basic overview of cancer - kalgenlab.files.wordpress.com ·...
TRANSCRIPT

basic overview of cancer
cell cycle, stem-cell, cancer stem-cell
Monday, January 30, 2012

• useful refs:
• harvey lodish Molecular Cell Biology
• robert weinberg Biology of Cancer
• useful weblinks:
• http://mydsn.org/docs-tools/BIO240/Chapter-11.ppt
• http://www.hugo-international.org/resources/Isik_Yulug_Molecular_Basis_of_cancer.ppt
Monday, January 30, 2012

hallmarks of cancer
Monday, January 30, 2012

Fig. 25-1:
Blood supply necessary for the tumor to grow and spread. Intravasation and extravasation.
Requires protease activity to penetrate basement membranes and the extracellular matrix.
Necessary for continued proliferation.
Necessary for continued proliferation.
Otherwise the tumor cells will die,
Otherwise the tumor will not grow.
Six characteristics of metastatic (malignant) tumor cellsBenign tumor cells are not metastatic.
Inhibition of one or more of these characteristics can inhibit cancer progression.
Monday, January 30, 2012

CANCERcell cycle dysregulation, proliferative disorder
Monday, January 30, 2012

Cell Cycle
Monday, January 30, 2012

Progress in Cell Cycle is regulated by Genes
✤ (Keep in mind what would happen in cell cycle in cancer cells)
Monday, January 30, 2012

Figure 8.9 The Biology Of Cancer (© Garland Science 2007)
Proliferative driver of cell cycle: Cyclins
Expression of cyclins fluctuates at certain phases of the cell cycle
Monday, January 30, 2012
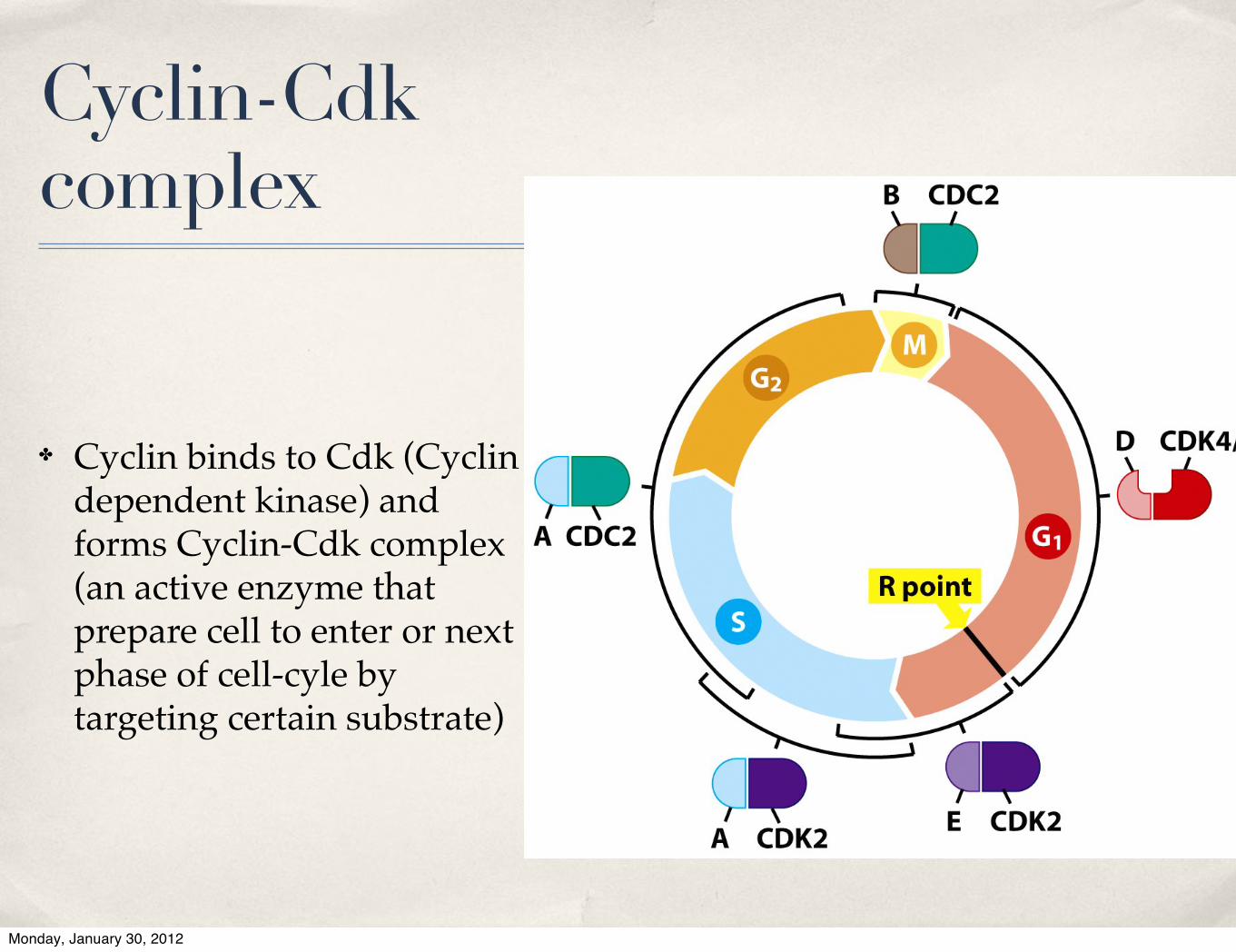
Cyclin-Cdk complex
✤ Cyclin binds to Cdk (Cyclin dependent kinase) and forms Cyclin-Cdk complex (an active enzyme that prepare cell to enter or next phase of cell-cyle by targeting certain substrate)
Monday, January 30, 2012

Figure 8.10 The Biology of Cancer (© Garland Science 2007)
Restric(on Point (R): Decision making point the late G1 phase of the cell cycle at which a cell commits itself to comple(ng the remaining phases of the cell cycle, remaining in G1, or exi(ng the ac(ve cell cycle and entering into Go
Monday, January 30, 2012

Table 8.1 The Biology of Cancer (© Garland Science 2007)
UPREGULATION OF CYCLIN D BY RECEPTORS
Monday, January 30, 2012

drug ther apy
n engl j med 358;11 www.nejm.org march 13, 2008 1161
exclusively and are therefore highly selective for this receptor. In addition, various small-molecule EGFR tyrosine kinase inhibitors can block differ-ent growth factor receptor tyrosine kinases, in-cluding other members of the EGFR family, or the vascular endothelial growth factor receptor. Vari-ous irreversible EGFR tyrosine kinase inhibitors
are now in early stages of clinical develop-ment.4,5,12 The mechanism (or mechanisms) of action, pharmacologic effects, and spectrum of activity of anti-EGFR monoclonal antibodies and small-molecule EGFR tyrosine kinase inhibitors have differences that may be relevant for clinical activity (Table 1 and Fig. 2 and 3).13
COLOR FIGURE
Figure!1.!Signal!Transduction!Pathways!Controlled!by!the!Activation!of!EGFR.!
Three steps can be schematically defined in the activation of EGFR-dependent intracellular signaling.2-17 First, the binding of a receptor-specific ligand occurs in the extracellular portion of the EGFR or of one of the EGFR-related receptors (HER2, HER3, or HER4). Second, the formation of a functionally active EGFR-EGFR dimer (homodimer) or of an EGFR-HER2, EGFR-HER3, or EGFR-HER4 dimer (heterodi-mer) causes the ATP-dependent phosphorylation of specific tyrosine residues in the EGFR intracellular domain. Third, this phosphoryla-tion triggers a complex program of intracellular signals to the cytoplasm and then to the nucleus. The two major intracellular pathways activated by EGFR are the RAS–RAF–MEK–MAPK pathway, which controls gene transcription, cell-cycle progression from the G1 phase to the S phase, and cell proliferation, and the PI3K–Akt pathway, which activates a cascade of anti-apoptotic and prosurvival signals. bFGF denotes basic fibroblast growth factor, HB-EGF heparin-binding EGF, MAPK mitogen-activated protein kinase, P phosphate, PI3K phosphatidylinositol 3,4,5-kinase, TGF! transforming growth factor !, and VEGF vascular endothelial growth factor. For more detailed information, see Figure 1 in the Supplementary Appendix (available with the full text of this article at www.nejm.org).
The New England Journal of Medicine Downloaded from www.nejm.org on October 14, 2010. For personal use only. No other uses without permission.
Copyright © 2008 Massachusetts Medical Society. All rights reserved.
Monday, January 30, 2012

Dysregulation of cell cycle causes cancer
Monday, January 30, 2012

Oncogenic mutation causes cancer
normal cells
All found in cancer cells
Monday, January 30, 2012

Table 8.3 The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012
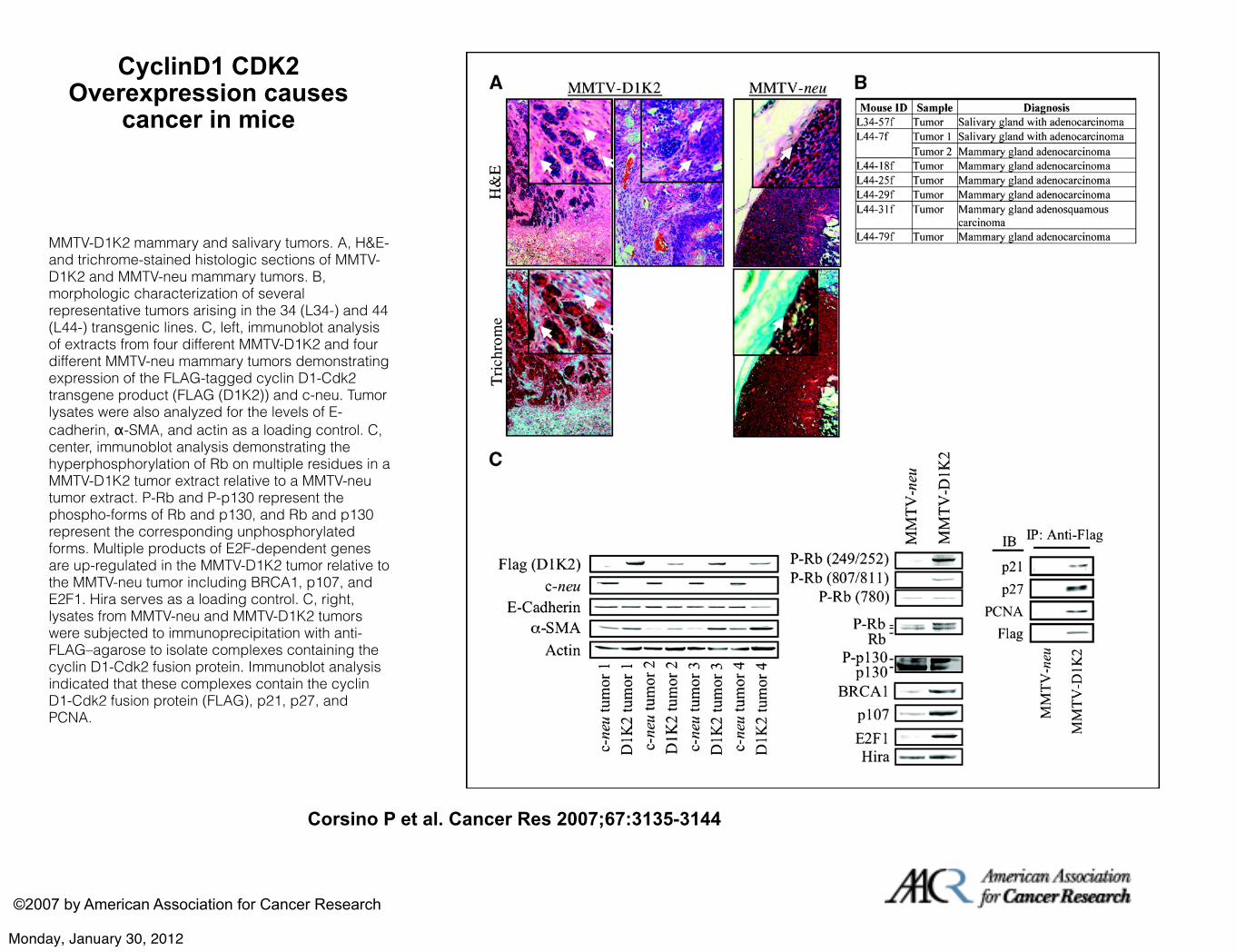
CyclinD1 CDK2 Overexpression causes
cancer in mice
Corsino P et al. Cancer Res 2007;67:3135-3144
©2007 by American Association for Cancer Research
MMTV-D1K2 mammary and salivary tumors. A, H&E- and trichrome-stained histologic sections of MMTV-D1K2 and MMTV-neu mammary tumors. B, morphologic characterization of several representative tumors arising in the 34 (L34-) and 44 (L44-) transgenic lines. C, left, immunoblot analysis of extracts from four different MMTV-D1K2 and four different MMTV-neu mammary tumors demonstrating expression of the FLAG-tagged cyclin D1-Cdk2 transgene product (FLAG (D1K2)) and c-neu. Tumor lysates were also analyzed for the levels of E-cadherin, α-SMA, and actin as a loading control. C, center, immunoblot analysis demonstrating the hyperphosphorylation of Rb on multiple residues in a MMTV-D1K2 tumor extract relative to a MMTV-neu tumor extract. P-Rb and P-p130 represent the phospho-forms of Rb and p130, and Rb and p130 represent the corresponding unphosphorylated forms. Multiple products of E2F-dependent genes are up-regulated in the MMTV-D1K2 tumor relative to the MMTV-neu tumor including BRCA1, p107, and E2F1. Hira serves as a loading control. C, right, lysates from MMTV-neu and MMTV-D1K2 tumors were subjected to immunoprecipitation with anti-FLAG–agarose to isolate complexes containing the cyclin D1-Cdk2 fusion protein. Immunoblot analysis indicated that these complexes contain the cyclin D1-Cdk2 fusion protein (FLAG), p21, p27, and PCNA.
Monday, January 30, 2012

Summary I
✤ Proliferative driver by Proto-oncogenes
✤ Alteration of proto-oncogene becoming oncogenes drives malignancy (amplification, translocation) by pushing overexpression of cyclins
✤ Proto-oncogene: EGFR/HER1, HER2, MYC
✤ Oncogenes: EGFR Amplification, EGFR mutation, HER2 amplification, C-MYC amplification
Monday, January 30, 2012

Oncogenic mutations alone are not sufficient
Monday, January 30, 2012

Figure 7.1 The Biology of Cancer (© Garland Science 2007)
History 1970s
• Sir Henry Harris asked:• Is malignancy
–Dominant, or–Recessive?
• Performed cell fusion
Monday, January 30, 2012

Harris Cell fusion experiment
• the phenotype of fused cells showed loss of tumorigenicity
• implied the presence of tumor supressor genes
• in 1989 the first tumor supressor gene ie Retinoblastoma (RB) is cloned
Monday, January 30, 2012

Retinoblastoma Gene Mutation in both Alleles causes cancer
in mice and men
medullary tumor of thyroid cancer in Rb ko miceretinoblastoma in early childhood
Monday, January 30, 2012

Loss of Heterozygosity
(LOH) is a genetic
mechanism to inactivate tumor supressor gene
(TSG)
Monday, January 30, 2012

Restriction Point (R):
Decision making point the late G1 phase of the cell cycle at which a cell commits itself to comple(ng the remaining phases of the cell cycle, remaining in G1, or exi(ng the ac(ve cell cycle and entering into Go
where does tumor supressor gene belong in cell cycle?
Monday, January 30, 2012

RB Phosphorylation
Figure 8.19 The Biology of Cancer (© Garland Science 2007)Figure 8.19 The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012

Figure 8.22 The Biology of Cancer (© Garland Science 2007)
RB is a key gatekeeper of G1/S transition at R pointLoss of RB in precancerous cells relaxes G1/S and promotescellular proliferation
Monday, January 30, 2012
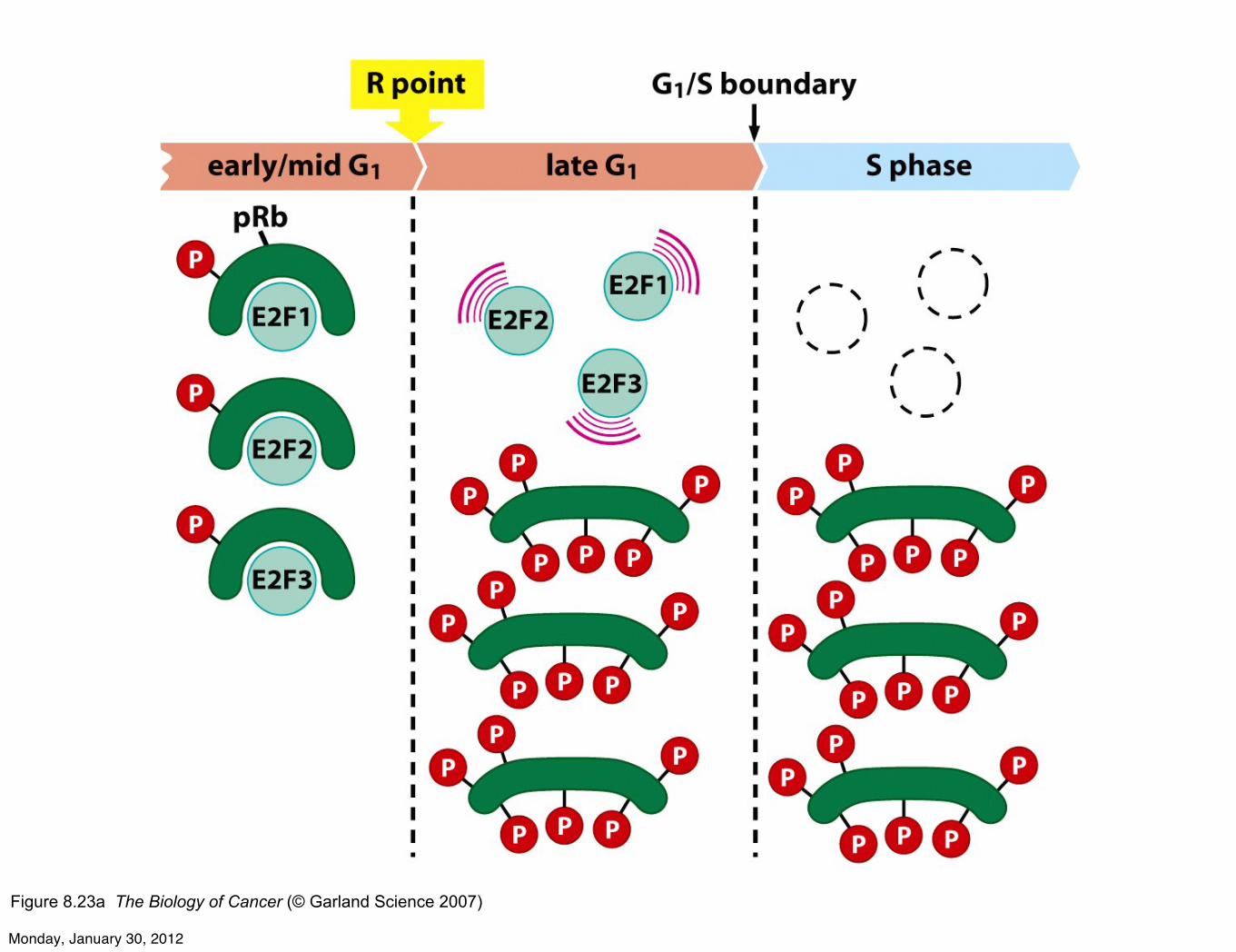
Figure 8.23a The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012

Figure 8.29 The Biology of Cancer (© Garland Science 2007)
Experimental evidence of C-MYC override TSG function of RB:
Artificial C-MYC activationcauses S-phase entry in quiescent cells
Monday, January 30, 2012

Date
E2F is controlled by RBLoss of RB function (by losing both alleles due to somatic mutation and Loss of Heterozygosity) causes unregulated entry to cell cycle
Monday, January 30, 2012

Summary
• RB gene is an important tumor suppressor gene that regulates entry into cell cycle
• Passing or overriding Restriction Point to start cell cycle is achieved by phosphorylating RB
Monday, January 30, 2012

Quality Control in Cell Cycle?
live with (genomic) integrity or die (apoptosis)
Monday, January 30, 2012

Cell Cycle CheckpointsPurpose:
To maintain DNA integrityMechanisms:
Cell cycle arrest (buying time for DNA Repair)Activation of Apoptotic cell death (catastrophic DNA damage)Experimental evidence:
Using yeast model, cell cycle is prolonged when yeasts are challenged with damaging agents. DNA analyses showed arrest at specific phase of cell cycleFigure 8.4 The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012

Monday, January 30, 2012

Monday, January 30, 2012
Monday, January 30, 2012
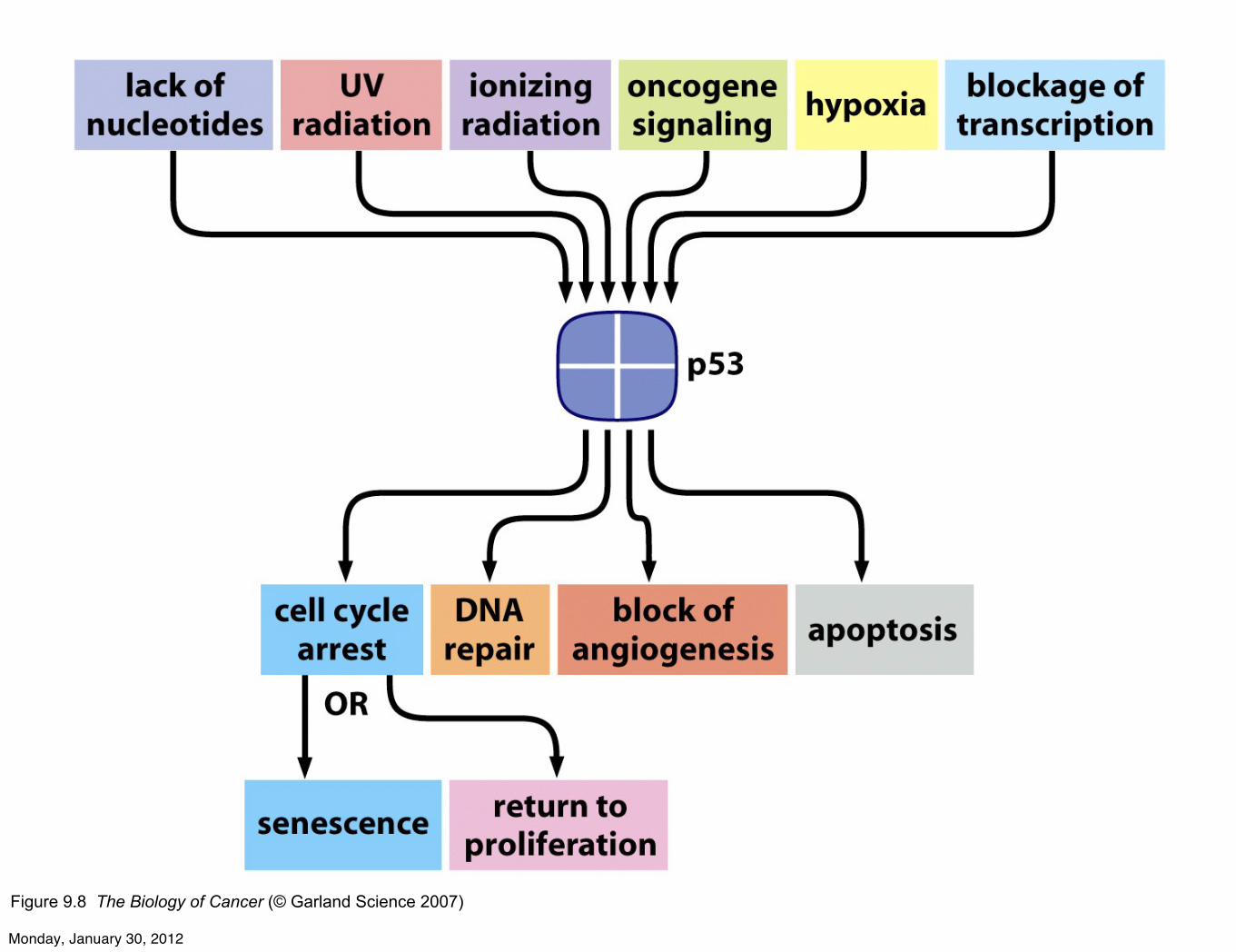
Figure 9.8 The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012

Date
Hair loss due to cyclophosphamideAnything to do with genes? which gene is responsible?
Monday, January 30, 2012

Fig. 2. p53 knockout mice treated by cyclophosphamide do not display hair loss. Hair cycle was induced in the back skin of 8-week-old wild-type (n! 25) and p53-null mice (n! 25)by depilation, and cyclophosphamide was administered 9 days after. Skin was harvested at day 7 after cyclophosphamide administration (day 16 postdepilation), and cryosections (8!m thick) were processed for the histoenzymatic detection of alkaline phosphatase (B and C) and double immunovisualization of proliferative marker Ki-67 and TUNEL (D and E).Cell nuclei in D and E were visualized by TO-PRO-3 staining. A, severe hair loss over the entire back of the wild-type mice treated by cyclophosphamide, with no hair loss visiblein the p53 knockout mice. B, shortening of the length and reduction in the volume of proximal hair bulb in the wild-type HF (arrows) is associated with dramatic decrease of skinthickness after cyclophosphamide treatment. C, large volume of the proximal hair bulb (arrows) and dermal papilla (red color) in the p53 null HF after cyclophosphamide treatment.D, numerous TUNEL-positive cells (green fluorescence, arrows) and single Ki-67-positive cells (red fluorescence, arrowhead) in the regressing HF compartments of wild-type skin.E, numerous Ki-67-positive cells in the HF matrix (red fluorescence, arrowheads) and single TUNEL-positive granules in the precortical zone (arrow). Some TUNEL-positive cellsare also visible in the subcutis around HF (green fluorescence). Bars, 100 !m.
5004
P53 AND CHEMOTHERAPY-INDUCED HAIR LOSS
American Association for Cancer Research Copyright © 2000 on January 13, 2011cancerres.aacrjournals.orgDownloaded from
Note: p53 gene is knocked out in the germline using embryonic stem cell homologous recombination technique
Monday, January 30, 2012

Summary II: Cell Cycle and TSGs RB p53
• RB and p53 have unique genetic and biochemical roles on controlling cell cycle
• Activation of proliferation (by removing RB) also activates apoptotic machinery. Therefore, cancer cells also inactivate wild type p53 to survive
Monday, January 30, 2012

CANCERgenetic disorder
Monday, January 30, 2012

gen sana in corpore sano
• majority of cancer patients harbor somatic genetic mutations
• minority of cancer patients have inherited germline genetic mutations
• enviromental risks will invariably lead to somatic genetic changes
• eat healthy keep mutations away (assuming your dna repair genes are intact)
Monday, January 30, 2012

CANCERstem cell
Monday, January 30, 2012

Monday, January 30, 2012

stem cell dna repair juice
Monday, January 30, 2012

Monday, January 30, 2012

Monday, January 30, 2012

How Do Cancer Stem Cells Arise? The molecular pathways that maintain "stem-ness" in stem cells are also active in numerous cancers. This similarity has led scientists to propose that cancers may arise when some event produces a mutation in a stem cell, robbing it of the ability to regulate cell
division. This figure illustrates 3 hypotheses of how a cancer stem cell may arise: (1) A stem cell undergoes a mutation, (2) A progenitor cell undergoes two or more mutations, or (3) A fully differentiated cell undergoes several mutations that drive it back to a stem-like state. In all 3 scenarios, the resultant
cancer stem cell has lost the ability to regulate its own cell division.
http://stemcells.nih.gov/info/2006report/2006chapter9.htm
Monday, January 30, 2012

How Does Tumor Develop?
Monday, January 30, 2012

CSC HIERARCHY: Are all cancer cells created equal?
Dirks, P Monday, January 30, 2012

Monday, January 30, 2012
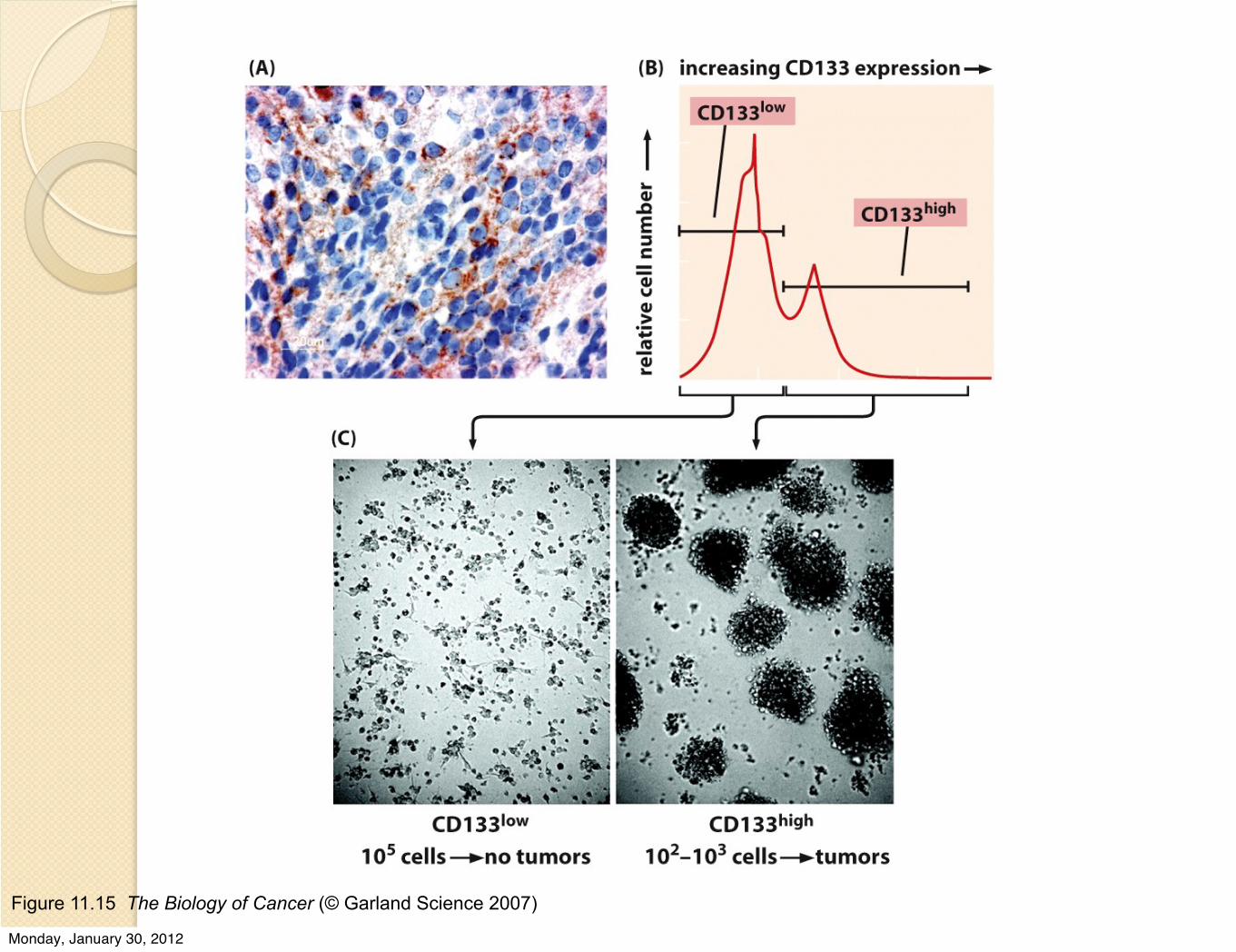
Figure 11.15 The Biology of Cancer (© Garland Science 2007)
Monday, January 30, 2012

properties of cancer stem cells
Monday, January 30, 2012

cancer recurrence
Monday, January 30, 2012
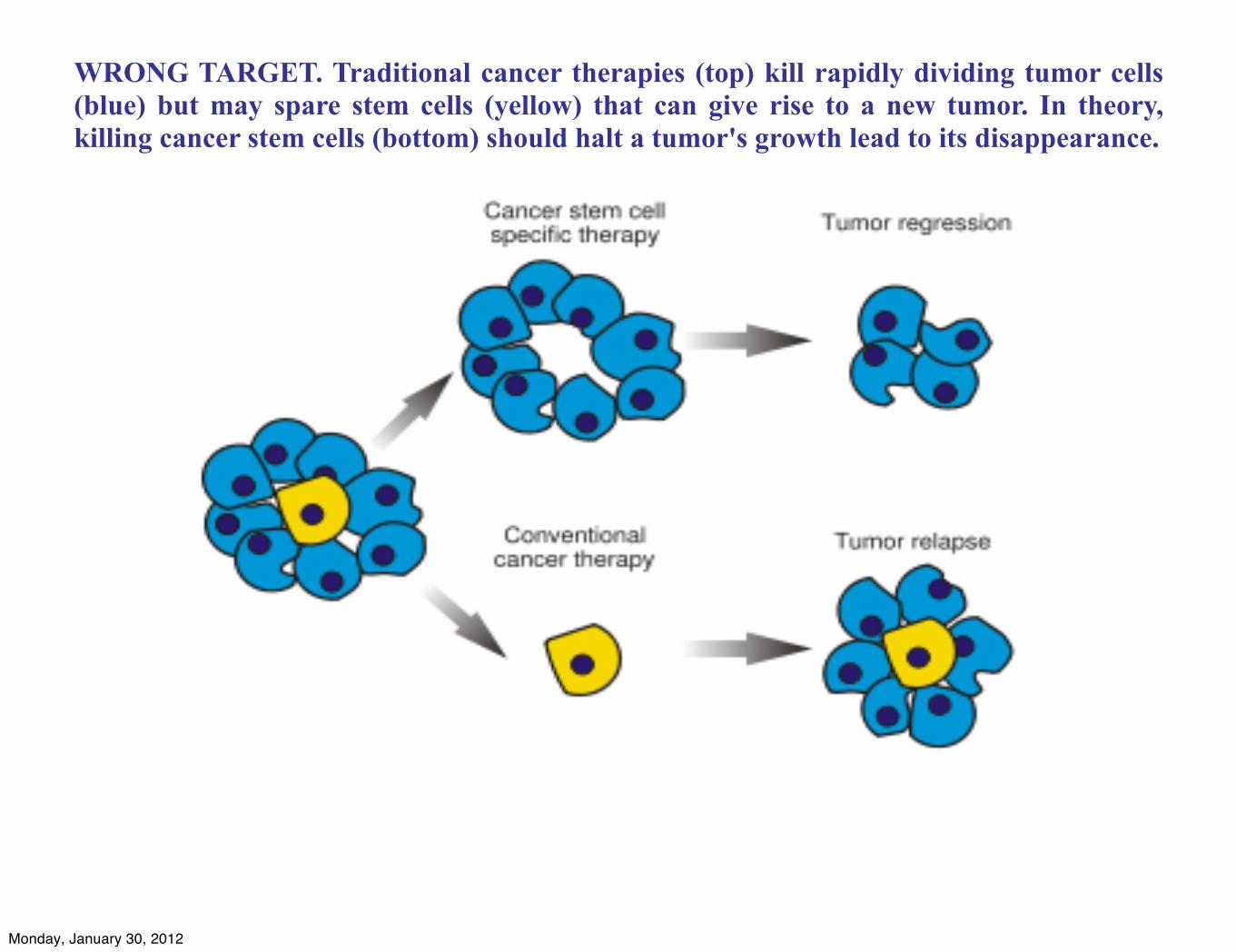
WRONG TARGET. Traditional cancer therapies (top) kill rapidly dividing tumor cells (blue) but may spare stem cells (yellow) that can give rise to a new tumor. In theory, killing cancer stem cells (bottom) should halt a tumor's growth lead to its disappearance.
Monday, January 30, 2012

intervention opportunity: targeting cancer stem
cell
Monday, January 30, 2012

Monday, January 30, 2012