biopharmacy and pharmacokinetics ciobanu cristina, dr
TRANSCRIPT

BIOPHARMACY AND PHARMACOKINETICS Ciobanu Cristina,
Dr., Associate professor
LECTURE 5 :

DRUG ABSORPTION
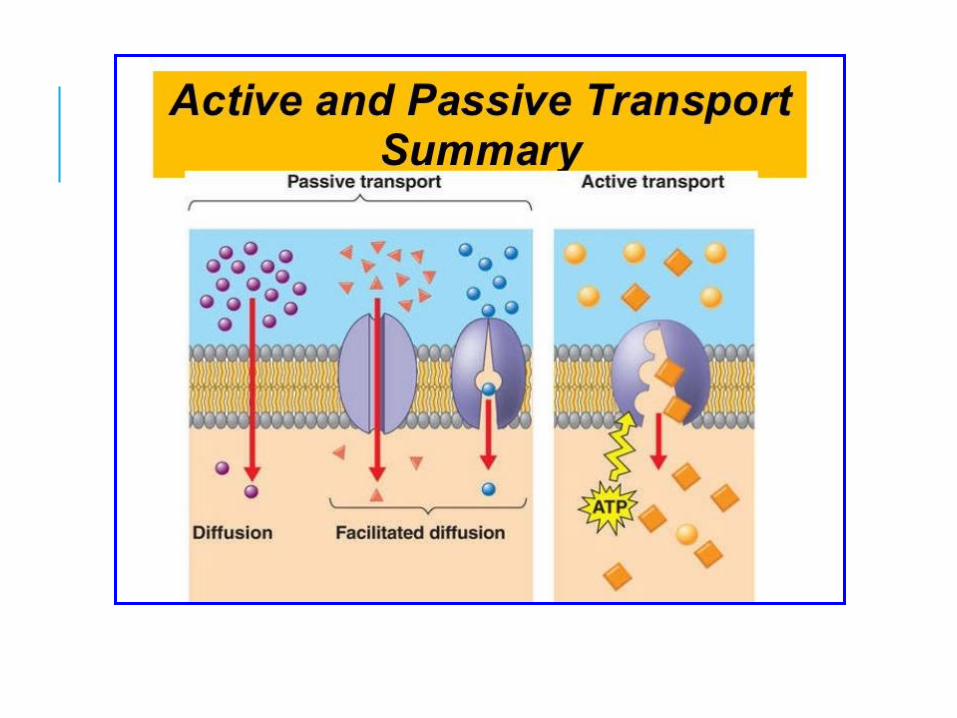
DRUG ABSORPTION is the movement of a drug from its site of application into the bloodstream. Unless a drug is directly applied to, or in the vicinity of, the target site, absorption must occur for a drug to exert its therapeutic effect.
For drugs applied at their target, such as local anesthetics, absorption often terminates the therapeutic effect. As drug absorption usually occurs by passive diffusion across membranes, the basic principles governing absorption are similar to those governing distribution.
Because biological membranes contain a lipid core, they represent a lipophilic environment. Consequently, the rate at which drugs diffuse through membranes is directly related to their relative degree of lipid solubility. Thus, lipid soluble agents usually pass readily through membranes, and more water-soluble drugs do so more slowly, if at all. As a result, lipid soluble drugs tend to be absorbed more quickly than water-soluble agents.

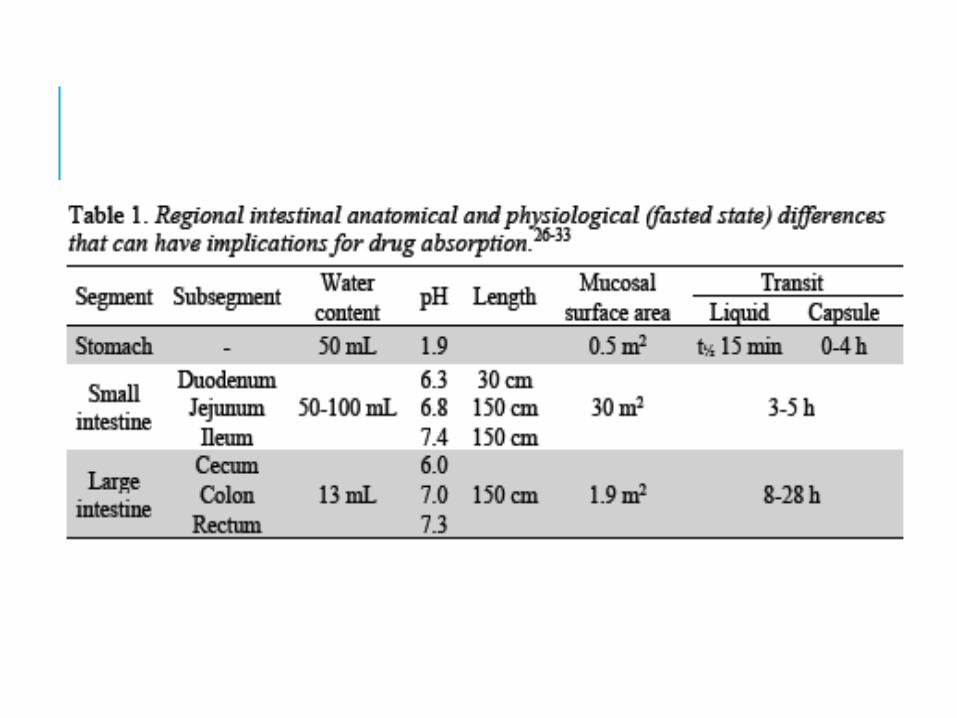
Oral drug absorption is the movement of the drug from its site of administration, gastrointestinal (GI), into the bloodstream.
The oral absorption of the drug in solid dosage form from the GI is largely controlled by (1) dissolution rate and solubility, which determine how fast a drug reaches maximum concentration in the GI fluid; and (2) intestinal permeability, which relates to the rate at which a dissolved drug crosses the intestinal wall to reach the portal blood circulation.
The determination of the dissolution, solubility, and permeability properties of drugs can thus provide information about drug absorption.

29.09.2017 LECTORI-PROFESOR UNIV. EUGEN DIUG; DR. CRISTINA CIOBANU
Biofarmacie șI farmacocinetică


Functional anatomy of the GI tract
The GI tract is a continuous, muscular and hollow tube that stretches from the mouth to the anus.
It is divided into regions that each have their own morphology, physiology, and function. First are the pharynx and esophagus, followed by the stomach, small intestine, and large intestine.
The small intestine is in turn subdivided into the duodenum, jejunum, and ileum, and the large intestine into the colon and rectum.
The total length of the human intestines during normal muscle tonus is about 5 meters, but it can double in length post mortem

SUBLINGUAL AND BUCCAL MEDICATION ADMINISTRATION
Sublingual and buccal medication administration are two different ways of giving medication by mouth. Sublingual administration involves placing a drug under your tongue to dissolve and absorb into your blood through the tissue there.
Buccal administration involves placing a drug between your gums and cheek, where it also dissolves and is absorbed into your blood. Both sublingual and buccal drugs come in tablets, films, or sprays.
The oral mucosa has a thin epithelium and rich
vascularity, which favor absorption; however, contact
is usually too brief for substantial absorption. A drug
placed between the gums and cheek (buccal
administration) or under the tongue (sublingual
administration) is retained longer, enhancing
absorption.

Buccal administration drugs under any of the following circumstances:
the drug needs to get into your system quickly
you have trouble swallowing medication
the medication doesn’t absorb very well in the stomach
the effects of the drug would be decreased by digestion
The cheek and area under the tongue have many capillaries, or tiny blood vessels. There, drugs can be absorbed directly into the bloodstream without going through your digestive system.

ADVANTAGES
Sublingual or buccal forms of drugs have their advantages. Because the medication absorbs quickly, these types of administration can be important during emergencies when you need the drug to work right away, such as during a heart attack.
Further, these drugs do not go through the digestive system, so they aren’t metabolized through your liver. This means you may be able to take a lower dose and still get the same results.
Another advantage is that you don’t have to swallow the drug. Drugs that are absorbed under the tongue or between the cheek and gum can be easier to take for people who have problems swallowing pills.

DISADVANTAGES
On the other hand, sublingual and buccal drugs also have some disadvantages.
Eating, drinking, or smoking, can affect how the drug is absorbed and how well it works.
Also, these forms don’t work for drugs that need to be processed slowly by your system, such as extended-release formulations.
Any open sores in your mouth can also become irritated by the medication.

Isosorbide concentrations after a 5 mg oral or sublingual dose.Data from: Assinder et al. J Pharm Sci 66:775, 1977.
Oral
Sublingual
0
2
4
6
8
10
12
14
5 15 30 45 60 90 120
Time (min)
Iso
so
rbid
e C
on
c (
ng
/ml)

Stomachabsorption
The stomach has a relatively large epithelial surface, but its thick mucous layer and short transit time limit absorption.
Because most absorption occurs in the small intestine, gastric emptying is often the rate-limiting step. Food, especially fatty food, slows gastric emptying (and rate of drug absorption), explaining why taking some drugs on an empty stomach speeds absorption.
Drugs that affect gastric emptying (eg, parasympatholytic drugs) affect the absorption rate of other drugs. Food may enhance the extent of absorption for poorly soluble drugs (eg, griseofulvin), reduce it for drugs degraded in the stomach (eg, penicillin G), or have little or no effect.

BALANCING THE IONIZED (I) AND NON -IONIZING (NI) FORMS OF ACETYLSALICYLIC ACID (ACIDIC CHARACTER; PKA = 3.5) IN THE STOMACH AND BLOOD AND CROSSING THE BIOLOGICAL MEMBRANE
0,99%
COO−
OCOCH3
99,01%
COOH
OCOCH3
99,98%
COO−
OCOCH3
0,02%
COOH
OCOCH3
pH=7,4 pH=1,5 StomacSânge
NINI
I I
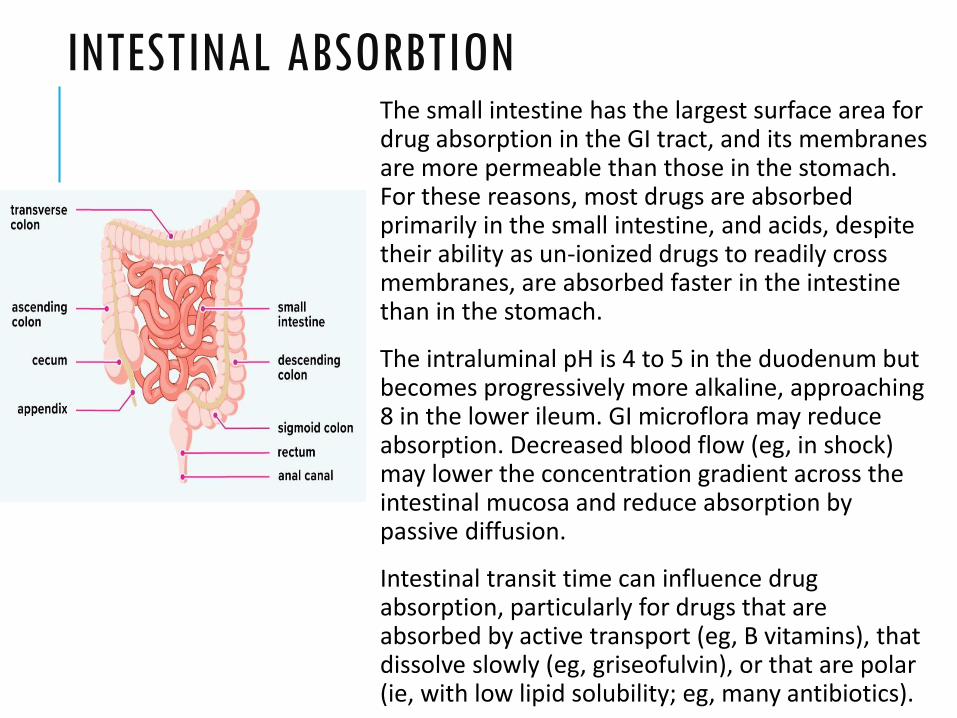
INTESTINAL ABSORBTIONThe small intestine has the largest surface area for drug absorption in the GI tract, and its membranes are more permeable than those in the stomach. For these reasons, most drugs are absorbed primarily in the small intestine, and acids, despite their ability as un-ionized drugs to readily cross membranes, are absorbed faster in the intestine than in the stomach.
The intraluminal pH is 4 to 5 in the duodenum but becomes progressively more alkaline, approaching 8 in the lower ileum. GI microflora may reduce absorption. Decreased blood flow (eg, in shock) may lower the concentration gradient across the intestinal mucosa and reduce absorption by passive diffusion.
Intestinal transit time can influence drug absorption, particularly for drugs that are absorbed by active transport (eg, B vitamins), that dissolve slowly (eg, griseofulvin), or that are polar (ie, with low lipid solubility; eg, many antibiotics).

Celuleabsorbante
Lumen
Lamina propria
Celuleendocrine Mucoasa musculară
Penel demarcator
Microvili


The tablet in the
stomachThe tablet at different stages in the intestine

ANATOMICAL AND PHYSIOLOGICAL FEATURES OF THE RECTUM
The human rectum is the terminal of GIT.
It is 10 – 15 cm long slightly dilated part of thelarge intestine.
In the resting position the rectum does not haveany active motility.
Normally the rectum is empty & contains 2-3 mlof inert mucus fluid. (pH 7-8), which is secretedby the goblet cells forming simple tubular glandsin mucosal layer.
This mucus has no enzymatic activity or bufferingcapacity. There are no villi or microvilli on therectal mucosa & thus a very limited surface area(200 – 400 cm 2) is available for absorption.
And this surface area is sufficient to absorbdrug,

PHISIOLOGY OF HUMAN RECTUM:
Fate of drug absorbed from rectum isdepends upon the position of it in therectum.
Both blood & lymphatic vessels areabundant in the sub mucosal regionof rectal wall.
Upper hemorrhoidal vein drains intothe portal circulation, so the drugabsorbed in the upper region willpass through the liver beforeentering the systemic circulation.
While the lower & middlehamorrhoidal veins drain directlyinto the inferior vena cava.
So the drugs absorbed in the lowerregion of the rectum will directlyenter into the systemic circulation.

PHARMACOLOGICAL USES:
1) LOCAL ACTION
Rectal suppositories intended for localized action are most frequently used to relieve constipation or pain, irritation, itching, and inflammation associated with hemorrhoids or other anorectal conditions
Ex: tetracycline, hydrocortisone, novocaine, anaesthesine, benzocaine, dermatole, propolis, zinc oxide etc.
A popular laxative, glycerin suppositories promote laxation by the local irritation of the mucous membranes.
2) SYSTEMIC ACTION
For systemic effects, the mucous membranes of the rectum and vagina permit the absorption of many soluble drugs.
Although the rectum is used frequently as the site for the systemic absorption of drugs
Ex: paracetamol, diclofenac, indomethacin, pentoxil, drotaverine.

PARENTERAL DRUG ADMINISTRATION
A parenteral dosage form can be defined as a sterile drug product, which is presented in the form of solution, suspension, emulsion, or reconstituted lyophilized powder, suitable for administration by injection. Typical routes of administration of a parenteral dosage form include subcutaneous, intramuscular, and intravenous delivery. Occasionally, parenteral dosage forms can also be administered via intrathecal, intracisternal, intraarterial, intraspinal, intraepidural, and intradermal routes to achieve local or systemic effects. Although a major drawback of parenteral delivery is the pain and discomfort associated with needle injection, significant usage has been maintained in hospital environments. This is due to the unique attributes of the parenteral dosage form: rapid absorption and distribution, high bioavailability, zero enzymatic degradation in the gastrointestinal tract, and an ability to be administered to unconscious patients.


Advantages
Can be used for drugs that are poorly absorbed, inactive or ineffective if given orally
The IV route provides immediate onset of action
The intramuscular and subcutaneous routes can be used to achieve slow or delayed onset of action
Patient concordance problems can be avoided
Disadvantages
Staff need additional training and assessment
Can be costly
Can be painful
Aseptic technique is required
May require additional equipment, for example programmable infusion devices

Intramuscular and subcutaneousIn general, IM and SC injection of drugs establishes a deposit or “depot” that will
be released gradually into the systemic circulation. The drug’s formulation will
influence the period over which it is released; for example, the formulation of
antipsychotic agents such as flupentixol in oil allows them to be administered once
a month or every three months.
Intravenous
The IV route carries the greatest risk of any route of drug administration. By
administering directly into the systemic circulation, either by direct injection or
infusion, the drug is instantaneously distributed to its sites of action. This route of
administration can be complex and confusing. It may require dose calculations,
dilutions, information to be gathered on administration rates and compatibilities
with other IV solutions, as well as the use of programmable infusion devices.
The preparation of IV medicines requires the use of an aseptic technique, often in a
ward environment that is unsuited to such work. To minimise the risk of errors, it is
imperative that practitioners can demonstrate competence to practise safely, and
have access to expert information and advice.


29.09.2017


OPHTHALMIC ADMINISTRATION

Ophthalmic drug forms have been one of the most important and widely developed areas of pharmaceutical technology for dozens of years.
The main reason of continuingly strong interest of scientists in these drug forms is the problem of a low bioavailability of medicinal substance after the application to the eyeball. It is caused by, amongst other reasons, the complicated anatomical structure of the eye, small absorptive surface and low transparency of the cornea, lipophilicity of corneal epithelium, metabolism, enzymolysis, bonding of the drug with proteins contained in tear fluid, and defence mechanisms, that is, tear formation, blinking, and flow of the substance through nasolacrimal duct.
Low capacity of conjunctival sac, that is, approximately 30 L without blinking, and the aforementioned defence mechanisms cause decrease in drug concentration in the place of application and shorten the time during which the active ingredient stays in the place of absorption. T
he primary purpose for the development of ophthalmic drug forms is to achieve the required drug concentration in the place of absorption and sustaining it for appropriately long time, which in turn contributes to smaller application frequency

TOPICAL OPHTHALMIC DRUG FORMS
Liquid Ophthalmic Drug Forms
Eye Drops
Ophthalmic Solutions
Microemulsions
Semisolid Ophthalmic Drug Forms
In Situ Gels (or Sol-to-Gel Systems)
Eye Ointments
Solid Ophthalmic Drug Forms
Contact Lenses Coated with Drugs
Ocular Inserts
Minidiscs/OTS (Ocular Therapeutic System)
NODS (New Ophthalmic Delivery System)
Minitablets

NASAL ADMINISTRATIONAdvantages with nasal systemic drug delivery
The nasal cavity is covered by a thin mucosa which is well vascularised.[1] Therefore, a drug molecule can be transferred quickly across the single epithelial cell layer directly to the systemic blood circulation without first-pass hepatic and intestinal metabolism. The effect is often reached within 5 min for smaller drug molecules.
Nasal administration can therefore be used as an alternative to oral administration, by crushing or grinding tablets or capsules and snorting or sniffing the resulting powder, providing a rapid onset of effects. If a fast effect is desired or if the drug is extensively degraded in the gut or liver,drugs which are poorly absorbed orally can also be given by this route.
Limitations with nasal systemic drug delivery
Nasal administration is primarily suitable for potent drugs since only a limited volume can be sprayed into the nasal cavity. Drugs for continuous and frequent administration may be less suitable because of the risk of harmful long-term effects on the nasal epithelium.
Nasal administration has also been associated with a high variability in the amount of drug absorbed. Upper airway infections may increase the variability as may the extent of sensory irritation of the nasal mucosa, differences in the amount of liquid spray that is swallowed and not kept in the nasal cavity and differences in the spray actuation process. However, the variability in the amount absorbed after nasal administration should be comparable to that after oral administration.


Vaginal absorbtion
Introduction : The vagina, in addition to being a genital organ with functions related to conception it serves as a potential route for drug administration.
Mainly used for local action in the vaginal region. It has the potential of delivering drugs for systemic effects and uterine targeting
Anatomy of Vagina human vagina is often described as slightly S-shaped fibromuscular tubes between 6-10 cm long and 2 cm wide extending from cervix of the uterus to the vestibule.
The vaginal wall consists of three layers: the epithelial layer, the muscular coat and the tunica adventia
Normal pH of the vagina in premenopausal women ranges from 4 to 5, and rises to almost 7 in the post menopausal female.

Types of vaginal dosage forms vaginal semisolids: Creams Gels Ointments Suppositories vaginal liquids : Solutions Suspensions vaginal aerosols vaginal controlled release formulations Vaginal rings
Advantages of vaginal drug delivery
Vaginal administration permits use of prolonged dosing, with continuous release of medicaments. Avoiding the fluctuations resulting from daily intake may also lower the incidence of side effects. Alleviating the inconvenience caused by pain, tissue damage and probable infection, it serves as a better alternative to parenteral route. Vaginal route can be used for local as well as systemic effect
Advantages of vaginal drug delivery avoidance of first pass effect , avoidance of enzymatic deactivation in GIT, large permeation area ,rich vascularization and relatively low enzymatic activity. Non-invasive, self-insertion and removal of the dosage form like vaginal films, gel, pessaries, etc.
Disadvantages of vaginal drug delivery cultural sensitivity personal hygiene gender specificity local irritation influence of sexual intercourse Menstrual cycle-associated vaginal changes Sometimes leakage of drugs from vagina and wetting of under garments

Factors affecting the vaginal absorption of drugs
1- Physiological factors - cyclic changes affect in thickness of vaginal epithelium fluid volume, viscosity and composition , pH , sexual arousal
Effect of pH changes on absorption pH may change degree of ionization of drug and affect their the absorption
2- Physicochemical properties of drugs Molecular weight , lipophilicity ,ionization surface charge , chemical nature Ex : low molecular weight lipophilic drugs are likely to be absorbed more than large molecular weight lipophilic or hydrophilic drugs.
3- Vaginal enzymes - proteases are likely to be the prominent barrier for the absorption of intact peptide and protein drugs into the systemic circulation The human genital tract has lower enzymatic activity leading to less degradation of protein and peptide drugs in the vagina than the gastrointestinal tract sheep > guinea pig > rabbit ≥ human ≥ rat

The uterus is located within the pelvic region immediately behind and almost overlying the bladder, and in front of the sigmoid colon. The human uterus is pear-shaped and about 7.6 cm (3.0 in) long, 4.5 cm (1.8 in) broad (side to side), and 3.0 cm (1.2 in) thick.
• Mirena is a hormonal intrauterine
device classified as a long-acting
reversible contraceptive method.
• T-shaped polyethylene frame (T-
body) with a steroid reservoir
(hormone elastomer core) made of a
mixture of levonorgestrel and silicone
(polydimethylsiloxane), containing a
total of 52 mg levonorgestrel around
the vertical stem.
• The device releases the hormone at
an initial rate of 20 μg/day and
declines to a rate of 14 μg after 5
years, which is still in the range of
clinical effectiveness.