Über die denaturierungstemperatur des enzyms
TRANSCRIPT

Über die Denaturierungstemperatur des Enzyms Glukoseoxidase als Maß seiner Funktionsstabilität sowie
ihre nano-kalorimetrische Bestimmung
Dissertation zur Erlangung des Doktorgrades
der Fakultät für Angewandte Wissenschaften
der Albert-Ludwigs-Universität Freiburg
Thomas Weiß
September 2006

2
Dekan: Prof. Dr. B. Nebel
Prüfungsausschuss:
Gutachter: Prof. Dr. G. Urban
Zweitgutachter: Prof. Dr. R. Schubert
Vorsitz: Prof. Dr. T. Stieglitz
Beisitz: Prof. Dr. J. Rühe
Datum der Prüfung: 15.02.2007

____________________________________________________________________Abstract
3
Abstract
Bioartificial hydrogels are hybrid materials comprising water, synthetic polymers
and enzymes. They are used in biosensors as sensing elements.
The stability of the enzyme structure and function depends on the
physicochemical properties of the embedding matrix. Moreover, the stability of
the catalytically active structures is temperature depending. In addition, a phase
transition to a random coil occurs at a specific temperature. This phase transition
can be characterized by the temperature of the mid-point of the transition, Tm.
Several techniques are capable of characterizing the transition, e.g.
fluorescence spectroscopy, NMR, circular dicroism, measurement of the
enzymatic activity and differential scanning Calorimetry (DSC). The latter two
were used in this work.
Some different matrices, including solutions and hydrogels with additional
stabilizing co-solvents were characterized with DSC and activity assay with
respect to the stabilization of the enzyme Glucoseoxidase (Gox).
A chip-calorimeter has been invented and proofed to be capable of determining
the Tm of the enzyme GOx.

____________________________________________________________Inhaltsverzeichnis
Inhaltsverzeichnis
1 Einleitung 14
2 Grundlagen 19
2.1 Biomimetische Hydrogele 19
2.2 Proteinstabilität 21
2.3 Phasenumwandlungen von Proteinen 23
2.4 Die Überschuss-Wärmekapazität von Proteinen
in Lösung zwischen 25°C und 100°C 27
2.5 Einfluss der Mikroumgebung auf die Proteinstabilität 32
2.5.1 Die Wyman-Gleichung und das Konzept
der bevorzugten Wechselwirkung 33
2.5.2 Stabilisierungsmechanismen 49
2.5.2.1 Sterischer Ausschluss 49
2.5.2.2 Oberflächenspannung 51
2.5.2.3 Solvophobie 52
2.5.2.4 Kompensation von Ladungen
auf der Proteinoberfläche 54
2.6 Funktionsstabilität 57
2.7 Diffusionswiderstand und Thiele-Modulus 58
2.8 Einfluss von Aggregation und Reversibilität auf die
Langlebigkeit von Enzym Präparationen 66
2.9 Die Tm als Maß für die Langlebigkeit von
Enzym-Präparationen 68
3 Materialien und Methoden 74
3.1 Bestimmung der enzymatischen Aktivität 74
3.1.1 Reaktionsgeschwindigkeit und Unit-Definition 75

____________________________________________________________ Inhaltsverzeichnis
5
3.1.2 Photometrie 76
3.1.3 Michaelis-Menten Kinetik 79
3.1.4 Direct-Plot 79
3.1.5 Nicht-lineare Kurvenanpassung 80
3.1.6 µ-Gele 83
3.1.7 Enzymkonzentration 83
3.1.8 Thermische Deaktivierung der Enzymfunktion 84
3.1.9 Stress-Aktivitätstest/Halbwertszeit 86
3.2 Dynamische Differenzialkalorimetrie 87
3.2.1 DTA, Differenzthermoanalyse 87
3.2.2 DSC, Dynamische Differenzialkalorimetrie 87
3.2.2.1 Leistungskompensations-DSC 88
3.2.2.2 Wärmestrom-DSC 89
3.2.3 Heizgeschwindigkeit 89
3.2.4 Vergleich der Messprinzipien DTA und DSC 90
3.2.4.1 Klassische DTA 92
3.2.4.2 Leistungskompensations-DSC 93
3.2.4.3 Wärmefluss-DSC 93
3.2.5 Mikrokalorimetrie 94
3.2.6 Probenvorbereitung DSC 96
3.2.7 Nano-DSC 99
3.2.7.1 Zur Auswahl der Kalibrierungssubstanzen 100
3.2.7.1.1 Flüssigkristalle 101
3.2.7.1.2 Phospholipidvesikel 102
3.2.7.2 Set-Up des Nano-Kalorimeters 104
3.2.7.3 Temperaturmessung mit Ge-Thermistor 108
3.2.7.4 Differenztemperaturmessung 110

____________________________________________________________Inhaltsverzeichnis
6
3.2.7.5 Versiegelung der Probenkammer 113
3.2.7.6 Zeitkonstante 114
3.2.7.7 Empfindlichkeit 117
4 Ergebnisse 121
4.1 GOx-Aktivität 122
4.1.1 Kinetische Konstanten GOx 122
4.1.2 Abschätzung Thiele Modulus 122
4.1.3 Halbwertszeiten 126
4.1.3.1 GOx in Puffer 128
4.1.3.2 GOx + NaCl 129
4.1.3.3 GOx + PEG 130
4.1.3.4 GOx + Sorbitol 131
4.1.3.5 GOx + Xylitol 132
4.1.3.6 GOx in pAA-Hydrogel 133
4.1.3.7 GOx im pAA-Hydrogel+ NaCl 134
4.1.3.8 GOx im pAA-Hydrogel + PEG 135
4.1.3.9 GOx im pAA-Hydrogel + Ficoll 137
4.2 Denaturierungstemperaturen (konventionelle DSC) 139
4.2.1 Einfluss der Heizrate 139
4.2.2 Einfluss der Konzentration des Enzyms 141
4.2.3 GOx in Phosphatpuffer 141
4.2.4 GOx + GndHCl 142
4.2.5 GOx + NaCl 142
4.2.6 GOx + PEG 143
4.2.7 GOx + Dextran 144
4.2.8 GOx + Monomer Acrylamid 144
4.2.9 GOx in pAA-Hydrogel 145

____________________________________________________________ Inhaltsverzeichnis
7
4.2.10 GOx + Monomer HEMA 146
4.2.11 GOx in pHEMA-Hydrogel 147
4.2.12 GOx in pHEMA-Hydrogel + NaCl 148
4.2.13 GOx in pAA-Hydrogel + PEG 149
4.2.14 GOx in pAA-Hydrogel + Ficoll 150
4.2.15 Vermischtes 151
4.3 Thermogramme (Nano-Kalorimeter) 151
4.3.1 Flüssigkristall MH24 151
4.3.2 DMPC (Dimyristoyl–Lecithin) 153
4.3.3 GOx in Lösung und im Hydrogel 154
4.4 Zusammenfassung der Halbwertszeitmessungen
und der Denaturierungstemperaturen 155
5 Diskussion 157
6 Ausblick 164
7 Literaturverzeichnis 168

____________________________________________________ Abkürzungen und Symbole
8
Abkürzungen und Symbole
1 Haupt-Solvens (Wasser)
2 Protein
3 Ko-Solvens
A Debye-Screening Parameter [M–1/2]
A Extinktion
A enzymatische Aktivität [mU]
A Fitparameter in der Steuergleichung des Thermistors [kΩ]
AA Aminosäure
(p)AA (Poly-)Acrylamid
ABTS 2, 2´-Azino-bis 8-ethylbenzthiazolin-6-sulfonsäure
ai Aktivität der Komponente i [M]
B Jones-Dole-Koeffizient [M-1]
Bi Anzahl der Moleküle der Komponente i auf der
Proteinoberfläche
C empirischer Parameter [kJ∗mol-1]
C.U. kooperative Einheit
<∆C> Überschuss-Wärmekapazität [J∗mol-1∗K-1]
<C> Wärmekapazität des Proteins [J∗mol-1∗K-1]
<Ctr> Überschuss-Wärmekapazität des Übergangs [J∗mol-1K-1]
<Cbl> Überschuss-Wärmekapazität der Basislinie [J∗mol-1∗K-1]
CN Wärmekapazität des N-Zustands [J∗mol-1∗K-1]
CD Wärmekapazität des D-Zustands [J∗mol-1∗K-1]
∆DNC Differenz der Wärmekapazitäten von D und N-Zustand
[J∗mol-1∗K-1]
c Konzentration [M]

_____________________________________________________Abkürzungen und Symbole
9
c∞ij Löslichkeit der Komponente i in Medium j [M]
cosh(x) Kosinus-Hyperbolicus
d Dicke eines absorbierenden Mediums [cm]
D denaturiertes Protein
DSC Dynamische Differential Kalorimetrie
DMPC Di-Myristoyl-Phosphatidylcholine
E Energie [J]
E Konzentration aktiven Enzyms [M]
Ed Deaktivierungsenergie [J∗mol-1]
εm millimolarer Absorptionskoeffizient [ml∗µmol-1∗cm-1]
F empirischer Parameter [kJ∗mol-1∗min-1]
FAD Flavin Adenin Dinucleotid
FWHM full width at half maximum
GOx Glucoseoxidase (EC 1.1.3.4)
∆G Gibb´sche Freie Enthalpie [J∗mol-1]
∆Gi(Tm) Freie Energie des Zustand i bei Tm [J∗mol-1]
∆DNG Freie Energie des Übergangs [J∗mol-1]
∆GB Freie Energie der Bindung [J∗mol-1]
∆GS Freie Energie der Solvatisierung [J∗mol-1]
Γ3 Oberflächenkonzentration des Ko-Solvens auf dem Protein
[mol∗m-2]
γi Aktivitätskoeffizient der Komponente i
(∂g1/∂g2)T,P,µ3 Parameter der bevorzugten Hydratation [g∗g-1]
GD gleitender Durchschnitt
GndHCl Guanidiniumhydrochlorid
H Enthalpie [J∗mol-1]
∆DNH Enthalpie des Übergangs [J∗mol-1]

____________________________________________________ Abkürzungen und Symbole
10
∆HvH Van´t Hoff-Enthalpie [J∗mol-1]
<∆H > Überschuss- Enthalpie [J∗mol-1]
η0 dynamische Viskosität des Wassers [Pa∗s]
(p)HEMA (Poly-)Hydroxyethylmethacrylat
I relative Intensität eines monochromatischen Lichtstrahls
ITC Isotherme Titrationskalorimetrie
K Gleichgewichtskonstante
KM Michaelis-Menten Konstante [mM]
KB Bindungskonstante [M-1]
Kex Austauschkonstante
KS Ko-Solvens
k Boltzmannkonstante [J∗K-1]
k3 Setschenow-Koeffizient des Ko-Solvens [M-1]
kcat Überführungszahl [s-1]
kd Geschwindigkeitskonstante der Deaktivierung [s-1]
kD Geschwindigkeitskonstante der Denaturierung [s-1]
kF Geschwindigkeitskonstante der Faltung [s-1]
l flüssig
l Länge der Kalibrierungsmarke in der Aufzeichnung [mm]
Mr Molekulargewicht [kDa]
MH24 4´-Oktyloxy-biphenyl-4-carbonitril
MPD 2-Mthyl-2,4-Pentandiol
mi Molalität der Komponente i [mol∗kg-1]
µi chemisches Potential der Komponente i [J∗mol-1]
∆µ2,Tr Freie Transfer-Energie des Proteins [J∗mol-1]
(∂m1/∂m2)T,P,µ3 Parameter der bevorzugten Hydratation [mol∗mol-1]
(∂m3/∂m2)T,P,µ3 Parameter der bevorzugten Bindung [mol∗mol-1]

_____________________________________________________Abkürzungen und Symbole
11
(∂µ3/∂m3)T,P,m2 Selbst-Interaktionsparameter [J∗g-1]
(∂µ2/∂m3)T,P,m2 Interaktionsparameter [J∗g-1]
ni Anzahl an Molekülen im i-Zustand
N natives Protein
N Anzahl der Aminosäuren eines Proteins
Ntotal Gesamtzahl von Molekülen
n nematisch
∆n Anzahl der im Zuge der Denaturierung neu ausgebildeter
Bindungsstellen
NEM N-Ethyl-Maleimid
NMR magnetische Kernresonanzspektroskopie
P (Heiz-)Leistung [W]
PD Bruchteil des Proteins im denaturierten Zustand
PEG Polyethylenglykol
R molare Gaskonstante [J∗mol-1∗K-1]
R elektrischer Widerstand [Ω]
R0 Fitparameter in der Steuergleichung des Thermistors [kΩ]
RP hydrodynamischer Radius des Proteinmoleküls [nm]
ρ1 Dichte des Haupt-Solvens [g∗cm-3]
s molekulare Oberfläche des Proteins [m2]
σ Oberflächenspannung [J∗m-2]
∆DNS Entropie des Übergangs [J∗mol-1∗K-1]
T1, T2 Membran-Thermistoren
T3 Thermistor auf dem Chipkörper
T absolute Temperatur [K]
Tm Temperatur des Übergangs [K]
TMax Temperatur maximaler Stabilität [K]

____________________________________________________ Abkürzungen und Symbole
12
τ Zeitkonstante [ms]
τ1/2 Halbwertszeit der biologischen Aktivität [min]
t Zeit [s]
t0 Fitparameter in der Steuergleichung des Thermistors [°C]
U Unit (Maß der enzymatischen Aktivität)
U elektrische Spannung [V]
U0 Versorgungsspannung der Messbrücke [V]
Ui Brückenspannung [V]
V Volumen [l]
Vex Ausschlussvolumen [m3]
vMax maximale Geschwindigkeit einer enzymatischen Reaktion [mM∗min-1]
v Reaktionsgeschwindigkeit [µmol∗min-1]
vd Geschwindigkeit der Enzymdeaktivierung [µmol∗min-1]
smA smektisch A
TKR Temperaturkoeffizient des Widerstandes [K-1]
θ Langmuir-Isotherme [∅]
θex Wahrscheinlichkeit der Belegung einer Bindungsstelle [∅]

___________________________________________________________Acknowledgements
13
Acknowledgements
I like to thank Prof. Urban for giving me the opportunity to perform this research
in his versatile interdisciplinary group (thanks to all of them) and for supervising
and promoting this thesis all along.
I thank Dr. Moser for introducing me into the novel and challenging field of
biosensor research, the helpful discussions and for proofreading parts of the
manuscript.
In particular I wish to thank G. Igel for the technical advice and the numerous
valuable discussions. Without his experienced patience and educational skills
the set-up would not have been kept that straightforward.
P. Letzkus (stress-activity test), N. Prabhakar (DSC) und M. Hofferberth (shelf
lives) were active and pleasant collaborators. I could not have missed their
assistance.
Last but not least I am indebted to the Deutsche Forschungsgemeinschaft for
financial support and for providing the infrastructure of the Sonderforschungs-
bereich 428, “Makromolekulare Netzwerke”.

________________________________________________________________ 1. Einleitung
14
1. Einleitung Das Cytosol der Zelle ist eine hochkonzentrierte Lösung mit mehreren hundert
Gramm an Gelöstem pro Liter. Diese Umgebung ist deutlich verschieden von den
verdünnten Lösungen, die normalerweise den Hintergrund für biochemische
Prozesse in vitro bilden. In der zellulären Umgebung ist die Diffusion vor allem
größerer Moleküle herabgesetzt, während die Neigung zur Selbstassoziation
verstärkt und die Proteinstabilität erhöht ist [1]. Hydrogele aus Wasser, einem
vernetzten quellbaren Polymer und darin eingebetteten katalytisch aktiven Proteinen
können als einfaches biomimetisches Cytosol angesehen werden, in denen ähnliche
Bedingungen wie im Zellinneren vorliegen [2]. Membranen aus biomimetischen
Hydrogelen finden in der Bioanalytik Verwendung in amperometrischen Biosensoren:
Produkte der enzymatischen Reaktion, die in dem Hydrogel, das in Kontakt steht mit
einer Analyt-Lösung von physiologisch relevanten Enzymsubstraten, akkumulieren,
können an unterlegten µ-Elektroden elektrochemisch nachgewiesen werden [3,4].
Enzyme dienen ihrer besonderen Selektivität wegen auch als heterogene
Katalysatoren in Bioreaktoren in der enantiomerenreinen Synthese einiger
Feinchemikalien [5,6]. Ist der Einsatz von Enzymkatalysatoren durch die Stabilität der
Enzymfunktion begrenzt, z.B. bei ihrem Einsatz bei erhöhter Temperatur oder im
Langzeiteinsatz, stellt sich die Frage, ob und wie das Enzym in dem Hydrogel
stabilisiert werden kann.
Ziel dieser Arbeit war es, eine physiko-chemische Beschreibung der Mechanismen
zu geben, die zur Stabilisierung bzw. Destabilisierung der Enzyme in Hydrogelen
führen. Im Zentrum stand dabei die Frage, ob oder unter welchen Voraussetzungen
die Denaturierungstemperatur Tm des Enzyms Glukoseoxidase (GOx) ein Maß für
deren Funktionsstabilität sein kann, die ein bedeutendes Qualitätsmerkmal von
Biosensoren ist [7]. Dazu wurden Messwerte der Denaturierungstemperatur des

________________________________________________________________ 1. Einleitung
15
Enzyms GOx in Beziehung gesetzt zu Messwerten der Lebensdauer der
Katalysefähigkeit bei erhöhter Temperatur.
Die Bestimmung der Denaturierungstemperatur Tm erfolgte zunächst mit
konventionellen DSC-Geräten (Perkin Elmer DSC7 oder Netzsch DSC 204).
Gleichzeitig wurde untersucht, ob es möglich ist, die Denaturierungstemperatur mit
einem dynamischen Nano-Kalorimeter, dessen Herzstück ein Membran-Chip bildet,
zu bestimmen, insbesondere, wenn das Enzym sich nicht in wässriger Lösung
sondern in einer Hydrogelmatrix befindet.
Hydrogele sind Materialien, die in ihrem Aggregatzustand zwischen fest und flüssig
angesiedelt sind. Sie sind fest, weil sie aufgrund der Vernetzung des Polymers nicht
fließen, und sie sind flüssig, weil sie weich sind [8]. Hochempfindliche Mikro-
Kalorimeter (MicroCal™, CSC™, Setaram™) können zur Untersuchung von
Hydrogelen nicht eingesetzt werden, weil diese aufgrund ihrer Bauart ausschließlich
für fließfähige Proben ausgelegt sind. Daher musste die Untersuchung der Energetik
der thermischen Denaturierung von Enzymen in quasi-festen Hydrogelen mit
konventionellen DSC-Geräten erfolgen und auf die Bestimmung der
Denaturierungstemperatur beschränkt bleiben. State-of-the-art Mikro-Kalorimeter
ermöglichen hingegen die vollständige Beschreibung der Thermodynamik der
Denaturierung, für die neben der Denaturierungstemperatur die Bestimmung der
Enthalpie des Übergangs ∆DNH und die Differenz der Wärmekapazitäten zwischen
nativem und denaturiertem Enzym ∆DNC nötig ist. Mit diesem Datensatz lässt sich die
temperaturabhängige Stabilitätskurve ∆DNG(T) des Enzyms im Hinblick auf die
Denaturierung bestimmen [9].
In der DSC von Enzymen mittels konventioneller DSC wird die Probenmenge zum
kritischen Faktor: Reihenuntersuchungen erfordern die Verfügbarkeit des Enzyms bis
in den Gramm-Bereich hinein. Solch große Mengen an gereinigtem Enzym stehen

________________________________________________________________ 1. Einleitung
16
jedoch nur ausnahmsweise zur Verfügung, wie hier die GOx, die in industriellem
Maßstab hergestellt wird.
Für diese Untersuchungen ist z.Z. die Nano-Kalorimetrie die Methode der Wahl.
Die katalytische Stabilität des Enzyms wurde mittels eines Stress-Aktivitätstests bei
60°C in unterschiedlichen Medien untersucht. Die so bestimmten Halbwertszeiten der
enzymatischen Aktivität dienten als Maß für die Funktionsstabilität der GOx.
Enzyme sind Makromoleküle aus Aminosäuren. Sie falten sich in wässriger Lösung
spontan zu hochspezifischen Strukturen von 1–10 nm räumlicher Ausdehnung
[10,11]. An diesen Strukturen verlaufen spezifische chemische Reaktionen
katalysiert. Die für das jeweilige Enzym charakteristische Struktur ist in der Sequenz
der Aminosäuren festgelegt. Eine Reihe inter- und intramolekularer
Wechselwirkungen trägt zur Aufrechterhaltung dieser Strukturen bei und macht sie
ca. 50 kJ/mol stabil gegenüber einer zufälligen Anordnung der Segmente der
Polypeptidkette [12,13]. Diese Energie reicht aus, das Polypeptid bis nahe an eine
charakteristische Temperatur Tm in der nativen, gefalteten Konformation zu halten.
Diese Tm liegt für typische Proteine bei 50-70°C. An der Aufrechterhaltung dieser
Strukturen sind neben intrinsischen Wechselwirkungen innerhalb des
Makromoleküls, wie van-der-Waals-Kräfte, intermolekulare Wasserstoffbrücken,
Salzbrücken und Disulfidbrücken, auch solche mit Lösungsmittelmolekülen und, so
vorhanden, mit Ko-Solventien in der Lösung beteiligt.
Thermophile, einzellige Organismen, die ihr Wachstumsoptimum bei annähernd 100
°C haben, müssen über besondere Mechanismen verfügen, die für ihren
Stoffwechsel notwendigen Enzyme zu stabilisieren. Bei derart hohen Temperaturen
sind die Enzyme von mesophilen homologen Organismen, die ihr
Wachstumsoptimum bei physiologischen Temperaturen haben, inaktiv, weil die

________________________________________________________________ 1. Einleitung
17
native gefaltete Konformation in die denaturierte statistische gewechselt hat [14].
Wodurch unterscheiden sich intrinsisch stabilisierte d.h. in der Primärstruktur
veränderte Proteine von weniger stabilen? Ein Vergleich der Sequenzen von
thermophilen Enzymen mit ihren Homologen aus mesophilen Organismen zeigt
einige Besonderheiten der thermophilen Enzyme wie die relative Häufung bestimmter
Aminosäuren (Lysin, Prolin) und die Verkleinerung von Schlaufen in der gefalteten
Struktur [15]. Generell sind die Sequenzen aber sehr ähnlich und unterschiedliche
Stabilitäten sind eher auf spezifische Änderungen einzelner Aminosäuren als auf
allgemeine Sequenzmuster zurückzuführen. Dieses Ergebnis wird dadurch bestärkt,
dass die Netto-Stabilität, die sich aus allen stabilisierenden und destabilisierenden
Wechselwirkunken, die jeweils im Bereich von einigen MJ/mol liegen,
zusammensetzt, nur ca. 50 kJ/mol beträgt [13,35]. Das entspricht der Energie einiger
weniger schwacher Wechselwirkungen, z.B. zweier Wasserstoffbrückenbindungen.
Die Stabilität von Enzymen, charakterisiert durch die Denaturierungstemperatur, lässt
sich durch Mutationen einzelner entscheidender Aminosäuren steigern unter
Verschiebung der Tm um mehr als 10°C [14]. Der Evolutionsdruck liegt in vivo aber
nicht nur bzw. immer auf Stabilitätserhöhung, ist doch die materielle Substanz
biologischer Systeme in ständigem Umbau begriffen, sondern eher auf Erhalt und
Optimierung der Funktion, die an die molekulare Dynamik der makromolekularen
Strukturen gekoppelt ist [16-18]. Das lässt Spezialisten unter den Organismen Raum,
ihr biochemisches Inventar an anspruchsvolle Umweltbedingungen wie
Temperaturstress in heißen Quellen oder hohen Salzgehalt anzupassen.
Eine solche Anpassungsleistung ist hier besonders interessant, weil sie einen
alternativen Weg aufzeigt, die Stabilität von Enzymen zu erhöhen: nicht intrinsisch
durch Mutagenese, sondern durch solvent engineering, also durch gezielte
Beeinflussung der Mikroumgebung der Enzyme.

________________________________________________________________ 1. Einleitung
18
Das Cytosol der Zelle ist der Reaktionsraum für eine Vielzahl von enzymatisch
katalysierten chemischen Reaktionen. Darin ist ein Großteil des Volumens von
Makromolekülen eingenommen und somit unzugänglich für andere Bestandteile der
hochkonzentrierten Lösung. Zur Erklärung des Effektes hoher Konzentrationen von
Ko-Solventien auf die Stabilität von Proteinen im Hinblick auf thermische
Denaturierung, wurde eine Wechselwirkung neuen Typs, die excluded volume
interaction, vorgeschlagen. Diese Wechselwirkung beruht auf dem Ausschluss von
größeren Molekülen durch kleinere an der Kontaktfläche Enzym/Lösung und wird als
molecular confinement oder molecular crowding bezeichnet [19-23]. Dieser
Ausschluss ist energetisch unvorteilhaft, weil er Konzentrationsgradienten erzeugt,
und er nimmt mit der molekularen Oberfläche des Enzyms proportional zu. Der
Wechsel der bei tiefen Temperaturen stabilen katalytisch aktiven Konformation zur
denaturierten inaktiven Form ist mit einer Vergrößerung der
lösungsmittelzugänglichen Oberfläche verbunden, die zur Verstärkung der
unvorteilhaften Wechselwirkung führt. Nach dem Prinzip des kleinsten Zwanges wird
deshalb durch voluminöse bzw. verdrängte Ko-Solventien die konformative Stabilität
des nativen Enzyms erhöht [24]. Bedenkt man, dass Wasser eines der kleinsten
Moleküle überhaupt ist (< 0,2 nm), kann man erahnen, dass die Ausschlussvolumen-
Wechselwirkung in wässriger Lösung und damit in biologischen Systemen ubiquitär
ist.
In einigen Organismen werden zur Anpassung an Stressfaktoren darüber hinaus
kleine Moleküle (compatible osmolytes) in hohen Konzentration akkumuliert [25-28].
Diese Osmolyten erhöhen die Oberflächenspannung des Wassers und sind von der
molekularen Oberfläche des Proteins verdrängt, d.h. die Konzentration der
Osmolyten an der Oberfläche des Proteins ist niedriger als in der umgebenden
Lösung.

_______________________________________________________________
19
2. Grundlagen
2. Grundlagen
2.1 Biomimetische Hydrogele
Biomimetische Hydrogele sind wassergequollene weiche Materialien, die biologische
Funktionen nachahmen. Die Funktion kann z.B. die Kontrolle des Blutflusses durch
die Venenklappen sein: ein Paar pH-sensitiver Hydrogelstreifen kann durch
Änderungen des lokalen pH-Wertes aktiviert werden als Folge von reversiblen
Volumen- oder Formänderungen [29]. Eine weitere Funktion, die von biomimetischen
Hydrogelen nachgeahmt wird, ist die Faktor VIII-induzierte Gerinnung von Blutzellen,
ein wichtiger Schritt in der Wundheilung. Der Faktor VIII, eine spezielle
Transglutaminase, kann benutzt werden, um spezielle biomimetische synthetische
Polymere (glutamin-derivatisiete PEGs und lysinreiche Polypeptide) Ca2+-abhängig
reversibel zu vernetzen, um so gezielt in situ biokompatible Materialien zu erzeugen.
Hier gibt es medizinische Anwendungen in der Wundheilung, in der Einführung
knochenbildenden Materials und im drug bzw. gene delivery [30]. Selbst die
Katalyse-Funktion der Enzyme kann durch biomimetische Hydrogele nachgebildet
werden: Sogenannte polymer imprints sind Hydrogelnetzwerke, die, ähnlich den
Enzymen, molekulare Taschen bilden (durch Co-Polymerisation eines das
Enzymsubstrat imitierenden templates). Nach Herauslösen des Templates verbleiben
in dem Hydrogel katalytisch aktive Stellen zur Aufnahme und Umwandlung des
Substrates. Durch die Möglichkeit zusätzlich das Quellverhalten dieser Hydrogele
durch externe Stimuli zu beeinflussen, können solche mimetischen Enzyme ein- und
ausgeschaltet werden, wodurch sowohl die Katalysefähigkeit als auch die
Regulierbarkeit der nativen Enzyme nachgebildet ist [31]. Mimetische Enzyme zeigen
sich häufig als robuster gegenüber ihren nativen Vorbildern, was für ihren Einsatz in
Bioreaktoren und Biosensoren von Vorteil ist [1].

_______________________________________________________________2. Grundlagen
20
Hydrogele aus synthetischen Polymeren und darin eingebetteten Enzymen können
als einfaches mimetisches Cytosol bzw. als mimetisches Gewebe angesehen werden
insofern sie nicht nur die limitierte Diffusion in der hochkonzentrierten Lösung im
Zellinneren nachahmen und als Modell desselben dienen, sondern es können auch
die biologischen Stabilisierungsmechanismen der nativen Enzymstruktur in vivo in
vitro nachgebildet werden. Diese Stabilisierungsmechanismen, die als molecular
crowding bezeichnet werden und die Begrenztheit des freien Lösungsmittelvolumens
in hochkonzentrierten Polymer- bzw. Elektrolyt-Lösungen zur Grundlage haben,
wurden durch die thermodynamische Beschreibung der Mehr-Komponenten-
Lösungen verständlich [32] (s. Kapitel 2.5). Mimetische Gewebe, die mit den
mimetischen Enzymen eng verwandt sind, dienen in Bioreaktoren und Biosensoren
als Reaktionsraum. Die spezifische Beschaffenheit des synthetischen
Polymergerüsts und von stabilisierenden Ko-Solventien ist durch Implementierung
von Strukturcharakteristika der in vivo gefundenen polymeren oder monomeren
Stabilisatoren an die Erfordernisse des jeweiligen zu stabilisierenden Enzyms
adaptierbar (solvent engineering).
Biomimetischen Hydrogele, in denen das Enzym in einem vernetzten Polymer
eingeschlossen ist, können erzeugt werden, in dem die reaktive Mischung des
Monomers, des Enzyms und der Ko-Solventien in Wasser durch Zugabe eines
Initiators (häufig ein Radikal) oder durch Bestrahlung mit Licht geeigneter
Wellenlänge zur Vernetzung gebracht wird. Diese Gele bieten dem Enzym einen
großen Diffusionswiderstand, so dass sie nicht oder nur wenig ausgewaschen
werden, wenn das Hydrogel in Kontakt mit einer Analyt-Lösung gebracht wird.
Umgekehrt können die wesentlich kleineren Substrate und Produkte der Reaktion,
hier Glukose und Wasserstoffperoxyd, in das Gel und aus diesem heraus
diffundieren. Allerdings bestehen auch für diese Diffusionswiderstände, die mit dem
Abstand zur Lösung zunehmen.

_______________________________________________________________
21
2. Grundlagen
2.2 Proteinstabilität
Die Stabilität von Biosensoren ist eng gekoppelt an die Stabilität der verwendeten
katalytischen Proteine, der Enzyme [33].
Ein Enzym als reaktive Substanz ist in Wasser einer Reihe von Abbaureaktionen, vor
allem der Derivatisierung durch oxidative Deamidation, dem Austausch und der
Modifizierung von Disulfid-Gruppen, der Proteolyse, dem Verlust von Co-Faktoren
sowie dem Einfluss von reaktiver Strahlung ausgesetzt [34,35,36]. Des weiteren
besteht ein Gleichgewicht zwischen der gefalteten nativen und der denaturierten
Konformation sowie zwischen einzelnen Proteinmolekülen und Aggregaten
derselben. Sind die Geschwindigkeiten der abbauenden Prozesse hinreichend
langsam, kann das Protein als stabile chemisch einheitliche Substanz betrachtet
werden, das sich im Gleichgewicht zwischen der nativen und denaturierten
Konformation befindet. Die Funktion der enzymatischen Proteine als Katalysator
biochemischer Reaktionen wird gesteuert durch biochemische Liganden (Aktivatoren,
Inhibitoren, Ko-Solventien) und ist empfindlich gegen Blockierung („Vergiftung“) der
reaktiven Zentren z.B. durch Bindung von Schwermetall-Ionen oder von toxischen
Gasen. Ist diese Bindung nicht-kovalent, etwa koordinativ, ist die resultierende
Inhibierung reversibel und das native Proteine kann rekonstituiert werden.
Andernfalls muss das Proteinmolekül als derivatisiert gelten.
Die chemische Funktion von Enzymen im Organismus ist ihre regulierbare
katalytische Aktivität. Sie ist an die native Konformation gekoppelt, so dass unter
Vorgabe kontrollierter Reaktionsbedingungen und niedriger
Reaktionsgeschwindigkeit der abbauenden Reaktionen die enzymatische Aktivität als
Maß für den Bruchteil des Enzyms gelten kann, der in der nativen Konformation
vorliegt. Kurz gesagt: Was aktiv ist, ist auf jeden Fall nativ. Das Umgekehrte gilt nur
eingeschränkt. Was nativ ist, ist aktiv, es sei denn, es ist inhibiert.

_______________________________________________________________2. Grundlagen
22
Abbildung 1: Verschiedene Aspekte der Stabilität eines Enzyms. Die thermodynamische Beschreibung
der Stabilität konzentriert sich auf die Lage des Gleichgewichtes zwischen der nativen und
denaturierten Konformation des Proteins.
Die Lage des Gleichgewichts zwischen der nativen und der denaturierten
Konformation in Abhängigkeit von der Ko-Solvens-Konzentration kann durch
dynamische Differenzialkalorimetrie bestimmt werden. Die technisch bedeutsamere
Halbwertszeit τ1/2 der enzymatischen Aktivität lässt sich durch Messung der
Deaktivierung unter Stressbedingungen wie z.B. erhöhter Temperatur bestimmen.
Bei erhöhter Temperatur ist die Deaktivierung schneller und die Bestimmung der
Halbwertszeiten ist genauer als bei 37°C [37]. Die Funktion des Enzyms ist bei
Zusatz hoher Konzentrationen extrinsisch stabilisierender Ko-Solventien stabiler d.h.
die Halbwertszeit τ1/2 ist verlängert und Tm ist häufig erhöht. Gleichzeitig ist die
spezifische Aktivität in Anwesenheit von Ko-Solventien häufig niedriger als in Wasser
[38].
Die Methoden zur Stabilisierung lassen sich in intrinsische und extrinsische
unterscheiden. Quellen der intrinsischen Stabilität von gefalteten Proteinen sind
elektrostatische Wechselwirkungen, Wasserstoffbrückenbindungen und die
hydrophobe Wechselwirkung [39]. Die intrinsische Stabilisierung verändert einzelne

_______________________________________________________________
23
2. Grundlagen
Bereiche des biologischen Makromoleküls durch Mutagenese, d.h. durch gezielten
Austausch einzelner Aminosäuren, oder durch Allosterie, d.h. durch Bindung von
Liganden an bestimmten Bindungsstellen. Die extrinsische Stabilisierung betrifft das
Makromolekül als Ganzes über seine Kontaktfläche zur Mikroumgebung. Häufig
führen Mutationen, die zur globalen Stabilisierung der nativen Konformation führen,
ablesbar an einer höheren Denaturierungstemperatur Tm zu einer verringerten
enzymatischen Aktivität. Daraus wurde eine entkoppelte Evolution der globalen
Stabilität einerseits und lokal erhöhter Flexibilität als Grundlage der Katalysefähigkeit
gefolgert [40-43].
2.3 Phasenumwandlungen von Proteinen
Unter dem Begriff Proteinfaltung versteht man den Prozess, in dem eine
Polypeptidkette in wässriger Lösung spontan ihre biologisch aktive dreidimensionale
Struktur annimmt [10]. Katalytisch aktiv ist die strukturiert gefaltete Konformation.
Häufig wird der physikochemischen Beschreibung der Denaturierung ein Modell
zugrunde gelegt, das die Phasenumwandlung als Balance zwischen nur zwei
Zuständen, einem geordneten gefalteten und einem ungeordneten statistischen
beschreibt. In diesem Modell befindet sich ein Proteinmolekül im Gleichgewicht
zwischen der nativen, gefalteten Konformation N und der denaturierten, inaktiven
Form D.
Formel 1
Für dieses Modell der Proteinfaltung lässt sich die Gleichgewichtskonstante K
definieren und mit Hilfe der Gibbs-Gleichung die thermodynamische Stabilität ∆G
eines nativen Proteins gegenüber seinem entfalteten Zustand angeben:

_______________________________________________________________2. Grundlagen
24
[ ][ ] D
F
kk
DNK ==
(1)
KTRGDN ln⋅⋅−=∆
(2)
Dabei sind kF und kD die mikroskopischen Geschwindigkeitskonstanten 1. Ordnung
von Faltung und Denaturierung, R die Gaskonstante und T die absolute Temperatur
in Kelvin. Strategien zur Verschiebung des Gleichgewichts hin zum nativen Zustand
können entweder an der Erhöhung der Faltungsgeschwindigkeit kF oder der
Erniedrigung der Denaturierungsgeschwindigkeit kD ansetzen.
Die Proteinfaltung und -entfaltung gehorcht der thermodynamischen Grundgleichung:
STHG DN
DN
DN ∆−∆=∆
(3)
Wechselt ein Protein bei konstanter Temperatur von einem Zustand in einen
anderen, N→D, ergeben sich die Änderungen der Enthalpie und der Entropie direkt
aus der integralen Form ihrer Definitionen:
( )∫ ∆+⋅∆=−=∆T
Tref
DN
DNND
DN
ref
THdTCHHH
(4a)
( )refDN
T
T
DN
NDDN TSdT
TCSSS
ref
∆+⋅∆
=−=∆ ∫
(4b)
Dabei ist
NDDN CCC −=∆
(5)
die Differenz zwischen der Wärmekapazität von Protein im nativen Zustand N und
dem denaturierten Zustand D bei der gegebenen Temperatur. Besteht also eine

_______________________________________________________________
25
2. Grundlagen
Wärmekapazitätsdifferenz zwischen den beiden Zuständen, sind sowohl ∆DNH als
auch ∆DNS temperaturabhängig [12,13].
Abbildung 2: Temperaturabhängigkeit der thermodynamischen Parameter für die Denaturierung eines
hypothetischen Modelproteins, berechnet nach einem 2-Zustandsmodell (Gleichungen 6a, 6b und 8)
mit Tm= 333 K, ∆DNH= 500 kJ mol-1, einem temperaturunabhängigen ∆D
NC von 7 kJ mol-1 K-1 (≅0,044
µJ/µg/K) und ∆DNS=1,5 kJ mol-1 K-1. Die Denaturierungstemperatur Tm wird markiert durch den
Schnittpunkt der ∆DNH - bzw. T∆D
NS-Linien. Bei dieser Temperatur schneidet ∆DNG die Nulllinie.
Für einen begrenzten Temperaturbereich ist es eine sinnvolle Näherung ∆DNC
zunächst als Konstante anzusehen, so dass die obigen Gleichungen integriert
werden können [13]:
)()()( refDNref
DN
DN TTCTHTH −⋅∆+∆≅∆
(6a)

_______________________________________________________________2. Grundlagen
26
⎟⎟⎠
⎞⎜⎜⎝
⎛⋅∆+∆≅∆
ref
DNref
DN
DN T
TCTSTS ln)()(
(6b)
Tatsächlich ist ∆DNC etwas temperaturabhängig: die Wärmekapazitätsdifferenz ∆D
NC
nimmt bis ca. 50 °C leicht zu, nimmt bei höheren Temperaturen ab und geht bei ca.
140 °C gegen Null. Die hydrophobe Wechselwirkung ist eine der Quellen der
Stabilität von Proteinen. Sie bestimmt auch die Löslichkeiten von unpolaren
Kohlenwasserstoffen in Wasser. Der hydrophobe Effekt hat seine obere Grenze
ebenfalls bei ca. 140°C und ein Zusammenhang zwischen dem ∆DNC und dem
hydrophoben Effekt wird in der Literatur diskutiert [44,45].
In der Beschreibung der Proteindenaturierung wird häufig der Mittelpunkt des
Phasenübergangs Tm als Referenztemperatur benutzt. Tm ist definiert als die
Temperatur, an der ∆DNG zu Null wird, und es gilt:
( ) ( )mDN
m
mDN TST
TH ∆=∆
(7)
ist. Daraus ergibt sich für die Temperaturabhängigkeit von ∆DNG unter der Annahme,
dass sich die Wärmekapazitätsdifferenz ∆DNC zwischen N und D mit der Temperatur
nicht ändert, Gleichung 8:
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⋅+−∆−⎟⎟
⎠
⎞⎜⎜⎝
⎛−∆=∆
mm
DN
mm
DN
DN T
TTTTCTTTHTG ln1)()(
(8)
Tm ist der Schmelzpunkt des Proteins, ∆DNH die thermische Umwandlungs-Enthalpie
und ∆DNC der Unterschied zwischen der Wärmekapazität des nativen und des
denaturierten Proteins. Diese Gleichung wird als Gibbs-Helmholtz-Gleichung
bezeichnet. Die drei Parameter Tm, ∆DNH und ∆D
NC können allesamt durch
kalorimetrische Messungen experimentell bestimmt werden. Die Auftragung von

_______________________________________________________________
27
2. Grundlagen
∆DNG gegen T liefert für alle Proteine, für die diese Daten vorliegen, eine
hyperbolisch gekrümmte Kurve (Abbildung 2). Sie wird als Stabilitätskurve des
Proteins bezeichnet [9]. Eine interessante Konsequenz dieser hyperbolischen
Temperaturabhängigkeit von ∆DNG(T) ist die Tatsache, dass neben dem
Schmelzpunkt auch einen Kältedenaturierungspunkt (kaltes „Schmelzen“) existiert
[46], und dass es für jedes Protein eine Temperatur maximaler thermodynamischer
Stabilität T´ gibt, die für die meisten Enzyme in der Nähe der Raumtemperatur (283-
293 K) liegt [47].
2.4 Die Überschuss-Wärmekapazität von Proteinen in Lösung
zwischen 25°C und 100°C.
Die Wärmekapazität einer Probe spiegelt die Fähigkeit des Materials wieder,
Wärmeenergie zu absorbieren ohne dabei seine Temperatur zu erhöhen. Dies ist
zentral für DSC-Messungen von verdünnten Proteinlösungen und die
zugrundeliegende fundamentale Thermodynamik. Flüssiges Wasser hat eine relativ
hohe Wärmekapazität (≅ R/2) aufgrund seines ausgedehnten Netzwerkes aus
Wasserstoffbrücken. Aufgenommene Wärmeenergie kann in das Aufbrechen dieser
Wasserstoffbrückenbindungen verteilt werden, ohne die kinetische Energie der
Wassermoleküle, d.h. ihre Temperatur zu erhöhen. Organische Materie, Proteine und
Nukleinsäuren eingeschlossen, hat eine niedrigere Wärmekapazität als Wasser,
außer es findet in ihr ein Prozess statt, der in irgendeiner Weise mit dem Bruch von
Bindungen verbunden ist. Die Wärmekapazität von verdünnten Lösungen von
Biomolekülen ist demnach hauptsächlich von der Wärmekapazität des Wassers
bestimmt.

_______________________________________________________________2. Grundlagen
28
Das erste Resultat eines DSC Experimentes ist die partielle spezifische
Wärmekapazität des Proteins <C(T)>. Diese Größe enthält wichtige Informationen
über den Zustand des Proteins. Für kleine, globuläre Proteine liegen die Werte der
spezifischen partiellen Wärmekapazität <C> bei 25°C im Bereich zwischen 1,2 und
2,3 Jg-1K-1, und nehmen linear mit der Temperatur mit einer Steigung von
5-7 mJK-2g-1 zu. Ist die Wärmekapazität bei 25°C signifikant höher als 2,3 Jg-1K-1,
kann das ein Hinweis darauf sein, dass das Protein nicht vollständig gefaltet ist. Die
Wärmekapazität des denaturierten Zustands ist um ca. 0,3-0,7 JK-1g-1 höher als die
des nativen und kann durch eine quadratische Funktion der Temperatur angenähert
werden [13].
Die entscheidende Größe der thermodynamischen Beschreibung des thermischen
Denaturierens eines Proteins ist die Überschuss-Wärmekapazität <∆DNC>. Diese ist
zugänglich durch Substraktion der extrapolierten temperaturabhängigen
Wärmekapazität des nativen Zustands CN von der kontinuierlich gemessenen
Wärmekapazität des Proteins <C> :
NDN CCC −=∆
(9)
Der Bruchteil des Proteins im denaturierten Zustand PD ist ein Maß für den Fortschritt
der Umwandlung [48]. Bei niedrigen Temperaturen, fern der
Umwandlungstemperatur Tm, ist er verschwindend klein, jenseits der
Umwandlungstemperatur nahezu 1.
[ ]
[ ] [ ]NDDPD +
=
(10)
[D] ist die Konzentration des denaturierten Proteins, [N] die des Nativen. [N]+[D] ist
die Gesamtenzymkonzentration.

_______________________________________________________________
29
2. Grundlagen
Mithilfe von PD kann die Überschuss-Enthalpie für den Zwei-Stufenprozess NºD
definiert werden:
HPH DND
DN ∆=∆
(11a)
Die Überschusswärmekapazität wird analog definiert:
CPC DND
DN ∆=∆
(11b)
Wegen:
D
D
PPK−
=1
(12)
und
( ) ( ) ( ) dTdP
PPdTdPP
dTdPP
RTH
TK D
DD
DD
DD
DN
−=−+=
∆=
∂∂
11111ln
2
(13)
ergibt sich für die Temperaturabhängigkeit von PD:
( )DD
DND PP
RT
HdTdP
−∆
= 12
(14)
_______________________________________________________________2. Grundlagen
30
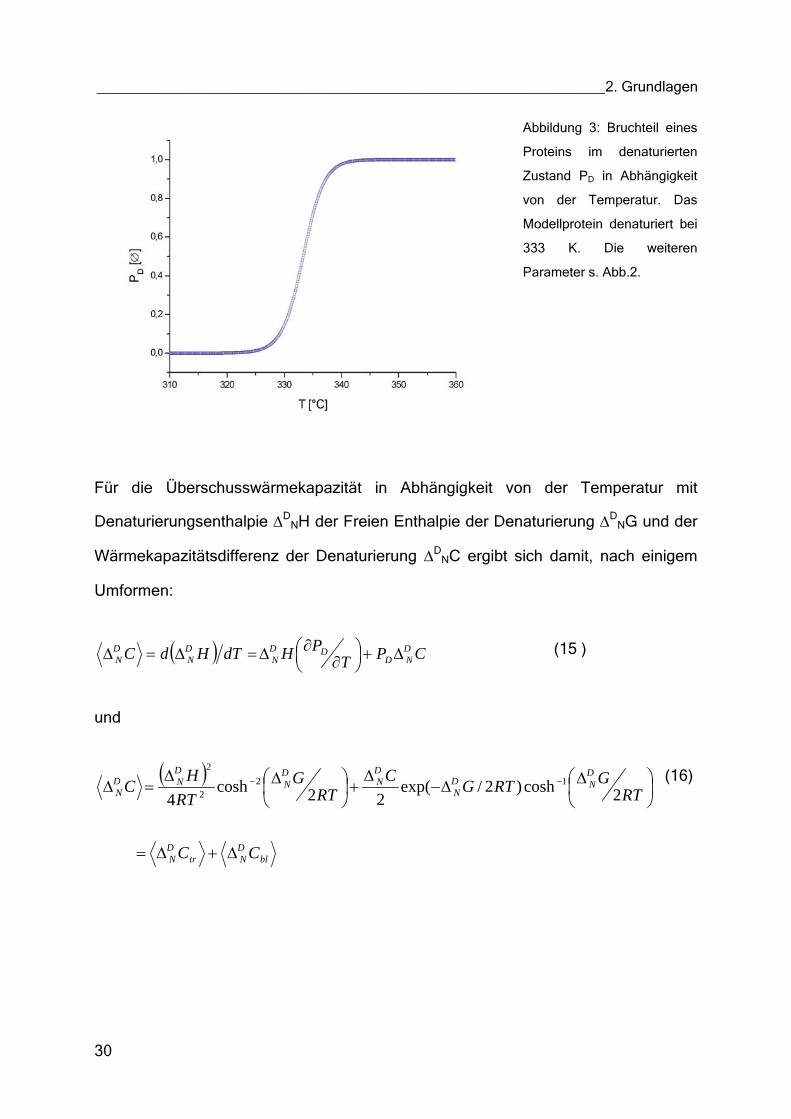
Abbildung 3: Bruchteil eines
Proteins im denaturierten
Zustand PD in Abhängigkeit
von der Temperatur. Das
Modellprotein denaturiert bei
333 K. Die weiteren
Parameter s. Abb.2.
Für die Überschusswärmekapazität in Abhängigkeit von der Temperatur mit
Denaturierungsenthalpie ∆DNH der Freien Enthalpie der Denaturierung ∆D
NG und der
Wärmekapazitätsdifferenz der Denaturierung ∆DNC ergibt sich damit, nach einigem
Umformen:
( ) CPTPHdTHdC D
NDDD
NDN
DN ∆+⎟
⎠⎞⎜
⎝⎛
∂∂∆=∆=∆
(15 )
und
( )
blDNtr
DN
DND
N
DN
DN
DND
N
CC
RTGRTG
CRT
GRT
HC
∆+∆=
⎟⎠⎞
⎜⎝⎛ ∆∆−
∆+⎟
⎠⎞
⎜⎝⎛∆∆
=∆ −−
2cosh)2/exp(22cosh
412
2
2(16)

_______________________________________________________________
31
2. Grundlagen
Abbildung 4: Überschuss-Wärmekapazität eines Protein, berechnet nach Gleichung 16. Die Parameter
des Übergangs sind dieselben wie in Abbildung 2.
Der erste Term auf der rechten Seite, <∆DNCtr>, ist die Funktion der Überschuss-
Wärmekapazität des Übergangs und definiert den charakteristischen Peak in der
Funktion der Wärmekapazität. Dieser Anteil der Exzess-Wärmekapazität ist eine
Folge der erhöhten Enthalpie-Fluktuationen des Systems, wenn es während der
Denaturierung von einem Zustand in den anderen wechselt [49].
Der zweite Term der rechten Seite, <∆DNCbl>, beschreibt den sigmoidalen Verlauf der
Basislinie, wie er üblicherweise mit der Proteindenaturierung oder anderen
Prozessen, die mit positivem ∆C einhergehen, verbunden ist.

_______________________________________________________________2. Grundlagen
32
,
,
2.5 Einfluss der Mikroumgebung auf die Proteinstabilität
„... if a biological process i.e., a biochemical reaction,
is controlled by some factor, x,
(which we may call the allosteric effector,
cofactor, coenzyme, cosolvent etc.),
then the extent of binding of x will change
during the course of the reaction; i.e.,
binding will be different to the reactant and to the product.“
Serge N. Timasheff, 1998
Die Stabilität von Proteinen kann durch Zusatz von Ko-Solventien in hohen
Konzentrationen (typischerweise zwischen 0,3 M und 10 M) modifiziert werden [50].
Die benötigten hohen Konzentrationen zeigen, dass die Wechselwirkungen schwach
sind, charakterisiert durch eine kaum von Null verschiedene Änderung der Freien
Energie. Zu den am häufigsten zur Stabilisierung verwendeten Substanzen gehören
Zucker und Polyole, sowie gute „salting-out“ Salze (z.B. Na2SO4, s.a. Kapitel 2.5.2.3
und 2.5.2.3). Die gebräuchlichsten Denaturierungsmittel sind Harnstoff und
Guanidiniumhydrochlorid [51].
Die Wirkmechanismen dieser Ko-Solventien auf die Löslichkeit und Stabilität von
Proteinen sind direkte Auswirkungen der bevorzugten Wechselwirkung des Proteins
[52]. Die beobachtete Wechselwirkung kann bevorzugte Bindung oder bevorzugte
Verdrängung, auch bevorzugte Hydratation genannt, sein. Bevorzugte Bindung
beschreibt einen Überschuss an Ko-Solvens in der Sphäre des Proteins gegenüber
der Lösung. Bevorzugte Hydratation beschreibt die Verarmung an Ko-Solvens in der
Proteinsphäre. Bevorzugte Bindung und bevorzugte Hydratation verhalten sich

_______________________________________________________________
33
2. Grundlagen
komplementär zueinander, weil freibleibende Stellen auf der Oberfläche nicht
existieren.
2.5.1 Die Wyman-Gleichung und das Konzept der bevorzugten
Wechselwirkung
Der Effekt eines Liganden auf ein chemisches Gleichgewicht wie die Denaturierung
wird durch die Wyman-Gleichung, Gleichung 17, beschrieben [53].
Für eine monomolekulare Gleichgewichtsreaktion wie die Denaturierung eines
Proteins, N º D, die von einem Ko-Solvens beeinflusst ist, besteht ein
Zusammenhang zwischen der Bindung des Ko-Solvens´ an die beiden
Konformationen des Proteins und der Gleichgewichtskonstanten der
Umwandlungsreaktion von N nach D.
Die sogenannte bevorzugte Wechselwirkung ist Ausdruck einer Neuverteilung der
Lösungsmittelzusammensetzung in der Umgebung eines Proteinmoleküls. Die Größe
(∂m3/∂m2)T,P,µ3 ist die Störung der Ko-Solvens-Konzentration m3 bei Änderung der
Proteinkonzentration m2 unter Bedingungen konstanten chemischen Potentials des
Ko-Solvens µ3. Sie wird im folgenden als thermodynamische oder bevorzugte
Bindung bezeichnet.
Ist der Parameter der bevorzugten Bindung (∂m3/∂m2)T,P,µ3 positiv, ist das Ko-Solvens
in der Sphäre des Proteins angereichert oder bevorzugt gebunden. Ist der Parameter
der bevorzugten Bindung negativ und der Parameter der bevorzugten Hydratation
(∂m1/∂m2)T,P,µ3 positiv, ist das Ko-Solvens in der Sphäre des Proteins relativ zur
umgebenden Lösung abgereichert oder bevorzugt verdrängt.
Nach der Scatchard-Notation, die in der Literatur für die Beschreibung der
Thermodynamik von Drei-Komponenten-Lösungen gebraucht wird, symbolisiert 1
Wasser als Solvens, 2 das Protein bzw. Enzym und 3 das Ko-Solvens [54,55]. Die
rechte Seite der Wyman-Gleichung zeigt, dass die Reaktion (hier die Denaturierung)
kontrolliert ist durch die Änderung der bevorzugten Bindung zwischen dem

_______________________________________________________________2. Grundlagen
34
reagierenden System (Protein) und dem Ko-Solvens. Die beiden Prozesse, das
Gleichgewicht N º D und die Bindung des Ko-Solvens, sind miteinander verknüpft
d.h. eine Störung des einen Prozesses wird notwendigerweise von einer Störung des
anderen begleitet [56]. Wenn ∆DN(∂m3/∂m2)T,P,µ3 größer als Null ist, verschiebt das
Ko-Solvens das Gleichgewicht N º D auf die rechte Seite und wirkt als Destabilisator.
Ist ∆DN(∂m3/∂m2)T,P,µ3 kleiner als Null, verschiebt das Ko-Solvens das Gleichgewicht
nach links und wirkt als Stabilisator:
N
PT
D
PTPT
DN
mPT
DN
mPTmm
mm
mmG
aK
33322 ,,2
3
,,2
3
,,2
3
,,3,,3lnln
µµµµ ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
−⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∆=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∆∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
(17)
Darin ist ∆DNG die Änderung der Standard Freien Energie der Reaktion N º D, µ ist
das chemische Potential der Komponente i, µi=µi+RTlnai, ai ist die Aktivität der
Komponente i, ai=miγi, γi ihr Aktivitätskoeffizient, T die thermodynamische
Temperatur. Diese Gleichung stellt die Verbindung her zwischen der Betrachtung der
monomolekularen Entfaltung eines Polypeptids im Hinblick auf eine günstige
Energetik charakterisiert durch eine negative Gibb´sche Freie Reaktionsenthalpie
∆DNG bei veränderlichen Konzentrationen des Ko-Solvens´ a3 und der Betrachtung
der nativen und der denaturierten Konformation separat im Hinblick auf die relative
Anzahl der auf der Oberfläche des Proteins befindlichen Wasser- bzw. Ko-Solvens-
Moleküle m1 und m3.

_______________________________________________________________
35
2. Grundlagen
Abbildung 5: Bein Wechsel zwischen nativer (N) und denaturierter Konformation (D) nimmt
die lösungsmittelzugängliche Oberfläche des Proteins zu. Sind die Wechselwirkungen des
Ko-Solvens mit dem Protein ungünstig und ist das Protein infolgedessen bevorzugt
hydratisiert ist (∂m3/∂m2)T,P,µ3 negativ sowohl für N als auch für D; stärker negativ aber für D.
∆DN(∂m3/∂m2)T,P,µ3 wird dann ebenfalls negativ. Das ist gleichbedeutend mit einer
Verkleinerung der Gleichgewichtskonstanten der Denaturierung K bei Erhöhung der Ko-
Solvens-Konzentration a3 d.h. mit einer Stabilisierung der nativen Konformation.
Während der Denaturierung verliert sich die Struktur des Proteins und die
Wechselwirkungen Protein/(Ko-)Solvens verändern sich. Verschiedene generelle
Typen können unterschieden werden:
Klasse A: Das Ko-Solvens ist aus der Sphäre des Proteins sowohl im nativen wie im
denaturierten Zustand vorzugsweise verdrängt. Der Mechanismus ist unspezifisch,
d.h. das Ko-Solvens ist vollständig indifferent gegenüber chemischen Charakteristika
der Proteinoberfläche. Weil im Zuge der Denaturierung die Protein/Solvens
Kontaktfläche anwächst, nehmen auch die ungünstigen Wechselwirkungen zu und
damit auch die Verdrängung. Die Wechselwirkung ist in jedem Falle ungünstig, für
den denaturierten wegen der größeren Kontaktfläche aber noch ungünstiger als für
den nativen. Die weiteren Klassen können als Varianten dieses allgemeinen Falles
betrachtet werden [56,57].

_______________________________________________________________2. Grundlagen
36
Klasse B: Das Ko-Solvens ist vom nativen Protein vorzugsweise verdrängt, vom
denaturierten Protein aber vorzugsweise gebunden. Das Gleichgewicht von Formel 1
wird auf die rechte Seite in Richtung Denaturierung verschoben. Dies wurde für die
Wechselwirkung einiger Proteine mit Polyethylenglykol (PEG) beobachtet [58-61].
Klasse C: Das Ko-Solvens (Harnstoff, Guanidiniumhydrochlorid) bindet nur schwach
oder überhaupt nicht an das native Protein aber intensiv an das denaturierte.
∆(∂m3/∂m2)T,P,µ3 ist hier positiv und die Denaturierung wird durch die günstige
Wechselwirkung des Ko-Solvens mit dem denaturierten Protein angetrieben [62].
Die ersten beiden Fälle sind in Abbildung 6 durch ein chemisches Potential-
Diagramm illustriert. Beide, Stabilisatoren und Destabilisatoren, erhöhen die Energie
des nativen Zustands gegenüber reinem Wasser als Referenz. Ein Stabilisator erhöht
gleichzeitig die Energie des denaturierten Zustands. Ist diese Erhöhung stärker als
die des nativen Zustands resultiert daraus eine Vergrößerung der Energiedifferenz
∆DNG zwischen den beiden Zuständen. Das ist gleichbedeutend mit einer
Stabilisierung der nativen Konformation.
Ein Destabilisator wie z.B. Guanidiniumhydrochlorid oder Harnstoff bindet an den
denaturierten Zustand und senkt damit dessen Energie. Dadurch wird die
Energiedifferenz zwischen der nativen und denaturierten Konformation gesenkt und
der native Zustand destabilisiert.

_______________________________________________________________
37
2. Grundlagen
Abbildung 6: Energieschema des
Effektes von Ko-Solventien auf die
Proteinstabilität. Ein Stabilisator
erhöht die Energiedifferenz
zwischen nativem Protein N und
denaturiertem Protein D, ein
Destabilisator senkt sie. Typische
Energien sind durch die Zahlen
angedeutet.
Dieser generelle Mechanismus macht deutlich, wie thermodynamisch ungünstige
Wechselwirkungen (Verdrängung bzw. Anhebung der Energie) zwischen einem
Protein und einem Ko-Solvens zu einer Stabilisierung des Proteins, genauer, der
Struktur des nativen Proteins, führen kann [63]. Plausibel wird das, wenn man
beachtet, dass die Wechselwirkungen, günstige wie eben auch ungünstige, mit der
lösungsmittelzugänglichen Oberfläche des Proteins zu- oder abnehmen. Die
Oberfläche der kompakteren nativen Konformation ist aber um 10-20% geringer als
die der denaturierten [64,65].
Tabelle 1: Übersicht über die Klassifizierung von Ko-Solventien im Hinblick auf ihre
protein(de)stabilisierende Wirkung.
Klasse (∂m3/∂m2)D
T,P,µ3 (∂m3/∂m2)N
T,P,µ3 ∆(∂m3/∂m2)T,P,µ3 Effekt Beispiel
A - - - Stabilisator Sucrose
B + - + Destabilisator PEG
C + +/- + Destabilisator GndHCl
D +/- + - Stabilisator (NaCl)
klassischer
Ligand
+/- ++ - Stabilisator Neuro-
transmitter

_______________________________________________________________2. Grundlagen
38
Auflösen des trockenen Proteins in Wasser führt notwendigerweise zur Besetzung
der Bindungsstellen der gesamten Oberfläche des Proteins mit Wassermolekülen
(Reaktion 1, Abbildung 7). Für eine einzelne Bindungsstelle ist damit eine Freie
Energie des Kontakts ∆G12 verbunden (Gleichung 19).
P + m H2O º P*(H2O)m (Reaktion 1)
( )[ ]
[ ] [ ]mm
OHPOHP
K2
21
⋅
⋅=
(18)
112 ln KRTG −=∆ (19)
Wird umgekehrt in einem hypothetischen Prozess das pure Ko-Solvens zu dem
trockenen Protein gegeben (Reaktion 2), wird pro Bindungsstelle die Energie ∆G32
frei.
P + KS ºP*KS (Reaktion 2)
[ ][ ] [ ]KSP
KSPK⋅⋅
=3
(20)
332 ln KRTG −=∆ (21)
Wird das Ko-Solvens zu einer Lösung des Proteins in Wasser gegeben und soll es zu
einer Bindung kommen, müssen zunächst die Wassermoleküle in Kontakt mit der
Bindungsstelle abgelöst werden unter Verlust der Wechselwirkungsenergie ∆G12.
Erst dann kann das Ko-Solvens mit der Energie ∆G32 (Reaktion 3) binden. Die
resultierende Gesamtenergie des Austauschs ist demnach:
exex KRTKKRTGGG lnln
1
31232 −=⎟⎠⎞⎜
⎝⎛−=∆−∆=∆
(22)

_______________________________________________________________
39
2. Grundlagen
Die gesamte Bindungsgleichgewicht kann folgenderweise dargestellt werden:
P•(H2O)n + KS º P•KS + n H2O (Reaktion 3)
Und die gemessene Bindungskonstante KB ist tatsächlich eine Austauschkonstante
Kex.
1
3
KK
Kex ≡
(23)
Ist die Affinität für den Liganden größer als für Wasser, wird im zeitlichen Mittel die
Stelle häufiger mit Ko-Solvens als mit Wasser besetzt sein. Das wird deutlich aus
Gleichung 24, die den Zusammenhang zwischen dem Interaktionsparameter und der
Bindung herstellt .
2233 ,,3
2
,,3
3
,,2
3
,,2
3
mPTmPTmPTPT mmmm
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
µµµµ
µ
(24)
Die Größe (∂µ2/∂m3)T,P,m2=(∂µ3/∂m2)T,P,m3 ist Ausdruck der wechselseitigen
Beeinflussung von Protein und Ko-Solvens und wird als Parameter der bevorzugten
Wechselwirkung bezeichnet.
Abbildung 7: Schematische
Darstellung der Wechselwirkungen
an der Oberfläche eines Proteins (2)
in einem gemischten Solvens.
Wassermoleküle (1) werden an der
Bindungsstelle durch Ko-Solvens-
Moleküle (3) ausgetauscht [24].

_______________________________________________________________2. Grundlagen
40
Der Nenner auf der rechten Seite ist der Selbst-Interaktionsparameter des Ko-
Solvens´[59]:
⎟⎟⎠
⎞⎜⎜⎝
⎛∂
∂+=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
3
3
3,,3
3 ln1
2mm
RTm
mPT
γµ
(25)
Der Selbstinteraktionsparameter (∂µ3/∂m3)T,P,m2 ist in der Regel positiv und nimmt mit
der Temperatur (leicht) ab. Deshalb haben der Parameter der bevorzugten
Wechselwirkung (∂µ2/∂m3)T,P,m2 und der Parameter der bevorzugten Bindung
(∂m3/∂m2)T,P,µ3 stets gegensätzliches Vorzeichen, d.h. Ko-Solvens, welches günstiger
mit dem Protein interagiert als Wasser, wird bevorzugt gebunden, andernfalls
bevorzugt verdrängt .
Abbildung 8: Selbstinteraktions-
Parameter SIP≡(∂µ3/∂m3)T,P,m2 von
PEG unterschiedlichen Molekular-
Gewichts [59].
Bevorzugte Verdrängung des Ko-Solvens ist gleichbedeutend mit bevorzugter
Hydratation der Bindungsstelle, d.h. (∂m1/∂m2)T,P,µ3>0. Den Zusammenhang
zwischen der Bindung des Ko-Solvens und der Hydratation gibt Gleichung 26. Ko-

_______________________________________________________________
41
2. Grundlagen
Solvens-Bindung und Hydratation sind komplementär. Der Austausch-Faktor ist
gegeben durch den Molenbruch von Haupt- und Ko-Solvens.
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
3
2
3
1
,,2
3
3
1
,,2
1
33µµ
µµmm
mm
mm
mm
PTPT
(26)
Der Bindungsparameter (∂m3/∂m2)T,P,µ3 ist Resultat der Balance der
Wechselwirkungen des Proteins mit Ko-Solvens und mit Wasser. Der Parameter
kann in individuelle Beiträge Bi der Lösungsmittelbestandteile zerlegt werden:
11
33
,,2
3
3
Bmm
Bmm
PT
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
µ
(27)
B1 und B3 sind Zahlen in der Größenordnung von einigen hundert. Sie geben an, wie
viele Wasser- bzw. Ko-Solvens-Moleküle sich effektiv an Stellen auf der
Proteinoberfläche befinden.
Aus der Form dieser Gleichung ist ersichtlich, dass für den Fall, dass das Verhältnis
B3/B1 den Verhältnissen in der Lösung entspricht, also
1
3
1
3m
mB
B =,
(28)
die thermodynamische Bindung (∂m3/∂m2)T,P,µ3 gleich Null ist, obwohl Stellen auf der
Oberfläche des Proteins mit Ko-Solvens (B3) besetzt sind. Die thermodynamische
oder die bevorzugte Bindung ist also nicht gleichbedeutend mit der Anzahl von
Bindungsstellen, die mit Ko-Solvens besetzt sind, sondern beschreibt die Balance
zwischen der Besetzung mit Ko-Solvens und Wasser relativ zur Zusammensetzung
der Lösung im zeitlichen Mittel. Diese Balance kann positiv, negativ oder
ausgeglichen sein. Für schwach und sehr schwach wechselwirkende Ko-Solventien
sind die Affinitäten von Wasser und Ko-Solvens von derselben Größenordnung, d.h.

_______________________________________________________________2. Grundlagen
42
∆G12 ≈ ∆G32, und die Wechselwirkung mit Wasser muss explizit berücksichtigt
werden [64].
Die thermodynamische Beschreibung kann sich generell auf zwei unterschiedliche
experimentelle Verfahren stützen:
Einerseits kann der Energieunterschied des Wechsel von einem Medium in das
andere z.B. von purem Wasser in die Lösung des Ko-Solvens in Wasser für beide
Konformationen des Proteins separat gemessen werden. Dies ist die Freie Transfer-
Energie ∆µ2,TR. Geeignete Techniken hierfür sind die Gleichgewichtsdialyse, NMR,
ITC, und dynamische Lichtstreuung [66].
Andrerseits kann der Konformationswechsel von N nach D in verschiedenen Medien
untersucht werden. Die entsprechende Technik ist die DSC.
Überträgt man die Reaktion des Proteins aus Wasser in das Ko-Solvens, ändert sich
die Freie Reaktionsenergie ∆DNG:
)1()3( GGG D
NDN
DN ∆−∆=∆∆ (29)
Auch verändern sich die Wechselwirkungen der Lösungsmittelkomponenten mit dem
Protein im Zuge der Denaturierung. Die Freie Standard Transfer-Energie ∆µ2,Tr ist die
Änderung des chemischen Potentials des Proteins, wenn es aus Wasser in das Ko-
Solvens/Wasser System übertragen wird.
)1()3( 22,2 µµµ −=∆ Tr (30)
Weil der Effekt von Ko-Solventien relativ zu Wasser durch ∆∆µ2,Tr definiert ist, ist es
nötig, den Einfluss des Ko-Solvens´ auf beide Zustände der Reaktion N und D zu
kennen.
)()( ,2,2,2 ND TrTrTr µµµ ∆−∆=∆∆
(31)

_______________________________________________________________
43
2. Grundlagen
Der Zwei-Stufen-Prozess vom Ausgangszustand, natives Protein in Wasser (N1),
zum veränderten Zustand, denaturiertes Protein in Ko-Solvens-Lösung (D3), kann
auf zweierlei Wegen erfolgen:
Denaturierung in Wasser, gefolgt von Transfer in die Ko-Solvens-Lösung, oder
Transfer des nativen Proteins aus Wasser in die Ko-Solvens-Lösung, gefolgt von
dessen Denaturierung.
Abbildung 9: Thermodynamische Box für die ko-
solvens-abhängige Denaturierung: Die beiden
Pfade N1 nach D3 sind thermodynamisch
äquivalent [65]. Das Konzept der bevorzugten
Bindung wird erhärtet dadurch, dass die
thermodynamischen Boxen der bestuntersuchten
Proteine, die auf Messwerten unterschiedlicher
Methodik (z.B. DSC und NMR) basieren, im
Rahmen des Messfehlers geschlossen sind [58].
Für die (ko-)solvensabhängige Denaturierung gilt :
TrN
TRD
TRDN
DN GG ,2,2,213 µµµ ∆∆=∆−∆=∆−∆
(32)
Die Freie Transfer-Energie eines Proteins, das aus reinem Wasser in ein Ko-
Solvens/Wasser-Gemisch überführt wird, ist der Wert der Änderung der gesamten
Freien Energie der Proteinlösung, wenn der zunächst ausschließlich wässrigen
Lösung das Ko-Solvens hinzugefügt wird. Deshalb steht sie in Beziehung zur
Änderung der bevorzugten Wechselwirkung bei Zugabe von Ko-Solvens [59].
30 ,,3
2,2
3
2
dmm
m
mPTTR ∫ ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
=∆µµ
(33)
Die Änderung der Freien Transfer-Energie ∆∆µ2,Tr kann durch Integration der
Wyman-Gleichung, Gleichung 17, erhalten werden:

_______________________________________________________________2. Grundlagen
44
30 ,,3
,2
3
2
ln dmm
KRTm
mPTTR ∫ ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂
∂=∆∆µ
(34)
Diese Gleichungen zeigen, dass die Kenntnis der bevorzugten Wechselwirkung bei
einer einzelnen Konzentration des Ko-Solvens´ zur vollständigen Charakterisierung
des Einflusses des Ko-Solvens´ auf das Denaturierungsgleichgewicht nicht ausreicht.
Der Parameter der bevorzugten Wechselwirkung ist ein Maß für die Änderung von
∆µ2,Tr d.h. für die Änderung des thermodynamischen Zustands des Proteins, wenn
die Konzentration des Ko-Solvens um einen infinitesimalen Betrag verringert wird.
Deshalb ist es durchaus möglich, dass die beiden Parameter gegenläufiges
Verhalten zeigen und auch unterschiedlichen Vorzeichens sein können, auch wenn
das zunächst paradox erscheint [57].
Ist der Kontakt mit dem Protein thermodynamisch ungünstig, d.h. ist (∂µ2/∂m3)T,P,m2
positiv, wie bei einer Vielzahl von stabilisierenden „salting-out“ Ko-Solventien, wird
das Ko-Solvens aus der Hydratationssphäre von nativen globulären Proteinen
vorzugsweise verdrängt. Umgekehrt ist der Kontakt von Denaturierungsmitteln wie 6
M Guanidiniumhydrochlorid (GndHCl) oder 8 M Harnstoff mit dem denaturierten
Zustand des Proteins vorteilhaft und (∂µ2/∂m3)T,P,m2 ist negativ.
Die Änderung des Parameter der bevorzugten Bindung ∆(∂m3/∂m2)T,P,µ3 entscheidet
darüber, in welche Richtung ein Gleichgewicht durch ein Ko-Solvens verschoben
wird. Dieser Parameter ist bei schwach wechselwirkenden Systemen nicht
gleichzusetzen mit der Änderung der Belegung von Bindungsstellen. Die klassische
Bindungstheorie erweist sich bei schwach bindenden Ko-Solventien als nicht
vollständig. In der klassischen Bindungstheorie [68-70] wird Bindung als Besetzung
einer Bindungsstelle des Proteins, 2, mit Ko-Solvens, 3, unter Bildung eines
Komplexes 2*3 definiert.

_______________________________________________________________
45
2. Grundlagen
3232 ⋅↔+ (Reaktion 2)
Für dieses Gleichgewicht ist die Gleichgewichtskonstante wie folgt definiert:
32
233 aa
aK⋅
=
(35)
a23 ist die Konzentration von Bindungsstellen, die mit einem Ko-Solvens-Molekül
belegt sind, a2 die Konzentration freier Bindungsstellen und a3 die Ko-Solvens-
Konzentration.
Die Wahrscheinlichkeit θ, eine Stelle mit Ko-Solvens belegt zu finden, ist durch die
Standard Langmuir-Isotherme gegeben [71]:
33
33
1 aKaK⋅+
⋅=θ
(36)
Klassische biochemischen Liganden, z.B. Enzymsubstrate oder Inhibitoren, haben
Bindungskonstanten >100 M-1 und die Bindungsstelle ist auch in verdünnter Lösung
des Liganden nahezu vollständig und zeitüberdauernd besetzt. Hier sind Bindung
und Besetzung identisch, weil die Wahrscheinlichkeit einer rein statistischen
Besetzung vernachlässigbar ist. Die Bindungsenergie pro Bindungstelle ist dann [65]:
( )331ln aKRTGB ⋅+−=∆
(37a)
Für die Denaturierung, ergibt sich daraus, wenn man annimmt, dass alle ∆n im Zuge
der Reaktion neu präsentierten Bindungsstellen thermodynamisch gleichwertig sind:
( )331ln aKnRTGB ⋅+∆−=∆∆ (37b)
∆n wird häufig identifiziert mit der Steigung des Wyman-Plots (Gleichung 17).

_______________________________________________________________2. Grundlagen
46
Die Gleichungen der klassischen Bindungstheorie sind gültig für Spezialfälle von Ko-
Solventien, etwa für ihre Bindung an wenige unabhängige Bindungsstellen mittlerer
und starker Affinität. Sie vermögen, die Stabilisierung von Enzymen durch den
Einfluss stark-bindender Substrate, Modulatoren oder Inhibitoren zu beschreiben
[72]. Werden sie jedoch auf schwach wechselwirkende Ko-Solventien angewendet,
führen sie zu Inkonsistenzen und die Definition von Bindung selbst wird fragwürdig.
Ist ein Ko-Solvens-Molekül in einer Bindungsstelle gebunden oder nicht?
Thermodynamisch ist es nur dann gebunden, wenn die Belegung das Maß
übersteigt, welches aufgrund der Lösungsmittelzusammensetzung zu erwarten ist. In
der klassischen stöchiometrischen Bindungstheorie hingegen gilt ein Ko-Solvens-
Molekül, das sich nur an einer Bindungsstelle befindet, per Definition schon als
gebunden.
Die Theorie der bevorzugten Bindung trägt der Tatsache Rechnung, dass ein Ko-
Solvens-Molekül, will es mit dem Protein in Kontakt treten, Wassermoleküle
verdrängen muss. Den Ausgangspunkt bildet das folgende Austauschgleichgewicht:
132312 ⋅+⋅↔+⋅ mm (Reaktion 3)
2*1m ist das Protein im Komplex mit m Wassermolekülen.
Die Gleichgewichtskonstante für den Austausch eines Wassermoleküls gegen ein
Ko-Solvens-Molekül ist analog zu Gleichung 35:
321
123
1
3
aaaa
KK
Km
ex ⋅⋅
==
(39)
und
exex KRTG ln−=∆
(40)

_______________________________________________________________
47
2. Grundlagen
Die gesamte Freie Energie der Solvatisierung ∆GS des unsolvatisierten Proteins im
gemischten Medium ist [52,73]:
( )33111ln aKaKRTGS ++−=∆ (41)
ai ist die Aktivität der Komponente i (die Konzentrationen in K ausgedrückt in
Molenbrüchen). Der Vergleich mit Gleichung 37a zeigt, dass ein Term für die
Bindung von Wasser (K1a1) explizit enthalten ist. Im realen System befindet sich das
Protein zunächst in reinem Wasser und alle Bindungsstellen sind notwendigerweise
mit Wassermolekülen belegt. Die Änderung der Freien Energie aufgrund des Eintrags
von Ko-Solvens in die wässrige Lösung ∆GB erhält man durch Substraktion des
Terms für die Wechselwirkung des unsolvatisierten Proteinmoleküls mit Wasser von
Gleichung 41:
( )31ln aKaRTG ExB +−=∆ (42)
Die Schwierigkeit der klassischen Betrachtungsweise liegt in der Gleichsetzung von
∆GB in Gleichung 22 mit ∆G32 unter der Annahme, dass ∆G12=0 ist.
Den Zusammenhang zwischen dem Parameter der bevorzugten Bindung und der
Behandlung der Wechselwirkung mit Wasser und Ko-Solvens mit Hilfe der
Austauschkonstanten Kex hat Schellman im Jahre 1990 hergestellt [73]:
exEx
Ex
Ex
ExstproBind
PT KmK
mK
mmK
mm
θµ
⋅⎟⎠⎞⎜
⎝⎛ −
=+
⎟⎠⎞⎜
⎝⎛ −
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂ 1
3
31
..
,,2
3
1
1
1
3
(43)
Kex in molalen Aktivitäten. Die Wahrscheinlichkeit, dass eine Bindungsstelle mit Ko-
Solvens besetzt ist, ist gegeben durch:
3
3
1 mKmK
Ex
Exex +
=θ
(44)

_______________________________________________________________2. Grundlagen
48
Gleichung 44 hat die Form einer Langmuir-Isotherme (Gleichung 36). Die übliche
Bindungskonstante darin ist durch die Austauschkonstante Kex ersetzt.
Die Bindung biochemischer Ko-Solventien (klassische Liganden) ist normalerweise
stark (KL>104 M-1). Bei geringen Ligand-Konzentrationen, m3<10-4, und mit m1=55,56
wird der zweite Term von Gleichung 27 vernachlässigbar und
3,,2
3
3
Bmm
PT
≅⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
µ
(45)
Bei der Solvenszusammensetzung, bei der die Belegung auf dem Protein dem
Konzentrationsverhältnis in der Lösung entspricht, ist die bevorzugte
(thermodynamische) Bindung gleich Null und das Protein infinitesimalen Änderungen
der Konzentration des Ko-Solvens gegenüber indifferent. (∂µ2/∂m3)T,P,m2 ist gleich
Null, obwohl die Belegung der Proteinoberfläche mit Ko-Solvens-Molekülen hoch ist
und die Gesamtheit der Wechselwirkungen zwischen Protein und Ko-Solvens durch
einen großen Beitrag an Freier Energie charakterisiert ist.
Das Protein ist vollständig indifferent gegenüber den Lösungsmittelkomponenten,
wenn für alle Konzentration des Ko-Solvens m3 Gleichheit der Belegung auf dem
Protein und der Solvenszusammensetzung herrscht:
11
33 Bm
mB =
(46)
Dann haben (∂m3/∂m2)T,P,µ3, (∂µ2/∂m3)T,P,m2 und ∆µ2,TR allesamt den Wert Null und die
Belegung erfolgt aus rein statistischen Gründen.
In der Regel ist der Ursprung der verschwindenden Bindung aber die Balance der
Wechselwirkungen des Proteins mit Wasser und Ko-Solvens bei einer bestimmten
Konzentration des Ko-Solvens m3 definiert durch Gleichung 22 und 27. In diesem
Punkt verschwinden sowohl (∂m3/∂m2)T,P,µ3 als auch (∂µ2/∂m3)T,P,m2. Die Freie

_______________________________________________________________
49
2. Grundlagen
Transfer-Energie ∆µ2,TR aber nimmt einen von Null verschiedenen Wert an. Einen
negativen, wenn es sich um ein günstig wechselwirkendes Ko-Solvens handelt, oder
einen positiven, wenn es sich um eine ungünstige Wechselwirkung handelt.
2.5.2 Stabilisierungsmechanismen
Aus dem vorherigen wird ersichtlich, dass die Verdrängung von Ko-Solventien aus
der Sphäre des Proteins bzw. die daraus resultierende bevorzugte Hydratation die
kompaktere native Konformation stabilisiert. Diese Verdrängung ist durch
Gleichgewichtsdialysemessungen, durch Messung der Löslichkeit sowie durch
Densiometrie für viele stabilisierende Ko-Solventien bzw. Osmolyten experimentell
bestätigt [74-76].
Warum aber werden bestimmte Ko-Solventien bevorzugt vom Protein verdrängt?
Die Untersuchung einer großen Zahl bevorzugt verdrängter Ko-Solventien hat zu
ihrer Klassifikation in zwei generelle Kategorien geführt: solche, deren Verdrängung
von Kräften herrührt, die vollständig unabhängig von der chemischen Beschaffenheit
der Proteinoberfläche sind, d.h. Protein und Ko-Solvens sind chemisch inert
füreinander. Und solchen, die spezielle chemische Charakteristika auf der
Proteinoberfläche erkennen. In der ersten sind hauptsächlich zwei Mechanismen zu
unterscheiden, sterischer Ausschluss und Anhebung der Freien Oberflächenenergie
des Wassers durch das Ko-Solvens (Oberflächenspannungseffekt) [77].
2.5.2.1 Sterischer Ausschluss
Sterischer Ausschluss als Ursache für Wasserüberschuss auf dem Protein in
gemischten Solventien (bevorzugte Hydratation) wurde erstmalig von Kauzman im
Jahre 1949 vorgeschlagen und wird heute als molecular crowding bezeichnet [78,79].
Der Effekt ist schematisch dargestellt in Abbildung 10. Sie zeigt ein Proteinmolekül

_______________________________________________________________2. Grundlagen
50
mit einem Radius RP in Kontakt mit einem Ko-Solvensmolekül mit Radius RKS, das
wesentlich größer als Wasser ist. Weil das Ko-Solvens inert gegenüber dem Protein
ist, können sie sich bis zu einem Abstand RP+RKS annähern. Weil das Ko-Solvens
aber wesentlich größer als Wasser ist, wird eine Schale um das Protein ausgebildet,
die zugänglich für Wasser, aber nicht für das Ko-Solvens ist. Der Nettoeffekt dieses
Ausschlusses (Verdrängung) von Ko-Solvens ist bevorzugte Hydratation [80].
Abbildung 10: Sterischer Ausschluss: Der Radius
RKS des Ko-Solvens ist wesentlich größer als der
Radius von Wassermolekülen (nicht gezeigt). Es
bildet sich um das Protein eine Schale mit einem
Volumen Vex, die ausschließlich für die kleineren
Wassermoleküle zugänglich ist. Das größerer Ko-
Solvens ist aus dieser Schale verdrängt. Die
Folge ist bevorzugte Hydratation [64].
In diesem einfachen Model wird das Ausschlussvolumen Vex mit dem Volumen der
bevorzugten Hydratation (∂g1/∂g2)T,P,µ3 ausgedrückt in Gramm Ko-Solvens pro
Gramm Protein identifiziert [60,65]:
3,,2
1
1
2
µρ PTex g
gMV ⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛=
(47)
ρ1 ist die Dichte des Wassers.
Mit Gleichung 24 und 26 wird daraus:
23 ,,3
31
3
..
,,3
2
1000mPT
ex
Aussster
mPTm
Vmm ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
⋅⋅⋅=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂ µρµ
(48)
Weil per Definition das Ausschlussvolumen einen Überschuss an Wasser enthält, ist
(∂g1/∂g2)T,P,µ3 und Vex positiv und deshalb ist, weil, s.o., der SIP positiv ist, auch der

_______________________________________________________________
51
2. Grundlagen
Parameter der bevorzugten Wechselwirkung positiv. Dieser Mechanismus wird der
bevorzugten Verdrängung von Polyethylen-Glykol (PEG) durch einige Proteine
zugrunde gelegt [58-61].
2.5.2.2 Oberflächenspannung
Der Ausschluss oder die Verdrängung aufgrund einer Vergrößerung der Freien
Oberflächenenergie des Wassers folgt direkt aus der Gibbs´schen Adsorptions-
Isotherme. Sie beschreibt die Konzentrationsänderung an einer Grenzfläche. An
einer Lösung/Protein Grenzschicht gilt [56,81,82]:
PTTaRT
asn
,3
3
3
33 ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−==Γσ
µσ
(49)
PT
spannOberfl
PT aRTsas
mm
,3
33
..
,,2
3
3
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−=Γ⋅=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂ σ
µ
(50)
Γ3 ist die Oberflächenkonzentration des Ko-Solvens, s ist die molare Oberfläche des
Proteins und σ die Oberflächenspannung.
Offensichtlich wird die Grenzschicht an Ko-Solventien, welche die
Oberflächenspannung des Wassers erhöhen, verarmen. Die experimentelle
Beobachtung wird bevorzugte Verdrängung des Ko-Solvens sein.

_______________________________________________________________2. Grundlagen
52
Abbildung 11: Lösungsmittelzusammensetzung an einer Grenzfläche aufgrund der Veränderung der
Oberflächenspannung des Wasser σ durch ein Ko-Solvens. Wenn das Ko-Solvens die Oberflächen-
spannung erhöht, wird es in der Oberflächengrenzschicht verarmen. Die Oberfläche ist dann bevorzugt
hydratisiert. Wenn es die Oberflächenspannung senkt, wird es in der Oberflächenschicht
akkumulieren. a3 ist die Aktivität des Ko-Solvens´ [64].
Zucker, die meisten Aminosäuren und die meisten Salze, vor allem vom salting-out-
Typ, erhöhen die Oberflächenspannung des Wassers. Deshalb werden sie bevorzugt
verdrängt, sowohl von nativen globulären Proteinen als auch von denaturierten.
Offensichtlich ist dies die vorherrschende Quelle für bevorzugte Verdrängung durch
Hofmeistereffekte [83-86].
2.5.2.3 Solvophobie
Die zweite Kategorie, in der die Ko-Solvens Verdrängung mit chemischer Erkennung
spezieller Stellen eines Proteins verbunden ist, umfasst alle die Situationen, in der
die Affinität des Proteins für Wasser größer ist als für das Ko-Solvens oder in denen
das Ko-Solvens abgestoßen wird, z.B. durch Ladungen. Der herausstechendste Fall
solcher Art Verdrängung ist der Effekt der Solvophobie. Solvophobe Substanzen wie
Glycerol und Polyole wie Sorbitol und Mannitol beeinflussen die Wechselwirkungen

_______________________________________________________________
53
2. Grundlagen
zwischen Lösungsmittelmolekülen in solcher Weise, dass die Aktivität von Wasser an
nicht-polaren Regionen des Proteins über das Maß hinaus erhöht wird, das durch die
Effekte des Ko-Solvens auf das Ausschlussvolumen und die Oberflächenspannung
gegeben ist. Solche Ko-Solventien wandern von der Proteinoberfläche ab, um die
ungünstige positive Freie Energie zu reduzieren. Netto ergibt sich zusätzliche
bevorzugte Hydratation [64]. Der Hydrophobe Effekt ist eine der Grundlagen der
Proteinstabilität [44,87,88]. Er wird durch solvophobe Substanzen und Ionen reguliert
(Hofmeister-Effekt). Die Ionen können in Kosmotrope (Wasserstrukturbildner) und
Chaotrope (Wasserstrukturbrecher) eingeteilt werden [89,90]. NaCl gilt als mild
Kosmotrop. Die Beeinflussung der Wasserstruktur durch Ionen betrifft vor allem zwei
Eigenschaften: die Viskosität und die Entropie der Solvatation der Ionen [91]. Die
Abhängigkeit der Viskosität η von der Salzkonzentration c wird durch die Jones-Dole
Gleichung beschrieben:
...1 21
0+++= BcAcη
η
(51)
η0 ist die Viskosität reinen Wassers, A eine Konstante, die das Debye Screening
beschreibt. Die Konstante B, die als Jones-Dole Koeffizient bekannt ist, beschreibt
den Einfluss auf die Wasserstruktur. B ist positiv für Kosmotrope und negativ für
Chaotrope. Weiter haben Kosmotrope einen negativen Hydratationsanteil der
Solvatationsentropie, d.h. sie ordnen die benachbarten Wassermoleküle, während für
Chaotrope diese Entropie positiv ist.
Hofmeister hatte die Löslichkeiten von Proteinen in Lösungen unterschiedlicher Salze
untersucht und sie in einer Reihe angeordnet entsprechend ihrem Vermögen die
Löslichkeit zu erhöhen oder zu erniedrigen [83]. Dies wurde schon früh interpretiert
als Modulation des hydrophoben Effekts, werden doch auch die Löslichkeiten von
einfachen Kohlenwasserstoffen wie z.B. Benzol auf ähnliche Weise durch
Salzzugabe verändert. Der Effekt korreliert mit der Ladungsdichte der Ionen: kleine

_______________________________________________________________2. Grundlagen
54
Ionen neigen zum Einsalzen, zur Erhöhung der Löslichkeit nicht-polarer
Verbindungen, während große zum Aussalzen, also zu Herabsetzung der Löslichkeit
nicht-polarer Verbindungen tendieren. Allerdings ist die Korrelation mit der
Ladungsdichte nicht vollkommen: obwohl Lithium kleiner als Natrium ist, hat es einen
kleineren Hofmeister-Effekt. Der Hofmeister-Effekt ist direkt proportional zur
Salzkonzentration c3 und wird durch die Setschenow-Gleichung beschrieben.
( ) 332123ln ckcc −=∞∞
(52)
c∞21 und c∞
23 sind die Löslichkeiten des Proteins in Wasser und Salzlösung, c3 die
molare Konzentration des Salzes (Ko-Solvens) und k3 der Setschenow-Koeffizient
des Ko-Solvens (Salz). Der experimentelle Befund, dass kleine starke Aussalz-Ionen
generell aus der ersten Hydratationssphäre einer nicht-polaren gelösten Substanz
verdrängt sind, wird mittlerweile auch durch Simulationen der Verhältnisse in der
Lösung gestützt [92,93]. Man könnte sagen, ein Wassermolekül „bindet“ an ein
kleines Ion stärker als an benachbarte Wassermoleküle, während Wassermoleküle in
Nachbarschaft eines großen chaotropen Ions beweglicher sind als bulk Wasser.
Der Ursprung des chaotropen Effektes, d.h. der über die Änderung der
Wasserstruktur vermittelte Änderung der Löslichkeit, der Aggregatstabilität sowie der
Destabilisierung der nativen Struktur von Proteinen ist weiter Gegenstand der
Forschung [89].
2.5.2.4 Kompensation von Ladungen auf der Proteinoberfläche
Intrinsisch wird die dreidimensionale Struktur eines Proteins neben der extrinsischen
Stabilisierung durch die hydrophobe Wechselwirkung, den Effekt des
Ausschlussvolumens und durch den Oberflächenspannungseffekt durch eine Vielzahl
nicht-kovalenter Wechselwirkungen, wie Wasserstoff-Brückenbindungen, van der

_______________________________________________________________
55
2. Grundlagen
Waals-Wechselwirkungen und elektrostatischen Wechselwirkungen stabilisiert [94].
Ein wichtiger Faktor ist die ionische Zusammensetzung der Aminosäuren. Bei den
meisten Proteinen sind die Ladungen auf der Oberfläche des Proteins angeordnet.
Und zwar so, dass mehr attraktive als abstoßende Wechselwirkungen ausgebildet
werden. Die vorherrschenden Methoden, um Oberflächenladungen zu kompensieren
sind Mutagenese, chemische Modifizierung von Aminosäuren und spezifische und
nicht-spezifische Ionen-Bindung [95,96].
GOx ist ein saures Protein. Das Enzym ist bei physiologischem pH-Wert vorwiegend
negativ geladen. Das Verhältnis von sauren zu basischen Aminosäuren beträgt 3,4.
Bei einem pH-Wert von 7 trägt das Enzymmolekül eine Ladung von –77e, zumeist
hervorgerufen von deprotonierten Carboxylat-Gruppen [97]. Diese hohe
Ladungsdichte führt zu abstoßenden Wechselwirkungen auf der Oberfläche, die das
Enzym in einer eher geöffneten Variante der nativen Konformation halten.
Anlagerung monovalenter Kationen kann zu einer partiellen Kompensation der sich
abstoßenden negativen Ladungen in einer dichteren Variante der nativen
Konformation führen. Dadurch können chemische Eigenschaften verändert sein. Die
kompaktere kationen-stabilisierte Variante der nativen GOx ist z.B. gegen
proteolytischen Abbau weniger anfällig als die nicht kationen-stabilisierte
Konformation. Die dichtere Konformation zeigt sich in der DSC als stabiler gegenüber
thermischem Denaturieren. 2 M NaCl führt zu einer Tm-Verschiebung von 67°C nach
74°C. Kalium stabilisiert stärker als Natrium, während Lithium kaum stabilisierend
wirkt, die stabilisierende Wirkung geht auch hier parallel mit der Hofmeister Serie der
Kationen: Cs+>K+>Na+>Li+>Mg2+>Ca2+>Ba2+ [98].
Diese beiden Arbeiten argumentieren nicht mit den Begriffen der bevorzugten
Wechselwirkung sondern sprechen explizit von Ionen-„Bindung“ und Ionen-
„Bindungsstellen“. Timasheff berücksichtigte Ionen-Bindung in seinem Konzept der
bevorzugten Wechselwirkung als Modifikation [64].

_______________________________________________________________2. Grundlagen
56
In Abbildung 12 ist das Verhältnis zwischen Freier Transfer Energie ∆µ2,tr und dem
Parameter der bevorzugten Wechselwirkung (∂µ2/∂m3)T,P,m2 als Funktion der Ko-
Solvens-Konzentration gezeigt. Im Fall 1 (oben) bleibt der Parameter der
bevorzugten Wechselwirkung über einen größeren Temperaturbereich positiv und
konstant. Deswegen steigt die Freie Transfer Energie mit konstanter Rate monoton
an. Im Fall 2 (unten) nimmt der Parameter der bevorzugten Wechselwirkung
kontinuierlich ab, von hohen positiven Werten bei niedriger Ko-Solvens-
Konzentration zu negativen Werten bei hoher Ko-Solvens-Konzentration
Abbildung 12: Zum Verhältnis zwischen Freier
Transferenergie ∆µ2,tr und dem Parameter der
bevorzugten Wechselwirkung (∂µ2/∂m3)T,P,m2 als
Funktion der Ko-Solvens-Konzentration m3 [65].
Die Freie Transfer-Energie ist bei allen Konzentrationen positiv. Sie hat ein Maximum
bei der Konzentration, bei der (∂µ2/∂m3)T,P,m2 zu Null wird, und nimmt danach stetig
ab, bleibt aber positiv d.h. thermodynamisch ungünstig gegenüber Wasser. Im
oberen Teil der Abbildung 12 repräsentieren die durchgezogenen Linien das Protein
in Lösung und die gestrichelten Linien das Protein im Niederschlag. Das Verhalten
der thermodynamischen Funktionen bei niedrigen Ko-Solvens-Konzentrationen stellt

_______________________________________________________________
57
2. Grundlagen
die salting-in-Region des Ko-Solvens bei niedriger Konzentration dar. Bei höheren
Ko-Solvens-Konzentrationen wurde im Fall 2 dem salting-out extensive Ionen-
Bindung überlagert und eine weniger ausgeprägte Ionen-Bindung im Fall 1. Laut
Timasheff sieht so das Bindungsmuster eines jeglichen schwachbindenden Ko-
Solvens´ aus [64,65].
2.6 Funktionsstabilität
Die meisten biochemischen Reaktionen würden ohne Enzyme nur mit
vernachlässigbarer Geschwindigkeit ablaufen. Die katalytische Wirksamkeit eines
Enzyms beruht einzig auf seiner Fähigkeit, die Aktivierungsenergie einer chemischen
Reaktion zu senken. Dies kann geschehen durch Stabilisierung des
Übergangszustands, durch Orientierung und Annäherung der Substrate, durch
Allgemeine Säure-Basen-Katalyse, durch kovalente Katalyse mithilfe von Ko-
Faktoren oder durch Metallionen-Katalyse.
Die Reaktionsgeschwindigkeit einer enzymatischen Reaktion hängt neben den
Reaktionsbedingungen wie Temperatur, Salzkonzentration und pH der Lösung von
den Konzentrationen des Enzyms, der Substrate und Produkte sowie von Effektoren
(Aktivatoren, Inhibitoren und Ko-Solventien) ab. Die Enzymaktivität gibt an wie viel
aktives Enzym sich in einer Enzympräparatiion befindet. Die Einheit der
Enzymaktivität ist das Unit (U) und ist definiert als diejenige Enzymmenge, die ein
Mikromol Substrat (etwa 180 µg Glukose) pro Minute umsetzt. Werden Enzyme in der
Bioanalytik oder in Bioreaktoren eingesetzt, ist die Stabilität der enzymatischen
Aktivität ein bedeutsames Qualitätsmerkmal. Häufig verläuft die Deaktivierung der
enzymatischen Funktion nach einem Exponentialgesetz (s. Kapitel 3.1.8), das die
Halbwertszeit der katalytischen Aktivität τ1/2 bestimmt.
Die Halbwertszeit der enzymatischen Aktivität τ1/2 ist eine intensive Größe, die nicht
abhängig ist von der in Hydrogelen schwer zu bestimmenden Gesamt-

_______________________________________________________________2. Grundlagen
58
Enzymkonzentration [E]t und dient hier neben der Denaturierungstemperatur Tm als
zweiter Parameter der Enzymstabilität.
Die Untersuchung eines möglichen Zusammenhangs zwischen der Halbwertszeit der
enzymatischen Aktivität τ1/2 und der Denaturierungstemperatur Tm kann von großem
praktischen Nutzen sein, weil die Bestimmung der kinetischer Parameter
experimentell aufwendig ist, zumal bei Vorliegen von Einflüssen durch Diffusion,
Quellung und Inaktivierung durch nicht umgesetztes Monomer. Der Zeitverbrauch ist
hoch und die Reproduzierbarkeit nur schwer herstellbar, vor allem wenn zwischen
den Messungen längere Zeitspannen liegen.
2.7 Diffusionswiderstand und Thiele Modulus
Das Verhalten von Enzymen, die in einer Hydrogelmatrix eingebettet sind, kann sich
vom Verhalten desselben Enzyms in verdünnter wässriger Lösung unterscheiden.
Die Änderungen können die kinetischen Parameter Km und kcat, die pH- und
Temperatur-Aktivitätsprofile, sowie die Stabilität gegenüber denaturierenden Ko-
Solventien oder erhöhter Temperatur betreffen. Die Änderungen des Verhaltens
heterogener Enzyme kann zwei Hauptursachen zugeschrieben werden. Die erste
betrifft Änderungen im Enzymmolekül selbst und seiner unmittelbaren Nachbarschaft.
Diese führen zu Konformationsänderungen und/oder Einschränkungen in der
Erreichbarkeit der reaktiven bzw. allosterischen Zentren. Ebenfalls in diese Kategorie
fallen direkte Kontakte zwischen funktionellen Gruppen der Hydrogelmatrix und dem
Enzymmolekül. Der zweite Grund rührt aus der heterogenen Natur der
Mikroumgebung des Enzyms her, in der sich die Konzentrationen an Substraten,
Produkten und Effektoren deutlich von denen in der umgebenden Lösung, in der der
Reaktionsverlauf verfolgt wird, unterscheiden können.
Findet eine enzymatische Reaktion in einer gut gerührten homogenen Lösung statt,
sind die Konzentrationen aller Reaktionspartner uniform. In der Mikroumgebung in

_______________________________________________________________
59
2. Grundlagen
unmittelbarer Nähe des Enzymmoleküls sind die Konzentrationen der
Reaktionspartner verschieden von denjenigen in der umgebenden Lösung, die als
Makroumgebung bezeichnet wird.
Die Untersuchung der Kinetik von in Hydrogelen eingeschlossenen Enzymen erfolgt
durch Messung der Reaktionsprodukte in der flüssigen Phase, die mit dem Hydrogel
in Kontakt steht. Während die Aktivität des Enzyms durch die Konzentrationen in der
Mikroumgebung, dem Hydrogel, bestimmt wird, sind experimentell zugänglich nur die
Änderungen in der die Makroumgebung. Hydrophobe und elektrostatische
Wechselwirkungen zwischen dem das Hydrogel bildenden Polymer und den
Substraten, Produkten und eventuell vorhandenen Ko-Solventien führen zu einer
ungleichen Verteilung dieser Substanzen zwischen Mikro- und Makroumgebung.
Dies wird als Partitionseffekt bezeichnet.
Abbildung 13: Schematische
Darstellung eines Hydrogels mit
eingebetteten Enzymmolekülen.
Jedes Enzymmolekül befindet
sich in seiner Mikroumgebung,
die von Partitionseffekten und
durch Diffusionswiderstände
charakterisiert ist.
Ist ein Enzymmolekül in einem Hydrogel eingeschlossen, diffundiert das Substrat aus
der Umgebungslösung zu den aktiven Zentren des Enzyms, und das Produkt
diffundiert zurück in die Umgebungslösung. Daran ist sowohl molekulare wie auch
konvektive Diffusion beteiligt. Die niedrigen Diffusionsgeschwindigkeiten der
Substrate/Produkte in wässriger Lösung und in Gelen sowie die hohe katalytische
Aktivität der Enzyme führt häufig zu signifikanten Diffusionswiderständen.

_______________________________________________________________2. Grundlagen
60
Es bilden sich Konzentrationsgradienten in der Umgebung des Enzymmoleküls aus,
sodass sich die Konzentrationen in der Mikro- bzw. Makroumgebung unterscheiden.
In Abbildung 14 sind mögliche Konzentrationsprofile für das Substrat und das
Produkt in einem Hydrogelfaden im Querschnitt gezeigt. Partition dieser Spezies
zwischen dem Hydrogel und der flüssigen Phase augrund von elektrostatischen und
hydrophoben Wechselwirkungen führt zu einem Konzentrationssprung an der
Grenzfläche zwischen Hydrogel und Lösung. Auf der anderen Seite führen die
Diffusionswiderstände zu Konzentrationsgradienten in der Lösung und im Inneren
des Hydrogels.
Abbildung 14: Konzentrationsprofile (nicht
maßstabgetreu) des Substrates und des
Produktes innerhalb und außerhalb eines
enzymbeladenen Hydrogelfadens. Oben:
die Spezies verteilen sich aufgrund von
Partitionseffekten. Die Reaktion bleibt
kinetisch kontrolliert. Mitte: es existieren
Diffusionswiderstände für beide Spezies
ohne Partitionseffekte. Unten: es treten
sowohl Partition als auch Diffusions-
widerstände auf [99].
Das kinetische Verhalten eines Enzyms ist charakterisiert durch seine intrinsischen
kinetischen Parameter. Diese können jedoch aus Messungen in der Makroumgebung
direkt nur ermittelt werden, wenn gesichert ist, dass die Konzentrationen aller die
Reaktionsgeschwindigkeit beeinflussenden Spezies (Substrate, Produkte, Ko-
Solventien) in der Mikroumgebung des Enzyms die gleichen sind wie in der
Makroumgebung, was normalerweise nicht der Fall ist.

_______________________________________________________________
61
2. Grundlagen
Die inhärente Reaktionsgeschwindigkeit ist die Geschwindigkeit, die sich ohne
Diffusionswiderstände ergäbe, d.h. wenn der Transport der Substrate und Produkte
zwischen der enzymatischen Mikroumgebung und der Makroumgebung unendlich
schnell wäre. Experimentell zugänglich sind die inhärenten kinetischen Parameter mit
sehr dünnen Membranen, geringer enzymatischer Aktivität (geringe
Enzymkonzentration) und ausreichendem Rühren der Umgebungslösung. v´max in
Gleichung 55 ist die inhärente Geschwindigkeit des Hydrogelkatalysators bei
Substratsättigung. Unterschiede zwischen den inhärenten und intrinsischen
kinetischen Parametern rühren von der Partition der Reaktionspartner zwischen
Mikro- und Makroumgebung her.
Abbildung 15: Zum Unterschied zwischen intrinsischer, inhärenter und effektiver
Reaktionsgeschwindigkeit [99].
Die effektive Reaktionsgeschwindigkeit wird beobachtet, wenn Diffusionswiderstände
in Anwesenheit oder Abwesenheit von Partitionseffekten auftreten. Nur die effektiven
kinetischen Parameter sind aus der Gesamtreaktionsgeschwindigkeit, die über

_______________________________________________________________2. Grundlagen
62
Konzentrationsänderungen in der Makroumgebung gemessen werden kann,
extrahierbar. Aufgrund der langsamen Diffusionsgeschwindigkeit biochemischer
Substanzen in wässriger Lösung und besonders in Hydrogelen und der hohen
Katalyserate von Enzymen, treten in heterogenen Enzymkatalysatoren regelmäßig
signifikante Diffusionswiderstände auf. Im Allgemeinen gilt die Michaelis-Menten
Kinetik nicht, wenn Diffusionswiderstände ein Rolle spielen.
Das Modell zur semi-quantitativen Beschreibung des Wechselspiels zwischen
Reaktion und interner Diffusion in heterogenen Enzymkatalysatoren bildete meist
eine Membran mit uniform darin verteiltem Enzym, für das Michaelis-Menten
kinetisches Verhalten angenommen wurde. Es wurde gezeigt, dass die
grundlegenden Zusammenhänge nicht von der genauen Geometrie (sphärische
Partikel, Scheibchen, Fäden oder Membranen) abhängen. Es ist lediglich die
Kenntnis einer das System beschreibenden charakteristischen Länge nötig, etwa die
Membrandicke oder der Fadendurchmesser [100].
Die Gesamtreaktionsgeschwindigkeit heterogen katalysierter Reaktionen zeigt zwei
limitierende Fälle. Bei hinreichend niedriger Enzymaktivität sind die Konzentrationen
der Substrate und Produkte im Inneren des Katalysators im wesentlichen die
gleichen wie in der umgebenden Lösung. Unter diesen Umständen ist die Reaktion
kinetisch kontrolliert und unbeeinflusst von Transporteigenschaften. Bei hinreichend
hoher Enzymaktivität werden alle Substratmoleküle schon an der Oberfläche des
heterogenen Katalysators umgesetzt, noch bevor sie ins Innere diffundieren können.
In diesem Fall ist die Reaktionsgeschwindigkeit nur abhängig von der Diffusion des
Substrats in der Umgebungslösung. In der Regel beeinflusst aber die interne
Diffusion die Geschwindigkeit der enzymatischen Reaktion. Wie aus Abbildung 14
und 16 (s.u.) ersichtlich, nimmt die Substratkonzentration von der Umgebungslösung
zum Inneren des Katalysators hin ab. Der relative Einfluss von internen und externer

_______________________________________________________________
63
2. Grundlagen
Diffusion kann durch die Massenerhaltung an der Grenzfläche zwischen Lösung und
Katalysator abgeschätzt werden:
i
effS
eS dX
dSDdxdSD ⎟
⎠
⎞⎜⎝
⎛−=⎟⎠
⎞⎜⎝
⎛−
(53)
Die Ableitungen drücken die Konzentrationsgradienten des Substrats S an der
Grenzfläche nach der Seite der Umgebungslösung hin, (dS/dx)e, und nach dem
Inneren des Katalysators, (dS/dX)i aus. DSeff ist das effektive Diffusionsvermögen des
Substrates innerhalb des biomimetischen Gewebes, das ausgedrückt werden kann
durch das Diffusionsvermögen in der Lösung:
τε Seff
SD
D⋅
=
(54)
ε ist der Leeranteil eines porösen Katalysators und entspricht in Hydrogelen dem
Gehalt an Wasser, das die Diffusion vermittelt. Der Faktor τ ist ein Maß ist für die
Gewundenheit der Hohlräume (der Porengeometrie) und ist meist größer als 1 ist. Er
ist definiert als Verhältnis der Wegstrecke, die ein Molekül bei der Passage durch
einen Film zurückzulegen hat, und der Filmdicke. Weil das Diffusionsvermögen im
Inneren des Hydrogels kleiner ist als in der umgebenden Lösung, muss der Gradient
dort größer sein, damit die Gleichung 53 erfüllt ist. Deshalb ist für größere
Schichtdicken die Verarmung an Substrat im Inneren des Hydrogel-Katalysators
bedeutsamer als in der Außenlösung. Werden die Hydrogelfäden in der Außenlösung
hinreichend bewegt, ist die Oberflächenkonzentration praktisch gleich der
Lösungskonzentration. Unter diesen Umständen ist die enzymatische Reaktion nur
von internen Diffusionswiderständen beeinflusst. Im Fließgleichgewicht kann die
Abnahme der Substratkonzentration von der Oberfläche zum Inneren der
Hydrogelmembran durch folgende Differentialgleichung beschrieben werden:

_______________________________________________________________2. Grundlagen
64
SKSv
dxSdD
m
effS ⋅
⋅=⎟⎟
⎠
⎞⎜⎜⎝
⎛ max2
2
(55)
vmax ist die inhärente Sättigungsgeschwindigkeit pro Katalysatorvolumen und x die
Distanz zur Oberfläche des Hydrogelfadens. Einfacher kann Gleichung 55 in
dimensionsloser Form geschrieben werden:
⎟⎟⎠
⎞⎜⎜⎝
⎛+
=β
βφβ1
22
2
dzd
(56)
β=S/KM ist die auf KM normierte dimensionslose Konzentration des Substrates
innerhalb des Hydrogelfadens, z ist die Position in der Membran, z=x/l, l die Dicke der
Membran und φ der Modulus für die interne Diffusion (Thiele-Modulus):
( ) 21
maxeffSM DK
Vl⋅
⋅=ϕ
(57)
l ist die Schichtdicke der Hydrogelmembran, KM die Michaelis-Menten Konstante der
GOx für Glukose, vmax die inhärente Sättigungsgeschwindigkeit ohne
Diffusionswiderstände und DeffS der effektive Diffusionskoeffizient des Substrates
(Glukose) im Hydrogel.
Numerische Integration mit den Randbedingungen β=β0, für z=0 und dβ/dz=0 für z=1
ergibt das Profil der Substratkonzentration im Inneren des Hydrogels. Im Abbildung
16 ist das für βe=1 und verschiedene Werte für den Thiele-Modulus φ gezeigt:
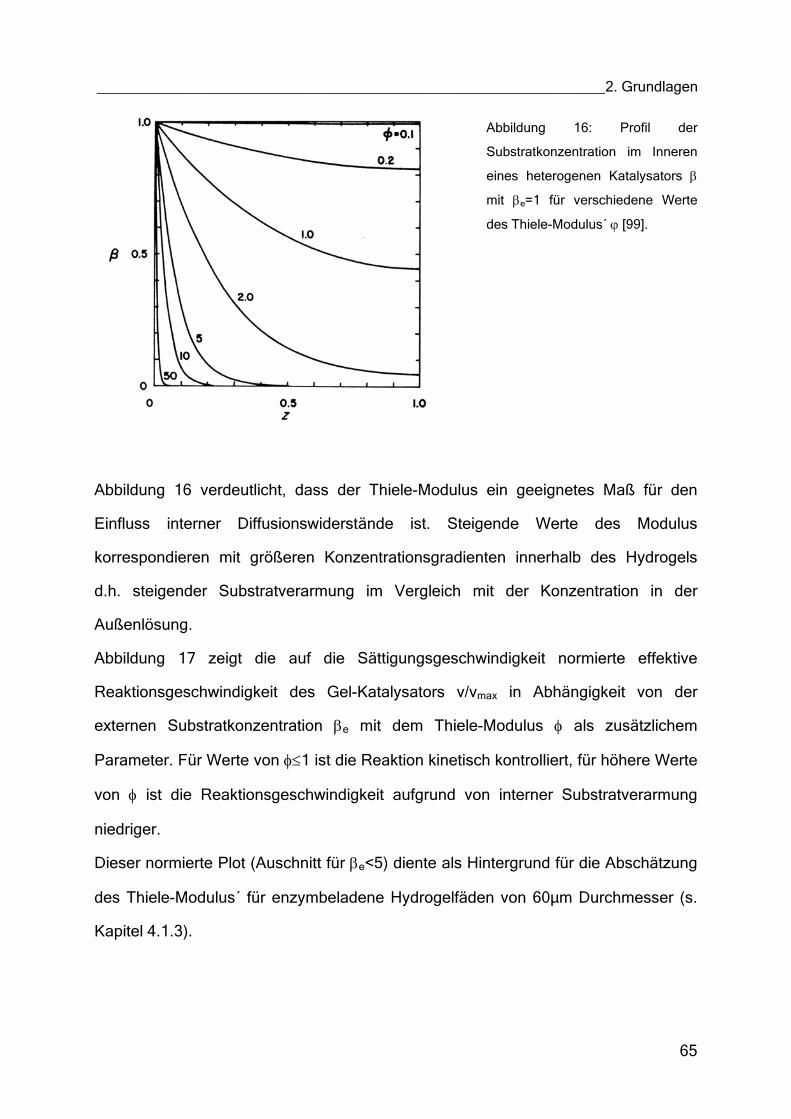
_______________________________________________________________
65
2. Grundlagen
Abbildung 16: Profil der
Substratkonzentration im Inneren
eines heterogenen Katalysators β
mit βe=1 für verschiedene Werte
des Thiele-Modulus´ ϕ [99].
Abbildung 16 verdeutlicht, dass der Thiele-Modulus ein geeignetes Maß für den
Einfluss interner Diffusionswiderstände ist. Steigende Werte des Modulus
korrespondieren mit größeren Konzentrationsgradienten innerhalb des Hydrogels
d.h. steigender Substratverarmung im Vergleich mit der Konzentration in der
Außenlösung.
Abbildung 17 zeigt die auf die Sättigungsgeschwindigkeit normierte effektive
Reaktionsgeschwindigkeit des Gel-Katalysators v/vmax in Abhängigkeit von der
externen Substratkonzentration βe mit dem Thiele-Modulus φ als zusätzlichem
Parameter. Für Werte von φ≤1 ist die Reaktion kinetisch kontrolliert, für höhere Werte
von φ ist die Reaktionsgeschwindigkeit aufgrund von interner Substratverarmung
niedriger.
Dieser normierte Plot (Auschnitt für βe<5) diente als Hintergrund für die Abschätzung
des Thiele-Modulus´ für enzymbeladene Hydrogelfäden von 60µm Durchmesser (s.
Kapitel 4.1.3).

_______________________________________________________________2. Grundlagen
66
Abbildung 17: Auf die
Sättigungsgeschwindigkeit normierte
inhärente Reaktionsgeschwindigkeit
v/vmax eines enzymbeladenen
Hydrogels in Abhängigkeit von der
externen auf Km normierten
Substratkonzentration βe [99].
2.8 Einfluss von Aggregation und Reversibilität auf die
Langlebigkeit von Enzym-Präparationen
Die thermische Reversibilität der Denaturierung ist neben der
Denaturierungstemperatur Tm ein wichtiger Faktor zur Beschreibung der
Langzeitstabilität wässriger Proteinpräparationen. Die Aggregation denaturierter
Proteinmoleküle ist eine der Ursachen irreversibler Inaktivierung und ist mit der
thermischen Reversibilität negativ korreliert [101]. Viele wässrige Proteinlösungen
zeigen keine oder nur sehr geringe Reversibilität. Im DSC Experiment zeigt sich das
darin, dass der Denaturierungspeak bei einer wiederholten Aufheizung der Probe
wesentlich weniger ausgeprägt ist oder ganz ausbleibt. Die irreversible Aggregation
stellt eine Nebenreaktion des Gleichgewichts zwischen der nativen und der
denaturierten Form des Protein dar. Tm allein ist in solchen Fällen kein ausreichendes
Stabilitätskriterium. Der rekombinante humane Flt3 Ligand zum Beispiel erreicht bei
pH 6 ein Tm-Plateau. Weitere Erhöhung des pH -Wertes verändert Tm kaum noch. Die
Neigung zur irreversiblen Aggregation nimmt hingegen von pH 6 bis pH 9 noch weiter

_______________________________________________________________
67
2. Grundlagen
zu und die Langzeitstabilität der Bioaktivität entsprechend ab. Im gleichen pH-
Bereich nimmt die thermische Reversibilität entsprechend ab [102].
Polyole wie Sorbitol oder Xylitol unterstützen die Aufrechterhaltung der
enzymatischen Aktivität des Modelenzyms Lysozym während der Inkubation bei
60°C und diese Eigenschaft von Polyolen, die enzymatische Aktivität zu
konservieren, korreliert mit der Eigenschaft, die Schmelztemperatur des Enzyms zu
erhöhen [28/9]. Hier, wie allgemein, gilt: weil die N-Konformation stabilisiert ist, ist die
Denaturierung weniger irreversibel, weil die Aggregation denaturierter Moleküle
stärker irreversibel ist als die Aggregation nativer Proteinmoleküle. Daraus ist die
Konsequenz gezogen worden und es wurde demonstriert, dass in einer Lösung eines
Denaturierungsmittels in milden Konzentrationen (z.B. 1 M GndHCl) unter
Inkaufnahme einer damit induzierten Senkung der Denaturierungstemperatur um ca.
10°C die biologische Aktivität von Lysozym als Modelenzym nach Inkubation bei
60°C für 24 h zu annähernd 100 % erhalten blieb, wenn die Lösung zusätzlich Polyol-
Stabilisatoren in hohen Konzentrationen (z.B. 2 M Sorbitol oder 1 M Trehalose)
enthielt. Das Denaturierungsmittel hat dabei die Aufgabe, die irreversible Aggregation
zu verhindern [103].
Natürlich auftretende Poly-Amine wie Spermidin (NH2(CH2)3NH(CH2)4NH2) können
sehr effektiv die Aggregatbildung verhindern und die thermische Reversibilität
deutlich erhöhen. Lysozym in Lösung mit einer Konzentration von 0, 2 mg/ml Protein
zeigt in Anwesenheit von100 mM Spermidin noch 50% Bioaktivität nach einer
thermischen Behandlung von 30 min bei 98°C, während die Kontrolle nach dieser
Behandlung nur noch zu 1% biologisch aktiv ist. Bei einer Konzentration von 1 mg/ml
ist der Prozentsatz aktiven Enzyms nach 30 min bei 98°C ohne Spermidin-Zusatz
20%, der der Probe mit Spermidin 90% [104].
Glycerol ist ein Proteinstabilisator und hilft die irreversible Aggregation zu verhindern.
Meng et al. zeigten für das Enzym Kreatin-Kinase, dass die Verschiebung der Tm hin

_______________________________________________________________2. Grundlagen
68
zu höheren Temperaturen und die Verlängerung der Halbwertszeit der biologischen
Aktivität einen annähernd linearen Zusammenhang haben [105].
Wird die irreversible Aggregation dadurch verhindert, dass intermolekulare Kontakte
der Proteinmoleküle durch chemische Bindung mit einem festen Träger verringert
werden, kann die Reversibilität erhöht werden, wenn die Bindung an einer einzigen
Stelle des Proteinmoleküls, etwa am C-Terminus, erfolgt. So voneinander getrennte
Moleküle bilden thermodynamisch geschlossene Systeme und die Denaturierung ist
nahezu vollständig reversibel [106].
2.9 Die Tm als Maß für die Langlebigkeit von Enzym-
Präparationen
Die Analyse der thermodynamischen Stabilität einer größeren Anzahl von Proteinen
lässt einen Trend sichtbar werden: Proteine, die bei höherer Temperatur
denaturieren, tendieren dazu, eine größere Stabilität bei der Temperatur maximaler
Stabilität T´ zu besitzen [107]. Die Temperatur der maximalen Stabilität T´ für die von
Rees et al. analysierten globulären wasserlöslichen Proteine ist in erster Näherung
unabhängig von der Denaturierungstemperatur Tm und liegt bei ca. 283 K. Diese
Tendenzen legen nahe, dass im Allgemeinen die Erhöhung der thermodynamischen
Stabilität durch Anhebung der Stabilitätskurve erreicht wird (Abbildung 18).
Einige Mikroorganismen, sogenannte Thermophile, haben ihr Wachstumsoptimum
bei nahezu 100°C. Die Proteine dieser Organismen haben eine deutlich höhere
Temperaturstabilität als ihre mesophilen Verwandten. Sie bieten daher eine
Möglichkeit zur Untersuchung des physiko-chemischen Ursprungs der Stabilität von
Proteinen im Allgemeinen [108]. Häufig beruht die erhöhte Stabilität auf Mutationen in
einigen wenigen oder gar nur einer einzelnen Aminosäure, wie von Perl et al. am
Beispiel R3E Bc-Csp, einem mutierten cold shock Protein aus dem Thermophilen B.
caldolyticus, gezeigt wurde (Abbildung 19) [109].

_______________________________________________________________
69
2. Grundlagen
Abbildung 18: Die thermodynamische Stabilität
eines Proteins (a) kann formal durch Anhebung
der Stabilitätskurve (b), durch Verschieben der
Stabilitätskurve (c) oder durch Weitung der
Stabilitätskurve (d) erhöht werden [107].
Dieses Protein, bei dem die Aminosäure 3, Arginin (R), durch Glutamat (E)
ausgetauscht wurde, ist genauso stabil wie sein mesophiles Pendant, mit dem es zu
80 % sequenzhomolog ist. Das unmutierte thermophile Protein Bc-Csp ist etwa 2
kcal/mol bei 20°C und etwa 4 kcal/mol bei 70 °C stabiler als sein mesophiles
Homolog. Die reverse Mutation des mesophilen Proteins ergibt eine ungefähr gleich
stark stabilisierte Variante. Dies wird erklärt durch Ausbildung einer einzigen
zusätzlichen Salzbrücke. Als Salzbrücke werden die attraktiven Wechselwirkungen
zwischen unterschiedlich geladenen Ionen im Abstand von weniger als o,5 nm
bezeichnet. Die Ausbildung einer einzelnen Salzbrücke führt nach dem
Coulomb´schen Gesetz zu einer Stabilisierung von 1 kcal/mol. Im thermophilen
stabilisierten Protein ist die positiv geladene Aminosäure 3, Arginin, dicht benachbart
der Aminosäure 46, Glutamat, und der C-terminalen Aminosäure 66, Leucin. In der
R3E-Mutation fehlt diese günstige Salzbrücke und deshalb ist die Stabilität erniedrigt.
Dies wird unterstützt durch die Tatsache, dass der Stabilitätsunterschied in 2 M NaCl
aufgrund von Debye-Hückel-Screening wesentlich geringer ist. Der Vergleich vieler
thermophiler und mesophiler Proteine zeigt, dass der Evolutionsdruck eher auf der
Vermeidung von ungünstigen Wechselwirkungen liegt und nicht auf der
Hervorbringung von günstigen. Eine weitere Eigentümlichkeit thermophiler Proteine
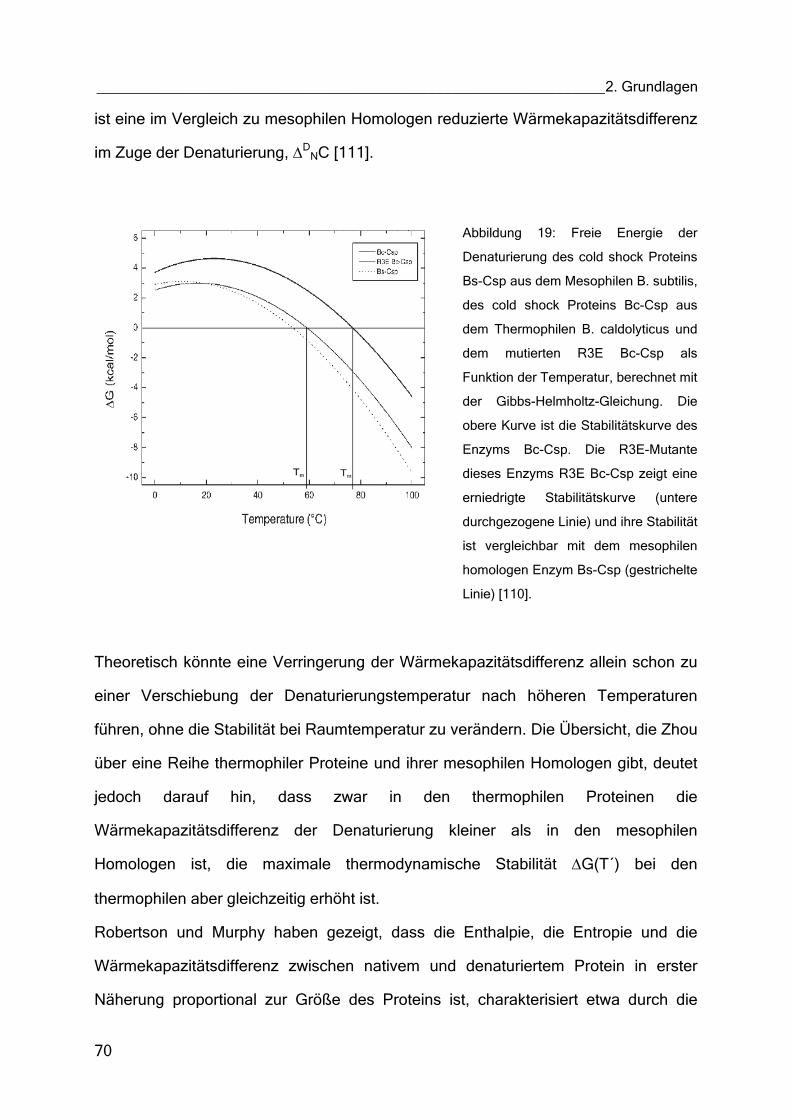
_______________________________________________________________2. Grundlagen
70
ist eine im Vergleich zu mesophilen Homologen reduzierte Wärmekapazitätsdifferenz
im Zuge der Denaturierung, ∆DNC [111].
Abbildung 19: Freie Energie der
Denaturierung des cold shock Proteins
Bs-Csp aus dem Mesophilen B. subtilis,
des cold shock Proteins Bc-Csp aus
dem Thermophilen B. caldolyticus und
dem mutierten R3E Bc-Csp als
Funktion der Temperatur, berechnet mit
der Gibbs-Helmholtz-Gleichung. Die
obere Kurve ist die Stabilitätskurve des
Enzyms Bc-Csp. Die R3E-Mutante
dieses Enzyms R3E Bc-Csp zeigt eine
erniedrigte Stabilitätskurve (untere
durchgezogene Linie) und ihre Stabilität
ist vergleichbar mit dem mesophilen
homologen Enzym Bs-Csp (gestrichelte
Linie) [110].
Theoretisch könnte eine Verringerung der Wärmekapazitätsdifferenz allein schon zu
einer Verschiebung der Denaturierungstemperatur nach höheren Temperaturen
führen, ohne die Stabilität bei Raumtemperatur zu verändern. Die Übersicht, die Zhou
über eine Reihe thermophiler Proteine und ihrer mesophilen Homologen gibt, deutet
jedoch darauf hin, dass zwar in den thermophilen Proteinen die
Wärmekapazitätsdifferenz der Denaturierung kleiner als in den mesophilen
Homologen ist, die maximale thermodynamische Stabilität ∆G(T´) bei den
thermophilen aber gleichzeitig erhöht ist.
Robertson und Murphy haben gezeigt, dass die Enthalpie, die Entropie und die
Wärmekapazitätsdifferenz zwischen nativem und denaturiertem Protein in erster
Näherung proportional zur Größe des Proteins ist, charakterisiert etwa durch die

_______________________________________________________________
71
2. Grundlagen
Anzahl der Aminosäuren N. Ihr Datensatz von kleinen globulären Proteinen führte sie
zu folgender Parametrisierung [112]:
∆DNC ≅ 0,058 N kJ/molAA/K
∆DNH(333 K) ≅ 2,92 N kJ/molAA
∆DNS(333 K) ≅ 0,0088 N kJ/molAA/K
Mit diesen Parametern werden die experimentellen Werte für ∆DNC, ∆D
NH(333 K) und
∆DNS (333 K) ihres Datensatzes mit Korrelationsfaktoren von 0,86, 0,77 bzw. 0,74
reproduziert. Untersuchungen jüngeren Datums geben ähnliche Parametrisierungen
[113].
Die Stabilitätskurve eines Proteins (Gibbs-Helmholtz-Gleichung und Abbildung 2)
lässt sich als quadratische Funktion der Temperatur annähern [114]. Damit lässt sich
die Freie Energie bei der Temperatur maximaler Stabilität T´ angeben als:
( ) ( )p
DNm
DND
N CTH
TG∆⋅
∆≅′∆
2
2
(58)
und die Temperatur maximaler Stabilität T´:
KCH
TTp
DN
DN
m 283≅∆∆
−≅′
(59)

_______________________________________________________________2. Grundlagen
72
Abbildung 20: Zusammenhang zwischen der Tm und ∆G(T´): Kreuze: Datensatz von Robertson und
Murphy. Dreiecke: Hyperthermophile, Sterne: Mutationen von T4-Lysozym. Die durchgezogene Linie
ist die Parametrisierung nach Gleichung 60 [112].
Wie sind die Stabilitätskurve und die Verschiebung der Denaturierungstemperatur Tm
miteinander gekoppelt? Werden die Parameter von Robertson und Murphy in
Gleichung 58 eingesetzt, erhält man einen empirischen Zusammenhang zwischen
der Freien Energie der Denaturierung bei der Temperatur maximaler Stabilität
∆NDG(T´) und der Denaturierungstemperatur [107]:
][)6,282(0290,0)( 2 molkJTT
NTG mm
DN −−=′∆
(60)
Dieser Zusammenhang ist in genereller Übereinstimmung mit experimentellen Daten
[107-114] sowohl für ein und dasselbe Protein in unterschiedlichen Medien als auch
für unterschiedliche Proteine.

_______________________________________________________________
73
2. Grundlagen
Das generelle Verhalten der Stabilitätskurven von Proteinen legt nahe, dass eine
Erhöhung der Tm eines Proteins einhergeht mit einer Zunahme der Freien Energie
bei maximaler Stabilität ∆G(T´). Und weiter, dass die Temperatur maximaler Stabilität
T´ annähernd konstant bei 283 K liegt. Diese Beobachtungen sind in einem weiteren
Zusammenhang zusammengefasst worden [107]:
( ) ( )( )
⎥⎦⎤
⎢⎣⎡
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛−−−
−−+=∆
molkJ
TTTTN
TTT
TNNTG
mm
m
mm
DN
ln1058,0
333058,092,2
(61)
Gleichung 61 ermöglicht die näherungsweise Berechnung der temperaturabhängigen
thermodynamischen Stabilität (Stabilitätskurve) mit Hilfe nur zweier Parameter: der
Denaturierungstemperatur Tm und der Anzahl der Aminosäuren N.
An dieser Stelle stellt sich die Frage, ob ein empirischer Zusammenhang zwischen
∆G(T´) und τ½ hergestellt werden kann. Weil die Analyse einer größeren Zahl
unterschiedlicher Proteine gezeigt hat, dass T´ in erster Näherung für alle Proteine
bei ca. 283K liegt, bietet sich ∆G(T´) als allgemeines Maß der Stabilität eines
Proteins an. Gleichzeitig zeigen die Daten, dass im Allgemeinen Stabilisierung unter
Anhebung der Stabilitätskurve erfolgt. Aus der Kombination dieser beiden Tendenzen
folgt, dass die Stabilität bei T´ direkt aus der Tm bestimmt werden kann (Gleichung
60) und dass darüber hinaus zur Konstruktion der vollständigen Stabilitätskurve die
Bestimmung der Tm ausreicht (Gleichung 61). Nimmt man weiter an, dass die
Halbwertszeit τ½ bei jedweder Temperatur ebenfalls ein universelles Maß für die
Stabilität bei anderen Temperaturen darstellt, kann man versuchen, τ ½ und ∆G(T´)
parallel zu führen. Dies wäre sicher unzulässig, wenn Stabilisierung unter
Verschiebung der Stabilitätskurve erfolgte, da dann Erhöhung der Stabilität bei hohen
Temperaturen mit einer Verringerung der Stabilisierung bei niedrigen Temperaturen

_______________________________________________________________2. Grundlagen
74
einherginge. Hat man sich aber einmal von der Anhebung der Stabilitätskurve als
dominierendem Kennzeichen der Stabilisierung überzeugt, kann versucht werden,
durch einfache mathematische Operationen die Halbwertszeiten so in Werte von
∆G(T´) umzuwandeln, dass sie sich in das allgemeine Bild einfügen.
Einen solchen Versuch der Parallelführung der Halbwertszeit τ½ und der
Denaturierungstemperatur Tm stellt Gleichung 62 dar:
( ) [ ]molkJCFNTG 602
160 +⋅=′∆ τ
(62)
Die Kombination von Gleichung 60 und 62 liefert:
[ ]min)6,282(0290,0160
2
6021 ⎟⎟
⎠
⎞⎜⎜⎝
⎛−−= CT
TF mm
τ
(63)
Die Evaluierung dieses ad-hoc-Zusammenhangs für das Enzym GOx erforderte die
Messung der Halbwertszeit der biologischen Aktivität bei 60°C sowie die Bestimmung
ihrer Denaturierungstemperatur in unterschiedlichen Medien.
3. Materialien und Methoden
3.1 Bestimmung der enzymatischen Aktivität
Neben den Parametern der thermodynamischen Stabilität, die aus DSC-Messungen
gewonnen werden können, kann das Enzym GOx durch die Stabilität der kinetischen
Eigenschaften charakterisiert werden.
GOx (Glukoseoxidase, EC 1.1.3.4) ist ein homo-dimeres Flavo-Protein (Mr
(Dimer)160000), das 1 mol FAD pro Untereinheit enthält. Das dimere Enzym besteht
aus 1208 Aminosäuren (N=1208) [115,116]. Es katalysiert die Oxidation von β-D-
Glukose zu δ-Glukonolakton, das anschließend spontan zu Glukonsäure hydrolysiert,
verbunden mit der Reduktion von molekularem Sauerstoff zu Wasserstoffperoxyd.
GOx arbeitet nach einem ping-pong Mechanismus [117,118] .

___________________________________________________
75
3. Materialien und Methoden
1. Teilschritt
tonGlukonolacEGlukoseE redox +↔+ (Reaktion 4a)
2. Teilschritt
2220 OHEE oxred +↔+
(Reaktion 4b)
Für die reduktive Halbreaktion wurden zwei Mechanismen vorgeschlagen. Nach
ersterem findet ein Hydridtransfer vom C-1 des Substrats zum N-5 des Flavins statt.
Gemäß dem alternativen Mechanismus findet zunächst eine nucleophile Addition der
–OH Gruppe des C-1 des Zuckers an der C-4a Position des Flavins gefolgt von einer
Protonenabstraktion von C-1 des Zuckers statt. Beiden Mechanismen ist gemeinsam,
dass sie unter allgemeiner Basenkatalyse stehen. Im aktiven Zentrum des Enzyms
findet sich oberhalb des Isoalloxazin-Rings in weniger als 0,4 nm Entfernung ein
potentieller Protonen-Akzeptor (Nε von His516). Als Resultat der reduktiven
Halbreaktion wurden zwei Elektronen und zwei Protonen zum Enzym transferiert,
vorzugsweise am Ort des reduzierten Flavins (2 Elektronen und 1 Proton) und am
protonierten Histidin 516 (1 Proton). In der oxidativen Halbreaktion werden die
Elektronen und Protonen auf molekularen Sauerstoff übertragen unter Bildung von
Wasserstoffperoxyd.
3.1.1 Reaktionsgeschwindigkeit und Unit Definition
Die Geschwindigkeit einer chemischen Reaktion v´ kann definiert werden als der
Stoffumsatz pro Zeiteinheit ∆N/∆t:
v´=∆N/∆t [µmols-1] (64a)

___________________________________________________
76
3. Materialien und Methoden
Ein Unit U ist diejenige Enzymmenge, die ein µmol Substrat pro Minute umsetzt.
Häufig ist es aber sinnvoll die Reaktionsgeschwindigkeit als Konzentrationsänderung
in der Zeit zu definieren. Dann ist ihre Dimension z.B. mMs-1 und wird mit v
symbolisiert.
v=v´/V=∆N/(∆t*V) [mMs-1] (64b)
V ist das Volumen des Reaktionsansatzes. Die Reaktionsgeschwindigkeit v in mMs-1
stimmt numerisch mit der mit 60 multiplizierten Anzahl der Units überein.
v=∆N/(∆t*V)[mMs-1]
für V=1 ml (Testvolumen) gilt:
1 mMs-1≙1 µmols-1=60 U
(64c)
Die Sättigungsgeschwindigkeit vmax einer enzymatisch katalysierten Reaktion ergibt
sich aus der Enzymeinwaage und dessen Verdünnung. Die katalytische Konstante
kcat (Wechselzahl (s-1) gibt an, mit welcher Frequenz die Reaktion am Enzym abläuft.
3.1.2 Photometrie
Die Bestimmung der funktionellen Aktivität des Enzyms Glukoseoxydase als
Katalysator der Glukoseoxidation kann mithilfe eines photometrischen Aktivitätstest
erfolgen. Das bei der enzymatischen Reaktion als Nebenprodukt gebildete
Wasserstoffperoxyd reagiert mit dem Chromogen ABTS (2,2´-Azino-bis-8-
ethylbenzthiazolin-6-sulfonsäure) unter Mitwirkung des Enzyms Peroxydase (EC
1.11.1.6) zu einem Farbstoff (ABTS+), dessen Absorption im Photometer bei 420 nm
gemessen wird.
D-Glukose + O2 → 2-Keto-D-Glukose + H2O2 (Reaktion 5a)

___________________________________________________
77
3. Materialien und Methoden
2 ABTS + H2O2 + 2 H+ → 2 ABTS+ + 2 H2O (Reaktion 5b)
Die Intensität eines monochromatischen Lichtstrahls I0 wird beim Durchtritt durch ein
absorbierendes Medium der Dicke d und der Konzentration c des absorbierenden
Moleküls exponentiell geschwächt:
kdceII −⋅= 0 (65)
Die Größe
0
logIIA −=
(66)
wird Extinktion, A, genannt. Das Lambert-Beersche Gesetz,
dcA m ⋅∆⋅=∆ ε (67)
stellt einen linearen Zusammenhang zwischen der Änderung der Extinktion und der
Änderung der Konzentration des absorbierenden Stoffes her. εm ist der molekulare
Absorptionskoeffizient des Farbstoffes.
Zur Messung der Enzymaktivität wurde die Änderung der Extinktion der
enzymhaltigen Probenlösung in der Zeit ∆A/∆t bestimmt. Daraus wurde die
entsprechende Teilchenzahländerung des Farbstoffs in der Zeit ∆N/∆t errechnet
(Gleichung 68).
[ ]1−
∆⋅⋅⋅∆
=∆
⋅∆=
∆∆ mols
tdVA
tVc
tN
m
µε
(68)
Der millimolare Extinktionskoeffizient εm ist numerisch gleich der Extinktion einer 1
millimolaren Lösung bei d=1 cm Lichtweg. Für ABTS+ gilt [119]:

___________________________________________________
78
3. Materialien und Methoden
[ ]117,34)( −−+ = cmmolmlABTSm µε
Weil H2O2 zwei Moleküle ABTS oxidiert, muss hier noch ein Faktor ½ eingeführt
werden:
[ ]1
2−
∆⋅⋅⋅⋅∆
=∆∆ mols
tdVA
tN
m
µε
(69)
Die auf das Testvolumen V = 1 ml bezogene Reaktionsgeschwindigkeit v ist gegeben
durch:
[ ]1
2−
∆⋅⋅⋅∆
=⋅∆
∆= mMs
tdA
VtNv
mε
(70)
v ist eine Konzentrationsänderung in der Zeit. Für die Abschätzung des Thiele-
Modulus wurde diese Größe verwendet, weil die Einheit der KM darin auch die
Konzentration (mM) ist.
Die zeitliche Entwicklung der Absorption ∆A/∆t der enzymhaltigen Testansätze nach
Zugabe des Substrates (Glukose) wurde im Spektrophotometer gemessen. Die
Enzympräparate wurden vor der Messung unterschiedlich lange bei 60°C inkubiert
(jeweils drei Proben). Zur Berechnung der Aktivität nach Gleichung 68 wurde die
Änderung der Extinktion im Intervall zwischen der ersten und zweiten Minute
herangezogen. Die Enzymmenge wurde mit ca. 5 mU/ml so gewählt, dass die
Änderung der Absorption wenigstens 5 min linear blieb (initial-state) und die
Extinktion in der Messzeit den Wert 2 (99% Absorption) nicht überstieg. Eine zu hohe
Enzymkonzentration führt zu Substratverarmung innerhalb der Küvette und äußert
sich in einer Abflachung der Absorptionszunahme, während bei zu geringer
Konzentration der Messfehler (Pipettierfehler, Inhomogenitäten, etc.) zu groß wird.

___________________________________________________
79
3. Materialien und Methoden
3.1.3 Michaelis-Menten Kinetik
Die kinetischen Eigenschaften eines (Enzym-)Katalysators werden durch seine
kinetischen Konstanten Km und vmax bzw. kcat beschrieben; die Abhängigkeit der
Reaktionsgeschwindigkeit von der Substratkonzentration durch die Michaelis-
Menten-Gleichung:
[ ][ ]SKSvv
M +⋅
= max
(71)
Diejenige Substratkonzentration, bei der eine Reaktionsgeschwindigkeit erreicht wird,
die gerade halb so groß ist wie ihr theoretischer Maximalwert vmax bei
Substratsättigung, wird als Michaelis-Menten-Konstante KM bezeichnet. Je höher KM
ist, desto höher muss die Substratkonzentration sein, damit die Reaktion (bei
gegebener Enzymkonzentration) mit halb-maximaler Geschwindigkeit abläuft. Bei
hohen Substratkonzentrationen ist der geschwindigkeitsbestimmende Schritt der
enzymkatalysierten Reaktion derjenige Schritt, in dem sich der Enzym-Substrat-
Komplex in den Enzym-Produkt-Komplex umwandelt. Weil vmax die Geschwindigkeit
bei unendlich großer Substratkonzentration ist, ist sie die Geschwindigkeit dieses
(geschwindigkeitsbestimmenden) Schritts. Damit ist sie ein direktes Maß für die
Fähigkeit eines Enzyms, ein gebundenes Substrat in das Produkt zu überführen.
3.1.4 Direct-Plot
KM und vmax kann durch Messung der Enzymaktivität bei unterschiedlichen Substrat-
Konzentrationen bestimmt werden. Die Analyse der Messwerte erfolgt nach einer
linearisierten Form der Michaelis-Menten Gleichung [120]:
[ ] mKSvvv ⋅+=max (72)

___________________________________________________
80
3. Materialien und Methoden
Die kinetischen Konstanten werden dabei als Variable aufgefasst. Für alle
Einzelmessungen wird –[S] auf der x-Achse und v auf der y-Achse aufgetragen und
eine Gerade durch die Punkte –[S]/0 und 0/v gezogen. Die x-Komponente des
Schnittpunkts der Kurvenschar liefert Km, die y-Komponente vMax.
Abbildung 21: Zur Ermittlung der kinetischen Konstanten Km und vMax mit Hilfe des direct plot
(Gleichung 72).
3.1.5 Nicht-lineare Kurvenanpassung
Alternativ wurden die Daten aus den Aktivitätsmessungen durch nichtlineare
Kurvenanpassung mithilfe des Programms Origin (MicroCal™) ausgewertet. Dazu
wurden die gemessenen Reaktionsgeschwindigkeiten gegen die
Substratkonzentration aufgetragen und KM und vmax durch Anpassung der Werte an
die Funktion

___________________________________________________
81
3. Materialien und Methoden
xKxvy
M +⋅= max
(73)
bestimmt.
Abbildung 22: Zur Ermittlung der kinetischen Parameter Km und vMax durch nichtlineare
Kurvenanpassung an die Messwerte der Reaktionsgeschwindigkeit der Peroxyd-Bildung in
Abhängigkeit von der Konzentration des Substrates der enzymatischen Reaktion Glukose.
Die Literaturwerte für Km des aus Aspergillus niger isolierten Enzyms Glukoseoxidase
variieren zwischen 22 mM und 37 mM [121]. Ein Literaturwert für kcat ≡vMax/[E]T ist 337
s-1 [122].
Glukoseoxidase (GOx) wurde von Biozyme Laboratories bezogen und ohne weitere
Reinigung eingesetzt. Die Herstellerangabe über die spezifische Aktivität des
Enzympräparats waren 342 U/mg Protein bzw. 270 U/mg Material. Dieser Wert
impliziert eine Wechselzahl kcat von 720 s-1. Die Angabe über den Katalase-Anteil war

___________________________________________________
82
3. Materialien und Methoden
<0,05% und über das GOx/Katalase-Verhältnis >25000. Die gemessenen
spezifischen Aktivitäten unter Standardbedingungen (25°C , pH 7) waren mit ca. 140
U/mg bzw. kcat von 370 s-1 etwa um die Hälfte niedriger als die Angaben des
Herstellers in guter Übereinstimmung mit dem Literaturwert [122].
Die Bestimmung der enzymatischen Aktivität erfolgte in 1 ml Küvetten in einem
Spektro-Photometer (Perkin-Elmer Lambda).
Der Testansatz hatte folgende Zusammensetzung:
780 µl 100 mM KH2PO4–Puffer,
pH 7,0 mit 2,56 mM
ABTS
100 µl Glukose 1 bis 500 mM
10 µl Peroxidase (1 mg/ml)
10 µl GOx (Verdünnung s.
Tab.: 2)
1000 µl Testansatz
Chemische Charakteristika der Immobilisierungsmatrix nehmen Einfluss auf die
Stabilität der Funktion des Enzyms [123]. Als Modellsystem wurden Gele bestehend
aus Poly-Acrylamid und Wasser verwendet, die mit Bis-Acrylamid vernetzt waren und
durch das Initiatorsystem APS (Ammoniumperoxodisulfat)/Temed
(Tetramethylethylendiamin) zur Polymerisation gebracht worden waren. Den
Hydrogelen waren außerdem unterschiedliche Mengen an Ko-Solventien (NaCl,
PEG, Dextran, etc.) beigesetzt.

___________________________________________________
83
3. Materialien und Methoden
3.1.6 µ-Gele
Um Diffusionseffekte und den Materialverbrauch so gering wie möglich zu halten,
wurden Gele mit 60 µm Durchmesser in Mikrokapillaren von 10 oder 20 µl Volumen
hergestellt. Die Herstellung der µ-Gele in Kapillaren erwies sich auch deshalb als
günstig, weil der Sauerstoffzutritt während der Polymerisation stark herabgesetzt ist
und die Arbeiten deshalb in normaler Laboratmosphäre durchgeführt werden konnten
(evt. nach vorherigem Entgasen der verwendeten Lösungen). Nach 10-15 min war
die Polymerisation abgeschlossen und die Gele blieben bis zur Verwendung in den
an den Enden versiegelten Kapillaren im Kühlschrank bei 4°C.
Gel-Zusammensetzung :
48 µl 100 mM KH2PO4-Puffer, pH 7,0
10 µl GOx 1 mg/ml in 100 mM KH2PO4-
Puffer, pH 7,0
40 µl Acrylamid Stammlösung; 40%
(w/w) Acrylamid, 2,4% Bis-
Acrylamid
1 µl Ammoniumperoxodisulfat (APS)
5% (w/w)
1 µl Temed 6,6% (v/v)
100 µl Gel
3.1.7 Enzymkonzentration
Die Konzentration der Stammlösung des Enzyms betrug 1 mg/ml (≈130 U/ml).
Für Messungen in Lösung wurde die Stammlösung im Küvettenansatz auf 1:25000
(ca. 5 mU/ml) verdünnt. In den Gelen war die eingesetzte Enzymmenge mit
Rücksicht auf eine scheinbar verringerte Aktivität aufgrund von Diffusionslimitierung
10-fach höher (ca. 50 mU/ml).

___________________________________________________
84
3. Materialien und Methoden
3.1.8 Thermische Deaktivierung der Enzymfunktion
Temperaturerhöhung führt in enzymatisch katalysierten Reaktionen zu zwei
gegenläufigen Effekten: 1. Erhöhung der Reaktionsgeschwindigkeit, aufgrund der
Temperaturabhängigkeit der Enzymaktivität. 2. Abnahme der Stabilität des Enzyms
im Hinblick auf thermische Denaturierung. Die Deaktivierungsenergie kann durch
Messung der verbliebenen Aktivität nach Inkubation für verschiedene Zeiten bei
verschiedenen Temperaturen errechnet werden [125,125].
Tabelle 2: Verwendete Enzymkonzentrationen
Stamm in 1 ml
Testansatz
in 10 µl Hydrogel
(pro 1 ml Testansatz)l
m2 [gL-1] 1 40*10-6 40*10-3 (0,4*10-3)
m2 [M] 6,25
*10-6
0,25*10-9 250 * 10-9 (2,5*10-9)
Verdünnung 1:1 1:25000 1:25 (1:2500)
Aktivität [Uml-1] 130 0,005 5 (0,05)
vmax [mMs-1] 2,2 83*10-6 0,083 (830*10-6)
Ist die Geschwindigkeit der Enzymdeaktivierung vd erster Ordnung in Bezug auf die
Enzymkonzentration, gilt:
Ekv dd ⋅−=
(74)
Die Deaktivierungskonstante kd ist eine Funktion der Temperatur. Ihre
Temperaturabhängigkeit wird durch die Gleichung von Arrhenius beschrieben:

___________________________________________________
85
3. Materialien und Methoden
⎟⎠⎞
⎜⎝⎛
⋅−
⋅= TRE
dd
d
ekk 0
(75)
Ed ist die Deaktivierungsenergie, R ie Gaskonstante und T die absolute Temperatur.
Die Abnahme der Konzentration aktiven Enzyms kann also folgenderweise
beschrieben werden:
EkdtdE d ⋅−=
(76)
Integration dieser Gleichung mit der Randbedingung E=0 für t=0 liefert:
tk deEE ⋅−⋅= 0
(77)
mit E0 als Anfangsenzymkonzentration. Weil die enzymatische Aktivität A direkt
proportional zur Konzentration aktiven Enzyms E ist, gilt außerdem:
00 EEAA = (78)
und damit
tk
rdeAA ⋅−⋅= 0 (79)
Dieses Ergebnis ist bekannt als Modell exponentiell abnehmender Enzymaktivität.
Auftragen der verbleibenden Aktivität als Funktion der Zeit liefert die
Deaktivierungskonstante kd. Aus der Arrhenius-Gleichung folgt, und das wird
experimentell bestätigt, dass die Deaktivierungskonstante mit steigender Temperatur
zunimmt. Auftragen von log(kd) gegen die reziproke absolute Temperatur liefert die
Deaktivierungsenergie Ed [126].
Ein weiterer wichtiger Parameter hinsichtlich der Enzymstabilität ist die Halbwertszeit
τ1/2 der enzymatischen Aktivität. Sie gibt an, wie lang die Zeitperiode ist, bis die
enzymatische Aktivität auf die Hälfte der ursprünglichen Aktivität abgesunken ist.

___________________________________________________
86
3. Materialien und Methoden
( ) dd kk 693,05,0ln
21 =−=τ
(80)
Die Deaktivierungsenergie Ed für die meisten Enzyme liegt zwischen 200 und 400
kJ/mol [127].
3.1.9 Stress-Aktivitätstest/Halbwertszeit
Als alternatives Maß für die (thermische) Stabilität des Enzyms wurde neben der
Denaturierungstemperatur Tm die Halbwertszeit der enzymatischen Aktivität τ1/2 bei
60°C gewählt. Zu deren Bestimmung wurden enzymbeladene Hydrogele in
Kapillaren in einem Wasserbad unterschiedlich lange bei 60 °C inkubiert und danach
bei 4°C verwahrt. Vor der Messung bei 25°C wurden die Kapillaren 10 min bei 25°C
temperiert. Um das Ausdiffundieren des Enzyms aus dem Gel zu vermeiden, wurde
auf eine Equilibrierung der Quellung vor der Messung verzichtet, und die
Hydrogelfäden aus der Kapillare in die Küvette mit der Testlösung gepresst und
sofort mit der Messung begonnen. Um die Ausbildung von Konzentrationsgradienten
an der Oberfläche der Hydrogelfäden zu vermeiden, wurde der Testansatz manuell
geschwenkt, und die Änderung der Adsorption erfolgte deshalb diskontinuierlich
[128].

___________________________________________________
87
3. Materialien und Methoden
3.2 Dynamische Differenzialkalorimetrie
3.2.1 DTA, Differenzthermoanalyse
Diese älteste Messanordnung der Thermoanalytik wurde 1887 durch LeChatelier und
Roberts-Austin (1889) erfunden. Dabei werden Probe und Referenzsubstanz einem
gemeinsamen Temperaturprogramm unterworfen. Die Temperaturdifferenz zwischen
Probe und Referenz wird durch zwei gegeneinander geschaltete in die Probe bzw. in
die Referenz eintauchende Thermoelemente gemessen. Erfolgt in der Probe eine
endotherme Reaktion, wodurch die Probentemperatur im Vergleich zur
Referenztemperatur nachhinkt, wird eine entsprechende Temperaturdifferenz
gemessen. Analog tritt eine umgekehrte Temperaturdifferenz bei exothermen
Effekten auf. Diese klassische DTA-Anordnung erlaubt das Erkennen thermischer
Effekte, nicht aber die quantitative Bestimmung des Wärmeumsatzes, da der
Wärmestrom vom Wärmewiderstand der Probe abhängig ist, sodass Peakflächen
nicht proportional dem Wärmeumsatz sind. Boersma ebnete durch Einbau eines
definierten Wärmewiderstandes den Weg zur Wärmestrom-DSC [129].
3.2.2 DSC, Dynamische Differenzkalorimetrie
Bei der DSC, nach DIN1005 Dynamische Differenzkalorimetrie (DDK) genannt, wird
der Wärmestrom zur Probe gemessen. Der Wärmestrom, eigentlich eine
Wärmeleistung, hat im SI System die Einheit Watt bzw. Milliwatt. Durch Integration
des Wärmestroms über die Zeit erhält man den Wärmeumsatz oder die
Enthalpieänderung einer Probe in mJ. DSC-Messungen werden meistens mit einem
dynamischen Temperaturprogramm durchgeführt, mit dem ein interessierender
Temperaturbereich überstrichen wird. Daneben sind aber auch Messungen bei
konstanter Temperatur (isotherm) üblich. Die Hauptanwendungen der DSC liegen in

___________________________________________________
88
3. Materialien und Methoden
der Bestimmung der spezifischen Wärme, thermischer Effekte, der Reinheit, der
Polymorphie, der Glasumwandlung, der Oxidationsstabilität, der Wärmetönung
chemischer Reaktionen, der Reaktionskinetik, des Schmelzverhaltens und der
Kristallisation. Hochempfindliche DSC wird derzeit immer stärker auch in den Life
Sciences eingesetzt zur Bestimmung der Denaturierung von Proteinen,
Nukleinsäuren und Phospholipidmembranen, sowie zur Untersuchung von
Biopolymer/Biopolymer-, bzw. Biopolymer/Ligand-Wechselwirkungen
Damit einer Fläche unter der DSC-Kurve einem Wärmeumsatz entspricht, muss die
Kurve gegen die Zeit aufgetragen werden (Wärmemenge=Wärmestrom mal Zeit). Um
aber bei dynamischen Messungen auch die Temperaturinformation anzugeben, wird
üblicherweise eine zeitproportionale Temperatur auf der Abszisse angegeben. Es
kann sich dabei allerdings nicht um die Probentemperatur handeln, da diese ja z.B.
beim Schmelzen einer Reinsubstanz konstant bleibt, sondern es ist, je nach
Hersteller, die aus Starttemperatur und Heizgeschwindigkeit errechnete
Programmtemperatur oder die Temperatur der inerten Referenz. Es haben sich
hauptsächlich zwei Messprinzipien durchgesetzt:
3.2.2.1 Leistungskompensations-DSC
Diese nach DIN 1005 Dynamische Differenz-Leistungskalorimetrie (DDLK) genannte
Messanordnung wurde 1963 von Watson und O´Neill erfunden [130]. Die
Probenmessstelle und die Vergleichs-(Referenz-) Messstelle verfügen über je einen
Temperaturfühler und eine Heizvorrichtung. Die beiden Seiten werden durch
getrennte Regler so mit Heizleistung versorgt, dass ihre Temperatur einem
gewünschten Temperaturprogramm folgt. Die Leistungsaufnahme bzw. -abgabe der
Probe wird dabei durch äquivalentes Vergrößern oder Vermindern der Heizleistung
eines Differenzheizkreises kompensiert. Der dazu nötige Heizleistungsunterschied
wird in eine proportionale elektrische Spannung umgewandelt, welche ihrerseits dem

___________________________________________________
89
3. Materialien und Methoden
Wärmestrom zur Probe proportional ist. Um ein gutes Abkühlverhalten zu
gewährleisten, wird die Umgebung der beiden Messstellen mittels Thermostat z.B.
bei 20°C stabil gehalten. Naturgemäß steigt die durch Strahlung, Konvektion und
Leitung an die Umgebung der beiden Messstellen abgegebene Heizleistung mit
steigender Temperatur stark an. Schon geringfügige Unterschiede der Wärmeabgabe
führen dabei zu einem merklich verschiedenen Leistungsbedarf und damit zu einer
Drift der Blindkurve (Basislinie).
3.2.2.2 Wärmestrom-DSC
Diese schon 1955 von Boersma [131] beschriebene Messanordnung wird nach
DIN51005 Dynamische Differenz-Wärmestrom-Kalorimetrie (DDWK) genannt. Hier
handelt es sich um eine Zwillingsanordnung von Probe und Vergleichsmessstelle in
einem einzigen Ofenkörper. Der Wärmestrom fließt vom Ofen über einen definierten
Wärmewiderstand zur Probenseite bzw. zur Referenzseite. Die treibende Kraft für
den Wärmestrom ist der Temperaturunterschied über dem Wärmewiderstand. Der
Wärmestrom zur Probe selbst entspricht dem Unterschied der beiden Wärmeströme.
Bei der Wärmestrom-DSC ist der zur Messung verwendete Wärmewiderstand
temperaturabhängig. In mikroprozessorgesteuerten Geräten wird diese
Temperaturabhängigkeit vom Auswertprogramm automatisch kompensiert.
3.2.3 Heizgeschwindigkeit
Die Heizgeschwindigkeit (Temperaturprogramm) spielt eine wichtige Rolle bei
thermoanalytischen Messungen. Für Vergleichszwecke soll möglichst immer die
gleiche Heizgeschwindigkeit verwendet werden. Exaktes thermodynamisches
Gleichgewicht ist nur bei isothermen Messungen möglich. Thermische Stabilität und
chemische Reaktionen werden oft isotherm gemessen. Da bei den üblicherweise

___________________________________________________
90
3. Materialien und Methoden
kleinen Proben, welche bei thermoanalytischen Messungen eingesetzt werden
(typisch 10 mg), kaum Temperatur- und Konzentrationsgradienten auftreten, ergibt
z.B. bei der DSC-Reinheitsbestimmung auch eine Heizgeschwindigkeit von 2°C/min
keine Verfälschungen. Chemische Reaktionen werden üblicherweise bei 5°C/min
gemessen. Kleine Heizgeschwindigkeiten helfen, eng benachbarte physikalische
Umwandlungen voneinander zu trennen, d.h. die Temperaturauflösung steigt mit
sinkender Heizrate. Da chemische Reaktionen mit höheren Heizgeschwindigkeiten
zu höheren Temperaturen verschoben werden, Phasenumwandlungen dagegen
kaum beeinflusst werden, gelingt deren Trennung hier oft besser bei höheren
Heizgeschwindigkeiten, z.B. 20°C/min. Werden nur geringe Ansprüche an die
Temperaturgenauigkeit gestellt, d.h. wird bei der DSC nur die Wärmetönung oder bei
der Thermogravimetrie nur der Gewichtsverlust gemessen, können auch
Heizgeschwindigkeiten von 50 oder gar 100°C/min angewendet werden.
Physikalische Umwandlungen zeigen bei negativen Heizgeschwindigkeiten häufig
starkes Unterkühlen, z.B. gefrieren 10 mg Wasser beim Abkühlen mit 10°C/min erst
bei ca. –15°C.
3.2.4 Vergleich der Messprinzipien von DTA und DSC
In Abbildung 23 ist ein schematisches Diagramm eines Gerätes für Differenzial-
Thermoanalyse zusammen mit den Definitionen der Begriffe, die für einen Vergleich
von klassischer DTA, Wärmefluss-DSC und Leistungskompensations-DSC gebraucht
werden, gezeigt.

___________________________________________________
91
3. Materialien und Methoden
Abbildung 23: Schema eines Messgerätes zur Differenzial-Thermo-Analyse [132]. Links die
Messstelle, rechts die Referenzmessstelle. Th: Temperatur der Wärmequelle, Ts: aktuelle
Probentemperatur, Tsm: gemessene Probentemperatur, Cs: Wärmekapazität der Probe plus
Pfännchen, Csm: Wärmekapazität der Proben-Messstelle, Tr: aktuelle Referenztemperatur, Trm:
gemessene Referenztemperatur, Cr: Wärmekapazität der Referenz plus Pfännchen, Crm:
Wärmekapazität der Referenz-Messstelle, R´s, Rs, R´r, Rr: Thermische Widerstände, dq/dt:
Wärmefluss.
In einem idealen Zwillingsgerät sind die Wärmekapazitäten und thermischen
Widerstände gematched, d.h. Crm=Csm; Rr=Rs=R und R´r=R´s=R´. Zu beachten ist,
dass R≠R´ und Cs≠Cr.
Der Wärmefluss wird durch Newtons Gesetz beschrieben:
TRdt
dq∆⎟
⎠⎞
⎜⎝⎛=
1
(81)
Der Wärmefluss zur Probe heizt sowohl die Probenmessstelle als auch die Probe
selbst:
⎟⎠⎞
⎜⎝⎛⋅+⎟
⎠⎞
⎜⎝⎛⋅=
dtdT
Cdt
dTC
dtdq s
ssm
sms
(82)

___________________________________________________
92
3. Materialien und Methoden
und es gilt weiterhin:
⎟⎠⎞
⎜⎝⎛⋅=
dtdT
Cdt
dq ss
s/
(83)
also:
dtdq
dtdT
Cdt
dq ssmsm
s/
+⎟⎠⎞
⎜⎝⎛⋅=
(84)
Newtons Gesetz angewendet:
( )smhs TT
Rdtdq
−⋅⎟⎠⎞
⎜⎝⎛=
1
(85)
und
( )ssms TT
Rdtdq
−⋅⎟⎠⎞
⎜⎝⎛= /
/ 1
(86)
Ähnliche Ausdrücke ergeben sich für die Referenzseite.
3.2.4.1 Klassische DTA
Inder klassischen DTA befinden sich die Temperatursensoren, meist Thermosäulen,
direkt innerhalb der Probe und der Referenz, so dass Tsm=Ts und Trm = Tr, d.h. R´=0.
Das Signal berechnet sich dann zu:
( )rs CCdtdTRT −⋅⎟
⎠⎞
⎜⎝⎛⋅=∆
(87)

___________________________________________________
93
3. Materialien und Methoden
und hängt von der Differenz der Wärmekapazitäten von Probe und Referenz, der
Heizrate und dem thermischen Widerstand R ab. R ist nur schwierig experimentell
zugänglich und hängt sowohl von den Eigenschaften des Messgerätes ab, wie auch
von den Eigenschaften von Probe und Referenz.
3.2.4.2 Leistungskompensations-DSC
In der Leistungskompensations-DSC wird die Leistung so geregelt, dass Tsm=Trm.
Also ist Th=Tsm=Trm und R=0, d.h. es besteht kein thermischer Widerstand zwischen
der Wärmequelle und der Messstelle. Das Signal errechnet sich dann zu:
( )rs CCdtdT
dtdq
−⋅⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛∆
(88)
3.2.4.3 Wärmefluss-DSC
Die Bedingungen hier lauten: Th ≠ Tsm ≠ Ts und R ≠ R´≠ 0.
Das Signal errechnet sich zu:
( )rssmrm CCdtdTRTTT −⋅⎟
⎠⎞
⎜⎝⎛⋅=−=∆
(89)
Dieser Ausdruck ist dem für die klassische DTA sehr ähnlich, außer dass der
Widerstand R ausschließlich vom Messgerät und nicht von den Eigenschaften der
Probe abhängt. R ist eine Funktion der Temperatur und der Zusammenhang
zwischen R und T muss durch Kalibrierung des Messgeräts ermittelt werden. Manche
Hersteller von DSC-Geräten haben eine elektronische Kompensation für diese
Temperaturabhängigkeit und für die Umwandlung des Signals in Leistungseinheiten
(mW) im Kontrollmodul ihrer Geräte eingebaut.

___________________________________________________
94
3. Materialien und Methoden
3.2.5 Mikrokalorimetrie
Die kalorimetrische Untersuchung von Änderungen des konformativen Zustands von
Proteinen ist die einzige Methode, die Enthalpieänderung der Denaturierung direkt zu
bestimmen. Alle temperaturinduzierten Änderungen in makroskopischen Systemen
sind mit Ethalpieänderungen verbunden. Wird der Prozess durch
Temperaturerhöhung ausgelöst, wie die Denaturierung von Proteinen, ist der Prozess
endotherm, andernfalls, wenn der Prozess durch Temperaturerniedrigung ausgelöst
wird, wie das Kristallisieren oder die Faltung von Proteinen, ist er mit
Wärmefreisetzung verbunden, also exotherm.
Die Temperaturabhängigkeit der Enthalpie H kann experimentell durch
kalorimetrische Messung der Wärmekapazität C(T) der zu untersuchenden Systeme
über den interessierenden Temperaturbereich bestimmt werden:
∫ ∆+=∆T
Tm
DN
DN
DN
m
THdTTCTH )()()(
(90)
Einzelne Proteinmoleküle sind so groß, ihre molekulare Masse ist, mehreren
Tausend Atomen entsprechend, größer als 10.000 kDa, dass sie als makroskopische
Objekte anzusehen sind. Erst bei Konzentration < 10-4 M sind die intramolekularen
Wechselwirkungen eines biologischen Makromoleküls in Lösung vernachlässigbar. In
solch verdünnten Lösungen ist der Einfluss des Makromoleküls auf die thermischen
Eigenschaften der gesamten Probe entsprechend klein. In mikrokalorimetrischen
Messungen übersteigt die Wärmekapazität des Proteins nicht 0,03% und die
Überschuss-Wärmekapazität des Peak nicht 1% der Wärmekapazität der gesamten
Lösung.
Eine weitere Schwierigkeit bereitet der Biokalorimetrie die oft schlechte
Zugänglichkeit biologischer Makromoleküle aufgrund erschwerter Gewinnung,

___________________________________________________
95
3. Materialien und Methoden
Isolierung und Reinigung, sodass üblicherweise höchstens einige Milligramm der
Substanzen vorliegen.
Dynamischen Mikrokalorimeter messen keine absoluten Wärmekapazitäten, sondern
Wärmekapazitätsdifferenzen bzw. Überschusswärmekapazitäten, und sie haben ein
Zwillingsdesign mit zwei identischen Messzellen, eine für die Probe und eine für eine
Referenzlösung, meist Puffer. Die Wärmekapazitätsdifferenz wird normalerweise
nach der Kompensationsmethode bestimmt: ein Controller steuert die Heizleistung in
den beiden elektrischen Heizern jeder Messzelle so, dass die Temperaturdifferenz
zwischen Proben- und Referenzzelle konstant bei Null gehalten wird. Die Differenz
der Heizleistungen wird kontinuierlich über der Temperatur aufgezeichnet.
Das freie Volumen bereitet insofern Schwierigkeiten, als Wasser eine hohe
Verdampfungs-Enthalpie besitzt (2 kJ/g).
Die Auflösung eines Messgerätes hängt nicht nur vom Signal/Rausch-Verhältnis des
Messgerätes ab, sondern ebenso von der Form des Signals. Im Falle der
Mikrokalorimetrie von biologischen Makromolekülen liegt das Spektrum möglicher
Signale zwischen 0,1°C (beim hochkooperativen Schmelzen von Homo-Polymeren
und Phospholipid-Doppelschichten) und bis zu 100°C bei wenig kooperativen
Übergängen mit geringer Umwandlungs-Enthalpie.
Die Heizraten in mikrokalorimetrischen Untersuchungen von Makromolekülen in
Lösung sollte 5 K/min nicht übersteigen und ist bevorzugt niedriger (typischerweise 1
K/min).
Die thermische Denaturierung begleitende intramolekulare Reaktionen, wie die
Aggregation, chemische Modifizierung und intermolekulares Vernetzen führen zu
(partieller) Nicht-Reversibilität des Phasenwechsels [133]. Deshalb sollten
Bedingungen ausgewählt werden, die den intramolekularen Kontakt auf ein Minimum
reduzieren: durch möglichst niedrige Konzentration des Proteins, durch Wahl des

___________________________________________________
96
3. Materialien und Methoden
geeigneten pH-Werts (entfernt vom iso-elektrischen Punkt) und Einstellen der Ionen-
Stärke.
Grundsätzlich erhöht sich die Empfindlichkeit eines Dynamischen Differenz-
Kalorimeters mit steigender Heizrate. Eine steigende Heizrate führt zu einer
Verringerung der Uniformität des Temperaturfeldes in der Probe, was zu einer
Verbreiterung des thermischen Effekts führt. Die Geschwindigkeitskonstante einer
typischen temperatur-induzierten Phasenumwandlung eAbbildung 23ines Proteins
liegt im Bereich von 103 s-1. Besondere kleine Proteine falten sich aber schon in
wenigen µs [134]. Dynamische Differential Mikro-Kalorimeter, z.B. VP-DSC,
MicroCal, Inc., sind für die Untersuchung von Wärmekapazitätsänderungen in
Flüssigkeiten ausgelegt (Kapillare, 300 µl -1500 µl Zelle, einfacher Einlass). Die
Untersuchung von festen Proben oder von Hydrogelen ist mit diesen Geräten nicht
möglich.
Abbildung 24: Dynamisches Differential Mikrokalorimeter (VP-DSC, MicroCal, Inc.) mit zwei identische
Messzellen (1,5-0,3 ml) [135].

___________________________________________________
97
3. Materialien und Methoden
Zur Bestimmung der Wärmekapazitätsänderung ist eine einzelne Messung nicht
ausreichend. Zuerst wird eine Gerätebasislinie aufgezeichnet mit Puffer in beiden
Messzellen. Diese Basislinie ist auch für hochempfindliche Kalorimeter nicht linear
oder gar horizontal, da die beiden Messzellen nie völlig gleich hergestellt werden
können. Allerdings ist in Präzisionsinstrumenten diese Basislinie stabil und
reproduzierbar, sodass die einmal aufgezeichnete Basislinie zur Korrektur folgender
Messungen verwendet werden kann.
Zur Messung der Überschuss-Wärmekapazität wird die Messzelle mit Proteinlösung
gefüllt, die Vergleichszelle mit Puffer. Nach Abzug der Basislinie erhält man so die
Überschuss-Wärmekapazität des Proteins. Die Differenz ist aber eine zwischen zwei
aufeinander folgende Messungen und die Vergleichsmesszelle dient nur als interne
Referenz. Dadurch wird den Nicht-Identitäten der beiden Messzellen Rechnung
getragen.
Zur Bestimmung der Wärmekapazität der Probe muss das Gerät kalibriert werden.
Das kann nicht durch bekante Wärmekapazitäten von Eichsubstanzen geschehen,
da deren absoluten Wärmekapazitäten niemals mit der erforderlichen Genauigkeit
zur Kalibrierung von Dynamischen Differenzkalorimetern bestimmt worden sind.
Daher erfolgt die Kalibrierung elektrisch durch Applizierung einer definierten
Heizleistung P in eine der Zellen, wodurch der Wärmekapazitätswechsel in der Probe
immitiert wird.
Wird die Kalibrierungsleistung für eine Zeitdauer von t Sekunden angelegt, wird eine
Energie von
tPE ⋅= (91)
übertragen und diese entspricht der Fläche S unter der Kalibrierungsmarke.
Die apparente Wärmekapazitätsänderung ist gegeben durch Gleichung 92:

___________________________________________________
98
3. Materialien und Methoden
1−
⎟⎠⎞
⎜⎝⎛⋅=∆
dtdTPC
(92)
(dT/dt) ist die Heizrate. Durch Dividieren des aufgezeichneten Signals <∆C> durch l,
der Stufenhöhe der Kalibrierungsmarke der Aufzeichnung, erhält man den Wert der
Wärmekapazität einer Einheit der Aufzeichnung [13,136].
1−
⎟⎠⎞
⎜⎝⎛
⎟⎟⎠
⎞⎜⎜⎝
⎛=∆
dtdT
lPlC
(93)
Ändert sich die Heizrate während der Messung, wird eine zeit- bzw.
temperaturabhängige Kalibrierkurve erstellt. Dazu wird eine periodische Heizleistung
während des gesamten Messung appliziert und die Stufenhöhe des Antwortsignals in
Abhängigkeit von der Temperatur (bzw. der Zeit) analysiert. Die Differenz der
Wärmekapazität zwischen einer Lösung eines biologischen Makromoleküls und dem
Lösungsmittel Wasser ist immer negativ, weil die Wärmekapazität von
Makromolekülen kleiner ist als die des Wassers gleichen Volumens.
3.2.6 Probenvorbereitung DSC
Am Institut für Makromolekulare Chemie und am Institut für Experimentelle
Polymerphysik bestand die Möglichkeit, Messungen der Denaturierung der GOx auf
herkömmlichen DSC-Maschinen (Perkin-Elmer DSC7) durchzuführen, und einige
Aspekte der Denaturierung der GOx und einige Ko-Solvens Einflüsse konnten
charakterisiert werden.

___________________________________________________
99
3. Materialien und Methoden
Abbildung 25: Schema einer konventionellen Leistungskompensations-DSC (Perkin-
Elmer DSC 7).
In der konventionellen DSC wird die Probe, fest, flüssig oder gelartig, in kleine
Aluminiumtiegel von 40 µl Fassungsvermögen eingewogen und diese dann mit
einem Deckel in einer Presse kaltverschweißt. Für dampfdruckentwickelnde Proben
gibt es die Möglichkeit, den Tiegel mit einem Deckel mit einem winzigen Loch zu
versiegeln, sodass zwar der Druckaufbau verringert wird, die Materialverluste aber
gering bleiben. Druck in den Tiegeln führt zu einer Verwölbung des Tiegelbodens,
was zu Störungen der Messung führen kann. Bei nichtflüssigen Proben ist evt. zu
beachten, dass guter thermischer Kontakt zum Tiegelboden hergestellt wird, z.B.
durch Benetzung mit Silikonöl. Der Tiegel wird dann auf die Messstelle des
Zwillingsinstruments eingesetzt (Abbildung 25); auf die Referenzstelle wird ein
Leertiegel eingesetzt. Die Heizrate betrug bei allen Messungen 5 K/min.
3.2.7 Nano-DSC
Traditionelle Kalorimeter gewährleisten hohe Auflösung und Präzision in der
Bestimmung von Wärmemengen nur, wenn die Probenmasse und die Heizrate nicht
zu klein sind. Die Reduzierung der thermischen Masse des Messgerätes (Addenda)
in Kombination mit verbesserten themischen Eigenschaften kann dazu beitragen, die

___________________________________________________
100
3. Materialien und Methoden
Auflösung der Temperaturmessung zu verbessern und das Signal-Rausch-Verhältnis
soweit zu erhöhen, dass auch kleine Proben mit kleinen Heizgeschwindigkeiten
untersucht werden können [158].
3.2.7.1 Zur Auswahl der Kalibrierungssubstanzen
Die Demonstration der Eignung des vorgestellten Chip-Kalorimeters neuen Typs zur
thermischen Detektion von Phasenumwandlungen geringer Wärmetönung erfolgte in
drei Schritten. Im ersten Schritt wurde die geforderte Empfindlichkeit anhand der
Phasenumwandlungen von Flüssigkristallen demonstriert. Im zweiten Schritt wurde
die Detektion einer Phasenumwandlung in wässrigem Milieu gezeigt. Aufgrund der
hohen Wärmekapazität des Wassers als Medium war mit einer starken Dämpfung
des thermischen Signals in diesem für die Bio-Kalorimetrie obligatorischen Medium
zu rechnen. Darüber hinaus hat Wasser eine hohe Verdampfungsenhalpie, sodass
die Verdunstung während des Aufheizens zu vermeiden war. Als weitere
Testsubstanz wurde deshalb eine verdünnte wässrige Lösung des Phospholipids
DMPC gewählt. Die in solchen Lösungen z.B. nach Ultraschallbehandlung sich
spontan bildenden Vesikel, bestehend aus Lipid-Doppelschichten, zeigen, ähnlich
Flüssigkristallen, kooperative Ordnungs/Unordnungs-Übergänge kleiner
Wärmetönung. Die Energie des Übergangs kann darüber hinaus durch die Wahl der
Konzentration des Phospholipids eingestellt werden.
Im dritten Schritt wurde das thermische Denaturieren des Enzyms GOx, dessen
Denaturierungstemperatur Tm im Bereich zwischen 70°C und 80°C liegt, in Lösung
und eingebettet in eine Hydrogelmatrix, detektiert.
Flüssigkristalle wie auch Phospholipide haben als Kalibriersubstanzen den Vorteil,
dass ihre Phasenumwandlungen, anders als die der meisten Enzyme, vollständig
reversibel sind und deshalb mehrere Messungen mit ein und derselben Probe
durchgeführt werden können.

___________________________________________________
101
3. Materialien und Methoden
3.2.7.1.1 Flüssigkristalle
Flüssigkristalle sind Vertreter einer speziellen Klasse von Materialien. Sie sind
Flüssigkeiten, weil sie fließfähig sind und sie sind Kristalle, weil sie in wenigstens
einer Raumrichtung geordnet sind [137-139]. Diese Materialien befinden sich in
einem Zustand zwischen fest und flüssig. Die am einfachsten gebauten
Flüssigkristalle (Mesogene) bestehen aus einem einzelnen semirigiden oder
mesogenen Kern, verbunden mit einer oder zwei terminalen Alkylketten. Das
flüssigkristalline Verhalten resultiert aus der anisotropen Wechselwirkung dieser
Kerne, die meist von Phenylringen gebildet werden. Die Strukturformel des
Flüssigkristalls MH24 ist in Abbildung 26 dargestellt [140].
Abbildung 26: Strukturformel des Mesogens MH24 (4´-Oktyloxy-biphenyl-4-carbonitril)
Der Flüssigkristall MH24 von Merck™ zeigt neben dem Schmelzen zwei
Umwandlungen zwischen flüssigkristallinen Subphasen. Bei 67,1 °C wandelt sich in
einem endothermen Prozess die smektische Subphase A (smA) in die ebenfalls
flüssigkristalline nematische Subphase (n) um, verbunden mit einer
Phasenumwandlungsenergie von 0,29 J/g. Diese wiederum wandelt sich bei 80,1 °C
mit einer Energie von 2,2 J/g in die isotrope Flüssigkeit um [140].
Das Augenmerk für die Demonstration der Empfindlichkeit lag dabei vor allem auf der
Umwandlung mit der kleineren Energie (smA↔n).

___________________________________________________
102
3. Materialien und Methoden
3.2.7.1.2 Phospholipidvesikel
Die Gel/Flüssigkristall-Phasenumwandlung von Phospholipid-Doppelschichten ist gut
geeignet, Phasenumwandlungen in Wasser zu untersuchen [141]. Phospholipide
sind amphiphile Moleküle, d.h. sie haben sowohl polare als auch nichtpolare
Segmente: hydrophile Kopfgruppen und hydrophobe Schwänze. In wässriger Lösung
aggregieren Phospholipide spontan zu Doppelschichtstrukturen. Die hydrophilen
Kopfgruppen auf der Oberfläche sind dem Lösungsmittel Wasser zugewandt, die
hydrophoben Schwänze sind inwärts gerichtet. Die Selbstassoziation ist
entropiegetrieben durch die Freisetzung von Wassermolekülen aus der
Hydratationssphäre des Phospholipids im Zuge der Selbstassoziation. Wenn sich die
Doppelschicht um eine wassergefüllte Kavität schließt, entstehen Vesikel.
Abbildung 27: Vesikel, geformt aus einer Phospholipid-Doppelschicht.
Der Phasenübergang des Phospholipids DMPC erfolgt bei niedrigen Temperaturen
(24°C), wodurch die Versuchsdurchführung erleichtert ist. Zur Untersuchung wurden
Multilagen-Vesikel von 100 nm - 5 µm im Durchmesser (nicht gemessen) verwendet,
die durch Ultraschall-Behandlung einer Dispersion des Phospholipids in Wasser
hergestellt worden waren. In den Vesikeln sind die Phospholipidmoleküle dicht
gepackt und über starke van-der-Waals-Kräfte miteinander verbunden. Beim
Einsetzen der Phasenumwandlung beginnen die Moleküle kooperativ zu schmelzen.
In der resultierenden flüssigkristallinen Phase sind die Moleküle nicht mehr dicht

___________________________________________________
103
3. Materialien und Methoden
gepackt, sondern nur lose assoziiert durch gelockerte van-der-Waals-Kräfte zwischen
den Alkyl-Ketten [142-144]. Der Gel/Flüssigkristall-Übergang ist hochkooperativ. In
kooperativen Übergängen beginnen sich die Moleküle, schon bevor der eigentliche
Übergang einsetzt, zu reorganisieren und gleichförmig zu bewegen. Die Moleküle
kooperieren miteinander, um größere Beweglichkeit zu erreichen. Nahe der
Phasenumwandlung bilden sich erkennbare Inseln stärker beweglicher Moleküle
neben Bereichen, die noch starr und dicht gepackt sind. Die Anzahl der Moleküle, die
eine solche Insel bilden, wird als kooperative Einheit (C.U.) bezeichnet. Je größer die
kooperative Einheit, desto schmaler ist der Temperaturbereich des Übergangs. Der
Gel/Flüssigkristall-Übergang ist erster Ordnung mit einigen Aspekten eines
Übergangs zweiter Ordnung. Übergänge erster Ordnung zeichnen sich durch eine
abrupte Enthalpie- und Volumenänderung bei der Phasenumwandlungstemperatur
aus. Die Phasenumwandlung erster Ordnung ist vollständig kooperativ, alle Moleküle
wandeln sich gemeinsam um. Phasenumwandlungen zweiter Ordnung zeigen keine
Enthalpie- oder Volumenänderung bei der Phasenumwandlungstemperatur. Ein
Phasenübergang erster Ordnung ist theoretisch unendlich scharf, während in
Übergängen zweiter Ordnung der Peak durch die Ausbildung von kooperativen
Bereichen von begrenzter Reichweite vor der eigentlichen Umwandlung verbreitert
wird. In Thermogrammen synthetischer Phospholipide ist deshalb ein kleiner Peak
bei niedrigerer Temperatur als der eigentlichen Umwandlungstemperatur beobachtet
worden, die sogenannte pre-transition, wie auch eine Verbreiterung der
Hauptumwandlung [142].
Je größer die kooperative Einheit, desto schmaler ist der Übergang und desto größer
ist die van´t Hoff-Enthalpie ∆HvH. Die Peakbreite kann durch die Halbwertsbreite ∆T1/2
(FWHM full width at half maximum) ausgedrückt werden. Breitere Peaks
korrespondieren mit weniger kooperativen Übergängen. Diese Überlegungen gelten
auch für die Phasen-Übergänge von Proteinen.

___________________________________________________
104
3. Materialien und Methoden
Tabelle 3: Energien und Temperaturen der untersuchten Phasenübergänge.
Substanz Übergang Temperatur [°C] Energie [Jg-1]
MH24 smA↔n
n↔l
67,1
80,1
0,29
2,2
DMPC (10% w/v) gel↔flüssigkristallin 24,2 3,8
GOx (5% w/v) nativ↔denaturiert 77 0,85
3.2.7.2 Set-up Nano-Kalorimeter
Die chip-basierte Nano-Kalorimetrie bedient sich einer neuen Generation von
Kalorimetern, die in ihren Abmessungen um ein Vielfaches reduziert sind gegenüber
ihren mikrokalorimetrischen Vorläufern. Diese Kalorimeter sind in Dünnschichttechnik
hergestellte Mikrosysteme mit einer temperatursensortragenden Membran als
gemeinsamen Konstruktionsmerkmal. Die Membran inclusive einer darauf platzierten
Probe kann als quasi-adiabatisch angesehen werden, d.h. der thermische Kontakt
zur Umgebung ist auf ein Minimum reduziert. Dadurch ist die Empfindlichkeit in der
Bestimmung von Wärmemengen erhöht bis in den nJ-Bereich hinein [147]. Als
Temperatursensoren kommen zumeist Thermosäulen oder Widerstands-
Temperatursensoren zum Einsatz [146-148,174-179]. Die Empfindlichkeit der
Temperaturauflösung beträgt <100 µK. Das Probenvolumen reicht von 60 pL
[147,148] bis 50 µl [146]. Die Leistungsempfindlichkeit hat den nW-Bereich erreicht
[147,148,174]. In der Regel handelt es sich, wie auch in den klassischen DSC-
Geräten, um Zwillingsinstrumente, mit einer Proben- und einer Referenzmessstelle.
Erstmalig konnte mit Nano-Kalorimetern die größenabhängige
Schmelzpunkterniedrigung von metallischen Nano-Partikeln beobachtet werden

___________________________________________________
105
3. Materialien und Methoden
[179]. Ein kalorimetrisches Mikrophysiometer wurde zur Bestimmung der Zellaktivität
durch Wärmemessung eingesetzt [146] und es wurden Nano-Kalorimeter zur
Bestimmung der Phasenumwandlung von Proteinen eingesetzt [174,177].
Das hier vorgestellte Nano-Kalorimeter besteht aus einer Nitridmembran von 0,25
mm2 Fläche mit darin eingebetteten Widerstands-Temperatursensoren aus
Germanium mit hohem negativen Temperaturkoeffizienten des Widerstands. Mit
diesem Chip sind Temperaturmessungen <100 µK möglich und eine
Leistungsempfindlichkeit von <50 nW [174].
Abbildung 28: Set-Up des Nano-Kalorimeters. Das Brückensignal wurde von einem IC-Verstärker (INA
110 KP) verstärkt und von einer 16 bit Messkarte (Computerboards DAS) aufgezeichnet.

___________________________________________________
106
3. Materialien und Methoden
Abbildung 29: links:Mikrograph des Nano-Kalorimeters (Rückansicht). Die dunkler erscheinende
Fläche in der Mitte ist die Membran (0,25 mm2). Der zentrale der 5 Thermistoren ist von einem
Chromheizelement umgeben. In der unteren Hälfte sieht man die Kontaktierungen. Rechts: der ganze
Chip mit Kontaktierung. Die Membran erscheint als hellere Fläche in der Mittte.
Der Chip (Abbildung 29) ermöglicht die simultane Temperaturmessung an 5
Positionen symmetrisch um den Heizer. Er wurde aus einem 500 µm Silizium-Wafer
hergestellt. Die Temperatursensoren sind mikrostrukturierte Halbmetallwiderstände
aus Germanium, metallisch kontaktiert und eingebettet in eine 1 µm Schicht
Siliziumnitrid. Der zentrale Bereich des Sensorarrays ist eine Kavität mit einer
Siliziumnitridmembran als unterer Begrenzung. Diese Kavität dient der Aufnahme
des Probenguts. Während der Aufheizung des Chips werden die Temperaturen an
den verschiedenen Positionen gemessen. Chemische Reaktionen innerhalb der
Probe, die mit der Freisetzung oder der Aufnahme von Wärme verbunden sind,
führen zu einem Temperaturunterschied zwischen den zentralen Membran-
Thermistoren, die in nahezu unmitelbarem Kontakt mit dem Probenmaterial stehen,
und den Thermistoren, die mit dem Siliziumkörper in Kontakt stehen.
Um die Temperatur des Einsetzens einer thermisch induzierten Reaktion mittels des
Nano-Kalorimeters zu bestimmen ist zweierlei notwendig: erstens die kontinuierliche

___________________________________________________
107
3. Materialien und Methoden
Messung der Temperatur der Probe während des Aufheizens, zweitens die ebenfalls
kontinuierliche Messung der Temperaturdifferenz zwischen der Probe und dem
Chipkörper. Erstere ist hinreichend genau bestimmt durch Angabe der
Umwandlungstemperatur plus/minus 0,1°C.
Da die Energien der untersuchten Reaktionen aber sehr gering sind, < 1 µJ, sind die
Anforderungen an die Differenztemperaturmessung jedoch wesentlich höher. Hier ist
eine Temperaturauflösung < 1 mK notwendig.
Diese geforderte hohe Sensitivität wird erreicht durch die Wahl des Halbleiters
Germanium als Sensormaterial. Die Dünnschichtwiderstände aus Germanium zeigen
einen hohen negativen Temperaturkoeffizienten des elektrischen Widerstandes
(TKR) von –2% und können sehr hochohmig hergestellt werden. Der Widerstand
eines Germanium-Thermistors von 1 MΩ sinkt bei einer Temperaturerhöhung um ein
Tausendstelgrad um 20 Ω oder 20 ppm.
Abbildung 30: Detailansicht des Nano-Kalorimeters. Der Thermistor TS und befindet sich auf der
Membran, der Thermistor TR auf dem Chipkörper. Um den zentralen Thermistor auf der Membrann
ist das Heizelement aus Chrom zur elekrischen Kalibration gewunden.

___________________________________________________
108
3. Materialien und Methoden
3.2.7.3 Temperaturmessung mit Ge-Thermistor
Die Zuordnung der gemessenen Widerstandsänderung zur Temperatur des Sensors
erfolgte durch vorhergehende Kalibrierung im Bereich zwischen 25 und 100°C. Dazu
wurde der Chip in einem Ofen auf eine bestimmte Temperatur gebracht und die
Widerstände der Thermistoren gemessen.
Diese Kalibrierung erfolgte für jeden Chip und jeden individuellen Thermistor. In
Abbildung 31 ist exemplarisch der Gang eines Germanium-Widerstandes in
Abhängigkeit von der Temperatur dargestellt. Ersichtlich ist eine gewisse Krümmung
der R/T-Kennlinie, was auf eine Temperaturabhängigkeit des
Temperaturkoeffizienten hinweist.
Die Messwerte lassen sich durch Gleichung 94 annähern:
00)( t
TeARTR
−⋅+=
(94)
Abbildung 31: Verhalten des Thermistorwiderstands in Abhängigkeit von der Temperatur. Fit nach
Gleichung 94; Fitparameter: R0= -19,877 kΩ; A= 1500.273 kΩ; t0= 50,304°C.

___________________________________________________
109
3. Materialien und Methoden
Somit erhält man einen approximierten Zusammenhang zwischen der gemessenen
Widerstandsänderung, R(T)-R0, und der Temperatur (Gleichung 94) und die
Temperaturmessung der Probe mit der geforderten Genauigkeit von +/- 0,1 °C ist
ohne weiteres möglich (Gleichung 95):
( )( )ARTRtT 00 )(ln −⋅−=
(95)
Zur einfachen Messung der Temperatur der Probe wird einer der Membran-
Thermistoren mit 3 Festwiderständen in einer Wheastone-Brücke integriert und die
Diagonalspannung ∆Ui aufgezeichnet.
Abbildung 32: Messbrücke mit einem einzelnen temperaturabhängigen Wiederstand (Thermistor)
und 3 Festwiederständen. Die Betriebsspannung U0 betrug typischerweise 12 V. Aus der
Brückenspannung Ui lässt sich nach Gleichung 96 die Temperatur am Ort des Thermistors
bestimmen.
Aus der Diagonalspannung lässt sich mit Gleichung 96 die Temperatur des
Thermistors unter Verwendung der Kenngrößen t0, A und R0 in Abhängigkeit von der
Betriebsspannung U0 und dem Widerstand R2 berechnen:
( )⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟
⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛+⋅⋅⎟
⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛+−+⋅=
−1
000200 1
211
21ln
UUA
UURRRtT ii
(96)

___________________________________________________
110
3. Materialien und Methoden
Abbildung 33: Zur Berechnung der Temperatur aus der Diagonalspannung Ui nach Gleichung 96 einer
Brücke mit drei Festwiderständen 900 kΩ und einem Germanium-Thermistor von 900 kΩ bei 25°C.
Versorgungsspannung 12 V.
3.2.7.4 Differenztemperaturmessung
Die Temperaturdifferenz zwischen der Probe und dem Chipkörper muss zur
Detektion von Phasenumwandlungen geringer Wärmetönung wesentlich genauer
bestimmt werden. Dazu muss das Signal verstärkt werden, bis 800-fach. Das ist
technisch möglich, wenn das Differenzsignal unabhängig gemacht werden kann von
der globalen Temperaturerhöhung während des Aufheizens des Chips. Dies gelingt
weitgehend durch Integration zweier Thermistoren in eine Halbseite einer
Wheatstone-Brücke, eines Membran-(Proben-)Thermistors und eines Thermistors
vom Chipkörper.

___________________________________________________
111
3. Materialien und Methoden
Abbildung 34: Zur Berechnung der Temperaturdifferenz aus der Diagonalspannung Ui nach Gleichung
103 einer Brücke mit zwei Festwiderständen (900 kΩ) und zwei Germanium-Thermistoren von 900 kΩ
bei 25°C. Thermistor TS befindet sich auf der Membran, Thermistor TR auf dem Chipkörper.
Versorgungsspannung 12 V. Temperaturänderungen des gesamten Chip lassen Ui unbeeinflusst.
( ) ⎟⎠⎞
⎜⎝⎛ −+⋅=
21)()()(0 TTTTTTUU RSSi
(97)
Den Zusammenhang zwischen der Diagonalspannung Ui und den beiden
temperaturabhängigen Widerständen TS(T) und TR(T) gibt Gleichung 97 wieder.
Eingesetzt die Ausdrücke für die temperaturabhängigen Widerstände gibt:
⎟⎟⎠
⎞⎜⎜⎝
⎛−⋅++⋅+⋅+⋅=
−−−
210
00
00
00RtRT
RRStST
SSStST
SSi eAReAReARUU
(98)
Unter der Voraussetzung gleichen Temperaturverhaltens bzw. gleicher Kenngrößen
für beide Thermistoren, also mit AS=AR=A und R0S=R0R=R0 und t0S=t0R=t0, ergibt sich:
0
0
0
0
0
00
2121 t
RTtST
tST
i eRAe
RAe
RA
UU −−−
⋅+⋅+⋅+=+
(99)
Und mit TR=TS+∆T und einigen Umformungen:
⎟⎟⎠
⎞⎜⎜⎝
⎛+++=+
∆
012121
0
tT
i eFFUU
(100)

___________________________________________________
112
3. Materialien und Methoden
Umgestellt nach ∆T
⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛+−=∆ i
tST
i UUeARUUtT 2212ln 0
0000
(101)
Mit
0
0
tST
eRAF
−
=
(102)
⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛ +−=∆ ii UU
FUUtT 2212ln 000
(103)
Der Chip eröffnet mit seinen 5 Thermistoren die Möglichkeit, neben der einfachen
Temperaturmessung (mit einem einzelnen Thermistor), die Differenztemperatur
zweifach, rechts und links (mi je zwei Thermistoren), zu messen. Diese Redundanz
erwies sich bei der Suche nach sehr kleinen Peaks bei der Messung der
Denaturierung der GOx als sehr hilfreich, weil Störungen nicht selbstverständlich
beide Seiten des Chip gleichermaßen betreffen. Bei den GOx-Messungen wurde als
Verstärkung der beiden Differenztemperatursignale 500- bzw. 800-fach gewählt.

___________________________________________________
113
3. Materialien und Methoden
Abbildung 35: Zur Berechnung der Temperaturdifferenz zwischen zwei Thermistoren nach Gleichung
103 aus der Diagonalspannung einer Brücke mit zwei Thermistoren (TS und TR). Die
Versorgungsspannung betrug 12. Die Temperaturdifferenz ist nahezu linear zum Brückensignal mit
einer Steigung von 18 K/V. Die Empfindlichkeit ist im Messbereich zwischen Raumtemperatur und
100°C annähernd konstant.
3.2.7.5 Versiegelung der Probenkammer:
Die Verdunstung der wässrigen Probe während des Aufheizens auf 100°C zu
verhindern, ist unerlässlich wegen des großen Wärmeeffekts der Verdunstung von
Wasser.
Der Verschluss mit einer hydrophoben Flüssigkeit wie Öl war nicht
erfolgversprechend. Die Kavität wurde deshalb mechanisch versiegelt.

___________________________________________________
114
3. Materialien und Methoden
3.2.7.6 Zeitkonstante
Einer der Vorzüge eines mikrotechnisch hergestellten Kalorimeters ist die aufgrund
der geringen thermischen Massen (Addenda) kurze Ansprechzeit des Kalorimeters.
Selbst modernste Mikrokalorimeter (z.B. CSC™, N-DSC III, oder MicroCal™ VP-DSC)
haben Systemzeitkonstanten >1 s.
Tabelle 4: Analogie elektrischer und thermischer Größen
Elektrische Größen Thermische Größen
U Spannung [V] ∆T Temperaturdifferenz [K]
dQel/dt≡I Strom [A] dQth/dt Wärmestrom [W]
R Widerstand [Ω] Rth Thermischer Widerstand
[KW-1]
C Kapazität [F] C Wärmekapazität [JK-1]
Ohm´sches
Gesetz
U=R*I Ohm´sches
Gesetz der
Wärmelehre
∆T*=Rth*(dQth/dt)
Laden U= U0(1-e-t/τ) [V] Heizen ∆T=∆T0 (1-e-t/τ) [mK]
Zeitkonstante τ= R*C Zeitkonstante τ= Rth*C [ms]
Die thermische Zeitkonstante τ ist, analog zur elektrischen Zeitkonstanten eines RC-
Glieds, definiert als die Zeit, die das System benötigt, um als Reaktion auf ein
elektrisches oder thermisches Signal 63% (1-1/e) des maximalen Ladezustands zu
erreichen. Es besteht eine Analogie zwischen elektrischen und thermischen Größen
(Tabelle 1).
CRth ⋅=τ
(104)
Der thermische Widerstand der Membran zwischen dem Heizelement und der
Position TS konnte zu 8250 K/W (s.o.) bestimmt werden. Die Wärmekapazität dieses
Membransegments liegt im Bereich von 10-6 JK-1. Damit lässt sich eine Zeitkonstante
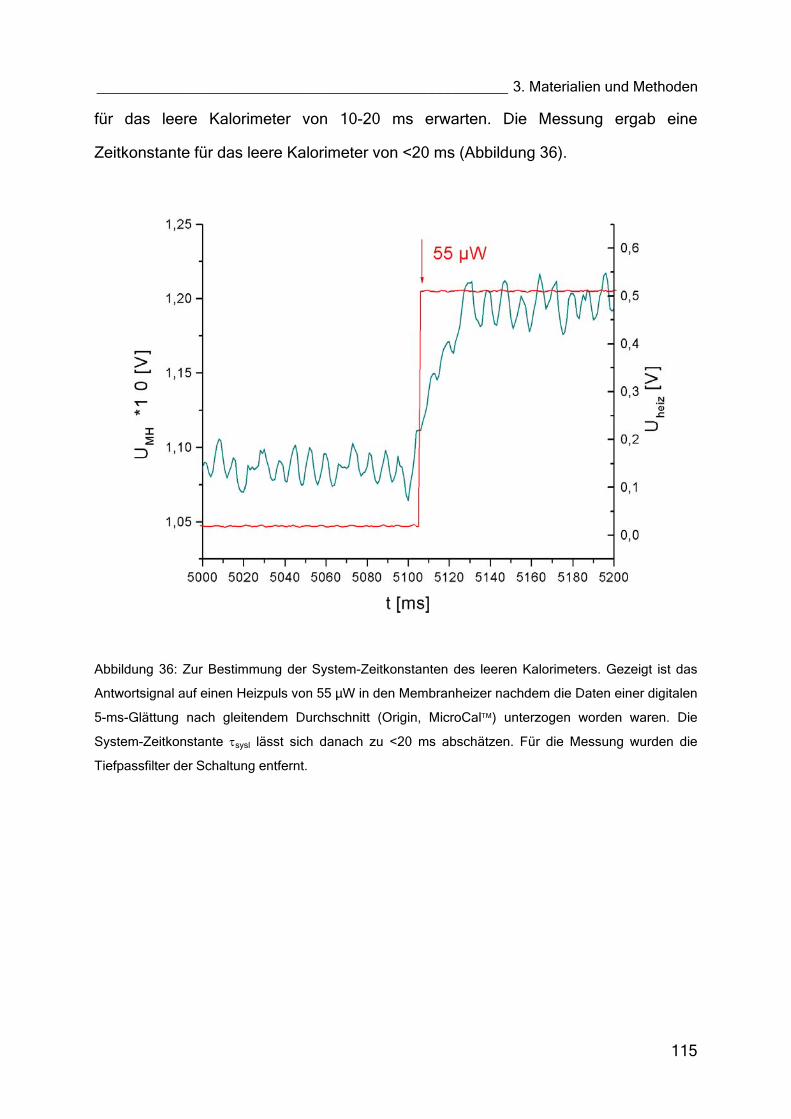
___________________________________________________
115
3. Materialien und Methoden
für das leere Kalorimeter von 10-20 ms erwarten. Die Messung ergab eine
Zeitkonstante für das leere Kalorimeter von <20 ms (Abbildung 36).
Abbildung 36: Zur Bestimmung der System-Zeitkonstanten des leeren Kalorimeters. Gezeigt ist das
Antwortsignal auf einen Heizpuls von 55 µW in den Membranheizer nachdem die Daten einer digitalen
5-ms-Glättung nach gleitendem Durchschnitt (Origin, MicroCal™) unterzogen worden waren. Die
System-Zeitkonstante τsysl lässt sich danach zu <20 ms abschätzen. Für die Messung wurden die
Tiefpassfilter der Schaltung entfernt.

___________________________________________________
116
3. Materialien und Methoden
Abbildung 37: Zur Abschätzung der Zeitkonstante des auf einer Seite der Memmbran mit Wasser
gefüllten Nanokalorimeters. Gezeigt ist das Antwortsignal auf einen Heizpuls von 55 µW in den
Membranheizer nachdem die Daten einer digitalen 100-ms-Glättung nach gleitendem Durchschnitt
(Origin, MicroCal™) unterzogen worden waren. Die Zeitkonstante τcal lässt sich so zu <100 ms
abschätzen. Für die Messung wurden die Tiefpassfilter der Schaltung entfernt.
Die Temperaturbestimmung aus der Brückenspannung einer mit einem einzelnen
Thermistor bestückten Messbrücke lässt sich für einen engeren Bereich (-2 bis 2 V)
gut linear annähern. Der Faktor ist abhängig von der Spannung U0 (nicht linear) und
beträgt für 12 V 18 [K/V]. Der Signalhub des gezeigten 55 µW Heizpulses ist 2,2 mV
oder 40 mK. Die gemittelte Heizrate des internen Aufheizens ist 15 K/min.

___________________________________________________
117
3. Materialien und Methoden
3.2.7.7 Empfindlichkeit
Die Empfindlichkeit des Nano-Kalorimeters ist vom TKR der Thermistoren, der
thermischen Masse der Addenda und der Rauschgrenze der Elektronik bestimmt.
Das Rauschniveau der Schaltung lag nach 800-facher Verstärkung (INA110 KP) bei
10-20 mV, entsprechend ca. 50 nW.
Abbildung 38: Demonstration der Empfindlichkeit des Nano-Kalorimeters. Erhöhung der Heizleistung
im Heizelement auf der Membran in 50 nW Stufen.
Das Nano-Kalorimeter ist zwar ein differenzielles Instrument und dient der Detektion
des Temperaturunterschieds zwischen der Probe (Membran) und einer
Referenzstelle (Chipkörper). Die beiden Messorte sind weder identisch noch
vollständig isoliert voneinander. Ein thermisches Übersprechen beim Eintragen von
Heizleistung am Heizer auf der Membran führt zu einer Temperaturerhöhung auch
der Referenzstelle durch Wärmeleitung durch die Membran. Die
Temperaturerhöhung aufgrund von Wärmeeintrag in die Membran an Position TS ist

___________________________________________________
118
3. Materialien und Methoden
8250 [KW-1 ] und im Abstand von ca. 600 µm im Innern des Siliziumträgers, an
Position TR 0,07 [KW-1] (Daten nicht gezeigt). Die Wärmeleitung durch die Membran
vom Membranheizer zum Chipträgers ist aufgrund der geringen Dicke der Membran
(1 µm) sehr gering und so ist die Temperaturerhöhung auf dem Chipkörper nur 10-3 %
derer auf der Membran, wenn die Heizleistung direkt in die Membran appliziert wird.
Die Membran gegenüber dem Chip kann demnach als quasi-adiabatisch angesehen
werden.
Befindet sich Probenmaterial ober- und/oder unterhalb der Membran ist dieses
thermische Übersprechen größer, weil Wärme nicht mehr nur über die Membran zur
Wärmesenke fließen kann sondern ebenso durch direkten Kontakt der
wärmefreisetzenden Probe mit dem Chipkörper. Durch die Probe auf der Membran
wird der thermische Widerstand zwischen Membran und Chip verringert. Befindet
sich Wasser ober- und unterhalb der Membran ist die Temperaturerhöhung aufgrund
von Wärmeeintrag in den Membranheizer an Position TS 504 [KW-1] und im Abstand
von ca. 600 µm im Innern des Siliziumträgers, an Position TR, 24,1 [KW-1]. oder ca.
5% des Wertes an Position TS.
Um den Thermistor auf dem Chipkörper als Referenz im dynamischen
Aufheizvorgang einsetzten zu können, ist es notwendig das Ausmaß der
Temperaturerhöhung auf dem Chip als Antwort auf Energiefreisetzung im
Probenvolumen anstelle der quasi-punktförmigen Heizung auf der Membran zu
bestimmen. Dazu kann die Phasenumwandlungswärme beim Schmelzen bzw.
Kristallisieren von Biphenyl genutzt werden.
Biphenyl schmilzt/kristallisiert mit relativ hoher Schmelzenthalpie von 120,4 Jg-1 bei
68,93°C [149].
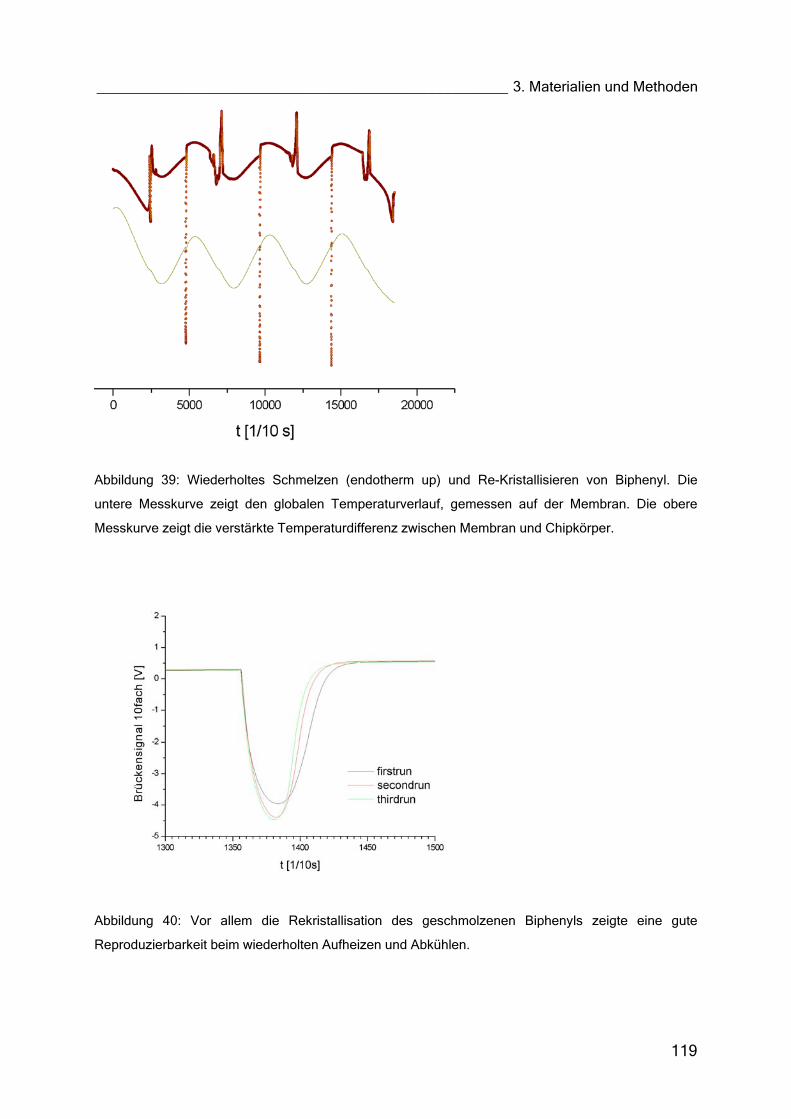
___________________________________________________
119
3. Materialien und Methoden
Abbildung 39: Wiederholtes Schmelzen (endotherm up) und Re-Kristallisieren von Biphenyl. Die
untere Messkurve zeigt den globalen Temperaturverlauf, gemessen auf der Membran. Die obere
Messkurve zeigt die verstärkte Temperaturdifferenz zwischen Membran und Chipkörper.
Abbildung 40: Vor allem die Rekristallisation des geschmolzenen Biphenyls zeigte eine gute
Reproduzierbarkeit beim wiederholten Aufheizen und Abkühlen.
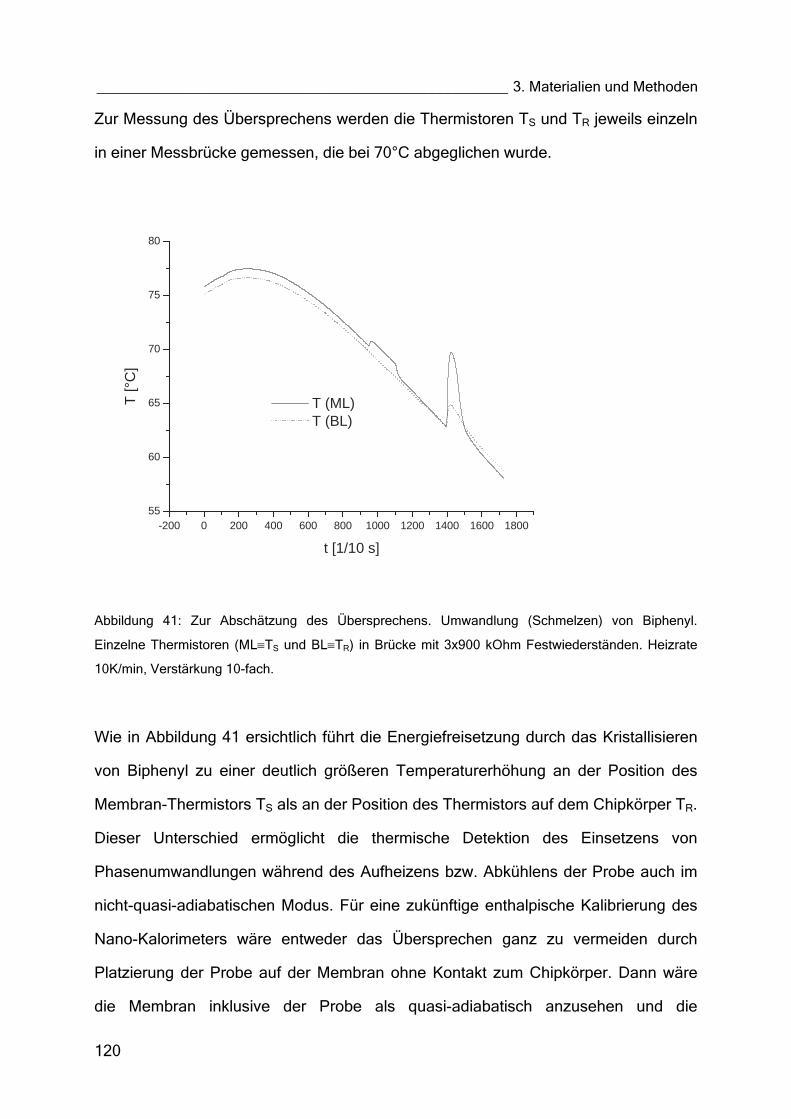
___________________________________________________
120
3. Materialien und Methoden
Zur Messung des Übersprechens werden die Thermistoren TS und TR jeweils einzeln
in einer Messbrücke gemessen, die bei 70°C abgeglichen wurde.
-200 0 200 400 600 800 1000 1200 1400 1600 180055
60
65
70
75
80
T (ML) T (BL)
T [°
C]
t [1/10 s]
Abbildung 41: Zur Abschätzung des Übersprechens. Umwandlung (Schmelzen) von Biphenyl.
Einzelne Thermistoren (ML≡TS und BL≡TR) in Brücke mit 3x900 kOhm Festwiederständen. Heizrate
10K/min, Verstärkung 10-fach.
Wie in Abbildung 41 ersichtlich führt die Energiefreisetzung durch das Kristallisieren
von Biphenyl zu einer deutlich größeren Temperaturerhöhung an der Position des
Membran-Thermistors TS als an der Position des Thermistors auf dem Chipkörper TR.
Dieser Unterschied ermöglicht die thermische Detektion des Einsetzens von
Phasenumwandlungen während des Aufheizens bzw. Abkühlens der Probe auch im
nicht-quasi-adiabatischen Modus. Für eine zukünftige enthalpische Kalibrierung des
Nano-Kalorimeters wäre entweder das Übersprechen ganz zu vermeiden durch
Platzierung der Probe auf der Membran ohne Kontakt zum Chipkörper. Dann wäre
die Membran inklusive der Probe als quasi-adiabatisch anzusehen und die

___________________________________________________
121
3. Materialien und Methoden
Kalibrierung ohne Model mithilfe der elektrischen Heizung möglich. Oder das
Übersprechen wird in Kauf genommen und charakterisiert. Dieleztere Version ist
jedoch technisch wesentlich einfacher, d.h. die ohnehin vorhandene Kavität dient
direkt als Probenkammer, die weiterhin alle Freiheiten an den Aggregatzustand der
Probe lässt. Der Verlust an Signalintensität durch das Übersprechen kann aufgrund
der hohen Auflösung der Temperaturmessung und die bessere Handhabbarkeit
ausgewogen werden. Durch die Modellierung der thermischen Verhältnisse des
Chips inklusive Probe mittels FEM bzw. PSpice sollte zudem die enthalpische
Kalibrierung dieser Version möglich sein.
4. Ergebnisse Um aufzuklären, in welchem Zusammenhang die Lebensdauer der enzymatischen
Aktivität (katalytische Stabilität) des Enzyms GOx zur Denaturierungstemperatur
dieses Enzyms steht, wurden einerseits Messungen der Halbwertszeit τ1/2 der
enzymatischen Aktivität bei erhöhter Temperatur (60°C) mithilfe eines
Stess/Aktivitätstests für die Untersuchung fadenförmiger Mikrogele durchgeführt und
andererseits wurde die Denaturierungstemperatur des Enzyms in den Hydrogelen
mittels Dynamischer Differenzialkalorimetrie bestimmt. Im Vordergrund stand dabei
die Frage nach der Besonderheit der Mikroumgebung, wie sie sich dem Enzym
innerhalb eines vernetzten Hydrogels darbietet. Die empirische Tatsache, dass sich
Enzyme in Hydrogelen, z.B. in Biosensoren, als stabiler gegenüber irreversibler
Inaktivierung erweisen, macht diese Frage auch technologisch relevant.
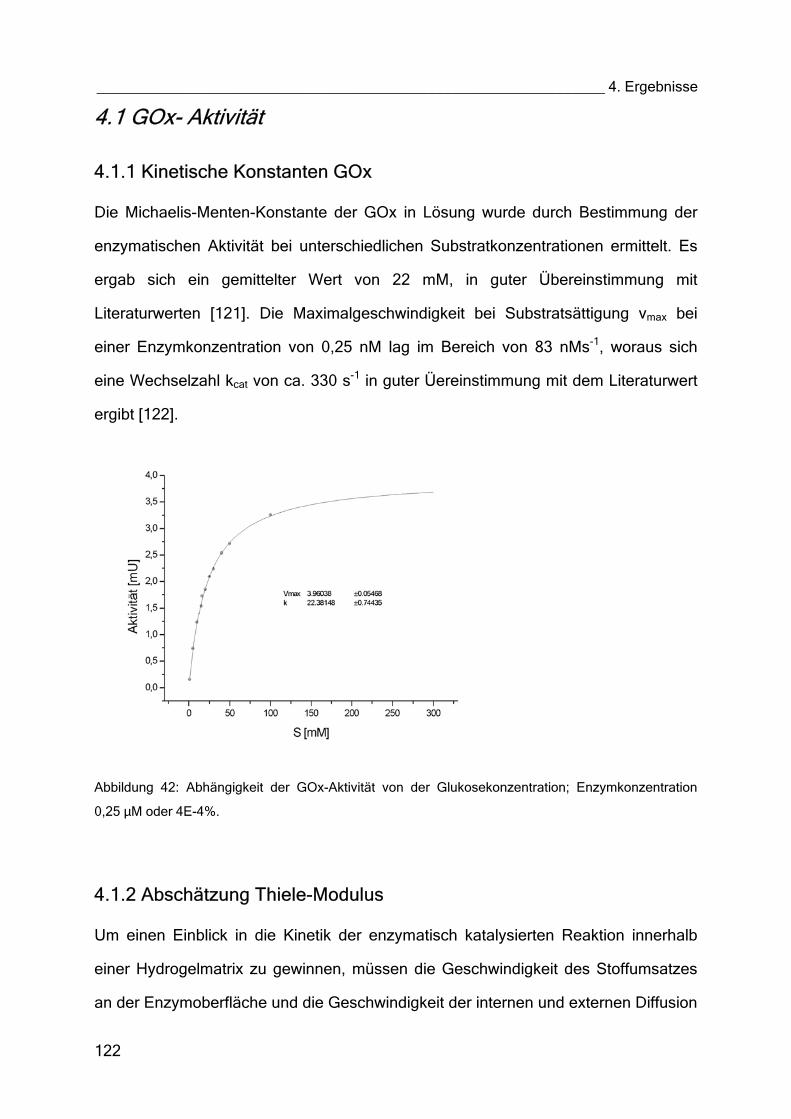
_______________________________________________________________
122
4. Ergebnisse
4.1 GOx- Aktivität
4.1.1 Kinetische Konstanten GOx
Die Michaelis-Menten-Konstante der GOx in Lösung wurde durch Bestimmung der
enzymatischen Aktivität bei unterschiedlichen Substratkonzentrationen ermittelt. Es
ergab sich ein gemittelter Wert von 22 mM, in guter Übereinstimmung mit
Literaturwerten [121]. Die Maximalgeschwindigkeit bei Substratsättigung vmax bei
einer Enzymkonzentration von 0,25 nM lag im Bereich von 83 nMs-1, woraus sich
eine Wechselzahl kcat von ca. 330 s-1 in guter Üereinstimmung mit dem Literaturwert
ergibt [122].
Abbildung 42: Abhängigkeit der GOx-Aktivität von der Glukosekonzentration; Enzymkonzentration
0,25 µM oder 4E-4%.
4.1.2 Abschätzung Thiele-Modulus
Um einen Einblick in die Kinetik der enzymatisch katalysierten Reaktion innerhalb
einer Hydrogelmatrix zu gewinnen, müssen die Geschwindigkeit des Stoffumsatzes
an der Enzymoberfläche und die Geschwindigkeit der internen und externen Diffusion

_______________________________________________________________
123
4. Ergebnisse
des Substrats und des Produkts der Reaktion getrennt behandelt werden, falls
letztere nicht vernachlässigbar sind. Ob sie vernachlässigbar sind, oder ob eventuell
noch weitere Einflüsse, Inhibierung des Enzyms durch das Raktionsprodukt etwa,
eine Rolle spielen, wurde durch Abschätzung des Thiele-Modulus´, der eine
kompakte Charakterisierung des Einflusses auf die Raktionsgeschwindigkeit
heterogen katalysierter Reaktionen durch interne Diffusion darstellt, versucht zu
klären. Sollten die Voraussetzungen, unter denen der Thiele-Modulus eine
brauchbare Charakterisierung des kinetischen Verhaltens des heterogenen
Enzymkatalysators ist (homogene Verteilung des Enzyms, Konstanz von
Inhibitorkonzentrationen, Michaelis-Menten-Verhalten des Enzyms auch in der
Immobilisierungsmatrix, Vernachlässigbarkeit von externer Diffusion) zutreffen,
ließen sich die Messwerte der Aktivität (Reaktionsgeschwindigkeit) in Abhängigkeit
von der Substratkonzentration im normierten Plot durch den Thiele-Modulus
charakterisieren (Abbildung 17). Aus den Abildungen 43-45 ist ersichtlich, dass dies
für die untersuchten Hydrogele nicht der Fal war.
Die Hydrogelfäden wurden in Kapillaren von 60 µm Durchmesser hergestellt. Als
charakteristische Länge l zur Abschätzung des Thiele-Modulus´ nach Gleichung 57
wurde der halbe Durchmesser gewählt. Die inhärente Sättigungsreaktions-
geschwindigkeit vmax ergibt sich aus der Einwaage des Enzyms.
( )02,0
107,62210830103
21
27
63
21
22
1max
=
⎥⎥
⎦
⎤
⎢⎢
⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛⋅⋅⋅⋅
⋅⋅⋅⋅=
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛
⋅⋅⋅⋅
⋅⋅= −
−−
⋅
′
cmmMssmMcm
cmmMssmMcml eff
SM DKvϕ
(57)

_______________________________________________________________
124
4. Ergebnisse
Tabelle 5: Theoretische Abschätzung des Thiele-Modulus für einen Hydrogelfaden von 60 µm
Durchmesser und einer Enzymbeladung von 5 U/ml nach Gleichung 57. Der Diffusionskoeffizient der
Glukose in dem Hydrogel wurde mit 10% bzw. 0,1% des Wertes in Wasser angesetzt.
Symbol Dimension Abschätzung 1 Abschätzung 2
Radius Hydrogelfaden l [cm] 3*10-3 3*10-3
Michaelis-Menten
Konstante GOx
KM [mM] 22 22
Maximalgeschwindigkeit Vmax [mMs-1] 830*10-6 830*10-6
Diffusionskoeffizient
Glukose in Lösung
DSeff [cm2s-1] 6,7*10-6 -
Diffusionskoeffizient
Glukose im Hydrogel
DSeff [cm2s-1] 6,7*10-7 6,7*10-9
Thiele-Modulus ϕ ∅ 0,02 0,2
Abbildung 43: Normierter Plot der
Reaktionsgeschwindigkeit in
Abhängigkeit von der
Substratkonzentration für 10 µl
Hydrogel (17% AA; 1% BIS) mit
54 mU GOx. Thiele–Modulus 20-
30.

_______________________________________________________________
125
4. Ergebnisse
Abbildung 44: Normierter Plot der
Reaktionsgeschwindigkeit in
Abhängigkeit von der
Substratkonzentration für 10 µl
pAA-Hydrogel (10% AA; 0,3%
BIS) mit 3,6 bzw. 36 mU GOx.
Thiele–Modulus 10-20 bzw. 20-50.
Abbildung 45: Normierter Plot der
Reaktionsgeschwindigkeit in
Abhängigkeit von der
Substratkonzentration für 10 µl
pAA-Hydrogel (10% AA; 0,5%
BIS) mit 3,6, 36 und 360 mU GOx.
Thiele–Modulus 5-10, 20-30 und
30—70.
Der Thiele-Modulus hängt von der Quadratwurzel der Enzymkonzentration ab
(Gleichung 57). Eine Verhundertfachung der Enzymkonzentration sollte demnach zu
einem um den Faktor 10 vergrößerten Thiele-Modulus führen. Für das Gel mit 3,6
mU kann man einen Wert des Thiele–Modulus von 5-10 abschätzen, für das Gel mit
360 mU einen Wert von 50-70, d.h. der Faktor 10 wurde annähernd gefunden.

_______________________________________________________________
126
4. Ergebnisse
Zur Aufklärung der Diskrepanz zwischen dem erwarteten (<<1) und abgeschätzten
Thiele-Modulus (>5) für die Hydrogelfäden wurden keine weiteren Versuche
unternommen. Vor allem bei höheren Enzymkonzentrationen im Hydrogelfaden
zeigte sich eine deutlich geringere Abhängigkeit der Produktbildung von der
Substatkonzentration als aufgrund des erwarteten Thiele-Modulus´ zu erwarten
gewesen wäre. Möglicherweise macht sich hier die Quellung des Gelfadens während
der Messung bemerkbar. Allerdings kann höchstens mit einer Verdoppelung des
Durchmessers und somit des Thiele-Modulus´ gerechnet werden. Die inhibitorische
Wirkung des Wasserstoffperoxyds, das im Hydrogel gebildet wird, auf die GOx-
Aktivität hingegen kann zu signifikant niedrigerer Poduktbildungsgeschwindigkeit v
und damit zu der beobachteten Abflachung der v/vmax-gegen-βe-Plots führen, wenn
vmax, wie hier, aus der Einwaage berechnet wird, und nicht aus der tatsächlich
vorliegenden Aktivität während der Messung (Produktinhibierung) [150]. Der Thiele-
Modulus allein reicht hier offenbar nicht aus, um das kinetische Verhalten der
biomimetischen Hydrogele zu beschreiben.
4.1.3 Halbwertszeiten
Die Halbwertszeit der katalytischen Aktivität der Hydrogelfäden, also die Zeitspanne,
die vergehen muss, bis ihre Fähigkeit zur Katalyse der Oxidation von Glukose mithilfe
von Sauerstoff, auf die Hälfte des ursprünglichen Niveaus abgesunken ist, ist in
erster Näherung von Diffusionseffekten unbeeinflusst. Auch eine fortschreitende
Produktinhibierung während der Messung, die Aussagen über den Aktivitätszustand
vor der Messung stören würde, schmälert die Aussagekraft der Halbwertszeit als
Stabilitätskriterium nicht. Die Stabilität wird durch die Angabe der Halbwertszeit eher
unterschätzt, denn ein sehr stabiles Enzympräparat wäre in Abwesenheit von
Produktinhibierung noch länger messbar stabil.

_______________________________________________________________
127
4. Ergebnisse
Gleichwohl reflektiert die Halbwertszeit im Gegensatz zur Denaturierungstemperatur
unmittelbar auf die Funktion des Enzyms, eben seine Fähigkeit zur Katalyse. Weil in
Biosensoren die Funktion des Enzyms (und nicht seine Struktur) genutzt wird, ist die
Halbwertszeit ein technologisch relevantes Kriterium.
Die Halbwertszeit lag für die unterschiedlichen Präparate bei 60°C zwischen einigen
Minuten und einigen Stunden, während sie bei Raumtemperatur im Bereich von
Wochen und Monaten liegt und bei 70°C und 80°C zu kurz war, um mit dem
verwendeten Testsystem (4 Minuten Messzeit) noch bestimmt werden zu können.
Für folgende Systeme wurden die Halbwertszeiten der enzymatischen Aktivität
deshalb bei 60 °C bestimmt:
- GOx in Puffer,
- GOx in 1 M NaCl,
- GOx in Puffer mit 150 bzw. 300 mg/ml Polyethylenglykol (PEG),
- GOx in Puffer mit 150 mg/ml Xylit bzw. Sorbit,
- GOx in Puffer mit Monomer Acrylamid
- GOx in Puffer mit Monomer HEMA
- GOx in Poly-Acrylamid Hydrogel
- GOx in Poly-Acrylamid Hydrogel mit 1 M NaCl
- GOx in Poly-Acrylamid Hydrogel mit PEG
- GOx in Poly-Acrylamid Hydrogel mit 50, 100, 150 mg/ml Ficoll™
- GOx in Poly-HEMA Hydrogel

_______________________________________________________________
128
4. Ergebnisse
4.1.3.1 GOx in Puffer
Abbildung 46: Relative Aktivität von GOx in 100 mM KH2PO4-Puffer in Abhängigkeit von der
Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der GOx in Lösung wurde mehrmals bestimmt (jeweils
durch Dreifachmessung aller Werte). Der gemittelte Wert lag bei 13±5 min.
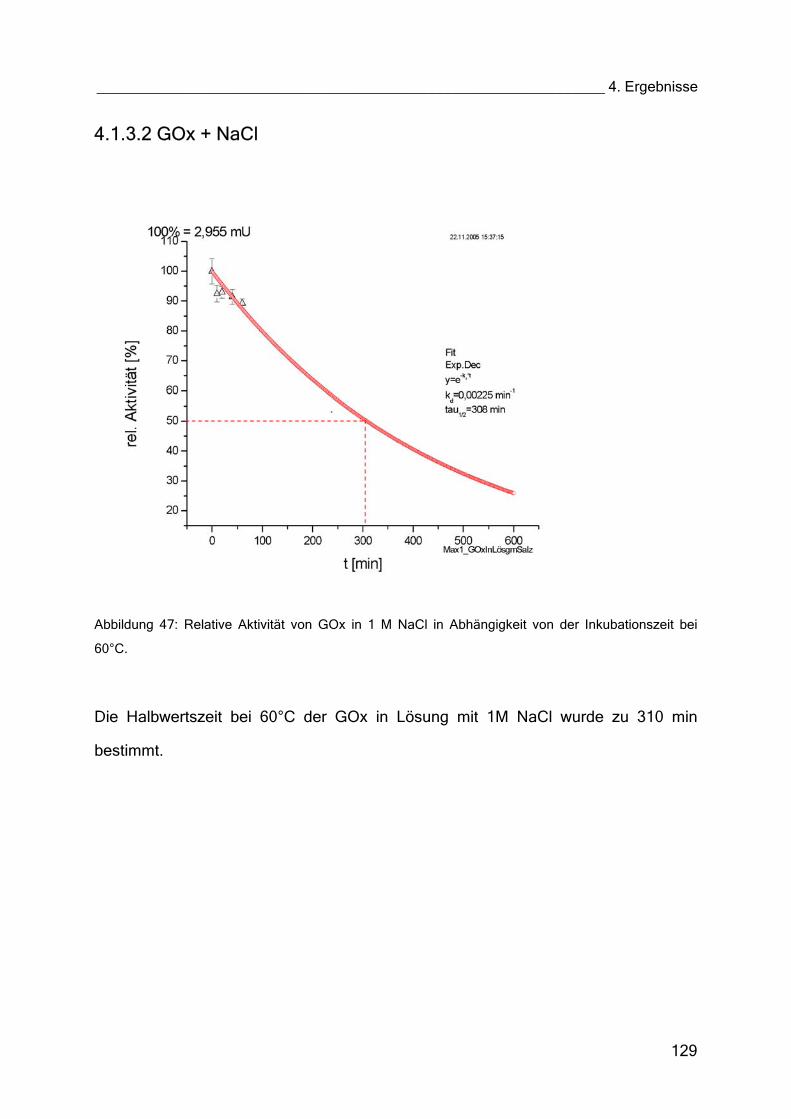
_______________________________________________________________
129
4. Ergebnisse
4.1.3.2 GOx + NaCl
Abbildung 47: Relative Aktivität von GOx in 1 M NaCl in Abhängigkeit von der Inkubationszeit bei
60°C.
Die Halbwertszeit bei 60°C der GOx in Lösung mit 1M NaCl wurde zu 310 min
bestimmt.
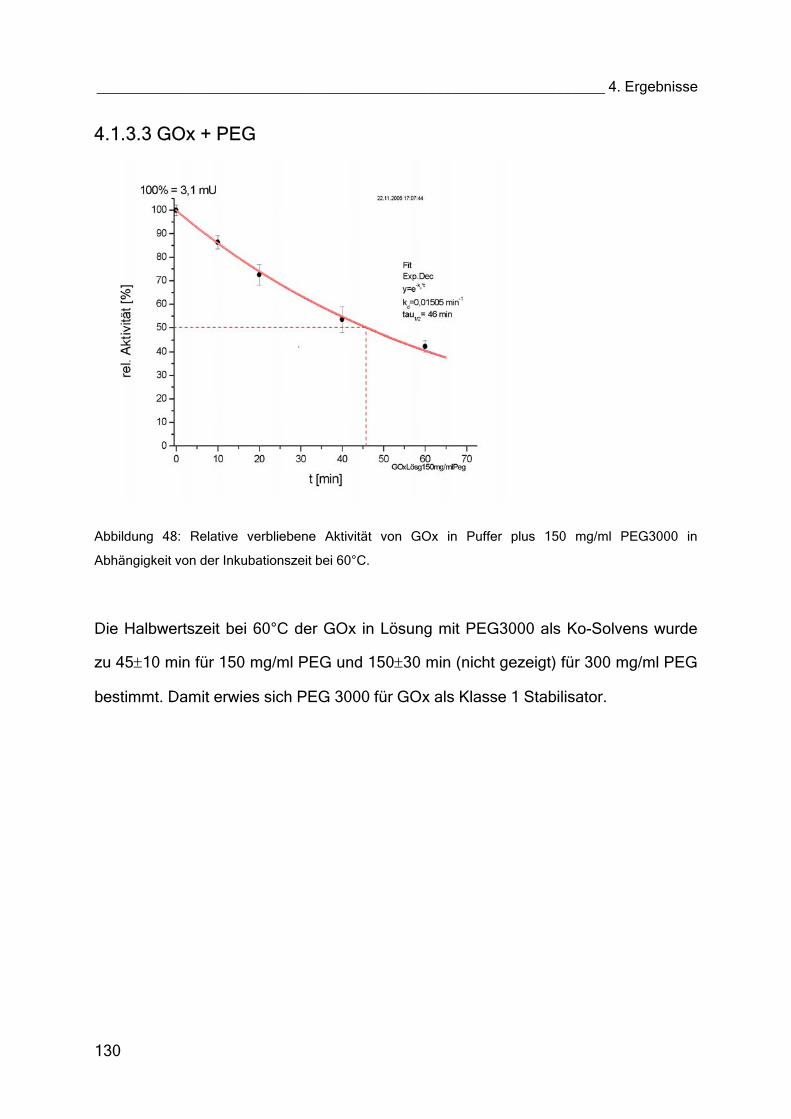
_______________________________________________________________
130
4. Ergebnisse
4.1.3.3 GOx + PEG
Abbildung 48: Relative verbliebene Aktivität von GOx in Puffer plus 150 mg/ml PEG3000 in
Abhängigkeit von der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der GOx in Lösung mit PEG3000 als Ko-Solvens wurde
zu 45±10 min für 150 mg/ml PEG und 150±30 min (nicht gezeigt) für 300 mg/ml PEG
bestimmt. Damit erwies sich PEG 3000 für GOx als Klasse 1 Stabilisator.

_______________________________________________________________
131
4. Ergebnisse
4.1.3.4 GOx + Sorbitol
Abbildung 49: Relative verbliebene Aktivität von GOx in Puffer plus 1M Sorbitol in Abhängigkeit von
der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der GOx in Lösung mit 1M Sorbitol als Ko-Solvens wurde
zu 18±5 min bestimmt. Dies ist gegenüber dem Wert für Puffer (13±5 min) kaum
verlängert.

_______________________________________________________________
132
4. Ergebnisse
4.1.3.5 GOx + Xylitol
Abbildung 50: Relative verbliebene Aktivität von GOx in Puffer plus 1M Sylitol in Abhängigkeit von der
Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der GOx in Lösung mit 1M Sorbitol als Ko-Solvens wurde
zu 15±5 min bestimmt. Dies ist gegenüber dem Wert für Puffer (13±5 min) kaum
verlängert.

_______________________________________________________________
133
4. Ergebnisse
4.1.3.6 GOx in pAA-Hydrogel
Abbildung 51: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel in Abhängigkeit von
der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel wurde
mehrmals bestimmt (jeweils durch Dreifachmessung aller Werte). Der gemittelte Wert
lag bei 40±20 min.
Die Halbwertszeit in einem pHEMA-Hydrogel von vergleichbarer Konzentration lag
bei 30 min.
pAA- bzw. pHEMA-Hydrogele zeigen demnach einen milden Stabilisierungseffekt auf
die GOx.

_______________________________________________________________
134
4. Ergebnisse
4.1.3.7 GOx im pAA-Hydrogel + NaCl
Abbildung 52: Relative verbliebene Aktivität von GOx in 20%igem PolyAcrylamid-Hydrogel plus
1 M NaCl in Abhängigkeit von der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 1 M NaCl
wurde zu >500 min abgeschätzt.
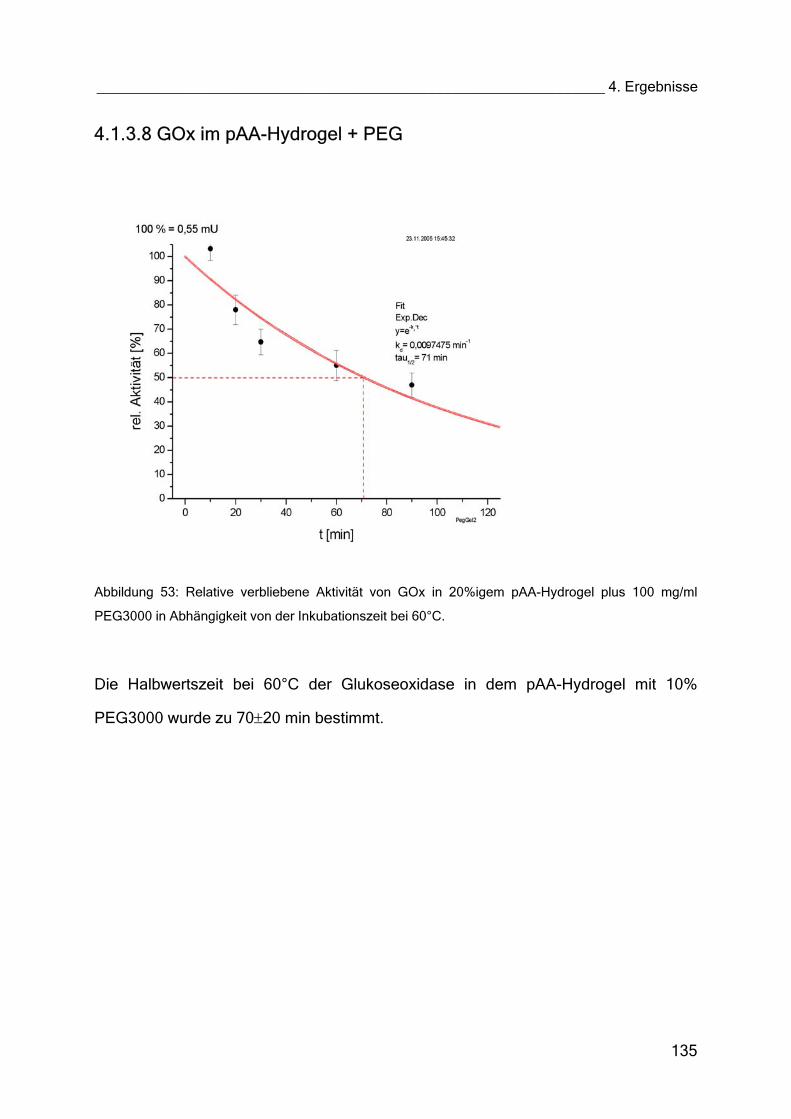
_______________________________________________________________
135
4. Ergebnisse
4.1.3.8 GOx im pAA-Hydrogel + PEG
Abbildung 53: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel plus 100 mg/ml
PEG3000 in Abhängigkeit von der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 10%
PEG3000 wurde zu 70±20 min bestimmt.

_______________________________________________________________
136
4. Ergebnisse
Abbildung 54: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel plus 292 mg/ml
PEG3000 in Abhängigkeit von der Inkubationszeit bei 60°C.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 30%
PEG3000 wurde zu >400 min bestimmt.
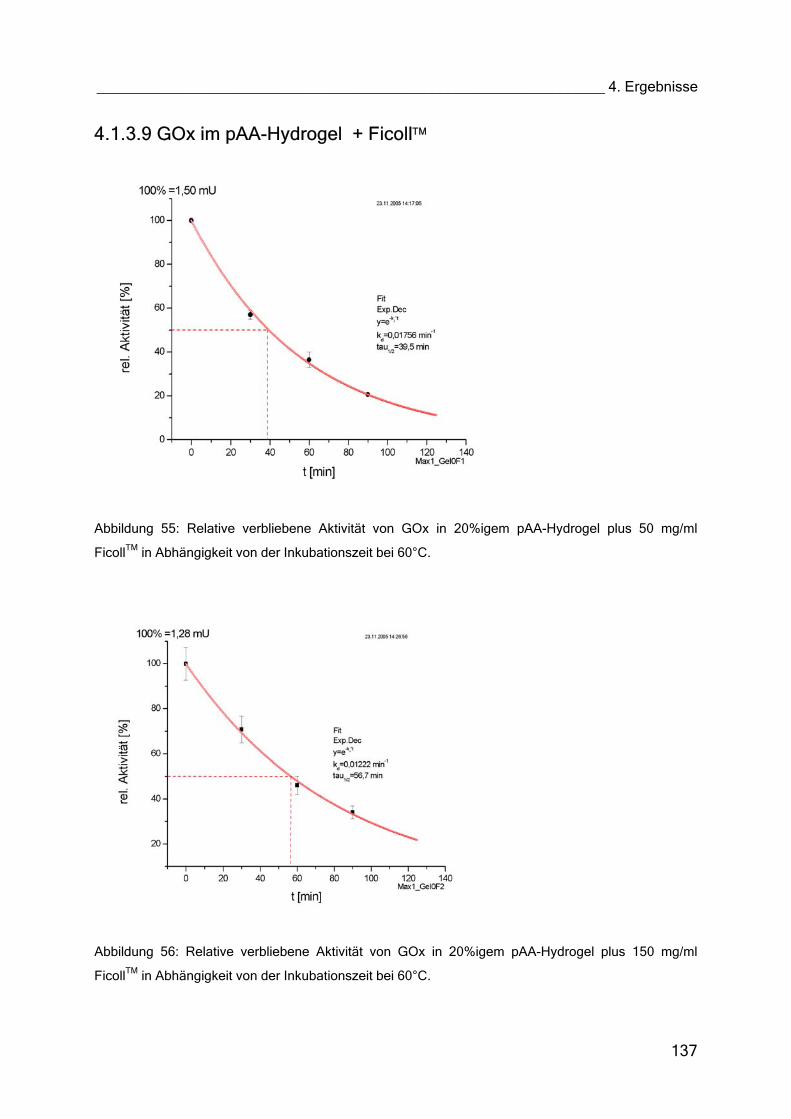
_______________________________________________________________
137
4. Ergebnisse
4.1.3.9 GOx im pAA-Hydrogel + Ficoll™
Abbildung 55: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel plus 50 mg/ml
FicollTM in Abhängigkeit von der Inkubationszeit bei 60°C.
Abbildung 56: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel plus 150 mg/ml
Ficoll in Abhängigkeit von der Inkubationszeit bei 60°C.TM

_______________________________________________________________
138
4. Ergebnisse
Abbildung 57: Relative verbliebene Aktivität von GOx in 20%igem pAA-Hydrogel plus 250 mg /ml
FicollTM in Abhängigkeit von der Inkubationszeit bei 60°C
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 5% FicollTM
wurde zu 40 ±5 min bestimmt.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 15%
FicollTM wurde zu 57 ±5 min bestimmt.
Die Halbwertszeit bei 60°C der Glukoseoxidase in dem pAA-Hydrogel mit 25%
FicollTM wurde zu 60 ±5 min bestimmt.
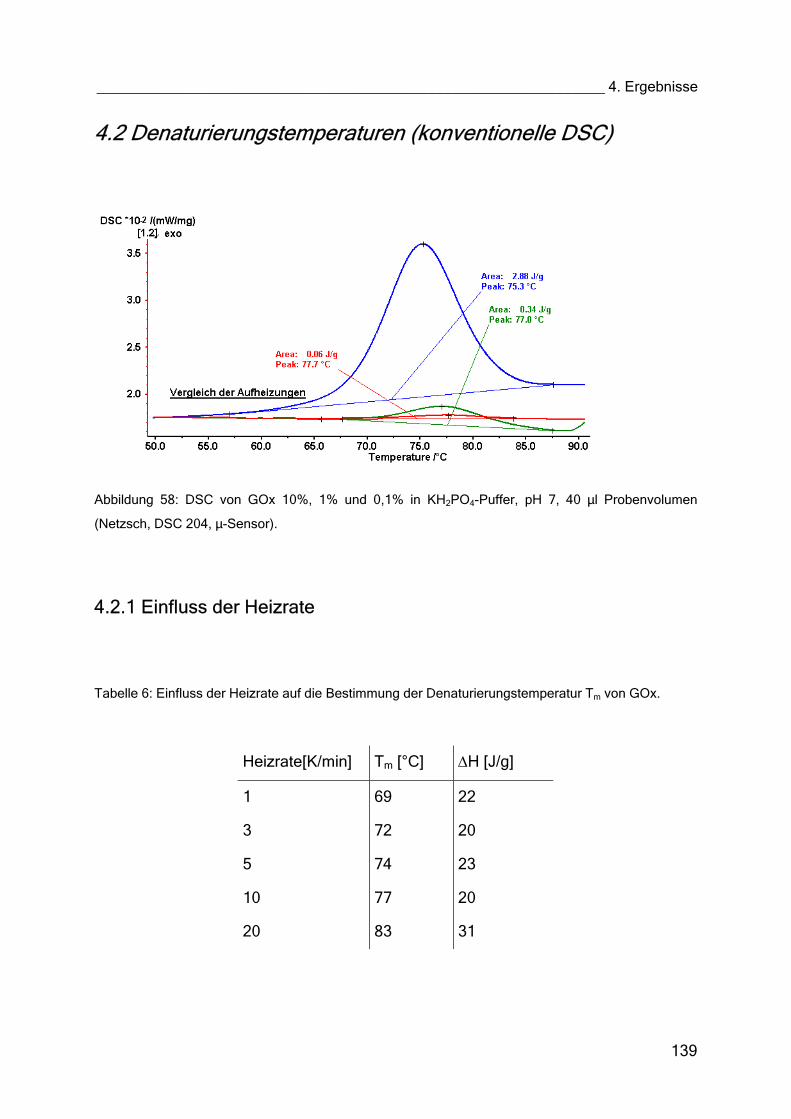
_______________________________________________________________
139
4. Ergebnisse
4.2 Denaturierungstemperaturen (konventionelle DSC)
Abbildung 58: DSC von GOx 10%, 1% und 0,1% in KH2PO4-Puffer, pH 7, 40 µl Probenvolumen
(Netzsch, DSC 204, µ-Sensor).
4.2.1 Einfluss der Heizrate
Tabelle 6: Einfluss der Heizrate auf die Bestimmung der Denaturierungstemperatur Tm von GOx.
Heizrate[K/min] Tm [°C] ∆H [J/g]
1
3
5
10
20
69
72
74
77
83
22
20
23
20
31

_______________________________________________________________
140
4. Ergebnisse
Abbildung 59: Thermogramme von GOx (40 µl, 5% (w/v), Perkin-Elmer DSC 7), aufgezeichnet mit
unterschiedlicher Heizrate. Es ist ersichtlich, wie sich die Denaturierungstemperatur Tm zu höheren
Werten verschiebt.
Abbildung 60: Einfluss der Heizrate (1, 3, 5, 10, 20 K/min) auf die Bestimmung der
Denaturierungstemperatur Tm von GOx (5% in 100 mM Phosphatpuffer).

_______________________________________________________________
141
4. Ergebnisse
4.2.2 Einfluss der Konzentration des Enzyms
Tabelle 7: Einfluss der Konzentration der GOx auf seine Denaturierungstemperatur.
[GOx] [%(w/v] Tm [°C] ∆H [J/g]
1
10
20
201
73
76
78
79
10
13
18
22
1 andere Messserie
4.2.3 GOx in Phosphatpuffer
Tabelle 8: Denaturierungstemperatur der GOx in Phosphatpuffer, Mehrfachmessung.
Probe Messung Tm [°C] ∆H [J/g]
5% GOx 1, 2
5% GOx
5% GOx
5% GOx
5% GOx
1
2
3
4
5
Mittelwert
72,0
72,0
72,2
73,0
71,9
72,2
18
26
19
25
31
24
1 (w/v), 2 in 100 mM KH2PHO4, pH 7.

_______________________________________________________________
142
4. Ergebnisse
4.2.4 GOx + GndHCl
Tabelle 9: Denaturierungstemperatur Tm von GOx in Abhängigkeit von der Konzentration von GndHCl;
Puffer 100 mM KH2PHO4; GOx 5% (w/v).
[GdnHCl] [M] Tm [°C] ∆H [J/g]
0
1
2
3
5
72
75
68
66
59
18
12
9
8
<1
4.2.5 GOx + NaCl
Tabelle 10: Denaturierungstemperatur Tm von GOx in Abhängigkeit von der Konzentration von NaCl;
Puffer 100 mM KH2PHO4; GOx 5% (w/v).
[NaCl] [M] Tm [°C] ∆H [J/g]
0 M
0,5M
1,0M
2,0M
72
81
82
83
24
24
15
16

_______________________________________________________________
143
4. Ergebnisse
4.2.6 GOx + PEG
Tabelle 11: Denaturierungstemperatur Tm von GOx in Anwesenheit von 20% Polyethylenglykol 3000;
Puffer 100 mM KH2PHO4; GOx 5% (w/v).
PEG [%] Tm [°C] ∆H [J/g]
0
20
72
85
24
31
Tabelle 12: Denaturierungstemperatur Tm von GOx in Anwesenheit von 30% Polyethylenglykol 3000,
Mehrfachmessung.
Probe Messung Tm [°C] ∆H [J/g]
5% GOx 1,2 Mittelwert 72,2 24
5% GOx + PEG 3
5% GOx + PEG
5% GOx + PEG
1
2
3
84,5
75,0
84,5
32
16
31
5% GOx + PEG Mittelwert 81,3 26
5% GOx + PEG Mittelwert4 84,5 32
1 (w/v), 2 in 100 mM KH2PHO4-Puffer, 330% (w/v), 4ohne Messung 2 (Ausreißer)

_______________________________________________________________
144
4. Ergebnisse
4.2.7 GOx + Dextran
Tabelle 13: Denaturierungstemperatur Tm von GOx in Anwesenheit des polymeren Zuckers Dextran;
Puffer 100 mM KH2PHO4; GOx 5% (w/v).
Dextran [%(w/v)] Tm [°C] ∆H [J/g]]
0
1
5
10
20
30
40
72
75
75
76
90
90
93
18
17
14
12
9
17
12
4.2.8 GOx + Monomer Acrylamid
Tabelle 14: Denaturierungstemperatur Tm von GOx in Anwesenheit des Monomers Acrylamid,
Messung erfolgte nach 20 h Inkubationszeit bei 4°C
AA [%(w/v)] Tm [°C] ∆H [J/g]
0
10
20
72
54
n.b1.
13
6
n.b.
1 nicht bestimmbar (kein deutlicher Peak)

_______________________________________________________________
145
4. Ergebnisse
4.2.9 GOx in pAA-Hydrogel
Tabelle 15: Denaturierungstemperatur Tm von GOx in pAA-Hydrogel unterschiedlichen Polymergehalts
pAA [%(w/v)] Tm [°C] ∆H [J/g]
0
10
20
30
40
50
50
72
77
65
n.b.1
80
76
77
13
10
14
n.b.
3
8
7
1 nicht bestimmbar (kein deutlicher Peak)
Tabelle 16: Denaturierungstemperatur Tm von GOx in 20% pAA-Hydrogel, Mehrfachmessung.
Probe Messung Tm [°C] ∆H [J/g]
5%1 GOx in pAA 2
5% GOx in pAA
5% GOx in pAA
5% GOx in pAA
5% GOx in pAA
5% GOx in pAA
5% GOx in pAA
5% GOx in pAA
1
2
3
4
5
6
7
8
77,0
75,9
74,2
75,5
74,8
72,5
72,4
75,7
17
17
17
17
19
20
24
16
5% GOx in pAA Mittelwert 75 18
1 (w/v), 2 20% (w/v)
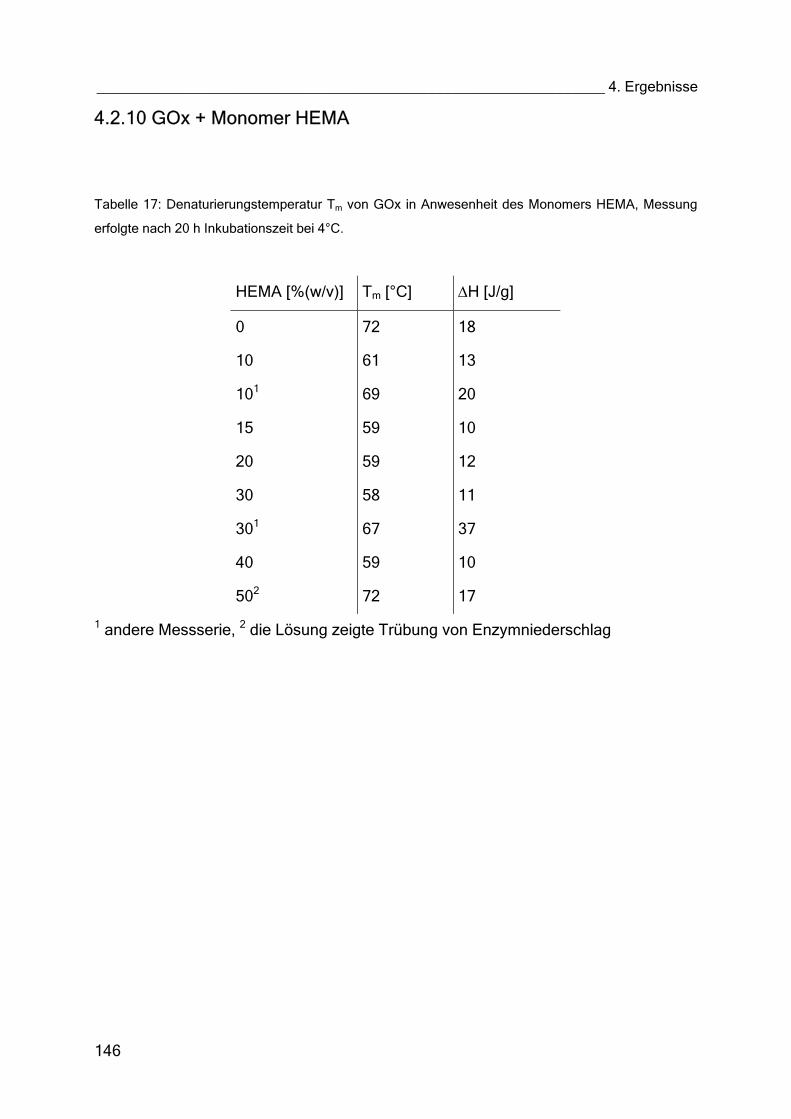
_______________________________________________________________
146
4. Ergebnisse
4.2.10 GOx + Monomer HEMA
Tabelle 17: Denaturierungstemperatur Tm von GOx in Anwesenheit des Monomers HEMA, Messung
erfolgte nach 20 h Inkubationszeit bei 4°C.
HEMA [%(w/v)] Tm [°C] ∆H [J/g]
0
10
101
15
20
30
301
40
502
72
61
69
59
59
58
67
59
72
18
13
20
10
12
11
37
10
17
1 andere Messserie, 2 die Lösung zeigte Trübung von Enzymniederschlag

_______________________________________________________________
147
4. Ergebnisse
4.2.11 GOx in pHEMA-Hydrogel
Tabelle 18: Denaturierungstemperatur Tm von GOx in pHEMA-Hydrogel unterschiedlichen
Polymergehalts.
pHEMA [%(w/v)] Tm [°C] ∆H [J/g]
0
10
15
20
25
30
35
40
45
501,2
72
77
75
72
69
69
67
70
68
79
18-24
13
12
9
8
10
8
11
9
16
1 andere Messserie,2 Gel zeigte Trübung von ausgefallenem Enzym

_______________________________________________________________
148
4. Ergebnisse
4.2.12 GOx in pHEMA-Hydrogel + NaCl
Tabelle 19: Denaturierungstemperatur Tm von GOx in pHEMA-Hydrogel (25%) unterschiedlichen
Gehalts an NaCl.
NaCl [M] Tm [°C] ∆H [J/g]
0
0,5
1
2
72
79
83
82
18-24
16
15
13

_______________________________________________________________
149
4. Ergebnisse
4.2.13 GOx in pAA-Hydrogel + PEG
Tabelle 20: Denaturierungstemperatur Tm von GOx in pAA-Hydrogel +
30% PEG3000.
Probe Messung # Tm [°C] ∆H [J/g]
5%1 GOx2 Mittelwert 72 17
5%1 GOx2 in pAA3 Mittelwert 75 18
5%1 GOx2 in pAA 3 + PEG 4
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
5% GOx in pAA + PEG
1
2
3
4
5
6
7
8
9
10
86,2
85,4
85,0
85,2
85,2
85,0
85,0
86,2
85,2
85,2
6
7
7
7
7
5
5
8
3
4
5% GOx in pAA + PEG Mittelwert 85,4 6
1 (w/v), 2 in 100 mM KH2PHO4-Puffer, pH 7, 3 20%(w/v), 4 30% (w/v)

_______________________________________________________________
150
4. Ergebnisse
4.2.14 GOx in pAA-Hydrogel + Ficoll™
Tabelle 21: Denaturierungstemperatur Tm von GOx in pAA-Hydrogel in Anwesenheit von 250 mg/ml
Ficoll™.
Probe Messung # Tm [°C] ∆H [J/g]
5%1 GOx2 Mittelwert 72 17
5%1 GOx2 in pAA3 Mittelwert 75 18
5%1 GOx2 in pAA3 + Ficoll4
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
5% GOx in pAA + Ficoll
1
2
3
4
5
6
7
8
81,4
82,0
82,7
81,7
80,8
81,4
81,9
80,4
6
9
11
3
8
7
5
9
5% GOx in pAA + Ficoll Mittelwert 81,5 7
1(w/v), 2in 100 mM KH2PHO4-Puffer, pH 7, 320%, 4250 mg/ml.

_______________________________________________________________
151
4. Ergebnisse
4.2.15 Vermischtes
Tabelle 22: Denaturierungstemperatur Tm von GOx in Pulverform1 bzw. mit Restfeuchte2 sowie dreier
weiterer Enzyme zum Vergleich.
Probe Tm [°C] ∆H [J/g]
GOx Lyophilisat1 + Siliconöl 58,2 1,7
Siliconöl - -
GOx im Tiegel eingetrocknet2 80 5
Myoglobin 78 16
Katalase 93 18
Lysozym 5% pH 5,2 78 25
4.3 Thermogramme (Nano-Kalorimeter)
4.3.1 Flüssigkristall MH24
Abbildung 61. Nano-Thermogramm MH 24. Rohdaten. Der erste Peak (nach der Kalibrierungsmarke
von 7,5 µW) bei ca. 4000 Pt (1pt=1/10 s) ist das Schmelzen des bei Raumtemperatur festen
Flüssigkristalls, dessen Energie den Rahmen der Aufzeichnung überschreitet. Die weiteren Peaks
zeigen die Umwandlung des smektischen Flüssigkristalls in eine nematische Phase bei 67°C und
deren weitere Umwandlung in die isotrope Flüssigkeit bei 80°C. Gezeigt sind zwei Aufheiz-
Abkühlungs-Zyklen.

_______________________________________________________________
152
4. Ergebnisse
Abbildung 62: Thermogramm des Flüssigkristalls 4´-Oktyloxy-biphenyl-4-carbonitril (MH-24) im
Abkühlmodus. Umwandlungs-Enthalpie flüssig nach nematisch bei 80°C ∆H = 220 µJ/100 nl eff. und
nematisch nach smektisch A bei 67°C ∆H =29 µJ/ 100 nl eff. Der dritte Peak ist die Kalibrierungsmarke
von 7,5 µW.
Tabelle 23: Enthalpie der Phasenumwandlungen von MH 24. In Klammern die Erwartungswerte für
100 nl Probe (Veff.). Zum Vergleich die Enthalpie der Denaturierung von 1 µg GOx und des
Schmelzens von 1 µg Wasser.
Impuls Fläche [µJ]
Peak 1 (l→n) 227,5 (220)
Peak 2 (n→smA) 14,9 (29)
Peak 3 (Kal.) 75
1 µg Protein (N→D) 30
1 µg Wasser (s→l) 334
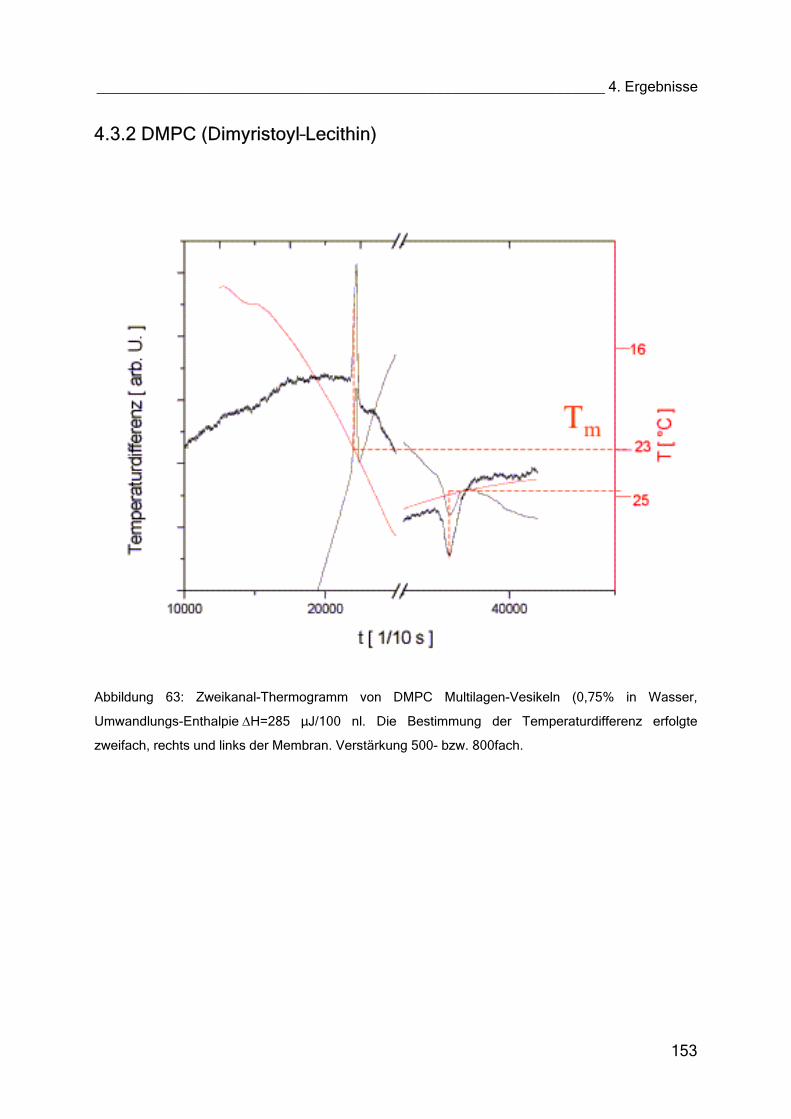
_______________________________________________________________
153
4. Ergebnisse
4.3.2 DMPC (Dimyristoyl–Lecithin)
Abbildung 63: Zweikanal-Thermogramm von DMPC Multilagen-Vesikeln (0,75% in Wasser,
Umwandlungs-Enthalpie ∆H=285 µJ/100 nl. Die Bestimmung der Temperaturdifferenz erfolgte
zweifach, rechts und links der Membran. Verstärkung 500- bzw. 800fach.

_______________________________________________________________
154
4. Ergebnisse
4.3.3 GOx in Lösung und im Hydrogel
Abbildung 64: Zweikanal-Thermogramm von GOx (3%) in 100 mM KH2PO4 Puffer, pH 7,2.
Umwandlungs-Enthalpie 50 µJ/100 nl (3 µg Enzym). Die Bestimmung der Temperaturdifferenz erfolgte
zweifach, rechts und links der Membran. Verstärkung 500- bzw. 800fach.
Abbildung 65: Zweikanal-Thermogramm von GOx (4%) in pAA-Hydrogel. Umwandlungs-Enthalpie 68
µJ/100 nl (4 µg Enzym). Die Bestimmung der Temperaturdifferenz erfolgte zweifach, rechts und links
der Membran. Verstärkung 500- bzw. 800fach.

_______________________________________________________________
155
4.4 Zusammenfassung der Halbwertszeitmessungen und der
Denaturierungstemperaturen
4. Ergebnisse
Die verwendeten Ko-Solventien lassen sich qualitativ im Hinblick auf ihre GOX-
stabilisierende Wirkung in folgender Reihe ordnen:
DextranNaClpAAPEGpAANaClPufferFicollpAAPEGPufferpAA
pHEMASorbitXylitPufferPrecursorGndHCl
<+<+≈+<+<+<≈
<≈≈<<
Abbildung 66: Zwischen der Denaturierungstemperatur Tm und der Halbwertszeit der enzymatischen
Aktiviät τ1/2 des Enzyms GOx in unterschiedlichen Ko-Solventien zeigte sich eine positive Korrelation.
Die durchgezogenen Linien entsprichen den Werten im Referenzmedium 100 µM Phosphat-Puffer, pH
7,2.

_______________________________________________________________
156
4. Ergebnisse
Tabelle 24: Halbwertszeiten und Denaturierungstemperaturen von GOx in unterschiedlichen Medien. 110% (w/v), 230% (w/v), 3100 mM KH2PHO4, pH 7.
Ko-Solvens τ1/2 [min] Tm [°C]
GndHCl 5M - 59
Monomer AA1 - 54
Monomer HEMA2 - 58
Wasser3 13 72
Xylit 1M 15 -
Sorbit 1M 18 -
pHEMA 20% 30 72
pAA 20% 40 75
pAA+Ficoll™ 250 mg/ml 61 82
PEG 30% 150 85
NaCl 1M 300 83
pAA+PEG 30% >400 85
pAA+NaCl 1M >500 82 (pHEMA)
Dextran 40% - 93

_______________________________________________________________
157
5. Diskussion
5. Diskussion Zur Charakterisierung des GOx-Präparates wurden zunächst die kinetischen
Konstanten des Enzyms bestimmt. Dazu wurde die Reaktionsgeschwindigkeit der
Glukoseoxidation für unterschiedliche Glukosekonzentrationen im Bereich zwischen
10 und 100 mM bestimmt. Es ergab sich eine Michaelis-Menten Konstante für
Glukose von 22 mM, in guter Übereinstimmung mit den Literaturwerten. Die
spezifische Aktivität betrug ca. 130 U/mg. Die Abschätzung des Thiele Modulus für
die Hydrogelfäden mit 60 µm Durchmesser und einer Enzymbeladung von 50 mU/10
µl ergab Werte für den Thiele-Modulus von 0,02-0,2 je nach Diffusionskoeffizient im
pAA-Hydrogel für Glukose, der mit 10% bzw. 0,1% des Wertes in Wasser angesetzt
wurde [151-154]. Für einen Thiele-Modulus < 1 sind Diffusionseinflüsse
vernachlässigbar und die Reaktion kann als kinetisch kontrolliert angesehen werden.
Experimentell zeigten die Hydrogele aber durchweg einen deutlich höheren Thiele-
Modulus zwischen 10 und 50 (Abbildung 43-45). Die Diskrepanz könnte ein Hinweis
auf die Akkumulation des Reaktionsproduktes und GOx-Inhibitors
Wasserstoffperoxyd zurückzuführen sein. Nicht berücksichtigt wurde ferner, dass bei
Vorliegen von Diffusionseinflüssen mit einer scheinbaren Verlängerung der
Halbwertszeit zu rechnen ist [155].
Die Durchführung des Stress-Aktivitätstests bei 60°C erlaubt eine kürzere Messzeit
und ermöglicht reproduzierbarere Messwerte als bei Raumtemperatur oder 37°C. Die
Aussagen des Stress-Aktivitätstests (Halbwertszeiten bei 60°C) über die katalytische
Stabilität der GOx sollten, als relative Größen verstanden, von eventueller
Inhibierung durch Wasserstoffperoxyd weniger beeinflusst sein, wenn angenommen
werden kann, dass diese unter den Bedingungen des Stress-Aktivitätstests
hinreichend konstant sind.
In hohen Konzentrationen (1-5%) ist die Bestimmung der Denaturierungstemperatur
Tm auch mit herkömmlichen DSC Geräten möglich. Eines der modernsten

_______________________________________________________________ 5. Diskussion
158
herkömmlichen DSC Geräte ist die DSC 204 von Netzsch, die mit dem µ-Sensor eine
kalorimetrische Empfindlichkeit von 40-65 mV/W hat. Das entspricht in der
Bestimmung der Wärmekapazität einer Empfindlichkeit von 30 mJg-1K-1 [156]. Die
Messungen zeigten, dass GOx in 100 mM Phosphatpuffer, pH 7,0 bei 75-77 °C
denaturiert mit einer Denaturierungsenthalpie von 28-34 Jg-1 (Abbildung 58). Der
Wert der Denaturierungsenthalpie liegt damit im Erwartungsbereich für globuläre
Proteine, der 30-40 Jg-1 beträgt. Die Messungen zeigten weiter, dass die
Denaturierung der GOx im DSC Experiment zumindest bei hohen Konzentrationen
ein kinetisch kontrollierter Prozess ist, d.h. dass die Tm-Bestimmung abhängig ist von
der Heizrate und dass der Prozess wenigstens teilweise irreversibel ist (Abbildung 34
und 35). Trotz dieser Irreversibilität können aus den DSC Messungen
thermodynamische Parameter gewonnen werden [133,157]. Des weiteren machen
sie deutlich, dass die Empfindlichkeit in DSC-Experimenten von der Heizrate
abhängig ist. Das kommt der Nano-Kalorimetrie mit Membran-Chips zugute, die im
quasi—adiabatischen Modus Heizraten von bis zu 104 Ks-1 ermöglicht [158-163].
Am Frauenhofer-Institut für Physikalische Messtechnik in Freiburg wurde ebenfalls
ein hochempfindliches, schnelles Differenzial-Nano-Kalorimeter zur Untersuchung
biologischer Systeme entwickelt. Es handelt sich um ein Kapillar-Mikrosystem,
welches somit auf die Untersuchung fließfähiger Proben begrenzt ist. Mit diesem
Kalorimeter ist die Detektion des Schmelzens von Lipidmembranen demonstriert
worden [159], nicht aber das Denaturieren eines Proteins.
In Nano-DSC Anwendungen zeigt sich das Schmelzen als oberflächen-initiierter
Prozess. Durch die hohe Auflösung ist die Monolagenempfindlichkeit erreicht und
frühe Phasen der Filmbildung konnten anhand der Schmelzpunkterniedrigung von
metallischen Nano-Partikeln (Indium-Clustern von 2—4 nm Durchmesser) mit sehr
hohen Heizraten (106 Ks-1) beobachtet werden Das von Efremov et al. vorgestellte
Kalorimeter bestand aus einem einfachen Nickelstreifen, der gleichzeitig als Heizer

_______________________________________________________________
159
5. Diskussion
und als resistiver Temperatursensor diente. Die Indium-Cluster zeigten periodische
Maxima in der gemessenen Wärmekapazität entsprechend der Partikelgröße, die
sich durch Zuwachs einer Monolage von 0,24 nm unterschied [160]. Dieselbe
Arbeitsgruppe hat gezeigt, dass die Größe von Wassertropfen von 1- 100 nl durch die
Verdunstungswärme mit ihrem Nano-Kalorimeter messbar ist [161].
Denlingers frühes Nano-Kalorimeter hat eine Addenda von nur 4*10-6 JK-1 und er
konnte damit Proben von wenigen µg messen [162].
Die physikalische Grenze der Temperaturmessung wird bestimmt durch das Nyquist-
Rauschen, das ca. 10 µK beträgt [163].
Für viele Proteine ist gezeigt, dass stabilisierende Ko-Solventien aus der Sphäre des
Proteins verdrängt und nicht etwa gebunden sind. Im Konzept der bevorzugten
Wechselwirkung und der Verdrängung wird Ionen-Bindung als modifizierender Faktor
berücksichtigt [56]. Der ausgesprochen saure Charakter der GOx mit seiner
ausgeprägt negativen Oberfläche mag dazu führen, dass Ionen-Bindung zum
dominierenden Faktor in der Stabilisierung durch Salze wird. Zum Beleg sind aber
Gleichgewichtsdialysemessungen, NMR- oder Lichtstreuungsmessungen nötig.
Diese Messmethoden sprechen direkt auf Kontakte zwischen Protein und Ko-Solvens
an [66].
Gouda et al. haben die thermische Deaktivierung von GOx in Anwesenheit von NaCl,
K2SO4 und dem basischen Enzym Lysozym untersucht und geben Werte für die
Halbwertszeit bei 60°C und die Deaktivierungsenergien an: native GOx: τ1/2 13 min;
Ed 250 kJ/mol; 1 M NaCl stabilisierte GOx: τ1/2 434 min; Ed 320 kJmol-1 [164]. Diese
Werte stimmen mit den von uns gemessenen Werten von 13±5 min für das
unstabilisierte und 310 min für das NaCl stabilisierte gut überein.
Gulla et al. untersuchten den Zusammenhang zwischen der
Denaturierungstemperatur und der Halbwertszeit der biologischen Aktivität von

_______________________________________________________________ 5. Diskussion
160
Glutardialdehyd vernetzen GOx-Membranen in Anwesenheit verschiedener
Stabilisatoren, darunter NaCl, K2SO4 und dem positiv geladenen Aminoglycosid-
Antibiotikum Kanamycin. Des weiteren zeigten sie, dass NEM-Derivatisierung von
Thiol-Gruppen in der GOx stabilisierend wirkt. Für diese Stabilisatoren und
Kombinationen von ihnen bestimmten sie die Tm und die Halbwertszeit bei 70°C.
Tm-und τ½-Werte lagen im Bereich von 75°C und 270 min für das immobilisierte
unstabilisierte Enzym, 78°C und 900 min für das NaCl stabilisierte und 84°C und
6000 min für das immobilisierte thiol-modifizierte und K2SO4 stabilisierte Enzym. Die
beiden Parameter Tm und τ½ zeigten auch hier einen positiven Zusammenhang [165].
Polyethylenglykol, PEG ([CH2CH20]nOCH2CH2OH), zeigt ungewöhnliche
Eigenschaften in der Wechselwirkung mit Wasser. PEG hat eine ungewöhnlich hohe
Löslichkeit in Wasser, während die Homologen mit einer Methylengruppe mehr,
Polypropylenoxid, bzw. einer Methylengruppe weniger, Polymethylenoxid, hydrophob
sind und in Wasser unlöslich. PEGs haben also einen gewissen hydrophoben
Charakter, der sich z.B. darin zeigt, dass PEGs wie Amphiphile an der Wasser/Luft
Grenzfläche spontan Monolagen ausbilden. PEGs gelten nichts desto trotz als
hydrophile Polymere, die von Proteinen abgestoßen werden und vielfach eingesetzt
werden als proteinabweisende Überzüge von biomimetischen Oberflächen [166].
Proteine, die mit PEGs kovalent modifiziert wurden, zeigen eine erhöhte
Lebensdauer und erniedrigte Antigenizität in vivo, was auf eine Verringerung der
Wechselwirkung mit anderen biologischen Makromolekülen und ihrer Aggregation
zurückgeführt wurde [167]. PEGs unterschiedlichen Molekulargewichts (200-20000)
stabilisieren GOx gegen thermische Deaktivierung. Für Konzentrationen von 300
mg/ml wurden Verlängerungen der Halbwertszeit auf das 20-fache beobachtet [91].
In Begriffen der bevorzugten Wechselwirkung wird die Wirkungsweise von PEG auf
die Proteinstabilität folgenderweise erklärt: Polyethylenglykol ist als voluminöses Ko-
Solvens vom nativen Protein vorzugsweise verdrängt, aufgrund seines partiell

_______________________________________________________________
161
5. Diskussion
hydrophoben Charakters vom denaturierten Protein aber vorzugsweise gebunden.
Das Gleichgewicht zwischen nativer und denaturierter Form wird auf die Seite der
Denaturierung verschoben (Klasse 2 Destabilisator) [58-61].
Dem stark geladenen, stark hydrophilen Protein GOx gegenüber zeigte sich PEG in
der vorliegenden Untersuchung als Klasse1 Stabilisator. Für weniger stark geladene
Proteine macht sich aufgrund von sterischer Verdrängung die Bindung an die
hydrophobere denaturierte Konformation bemerkbar, so dass PEG wegen seiner
partiellen Hydrophobizität im Allgemeinen ein Klasse 2 Destabilisator ist.
GndHCl ist ein Klasse 3 Destabilisator. Durch Zugabe von 2 M GndHCl wird die
Denaturierungstemperatur von GOx um ca. 7°C gesenkt [62,168]. Ähnliches zeigte
sich auch in der vorliegenden Untersuchung: 2M GndHCl senkte die Tm um 4°C. 5 M
GndHCl senkte die Tm von GOx um 13°C. GndHCl kann benutzt werden, um den
Temperaturbereich der Denaturierung hochschmelzender Proteine zu senken, was in
der Nano-Biokalorimetrie von Vorteil ist.
Für die Enzyme α-Chymotrypsin, Chymotrypsinogen und RNAse, die zum
Standardrepertoire der physikalischen Biochemie gehören, wurde gezeigt, dass ihre
Denaturierung in Sucrose-Lösungen unterschiedlicher Konzentrationen bei
annähernd derselben Oberflächenspannung einsetzt, bei veränderter Tm [56].
Polyole sind bekannt für ihre Eigenschaft, die Schmelztemperatur von Proteinen zu
erhöhen [169]. Sorbitol von 2 molarer Konzentration erhöht die Schmelztemperatur
von Lysozym bei pH 7,0 um 12 °C, während Xylitol bei derselben Konzentration die
Tm um 8°C erhöht [170,171].
Es konnte gezeigt werden, dass die stabilisierende Wirkung mit der Anzahl der –
CHOH Gruppen zunimmt. Polyole führen zu bevorzugter Hydratation der Proteine.
Diese bevorzugte Hydratation geht auf die Erhöhung der Oberflächenspannung des
Wassers durch diese Substanzen zurück. Der Oberflächenspannungseffekt wird als
ein dominierender Faktor der Stabilitätsbeeinflussung durch Lösungsmittel

_______________________________________________________________ 5. Diskussion
162
beschrieben. Inositol erhöht Tm stärker als Sorbitol/Xylitol. Ebenso ist der Trend bei
der Bewahrung der Aktivität. Es deutet sich eine Korrelation an. Die Bewahrung der
Aktivität in Abhängigkeit von der Konzentration legt nahe, dass diese Substanzen in
unspezifischer Weise ihre Wirkung entfalten und das der Effekt wesentlich
wasservermittelt ist.
Polyhydroxy-Verbindungen haben einen stabilisierenden Einfluss auf GOx. Vor allem
für Polyole mit 4 (Erythritol), 5 (Xylitol) und 6 (Sorbitol) Hydroxyl-Gruppen wurde
dieser konzentrationsabhängige Stabilisierungseffekt gezeigt. Für 4 M Xylitol wurde
ein Verlängerung der Halbwertszeit auf das 20- bis 25-fache beobachtet. Während
Ethylenglykol mit nur 2 Hydroxyl-Gruppen keinen Stabilisierungseffekt zeigt [98].
In unseren Messungen konnte für Xylit und Sorbit in Konzentrationen von 1 M keine
Stabilisierung der GOx-Aktivität beobachtet werden.
Dextran zeigte den stärksten Einfluss auf die Denaturierungstemperatur Tm. von
GOx. In 40% Dextran betrug die Tm-Erhöhung 21°C. Nach Gleichung 63 bedeutet
dies eine 14-fache Lebensdauer.
Die Möglichkeit einer Parallelführung der beiden Messgrößen Tm und τ½ ist eine ad
hoc Annahme, die ihrer theoretischen Grundlage noch harrt. Allerdings sprechen
verschiedene experimentelle Ergebnisse der letzen Jahre, vor allem Untersuchungen
von Proteinen aus Thermophilen, erste theoretische Ansätze (Abbildung 20) sowie
die vorgestellten eigenen Messungen der beiden Größen für die GOx (Abbildung 67)
für einen Zusammenhang von Tm und τ½:
Die Parametrisierung von Robertson und Murphy hat zum Ergebnis, dass es eine
Temperatur maximaler Stabilität für alle Enzyme gibt und das diese T´ bei 10°C bzw.
20°C liegt, je nachdem, ob der Datensatz kälte-adaptierte Proteine beinhaltet oder
nicht [172]. Diese Beobachtung hat einige Implikationen. Augenscheinlich ist, dass
Proteine mit höherer Tm dazu neigen, eine größere Stabilität bei der Temperatur
maximaler Stabilität ∆G(T´) zu besitzen (Abbildung 12). Dies ist zwar naheliegend,
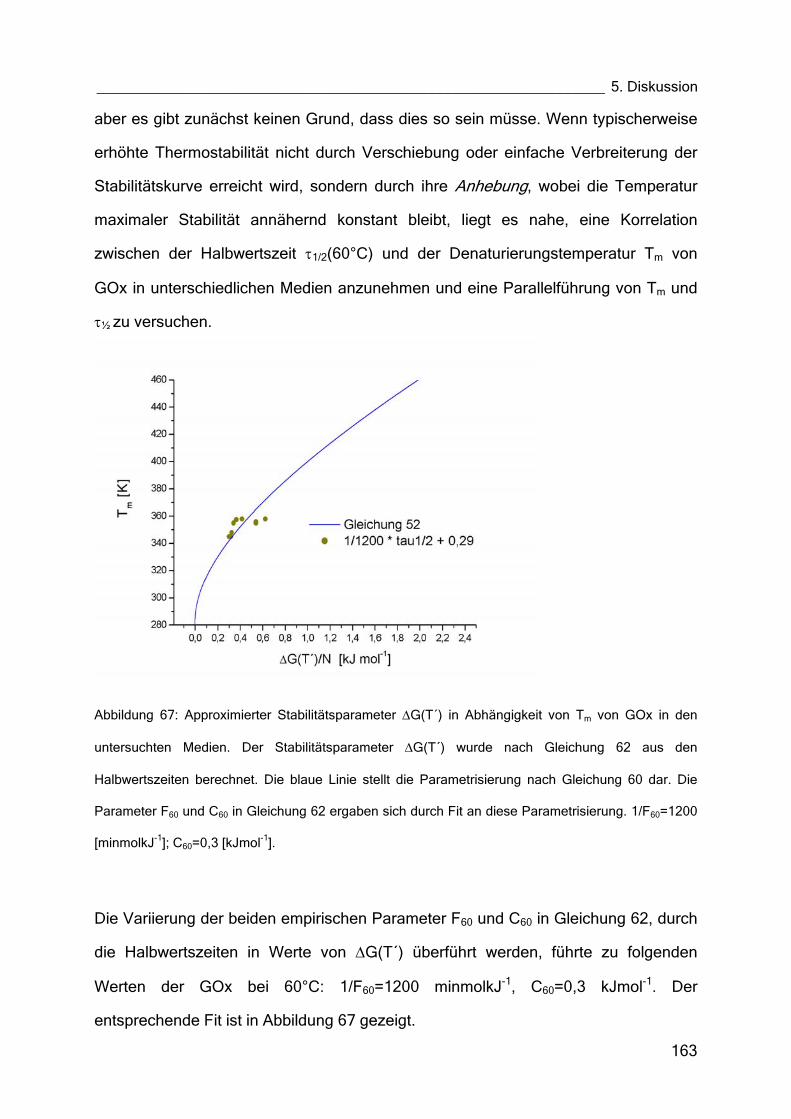
_______________________________________________________________
163
5. Diskussion
aber es gibt zunächst keinen Grund, dass dies so sein müsse. Wenn typischerweise
erhöhte Thermostabilität nicht durch Verschiebung oder einfache Verbreiterung der
Stabilitätskurve erreicht wird, sondern durch ihre Anhebung, wobei die Temperatur
maximaler Stabilität annähernd konstant bleibt, liegt es nahe, eine Korrelation
zwischen der Halbwertszeit τ1/2(60°C) und der Denaturierungstemperatur Tm von
GOx in unterschiedlichen Medien anzunehmen und eine Parallelführung von Tm und
τ½ zu versuchen.
Abbildung 67: Approximierter Stabilitätsparameter ∆G(T´) in Abhängigkeit von Tm von GOx in den
untersuchten Medien. Der Stabilitätsparameter ∆G(T´) wurde nach Gleichung 62 aus den
Halbwertszeiten berechnet. Die blaue Linie stellt die Parametrisierung nach Gleichung 60 dar. Die
Parameter F60 und C60 in Gleichung 62 ergaben sich durch Fit an diese Parametrisierung. 1/F60=1200
[minmolkJ-1]; C60=0,3 [kJmol-1].
Die Variierung der beiden empirischen Parameter F60 und C60 in Gleichung 62, durch
die Halbwertszeiten in Werte von ∆G(T´) überführt werden, führte zu folgenden
Werten der GOx bei 60°C: 1/F60=1200 minmolkJ-1, C60=0,3 kJmol-1. Der
entsprechende Fit ist in Abbildung 67 gezeigt.

_______________________________________________________________ 5. Diskussion
164
Kumar et al. haben auf der Grundlage von Stabilitätskurven einer Auswahl von 31
Proteinen ebenfalls eine positive Korrelation zwischen der Freien Enthalpie bei der
Temperatur maximaler Stabilität ∆G(T´), die in ihrem Datensatz nicht besonders an
Kälte adaptierter Proteine im Mittel bei 293 ±8 K lag, und der
Denaturierungstemperatur Tm gefunden. Die Steigung des linearen Fits betrug 7,5
Jmol-1K-1. Dieser Wert ist gut vergleichbar mit der linearen Korrelation des
Datensatzes von Rees et al., der 8,4 Jmol-1K-1 beträgt [107,172].
Unter der praktischen Vorgabe, dass die Freie Energie bei der Temperatur maximaler
Stabilität ∆G(T´) ein Mass für die Lebensdauer der katalytischen Stabilität sein soll,
die sicher nur bei bekannten Inhibitorkonzentrationen sowie hinreichender Stabilität
gegen irreversiblen Aktivitätsverlust durch Aggregation etc., geeignet ist, kann diese
Korrelation zur Berechnung der Lebensdauer der katalytischen Aktivität aus der
Denaturierungstemperatur mittels Gleichung 63 benutzt werden. Es ergibt sich als
rule of thumb, dass eine Erhöhung der Denaturierungstemperatur Tm von GOx, die
unstabilisiert in Lösung eine Halbwertszeit bei 60°C von 13±5 min hat, um 1°C mit
einer Verlängerung der Halbwertszeit der katalytischen Funktion um ca. 10 min
einhergeht.
6. Ausblick Die Bestimmung der inhärenten kinetischen Konstanten von GOx in den Hydrogelen
wurde nicht durchgeführt, obwohl Einflüsse der Mikroumgebung auch auf Km und kcat
der GOx zu erwarten waren. Die experimentelle Bestimmung der inhärenten
kinetischen Eigenschaften eines heterogen immobilisierten Enzyms könnte zukünftig
durch die Benutzung einer Diffusionszelle gelingen [173].
Die Miniaturisierung und damit verbunden der geringe Materialbedarf sind in der
Biokalorimetrie ein großer Vorzug [174,175]. Weiterführende Arbeiten könnten der

_________________________________________________________________ 6. Ausblick
165
Kalibrierung des Nano-Kalorimeters dienen. Dabei könnte die Besonderheit des
vorgestellten Nano-Kalorimeters genutzt werden, die Temperaturmessung an fünf
verschiedenen Positionen des Chip zu ermöglichen. Die Möglichkeit zur dreifachen
entkoppelten Temperaturmessung ist möglicherweise schon ausreichend, um ein
einfaches thermisches Ersatzschaltbild zu parametrisieren und so ein berechenbares
Model als Grundlage der Kalibrierung des Nano-Kalorimeters auch im nicht-
adiabatischen Modus zu gewinnen. Diese Möglichkeit besteht bei Nano-
Kalorimetern, die Thermopiles als Temperatursensoren verwenden, so einfach nicht,
weil hier die Temperaturmessung stets differenziell ist [174-179].
Die Aufklärung der Mechanismen der Stabilisierung von GOx durch Ko-Solventien
wie NaCl oder PEG wird durch unterschiedliche Techniken gefördert: Dynamische
Lichtstreuung, NMR, Gleichgewichtsdialyse, Densiometrie und Thermoanalyse.
Privalov und Timasheff haben die Anzahl von auf der Oberfläche von Proteinen
gebundenen Wasser- und Ko-Solvensmolekülen mittels DSC und
Gleichgewichtsdialyse mit einer Genauigkeit von etwa einem Molekül bestimmt.
Beide Methoden ergaben für ausgewählte Proteine nahezu identische Werte [180-
184].
Eine vollständige Beschreibung der Thermodynamik der bevorzugten
Wechselwirkung von NaCl bzw. PEG und GOx, die durch Miniaturisierung der DSC-
Technik erleichtert ist, kann zur Aufklärung der Frage beitragen, ob PEG3000 auf der
Oberfläche von GOx gebunden ist oder ob diese Bindung aufgrund deren
ausgeprägter Hydrophilie verhindert ist und ob es Bindungsstellen für monovalente
Kationen wie z.B. Na-Ionen auf dieser Oberfläche gibt (Abbildung 69 und 70). Die
Analyse der DSC-Daten konzentriert sich auf die energetische Distanz zwischen der
nativen und der denaturierten Konformation, während etwa die Gleichgewichtsdialyse
die absoluten Werte der bevorzugten Bindung und damit die chemischen Potentiale
jeweils für das native und das denaturierte Protein separat liefert.

__________________________________________________________________ 6 Ausblick
166
Abbildung 68: Ist
PEG3000 an GOx
spezifisch gebunden?
Abbildung 69: Sind Na-
Ionen auf der Oberfläche
von GOx spezifisch
gebunden?
Chipbasierte Kalorimeter von der vorgestellten Bauart mit einer
temperatursensortragenden Membran können in der Untersuchung der
Thermodynamik biologischer Makromoleküle ihren augenscheinlichsten Vorzug zur
Geltung bringen, ihre Winzigkeit. Nur wenige Substanzen sind von so großem

_________________________________________________________________ 6. Ausblick
167
Interesse für die Forschung bei gleichzeitig so begrenzter Verfügbarkeit wie einige
Proteine bzw. Enzyme sowie ihre Liganden. Enormer Aufwand ist nötig um z.B.
einige Mikrogramm eines Transkriptionsfaktors aus Zellkernen von pflanzlichen oder
tierischen Zellen in hoher Reinheit zu isolieren. Wenn erst die Mikrotechnik die
benötigten Messgeräte en miniature bereitstellt, lassen sich aufgrund der
umfassenden Informationen, die sich aus der thermodynamischen Charakterisierung
der Bindung von biologischen Makromolekülen untereinander, z.B. der Bindung eines
Fettsäuremoleküls an einen Transkriptionsfaktor und dieses Komplexes wiederum an
seine spezielle DNA-Sequenz, ergeben, Einblicke in die Komplexität biologischer
Regulation (Metabolomik) gewinnen.

________________________________________________________ 7. Literaturverzeichnis
168
7. Literaturverzeichnis
(1) Asaad, N., Engberts, B. F. N. (2003). "Cytosol-Mimetic Chemistry: Kinetics of
the Trypsin-Catalyzed Hydrolysis of p-Nitrophenyl Acetate upon Addition of
Polyethylene Glycol and N-tert-Butyl Acetoamide." J. Am. Chem. Soc. 125:
6874-6875.
(2) Karmalkar, R. N., Premnath, V., Kulkarni, M. G., Mashelkar, R.A. (2000).
"Switching biomimetic hydrogels." Proc. R. Soc. Lond. A 456: 1305-1320.
(3) Moser, I., G. Jobst, et al. (1997). "Rapid Liver Enzyme Assay With Miniaturized
Liquid Handling System Comprising Thin-Film Biosensor Array." Sensors and
Actuartors B 44: 377-380.
(4) Jobst, G., I. Moser, et al. (1996). "Thin-Film Microbiosensor For Glucose-
Lactate Monitoring." Anal. Chem. 68: 3173-3179.
(5) Schmid, A., Dordick, J.S., Hauer, B., Kiener, A., Wubbolts, M., Withold, B.
(2001). "Industrial Biocatalysis Today and Tomorrow." Nature 409: 258-268.
(6) Ramachandran, S., Fontanille, P., Pandey, Larroche, C. (2006). "Gluconic
Acid: Properties, Application and Microbial Production." Food Technol.
Biotechnol. 44: 185-195.
(7) Sarath Babu, V. R., Kumar, M.A., Karanth, N. G., Thakur, M. S. (2004).
"Stabilization of Immobilized Glucose Oxidase against Thermal Inactivation by
Silanization for Biosensor Applications." Biosensors and Bioelectronics 19:
1337-1341.
(8) Witten, T., (1999). "Insights from Soft Condensed Matter." Rev. Mod. Phys. 71:
367-373.
(9) Becktel, W., J. and J. A. Schellman (1987). "Protein Stability Curves."
Biopolymers 26: 1859-1877.
(10) Anfinsen, C. B. (1973). "Principles That Govern The Folding of Protein
Chains." Science 181: 223-230.

________________________________________________________ 7. Literaturverzeichnis
169
(11) Kauzman, W. (1959). "Some Factors in The Interpretation of Protein
Denaturation." Adv. Protein Chem. 14: 1-63.
(12) Privalov, P. L. (1974). "A Thermodynamic Approach to the Problem of
Stabilization of Globular Protein Structure: A Calorimetric Study." J. Mol. Biol.
86: 665-684.
(13) Privalov, P. L. and S. A. Potekhin (1986). "Scanning Microcalorimetry in
Studying Temperature-Induced Changes in Proteins." Methods Enzymol. 131:
4-51
(14) Hollien, J., Marqusee, S. (1999). "A Thermodynamic Comparison of
Mesophilic and Thermophilic Ribonuclease H." Biochemistry 38: 3831-3836.
(15) Razvi, A., Scholtz, M. J. (2006). "Lessons in Stability from Thermophilic
Proteins." Protein Science 15: 1569-1578.
(16) Collins, T., M.-A. Meuvis, et al. (2003). "Activity, Stability and Flexibility in
Glycosidase Adapted to Extreme Thermal Environments." J. Mol. Biol. 328:
419-428.
(17) Jaenicke, R. (2000). "Do Ultrastable Proteins from Hyperthermophiles Have
High or Low Conformational Rigidity." Proc. Natl. Ac. Sc. 97: 2962-2964.
(18) Shoichet, K., W. A. Baase, et al. (1995). "A Relationship Between Protein
Stability and Protein Function." Proc. Natl. Ac. Sc. 92: 452-456.
(19) Minton, A. P.(2005). "Models for Excluded Volume Interaction between an
Unfolded Protein and Rigid Macromolecular Cosolutes: Macromolecular
Crowding and Protein Stability Revisited." Biophys. J. 88: 971-985.
(20) Schnell, S., Turner, T. E. (2004). "Reaction Kinetics in Intracellular
Environments with Macromolecular Crowding: Simulation and Rate Laws."
Progress in Biophysics & Molecular Biology 85: 235-260.
(21) Eggers, D. K., Valentine, J. S. (2001). "Molecular Confinement Influences
Protein Structure and Enhances Thermal Protein Stability." Protein Sciences
10: 250-261.

________________________________________________________
170
7. Literaturverzeichnis
(22) Minton, A. P. (2001). "The Influence of Macromolecular Crowding and
Macromolecular Confinement on Biochemical Reactions in Physiological
Media." J. Biol. Chem 276: 10577-10580.
(23) Minton, A. P. (2001). "Effect of a Concentrated “inert” Macromolecular
Cosolute on the Stability of a Globular Protein with Respect to Denaturation by
Heat and by Chaotropes: A Statistical-Thermodynamic Model." Biophys. J. 78:
101-109.
(24) Timasheff, S. N. (1998). "Control of Protein Stability and Reactions by Weakly
Interacting Cosolvents: The Simplicity Of The Complicated." Advances in
Protein Chemistry 51: 355-432.
(25) Bolen, W. D. (2001) "Protein Stabilization by Naturally Occurring Osmolytes."
Methods in Molecular Biology 168: 1-15.
(26) Galinski, E. A., Stein, M., Amendt, B., Kinder, M. (1997). "The Kosmotropic
(Structure-Forming) Effect of Compensatory Solutes." Comp. Biochem.
Physiol. 117A: 357-365.
(27) Galinski, E. A. (1993). "Compatible Solutes of Halophilic Eubacteria: Molecular
Principles, Water-Solute Interaction, Stress Protection." Experimentia 49: 487-
496.
(28) Somero, G. N. (1986). "Protons, Osmolytes, and Fitness of Internal Milieu for
Protein Function." Am. J. Physiol. Regul. Integr. Comp. Physiol. 251: R197-
R213.
(29) Yu, Q., Bauer, J. M., Moore, J. S. (2001). "Responsive Biomimetic Valve for
Microfluidics." Appl. Phys. Lett. 78: 2589-2591.
(30) Sandborn, T. J., Messersmith, P. B., Barron, A. E. (2002). "In Situ Crosslinking
of a Biomimetic Peptide-PEG Hydrogel via Thermally Triggered Activation of
Factor VIII." Biomaterials 23: 2703-2710.
(31) Wang, G., K. Kuroda, et al. (2000). "Gel Catalyst That Switch On and Off."
Proc. Natl. Ac. Sc. 97: 9861-9864.
(32) Casassa, E. F., Eisenberg, H. (1948). "Thermodynamic Analysis of
Multicomponent Solutions." Adv. Prot. Chem. 4: 287-395.

________________________________________________________ 7. Literaturverzeichnis
171
(33) Chaniotakis, N. A. (2004). "Enzyme Stabilization Strategies Based on
Electrolytes and Polyelectrolytes for Biosensor Applications." Anal. Bioanal.
Chem. 378: 89-95.
(34) Zale, S. E. and A. M. Klibanov (1986). "Why Does Ribonuclease Irreversibly
Inactivate at High Temperatures?" Biochemistry 25: 5432-5444.
(35) Daniel, R. M., M. Dines, et al. (1996). "The Denaturation and Degradation of
Stable Enzymes at High Temperatures." Biochem. J. 317: 1-11.
(36) Singh. S., Singh, J. (2003). "Effect of Polyols on the Conformational Stability
and Biological Activity of a Model Protein Lysozyme." AAPS Pharm. Sci. Tech.
4: 1-9.
(37) Anderson, E. Hahn-Hägerdal, B. (1987). "Enzyme Action in Polymer and Salt
Solutions. I. Stability of Penicillin Acylase in Poly(ethylene glycol) and
Potassium Phosphate Solutions in Relation to Water Activity ." Biochim.
Biophys. Acta 912: 317-324.
(38) Anderson, E. Hahn-Hägerdal, B. (1987). "Enzyme Action in Polymer and Salt
Solutions. II. Activity of Penicillin Acylase in Poly(ethylene glycol) and
Potassium Phosphate Solutions in Relation to Water Activity ." Biochim.
Biophys. Acta 912: 325-328.
(39) Nölting, B. (1999). Protein Folding Kinetics: Biophysical Methods. Berlin,
Heidelberg, Springer-Verlag, 17-31.
(40) Shu, Q. and C. Frieden (2005). "Relation of Enzyme Activity to Local/Global
Stability of Murine Adenosine Deaminase:19F-NMR Studies." J. Mol. Biol. 345:
599-610.
(41) Fields, P., A., Walstrand, B. D., Somero, G. N. (2001). "Intrinsic versus
Extrinsic Stabilization of Enzymes. The Interaction of Solutes and Temperature
on A4-Lactate Dehydrogenase Orthologs from Warm-adapted and Cold-
Adapted Marine Fishes." Eur. J. Biochem. 268: 4497-4505.
(42) D´Amico, M. S., J. C., et al. (2003). "Activity-Stability Relationship in
Extremophile Enzymes." J. Biol. Chem. 278: 7891-7896.

________________________________________________________
172
7. Literaturverzeichnis
(43) D´Amico, S., C. Geray, et al. (2003). "Temperature Adaptation of Proteins:
Engineering Mesophilic-like Activity and Stability in a Cold-adapted a-
Amylase." J. Mol. Biol. 332: 981-988.
(44) Privalov, P. L., Gill, S. J. (1989). "The Hydrophobic Effect: A Reappraisal."
Pure & Applied Chem. 61: 1097-1104.
(45) Privalov, P. L., E. I. Tiktopulo, et al. (1989). "Heat Capacity and Conformation
of Proteins in the Denatured State." J. Mol. Biol. 205: 737-750.
(46) Franks, F. (1995) "Protein Destabilization at Low Temperatures." Adv. Prot.
Chem. 46: 105-139.
(47) Kumar, S., C. J. Tsai, et al. (2003). "Temperature Range of Thermodynamic
Stability for The Native State of Reversible Two-State Proteins." Biochemistry
42: 4864-4873.
(48) Freire, E. (1995). "Thermal Denaturation Methods in the Study of Protein
Folding." Methods Enzymol. 259: 144-168.
(49) Jelesarov, I. and H. R. Bosshard (1999). "Isothermal Titration Calorimetry and
Differential Scanning Calorimetry as Complementary Tools to Investigate the
Energetics of Biomolecular Recognition." Journal of molecular recognition 12:
3-18.
(50) Schellman, J. A. (1987). "The Thermodynamic Stability of Proteins." Ann. Rev.
Biophys. Biophys. Chem 16: 115-137.
(51) Schellman, J. A. (1978). "Solvent Denaturation." Biopolymers. 17: 1081305-
1322.
(52) Schellman, J. A. (1987). "Selective Binding and Solvent Denaturation."
Biopolymers 26: 49-559.
(53) Wyman, J. Jr. (1948). "Heme Proteins." Adv. Prot. Chem. 4: 407-531.
(54) Scatchard, G. (1946). "Physical Chemistry of Protein Solutions. I. Derivation of
the Equation for the Osmotic Pressure." J. Am. Chem. Soc.68: 2315-2319.
(55) Wyman, J. Jr. (1964). "Linked Functions and Reciprocal Effects in
Hemoglobin: a Second Look." Adv. Prot. Chem. 19: 223-286.

________________________________________________________ 7. Literaturverzeichnis
173
(56) Lee, J. C. and N. Timasheff (1981). "The Stabilization of Protein by Sucrose."
J. Biol. Cem. 256: 7193-7201].
(57) Arakawa, T., R. Bhat, et al. (1990). "Why Preferential Hydration Does Not
Always Stabilize the Native Structure of Gloular Proteins?" Biochemistry 29:
1924-1931.
(58) Lee, L. Y. and J. C. Lee (1987). "Thermal Stability of Proteins in the Presence
of Poly(ethylene glycols)." Biochemistry 26: 7813-7819.
(59) Arakawa, T. and N. Timasheff (1985). "Mechanism of Poly (ethylene Glycol)
Interaction with Proteins." Biochemistry 24: 6756-6762.
(60) Bhat, R., Timasheff, S. N. (1992). "Steric Exclusion is the Principal Source of
the Preferential Hydration of Proteins in the Presence of Polyethylene
Glycols." Protein Science 1: 1133-1143.
(61) Lee, L. Y. and J. C. Lee (1979). "Interaction of Calf Brain Tubulin with
Poly(ethylene glycols)." Biochemistry 18: 5518-5526.
(62) Arakawa, T. and S. Timasheff, N. (1984). "Protein Stabilization and
Destabilization by Guanidinium Salts." Biochemistry 23: 5924-5929.
(63) Schellman, J. A. (2003). "Protein Stability in Mixed Solvents: a Ballance of
Contact Interaction and Excluded Volume." Biophys. J. 85: 108-125.
(64) Timascheff, S. N. (1993). "The Control of Protein Stability and Association by
Weak Interactions with Water: How do Solvents Affect these Processes." Ann.
Rev. Biophys. Biomol. Struct. 22, 67-97.
(65) Timasheff, S. N. (1995). "Preferential Interactions of Water and Cosolvents
with Proteins." In: Gregory, R. B. (Ed.) Protein Solvent Interactions. Marcel
Dekker, New York, 445-482.
(66) Winter, R., Noll, F., Methoden der Biophysikalischen Chemie, Täubner,
Stuttgart, 1998.
(67) Gekko, K. and N. Timasheff (1985). "Mechanism of Protein Stabilization by
Glycerol: Preferential Hydration in Glycerol-Water Mixtures." Biochemistry 20:
4667-4676.

________________________________________________________
174
7. Literaturverzeichnis
(68) Cooper, A. (1999) "Thermodynamics of Protein Folding and Stability". In :
Allen, G. (Ed.), JAI Press Inc., Protein: A Comprehensive Treatise, Vol. 2, 217-
270.
(69) Barone, G., Catanzano, F., Del Vecchio, P., Graziano, G. (1995) "Differential
Scanning Calorimetry as a Tool to Study Protein-Ligand Interactions". Pure &
Appl. Chem. 67, 1867-1872.
(70) Tanford, C. (1969). "Extension of the Theory of Linked Functions to
Incorporate the Effects of Protein Hydration" . Mol. Biol. 39: 539-544.
(71) Langmuir, I. (1916). "The Constitution and Fundamental Properties of Solids
and Liquids. J. Am. Chem. Soc. 38: 2221-2295.
(72) Michnik, A. (2003). "Thermal Stability of Bovine Serum Albumin. DSC Study."
Journal of Thermal Analysis and Calorimetry 71: 509-519.
(73) Schellman, J. A. (1990). "A Simple Model of Solvation in Mixed Solvents.
Applications to the Stabilization and Destabilization of Macromolecular
Structures." Biophys. Chem. 37: 1081305-1322.
(74) Liu, Y., Bolen, D. W. (1995). "The Peptide Backbone Plays a Dominant Role in
Protein Stabilization by Naturally Occurring Osmolytes." Biochemistry 34,
12884-12891.
(75) Arakawa, T., Bhat, R., Timasheff, N. (1990). "Preferential Interaction
Determine Protein Solubility in Three-Component Solutions: The MgCl2
System." Biochemistry 29: 1914-1923.
(76) Arakawa, T. (1990). "Mechanism of Protein Salting In and Salting Out by
Divalent Cation Salts: Balance between Hydration and Salt Binding."
Biochemistry 23: 5912-5923.
(77) Davis-Searles, P. R., Saunders, A. J., Erie, D. A., Winzor, D. J., Pielak, G. J.
(2001) "Interpreting the Effects of Small Uncharged Solutes on Protein-Folding
Equilibria." Ann. Rev. Biophys. Biomol. Struct. 30: 271-306.
(78) Fußnote in: Schachman, H. K., Lauffer, M. A. (1949). "The Hydration, Size and
Shape of Tobacco Mosaic Virus." J. Am. Chem. Soc. 71: 536-541.

________________________________________________________ 7. Literaturverzeichnis
175
(79) Chebotareva, N. A., B. I. Kurganov, et al. (2004). "Biochemical Effects of
Molecular Crowding." Biochemistry (Moscow) 69: 1522-1536.
(80) Jacobsen, M. P., Wills, P. R., Winzor, D. J. (1996). "Thermodynamic Analysis
of the Effects of Small Inert Cosolutes in the Ultracentrifugation of
Noninteracting Proteins." Biochemistry 35: 13173-13179.
(81) Gibbs, J. W. (1878). "On the Equilibrium of Heterogeneous Substances."
Trans. Conn. Acad. 3: 343-524.
(82) Lin, T.-Y., Timasheff, S. N. (1996). "On the Role of Surface Tension in the
Stabilization of Globular Proteins." Protein Science: 372-381.
(83) Hofmeister, F. (1888). "On the Understanding of the Effects of Salts. II. On
Regularities in the Precipitating Effect of the Salts and the relationship of
theese to their physiological behaviour." Arch. Experim. Path. Pharmak. 24:
247-260.
(84) Collins, K. D. and M. W. Washabaugh (1985). "The Hofmeister Effect and
Water at Interfaces." Quarterly Reviews of Biophysics 18: 323-422.
(85) Neagu, A., Neagu, M., Der, A. (2001). "Fluctuations and the Hofmeister
Effect." Biophys. J. 81: 1285-1294.
(86) Ramos, C. H. I. and R. L. Baldwin (2002). "Sulfate Anion Stabilization of Native
Ribonuclease A Both by Anion Binding and by the Hofmeister Effect." Protein
Science 11: 1771-1778.
(87) Yaacobi, M. and A. Ben-Naim (1974). "Solvophobic Interaction." J. Phys.
Chem. 78: 175-178.
(88) Dill, K. A., Alonso, D. O. V., Hutchinson, K. (1989) "Thermal Stabilities of
Globular Proteins." Biochemistry 18: 5439-5449.
(89) Moelbert, S., Normand, B., De Los Rios, P. (2004). "Kosmotropes and
Chaotropes: Modeling Preferential Exclusion, Binding and Aggregate
Stability." Biophys. Chem. 112: 45-57.
(90) Moelbert, S. and P. De Los Rios (2003). "Chaotropic Effect and Preferential
Binding in a Hydrophobic Interaction Model." J. Chem. Phys. 119: 7988-8001.

________________________________________________________
176
7. Literaturverzeichnis
(91) Hribar. (2002). "How Ions Affect the Structure of Water." J. Am. Chem. Soc.
12302-12311.
(92) Smith, P. E. J. (1999). "Computer Simulation of Cosolvent Effects on
Hydrophobic Hydration." J. Phys. Chem. B. 103: 525-534.
(93) Kalra, A.,Tugcu, N., Cramer, S., Garde, S. J. (2001). "Salting-In and Salting-
Out of Hydrophobic Solutes in Aqueous Salt Solutions." J. Phys. Chem. B.
105: 6380-6386.
(94) Dill, K. A. (1990). "Dominant Forces in Protein Folding." Biochemistry 29:
7133-7155.
(95) Di Stasio, E. (2004). "Ionic Regulation of Proteins." Ital. J. Biochem. 53: 112-
119.
(96) Record Jr., M. T., Anderson, C. F., Lohman, T. M. (1978). " Thermodynamic
Analysis of Ion Effects on the Binding and Conformational Equilibria of
Proteins and Nucleic Acids: the Roles of Ion Association or Release,
Screening, and Ion Effects on Water Activity. " Q. Rev. Biophys. 11: 103-178.
(97) Atta, A., M. S. Akhtar, et al. (2001). "Monovalent Cation-Induced
Conformational Change in Glucoseoxidase Leading to Stabilization of the
Enzyme." Biochemistry 40: 1945-1955.
(98) Ye, W. N., D. Combes, et al. (1988). "Influence of Additives on the
Thermostability of Glucoseoxidase." Enzyme Microb. Technol. 10: 498-502.
(99) EngasserJ.-M., Horvath, C. (1976). "Diffusion and Kinetics with Immobilized
Enzymes." Applied Biochemistry and Bioengineering 1: 127-220.
(100) Aris, R. (1957). "On Shape Factors for Irregular Particles - I. The Steady State
Problem." Chem. Eng. Sci. 6: 262-268.
(101) Mishra, R., Seckler, R., Bhat, R. (2005). "Efficient Refolding of Aggregation-
Prone Citrate Synthase by Polyol Osmolytes." J. Biol. Chem. 280: 15553-
15560.
(102) Remmele, R. L., S. D. Bhat, et al. (1999). "Minimization of Recombinant
Human Flt3 Ligand Aggregation at the Tm Plateau: A Matter of Thermal
Reversibility." Biochemistry: 5241-5247.

________________________________________________________ 7. Literaturverzeichnis
177
(103) Sangeeta Devi, Y., B. N. Usha, et al. (1998). "Effect of a Mixed Stabilizer
Denaturant System on the Stability of Enzyme Activity at High Temperatures:
Lysozyme as Model", in: Ballesteros, A., Plou, F. J., Iborra, J. L., Halling, P. J.
(Editors) Stability and Stabilization of Biocatalysts, Elsevier.
(104) Kudou, M., K. Shiraki, et al. (2003). "Prevention of Thermal Inactivation and
Aggregation of Lysozyme by Polyamines." Eur. J. Biochem. 270: 4547-4554.
(105) Meng, F.-G., Y.-K. Hong, et al. (2004). "Osmophobic Effect of Glycerol on
Irreversible Thermal Denaturation of Rabbit Creatine Kinas." Biophys. J. 87:
2247-2254.
(106) Iwakura, M., Nakamura, D., Takenawa, T., and Mitsuishi, Y. (2001). "An
Approach for Protein to Be Completely Reversible to Thermal Denaturation
Even at Autoclave Temperatures." Protein Engineering, 14, 583-589.
(107) Rees, D., C. and A. D. Robertson (2001). "Some Thermodynamic Implications
for the Thermostability of proteins." Protein Science 10: 1187-1194.
(108) Kumar, S., Nussimov, R. (2001). "How do Thermophilic Proteins Deal with
Heat?" Cell. Mol. Life Sci. 58: 1216-1233.
(109) Perl, D., Mueller, U., Heinemann, U., Schmid, F. X. (2000). "Two Exposed
Amino Acid Residues Confer Thermostability on a Cold Shock Protein." Nature
Struct. Biol. 7: 380-383.
(110) Pace, C. N. (2000). "Single Surface Stabilizer." Nature Struct. Biol. 7: 345-346.
(111) Zhou, H.-X. (2002). "Toward the Physical Basis of Thermophilic Proteins of
Enriched Polar Interactions and Reduced Heat Capacity." Biophys. J. 83:
3126-3133.
(112) Robertson, A. D., Murphy, K. P. (1997). "Protein Structure and Energetics of
Protein Stability." Chem. Rev. 97: 1251-1267 und Referenzen darin.
(113) Chen, J., Lu, Z., Sakon, J., Stites, W. E. (2000) "Increasing the Thermostability
of Staphylococcus Nuclease: Implications for the Origin of Protein Stability." J.
Mol. Chem. 303: 125-130.
(114) Zipp, A., Kauzman, W. (1973) "Pressure Denaturation of Metmyoglobin."
Biochemistry 12: 4217-4228.

________________________________________________________
178
7. Literaturverzeichnis
(115) Swoboda, B. E. P. and V. Massey (1965). "Purification and Properties of the
Glucose Oxidase from Aspergillus Niger." J. Biol. Chem. 240: 2209-2215.
(116) Frederick, R. K., J. Tung, et al. (1990). "Glucose Oxidase from Aspergillus
Niger. Cloning, Gene Sequence, Secretion from Saccharomyces Cerevisiae
and Kinetic Analysis of a Yeast-derived Enzyme." J. Biol. Chem. 265: 3793-
3802.
(117) Gibson, Q. H., B. E. P. Swoboda, et al. (1964). " Kinetics and Mechanism of
Action of Glucose Oxidase." J. Biol. Chem. 239: 3927-3934.
(118) Weibel, K. M. and H. J. Bright (1971). "The Glucose Oxidase Mechanism.
Introduction of the pH-Dependence." J. Biol. Chem. 246: 2734-2744.
(119) Kirstin Seidel, Diplomarbeit, Universität Freiburg, 2003.
(120) R. Eisenthal & A. Cornish-Bowden (1974). “The direct linear plot.” Biochem. J.
139: 715–720.
(121) Constantinides, A.(1973). “Characterization of Glucose Oxidase Immobilized
on Collagen.” Mol. Cell. Biochem. 1:127-33.
(122) Nakamura, S.; Hayashi, S.; Koga, K. (1976). "Effect of periodate oxidation on
the structure and properties of glucose oxidase." Biochim. Biophys. Acta 445,
294-308.
(123) Antonson, K. P., Gombotz, W. R., Hoffman, A. S. (1994). "Attempts to Stabilize
a Monoclonal Antibody with Water Soluble Synthetic Polymers of Varying
Hydrophobicity." J. Biomater. Sci. Polymer Edn. 6, 55-65.
(124) Bassetti, F. J., Bergamasco, R., Moraes, F. F., Zanin, G. M. (2000). "Thermal
Stability and Deactivation Energy of Free and Immobilized Invertase." Braz. J.
Chem. Eng. 17: no. 4-7, 867-872.
(125) Nath, S., G. R. Satpathy, et al. (1998). "Thermal Stability of Alcohol-
Dehydrogenase Enzyme Determined by Activity Assay and Calorimetry."
Thermochimica Acta 309: 193-196.
(126) Sanchez-Ruiz, J. M., J. L. Lopez-Lacomba, et al. (1988). "Differential Scanning
Calorimetry of the Irreversible Thermal Denaturation of Thermolysin."
Biochemistry 27: 1648-1652.

________________________________________________________ 7. Literaturverzeichnis
179
(127) Haouz, A., J. M. Glandieres, et al. (2001). "Involvement of Protein Dynamics in
Enzyme Stability. The Case of Glucoseoxidase." FEBS Letters 506: 216-220.
(128) Cinar, P. et al., unveröffentlicht
(129) Höhne, G., Hemminga, W., Flammersheim, H.-J., Differential Scanning
Calorimetry. An Introduction for Practitioners, 1. Ed. 1996.
(130) O`Neill, M. J. (1975) "Measurement of Exothermic Reactions by Differential
Scanning Calorimetry. " Analytical Chemistry 47: 630.
(131) Boersma, S. L., J. A. (1955) "Theory of Differential Thermal Analysis and New
Methods of Measurement and Interpretation. " Am. Ceram. Soc. 8: 281.
(132) Mraw, S. C. (1982) " Mathematical Treatment of Heat Flow in Differential
Scanning Calorimetry and Differential Thermal Analysis Instruments. " Rev.
Sci. Inst. 53: 228-231.
(133) Vogl, T., Jatzke, C., Hinz, H.-J. (1997). "Thermodynamic Stability of Annexin V
E17G: Equilibrium Parameters from an Irreversible Unfolding Reaction."
Biochemistry 36: 1657-1668.
(134) Arora, P., Oas, T., Myers, J. (2004). "Fast and Faster: A Designed Variant of
the B-Domain of Protein A folds in 3 µsec." Protein Science 13: 847-853.
(135) Cooper, A., Nutley, A., Wadood, A. (2000). "Differential Scanning
Microcalorimetry." In Harding, S. E., Chowdhry, B. Z. (Eds.) Protein-Ligand
Interactions: Hydrodynamics and Calorimetry, Oxford University Press, Oxford
New York, 287-318.
(136) Privalov, G. P. and P. L. Privalov (2000). "Problems and Prospects in
Microcalorimetry of Biological Macromolecules." Methods Enzymol. 323: 31-
62.
(137) Sun, S, F. (2004). "Liquid Crystals." In: Physical Chemistry of Macromolecules:
Basic Principles and Issues, 2nd Edn., Wiley & Sons, 127-149.
(138) Shinoda, T., Y. Maeda, et al. (1974). "Thermodynamic Properties of N-(p-
methoxybenzylidene)-p-n-butyl-aniline (MBBA) from 2 K to its Isotropic-Liquid
Phase." J. Chem. Thermodynamics 6: 921-934.

________________________________________________________
180
7. Literaturverzeichnis
(139) Spratte, W. and G. M. Schneider (1976). "Differential Thermal Analysis (DTA)
under High Pressure VI: Pre Transitions of Some Liquid Crystals up to 3 kbar."
Berichte der Bunsengesellschaft 80(9): 886-891.
(140) Höhne, G. W. H., J. Schawe, et al. (1993). "Temperature Calibration on
Cooling Using Liquid Crystal Phase Transition." Thermochimia Acta 221: 129-
137.
(141) Ohline, S. M., Campbell, M. L., Turnbull, M. T., Kohler, S. J. (2001).
"Differential Scanning Calorimetry Study of Bilayer Membrane Phase
Transitions: A Biophysical Chemistry Experiment." J. Chem. Ed. 78: 1251-
1256.
(142) Heimburg, T. (2000). "A Model for the Lipid Pretransition: Coupling of Ripple
Formation With The Chain-Melting Transition." Biophys. J. 78: 1154-1165.
(143) Ali, S., S. Minchey, et al. (2000). "A Differential Scanning Calorimetry Study of
Phosphocholines Mixed with Paclitaxel and It´s Bromoacylated Taxanes."
Biophys. J. 78: 246-256.
(144) Schrader, W., Ebel, Grabitz, P., Hanke, E., Heimburg, T., Hoeckel, M., Kahle,
M., Wente, F., Kaatze, U. (2002). “Compressibility of Lipid Mixtures Studied by
Calorimetry and Ultrasonic Velocity Measurements.“ J. Phys. Chem. B. 106:
6581-6586.
(145) Geankoplis, C. J. (2003). "Transport Processes and Separation Process
Principles." 4th ed., Prentice Hall, PTR Upper Saddle River, NJ.
(146) Verhaegen, K., K. Baert, et al. (2000). "A high-throughput silicon
microphysiometer." Sensors and Actuators 82: 186-190.
(147) Johannessen, E. A., J. M. R. Weaver, et al. (2002). "A Suspended Membrane
Nanocalorimeter for Ultralow Volume Bioanalysis." IEEE Transactions on
Nanobioscience 1(1): 29-36.
(148) Johannessen, E. A. and J. M. R. Weaver (2003). "Heat Conduction
Nanocalorimeter For pl-Scale Single Cell Measurement." Applied Physics
Letters 80: 2029-2031.
(149) CRC Handbook of Chemistry and Physics, 80th Edn.

________________________________________________________ 7. Literaturverzeichnis
181
(150) Kleppe, K. (1966). “The Effect of Hydrogen Peroxide on Glucose Oxidase from
Aspergillus Niger“. Biochemistry 5, 139-143.
(151) Van Stroe-Biezen, S. A. M., Everaerts, F. M., Janssen, L. J.J ., Tacken, R. A.
(1993) "Diffusion Coefficients of Oxygen Hydrogen Peroxide and Glucose in a
Hydrogel." Analytica Chimica Acta 273: 533-560.
(152) Yankov, D. (2004). "Diffusion of Glucose and Maltose in Poly-Acrylamid gel."
Enzyme and Microbial Technol. 34: 603-610.
(153) Murphy, S. M., C. J. Hamilton, et al. (1988). "Synthetic Hydrogels: 5. Transport
Processes in 2-Hydroyethyl Methacrylate Copolymers." Polymer 29: 1887-
1893.
(154) Handrikova, G., V. Stefuca, et al. (1996). "Determination of effective diffusion
coefficient of substrate in gel particles with immobilized biocatalyst." Enzyme
and Microbial Technol. 18: 581-5584.
(155) Korus, R. A. and O´Driscoll (1975). "The Influences of diffusion on the
Apparent Rate of Denaturation of Gel Entrapped Enzymes." Biotechnology
and Bioengineering 17: 441-444.
(156) Dr. M. Grüner, Netzsch-Gerätebau GmbH, persönliche Mitteilung
(157) Sinha, A., Yadav, S., Ahmad, R., Ahmad, F. (2000). "A Possible Origin of
Differences between Calorimetric and Equilibrium Estimates of Stability
Parameters of Proteins." Biochem. J. 345: 711-717.
(158) Winter, W. and W. H. Höhne (2003). "Chip-calorimeter for small samples."
Thermochimica Acta 403: 43-53.
(159) Jägle, M. (2003). "Hochempfindliches, schnelles Nanokalorimeter."
Technisches Messen 12: 557-560.
(160) Efremov, M.Yu., et al. (2000). "Discrete Periodic Melting Point Observations
for Nanostructure Ensembles." Physical Review Letters 85: 3560-3563.
(161) Olson, E. A., M. Efremov, Y., et al. (2000). "Scanning Calorimeter for
Nanoliter-Scale Liquid Samples." Applied Physics Letters 77: 2671-2673.

________________________________________________________
182
7. Literaturverzeichnis
(162) Denlinger, D. W., E. N. Abarra, et al. (1994). "Thin Film Microcalorimeter for
Heat Capacity Measurements from 1,5 to 800K." Rev. Sci. Instrum. 65(4): 946-
959.
(163) Köhler, J. M., Baier, V., Dillner, U., Zieren, M. (1999). "Thermal
Microtransducers in Molecule Processing Chips." Sensor 99 Proceeding II,
P3.20: 583-588.Gouda, M. D., S. A. Singh, et al. (2003). "Thermal Inactivation
of Glucoseoxidase." J. Biol. Chem 278: 24324-24333.
(164) Gulla, K. C., M. D. Gouda, et al. (2003). "Enhancement of Stability of
Immobilized Glucoseoxidase by Modification of Free Thiols Generated by
Reducing Disulfide Bonds and Using Additives." Biosensors & Bioelectronics
19: 621-625.
(165) Israelachvili, J. (1997). "The Different Faces of Poly(Ethylene Glycol)." Proc.
Natl. Ac. Sc. 94: 8378-8379.
(166) Stone, M., S. B. Harvey, et al. (2002). "Unusual Benefits of Macromelecular
Shielding by Polyethylene Glycol for Reactions at the Diffusional Limit: The
Case of Factor VIIai and Tissue Factor." Biochemistry 41: 15820-15825.
(167) Akhtar, M. S., A. Atta, et al. (2002). "Guanidinium Chloride- and Urea-Induced
Unfolding of the Dimeric Enzyme Glucose Oxidase." Biochemistry 41: 3819-
3827.
(168) Back, J. F., Oakenfull, D., Smith, M. B. (1979). "Increased Thermal Stability of
Proteins in the Presence of Sugars and Polyols" Biochemistry 18: 5191-5196.
(169) Kaushik, J. K. and R. Bhat (1998). "Thermal Stability of Proteins in Aqueous
Polyol Solutions: Role of the Surface Tension of Water in the Stabilizing Effect
of Polyols." J. Phys. Chem. B. 102: 7058-7066.
(170) Kaushik, J. K. and R. Bhat (2003). "Why is Trehalose an Exceptional Protein
Stabilizer?" J. Biol. Chem. 278: 26458-26465.
(171) Kumar, S., C.-J. Tsai, et al. (2002). "Maximal stabilities of reversible two-state
proteins." Biochemistry 41: 5359-5374.
(172) van Stroe-Biezen, S. A. M., J. M. H. van der Loo, et al. (1996). "Determination
of the inherent kinetics of immobilized glucose oxidase using a diffusion cell."
Bioprocess Engineering 15: 87-94.

________________________________________________________ 7. Literaturverzeichnis
183
(173) Wang, L., Lin, Q. (2005). "A Polymer-Based MEMS Sensor for Biocalorimetric
Measurements." 9th International Conference on Miniaturized Systems for
Chemistry and Life Sciences (microTAS 2005), Boston., Ma.
(174) Ernst, H. (2001). "High-Resolution Thermal Measurements in
Fluids."Dissertation, Universität Freiburg,.
(175) Chateau, E., J. L. Garden, et al. (2005). "Physical Kinetics and
Thermodynamics of Phase Transitions Probed by Dynamic Nanocalorimetry."
Applied Physics Letters 86: 151913.
(176) Garden, J. L., E. Chateau, et al. (2004). "Highly Sensitive AC Nanocalorimeter
for Microliter-Scale Liquids or Biological Samples." Applied Physics Letters 84:
3597-3599.
(177) Chancellor, E. B., J. P. Wikswo, et al. (2004). "Heat Conduction Calorimeter
for Massively Parallel High Throughput Measurements with Picoliter Sample
Volumes." Applied Physics Letters 85: 2408-2410.
(178) Zhang, M., M. Y. Efremov, et al. (2000). "Size-dependent Melting Point
Depression of Nanostructures: Nanocalorimetric Measurements,." Physical
Review B 62: 547-557.
(179) Makhatadze, G. I. and P. L. Privalov (1992). "Protein Interactions with Urea
and Guanidinium Chloride: A Calorimetric Study." J. Mol. Biol. 226: 491-505.
(180) Timasheff, N. (2002). "Thermodynamic Binding and Site Occupancy in The
Light of The Schellman Exchange Concept." Biophys. J. 101-102: 99-111.
(181) Schellman, J., Gassner, N. C. (1996). "The Enthalpy of Transfer of Unfolded
Proteins into Solutions of Urea and Guanidinium Chloride." Biophys. Chem.
59: 259-275.
(182) Wyman, J., Gill, S. J. (1990). Binding and Linkage. Functional Chemistry of
Biological Macromolecules. University Science Books, Mill Valley, Ca.
(183) Schellman, J. A. (1994). "The Thermodynamics of Solvent Exchange."
Biopolymers 34: 1015-1028.