axin2 as regulatory and therapeutic target in newborn...
TRANSCRIPT

nature neurOSCIenCe VOLUME 14 | NUMBER 8 | AUGUST 2011 1009
a r t I C l e S
Oligodendrocytes are the myelinating cells of the CNS that enable formation of myelin and saltatory nerve conduction. In humans, pre-myelinating oligodendrocytes are most abundant at the gestational age of 23–32 weeks and are thought to be selectively vulnerable during injury in the neonatal brain1. Hypoxic ischemic encephalopathy (HIE) causes neuronal apoptosis as well as diffuse primary damage to sub-cortical white matter in the term infant brain, whereas periventricular leukomalacia (PVL) comprises focal injury to white matter tracts as well as diffuse gliotic lesions, typically in premature infants2. Such diffuse and focal injuries, collectively known as white matter injury (WMI) in the newborn brain, can result in cerebral palsy and cogni-tive disability. Indeed, WMI is the most reliable prognostic indica-tor of development of severe cerebral palsy in premature infants3. In multiple sclerosis, the most common cause of neurological disability in young adults, myelin sheaths are lost through the injury or death of mature oligodendrocytes that results from autoimmune damage4.
In these conditions, myelin sheaths can be regenerated by OLPs that are recruited to lesions and differentiate in a process called remy-elination. Conversely, inhibition of remyelination may contribute to ongoing neurological dysfunction, axonal loss and disease progres-sion5,6. Although oligodendrocytes are thought to be cellular targets of excitotoxic damage in newborn brain injuries2,7, several lines of evidence indicate that failure of remyelination contributes to fixed demyelinated lesions8,9. Indeed, non-myelinating OLPs can be found in both PVL8,9 and multiple sclerosis5,6 lesions.
The molecular mechanisms that might dysregulate myelination in neonatal brain injury are unknown. Previously, we found that active Wnt signaling can act to inhibit OLP differentiation during remy-elination in the adult rodent CNS10, and several studies have shown that Wnt activation inhibits developmental myelination10–12. In the absence of Wnt ligands, β-catenin levels are regulated through a phosphorylation/degradation complex13,14 containing glycogen syn-thase kinase 3β (GSK-3β) and the scaffolding proteins Axis inhibition protein 1 (Axin1), adenomatous polyposis coli (APC) and disheveled (Dsh). The presence of Wnt ligand results in stabilization of β-catenin and its translocation to the nucleus, where it forms a nucleoprotein complex with Tcf/LEF transcription factors to activate or repress the expression of target genes15,16. Axin1 is expressed ubiquitously, whereas Axin2 (also known as conductin) is a transcriptional target of active Wnt signaling that also serves to autoregulate and repress the pathway by promoting β-catenin degradation in many different tissues and sys-tems14,17–19. Dual roles for signaling targets that also serve as feedback repressors are seen in other pathways, such as Sonic hedgehog (Shh), which activates the repressors Patched and Gli3 (ref. 20).
We observed expression of AXIN2 mRNA transcripts in situ in OLPs in WMI associated with human neonatal HIE and PVL, as well as in active multiple sclerosis lesions in adults. Although Axin2 function has been reported as being dispensable during brain development21, we found Axin2 function to be essential for normal myelination and remyelination in mice. Moreover, Axin2 levels can be manipulated
1Departments of Pediatrics and Neurosurgery, Eli and Edythe Broad Institute for Stem Cell Research and Regeneration Medicine and Howard Hughes Medical Institute, University of California, San Francisco, California, USA. 2Medical Scientist Training Program, University of California, San Francisco, California, USA. 3MRC Centre for Stem Cell Biology and Regenerative Medicine and Department of Veterinary Medicine, University of Cambridge, Cambridge, UK. 4Department of Neurology, University of California, San Francisco, California, USA. 5Department of Pathology, University of California, San Francisco, California, USA. 6Department of Developmental Biology and Howard Hughes Medical Institute, Stanford University, Stanford, California, USA. 7Division of Neonatology, University of California, San Francisco, California, USA. Correspondence should be addressed to D.H.R. ([email protected]).
Received 16 March; accepted 3 May; published online 26 June 2011; doi:10.1038/nn.2855
Axin2 as regulatory and therapeutic target in newborn brain injury and remyelinationStephen P J Fancy1, Emily P Harrington1,2, Tracy J Yuen1, John C Silbereis1, Chao Zhao3, Sergio E Baranzini4, Charlotte C Bruce3, Jose J Otero1,5, Eric J Huang5, Roel Nusse6, Robin J M Franklin3 & David H Rowitch1,7
Permanent damage to white matter tracts, comprising axons and myelinating oligodendrocytes, is an important component of brain injuries of the newborn that cause cerebral palsy and cognitive disabilities, as well as multiple sclerosis in adults. However, regulatory factors relevant in human developmental myelin disorders and in myelin regeneration are unclear. We found that AXIN2 was expressed in immature oligodendrocyte progenitor cells (OLPs) in white matter lesions of human newborns with neonatal hypoxic-ischemic and gliotic brain damage, as well as in active multiple sclerosis lesions in adults. Axin2 is a target of Wnt transcriptional activation that negatively feeds back on the pathway, promoting -catenin degradation. We found that Axin2 function was essential for normal kinetics of remyelination. The small molecule inhibitor XAV939, which targets the enzymatic activity of tankyrase, acted to stabilize Axin2 levels in OLPs from brain and spinal cord and accelerated their differentiation and myelination after hypoxic and demyelinating injury. Together, these findings indicate that Axin2 is an essential regulator of remyelination and that it might serve as a pharmacological checkpoint in this process.
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

1010 VOLUME 14 | NUMBER 8 | AUGUST 2011 nature neurOSCIenCe
a r t I C l e S
pharmacologically in OLPs to promote accelerated differentiation, suggesting that Axin2 serves as a therapeutic target in situations in which OLP differentiation is delayed or stalled.
RESULTSAXIN2 mRNA marks OLP in human neonatal WMIPreviously, we found Wnt pathway activation using the transgenic BAT-gal reporter (β-catenin–activated transgene driving lacZ) in developing OLPs in vivo and that forced activation of the canonical Wnt pathway during development of transgenic Olig2-cre; DA-cat mice (see Online Methods) inhibits differentiation of OLPs to mature oligodendrocytes10. Expression profiling of postnatal day 4 (P4) spinal cord from these mice revealed significant (P < 0.00006) downregula-tion of multiple genes associated with oligodendrocyte maturation (Supplementary Table 1 and Supplementary Fig. 1), including Mbp, Plp, Cnp, Mog, Mag, Mobp, Fa2h, Mal and the transcription factor Mrf 22. This analysis also identified known Wnt-activated targets, such as Nkd1, Notum and Axin2, as being significantly upregulated (P < 0.0002). In contrast, changes in expression of the Wnt pathway antagonist gene adenomatous polyposis coli (Apc, also known as CC1) were not significant.
Using expression of AXIN2 mRNA transcripts to mark Wnt-activated white matter cells in situ, we investigated two types of human neona-tal brain injury that are characterized by white matter damage and injury to oligodendrocyte lineage cells, resulting in hypomyelination (Supplementary Fig. 2). AXIN2 mRNA was solely expressed in Olig2-positive cells in affected white matter in neonatal HIE and PVL (note
that staining in PVL localized adjacent to the cystic core of lesion; Fig. 1a–c), but not white matter in age-matched controls (Fig. 1a,c). In the HIE samples, AXIN2 mRNA was expressed in a subset of the Tcf4-positive cells (Fig. 1d), consistent with activation of canonical Wnt signaling. AXIN2 mRNA segregated from the mature oligodendrocyte marker PLP in situ (Fig. 1e) and segregated completely from GFAP (Fig. 1f). Similarly, the independent Wnt-
activated target Naked1 (Nkd1) was expressed in PDGFRα-positive OLPs, but not in astroglia (Fig. 1g,h and Supplementary Fig. 3). The expression of these Wnt targets provides strong evidence that the Wnt pathway is activated specifically in immature, pre-myelinating OLPs in human neonatal WMI (Supplementary Fig. 4), and that they can be used to assess human neuropathological specimens (Supplementary Fig. 5).
Axin2 function is required for OLP differentiationAxin2 mRNA serves as a marker for pathway activation, whereas Axin2 protein functions as a negative feedback mechanism to control activa-tion of the Wnt pathway via β-catenin degradation17,18. Using wild-type and Axin2-lacZ heterozygous mice18, we characterized Axin2 mRNA and reporter gene expression during developmental myelina-tion (Fig. 2a–d). Axin2 mRNA was expressed in immature Nkx2.2-positive OLPs, but not CC1-positive differentiated oligodendrocytes (Fig. 2b). In contrast, β-galactosidase was first detectable at the later CC1-positive stage (Fig. 2c,d and Supplementary Fig. 6).
Axin2-lacZ homozygous null mice showed a significant (P = 0.007) delay in OLP differentiation during developmental myelination (Fig. 2e,f and Supplementary Fig. 7). Moreover, as indicated by coex-pression of β-galactosidase with PDGFRα (Fig. 2g), the reduction in mature oligodendrocyte numbers reflects a delay in OLP maturation rather than decreased OLP numbers. Indeed, there was no difference in apoptosis or proliferation of the OLP pool (Supplementary Fig. 8). We also observed a significant (P < 0.005) impairment of Axin2−/− oligodendrocyte differentiation in vitro (Fig. 2h,i), and strong acti-vation of the independent Wnt target Notum (Fig. 2j). Thus, Axin1
Human newbornbrain injury
PVL
100
0Axi
n2 m
RN
A–e
xpre
ssin
gce
lls p
er m
m2
PVL
HIE
HIE
HIE
HIEHIE HIEHIE
Control
HIEHIE
HIE HIE HIE HIE
HIE
Control
AXIN2 AXIN2
AXIN2
AX
IN2
AXIN2 Olig2
AXIN2 Tcf4
AXIN2 GFAP DAPI Nkd1 GFAP DAPI Nkd1 PDGFRα DAPI Nkd1 PDGFRα DAPI
Olig2 DAPI
AXIN2 PLP AXIN2 PLP
Olig2
AXIN2a
b
c d e
f g h
Figure 1 AXIN2 mRNA expression identifies Wnt pathway activation in immature oligodendrocytes in neonatal human WMI. (a) AXIN2 mRNA was expressed in areas of affected subcortical white matter in human pediatric cases of HIE and also adjacent to the cystic core of a periventricular lesion of PVL but was not seen in age-matched controls. (b) AXIN2 mRNA was expressed solely in Olig2-positive cells in affected white matter in neonatal HIE. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization. (c) Quantification of the number of AXIN2 mRNA–expressing cells in areas of white matter from subjects with HIE and control subjects. Error bars represent s.d. (d,e) In the subjects with HIE, AXIN2 mRNA was expressed in a subset of the Tcf4-positive cells (d), which segregated from the mature oligodendrocyte marker PLP in situ (e). (f) AXIN2 mRNA expression in HIE was completely separate from GFAP expression. (g,h) The independent Wnt-activated target Naked1 (Nkd1) was upregulated in neonatal HIE; proteins were expressed cytoplasmically in OLPs expressing PDGFRα (h) but was not expressed in cells expressing GFAP proteins (g). Scale bars represent 10 µm.
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

nature neurOSCIenCe VOLUME 14 | NUMBER 8 | AUGUST 2011 1011
a r t I C l e S
is unable to compensate for the loss of early Axin2 functions in oligodendrocyte differentiation in vitro or in vivo.
Axin2 expressed in multiple sclerosis and functions in myelin repairPrevious studies have found that lesions of both neonatal PVL and adult multiple sclerosis have OLPs that are evidently ‘stalled’ in their differentiation5,6,8,9, which has been proposed to result in defec-tive repair and fixed demyelinated lesions23. Thus, we focused on regulatory functions of Axin2 in the context of primary damage to oligodendrocytes. We confirmed that AXIN2 mRNA transcripts are expressed in OLP in active multiple sclerosis lesions (Fig. 3a,b), in which oligodendrocytes are targeted for autoimmune attack. Axin2 mRNA–expressing cells were not seen in areas of normal-appearing white matter or in chronic silent plaques (Fig. 3a). A similar pattern of
expression was seen for the independent Wnt-activated target Naked1 (Nkd1). Naked1-expressing cells were seen at active multiple sclerosis lesion edges (Fig. 3c and Supplementary Fig. 5), with the simple bipolar morphology that is characteristic of OLP (Fig. 3c), and at a similar cellular density as Axin2 mRNA–expressing cells (Fig. 3c). Remyelination can be investigated after adult murine lysolecithin injury (Fig. 4), which kills resident oligodendrocytes while leav-ing axons largely intact. Such lesions in spinal cord ventral or dor-sal white matter have been extensively characterized24 and revealed OLP recruitment (5 days post lesion, dpl), differentiation (10 dpl) and myelination (14 dpl) with stereotyped timing in young adult animals (Fig. 4a), allowing precise assessment of remyelination kinetics.
In adult animals, we observed robust expression of the β-galac-tosidase reporter in Axin2-lacZ heterozygote mice 10 d after
Figure 2 Axin2 functions as a negative regulator of Wnt signaling in OLPs and promotes differentiation. (a) Schematic for Axin2 mRNA expression in wild-type (WT) mice and β-catenin (β-cat)/Tcf4-driven reporter expression in heterozygous Axin2-lacZ mice. (b) During developmental myelination of corpus callosum (CC), Axin2 mRNA was confined to earlier stage immature OLPs that expressed Nkx2.2 protein and was not seen in mature oligodendrocytes expressing APC (CC1) protein. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization. (c,d) In Axin2-lacZ heterozygous mice during developmental myelination, β-galactosidase (β-gal) proteins were first detected at a stage of the oligodendrocyte lineage in P9 corpus callosum that expressed the mature marker APC (CC1) but were not detected in cells expressing the OLP markers PDGFRα (c) and Nkx2.2 (d), suggesting that the kinetics of reporter expression lagged behind that of Axin2 mRNA during developmental myelination. Tcf4 expression was observed in only a subset of β-galactosidase–positive cells (d). Scale bars represent 10 µm (b–d). (e) Axin2-lacZ homozygous null animals demonstrated a significant reduction in mature oligodendrocytes expressing PLP mRNA (and MBP protein, inset) at P9 during developmental myelination of the corpus callosum, despite having normal numbers of OLPs expressing Nkx2.2 protein. Scale bars represent 600 µm, 100 µm (MBP inset) and 15 µm (Nkx2.2 inset). (f) Quantification during developmental myelination of the reduced number of (t test, P = 0.007) mature PLP-expressing oligodendrocytes in P9 Axin2−/− corpus callosum (black bars) compared with heterozygous littermates (gray bars), despite normal numbers of Nkx2.2-expressing OLPs. (g) Although β-galactosidase was not coex-pressed with OLP markers in Axin2-lacZ heterozygotes (c,d), it was coexpressed with PDGFRα in Axin2−/− mice as OLPs were delayed in their differentiation. (h,i) There was a marked impairment of Axin2−/− oligodendrocyte differentiation in vitro, as evidenced by a significant (t test, P < 0.005) reduction in the percentage of Olig2-positive cells expressing MBP at 60 h post differentiation in culture. Scale bar represents 25 µm. (j) There was a strong activation of Notum mRNA, an independent Wnt target, in Axin2−/− OLP cultures as compared with wild type, indicating that loss of Axin2 leads to an increase in Wnt pathway activity in these cultures.
a b
c d
e
g
h i j
β-cat
β-gal
β-gal Nkx2.2
β-gal β-gal
CC1PDGFRα
β-gal PDGFRα β-gal Tcf4
β-cat
Axin2 mRNA P9
P9P9P9 P9
CC
P9 P9
Per
cent
age
Olig
2+ c
ells
expr
essi
ng M
BP
Fol
d ch
ange
(Axi
n2 n
ull /
WT
)
P9P9
Nkx2.2
Nkx2.2
MBP
MBP
MBP Olig2 MBP Olig2
CC
CC
*
WT
WT
50
0
OLP
cul
ture
sCCCCCC
PLP PLP
CC
CC
P9
Axin2 mRNA Nkx2.2 Axin2 mRNA APC (CC1)
β-gal APC (CC1)
Tcf4
Tcf4
Axin2
Axin2-lacZ
Axi
n2+
/–
Axi
n2–/
–
Axin2–/–
Axin2–/–
Axi
n2–/
–
f
300
*
Cel
ls p
er s
ectio
nP
9 co
rpus
Cel
ls p
er s
ectio
nP
9 co
rpus
0
PLP
PLP
Axin2+/– Axin2–/–
600
0
Nkx
2.2
Nkx
2.2
Axin2+/– Axin2–/–
Notum
4.0
2.0
0
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

1012 VOLUME 14 | NUMBER 8 | AUGUST 2011 nature neurOSCIenCe
a r t I C l e S
demyelination (Fig. 4b). Although mature oligodendrocyte numbers normalized by 8 weeks of age in Axin2−/− animals (Fig. 4f and Supplementary Fig. 9), Axin2−/− mice showed significantly delayed remyelina-tion (P < 0.05) compared with wild-type littermates (Fig. 4c and Supplementary Fig. 10) as a result of a delay in Nkx2.2-positive OLP differentiation (with normal OLP recruitment) in lesions at 10 and
14 dpl (Fig. 4c–f). The inflammatory cell and astrocyte response in lesions was not affected in Axin2−/− mice (Supplementary Fig. 11), suggesting that the delay in remyelination in Axin2−/− animals is attributable to a cell-autonomous requirement in OLPs.
Axin2 protein stabilization promotes OLP differentiationWe next investigated whether enhanced Axin2 activity might pro-mote accelerated oligodendrocyte maturation. Axin2 and Axin1 were recently identified as substrates for the poly-ADP-ribosylating enzymes tankyrase 1 and 2 (ref. 25), which promote Axin degrada-tion through the ubiquitin-proteosome pathway. XAV939 inhibited tankyrase activity in cell lines at a concentration of 5 µM, resulting in stabilized Axin protein levels25.
LFB LN3
Human adult brain: MS Active MS lesions
LFB LN3
Naked1
Nkd
1- a
nd A
xin2
- ex
pres
sing
cells
per
mm
2
Olig2
Naked1 Naked1 DAPI
AXIN2
AXIN2
AXIN2 Olig2
NAWM
a
c
b
Chronic plaque
Active plaque
b
c
Nak
ed1
125Active MS lesions
0
AX
IN2
Figure 3 AXIN2 is expressed in OLPs in active multiple sclerosis lesions. (a) Multiple sclerosis (MS) lesions were characterized as described previously33, using Luxol Fast Blue (LFB) to assess demyelination and LN3 immunohistochemistry to assess inflammatory cell activity. AXIN2 mRNA was expressed in cells in active multiple sclerosis lesions but not in normal-appearing white matter (NAWM) or chronic plaques. Scale bar represents 100 µm. (b) AXIN2 mRNA expression in active multiple sclerosis lesions was specific to the oligodendrocyte lineage, where it colocalized with Olig2 proteins. Scale bar represents 10 µm. (c) The independent Wnt-activated target Nkd1 was also upregulated in active multiple sclerosis lesions (Supplementary Fig. 5). Nkd1 proteins were expressed in the cytoplasm of OLPs with characteristic simple bipolar morphology. The density of cells expressing Nkd1 proteins in active multiple sclerosis lesions was similar to the density of cells expressing AXIN2 mRNA. Scale bars represent 10 µm. Error bars represent s.d.
Kinetics of remyelination in mouse
Day 0
Myelin injury
Axi
n2-la
cZ/+
Axi
n2-la
cZ/la
cZA
xin2
+/+
Axi
n2–/
–
β-gal β-gal APC (CC1) β-gal Nkx2.2
β-gal Nkx2.2 WT versus Axin2–/–
PLP mRNA–expressingcells
WT versus Axin2–/–
Nkx2.2 protein–expressingcells
OLP recruitment OL differentiation Remyelination
5 dpl 10 dpl
10 dpl
Lesion SC
DAPI10 dpl
10 dpl
10 dpl
SC
SC
SC
1,200 1,800
1,200
600
0
800
400
0
Unlesio
ned
Unlesio
ned
14 d
pl
10 d
pl5
dpl
14 d
pl
10 d
pl5
dpl
PLP Nkx2.2
14 dpl
***
a
b
c
d e f
Figure 4 Axin2 function is essential for timely myelin repair. (a) Schematic showing use of adult murine lysolecithin injury for investigating remyelination kinetics. Such lesions in spinal cord (SC) showed OLP recruitment (5 dpl), differentiation (10 dpl) and myelination (14 dpl) with stereotyped timing in young adult animals, allowing precise assessment of remyelination kinetics. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization. (b) Following demyelination of adult Axin2-lacZ heterozygote animals with lysolecithin in the spinal cord, β-galactosidase proteins were observed in the lesion in mature oligodendrocytes (coexpressing APC) at 10 dpl but not in cells expressing the OLP marker Nkx2.2. Scale bars represent 80 µm (left) and 10 µm (right). (c) Axin2−/− mice showed delayed repair compared with wild-type littermates. This was a result of reduced (t test, P = 0.03 at 10 dpl, P = 0.02 at 14 dpl) OLP differentiation into mature oligodendrocytes expressing PLP mRNA in lesions at 10 dpl, despite a normal recruitment of Nkx2.2-expressing OLPs into lesions. Scale bar represents 80 µm. (d) OLPs with dysregulated Wnt signaling in Axin2−/− (Axin2-lacZ homozygote) mice at 10 dpl following demyelination showed abnormal kinetics of mature marker acquisition and β-galactosidase proteins colocalized with the OLP marker Nkx2.2, in contrast with the Axin2-lacZ heterozygote mice (b). Scale bar represents 10 µm. (e,f) Quantification of mature oligodendrocytes (expressing PLP mRNA, e) and OLPs (expressing Nkx2.2 protein, f) in unlesioned and 5 dpl, 10 dpl and 14 dpl demyelinated spinal cord of Axin2−/− animals (black) and wild-type littermates (gray). *P = 0.03, **P = 0.02. Error bars represent s.d.
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

nature neurOSCIenCe VOLUME 14 | NUMBER 8 | AUGUST 2011 1013
a r t I C l e S
Tankyrase was expressed in the rodent oligodendrocyte lineage during development, commencing at a mature stage (Fig. 5a,b), and continued to be expressed in oligodendrocytes in adult white matter (S.P.J.F. and D.H.R., unpublished observations). Tankyrase was also expressed in early differentiating oligodendrocytes following demyeli-nation in the adult spinal cord white matter of the mouse (Fig. 5c).
Given these findings, we tested the effects of XAV939 on stabilizing Axin protein levels in OLPs in vitro. Treatment with XAV939 (0.01 or 0.1 µM) for 24 h produced increases in the levels of both Axin2 and Axin1 proteins in OLPs versus vehicle controls, and increased protein levels of tankyrase 1 and 2, presumably as a result of a reduc-tion in their autoparsylation and self-induced degradation (Fig. 5d)25. XAV939 treatment of OLPs resulted in increased levels of phospho-β-catenin and β-catenin degradation, as well as reductions in the levels of Axin2 mRNA transcripts (Fig. 5e) and the activity of a transduced Topflash reporter (data not shown), indicating that the Wnt pathway was inhibited. Moreover, in addition to these early effects on Wnt pathway per se, XAV939 treatment promoted precocious oligodendro-cyte differentiation (Fig. 5f–h), as evidenced by increased expression of the mature oligodendrocyte marker myelin basic protein (MBP).
We next confirmed tankyrase protein expression in the human oligo-dendrocyte lineage during neonatal WMI. Tankyrase was expressed in later stage oligodendrocyte cells that expressed NOGO-A and cytoplas-mic Olig1 (Fig. 5i). Conversely, tankyrase expression was not observed in inflammatory macrophages, microglia or reactive astrocytes in gliotic areas of white matter damage. These data suggest that pharmacological manipulation of Axin2 levels in OLPs through tankyrase inhibition has therapeutic potential in human neonatal WMIs.
To address this question, we investigated the effects of XAV939 on OLPs in mouse cerebellar slice cultures (Fig. 6). In this system, slices of P0–1 neonatal cerebellum are cultured in the presence of factors that can affect remyelination of cerebellar white matter26. We exam-ined the ratio of neurofilament 200 kDa (NFH)-positive axons that coexpressed myelin basic protein (MBP) and measured the density of the nodes of Ranvier, indicated by Caspr-positive paranodes. XAV939 treatment (0.01 µM) significantly increased myelination compared with controls (P < 0.001; Fig. 6a). Moreover, we observed that acute hypoxic exposure (2% oxygen for 24 h) reduced myelination to levels significantly below controls (P < 0.01; Fig. 6a,b). We suggest that the effect of hypoxia in this system is primarily a result of inhibition of
Figure 5 Axin protein stabilization through small molecule tankyrase inhibition promotes OLP differentiation in vitro. (a) Tankyrase proteins were detected in the cytoplasm of mouse Olig2-positive cells but not at the PDGFRα-positive (OLP) stage, at P9 during developmental myelination in the spinal cord. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization. (b) The onset of tankyrase expression coincided with expression of β-galactosidase in Axin2-lacZ heterozygous reporter mice, at approximately the CC1-positive stage of oligodendrocyte development. (c) Tankyrase was also expressed in the oligodendrocyte lineage at 10 dpl following demyelination with lysolecithin in the adult spinal cord white matter of the mouse, colocalizing with mature oligodendrocyte marker cytoplasmic Olig1 (inset). Scale bars represent 10 µm (a–c). (d) XAV939 treatment of mouse OLPs in vitro with 0.01 or 0.1 µM for 24 h produced marked increases in the protein levels of both Axin2 and Axin1 versus vehicle controls, leading to an increase in the activity of the β-catenin degradation complex, as evidenced by an increase in degraded phospho–β-catenin protein levels. (e) At 0.01 µM, XAV939 treatment effectively inhibited the Wnt pathway in OLP in vitro after 96 h, as evidenced by a reduction in Axin2 mRNA levels. (f,h) In vitro OLP differentiation assays revealed a significant increase in the proportion of Olig2-positive cells expressing the mature oligodendrocyte marker MBP in the presence of either 0.1 or 0.01 µM XAV939 at both 48 and 60 h post-differentiation compared with vehicle control treatment. Scale bar represents 40 µm. **P < 0.001. Error bars represent s.d. (g) At 60 h post differentiation of OLP in vitro in the presence of 0.01 µM XAV939, there was a significant increase in the quantity of MBP protein harvested from the culture compared with vehicle control. (i) Tankyrase protein was expressed in oligodendrocyte lineage in human pediatric HIE WMI, where it was expressed in Olig2-positive, NOGO-A–positive and cytoplasmic Olig1-positive cells but not in Iba1-positive macrophages/microglia or GFAP-positive astrocytes. Scale bar represents 10 µm.
a
d
e
i
g h
f
b cTankyrase
Tankyrase
Axin1
Phos-β-cat
β-actin
β-actin
Axin2
Axin2 MBP
Per
cent
age
Olig
2+
cells
exp
ress
ing
MB
P
Cyclophilin
96 h mRNA 60 h protein
Tankyrase Tankyrase Tankyrase Olig2
Tank
DAPIOlig2
P9
SC
P9
SC
P1
SC
10 dpl
Lesion SC
Olig1
MBP Olig2
PDGFRα
24 h protein
60 h
diff
eren
tiatio
n48
h d
iffer
entia
tion
DMSO
DMSO
Tankyrase Olig1 DAPI Tankyrase lba1 DAPI Tankyrase GFAP DAPITankyrase NOGO-A DAPI
DMSO
DMSO
DMSO
XAV
XAV
XAV XAV
XAV0.01 µM
0.01 µMXAV
XAV 0.01 µM
XAV 0.1 µM60
40
20
048 h MBP 60 h MBP
****
**
**0.01 µM
0.1 µM
0.01 µM 0.1 µM
β-gal
Tankyrase Olig2
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

1014 VOLUME 14 | NUMBER 8 | AUGUST 2011 nature neurOSCIenCe
a r t I C l e S
OLP maturation, as Nkx2.2 and Olig2 double-positive OLP num-bers were significantly increased without detectable reduction in the number of Olig2-positive cells (P < 0.001; Fig. 6b). Treatment with XAV939 reversed the effect of hypoxia and increased the extent of myelinated axons to well above control levels. To determine whether XAV939 treatment can act on cerebellar OLPs to promote remyelina-tion, we also tested its effects after addition of 0.5% lysolecithin to culture medium to induce toxic injury to oligodendrocytes (Fig. 6c). In this procedure, XAV939 also promoted a significant enhancement of myelin regeneration (P < 0.001; Fig. 6c).
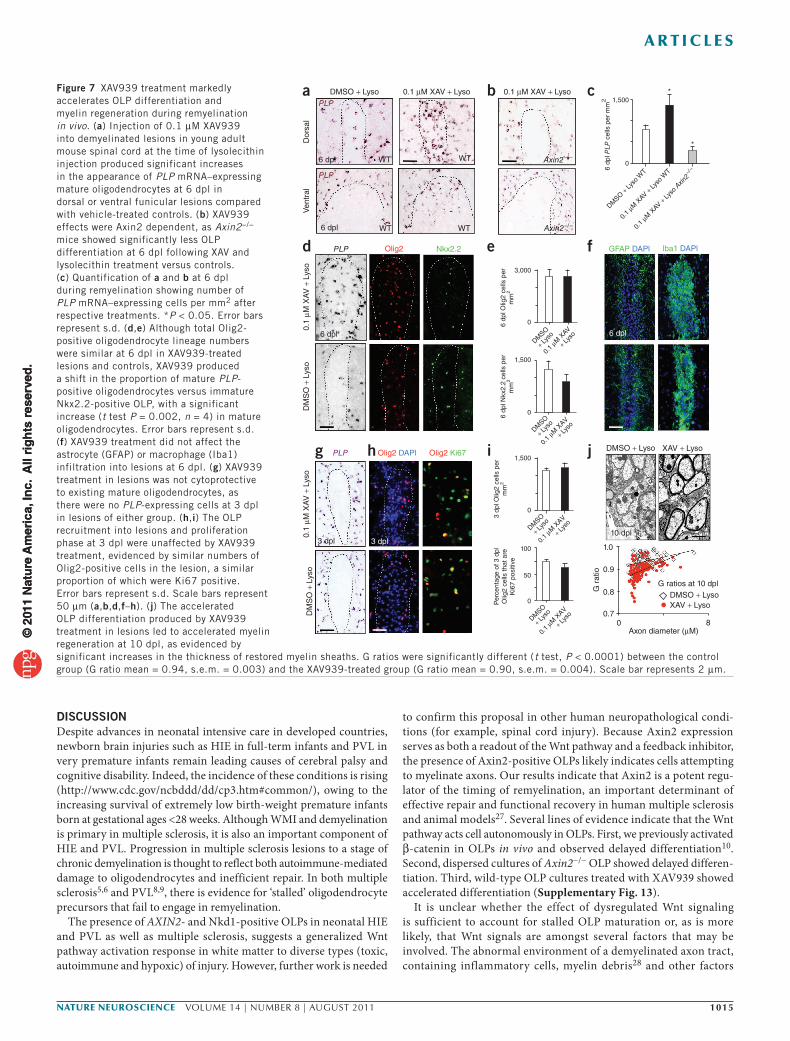
As illustrated above (Fig. 4a), the kinetics of remyelination in vivo in focal toxic injury models is tightly regulated and, to the best of our knowledge, no drugs have been reported that acceler-ate OLP differentiation. To test whether pharmacologic stabilization of Axin might promote OLP differentiation during remyelination in vivo, we co-injected young adult (8–10 weeks old) mouse spinal cord lysolecithin lesions with XAV939 (0.1 µM; Fig. 7). We observed a notable increase in PLP-positive differentiated oligodendrocytes as early as 6 dpl in XAV939-treated dorsal and ventral lesions compared with vehicle-treated controls (Fig. 7a,c). Moreover, such XAV939
effects were Axin2 dependent, as Axin2−/− mice showed markedly less OLP differentiation at 6 dpl following lysolecithin treatment versus controls (Fig. 7b,c). This increase in mature oligodendrocyte numbers at 6 dpl was a result of a precocious differentiation of recruited OLP rather than an increased survival of existing oligodendrocytes (Fig. 7g). Although total Olig2-positive cells were similar at 6 dpl in XAV939-treated lesions and controls, XAV939 produced a shift in the proportion of Olig2-positive cells that were mature PLP-positive oligodendrocytes versus immature Nkx2.2-positive OLPs (Fig. 7d,e). XAV939 treatment did not affect the inflammatory cell or astrocyte infiltration into lesions (Fig. 7f), nor early OLP recruitment or prolif-eration in the lesion (Fig. 7h,i). Such precocious OLP differentiation in XAV939-treated lesions was associated with significantly thicker (P < 0.0001) myelin sheaths (smaller g ratios) than in controls at 10 dpl, when myelin sheath formation is on-going (Fig. 7j). Myelin thickness in XAV939-treated remyelinated lesions was similar to con-trols when lesions in both groups were fully remyelinated at 28 dpl (Supplementary Fig. 12). These findings indicate that XAV939 treat-ment substantially accelerates the processes of OLP differentiation and compact myelin formation in vivo.
Myelination Hypoxia
Factors added
0 DIV
DMSO
a b c
MBP NFH
MBPNFHCaspr
Caspr
0.01 µM XAV 0.01 µM XAV + hypoxia 0.01 µM XAVDMSO + hypoxia
12 DIV 12 DIV
DMSO
3 DIV0 DIV 28 DIV14 DIV0 DIV2 DIV
Factors added Factors added
Myelinationculture endpoint
Remyelinationculture endpoint
Nkx
2.2
and
Olig
2 do
uble
-po
sitiv
e ce
lls:O
lig2-
posi
tive
cells
Num
ber
of O
lig2-
posi
tive
cells
Cas
pr:N
FH
(%
)
Cas
pr:N
FH
(%
)
0.10
0.15
0.10
0.05
0
600
400
200
0
0.08
0.10
0.06
0.04
0.02
0
***
****** ***
** **
0.08
0.06
0.04
0.02
0
Contro
lControl HypoxiaControl Hypoxia
DMSO
Contro
l
DMSO
0.01
µM X
AV
0.01
µM X
AV
Contro
l hyp
oxia
DMSO +
hyp
oxia
0.0
1 µM
XAV +
hypo
xia
Remyelination
Demyelinationwith LPC (18 h)
Hypoxia culture endpoint
Sliceculture
Sliceculture
Sliceculture
Acutehypoxia(24 h)
Figure 6 XAV939 treatment increases myelination, myelination following hypoxia and remyelination in ex vivo mouse cerebellar slice cultures. (a) XAV939 promoted developmental myelination. Axons (NFH) are shown in red, myelin (MBP) is shown in green and paranodes with compacted myelin sheaths (Caspr) are shown in white. Quantification of myelination using the ratio of the area stained for Caspr to the area stained for NFH (%) is shown below. Values shown are mean ± s.d. and the data were analyzed by one-way ANOVA with Dunnett’s multiple comparison test (**P < 0.01 and ***P < 0.001). (b) XAV939 promoted myelination and recovery following acute hypoxic insult. Data are presented as in a. Acute hypoxia impeded differentiation of OLPs in cerebellar slice cultures. Data were analyzed by unpaired t test. (c) XAV939 promoted remyelination following demyelination by lysolecithin. Data are presented as in a. Data were analyzed by one-way ANOVA with Dunnett’s multiple comparison test. Scale bars represent 50 µm (MBP/NFH panels) and 25 µm (Caspr panels). Three independent experiments were conducted per condition tested and 5–10 separate slices were counted per experiment. LPC; lysophosphatidylcholine/lysolecithin.
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
nature neurOSCIenCe VOLUME 14 | NUMBER 8 | AUGUST 2011 1015
a r t I C l e S
DISCUSSIONDespite advances in neonatal intensive care in developed countries, newborn brain injuries such as HIE in full-term infants and PVL in very premature infants remain leading causes of cerebral palsy and cognitive disability. Indeed, the incidence of these conditions is rising (http://www.cdc.gov/ncbddd/dd/cp3.htm#common/), owing to the increasing survival of extremely low birth-weight premature infants born at gestational ages <28 weeks. Although WMI and demyelination is primary in multiple sclerosis, it is also an important component of HIE and PVL. Progression in multiple sclerosis lesions to a stage of chronic demyelination is thought to reflect both autoimmune-mediated damage to oligodendrocytes and inefficient repair. In both multiple sclerosis5,6 and PVL8,9, there is evidence for ‘stalled’ oligodendrocyte precursors that fail to engage in remyelination.
The presence of AXIN2- and Nkd1-positive OLPs in neonatal HIE and PVL as well as multiple sclerosis, suggests a generalized Wnt pathway activation response in white matter to diverse types (toxic, autoimmune and hypoxic) of injury. However, further work is needed
to confirm this proposal in other human neuropathological condi-tions (for example, spinal cord injury). Because Axin2 expression serves as both a readout of the Wnt pathway and a feedback inhibitor, the presence of Axin2-positive OLPs likely indicates cells attempting to myelinate axons. Our results indicate that Axin2 is a potent regu-lator of the timing of remyelination, an important determinant of effective repair and functional recovery in human multiple sclerosis and animal models27. Several lines of evidence indicate that the Wnt pathway acts cell autonomously in OLPs. First, we previously activated β-catenin in OLPs in vivo and observed delayed differentiation10. Second, dispersed cultures of Axin2−/− OLP showed delayed differen-tiation. Third, wild-type OLP cultures treated with XAV939 showed accelerated differentiation (Supplementary Fig. 13).
It is unclear whether the effect of dysregulated Wnt signaling is sufficient to account for stalled OLP maturation or, as is more likely, that Wnt signals are amongst several factors that may be involved. The abnormal environment of a demyelinated axon tract, containing inflammatory cells, myelin debris28 and other factors
Figure 7 XAV939 treatment markedly accelerates OLP differentiation and myelin regeneration during remyelination in vivo. (a) Injection of 0.1 µM XAV939 into demyelinated lesions in young adult mouse spinal cord at the time of lysolecithin injection produced significant increases in the appearance of PLP mRNA–expressing mature oligodendrocytes at 6 dpl in dorsal or ventral funicular lesions compared with vehicle-treated controls. (b) XAV939 effects were Axin2 dependent, as Axin2−/− mice showed significantly less OLP differentiation at 6 dpl following XAV and lysolecithin treatment versus controls. (c) Quantification of a and b at 6 dpl during remyelination showing number of PLP mRNA–expressing cells per mm2 after respective treatments. *P < 0.05. Error bars represent s.d. (d,e) Although total Olig2-positive oligodendrocyte lineage numbers were similar at 6 dpl in XAV939-treated lesions and controls, XAV939 produced a shift in the proportion of mature PLP-positive oligodendrocytes versus immature Nkx2.2-positive OLP, with a significant increase (t test P = 0.002, n = 4) in mature oligodendrocytes. Error bars represent s.d. (f) XAV939 treatment did not affect the astrocyte (GFAP) or macrophage (Iba1) infiltration into lesions at 6 dpl. (g) XAV939 treatment in lesions was not cytoprotective to existing mature oligodendrocytes, as there were no PLP-expressing cells at 3 dpl in lesions of either group. (h,i) The OLP recruitment into lesions and proliferation phase at 3 dpl were unaffected by XAV939 treatment, evidenced by similar numbers of Olig2-positive cells in the lesion, a similar proportion of which were Ki67 positive. Error bars represent s.d. Scale bars represent 50 µm (a,b,d,f–h). (j) The accelerated OLP differentiation produced by XAV939 treatment in lesions led to accelerated myelin regeneration at 10 dpl, as evidenced by significant increases in the thickness of restored myelin sheaths. G ratios were significantly different (t test, P < 0.0001) between the control group (G ratio mean = 0.94, s.e.m. = 0.003) and the XAV939-treated group (G ratio mean = 0.90, s.e.m. = 0.004). Scale bar represents 2 µm.
a
g h i
e f
b cDMSO + Lyso
6 dpl
6 dpl
6 dpl
Olig2
Olig2 DAPl
3 dpl 3 dpl
DMSO
+ Lyso
DMSO
+ Lyso
DMSO
+ Lyso
0.1
µM X
AV
+ Lyso
0.1
µM X
AV
+ Lyso
Axon diameter (µM)
G ratios at 10 dpl
0.9
1.0
0.8
0.70 8
DMSO + LysoXAV + Lyso
0.1
µM X
AV
+ Lyso
Olig2 Ki67
Nkx2.2
6 dp
l Olig
2 ce
lls p
erm
m2
6 dp
l PLP
cel
ls p
er m
m2
6 dp
l Nkx
2.2
cells
per
mm
2
3 dp
l Olig
2 ce
lls p
erm
m2
Per
cent
age
of 3
dpl
Olig
2 ce
lls th
at a
reK
i67
posi
tive
G r
atio
3,000
1,500
0
GFAP DAPl lba1 DAPl
0
1,500
0
1,500
0
100
50
0
DMSO
+ Lyso
DMSO + Ly
so W
T
0.1
µM X
AV
+ Lyso
0.1
µM X
AV + Lyso
WT
0.1
µM X
AV + Lyso
Axin
2−/
−
PLP
PLP
WT
WT WT
10 dpl
XAV + LysoDMSO + Lyso
WT
PLP
PLP
Axin2−/−
Axin2−/−
Dor
sal
Ven
tral
0.1 µM XAV + Lyso
0.1
µM X
AV
+ L
yso
0.1
µM X
AV
+ L
yso
DM
SO
+ L
yso
DM
SO
+ L
yso
0.1 µM XAV + Lyso
*
*
j
d
6 dpl
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

1016 VOLUME 14 | NUMBER 8 | AUGUST 2011 nature neurOSCIenCe
a r t I C l e S
(for example, Lingo-1 (ref. 29), Notch30 signaling), appear to also inhibit remyelination. Delayed remyelination kinetics might lead to chronic demyelination23, in keeping with recent studies showing that oligodendrocytes initiate new myelin segments only during a temporally restricted developmental window31.
Despite probably complex roles of multiple factors in remyelina-tion, treatment with XAV939 alone was sufficient to stabilize Axin proteins and promote markedly accelerated oligodendrocyte differ-entiation after demyelinating injury. Our results from Axin2−/− mice indicate that XAV939 acts specifically through its effects on Axin2, and our in vitro findings suggest that this acts to inhibit the canoni-cal Wnt pathway through β-catenin phosphorylation/degradation. XAV939 was non-toxic to OLPs at early stages of recruitment and proliferation and acted to accelerate differentiation and formation of compact myelin. These findings indicate that Wnt pathway activity is dispensable for myelin regeneration.
Our therapeutic approach employed targeted injection of XAV939 into spinal cord lesions. Although this approach might seem to present technical challenges, we note that non-invasive imaging currently allows for precise neurosurgical placement of devices, neural progeni-tor cells and drugs directly into white matter tracts of the brain. In preliminary studies, we observed upregulation of Tcf4, Axin2, Nkd1 and Notum in the white matter of neonatal mice exposed to chronic hypoxia, and Axin2−/− animals sustained permanent deficits in white matter after such injury (J.C.S., E.P.H., S.P.J.F. and D.H.R., unpub-lished observations). XAV939 treatment promoted robust myeli-nation of axons after cerebellar cultures were exposed to hypoxia, highlighting the efficacy of XAV939 to act on OLPs from the brain to promote myelination after a developmental hypoxic insult. Further study of systemic XAV939 (and similar agents that stabilize Axin2) administration is needed to determine possible toxicity and efficacy in promoting remyelination in pre-clinical models of human neonatal hypoxic WMI and adult demyelinating diseases32.
METHODSMethods and any associated references are available in the online version of the paper at http://www.nature.com/natureneuroscience/.
Accession codes. Microarray data are available on the GEO website, GSE19403.
Note: Supplementary information is available on the Nature Neuroscience website.
AcknowledgmenTSWe thank W. Birchmeier for Axin2-lacZ mice. This work was supported by a Promise 2010 grant from the National Multiple Sclerosis Society (to R.J.M.F. and D.H.R.), the United Kingdom Multiple Sclerosis Society (to R.J.M.F.), the US National Institutes of Health (NS040511 and NS047572 to D.H.R.) and the Medical Scientist Training Program at the University of California, San Francisco (to E.P.H.). S.E.B. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. R.N. and D.H.R. are Howard Hughes Medical Institute Investigators.
AUTHoR conTRIBUTIonSS.P.J.F. helped conceive the project and performed all experiments and analysis, with the exception of the following. E.P.H. performed and analyzed all experiments related to in vitro OLP cultures. T.J.Y. performed and analyzed the ex vivo cerebellar slice cultures. J.C.S. helped analyze Wnt pathway activation in murine hypoxic injury. C.Z. performed the electron microscopy and C.C.B. performed the G ratio analysis. S.E.B. performed bioinformatics. J.J.O. and E.J.H. procured human brain developmental tissue. R.J.M.F. and D.H.R. conceived the experiments and oversaw all aspects of the analysis. The paper was written by S.P.J.F., R.N., R.J.M.F. and D.H.R.
comPeTIng FInAncIAl InTeReSTSThe authors declare no competing financial interests.
Published online at http://www.nature.com/natureneuroscience/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
1. Back, S.A. et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 22, 455–463 (2002).
2. Khwaja, O. & Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 93, F153–F161 (2008).
3. Woodward, L.J., Anderson, P.J., Austin, N.C., Howard, K. & Inder, T.E. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 355, 685–694 (2006).
4. Compston, A. & Coles, A. Multiple sclerosis. Lancet 372, 1502–1517 (2008).5. Chang, A., Tourtellotte, W.W., Rudick, R. & Trapp, B.D. Premyelinating
oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 346, 165–173 (2002).
6. Kuhlmann, T. et al. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131, 1749–1758 (2008).
7. Káradóttir, R., Cavelier, P., Bergersen, L.H. & Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166 (2005).
8. Billiards, S.S. et al. Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 18, 153–163 (2008).
9. Segovia, K.N. et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 63, 520–530 (2008).
10. Fancy, S.P.J. et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 23, 1571–1585 (2009).
11. Ye, F. et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin–TCF interaction. Nat. Neurosci. 12, 829–838 (2009).
12. Feigenson, K., Reid, M., See, J., Crenshaw, E.B. III & Grinspan, J.B. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell. Neurosci. 42, 255–265 (2009).
13. Ikeda, S. et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta–dependent phosphorylation of beta-catenin. EMBO J. 17, 1371–1384 (1998).
14. Behrens, J. et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 280, 596–599 (1998).
15. Molenaar, M. et al. XTcf-3 transcription factor mediates beta-catenin–induced axis formation in Xenopus embryos. Cell 86, 391–399 (1996).
16. Behrens, J. et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638–642 (1996).
17. Jho, E.H. et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 (2002).
18. Lustig, B. et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 22, 1184–1193 (2002).
19. Zeng, Y.A. & Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 6, 568–577 (2010).
20. Dessaud, E., McMahon, A.P. & Briscoe, J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489–2503 (2008).
21. Dao, D.Y. et al. Axin2 regulates chondrocyte maturation and axial skeletal development. J. Orthop. Res. 28, 89–95 (2010).
22. Emery, B. et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185 (2009).
23. Franklin, R.J.M. & ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855 (2008).
24. Arnett, H.A. et al. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 306, 2111–2115 (2004).
25. Huang, S.M. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signaling. Nature 461, 614–620 (2009).
26. Huang, J.K. et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat. Neurosci. 14, 45–53 (2011).
27. Duncan, I.D., Brower, A., Kondo, Y., Curlee, J.F. Jr. & Schultz, R.D. Extensive remyelination of the CNS leads to functional recovery. Proc. Natl. Acad. Sci. USA 106, 6832–6836 (2009).
28. Kotter, M.R., Li, W.W., Zhao, C. & Franklin, R.J.M. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 26, 328–332 (2006).
29. Mi, S. et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 8, 745–751 (2005).
30. Zhang, Y. et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl. Acad. Sci. USA 106, 19162–19167 (2009).
31. Watkins, T.A., Emery, B., Mulinyawe, S. & Barres, B.A. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron 60, 555–569 (2008).
32. Silbereis, J.C., Huang, E.J., Back, S.A. & Rowitch, D.H. Towards improved animal models of neonatal white matter injury associated with cerebral palsy. Dis. Model Mech. 3, 678–688 (2010).
33. Lock, C. et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 8, 500–508 (2002).
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

nature neurOSCIenCedoi:10.1038/nn.2855
ONLINE METHODSTransgenic mice and procedures. Animal husbandry and procedures were per-formed according to University of California, San Francisco guidelines and the protocols were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. The Axin2-lacZ mouse has been described previously18. Insertion of the β-galactosidase gene into the Axin2 locus (Axin2-lacZ) provides a useful tool for visualizing cells that actively respond to Wnt in vivo. The lacZ insert mimics the expression pattern of Axin2, but does not lead to detectable phenotype in the heterozygous state. In the homozygous state, this effectively acts as an Axin2−/− animal.
A multi-functional mouse line (Olig2-tva-cre) was constructed previously34. Olig2-cre allows for Cre-mediated activity in oligodendrocyte lineage cells.
The DA-cat mouse was produced previously35 and has exon 3 of the mouse β-catenin gene located between loxP sequences. Cre recombinase–mediated dele-tion of exon 3 produces dominant-stable mutant β-catenin protein.
Induction of demyelination with lysolecithin in mouse spinal cord. Demyelinated lesions were produced in ventrolateral spinal cord white mat-ter of 8–10-week-old Axin2−/− and wild-type littermate mice. The method has been described previously10. Mice were killed at three survival time points: 5 dpl (representing peak OLP recruitment), 10 dpl (representing onset of OLP differentiation) and 14 dpl (representing new myelin sheath formation) (n = 4 for each survival time).
Immunohistochemistry. The method has been described previously10. For primary antibodies, we used antibodies to Tcf4 (mouse monoclonal 6H5-3, Upstate), Olig2 (rabbit polyclonal from C.D. Stiles, Harvard), Nkx2.2 (mouse monoclonal, Developmental Studies Hybridoma Bank), PDGFRα (rat 558774, BD Biosciences), APC (CC1, mouse monoclonal OP80, Calbiochem), NOGO-A (rabbit, Millipore), β-galactosidase (rabbit 55976, MP), tankyrase (mouse mono-clonal 19A449, Abcam), Iba1 (rabbit, Wako), CD3 (rabbit, Dako), GFAP (mouse, Sigma), Nkd1 (rabbit, Cell Signaling Technology) and Notum (rabbit, Abcam). Secondary fluorescent antibodies from Alexa were used.
cell counts and statistical measures. For all cell counts, at least four animals per genotype were used for each time point indicated. Cells were counted on three or more non-adjacent sections per mouse and presented as the average ± s.d. Statistical comparisons of cell counts were established using Student’s t test.
Human multiple sclerosis tissue. Human post-mortem tissue blocks were pro-vided by the UK Multiple Sclerosis Tissue Bank at Imperial College London. All tissues were collected following fully informed consent by the donors via a prospective donor scheme following ethical approval by the London Multicentre Research Ethics committee (MREC 02/2/39). Multiple sclerosis lesions were char-acterized as described previously33, using LFB to assess demyelination, SMI-31 immunohistochemistry to assess preservation of axons, and LN3 immunohisto-chemistry to assess inflammatory cell activity. Lesions with florid parenchymal and perivascular inflammatory cell infiltration, myelin fragmentation, and demy-elination with indistinct margins were classified as active plaques. Chronic active plaques were classified as those with extensive demyelination, well-demarcated borders and abundant inflammatory cells at the lesion edge. Chronic plaques were classified as those with extensive demyelination and well-demarcated borders, but few or no inflammatory cells in any part of the demyelinated area.
Human developmental tissue. All human tissue was collected in accordance with guidelines established by the University of California San Francisco Committee on Human Research (H11170-19113-07). Immediately after procurement, all brains, except no. 1, were immersed in 4% phosphate-buffered formalin (wt/vol) for 1 week at room temperature (20–25 °C). Brain no. 1 was immersed in phosphate-buffered saline (PBS) with 4% paraformaldehyde (wt/vol) for 3 d. On day 3, brains were cut in coronal plane at level of mamillary body and immersed in fresh 4% paraformaldehyde/PBS for additional 3 d. Post fixation, all tissue samples were equilibrated in PBS with 30% sucrose (wt/vol) for at least 2 d, before optimum cut-ting temperature medium embedding. The diagnosis of HIE requires clinical and pathological correlations. With respect to the pathological features, all HIE cases in study showed consistent evidence of diffuse WMI, including astrogliosis and macrophage infiltration. These findings were confirmed by increase in number
and staining intensity of GFAP- or CD68-positive cells, respectively. The HIE cases also showed evidence of neuronal injury, including presence of ischemic neurons and variable degrees of neuronal loss, in cerebral cortex, hippocampus and basal ganglia. Representative images in Supplementary Figure 2 revealed that GFAP immunoreactivity was most intense in layer 1 of cerebral cortex and in the subcortical white matter.
Subjects 1–5 demonstrated clinical evidence of HIE and pathological evidence of HIE and low output state in multiple organs on post-mortem examination. More specific findings in subject 1 (8 weeks old, born at full term) included hypoplastic left heart and diffuse WMI in brain with focal neuron dropout in cerebral and cerebellar cortices. Subject 2 (5 d old, born full term) with a clinical diagnosis of severe HIE who underwent therapeutic hypothermia showed diffuse WMI on post-mortem evaluation. Subject 3 (5 months old, born preterm at 26 weeks gestation) clinically showed failure to thrive, enterocolitis, hypoxemia and ventriculomegaly. Hypomyelination relative to the patient’s age, diffuse white matter gliosis and focal loss of cortical neurons were noted in this patient. Subject 4 (6-d-old male born at 35 weeks and 5 d) with in utero polyhydramnios showed neuropathological findings of HIE and intraventricular hemorrhage. Subject 5 (6-d-old female born at 37 weeks and 4 d) with intrauterine growth retardation showed HIE characterized by widespread neuronal death in the putamen, thalamus, hippocampus, diffuse hypoxic white matter changes and diffuse cerebral edema. Subject 6 (12 weeks old, born at 35 weeks and 3 d) with congenital diaphragmatic hernia, hypoplastic heart and coarctation of aortic arch showed PVL adjacent to the lateral ventrical. Subject 7 (1-d-old term infant) with midgut volvulus with extensive hemorrhagic necrosis of small bowel was not found to have significant neuropathological findings of HIE. Subject 8 (1-d-old female, born at 37 weeks) with hypoplastic left heart complex died shortly after birth and showed no evidence of white matter damage. Subject 9 (4-month-old male born at 38 weeks and 5 d) with congenital diaphragmatic hernia and hypoplastic aortic arch was also affected by uncontrolled respiratory syncytial virus pneumonia. Neuropathological evaluation of subject 9 did not show diffuse WMI.
In vitro olP culture. OLPs were isolated by immunopanning P7 CD-1 mouse cerebral cortices as previously described22,36. OLPs were plated on poly-d-lysine–coated plates and coverslips and maintained in proliferation media in a 10% CO2 incubator at 37 °C with 50% media changes every 2 d. Proliferation media has been described previously36. For differentiation, OLP proliferation media was replaced with differentiation media: OLP mitogens in proliferation media were replaced with ciliary neurotrophic factor (10 ng ml−1) and triiodothyronine (40 ng ml−1).
XAV939 treatment of olP cultures and differentiation assays. After immuno-panning, OLPs were plated at a density of 5,000 cells per coverslip in proliferation media and allowed to recover for 24 h. XAV939 (Tocris) or DMSO were added to the media for 24 h. Differentiation was induced by replacement of media with dif-ferentiation media including pre-treatment XAV939 concentration or DMSO. For western blots, OLPs were plated at a density of 0.5 × 106 cells per 15-cm plate and maintained until 75% confluent ~4 d. XAV939 or DMSO was added to media for the indicated time period. For differentiation, cell counts of at least three coverslips and three separate experiments (n = 9) and n = 300–1,000 cells for each test group were imaged and counted. Comparisons of the means between two groups were analyzed with unpaired t test and statistical significance was set at P < 0.01.
western blot of olPs. Western blot was performed as previously described36. We used primary antibodies to beta-actin (1:5,000, Cell Signaling 4970), axin1 (1:250, Cell Signaling 2087), axin2 (1:250, Cell Signaling 2151), phospho–beta-catenin (1:1,000, Cell Signaling 9561), tankyrase (1:1,000, Abcam ab13587) and MBP (1:10,000, Sternberger SMI-94).
XAV939 treatment in vivo in mouse spinal cord demyelination model. XAV939 (Tocris) was dissolved in DMSO to a concentration of 10 mM, then diluted in sterile water to a concentration of 10 µM. This stock was then added to 1% lysol-ecithin (wt/vol) immediately before lesioning to a final concentration of 0.1 µM XAV939. DMSO alone was added for control animals. Wild-type young adult mice (8–10 weeks old) were then co-injected with the mixture in dorsal or ventral funiculus spinal cord white matter as described above, and harvested at the times described post-lesioning.
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.

nature neurOSCIenCe doi:10.1038/nn.2855
expression profiling of Olig2-cre; DA-cat versus wild-type spinal cord at P4. Whole spinal cords were harvested, homogenized in Trizol and then RNA was extracted using RNeasy kit (Qiagen). Microarray analysis was performed at the Genetics Core Facility of the Gladstone Institute. For each of P4 wild-type and Olig2-cre/DA-cat, RNA from three animals was pooled for use in each array chip, and three Affymetrix Mouse Genome 430 2.0 chips were run for each group, for a total of nine animals for each group of wild type and Olig2-cre/DA-cat mice.
Ex vivo cerebellar slice cultures. Following decapitation, the brains of new-born CD1 wild-type mice (P0–1) were dissected out into cold MEM (MEM supplemented with penicillin/streptomycin, Invitrogen) and l-glutamine (4 mM). Excess tissue was trimmed around the brain, ensuring that the cerebel-lum remained attached to the underlying piece of hindbrain. Using a McIlwain tissue chopper, 350-µm sagittal slices of the cerebellum were cut and plated on Millicell-CM organotypic culture inserts (Millipore, 0.4 µm) in slice culture medium containing 50% MEM with Earle’s salts, 35% Earle’s balanced salt solu-tion, 25% heat-inactivated horse serum, glutamax, fungizone, penicillin-strep-tomycin (each from Invitrogen) and glucose (Sigma).
For the myelinating slice cultures, XAV or DMSO was added at the time of plating. Cultures were maintained at 37 °C and 7.5% CO2. Membranes were transferred into fresh medium supplemented with tested factors every 2 d. The hypoxic slice cultures were exposed to 2% O2 conditions for 24 h between 2–3 d in culture and then returned to normal culture conditions. XAV or DMSO were added following hypoxia exposure. Both the myelinating and hypoxic slices were fixed at 12 d in vitro. Remyelinating slice cultures were adapted from previously described methods37. After culturing slices in medium with no factors added for 14 d, demyelination was induced by the addition of 0.5% lysolecithin (Sigma) to the slice culture medium for 18 h. Following demyelination, slices were then transferred to medium containing XAV or DMSO and maintained for an addi-tional 14 d to allow remyelination to occur.
Immunostaining of slice cultures. Slices were immersed in 4% paraformal-dehyde for 1 h while attached to membranes at the desired time points. Slices were then rinsed in PBS, blocked with 3% heat-inactivated horse serum, 2% bovine serum albumin (wt/vol) and 0.25% Triton X-100 (wt/vol) in PBS, then incubated overnight at 4 °C in primary antibody diluted in block solution. For primary antibodies, we used chicken polyclonal antibody to NFH (Encor Biotech), rat monoclonal antibody to MBP (Serotec) and rabbit polyclonal antibody to Caspr (AbCam).
Slice culture imaging and quantification. Confocal z stacks were acquired (12 slices) using Leica SP5 confocal microscope. On examination of staining, sections were chosen based on their intact cytoarchitechture and the formation of parallel myelinated tracts in the cerebellum. Any slices that did not meet these crite-ria were not analyzed. Imaging was focused on the areas of the cerebellum that contained parallel tracts of myelinated axons. Obtained images were processed and analyzed using NIH Image (http://rsb.info.nih.gov/nih-image/). Following compression of images into one z stack, NIH Image was used to threshold area of staining for Caspr and NFH. Myelination was then quantified by a ratio of percent area stained for Caspr to percent area stained for NFH. All imaging and subsequent data analysis was done blinded to conditions being tested. Three inde-pendent experiments were conducted and analyzed. The data were analyzed by one-way ANOVA with Dunnett’s multiple comparison test (GraphPad Prism).
34. Schüller, U. et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14, 123–134 (2008).
35. Harada, N. et al. Intestinal polyposis in mice with a dominant-stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942 (1999).
36. Harrington, E. et al. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann. Neurol. 68, 703–716 (2010).
37. Birgbauer, E., Rao, T.S. & Webb, M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J. Neurosci. Rev. 78, 157–166 (2004).
© 2
011
Nat
ure
Am
eric
a, In
c. A
ll ri
gh
ts r
eser
ved
.©
201
1 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.