binary encoding and gene rearrangement analysis jijun tang tianjin university university of south...
TRANSCRIPT

Binary Encoding and Gene Rearrangement Analysis
Jijun Tang
Tianjin UniversityUniversity of South Carolina
[email protected](803) 777-8923

Outline
Backgrounds
Maximum Likelihood Methods for Phylogenetic Reconstruction
Maximum Likelihood Methods for Ancestral Genome Inferrence
Conclusions
Phylogenetic Reconstruction

Data Type
Sequence Data
DNA/RNA/Protein Sequences
String on an alphabet of 4 or 20 characters
Gene-Order Data


Simple Rearrangements

Rearrangement Phylogeny



Median Problem
Goal: find M so that DAM+DBM+DCM is minimized
NP hard for most metric distances

Binary Encoding

Biased Model
Model of evolution:Duplications, insertions and deletions of syntenic blocksRearrangements: inversions, translocations, fusions, fissions
Binary sequences: 1(presence) vs. 0(absence)Adjacency: Pr (1 ->0) vs. Pr (0 -> 1)Gene content: Pr (1 -> 0) vs. Pr (0 -> 1)
Strong bias:Pr (1 ->0) >> Pr (0 ->1) for adjacencyLose an existing adjacency: Pr (1->0) 1/O(n)Gain a new adjacency: Pr (0 -> 1) 1/O(n2)
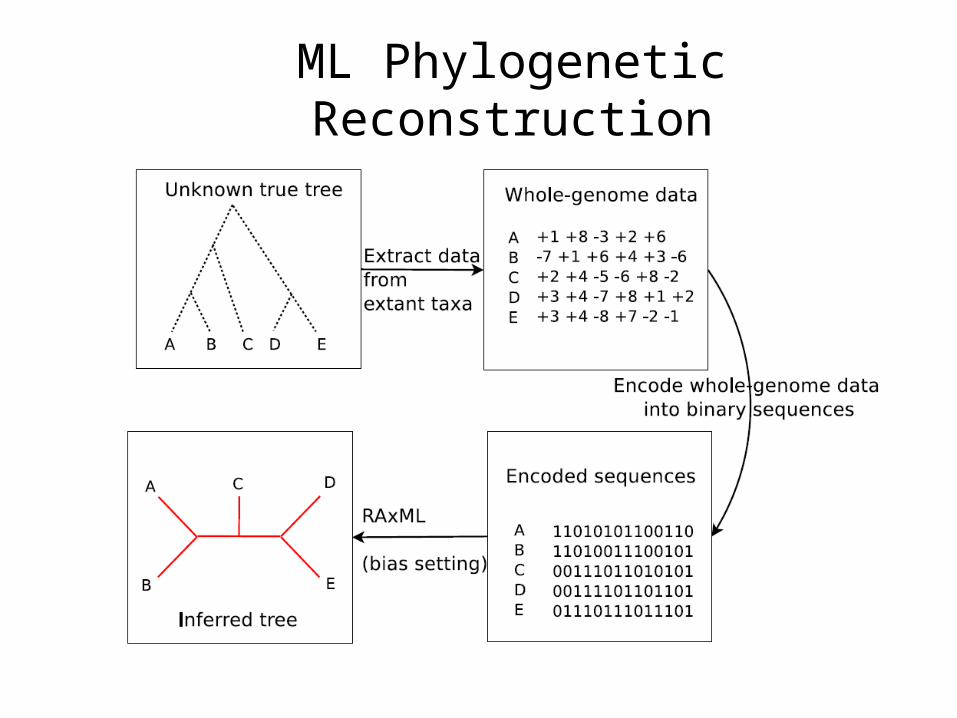
ML Phylogenetic Reconstruction

Simulated Results

Ancestral Inference
Step 1. Encoding gene orders into binary sequences.
Step 2. Setup the biased transition model.
Step 3. Arrange target ancestor to the root, and calculate the probabilities of character states for each character in the root.
Step 4. Building the adjacency graph and use a greedy heuristic to assemble adjacencies into valid gene order for the target ancestor.

Probabilities are calculated with a bottom-up recursive manner, so the target ancestor is placed to the root to prevent information loss.
Step 3 – Root Tree

Likelihood of a tree given sequence data at leaves can be computed (Felsenstein1981)
X Y ZW 1 1 00
01 01
01
X Y ZWPick one
tree
Pick one
site
Step 3 –Probabilities of Adjacencies

Posterior probabilities of character states (0 and 1) can be calculated according to Yang (Yang1995).
This is calculated by summing over all other ancestral states except root
8 histories8 histories 4 histories +4 histories +4 histories4 histories
Step 3 –Probabilities of Adjacencies

Independent adjacencies are assembled into valid gene order permutations by a greedy heuristic proposed by Jian Ma (Ma2007).
Sort the edges by weight.
Add the current heaviest edge to the path until a cycle is formed, then repeat the process until all vertices are traversed.
Remove the lightest edge in each cycle.
(1 -4 -3 5 2)(1 -4 -3 5 2)
Step 4 – Assemble Adjacencies

Transition model and reroot procedure are necessary
Simulation Result

PMAG was compared with InferCarsPro (Ma2011) and GRAPPA_DCJ(Xu2008)
Results-2

Tests on Large Scale Dataset

ML on Binary Encoding is more accurate and thousands of times faster than other methods
Binary encoding reduces the complexity and allows us to using existing methods for sequence data
Biased transition model and rerooting procedure are very useful
Future work:Extend PMAG to handle a more general model of evolution, including gene indel and duplication
Missing Adjacencies?
Conclusions

Thank You!