet dissolved in orange juice/fed The study was of an appropriate design and the test method used to determine the levels of sapropterin in the subject plasma samples was satisfactorily validated for this purpose. The major Phase III clinical efficacy formulation was used.
Table 1: Pharmacokinetic results from Study PKU-005
The results of study PKU-005 (Table 1) indicate that: - Dissolving in orange juice rather than water does not change the bioavailability when
both are administered in the fasted state (bioequivalence was observed). - Food increases the bioavailability by ~85% when the product is given dissolved in water
and by ~50% when the product is dissolved in orange juice.
Justification for Not Providing Bioavailability Data
The sponsor did not perform a bioavailability study to determine the absolute bioavailability in humans. Therefore the evaluator did not view any assessment of the pharmacokinetic
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 8 of 123

Therapeutic Goods Administration
parameters of clearance or volume of distribution. The sponsor noted that in animals the absolute bioavailability was 10%. This was brought to the attention of the Delegate. Consideration by PSC Details of this submission were presented at the 131st meeting of the Pharmaceutical Subcommittee (PSC) of the Advisory Committee on Prescription Medicines (ACPM) in March 2010. The PSC: - Concluded that an absolute bioavailability study was not required for this product. - Concluded that it should be recommended that all dosing occur with food. This is the
case. Quality Summary and Conclusions Approval of this submission was not recommended with respect to the chemistry and quality control pending review of the limit for the synthetic impurity S-tetrahydrobiopterin by the Delegate. Food increases the bioavailability by ~85% when the product is given dissolved in water and by ~50% when the product is dissolved in orange juice. It was recommended that the PI states that all dosing occur with food and this is indeed the case.
The formulation proposed for supply can be considered bioequivalent to the formulation used in the majority of the Phase III clinical efficacy studies.
The justification for the lack of an absolute bioavailability study has been accepted but this means that details relating to clearance and volume of distribution have not been included in the PI.
III. Nonclinical Findings Introduction This data package consisted of studies conducted in the 1980s, as well as a few more recent studies. The number of nonclinical primary pharmacology studies was limited. A recent review of the nonclinical literature had not been conducted by the sponsor, so important publications relevant to the primary and secondary pharmacology were not included with this submission. Studies submitted included safety pharmacology studies, single dose and repeat dose toxicity studies, genotoxicity studies, mouse and rat carcinogenicity studies, reproductive toxicity studies in rats and rabbits and juvenile toxicity studies. Most of the pivotal toxicity studies were Good Laboratory Practice (GLP) compliant, although not all, and individual animal data were provided in all pivotal studies. There were no concurrent toxicokinetic data in the pivotal toxicity studies. Moreover, there were extremely limited pharmacokinetic data available for marmosets, one of the pivotal species in the repeat dose toxicity studies, and pharmacokinetic data were only available for one strain of rat, whereas several different strains of rat were used in pivotal toxicity studies. Pharmacokinetic data in children were not provided, making exposure ratios for the paediatric population extremely difficult to estimate.
The sponsor provided the European Medicines Authority (EMA) European Public Assessment Report (EPAR) and an edited FDA report. These were consulted in the preparation of the nonclinical evaluation report.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 9 of 123

Therapeutic Goods Administration
Pharmacology Primary pharmacodynamics
Sapropterin is indicated for the treatment of hyperphenylalaninaemia (HPA) in two patient populations: adult and paediatric patients with tetrahydrobiopterin (BH4) deficiency and sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU). HPA occurs when there is a low level of phenylalanine (Phe) hydroxylase activity; because this is the major clearance pathway for Phe (about 75% of Phe is usually converted to tyrosine). High plasma levels of Phe result in high brain levels of Phe because Phe is transported across the blood brain barrier by a transporter that transports neutral amino acids. Because Phe hydroxylase requires the cofactor BH4, this enzyme has low activity both when the enzyme itself has been mutated, and when there are low levels of cofactor present. Sapropterin administration in BH4-deficient patients is a replacement therapy, whereas sapropterin administration in PKU patients is not a replacement therapy.
BH4 deficiency The usual dietary intake of BH4 is insignificant, with daily requirements met by synthesis and regeneration. Five enzymes are involved in BH4 synthesis and regeneration, and defects in both copies of the gene for one of these enzymes results in BH4 deficiency. In addition to Phe hydroxylase, two other amino acid hydroxylases also require this cofactor: tyrosine hydroxylase (present in the central nervous system [CNS], peripheral sympathetic neurons and the adrenal medulla) and tryptophan hydroxylase. Tryptophan hydroxylase 1(TPH1) is expressed mainly in the intestinal enterochromaffin cells and the pineal body, with lower amounts in adult brain; TPH2 is mainly expressed in the serotonergic neurons of the raphe nuclei in the brain and serotonergic neurons of the gut. Tryptophan hydroxylation is the rate limiting step in the production of the neurotransmitter serotonin and tyrosine hydroxylation is the rate limiting step in the production of the neurotransmitters dopamine, noradrenaline and adrenaline. Defective activity of tryptophan and tyrosine hydroxylases explains the neurological deterioration in patients with BH4 deficiency even when Phe levels are controlled. Because BH4 is required in the brain where it is usually synthesised, the distribution of plasma BH4 to the brain is relevant, and in adults is low. Thus concomitant treatment with neurotransmitters is usually required in BH4 deficient patients. In addition to the amino acid hydroxylases, the three isoforms of nitric oxide synthase (nNOS, iNOS and eNOS) require BH4 as a cofactor, as does glyceryl-ether monooxygenase (involved in lipid metabolism for which physiological relevance has not yet been established). Several animal models for BH4 deficiency exist. The sponsor submitted two studies in which animals (rats and guinea pigs) received DAHP (2,4-diamino-6-hydroxy-pyrimidine), an inhibitor of GCH1 (GTP cyclohydrolase 1, the first and rate limiting step in the biosynthetic pathway for BH4). In the rat study sapropterin (at all doses tested, ≥5 mg/kg) restored plasma Phe levels to control levels, whereas in the guinea pig 20 mg/kg sapropterin partially restored plasma Phe levels. Phe levels in the fetuses of pregnant guinea pigs were also lowered. The majority of BH4 deficient patients do not have mutations in GCH1. About 90% have mutations that affect either the PTPS gene (PTPS: 6-pyruvoyl-BH4 synthase, the second enzyme in the biosynthetic pathway) or the dihydropteridine reductase gene (DHPR, which affects part of the recycling pathway). Thus the studies involving the inhibition of GCH1 are not an ideal animal model for BH4 deficiency in patients, although the general principle of restoring plasma Phe levels by replacing the missing cofactor has been demonstrated. In addition to the studies submitted by the sponsor, there are studies in the literature that have used BH4 deficient mice with a knock-out of PTPS, the most common deficiency in patients.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 10 of 123

Therapeutic Goods Administration
PTPS-deficient mice died within 48 hours of birth unless BH4 treatment occurred.3
In conclusion, sapropterin administration at sufficiently high doses can result in plasma Phe levels similar to controls in BH4-deficient animals. However, because little sapropterin crosses the blood-brain barrier in adult animals, sapropterin administration alone is insufficient to overcome the full effects of this deficiency.
Groups treated with 10 or 60 mg/kg/day BH4 orally (PO) (in combination with L-dopa, serotonin, carbidopa, ascorbic acid and N-Acetyl-cysteine) still had higher than control levels of plasma Phe and died within the first 40 days. Treatment with 122 mg/kg BH4 PO (+ L-dopa, serotonin, carbidopa, ascorbic acid and N-Acetyl-cysteine) resulted in plasma Phe levels the same as wild type. These treated mice were healthy at 6 weeks of age, although growth had stopped at 23 days and brain dopamine and serum IGF-1 levels were low. These knockout mice clearly had a far greater deficiency in BH4 than BH4-deficient patients. Plasma Phe levels could be restored in these mice but at much higher doses than the proposed clinical doses. This animal model is not ideal because the BH4 deficiency in these animals is total, whereas it is likely that BH4-deficient patients have some residual enzyme activity. Nevertheless, these studies have shown that BH4 replacement therapy, in combination with neurotransmitter precursors, can prevent HPA and mortality in these animals.
Phenylketonuria (PKU) Phenylketonuria is an autosomal recessive disease for which 75% of patients are compound heterozygotes. It is associated with more than 500 identified mutations in the Phe hydroxylase gene, with about two thirds of these being missense mutations. The original evidence for the efficacy of sapropterin administration in PKU was clinical. However, not all patients with PKU respond to sapropterin which is why the proposed indication is for “sapropterin-responsive” patients with PKU.
A number of hypotheses have been proposed to explain how increased cofactor concentrations result in an increase in Phe hydroxylase activity. The natural BH4 concentration (5-9 mM in liver) appears to be subsaturating in vivo both with respect to the enzyme concentration (about 9 mM) and the cofactor Km (about 20 mM).4 However, although the classical view of this disease is as a loss of enzyme function, sapropterin-responsive PKU is now being viewed as a protein misfolding disease. Phe hydroxylase is a three domain protein that is organised as an asymmetric dimer of dimers. This complicated structure permits fine-tuned regulation involving substrate activation, modulation of oligomerisation and modulation of the affinity for the substrate and the cofactor. However, this structural flexibility may render Phe hydroxylase particularly susceptible to protein misfolding. Using the available partial crystal structures of Phe hydroxylase, the energetic impact (DDG) of all missense PKU mutations was calculated.5
3 Elzaouk L, Leimbacher W, Turri M, Ledermann B, Bürki K, Blau N, Thöny B. Dwarfism and low insulin-like
growth factor-1 due to dopamine depletion in Pts-/- mice rescued by feeding neurotransmitter precursors and H4-biopterin. J Biol Chem 2003; 278: 28303-28311.
There was a good correlation between the calculated structural effects on the Phe hydroxylase native state with the severity of the phenotype. This correlation was better than the correlation of residual enzyme activity in vitro and phenotype severity.
4 Teigen K, McKinney JA, Haavik J, Martinez A. Selectivity and affinity determinants for ligand binding to the aromatic acid hydroxylases. Curr Med Chem 2007; 14: 455-467.
5 Pey AL, Stricher F, Serrano L, Martinez A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome of phenylketonuria, a paradigm of misfolding disease. Am J Hum Gen 2007; 81: 1006-1024.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 11 of 123

Therapeutic Goods Administration
The effects of about 100 mutations on Phe hydroxylase function and stability have been identified by using in vitro expression (compiled in the PAH locus knowledgebase, PAHdb). A large proportion of all missense mutations studied, including most of the common clinical mutations display stability and folding defects when expressed in vitro. In a more detailed study, ten sapropterin-responsive mutations were investigated by expression as fusion proteins with MBP (maltose binding protein) in E coli.6
The sponsor indicated that none of the three available mouse models of PKU (Pahenu1, Pahenu2, Pahenu3) were suitable for pharmacodynamic studies of sapropterin in PKU. However, in a recent publication, the effect of sapropterin administration to the Pahenu1 mouse was investigated.
The mutant enzymes still had considerable residual activity and the Km for BH4 was increased (3-fold) for only one of the ten variants. However, allostery was altered in nine of ten variants and oligomerisation was affected in five of the ten variants with a greater proportion of dimers, monomers or aggregates than the wild-type protein. Increased susceptibility to proteolysis with proteinase K was observed for nine of the ten mutants and the results of thermal denaturation experiments showed that there was substantial distortion in the protein conformation, with particular impact on the regulatory domain. All ten of these sapropterin-responsive variants either caused protein misfolding or caused milder conformational changes that still resulted in deleterious effects on enzyme function.
7
Secondary pharmacodynamics
The Pahenu1 mouse has a mutation in the regulatory domain of Phe hydroxylase (V106A) that leads to mild hyperphenylalaninaemia. The sponsor did not consider this mild phenotype to be sufficient to provide a suitable model for PKU. However, in the recent paper it has been shown that the V106A mutation leads to a complete loss of allostery. The mutant was more hydrophobic than the wild-type protein but this hydrophobicity was significantly reduced in the presence of BH4. BH4 stabilised both the mutant and the wild-type protein at high Phe concentrations. In the Pahenu1 mouse, BH4 intraperitoneally (IP) (40 mg/kg/day for 3 days) significantly decreased plasma Phe levels. Thus, this is an animal model of PKU in which sapropterin administration did reduce hyperphenylalaninaemia. However, the route was IP not oral and the hyperphenylalaninaemia being corrected was considered mild. Hence, the nonclinical data was consistent with the efficacy of sapropterin for treating some cases of PKU but evidence for efficacy relied on clinical data.
The sponsor briefly reviewed some of the literature concerning secondary pharmacodynamic effects of sapropterin in multiple animal models of cardiovascular diseases with underlying endothelial dysfunction (associated with BH4 deficiency). In these models BH4 administration appeared to restore endothelial function and reduce oxidative stress, as would be expected from the stabilising effect that BH4 has on eNOS. However, it is important to note that data relevant to such indications have not been fully evaluated in this report. There is nonclinical evidence that BH4 and nitric oxide increase neuropathic pain but have little effect on acute pain. Mice deficient in NO-sensitive guanyl cyclase (GC-KO mice) exhibited considerably reduced nociceptive behaviour in inflammatory and neuropathic pain
6 Gersting SW, Kemter KF, Staudigl M et al. Loss of function in phenylketonuria is caused by impaired
molecular motions and conformational instability. Am J Hum Gen 2008; 83: 5-17. 7 Gersting SW, Lagler FB, Eichinger A et al. Pahenu1 is a mouse model for tetrahydrobiopterin-responsive
phenylalanine hydroxylases deficiency and promotes analysis of the pharmacological chaperone mechanism in vivo. Hum Mol Genet 2010; 19: 2039-2049.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 12 of 123

Therapeutic Goods Administration
models (Schmidtko et al, 2008).8
· Mechanical and cold pain hypersensitivity were reversed following spared nerve injury surgery;
Inhibiting BH4 synthesis by two different means (inhibition of GTP cyclohydrolase 1 or sepiapterin reductase) resulted in similar effects on pain:
· Analgesia was induced in the chronic constriction injury and spinal ligation models of peripheral neuropathic pain;
· Heat hyperalgaesia was reduced following complete Freund’s adjuvant-induced paw inflammation;
· Mechanical and cold allodynia were reduced; · Thermal hyperalgaesia was reduced in an inflammation model.
In addition, BH4 enzymes were shown to be upregulated in rats following spared nerve injury, with a corresponding increase in BH4 levels. Intrathecal administration of BH4 caused a rapid and long-lasting increase in the response to noxious radiant heat and increased pain sensitivity in both the spared nerve injury and complete Freund’s adjuvant models of neuropathic and inflammatory pain (Naylor et al, 2010 and Tegeder et al, 2006).9,10
In addition to the above nonclinical evidence for the increased pain caused by higher intrathecal levels of BH4 there is also clinical evidence. A GTP cyclohydrolase 1 pain protective haplotype has been discovered in humans (Tegeder et al, 2006). Constitutive expression of GTP cyclohydrolase and BH4 production was equivalent in carriers and non-carriers of the pain-protective haplotype but forskolin-evoked upregulation was significantly smaller in carriers of the pain-protective haplotype. These carriers had significantly less pain following diskectomy for persistent radicular low back pain and healthy carriers had reduced experimental pain sensitivity.
Thus, there is considerable evidence that increased intrathecal levels of BH4 result in increased levels of neuropathic pain in animals and humans. A risk of sapropterin treatment, therefore, is increased levels of neuropathic and inflammatory pain. In support of this is the observation that headaches were a very common adverse event in clinical trials. However, the nonclinical evidence indicates that sapropterin is not markedly distributed to the brain in adult animals. In contrast, sapropterin is distributed to the brain to a much greater extent in juvenile rats. Thus, as the blood-brain barrier is not fully developed until 12 months of age, the risk of increased pain is higher in infants. Safety pharmacology
Safety pharmacology studies were conducted with adult animals. In mice, 300 mg/kg PO prolonged thiopental-induced sleep and at doses ranging from 100 to 600 mg/kg PO sapropterin there were antidepressive, anxiolytic and ameliorative effects on memory and learning disorder behavioural tests in rodents. The effects of BH4 levels on brain development and function in juvenile animals have not been investigated and are therefore unknown. 8 Schmidtko A, Gao W, König P et al. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neuroscience 2008; 28: 8568-8576. 9 Naylor AM, Pojasek KR, Hopkins AL, Blagg J. The tetrahydrobiopterin pathway and pain. Curr Op Invest Drugs 2010; 11: 19-30. 10 Tegeder I, Costigan M. Griffin RS et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med 2006; 12:1269-1277.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 13 of 123

Therapeutic Goods Administration
Sapropterin did not inhibit the hERG potassium channel in an in vitro study and had no effect on dog cardiovascular parameters at plasma concentrations up to 8663 ng/mL, 88-fold the maximal plasma concentration (Cmax) in humans (99 ng/mL). Pharmacodynamic interactions
BH4 stabilises eNOS, which produces NO. Nitric oxide binds to the haem group of guanylate cyclase and increases cGMP production. Sildenafil citrate inhibits phosphodiesterase 5 (PDE5), which catabolises cGMP to GMP. There is a theoretical possibility that sapropterin might have an additive or synergistic effect with sildenafil leading to high cGMP levels. cGMP can induce systemic and pulmonary vasorelaxation. An additive effect might lead to hypotension and an unsafe reduction in blood pressure. To investigate this possibility the effect of combining sapropterin and sildenafil on respiratory parameters was studied in rats and, on cardiovascular parameters, in dogs. In these studies, sapropterin did not exhibit any effects on respiratory or cardiovascular parameters in the presence or absence of sildenafil. Pharmacokinetics Measuring BH4 concentrations is difficult because the redox ratio reflects how much is in the oxidised form and how much in the reduced form. Therefore all BH4 concentrations have to be calculated using this ratio. In addition, all recent measurements (including those in humans) have been conducted using liquid chromatography/mass spectrometry (LC/MS/MS), whereas the older experiments (mostly in rats) used high pressure liquid chromatography (HPLC) and a fluorometric detection method. Comparing measurements using the modern method with measurements using the older method is likely to have resulted in a degree of error.
The pharmacokinetic data provided were limited. Bioavailability data were available for juvenile and adult rats and cynomolgus monkeys but not in any other species, including humans. Bioavailability was similar between adult rats and monkeys (about 10%) but was quite different for juvenile rats (about 50%). Data for the area under the plasma concentration time curve (AUC) is available for mice, rats, cynomolgus monkeys and human adults but not for marmosets (the non-rodent species in repeat dose toxicity studies) or in children. In addition, the AUC data that is available was not collected concurrently with toxicity studies, this was only available for one strain of rat (Sprague-Dawley), whereas the 52-week repeat dose toxicity study, carcinogenicity study and reproductive toxicity studies were all conducted with different strains. Hence the exposure ratios have been calculated using estimates of the AUC values, not AUC values measured in the correct strain at the same time as the study was conducted.
Sapropterin did not bind to plasma proteins to any great extent. Despite endogenous BH4 levels being high in a number of tissues, administered sapropterin levels accumulated mostly in the kidney and liver, and slightly in the adrenal glands and spleen in rats. A number of studies investigated the distribution of sapropterin to the brain. Sapropterin and/or its metabolites were distributed to the brain in juvenile rats but little sapropterin was demonstrated to be distributed to the brain in adult rats.
The metabolism of BH4 is similar in rats and humans. The sponsor provided diagrams of the established pathways. In addition, a study was conducted to quantify the metabolites excreted in rat urine after intravenous (IV) administration of sapropterin. No attempt was made to determine the metabolic pathway in marmosets. In marmosets a bright yellow compound was found on the cage tray paper. A similar coloured compound was also found on the tray paper in a study with 3-week old rats. The sponsor’s response to a TGA enquiry suggested that the
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 14 of 123

Therapeutic Goods Administration
bright yellow compound was not a metabolite but a degradation product, given that the daily clinical observations occurred several hours after dosing.
While most of the orally administered sapropterin is excreted in the faeces, the absorbed sapropterin is either excreted in the urine unchanged, or metabolised and then excreted in the urine. Relative exposure
Adequate toxicokinetic data were not collected in any study. The pharmacokinetic data for mice and pregnant rabbits were measured using the same method as that used for humans (LC-MS-MS, with the redox ratio taken into account). However, the limited pharmacokinetic data available for rats used an older method (fluorometric HPLC), the redox ratio was not been taken into account. (The redox ratio according to the HPLC method differed from that according to the LC-MS-MS method.). In addition, the strain of rat used to collect the pharmacokinetic data (Sprague Dawley) differed from that used in the carcinogenicity studies (F-344) and the reproductive toxicity studies (Wistar). No data were collected using pregnant rats.
It is considered that the estimated AUC values give a more accurate estimate of the animal:human exposure ratios than the body surface area (BSA) estimates. This is because, for both mice and rats, the estimated AUC values give considerably higher exposure ratios than the BSA calculations. The mouse estimate used the correct strain of mouse and the same method that was used clinically. The toxicokinetic data available for marmosets were very limited so no AUC values can be calculated. Thus, the animal:human exposure ratios for the marmosets are based on BSA. The body weight of the marmosets was about 360 g, so a conversion factor of 6 kg/m2 is appropriate for calculating exposure based on body surface area. The calculated exposure ratios are summarised in Table 2. No toxicokinetic data were collected in the rabbit embryofetal development study. The only relevant pharmacokinetic data come from a study conducted almost two decades later in a different laboratory. One pregnant rabbit receiving 600 mg/kg/day in the pharmacokinetic study lost a considerable amount of weight during the dosing period and this rabbit had a higher exposure to sapropterin at the end of the study than the other rabbits. Body weight loss was not observed in the embryofetal development study so this animal, at gestation day (GD) 18, is considered to have been exposed to higher levels of sapropterin than were achieved in the earlier study. In order to estimate exposure to sapropterin in the earlier study, it is therefore considered more appropriate to use the lower concentrations recorded at the first time point (GD 6). This gives a conservative estimate of exposure because sapropterin levels at the high dose (600 mg/kg/day) at the end of the dosing period were similar to or higher than the levels achieved after the first dose.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 15 of 123
Therapeutic Goods Administration
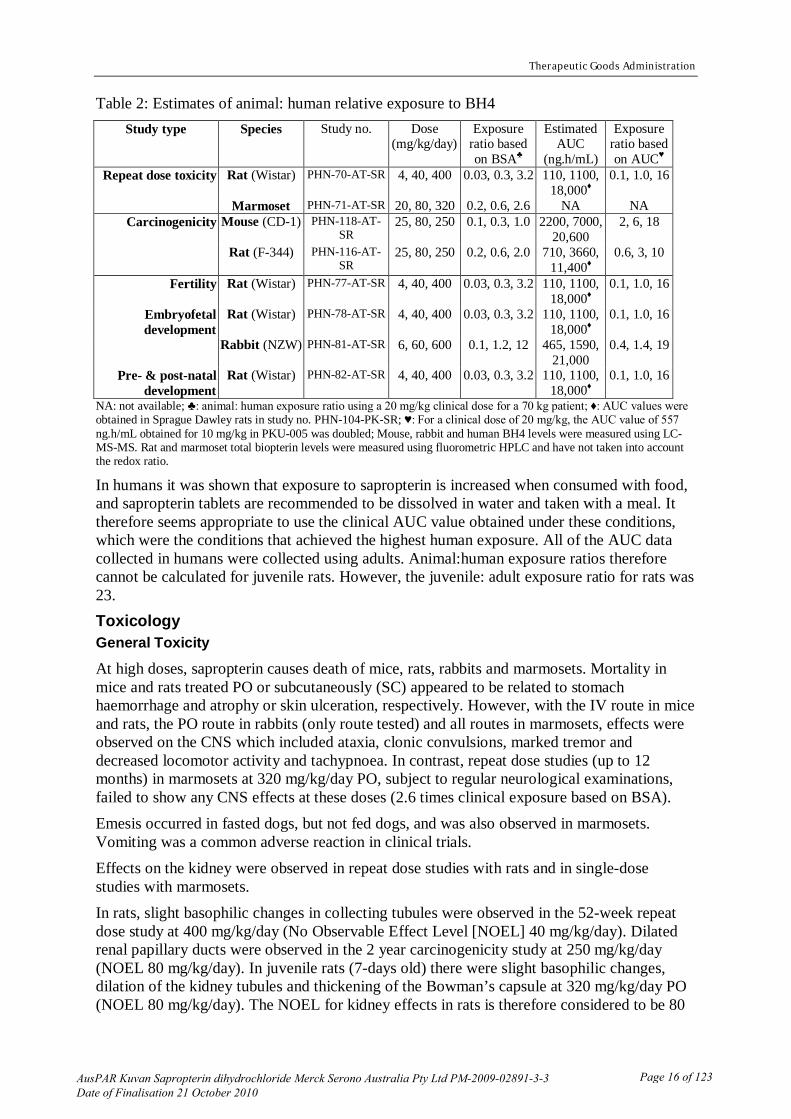
Table 2: Estimates of animal: human relative exposure to BH4 Study type Species Study no. Dose
(mg/kg/day) Exposure
ratio based on BSA♣
Estimated AUC
(ng.h/mL)
Exposure ratio based on AUC♥
Repeat dose toxicity Rat (Wistar) PHN-70-AT-SR 4, 40, 400 0.03, 0.3, 3.2 110, 1100, 18,000♦
0.1, 1.0, 16
Marmoset PHN-71-AT-SR 20, 80, 320 0.2, 0.6, 2.6 NA NA Carcinogenicity Mouse (CD-1) PHN-118-AT-
SR 25, 80, 250 0.1, 0.3, 1.0 2200, 7000,
20,600 2, 6, 18
Rat (F-344) PHN-116-AT-SR
25, 80, 250 0.2, 0.6, 2.0 710, 3660, 11,400♦
0.6, 3, 10
Fertility Rat (Wistar) PHN-77-AT-SR 4, 40, 400 0.03, 0.3, 3.2 110, 1100, 18,000♦
0.1, 1.0, 16
Embryofetal development
Rat (Wistar) PHN-78-AT-SR 4, 40, 400 0.03, 0.3, 3.2 110, 1100, 18,000♦
0.1, 1.0, 16
Rabbit (NZW) PHN-81-AT-SR 6, 60, 600 0.1, 1.2, 12 465, 1590, 21,000
0.4, 1.4, 19
Pre- & post-natal development
Rat (Wistar) PHN-82-AT-SR 4, 40, 400 0.03, 0.3, 3.2 110, 1100, 18,000♦
0.1, 1.0, 16
NA: not available; ♣: animal: human exposure ratio using a 20 mg/kg clinical dose for a 70 kg patient; ♦: AUC values were obtained in Sprague Dawley rats in study no. PHN-104-PK-SR; ♥: For a clinical dose of 20 mg/kg, the AUC value of 557 ng.h/mL obtained for 10 mg/kg in PKU-005 was doubled; Mouse, rabbit and human BH4 levels were measured using LC-MS-MS. Rat and marmoset total biopterin levels were measured using fluorometric HPLC and have not taken into account the redox ratio.
In humans it was shown that exposure to sapropterin is increased when consumed with food, and sapropterin tablets are recommended to be dissolved in water and taken with a meal. It therefore seems appropriate to use the clinical AUC value obtained under these conditions, which were the conditions that achieved the highest human exposure. All of the AUC data collected in humans were collected using adults. Animal:human exposure ratios therefore cannot be calculated for juvenile rats. However, the juvenile: adult exposure ratio for rats was 23. Toxicology General Toxicity
At high doses, sapropterin causes death of mice, rats, rabbits and marmosets. Mortality in mice and rats treated PO or subcutaneously (SC) appeared to be related to stomach haemorrhage and atrophy or skin ulceration, respectively. However, with the IV route in mice and rats, the PO route in rabbits (only route tested) and all routes in marmosets, effects were observed on the CNS which included ataxia, clonic convulsions, marked tremor and decreased locomotor activity and tachypnoea. In contrast, repeat dose studies (up to 12 months) in marmosets at 320 mg/kg/day PO, subject to regular neurological examinations, failed to show any CNS effects at these doses (2.6 times clinical exposure based on BSA). Emesis occurred in fasted dogs, but not fed dogs, and was also observed in marmosets. Vomiting was a common adverse reaction in clinical trials. Effects on the kidney were observed in repeat dose studies with rats and in single-dose studies with marmosets. In rats, slight basophilic changes in collecting tubules were observed in the 52-week repeat dose study at 400 mg/kg/day (No Observable Effect Level [NOEL] 40 mg/kg/day). Dilated renal papillary ducts were observed in the 2 year carcinogenicity study at 250 mg/kg/day (NOEL 80 mg/kg/day). In juvenile rats (7-days old) there were slight basophilic changes, dilation of the kidney tubules and thickening of the Bowman’s capsule at 320 mg/kg/day PO (NOEL 80 mg/kg/day). The NOEL for kidney effects in rats is therefore considered to be 80
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 16 of 123

Therapeutic Goods Administration
mg/kg/day (exposure ratio of 3 based on AUC). Nephrosis of the distal and proximal convoluted tubule was observed at 2000 mg/kg PO sapropterin in marmosets, and dilation of the distal tubule occurred at 1000 mg/kg PO. However, no remarkable histopathology of the kidney was observed in the 52 week repeat dose toxicity study in marmosets at 320 mg/kg/day PO. Although the Lowest Observable Effect Levels (LOELs) for these effects occurred at reasonable exposure margins, the exposure margins for the NOELs are small. The sponsor’s proposal to place a warning statement about nephrotoxicity in the Product Information document is considered appropriate. However, the renal damage was slight in lifelong rodent studies so, for adult patients, the risk is considered minimal. For young infants the risk of toxicity is considered to be higher. This is discussed in the Use in children section below. Genotoxicity
The original genotoxicity studies were conducted in the 1980s. Following advice from the EMA, the sponsor conducted additional in vitro genotoxicity studies in 2007. All of the in vitro genotoxicity studies were GLP-compliant. Sporadic effects were obtained in all in vitro genotoxicity studies conducted. In the recent bacterial reverse mutation assays there were positive results in two strains in the absence of metabolic activation but negative results in the presence of metabolic activation. In contrast, an increase in chromosomal aberration was observed in the presence of metabolic activation in both Chinese hamster ovary and lung cells, whereas changes, in the absence of metabolic activation, were only observed in Chinese hamster lung cells.
The sponsor and study authors claimed that the results of the human lymphocyte assay were negative. The study authors claimed that the only statistically significant result was a statistical anomaly due to there being 0% cells with chromosomal aberrations in the vehicle controls. However, the historical control data supplied with the report indicate that a 0% value is quite common. As the positive result obtained (4.5%) is more than two standard deviations above the maximum recorded level for a negative control, this single result is therefore considered to be positive. The combined results of this assay are considered equivocal.
Overall, there were no clear negative results in any of the in vitro genotoxicity tests conducted. As an explanation for the positive results observed in the in vitro genotoxicity assays the sponsor cited Thoeni et al 2004.11
11 Thoeni G, Werner ER, Werner-Felmayer G. Tetrahydopteridines suppress gene expression and induce
apoptosis of activated RAW264.7 cells via formation of hydrogen peroxide. Free Radical Biol Med 2004; 37: 375-384.
In this paper, BH4 was added to mouse macrophage cell cultures (RAW264.7 cells) and exhibited a dose-dependent cytotoxic (apoptotic) effect. This apoptotic effect did not occur when sepiapterin was added. Extracellular sepiapterin resulted in a much higher increase in intracellular BH4 than did extracellular BH4. It was therefore concluded that the cytotoxicity was caused by extracellular BH4, not intracellular BH4. Extracellular BH4 also dose-dependently inhibited the induction by LPS (lipopolysaccharide) of nitrite and nitrate (the stable end products of NO formation) by RAW264.7 cells and quantification of mRNA levels showed that BH4 decreased iNOS and TNF-a gene expression in LPS-activated cells. The addition of catalase to the cell culture abolished these effects. In addition, extracellular hydrogen peroxide had similar effects to those of BH4. In this particular in vitro cell culture system, the effects observed with extracellular BH4 appear to be a result of the autoxidation of BH4 to produce hydrogen peroxide.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 17 of 123

Therapeutic Goods Administration
Given the precedent described above, the sponsor has proposed that it is likely that reactive oxygen species were formed from sapropterin in the cell-culture medium of the bacteria and Chinese hamster cell lines and that it was these reactive oxygen species that resulted in the positive genotoxicity results. The mostly negative results in the human peripheral blood lymphocyte cultures might reflect more efficient protective mechanisms in these cells and higher catalase content from co-cultured erythrocytes. This explanation seems plausible. However, it is considered to be a deficiency in this application that experiments were not carried out adding catalase to positive genotoxicity assays to verify that this mechanistic explanation is correct. There was no evidence for genotoxicity in two in vivo mouse micronucleus assays. In order for a negative result in the micronucleus assay to be relevant the drug must be distributed to the bone marrow; however, the tissue distribution studies indicate that sapropterin is not significantly distributed to the bone marrow in mice and there are already high endogenous levels of BH4 in bone marrow. The results of these micronucleus studies therefore do not significantly contribute to the understanding of the potential genotoxicity and carcinogenicity of sapropterin. Of more relevance are the rodent carcinogenicity studies that have also been conducted (see below). Given the results of the carcinogenicity studies and the plausible explanation for the genotoxicity observed in the in vitro studies, the weight of evidence suggests that sapropterin does not present a genotoxic risk. Carcinogenicity
Two GLP-compliant rodent carcinogenicity studies were conducted. There was an increase in the incidence of malignant lymphoma in male mice at ≥80 mg/kg/day in a 78 week study. This apparent increase in treated male mice may have been because of the low incidence of malignant lymphoma in the control and 25 mg/kg/day groups (0/60 and 1/60 respectively). Given that this finding did not occur in female mice or in either sex of rats in a 104 week study it is not considered toxicologically relevant. There was a dose-dependent increase in pituitary gland hyperplasia in female rats in the 52 week repeat dose toxicity study. However, in the 2 year rat carcinogenicity study the incidence of pituitary gland adenoma was not seen in the treatment group and pituitary gland adenoma was not observed in the mouse carcinogenicity study. Thus, pituitary gland hyperplasia is not considered to be a concern.
There was a dose-dependent increase in adrenal phaeochromocytoma in male rats but not in female rats or in mice. An increase in these tumours is consistent with the distribution of sapropterin to the adrenal glands and the location of enzymes that use BH4 in the adrenal medulla. However, male rats are known to be particularly susceptible to phaeochromocytoma (Tischler et al, 2004).12
Sapropterin is a cofactor for nitric oxide synthase (NOS). The role of this enzyme in cancer is still under investigation with evidence for roles in both tumourigenesis and tumour cytotoxicity. Certainly, in the sapropterin rodent studies there was no marked increase in the general incidence or progression of neoplasms. Indeed, inducers of NOS have been suggested as possible anti-cancer agents. However, there is evidence that some cancers contain significant levels of NOS and for patients with these particular cancers it is a theoretical possibility that sapropterin might enhance tumour growth.
An increase in adrenal hyperplasia was not observed in the rodent carcinogenicity studies or in the chronic studies in rats and marmosets. Therefore, these tumours are not considered to be of clinical relevance.
12 Tischler AS, Powers JF, Alroy J. Animal models of phaeochromocytoma. Histol Histopath 2004; 19: 883-
895.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 18 of 123

Therapeutic Goods Administration
Reproductive toxicity
After IV administration, sapropterin and/or its metabolites crossed the placenta in rats and, after oral administration (20 mg/kg), in guinea pigs. At 10 mg/kg sapropterin and/or its metabolites were expressed in rat milk after administration by the IV route but not the oral route but the plasma levels would have been considerably higher with the IV route. Sapropterin and/or its metabolites were shown to concentrate in the mammary gland of rats in a distribution study. Given that the rat was the animal used in the majority of the reproductive studies, pharmacokinetic evidence that the rat was a suitable model for investigating these toxicities would have been helpful.
No toxicokinetic data were collected in the reproductive toxicity studies but distribution data (but not AUC data) were collected in pregnant rats. Pharmacokinetic data were collected in pregnant rabbits but this data was in a separate study conducted almost two decades after the rabbit embryofetal development study. This lack of toxicokinetic data is a deficiency in that the level of exposure achieved in these studies is uncertain. Studies indicated that sapropterin had no effect on the fertility of rats (male or female), embryofetal development or postnatal development at oral doses of up to 400 mg/kg/day. In a rabbit embryofetal development study, the FDA considered an increase in the incidence of holoprosencephaly to be noteworthy. However, if the incidence of litters with holoprosencephaly is considered, one litter in the control group and two litters in the high dose group (600 mg/kg/day) contained fetuses with this external malformation. Thus, when comparing the litter incidence, the incidence in the 600 mg/kg/day group is not markedly greater than that in the control group. According to the TGA-adopted EU guideline on reproductive toxicity, when employing inferential statistics the litter, not the fetus, should be used as the basic unit of comparison.13
In summary, although toxicokinetic data in the reproductive studies would have been useful, the reproductive toxicity of sapropterin has been adequately investigated and no reproductive or developmental toxicity was observed.
Although statistical analysis of these results have not been conducted, it is considered appropriate to use the litter, not the fetus, as the basic unit of comparison and, thus, the incidence of holoprosencephaly is considered unlikely to have been related to treatment. This interpretation of these results is consistent with that of the EMA.
Pregnancy classification
A pregnancy category of B1 is considered to be the most appropriate:14
Drugs which have been taken by only a limited number of pregnant women and women of child-bearing age without an increase in the frequency of malformation or other direct or indirect harmful effects on the human fetus having been observed. Studies in animals have not shown evidence of an increased occurrence of fetal damage.
Use in children
Sapropterin is indicated for use in paediatric patients with phenylketonuria or BH4 (BH4) deficiency. For the patients with BH4 deficiency it appears to be clinically important for this treatment to start as early as possible. However, for infants with phenylketonuria, this disease
13 pp. 25 - 44 of the Rules Governing Medicinal Products in the European Union - EudraLex - Medicinal products for human use, 1998 Edition: Volume 3B - Safety and the Environment - 3BS4A. Detection of Toxicity to Reproduction for Medicinal Products Including Toxicity to Male Fertility. 14 Prescribing medicines in pregnancy - 4th edition - An Australian categorisation of risk of drug use in
pregnancy”, 1999.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 19 of 123

Therapeutic Goods Administration
can be controlled by dietary means relatively easily in the first year of life, although this becomes progressively difficult as the child grows older. With reduced benefits in early years, the safety of sapropterin use in infants with phenylketonuria needs to be carefully considered; for these patients the risk/benefit ratio differs from those with BH4 deficiency for whom there is significant benefit of treatment soon after birth. In juvenile rats (≤ 2 weeks of age) the bioavailability of sapr opterin was considerably higher (about 50% as opposed to about 10%) than in adult rats. In addition, the juvenile: adult exposure ratio in rats was 23. Therefore, increased bioavailability only partially accounts for the observed increase in exposure; decreased clearance must also occur in juvenile rats. This has implications for the proposed starting dose for young children. Currently the proposed starting dose for children and adults is the same on a mg/kg basis. The median lethal dose (LD50) values for juvenile rats were about half those in adult rats with this being attributed to the higher absorption in young rats. Likewise, the renal toxicity observed in young rats (treatment starting at 7 days) at 320 mg/kg/day (NOEL 80 mg/kg/day) was similar to that in adult rats at 400 mg/kg/day (NOEL 40 mg/kg/day). The difference was that the renal toxicity was observed after 2 weeks instead of 52 weeks. Thus the risk of renal toxicity in infants is considerably greater than in adults. Exposure ratios cannot be estimated because of the lack of pharmacokinetic data in children. No renal damage was observed in a 4 week study with 3-week old rats, indicating that the risk of kidney damage in older children is less than in infants.
Whereas in adult rats (6 or 9 weeks old) sapropterin administration only slightly increased total biopterin levels in the brain, in young rats (3 days, 7 days, 2 and 3 weeks) brain levels of total biopterin were markedly increased. A time course study was conducted for 2 week old rats, and after 24 hours total biopterin levels were still higher than the starting concentration. Thus, in young rats, sapropterin administration results in a much higher distribution of sapropterin and/or its metabolites to the brain than in adult rats. As the blood-brain barrier is not fully developed in infants until twelve months, infants are likely to get higher cerebral exposure to sapropterin than adults.
There is nonclinical evidence that higher levels of sapropterin in the brain increase pain levels (Secondary pharmacodynamics). Given that a very common adverse event in clinical trials was headaches; this indicates, even in adults, cerebral concentrations can become too high. In addition, dizziness can occur in humans with overdosage. With higher distribution to the brain it would be expected that infants are more likely than adults to experience pain as a result of sapropterin administration. Although behavioural tests were conducted in the rat postnatal development study they have not been conducted with juvenile rats dosed directly with sapropterin. Thus, the effects of increased BH4 levels on brain development and function have not been fully investigated and pose an unknown risk to infants. Whereas these risks would be acceptable for BH4-deficient infants, who have dysfunctional brain enzymes because of the lack of BH4 cofactor, for infants with phenylketonuria there is no advantage to higher BH4 brain levels that might outweigh the risks.
In conclusion, serious consideration should be given to the justification for the current proposed starting dose in infants and children because animal data indicate that exposure to sapropterin could be considerably higher in children than in adults. In addition, in infants less than 12 months old the brain levels of sapropterin are likely to be considerably higher than in adults. This might be an advantage for BH4-deficient infants, but it has no advantages, and poses several risks for infants with phenylketonuria. The nonclinical data therefore do not support the registration of sapropterin to infants under 12 months of age with phenylketonuria because of a change in the risk/benefit ratio.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 20 of 123

Therapeutic Goods Administration
Immunotoxicity
Sapropterin did not demonstrate allergenic or antigenic potential in mouse or guinea pig models designed to detect immunoglobulin E and anaphylactoid-type responses. Impurities
The quality evaluator requested advice on the qualification of three impurities: dihydrobiopterin, biopterin and S-BH4. Dihydrobiopterin is a major metabolite of plasma sapropterin in both rats and humans and so the proposed limit is qualified. Biopterin is a minor metabolite of plasma sapropterin in rats (about 1% of total dose administered IV) and humans. Mice that received 45 mg/kg 14C-biopterin IV almost completely eliminated 14C-biopterin in the urine after 30 minutes, in contrast to the tissue accumulation observed for 14C-BH4 (Hoshiga et al, 1993).15
S-BH4 is not a metabolite of 6R-tetrabiopterin but a synthetic impurity. S-BH4 is an enantiomer of sapropterin but there is no interconversion in vivo. In the TGA-adopted EU guideline, it states that the enantiomeric purity of active substances used in nonclinical studies should be defined.
Assuming that the relative metabolism of sapropterin in rats and humans is similar and that the absorption of biopterin is similar to that of sapropterin, and given that the rate of excretion of biopterin is rapid in mice, the expected increases in plasma levels of biopterin are only expected to be transient and are considered acceptable for biopterin. Biopterin is therefore considered qualified.
16
In conclusion the metabolites dihydrobiopterin and biopterin are each considered qualified in the sapropterin drug product at expiry. Initially the impurity S-BH4 was not considered qualified in the sapropterin drug substance, however following discussion with the sponsor this issue was resolved (see Overall Conclusion and Risk/Benefit Assessment).
With an unknown amount of S-BH4 present in the nonclinical studies, these studies cannot be used to qualify this impurity.
Nonclinical Summary and Conclusions Limited primary pharmacology data was provided by the sponsor. Some additional data was also available in the literature. The nonclinical data support the efficacy of sapropterin treatment in BH4-deficient patients and are consistent with efficacy in PKU patients. There is nonclinical evidence that BH4 increases neuropathic pain. This is consistent with the common adverse effect of headache. Safety pharmacology studies were conducted with adult animals. There were some effects on the CNS system of adult rodents but none of these effects led to concerns. The effect of sapropterin on the CNS of juvenile animals is unknown. Adequate nonclinical cardiovascular safety pharmacology studies were conducted and, with oral administration of sapropterin, no effects were observed on cardiovascular parameters in these studies.
The bioavailability of sapropterin in juvenile rats (about 50%) was about 5-fold higher than in adult rats (about 10%) and the juvenile: adult exposure ratio in rats was 23.
15 Hoshiga M, Hatakeyama K, Watanabe M, Shimada M and H Kagamiyama (1993) J Pharmacol Expt Therap
267: 971-978 “Autoradiographic distribution of [14C]tetrahydrobiopterin and its developmental change in mice”
16 Investigation of Chiral Active Substances, pp. 381 - 391 of Rules (3CC29a) Governing Medicinal Products in the European Union - EudraLex - Medicinal products for human use, 1998 Edition: Volume 3C - Efficacy and information on the medicinal product
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 21 of 123

Therapeutic Goods Administration
Sapropterin was mainly distributed to the liver and kidney in adult rats. Sapropterin does not readily cross the blood brain barrier in adult rats but in juvenile rats sapropterin and/or its metabolites crossed the blood brain barrier and remained at high levels for 24 hours. The BH4 metabolic pathways are similar in rats and humans and have been extensively studied. The majority of oral administered sapropterin is not absorbed and so is excreted in the faeces. The absorbed sapropterin is excreted directly into the urine or metabolised and then excreted into the urine. At high doses, sapropterin caused mortality in single-dose studies in mice, rats, rabbits and marmosets. Effects on the nervous system in single-dose studies occurred in rodents with the IV route and in all routes tested in rabbits and marmosets. In repeat dose toxicity studies in marmosets no effects on the nervous system were observed (exposure ratio based on BSA of 2.6).
Effects on the kidney (characterised as slight) were observed in rat repeat dose toxicity studies (NOEL exposure ratio based on AUC of 3) and in marmoset single dose toxicity studies. However, there was no renal damage in the 52 week marmoset study (exposure ratio based on BSA of 2.6). The risk of renal damage in adult patients is considered minimal.
The results of the in vitro genotoxicity assays ranged from positive to equivocal. The sponsor has proposed a plausible mechanism for these results. The mouse in vivo micronucleus assay was negative but sapropterin is not distributed significantly to bone marrow in mice. Results from the rodent carcinogenicity studies (see below) add to the weight of evidence indicating that sapropterin is unlikely to be genotoxic. A deficiency in this application is the lack of a study providing direct evidence for the mechanistic explanation provided. Two GLP-compliant carcinogenicity studies were conducted. The positive findings (malignant lymphoma in male mice and phaeochromocytoma in male rats) are considered unlikely to be of clinical relevance. However, in patients with cancers containing high levels of NOS (nitric oxide synthase) there is a theoretical possibility that sapropterin (a cofactor of NOS) might enhance tumour growth.
The reproductive toxicity of sapropterin was adequately investigated in rats and rabbits. No evidence for reproductive or developmental toxicity was obtained.
Sapropterin had a lower LD50 in juvenile rats than in adult rats. In addition, kidney damage occurred after 2 weeks of oral administration to 7-day old rats. Thus, the risk of kidney damage in infants is greater than that in adults. No specific studies were conducted to investigate the effects of sapropterin administration to juvenile animals on brain development and function. As sapropterin crosses the blood brain barrier in juvenile rats and is expected to affect brain function it is considered a deficiency in this application that these studies have not been conducted. The nonclinical data support the efficacy of sapropterin treatment of hyperphenylalaninaemia in BH4-deficient patients, although the nonclinical data indicate that this treatment alone is unlikely to avert all the effects of this deficiency in patients. Evidence for the efficacy of sapropterin in sapropterin-responsive PKU relies on clinical data. The nonclinical data are consistent with efficacy.
The nonclinical data lacked concurrent toxicokinetic data. However, data were available to indicate that sufficient exposure was achieved in the nonclinical studies such that most of the toxicological effects observed are unlikely to be of clinical relevance. However, if sapropterin
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 22 of 123

Therapeutic Goods Administration
were to be proposed for an indication that affects a larger proportion of the population, some of the deficiencies within this nonclinical package would need to be addressed.
Sapropterin is proposed for the treatment of adult and paediatric patients with BH4 deficiency. The benefits of this treatment for these patients are likely to outweigh the risks. Therefore, there are no nonclinical objections to the registration of sapropterin for the treatment of adult and paediatric patients of all ages with BH4 deficiency.
The bioavailability of oral sapropterin in juvenile rats was about 5-fold higher than in adult rats. The juvenile:adult exposure ratio in rats was 23. This has implications for the proposed starting dose for young children which is currently the same as for adults on a mg/kg basis. Therefore, the Delegate should consider whether it is appropriate to recommend a lower starting dose for young children. Renal damage occurred in young rats after 2 weeks of administration of oral sapropterin to 7-day old rats (NOEL = 80 mg/kg/day). Because of a lack of pharmacokinetic data in young children, an exposure ratio cannot be estimated. Therefore the risk of kidney damage to infants is unknown. However, this increased risk in comparison to adults indicates that it would be advisable to use a conservative starting dose in BH4-deficient infants so that the kidneys are not unnecessarily challenged. Sapropterin is also proposed for the treatment of adult and paediatric patients with sapropterin responsive phenylketonuria. For patients under the age of one year, for whom the blood-brain barrier has not fully developed, the benefits of sapropterin treatment may not outweigh the risks. In patients under one year of age, phenylketonuria is relatively easily controlled with dietary control, so the benefits of sapropterin are reduced. In addition, the risks involved in sapropterin use are increased. A very common adverse effect of sapropterin in patients is headache; an effect that an infant might have difficulty in communicating. There is nonclinical evidence that higher levels of sapropterin in the brain increase the levels of perception of pain.
Absorption of oral sapropterin was higher in juvenile rats than adult rats. Total biopterin levels increased markedly in the brains of young rats following oral sapropterin. Infants are likely to subject to higher cerebral exposure of sapropterin than adults as the blood-brain barrier is not fully developed in infants until twelve months. The effects of increased BH4 levels on brain development and function have not been fully investigated and therefore pose a risk to infants. Whereas this risk would be acceptable for BH4-deficient infants, who have dysfunctional brain enzymes because of the lack of BH4 cofactor, for infants with phenylketonuria there is no advantage to higher BH4 brain levels.
In conclusion, the registration of sapropterin for use in infants under 12 months of age with phenylketonuria is not supported on nonclinical grounds because the risk/benefit ratio differs between infants and adults with PKU. However, there are no nonclinical objections to the registration of sapropterin for use in adult and paediatric BH4-deficient patients.
IV. Clinical Findings Introduction Clinical data in support of this application consisted of company-sponsored studies, clinical results for a granule formulation of sapropterin (Biopten), which formed the basis for registration in Japan in 1992, and data from a systematic review of the literature to support treatment in phenylketonuria (PKU) children less than 4 years old and in patients with BH4 deficiency, an extremely rare condition (estimated 16-32 cases in Australia in 2006).
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 23 of 123

Therapeutic Goods Administration
The two pivotal Kuvan studies were the first randomised, placebo-controlled Phase III studies to be undertaken for the evaluation of efficacy and safety of a pharmaceutical agent in patients with PKU. The clinical development program was reviewed and agreed to by the EMA. The reduction in blood phenylalanine (Phe) levels as compared to placebo control was accepted as a clinical surrogate efficacy endpoint for the prevention of HPA-related neurotoxicity. The number of patients selected for each study was considered acceptable given the rare nature of the disease. Clinical Development Program
The clinical development program for sapropterin in HPA due to PKU was intended to meet two primary and three additional efficacy objectives. The two primary efficacy endpoints were to:17
1. Evaluate the efficacy of once-daily treatment with sapropterin tablets to reduce blood Phe in subjects with HPA due to PKU.
2. Evaluate the efficacy of once-daily treatment with sapropterin tablets to increase Phe tolerance in subjects with PKU with Phe levels controlled on a Phe-restricted diet.
The additional efficacy objectives were to: 1. Evaluate the efficacy of treatment with once-daily sapropterin tablets to reduce blood Phe levels to <600 µmol/L in subjects with PKU and a pre-treatment blood Phe ≥600 µmol/L. 2. Compare the effects of three dose levels of once-daily sapropterin tablets (5, 10 and 20 mg/kg/day) on reduction of blood Phe in HPA due to PKU. 3. Evaluate persistence of effect of once-daily sapropterin tablets to reduce blood Phe in HPA due to PKU. The clinical development program for sapropterin included two pivotal, randomised, placebo-controlled Phase III studies: PKU-003 and PKU-006 (Part 2). For these trials, reduction in blood Phe levels as compared to placebo control was used as a clinical surrogate efficacy endpoint for prevention of HPA-related neurotoxicity. Both pivotal studies were preceded by short-term open-label treatment response studies to identify responders to sapropterin treatment, thereby reducing the likelihood of not detecting a true response due to the rarity of the condition (PKU-001 and PKU-001 SS01). Responders were defined by the criterion of a ≥30% decrease in blood Phe from baseline after 8 days of daily sapropterin at the same dose used in the consequent, respective pivotal trial.
Age ranges included in the clinical development program were: ≥8 years old for PKU -001, PKU-003 and PKU-004 and 4 to 8 years old for PKU-006.
The initial plan for PKU-006 was an open-label study with 20 subjects, 4 to 8 years old, whereas the final protocol conducted within the clinical development program included 46 subjects, 4 to 12 years old, inclusive, selected as responders from an initial enrolled population of 90 and randomised 3:1 to active treatment versus placebo. The final design of PKU-006 was intended to demonstrate the potential benefit of sapropterin treatment to increase dietary Phe tolerance and thereby allow a less restrictive and more palatable diet for patients with PKU. The final clinical development program included investigations of a 10 mg/kg/day dose in PKU-003, 20 mg/kg/day dose in PKU-006 and doses of 5, 10, and 20 mg/kg/day in PKU-
17 EMA. Kuvan EPAR, EMEA/604757/2008
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 24 of 123

Therapeutic Goods Administration
004. Thus, the dose ranges used for the Phase III program are appropriate to assess efficacy and safety of the range of weight-based doses proposed for the product.
The 6-week treatment period for PKU-003 was not extended to 3 months due to ethical concerns regarding maintenance of placebo treatment for an extended period, particularly since this study involved children. Instead, additional safety data were collected during the 22-week treatment period of the Phase III, open-label, extension study, PKU-004, and also are being collected in PKU-008, an ongoing Phase IIIb safety trial in which sapropterin tablets are provided in acknowledgement of the absence of alternative drug product.
PKU-004 was further re-designed to include exposure across the intended treatment dose range of 5 to 20 mg/kg/day, and included an initial forced-dose titration study.
The studies submitted for evaluation were conducted in accordance with Good Clinical Practice guidelines. Pharmacology Introduction
In the literature it has been shown that, in patients with PKU, sapropterin enables endogenous PAH activity and can partially restore oxidative metabolism of Phe, resulting in decreased blood Phe levels in PKU patients (Kure et al., 1999; Muntau et al., 2002).18,19
Sapropterin is also the active pharmaceutical ingredient in the drug product Biopten Granules 2.5% developed by Daiichi Suntory Pharma (DSP) (now known as Asubio Pharma), which was approved in Japan in 1992 as a treatment for primary BH4 deficiency. A 10% granule formulation was developed containing 100 mg of sapropterin formulated with other excipients using a wet granulation process.
In patients with BH4 deficiency, sapropterin restores endogenous PAH activity by providing an exogenous source of the missing cofactor.
Formulation development
The current 100 mg sapropterin tablet formulation studied in the clinical trials evolved from the granule formulation (marketed by Asubio Pharma in Japan). The granule formulation was modified into an immediate release tablet dosage form.
The pharmacokinetics (PK) of sapropterin has been evaluated in healthy subjects in four Phase I studies, three studies with sapropterin granules, and one Phase I study with sapropterin tablets. The PK of sapropterin has also been independently reported in two publications using a non-pharmaceutical grade preparation (Fiege et al., 2004; Zurfluh et al., 2006).20,21
18 Kure S, Hou D-C, Ohura T et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatrics 1999; 135: 375-378.
The initial Phase I pharmacokinetic study of the 2.5% and 10% granule formulations (study number P1501), was conducted by Suntory in 1985, according to the acceptable standards appropriate at that time. All other sponsored studies with granule formulations (FB1602, FB1701) were conducted in accordance with the Japanese good clinical practice regulations in effect at that time, and the more recent study with sapropterin
19 Muntau AC, Beblo S, Koletzko B. Phenylketonurie und hyperphenylananinamie in Padiatrie Upgrade 2002: Weiter- und Fortbildung by Koletzko B, Reinhardt D, Stvckler-Ipsiroglu S. 20 Fiege B, Ballhausen D, Kierat L et al. Plasma tetrahydrobiopterin and its pharmacokinetic following oral administration. Molec Genetics Metab 2004; 81: 45-51. 21 Zurflüh MR, Fiori L, Fiege B. Pharmacokinetics of orally administered tetrahydrobiopterin in patients with phenylalanine hydroxylase deficiency. J Inherit Metab Dis 2006; 29: 725–731.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 25 of 123
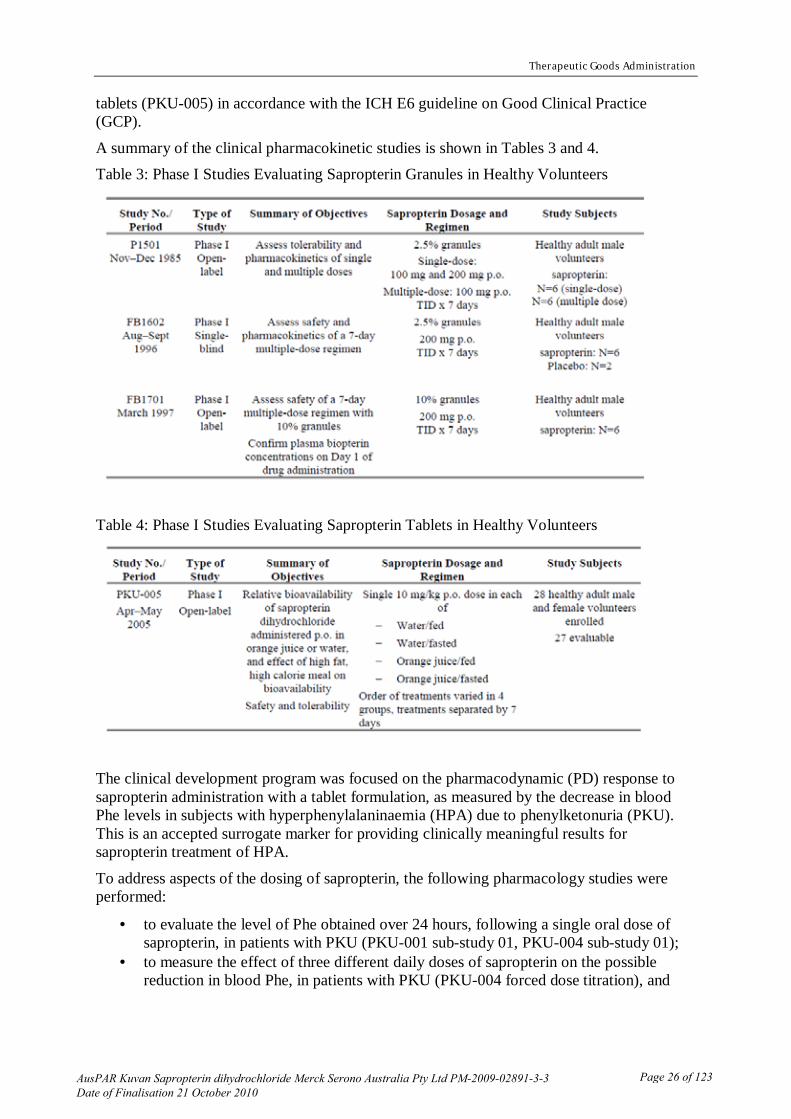
Therapeutic Goods Administration
tablets (PKU-005) in accordance with the ICH E6 guideline on Good Clinical Practice (GCP).
A summary of the clinical pharmacokinetic studies is shown in Tables 3 and 4. Table 3: Phase I Studies Evaluating Sapropterin Granules in Healthy Volunteers
Table 4: Phase I Studies Evaluating Sapropterin Tablets in Healthy Volunteers
The clinical development program was focused on the pharmacodynamic (PD) response to sapropterin administration with a tablet formulation, as measured by the decrease in blood Phe levels in subjects with hyperphenylalaninaemia (HPA) due to phenylketonuria (PKU). This is an accepted surrogate marker for providing clinically meaningful results for sapropterin treatment of HPA.
To address aspects of the dosing of sapropterin, the following pharmacology studies were performed:
· to evaluate the level of Phe obtained over 24 hours, following a single oral dose of sapropterin, in patients with PKU (PKU-001 sub-study 01, PKU-004 sub-study 01);
· to measure the effect of three different daily doses of sapropterin on the possible reduction in blood Phe, in patients with PKU (PKU-004 forced dose titration), and
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 26 of 123

Therapeutic Goods Administration
· to assess the blood level of 6R-BH4 over 24 hours, following a single oral dose of sapropterin, in patients with PKU (PKU-004 sub-study 02).
A summary of the clinical pharmacology studies is shown in Table 5. Table 5: Pharmacology Studies evaluating Sapropterin in subjects with HPA due to PKU
Pharmacokinetics In PK studies, BH4 is usually measured as L-biopterin, a metabolite of BH4, following sample treatment with iodide to facilitate the in vitro conversion of BH4 to biopterin. This conversion is incomplete; therefore, BH4 levels are calculated by adjusting the measured L-biopterin concentration for the estimated conversion of BH4 to L-biopterin and results are expressed as concentrations of BH4. Sapropterin granule Phase I studies
Sapropterin (2.5% and 10% granules) was evaluated for safety and PK in three Phase 1 studies. Twenty-four healthy adult male volunteers were treated orally (PO). The studies were conducted using either a single-dose format consisting of a 100 mg dose followed by a 200 mg dose, with one week between the two doses (N=6), or a multiple-dose format consisting of 100 mg three times daily (TID) for seven days (N=6) or 200 mg TID for seven days (N=12). A summary of the study objectives and dosage regimens is provided in Table 6 below.
Table 6: Phase 1 Studies Evaluating Sapropterin Granules in Healthy Males
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 27 of 123

Therapeutic Goods Administration
Single-dose administration In Study P1501, 6 subjects received single 100 mg and 200 mg doses of the sapropterin 2.5% granule formulation, with one week between the two doses. The endogenous plasma total biopterin concentration was 3.3 ± 0.3 ng/mL at 8 am, after which it increased gradually to 4.7 ± 0.2 ng/mL at 5 pm, which suggested a circadian rhythm. The maximum plasma total biopterin concentration above the endogenous level was 6.8 ng/mL and 12.5 ng/mL after the 100 mg and 200 mg sapropterin doses, respectively. The maximum plasma concentration was reached 3.1 and 2.6 hours after 100 and 200 mg dosing respectively. Total biopterin levels decreased gradually (half-life [t1/2] ~3.5 hours) and returned to nearly endogenous levels (3-3.5 ng/mL) 12 hours after dosing. Change in the maximum plasma concentration (ΔCmax) and ΔAUC0–24 the area under the concentration-time curve (ΔAUC0–24) based on plasma concentration minus endogenous concentration for total biopterin rose in reasonable proportion to the dose. The ratio of reduced biopterin to total biopterin in plasma ranged from 60% to 70%. Overall, the ratio did not appear to be dependent on dose. Amounts equivalent to 1.12% and 1.18% of the 100 mg and 200 mg sapropterin doses, respectively, were excreted in urine as total biopterin in the 24 hours after administration. The ratio of reduced biopterin to total biopterins in urine was 48%–68%.
Multiple-dose administration Administration of 2.5% Formulation Multiple-dose oral administration of the 2.5% sapropterin formulation over a 7 day period was evaluated in Study P1501 at a dosage of 300 mg/day (100 mg three times daily [tds]) and Study FB1602 at a dosage of 600 mg/day (200 mg tds). On Day 1 and Day 7, ΔCmax for total biopterin was 13.6 ng/mL and 17.1 ng/mL, respectively, during administration at 100 mg tds, and 36.6 ng/mL and 39.4 ng/mL, respectively, during administration at 200 mg tds. Plasma total biopterin concentration increases were slightly greater than dose proportional; however, the small sample size precluded reaching firm conclusions about linearity. Change in reduced biopterin plasma concentration over time was similar on Day 1 and Day 7, and no observed change was attributable to repeat administration.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 28 of 123

Therapeutic Goods Administration
With both the 100 mg tds and 200 mg tds sapropterin dosages, the total daily amount of biopterin excreted in urine was almost constant throughout the 7-day administration period, with an average 24-hour excretion rate of 1.02% reported in Study P1501. No observed drug accumulation effect was attributable to repeated administration.
Administration of 10% Formulation Multiple-dose oral administration of the 10% sapropterin formulation over a 7-day period was evaluated in Study FB1701 at a dosage of 600 mg/day (200 mg tds). Cmax for total biopterin was approximately 40 ng/mL for all subjects. Cmax for reduced biopterin was approximately 35 ng/mL. The results were consistent with those obtained with the 2.5% sapropterin formulation. Sapropterin tablet Phase 1 studies
Absorption The PK of sapropterin in individuals with PKU was examined in PKU-004 Substudy 02 using a sparse sampling strategy and population-based non-linear mixed effect modelling, with inclusion of a term for endogenous BH4. Of the 80 subjects enrolled in PKU-004, 78 participated in PKU-004 Substudy 02, which was conducted during the fixed-dose period (Week 16 to Week 22) when sapropterin doses were prescribed at either 5, 10 or 20 mg/kg. The results indicate that sapropterin is rapidly absorbed, with a bi-exponential decline following attainment of peak levels, with PK following a 2-compartment, first-order input model with first-order elimination. The mean terminal half-life was 6.69 hours (range 3.91 to 16.6 hours). Assuming 4 half-lives for clearance, coverage is estimated to be 26.8 hours, which supports once-daily dosing. In addition, stochastic simulations revealed no evidence of accumulation for 5 daily doses at 5, 10 or 20 mg/kg/day doses.
These results compare favourably with PK data collected for Biopten 2.5% granules in three Phase I trials in healthy adult volunteers, FB1501 (2 parts) and FB1602, as presented in Table 7. In Study FB1501, single and multiple dose regimens were tested. An additional study using sapropterin 10% granules 200 mg tds (FB1701) gave results similar to FB1602. These PK studies for Biopten did not use weight-based dosing.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 29 of 123

Therapeutic Goods Administration
Table 7: Biopten Phase I Pharmacokinetic Studies
Formulation development The current 100 mg sapropterin tablet formulation studied in the reported clinical trials evolved from the granule formulation (marketed by Asubio Pharma in Japan). The tablet formulation was further developed to eliminate excipients of animal origin, to leverage excipient compatibility data, to optimise excipient quantities for an immediate release tablet with similar solubility characteristics as the granules, to decrease the amount of mannitol in order to minimise the potential for gastrointestinal symptoms, to improve tablet disintegration by adding crospovidone, and to improve tablet manufacturing by adding sodium stearyl fumarate as a lubricant.
In vitro dissolution profiles of the tablet batches, used in the clinical studies and that proposed for marketing, were used to assess the potential impact of the minor formulation changes and the different tablet production processes on the drug product quality attributes. The dissolution data indicate that the various tablet formulations (tablet production method and manufacturer) have very similar dissolution profiles, and consequently the sponsor has argued that it is not justified that comparative bioavailability data be required. The differences observed between the profiles of the direct compression and the roller compression batches do not have a clinical impact, since the tablets are dissolved prior to administration.
Bioavailability and influence of food PKU-005 A Phase I study was performed in healthy volunteers to evaluate the safety and relative bioavailability of sapropterin tablets when administered as an oral solution dissolved in either water or orange juice, and administered to subjects who were in a fed or fasted state. This study is discussed in Section II. The results indicated that administration of sapropterin to fed subjects dissolved in either water or orange juice led to increases in mean plasma concentrations and in mean values for Cmax, the AUC over a dosing period (AUC0-t), and the AUC from time zero to infinity (AUC∞) compared to administration to fasted subjects in the same vehicle. The effect of food was less pronounced for orange juice than for water, though this was not statistically significant. It was concluded that, for either vehicle, sapropterin was more readily absorbed by subjects, after a high fat, high calorie meal than while fasting (41-84% higher with a Cmax at 4-5 hours after administration). However, the high fat, high calorie meal is not appropriate for patients with HPA who have to maintain a strict controlled diet. To avoid fluctuations in
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 30 of 123

Therapeutic Goods Administration
bioavailability due to dietary intake, it is recommended that patients with HPA take sapropterin at the same time every day. Population pharmacokinetic analyses
Population PK analyses were conducted in the PKU-004 Sub-study 02 to characterise the PK and pharmacokinetic variability of sapropterin, and to identify and characterise factors that influence the PK and pharmacokinetic variability of sapropterin in subjects with PKU during their participation in PKU-004. A sparse sampling strategy was employed in this sub-study with population based modelling methods (non-linear mixed effects modelling). PKU-004 was an open-label study designed to evaluate the long-term. During the 12 week fixed dose period of the study (from Week 10 visit until Week 22 visit), each subject’s daily dose of sapropterin was fixed at 5, 10, or 20 mg/kg/day based on the subject’s Phe levels during the forced dose titration period. The 78 subjects who completed the 12 weeks of treatment during the fixed dose period were included in the population PK analysis.
With all doses tested (5 mg, 10 mg, and 20 mg/kg/day), following administration of sapropterin, there was a short lag time prior to the appearance of measurable biopterin concentrations. Biopterin was rapidly absorbed after this initial lag. After reaching peak concentrations, measurable levels of biopterin declined in a bi-exponential fashion. This characteristic, suggests that the PK of sapropterin can be described by a two-compartment, first-order input model with first-order elimination.
Total body weight was the only significant covariate identified in this evaluation. Other measures of body size such as body surface area (BSA) were evaluated but were not found to be as predictive of PK variability as weight. The inclusion of weight on both clearance (CL) and Vc/F substantially improved the model’s ability to describe the data. These results suggest that individual exposure is similar across a wide range of body weights when dosage is adjusted on the basis of body weight.
There was a small effect of creatinine clearance (CrCl) on biopterin clearance; however, this effect was not sufficiently large to include in the final model. It should be noted that the range of CrCl values in the database only covered the range of normal renal function (CrCl greater than 80 mL/min) and mild renal impairment (CrCl greater than 50 and less than or equal to 80 mL/min). There was only one subject that had a CrCl of 47 mL/min. Consequently, the PK of biopterin in subjects with moderate or severe renal impairment has not been evaluated.
In addition, the effects of hepatic impairment have not been evaluated as the majority of subjects in the study database had liver enzyme levels that were at or near normal levels.
Estimating half-life values using the final PK model produced alpha half-life values that ranged from approximately 0.578 to 2.94 hours. In the terminal elimination phase, the mean half-life was approximately 6.69 hours (range: 3.91 to 16.6 hours). Assuming approximately 4 half lives is required to achieve clearance, coverage would be estimated to last 26.8 hours on average, following dosing, which supports once-daily dosing. Stochastic simulations based on the final PK model for 5 daily doses of 5, 10 and 20 mg/kg were generated to explore the expected concentration time profiles for these doses and to evaluate the potential, if any, for accumulation. Results showed that there is little accumulation with daily doses, even at the highest dose. Drug metabolism
No special metabolic studies were performed. Sapropterin is a synthetic version of the naturally occurring cofactor, 6R-BH4, and the metabolic fate is assumed to be as for the naturally occurring cofactor. The metabolic fate of exogenously administered BH4 in humans
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 31 of 123

Therapeutic Goods Administration
has been only partially defined. It is presumed that a large proportion of the BH4 serving as a cofactor for aromatic amino acid hydroxylases enters the BH4 regeneration pathway. Consistent with this assumption, following oral administration of BH4 in healthy individuals, <10% is recovered as pterin or lumazine metabolites in urine and faeces, implying substantial regeneration and/or tissue uptake and metabolism. Forced dose titration
Study PKU-004 was designed to evaluate the long-term safety and tolerability of the long term use of sapropterin in PKU patients. This study enrolled 80 of the 89 subjects who received at least 1 dose of study drug in PKU-003 and 79 subjects completed the study.
For each of the 3 doses (5, 10 and 20 mg/kg/day sapropterin tablets), there was a mean decrease in blood Phe level from Week 0 (pre-treatment). The mean reduction in blood Phe level from Week 0 for each of the sapropterin groups was 100.1 ± 295.2 µmol/L for the 5 mg/kg/day dose (Week 2), 204.1 ± 303.0 µmol/L for the 10 mg/kg/day dose (Week 6), and 263.3 ± 318.2 µmol/L for the 20 mg/kg/day dose (Week 4), indicating a dose-related response for the 3 doses of sapropterin evaluated during the forced dose titration period of PKU-004. These results support the recommended dose range of 5 to 20 mg/kg/day for sapropterin treatment of HPA due to PKU. Interactions
Formal drug interaction studies were not performed and were not feasible due to the rarity of HPA. Sapropterin is a synthetic formulation of the naturally occurring co-factor, 6R-BH4 and, as such, is expected to have similar metabolism. In pre-clinical repeat dose studies in rats, no induction of hepatic CYP450 enzymes was observed. Folic acid and vitamin B12 may increase BH4 levels; although the mechanisms are not completely defined (Hamon et al., 1986; d’Uscio et al., 2003).22,23 Ascorbic acid has been shown to prevent the auto-oxidation of BH4 to BH2 that occurs in aqueous solution, (Davis et al., 1988; Toth et al., 2002).24,25
Inhibitors of dihydrofolate reductase (DHFR) may also inhibit the activity of dihydropyridine reductase (DHPR) and theoretically prevent salvage of BH4 (Millot et al., 1992, Sakai 1998). DHFR inhibitors include methotrexate, aminopterin and trimethoprim.
26,27
BH4 acts to enhance nitric oxide synthetase activity and could have a synergistic effect with PDE5 inhibitors, such as sildenafil, on vasorelaxation. Animal studies however have not indicated interaction between sildenafil and sapropterin. Synergism may also theoretically occur with antihypertensive agents, such as minoxidil, that act as a nitric oxide agonist as part of their mechanism of action; potential interactions of these agents with BH4 have not been investigated.
22 Hamon CGB, Blair JA, Barford PA. The effect of tetrahydrofolate on tetrahydrobiopterin metabolism. J Intellect Disabil Res 1986; 30: 179-183. 23 d’Uscio LV, Milstien S, Richardson D, Smith L, Katusic ZS. Long-term vitamin C treatment increases vascular tetrahydrobiopterin levels and nitric oxide synthase activity. Circulation Research. 2003; 92: 88. 24 Davis MD, Kaufman S, Milstien S. The auto-oxidation of tetrahydrobiopterin. Eur J Biochem 1988; 173: 345-351.
25 Toth M. Role of tetrahydrobiopterin in the regulation of activity of human placental nitric oxide synthase in normal and pre-eclamptic pregnancies. Orv Hetil 2002; 143: 391-8. 26 Sakai T, Antoku Y, Matsuishi T. Method for treating spinocerebellar degeneration. US Patent 5,753,656, 1998. 27 Millot F, Dhondt JL, Hayte JM et al. Impairment of cerebral biogenic amine synthesis in a patient receiving high-dose methotrexate. Am J Pediatric Haematol Oncol 1992; 14: 276-278.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 32 of 123

Therapeutic Goods Administration
Evaluator’s overall conclusions on pharmacokinetics
A limitation of the data submitted is that there were no studies that directly compared the granules with the tablet preparation proposed for registration and marketing in Australia. However data were submitted that showed that the various tablet formulations have very similar dissolution profiles. It is therefore considered that the sponsor’s justification that comparative bioavailability data are not required is acceptable. The differences observed between the dissolution profiles are not likely to have a clinical impact.
In the dataset submitted for evaluation, the PK of sapropterin hydrochloride in subjects with moderate or severe renal impairment has not been evaluated. Similarly, the effects of hepatic impairment have not been evaluated as the majority of subjects in the study database had liver enzyme levels that were at or near normal levels. Pharmacodynamics PKU is an autosomal recessive condition caused by deficient activity of PAH, the enzyme that metabolises Phe to tyrosine. Affected individuals inherit a defective copy of the PAH gene from each parent, while heterozygous carriers are clinically and biochemically unaffected. It has been estimated that 75% of individuals with PKU are compound heterozygotes, inheriting a different mutation from each parent. Blood Phe levels in non-PKU populations have an approximate range between 25 and 100 μmol/L, with variability related to age, Phe intake and assay methodology. In individuals with PKU, blood Phe levels can range above 2,000 μmol/L, depending on the degree of PAH deficiency and dietary Phe intake. Adequate treatment of HPA in PKU depends on limitation of whole protein intake to achieve control of blood Phe levels, and supplementation with Phe-free protein supplements to meet daily amino acid (protein) requirements.
BH4 deficiency, a very rare inborn error of metabolism, is estimated to account for 1-2% of cases of HPA. BH4 deficiency can result from mutations or deletions of any of the five different enzymes involved in BH4 synthesis and regeneration. BH4 is a necessary co-factor for PAH. Therefore, BH4 deficiency impairs PAH activity leading to a biochemical situation similar to PKU, with HPA resulting from deficient conversion of Phe to tyrosine. Mechanism of action
Sapropterin is a synthetic version of the naturally occurring 6R-BH4, which is a co-factor of the hydroxylases for phenylalanine, tyrosine and tryptophan. The rationale for administration of sapropterin in patients with BH4-responsive PKU is to enhance the activity of the defective Phe hydroxylase and thereby increase or restore the oxidative metabolism of Phe sufficient to reduce or maintain blood Phe levels, prevent or decrease further Phe accumulation, and increase tolerance to Phe intake in the diet. The rationale for administration of sapropterin in patients with BH4 deficiency is to replace the deficient levels of BH4. Relationship between plasma concentration and effect
The once a day dosing interval for the clinical studies for sapropterin was based on the observed PD effect on blood Phe reduction, and was further supported by the results of the population PK study (PKU-004 sub-study 02). PD activity over 24 hours after once daily administration has been widely reported in literature. This relates to the ability of the drug to enable or to enhance overall PAH activity and maintain reduced blood Phe levels over a longer period than anticipated from the half-life of sapropterin. Evaluations of multiple blood
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 33 of 123

Therapeutic Goods Administration
Phe levels taken over a 24-hour period were conducted as PKU-001 Sub-study 01 and PKU-004 Sub-study 01.
The primary objective of both sub-studies was to evaluate the ability of sapropterin, when given as a single daily dose, to sustain stable blood Phe levels during a 24 hour period in PKU subjects. In Study PKU-001 Sub-study 01, participating subjects received sapropterin 10 mg/kg/day for a total of 8 days, according to the PKU-001 study protocol. Subjects were instructed to continue their usual diet without modification (that is, no change in daily Phe ingestion). Originally it was planned to enrol up to 40 patients, assuming a total of 10 responders and 30 non-responders. The sub-study was initiated, however only 11 patients could be recruited and of the 11 subjects who participated, only one was subsequently found to have met the protocol-defined response criteria of ≤30% reduction in blood Phe levels from Day 1 (pre -treatment) to Day 8. Therefore, 10/11 subjects could not be included in the analysis of PKU-001 Sub-study 01.
Mean blood Phe levels remained stable over the course of 24 hours. The Phe levels increased slightly, with a peak at 10:00 pm, and had returned to starting levels the following morning. Change in blood Phe between the 8:00 am on Day 8 and subsequent time points showed mean standard deviation (SD) changes ranging from 37.0 ± 118.2 µmol/L at 4:00 pm to -5.9 ± 186.1 µmol/L at 12:00 midnight. No consistent relationship to meals was observed. A planned comparison between subjects who were responsive to sapropterin and those who were non-responsive could not be performed in this study because only one subject was classified as a responder. However, among the 11 subjects studied, mean blood Phe levels remained stable over the course of 24 hours. This study has limited value as the majority of the patients did not respond to sapropterin.
The PKU-004 Sub-study 01 enrolled subjects from PKU-004 and was conducted during the dose analysis period of PKU-004 (Week 6 visit to Week 10 visit) when all subjects (N=12) were treated with sapropterin at a dose of 10 mg/kg/day. During this time each PKU-004 Sub-study 01 subject was admitted to a study site hospital for a 24-hour period for determination of blood Phe levels. Mean blood Phe levels remained relatively stable over the course of 24 hours after a single dose of sapropterin. The mean ± SD pre-dose (8:00 am) blood Phe level was 661.1 ± 432.8 µmol/L, and at 24 hours post-dose, the mean ± SD blood Phe was 631.0 ± 453.9 µmol/L; the lowest mean ± SD blood Phe level was at 16 hours post-dose (12:00am) and was 477.3 ± 241.3 µmol/L. The mean ± SD change in blood Phe levels from pre-dose values ranged from - 30.1 ± 173.4 µmol/L at 24 hours post-dose to – 74.1 ± 152.4 µmol/L at 8 hours post-dose (4:00pm).
Of the 12 subjects in the sub-study, 9 had lower blood Phe levels throughout the 24-hour observation period than they had at Week 0 (baseline) in Study PKU-004. A subset of patients (n=8) had a blood Phe level at the first time point for this sub-study (8:00 am) that was at least 30% lower than the PKU-004 Week 0 (baseline) blood Phe level. In this subset of subjects the mean ± SD blood Phe level declined to the lowest level at 4:00 pm, remained relatively unchanged until midnight, and returned to a mean level almost identical to the one observed at 8:00 am of the prior day. Although variability in blood Phe levels between subjects was observed, the intra-subject variations in blood Phe levels are not likely to be clinically significant. No apparent relationship was seen in the timing of changes in blood Phe levels and ingestion of meals.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 34 of 123

Therapeutic Goods Administration
Evaluator’s overall conclusions on pharmacodynamics
Overall, data from PKU-004 Sub-study 01 suggest that once daily sapropterin dosing at 10 mg/kg is associated with stable reduction in blood Phe levels over a 24-hour period in PKU subjects. Efficacy Introduction
Clinical efficacy in HPA due to PKU in patients ≥4 years of age The clinical program to evaluate the efficacy and safety of sapropterin tablets in the treatment of PKU of adult patients included the following multicentre, multinational studies: PKU-001, PKU-003, PKU-004, PKU-006 and PKU-008. The pivotal efficacy data that are most relevant to the proposed indication in the application are derived from studies PKU-003 and PKU-006.
The efficacy and safety of sapropterin tablets in the treatment of PKU of adult patients were evaluated in the multicentre, multinational 8-day study (PKU-001) in subjects with uncontrolled HPA due to PKU to determine the percent responders to sapropterin tablets, 10mg/kg/day, and the resulting safety profile. Subjects who met the protocol definition of responder were eligible for enrolment in PKU-003. There was also an 8-day study (PKU-006 Part 1) in subjects with diet-controlled HPA due to PKU to determine the percent responders to sapropterin tablets, 20 mg/kg/day, and the resulting safety profile. Subjects who met the protocol definition of responder were eligible for enrolment in PKU-006 Part 2. The pivotal, placebo controlled, Phase III trial, PKU-003, was designed to evaluate the safety and efficacy of sapropterin tablets, 10 mg/kg/day, to decrease blood Phe levels in subjects, ≥ 8 years old, with uncontrolled HPA due to PKU.
The pivotal, placebo controlled, Phase III trial, PKU-006 Part 2, was designed to evaluate the safety and efficacy of sapropterin tablets, 20 mg/kg/day, to increase dietary Phe tolerance in subjects with HPA due to PKU, 4 to 12 years old (inclusive), with blood Phe levels controlled on a Phe-restricted diet.
The Phase III, open label, 22-week, extension study, PKU-004, was designed to assess longer term exposure to sapropterin tablets, dose-relationship to reduction in blood Phe and persistence of effect to reduce blood Phe levels in subjects previously enrolled in PKU-003. The ongoing Phase IIIb, open-label extension study, PKU-008, was designed to assess longer term safety of exposure to sapropterin tablets in subjects previously enrolled in PKU-004 and PKU-006.
Figure 1 shows the clinical program for sapropterin tablets in the treatment of PKU, including the flow of the studies and the flow of subjects enrolled in PKU-001, PKU-003, PKU-004, PKU-006 and PKU-008. Study PKU-008 is ongoing and efficacy results will not be discussed in this evaluation report.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 35 of 123

Therapeutic Goods Administration
Figure 1: Flow of Subjects Enrolled in PKU-001, PKU-003, PKU-004, PKU-006 and PKU-008
Clinical efficacy in HPA due to PKU in patients 0-4 years of age A systematic review of the published literature was performed and articles selected for inclusion were used to support the clinical efficacy of sapropterin tablets in the treatment of PKU for patients in the 0 to 4 year old age group. The primary studies that contribute to the efficacy assessment of BH4 treatment in PKU patients aged 0 to 4 years include Baldellou Vazquez et al.,28 Belanger-Quintana et al.,29 Boneh et al.,30 Burlina et al.,31 Feillet et al.,32
28 Baldellou Vázquez A, Salazar García-Blanco MI, Ruiz-Echarri Zalaya MP et al.
Tratamiento de la hiperfenilalaninemia por déficit de fenilalanina hidroxilasa con tetrahidrobiopterina.¿ Cuándo y cómo? Anales de Pediatría, 2006.
29 Belanger-Quintana A et al. Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: Evolution of seven patients on long-term treatment with tetrahydrobiopterin. Mol Genetics Metab 2005; 86: 61-66. 30 Boneh A et al. Three-year audit of the hyperphenylalaninaemia/phenylketonuria spectrum in Victoria. J Paed Child Health 2006; 42: 486-498. 31 Burlina A, Blau N. Effect of BH4 supplementation on phenylalanine tolerance. J Inher Metabol Dis 2009; 32:
40-45. 32 Feillet F et al. Evaluation of neonatal BH4 loading test in neonates screened for hyperphenylalaninaemia. Early Human Devel 2008; 84: 561-567.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 36 of 123

Therapeutic Goods Administration
Hennermann et al.,33 Lambruschini et al.,34 Shintaku et al.,35 Steinfeld et al.,36,37 Trefz et al.38 and Ye et al.;39
Publications providing only short-term data add to the support of treatment in PKU patients aged 0 to 4 years, in particular those with high numbers of patients: Bernegger et al.(N=1730),
all of which treated patients for ≥ 2 months, for a combined total of at least 54 completed patients ≤4 years old.
40 Fiege et al. (N=557)41 and Fiori et al. (N=107).42
Clinical efficacy in HPA due to BH4 deficiency
A total of 2,714 patients with PKU aged ≤4 years completed the short-term only studies.
BH4 deficiency is an exceedingly rare disease, estimated to account for only 1-2% of cases of clinically significant HPA. In this submission safety and effectiveness for sapropterin in the treatment of HPA due to BH4 deficiency were supported by literature-based data as follows: (1) the study performed for registration of sapropterin 2.5% granules (Biopten) in Japan
(2) a 10-year post-marketing surveillance study for sapropterin 2.5% granules reported by Daiichi Suntory Pharma (DSP) in Japan
(3) the results of an interim analysis at Week 10 for clinical trial PKU-007, designed to further extend the existing safety and efficacy data in this very rare disease
(4) 11 published studies identified via a systematic review, reporting long-term data (5) 5 published studies identified via a systematic review, reporting short-term data. Main clinical studies
Study PKU-003 PKU-003 was a Phase III, multicentre, multinational, randomised, double-blind, placebo-controlled study to evaluate single daily doses of sapropterin tablets, 10 mg/kg/day, administered for 6 weeks to subjects with PKU, who satisfied responder criteria in PKU-001 and had elevated blood Phe levels at screening (defined originally as blood Phe ≥600 µmol/L or ≥450 µmol/L after Protocol Amendment No.2). Subjects were randomly assigned in a 1:1
33 Hennermann JB, Buhrer C, Blau N, Vetter B, Monch E. Long-term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuria. Mol Genetics Metab 2005; 86: 86-90. 34 Lambruschini N, Pérez-Dueñas B, Vilaseca MA et al. Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genetics Metab 2005; 86: 54-60. 35 Shintaku H, Kure S, Ohura T et al, Long-term treatment and diagnosis of tetrahydrobiopterin-responsive
hyperphenylalaninemia with a mutant phenylalanine hydroxylase gene." Pediatr Res 2004; 55: 425-430. 36 Steinfeld R A. Kohlschutter K Ullrich K, Lukacs Z. A hypothesis on the biochemical mechanism of BH(4)-
responsiveness in phenylalanine hydroxylase deficiency. Amino Acids 2003; 25: 63-8. 37 Steinfeld R A. Kohlschutter K Ullrich K, Lukacs Z. Efficiency of long-term tetrahydrobiopterin monotherapy
in phenylketonuria. J Inher Metab Dis 2004; 27: 449-453. 38 Trefz F K, Blau N. Potential role of tetrahydrobiopterin in the treatment of maternal phenylketonuria."
Pediatrics 2003; 112: 1566-1569. 39 Ye J, Qiu WJ, Han LS et al. Diagnosis, treatment and long-term following up of 223 patients with
hyperphenylalaninemia detected by neonatal screening programs. Zhonghua Yu Fang Yi Xue Za Zhi 2007; 41: 189-92. 217-221
40 Bernegger C, Blau N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: A study of 1919 patients observed from 1988 to 2002. Mol Genet Metab 2002; 77: 304-313.
41 Fiege B, Blau N. Assessment of tetrahydrobiopterin (BH(4)) responsiveness in phenylketonuria. J Pediatr 2007; 150: 627-630.
42 Fiori L, Fiege B, Riva E, Giovannini M. Incidence of BH4-responsiveness in phenylalanine-hydroxylase-deficient Italian patients. Mol Genet Metab 2005; 86: S67-S74.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 37 of 123

Therapeutic Goods Administration
ratio to receive either sapropterin tablets 10 mg/kg/day or placebo tablets once daily for 6 weeks. Subjects were instructed to continue their usual diet without modification.
The primary efficacy endpoint was the mean change from Baseline (pre-treatment) to the Week 6 visit in blood Phe levels. Secondary efficacy endpoints included mean change in weekly blood Phe levels during the 6 weeks of treatment and the proportion of subjects who had blood Phe levels ≥600 µmol/L at Week 6. Safety data were obtained through medical history, monitoring of adverse events, physical examinations, measurement of vital signs, and clinical laboratory testing (haematology, blood chemistry, thyroid function tests, and urinalysis). Subjects in PKU-001 who met the protocol definition of responder [achieved a ≥30% reduction in blood Phe on Day 8 vs Day 1 (pre-treatment)] were eligible to enrol in PKU-003, contingent upon satisfying the PKU-003 entry criteria. In addition, 6 subjects in PKU-001 with a ≥28% but <30% reduction in blood Phe on Day 8 vs Day 1 (pre-treatment) were granted an exemption and were permitted to enrol in PKU-003.
Primary efficacy endpoints: 1) Reduction in Blood Phe Levels
Reduction in blood Phe compared to placebo was accepted as the surrogate clinical efficacy endpoint for the pivotal trials within the clinical development program and was incorporated in the design of PKU-003 and PKU-006 Part 2. Reduction in blood Phe levels was also an efficacy endpoint for each of the 3 open label trials, PKU-001, PKU-004, and PKU-006 Part 1. Blood Phe levels are universally used in the diagnosis and clinical management of patients with HPA due to PKU and BH4 deficiency. The normal level of blood Phe in individuals without PKU is approximately 60 μmol/L (1 mg/dL) and varies inversely with age. Reference ranges listed by Mayo Medical Laboratories (primary reference laboratory for the clinical program), as determined by ion exchange chromatography, are listed in Table 8. Table 8: Blood Phe Reference Range by Mayo Medical Laboratories*
In individuals with PKU, blood Phe levels may be 20-fold or more above the normal range with normal dietary Phe intake, although the phenotype varies considerably, with some affected individuals having levels that are only modestly elevated. In addition, blood Phe levels may show wide fluctuations, particularly in relation to dietary intake of Phe-free protein supplements. Current management guidelines for HPA due to PKU do not aim for normalisation of blood Phe levels. Instead, the goal is reduction of blood Phe levels into selected therapeutic ranges as summarised in Table 9.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 38 of 123

Therapeutic Goods Administration
Table 9: Therapeutic Targets for Blood Phe Levels (μmol/L)
2) Increase in Dietary Phe Tolerance
The second primary efficacy objective for the clinical development program was to demonstrate the ability of sapropterin tablets to increase the dietary Phe tolerance in subjects with HPA due to PKU, controlled on a Phe-restricted diet. This endpoint was evaluated in PKU-006 Part 2.
Current management of HPA is based on restriction of dietary protein to reduce Phe intake to levels that will allow maintenance of target Phe levels. The level of dietary Phe that allows achievement and maintenance of target Phe levels varies between patients and is referred to as the dietary Phe tolerance. In addition, current management includes ingestion of Phe-free protein supplements to maintain daily protein requirements. Patients with HPA due to PKU may tolerate only a small fraction of the usual daily intake of protein and must meet >80% of daily protein requirements using Phe-free protein supplements (Blau and Burgard 2005; MacDonald et al., 2004).43,44
In addition to the two primary efficacy objectives, three additional efficacy objectives were assessed within the clinical development program:
Maintenance of this degree of protein restriction is impractical in daily life. In addition, the Phe-free protein supplement is widely known to be unpalatable. Poor compliance with diet management is a well-known occurrence in children and adults with HPA due to PKU. Thus, increase in dietary Phe tolerance is included as a primary efficacy objective.
43 Blau N, Burgard P. Disorders of phenylalanine and tetrahydrobiopterin. Physician's Guide to the Treatment
and Follow-up of Metabolic Diseases. N. Blau, G. Hoffmann, J. Leonard and J. Clarke. Heidelberg, Springer, 2005.
44 MacDonald A, Asplin D. Phenylketonuria: practical dietary management." J Fam Health Care 2006; 16: 83-5.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 39 of 123

Therapeutic Goods Administration
1) Reduction in Blood Phe to <600 μmol/L Demonstrating the ability of sapropterin tablets to decrease the blood Phe level to <600 μmol/L at the end of study in patients with a blood Phe level ≥600 μmol/L prior to treatment was an additional efficacy objective evaluated in PKU-003. A secondary analysis for this efficacy objective was also performed for the open-label, short-term study, PKU-001. The boundary blood Phe level of 600 μmol/L was selected as being consistent with commonly used therapeutic guidelines for control of blood Phe in individuals with HPA due to PKU. Internationally agreed consensus guidelines for target blood Phe levels for treatment of HPA are not available. Infants with HPA are typically identified in national newborn screening programs (Loeber 2007).45 For Germany, the level of HPA required for initiation of treatment of infants involves persistent levels of blood Phe >600 μmol/L on normal intake, based in part on the data of Weglage et al. in 31 adults with mild PKU,46 who had apparently normal neurocognitive outcomes with childhood blood Phe levels of 360 to 600 μmol/L on unrestricted diets (Burgard et al., 1999; Weglage, Pietsch, et al. 2001.46,47 The UK consensus recommendation is to treat infants with confirmed blood Phe >400 to 600 μmol/L, as measured for several days on normal protein intake (Smith, Beasley, et al. 1990).48
Upon review of the various national guidelines and consultation with experts, achievement of a blood level of <600 μmol/L in subjects with a pre-treatment blood Phe level of ≥600 μmol/L was chosen as representative of a typical clinical therapeutic goal.
2) Comparison of 3 Daily Dose Levels (5, 10 and 20 mg/kg/day) on Reduction of Blood Phe
This evaluation was performed during the forced dose titration period in PKU-004 in support of the intended dose range.
3) Persistence of Effect of Sapropterin to reduce Blood Phe Levels This evaluation was performed over the 22-week treatment period in PKU-004. A supportive analysis, compared to placebo, was performed for PKU-003. Study population
Figure 2 displays the disposition of subjects enrolled in PKU-001, PKU-003, PKU-004, PKU-006, and PKU-008.
In the overall clinical program, 13 subjects withdrew from the clinical trials: 4 subjects in the placebo groups, 0 subjects in a 5 mg/kg/day sapropterin treatment group, 3 subjects in a 10 mg/kg/day sapropterin treatment group, and 6 subjects in a 20 mg/kg/day sapropterin treatment group. In addition, 2 subjects (1 subject in the 10 mg/kg/day sapropterin treatment group and 1 subject in a 20 mg/kg/day sapropterin treatment group) did not have a blood Phe level on Day 8 (end of study) in PKU-001 and PKU-006, respectively.
During PKU-003, a total of 89 subjects were randomised and 88 subjects received at least one dose of either sapropterin tablets (41 subjects) or placebo tablets (47 subjects). The majority of the 88 treated subjects had a screening blood Phe level ≥600 μmol/L. Nine subjects in the placebo group and 7 subjects in the sapropterin group were entered under Protocol
45 Loeber JG. Neonatal screening in Europe; the situation in 2004. J Inher Metabol Dis 2007; 30: 430-438. 46 Weglage J, Pietsch M, Feldmann R et al. Normal clinical outcome in untreated subjects with mild
hyperphenylalaninaemia. Pediatr Res 2001; 49: 532-536. 47 Burgard P, Bremer HJ, Burhdel P et al. Rationale for the German recommendations for phenylalanine level
control in phenylketonuria 1997. Eur J Pediatr 1999; 158: 46-54. 48 Smith I, Beasley MG, Ades AE. Intelligence and quality of dietary treatment in phenylketonuria. Arch Dis
Child 1990; 65: 472-478.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 40 of 123

Therapeutic Goods Administration
Amendment 2 and had a screening blood Phe level ≥450 μmol/L, but <600 μmol/L. The 88 subjects who received at least one dose of study drug were included in the efficacy analyses.
Figure 2: Disposition of Subjects Enrolled in PKU-001, PKU-003, PKU-004, PKU-006, and PKU-008
Baseline data of the study population Baseline physical and physiological parameters were similar in the two treatment groups. For PKU-003, the mean ± SD age of subjects at screening was 19.5 ± 9.8 years old in the placebo group and 21.5 ± 9.5 years old in the sapropterin group, with 61.7% and 65.9% of subjects, respectively, from sites in the EU. The majority of subjects were Caucasian (100.0% of the placebo group and 95.1% of the sapropterin group); 51.1% of the placebo group and 65.9% of the sapropterin group were male. The mean ± SD body mass index (BMI) was 24.98 ± 6.80 kg/m2 in the placebo group and 22.97 ± 4.16 kg/m2 in the sapropterin group. Overall, the
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 41 of 123
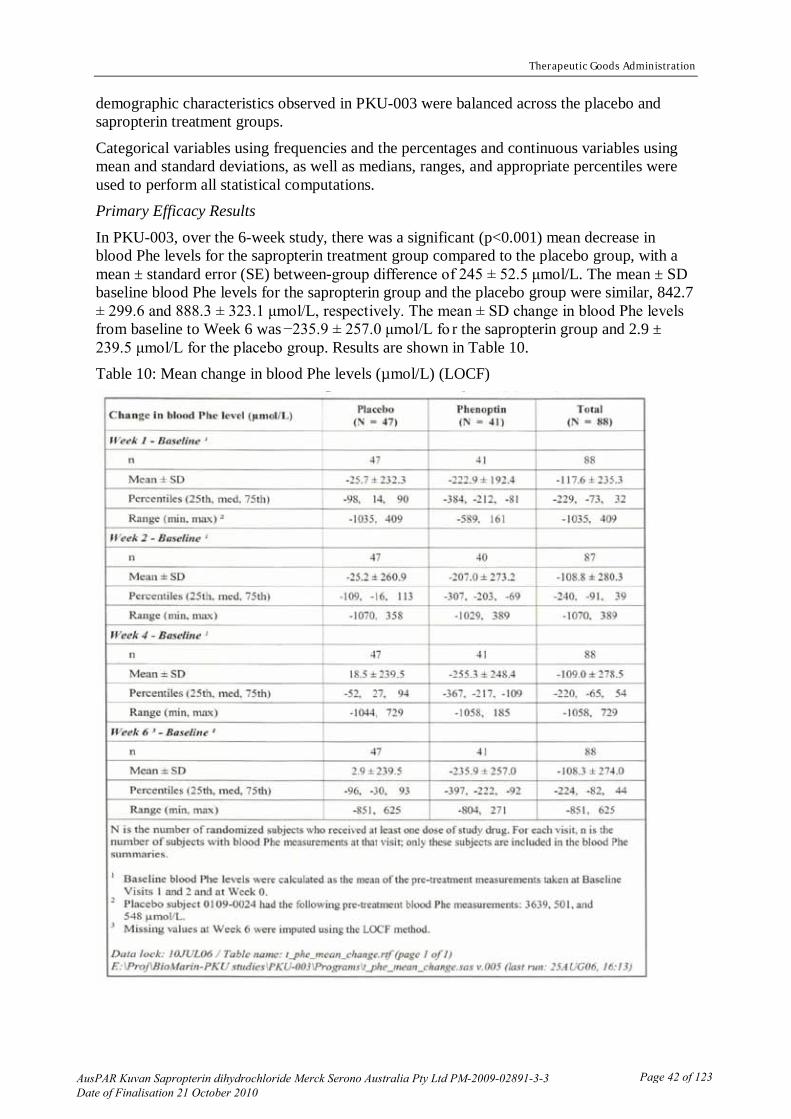
Therapeutic Goods Administration
demographic characteristics observed in PKU-003 were balanced across the placebo and sapropterin treatment groups.
Categorical variables using frequencies and the percentages and continuous variables using mean and standard deviations, as well as medians, ranges, and appropriate percentiles were used to perform all statistical computations. Primary Efficacy Results
In PKU-003, over the 6-week study, there was a significant (p<0.001) mean decrease in blood Phe levels for the sapropterin treatment group compared to the placebo group, with a mean ± standard error (SE) between-group difference of 245 ± 52.5 μmol/L. The mean ± SD baseline blood Phe levels for the sapropterin group and the placebo group were similar, 842.7 ± 299.6 and 888.3 ± 323.1 μmol/L, respectively. The mean ± SD change in blood Phe levels from baseline to Week 6 was −235.9 ± 257.0 μmol/L fo r the sapropterin group and 2.9 ± 239.5 μmol/L for the placebo group. Results are shown in Table 10. Table 10: Mean change in blood Phe levels (µmol/L) (LOCF)
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 42 of 123

Therapeutic Goods Administration
Secondary Efficacy Results In PKU-003, the mean ± SD blood Phe levels for the sapropterin group decreased from 842.7 ± 299.6 μmol/L at baseline to 619.9 ± 354.7 μmol/L at Week 1 and remained below this level for the duration of treatment. For the placebo group, blood Phe levels fluctuated only slightly from the mean ± SD baseline level of 888.3 ± 323.1 μmol/L, reaching a Week 1 mean ± SD level of 862.2 ± 345.6 μmol/L and remaining above this level for the duration of the study. The mean ± SD change in blood Phe levels from baseline to Week 1 was − 222.9 ± 192.4 μmol/L for the sapropterin group and − 25.7 ± 232.3 μmol/L for the placebo group. The 95% confidence intervals for the mean change from baseline blood Phe level for the placebo group is completely above that for the sapropterin group at each post-baseline visit, indicating that on average, the subjects in the sapropterin group had at each of these visits significantly greater decreases from their baseline blood Phe level than the placebo subjects.
A longitudinal model with weekly blood Phe measurements as the response variable and treatment group, visit, treatment group by visit interaction, and baseline blood Phe levels as covariates was used to assess the mean blood Phe levels over time. The treatment group by visit interaction term was not significant; so it was dropped from the model. Using the model without the interaction term, a significant difference exists between the two treatment groups (p<0.001). Thus, it can be concluded that the effect of sapropterin relative to placebo was sustained and unchanged throughout the treatment period. The estimated difference in mean ± SE blood Phe level between the two treatment groups (sapropterin − placebo) was −230 ± 43.4 μmol/L at Week 6. Fifty four percent of subjects in the sapropterin group and 23% of subjects in the placebo group had Week 6 blood Phe levels <600 μmol/L (p=0.004). In the subgroup of subjects whose baseline blood Phe levels had been ≥600 μmol/L, 42% of those in the sapropterin group and 13% of those in the placebo group had Week 6 blood Phe levels <600 μmol/L (p=0.012).
Additional Efficacy Results A post hoc analysis of the proportion of subjects who had blood Phe levels <360 μmol/L at Week 6 (end-of-study) was performed. No subject had a screening blood Phe level <360 μmol/L. At Week 6, 13 of 41 subjects (32%) in the sapropterin group and 1 of 47 subjects (2%) in the placebo group had blood Phe levels <360 μmol/L (p<0.001). For subjects with screening blood Phe levels ≥600 μmol/L, 26% of subjects in the sapropterin group and 3% of subjects in the placebo group had Week 6 blood Phe levels <360 μmol/L. Genotype Analysis
All 88 subjects included in the efficacy analyses for PKU-003 had blood samples for PAH genotype analysis obtained during their participation in study PKU-001. However, a full PAH genotype could not be determined for 16 samples due to technical limitations. Therefore, a total of 72/88 (82%) of subjects in PKU-003 had full PAH genotype analysis. The PAH genotype patterns were similar between the placebo and sapropterin treatment groups; however, detailed analysis was not possible due to the large number of observed mutations and the fact that the majority of subjects had compound heterozygosity for PAH mutations.
Study PKU-006 PKU-006 was a two-part Phase III study that involved once-daily administration of sapropterin tablets, 20 mg/kg/day, to subjects with HPA due to PKU, 4 to 12 years old (inclusive), who were following a strict Phe restricted diet and who had blood Phe levels of ≤480 μmol/L at screening.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 43 of 123

Therapeutic Goods Administration
In the first part of the study, PKU-006 Part 1, all subjects were to receive sapropterin tablets, 20 mg/kg/day, for 8 days. According to the enrichment design of the program, only those subjects whose blood Phe levels were reduced by ≥30% from Day 1 to Day 8 and whose blood Phe level was ≤300 μmol/L on Day 8 were to be eligible for enrolment in the second part of the study, PKU-006 Part 2. PKU-006 Part 2 was a randomised, double-blind, placebo-controlled study in which subjects were randomly assigned in a 3:1 ratio to receive either single daily administrations of 20 mg/kg sapropterin tablets or daily placebo tablets, respectively, for 10 weeks. Subjects were instructed to make no changes in their normal diet during the 10-week study period. At Week 3, subjects’ dietary Phe intake was increased by 5 mg/kg/day by adding a quantity of Phe containing powder if their blood Phe level at Week 2 was ≤300 μmol/L. Dietary intake of Phe was also modified at Weeks 5, 7, and 9 based on the preceding week blood Phe levels (that is, from Weeks 4, 6, and 8, respectively), with Phe supplement increased or decreased by 0, 5, 10 or 15 mg/kg /day according to the defined algorithm below.
Dietary Phe intake was increased by adding a specified quantity of Phe-containing powder to the diet (that is, non-fat dry milk or dried egg white powder). The primary efficacy endpoint for PKU-006 Part 2 was the amount of Phe supplement prescribed while maintaining adequate blood Phe control (blood Phe level <360 μmol/L). The two secondary efficacy endpoints in PKU-006 Part 2 were: (1) the difference in blood Phe levels in the sapropterin group between the Week 0 visit prior to dosing and the Week 3 visit prior to Phe supplementation and (2) the comparison of the sapropterin and placebo treatment groups in amount of Phe supplement tolerated. For both parts 1 and 2 of Study PKU-006, safety data were obtained through medical history, monitoring of adverse events, physical examinations, measurement of vital signs, and clinical laboratory testing (haematology, blood chemistry and urinalysis).
Subjects who participated in PKU-004 or PKU-006 were eligible to enrol in PKU-008 (see Figure 1 for flow of studies), contingent upon satisfying the PKU-008 entry criteria. Protocol PKU-008 was a Phase IIIb, open-label, extension safety study enrolling subjects from 1) PKU-004 after the Week 22 visit in PKU-004, and 2) PKU-006 after the Week 10 visit,
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 44 of 123

Therapeutic Goods Administration
contingent upon satisfying the PKU-008 entry criteria. PKU-008 is not summarised in this report as this study was ongoing at the time of evaluation.
Categorical variables using frequencies and percentages and continuous variables using means and standard deviations, as well as medians, ranges, and appropriate percentiles were presented. Study population
Figure 2 displays the disposition of subjects enrolled in PKU-001, PKU-003, PKU-004, PKU-006, and PKU-008.
Ninety subjects were enrolled and received at least one dose of sapropterin in PKU-006 Part 1. With the identification of 50 subjects who met eligibility criteria for PKU-006 Part 2, 46 were randomised 3:1 to sapropterin or placebo treatment, and 45 received at least one dose of study drug (33 subjects received sapropterin tablets, 12 subjects received placebo). Forty one subjects (32 sapropterin, 9 placebo) completed Part 2 of the study by attending the Week 10 visit.
Data for 89 of the 90 subjects (99%) enrolled in Part 1 of the study were used in the efficacy analyses. One subject was excluded from the analyses due to lack of a Day 8 blood Phe measurement. For PKU-006 Part 2, all 45 subjects who received at least one dose of study drug were included in the efficacy analyses.
Primary Efficacy Results PKU-006 Part 1
Of the 89 subjects who received at least one dose of study drug and for which there were both Day 1 (pre-treatment) and Day 8 blood Phe level data, 50 (56%) met the protocol definition of responder (reduction in blood Phe level ≥30% from Day 1 to Day 8 and blood Phe level ≤300 μmol/L on Day 8), as defined for the enrichment design of the program. For the responders, the mean ± SD blood Phe level at Day 8 was 108.1 ± 70.2 μmol/L, compared to 317.0 ± 173.2 μmol/L at Day 1, with a mean ± SD change and percent change in blood Phe levels of –209.0 ± 138.6 μmol/L and –64.0 ± 17.5%, respectively. PKU-006 Part 2
In PKU-006 Part 2, the Phe supplement at the Week 0 visit of PKU-006 Part 2 was 0 mg/kg/day; Phe supplement was not started until the Week 3 visit. The amount of Phe supplement tolerated (that is, Phe supplement prescribed while maintaining adequate blood Phe control) was defined as the cumulative increase/decrease in Phe supplement prescribed at the visit prior to the last visit when the subject’s blood Phe level was <360 μmol/L. Over the 10 week study period, the mean ± SD Phe supplement tolerated by subjects in the sapropterin group, 20.9 ± 15.4 mg/kg/day, was significantly different from zero (p<0.001). Of the 33 subjects who received sapropterin, 11 (33.3%) tolerated a Phe supplement of 31 to 50 mg/kg/day (50 mg/kg/day was the maximum amount of Phe supplementation allowed by the protocol), 10 (30.3%) tolerated 11 to 30 mg/kg/day, and 7 (21%) tolerated 1 to 10 mg/kg/day; 5 subjects (15%) were unable to tolerate any Phe supplementation. Of the 12 subjects in the placebo group, none were able to tolerate Phe supplementation above 10 mg/kg/day.
Secondary Efficacy Results There were no secondary efficacy endpoints for Part 1 of PKU-006.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 45 of 123

Therapeutic Goods Administration
In PKU-006 Part 2, for subjects treated with sapropterin, the mean ± SD change in blood Phe from the Week 0 visit to the Week 3 visit was 148.5 ± 134.2 μmol/L, which was a significant decrease from the Week 0 visit (p<0.001). Another secondary efficacy endpoint, comparison between treatment groups of the amount of Phe supplement tolerated, was analysed using a two way analysis of variance (ANOVA) model with effects for blood Phe level stratum and treatment group. The adjusted mean ± SE Phe supplement tolerated was 21.0 ± 2.3 mg/kg/day for subjects in the sapropterin group and 3.3 ± 3.9 mg/kg/day for subjects in the placebo group. The difference between the two treatment groups was statistically significant (p<0.001). Supportive studies
Study PKU-001 The primary objective of this Phase II, multicentre, open-label study was to evaluate the degree and frequency of response to sapropterin tablets, as measured by a reduction in blood Phe level, in PKU subjects with pre-treatment elevated blood Phe levels (≥600 µmol/L initially and then ≥450 µmol/L per Amendment No. 2). Subjects were to receive single daily oral administrations of 10 mg/kg/day of sapropterin for 8 days. A total of 490 subjects ≥8 years of age were enrolled, and 485 had blood Phe measurements at both Days 1 and 8.
A total of 490 subjects with clinically diagnosed PKU were enrolled in PKU-001 and 489 subjects received at least one dose of sapropterin tablets. The 485 subjects who received at least one dose of study drug and had blood Phe level data available at Day 1 (pre-treatment) and Day 8 were included in the efficacy analyses.
Primary Efficacy Results Of the 485 subjects in PKU-001 who received at least one dose of study drug and who had blood Phe level measurements at both Day 1 and Day 8, 96 (19.8%) were responders, as defined for the enrichment design of the program (that is, ≥30% reduction in blood Phe level from Day 1 to Day 8). The mean ± SD change and mean ± SD percent change in blood Phe levels from Day 1 to Day 8 for responders were −391.8 ± 185.3 μmol/L and −50.0 ± 16.0%, respectively. When changes in blood Phe levels from Day 1 to Day 8 for protocol-defined responders were examined by baseline blood Phe level (<600 μmol/L, n=31; ≥600 μmol/L; n=65), the mean (± SD) changes were −286.3 ± 84.0 μmol/L for subjects with baseline levels <600 μmol/L and −442.0 ± 199.2 μmol/L for subject with baseline levels ≥600 μmol/L, while the mean ± SD percent changes were −55.3 ± 16.7% and −47.5 ± 15.2%, respectively. Additional Efficacy Results
For the 485 subjects in PKU-001 who received at least one dose of study drug and had both Day 1 and Day 8 blood level results, the mean ± SD change and mean ± SD percent change in blood Phe levels from Day 1 to Day 8 were −99.1 ± 219.8 μmol/L and −11.4 ± 25.5%, respectively. When changes in blood Phe level from Day 1 to Day 8 were examined by baseline blood Phe levels (<600 μmol/L, n=57; ≥600 μmol/L; n=428) , subjects with baseline blood Phe levels <600 μmol/L had a greater mean ± SD percent change than subjects whose baseline level was ≤600 μmol/L, 25.4 ± 39.3% and −9.6 ± 22.5%, respectively. The magnitude of the mean ± SD change in blood Phe was also greater for the group with lower baseline blood Phe levels: −134.4 ± 199.9 μmol/L for subjects with baseline levels <600 μmol/L and −94.4 ± 222.1 μmol/L for subject with baseline levels ≥600 μmol/L.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 46 of 123

Therapeutic Goods Administration
Genotype Analysis Subjects enrolled in PKU-001 were invited to participate in a study involving analysis of PAH genotype. Of the 485 subjects who completed PKU-001, 400/485 (82.5%) had full PAH genotype analysis. An additional 85 subjects had indeterminate PAH genotypes, probably due to technical limitations rather than misdiagnosis. Among the 400 fully genotyped subjects, 57/400 (14.3%) were homozygous for a single PAH mutation and 343/400 (85.7%) were compound heterozygotes. A total of 118 different PAH gene mutations were identified, including 8 that had not been previously reported to PAH Locus Knowledgebase, an international online dataset of PAH gene mutations. The majority of subjects in this study had not been previously genotyped and were not included in PAH database (PAHdb). The range of observed PAH genotypes was otherwise consistent with the range of PAH genotypes listed in PAHdb. Comparison of PAH genotypes to the response of blood Phe to sapropterin treatment in PKU-001 did not reveal any consistent patterns. In addition, several subjects who responded to sapropterin treatment had PAH genotypes that had previously been postulated to be unresponsive to this treatment. Therefore, it was concluded that genotype alone is not a conclusive indicator of sapropterin treatment response.
Study PKU-004 Subjects who had received at least 80% of the scheduled doses in PKU-003, unless they were removed from PKU-003 because of high levels of blood Phe, were eligible to enrol in PKU-004. PKU-004 was a Phase III, open-label, 22-week extension study. A forced dose titration study was conducted during the first 6 weeks, in which subjects received 3 consecutive 2-week courses of once-daily sapropterin tablets in the following order: 5 mg/kg/day, 20 mg/kg/day, and 10 mg/kg/day. During the dose-analysis period, starting at the Week 6 visit and continuing to the Week 10 visit, subjects continued to receive once-daily sapropterin tablets, 10 mg/kg/day, during analysis of the blood Phe results from the forced-dose titration, 6-week study. Starting at the Week 10 visit and continuing until the 22-week visit, subjects were assigned to a fixed dose of 5, 10 or 20 mg/kg/day (fixed dose period) based on an algorithm utilising the blood Phe results measured at the Week 2 and Week 6 visit. The primary efficacy endpoint was blood Phe levels at the Weeks 10, 12, 16, 20 and 22 visits. The secondary efficacy endpoint was mean blood Phe levels after each 2-week dosing period (5, 20, and 10 mg/kg/day), which was used to estimate the effect of dose on reduction of blood Phe levels. PKU-004 included 2 substudies: PKU-004 Substudy 01 and PKU-004 Substudy 02.
Eighty subjects were enrolled in PKU-004; 39 subjects had received sapropterin tablets and 41 subjects had received placebo in PKU-003. All enrolled subjects received at least one dose of sapropterin in PKU-004. All enrolled subjects completed the study through the Week 10 visit and 79 subjects completed the study through the Week 22 visit. One subject was withdrawn at Week 16 due to non-compliance with study procedures. All subjects who enrolled in the study were included in the efficacy analyses.
Primary Efficacy Results During the fixed dose period of PKU-004 (Week 10 visit to Week 22 visit), each subject’s optimal sapropterin dose was based on his/her Week 2 and Week 6 blood Phe levels. Of the 80 subjects, 6 (8%) received 5 mg/kg/day of sapropterin, 37 (46%) received 10 mg/kg/day, and 37 (46%) received 20 mg/kg/day during the fixed dose period of PKU-004. The mean ± SD blood Phe levels at the Weeks 12 - 22 visits ranged between 619.8 ± 371.0 and 652.2 ± 382.5 μmol/L. On average, subjects maintained a stable reduction in Phe levels. The 95%
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 47 of 123

Therapeutic Goods Administration
confidence interval for the mean change from baseline blood Phe level at the first visit after subjects started using their optimal sapropterin dose, Week 12, was (-296.8 μmol/L, –151.6 μmol/L), and each of the 95% confidence intervals for the mean change from baseline blood Phe level at Weeks 16, 20 and 22 overlap with this interval indicating persistence of the effect of sapropterin treatment. The confidence intervals at Weeks 16, 20 and 22 of the mean change from baseline blood Phe level were (–291.4 μmol/L, –121.0 μmol/L), (–299.3 μmol/L, –140.5 μmol/L), and (–270.2 μmol/L, –110.8 μmol/L), respectively. Subjects who received a fixed sapropterin dose of 10 or 20 mg/kg/day had comparable mean blood Phe levels at Weeks 12, 16, 20, and 22 as they had previously had on the same dose in the forced dose-titration period. Subjects who received the fixed 5 mg/kg/day dose of sapropterin did not have as low of a mean blood Phe level as observed for the 5 mg/kg/day dose during the forced-dose titration period.
Secondary Efficacy Results During the forced-dose titration period (Week 0 visit until Week 6 visit), the mean blood Phe level observed at the end of each 2 week dosing period was inversely related to the dose of sapropterin. At baseline (Week 0), the mean ± SD blood Phe level was 844.0 ± 398.0 μmol/L. After sapropterin dosing at 5 mg/kg/day (Week 2), 10 mg/kg/day (Week 6), and 20 mg/kg/day (Week 4), mean ± SD blood Phe levels were 743.9 ± 384.4 μmol/L, 639.9 ± 381.8 μmol/L, and 580.8 ± 398.8 μmol/L, respectively. For each pair-wise comparison of the dose levels, the mean change in blood Phe levels differed significantly (p<0.01).
From the Week 6 visit to the Week 10 visit, during which all subjects received 10 mg/kg/day of sapropterin, subjects maintained blood Phe levels similar to those that occurred on the same dose during the dose titration period (Weeks 4 to 6), with a mean ± SD blood Phe level at Week 10 of 645.2 ± 393.4 μmol/L. Comparison of efficacy results of all studies
This section summarises results for the following clinical efficacy objectives for sapropterin treatment of HPA due to PKU: 1) reduction of blood Phe levels, 2) increase in Phe tolerance, and 3) decreases in blood Phe levels from ≥600 μmol/L to <600 μmol/L. Primary analyses for these efficacy objectives are based on results of the pivotal trials, PKU-003 and PKU-006 Part 2. Relevant supportive analyses are from the open label trials, PKU-001, PKU-004 and PKU-006 Part 1.
1) Reduction of Blood Phe Levels Data from PKU-003 and PKU-006 Part 2 are the primary focus due to their placebo controlled study designs. In PKU-001, the mean ± SD decrease in blood Phe level from Day 1 to Day 8 was 99.1± 219.8 μmol/L. In PKU-006 Part 1, the mean ±SD decrease in blood Phe level from Day 1 to Day 8 was 104.5 ± 175.5 μmol/L. In PKU-003, there was a significant (p<0.001) mean decrease in blood Phe levels for the sapropterin group compared to the placebo group. The mean ±SD change in blood Phe levels from baseline to Week 6 was −235.9 ± 257.0 μmol/L for the sapropterin group and 2.9 ± 239.5 μmol/L for the placebo group. Comparable results were observed when considering the change from the Week 0 (last observation prior to treatment) blood Phe level. Treatment with sapropterin tablets caused a significant reduction in blood Phe levels in subjects with uncontrolled HPA due to PKU. In PKU-006 Part 2, the mean ± SD Week 0 (last observation prior to treatment) blood Phe levels were 275.7 ± 135.2 μmol/L for the placebo group. The mean ± SD decrease in blood Phe level from Week 0 to the Week 3 visit (prior to addition of Phe supplement) was 96.6 ±
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 48 of 123

Therapeutic Goods Administration
243.6 μmol/L for the placebo group (p=0.20) and 148.5 ± 134.2 μmol/L for the sapropterin group (p<0.001). Treatment with sapropterin tablets caused a significant reduction in blood Phe levels in subjects with HPA due to PKU controlled on a Phe-restricted diet. While there was not a statistically significant difference between the two treatment groups in change from Week 0 blood Phe level at Week 3, a significant difference was observed in the treatment groups with respect to blood Phe levels (p<0.001). Since all subjects in PKU-006 Part 2 had blood Phe levels controlled on a stable Phe-restricted diet, the reduction in blood Phe levels with sapropterin treatment from the Week 0 to Week 3 visit was consistent with observation of a significant increase in dietary Phe tolerance observed during the subsequent study period in PKU-006 Part 2.
Evaluator’s comment Overall the results from both pivotal trials, PKU-003 and PKU-006 Part 2, demonstrated efficacy of sapropterin tablets to reduce blood Phe levels in patients with (uncontrolled or diet-controlled) HPA due to PKU.
It is important to note that in the pivotal efficacy studies only patients who satisfied criteria for response to BH4 were enrolled. The indication in the Product Information reflects this, that is, sapropterin should only be indicated for patients responsive to such treatment.
2) Increase in Dietary Phe Tolerance The mean ± SD daily dietary Phe intake at the Week 0 visit (pre-treatment before treatment with sapropterin) was 14.713 ± 7.471 mg/kg/day for the placebo group and 15.660 ± 7.159 mg/kg/day for the sapropterin group. The mean ± SD daily dietary Phe intake (excluding Phe supplement) while subjects were maintaining adequate blood Phe control was 17.709 ± 8.331 mg/kg/day for the placebo group and 19.154 ± 13.480 mg/kg/day for sapropterin group. There was not a statistically significant (p>0.384) treatment effect on the change from pre-treatment daily dietary Phe intake, excluding Phe supplement, when subjects were maintaining adequate blood Phe control, that is, the dietary Phe intake excluding Phe supplement was not statistically different between the two treatment groups. The mean ± SD change in dietary Phe intake, excluding Phe supplement, from Week 0 was 1.817 ± 4.857 for the placebo group compared to 0.574 ± 3.847 mg/kg/day for the sapropterin treatment group. The mean ± SD Phe supplement tolerated by subjects in the sapropterin treatment group was 20.9 ± 15.4 mg/kg/day, which was significantly different from 0 (p<0.001), as compared to 2.9 ± 4.0 mg/kg/day for the placebo group. There was a significant difference between the effect on Phe supplement tolerated of these 2 treatments (p<0.001, sapropterin vs placebo). It should be noted however, when subjects were maintaining adequate blood Phe control, the mean total daily dietary Phe intake, including study-prescribed Phe supplement, was 18.889 ± 9.698 mg/kg/day for the placebo group and 38.406 ± 21.606 mg/kg/day for the sapropterin group. In addition, the change from pre-treatment total dietary Phe intake in the placebo group was significantly different from the change in the sapropterin treatment group. The mean ± SD change from pre-treatment total daily dietary Phe intake, including study prescribed Phe supplement when subjects were maintaining adequate blood Phe control was 3.259 ± 5.291 for the placebo group compared to 17.513 ± 13.628 mg/kg/day for the sapropterin treatment group.
Evaluator’s comment Results supported that treatment with sapropterin resulted in a significant increase in dietary Phe tolerance in individuals with HPA due to PKU controlled on a Phe restricted diet.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 49 of 123

Therapeutic Goods Administration
3) Reduction in Blood Phe to <600 μmol/L For this efficacy endpoint, PKU-003 is the primary focus due to its placebo controlled study design. In PKU-003, 38/47 subjects in the placebo group and 31/41 subjects in the sapropterin treatment group had a baseline blood Phe level ≥600 μmol/L (baseline blood Phe level was defined as the mean of blood Phe levels after the screening visit and before the first dose of study drug). At the end of the 6 week treatment period, 5/38 (13.2%) of these placebo subjects and 13/31 (41.9%) of these sapropterin treated subjects had a blood Phe level <600 μmol/L (p=0.012, sapropterin vs placebo).
In PKU-001, 428 subjects had a baseline (day 1, prior to treatment) blood Phe level ≥600 μmol/L. Of this group, 68/428 (15.9%) achieved a blood Phe levels of <600 μmol/L after 8 days of treatment with sapropterin tablets, 10 mg/kg/day.
Evaluator’s comment These results from PKU-003 and PKU-001 indicate that a large proportion of subjects with uncontrolled HPA due to PKU (blood Phe level ≥600 μmol/L) can achieve a blood Phe level within a selected therapeutic range (<600 μmol/L) during treatment with sapropterin tablets. Results in subpopulations
Age Based on available data there was no evidence for an age-related effect of sapropterin to reduce Phe levels in patients with HPA due to PKU.
Gender In PKU-001, PKU-003, and PKU-006, overall, there were no major differences between male and female subjects in reductions in mean blood Phe during treatment with sapropterin 10 or 20 mg/kg/day. In PKU-003 there was a significant difference between placebo and sapropterin 10 mg/kg/day for male subjects only (p=0.016). Clinical efficacy in HPA due to PKU (0 to 4 years of age)
The application contained literature reports to support use of the product in the following patient populations:
· Patients with Hyperphenylalaninaemia due to PKU 0-4, years old, and · Patients with Hyperphenylalaninaemia due to BH4 deficiency. A methodology was prepared and approved by the sponsor before any search was run. The search strategies were developed by a trained information retrieval specialist and agreed to with the TGA’s Office of Prescription Medicines.
Two searches were conducted separately for the two patient populations but were then consolidated into a single list of abstracts that covered both due to the closeness of the conditions and their possible overlap (some articles reported on patients with PKU and patients with BH4 deficiency).
A total of 706 abstracts were identified as a consolidated output. Abstract screening was performed according to pre-determined exclusion criteria and resulted in 177 articles selected for assessment. During assessment and consideration of the articles retrieved, the following criteria were documented:
· Presence of Efficacy data If present, are the outcome measures relevant to the proposed indication?
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 50 of 123

Therapeutic Goods Administration
· Presence of Safety data · Number of subjects · Study design · Level of evidence (MERGE) - As defined in Appendix-2 of the TGA Literature Based Submissions: Points to Consider · Ethics certification. The systematic review of the published literature was performed and articles selected for inclusion that were intended to provide support for the clinical efficacy of sapropterin tablets in the treatment of PKU for patients in the 0 to 4 year old age were reviewed; all of which treated patients for ≥2 months, for a combined total of at least 54 completed patients ≤4 years old. Publications providing only short-term data add to the support of treatment in PKU patients aged 0 to 4 years, in particular those with high numbers of patients. A total of 2,714 patients with PKU aged ≤4 years completed the short-term only studies.
Long-term data Only the studies providing the most important data are presented.
Baldellou Vazquez et al. reported a study of 20 patients with HPA due to PKU.28 Five of these patients were £4 years of age and underwent a combined single dose-loading test with Phe (100 mg/kg) and a non-pharmaceutical preparation of BH4 (20 mg/kg; Schircks Laboratories, Switzerland); 4 of these patients were classified as responders and received 7, 7.5 or 15 mg/kg/day as long term treatment (range: 2 to 15 months). The response to the test was considered to be positive if Phe levels decreased by 30% 8 hours or 50% 24 hours after the BH4 dose. Outcomes of interest included blood Phe levels and diet liberalisation.
In the short term group (N=5) results were as follows: Loading test: Blood Phe at diagnosis, % reduction at 8 hours and 24 hours post-load
Pt 5 (1 yr. old): 1800 nmol/mL, 9%; 32% Pt 12 (5 days old): 915 nmol/mL, 41%; 73% Pt 13 (14 mo. old): 803 nmol/mL, 65%; 90% Pt 14 (4 yrs. old): 787 nmol/mL, 60%; 92% Pt 20 (18 mo. old): 387 nmol/mL, 60%; 89% With long-term treatment (N=4) results were as follows:
Blood Phe level after treatment; diet liberalisation Pt 12: 200 nmol/mL; liberalised diet (treated 2 months) Pt 13: 186 nmol/mL; liberalised diet (treated 3 months) Pt 14: 240 nmol/mL; free diet (treated 15 months) Pt 20: 196 nmol/mL; liberalised diet (treated 3 months) Belanger Quintana et al. reported a study that included 14 patients with PKU during 2000-2005 and 36 patients with PKU who were observed in follow up care (total N = 50).29
Nineteen of these 50 patients responded to a single dose of a non-pharmaceutical preparation of BH4 (20 mg/kg; Schircks Laboratories, Switzerland), and 8 of those 19 were £4 years of age; 3 of these patients received this same preparation of BH4 10, 15, or 20 mg/kg/day as long term treatment (range: 5 to 10 months). Response to the BH4 challenge was considered to be positive if Phe levels decreased by >30% 8 hours after the BH4 dose (patients were
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 51 of 123

Therapeutic Goods Administration
considered slow responders when >30% decrease occurred 12-16 hours after the BH4 dose). Outcomes of interest included blood Phe levels and Phe tolerance.
In the short term group (N=8) results were as follows: Loading test: Blood Phe at diagnosis, % reduction at 8 hours post-load
Pt 18930 (neonate): baseline 250 mM, 70% Pt 18811 (neonate): baseline 258 mM, 78% Pt 18801 (neonate): baseline 300 mM, 87% Pt 18447 (neonate): baseline 300 mM, 39% Pt 20115 (neonate): baseline 840 mM, 41% Pt 20136 (neonate): baseline 870 mM, 34% Pt 18236 (neonate): baseline 1130 mM, 29% Pt 16271 (3 years old): baseline 1140 mM, 37%. With long-term treatment (N=3) results were as follows:
Blood Phe level and Phe tolerance before and after BH4 and diet liberalisation
Pt 20115 (8 months old): Phe level: 200 mM to 300 mM; Phe tolerance: 45 to 133 mg/kg/day (treated 5 months) Pt 20136 (8 months old): Phe level: 200 mM to 145 mM; Phe tolerance: 45 to 90 mg/kg/day (treated 5 months) Pt 16271 (3 years old): Phe level: 300 mM to 280 mM; Phe tolerance: 30 to 75 mg/kg/day (treated 10 months). Boneh et al. reported a study that included 10 newborn patients with HPA due to PKU during a 3-year audit.30 All patients underwent a single dose loading test of a non-pharmaceutical preparation of BH4 (20 mg/kg; Schircks Laboratories, Switzerland); 3 of these patients received this same preparation of BH4 7.5 mg/kg/day initially then, 11 or 16 mg/kg/day as long term treatment (for up to 3 years). A >35% decrease in blood Phe level after BH4 load was considered significant. Outcomes of interest included blood Phe levels and Phe tolerance. Short term (N=10) results:
Loading test: Blood Phe at diagnosis; response 24 hours post-load
600-1200 mmol/L; 3/10 babies had >35% decrease Long term (N=3) results:
Controlled at Phe-to-tyrosine ratio less than 10 Pt 1: Natural protein intake 1.2 g/kg/day Pts 2 and 3: 2-3 g protein/day
Burlina et al. reported a study that included 8 patients £4 years of age with PKU who were not fully compliant with a Phe-restricted diet.31 All patients underwent a single dose loading test of a non-pharmaceutical preparation of BH4 (20 mg/kg; Schircks Laboratories, Switzerland); all 8 patients then received this same preparation of BH4 20 mg/kg/day (in 2 doses) as long term treatment (range: 6 months-6 years). Response to the BH4 challenge was considered to be positive if Phe levels decreased by ≥30% 8 hours after the BH4 dose. Outcomes of interest included blood Phe levels and Phe tolerance. Short term (N≥ 8) results:
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 52 of 123

Therapeutic Goods Administration
Data for loading test specifically in all ≤ 4 years old not reported. Overall 63% patients were responders. Loading test in 8 responders ≤ 4 years old: Blood Phe before test; % reduction at 24 hours
Pt 1: 561 mmol/L; 37% Pt 2: 502 mmol/L; 66% Pt 3: 564 mmol/L; 39% Pt 6: 433 mmol/L; 73% Pt 7: 605 mmol/L; 39% Pt 8: 1215 mmol/L; 37% Pt 10: 649 mmol/L; 54% Pt 12: 716 mmol/L; 44%.
Long term (N=8) results (treated for 6 months to 6 years: mean 3.3 years): Phe tolerance before BH4; on BH4:
Pt 1: 400; 1000 mg/day Pt 2: 650; 2700 mg/day Pt 3: 350; 1400 mg/day Pt 6: 370; 1600 mg/day Pt 7: 400; 1000 mg/day Pt 8: 550; 800 mg/day Pt 10: 500; 1200 mg/day Pt 12: 350; 1200 mg/day Feillet et al. reported a study that included 9 neonate patients with HPA due to PKU who underwent a single dose loading test of a non-pharmaceutical preparation of BH4 (20 mg/kg; Schircks Laboratories, Switzerland); 1 patient then received this same preparation of BH4 20 mg/kg/day as long term treatment (3 years).32 Response to the BH4 challenge was considered to be positive if Phe levels decreased by ≥30% 24 hours after the BH4 dose. Outcomes of interest included blood Phe levels and Phe tolerance. Short term (N=9) results:
Loading test: Blood Phe at diagnosis; % change at 24 hours after loading test
Pt 1: 714 mmol/L; - 7.6% Pt 2: 1080 mmol/L; - 42.8% Pt 4: 1518 mmol/L; - 13.0% Pt 5: 834 mmol/L; - 95.0% Pt 6: 1800 mmol/L; + 14.7% Pt 7: 2160 mmol/L; -72.2% Pt 8: 1560 mmol/L; + 3.8% Pt 9: 1146 mmol/L; - 39.3% Pt 10: 1824 mmol/L; - 40.1%
Long term (N=1) results: Phe tolerance: 1500 mg/day
Hennermann et al. reported a study investigating responsiveness and long-term treatment of 40 infants with PKU.33 Initially, a single dose BH4 loading test (20 mg/kg; Schircks Laboratories, Switzerland) was performed. Five patients who were BH4-responsive, with initial blood Phe >1’000 mmol/L and low Phe tolerance (<20 mg/kg/day) received 10 mg/kg
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 53 of 123

Therapeutic Goods Administration
bid (that is, 20 mg/kg qd) as long term treatment (range: 5.5 to 29 months). Response to the BH4 challenge was considered to be positive if Phe levels decreased by >30% 8-24 hours after the BH4 dose. Outcomes of interest included blood Phe levels and Phe tolerance. Genotype findings were also presented.
Short term (N=40) results: Loading test: Proportion of patients responsive; % reduction at 8 hours and 24 hours post-load 18/40 patients were BH4 responsive; defined as >30% reduction in blood Phe 8 to 24 hours after BH4.
At 8 hours: BH4 responsive patients (N=18) had a median blood Phe reduction of 46% (range: 12.3 to 82.6%)
At 24 hours: BH4 responsive patients (N=18) had a median blood Phe reduction of 46% (range: 31.6 to 59.6%)
Loading test: Baseline Blood Phe and % reduction for patients who went on to long-term treatment
Pt 1: 1816 mmol/L, 46.2% (8h) Pt 2: 1459 mmol/L, 34.2% (8h) Pt 3: 1316 mmol/L, 25.4% (8h); 43.8% (24h) Pt 4: 1150 mmol/L, 22.1% (8h); 58.4% (24h) Pt 5: 1077 mmol/L, 82.2% (8h) Long term (N=5) results:
Phe tolerance pre-BH4, on-BH4, without BH4; Median Blood Phe pre-BH4, on-BH4, without BH4
Pt 1 (18 mo. old): 19, 35, 16 mg/kg/day; 143, 299, 309 mmol/L (treated 24 months) Pt 2 (1.2 mo. old): 19, 80, 12 mg/kg/day; 77, 314, 335 mmol/L (treated 29 months) Pt 3 (0.5 mo. old): not done, 40, 17 mg/kg/day; not done, 293, 327 mmol/L(treated 8 months) Pt 4 (0.5 mo. old): not done, 30, 17 mg/kg/day; not done, 190, 246 mmol/L(treated 5.5 months) Pt 5 (42 mo. old): 18, 120, 20 mg/kg/day; 208, 249, 340 mmol/L(treated 24 months). Lambruschini et al. reported a study that investigated BH4 responsiveness in 73 patients with PKU.34 Five of these patients were £4 years of age and responded to a single dose BH4 loading test (20 mg/kg; Schircks Laboratories, Switzerland). Following the single dose loading test, these patients then received this same non-pharmaceutical preparation, 5 mg/kg/day, for 1 year. In this report, one patient treated with BH4 had liberalisation of the Phe-restricted diet by addition of 200 mg Phe per day every week for 2 months, while Phe-free protein supplement was gradually reduced and finally discontinued. A ‘good response’ to the BH4 loading test was defined as a decrease of ≥45% in plasma Phe. Outcomes of interest included blood Phe levels and Phe tolerance. Anthropometric, nutritional, and neuropsychological evaluations were also a part of this study, and genotype findings were presented.
Short term (N=5) results: Loading test: Blood Phe at 0 hours and 21 hours post-load
Pt 1 (0.9 years old): 612 mmol/L; 37 mmol/L Pt 2 (2.2 years old): 950 mmol/L; 153 mmol/L Pt 4 (1.6 years old): 672 mmol/L; 148 mmol/L
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 54 of 123

Therapeutic Goods Administration
Pt 6 (0.8 years old): 668 mmol/L; 185 mmol/L Pt 11(0.2 years old): 737 mmol/L; 365 mmol/L
Long term (N=5) results: Diet liberalisation and median blood Phe level
All 5 patients on Phe-liberalised or free diets. Median blood Phe at or below 360 mmol/L for all 5 patients. Shintaku et al. reported a study regarding diagnosis and treatment of 12 patients with HPA due to PKU.35 Eleven of these patients were £4 years of age. Three different BH4 loading tests (Biopten 2.5% granules) were employed in this study: 1) a single 10 mg/kg dose BH4 loading test, 2) a 4-dose BH4 loading test (10 mg/kg, 10 mg/kg, 5 mg/kg, 5 mg/kg) over a 48-hour period, and 3) a multiple dose loading test (20 mg/kg/day [divided into 3 doses] administered for 1 week). Nine of these patients received 10 to 20 mg/kg/day [divided into 3 doses] as long term treatment (range: 3 to 56 months). Outcomes of interest included blood Phe levels and diet liberalisation.
Short term (N=11) results:
· Proportion of patients with reduction in Blood Phe · Patients 14 to 50 days old at single dose test · Single dose: 10/11 patients had >20% decline · 4 doses over 48h: 6/7 patients had >30% decline · Dosing over 1 week: 5/6 patients had ³50% decline.
Long term (N=9) results:
· Diet liberalisation or cessation in 7/8 patients
Steinfeld et al. reported a study that included 6 patients £4 years of age with BH4-responsive PKU who underwent a single dose loading test of BH4 (20 mg/kg; source unspecified); 2 patients then received this same preparation of BH4 10 or 20 mg/kg/day as long term treatment (2 years).36,37 A blood Phe level decrease cut-off for determination of BH4 responsiveness was not specified. The outcome of interest was blood Phe levels.
Short term (N=6) results: Loading test: Responsiveness: Demonstrated in all 6 patients.
Long term (N=2) results: Blood Phe level at baseline; during BH4 treatment/free diet:
Pt 1: >600 mmol/L (birth); maintained within desired range (60-360 mmol/L) Pt 2: >600 mmol/L (6 months); maintained within desired range (60-360 mmol/L). Ye et al. reported a study of long term treatment in 3 patients with BH4-responsive PKU who received 10 mg/kg/day BH4 (source unspecified) for up to a year.39 Previously, 29 patients with PKU received a single loading dose and were considered BH4 responsive if their blood Phe levels decreased by more than 30% within 24 hours after administration of BH4. 24 BH4-responsive PKU patients received 10 mg/kg/day BH4 for 1 week and 3 of those continued on treatment. Outcomes of interest included blood Phe levels and mental development.
Short term (N=129) results: Loading test: Responsiveness (N=129 PKU):
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 55 of 123

Therapeutic Goods Administration
· 64 (49.6%) were diagnosed as BH4 responsive (that is, more than 30% decrease in blood Phe levels).
· Blood Phe level at baseline; after 1 week BH4 treatment (N=24): 6.8 ± 3.3 mmol/L to 2.9 ± 2.1 mmol/L.
Long term (N=3) results:
· After long-term BH4 treatment (N=3): Blood Phe controlled at a satisfactory level with normal mental development.
Short-term data
The following studies provide short-term data only (single-dose) in >100 patients aged 0 to 4 years: Bernegger and Blau reported a retrospective analysis of 1730 patients with HPA, aged 1 week – 4.6 years (median 3 weeks).40 All patients were loaded with a single dose of a non-pharmaceutical preparation of BH4 (Schircks Laboratories, Switzerland), 20 mg/kg, either the 6R,S-BH4 form (N=1452) or the 6R-BH4 form - which is the subject of this application - in 278 patients. The outcome of interest was response to BH4, determined by the slope of the Phe hydroxylation rate. Loading test: Blood Phe at diagnosis; % positive loading test after 8 hours
Patients aged 1 week – 4.6 years old (median 3 weeks old) Median 1040 mmol/L; In group loaded with 6R-BH4 significantly positive response in 99 patients:
Initial 120-400 mmol/L; 65% Initial 400-800 mmol/L; 74% Initial 800-1200 mmol/L; 33% Initial 1200-1600 mmol/L; 17% Initial 1600-2200 mmol/L; 0% Initial >2200 mmol/L; 10% Fiege and Blau reported a study that investigated BH4 responsiveness in 557 newborns and children with HPA due to PKU by administering a single dose of a non-pharmaceutical preparation of BH4, 20 mg/kg.41 The outcome of interest was blood Phe level. Different cut-offs of 20 to 50% Phe reduction were used to define BH4 responsiveness. Loading test: Proportion of patients responsive at 8 hours and 24 hours post-load
557 patients identified as newborns and children (ages: 1 week to 7 years; median of 2 weeks)
At 8 hours: 48% of patients had a >20% decline; 38% of patients had a >30% decline At 24 hours: 55% of patients had a >20% decline; 46% of patients had a >30% decline.
Fiori et al. reported a study that investigated BH4 responsiveness in 107 infants with HPA due to PKU by administering a single dose of a non-pharmaceutical preparation of BH4, 20 mg/kg.42 The outcome of interest was blood Phe level. Response to the BH4 challenge was considered to be positive if Phe levels decreased by ≥30% 8 hours after the BH4 dose.
Loading test: Proportion of patients responsive 107 patients identified through neonatal screening
· At 8 hours: 91/107 patients were BH4 responsive (³30% decline at 8hs after BH4)
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 56 of 123

Therapeutic Goods Administration
· At 8 hours: 97/107 had >10% decline in Phe At 24 hours: 53/61 had ³30% decline in Phe (Publication contains listing of individual data for all 107 patients.)
Evaluator’s comment The methodology used in the literature search was appropriate. In the literature reports described above, approximately 240 patients were in the ≤4year age group (ages were not clearly stated in all clinical papers). Clinical Efficacy in HPA due to BH4 deficiency
BH4 deficiency is an exceedingly rare disease, estimated to account for only 1-2% of cases of clinically significant HPA. Based on the prevalence data from the Orphan Drug designation application, 16 to 32 patients are estimated to suffer from this disease in Australia. Demonstration of safety and effectiveness for sapropterin in the treatment of HPA due to BH4 deficiency is based on the following:
(1) the study performed for registration of sapropterin 2.5% granules (Biopten) in Japan, (2) a 10-year post-marketing surveillance study for sapropterin 2.5% granules reported by DSP in Japan, (3) the results of an interim analysis at Week 10 for clinical trial PKU-007, designed to further extend the existing safety and efficacy data in this very rare disease, (4) 11 published studies identified via a systematic review, reporting long-term data, (5) 5 published studies identified via a systematic review, reporting short-term data.
Long-term data (1) The Biopten study (D272) reported by Kitagawa et al. evaluated the effectiveness of sapropterin 2.5% granules (Biopten) to decrease blood Phe levels in patients with BH4 deficiency (PTPS, DHPR, or GCH1 deficiency).49
In the original Biopten study, sapropterin treatment began at a dose of 1.5 to 10 mg/kg/day, and the dose was increased or decreased based on clinical symptoms and examination results. Daily dosing ranged from 0.8 to 37 mg/kg/day, with maintenance dosing ranging from 2 to 5 mg/kg/day. Sapropterin was administered for a mean of 15.5 months; 15 subjects were treated for 10 to 20 months and 1 subject was treated for 4 months.
This open-label trial which was used to support Biopten (sapropterin 2.5% granules) registration in Japan, was conducted between 1987 and 1989, and enrolled and treated 16 subjects with BH4 deficiency. One subject was newborn and 5 subjects were under 5 years of age.
Assessment of clinical effectiveness was based on the degree of improvement in blood Phe levels, a meaningful increase in dihydrobiopterin (BH2; indicating oxidation of 6R-BH4) for patients with DHPR deficiency, and decrease of the urine neopterin to biopterin ratio, (indicating 6R-BH4 utilization) for patients with PTPS deficiency. Based on this global improvement rating, there was marked improvement in 87.5% (14/16) of patients and marked or moderate improvement in all 16 patients. When overall improvement, general safety, and other clinical assessments were taken into account, 87.5% (14/16) of patients had a score of very useful, and all 16 patients had a score of very useful or useful. These results are included in the package insert for Biopten 2.5% granules, which received marketing authorisation in Japan in 1992 for treatment of BH4 deficiency and are also reported in the literature. For all 16 subjects, blood Phe levels were lower after treatment with sapropterin and were maintained within the normal range (£3 mg/dL [£181.6 mmol/L]) during the treatment period.
49 Kitagawa T, Yamato T, Arashima S et al. Clinical results of using apropterin hydrochloride (R-
tetrahydrobiopterin) for atypical hyperphenylalaninemia. Jap J Pediatr Med 1990; 22: 1737-1750.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 57 of 123

Therapeutic Goods Administration
(2) A post-marketing surveillance study of the use of sapropterin granules (Biopten) in the treatment of BH4 deficiency was conducted. This study involved a 10-year data collection period, from March 1992 to March 2002, and was reported by DSP in 2004. This study enrolled 30 patients: 16 patients previously treated and evaluated in the original Biopten study. Three patients who started treatment during the pre-approval period for Biopten, and 11 patients newly treated following the approval of Biopten. Of the 30 patients treated, 27 patients were diagnosed with BH4 deficiency and 3 patients were diagnosed with BH4-responsive HPA (that is, PKU) after enrolment in the study. PKU is an off-label indication for Biopten; thus, the efficacy analysis focused on the 27 patients with BH4 deficiency. This post-marketing surveillance study focused primarily on safety and blood Phe reduction as clinical endpoints. In this post-marketing surveillance study, patients were treated with sapropterin 2.5% granules (Biopten) at doses of 2 to ~15 mg/kg/day. Nineteen of 27 patients (70%) received 2 to 5 mg/kg/day. All 30 patients were treated for at least 1 year, with 19 patients being treated for 10 to 20 years. The treatment duration ranged from 16 months to 20 years.
In the Biopten post-marketing surveillance study, global improvement based on blood Phe levels and physical/mental development before and during Biopten treatment was assessed as an efficacy endpoint. For the overall study population with BH4 deficiency, 93% (25/27) of patients were described as having a global improvement of “markedly improved”, “improved”, or “slightly improved.” One patient with DHPR deficiency was described as having a global improvement of “unchanged” and another was classified as “exacerbated”. These 2 patients had also developed neurological symptoms before treatment with sapropterin, and symptoms of epilepsy increased during the surveillance period. Overall, all 22 patients with PTPS deficiency demonstrated a global improvement of “markedly improved”, “improved”, or “slightly improved”, while 3 of the 5 patients with DHPR deficiency demonstrated a global improvement level of “markedly improved” or “improved”. Blood Phe levels were summarised separately for patients with PTPS deficiency (n=22) and DHPR deficiency (n=5). Of the 22 patients with PTPS deficiency, all but 1 patient had a mean decrease in blood Phe level into the normal range (defined in this study as £3.0 mg/dL, i.e. £181.6 mmol/L). Mean ± SD blood Phe levels before treatment were 1,198.7 ± 817.3 mmol/L (19.8 ± 13.8 mg/dL) and decreased to 84.8 ± 36.3 mmol/L (1.4 ± 0.6 mg/dL) at the last evaluation. During the surveillance period, 19 patients with PTPS deficiency maintained normal blood Phe levels, while 3 patients had a blood Phe level that exceeded the upper limit of normal for a least one time point; all 3 of these patients were cited as being noncompliant with treatment.
For the 5 patients diagnosed with DHPR deficiency, a mean ± SD decrease in blood Phe level was observed from 732.5 ± 526.7 mmol/L (12.1 ± 8.7 mg/dL) prior to treatment to 308.8 ± 78.7 mmol/L (5.1 ± 1.3 mg/dL) at last examination, including 1 patient who had an increase in blood Phe level. Although blood Phe levels decreased for 4 of the 5 patients at the last examination, this level did not reach the normal range; for these 4 patients, mean ± SD blood Phe levels were 853.6 ± 520.7 mmol/L (14.1 ± 8.6 mg/dL) before treatment and 296.6 ± 78.7 mmol/L (4.9 ± 1.3 mg/dL) at last examination. Blood Phe levels in some of the patients with DHPR deficiency decreased into the normal range at some point during treatment; however, these levels were not maintained during the entire treatment period.
(3) A clinical trial evaluating the efficacy and safety of sapropterin tablets in the treatment of BH4 deficiency (PKU-007) recently completed and results of an interim analysis at Week 10 were available. PKU-007 was a Phase II, multicentre, open-label study evaluating sapropterin
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 58 of 123

Therapeutic Goods Administration
tablets in subjects with HPA due to BH4 deficiency. The protocol included 3 parts and an extension study. Twelve subjects were enrolled in study PKU-007. Nine of the subjects had defects in enzymes of BH4 biosynthesis (the synthesis group) and three subjects had defects in enzymes involved in BH4 recycling. All subjects were included in the interim efficacy analyses. In Part 1 of PKU-007, subjects were followed for 2 weeks without modification of their baseline medical care (including BH4 treatment) or dietary care. In Part 2 of PKU-007, subjects who were receiving non-registered formulations of BH4 at enrolment suspend this treatment and within one day started sapropterin tablets at approximately the same dose as the non-registered BH4 formulation. Subjects not receiving BH4 at enrolment began treatment with sapropterin tablets at 5 mg/kg/day, divided into two doses, taken prior to the morning and evening meals. At the discretion of the investigator, the sapropterin dose could be adjusted up or down at the Week 6 visit, and at any visit during the extension period. The interim analysis reported data for patients who were treated for 8 weeks. In Part 3 of PKU-007 (Amendment 1), subjects received 10 mg/kg twice daily (bd) for 21 days, then 20 mg/kg once daily (QD) for 28 days. Upon completion of Part 2 and again (if applicable) upon completion of Part 3 of the study, subjects were then given the option to enter into the extension study. At the discretion of the investigator, the sapropterin dose could be adjusted up or down at any visit during the extension period.
For all parts of the study, the maximum allowed dose of sapropterin tablets was 20 mg/kg/day. The efficacy analysis of PKU-007 included assessments of the proportion of subjects whose blood Phe level was <360 mmol/L and the mean blood Phe levels at the Week 10 visit in Part 2 and selected time points for different dosing regimens in Part 3. Safety was assessed as the incidence of adverse events, clinically significant changes in laboratory test results (chemistry, haematology and urinalysis) and assessment of neurological symptoms such as seizures, changes in muscle tone and weakness. The interim analysis at Week 10 was provided for evaluation.
For the whole sapropterin-treated study population, mean ± SD blood Phe remained at levels similar to the baseline value (132.9 ± 134.7 µmol/L) at all study visits during sapropterin treatment (104.1 ± 66.7 µmol/L to 143.2 ± 147.3 µmol/L). The 95% CI for the mean change in blood Phe from baseline spanned 0 at all time points from the Week 4 through the Week 10 visits, indicating that blood Phe remained at levels comparable to baseline during sapropterin treatment. The upper limit of the 95% CI for mean blood Phe was lower at the Week 4, 6, and 8 visits than at baseline, and slightly higher at the Week 10 visit. The highest upper limit of the 95% CIs for mean blood Phe observed at scheduled visits was 236.8 μmol/L. This demonstrates that subjects’ blood Phe level remained below the target of <360 µmol/L during sapropterin treatment.
For the synthesis group, the group of subjects taking non-registered formulations of BH4 prior to switching to sapropterin treatment, mean ± SD blood Phe remained at levels comparable to those observed at baseline (72.2 ± 13.4 µmol/L) at all study visits following the switch to sapropterin treatment (66.6 ± 13.6 µmol/L to 78.2 ± 28.4 µmol/L). For individual subjects in the synthesis group, blood Phe values remained relatively stable during sapropterin treatment.
Mean blood Phe at baseline was substantially higher for subjects in the recycling group, who were not taking any non-registered BH4 formulations at study enrolment or during Part 1, than for subjects in the synthesis group. The response to sapropterin treatment varied in the
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 59 of 123

Therapeutic Goods Administration
recycling group. For the group as a whole, mean ± SD blood Phe was reduced from the baseline value of 315.0 ± 180.9 µmol/L to values ranging from 181.7 ±96.1 µmol/L to 272.7 ± 175.1 µmol/L) at the Week 4, 6, and 8 study visits but was somewhat higher than baseline at the Week 10 visit (347.7 ± 187.7 µmol/L). Because only 3 subjects were in the recycling group, statistics for this group should be interpreted with extra caution. For one subject in the recycling group, all blood Phe levels during sapropterin treatment (79 µmol/L to 345 µmol/L) were below the range reported for the subject prior to initiation of sapropterin treatment (378 µmol/L to 666 µmol/L) and below the target of <360 µmol/L. In the other two subjects, the level of response to sapropterin treatment in the recycling group varied.
Most subjects in the study remained below the blood Phe target of <360 µmol/L during sapropterin treatment. All 9 of the subjects in the synthesis group and 2 or 3 of the subjects in the recycling group (for a total of 11 or 12 of the 12 subjects in the total population) had blood Phe <360 µmol/L at each of the time points during sapropterin treatment.
(4) A systematic review of the published literature was performed and articles selected for inclusion were used to support the clinical efficacy of sapropterin tablets in the treatment of patients with BH4 deficiency. The methodology used for the systematic review was described previously. The primary studies that contribute to the efficacy assessment of BH4 treatment in patients with BH4 deficiency include 11 studies all of which treated patients long term, for a combined total of 198 enrolled and 171 completed patients.
Al Aqeel et al. conducted an open-label study of 10 patients with BH4 deficiency.50
Initial plasma Phe levels for patients ranged 0.5 to 3.0 mM (a normal reading was reported to be <0.12 mM). Levels remained normal or slightly above 0.12 mM whilst on BH4 therapy.
Nine patients were loaded with 20 mg/kg/day for 3-5 days and then treated with 20 mg/kg/day (in equal doses every 6 hours) non-pharmaceutical preparation of BH4 (Schircks Laboratories, Switzerland), for 5 to 24 months. Patients also received neurotransmitter precursors. The main outcomes of interest were blood Phe levels.
Cabalska et al. conducted an open-label study of 10 patients with BH4 deficiency.51
In 8/9 patients, there was a marked decrease in Phe levels after the BH4 loading test. After the start of treatment, in 6 (75%) cases, a marked increase in psychomotor development parameters was observed; in two cases, the score was normal and in four cases it was close to the lower limit of the normal range. In the remaining patients who received treatment, despite initiation of treatment at 5 months and 3 weeks, there was no progress in mental development or IQ.
Nine patients were loaded with an unspecified dose of BH4 and 8 were then treated with 1-5 mg/kg/day BH4 (divided into 3 to 4 courses a day) for 11 months to 18.5 years. Patients also received neurotransmitter precursors (DOPA + carbidopa and 5-hydroxytryptophan). The main outcome of interest for long-term treatment was IQ.
Chien et al. conducted a study from 1991 to 2000 in Taiwan. During this time period, 1,337,490 newborns were screened at the neonatal screening centre at the National Taiwan University Hospital; 31 patients were diagnosed with HPA, 10 with BH4 deficiency and 21
50Al Aqeel A et al, Biopterin-dependent hyperphenylalaninaemia due to deficiency of 6-pyruvoyl tetrahydropterin synthase, Neurology 1991; 41: 730-737. 51 Cabalska B, Nowacka M, Nowaczewska I, Zekanowski C, Zorska K. [Atypical phenylketonuria treatment
effectiveness]. Med Wieku Rozwoj 2002; 6: 193-202.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 60 of 123

Therapeutic Goods Administration
with PKU.52
Nine of 10 patients had a BH4 loading test performed as a diagnostic procedure. Blood Phe levels for these 9 patients were £600 mmol/L at 4 hours, and all 9 patients had normal blood Phe levels within 6 hours of receiving the BH4 dose. As described in the report, all patients had normal blood Phe levels for the first 2 years of life while treated with BH4. After 2 years of age, patients tended to have a mild elevation in blood Phe level during treatment with BH4, perhaps due to an increase in dietary protein. Physical growth and developmental milestones were normal, except for speech delay. IQ tests at 3 years of age showed a mean ± SD full-scale IQ (FSIQ) score of 76 ± 14 (range: 56 to 98), mean verbal IQ score (VIQ) of 75 ± 10 (range: 58 to 92), and performance IQ score (PIQ) of 80 ± 18 (range: 55-106).
A single dose of a non-pharmaceutical preparation of BH4 10 mg/kg was administered as part of the diagnostic process. Patients were started on a low Phe diet, and treatment was started with BH4 (starting dose 3.3 mg/kg/day) a levodopa preparation (200 mg levodopa and 50 mg benserazide per tablet, starting dose 10 mg/kg/day). In addition, 5-HTP was started later at 5 mg/kg/day (drug was not available at start of study). The main outcomes of interest were blood Phe levels, physical growth and development milestones, and IQ score.
IQ scores were also analysed by age at the start of combined BH4 and levodopa treatment. A significant negative correlation was found between younger age at start of BH4/levodopa treatment and PIQ score (PIQ score: r = 0.705, p=0.034), indicating that patients who started BH4/levodopa treatment earlier in life had higher PIQ scores. Dhondt et al. 1987 reported data from a registry started in 1981, where some patients (at least 13) received a single loading dose of BH4 (source unspecified) 7.5 mg/kg.53
The BH4 loading test was positive in all patients with protein-tyrosine-phosphatases (PTPS) and guanosine triphosphate cyclohydrolase GTPCH (N=unknown). The BH4 loading test was positive, with a smaller decrease in Phe levels, in 8/13 patients with DHPR and negative in 5/13 patients with DHPR. Treatment with BH4 led to an increase in Phe tolerance in 14/17 patients with PTPS deficiency and in all 18 patients with DHPR deficiency.
A total of 35 patients were treated with BH4 (source and duration unspecified). Outcomes of interest were blood Phe levels and Phe tolerance.
Jaggi et al. 2008 reported data from an international database of European patients on 33 patients with BH4 deficiency who received a loading dose of BH4 (source and dosage unspecified).54
Blood Phe at newborn screening was 303-2117 μmol/L for the PTPS patients. The BH4 loading test was positive in all 24 tested PTPS patients. Blood Phe at newborn screening was 151-1797 μmol/L for the DHPR patients. The BH4 loading test was positive in 8/9 and negative in 1/9 tested DHPR patients. Developmental delay was also assessed, in 23 PTPS patients and 4 DHPR patients. Of the 23 PTPS patients, 13 were developing according to their age, 3 experienced a slight delay and 7 a moderate delay. Of the 4 DHPR patients, 1 was developing according to their age and 3 were severely retarded. Among those treated with
A total of 27 patients were treated with BH4 (source unspecified) 2- 12 or 2-20 mg/kg/day long-term (range: 2 to 23 years). Outcomes of interest were blood Phe levels and development delay.
52 Chien YH, Chiang SC, Huang A et al. Treatment and outcome of Taiwanese patients with 6-
pyruvoyltetrahydropterin synthase gene mutations. J Inherit Metab Dis 2001; 24: 815-823. 53 Dhondt JL, Farriaux JP, Raux M, Paux E, Bouchery AM, Debersée J. Evaluation of the detection of phenylketonuria and hypothyroidism at the Lille Regional Center. Arch Fr Pediatr 1987; 44: 787-90. 54 Jaggi L, Zurfluh MR, Schuler A et al. Outcome and long-term follow-up of 36 patients with
tetrahydrobiopterin deficiency." Mol Genet Metab 2008; 93: 295-305.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 61 of 123

Therapeutic Goods Administration
BH4, IQ values were reported for 9 PTPS patients and ranged from 43 at 13 years to 118 at 8 years.
Kao et al. 2004 reported treatment of 12 patients (6 to 18 years of age) with BH4 responsive PTPS deficiency with BH4 (source unspecified) 2 mg/kg/day and neurotransmitter precursors.55
Lee et al. reported a retrospective analysis of 9 patients with BH4 deficiency, aged 8 months to 20 years 8 months at time of diagnosis.
The outcome of interest was blood Phe levels. Blood Phe levels were reported to be always kept under 2 mg/dL or 120 μmol/L.
56
Blood Phe levels at diagnosis were reported to be: 234-2340 μmol/L. Serum Phe levels in all 9 patients that completed the study were kept below 120 μmol/L without a Phe-restricted diet. The mean DQ/IQ measured at the first test in patients #4 to #8 was 45.40 ± 13.94, while the final full-scale intelligence quotient (FIQ) was 62.8 ± 3.26 (p=0.042), verbal intelligence quotient 68.4 ± 13.90, and performance intelligence quotient 60.4 ± 8.11. Among these five patients, 2 had reached borderline (FIQ 70–85), 2 mild (FIQ 51–69), and 1 moderate mental retardation (FIQ 35–50).
Patients were treated with BH4 (source unspecified) 2 mg/kg/day initially followed by a mean dose of 1.78 ± 0.85 mg/kg/day, and neurotransmitter precursors. They were followed at the hospital for over 15 years. Outcomes of interest were blood Phe levels and IQ.
Although patients #1 and #2 remained in a state of profound mental retardation, gradual improvements in neurological function were nevertheless observed. Patients with the moderate form of disease (#9 and #10) reached final IQs in the range of borderline mental retardation (FIQ 70–85).
Liu et al. reported a retrospective analysis of 12 patients with BH4 deficiency, aged 20.0 (6.3) days on average at time of diagnosis. Patients received an initial dose of 3-4 mg/kg/day BH4 (source unspecified) and neurotransmitter precursors.57
Excluding 2 patients diagnosed before birth, no correlation was found between initial Phe peak level and FIQ (Pearson r=−0.421; P=0.23). No correlation between age at time of treatment and FIQ was found either (BH4: Pearson r=0.170, P=0.6; 5-hydroxytryptophan: Pearson r=−0.021, P=0.95; levodopa: Pearson r=0.069, P=0.83). The authors repeated the analysis after adding the 10 patients from the Chien et al. 2001 study and this new analysis revealed correlations between age at onset of each medication and full-scale IQ (BH4, Pearson r=−0.655, P=0.001; 5-hydroxytryptophan, Pearson r=−0.780, P<0.001; levodopa, Pearson r=−0.645, P=0.002).
Mean duration of follow-up was 11.7 (3.3) years (range: 7.2- 19.0). Outcomes of interest were blood Phe levels and IQ.
Wang et al. conducted a study from 1992 to 2005 in northern China.58
55 Kao C D, Niu DM, Chen JT et al. Subtle brain dysfunction in treated 6-pyruvoyl-tetrahydropterin synthase
deficiency: relationship to motor tasks and neurophysiological tests. Brain Dev 2004; 26: 93-8.
During this time period, 618 patients with HPA were either diagnosed or treated in the investigators’ outpatient clinic; 38 of these patients were diagnosed with BH4 deficiency. Of these 38 patients, 27 were treated with BH4. A combined Phe and BH4 test was performed (100 mg/kg Phe with 20 mg/kg BH4 administered 3 hours later) as part of the diagnostic process.
56 Lee N C, Cheng LY, Liu TT et al Long-term follow-up of Chinese patients who received delayed treatment for 6-pyruvoyl-tetrahydropterin synthase deficiency." Mol Genet Metab 2006; 86: 128-134.
57 Liu KM, Liu TT, Lee NCF et al. Long-term follow-up of Taiwanese Chinese patients treated early for 6-pyruvoyl-tetrahydropterin synthase deficiency." Arch Neurol 2008; 65: 387-92.
58 Wang L, Yu W, Li X et al. Study on tetrahydrobiopterin deficiency in Northern Chinese population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2006; 23: 275-9. [Chinese]
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 62 of 123

Therapeutic Goods Administration
Following this procedure, patients with BH4 deficiency were treated with BH4 (starting dose: 1 to 5 mg/kg/day), levodopa (starting dose: 5 to 15 mg/kg/day), and 5-hydroxytryptophan (5-HTP; starting dose: 3 to 10 mg/kg/day). The primary outcome of interest was the full-scale development or intelligence quotient (DQ or IQ). Blood Phe levels were also measured over time.
Pre-treatment blood Phe levels were 181 to 2,045 mmol/L. Blood Phe levels were rapidly normalised during the single-dose diagnostic loading test (100 mg/kg Phe with 20 mg/kg BH4 administered 3 hours later), and blood Phe levels were maintained within the normal range for all subjects during subsequent chronic treatment. Depending on patient age, either developmental quotient (DQ) or intelligence quotient (IQ) were measured to assess treatment efficacy, with overall results expressed as DQ or IQ scores using equivalent scoring systems. The mean DQ or IQ was 52±16 before treatment and 79±15 after treatment, indicating improvement in this measure. The DQ or IQ score was also analysed by age at start of treatment. For patients who started treatment at <6 months of age, 94% (15/16) had a normal DQ or IQ score at follow up. For patients who started treatment at >6 months of age, 18% (2/11) had a normal DQ or IQ score at follow up. An inverse correlation was found between DQ or IQ and the age at start of treatment (r= 0.714, p<0.01), with patients who started before age 6 months having essentially normal follow up DQ or IQ. Ye et al. (2002) reported results for 9/11 patients with BH4 deficiency, 21 days to 6 years of age, studied in China.59
Blood levels 3 hours after Phe loading and 6 hours after BH4 loading decreased from 960 to 120 µmol/L in patient 4; from 720 to 360 µmol/L in patient 8; from >1200 to <120 µmol/L in patient 9 and from >1200 to 120 µmol/L in patient 11. On treatment, the blood Phe levels of the 5 reported patients with PTPS deficiency were between 120-240 µmol/L. IQ was reported in 3 patients with PTPS deficiency at last visit: 50 at 1 year and 81 at 4 years for patient 6; 71 at 2.5 years for patient 8 and 69 at 5 years for patient 10.
Four patients received an initial combined loading test of 7.5 mg/kg or 20 mg/kg BH4 (Schircks Laboratories, Switzerland) and Phe. Five patients were then treated with 1-2 mg/kg/day of the same preparation of BH4 for at least 3 years. Outcomes of interest were blood Phe levels and IQ.
Ye et al. (2007) reported a study of long term treatment in 17 patients with BH4 deficiency (PTPS) who received 1-2 mg/kg/day BH4 (source unspecified) in combination with DOPA and 5-hydroxytryptophan.39 Previously, 30 patients with BH4 deficiency received a single loading dose and were considered BH4 responsive if their blood Phe levels decreased by more than 30% within 24 hours after administration of BH4. Outcomes of interest included blood Phe levels and mental development.
Blood Phe levels decreased significantly 2 hours after the BH4 loading test and were down to target levels at 4 hours in 30 patients with PTPS deficiency. The average blood Phe level for the 17 patients with PTPS deficiency who received long-term treatment was 108 ± 78 µmol/L. 96.3 ± 9.7% of patients had satisfactory control of blood Phe levels and good treatment compliance.
Short-term data
5) The following studies provide short-term data in >100 patients with BH4 deficiency:
59 Ye J, Liu X, Xieqin M et al. Screening for tetrahydrobiopterin deficiency among hyperphenylalaninemia
patients in Southern China. Chinese Medical Journal 2002; 115: 217-221.:
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 63 of 123

Therapeutic Goods Administration
Bernegger and Blau reported a retrospective analysis of 189 patients with BH4 deficiency and a median age of 14 weeks.40 All patients were loaded with a single dose of a non-pharmaceutical preparation of BH4 (Schircks Laboratories, Switzerland), median 10 mg/kg, and either the 6R, S-BH4 or the 6R-BH4 form - which is the subject of this application. The outcome of interest was response to BH4, determined by the slope of the Phe hydroxylation rate.
Blau reported data from a registry (1975-1995), where 308 patients with BH4 deficiency were loaded with a single dose of BH4 (source unspecified), 20 mg/kg or 7.5 mg/kg.60
Comparison of results in subpopulations in HPA due to BH4 deficiency
The outcome of interest was blood Phe levels.
This section describes efficacy results by age as described in the Biopten post-marketing surveillance study at the interim analysis at Week 10 for clinical trial PKU-007 and the relevant publications identified by the systematic review that reported long-term data. The post-marketing surveillance study report included descriptive listings by starting age of treatment for patients with PTPS deficiency (22 patients) or DHPR deficiency (5 patients). 14 patients were aged <1 year and 5 patients were aged between 1 and <7 years at the commencement of treatment. The results of this study demonstrated that all 22 of the patients with HPA due to PTPS had normalisation of blood Phe levels and most patients had evidence of clinical improvement. This was particularly apparent for the patients who started treatment at a young age. Nine of 10 patients (90%) who started treatment at <1 year of age were growing normally, while the remaining patient <1 year of age had pre-existing conditions which gradually improved. The small number of patients with HPA due to DHPR limited conclusions for this group.
However, as with PTPS deficiency, age at start of treatment appeared to predict better clinical response. Overall, the efficacy observations within this post-marketing surveillance study confirmed the results from the registration trial (Study D272) for Biopten. Results from the literature are as follows:
1. Of the patients who received treatment, 4 were <1 year old and 5 were 1-4 years of age. Blood Phe levels remained normal or slightly above 0.12 mM whilst on BH4 therapy.50
2. For the patients who received treatment, 4 patients were <1 year old, 3 were between 1 and 3 years old and 1 was 7 years old at diagnosis.51 Of the 4 patients <1 year old, a marked increase in psychomotor development parameters was observed in two of them whilst the other two showed no progress in mental development or IQ. A marked increase in psychomotor development parameters was observed in the 3 patients aged between 1 and 3 years old and in the 1 patient who was 7 years old at diagnosis.
3. Chien et al. analysed IQ scores by age at the start of BH4 and levodopa treatment and age at the start of 5-HTP treatment.52 A significant negative correlation was found between younger age at start of BH4/levodopa treatment and PIQ score (PIQ score: r = -0.705, p=0.034), indicating that patients who started BH4/levodopa treatment earlier in life had higher PIQ scores. A significant negative correlation was also found between age at start of 5 HTP treatment and FSIQ score and PIQ score (FSIQ score: r = -0.714, p=0.031 PIQ score: r
60 Blau N, Barnes I, Dhondt JL. International database of tetrahydrobiopterin deficiencies. J Inherit Metab Dis
1996; 19: 8-14.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 64 of 123

Therapeutic Goods Administration
= -0.771, p=0.015), indicating that patients who started treatment with 5-HTP earlier in life had higher FSIQ and PIQ scores.
4. Blood Phe at newborn screening was 303-2117 µmol/L for the PTPS patients.53 21 of the 24 patients were <1 year old and 3 were 1 to 7 years of age. The BH4 loading test was positive in all patients. Blood Phe at newborn screening was 151-1797 µmol/L for the DHPR patients. 5 of 9 patients were <1 year old and 4 were between 1 and 2 years of age. The BH4 loading test was positive in 8/9 and negative in 1 (7 month old)/9 tested DHPR patients. Developmental delay was also assessed, in 23 PTPS patients and 4 DHPR patients. 20 of the 23 patients were <1 year old and 3 were 1 to 7 years of age at start of treatment. 12 were developing according to their age (1 of which was 6 years old at start of treatment), 3 experienced a slight delay (all <1 year old at start of treatment) and 6 a moderate delay (3 of which were 1 to 7 years of age at start of treatment). Of the 4 DHPR patients, all of whom were ≤1 year of age at start of treatment, 1 was developing according to their age (treatment started at 1 month) and 3 were severely retarded. Among those treated with BH4, IQ values were reported for 8 PTPS patients. 4 were <1 year of age at start of treatment and their IQ at the time of assessment were: 89 at 7 years, 118 (language free test) at 8 years, 71 at 10 years and 100 at 14 years. 5. Serum Phe levels in all 9 patients that completed the study were kept below 120 μmol/L without a Phe-restricted diet.56 The DQ/IQ for patients #4 and #5 who were ≤2 years of age measured at the first test was 33 and 44, respectively, while the final full-scale intelligence quotient (FIQ) was 53 and 64, verbal intelligence quotient (VIQ) 59 and 70, and performance intelligence quotient (PIQ) 53 and 58. Both patients reached mild mental retardation. The DQ/IQ for patients #6, #7 and #8 who were >4 years of age measured at the first test was 32, 65 and 53, respectively, while the final FIQ was 46, 75 and 76, VIQ 50, 81 and 82, and PIQ 53, 69 and 69. 1 patient reached moderate mental retardation. Patients #1 and #2 were ≤2 years of age and remained in a state of profound mental retardation, however gradual improvements in neurological function were observed. Patients with the moderate form of disease were ≥7 years of age (#9 and #10) and reached final IQs in the range of borderline mental retardation. Mental retardation: Borderline = FIQ 70–85; mild = FIQ 51–69; moderate = FIQ 35–50.
6. No correlation between age at time of treatment and FIQ was found (BH4: Pearson r=0.170, P=0.6; 5-hydroxytryptophan: Pearson r=−0.021, P=0.95; levodopa: Pearson r=0.069, P=0.83).57 The authors repeated the analysis after adding the 10 patients from the Chien et al 2001 study and this new analysis revealed correlations between age at onset of each medication and fullscale IQ (BH4, Pearson r=−0.655, P=0.001; 5 -hydroxytryptophan, Pearson r = − 0.780, P<0.001; levodopa, Pearson r=−0.645, P=0.002).
7. Wang et al. analysed DQ or IQ scores by age at start of treatment. For patients who started treatment at <6 months of age, 94% (15/16) had a normal DQ or IQ level.58 However, for patients who started treatment at >6 months of age, 18% (2/11) had a normal DQ or IQ level. A significant negative correlation was found between the development quotient and the age at start of treatment (r= 0.714, p<0.01), with subjects who started before age 6 months having essentially normal developmental or intelligence quotients.
8. Blood levels 3 hours after Phe loading and 6 h after BH4 loading were reported for 4 patients, all of which were <1 year old: levels decreased from 960 to 120 μmol/L in patient 4; from 720 to 360 μmol/L in patient 8; from >1200 to <120 μmol/L in patient 9 and from >1200 to 120 μmol/L in patient 11.59
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 65 of 123

Therapeutic Goods Administration
On treatment, the blood Phe levels of the 5 reported patients (four <1 year old and one 6 year old) with PTPS were between 120-240 μmol/L.
IQ was reported in 3 PTPS patients (all <1 year old at diagnosis) at last assessment visit: 50 at 1 year and 81 at 4 years for patient 6; 71 at 2.5 years for patient 8 and 69 at 5 years for patient 10. 9. All patients were 1 to 12 months of age at initiation of treatment.39 Following treatment, the average blood Phe level was 108 ± 78 µmol/L and 96.3 ± 9.7% of patients had satisfactory control of blood Phe levels and good treatment compliance. Analysis of clinical information relevant to dosing recommendations
Analysis of clinical information relevant to dosing recommendations in HPA due to PKU The proposed indication for sapropterin tablets is treatment of HPA due to PKU or BH4 deficiency. For HPA due to PKU, the recommended starting dose is 10 mg/kg/day, with subsequent adjustments most commonly between 5 and 20 mg/kg/day. The recommended dose interval is once daily. The maximum daily dose studied in the clinical development program was 20 mg/kg/day.
The effect of sapropterin to reduce blood Phe levels in subjects with HPA due to PKU was studied in the pivotal trial PKU-003. The results indicate that treatment with sapropterin tablets at a dose of 10 mg/kg/day can result in a significant reduction in blood Phe levels in subjects with HPA due to PKU.
The efficacy of sapropterin treatment, in the dose range of 5 to 20 mg/kg/day, to reduce blood Phe levels in HPA due to PKU was evaluated in PKU-004. The mean reduction in blood Phe level from Week 0 for each of the sapropterin groups was 100.1 ± 295.2 μmol/L for the 5 mg/kg/day dose (Week 2), 204.1 ± 303.0 μmol/L for the 10 mg/kg/day dose (Week 6), and 263.3 ± 318.2 μmol/L for the 20 mg/kg/day dose (Week 4), indicating a dose-related response of blood Phe reduction within the sapropterin dose range of 5 to 20 mg/kg/day. Safety of sapropterin at 20 mg/kg/day was demonstrated in the pivotal, placebo controlled trial PKU-006 Part 2. These results support the recommended dose range of 5 to 20 mg/kg/day for sapropterin treatment of HPA due to PKU. Once daily dosing of sapropterin for the treatment of PKU is supported by the results obtained from PKU-004 Substudy 01 and PKU-004 Substudy 02. In PKU-004 Substudy 01, all 8 subjects whose 8:00 am blood Phe level prior to a scheduled daily dose (during the period between the Week 6 and Week 10 visits) was at least 30% lower than at Week 0 (prior to treatment) in PKU-004 had a sustained reduction of blood Phe over the 24-hour period following a dose of sapropterin, 10 mg/kg/day. The study demonstrated a stable reduction of blood Phe over a 24-hour period following a daily oral dose of sapropterin, 10 mg/kg.
In PKU-004 Substudy 02, a population PK study, alpha half-life values were estimated at 0.578 to 2.94 hours. In the terminal elimination phase, the mean half-life was estimated to be 6.69 hours (range: 3.91 to 16.6 hours). Assuming 4 half-lives to achieve clearance, average coverage would be estimated at 26.8 hours for a single dose. Therefore, data from this population PK study support once-daily dosing. In addition, stochastic simulations revealed no evidence of accumulation with daily doses of sapropterin tablets over the 5 to 20 mg/kg dose range. The lack of accumulation with daily dosing is supported by PK studies for sapropterin granules in healthy volunteers (Studies P1501 and FB1602).
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 66 of 123

Therapeutic Goods Administration
Analysis of clinical information relevant to dosing recommendations in HPA due to BH4 deficiency For treatment of HPA due to BH4 deficiency, the dose recommendation for sapropterin is a starting dose of 2 to 5 mg/kg/day, given once daily. Doses may be adjusted up to 20 mg/kg/day and it may be necessary to divide the total daily dose into 2 or 3 doses to optimize the therapeutic response. Due to the extreme rarity of this condition, dosing recommendations for sapropterin tablets are based on 2 studies of Biopten treatment of BH4 deficiency (Biopten post-marketing surveillance study and the interim analysis at Week 10 for clinical trial PKU-007) and 11 studies in the published literature, identified by systematic review, which reported long-term data.
Evaluator’s comment It is considered that the data submitted adequately support the proposed dosage regimens for treatment of patients with HPA due to PKU and BH4 deficiency. Persistence of efficacy and/or tolerance effects in HPA due to PKU
The persistence of efficacy of sapropterin to reduce blood Phe levels in HPA due to PKU is supported by PKU-004 (22-week, open-label study), and is supported by PKU-003 (6-week, placebo controlled study). Evaluator’s overall conclusions on clinical efficacy
The results from the pivotal studies do support that sapropterin causes a dose-related reduction in Phe blood levels. Results in study PKU-003 demonstrated short term efficacy of sapropterin at a dose of 10 mg/kg/day in patients 8 to 49 years. In addition, results from PKU-006 demonstrated efficacy of sapropterin tablets to reduce blood Phe levels with blood Phe levels controlled on a Phe-restricted diet, in patients from 4 years old. Data from PKU-004 demonstrated that reduction in blood Phe in patients with BH-4-responsive PKU can be maintained for 22 weeks. The clinical development program included adults and children with HPA due to PKU who were ≥4 years old. Younger children were not included, in part due to the difficulty of maintaining a constant daily dietary Phe intake in children under the age of 4 years.
In the submission, patients treated for one week in open-label uncontrolled studies included 50 patients who were 4 to 8 years old, and 104 patients who were 8 to 12 years old. Twenty four patients aged 4 to 8 years, and 38 patients aged 8 to 12 years were included in double-blind placebo-controlled trials. Fifteen paediatric patients were treated for up to 22 weeks in open-label uncontrolled trials. Given the rarity of the clinical condition it is considered that short term efficacy has been demonstrated in paediatric patients aged 4 years and older.
For HPA due to BH4 deficiency, a very rare condition, the available data from the Biopten registration and post-marketing surveillance trials and published series, demonstrate efficacy of sapropterin treatment to reduce, and in most cases normalise, blood Phe levels, thereby reducing or eliminating the need for dietary Phe restriction. In addition, persistence of response to treatment with sapropterin was demonstrated in a 10 year post-marketing surveillance study. It should be noted that no long term efficacy data have been presented.
In conclusion, overall the data submitted for evaluation support that sapropterin is efficacious in reducing blood Phe levels and increasing dietary Phe tolerance in patients with HPA due to PKU or BH4 deficiency.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 67 of 123

Therapeutic Goods Administration
Safety Introduction
Safety data were presented separately for subjects with HPA due to PKU or HPA due to BH4 deficiency. Given the differences in these two populations, there was no pooling of the safety data obtained from studies in PKU and BH4-deficient subjects. Healthy volunteers
Exposure data for sapropterin tablets in healthy volunteers were reported from study PKU-005. In study PKU-005, 27 of 28 subjects enrolled were administered weight-related doses of sapropterin in all four assigned dosing periods. Safety data for the studies of sapropterin granules in healthy volunteers were reported in the individual report for studies FB1602, FB1701 and P1501/PHN-111. The most relevant and important safety data are from the studies in subjects with HPA, therefore this evaluation report will focus on safety data reported from those studies. PKU subjects
In the submission, the analysis of adverse effects (AEs) in HPA due to PKU focused on those events reported from the placebo-controlled trials, PKU-003 and PKU-006 Part 2, but also considered AEs reported from the open-label, short-term exposure studies, PKU-001 and PKU-006 Part 1, and from the open-label, long-term exposure study, PKU-004. Data were presented by individual study as well as combined (“pooled” data) according to study type (placebo-controlled or open-label) and duration of treatment (short or long-term). Consequently, data for the placebo-controlled trials, PKU-003 and PKU-006 Part 2, were combined and data for the open-label, short-term exposure studies, PKU-001 and PKU-006 Part 1, were combined. No pooling of data was done for study PKU-004.
In addition the sponsor submitted safety data from an ongoing study: PKU-008. Data from an interim study report were provided.
PKU-008, Interim Study Report This was an ongoing Phase IIIb, multicentre, open–label extension study performed to assess the long-term safety of sapropterin in subjects with PKU who participated in studies PKU-004 or PKU-006. An interim Clinical Study Report was available for the data collection period ending 30 May 2008. As the study was open-label and not yet complete at the time of evaluation, data from the report have not been integrated into the safety analysis and are presented separately.
Patient exposure PKU subjects randomised to sapropterin in the two placebo-controlled studies, PKU-003 and PKU-006 Part 2, were treated with sapropterin tablets 10 mg/kg/day for 6 weeks and sapropterin 20 mg/kg/day for 10 weeks, respectively.
In the open-label studies PKU-001 and PKU-006 Part 1 subjects were treated with sapropterin tablets for 8 days at doses of 10 mg/kg/day and 20 mg/kg/day, respectively. In the first part of study PKU-004, subjects underwent a forced dose-titration receiving sapropterin tablets in three consecutive 2-week courses of daily single oral doses of 5 mg/kg/day, followed by 20 mg/kg/day, and finally, 10 mg/kg/day, and were then treated for an additional 4 weeks at 10 mg/kg/day. In the second part of the study, subjects were treated with sapropterin tablets for 12 weeks at a fixed dose within the range of 5 to 20 mg/kg/day, based upon each subject’s Phe levels at the Week 2 and Week 6 visits during the forced dose-titration period.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 68 of 123

Therapeutic Goods Administration
Overall, 579 subjects have received one or more doses of sapropterin tablets in a PKU clinical study. Complete dosing data were available for 575 of the 579 subjects who received one or more doses of sapropterin tablets. Four subjects with incomplete dosing data are excluded from the analysis.
In accordance with the enrichment design of the studies, the majority of treated subjects (79.7% [458/575]) were exposed to sapropterin tablets for fewer than 10 days, with most of these subjects enrolled in study PKU-001. Of those who experienced a longer treatment period, only a small number were treated for either less than 60 days (1.2% [7/575]) or longer than 209 days (0.7% [4/575]). Thus, most subjects who continued treatment beyond studies PKU-001 and PKU-006 Part 1 (beyond 10 days), received between approximately 2 and 7 months (60 to 209 days) of treatment. Subjects who were treated for between approximately 2 and 7 months (18.4% [106/575]) had the highest total exposure to sapropterin, representing approximately 77% of the total subject-months of exposure and approximately 76% of total mg, or 79% of mg/kg, of sapropterin administered. Among the 106 most highly exposed subjects, those receiving between approximately 2 and 7 months of treatment, there was a relatively equal distribution of subject ages: 40 subjects aged ≥4 to <12 years, 31 subjects aged ≥12 to <18 years, and 35 subjects aged ≥18 years.
PKU-008, Interim Study Report During the study period included in the interim report, subjects had received up to 18 months of sapropterin treatment. All subjects received open-label sapropterin, regardless of whether they received sapropterin or placebo in previous studies. The dose of sapropterin was individualised for each subject within a range of 5 mg/kg/day to 20 mg/kg/day to control blood Phe concentrations; subjects previously enrolled in PKU-004 received a dose equivalent to that last prescribed (or optimised) in the previous study, whereas subjects previously enrolled in PKU-006 Part 2 began PKU-008 at 20 mg/kg/day.
Adverse events Treatment-emergent events were defined as those which began on or after the date of the first study treatment and up to 30 days after the date of the last study treatment, or prior to the first dose in a subsequent study, whichever came first. Because some subjects participated in multiple studies, each event was assigned to the study in which it first began and, even if it continued into a subsequent study, the event was counted just once.
In the placebo-controlled trials, common AEs were defined as those occurring in at least 3 subjects treated with sapropterin tablets and the most common AEs are defined as those events occurring in ≥5% of subjects treated with sapropterin tablets. Tables 11 and 12 summarise the most common treatment emergent adverse events for the combined analysis and for the individual placebo-controlled trials, respectively, the most common AEs that were also reported in excess over placebo, where “in excess over placebo” was defined as the proportion of sapropterin-treated subjects having reported the event exceeded the proportion of placebo subjects reporting the same event by ≥3 percentage points.
In the open-label studies, the most common AEs were defined as those reported by at least 3% of subjects. Additional summaries of the most common AEs in the open-label, short and long-term exposure, studies are provided in Tables 13 and 14 respectively. Table 11: Treatment-Emergent Adverse Events in the Combined Placebo-Controlled Trials, PKU-003 and PKU-006 Part 2, Occurring in ≥5% of Sapropterin-Treated Subjects and in Excess over Placebo
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 69 of 123

Therapeutic Goods Administration
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 70 of 123

Therapeutic Goods Administration
Table 12: Treatment-Emergent Adverse Events in the Placebo-Controlled Trials, PKU-003 or PKU-006 Part 2, Occurring in ≥5% of Sapropterin-Treated Subjects and in Excess over Placebo
Table 13: Treatment-Emergent Adverse Events occurring in ≥3% of Sapropterin-Treated Subjects in the Open-Label, Short-Term Exposure Studies, PKU-001 and PKU-006 Part 1, Combined and by Individual Study
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 71 of 123
Therapeutic Goods Administration
Table 14: Treatment-Emergent Adverse Events Occurring in ≥3% of Sapropterin-Treated Subjects in the Open-Label, Long-Term Exposure Study, PKU-004a,b
In the combined, placebo-controlled trials, the treatment-emergent adverse events which were reported by ≥5% of subjects treated with sapropterin tablets and at a rate which was in excess over placebo, included: rhinorrhoea, pharyngolaryngeal pain, diarrhoea, and contusion. Within the individual placebo-controlled studies, the treatment-emergent adverse events which were reported by at least three sapropterin-treated subjects and at a rate which was in excess over placebo in either one of the studies included: rhinorrhoea, pharyngolaryngeal pain, diarrhoea, headache, nasal congestion, vomiting and cough. In the open-label, short-term exposure studies, PKU-001 and PKU-006 Part 1, the most commonly reported events (≥3% of subjects) included headache, abdominal pain, diarrhoea, nausea and upper respiratory tract infection.
The events reported by four or more subjects (≥5%) in the open-label, long-term exposure study, PKU-004 included: headache, pharyngolaryngeal pain, nasopharyngitis, vomiting, diarrhoea, upper respiratory tract infection, cough, migraine, back pain, gastroenteritis and influenza.
Common adverse events reported by the sapropterin-treated subjects and in excess over placebo (that is, where the proportion of sapropterin-treated subjects reporting the event exceeding the proportion of placebo subjects reporting the same event by at least 3 percentage points) in either the combined or one of the individual placebo-controlled trials, included headache, rhinorrhoea, pharyngolaryngeal pain, vomiting, diarrhoea, nasal congestion, cough and contusion.
Across studies, nearly all reported events were mild or moderate in severity. The proportion of severe events was low in each study and no differences were seen between studies. Severe events were reported with the following Preferred Terms (PTs): headache, abdominal pain, diarrhoea, migraine, tooth abscess and vomiting. Two severe events of headache were reported. All other events represented single reports.
In most studies, the majority of events were assessed as unrelated to the study treatment by the investigator. In the combined placebo-controlled trials, PKU-003 and PKU-006 Part 2, 71.9% (110/153) of events in the sapropterin treatment group and 66.7% (80/120) of events
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 72 of 123

Therapeutic Goods Administration
in the placebo group were assessed as unrelated. Similarly, in the open-label, long-term exposure study, PKU-004, 68.1% (175/257) events were considered as unrelated. Across studies, the events that were most frequently reported as related to the study treatment included diarrhoea, abdominal pain and headache.
Headache and diarrhoea were more frequently reported among the sapropterin-treated subjects than the placebo subjects in the pooled analysis of the placebo-controlled trials, as previously described. Abdominal pain was more frequently reported by placebo subjects (8.5% [5/59]) versus subjects treated with sapropterin (5.4% [4/74]) in the pooled analysis. In study PKU-006 Part 2, a slightly higher percentage of sapropterin-treated subjects (9.1% [3/33]) versus placebo (8.3% [1/24]) reported abdominal pain, while in study PKU-003 the opposite occurred, abdominal pain was reported by 8.5% [4/47] of placebo subjects versus 2.4% [1/41] of sapropterin-treated subjects. Similar to the other gastrointestinal events, the observed abdominal pain may be associated with the acidity of the dissolved drug. PKU-008, Interim analysis
Seventy-nine of 111 subjects (71.2%) receiving sapropterin experienced at least 1 AE during the study report period. The incidence of AEs was highest in the Infections and Infestations System Organ Class (SOC): 133 AEs in 59 (53.2%) of 111 subjects who received any amount of sapropterin, Twenty seven AEs in the Infections and Infestations SOC reported for 11 (9.9%) of the 111 treated subjects were classified as possibly or probably related to study treatment.
The AE reported for the highest number of subjects was cough in 18 (16.2%) subjects, followed by pyrexia in 16 (14.4%) of subjects and nasopharyngitis in 15 (13.5%) of the subjects. These three events could be considered signs and symptoms of common communicable infections. This result would be expected in this relatively young subject population (16.4 ± 10.2 years) who were followed over the course of approximately 2 years. Most AEs were classified by the investigator as mild; the mild AEs were experienced in 43 of 110 subjects while receiving dissolved sapropterin and 15 of 54 subjects while receiving intact sapropterin. Severe AEs were experienced by three subjects while receiving dissolved tablets (testicular mass; broken ankle; and difficulty concentrating and mood swings) and one subject while receiving intact tablets (blood in the urine and lower abdominal pain). Only the difficulty concentrating and mood swings were classified as possibly related to study drug treatment; all other severe AEs were considered unrelated to study drug treatment.
Serious adverse events and deaths No deaths were reported in the studies of sapropterin tablets in PKU.
Among the placebo-controlled studies, there were two serious AEs (SAEs) reported, both of which occurred in PKU-006 Part 2:
− A 4 year-old female was hospitalised for a streptococcal infection of the throat (“strep throat”) approximately 2 months after starting treatment with sapropterin 20 mg/kg/day in Part 2 of PKU-006. This event was considered unrelated to study treatment. Treatment with sapropterin was continued.
− A 7 year-old female was hospitalised for acute appendicitis. This event was considered unrelated to study treatment. Treatment with sapropterin was continued.
In the open-label, short-term exposure studies, one SAE was reported which occurred in a subject who had participated in PKU-001:
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 73 of 123

Therapeutic Goods Administration
− A 14 year-old female was hospitalised for acute appendicitis. This event was considered unrelated to study treatment.
In the open-label, long-term exposure study PKU-004, three subjects each experienced one SAE during the study:
− A 15 year-old male was hospitalised for severe back pain (PT = spinal cord injury) during a physical exercise class, approximately 3.5 months after starting treatment in PKU-004. This event was considered unrelated to study treatment. Treatment with sapropterin was continued.
− A 13 year-old female was hospitalised for a urinary tract infection approximately 4 months after starting treatment in PKU-004. This event was considered unrelated to study treatment. Treatment with sapropterin was continued. − A 13-year-old male subject was hospitalised for a broken tibia secondary to a fall approximately 22 weeks after starting treatment in PKU-004. This event was considered unrelated to study treatment. The event occurred on the day of the subject’s last dose.
PKU-008, Interim analysis Four subjects reported SAEs and only one was considered to have an SAE possibly or probably related to study treatment - gastroesophageal reflux while taking dissolved tablets.
Laboratory findings Within the individual clinical studies, PKU-001, PKU-003, PKU-004 and PKU-006 Parts 1 and 2, no particular trend emerged with respect to laboratory abnormalities.
Within the placebo-controlled trials, PKU-003 and PKU-006 Part 2, a small percentage of subjects reported one or more AEs related to an abnormal laboratory result in both the placebo group (5.1% [3/59]) and the sapropterin-treated subjects (2.7% [2/74]). Each type of abnormality was reported just once in either treatment group and no particular pattern was seen in the events reported. AEs related to laboratory abnormalities were reported in a similar proportion of subjects treated with sapropterin in the open-label, short-term exposure studies, PKU-001 and PKU-006 Part 1. The following events were reported infrequently (< 1% of subjects): aspartate aminotransferase (AST) increased (0.7% [4/579]), alanine aminotransferase (ALT) increased (0.5% [3/579]), and blood cholesterol increased (0.5% [3/579]). All other AEs related to clinical laboratory abnormalities were reported by 2 or fewer subjects. Within subjects participating in the open-label, long-term exposure study, the incidence of AEs related to laboratory abnormalities was higher at 15.0% (12/80 subjects). In this study, events noted by 2 or more subjects included ALT increased (3.8% [3/80]), blood amino acid level increase (2.5% [2/80]), blood urine present (2.5% [2/80]) and neutrophil count decreased (2.5% [2/80]). All other AEs related to laboratory abnormalities were reported by only one subject.
PKU-008, Interim analysis No clinically significant results or clinically meaningful trends in change from baseline during treatment through the Month 18 visit were evident from the shift tables for the serum chemistry, haematology, or urinalysis through the Month 18 visit.
Evaluator’s comment Overall, analyses of clinical laboratory data did not reveal any adverse effects associated with treatment with sapropterin tablets. The potential signal for ALT and AST elevations observed in the early Phase I studies was not observed in the later clinical trials. The clinical trial data
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 74 of 123

Therapeutic Goods Administration
showed relatively constant values over time. No clinically meaningful changes were seen in the haematology values over time.
Specific safety issues Low blood Phe levels
Because sapropterin treatment has the potential to reduce blood Phe levels below the desired therapeutic target, an analysis was conducted across studies to determine the number of subjects experiencing Phe levels below the paediatric normal range (≤26 μmo l/L), below the German consensus lower target or adult normal range (≤40 μmol/L), and below the most common lower target (≤120 μmol/L). Hypophenylalaninaemia, defined as a blood Phe level at or below the lower limit of the normal range, which in this analysis was 26 μmol/L, was observed to occur more frequently in subjects treated with sapropterin versus placebo. Because the potential exists for Phe levels to drop below either the normal or desired therapeutic levels, careful monitoring may be necessary during therapeutic adjustments of sapropterin doses and dietary Phe levels.
PKU-008, Interim analysis Blood Phe concentrations fell below what was considered the lower limit of normal for healthy individuals (26 µmol/L) or for patients with PKU (120 µmol/L) only sporadically. None of these events were reported as AEs or were determined to be clinically significant; all resolved without intervention. BH4 Deficiency
Safety data in relation to subjects with HPA due to BH4 Deficiency were provided from studies PKU-007 (interim report), study D272 and from post-marketing surveillance data using Biopten 2.5% granules, a formulation of sapropterin.
Study PKU-007, Interim analysis at Week 10 Further data are derived from an interim Clinical Study Report of PKU-007, a study sponsored by BioMarin Pharmaceutical Inc. in California, USA. This was an open-label study performed to evaluate the safety and efficacy of sapropterin in subjects with HPA due to primary BH4 (BH4) deficiency. At Week 10, an interim analysis was performed and an interim Clinical Study Report was available.
Patient exposure Study D272 and post-marketing data
Most subjects (87.5%) in the BH4 deficiency study, D272, were treated with sapropterin granules at a dose of 1 to 5 mg/kg/day; 2 subjects (12.5%) received variable doses. The duration of study treatment varied widely, from 140 to 606 days. The mean duration of sapropterin granules treatment ( ±SD) was 458.3 ± 130.6 days (median 497.0 days). Most subjects in the study (93.8%) were treated for ≥210 days; 1 subject (6.3%) had a treatment duration of ≥60 to 169 days.
Study PKU-007, Interim analysis at Week 10 Twelve subjects received at least one dose of study drug. The administered mean daily dose of sapropterin was 6.6 ± 5.3 mg/kg/day (median: 5.4 mg/kg/day, range: 2.0 to 20.3 mg/kg/day). The mean duration of exposure ± SD throughout Week 10 (that is, through the 8 weeks of sapropterin treatment in Part 2) was 56.8 ± 0.8 days (median 57 days, range: 55 to 58 days). The most recent daily dose taken prior to Week 10 visit (the cut-off date for this interim report) was approximately 2.5 mg/kg/day for 4 subjects (33.3%), approximately 5
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 75 of 123

Therapeutic Goods Administration
mg/kg/day for 5 subjects (41.7%), approximately 10 mg/kg/day for 2 subjects (16.7%), and approximately 20 mg/kg/day for one subject (8.3%).
Adverse events Study D272
AEs were reported for 2 of the 16 subjects in this study: 1 report of moderate hypothermia, possibly or probably related to study treatment, for 1 subject, and 1 report of mild convulsion with an unknown relationship to study treatment in another subject. The incidence for each AE was 6.3%.
PKU-007, Interim Analysis at Week 10 In Week 10, adverse events (AEs) were reported for nine of the 12 enrolled subjects (75%). These AEs were distributed among 10 different SOCs and 19 different PTs. The highest incidence of AEs occurred in the SOCs Gastrointestinal Disorders (7 subjects, 58.3%), Infections and Infestations (4 subjects, 33.3%) and Respiratory, Thoracic and Mediastinal Disorders (3 subjects, 25.0%).
The most frequently reported adverse events were diarrhoea (4 subjects, 33.3%), vomiting (3 subjects, 25.0%) and sinusitis (2 subjects, 16.7%). All other adverse events were isolated events. AEs classified by the investigator as possibly or probably related to the study treatment were reported for 5 subjects (41.7%) and included diarrhoea (4 subjects, 33.3%) and vomiting (2 subjects, 16.7%). No other drug-related AE was reported for more than one subject.
Most adverse events were mild to moderate in severity. Only one adverse event (dystonia) was considered severe by the investigator but was assessed as not related to study treatment. It resolved within 6 days following treatment with concomitant medication. Two adverse events based on laboratory results were reported, both for the same subject: mild neutropenia and mild blood creatinine increased. Both AEs were considered possibly related to study treatment by the investigator and resolved during the study period without intervention. Transient severe dystonia and moderate dyskinesia (each reported as a single event for one subject and classified as unrelated to sapropterin treatment), and transient mild vertigo (reported as a single event for one other subject and classified as possibly related to sapropterin treatment) were the only AEs reported during this study period that were associated with the pre-specified signs and symptoms of primary BH4 deficiency. The dystonia and dyskinesia resolved after 6 days with treatment with concomitant medication, and the vertigo resolved during the study period without intervention.
Serious adverse events and deaths Study D272 and post-marketing surveillance
No deaths were reported in the clinical study D272 of sapropterin granules in BH4 deficiency. However, 2 deaths were reported in the pre-approval and post-marketing safety surveillance study of sapropterin granules in BH4 deficiency (see below). No SAEs were reported in study D272.
PKU-007, Interim Analysis at Week 10 No death and no serious adverse events were reported.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 76 of 123

Therapeutic Goods Administration
Laboratory findings Study D272
Although clinically significant results and clinically significant changes from screening were sporadically reported in this limited dataset, no specific trends in haematological or chemistry abnormalities were detected. PKU-007, Interim Analysis at Week 10
In PKU-007 no clinically significant changes of sapropterin on liver function were reported up to Week 10. One subject experienced clinically significant elevated percent lymphocytes and reduced percent neutrophils and absolute neutrophils at Week 10. This subject had normal values at baseline and at Week 4 visit. In PKU-007 one subject experienced clinically significant elevated creatinine at the Week 4 visit. The change was transient and resolved without intervention. The subject had a normal creatinine level at baseline and at Week 10. No other clinically significant results or trends in change from baseline during sapropterin treatment through to Week 10 were evident from the shift tables or any other haematology analyte.
Evaluator’s comment Although changes from screening were reported in this limited dataset, no specific trends in serum chemistry abnormalities were detected for study D272 and PKU-007. Safety in special populations
Withdrawal and rebound in HPA due to PKU Withdrawal and rebound were examined in terms of the safety profile during the post-treatment period as well as the effect on Phe values when subjects were withdrawn from treatment. The potential effects of drug withdrawal were evaluated in the context of the placebo-controlled studies, PKU-003 and PKU-006 Part 2, by examining the AEs that occurred following treatment discontinuation (“post-treatment period”). There was no discernable pattern to the events that emerged following discontinuation of sapropterin tablets and no safety concerns relevant to withdrawal effects were identified.
Rebound was examined in the context of study PKU-006 and the break in treatment that occurred between Parts 1 and 2. At Week 0 of Part 2, prior to re-treatment, 14 subjects had experienced an increase of >25% over their baseline Phe level (rebound), with 8 of these subjects having Phe levels exceeding 300 μmol/L. The treatment break ranged from 6 to 41 days, but there was no obvious pattern that emerged in terms of the degree of rebound (% of baseline) and the length of the treatment break.
Among the subjects who experienced a rebound, four subjects were randomised to placebo in Part 2 and 10 subjects were randomised to sapropterin 20 mg/kg/day. Following one week of re-treatment, all of sapropterin-treated subjects experienced significant decreases in their Phe levels, with all subjects achieving levels below 300 μmol/L and 8 of 10 achieving levels below their baseline values. Among the 4 rebounding subjects who were given placebo in Part 2, the Phe levels of 3 subjects remained above 300 μmol/L and above their baseline (Part 1, Day 1) values. These results support continued treatment with sapropterin as, even in subjects maintained on a Phe-restricted diet, Phe levels may rebound upon cessation of treatment. It appears that, in subjects experiencing rebound, resumption of treatment with sapropterin is effective in returning Phe levels to the desired therapeutic level.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 77 of 123

Therapeutic Goods Administration
Discontinuation due to adverse events
In the studies in subjects with HPA due to PKU, only one subject discontinued treatment due to an adverse event. A female subject with a positive pregnancy test stopped further treatment in study PKU-001. One subject in study PKU-008 withdrew due to difficulty concentrating, an AE that was classified by the investigator as severe. Another subject in the study withdrew due to intermittent diarrhoea, which was classified by the investigator as possibly related to study drug treatment. Post marketing experience
USA and Europe On 13 December 2007, sapropterin (Kuvan) was granted Marketing Authorisation for the treatment of HPA due to PKU in the USA and on 2 December 2008 in Europe (EU) for the treatment of HPA due to PKU or BH4 deficiency. Following the marketing authorisation of sapropterin (Kuvan) in the USA, and before the first marketing authorisation in Europe, there have been four periodic adverse experience submissions (PAES; NDA 022181) covering the following periods:
PAES 1, period from 13 December 2007 to 12 March 2008 PAES 2, period from 13 March 2008 to 12 June 2008 PAES 3, period from 13 June 2008 to 12 September 2008 PAES 4, period from 13 September 2008 to 12 December 2008.
No safety concerns were detected during the reporting period of PAES 1-4. No change to the US Package Insert due to safety-related reasons was performed.
Japan In Japan, sapropterin granules (Biopten 2.5% Granules) have been approved for the treatment of BH4 deficiency since 1992. On 16 July 2008, Biopten received supplemental indication approval from the Japanese Ministry of Health for the indication of:
1. Lowering of elevated serum Phe level in hyperphenylalaninaemia caused by deficiency of dihydrobiopterin synthase or dihydropteridine reductase (atypical hyperphenylalaninaemia)
2. Lowering of elevated serum Phe level in BH4 responsive hyperphenylalaninaemia (BH4 responsive hyperphenylalanaemia).
A pre-approval and 10-year post-marketing safety surveillance program was conducted by DSP in patients with atypical hyperphenylalaninaemia due to BH4 deficiency [DSP Report: Sapropterin Hydrochloride (Biopten) Post Marketing Surveillance Study (Study Period: 27th March, 1992 to 26 March, 2002), 3rd March, 2004].
The only adverse drug reactions (ADRs) reported in the pre-approval surveillance program were two cases of diarrhoea reported for one of the 19 patients (5.3%).
Thirty-two ADRs were reported for 11 of 30 patients (36.7%) in the post-marketing surveillance program. The major ADRs were convulsion and exacerbation of convulsion in three of the 30 patients (10.0%), and gamma-glutamyltransferase (GGT) increased in two of the 30 patients (6.7%). The annual number of ADRs reported was 0 to 8 ADRs reported per year for 0 to 3 patients per year (0.0% to 10.0% of patients in the surveillance program). In the post-marketing surveillance program, the frequency and incidence of ADRs categorised by body system was: 16 events of central and peripheral nervous system disorders in 5 patients (16.7%), 1 event of an autonomic nervous system disorder in 1 patient (3.3%), 6 events of vision disorders in 2 patients (6.7%), 3 events of psychiatric disorders in 2
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 78 of 123

Therapeutic Goods Administration
patients (6.7%), 1 event of a gastrointestinal system disorder in 1 patient (6.7%), and 5 events of liver and biliary system disorders in 4 patients (13.3%). None of the ADRs were considered serious. All ADRs were transient, and subjects recovered or improved without discontinuing sapropterin granules treatment, except for one subject in whom stammering, involuntary movement of lips, and ocular displacement were continuously reported from the sixth year of the program onward while sapropterin granules treatment was continued.
Two deaths, both considered to be unrelated to sapropterin granules treatment, were reported as part of the post-marketing surveillance program. One death was reported during the 10-year surveillance period and one was reported 5 days after completion of the surveillance period. One patient, a male, suffered an airway obstruction secondary to a large amount of sputum, developed severe complications, and died from sepsis at age 4 years and 5 months. The second patient, also a male, was found dead in his apartment 5 days after the surveillance period had ended, at age 24 years and 10 months, having apparently died “several days” earlier of complications related to his underlying BH4 deficiency with severe muscle stiffness. Autopsies were not performed in either case. No other serious AEs were identified in the study report for the surveillance study. Evaluator’s overall conclusions on clinical safety
Evaluation of the safety data submitted showed that few treatment emergent severe adverse events were observed during the clinical development program for sapropterin tablets in HPA due to PKU, or in the clinical studies of treatment of BH4-deficiency with sapropterin 2.5% granules. No serious adverse events were assessed to be related to treatment with sapropterin.
All other treatment emergent adverse events were assessed to be mild to moderate and were transient in nature. Two very common potential undesirable effects, headache and rhinorrhoea, and 7 common potential undesirable effects - diarrhoea, vomiting, abdominal pain, hypophenylalaninaemia, pharyngolaryngeal pain, nasal congestion and cough - were identified. In addition, rebound of blood Phe levels to above pre-treatment levels may occur upon cessation of treatment with sapropterin; therefore, it is recommended that cessation of treatment be conducted only with physician supervision and monitoring. Due to the rarity of the condition, the number of observations for each potential undesirable effect is small and cause and effect relationship to treatment cannot be assessed. Clinical Summary and Conclusions Pharmacology
A limitation of the pharmacokinetic data submitted was that there were no studies that directly compared the granules with the tablet preparation proposed for registration and marketing in Australia. However data were submitted that showed that the various tablet formulations have very similar dissolution profiles. It is therefore considered that the sponsor’s justification that comparative bioavailability data are not required is acceptable. In the dataset submitted for evaluation, the PK of sapropterin hydrochloride in subjects with moderate or severe renal impairment has not been evaluated. Similarly, the effects of hepatic impairment have not been evaluated as the majority of subjects in the study database had liver enzyme levels that were at or near normal levels. Overall, pharmacodynamic data from PKU-004 Sub-study 01 suggest that once daily sapropterin dosing at 10 mg/kg is associated with stable reduction in blood Phe levels over a 24-hour period in PKU subjects.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 79 of 123

Therapeutic Goods Administration
Efficacy
This section will summarise results for the following clinical efficacy objectives for sapropterin treatment of HPA due to PKU: 1) reduction of blood Phe levels, 2) increase in Phe tolerance, and 3) decreases in blood Phe levels from ≥600 μmol/L to <600 μmol/L. Primary analyses for these efficacy objectives are based on results of the pivotal trials, PKU-003 and PKU-006 Part 2. Relevant supportive analyses are from the open label trials, PKU-001, PKU-004 and PKU-006 Part 1.
1) Reduction of Blood Phe Levels In PKU-001, the mean ± SD decrease in blood Phe level from Day 1 to Day 8 was 99.1± 219.8 μmol/L. In PKU-006 Part 1, the mean ±SD decrease in blood Phe level from Day 1 to Day 8 was 104.5 ± 175.5 μmol/L. In PKU-003, there was a significant (p<0.001) mean decrease in blood Phe levels for the sapropterin group compared to the placebo group. The mean ±SD change in blood Phe levels from baseline to Week 6 was −235.9 ± 257.0 μmol/L for the sapropterin group and 2.9 ± 239.5 μmol/L for the placebo group. Comparable results were observed when considering the change from the Week 0 (last observation prior to treatment) blood Phe level. Treatment with sapropterin tablets caused a significant reduction in blood Phe levels in subjects with uncontrolled HPA due to PKU.
In PKU-006 Part 2, the mean ± SD Week 0 (last observation prior to treatment) blood Phe levels were 275.7 ± 135.2 μmol/L for the placebo group. The mean ± SD decrease in blood Phe level from Week 0 to the Week 3 visit (prior to addition of Phe supplement) was 96.6 ± 243.6 μmol/L for the placebo group (p=0.20) and 148.5 ± 134.2 μmol/L for the sapropterin group (p<0.001). Treatment with sapropterin tablets caused a significant reduction in blood Phe levels in subjects with HPA due to PKU controlled on a Phe-restricted diet.
Overall the results from both pivotal trials, PKU-003 and PKU-006 Part 2, demonstrated efficacy of sapropterin tablets to reduce blood Phe levels in patients with (uncontrolled or diet-controlled) HPA due to PKU.
2) Increase in Dietary Phe Tolerance In PKU-006 Part 2 the mean ± SD daily dietary Phe intake at the Week 0 visit (pre-treatment before treatment with sapropterin) was 14.713 ± 7.471 mg/kg/day for the placebo group and 15.660 ± 7.159 mg/kg/day for the sapropterin group. The mean ± SD daily dietary Phe intake (excluding Phe supplement) while subjects were maintaining adequate blood Phe control was 17.709 ± 8.331 mg/kg/day for the placebo group and 19.154 ± 13.480 mg/kg/day for sapropterin group. There was not a statistically significant (p>0.384) treatment effect on the change from pre-treatment daily dietary Phe intake, excluding Phe supplement, when subjects were maintaining adequate blood Phe control, that is, the dietary Phe intake excluding Phe supplement was not statistically different between the two treatment groups. The mean ± SD change in dietary Phe intake, excluding Phe supplement, from Week 0 was 1.817 ± 4.857 for the placebo group compared to 0.574 ± 3.847 mg/kg/day for the sapropterin treatment group. The mean ± SD Phe supplement tolerated by subjects in the sapropterin treatment group was 20.9 ± 15.4 mg/kg/day, which was significantly different from 0 (p<0.001), as compared to 2.9 ± 4.0 mg/kg/day for the placebo group. There was a significant difference between the effect on Phe supplement tolerated of these 2 treatments (p<0.001, sapropterin vs placebo). The evaluator noted, however, when subjects were maintaining adequate blood Phe control, the mean total daily dietary Phe intake, including study-prescribed Phe supplement, was 18.889 ± 9.698 mg/kg/day for the placebo group and 38.406 ± 21.606 mg/kg/day for the sapropterin group. In addition, the change from pre-treatment total dietary Phe intake in the placebo group was significantly different from the change in the sapropterin treatment group.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 80 of 123

Therapeutic Goods Administration
The mean ± SD change from pre-treatment total daily dietary Phe intake, including study prescribed Phe supplement when subjects were maintaining adequate blood Phe control was 3.259 ± 5.291 for the placebo group compared to 17.513 ± 13.628 mg/kg/day for the sapropterin treatment group.
Results supported that treatment with sapropterin resulted in a significant increase in dietary Phe tolerance in individuals with HPA due to PKU controlled on a Phe restricted diet.
3) Reduction in Blood Phe to <600 μmol/L In PKU-003, 38/47 subjects in the placebo group and 31/41 subjects in the sapropterin treatment group had a baseline blood Phe level ≥600 μmol/L (baseline blood Phe level was defined as the mean of blood Phe levels after the screening visit and before the first dose of study drug). At the end of the 6 week treatment period, 5/38 (13.2%) of these placebo subjects and 13/31 (41.9%) of these sapropterin treated subjects had a blood Phe level <600 μmol/L (p=0.012, sapropterin vs placebo). In PKU-001, 428 subjects had a baseline (Day 1, prior to treatment) blood Phe level ≥600 μmol/L. Of this group, 68/428 (15.9%) achieved a blood Phe levels of <600 μmol/L after 8 days of treatment with sapropterin tablets, 10 mg/kg/day.
These results from PKU-003 and PKU-001 indicate that a large proportion of subjects with uncontrolled HPA due to PKU (blood Phe level ≥600 μmol/L) can achieve a blood Phe level within a selected therapeutic range (<600 μmol/L) during treatment with sapropterin tablets. Safety
Common adverse events reported by the sapropterin-treated subjects and in excess over placebo included headache, rhinorrhoea, pharyngolaryngeal pain, vomiting, diarrhoea, nasal congestion, cough and contusion. Across studies, nearly all reported events were mild or moderate in severity. The proportion of severe events was low in each study and no important differences were seen between studies. Severe events were reported with the following preferred terms headache, abdominal pain, diarrhoea, migraine, tooth abscess, and vomiting. In most studies, the majority of events were assessed as unrelated to the study treatment by the investigator. Benefit risk assessment
Benefits Within the Phase III program, two pivotal, randomised, placebo-controlled trials, PKU-003 and PKU-006 Part 2 and a 22 week extension trial PKU-004 provided evidence that sapropterin tablets cause a significant, dose-related, persistent reduction in blood Phe levels in subjects with PKU, allowing some subjects with HPA to achieve blood Phe levels within a selected therapeutic guideline of <600 μmol/L. The studies submitted for evaluation have adequately demonstrated that the sapropterin tablet is efficacious in short term (up to 30 weeks) treatment of patients with HPA due to PKU. In addition, results from PKU-006 demonstrated efficacy of sapropterin tablets to reduce blood Phe levels with blood Phe levels controlled on a Phe-restricted diet, thereby enabling an increase in dietary Phe tolerance.
Results in study PKU-003 demonstrated short term efficacy of sapropterin at a dose of 10 mg/kg/day in patients 8 to 49 years. In addition, results from PKU-006 demonstrated efficacy of sapropterin tablets to reduce blood Phe levels with blood Phe levels controlled on a Phe-restricted diet, in patients from 4 years old. Data from PKU-004 demonstrated that reduction in blood Phe in patients with BH-4-responsive PKU can be maintained for 22 weeks.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 81 of 123

Therapeutic Goods Administration
The clinical development program included adults and children with HPA due to PKU who were ≥4 years old. Younger children were not included, in part due to the difficulty of maintaining a constant daily dietary Phe intake in children under the age of 4 years. In the submission, patients treated for one week in open-label uncontrolled studies included 50 patients who were 4 to 8 years old, and 104 patients who were 8 to 12 years old. Twenty four patients aged 4 to 8 years, and 38 patients aged 8 to 12 years were included in double-blind placebo-controlled trials. Fifteen paediatric patients were treated for up to 22 weeks in open-label uncontrolled trials. Given the rarity of the clinical condition it is considered that short term efficacy has been demonstrated in patients aged 4 years and older. For HPA due to BH4 deficiency, a very rare condition, the available data from the Biopten registration and post-marketing surveillance trials and published series, demonstrate efficacy of sapropterin treatment to reduce, and in most cases normalise, blood Phe levels, thereby reducing or eliminating the need for dietary Phe restriction.
Risks Given the rarity of HPA the number of patients evaluated for safety in the clinical program is considered acceptable. Review of the safety data for sapropterin treatment of HPA due to PKU or BH4 deficiency, revealed undesirable effects that may be reasonably associated with this treatment; headache, rhinorrhoea, diarrhoea, vomiting, abdominal pain, hypophenylalaninaemia, pharyngolaryngeal pain and nasal congestion. These effects were typically mild to moderate in severity, transient in nature, and did not prevent continued treatment with sapropterin.
Safety specification The Safety Specification forms part of the Risk Management Plan (Section V). In the opinion of the evaluator, nonclinical safety outcomes that have not been adequately addressed by clinical data or which are of unknown significance are listed below: 1) Repeat dose toxicity
Nephrotoxicity A chronic 52 week study in rats showed mild histological changes in the kidney. Nephrotoxicity in humans cannot be excluded. 2) Mechanisms for drug interactions
Sapropterin is expected to follow the same metabolic pathways as the naturally occurring enzyme co-factor, 6R-BH4. According to literature data, dihydrofolate reductase and dihydropteridine reductase are likely to be involved in the in vivo metabolism and recycling of sapropterin. Medicinal products inhibiting dihydrofolate reductase (for example, methotrexate, trimethoprim) may interfere with BH4 metabolism. Sapropterin has the potential to cause vasodilation through the nitrous oxide (NO) system. In clinical use, synergistic hypotensive effects with other medicinal products releasing NO or interfering with NO metabolism (for example, glycerol trinitrate, isosorbide dinitrate, sodium nitroprusside, molsidomin, phosphodiesterase type 5 and minoxidil) cannot be excluded. As sapropterin may increase the availability of tyrosine, a precursor to levodopa, sapropterin may cause increased excitability and irritability in patients receiving levodopa treatment. The evaluator commented that the safety specifications are considered appropriate and adequate.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 82 of 123

Therapeutic Goods Administration
Balance Overall the data submitted for evaluation support that sapropterin is efficacious in reducing blood Phe levels and increasing dietary Phe tolerance in patients with HPA due to PKU or BH4 deficiency. The data submitted for evaluation have demonstrated that when used at the recommended doses, sapropterin has a positive balance of benefits to risk for patients ≥4 years old with HPA due to PKU or BH4 deficiency. Information from the registration and post-marketing surveillance studies for sapropterin 2.5% granules provide evidence for a favourable safety and efficacy profile for sapropterin treatment of HPA due to BH4 deficiency. This is further supported by published literature for sapropterin treatment of BH4 deficiency, and the interim data from study, PKU-007.
Sapropterin satisfies two major clinical unmet needs for a substantial number of patients with HPA due to PKU or BH4 deficiency; that is, an easily administered, well-tolerated oral daily therapy that: (1) causes a clinically relevant reduction in blood Phe levels, that may enable many patients to achieve levels within individualised therapeutic targets, (2) increases tolerance of dietary Phe and, consequently, normal dietary protein, thereby enabling reduction or potential elimination of the need for Phe-free protein supplements. There is currently no approved pharmacological treatment for this orphan indication; therefore, sapropterin represents a major advance in the treatment of HPA. The evaluator was of the opinion that sapropterin has a positive benefit/risk profile for treatment of patients with HPA due to PKU or BH4 deficiency.
Conclusions It was recommended that the application to register sapropterin for treatment of patients with HPA due to PKU or BH4 deficiency should be approved.
It should be requested that children younger than 4 years of age with PKU should be studied in the post-approval period.
V. Pharmacovigilance Findings Risk Management Plan The sponsor submitted a Risk Management Plan (RMP) which was reviewed by the TGA’s Office of Medicines Safety Monitoring (OMSM). The sponsor identified the following identified and potential risks and areas of missing information: Important identified risks:
· Gastrointestinal disorders (vomiting, diarrhoea, abdominal pain). Most cases have been non-serious, reversible with dose reduction or discontinuation and of mild to moderate severity.
· Hypophenylalaninaemia. In the clinical trials non-serious hypophenylalaninaemia have been detected through routine blood monitoring. Monitoring of blood Phe levels and active management of dietary Phe and overall protein intake is required. Prolonged exposure to low blood Phe and tyrosine levels in infancy has been associated with impaired neurodevelopmental outcome.
· Rebound increase in blood Phe after drug discontinuation. In the clinical trials this drug withdrawal effect occurred in some patients and is reported as non-serious with cases resolving without sequelae.
· Drug interactions
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 83 of 123

Therapeutic Goods Administration
Important potential risks: · Nephrotoxicity. Identified as a potential risk in the context of an increased incidence of
histological changes found in the renal tubules of rats in high dose studies. The sponsor recommended regular monitoring of renal function.
Important missing information: · Size of safety database; · Long term exposure; · Limited data on patients with BH4 deficiency; · Pregnancy experience; · Experience in children <4 years old; · Hepatic insufficiency; · Renal insufficiency; and · Experience in the elderly population.
The OMSM reviewer noted that as a consequence of the low prevalence of the target diseases, the efficacy and safety data bases are not large. Ongoing safety concerns have been outlined and pharmacovigilance and risk minimisation activities proposed. The sponsor has identified an undertaking to engage in post-authorisation studies including a Kuvan patient registry in Europe. Kuvan has not been specifically studied in children <4 years old and as such it was recommended by the reviewer that the sponsor commit to active surveillance in this age group to more completely monitor AEs in this population. This could be achieved through monitoring at sentinel sites as the majority of these patients would be treated at specialist paediatric services.
Other specific recommendations by the OMSM reviewer included:
· Non-clinical studies revealed a statistically significant higher incidence of benign adrenal pheochromocytomas in rats and mice; the conclusion by the sponsor being that that this was related to the unusually low frequency of spontaneously occurring adrenal pheochromocytomas in the controls of the sapropterin experiment rather than an effect of treatment itself. This conclusion was based on a retrospective analysis of several carcinogenicity studies conducted at the testing facility. The sapropterin carcinogenic animal studies have not been repeated to support this hypothesis. In view of this, the sponsor was requested to comment about including malignancy/tumours/adrenal phaeochromocytoma in the potential safety concerns in the RMP.
The sponsor responded that they did not consider it appropriate to include malignancy/tumours/adrenal pheochromocytoma as potential safety concerns in the RMP. Based on results of in vivo studies, the fact that sapropterin dihydrochloride is a synthetic formulation of the naturally occurring co-factor (BH4) and the absence of hyperplastic/pre-neoplastic lesions in both rats and marmosets after chronic administration, it is not considered that sapropterin dihydrochloride pose a significant carcinogenic risk to humans. It was also noted that the TGA Nonclinical Evaluator concurred that these tumours are not considered to be of clinical relevance.
· In the limitations of the human database section in the RMP, the demographic data is presented in 3 age group ranges; ≥4 to <12 years, ≥12 to <18 years and ≥18 years. It appears that most of the paediatric exposure is in the >8 year old age group but this cannot be accurately ascertained from the presented data. The sponsor was requested to show a breakdown of age demographics to include children ≥4 to <8 years and ≥8 to <12 years.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 84 of 123

Therapeutic Goods Administration
The sponsor responded by providing the requested breakdown of age demographics and by including this level of detail in the updated RMP.
· The RMP indicates that patients with concurrent disease (including seizure disorder, oral steroid-dependent asthma or other condition requiring oral or parenteral corticosteroid administration, insulin dependent diabetes, history of organ transplantation and serious neuropsychiatric illness not under medical control) that may interfere with safety were excluded from two open label studies PKU008 and PKU007. The sponsor was requested to clarify whether these concurrent disease exclusion criteria were used in the other clinical trials, in particular the placebo controlled trials PKU003 and PKU006 and the PKU003 open label extension study (PK004). If these conditions were excluded from these trials it was recommended that this be reflected in the appropriate section in the proposed PI together with comment about such individuals potentially being more susceptible to adverse drug reactions when treated with Kuvan.
The sponsor confirmed that the concurrent disease exclusion criteria used in PKU007 and PKU008 were also used in the other clinical trials, including PKU003, PKU004 and PKU006. Patient populations with concurrent diseases are usually excluded in paediatric trials, as these conditions can interfere with the safety and efficacy analysis. Such patients are at high risk of acute decompensation of these concurrent diseases independently from treatment with Kuvan. These patients were not excluded because they were deemed to be more susceptible to Kuvan related adverse drug reactions.
· The sponsor was requested to provide projected post authorisation usage data for the Australian market, including estimates of usage in the 0 to 4 year old age group.
The sponsor estimated the projected usage in Australia, including both PKU and BH4 in all age groups, is between 150 and 250 patients. Within this total, the anticipated number less than 4 years of age is about 30 patients.
· The sponsor was requested to comment on the following safety concerns and the addition of these items to the Pharmacovigilance Plan (PV) Plan:
î Important identified risk: Respiratory disorders (Rhinorrhoea, pharyngolaryngeal pain, nasal congestion and cough). These were commonly reported. The objective for PV would include improving understanding of the mechanisms involved, risk factors, therapeutic options and preventability.
î Important missing information: Experience in patients with moderate to severe neurocognitive disability. Experience in patients with a history of seizures. Experience in patients with Psychiatric disorders.
The sponsor agreed to these changes. · The sponsor was requested to comment on the potential for overdose in neonates and
infants in the context of doses below 100 mg requiring the parent/guardian to determine the dose from a volume of solution and the possibility of higher absorption of Kuvan in this age group (identified in juvenile animal studies). Additionally, comment was requested on there being any potential for overdose, particularly in young children, using the rounding to the nearest multiple of 100 regimen to calculate the daily dose.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 85 of 123

Therapeutic Goods Administration
The sponsor response referred to appropriate statements being included in the PI and noting that treatment initiation and dosage adjustments in very young patients with PKU or BH4 deficiency will be made under close clinical supervision.
· The sponsor was requested to comment on the administration of Kuvan to infants, particularly whether the Kuvan-water solution can be added to the modified protein milk formulas. For young children who refuse to drink the Kuvan-water solution, can Kuvan be dissolved in other liquids without influencing drug composition and bioavailability?
The sponsor recommends that Kuvan is dissolved in water and administered with a meal as a single daily dose, at the same time each day, preferably in the morning.
· The sponsor was requested to outline if there have been any new safety signals identified from the safety data analysis of studies PKU-007 and PKU-008. These studies are reported as completed.
The sponsor confirmed that were no new safety signals apparent from PKU007 or PKU008.
· A study in healthy adults to evaluate the effects of sapropterin on QTc intervals was outlined in Annex 3 of the RMP as completed with an expected report in October 2009. The sponsor was requested to comment on whether QTc prolongation was detected as a safety concern.
The sponsor provided a summary of results from the QTc study. Oral sapropterin dissolved in liquid at a therapeutic dose of 20 mg/kg and at a supratherapeutic dose of 100 mg/kg was well tolerated and not associated with QTc prolongation or other repolarisation abnormalities in healthy adult subjects under fed conditions.
· Except for the limited BH4 deficiency data item, all the other ongoing safety concerns include the additional PV activity of data analyses from the post-authorisation studies (A, B and C). Regarding study B on the long term neurocognitive outcomes in children treated with Kuvan, the sponsor was asked to comment on whether the specific safety issues in the PV Plan are included as additional study objectives with defined outcome measures or if the data analyses will be based on spontaneous AE reporting from participating clinicians.
The sponsor confirmed that in all three post-authorisation safety studies, safety is either a primary or secondary objective and provided some relevant details from the study designs.
· With respect to the nephrotoxicity safety concern and the proposed additional PV action of data analyses of the 3 post-authorisation studies, the sponsor was asked to comment on whether there are standardised parameters and protocols in place in the study objectives to monitor renal function.
The sponsor responded that renal function is not a direct objective in the post-authorisation studies but safety, including laboratory investigations and renal function parameters, is an objective. There are standardised criteria for collecting information in relation to renal function specified in all three protocols and a brief summary was provided.
· The sponsor was requested to comment on what specifications are in place to detect and measure the potential drug interactions in the 3 post-authorisation studies.
The sponsor responded that in all three clinical trials, concomitant medications and adverse events will be recorded in the Case Report Form (CRF). A reported potential drug interaction will be recorded in an Alert Report Form which requests information on the concomitantly administered medications.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 86 of 123

Therapeutic Goods Administration
VI. Overall Conclusion and Risk/Benefit Assessment The submission was summarised in the following Delegate’s overview and recommendations: Quality In relation to bioavailability:
· Food was shown to increase the bioavailability by approximately 85% when the product was given dissolved in water and by approximately 50% when dissolved in orange juice. It was recommended that the PI should state that all dosing occur with food and this is indeed the case.
· The formulation proposed for supply can be considered bioequivalent to the formulation used in the majority of the Phase III clinical efficacy studies.
· The justification for the lack of an absolute bioavailability study was accepted. As pointed out by the pharmaceutical chemistry evaluator, this means that information relating to clearance and volume of distribution have not been included in the PI.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 87 of 123

Therapeutic Goods Administration
There was only one matter on which the quality evaluator could not reach agreement with the sponsor and this meant that the pharmaceutical chemistry evaluator was unable to recommend approval of the submission with respect to chemistry and quality control. Apart from this one issue, the submission was approvable. The issue concerned the limit in the drug substances specifications for the synthetic impurity S-BH461
In the relevant TGA-adopted EU guideline, it states that the enantiomeric purity of active substances used in non-clinical studies should be defined. During the evaluation, the sponsor was requested to provide data on the exact amounts of S-BH4 present in two batches of drug used in rat and mouse studies, not just the upper limit. In response, the sponsor stated that a Japanese investigator reported higher levels in the two studies. The mechanism for quantitation and the validation status of the method at the time of testing were not available to the sponsor of this application. Therefore the amount of S-BH4 in the batches is unknown. With an unknown amount of S-BH4 present in nonclinical studies, these studies cannot be used to qualify this impurity.
. The quality evaluator sought advice from the Medicines Toxicology Evaluation Section of the Office of Prescription Medicines and the advice of this section was that this limit was not considered qualified.
With the above toxicological advice, namely that the limit had not been qualified with toxicological data, the pharmaceutical chemistry evaluator then put it to the sponsor that the limit must be further tightened to the ICH qualification threshold of NMT 0.15%. The sponsor responded, arguing that adoption of this tighter limit was not possible due to the limit of quantification of the test method which was true according to the quality evaluator but, also according to this evaluator, the test method could be changed to increase the sensitivity. The sponsor also argued that there was clinical data qualifying the higher limit. This clinical data will be assessed by Delegate later in this overview. The Delegate has been in correspondence with the sponsor with regard to this issue and the sponsor has provided even more clinical data supporting the sponsor’s case. These extra clinical data will also be assessed later in this report. Finally, as a result of this discussion between the sponsor and the Delegate, the sponsor found a study reporting results of acute and subchronic toxicity testing with the 6R,S-BH4 enantiomeric mixture in mice. This study should have been part of the original submission. The Delegate requested that the sponsor provide the full article reporting the study so that it could be assessed by the nonclinical evaluator.
The quality evaluator’s final comment was that, if it is accepted that the clinical data qualifies the limit then the chemistry and quality control aspects of the submission could be accepted. In further communications between the Delegate and the sponsor, the latter has confirmed that, since the conduct of the relevant nonclinical studies, Merck Serono’s commercialisation partner, BioMarin, has validated the quantitation of S-BH4 as a commercial batch release test for the Kuvan drug substance and drug product so that levels can now be reliably quantified. The current specification limit is based on the capacity of this method. The sponsor also made the comment that the levels of S-BH4 in Kuvan appear to be lower, which is why the sponsor has not been able to supply evidence that batches used in the non-clinical and clinical studies conducted by BioMarin had levels greater than the proposed limit. Nonclinical Conclusions and recommendations of the nonclinical evaluator were as follows:
61 S-tetrahydrobiopterin is not a metabolite of 6R-tetrahydrobiopterin, but a synthetic impurity. S-
tetrahydrobiopterin is an enantiomer of sapropterin but there is no interconversion in vivo.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 88 of 123

Therapeutic Goods Administration
The nonclinical data support the efficacy of sapropterin treatment for hyperphenylalaninaemia in BH4-deficient patients, although the non-clinical data indicate that this treatment alone is unlikely to treat all the effects of this deficiency in patients. The benefits of the treatment for these patients were judged as likely to outweigh the risks and thus there were no non-clinical objections to the registration of sapropterin for the treatment of adult and paediatric patients of all ages with BH4 deficiency.
The bioavailability of oral sapropterin in juvenile rats was about 5-fold higher than in adult rats and the juvenile adult exposure ratio in rats was 23:1. The nonclinical evaluator noted that this may have implications for the proposed starting dose in young children, which is currently the same as for adults on a mg/kg basis. Therefore the nonclinical evaluator asked the Delegate to consider whether it is appropriate to recommend a lower starting dose in young children.
Renal damage occurred in young rats after 2 weeks of administration of oral sapropterin to 7-day old rats (NOEL = 80 mg/kg/day). Because of a lack of PK data in young children, an exposure ratio could not be estimated. Therefore the risk of renal damage in infants is unknown. The nonclinical evaluator considered that, on the basis of a possible increased risk of renal damage in comparison with adults, it would be prudent to advise the use of a conservative starting dose in BH4-deficient infants in order to avoid unnecessary overloading of the kidneys. With regard to sapropterin-responsive PKU, the nonclinical evaluator observed that, while the nonclinical data are consistent with efficacy, evidence for the latter will need to rely on the clinical data. The nonclinical evaluator expressed concern about patients under one year of age. For these patients, in whom the blood brain barrier is not fully developed, the benefits of sapropterin treatment may not outweigh the risks. In patients under one year of age, PKU is relatively easily controlled using diet, so the benefits of sapropterin are reduced. In addition the risks involved in sapropterin use are increased. As the nonclinical evaluator noted, a very common adverse effect of sapropterin reported by patients is headache, an effect that an infant would have difficulty in communicating. There is nonclinical evidence that higher levels of sapropterin in the brain increase pain levels. As noted earlier, absorption of oral sapropterin was higher in juvenile rats than in adult rats and total biopterin levels increased markedly in the brains of young rats following oral sapropterin. The nonclinical evaluator further observed that, as the blood brain barrier is not fully developed in infants until 12 months, infants are likely to have higher cerebral exposure to sapropterin than adults. The effect of such increased BH4 levels on brain development and function have not been fully investigated and therefore pose a potential risk to infants. The nonclinical evaluator argued that whereas this risk would be acceptable for BH4-deficient infants, who have dysfunctional brain enzymes because of the lack of BH4 cofactor, for infants with PKU there appears to be no advantage to higher BH4 brain levels.
Thus, based on the foregoing, the nonclinical evaluator was of the view that registration of sapropterin for use in infants under 12 months of age with PKU was not supported on nonclinical grounds because the risk/benefit ratio differs between infants and adults with PKU. As noted already, there were no nonclinical objections to the registration of sapropterin for use in adult and paediatric BH4-deficient patients. Clinical The clinical evaluator’s final recommendation was that the data adequately support the indication as follows:
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 89 of 123

Therapeutic Goods Administration
Kuvan is indicated for the treatment of hyperphenylalaninaemia (HPA) in sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency. Pharmacokinetics
The pharmacokinetics of sapropterin were evaluated in healthy subjects in four Phase I studies as well as in 78 individuals with PKU in PKU-004 Substudy 02. The results indicate that sapropterin is rapidly absorbed, with a bi-exponential decline following attainment of peak levels, indicative of a 2-compartment, first-order input model with first-order elimination.
The mean terminal half-life was 6.69 hours (range 3.91 to 16.6 hours). On the assumption of 4 half-lives for clearance, coverage was estimated to be for 26.8 hours which would support once-daily dosing. Simulations revealed no evidence of accumulation after 5 doses at 5, 10 or 20 mg/kg daily. These results compared favourably with the PK data collected for Biopten 2.5% granules in the three Phase I trials conducted in healthy volunteers. Population pharmacokinetic analyses demonstrated that total body weight was the only significant covariate. There was a small effect of creatinine clearance on biopterin clearance, but not sufficiently large to include in the final model. It should be noted that the pharmacokinetics of biopterin in subjects with moderate or severe renal impairment has not been evaluated. Likewise the effects of hepatic impairment have not been evaluated.
No special metabolic studies were performed. Sapropterin is a synthetic version of the naturally occurring co-factor, 6R-BH4 and the metabolic fate is assumed to be the same as for the naturally occurring co-factor. The metabolic fate of exogenously administered BH4 in humans has been only partially defined. It is assumed that a large proportion of the BH4 serving as a co-factor for aromatic amino acid hydroxylases enters the BH4 regeneration pathway (following oral administration in healthy individuals, less than 10% is recovered as metabolites in urine and/or faeces). Study PKU-004 had a forced dose titration period which revealed a dose-related response for the 3 daily doses evaluated – 5 mg/kg, 10 mg/kg and 20 mg/kg. No formal drug-drug interaction studies were performed due to the rarity of HPA. As noted above, sapropterin is a synthetic formulation of the naturally occurring co-factor, 6R-BH4 and therefore is expected to undergo similar metabolism. Folic acid and vitamin B12 may increase BH4 levels although the mechanisms are not completely defined. Inhibitors of dihydrofolate reductase, for example, methotrexate, aminopterin and trimethoprim, may also inhibit the activity of dihydropyridine reductase and theoretically prevent salvage of BH4. BH4 acts to enhance nitric oxide synthetase activity, which provides a theoretical basis for synergistic effects with PDE5 inhibitors such as sildenafil and drugs such as minoxidil which have nitric oxide agonist effects. While animal studies have not indicated any interaction with sildenafil, there have been no studies of interaction in humans with either type of agent. Pharmacodynamics
Data from PKU-004 Sub-study indicate that once daily sapropterin dosing at 10 mg/kg is associated with stable reduction in blood Phe levels over a 24-hour period in PKU subjects. Efficacy
There were 3 components to the clinical efficacy data:
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 90 of 123

Therapeutic Goods Administration
Clinical Efficacy in HPA due to PKU in patients ≥4 years of age The most important feature to note about the clinical program to evaluate the efficacy and safety of sapropterin in the treatment of PKU in patients aged at least 4 years of age is that there were two distinct populations studied. Population 1 consisted of those subjects aged at least 8 years in whom Phe intake was uncontrolled. Population 2 consisted of subjects aged between 4 and 12 years whose Phe intake was controlled.
The data were generated in the following multinational, multi-centre studies: PKU-001, PKU-003, PKU-004 and PKU-006 (Parts 1 and 2) of which two studies, PKU-003 and PKU-006 Part 2 were pivotal, randomised, placebo-controlled Phase III trials. PKU-003 evaluated the safety and efficacy of sapropterin tablets, 10 mg/kg/day, to decrease blood Phe levels in subjects ≥8 years old, with uncontrolled HPA due to PKU while PKU-006 Part 2 evaluated the safety and efficacy of sapropterin tablets, 20 mg/kg/day, to increase dietary Phe tolerance in subjects with HPA due to PKU, aged 4 to 12 years (inclusive), with blood levels controlled on a Phe-restricted diet.
The remaining studies were dose-response studies. Two short-term open-label treatment response studies, PKU-001 and PKU-006 Part 1 were performed to identify responders to sapropterin treatment. PKU-001 identified responders to go forward to PKU-003, the pivotal study for Population 1, while PKU-006 Part 1 identified responders to go forward to PKU-006 Part 2, the pivotal study for Population 2. In addition, PKU-004 was a long-term, open-label study looking at safety and tolerability of the 3 doses of 5, 10 or 20 mg/kg/day of sapropterin. PKU-004 enrolled subjects who completed PKU-003. Thus for Population 1, there was a progression of subjects (some not all) from PKU-001 to PKU-003 to PKU-004 and for Population 2, a progression of subjects (again some not all) from PKU-006 Part 1 to PKU-006 Part 2. There is an ongoing Phase IIIb, open-label extension study, PKU-008, to assess longer-term safety of sapropterin in subjects previously enrolled in PKU-004 and PKU-006, that is, involving subjects from both Populations 1 and 2.
Study PKU-003 (pivotal trial) PKU-003 was a Phase III, multicentre, multinational, randomised, double-blind, placebo-controlled study to evaluate single daily doses of sapropterin tablets, at a dose of 10 mg/kg/day, administered for 6 weeks to subjects with PKU. Subjects (n = 89) were randomised 1:1 to receive either sapropterin or placebo. In the placebo group, ages ranged from 8 years to 49 years with a mean of 19.5 ± 9.8 years while the corresponding age range in the sapropterin group was from 8 years to 42 years with a mean of 21.5 ± 9.5 years. For children aged from 4 to 12 years, there were 8 in the placebo group and 4 in the sapropterin group; for adolescents aged from 12 to 18 years, there were 16 in the placebo group and 14 in the sapropterin group and for adults over the age of 18 years, there were 23 in each group.
Over the 6-week study, there was a statistically significant (p < 0.001) mean decrease in blood Phe levels for the sapropterin treatment group compared with the placebo group, with a mean ± SE between-group difference of 245 ± 52.5 µmol/L. The mean ± SD baseline blood Phe levels for the sapropterin group and the placebo group were similar, 842.7 ± 299.6 and 888.3 ± 323.1 µmol/L, respectively. The mean ± SD changes in blood Phe levels from baseline to Week 6 were -235.9 ± 257.0 µmol/L for the sapropterin group and +2.9 ± 239.5 µmol/L for the placebo group. Statistically significant differences between the two groups were observed beginning at Week 1 and continued to be observed for the duration of treatment. 54% of subjects in the sapropterin group and 23% of subjects in the placebo group had Week 6 blood Phe levels <600 µmol/L (p = 0.004). In the subgroup of subjects whose baseline blood Phe levels had
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 91 of 123

Therapeutic Goods Administration
been ≥600 µmol/L, 42% of those in the sapropterin group and 13% of those in the placebo group had Week 6 Phe levels <600 µmol/L (p = 0.012).
Study PKU-006 (Part 2 was pivotal trial) PKU-006 was a two-part Phase III study that involved once-daily administration of sapropterin tablets, 20 mg/kg/day, to subjects with HPA due to PKU, 4 to 12 years old (inclusive), who were following a strict Phe-restricted diet and who had blood levels of ≤480 µmol/L at screening. In the first part of the study, subjects received treatment for 8 days after which only those subjects whose blood Phe levels were reduced by at least 30% from day 1 to day 8 and whose blood Phe level was ≤300 µmol/L on day 8 were to be eligible for enrolment in the second part of the study. The latter was a randomised, double-blind, placebo-controlled study in which subjects were randomly assigned in a 3:1 ratio to receive either single daily administration of 20 mg/kg sapropterin or placebo, respectively, for 10 weeks. Dietary Phe intake was also varied according to a rather complicated algorithm. In PKU-006 Part 2, the age range in the placebo group (n = 12) was from 4 to 10 years with a mean of 7.1 ± 2.0 years while the age range in the sapropterin group (n = 33) was from 4 to 12 years with a mean of 7.7 ± 2.8 years.
Of the 89 subjects who received at least one dose of study drug and for whom there were both day 1 (pre-treatment) and day 8 blood Phe level data, 50 (56%) met the protocol definition of responder for Part 1 of the study and were able to proceed to Part 2. For these responders, the mean ± SD blood Phe level at day 8 was 108.1 ± 70.2 µmol/L, compared to 317.0 ± 173.2 µmol/L at day 1, with an absolute mean ± SD change and percentage change in blood Phe levels of -209.0 ± 138.6 µmol/L and -64.0 ± 17.5%, respectively. These results are consistent with those from PKU-003. Over the 10 week study period, the mean ± SD Phe supplement tolerated by subjects in the sapropterin group, 20.9 ± 15.4 mg/kg/day, was significantly different from zero (p < 0.001). However, the Delegate was of the opinion that a more meaningful way of reporting the outcome of this study is that the mean ± SD increase in dietary Phe tolerance was 17.513 ± 13.268 mg/kg/day for the group treated with sapropterin 20 mg/kg/day, compared to 3.259 ± 5.291 mg/kg for the placebo group (p = 0.006). The latter result is more meaningful because the parameter, ‘increase in dietary Phe tolerance’, is of vital, practical interest to clinicians and because the result allows a direct comparison with the effect of placebo. Study PKU-001
This was a Phase II, multicentre, open-label study with a total of 490 subjects with clinically diagnosed PKU, of these 485 received at least one dose of study drug (38 aged from 4 to 12 years, 153 aged between 12 and 18 years and 294 at least 18 years of age). Of the 485 subjects who received at least one dose of study drug and who had blood Phe level measurements at both days 1 and 8, 96 (19.8%) were responders (with at least a 30% reduction in blood Phe level from day 1 to day 8). The mean ± SD absolute change and the mean ± SD relative change in blood Phe levels from day 1 to day 8 for responders were -391.8 ± 185.3 µmol/L and -50.0 ± 16.0%, respectively.
For the 485 subjects who received at least one dose of study drug and had both day 1 and day 8 blood level results, the mean ± SD absolute change and the mean ± SD relative change in blood Phe levels from day 1 to day 8 were -99.1 ± 219.8 µmol/L and -11.4 ± 25.5%, respectively.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 92 of 123

Therapeutic Goods Administration
Study PKU-004 Subjects who had received at least 80% of the scheduled doses in PKU-003, unless they were removed from PKU-003 because of high blood levels of Phe, were eligible to enrol in PKU-004 which was a Phase III, open-label, 22-week extension study.
A forced dose titration study was conducted during the first 6 weeks, in which subjects received 3 consecutive 2-week courses of once-daily sapropterin in the following order: 5 mg/kg/day, 20 mg/kg/day and 10 mg/kg/day. During the dose-analysis period, starting at Week 6 and continuing to Week 10, subjects continued to receive sapropterin, 10 mg/kg/day. Starting at Week 10 and continuing until Week 22, subjects were assigned a fixed dose of 5, 10 or 20 mg/kg/day (fixed-dose period), based on an algorithm utilising the blood Phe results from weeks 2 and 6. During the fixed-dose period (Week 10 to Week 22), each subject’s optimal sapropterin dose was based on his/her Week 2 and Week 6 blood Phe levels. Of the 80 subjects, 6 (8%) received 5 mg/kg/day of sapropterin, 37 (46%) received 10 mg/kg/day and 37 (46%) received 20 mg/kg/day during the fixed-dose period. The mean ± SD blood Phe levels at the visits between Week 12 and Week 22 ranged between 619.8 ± 371.0 and 652.2 ± 382.5 µmol/L. Subjects who received a fixed sapropterin dose of 10 or 20 mg/kg/day had comparable mean blood Phe levels at weeks 12, 16, 20 and 22 as they had previously on the same dose in the forced dose-titration period. Subjects who received the 5 mg/kg/day dose of sapropterin did not have as low a mean blood Phe level as observed for the 5 mg/kg/day dose during the forced-dose titration period. Comparison of efficacy results across all studies
The results from the pivotal trials, PKU-003 and PKU-006 Part 2, demonstrated efficacy of sapropterin compared with placebo to reduce blood Phe levels in patients both uncontrolled or diet-controlled HPA due to PKU. Results showed that treatment with sapropterin allowed a significant increase in dietary Phe tolerance in subjects with HPA due to PKU and controlled on a Phe-restricted diet. Results demonstrated that significant proportions of subjects with uncontrolled HPA due to PKU (blood levels Phe ≥600 µmol/L) were able to achieve blood levels within the selected therapeutic range of <600 µmol/L with treatment with sapropterin.
There appears to be no evidence of either an age-related effect or a gender-related effect of sapropterin on the drug’s ability to reduce Phe levels in patients with HPA due to PKU.
Clinical efficacy in HPA due to PKU in patients aged 0 – 4 years of age A systematic review of the published literature was performed and articles selected for inclusion were used to support the clinical efficacy and safety of sapropterin tablets in the treatment of PKU for patients aged up to 4 years.
From this review, there were ten articles judged by the clinical evaluator as having the most important long-term data. These studies included both short-and long-term data.
Concerning the very young, there were three studies which the Delegate could identify: 10 newborns, 3 of whom received the medication as long-term treatment for up to 3 years (Boneh);30 9 neonates, 1 of whom received the medication for 3 years (Feillet et al.)32 and 40 infants, 5 of whom were treated long-term, for up to 29 months (Hennermann et al.).33
All of the short-term results showed consistent reductions in blood Phe levels up to 24 hours after loading dose, even in the very young.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 93 of 123

Therapeutic Goods Administration
For the long-term treatments, there were a variety of outcomes reported, such as diet liberalisation, increase in Phe tolerance, median blood Phe levels pre- and post-BH4 treatment and maintenance of blood Phe levels within a desired range (60-360 µmol/L). Again, there was consistency of positive outcomes by these measures. None of the long-term studies appeared to assess such parameters as development or intelligence scores. From the review, there were 3 studies which each provided short-term data only (single-dose) in > 100 patients aged 0-4 years. These 3 studies looked at a total of 2,394 patients. Once more, there were consistent reductions in blood Phe levels at 8 and 24 hours post-load.
Clinical efficacy in HPA due to BH4 deficiency Data in this part of the submission was hybrid in nature, consisting of the following:
· the study performed for the registration of sapropterin 2.5% granules (Biopten) in Japan
· the results of a 10-year post-marketing study of the 2.5% granules in Japan · the results of an interim analysis at Week 10 for the trial PKU-007 · the results of 11 long-term studies and 5 short-term studies, all published and
identified via a systematic review. Long-term data
The Biopten study (D272) evaluated the effectiveness of sapropterin 2.5% granules to decrease blood Phe levels in 16 patients with BH4 deficiency. Of the 16 subjects, one was newborn and five were under 5 years of age. The Delegate requested that the sponsor provide information about the ages of the remaining 10 subjects. Daily dosing ranged from 0.8 to 37 mg/kg/day with maintenance dosing ranging from 2 to 5 mg/kg/day. The sponsor was asked to clarify this statement. Based on a rating of global improvement, there was moderate or marked improvement in all 16 subjects. Sapropterin was administered for a mean of 15.5 months. For all sixteen subjects, blood Phe levels were lower after treatment with sapropterin and were maintained within the normal range. A post-marketing surveillance study for Biopten was conducted in Japan and enrolled 30 patients with HPA, assessed at enrolment to have BH4 deficiency, with a data collection period of 10 years. The 30 subjects included the 16 subjects evaluated in the Biopten study and another 14 subjects. During the study, 3 of the 30 subjects were found to have HPA due to PKU. Thus there were 27 subjects with confirmed BH4 deficiency included in the primary efficacy analysis. With regard to age distribution, 14 patients were aged less than 1 year and 5 patients were aged between 1 and 7 years at the commencement of treatment. It was not known how many of the latter group of 5 were aged less than 4 years. The Delegate requested that the sponsor clarify this issue. All patients were treated with sapropterin 2.5% granules at doses between 2 and 15 mg/kg/day with 19/27 (70%) receiving doses between 2 and 5 mg/kg/day. All 30 patients were treated for at least 1 year, with 19 patients treated for 10-20 years. For the overall study population with BH4 deficiency, 25/27 (93%) achieved a global improvement of at least ‘slightly improved’. All 22 patients with PTPS deficiency demonstrated a global improvement of at least ‘slightly improved’ while 3 of the 5 patients with DHPR deficiency demonstrated a global improvement of at least ‘improved’. Of the 22 patients with PTPS deficiency, all but 1 patient had a mean decrease in blood Phe levels into the normal range. For the 5 patients diagnosed with DHPR deficiency, there were 4 patients for whom blood Phe levels decreased at the last examination – for these 4 patients, mean ± SD blood Phe levels were 853.6 ± 520.7 µmol/L before treatment and 296.6 ± 78.7 µmol/L at last examination.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 94 of 123

Therapeutic Goods Administration
There were 11 published studies identified via a systematic review, reporting long-term data. By the Delegate’s reckoning, there were 197 participants with BH4 deficiency in these 11 trials. Frequently, both the source and the dosage of the BH4 were not stated. All were open-label, most appear to have been prospective but at least 2 were retrospective. In almost all subjects, blood Phe levels fell significantly after the sapropterin single-dose loading test and in the majority of subjects, blood Phe levels remained at lower levels than baseline. In one study, Wang et al., 2006, which was conducted between 1992 and 2005 in Northern China in 38 patients with BH4 deficiency, the DQ and IQ scores were analysed by age at start of treatment.58 For Patients who started treatment at less than 6 months of age, 15/16 (94%) had a normal DQ or IQ at follow up. Patients who started treatment at more than 6 months of age, 2/11 (18%) had a normal DQ or IQ at follow up. An inverse correlation was found between DQ or IQ and the age at start of treatment (r = - 0.714, p < 0.01), with patients who started before age 6 months having essentially normal DQ or IQ. A similar negative correlation was found by Liu et al., 2008 in their analysis of 20 patients with BH4 deficiency (10 from Liu et al. and 10 from Chien et al.), that is, a significant negative correlation between age of onset of treatment with BH4 and full-scale IQ (Pearson r = -0.655, p = 0.001).57 Short-term data
Study PKU-007 was a Phase II, multicentre, open-label study evaluating sapropterin tablets in subjects with HPA due to BH4 deficiency. The results of an interim 10-week analysis were available. Twelve subjects were enrolled, 9 with defects in enzymes of biosynthesis (the synthesis group) and 3 with defects in enzymes involved in BH4 recycling. The Delegate was unable to find any information about the age distribution of the subjects and requested that the sponsor clarify this point. For the synthesis group which was the group of subjects taking non-registered formulations of BH4 prior to switching to sapropterin treatment, mean ± SD blood Phe levels remained comparable to those observed at baseline (72.2 ± 13.4 µmol/L) at all study visits following the switch to sapropterin treatment (66.6 ± 13.6 µmol/L to 78.2 ± 28.4 µmol/L). Mean blood Phe levels at baseline were substantially higher for subjects in the recycling group than for subjects in the synthesis group. The three subjects in the recycling group were not taking any non-registered BH4 formulations at study enrolment or during Part 1 of the study. The response to sapropterin treatment varied in the recycling group. All 9 subjects in the synthesis group and 2 or 3 of the subjects in the recycling group had, blood Phe levels less than the 360 µmol/L target at each of the time points during sapropterin treatment. The sponsor was asked to confirm whether 2 or 3 subjects in the recycling group achieved this target level. Out of the 5 published studies identified via a systematic review, reporting short-term data, there were 2 studies which each gave results in more than 100 patients with BH4 deficiency. The first of these 2 was a retrospective analysis of 189 patients while the second was an analysis of 308 patients whose data was maintained in a registry (1975-1995). Unfortunately, the clinical evaluator appears not to have reported the results of these studies. The Delegate requested that the sponsor, in its pre-ACPM response, give a brief summary of the results of these 2 studies (Bernegger and Blau 2002 and Blau et al., 1996).40,60 It was also uncertain whether there was any overlap in subjects between these two studies. The sponsor was asked to clarify whether this was the case.
Data for younger subjects with BH4 deficiency For all six of the younger subjects in the Biopten (D272) study, blood Phe levels were lower after treatment with sapropterin and were maintained within the normal range.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 95 of 123

Therapeutic Goods Administration
In the post-marketing Biopten study there were 14 patients aged less than 1 year and 5 patients aged between 1 and 7 years at the commencement of treatment (out of a total of 27 patients – 22 with PTPS deficiency and 5 with DHPR deficiency). All patients with HPA due to PTPS deficiency had normalisation of blood Phe levels. Nine of 10 patients (90%) who started treatment at less than 1 year of age were growing normally while the remaining patient less than 1 year of age had pre-existing conditions which gradually improved. How many of the younger patients were in the PTPS group and how many in the DHPR group was unknown and the sponsor was requested to clarify this point. By simple arithmetic, there must have been at least 9 (14-5) younger patients in the PTPS group and it follows that all 9 must have had normalisation of blood Phe levels. The Delegate also assumed that none of the younger patients was one of the 3 patients found to have HPA due to PKU (out of the original total of 30 subjects). Even of one assumes that all 3 of these patients were in fact younger patients, then it would still be the case that 6/14 younger patients would have had normalisation of blood Phe levels.
Of the 11 studies previously reported, results from 9 studies which reported findings for children less than 4 years of age were summarised. By the Delegate’s reckoning, there were 104 subjects less than 1 year of age and 19 who were aged 1-4 years (assuming for example that all 27 subjects reported by Wang et al., 2006 were less than 1 year of age at initiation of treatment and that of the 4 subjects aged ≤2 years in the Lee et al., 2006 study, none was aged less than 1 year). All studies demonstrated satisfactory control of blood Phe levels. There was reassuring positive evidence of either maintenance of or an increase in developmental or intelligence quotient scores. From at least 3 studies (Chien et al., 2001, Liu et al., 2008 and Wang et al., 2006) there was evidence that the earlier the medication was started, then the higher the chance of the subject having normal developmental or intelligence quotients.52,57,58
Dosing recommendations in both HPA due to PKU and in HPA due to BH4 deficiency For HPA due to PKU, the recommended starting dose is 10 mg/kg/day with subsequent adjustments to a maintenance dose between 5 and 20 mg/kg/day. The maximum dose studied in the clinical development program was 20 mg/kg/day. In PKU-003, the results indicated that treatment with sapropterin tablets at a dose of 10 mg/kg/day can result in significant reduction in blood Phe levels. Results from the studies PKU-004 and PKU-006 support the recommended dose range of 5 to 20 mg/kg/day. For the treatment of HPA due to BH4 deficiency, the dose recommendation for sapropterin is a starting dose of 2 to 5 mg/kg/day, given once daily. Doses may be adjusted up to a total of 20 mg/kg/day and it may be necessary to divide the total daily dose into 2 or 3 doses to optimize the therapeutic response. The clinical evaluator was of the opinion that the submitted data from the Biopten study (D272), the Biopten post-marketing surveillance study, the interim Week 10 analysis of study PKU-007 and the 11 long-term studies in the published literature all supported the proposed dosage regimen for HPA due to BH4 deficiency. Safety
Safety data were presented separately for subjects with HPA due to PKU or HPA due to BH4 deficiency.
Safety data for subjects with HPA due to PKU In the submission, the analysis of AEs in subjects with HPA due to PKU involved the placebo-controlled trials, PKU-003 and PKU-006 part 2, the open-label, short-term studies, PKU-001 and PKU-006 part 1 and the open-label, long-term study, PKU-004. As well there was interim data from an ongoing study, PKU-008.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 96 of 123

Therapeutic Goods Administration
Overall, 579 subjects have received one or more doses of sapropterin tablets in a PKU clinical study. Of these 579 subjects, complete dosing data were available for 575 subjects. There were 106 subjects exposed for between 2 and 7 months. In study PKU-008, subjects had received the drug for up to 18 months.
Common AEs reported by the sapropterin-treated subjects and at higher rates than for placebo-treated subjects were headache, rhinorrhoea, pharyngolaryngeal pain, vomiting, diarrhoea, nasal congestion, cough and contusion. Across studies, nearly all reported events were mild or moderate in severity. Severe events were reported with the following preferred terms: headache (2), abdominal pain (1), diarrhoea (1), migraine (1), tooth abscess (1) and vomiting (1). In most studies, the majority of AEs were assessed as unrelated to the study treatment. Across studies, the events that were most frequently reported as related to the study treatment were diarrhoea, abdominal pain and headache. In the interim analysis of PKU-008, 79/111 (71.2%) subjects receiving sapropterin experienced at least 1 AE. The most common AEs (cough, pyrexia and nasopharyngitis) are all signs of common communicable URTIs, consistent with the relatively young study population. Most AEs in PKU-008 were classified as mild. There was only one severe AE, namely difficulty concentrating and mood swings which was classified as possibly related to the study drug. In the efficacy and safety studies evaluated, only 1 subject discontinued treatment due to an AE. A female subject with a positive pregnancy test stopped further treatment in study PKU-001. In the study PKU-008, one subject withdrew due to difficulty concentrating, an AE classified as severe. Another subject in the study withdrew because of intermittent diarrhoea, classified as possibly related to study treatment.
There were no deaths reported. Of the six serious AEs, none was considered related to study treatment. For study PKU-008, out of the four subjects reporting SAEs, only one was considered to have an SAE possibly or probably related to treatment, namely gastroesophageal reflux while taking the dissolved tablets.
Analyses of clinical laboratory data did not reveal any significant patterns. The potential signal of ALT and AST elevations observed in the early Phase I studies was not replicated. The clinical trial data showed relatively constant values over time. No clinically meaningful changes were seen in haematology values over time.
Hypophenylalaninaemia, defined as a blood Phe level at or below the lower limit of the normal range, which in this analysis was 26 µmol/L, was observed to occur more frequently in subjects treated with sapropterin than in those treated with placebo (e.g. in PKU-006 part 2, 9/33 [27.3%] patients on 20 mg/kg/day sapropterin vs. 1/12 [8.3%] on placebo). It appeared to occur more commonly at the highest dosage level of 20mg/kg/day and in the younger patients, those aged less than 18 years.
Safety data for subjects with HPA due to BH4 deficiency Data for the safety of sapropterin in HPA due to BH4 deficiency were provided from the Biopten, D272 study, the data from the corresponding post-marketing surveillance study and the interim Week 10 results from study PKU-007.
In study D272, 14/16 (87.5%) subjects were treated with sapropterin granules at a dose of 1 to 5 mg/kg/day. The remaining 2/16 (12.5%) received variable doses. The duration of study treatment was from 140.0 to 606.0 days with mean ± SD = 458.3 ± 130.6 and median = 497.0 days. In study PKU-007, the range of doses administered was from 2.0 to 20.3 mg/kg/day with mean ± SD = 6.6 ± 5.3 mg/kg/day and median = 5.4 mg/kg/day. The range for the duration of exposure was from 55.0 to 58.0 days.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 97 of 123

Therapeutic Goods Administration
In study D272, AEs were reported for 2/16 subjects: 1 AE of moderate hypothermia, possibly or probably related to study treatment and 1 AE of mild convulsion with unknown relationship to study treatment. Over the 10 weeks in study PKU-007, there were 10 AEs reported for 9/12 enrolled subjects. The most frequently reported AEs were diarrhoea (4 subjects, 33.3%), vomiting (3 subjects, 25.0%) and sinusitis (2 subjects, 16.7%). All other AEs were isolated events. AEs classified by the investigator as possibly or probably related to the study treatment were reported for 5 subjects and included diarrhoea (4 subjects, 33.3%) and vomiting (2 subjects, 16.7%). No other drug-related AE was reported for more than one subject. Most AEs were mild to moderate in severity. Only 1 AE, dystonia, was considered severe but it was assessed as not related to study treatment.
No subject was withdrawn due to an AE in the interim analysis at Week 10 for PKU-007. No deaths and no SAEs were reported either in Study D272 or in Study PKU-007.
In Study D272, no specific trends in haematological or chemistry abnormalities were detected. In PKU-007, there were no clinically significant changes in LFTs, there was one subject who experienced clinically significant elevation of lymphocytes and reduction of neutrophils at Week 10 and who also experienced a clinically significant elevation in serum creatinine at Week 4 which resolved by Week 10. The Delegate requested that the sponsor clarify the outcome of the latter subject’s haematological parameters.
In those subjects who experienced a rebound in their Phe blood levels upon cessation of treatment, resumption of treatment with sapropterin was effective in returning Phe levels to the desired therapeutic level.
Post-marketing experience There were the first four 3-month periodic adverse event submissions to the US FDA, covering the year from 13 December 2007 to 12 December 2008. No safety signals of note were reported. Nor were there any significant safety signals arising from the pre-approval and 10-year post-marketing surveillance program conducted by the Japanese sponsor of sapropterin granules (Biopten 2.5% granules) in patients with atypical HPA due to BH4 deficiency.
The sponsor was requested, as part of the pre-ACPM response, to provide an up-to-date summary of the post-marketing experience in both the EU and the USA. Risk Management Plan The RMP Evaluation Report made a number of recommendations and/or requests to the sponsor, all of which the Delegate supported. Other issues
Clinical data in support of the limit of qualification of the impurity S-tetrahydrobiopterin (more formally 6S-tetrahydrobiopterin) The Delegate has already highlighted the difficulties encountered by both the quality evaluator and the nonclinical evaluator in setting an appropriate qualification limit for this impurity. The sponsor has submitted, at the request of the Delegate, some clinical evidence which, the sponsor maintains, supports the higher qualification limit.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 98 of 123

Therapeutic Goods Administration
In the first reference, Spaapen et al. the authors pointed out that much higher levels of S-BH4 have been used, up to 31%, with no apparent significant safety concerns.62
In the second reference, Bernegger et al. they retrospectively evaluated 1919 loading tests from 33 different countries performed in their laboratory between 1988 and 2002, that is, both pre- and post-1999.
Actually, on reading the article, it is apparent that issues of efficacy rather than safety were discussed. The authors noted that, since 1999 an increasing number of patients with Phe hydroxylase (PAH) deficiency had been to be able to decrease their plasma Phe concentrations after a BH4 challenge and they were wondering why this was the case. They argued that, for European patients, the answer was most probably found in the purity of the BH4 preparation. Before 1999 the BH4 tablets of Dr. Schircks Laboratories were composed of 69% of the natural 6R-BH4 and 31% of 6S-BH4. They also spoke about their own experience of testing patients with mild hyperphenylalaninaemia with a combined Phe/BH4 (69% 6R-BH4 and 31% 6S-BH4) loading test in the years before 1999. The patients tested in those years showed either no or only a slight decrease of plasma Phe within 8 hours of the BH4 challenge. Since 1999 the diastereoisomeric purity of the BH4 preparation has been improved to 99.5% 6R-BH4. It would appear that the (6R)-L-erythro-dihydroxypropyl side chain of the natural cofactor is critical for many aspects of the regulation of the PAH enzyme. The affinity of 6R-BH4 to rat liver PAH appears to be 2-3 times higher than that of the unnatural 6S-BH4. It has also been reported that 6S-BH4 causes an irreversible inactivation of rat liver PAH. According to the authors, extrapolating these findings to the human PAH makes it quite conceivable that the mixture of the 6R- and 6S-epimers (69:31) would not have enhanced in vivo PAH activity in potentially 6R-BH4-responsive PAH-deficient patients. Thus the issue would appear to have been one of efficacy. In the article, there was actually no direct comment made about the safety of the pre-1999 product although one would assume that it would have had wide usage.
63
Evidence of the use of BH4 from Dr. Schircks Laboratories in 10 patients with HPA due to BH4 deficiency was provided by Al Aqeel.
There were 278 loading tests performed with the purer 6R-BH4, which the authors commented was about 33% more active than the formerly used mixture of 6R,S-BH4, the latter used in the remaining 1641 patients. The authors speak of the same product from Dr Schircks Laboratories but refer to its content as 66.6% 6R-BH4 and 33.3% 6S-BH4. As noted by the sponsor this indicates wide usage of the pre-1999 formulation, at least for diagnostic if not treatment purposes. Once again safety was not specifically addressed but there were no negative comments about safety in the article. Out of the total of 1919 loading tests, there were, incidentally, 189 patients diagnosed as BH4-deficient.
50 This study has in fact been assessed by the clinical evaluator. Neurologic findings improved significantly in all patients after 5 to 24 months. There were no particular safety concerns commented upon in the article.
The final article, a review by Blau, provided the comment that that BH4 was available as tablets (10 or 50 mg) from Schircks Laboratories or from DSP, Japan as a granulate.60 In the final section of the article, the authors commented that BH4 had been used successfully for treatment of patients with BH4 deficiency for over 20 years and that based on the information from the BIODEF database (www.bh4.org/biodef.html), there were at that time no side effects reported. Furthermore, they commented that in 1998 a questionnaire had been sent to 62 Spaapen LJM et al, Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency, state of the art, Molecular Genetics and Metabolism 2003; 78: 93-99. 63 Bernegger C et al, High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninaemias: a study of 1919 patients observed from 1988 to 2002. Molecular Genetics and Metabolism 2002; 77: 304-313.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 99 of 123

Therapeutic Goods Administration
all clinics in Germany who used Schircks BH4 and the only AE report was one report of a transient rash in a child on 2.5-10.0 mg/kg BH4.
Dr. Schircks Laboratories has ceased worldwide supply of its BH4 product. Risk-Benefit Analysis Delegate Considerations
The bioavailability of oral sapropterin in juvenile rats was about 5-fold higher than in adult rats and the juvenile:adult exposure ratio in rats was 23:1. The nonclinical evaluator expressed concerns that this may have implications for the proposed starting dose in young children, which is currently the same as for adults on a mg/kg basis. The same concerns have been expressed by the RMP evaluator who has made some relevant recommendations for amendments to the proposed PI which will alert prescribers to this issue. The kidney was identified as a target organ of toxicity in the rat repeat-dose studies (mild tubular basophilia) at doses resulting in very low safety margins compared to the human exposure. The nonclinical evaluator made the comment that the risk of renal damage in infants is unknown, because of the lack of PK data in young children and urged caution in dose selection in infants.
Because of the putative higher absorption in infants and also the immaturity of the blood brain barrier in infants, especially under the age of 12 months, the nonclinical evaluator expressed concerns that infants are likely to have higher cerebral exposure to sapropterin than adults. The potential effects of the latter higher exposure have not been fully investigated. A very common AE of sapropterin is headache, an effect that an infant would have difficulty communicating. Also in infants, Phe intake is more easily controlled. Weighing up all these factors, the nonclinical evaluator argued that, whereas the unquantified risks of increased BH4 levels on brain development and function may be acceptable for BH4-deficient infants, such is not the case for infants with PKU. The Delegate acknowledged that all the concerns expressed by the nonclinical evaluator are valid concerns. Taking into account all of the information relating to the level of the impurity, S-BH4, in the tablet formulation proposed for registration, the Delegate was satisfied that the extra clinical data provided by the sponsor does qualify the limit for that impurity. There is a long history of prior usage by clinicians of a formulation of BH4 which clearly contained a much higher content of the impurity. While there is evidence that such high content may have had an effect upon efficacy, there is no compelling evidence that the difference in composition was translated into any difference in safety profile. The Delegate acknowledged the concerns of the quality evaluator. However, the argument is turning over differences in quality specifications of very small magnitude. In fact the sponsor’s view is that the product does meet the required quality specifications. On balance, the Delegate was satisfied that there are no significant clinical concerns arising from this specifications issue which is ultimately a matter of fine tuning of the specifications limit. The ACPM was asked for advice on this issue.
With regard to the clinical efficacy in HPA due to PKU in patients ≥4 years, the sponsor has, in the two Phase III studies, PKU-003 and PKU-006 Part 2, demonstrated a statistically significant dose related effect on Phe levels by sapropterin compared with placebo, which was maintained for the duration of the studies. Furthermore in PKU-006 Part 2, a statistically significant increase in Phe tolerance has been demonstrated for sapropterin compared with placebo. In this population, sapropterin has been shown to have an acceptable safety profile.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 100 of 123

Therapeutic Goods Administration
On this basis and because there were no major concerns relevant to this population from the nonclinical review, the Delegate proposed to recommend approval of the use of sapropterin for this population. With regard to clinical efficacy in HPA due to PKU in patients aged 0 – 4 years of age, the evidence was entirely from the literature; 10 published studies with a mix of short- and long-term data. While the short-term data was in a large number of patients, the numbers of patients for whom there was long-term data was relatively small, namely 43. Even though the condition is uncommon, even rare, there was a paucity of long-term data, especially in comparison with the short-term data available. The short-term data did demonstrate consistent reductions in Phe levels up to 24 hours after loading dose, even in the very young. For the long-term treatments, there was consistency in a variety of positive outcome measures, surrogate measures such as Phe blood levels, Phe tolerance, diet liberalisation and Phe to tyrosine ratio but no clinical outcome measures such as development or intelligence scores. Then there are the valid concerns of the nonclinical evaluator, particularly with regard to the use of sapropterin in infants aged less than 12 months and with regard to the issue of starting doses in the very young. While there was no evidence of any specific safety signals emerging from the literature sources, one always must be careful, as invariably the focus of the articles is on efficacy, unless the primary objective of the study is safety-related. Determination of the risk-benefit balance in this population is difficult. Perhaps it may be possible to mitigate this risk to an acceptable level by appropriate precautions and warnings in the PI but at this stage, the Delegate was of the view that the risk benefit balance is not acceptable enough in the direction of benefit and so would recommend rejection of the application for use in patients with HPA due to PKU who are less than 4 years of age. Nonetheless, the Delegate sought the specific advice from the ACPM on this issue.
With regard to clinical efficacy in HPA due to BH4 deficiency, there was evidence adduced in a smaller number of patients than in HPA due to PKU but this is a reflection of the fact that HPA due to BH4 deficiency is extremely rare. Overall, the information from the Biopten (D272) study, from the 10-year post-marketing surveillance study for Biopten, from the short-term study PKU-007 and from a range of published studies reporting both short- and long-term data, demonstrate that normalisation of blood Phe levels may occur at doses of sapropterin less than 10 mg/kg/day, particularly in the subgroup of patients with PTPS deficiency. The evidence for efficacy in DHPR deficiency was somewhat more equivocal. With regard to the evidence of efficacy in subjects aged less than 4 years, the evidence submitted was much more compelling than that for the same age group of patients with HPA due to PKU. The evidence collected from the literature was from an impressively large number of subjects – 104 under 1 year of age and 19 aged between 1 and 4 years and consisted of evidence of satisfactory control of blood Phe levels and also actual positive clinical outcome evidence. Again there are no specific significant safety signals emerging from the data. The medication appears to be well tolerated. By and large the reservations expressed by the nonclinical evaluator with regard to the use of sapropterin in younger subjects were not of the same degree when applied to younger subjects with BH4 deficiency. However, there were still valid concerns expressed about the need for caution in dose selection, particularly the starting dose. In the view of the Delegate, the latter can be managed via appropriate wording in the PI. Thus the Delegate proposed to recommend approval of the use of sapropterin for subjects with BH4 deficiency, both younger subjects aged less than 4 years and those above the age of four years.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 101 of 123

Therapeutic Goods Administration
Response from Sponsor
In its pre-ACPM submission, the sponsor noted that the Delegate’s conclusion appears not unreasonable but the sponsor outlined a number of reasons why this would not be an appropriate outcome for Australian healthcare professionals, patients and patient families striving to contain the effects of this potentially devastating inherited metabolic disorder. Patients in the first few years of life are those most of risk of permanent neurological impairment due to HPA. In the response, the sponsor discussed how Kuvan can be used safely in patients of all ages, irrespective of whether they suffer from primary BH4 deficiency or PKU, by reviewing the nature of sapropterin, how PKU is treated with specific reference to Australia, and how sapropterin is administered and monitored to ensure the benefits outweigh the risks.
The sponsor noted that an important aim of treatment for PKU is to maintain plasma Phe within the recommended range for age to prevent neurological damage. The mainstay of treatment for PKU is a Phe-restricted diet. Age at initiation and level of metabolic control clearly influence outcomes so effective treatment needs to be instituted early with regular monitoring of blood Phe levels. Compliance is crucial. Considering the known toxicity of elevated Phe, the risk particularly in young patients at these early developmental ages, and the potential benefit of Kuvan in helping to control Phe, it is only reasonable to suggest withholding Kuvan in patients less than 4 years old on the understanding that an alternative effective treatment is available. The Phe-restricted diet is designed in a way that allows a decrease in blood Phe concentration and provides sufficient Tyr (now an essential amino acid as it is not being synthesised) and other nutrients required for optimal growth and development of the child. This involves a measured allowance of natural protein in the diet to provide the Phe requirement for growth and health as guided by routine and regular blood Phe monitoring. All high protein foodstuffs such as meat, cheese, fish, and flour-based foods are excluded. Foods such as rice, potatoes and milk, which contain less protein, are allowed in measured amounts while others which have negligible amounts of natural protein, like most fruits and many vegetables, are allowed ‘freely’. Commercially available supplements of amino acids that lack Phe are to be taken on a daily basis. The diet itself is very restrictive in nature and the supplements used have an unpleasant taste and odour, presenting particular issues in young infants who cannot comprehend the essential need for supplements. Some progress has been achieved in recent years in these aspects with newer, better formulations and special foods (not all of which are available in Australia). In spite of these advances, there is still high potential for compromised quality of life as well as non-compliance associated with risks of nutritional deficiencies and neurological toxicity from uncontrolled Phe. According to Australian metabolic specialists consulted by the sponsor, Kuvan should be available for patients of all ages, from the neonatal period and onwards. Considering the natural development of taste and eating habits and the difficulty of transitioning childhood eating patterns, limiting Kuvan to patients 4 years and older represents a restriction that is not without its own clinical consequences. Experts describe for example a proportion of patients with elevated Phe levels, initially not high enough to warrant dietary management, but which increase above 350 μmol/L with the transition from breastfeeding to cow’s milk based formula and solids. Because these infants are not accustomed to the taste of PKU supplement formula, they can be difficult to manage. Should Kuvan be limited to patients 4 years or older, the converse situation would occur with children being managed with diet alone having the option to transition to Kuvan plus diet after their fourth birthday, then needing to be stabilised again on the new regime.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 102 of 123

Therapeutic Goods Administration
Although outside the scope of the sponsor’s submission, there is ongoing research into clinical benefits of improved metabolic control beyond maintaining Phe within the recommended range for age. This includes lower Phe to Tyr ratios and smaller fluctuations in blood Phe levels (i.e. smaller standard deviation or inter quartile range of Phe levels despite similar mean and median levels), both of which have been shown to improve cognitive outcome. Sapropterin offers improved metabolic control based on these parameters in patients treated for several years, starting from birth. The sponsor acknowledged the need to take safety findings from nonclinical studies into account when designing a clinical development program and planning for the post-marketing period. The Risk Management Plan for Kuvan has been evaluated by the TGA and a number of questions have been asked and answered relating to potential safety issues, including findings from the animal studies. All of the TGA concerns were addressed in the revised Kuvan RMP. In addition, there is no clinical evidence of a different safety profile in young infants (based on published experience) or of a relationship between dose and adverse effects within the therapeutic range (Kuvan study PKU-004, doses tested from 5 to 20 mg/kg/day). The absence of information about the effect of Kuvan on brain development and function needs to be balanced against the known effects of uncontrolled Phe on brain development and function. This is also not a question specific to patients less than 4 years old but needs to be studied further for all patients. The company has committed to conduct such a study. A concern was raised about the nonclinical evidence that sapropterin increases neuropathic pain, consistent with the common clinical adverse effect of headache, which might be difficult to detect in infants. However, there are means to detect pain in young infants including poor feeding, otherwise unexplained whining, irritability or crying, etc, which would be obvious to parent and physician.
This has not been seen with BH4 in young infants to date but will be looked for in the planned Phase IIIb study in this population (SPARK study). As described in the RMP responses, the sponsor suggested that rather than exclude patients less than 4 years old from treatment with Kuvan, the company establish a local registry study to include patients less than 4 years of age and invite the major Australian metabolic centres to participate. This will ensure that safety data, including any on which the TGA would like the sponsor to focus, will be collected and analysed centrally so that any actual safety concerns are identified and addressed in a timely manner. Requests from the Delegate and the Sponsor’s pre-ACPM Submission
The Delegate requested that the sponsor indicate in the pre-ACPM response when the following final study reports will be available to the sponsor and hence to the TGA. The provision of these reports as evaluable data within the context of corresponding category 1 submissions may form specific conditions of registration of the product in Australia:
· the final study report of the clinical safety, efficacy and pharmacokinetics trial of Kuvan in patients with PKU who are 4 years of age or younger at study entry (the trial for which the final study report was due to have been submitted to the FDA’s Center for Drug Evaluation and Research (CDER) by 14 June 2010)
The sponsor advised that this study is not within the scope of Merck Serono’s Commercial Agreement with BioMarin, and will not be available for Merck Serono to submit to the TGA. Instead, Merck Serono proposed to submit to TGA the results of a similar study being conducted to fulfil a European post-approval commitment for which the Final Report will be available 3 years from first patient enrolled (excluding extension period).
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 103 of 123

Therapeutic Goods Administration
· the final study report (and any released interim study reports) of the long-term study to assess growth and neurocognitive development with Kuvan in patients who are 8 years of age or younger at study entry (the trial for which the final study report is due for submission to CDER by 14 June 2017)
The sponsor advised that this study is not within the scope of Merck Serono’s Commercial Agreement with BioMarin, and will not be available for Merck Serono to submit to the TGA. Instead, Merck Serono proposed to submit to TGA the results of a similar study being conducted to fulfil a European post-approval commitment for which the Interim Reports will be available every 2 years, dated from first enrolment and the final report will be available 8 years after first enrolment.
· the final study report of the open-label extension study PKU-008 (the report which was due for submission to CDER by 30 March 2010)
The sponsor indicated that the PKU-008 final study report is available now and can be submitted to the TGA.
· each annual report of the proposed registry of patients with PKU being treated with Kuvan (the first annual report should already be available) and the final study report of the sub-study within that registry which will evaluate the effect of Kuvan on pregnancy and lactation
The sponsor indicated that the Interim Reports will be available annually, dated from first enrolment (December 2009 – first interim report will be available in July 2011) and the final report will be available 16 years after first enrolment.
· the final study report of the thorough QT study with Kuvan, the report of which was due for submission to CDER by 14 October 2009
The sponsor indicated that the QTC-001 final study report is available now and can be submitted to the TGA.
· the final study report of the analysis of the whole blood samples for Phe hydroxylase (PAH) gene mutations which were collected during the PKU-001 study (the final report of this analysis was to have been submitted to CDER by 14 December 2008)
The sponsor indicated that this was available in the submission
· the final study report of the open-label study PKU-007 in subjects with HPA due to primary BH4 deficiency
The sponsor indicated that the final study report for PKU-007 is expected in July 2010 and can be submitted to the TGA.
The Delegate also requested that the sponsor address the following issues in their Pre-ACPM response:
· An up-to-date summary of the post-marketing experience in both the EU and the USA
· Clarification of the nature of the literature evidence submitted to the EMA
Both of these issues were addressed · Comment on the delegate’s estimates of 231 subjects with short-term data and 43 with
long-term data for those aged 0 to 4 years of age with HPA due to PKU
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 104 of 123

Therapeutic Goods Administration
The sponsor indicated that after revisiting the publication by Ye et al, 2007 it was noted that the study actually reported on at least 138 PKU patients that received a BH4 loading test. This is incorrectly represented in the clinical evaluation report which refers to short term results for 129 patients.
There were a total of 168 hyperphenylalaninaemia (HPA) patients that received a BH4 loading test out of 223 patients detected with HPA by newborn screening. Assuming all 30 cases that were diagnosed with 6-pyruvoyl tetrahydropterin synthase deficiency (PTSD) received a BH4 loading test, results in at least 138 cases of patients with HPA due to PKU. Twenty four of the BH4-responsive patients went on to receive treatment of 10 mg/kg/day for one week and 3 of these patients continued on long term treatment.
The increased figure of 138 PKU patients in this group indicates that there were at least 240 subjects with short term data across the 10 published articles.
With regard to the remaining requests below, the Delegate required only very brief responses. In those cases, where the answer to the question is not available or not readily or easily available, please state that:
· Further information on the ages of patients in the Biopten study
· Clarification on the difference between the daily dosing range and the maintenance dosing range in the Biopten study
· For the post-marketing surveillance study of Biopten, clarification of how many of the children aged between 1 and 7 years at the commencement of treatment were under 4 years of age
· Clarification of the age distribution of patients in the study PKU-007
· Clarification of whether 2 or 3 subjects in the recycling group in PKU-007 achieved the target level
· A brief summary of the results of the 2 studies, Bernegger and Blau 2002 and Blau et al 1996, and whether there was any overlap in subjects between these two studies
· Of the 5 subjects less than 5 years of age in the Biopten study, is it known how many were less than 4 years of age
· Is it known how many of the younger patients in the post-marketing Biopten study were in the PTPS group and how many in the DHPR group
· Is the sponsor able to clarify the outcome of the subject who experienced a clinically significant elevation of lymphocytes and reduction of neutrophils at Week 10 in the Biopten study
The sponsor provided satisfactory answers to all of the above questions. Advisory Committee Consideration
The Delegate proposed to reject the submission for registration of the new chemical entity, sapropterin, for the indications proposed by the sponsor but instead approve the submission for the wording of the indications which appears in the EU Summary of Product Characteristics, namely:
Kuvan is indicated for the treatment of hyperphenylalaninaemia (HPA) in adult and paediatric patients of 4 years of age and over with phenylketonuria (PKU) who have been shown to be responsive to such treatment (see Dosage and Administration)
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 105 of 123

Therapeutic Goods Administration
Kuvan is also indicated for the treatment of hyperphenylalanaemia (HPA) in adult and paediatric patients with tetrahydrobiopterin (BH4) deficiency who have been shown to be responsive to such treatment (see Dosage and Administration). The Delegate directed the following questions to the ACPM:
· Does the ACPM agree with the Delegate that there is insufficient evidence to approve the registration of sapropterin for use in paediatric patients under 4 years of age with phenylketonuria (PKU) or is the committee of the view the submission could be approved with appropriate precautions and warnings in the PI, particularly with regard to its use in children less than 12 months of age?
· In the light of the concerns of the nonclinical evaluator, do the precautions and warnings in the PI need further strengthening with regard to initial dose selection in the very young?
· Does the ACPM agree with the delegate that the information in the Dosage and Administration section, particularly with regard to the situation of young children and infants, needs considerable revision?
· Does the ACPM agree with the Delegate that, from a clinical perspective, the specifications requirements for the enantiomeric impurity, S-BH4, are satisfied?
· Should the post-marketing commitments entered into by the US sponsor of this product with the US FDA also be imposed as conditions of registration here in Australia?
The ACPM, having considered the evaluations and the Delegate’s overview, as well as the sponsor’s response to these documents, recommended approval of the submission for the indication: For the treatment of Hyperphenylalaninaemia (HPA) in sapropterin-responsive adult and paediatric patients, with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency. In making this recommendation, the ACPM considered that an overall positive risk benefit profile for the amended indication was demonstrated for the small target population. The specific conditions of registration should include development and full implementation of an appropriate Risk Management Plan. Outcome Based on a review of quality, safety and efficacy, TGA approved the registration of Kuvan soluble tablets containing sapropterin dihydrochloride 100 mg, indicated for: Kuvan is indicated for the treatment of hyperphenylalaninaemia (HPA) in sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency (see Dosage and Administration for definition of sapropterin responsiveness). The following specific conditions of approval were included:
1. The full implementation of the Risk Management Plan, version 4.0, dated 1 June 2010 and of the Australian-specific patient registry, as agreed with the Office of Product Review (formerly the Office of Medicines Safety Monitoring). 2. The provision to the TGA of each annual report of the proposed global registry of patients with PKU being treated with Kuvan, as soon as that annual report becomes available.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 106 of 123

Therapeutic Goods Administration
3. The provision, as evaluable data as part of a category 1 submission and within a period of six (6) months from the date of registration of Kuvan, of the following three (3) studies together:
· The final study report for the open-label extension study PKU-008, · The final study report of the thorough QT study with Kuvan, and · The final study report of the open-label study PKU-007 in subjects with HPA
due to BH4 deficiency. 4. The provision, as evaluable data as part of category 1 submissions, of each of the following studies being conducted by Merck Serono to fulfil EMA registration commitments:
· The final study report of the clinical safety, efficacy and pharmacokinetics trial of Kuvan in patients with PKU who are 4 years of age or younger at study entry, the report to be submitted when it is available.
· The final study report (and any released interim study reports) of the long-term study to assess growth and neurocognitive development with Kuvan, the report(s) to be submitted when it is available.
Attachment 1. Product Information The following Product Information was approved at the time this AusPAR was published. For the current Product Information please refer to the TGA website at www.tga.gov.au.
AusPAR Kuvan Sapropterin dihydrochloride Merck Serono Australia Pty Ltd PM-2009-02891-3-3 Date of Finalisation 21 October 2010
Page 107 of 123

Version: A001-1010 Page 1 of 15 Supersedes: N/A
PRODUCT INFORMATION
KUVAN
Sapropterin dihydrochloride
NAME OF THE MEDICINE KUVAN (sapropterin dihydrochloride) 100 mg soluble tablets Structural formula:
Molecular formula: C9H15N503 · 2HCl CAS number: 69056–38–8 DESCRIPTION Sapropterin dihydrochloride is an off-white to light yellow crystalline powder. It melts with decomposition at 239-241C. The stereochemical configuration of sapropterin dihydrochloride has been demonstrated by single-crystal X-ray analysis. Sapropterin dihydrochloride is hygroscopic. At room temperature, sapropterin dihydrochloride is very soluble in water ( 1 g/mL). It is sparingly soluble in methanol (10 mg/mL) and ethanol (0.9 mg/mL), and practically insoluble ( 0.1 mg/mL) in aprotic solvents such as diethyl ether. The water/octanol partition coefficient greatly favours dissolution in water indicating that the compound is hydrophilic. The most basic pKa value of sapropterin is 9.20 and consequently it will be fully ionised at gastrointestinal pH. KUVAN is supplied as immediate release soluble tablets intended to be administered orally following dissolution. The tablets are off-white to light yellow. Each tablet contains 100 mg of sapropterin dihydrochloride (equivalent to 77 mg of sapropterin), mannitol (E421), calcium hydrogen phosphate anhydrous, crospovidone type A, ascorbic acid (E300), sodium stearylfumarate and riboflavin (E101). PHARMACOLOGY Pharmacodynamics/Mechanism of Action
Hyperphenylalaninaemia (HPA) is diagnosed as an abnormal elevation in blood phenylalanine (Phe) levels and is usually caused by autosomal recessive mutations in the genes encoding for the liver enzyme phenylalanine hydroxylase (in the case of

Version: A001-1010 Page 2 of 15 Supersedes: N/A
phenylketonuria, PKU) or for the enzymes involved in 6R-tetrahydrobiopterin (6R-BH4) biosynthesis or regeneration (in the case of BH4 deficiency). BH4 deficiency is a group of disorders arising from mutations or deletions in the genes encoding for one of the five enzymes involved in the biosynthesis or recycling of BH4. In both PKU and BH4 deficiency, Phe cannot be effectively transformed into the amino acid tyrosine, leading to increased Phe levels in the blood. However, in patients with BH4 deficiency there are other enzymes in addition to phenylalanine hydroxylase that cannot function properly. These include tryptophan and tyrosine hydroxylase (located in the brain and other tissues) and nitric oxide synthase. Sapropterin dihydrochloride is a synthetic version of the naturally occurring 6R-BH4, which is a cofactor of the hydroxylases for phenylalanine, tyrosine and tryptophan. The rationale for administration of KUVAN in patients with BH4-responsive PKU is to enhance the activity of the defective phenylalanine hydroxylase and thereby increase or restore the oxidative metabolism of Phe sufficient to reduce or maintain blood Phe levels, prevent or decrease further Phe accumulation, and increase tolerance to Phe intake in the diet. The rationale for administration of KUVAN in patients with BH4 deficiency is to replace the deficient levels of BH4, thereby restoring the activity of phenylalanine hydroxylase. In PKU patients who are responsive to BH4 treatment, blood Phe levels decrease within 24 hours after a single administration of KUVAN, although maximal effect on Phe level may take up to a month, depending on the patient. A single daily dose of KUVAN is adequate to maintain stable blood Phe levels over a 24-hour period. In a sub-study of the clinical trial described as ‘Study 3’ under CLINICAL TRIALS, blood Phe levels were measured multiple times over a 24-hour period in 12 patients taking 10 mg/kg/day. The blood Phe levels remained stable during the 24-hour observation period: mean (± Standard Deviation) was 661 (±433) µmol/L at pre-dose and 631 (±454) µmol/L at 24 hours post-dose; the lowest mean value during the 24-hour period was 477 (±241) µmol/L at 16 hours post-dose. No consistent relationship between meals and blood Phe levels was observed during the 24-hour period. Pharmacokinetics Absorption Sapropterin is absorbed after oral administration of the dissolved tablet and the maximum blood concentration (Cmax) is achieved 3 to 4 hours after dosing in the fasted state. The rate and extent of absorption of sapropterin is influenced by food. Compared to fasting, absorption is higher after a high-fat, high-calorie meal, resulting, on average, in 40-85% higher maximum blood concentrations achieved 4 to 5 hours after administration. Neither the absolute bioavailability nor the bioavailability after oral administration in humans is known. Distribution In non-clinical studies, sapropterin was primarily distributed to the kidneys, liver, adrenal glands and spleen as assessed by levels of total and reduced biopterin concentrations (see also PRECAUTIONS, Use in Lactation and Paediatric Use). Very small amounts of sapropterin

Version: A001-1010 Page 3 of 15 Supersedes: N/A
were distributed to the brain in adult rats but in juvenile rats total brain biopterin levels were significantly increased following sapropterin administration. Metabolism 6R-BH4 is primarily metabolised in the liver with dihydrobiopterin and dihydroxanthopterin as the main human metabolites. Since sapropterin is a synthetic version of the naturally occurring 6R-BH4, it can be reasonably anticipated to undergo the same metabolism, including 6R-BH4 regeneration. Folic acid and vitamin B12 may increase BH4 levels. Excretion The mean elimination half-life of KUVAN in PKU patients was approximately 6-7 hours. Following intravenous administration in rats, sapropterin is mainly excreted in the urine. Following oral administration it is mainly excreted in the faeces while a small proportion is excreted in urine. CLINICAL TRIALS Phenylketonuria (PKU) The efficacy and safety of KUVAN were evaluated in 4 clinical trials in patients with PKU ranging in age from 4 to 48 years old. Patients with significant concurrent diseases with potential to interfere with efficacy and safety analyses were excluded from the trials. The results of these studies demonstrate the efficacy of KUVAN to reduce blood Phe levels and to increase dietary Phe tolerance. Study 1 was a multicentre, open-label, uncontrolled clinical trial of 489 patients with PKU who had baseline blood Phe levels 450 µmol/L. Patients ranged in age from 8 to 48 years (38 patients were 8-11 years old and 451 were 12 years of age or older). Patients were to receive treatment with KUVAN 10 mg/kg/day for 8 days. For the purposes of this study, response to KUVAN treatment was defined as a 30% decrease in blood Phe from baseline. At Day 8, 96 patients (20%) were identified as responders. Study 2 was a multicentre, double-blind, placebo-controlled trial of patients with PKU who responded to KUVAN in Study 1. After a washout period from Study 1, patients were randomised equally for 6 weeks of treatment with KUVAN 10 mg/kg/day or placebo. Four (10%) of the 41 KUVAN-treated patients and 8 (17%) of the 47 placebo patients were 8-11 years old; all other treated patients were 12 years of age or older. Efficacy was assessed by the mean change in blood Phe level from baseline to Week 6 in the KUVAN–treated group as compared to the mean change in the placebo group. The results showed that KUVAN 10 mg/kg/day significantly reduced blood Phe levels as compared to placebo (See Figure 1). The baseline blood Phe levels for the KUVAN-treated group and the placebo group were similar, with mean (±SD) baseline blood Phe levels of 843 (±300) μmol/L and 888 (±323) μmol/L, respectively. The mean (±SD) decrease from baseline in blood Phe levels at the end of the 6 week study period was 236 (±257) μmol/L for the KUVAN treated group as compared to an increase of 3 (±240) μmol/L for the placebo group (p<0.001). For patients with baseline blood Phe levels 600 µmol/L, 42% (13/31) of

Version: A001-1010 Page 4 of 15 Supersedes: N/A
those treated with KUVAN and 13% (5/38) of those treated with placebo had blood Phe levels < 600 µmol/L at the end of the 6-week study period (p=0.012).
Displayed are mean blood Phe values for each treatment group at each visit and the associated 95% CIs. The numbers above
and below the lines are the number of subjects who have data at a given time-point. BL refers to Baseline Visit.
Study 3 was a multicentre, open-label, 22-week extension study in which 80 patients who responded to treatment in Study 1 and completed Study 2 were treated. During the first 6 weeks of Study 3, patients underwent forced dose-titration with 3 different doses of KUVAN. Treatment during this dose titration period consisted of 3 consecutive 2-week courses of KUVAN at doses of 5, then 20, and then 10 mg/kg/day. At baseline, mean (±SD) blood Phe was 844 (±398) µmol/L. At the end of treatment with 5, 10, and 20 mg/kg/day, mean (±SD) blood Phe levels were 744 (±384) µmol/L, 640 (±382) µmol/L, and 581 (±399) µmol/L, respectively. During the period from Week 6 to Week 10, patients were maintained on KUVAN 10 mg/kg/day pending analysis of their blood Phe results from the forced-dose titration period. Starting at the Week 10 visit, each patient was assigned to receive a fixed dose of 5, 10 or 20 mg/kg/day based on their blood Phe results measured at the Week 2 and Week 6 visit, then continued using this optimal KUVAN dose until the Week 22 visit. Of the 80 patients, 6 (8%) received 5 mg/kg/day, 37 (46%) received 10 mg/kg/day and 37 (46%) received 20 mg/kg/day KUVAN from Week 10 to Week 22. Patients who received 10 or 20 mg/kg/day at all time points between Week 10 and Week 22 had mean blood Phe levels during this time comparable to those obtained on the same dose during the forced dose-titration period. Patients treated with 5 mg/kg/day from Week 10 to Week 22 had mean blood Phe levels higher than during the forced dose-titration period. The mean (±SD) blood Phe levels at the Weeks 12-22 visits ranged between 620 (±371) and 652 (±383) µmol/L. On average, patients maintained a stable reduction in Phe levels. The 95% confidence interval for the mean change from baseline blood Phe level at the first visit after subjects started using their optimal dose was (-297 μmol/L, -152 μmol/L), and each of the 95% confidence intervals for the mean change from baseline blood Phe level at Weeks 16, 20 and 22 overlap with this interval indicating persistence of the effect of KUVAN treatment.
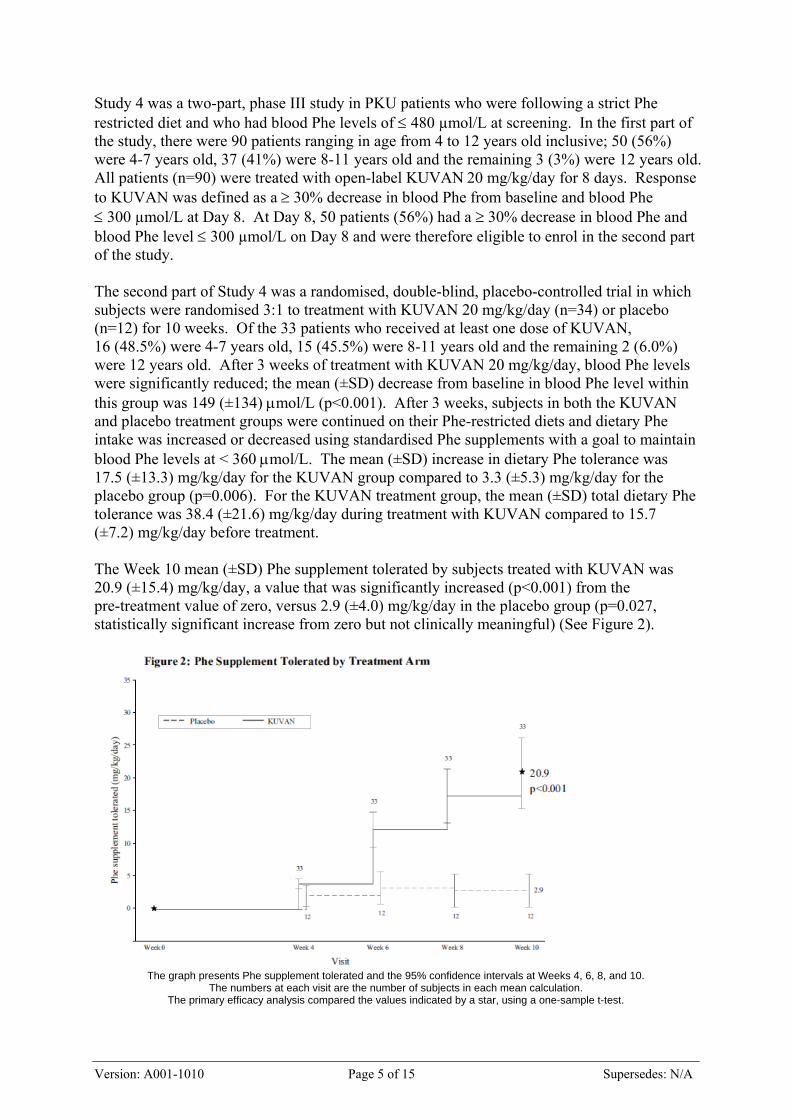
Version: A001-1010 Page 5 of 15 Supersedes: N/A
Study 4 was a two-part, phase III study in PKU patients who were following a strict Phe restricted diet and who had blood Phe levels of 480 µmol/L at screening. In the first part of the study, there were 90 patients ranging in age from 4 to 12 years old inclusive; 50 (56%) were 4-7 years old, 37 (41%) were 8-11 years old and the remaining 3 (3%) were 12 years old. All patients (n=90) were treated with open-label KUVAN 20 mg/kg/day for 8 days. Response to KUVAN was defined as a 30% decrease in blood Phe from baseline and blood Phe 300 µmol/L at Day 8. At Day 8, 50 patients (56%) had a 30% decrease in blood Phe and blood Phe level 300 µmol/L on Day 8 and were therefore eligible to enrol in the second part of the study. The second part of Study 4 was a randomised, double-blind, placebo-controlled trial in which subjects were randomised 3:1 to treatment with KUVAN 20 mg/kg/day (n=34) or placebo (n=12) for 10 weeks. Of the 33 patients who received at least one dose of KUVAN, 16 (48.5%) were 4-7 years old, 15 (45.5%) were 8-11 years old and the remaining 2 (6.0%) were 12 years old. After 3 weeks of treatment with KUVAN 20 mg/kg/day, blood Phe levels were significantly reduced; the mean (±SD) decrease from baseline in blood Phe level within this group was 149 (±134) mol/L (p<0.001). After 3 weeks, subjects in both the KUVAN and placebo treatment groups were continued on their Phe-restricted diets and dietary Phe intake was increased or decreased using standardised Phe supplements with a goal to maintain blood Phe levels at < 360 mol/L. The mean (±SD) increase in dietary Phe tolerance was 17.5 (±13.3) mg/kg/day for the KUVAN group compared to 3.3 (±5.3) mg/kg/day for the placebo group (p=0.006). For the KUVAN treatment group, the mean (±SD) total dietary Phe tolerance was 38.4 (±21.6) mg/kg/day during treatment with KUVAN compared to 15.7 (±7.2) mg/kg/day before treatment. The Week 10 mean (±SD) Phe supplement tolerated by subjects treated with KUVAN was 20.9 (±15.4) mg/kg/day, a value that was significantly increased (p<0.001) from the pre-treatment value of zero, versus 2.9 (±4.0) mg/kg/day in the placebo group (p=0.027, statistically significant increase from zero but not clinically meaningful) (See Figure 2).
The graph presents Phe supplement tolerated and the 95% confidence intervals at Weeks 4, 6, 8, and 10.
The numbers at each visit are the number of subjects in each mean calculation. The primary efficacy analysis compared the values indicated by a star, using a one-sample t-test.

Version: A001-1010 Page 6 of 15 Supersedes: N/A
Patients less than 4 years of age were not included in the KUVAN clinical trials described above. However, reports in published literature indicate that more than 2,700 children with PKU aged newborn to 4 years of age have been administered BH4, including at least 43 who received therapy for 2 months or longer. The maximum daily dose reported was 20 mg/kg body weight. BH4 Deficiency Evidence of the safety and effectiveness of KUVAN for the treatment of HPA due to BH4 deficiency is provided by the results of an interim analysis of data from a study conducted with KUVAN, results from studies conducted with sapropterin dihydrochloride granules registered in Japan for this indication, and published studies of clinical experience with BH4 identified via a systematic literature review. Clinical experience reported in published literature includes prospective and retrospective open-label studies, using both Phe blood levels and clinical outcomes (e.g. IQ and development measures), to determine efficacy. Approximately 120 patients were less than 4 years old at start of treatment, including 104 who started treatment when less than 1 year old. An open-label, multicentre clinical trial evaluating the efficacy and safety of KUVAN for the treatment of HPA due to BH4 deficiency enrolled 12 patients, 9 with defects in enzymes of BH4 biosynthesis and 3 with defects in enzymes involved in BH4 recycling. Patients ranged in age from 3 to 35 years, 1 (8%) less than 4 years, 3 (25%) between 4-7 years, 2 (17%) between 8-11 years, and the remaining 6 patients (50%) were 12 years of age or older. Patients receiving an unregistered formulation of BH4 prior to study entry started treatment with KUVAN at approximately the same daily dose as the prior BH4 dose; other patients commenced treatment at 5 mg/kg/day. Dose adjustment up or down to a maximum of 20 mg/kg/day was permitted at study Week 6. Mean (±SD) blood Phe remained at levels similar to baseline (133 ± 135 µmol/L) at all study visits covered by the interim analysis (104 ± 67 µmol/L to 143 ± 147 µmol/L). Most subjects (11/12, 91.7%) remained below the blood Phe target of < 360 µmol/L at all study visits covered by the interim analysis (weeks 4, 6, 8 and 10). A study with sapropterin dihydrochloride 2.5% granules was conducted in 16 patients with BH4 deficiency treated with 2-5 mg/kg/day for a mean of 15.5 months. Blood Phe levels were reduced by sapropterin dihydrochloride, and were maintained within normal range for the duration of treatment. Based on a rating of global improvement, there was moderate or marked improvement in all 16 subjects. Subjects from this study together with another 14 subjects were subsequently entered into a post-marketing surveillance study. Although patients were meant to have BH4 deficiency, 3 were subsequently found to have HPA due to PKU. All 30 patients were treated for at least one year, with 19 patients treated for 10-20 years. For the study population with BH4 deficiency, 25/27 (93%) achieved a global improvement rating of ‘markedly improved’, ‘improved’ or ‘slightly improved’. INDICATIONS KUVAN is indicated for the treatment of hyperphenylalaninemia (HPA) in sapropterin-responsive adult and paediatric patients with phenylketonuria (PKU) or tetrahydrobiopterin (BH4) deficiency (see DOSAGE AND ADMINISTRATION for definition of sapropterin responsiveness).

Version: A001-1010 Page 7 of 15 Supersedes: N/A
CONTRAINDICATIONS KUVAN is contraindicated in patients with hypersensitivity to sapropterin or to any of the excipients (See DESCRIPTION). PRECAUTIONS The safety and efficacy of KUVAN in paediatric patients less than 4 years of age have not been established in controlled clinical trials. Treatment with KUVAN should be directed by specialist physicians knowledgeable in the management of PKU and BH4 deficiency. KUVAN does not work in all patients with PKU or BH4 deficiency but only in those who have shown a definite response. Response to treatment cannot be predetermined by laboratory testing (e.g. genetic testing) but can only be determined by a therapeutic trial of KUVAN (see DOSAGE AND ADMINISTRATION). Patients treated with KUVAN must continue a restricted phenylalanine diet and undergo regular clinical assessment (such as monitoring of blood phenylalanine and tyrosine levels, nutrient intake, and psychomotor development). Sustained or recurrent dysfunction in the phenylalanine-tyrosine-dihydroxy-L-phenylalanine (DOPA) metabolic pathway can result in deficient body protein and neurotransmitter synthesis. Prolonged elevations in blood phenylalanine levels in patients with PKU and BH4 deficiency can result in severe neurologic damage, including severe mental retardation, microcephaly, delayed speech, seizures, and behavioural abnormalities. This may occur even if patients are taking KUVAN but not adequately controlling their blood phenylalanine levels within the recommended target range. Conversely, prolonged exposure to low blood phenylalanine and tyrosine levels during infancy has been associated with impaired neurodevelopmental outcome. Active management of dietary phenylalanine and overall protein intake while taking KUVAN is required to ensure adequate control of blood phenylalanine and tyrosine levels and nutritional balance. It is of primary importance to initiate KUVAN treatment as early as possible to avoid the appearance of non-reversible clinical manifestations of neurological disorders in paediatric patients and cognitive deficits and psychiatric disorders in adults due to sustained elevations of blood phenylalanine. Caution is advised when KUVAN is used in patients with predisposition to convulsions. In clinical studies of patients with BH4 deficiency treated with another preparation of sapropterin, convulsions and exacerbation of convulsions was observed. This has not been observed in clinical trials with KUVAN. KUVAN should be used with caution in patients who are receiving concomitant levodopa, as combined treatment may cause increased excitability and irritability.

Version: A001-1010 Page 8 of 15 Supersedes: N/A
Consultation with a physician is recommended during concomitant illness as blood phenylalanine levels may increase. There are limited data regarding the long-term use of KUVAN. Renal and Hepatic Impairment Safety and efficacy of KUVAN in patients with renal or hepatic insufficiency have not been established. Caution must be exercised when prescribing to patients with renal or hepatic insufficiency. An increased incidence of altered renal microscopic morphology (collecting tubule basophilia) was observed in rats following chronic oral administration of sapropterin dihydrochloride at doses higher than 80 mg/kg/day, i.e. at exposures (based on area under curve, AUC) about 3 times the exposure at the maximal recommended human dose. No kidney changes were seen in marmoset monkeys after chronic treatment at oral doses of up to 320 mg/kg/day, approximately 2.6-times the highest dose anticipated in humans, based on body surface area. Effects on Fertility Sapropterin dihydrochloride at oral doses up to 400 mg/kg/day (about 16 times the exposure in adults taking 20 mg/kg/day, based on AUC values) had no effect on the fertility of male or female rats. Use in Pregnancy (Pregnancy Category B1) For KUVAN, no clinical data on exposed pregnancies are available. Maternal blood phenylalanine levels must be strictly controlled before and during pregnancy. If maternal phenylalanine levels are not strictly controlled before and during pregnancy, this could be harmful to the mother and the fetus. Uncontrolled levels of phenylalanine, above 600 mol/L in pregnant women, have been associated with a very high incidence of neurological, cardiac, facial dysmorphism and growth anomalies in their infants. Physician-supervised restriction of dietary phenylalanine intake prior to and throughout pregnancy is the first choice of treatment in this patient group. In rats, following intravenous administration of radiolabelled sapropterin, radioactivity was found to be distributed in fetuses. No increase in total biopterin concentrations in fetuses was observed in rats after oral administration of 10 mg/kg sapropterin dihydrochloride. However, in pregnant guinea pigs there was a marked increase in sapropterin and/or its metabolites in the fetus after oral administration of 20 mg/kg sapropterin dihydrochloride. No clear evidence of teratogenic activity was found in rats or rabbits at doses of 400 and 600 mg/kg/day, corresponding to about 16 and 19 times, respectively, the exposure in adults at the maximum recommended human dose (based on AUC). Sapropterin dihydrochloride had no effect on parturition and postnatal development in rats at doses of 400 mg/kg/day.

Version: A001-1010 Page 9 of 15 Supersedes: N/A
The use of KUVAN during pregnancy should be considered only if strict dietary management does not adequately reduce blood phenylalanine levels. Caution must be exercised when prescribing to pregnant women. Use in Lactation It is not known whether sapropterin or its metabolites are excreted in human breast milk. KUVAN should not be used during breastfeeding. Excretion of total biopterin in milk occurred in rats when sapropterin dihydrochloride (10 mg/kg) was administered by the intravenous route. No increase in total biopterin concentrations in milk was observed in rats after oral administration of 10 mg/kg sapropterin dihydrochloride. There were no effects on the development of rat pups of dams given 400 mg/kg/day sapropterin dihydrochloride orally from gestation Day 17 to post-partum Day 20 (approximately 16 times the exposure in adults at the maximum recommended human dose, based on AUC). Paediatric Use Paediatric patients, 4 years of age and older, with HPA due to PKU and BH4 deficiency have been treated with KUVAN in clinical studies (see CLINICAL TRIALS). KUVAN has not been specifically studied in PKU children under 4 years of age. Published literature indicates that more than 2,700 children with PKU aged newborn to 4 years have been administered BH4, including at least 43 who received therapy for 2 months or longer. BH4 deficiency is an extremely rare condition but reports of published studies include at least 120 patients starting treatment when less than 4 years of age (see CLINICAL TRIALS). Data from toxicity studies in juvenile and adult rats are suggestive of an inverse relationship between age and the oral absorption rate of KUVAN. Microscopic changes occurred in kidneys in the early postnatal period at lower sapropterin doses than the ones causing similar effects in adult rats, most likely related to this absorption rate effect. In addition, sapropterin and/or its metabolites were distributed to the brain to a much greater extent in young rats compared to adult rats. Pharmacokinetic studies of KUVAN in children less than 4 years of age are not available. Prescribers should use caution when dosing children, particularly infants, as the absorption rate may be higher in this population. Frequent blood monitoring is recommended to maintain adequate blood phenylalanine levels as defined by the physician. Use in the Elderly The safety and efficacy of KUVAN in patients over 50 years of age, including adults who did not receive early dietary treatment, have not been established. Caution must be exercised when prescribing to elderly patients. Carcinogenicity In a 2-year rat oral carcinogenicity study there was a statistically significant increase in the incidence of benign adrenal phaeochromocytoma in male rats treated with 250 mg/kg/day

Version: A001-1010 Page 10 of 15 Supersedes: N/A
sapropterin dihydrochloride (about 10 times human exposure based on AUC). No evidence of a carcinogenic effect was evident in an abbreviated 78-week oral carcinogenicity study in mice at sapropterin dihydrochloride doses up to 250 mg/kg/day (18 times human exposure based on AUC). Genotoxicity Sapropterin had variable mutagenic effects in bacterial cells and elicited an increase in chromosome aberrations in Chinese hamster lung and ovary cells. The results of the in vitro genotoxicity test in human lymphocytes were equivocal. Sapropterin has been shown to produce hydrogen peroxide in at least one in vitro cell culture system, which may explain the positive results in these assays. Sapropterin was not genotoxic in in vivo mouse micronucleus tests. Interactions with other Medicines No specific drug-drug interaction studies have been performed. Although concomitant administration of inhibitors of dihydrofolate reductase (e.g. methotrexate, trimethoprim) has not been studied, such medicinal products may interfere with BH4 metabolism. Caution is recommended when using such agents during treatment with KUVAN. BH4 is a cofactor for nitric oxide synthetase. Caution is recommended during concomitant use of KUVAN with all agents that cause vasodilation by affecting nitric oxide (NO) metabolism or action, including classical NO donors (e.g. glyceryl trinitrate (GTN), isosorbide dinitrate (ISDN), sodium nitroprusside (SNP), molsidomine), phosphodiesterase type 5 (PDE-5) inhibitors and minoxidil. Caution should be exercised when prescribing KUVAN to patients receiving treatment with levodopa, as increased excitability and irritability has been reported during concomitant use. Effects on the Ability to Drive and Use Machines No studies on the effects on the ability to drive and use machines have been performed. ADVERSE EFFECTS Clinical Trials In clinical trials, KUVAN has been administered to 579 patients with PKU in doses ranging from 5 to 20 mg/kg/day for lengths of treatment ranging from 1 week to 18 months. Patients were aged 4 to 48 years old at study entry. The patient population was nearly evenly distributed in gender, and approximately 95% of patients were Caucasian. Approximately 35% of the 579 patients with PKU who received treatment with KUVAN in the clinical trials experienced adverse events. The overall incidence of adverse events in patients receiving KUVAN was similar to that reported with patients receiving placebo. The

Version: A001-1010 Page 11 of 15 Supersedes: N/A
most commonly reported adverse reactions for which a causal relationship is at least a reasonable possibility are headache and rhinorrhoea. Rebound, as defined by an increase in blood phenylalanine levels above pre-treatment levels, may occur upon cessation of treatment. Table 1 shows by preferred term the number and percentage of 74 patients with PKU who had treatment-emergent adverse events (regardless of relationship) that occurred in at least 4% of patients following exposure to KUVAN at doses of 10 to 20 mg/kg/day for 6 to 10 weeks in 2 double-blind, placebo-controlled clinical trials. Table 1: Treatment-emergent adverse events with an incidence 4% in patients following
exposure to KUVAN in controlled clinical studies
System Organ Class Preferred Term KUVAN n=74 n (%)
Placebo n=59 n (%)
Any adverse event 47 (64) 42 (71)
Nervous system disorders Headache 11 (15) 8 (14)
Infections and infestations Upper respiratory tract infection1
9 (12) 14 (24)
Respiratory disorders Rhinorrhoea 8 (11) 0 Pharyngolaryngeal pain 7 (10) 1 (2) Cough 5 (7) 3 (5) Nasal congestion 3 (4) 0
Gastrointestinal disorders Diarrhoea 6 (8) 3 (5) Vomiting 6 (8) 4 (7) Abdominal pain 4 (5) 5 (8)
General disorders and administration site conditions
Pyrexia1 5 (7) 4 (7)
Injury, poisoning and procedural complications
Contusion1 4 (5) 1 (2)
Skin and subcutaneous tissue disorders
Rash1 4 (5) 4 (7)
1 Causal association with KUVAN is considered unlikely In addition, hypophenylalaninaemia occurred in 2% patients treated with KUVAN (n=1) and in 12% patients treated with placebo (n=9). In open-label, uncontrolled clinical trials in which all patients received KUVAN in doses of 5 to 20 mg/kg/day, adverse reactions were similar in type and frequency to those reported in the double-blind, placebo-controlled clinical trials. Post-marketing Experience Limited post-marketing experience with KUVAN does not reveal any new safety findings.

Version: A001-1010 Page 12 of 15 Supersedes: N/A
A 10-year post-approval safety surveillance program of another formulation of the same active ingredient (sapropterin dihydrochloride granules) was conducted in Japan with 30 patients, 27 of these patients had BH4 deficiency and 3 had PKU. The most common adverse reactions identified during this program were convulsions and exacerbation of convulsions in 3 patients (see PRECAUTIONS) and increased gamma-glutamyltransferase (GGT) in 2 patients. DOSAGE AND ADMINISTRATION Treatment with KUVAN must be initiated and supervised by a physician experienced in the treatment of PKU and BH4 deficiency. KUVAN should be administered with a meal as a single daily dose, at the same time each day, preferably in the morning. Active management of dietary phenylalanine and overall protein intake while taking KUVAN is required to ensure adequate control of blood phenylalanine levels and nutritional balance. As HPA due to either PKU or BH4 deficiency is a chronic condition, once responsiveness is demonstrated, KUVAN is intended for long-term use. However, there are limited data regarding the long-term use of KUVAN. Cessation of treatment must be conducted with close physician observation and monitoring due to the possibility of rebound in blood phenylalanine levels above pre-treatment levels (see ADVERSE EFFECTS). Dosage KUVAN is provided as 100 mg tablets. For doses above 100 mg, the calculated daily dose based on body weight should be rounded to the nearest multiple of 100. For instance, a calculated dose of 401 to 450 mg should be rounded down to 400 mg corresponding to 4 tablets. A calculated dose of 451 mg to 499 mg should be rounded up to 500 mg corresponding to 5 tablets. For doses below 100 mg, one tablet should be dissolved in 100 mL of water and the volume of solution corresponding to the prescribed dose administered. An accurate measuring device with suitable graduations should be used to ensure administration of the appropriate volume of solution. Any unused portion should be discarded. PKU The starting dose of KUVAN in adult and paediatric patients with PKU is 10 mg/kg body weight once daily. The dose is adjusted to achieve and maintain adequate blood phenylalanine levels as defined by the physician. The recommended daily dose is between 5 and 20 mg/kg/day. BH4 deficiency The starting dose of KUVAN in adult and paediatric patients with BH4 deficiency is 2 to 5 mg/kg body weight once daily. The dose is adjusted to achieve and maintain adequate blood phenylalanine levels as defined by the physician. The recommended daily dose is

Version: A001-1010 Page 13 of 15 Supersedes: N/A
between 2 and 20 mg/kg/day. It may be necessary to divide the total daily dose into 2 or 3 administrations, distributed over the day, to optimise the therapeutic effect. Determination of Response Response to treatment is determined by a decrease in blood phenylalanine following treatment with KUVAN. Blood phenylalanine levels should be checked before initiating treatment and after 1 week of treatment with KUVAN at the recommended starting dose. If an unsatisfactory reduction in blood phenylalanine levels is observed, then the dose of KUVAN can be increased weekly to a maximum of 20 mg/kg/day, with continued weekly monitoring of blood phenylalanine levels over a one month period. The dietary phenylalanine intake should be maintained at a constant level during this period. A satisfactory response is defined as a ≥ 30 percent reduction in blood phenylalanine levels or attainment of the therapeutic blood phenylalanine goals defined for an individual patient by the treating physician. Patients who fail to achieve this level of response within the described one month test period should be considered non-responsive and should not receive further treatment with KUVAN. Once responsiveness to KUVAN has been established, the dose may be adjusted according to response to therapy within the therapeutic ranges specified under ‘Dosage’ above. Administration Compared to fasting, absorption of sapropterin is higher after a high-fat, high-calorie meal, resulting, on average, in 40-85% higher maximum blood concentrations achieved 4 to 5 hours after administration. To increase absorption, tablets should be administered as a single daily dose with a meal, at the same time each day preferably in the morning. The prescribed number of tablets should be placed in a glass or cup of water and stirred until dissolved. It may take a few minutes for the tablets to dissolve. To make the tablets dissolve faster they can be crushed. Small particles may be visible in the solution and will not affect the effectiveness of the medicinal product. The solution should be drunk within 15 to 20 minutes. Limited information is available on administering KUVAN in solutions other than water and no information is available on administering it in formula or milk. Because of the possibility that absorption may be affected, only water should be used to prepare and administer KUVAN. Adults The prescribed number of tablets should be placed in a glass or cup with 120 to 240 mL of water and stirred until dissolved. Paediatric Patients For doses above 100 mg, the prescribed number of tablets should be placed in a glass or cup with up to 120 mL of water and stirred until dissolved.

Version: A001-1010 Page 14 of 15 Supersedes: N/A
For doses below 100 mg, one tablet should be dissolved in 100 mL of water and the volume of solution corresponding to the prescribed dose administered. An accurate measuring device with suitable graduations should be used to ensure administration of the appropriate volume of solution. Any unused portion should be discarded. It is recommended that the prescriber, clinic nurse or pharmacist calculate and specify the volume of administration as well as the dose, in particular for young children, to reduce the risk of dosing errors. KUVAN tablets can be dissolved in smaller volumes should this be required for particular patients, e.g. young children. The minimum volume of solution required to dissolve each tablet is 20 mL, i.e. 1 tablet in 20 mL, 2 tablets in 40 mL, and so on. Patients should be advised not to swallow the desiccant capsule found in the bottle. Monitoring Treatment with KUVAN may decrease blood phenylalanine levels below the desired therapeutic level. Adjustment of the KUVAN dose or modification of dietary phenylalanine intake may be required to achieve and maintain blood phenylalanine levels within the desired therapeutic range. Blood phenylalanine and tyrosine levels should be tested, particularly in children, one to two weeks after each dose adjustment and monitored frequently thereafter, under the direction of the treating physician. If inadequate control of blood phenylalanine levels is observed during treatment with KUVAN, the patient’s adherence to the prescribed treatment, and diet, should be reviewed before considering an adjustment of the dose of KUVAN. Discontinuation of KUVAN treatment should be done only under the supervision of a physician. More frequent monitoring may be required, as blood phenylalanine levels may increase. Dietary modification may be necessary to maintain blood phenylalanine levels within the desired therapeutic range. OVERDOSAGE Headache and dizziness have been reported after the administration of KUVAN above the recommended maximum dose of 20 mg/kg/day. Treatment of overdose should be directed to symptoms. Contact the Poisons Information Centre on 131 126 for advice on management of overdosage. PRESENTATION AND STORAGE CONDITIONS KUVAN is supplied as soluble tablets. Each soluble tablet contains 100 mg of sapropterin dihydrochloride (equivalent to 77 mg of sapropterin), and is off-white to light yellow with “177” imprinted on one face.

Version: A001-1010 Page 15 of 15 Supersedes: N/A
Tablets are supplied in high-density polyethylene (HDPE) bottles with child-resistant closure. The bottles are sealed with an aluminium seal. Each bottle of KUVAN contains a small plastic tube of desiccant (silica gel). Each bottle is packaged in an individual carton and contains 30, 120 or 240# tablets. # Not all pack sizes are being distributed in Australia. Storage Store below 25C. Keep the bottle tightly closed in order to protect from moisture. Product should be used within two months after first opening the bottle. NAME AND ADDRESS OF THE SPONSOR Merck Serono Australia Pty Ltd 3-4/25 Frenchs Forest Road Frenchs Forest NSW 2086 POISON SCHEDULE OF THE MEDICINE S4 – Prescription Only Medicine TGA Approval Date: 21 October 2010 Registered Trademark of Biomarin Pharmaceutical Inc

Therapeutic Goods Administration PO Box 100 Woden ACT 2606 Australia
Email: [email protected] Phone: 1800 020 653 Fax: 02 6232 8605 www.tga.gov.au