basic principles of chromatographyanuragaja.staff.ipb.ac.id/files/2017/05/1_prinsip-dasar... ·...
TRANSCRIPT

27chapter
Basic Principlesof Chromatography
Baraem Ismail∗Department of Food Science and Nutrition, University of Minnesota,
St. Paul, MN 55108-6099, [email protected]
and
S. Suzanne NielsenDepartment of Food Science, Purdue University,
West Lafayette, IN 47907-2009, [email protected]
27.1 Introduction 47527.2 Extraction 475
27.2.1 Batch Extraction 47527.2.2 Continuous Extraction 475
27.2.3 Countercurrent Extraction 47527.3 Chromatography 475
27.3.1 Historical Perspective 47527.3.2 General Terminology 476
S.S. Nielsen, Food Analysis, Food Science Texts Series, DOI 10.1007/978-1-4419-1478-1_27,c© Springer Science+Business Media, LLC 2010
473

474 Part V • Chromatography
27.3.3 Gas Chromatography 47627.3.4 Liquid Chromatography 477
27.3.4.1 Paper Chromatography 47727.3.4.2 Thin-Layer
Chromatography 47827.3.4.2.1 General
Procedures 47827.3.4.2.2 Factors Affecting
Thin-LayerSeparations 478
27.3.4.3 Column LiquidChromatography 479
27.3.5 Supercritical Fluid Chromatography 48027.4 Physicochemical Principles of Chromatographic
Separation 48127.4.1 Adsorption (Liquid–Solid)
Chromatography 48127.4.2 Partition (Liquid–Liquid)
Chromatography 48227.4.2.1 Introduction 48227.4.2.2 Coated Supports 48327.4.2.3 Bonded Supports 483
27.4.3 Ion-Exchange Chromatography 48327.4.4 Size-Exclusion Chromatography 48527.4.5 Affinity Chromatography 488
27.5 Analysis of Chromatographic Peaks 48927.5.1 Separation and Resolution 490
27.5.1.1 Developing a Separation 49027.5.1.2 Chromatographic
Resolution 49127.5.1.2.1 Introduction 49127.5.1.2.2 Column
Efficiency 49227.5.1.2.3 Column
Selectivity 49427.5.1.2.4 Column Capacity
Factor 49427.5.2 Qualitative Analysis 495
27.5.3 Quantitative Analysis 49527.6 Summary 49627.7 Study Questions 49727.8 Acknowledgments 49827.9 References 498

Chapter 27 • Basic Principles of Chromatography 475
27.1 INTRODUCTION
Chromatography has a great impact on all areas ofanalysis and, therefore, on the progress of sciencein general. Chromatography differs from other meth-ods of separation in that a wide variety of materials,equipment, and techniques can be used. [Readers arereferred to references (1–19) for general and specificinformation on chromatography.]. This chapter willfocus on the principles of chromatography, mainlyliquid chromatography (LC). Detailed principles andapplications of gas chromatography (GC) will bediscussed in Chap. 29. In view of its widespreaduse and applications, high-performance liquid chro-matography (HPLC) will be discussed in a separatechapter (Chap. 28). The general principles of extrac-tion are first described as a basis for understandingchromatography.
27.2 EXTRACTION
In its simplest form, extraction refers to the trans-fer of a solute from one liquid phase to another.Extraction in myriad forms is integral to food anal-ysis – whether used for preliminary sample cleanup,concentration of the component of interest, or as theactual means of analysis. Extractions may be catego-rized as batch, continuous, or countercurrent pro-cesses. (Various extraction procedures are discussed indetail in other chapters: traditional solvent extractionin Chaps. 8, 18, and 29; accelerated solvent extractionin Chap. 18; solid-phase extraction in Chaps. 18 and29; and solid-phase microextraction and microwave-assisted solvent extraction in Chap. 18).
27.2.1 Batch Extraction
In batch extraction the solute is extracted from one sol-vent by shaking it with a second, immiscible solvent.The solute partitions, or distributes, itself between thetwo phases and, when equilibrium has been reached,the partition coefficient, K, is a constant.
K =Concentration of solute in phase l
Concentration of solute in phase 2[1]
After shaking, the phases are allowed to separate,and the layer containing the desired constituent isremoved, for example, in a separatory funnel. Inbatch extraction, it is often difficult to obtain a cleanseparation of phases, owing to emulsion formation.Moreover, partition implies that a single extraction isusually incomplete.
27.2.2 Continuous Extraction
Continuous liquid–liquid extraction requires specialapparatus, but is more efficient than batch separa-tion. One example is the use of a Soxhlet extractor forextracting materials from solids. Solvent is recycled sothat the solid is repeatedly extracted with fresh sol-vent. Other pieces of equipment have been designedfor the continuous extraction of substances from liq-uids, and different extractors are used for solvents thatare heavier or lighter than water.
27.2.3 Countercurrent Extraction
Countercurrent distribution refers to a serial extrac-tion process. It separates two or more solutes with dif-ferent partition coefficients from each other by a seriesof partitions between two immiscible liquid phases.Liquid–liquid partition chromatography (Sect. 27.4.2),also known as countercurrent chromatography, is adirect extension of countercurrent extraction. Yearsago the countercurrent extraction was done with a“Craig apparatus” consisting of a series of glass tubesdesigned such that the lighter liquid phase (mobilephase) was transferred from one tube to the next,while the heavy phase (stationary phase) remainedin the first tube (4). The liquid–liquid extractions tookplace simultaneously in all tubes of the apparatus,which was usually driven electromechanically. Eachtube in which a complete equilibration took placecorresponded to one theoretical plate of the chromato-graphic column (refer to Sect. 27.5.2.2.1). The greaterthe difference in the partition coefficients of varioussubstances, the better was the separation. A muchlarger number of tubes was required to separate mix-tures of substances with close partition coefficients,which made this type of countercurrent extractionvery tedious. Modern liquid–liquid partition chro-matography (Sect. 27.4.2) that developed from thisconcept is much more efficient and convenient.
27.3 CHROMATOGRAPHY
27.3.1 Historical Perspective
Modern chromatography originated in the late nine-teenth and early twentieth centuries from independentwork by David T. Day, a distinguished Americangeologist and mining engineer, and Mikhail Tsvet,a Russian botanist. Day developed procedures forfractionating crude petroleum by passing it throughFuller’s earth, and Tsvet used a column packed withchalk to separate leaf pigments into colored bands.

476 Part V • Chromatography
Because Tsvet recognized and correctly interpretedthe chromatographic processes and named the phe-nomenon chromatography, he is generally creditedwith its discovery.
After languishing in oblivion for years, chro-matography began to evolve in the 1940s due to thedevelopment of column partition chromatography byMartin and Synge and the invention of paper chro-matography. The first publication on GC appeared in1952. By the late 1960s, GC, because of its impor-tance to the petroleum industry, had developed intoa sophisticated instrumental technique, which wasthe first instrumental chromatography to be availablecommercially. Since early applications in the mid-1960s, HPLC, profiting from the theoretical and instru-mental advances of GC, has extended the area ofliquid chromatography into an equally sophisticatedand useful method. SFC, first demonstrated in 1962,is finally gaining popularity. Modern chromatographictechniques, including automated systems, are widelyutilized in the characterization and quality control offood raw materials and food products.
27.3.2 General Terminology
Chromatography is a general term applied to a widevariety of separation techniques based on the parti-tioning or distribution of a sample (solute) betweena moving or mobile phase and a fixed or stationaryphase. Chromatography may be viewed as a series
of equilibrations between the mobile and stationaryphase. The relative interaction of a solute with thesetwo phases is described by the partition (K) or distri-bution (D) coefficient (ratio of concentration of solutein stationary phase to concentration of solute inmobilephase). The mobile phase may be either a gas (GC) orliquid (LC) or a supercritical fluid (SFC). The station-ary phase may be a liquid or, more usually, a solid. Thefield of chromatography can be subdivided accordingto the various techniques applied (Fig 27-1), or accord-ing to the physicochemical principles involved in theseparation. Table 27-1 summarizes some of the chro-matographic procedures or methods that have beendeveloped on the basis of different mobile–stationaryphase combinations. Inasmuch as the nature of inter-actions between solute molecules and the mobile orstationary phases differ, thesemethods have the abilityto separate different kinds of molecules. (The reader isurged to review Table 27-1 again after having read thischapter.)
27.3.3 Gas Chromatography
Gas chromatography is a column chromatographytechnique, in which the mobile phase is gas and thestationary phase is either an immobilized liquid or asolid packed in a closed tube. GC is used to separatethermally stable volatile components of a mixture. Gaschromatography, specifically gas–liquid chromatogra-phy, involves vaporizing a sample and injecting it onto
27-1f igure
A scheme for subdividing the field of chromatography, according to various applied techniques.

Chapter 27 • Basic Principles of Chromatography 477
27-1table Characteristics of Different Chromatographic Methods
Method Mobile/Stationary Phase Retention Varies with
Gas–liquid chromatography Gas/liquid Molecular size/polarityGas–solid chromatography Gas/solid Molecular size/polaritySupercritical fluid
chromatographySupercritical fluid/solid Molecular size/polarity
Reversed-phasechromatography
Polar liquid/nonpolar liquid or solid Molecular size/polarity
Normal-phase chromatography Less polar liquid/more polar liquidor solid
Molecular size/polarity
Ion-exchange chromatography Polar liquid/ionic solid Molecular chargeSize-exclusion chromatography Liquid/solid Molecular sizeHydrophobic-interaction
chromatographyPolar liquid/nonpolar liquid or solid Molecular size/polarity
Affinity chromatography Water/binding sites Specific structure
Reprinted from (8), p. A21, with kind permission from Elsevier Science-NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam,
The Netherlands.
the head of the column. Under a controlled tempera-ture gradient, the sample is transported through thecolumn by the flow of an inert, gaseous mobile phase.Volatiles are then separated based on several prop-erties, including boiling point, molecular size, andpolarity. Physiochemical principles of separation arecovered in Sect. 27.4. However, details of the chro-matographic theory of separation as it applies specif-ically to GC, as well as detection and instrumentationof GC, are detailed in Chap. 29.
27.3.4 Liquid Chromatography
There are several liquid chromatography techniquesapplied in food analysis, namely paper chromatogra-phy, thin layer chromatography (TLC) (both of thesetechniques may be referred to as planar chromatogra-phy), and column liquid chromatography, all of whichinvolve a liquid mobile phase and either a solid or aliquid stationary phase. However, the physical form ofthe stationary phase is quite different in each case. Sep-aration of the solutes is based on their physicochemicalinteractions with the two phases, which is discussed inSect. 27.4.
27.3.4.1 Paper Chromatography
Paper chromatography was introduced in 1944. Inpaper chromatography the stationary phase and themobile phase are both liquid (partition chromatog-raphy, see Sect. 27.4.2). Paper generally serves asa support for the liquid stationary phase. The dis-solved sample is applied as a small spot or streakone half inch or more from the edge of a strip orsquare of filter paper (usually cellulose), which isthen allowed to dry. The dry strip is suspended in
a closed container in which the atmosphere is satu-ratedwith the developing solvent (mobile phase), andthe paper chromatogram is developed. The end closerto the sample is placed in contact with the solvent,which then travels up or down the paper by capillaryaction (depending on whether ascending or descend-ing development is used), separating the sample com-ponents in the process. When the solvent front hastraveled the length of the paper, the strip is removedfrom the developing chamber and the separated zonesare detected by an appropriate method.
The stationary phase in paper partition chro-matography is usually water. However, the supportmay be impregnated with a nonpolar organic solventand developed with water or other polar solvents orwater (reversed-phase paper chromatography). In thecase of complex sample mixtures, a two-dimensionaltechnique may be used. The sample is spotted in onecorner of a square sheet of paper, and one solvent isused to develop the paper in one direction. The chro-matogram is then dried, turned 90◦, and developedagain, using a second solvent of different polarity.Another means of improving resolution is the useof ion-exchange (Sect. 27.4.3) papers, that is, paperthat has been impregnated with ion-exchange resinor paper, with derivatized cellulose hydroxyl groups(with acidic or basic moieties).
In paper and thin-layer chromatography, compo-nents of a mixture are characterized by their relativemobility (Rf) value, where:
Rf =Distance moved by component
Distance moved by solvent[2]
Unfortunately, Rf values are not always constant fora given solute/sorbent/solvent, but depend on many

478 Part V • Chromatography
factors, such as the quality of the stationary phase,layer thickness, humidity, development distance, andtemperature.
27.3.4.2 Thin-Layer Chromatography
Thin-layer chromatography (TLC), first described in1938, has largely replaced paper chromatographybecause it is faster, more sensitive, and more repro-ducible. The resolution in TLC is greater than in paperchromatography because the particles on the plate aresmaller and more regular than paper fibers. Exper-imental conditions can be easily varied to achieveseparation and can be scaled up for use in columnchromatography, although thin-layer and column pro-cedures are not necessarily interchangeable, due todifferences such as the use of binders with TLC plates,vapor-phase equilibria in a TLC tank, etc. Thereare several distinct advantages to TLC: high samplethroughput, low cost, the possibility to analyze sev-eral samples and standards simultaneously, minimalsample preparation, and that a plate may be stored forlater identification and quantification.
TLC is applied in many fields, including envi-ronmental, clinical, forensic, pharmaceutical, food,flavors, and cosmetics. Within the food industry, TLCmay be used for quality control. For example, cornand peanuts are tested for aflatoxins/mycotoxins priorto their processing into corn meal and peanut but-ter, respectively. Applications of TLC to the analysisof a variety of compounds, including lipids, carbohy-drates, vitamins, amino acids, and natural pigments,are discussed in reference (5).
27.3.4.2.1 General Procedures TLC utilizes a thin(ca. 250µm thick) layer of sorbent or stationary phasebound to an inert support in a planar configura-tion. The support is often a glass plate (traditionally,20 cm × 20 cm), but plastic sheets and aluminum foilalso are used. Precoated plates, of different layerthicknesses, are commercially available in a wide vari-ety of sorbents, including chemically modified sili-cas. Four frequently used TLC sorbents are silica gel,alumina, diatomaceous earth, and cellulose. Mod-ified silicas for TLC may contain polar or nonpo-lar groups, so both normal and reversed-phase (seeSect. 27.4.2.1) thin-layer separations may be carriedout. High-performance thin-layer chromatography(HPTLC) simply refers to TLC performed using platescoated with smaller, more uniform particles. Thispermits better separations in shorter times.
If adsorption TLC is to be performed, the sor-bent is first activated by drying for a specified timeand temperature. Sample (in carrier solvent) is appliedas a spot or streak 1–2 cm from one end of the plate.
After evaporation of the carrier solvent, the TLCplate is placed in a closed developing chamber withthe end of the plate nearest the spot in the solventat the bottom of the chamber. Traditionally, solventmigrates up the plate (ascending development) bycapillary action and sample components are sepa-rated. After the TLC plate has been removed from thechamber and solvent allowed to evaporate, the sep-arated bands are made visible or detected by othermeans. Specific chemical reactions (derivatization),which may be carried out either before or after chro-matography, often are used for this purpose. Twoexamples are reaction with sulfuric acid to producea dark charred area (a destructive chemical method)and the use of iodine vapor to form a colored com-plex (a nondestructive method inasmuch as the col-ored complex is usually not permanent). Commonphysical detection methods include the measure-ment of absorbed or emitted electromagnetic radiation(e.g., fluorescence) by means of autoradiography andthe measurement of β-radiation from radioactivelylabeled compounds. Biological methods or biochemi-cal inhibition tests can be used to detect toxicologicallyactive substances. An example is measuring the inhi-bition of cholinesterase activity by organophosphatepesticides.
Quantitative evaluation of thin-layer chro-matograms may be performed (1) in situ (directly onthe layer) by using a densitometer or (2) after scrap-ing a zone off the plate, eluting compound from thesorbent, and analyzing the resultant solution (e.g., byliquid scintillation counting).
27.3.4.2.2 Factors Affecting Thin-Layer SeparationsIn both planar and column liquid chromatography,the nature of the compounds to be separated deter-mines what type of stationary phase is used. Separa-tion can occur by adsorption, partition, ion-exchange,size-exclusion, or multiple mechanisms (Sect. 27.4).Table 27-2 lists the separation mechanisms involved insome typical applications on common TLC sorbents.
Solvents for TLC separations are selected for spe-cific chemical characteristics and solvent strength(a measure of interaction between solvent and sor-bent; see Sect. 27.4.1). In simple adsorption TLC, thehigher the solvent strength, the greater the Rf valueof the solute. An Rf value of 0.3–0.7 is typical. Mobilephases have been developed for the separation of var-ious compound classes on the different sorbents [seeTable 7.1 in reference (15)].
In addition to the sorbent and solvent, severalother factors must be considered when perform-ing planar chromatography. These include the typeof developing chamber used, vapor phase condi-tions (saturated vs. unsaturated), development mode

Chapter 27 • Basic Principles of Chromatography 479
27-2table Thin-Layer Chromatography Sorbents and Mode of Separation
Sorbent Chromatographic Mechanism Typical Application
Silica gel Adsorption Steroids, amino acids, alcohols,hydrocarbons, lipids, aflatoxins, bileacids, vitamins, alkaloids
Silica gel RP Reversed phase Fatty acids, vitamins, steroids, hormones,carotenoids
Cellulose, kieselguhr Partition Carbohydrates, sugars, alcohols, aminoacids, carboxylic acids, fatty acids
Aluminum oxide Adsorption Amines, alcohols, steroids, lipids,aflatoxins, bile acids, vitamins, alkaloids
PEI cellulosea Ion exchange Nucleic acids, nucleotides, nucleosides,purines, pyrimidines
Magnesium silicate Adsorption Steroids, pesticides, lipids, alkaloids
Reprinted from (15) by permission of Wiley, New York.aPEI cellulose refers to cellulose derivatized with polyethyleneimine (PEI).
(ascending, descending, horizontal, radial, etc.), anddevelopment distance. For additional reading refer toreferences (5), (7), and (16).
27.3.4.3 Column Liquid Chromatography
Column chromatography is the most useful methodof separating compounds in a mixture. Fractionationof solutes occurs as a result of differential migrationthrough a closed tube of stationary phase, and analytescan be monitored while the separation is in progress.In column liquid chromatography, the mobile phaseis liquid and the stationary phase can be either solidor liquid supported by an inert solid. A system forlow-pressure (i.e., performed at or near atmosphericpressure) column liquid chromatography is illustratedin Fig. 27-2.
Having selected a stationary and mobile phasesuitable for the separation problem at hand, the ana-lyst must first prepare the stationary phase (resin, gel,or packing material) for use according to the sup-plier’s instructions. (For example, the stationary phaseoften must be hydrated or preswelled in the mobilephase). The prepared stationary phase then is packedinto a column (usually glass), the length and diameterof which are determined by the amount of sample tobe loaded, the separation mode to be used, and thedegree of resolution required. Longer and narrowercolumns usually enhance resolution and separation.Adsorption columns may be either dry or wet packed;other types of columns are wet packed. The most com-mon technique for wet packing involves making aslurry of the adsorbent with the solvent and pouringthis into the column. As the sorbent settles, excesssolvent is drained off and additional slurry is added.
This process is repeated until the desired bed heightis obtained. (There is a certain art to pouring uniformcolumns and no attempt is made to give details here.)If the packing solvent is different from the initial elut-ing solvent, the column must be thoroughly washed(equilibrated) with the starting mobile phase.
The sample to be fractionated, dissolved in a min-imum volume of mobile phase, is applied in a layerat the top (or head) of the column. Classical or low-pressure chromatography utilizes only gravity flow ora peristaltic pump to maintain a flow of mobile phase(eluent or eluting solvent) through the column. In thecase of a gravity-fed system, eluent is simply siphonedfrom a reservoir into the column. The flow rate is gov-erned by the hydrostatic pressure, measured as thedistance between the level of liquid in the reservoirand the level of the column outlet. If eluent is fed to thecolumn by a peristaltic pump (see Fig. 27-2), then theflow rate is determined by the pump speed and, thus,regulation of hydrostatic pressure is not necessary.
The process of passing the mobile phase throughthe column is called elution, and the portion thatemerges from the outlet end of the column is some-times called the eluate (or effluent). Elution may beisocratic (constant mobile-phase composition) or agradient (changing the mobile phase, e.g., increas-ing solvent strength or pH) during elution in orderto enhance resolution and decrease analysis time (seealso Sect. 27.5.1). As elution proceeds, components ofthe sample are selectively retarded by the stationaryphase based on the strength of interaction with thestationary phase, and thus they are eluted at differ-ent times.
The column eluate may be directed through adetector and then into tubes, changed at intervals by

480 Part V • Chromatography
27-2f igure
A system for low-pressure column liquid chromatography. In this diagram, the column effluent is being splitbetween two detectors in order to monitor both enzyme activity (at Right) and UV absorption (at Left). The twotracings can be recorded simultaneously by using a dual-pen recorder. [Adapted from (12), with permission.]
a fraction collector. The detector response, in theform of an electrical signal, may be recorded (thechromatogram), using either a chart recorder or a com-puterized software, and used for qualitative or quan-titative analysis, as discussed in more detail later. Thefraction collector may be set to collect eluate at speci-fied time intervals or after a certain volume or numberof drops has been collected. Components of the sam-ple that have been chromatographically separated andcollected then can be further analyzed as needed.
27.3.5 Supercritical Fluid Chromatography
SFC refers to chromatography performed above thecritical pressure (Pc) and critical temperature (Tc) ofthe mobile phase. A supercritical fluid (or compressedgas) is neither a liquid nor a typical gas. The com-bination of Pc and Tc is known as the critical point.A supercritical fluid can be formed from a conven-tional gas by increasing the pressure, or from a con-ventional liquid by raising the temperature. Carbondioxide frequently is used as a mobile phase for SFC;however, it is not a good solvent for polar and high-molecular-weight compounds. A small amount of apolar, organic solvent such as methanol can be addedto a nonpolar supercritical fluid to enhance solute solu-bility, improve peak shape, and alter selectivity. Other
supercritical fluids that have been used in food appli-cations include nitrous oxide, trifluoromethane, sulfurhexafluoride, pentane, and ammonia.
Supercritical fluids confer chromatographic prop-erties intermediate to LC and GC. The high diffu-sivity and low viscosity of supercritical fluids meandecreased analysis times and improved resolutioncompared to LC. SFC offers a wide range of selectiv-ity (Sect. 27.5.2) adjustment, by changes in pressureand temperature as well as changes in mobile phasecomposition and the stationary phase. In addition,SFC makes possible the separation of nonvolatile,thermally labile compounds that are not amenableto GC.
SFC can be performed using either packedcolumns or capillaries. Packed column materials aresimilar to those used for HPLC. Small particle, porous,high surface area, hydrated silica may serve as thestationary phase itself, or simply as a support for abonded stationary phase (Chap. 28). Polymer-basedpacking has been used, but is less satisfactory owingto long solute retention times. Capillaries are generallycoated with a polysiloxane (−Si−O−Si) film, whichis then cross-linked to form a polymeric stationaryphase that cannot be washed off by the mobile phase.Polysiloxanes containing different functional groups,such as methyl, phenyl, or cyano, may be used to

Chapter 27 • Basic Principles of Chromatography 481
vary the polarity of this stationary phase. Instrumen-tation for packed column SFC is similar to that usedfor HPLC with one major difference: A back pres-sure regulator is used to control the outlet pressureof the system. Without this device, the fluid wouldexpand to a low-pressure, low-density gas. Besides theadvantages of decreased analysis time and improvedresolution, SFC offers the possibility to use a widevariety of detectors, including those designed for GC.
SFC has been used primarily for nonpolar com-pounds. Fats, oils, and other lipids are compounds towhich SFC is increasingly applied. For example, thenoncaloric fat substitute, Olestra R©, was characterizedby SFC-MS (mass spectroscopy). Other researchershave used SFC to detect pesticide residues, study ther-mally labile compounds from members of the Alliumgenus, fractionate citrus essential oils, and character-ize compounds extracted from microwave packaging(3). Borch-Jensen and Mollerup (1) highlighted the useof packed column and capillary SFC for the analysis offood and natural products, especially fatty acids andtheir derivatives, glycerides, waxes, sterols, fat-solublevitamins, carotenoids, and phospholipids.
27.4 PHYSICOCHEMICAL PRINCIPLESOF CHROMATOGRAPHIC SEPARATION
Several physicochemical principles (illustrated inFig. 27-3) are involved in chromatography mecha-nisms employed to separate or fractionate variouscompounds of interest, regardless of the specific tech-niques applied (discussed in Sect. 27.3). The mecha-nisms described below apply mainly to liquid chro-matography; GC mechanisms will be detailed inChap. 29. Although it is more convenient to describeeach of these phenomena separately, it must be empha-sized that more than one mechanism may be involvedin a given fractionation. For example, many cases ofpartition chromatography also involve adsorption.
27.4.1 Adsorption (Liquid–Solid)Chromatography
Adsorption chromatography is the oldest form ofchromatography, originated with Tsvet in 1903 in theexperiments that spawned modern chromatography.In this chromatographic mode, the stationary phaseis a finely divided solid to maximize the surface area.The stationary phase (adsorbent) is chosen to per-mit differential interaction with the components ofthe sample to be resolved. The intermolecular forcesthought to be primarily responsible for chromato-graphic adsorption include the following:
• Van der Waals forces• Electrostatic forces• Hydrogen bonds• Hydrophobic interactions
Sites available for interactionwith any given substanceare heterogeneous. Binding sites with greater affinities,the most active sites, tend to be populated first, so thatadditional solutes are less firmly bound. The net resultis that adsorption is a concentration-dependent pro-cess, and the adsorption coefficient is not a constant(in contrast to the partition coefficient). Sample loadsexceeding the adsorptive capacity of the stationaryphase will result in relatively poor separation.
Classic adsorption chromatography utilizes sil-ica (slightly acidic), alumina (slightly basic), charcoal(nonpolar), or a few other materials as the station-ary phase. Both silica and alumina possess surfacehydroxyl groups, and Lewis acid-type interactionsdetermine their adsorption characteristics. The elutionorder of compounds from these adsorptive stationaryphases can often be predicted on the basis of their rela-tive polarities (Table 27-3). Compounds with the mostpolar functional groups are retained most stronglyon polar adsorbents and, therefore, are eluted last.Nonpolar solutes are eluted first.
One model proposed to explain the mechanismof liquid–solid chromatography is that solute andsolvent molecules are competing for active sites onthe adsorbent. Thus, as relative adsorption of themobile phase increases, adsorption of the solute mustdecrease. Solvents can be rated in order of theirstrength of adsorption on a particular adsorbent, suchas silica. Such a solvent strength (or polarity) scaleis called a eluotropic series. A eluotropic series foralumina is listed in Table 27-4. Silica has a similarrank ordering. Once an adsorbent has been chosen,solvents can be selected from the eluotropic seriesfor that adsorbent. Mobile phase polarity can beincreased (often by admixture of more polar solvents)until elution of the compound(s) of interest has beenachieved.
Adsorption chromatography separates aromaticor aliphatic nonpolar compounds, based primarily onthe type and number of functional groups present.The labile, fat-soluble chlorophyll and carotenoid pig-ments from plants have been studied extensively byadsorption column chromatography. Adsorption chro-matography also has been used for the analysis offat-soluble vitamins. Frequently, it is used as a batchprocedure for removal of impurities from samplesprior to other analyses. For example, disposable solid-phase extraction cartridges (see Chap. 29) containingsilica have been used for food analyses, such as lipidsin soybean oil, carotenoids in citrus fruit, and vitaminE in grain.

482 Part V • Chromatography
27-3f igure
Physicochemical principles of chromatography. [From Quantitative Chemical Analysis, 5th ed., by D.C. Harris.c© 1999 by W.H. Freeman & Co. Used with permission.]
27.4.2 Partition (Liquid–Liquid)Chromatography
27.4.2.1 Introduction
In 1941, Martin and Synge undertook an investiga-tion of the amino acid composition of wool, using acountercurrent extractor (Sect. 27.2.3) of 40 tubes withchloroform and water flowing in opposite directions.The efficiency of the extraction process was improvedenormously when a column of finely divided inertsupport material was used to hold one liquid phase(stationary phase) immobile, while the second liquid,an immiscible solvent (mobile phase), flowed overit, thus providing intimate contact between the two
phases. Solutes partitioned between the two liquidphases according to their partition coefficients, hencethe name partition chromatography.
A partition system is manipulated by changing thenature of the two liquid phases, usually by combina-tion of solvents or pH adjustment of buffers. Often, themore polar of the two liquids is held stationary on theinert support and the less polar solvent is used to elutethe sample components (normal-phase chromatogra-phy). Reversal of this arrangement, using a nonpolarstationary phase and a polar mobile phase, has cometo be known as reversed-phase chromatography (seeSect. 27.4.2.3).

Chapter 27 • Basic Principles of Chromatography 483
27-3table Compounds Class Polarity Scalea
FluorocarbonsSaturated hydrocarbonsOlefinsAromaticsHalogenated compoundsEthersNitro compoundsEsters ≈ ketones ≈ aldehydesAlcohols ≈ aminesAmidesCarboxylic acids
From (9), used with permission.aListed in order of increasing polarity.
27-4table Eluotropic Series for Alumina
Solvent
1-PentaneIsooctaneCyclohexaneCarbon tetrachlorideXyleneTolueneBenzeneEthyl etherChloroformMethylene chlorideTetrahydrofuranAcetoneEthyl acetateAnilineAcetonitrile2-PropanolEthanolMethanolAcetic acid
From (9), used with permission.
Polar hydrophilic substances, such as aminoacids, carbohydrates, and water-soluble plant pig-ments, are separable by normal-phase partition chro-matography. Lipophilic compounds, such as lipidsand fat-soluble pigments, and polyphenols may beresolved with reversed-phase systems. Liquid–liquidpartition chromatography has been invaluable to car-bohydrate chemistry. Column liquid chromatographyon finely divided cellulose has been used extensivelyin preparative chromatography of sugars and theirderivatives. Paper chromatography (Sect. 27.3.4.1) isa simple method for distinguishing between vari-ous forms of sugars (following normal-phase partitionchromatography) or phenolic compounds (follow-ing reverse-phase partition chromatography) presentin foods.
27.4.2.2 Coated Supports
In its simplest form, the stationary phase for parti-tion chromatography consists of a liquid coating ona solid matrix. The solid support should be as inertas possible and have a large surface area in order tomaximize the amount of liquid held. Some examplesof solid supports that have been used are silica, starch,cellulose powder, and glass beads. All are capableof holding a thin film of water, which serves as thestationary phase. It is important to note that mate-rials prepared for adsorption chromatography mustbe activated by drying them to remove surface water.Conversely, some of these materials, such as silica gel,may be used for partition chromatography if they aredeactivated by impregnation with water or the desiredstationary phase. One disadvantage of liquid–liquidchromatographic systems is that the liquid stationaryphase is often stripped off. This problem can be over-come by chemically bonding the stationary phase tothe support material, as described in the next section.
27.4.2.3 Bonded Supports
The liquid stationary phase may be covalentlyattached to a support by a chemical reaction. Thesebonded phases have become very popular for HPLCuse, and a wide variety of both polar and nonpolarstationary phases is now available. Especially widelyused is reversed-phase HPLC (see Chap. 28), with anonpolar bonded stationary phase (e.g., silica coveredwith C8 or C18 groups) and a polar solvent (e.g., water–acetonitrile). It is important to note that mechanismsother than partition may be involved in the sepa-ration using bonded supports. Bonded-phase HPLCcolumns have greatly facilitated the analysis of vita-mins in foods and feeds, as discussed in Chap. 3 ofreference (10). Additionally, bonded-phase HPLC iswidely used for the separation and identification ofpolyphenols such as phenolic acids (e.g., p-coumaric,caffeic, ferulic, and sinapic acids) and flavonoids (e.g.,flavonols, flavones, isoflavones, anthocyanidins, cate-chins, and proanthocyanidins).
27.4.3 Ion-Exchange Chromatography
Ion exchange is a separation/purification processoccurring naturally, for example, in soils and is uti-lized in water softeners and deionizers. Three typesof separation may be achieved: (1) ionic from non-ionic, (2) cationic from anionic, and (3) mixtures ofsimilarly charged species. In the first two cases, onesubstance binds to the ion-exchange medium, whereasthe other substance does not. Batch extraction meth-ods can be used for these two separations; however,chromatography is needed for the third category.
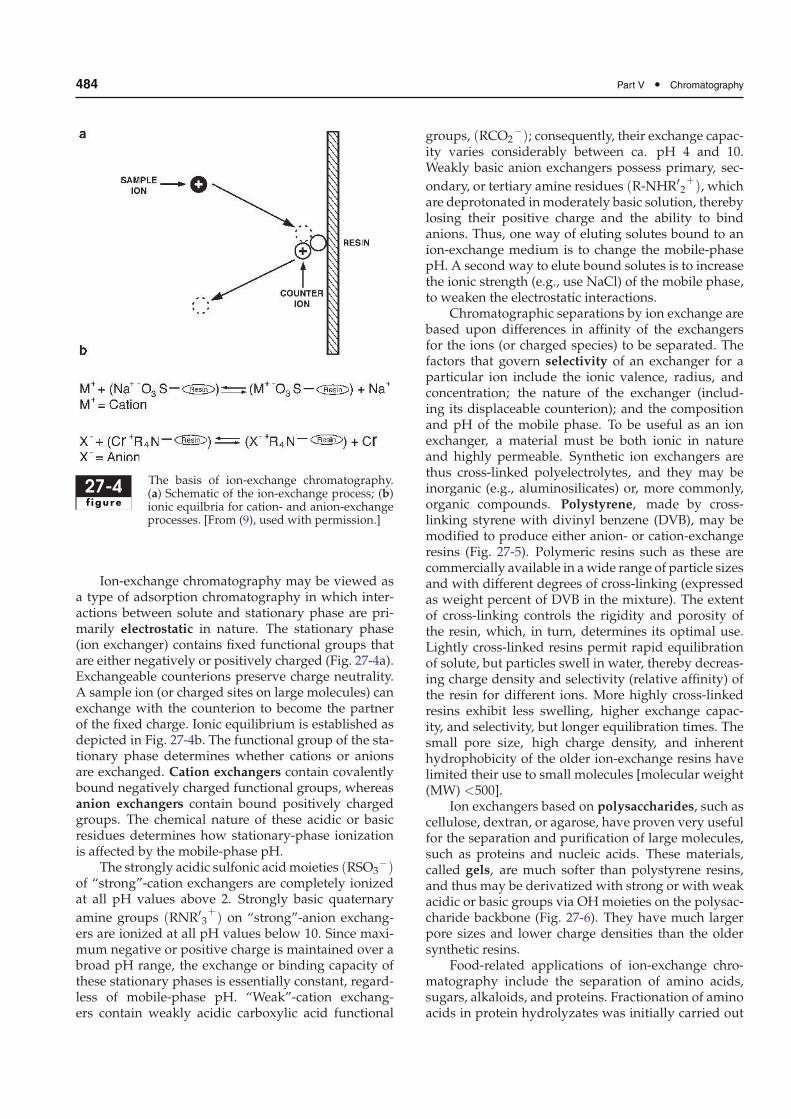
484 Part V • Chromatography
27-4f igure
The basis of ion-exchange chromatography.(a) Schematic of the ion-exchange process; (b)ionic equilbria for cation- and anion-exchangeprocesses. [From (9), used with permission.]
Ion-exchange chromatography may be viewed asa type of adsorption chromatography in which inter-actions between solute and stationary phase are pri-marily electrostatic in nature. The stationary phase(ion exchanger) contains fixed functional groups thatare either negatively or positively charged (Fig. 27-4a).Exchangeable counterions preserve charge neutrality.A sample ion (or charged sites on large molecules) canexchange with the counterion to become the partnerof the fixed charge. Ionic equilibrium is established asdepicted in Fig. 27-4b. The functional group of the sta-tionary phase determines whether cations or anionsare exchanged. Cation exchangers contain covalentlybound negatively charged functional groups, whereasanion exchangers contain bound positively chargedgroups. The chemical nature of these acidic or basicresidues determines how stationary-phase ionizationis affected by the mobile-phase pH.
The strongly acidic sulfonic acidmoieties (RSO3−)
of “strong”-cation exchangers are completely ionizedat all pH values above 2. Strongly basic quaternary
amine groups (RNR′3+) on “strong”-anion exchang-
ers are ionized at all pH values below 10. Since maxi-mum negative or positive charge is maintained over abroad pH range, the exchange or binding capacity ofthese stationary phases is essentially constant, regard-less of mobile-phase pH. “Weak”-cation exchang-ers contain weakly acidic carboxylic acid functional
groups, (RCO2−); consequently, their exchange capac-
ity varies considerably between ca. pH 4 and 10.Weakly basic anion exchangers possess primary, sec-
ondary, or tertiary amine residues (R-NHR′2+), which
are deprotonated inmoderately basic solution, therebylosing their positive charge and the ability to bindanions. Thus, one way of eluting solutes bound to anion-exchange medium is to change the mobile-phasepH. A second way to elute bound solutes is to increasethe ionic strength (e.g., use NaCl) of the mobile phase,to weaken the electrostatic interactions.
Chromatographic separations by ion exchange arebased upon differences in affinity of the exchangersfor the ions (or charged species) to be separated. Thefactors that govern selectivity of an exchanger for aparticular ion include the ionic valence, radius, andconcentration; the nature of the exchanger (includ-ing its displaceable counterion); and the compositionand pH of the mobile phase. To be useful as an ionexchanger, a material must be both ionic in natureand highly permeable. Synthetic ion exchangers arethus cross-linked polyelectrolytes, and they may beinorganic (e.g., aluminosilicates) or, more commonly,organic compounds. Polystyrene, made by cross-linking styrene with divinyl benzene (DVB), may bemodified to produce either anion- or cation-exchangeresins (Fig. 27-5). Polymeric resins such as these arecommercially available in awide range of particle sizesand with different degrees of cross-linking (expressedas weight percent of DVB in the mixture). The extentof cross-linking controls the rigidity and porosity ofthe resin, which, in turn, determines its optimal use.Lightly cross-linked resins permit rapid equilibrationof solute, but particles swell in water, thereby decreas-ing charge density and selectivity (relative affinity) ofthe resin for different ions. More highly cross-linkedresins exhibit less swelling, higher exchange capac-ity, and selectivity, but longer equilibration times. Thesmall pore size, high charge density, and inherenthydrophobicity of the older ion-exchange resins havelimited their use to small molecules [molecular weight(MW) <500].
Ion exchangers based on polysaccharides, such ascellulose, dextran, or agarose, have proven very usefulfor the separation and purification of large molecules,such as proteins and nucleic acids. These materials,called gels, are much softer than polystyrene resins,and thus may be derivatized with strong or with weakacidic or basic groups via OH moieties on the polysac-charide backbone (Fig. 27-6). They have much largerpore sizes and lower charge densities than the oldersynthetic resins.
Food-related applications of ion-exchange chro-matography include the separation of amino acids,sugars, alkaloids, and proteins. Fractionation of aminoacids in protein hydrolyzates was initially carried out

Chapter 27 • Basic Principles of Chromatography 485
27-5f igure
Chemical structure of polystyrene-based ion-exchange resins.
by ion-exchange chromatography; automation of thisprocess led to the development of commercially pro-duced amino acid analyzers (see Chap. 15). Manydrugs, fatty acids, and the acids of fruit, being ion-izable compounds, may be chromatographed in theion-exchange mode. For additional details on the prin-ciples and applications of ion chromatography pleaserefer to reference (18).
27.4.4 Size-Exclusion Chromatography
Size-exclusion chromatography (SEC), also knownas molecular exclusion, gel permeation (GPC), andgel-filtration chromatography (GFC), is probably theeasiest mode of chromatography to perform and tounderstand. It is widely used in the biological sciences
for the resolution of macromolecules, such as pro-teins and carbohydrates, and also is used for thefractionation and characterization of synthetic poly-mers. Unfortunately, nomenclature associated withthis separation mode developed independently in theliterature of the life sciences and in the field of polymerchemistry, resulting in inconsistencies.
In the ideal SEC system, molecules are sepa-rated solely on the basis of their size; no interactionoccurs between solutes and the stationary phase. Inthe event that solute/support interactions do occur,the separation mode is termed nonideal SEC. Thestationary phase in SEC consists of a column pack-ing material that contains pores comparable in sizeto the molecules to be fractionated. Solutes too largeto enter the pores travel with the mobile phase in
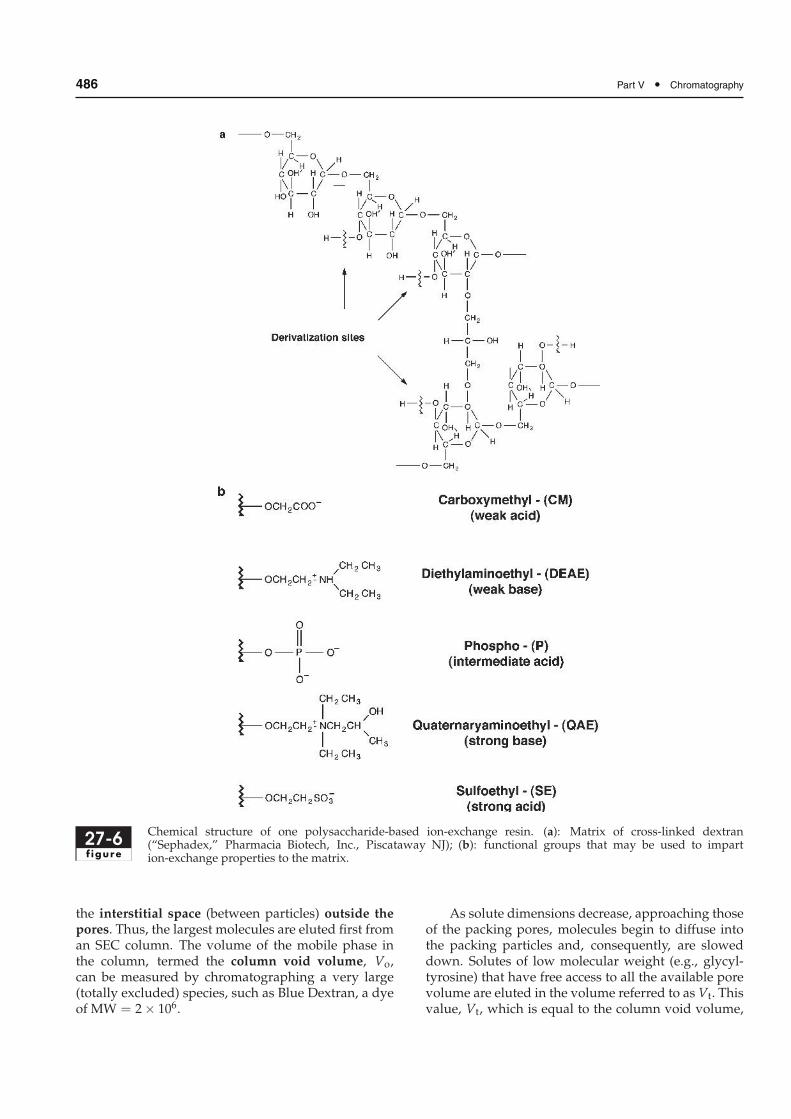
486 Part V • Chromatography
27-6f igure
Chemical structure of one polysaccharide-based ion-exchange resin. (a): Matrix of cross-linked dextran(“Sephadex,” Pharmacia Biotech, Inc., Piscataway NJ); (b): functional groups that may be used to impartion-exchange properties to the matrix.
the interstitial space (between particles) outside thepores. Thus, the largest molecules are eluted first froman SEC column. The volume of the mobile phase inthe column, termed the column void volume, Vo,can be measured by chromatographing a very large(totally excluded) species, such as Blue Dextran, a dyeof MW = 2× 106.
As solute dimensions decrease, approaching thoseof the packing pores, molecules begin to diffuse intothe packing particles and, consequently, are sloweddown. Solutes of low molecular weight (e.g., glycyl-tyrosine) that have free access to all the available porevolume are eluted in the volume referred to as Vt. Thisvalue, Vt, which is equal to the column void volume,

Chapter 27 • Basic Principles of Chromatography 487
Vo, plus the volume of liquid inside the sorbent pores,Vi, is referred to as the total permeation volume ofthe packed column (Vt = Vo + Vi). These relation-ships are illustrated in Fig. 27-7. Solutes are ideallyeluted between the void volume and the total liquidvolume of the column. Because this volume is lim-ited, only a relatively small number of solutes (ca. 10)can be completely resolved by SEC under ordinaryconditions.
The behavior of a molecule in a size-exclusion col-umn may be characterized in several different ways.Each solute exhibits an elution volume, Ve, as illus-trated in Fig. 27-7. However, Ve depends on columndimensions and the way in which the column waspacked. The available partition coefficient is used to
27-7f igure
Schematic elution profile illustrating some ofthe terms used in size-exclusion chromatogra-phy. [Adapted from (8), p. A271, with kindpermission from Elsevier Science – NL, SaraBurgerhartstraat 25, 1055KV Amsterdam, TheNetherlands.]
define solute behavior independent of these variables:
Kav = (Ve − Vo)/ (Vt−Vo) [3]
where:
Kav = available partition coefficient
Ve = elution volume of solute
Vo = column void volume
Vt = total permeation volume of column
The value of Kav calculated from experimental datafor a solute chromatographed on a given SEC columndefines the proportion of pores that can be occupiedby that molecule. For a large, totally excluded species,such as Blue Dextran or DNA, Ve = Vo and Kav = 0.For a small molecule with complete access to the inter-nal pore volume, such as glycyltyrosine, Ve = Vt andKav = 1.
For each size-exclusion packing material, a plotof Kav vs. the logarithm of the molecular weight fora series of solutes, similar in molecular shape anddensity, will give an S-shaped curve (Fig. 27-8). Inthe case of proteins, Kav is actually better related tothe Stokes radius, the average radius of the pro-tein in solution. The central, linear portion of thiscurve describes the fractionation range of the matrix,wherein maximum separation among solutes of sim-ilar molecular weight is achieved. This correlationbetween solute elution behavior andmolecular weight(or size) forms the basis for a widely used method forcharacterizing large molecules such as proteins andpolysaccharides. A size-exclusion column is calibratedwith a series of solutes of known molecular weight (orStokes radius) to obtain a curve similar to that shownin Fig. 27-8. The value of Kav for the unknown is thendetermined, and an estimate of molecular weight (orsize) of the unknown is made by interpolation of thecalibration curve.
27-8f igure
Relationship between Kav and log (molecular weight) for globular proteins chromatographed on a column ofSephadex G-150 Superfine. (Reproduced by permission of Pharmacia Biotech, Inc., Piscataway, NJ.)

488 Part V • Chromatography
Column packing materials for SEC can be dividedinto two groups: semirigid, hydrophobic media, andsoft, hydrophilic gels. The former are usually derivedfrom polystyrene and are used with organic mobilephases (GPC or nonaqueous SEC) for the separa-tion of polymers, such as rubbers and plastics. Softgels, polysaccharide-based packings, are typified bySephadex, a cross-linked dextran (see Fig. 27-6a).These materials are available in a wide range of poresizes and are useful for the separation of water-solublesubstances in the molecular weight range 1–2.5× 107.In selecting an SEC column packing, both the pur-pose of the experiment and size of the molecules to beseparated must be considered. If the purpose of theexperiment is group separation, where molecules ofwidely different molecular sizes need to be separated,a matrix is chosen such that the larger molecules, e.g.,proteins, are eluted in the void volume of the col-umn, whereas small molecules are retained in the totalvolume. A common example of group separation isbuffer exchange and desalting. When SEC is used forseparation of macromolecules of different sizes, themolecular sizes of all the components must fall withinthe fractionation range of the gel.
As discussed previously, SEC can be used, directly,to fractionatemixtures or, indirectly, to obtain informa-tion about a dissolved species. In addition to molecu-lar weight estimations, SEC is used to determine themolecular weight distribution of natural and syntheticpolymers, such as dextrans and gelatin preparations.Fractionation of biopolymer mixtures is probably themost widespread use of SEC, since the mild elu-tion conditions employed rarely cause denaturationor loss of biological activity. It is also a fast, effi-cient alternative to dialysis for desalting solutions ofmacromolecules, such as proteins.
27.4.5 Affinity Chromatography
Affinity chromatography is unique in that separationis based on the specific, reversible interaction betweena solute molecule and a ligand immobilized on thechromatographic stationary phase. While discussedhere as a separate type of chromatography, affin-ity chromatography could be viewed as the ultimateextension of adsorption chromatography. Althoughthe basic concepts of so-called biospecific adsorptionwere known as early as 1910, they were not perceivedas potentially useful laboratory tools until ca. 1968.
Affinity chromatography usually involves immo-bilized biological materials as the stationary phase.These ligands can be antibodies, enzyme inhibitors,lectins, or other molecules that selectively andreversibly bind to complementary analyte moleculesin the sample. Separation exploits the lock and key
binding of biological systems. Although both ligandsand the species to be isolated are usually biologi-cal macromolecules, the term affinity chromatographyalso encompasses other systems, such as separation ofsmall molecules containing cis-diol groups via phenyl-boronic acid moieties on the stationary phase.
The principles of affinity chromatography areillustrated in Fig. 27-9. A ligand, chosen based onits specificity and strength of interaction with themolecule to be isolated (analyte), is immobilized ona suitable support material. As the sample is passedthrough this column, molecules that are complemen-tary to the bound ligand are adsorbed while othersample components are eluted. Bound analyte is sub-sequently eluted via a change in the mobile-phasecomposition as will be discussed below. After reequi-libration with the initial mobile phase, the stationaryphase is ready to be used again. The ideal support for
27-9f igure
Principles of bioselective affinity chromatog-raphy. (a) The support presents the immobi-lized ligand to the analyte to be isolated. (b)The analyte makes contact with the ligand andattaches itself. (c) The analyte is recovered bythe introduction of an eluent, which dissoci-ates the complex holding the analyte to theligand. (d) The support is regenerated, readyfor the next isolation. [Reprinted from (8),p. A311, with kind permission from ElsevierScience NL, Sara Burgerhartstraat 25, 1055KVAmsterdam, The Netherlands.]

Chapter 27 • Basic Principles of Chromatography 489
affinity chromatography should be a porous, stable,high-surface-area material that does not adsorb any-thing itself. Thus, polymers such as agarose, cellulose,dextran, and polyacrylamide are used, as well ascontrolled-pore glass.
Affinity ligands are usually attached to the sup-port or matrix by covalent bond formation, and opti-mum reaction conditions often must be found empir-ically. Immobilization generally consists of two steps:activation and coupling. During the activation step, areagent reacts with functional groups on the support,such as hydroxyl moieties, to produce an activatedmatrix. After removal of excess reagent, the ligandis coupled to the activated matrix. (Preactivated sup-ports are commercially available, and their availabilityhas greatly increased the use of affinity chromatogra-phy.) The coupling reaction most often involves freeamino groups on the ligand, although other functionalgroups can be used. When small molecules such asphenylboronic acid are immobilized, a spacer arm(containing at least four to six methylene groups) isused to hold the ligand away from the support surface,enabling it to reach into the binding site of the analyte.
Ligands for affinity chromatographymay be eitherspecific or general (i.e., group specific). Specific lig-ands, such as antibodies, bind only one particularsolute. General ligands, such as nucleotide analogsand lectins, bind to certain classes of solutes. For exam-ple, the lectin concanavalin A binds to all moleculesthat contain terminal glucosyl and mannosyl residues.Bound solutes then can be separated as a group orindividually, depending upon the elution techniqueused. Some of the more common general ligands arelisted in Table 27-5. Although less selective, generalligands provide greater convenience.
Elution methods for affinity chromatography maybe divided into nonspecific and (bio)specific methods.Nonspecific elution involves disrupting ligand ana-lyte binding by changing the mobile-phase pH, ionicstrength, dielectric constant, or temperature. If addi-tional selectivity in elution is desired, for example, inthe case of immobilized general ligands, a biospecificelution technique is used. Free ligand, either identicalto or different from the matrix-bound ligand, is addedto the mobile phase. This free ligand competes forbinding sites on the analyte. For example, glycopro-teins bound to a concanavalin A (lectin) column can beeluted by using buffer containing an excess of lectin.In general, the eluent ligand should display greateraffinity for the analyte of interest than the immobilizedligand.
In addition to protein purification, affinity chro-matography may be used to separate supramolecularstructures such as cells, organelles, and viruses; con-centrate dilute protein solutions; investigate bindingmechanisms; and determine equilibrium constants.
27-5table
General Affinity Ligands and Their
Specificities
Ligand Specificity
Cibacron Blue F3G-A dye,derivatives of AMP,NADH, and NADPH
Certain dehydrogenases viabinding at the nucelotidebinding site
Concanavalin A, lentil lectin,wheat-germ lectin
Polysaccharides,glycoproteins, glycolipids,and membrane proteinscontaining sugar residuesof certain configurations
Soybean trypsin inhibitor,methyl esters of variousamino acids, D-aminoacids
Various proteases
Phenylboronic acid Glycosylated hemoglobins,sugars, nucleic acids, andother cis-diol-containingsubstances
Protein A Many immunoglobulinclasses and subclassesvia binding to the Fc
regionDNA, RNA, nucleosides,
nucleotidesNucleases, polymerases,
nucleic acids
Reprinted with permission from (17). Copyright 1985 American
Chemical Society.
Affinity chromatography has been useful especiallyin the separation and purification of enzymes andglycoproteins. In the case of the latter, carbohydrate-derivatized adsorbents are used to isolate specificlectins, such as concanavalin A, and lentil or wheat-germ lectin. The lectin thenmay be coupled to agarose,such as concanavalin A- or lentil lectin-agarose, to pro-vide a stationary phase for the purification of specificglycoproteins, glycolipids, or polysaccharides. Foradditional details on affinity chromatography pleaserefer to reference (19).
27.5 ANALYSIS OF CHROMATOGRAPHICPEAKS
Once the chromatographic technique (Sect. 27.3) andchromatographic mechanism (Sect. 27.4) have beenchosen, the analyst has to ensure adequate separationof constituents of interests from a mixture, in a rea-sonable amount of time. After separation is achievedand chromatographic peaks are obtained, qualitativeas well as quantitative analysis can be carried out.Basic principles of separation and resolution will bediscussed in the subsequent sections. Understand-ing these principles allows the analyst to optimizeseparation and perform qualitative and quantitativeanalysis.

490 Part V • Chromatography
27.5.1 Separation and Resolution
This section will discuss separation and resolution asit pertains mainly to LC; separation and resolutionoptimization as it pertains specifically to GC will bediscussed in Chap. 29.
27.5.1.1 Developing a Separation
There may be numerous ways to accomplish a chro-matographic separation for a particular compound. Inmany cases, the analyst will follow a standard labora-tory procedure or published methods. In the case ofa sample that has not been previously analyzed, theanalyst begins by evaluating what is known about thesample and defines the goals of the separation. Howmany components need to be resolved? What degreeof resolution is needed? Is qualitative or quantitativeinformation needed? Molecular weight (or molecu-lar weight range), polarity, and ionic character of thesample will guide the choice of chromatographic sep-aration mechanism (separation mode). Figure 27-10shows that more than one correct choice may be pos-sible. For example, small ionic compounds may beseparated by ion-exchange, ion-pair reversed-phase
(see Sect. 28.3.2.1), or reversed-phase LC. In this case,the analyst’s choice may be based on convenience,experience, and personal preference.
Having chosen a separation mode for the sam-ple at hand, one must select an appropriate stationaryphase, elution conditions, and a detection method.Trial experimental conditions may be based on theresults of a literature search, the analyst’s previousexperience with similar samples, or general recom-mendations from chromatography experts.
To achieve separation of sample components by allmodes except SEC, one may utilize either isocratic orgradient elution. Isocratic elution is the most simpleand widely used technique, in which solvent com-position and flow rate are held constant. Gradientelution involves reproducibly varying mobile phasecomposition or flow rate (flow programming) duringthe LC analysis. Gradient elution is used when sam-ple components possess a wide range of polarities, sothat an isocratic mobile phase does not elute all com-ponents within a reasonable time. The change maybe continuous or stepwise. Gradients of increasingionic strength are extremely valuable in ion-exchangechromatography (see Sect. 27.4.3). Gradient elution iscommonly used for desorbing large molecules, such as
27-10f igure
A schematic for choosing a chromatographic separation mode based on sample molecular weight and solubility.[From (11), used with permission.]
Chapter 27 • Basic Principles of Chromatography 491
proteins, which can undergo multiple-site interactionwith a stationary phase. Increasing the “strength” ofthe mobile phase (Sect. 27.4.1), either gradually or in astepwise fashion, shortens the analysis time.
Method development may begin with an iso-cratic mobile phase, possibly of intermediate solventstrength; however, using gradient elution for the initialseparation may ensure that some level of separation isachieved within a reasonable time period and nothingis likely to remain on the column. Data from this initialrun allow one to determine if isocratic or gradient elu-tion is needed, and to estimate optimal isocraticmobilephase composition or gradient range. The use of a gra-dient run does not presuppose that the final methodwill use gradient elution.
Once an initial separation has been achieved, theanalyst can proceed to optimize resolution. This gener-ally involves manipulation of mobile phase variables,including the nature and percentage of organic compo-nents, pH, ionic strength, nature and concentration ofadditives (such as ion-pairing agents), flow rate, andtemperature. In the case of gradient elution, gradientsteepness (slope) is another variable to be optimized.However, the analyst must be aware of the principlesof chromatographic resolution as will be discussed inthe following section.
27.5.1.2 Chromatographic Resolution
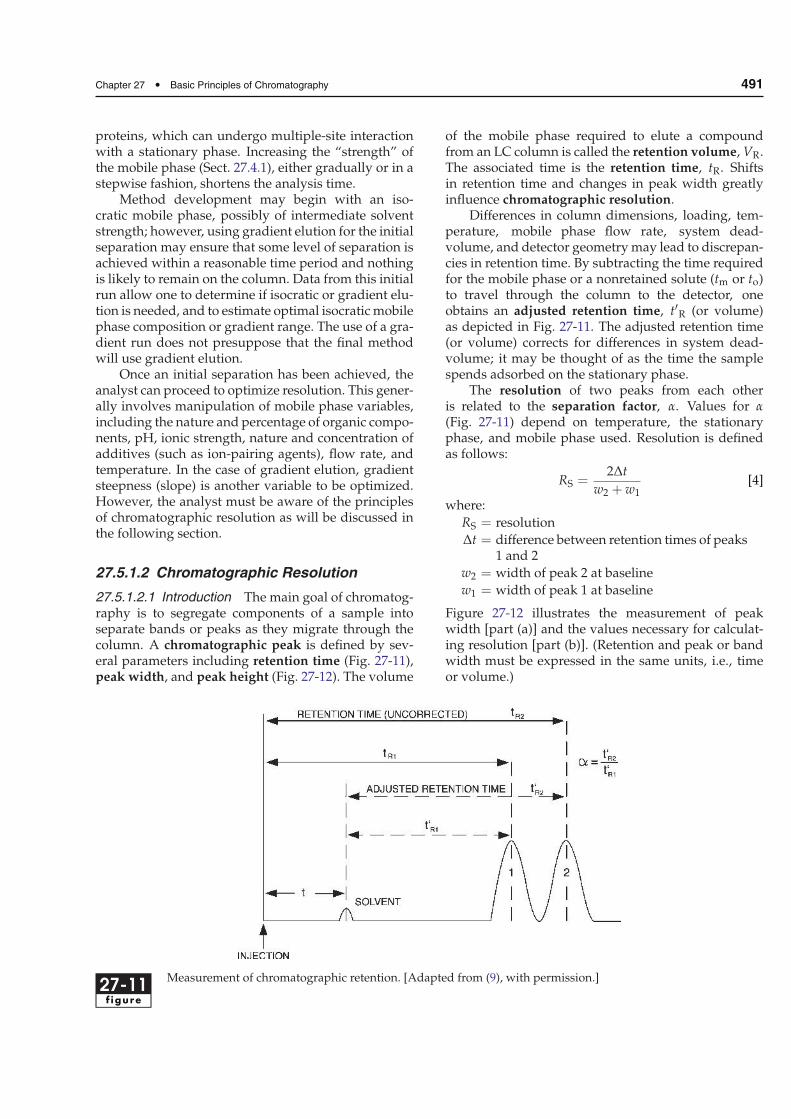
27.5.1.2.1 Introduction The main goal of chromatog-raphy is to segregate components of a sample intoseparate bands or peaks as they migrate through thecolumn. A chromatographic peak is defined by sev-eral parameters including retention time (Fig. 27-11),peak width, and peak height (Fig. 27-12). The volume
of the mobile phase required to elute a compoundfrom an LC column is called the retention volume, VR.The associated time is the retention time, tR. Shiftsin retention time and changes in peak width greatlyinfluence chromatographic resolution.
Differences in column dimensions, loading, tem-perature, mobile phase flow rate, system dead-volume, and detector geometrymay lead to discrepan-cies in retention time. By subtracting the time requiredfor the mobile phase or a nonretained solute (tm or to)to travel through the column to the detector, oneobtains an adjusted retention time, t′R (or volume)as depicted in Fig. 27-11. The adjusted retention time(or volume) corrects for differences in system dead-volume; it may be thought of as the time the samplespends adsorbed on the stationary phase.
The resolution of two peaks from each otheris related to the separation factor, α. Values for α
(Fig. 27-11) depend on temperature, the stationaryphase, and mobile phase used. Resolution is definedas follows:
RS =2∆t
w2 + w1[4]
where:
RS = resolution
∆t = difference between retention times of peaks1 and 2
w2 = width of peak 2 at baseline
w1 = width of peak 1 at baseline
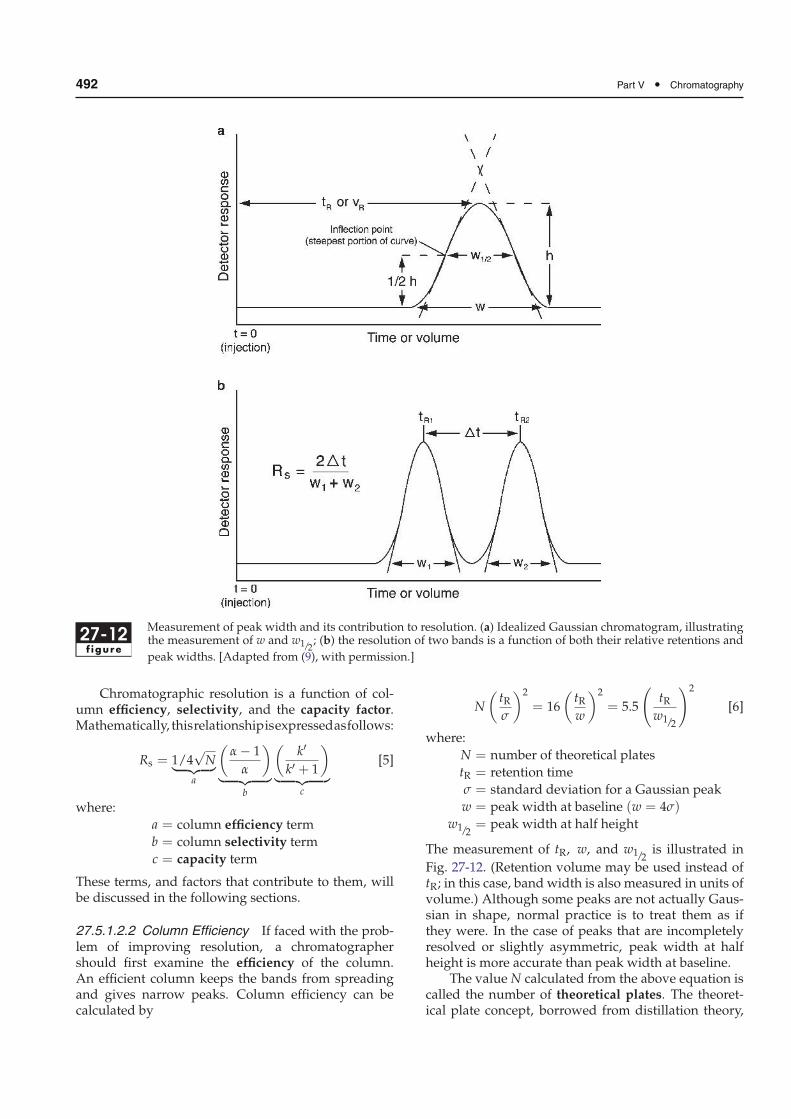
Figure 27-12 illustrates the measurement of peakwidth [part (a)] and the values necessary for calculat-ing resolution [part (b)]. (Retention and peak or bandwidth must be expressed in the same units, i.e., timeor volume.)
27-11f igure
Measurement of chromatographic retention. [Adapted from (9), with permission.]
492 Part V • Chromatography
27-12f igure
Measurement of peak width and its contribution to resolution. (a) Idealized Gaussian chromatogram, illustratingthe measurement of w and w1/2
; (b) the resolution of two bands is a function of both their relative retentions and
peak widths. [Adapted from (9), with permission.]
Chromatographic resolution is a function of col-umn efficiency, selectivity, and the capacity factor.Mathematically, thisrelationshipisexpressedasfollows:
Rs = 1/4√
N︸ ︷︷ ︸
a
(α − 1
α
)
︸ ︷︷ ︸
b
(k′
k′ + 1
)
︸ ︷︷ ︸
c
[5]
where:
a = column efficiency term
b = column selectivity term
c = capacity term
These terms, and factors that contribute to them, willbe discussed in the following sections.
27.5.1.2.2 Column Efficiency If faced with the prob-lem of improving resolution, a chromatographershould first examine the efficiency of the column.An efficient column keeps the bands from spreadingand gives narrow peaks. Column efficiency can becalculated by
N
(tRσ
)2
= 16
(tRw
)2
= 5.5
(
tRw1/2
)2
[6]
where:
N = number of theoretical plates
tR = retention time
σ = standard deviation for a Gaussian peak
w = peak width at baseline (w = 4σ)w1/2
= peak width at half height
The measurement of tR, w, and w1/2is illustrated in
Fig. 27-12. (Retention volume may be used instead oftR; in this case, band width is also measured in units ofvolume.) Although some peaks are not actually Gaus-sian in shape, normal practice is to treat them as ifthey were. In the case of peaks that are incompletelyresolved or slightly asymmetric, peak width at halfheight is more accurate than peak width at baseline.
The value N calculated from the above equation iscalled the number of theoretical plates. The theoret-ical plate concept, borrowed from distillation theory,

Chapter 27 • Basic Principles of Chromatography 493
can best be understood by viewing chromatography asa series of equilibrations between mobile and station-ary phases, analogous to countercurrent distribution.Thus, a column would consist of N segments (the-oretical plates) with one equilibration occurring ineach. As a first approximation, N is independent ofretention time and is therefore a useful measure ofcolumn performance. One method of monitoring col-umn performance over time is to chromatograph astandard compound periodically, under constant con-ditions, and to compare the values of N obtained. It isimportant to note that columns often behave as if theyhave a different number of plates for different solutesin a mixture. Different solutes have different partitioncoefficient and thus have distinctive series of equili-brations between mobile and stationary phases. Bandbroadening due to column deterioration will result ina decrease of N for a particular solute. Band broaden-ing is a result of an extended time for a solute to reachequilibrium between mobile and stationary phases.
The number of theoretical plates is generally pro-portional to column length. Since columns are avail-able in various lengths, it is useful to have a measureof column efficiency that is independent of columnlength. This may be expressed as follows:
HETP =L
N[7]
where:
HETP = height equivalent to a theoretical plate
L = column length
N = number of theoretical plates
The so-called HETP is sometimes more simplydescribed as plate height (H). If a column consisted ofdiscrete segments, HETP would be the height of eachimaginary segment. Small plate height values (a largenumber of plates) indicate good efficiency of separa-tion. Conversely, reduced number of plates results inpoor separation due to the extended equilibrium timein a deteriorating column.
In reality, columns are not divided into discretesegments and equilibration is not infinitely fast. Theplate theory is used to simplify the equilibration con-cept. Themovement of solutes through a chromatogra-phy column takes into account the finite rate at whicha solute can equilibrate itself between stationary andmobile phases. Thus, band shape depends on the rateof elution and is affected by solute diffusion. Anymechanism that causes a band of solute to broadenwill increase HETP and decrease column efficiency.The various factors that contribute to plate height areexpressed by the Van Deemter equation:
HETP = A +B
u+Cu [8]
where:
HETP = height equivalent to a theoretical plate
A, B, C = constants
u = mobile phase rate
The constants A, B, and C are characteristic for a givencolumn, mobile phase, and temperature. The A termrepresents the eddy diffusion or multiple flowpaths.Eddy diffusion refers to the different microscopic flow-streams that the mobile phase can take between parti-cles in the column (analogous to eddy streams aroundrocks in a brook). Sample molecules can thus take dif-ferent paths as well, depending on which flowstreamsthey follow. As a result, solute molecules spread froman initially narrow band to a broader area withinthe column. Eddy diffusion may be minimized bygood column packing techniques and the use of smalldiameter particles of narrow particle size distribution.
The B term of the Van Deemter equation, some-times called the longitudinal diffusion term, existsbecause all solutes diffuse from an area of high con-centration (the center of a chromatographic band) toone of low concentration (the leading or trailing edgeof a chromatographic band). In LC, the contribution ofthis term to HETP is small except at low flow rate ofthe mobile phase. With slow flow rates there will bemore time for a solute to spend on the column, thus itsdiffusion will be greater.
The C (mass transfer) term arises from the finitetime required for solute to equilibrate between themobile and stationary phases. Mass transfer is prac-tically the partitioning of the solute into the station-ary phase, which does not occur instantaneously anddepends on the solute’s partition and diffusion coef-ficients. If the stationary phase consists of porousparticles (see Chap. 28, Sect. 28.2.3.2, Fig. 28-3), asample molecule entering a pore ceases to be trans-ported by the solvent flow and moves by diffusiononly. Subsequently, this solute molecule may diffuseback to the mobile phase flow or it may interact withthe stationary phase. In either case, solute moleculesinside the pores are slowed down relative to thoseoutside the pores and band broadening occurs. Contri-butions to HETP from the C term can be minimized byusing porous particles of small diameter or pellicularpacking materials (Chap. 28, Sect. 28.2.3.2.2).
As expressed by the Van Deemter equation,mobile phase flow rate, u, contributes to plate heightin opposing ways – increasing the flow rate increases
the equilibration point (Au1/3 and Cu), but decreaseslongitudinal diffusion of the solute particles (B/u).A Van Deemter plot (Fig. 27-13) may be used todetermine the mobile phase flow rate at which plateheight is minimized and column efficiency is maxi-mized. Flow rates above the optimum may be usedto decrease analysis time if adequate resolution is still
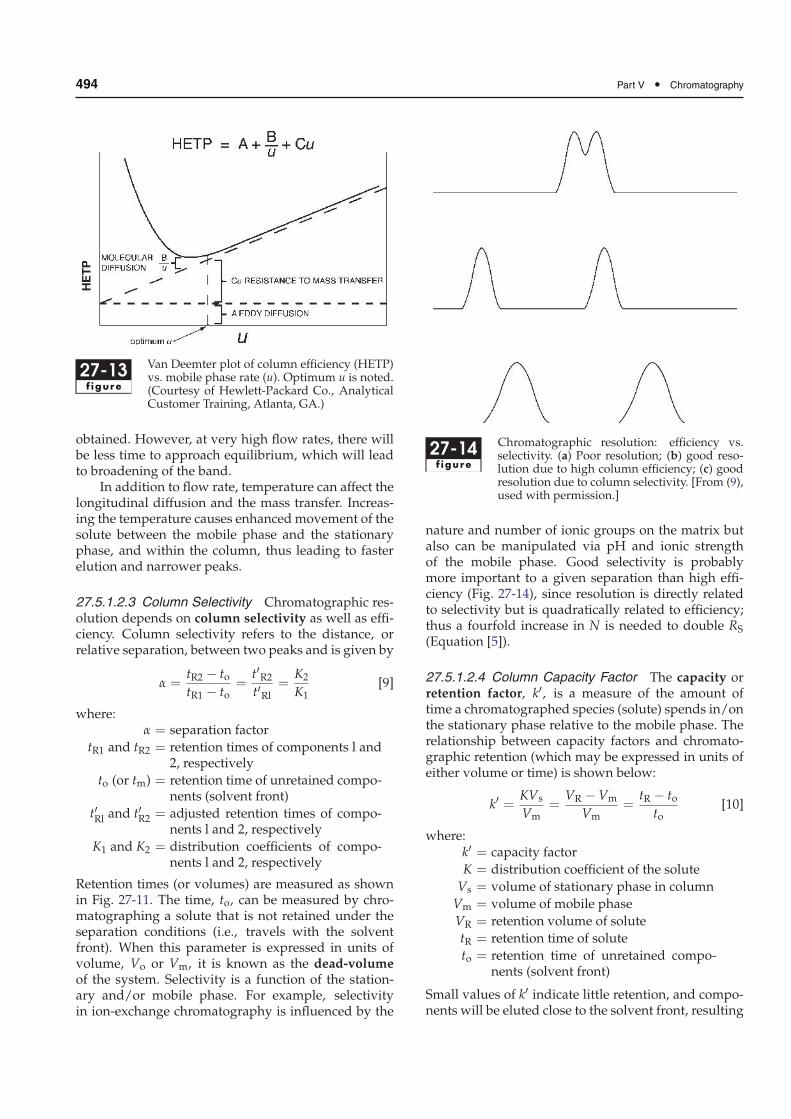
494 Part V • Chromatography
27-13f igure
Van Deemter plot of column efficiency (HETP)vs. mobile phase rate (u). Optimum u is noted.(Courtesy of Hewlett-Packard Co., AnalyticalCustomer Training, Atlanta, GA.)
obtained. However, at very high flow rates, there willbe less time to approach equilibrium, which will leadto broadening of the band.
In addition to flow rate, temperature can affect thelongitudinal diffusion and the mass transfer. Increas-ing the temperature causes enhancedmovement of thesolute between the mobile phase and the stationaryphase, and within the column, thus leading to fasterelution and narrower peaks.
27.5.1.2.3 Column Selectivity Chromatographic res-olution depends on column selectivity as well as effi-ciency. Column selectivity refers to the distance, orrelative separation, between two peaks and is given by
α =tR2 − totR1 − to
=t′R2t′Rl
=K2
K1[9]
where:α = separation factor
tR1 and tR2 = retention times of components l and2, respectively
to (or tm) = retention time of unretained compo-nents (solvent front)
t′Rl and t′R2 = adjusted retention times of compo-nents l and 2, respectively
K1 and K2 = distribution coefficients of compo-nents l and 2, respectively
Retention times (or volumes) are measured as shownin Fig. 27-11. The time, to, can be measured by chro-matographing a solute that is not retained under theseparation conditions (i.e., travels with the solventfront). When this parameter is expressed in units ofvolume, Vo or Vm, it is known as the dead-volumeof the system. Selectivity is a function of the station-ary and/or mobile phase. For example, selectivityin ion-exchange chromatography is influenced by the
27-14f igure
Chromatographic resolution: efficiency vs.selectivity. (a) Poor resolution; (b) good reso-lution due to high column efficiency; (c) goodresolution due to column selectivity. [From (9),used with permission.]
nature and number of ionic groups on the matrix butalso can be manipulated via pH and ionic strengthof the mobile phase. Good selectivity is probablymore important to a given separation than high effi-ciency (Fig. 27-14), since resolution is directly relatedto selectivity but is quadratically related to efficiency;thus a fourfold increase in N is needed to double RS
(Equation [5]).
27.5.1.2.4 Column Capacity Factor The capacity orretention factor, k′, is a measure of the amount oftime a chromatographed species (solute) spends in/onthe stationary phase relative to the mobile phase. Therelationship between capacity factors and chromato-graphic retention (which may be expressed in units ofeither volume or time) is shown below:
k′ =KVs
Vm=
VR − Vm
Vm=
tR − toto
[10]
where:k′ = capacity factor
K = distribution coefficient of the solute
Vs = volume of stationary phase in column
Vm = volume of mobile phase
VR = retention volume of solute
tR = retention time of solute
to = retention time of unretained compo-nents (solvent front)
Small values of k′ indicate little retention, and compo-nents will be eluted close to the solvent front, resulting

Chapter 27 • Basic Principles of Chromatography 495
in poor separations. Overuse or misuse of the columnmay lead to the loss of some functional groups, thusresulting in small k′ values. Large values of k′ result inimproved separation but also can lead to broad peaksand long analysis times. On a practical basis, k′ val-ues within the range of 1–15 are generally used. (In theequation for Rs, k′ is actually the average of k1
′ and k2′
for the two components separated.)
27.5.2 Qualitative Analysis
Once separation and resolution have been optimized,identification of the detected compounds can beachieved. (Various detection methods are outlinedin Chaps. 28 and 29.) Comparing VR or tR to thatof standards chromatographed under identical con-ditions often enables one to identify an unknowncompound. When it is necessary to compare chro-matograms obtained from two different systems orcolumns, it is better to compare adjusted retentiontime, t′R (see Sect. 27.5.2.2). Different compounds mayhave identical retention times. In other words, even ifthe retention time of an unknown and a standard areequivalent, the two compounds might not be identi-cal. Therefore, other techniques are needed to confirmpeak identity. For example:
1. Spike the unknown sample with a known com-pound and compare chromatograms of theoriginal and spiked samples to see which peakhas increased. Only the height of the peak ofinterest should increase, with no change inretention time, peak width, or shape.
2. A diode array detector can provide absorptionspectra of designated peaks (see Sects. 23.2.6and 28.2.4.1). Although identical spectra do notprove identity, a spectral difference confirmsthat sample and standard peaks are differentcompounds.
3. In the absence of spectral scanning capabil-ity, other detectors, such as absorption orfluorescence, may be used in a ratioing pro-cedure. Chromatograms of sample and stan-dard are monitored at each of two differentwavelengths. The ratio of peak areas at thesewavelengths should be the same if sample andstandard are identical.
4. Peaks of interest can be collected and subjectedto additional chromatographic separation usinga different separation mode.
5. Collect the peak(s) of interest and establishtheir identity by another analytical method(e.g., mass spectrometry, which can give a massspectrum that is characteristic of a particularcompound; see Chap. 26).
27.5.3 Quantitative Analysis
Assuming that good chromatographic resolutionand identification of sample components have beenachieved, quantification involves measuring peakheight, area, or mass and comparing these data withthose for standards of known concentration. Whenstrip chart recorders were commonly used, measure-ment of the peak height, area (usually using: peak area= width at half height × height) (see Fig. 27-12a), ormass was done manually. This was followed by stand-alone integrators that produced a chromatogram ona strip of paper and digitally integrated the peaksby area for a tabular report. Nearly all chromatogra-phy systems (especially GC and HPLC systems) nowuse data analysis software, which recognizes the start,maximum, and end of each chromatographic peak,even when not fully resolved from other peaks. Thesevalues then are used to determine retention times andpeak areas. At the end of each run, a report is gener-ated that lists these data and postrun calculations, suchas relative peak areas, areas as percentages of the totalarea, and relative retention times. If the system hasbeen standardized, data from external or internal stan-dards can be used to calculate analyte concentrations.Data analysis software to quantify peaks is not as com-mon in low-pressure, preparative chromatography,in which postchromatography analysis of collectedfractions is used to identify samples eluted. Exam-ples of postchromatography analysis include the BCA(bicinchoninic acid) protein assay (Chap. 9, Sect. 9.2.7)and the phenol-sulfuric acid assay for carbohydrate(Chap. 10, Sect. 10.3.2). After obtaining the absorbancereading on a spectrophotometer for such assays, theresults are plotted as fraction number on the x-axis andabsorbance on the y-axis, to determine which fractionscontain protein and/or carbohydrate.
Having quantified sample peaks, one must com-pare these data with appropriate standards of knownconcentration to determine sample concentrations.Comparisons may be by means of external or internalstandards. Comparison of peak height, area, or massof unknown samples with standards injected sepa-rately (i.e., external standards) is common practice.Standard solutions covering the desired concentra-tion range (preferably diluted from one stock solution)are chromatographed, and the appropriate data (peakheight, area, or mass) plotted vs. concentration toobtain a standard curve. An identical volume of sam-ple is then chromatographed, and height, area, or massof the sample peak is used to determine sample con-centration via the standard curve (Fig. 27-15a). Thisabsolute calibration method requires precise analyti-cal technique and requires that detector sensitivity beconstant from day to day if the calibration curve is toremain valid.
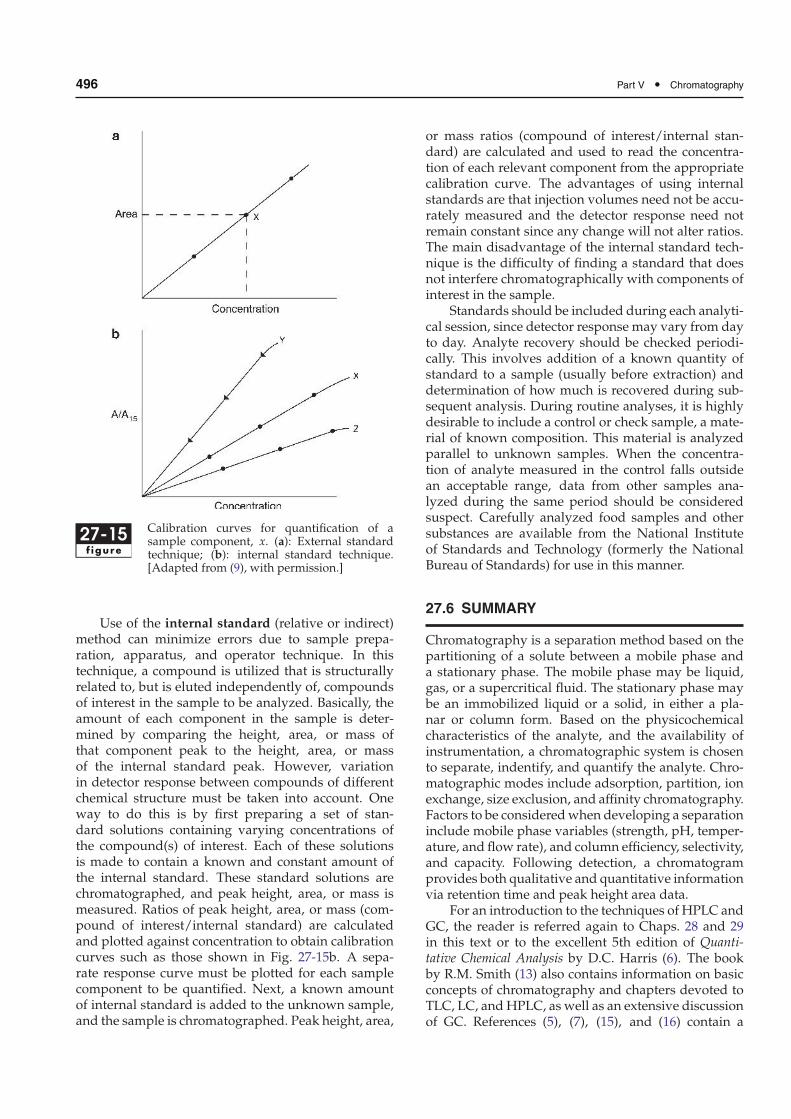
496 Part V • Chromatography
27-15f igure
Calibration curves for quantification of asample component, x. (a): External standardtechnique; (b): internal standard technique.[Adapted from (9), with permission.]
Use of the internal standard (relative or indirect)method can minimize errors due to sample prepa-ration, apparatus, and operator technique. In thistechnique, a compound is utilized that is structurallyrelated to, but is eluted independently of, compoundsof interest in the sample to be analyzed. Basically, theamount of each component in the sample is deter-mined by comparing the height, area, or mass ofthat component peak to the height, area, or massof the internal standard peak. However, variationin detector response between compounds of differentchemical structure must be taken into account. Oneway to do this is by first preparing a set of stan-dard solutions containing varying concentrations ofthe compound(s) of interest. Each of these solutionsis made to contain a known and constant amount ofthe internal standard. These standard solutions arechromatographed, and peak height, area, or mass ismeasured. Ratios of peak height, area, or mass (com-pound of interest/internal standard) are calculatedand plotted against concentration to obtain calibrationcurves such as those shown in Fig. 27-15b. A sepa-rate response curve must be plotted for each samplecomponent to be quantified. Next, a known amountof internal standard is added to the unknown sample,and the sample is chromatographed. Peak height, area,
or mass ratios (compound of interest/internal stan-dard) are calculated and used to read the concentra-tion of each relevant component from the appropriatecalibration curve. The advantages of using internalstandards are that injection volumes need not be accu-rately measured and the detector response need notremain constant since any change will not alter ratios.The main disadvantage of the internal standard tech-nique is the difficulty of finding a standard that doesnot interfere chromatographically with components ofinterest in the sample.
Standards should be included during each analyti-cal session, since detector response may vary from dayto day. Analyte recovery should be checked periodi-cally. This involves addition of a known quantity ofstandard to a sample (usually before extraction) anddetermination of how much is recovered during sub-sequent analysis. During routine analyses, it is highlydesirable to include a control or check sample, a mate-rial of known composition. This material is analyzedparallel to unknown samples. When the concentra-tion of analyte measured in the control falls outsidean acceptable range, data from other samples ana-lyzed during the same period should be consideredsuspect. Carefully analyzed food samples and othersubstances are available from the National Instituteof Standards and Technology (formerly the NationalBureau of Standards) for use in this manner.
27.6 SUMMARY
Chromatography is a separation method based on thepartitioning of a solute between a mobile phase anda stationary phase. The mobile phase may be liquid,gas, or a supercritical fluid. The stationary phase maybe an immobilized liquid or a solid, in either a pla-nar or column form. Based on the physicochemicalcharacteristics of the analyte, and the availability ofinstrumentation, a chromatographic system is chosento separate, indentify, and quantify the analyte. Chro-matographic modes include adsorption, partition, ionexchange, size exclusion, and affinity chromatography.Factors to be consideredwhen developing a separationinclude mobile phase variables (strength, pH, temper-ature, and flow rate), and column efficiency, selectivity,and capacity. Following detection, a chromatogramprovides both qualitative and quantitative informationvia retention time and peak height area data.
For an introduction to the techniques of HPLC andGC, the reader is referred again to Chaps. 28 and 29in this text or to the excellent 5th edition of Quanti-tative Chemical Analysis by D.C. Harris (6). The bookby R.M. Smith (13) also contains information on basicconcepts of chromatography and chapters devoted toTLC, LC, andHPLC, as well as an extensive discussionof GC. References (5), (7), (15), and (16) contain a

Chapter 27 • Basic Principles of Chromatography 497
wealth of information on TLC. SFC is discussed indetail by Caude and Thiébaut (2). Chromatograph (8),the standard work edited by E. Heftmann (2004 andearlier editions), is an excellent source of informa-tion on both fundamentals (Part A) and applications(Part B) of chromatography. Part B includes chapterson the chromatographic analysis of amino acids, pro-teins, lipids, carbohydrates, and phenolic compounds.In addition, Fundamental and Applications Reviews pub-lished (in alternating years) by the journal AnalyticalChemistry relate new developments in all branches ofchromatography, as well as their application to specificareas, such as food. Recent books and general reviewpapers are referenced, along with research articlespublished during the specified review period.
27.7 STUDY QUESTIONS
1. Explain the principle of countercurrent extraction andhow it stemmed into partition chromatography.
2. For each set of two (or three) terms used in chromatogra-phy, give a brief explanation as indicated to distinguishbetween the terms.
a. Adsorption vs. partition chromatography
Adsorption Partition
Nature of stationary phase
Nature of mobile phase
How solute interacts withthe phases
b. Normal-phase vs. reversed-phase chromatography
Normal-phase
Reversed-phase
Nature of stationary phase
Nature of mobile phase
What elutes last
c. Cation vs. anion exchangers
Cationexchanger
Anionexchanger
Charge on column
Nature of compoundsbound
d. Internal standards vs. external standards
Nature of Stds.
How stds. arehandled inrelation tosamples
What isplotted onstd. curve
Internal standard
External standard
e. TLC vs. column liquid chromatography
Thin layerColumnliquid
Nature and location ofstationary phase
Nature and location ofmobile phase
How sample is applied
Identification of solutesseparated
f. HETP vs. N vs. L (from the equation HETP = L/N)
3. State the advantages of TLC as compared to paperchromatography.
4. State the advantages of column liquid chromatography ascompared to planar chromatography.
5. Explain how SFC differs from LC and GC, including theadvantages of SFC.
6. What is the advantage of bonded supports over coatedsupports for partition chromatography?
7. You are performing LC using a stationary phase thatcontains a polar nonionic functional group. What typeof chromatography is this, and what could you do toincrease the retention time of an analyte?
8. You applied a mixture of proteins, in a buffer at pH 8.0, toan anion-exchange column. On the basis of some assaysyou performed, you know that the protein of interestadsorbed to the column.
(a) Does the anion-exchange stationary phase have apositive or negative charge?
(b) What is the overall charge of the protein of interestthat adsorbed to the stationary phase?
(c) Is the isoelectric point of the protein of interest(adsorbed to the column) higher or lower than pH 8.0?
(d) What are the two most common methods you coulduse to elute the protein of interest from the anion-exchange column? Explain how each method works.(See also Chap. 15.)
9. Would you use a polystyrene- or a polysaccharide-basedstationary phase for work with proteins? Explain youranswer.
10. Explain how you would use SEC to estimate the molecu-lar weight of a protein molecule. Include an explanationof what information must be collected and how it is used.
11. Explain the principle of affinity chromatography, why aspacer arm is used, and how the solute can be eluted.
12. What is gradient elution from a column, and why is itoften advantageous over isocratic elution?
13. A sample containing compounds A, B, and C is analyzedvia LC using a column packed with a silica-based C18
bonded phase. A 1:5 solution of ethanol and H2O wasused as the mobile phase. The following chromatogramwas obtained.
Time (min)
A B C
2 4 6 8 10 12 14 16 18 20

498 Part V • Chromatography
Assuming that the separation of compounds is basedon their polarity,
(a) Is this normal- or reversed-phase chromatography?Explain your answer.
(b) Which compound is the most polar?(c) How would you change the mobile phase so that
compound C would elute sooner, without changingthe relative positions of compounds A and B? Explainwhy this would work.
(d) What could possibly happen if you maintained anisocratic elution mode at low solvent strength?
14. Using the Van Deemter equation, HETP, and N, as appro-priate, explain why the following changes may increasethe efficiency of separation in column chromatography:
(a) Changing the flow rate of the mobile phase(b) Increasing the length of the column(c) Reducing the inner diameter of the column
15. State the factors and conditions that lead to poor resolu-tion of two peaks.
16. How can chromatographic data be used to quantify sam-ple components?
17. Why would you choose to use an internal standard ratherthan an external standard? Describe how you wouldselect an internal standard for use.
18. To describe how using internal standards works, answerthe following questions.
(a) What specifically will you do with the standards?(b) What do you actually measure and plot?(c) How do you use the plot?
27.8 ACKNOWLEDGMENTS
The authors of this chapter wish to acknowledgeDr. Bradley Reuhs, who was of great help in dis-cussions about reorganizing the chromatographychapters and in the editing of this chapter.
27.9 REFERENCES
1. Borch-Jensen C, Mollerup J (1996) Applications of super-critical fluid chromatography to food and natural prod-ucts. Semin Food Anal 1:101–116
2. Caude M, Thiébaut D (1999) Practical supercritical fluidchromatography and extraction. Harwood Academic,Amsterdam
3. Chester TL, Pinkston JD, Raynie DE (1996) Super-critical fluid chromatography and extraction (funda-mental review). Anal Chem 68:487R–514R
4. Craig LC (1943) Identification of small amounts oforganic compounds by distribution studies. Applicationto Atabrine. J Biol Chem 150:33–45
5. Fried B, Sherma J (1999) Thin-layer chromatography, 4thedn. Marcel Dekker, New York
6. Harris DC (1999) Quantitative chemical analysis, 5thedn. W.H. Freeman, New York
7. Hahn-Deinstrop E (2007) Applied thin-layer chromatog-raphy: best practice and avoidance of mistakes, 2nd edn.Wiley-VCH, Weinheim, Germany
8. Heftmann E (ed) (2004) Chromatography, 6th edn.Fundamentals and applications of chromatographyand related differential migration methods. Part A:fundamentals and techniques. Part B: applications.J Chromatogr Library Ser vols 69A and 69B. Elsevier,Amsterdam
9. Johnson EL, Stevenson R (1978) Basic liquid chromatog-raphy. Varian Associates, Palo Alto, CA
10. Lawrence JF (ed) (1984) Food constituents and foodresidues: their chromatographic determination. MarcelDekker, New York
11. Lough WJ, Wainer IW (eds) (1995) High performanceliquid chromatography: fundamental principles andpractice. Blackie Academic & Professional, Glasgow,Scotland
12. Scopes RK (1994) Protein purification: principles andpractice, 3rd edn. Springer-Verlag, New York
13. Smith RM (1988) Gas and liquid chromatography inanalytical chemistry, Wiley, Chichester, England
14. Snyder LR, Kirkland JJ (eds) (1979) Introduction to mod-ern liquid chromatography, 2nd edn. Wiley, New York
15. Touchstone JC (1992) Practice of thin layer chromatogra-phy. Wiley, New York
16. Wall PE (2005) Thin-layer chromatography: a modernpractical approach. The Royal Society of Chemistry,Cambridge, UK
17. Walters RR (1985) Report on affinity chromatography.Anal Chem 57:1099A–1113A
18. Weiss J (2004) Handbook of ion chromatography, 3rdedn. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
19. Zachariou M (2008) Affinity chromatography: methodsand protocols. Humana, Totowa, NJ