bleeding disorders vascular and platelet disorders ahmad shihada silmi msc,fibms iug medical...
Post on 22-Dec-2015
228 views
TRANSCRIPT

Bleeding DisordersVascular and Platelet Disorders
Ahmad Shihada Silmi Msc,FIBMS
IUG Medical Technology Dept

Bleeding Disorder Terms
Petechiae-pinpoint size/pinhead size hemorrhagic spots in skin Purpura-hemorrhage under the skin, varying in color and duration Ecchymosis-purplish patch caused by extravasation of blood into skin,
larger than petechiae Epistaxis-nosebleed Menorrhagia-excessive menses Hematuria-blood in urine Hemarthrosis-bleeding into joint Hematemesis-vomiting blood Hemoptysis-spitting blood Melena-blood in stool (occult blood)


Bleeding disorders
Vascular abnormalities
Platelet disordersClotting factorabnormalities
DIC

Vessel Defects Causing Bleeding
Begins with bleeding episode in presence of normal lab tests for coagulation function
Types divided into hereditary and acquired Symptoms are usually of the superficial ones. Usually these diseases are diagnosed by exclusion.
After ruling out PLT disorders, coagulation or fibrinolytic disorders in a patient who has bleeding symptoms.


Vascular Diseases
PLT count and screening tests for coagulation factors are usually normal.
PLT function tests such as bleeding time and other PLT function tests are also normal, but BT may be prolonged in some vascular diseases.

They are very rare, and while bleeding is a common symptom, hemostasis tests are NOT necessary for diagnosis.
Inherited vascular disorders

Hereditary Connective Tissue Defects
Defect affects ability to support vessel walls Examples
– Ehlers-Danlos Syndrome Lack of structural tissue support (collagen disorders) Skin elasticity and fragility. Hypermobility of joints Evidenced by bleeding/bruising Recurrent joint problems & scarring of the face. The most serious is deficient of type III collagen (blood
vessel type). Which leads to Acute & sever Internal bleeding & sudden death.

Hereditary Connective Tissue Defects
Pseudoxanthoma Elasticum– Autosomal recessive trait– Lack of skin elasticity– Some connective tissue calcified– Bleeding and bruising evident

Hereditary Vessel Disease
Hereditary Haemorrhagic telangiectasia (HHT) Inherited as autosomal dominant trait Defect of angiogenesis Involves bleeding from abnormally dilated vessels
“telangiectasias” Vessels involved do not contract normally and collapse easily Patient has pinpoint lesions (tiny areas of bleeding) Lesions occur on face, hands and feet May develop at all ages Blood loss may cause anemia Diagnosis based on physical appearance

Hereditary Vessel Disease
Kasabach-Merritt syndrome (Hemangioma)
Benign tumour of vascular tissue Grow rapidly to giant proportion. Threaten the function of neighbouring tissues. Mechanical injury may result in sever bleeding. May trigger a localized DIC with thrombocytopeia &
consumption coagulopathy, thereby worsening bleeding.
Tumor composed of many blood vessels (blood-filled)

Hereditary Vessel Disease
HHT or also called Cavernous Hemangioma Lesion may swell and bleed Tumor site may form clots, hemolyzed RBCs and
vessel obstruction Present at birth Treatment is by surgical removal, if possible,or
localized radiotherapy with injection of fibrinolytic inhibitors.

Are seen quite often.
Are characterized by bruising and petechiae
Acquired Vascular Disorders

Acquired Connective Tissue Defects
Vitamin C Deficiency (Scurvy) Caused deficient Vitamin C Vitamin C required for vessel collagen integrity Acts as “cement” holding endothelial cells together Lack of Vitamin C prevents proper collagen formation Result: bleed and vessel fragility Symptoms include gum bleeding, petechiae and
bleeding into tissues and muscles Treated with Vitamin C

Acquired Connective Tissue Defects
Senile PurpuraOccurs in elderly populationUsually benignCollagen degradation/loss affects vessel
integrityBruising on arms/handsNo treatment/therapy available

Due to abnormal proteins in the vascular system
Paraproteins are monoclonal Ig produced by a single clone of plasma cells.
Also called M component.
These paraproteins occur in MM, WM, and lymphoproliferative disorders.
Purpura due to Paraproteins

Symptoms: purpura, bleeding and thrombosis. The defect is related to multifactors:
– Qualitative PLT defects.– Acquired inhibitors.– Deficiency in coagulation factors.– Paraproteins binds Ca, Ca either will not be
available for coagulation or the Ca bounded paraproteins will interfere with coagulation.
– Thrombocytopenia.
Purpura due to Paraproteins
Cont’d

Bleeding symptoms include:– Epistaxis– Petechiae,– Purpura, and– Retinal bleeding.
Purpura due to Paraproteins
Cont’d

Amyloidosis Occur as primary disease or is associated
with paraproteinemias. Deposition of amyloid on skin and
vascular walls. Leads to fragility of vessel walls.
Purpura due to Paraproteins

Other Vascular Disorders
Self-produced “autoantibodies” damage to vessels Caused by drugs resulting in purpura Caused by allergic/immune disturbance
Evidenced by swelling, ulcers, purpura and lesions and other symptoms
Affects children Other allergic purpura-Hemoch-Schonlein variety
Accompanied by joint and abdominal pain Avoidance of allergen aids recovery

Other Vascular Disorders
Infectious purpura Observe petechiae and purpura Results from
Inflammatory response to agent Autoimmune/autoantibody response Bacterial products or toxins Injury caused by agent
Low platelets observed and DIC Cure is to treat infection

Bleeding disorders
Vascular abnormalities
***Platelet disorders
Clotting factorabnormalities
DIC

Platelet Disorders Classification:
Thrombocytosis
Thrombocytopathy
Thrombocytopenia
PLT Reference Range = 150 - 450 x109/L
Qualitative PLT Disorders
Quantitative PLT Disorders

Thrombocytosis
Thrombocytosis resulting from myeloproliferation– essential thrombocythemia– polycythemia vera– chronic myelogenous leukemia– myeloid metaplasia
Secondary (reactive) thrombocytosis– systemic inflammation– malignancy– iron deficiency– hemorrhage– postsplenectomy

Thrombocytopenia
Reduced platelet count.
The most common cause of excess or abnormal bleeding.
As anemia is a symptom of a disease as we have seen in Hematology #1, thrombocytopenia is also a symptom of a disease!

Do not forget to assure that the case you have is a real thrombocytopenia
and not PseudothrombocytopeniaPlatelet
clumping, or aggregation
Platelet Satelltism
Pseudothrombocytopenia

Platelet countSymptoms
50-100 X109/LProlonged bleeding following trauma
< 50X109/LEasy bruising
Purpura following minor trauma
< 20 X109/LSpontaneous bleeding
Petechiae
May suffer spontaneous internal and intracranial bleeding
Thrombocytopenia

Hemostatic Level
Hemostatic Platelet count level is more than 50 x109/L.
This means that normal hemostasis may occur ≥ 50 x109/L

Classification: platelet disorders

Thrombocytopenia
Causes
Impaired or DecreasedProduction
Distribution/Dilution Disorders
Increased Destruction
Megakaryocyte aplasia
BM Replacement
Ineffective poiesis
Immune Non-Immune

General characteristics of platelet disorders
characterized by variable mucocutaneous bleeding manifestations.
excessive hemorrhage may follow surgical procedures or trauma.
Platelet counts and morphology are normal. prolonged bleeding time, Abnormal Platelet aggregation and secretion
studies

Thrombocytopenia
Usually mucosal bleeding Epistaxis, menorrhagia, and GI bleeding is
common Trauma does not usually cause bleeding

Thrombocytopenia
Three mechanisms of Thrombocytopenia– Decreased production
Usually chemotherapy, myelophthisic disease, or BM effects of alcohol or thiazides
– Splenic Sequesteration Rare Results from malignancy, portal hypertension, or
increased Splenic RBC destruction ( hereditary spherocytosis, autoimmune hemolytic anemia)
– Increased Destruction

Thrombocytopenia
Immune thrombocytopenia– Multiple causes including drugs, lymphoma, leukemia,
collagen vascular disease– Drugs Include
Digitoxin, sulfonamindes, phenytoin, heparin, ASA, cocaine, Quinine, quinidine, glycoprotein IIb-IIa antagonists
– After stopping drugs platelet counts usually improve over 3 to 7 days
– Prednisone (1mg/kg) with rapid taper can shorten course

Thrombocytopenia
HIT– Important Immunologic Thrombocytopenia– Usually within 5-7 days of Initiation of Heparin
Therapy but late onset cases are 14-40 days– Occurrence 1-5% with unfractionated heparin
and less than 1% with low molecular-weight heparin
– Thrombotic complications in up to 50% of HIT with loss of limb in 20% and mortality up to 30%

ITP
Diagnosis of exclusion Associated with IgG anti-platelet antibody Platelet count falls to less that 20,000
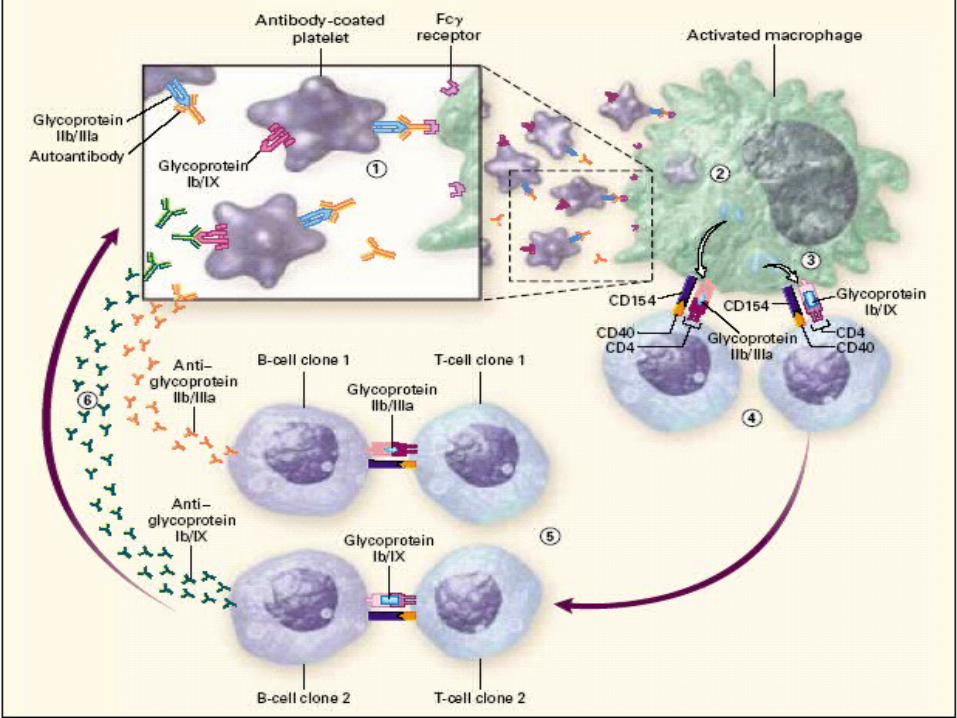

ITP
Acute Form– Most common in children 2 to 6 years– Viral Prodrome common in the 3 weeks prior– Self Limited and > 90% remission rate– Supportive Treatment– Steroids are not helpful

ITP
Chronic Form– Adult disease primarily– Women more often than men– Insidious onset with no prodrome– Symptoms include: easy bruising, prolonged
menses, mucosal bleeding– Bleeding complications are unpredictable– Mortality is 1%– Spontaneous remission is rare

ITP
Chronic Form– Hospitalization common because of a complex
differential diagnosis– Multiple treatments– Platelet transfusions are used only for life
threatening bleeding– Life threatening bleeding is treated with IV
Immune globulin (1g/kg)

TTPHUS
Exist on a continuum and are likely the same disease
Diagnosed by a common pentad– Microangiopathic Hemolytic Anemia: Schistocytes
membranes are sheared passing through microthrombi– Thrombocytopenia: More sever in TTP– Fever– Renal Abnormalities: More prominent in HUS: include Renal
insufficiency, azotemia, proteinuria, hematuria, and renal failure
– Neurologic Abnormalities: hallmark of TTP 1/3 of HUS: Sx of HA, confusion, CN palsies, seizure,coma

TTPHUS
Labs– PT, PTT, and fibrinogen are within reference
range– Helmet Cells (Shistocytes) are common

TTPHUS
HUS– Most common in infants and children 6mo - 4
years– Often associated with a prodromal diarrhea– Strongest association to E. coli O157:H7 but also
associated with SSYC as well as multiple virus– Prognosis
Mortality 5-15% Younger patients do better

TTPHUS
HUS– Treatment
Mostly supportive Plasma exchange reserved for sever cases Treat hyperkalemia Avoid antibiotics with Ecoli
– May actually increase verotoxin production.– May be helpful with cases of Shigella dysenteriae

TTPHUS
TTP– More common in adults– Untreated mortality rate of 80% 1 to 3 months
after diagnosis– Aggressive plasma exchange has dropped the
mortality to 17%– Splenectomy, immune globulin, vincristine all play
a role in therapy

TTPHUS
AVOID PLATELET TRANSFUSION– May lead to additional microthrombi in circulation– Transfuse only with life threatening bleeding

Dilutional Thrombocytopenia
PRBC are platelet poor Monitor platelet count with every 10 u PRBC (for each 8-10 units of PRBC, 2 units of FFP
& 5 units of platelet concentrate are given) Transfuse when count below 50,000 Get them upstairs before you transfuse 10
units PRBC

Bleeding Clinical Correlation - PLT Numbers
PLT CountSpontaneous BleedPost Trauma Bleed
>50K/lNoRare
30 – 50K/lRareOccasional
10 – 30K/lOccasionalAlways
10K/lFrequentAlways
BT prolongation proportional to PLT count IF no complicating factors.

Significant Lab Data in Defects of Primary Hemostasis
TestVascular DisorderThrombocytopeniaPLT Dysfunction
PLT CountNDN
PTNNN
APTTNNN
BTN or ABNABNN or ABN


Hereditary Platelet Function DefectsHereditary Platelet Function Defects
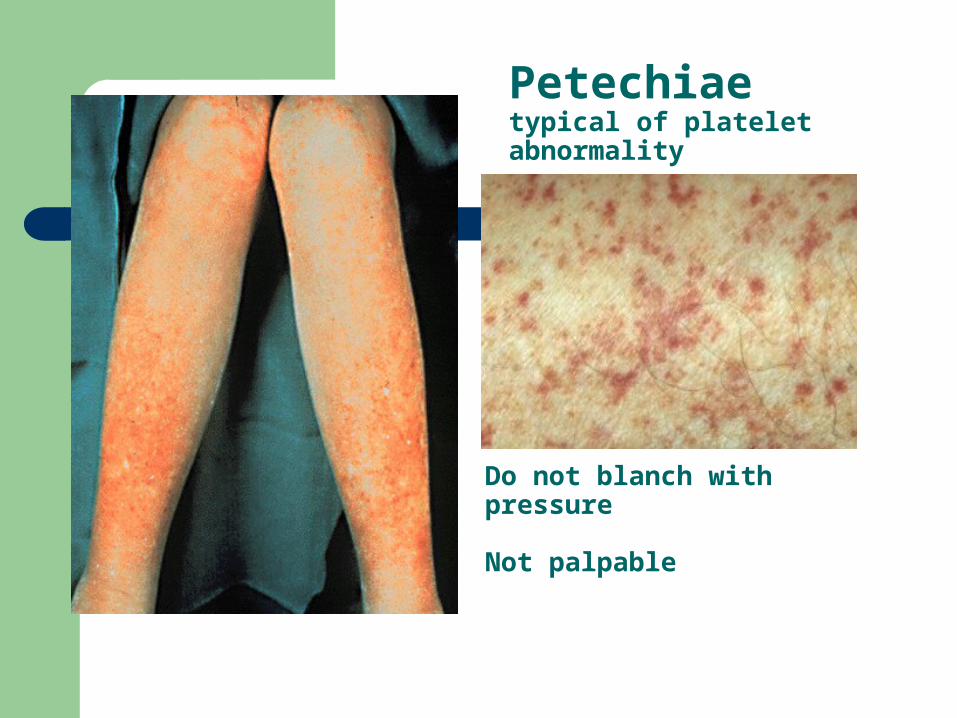
Petechiaetypical of platelet abnormality
Do not blanch with pressure Not palpable


Bernard-Soulier Syndrome
First described in 1948 by Jean Bernard and Jean-Pierre Soulier; French hematologists
Bernard J, Soulier JP: Sur une nouvelle variete de dystrophie thrombocytaire hemarroagipare congenitale. Sem Hop Paris 24:3217, 1948
AR; characterized by moderate to severe thrombocytopenia, giant platelets, and perfuse/spontaneous bleeding
Basis for the disease is deficiency or dysfunction of the GP Ib-V-IX complex

Bernard-Soulier Syndrome
Decreased GP Ib-V-IX leads to decreased platelet Decreased GP Ib-V-IX leads to decreased platelet adhesion to the subendothelium via decreased binding of adhesion to the subendothelium via decreased binding of vWFvWF
Approximately 20,000 copies of GP Ib-V-IX per plateletApproximately 20,000 copies of GP Ib-V-IX per platelet GP 1b: heterodimer with an alpha and beta subunitGP 1b: heterodimer with an alpha and beta subunit The gene for GP Ib alpha is located on chromosome 17; The gene for GP Ib alpha is located on chromosome 17;
GP Ib beta: chromosome 22; GPIX and V: chromosome 3GP Ib beta: chromosome 22; GPIX and V: chromosome 3 Most mutations are missense or frameshifts resulting in Most mutations are missense or frameshifts resulting in
premature stop codonspremature stop codons Most mutations involve GP Ib expression (rare GP IX Most mutations involve GP Ib expression (rare GP IX
mutations have been described; no mutations in GP V)mutations have been described; no mutations in GP V)


Diagnosis
Prolonged bleeding time, thrombocytopenia Prolonged bleeding time, thrombocytopenia (plt>20 K), peripheral smear shows large (plt>20 K), peripheral smear shows large platelets (mean diameter >3.5 microns)platelets (mean diameter >3.5 microns)


Diagnosis
Platelet aggregation studies show normal aggregation in response to all agonists except Ristocetin (opposite pattern than thrombasthenia)
Flow cytometry: decreased expression of mAbs to CD 42b (GPIb), CD42a(GPIX), CD42d(GPV)

Platelet Aggregometer

Light focused on sample cuvette contained PRP
PRP stirred and recorder identified baseline – 0% transmittance
Agonist added Transmitted light changes
proportionally in response to degree PLT shape changes
Change in light transmission continuously monitored and recorded
As PLT aggregates form, recorder moves towards 100% transmittance
Abnormalities– Diminished or absent shape
change– Diminished aggregation
Graphic accessed URL http://evolvels.elsevier.com/section/default.asp?id=1138_ccalvo7_0001, 2008.
Platelet-rich plasma in an optical aggregometer. Platelet count is approximately 200 × 109/L, and platelets are maintained in suspension by a magnetic stir bar turning at 1000 rpm. (Courtesy of Kathy Jacobs, Chronolog, Inc., Havertown, Penn.)



Differentiation between vWD & BSD
Measuring the platelet aggregation response to ristocetin following the addition of PPP.
A normal aggregation response suggests the plasma defect, which is typical of vWD.
The persistence of defective response suggests the presence of a platelet defect which is typical of BSD.


Glanzmann’s ThrombastheniaGlanzmann’s Thrombasthenia
Eduard Glanzmann (1887-1959), Swiss Eduard Glanzmann (1887-1959), Swiss pediatricianpediatrician
Reported a case of a bleeding disorder starting Reported a case of a bleeding disorder starting immediately after birthimmediately after birth
W. E. Glanzmann:W. E. Glanzmann:Hereditäre hämorrhägische Hereditäre hämorrhägische Thrombasthenie. Ein Beitrag zur Pathologie Thrombasthenie. Ein Beitrag zur Pathologie der Blutplättchen.der Blutplättchen.Jahrbuch für Kinderheilkunde, 1918; 88: 1-Jahrbuch für Kinderheilkunde, 1918; 88: 1-42, 113-14142, 113-141. .


Glanzmann’sGlanzmann’s
IIbIIIa most abundant platelet surface receptor IIbIIIa most abundant platelet surface receptor (80,000 per platelet)(80,000 per platelet)
IIbIIIa complex is a Ca++ dependent heterodimerIIbIIIa complex is a Ca++ dependent heterodimer Genes for both subunits are found on Genes for both subunits are found on
Chromosome 17Chromosome 17 Disease is caused by mutations (substitution, Disease is caused by mutations (substitution,
insertion, deletion, splicing abnormalities) in insertion, deletion, splicing abnormalities) in genes encoding for IIb or IIIa resulting in genes encoding for IIb or IIIa resulting in qualitative or quantitative abnormalities of the qualitative or quantitative abnormalities of the proteinsproteins
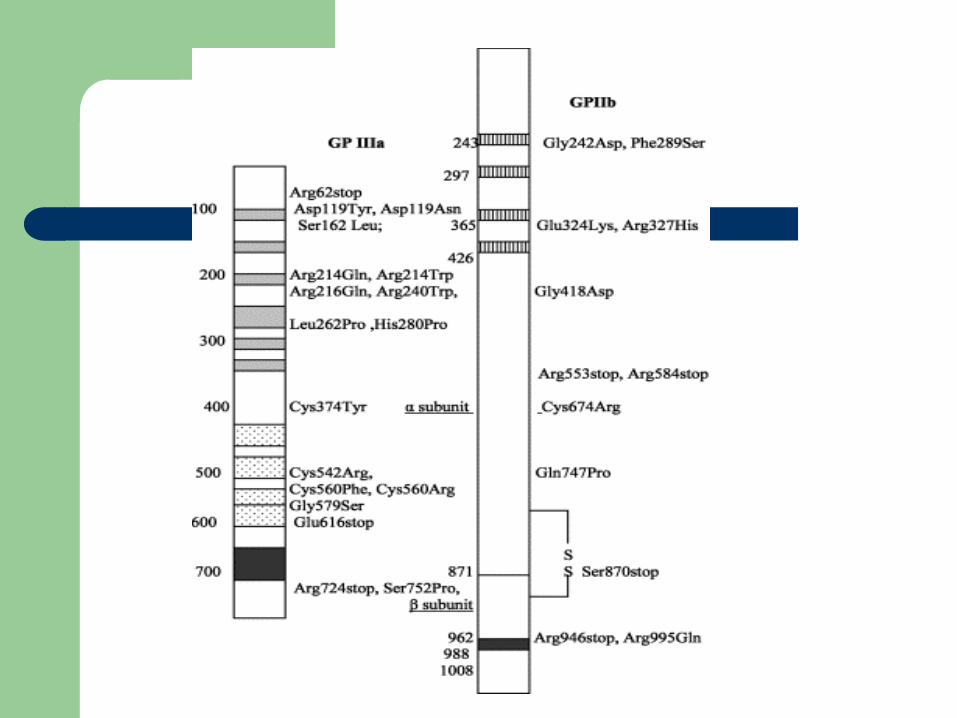

Glanzmann’sGlanzmann’s
Fundamental defect of thrombasthenic Fundamental defect of thrombasthenic patients is the inability of the platelets to patients is the inability of the platelets to aggregateaggregate
Other problems: platelets do not spread Other problems: platelets do not spread normally on the subendothelial matrix (due to normally on the subendothelial matrix (due to lack of IIbIIIa – vWF/fibronectin interaction)lack of IIbIIIa – vWF/fibronectin interaction)
Also, alpha granule fibrinogen is decreased Also, alpha granule fibrinogen is decreased to absentto absent

Glanzmann’sGlanzmann’s
AR inheritance Patients present with wide spectrum of disease Like thrombocytopenic bleeding: skin, mucous
membrane (petichiae, echymoses), recurrent epistaxis, GI hemorrhage, menorrhagia, and immediate bleeding after trauma/surgery
ICH, joint, muscle bleeding uncommon

Glanzmann’s patients are stratified into three groups based on complex expression:– Type I less than 5 percent GPIIbIIIa, absent alpha
granule fibrinogen
Usually as a result of IIb gene mutation– Type II >20 percent, fibrinogen present– Type III >50 percent; “variant” thrombasthenia;
qualitative disorder

Diagnosis
Platelet count and morphology are normal Bleeding time prolonged The hallmark of the disease is severely
reduced or absent platelet aggregation in response to multiple agonists ie ADP, thrombin, or collagen (except Ristocetin)
Flow cytometry: decreased mAb expression of CD41 (GPIIb) and CD61 (GPIIIa)


Platelet Aggregation Studies
Platelet-rich plasma (PRP) is prepared from citrated whole blood by centrifugation
Inactive platelets impart a characteristic turbidity to PRP
When platelets aggregate after injection of an agonist, the turbidity falls, and light transmission through the sample increases proportionally
The change in light transmission can be recorded on an aggregometer


Agonists
Different concentrations of each agonist are used
ADP: biphasic pattern: First wave: low concentration, reversible Second wave: high concentration, irreversible


Other agonists
Epinephrine: triphasic (resting platelets, primary aggregation, secondary aggregation)


Other agonists
Collagen, arachidonic acid, Calcium ionophore, PAF are potent agonists and induce a single wave of irreversible aggregation
Ristocetin (antibiotic): aggregation can be reproduced with metabolically inert, formalin-fixed platelets
Defective risto-induced aggregation is characteristic of Bernard-Soulier
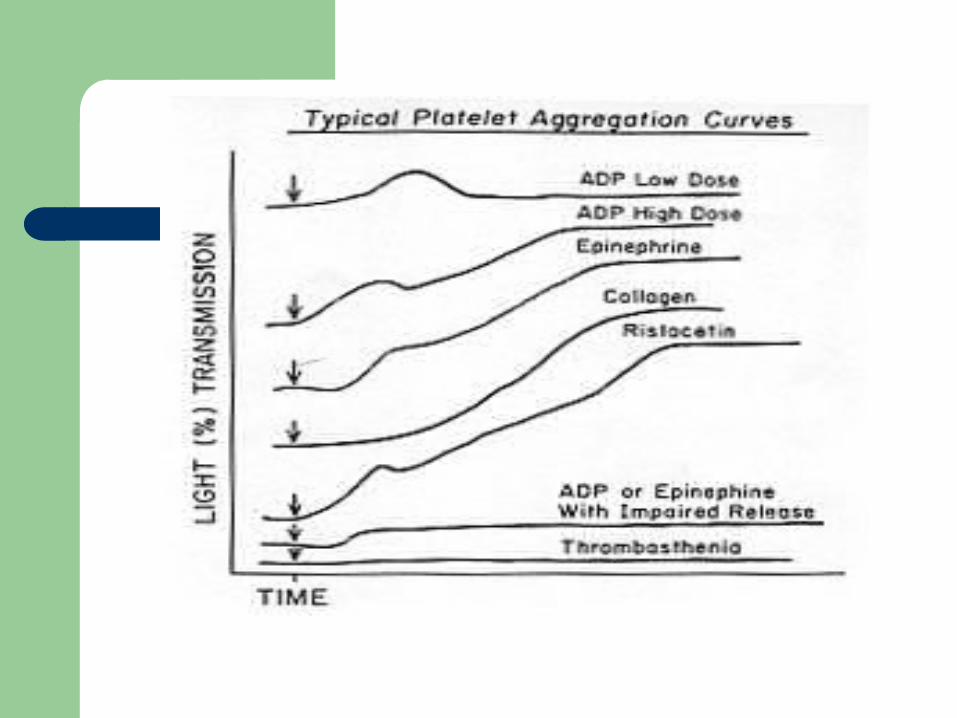

Problems with platelet aggregation Problems with platelet aggregation studiesstudies
Numerous variables affect aggregation:Anticoagulant (sodium citrate best)Plt count in PRPPlt size distributionTime of dayTemporal relation to meals and physical
activity

Storage Pool Defects
Classified by type of granular deficiency or secretion defect.
Dense body deficiency, alpha granule deficiency (gray platelet syndrome), mixed deficiency, Factor V Quebec

Defects in secondary aggregationDeficiency of contents of one of granulesInheritance is variable (heterogeneous group)Bleeding is usually mild to moderate but can
be exacerbated by aspirinClinical: easy bruising, menorrhagia, and
excessive postpartum or postoperative bleeding

Dense body deficiency
decreased dense bodies (ADP, ATP, calcium, pyrophosphate, 5HT)
Normal platelet contains 3-6, 300 micron dense bodies

Described in inherited disorders ie Hermansky-Pudlak syndrome, Wiskott-Aldrich syndrome, Chediak-Higashi syndrome, and Thrombocytopenia with absent radius (TAR) syndrome

Wiskott-Aldrich
X-linked, genetic defect in WASp (protein responsible for actin cytoskeleton formation in hematopoetic cells)
characterized by thrombocytopenia (with platelet storage pool defect), eczema, and recurrent infections


Hermansky-Pudlak
Described in 1959 by Hermansky and Pudlak AR, tyrosinase-positive oculocutaneous albinism,
ceroid-like deposition in lysosomes of the RES and marrow
Highest prevalence in Puerto Rico May be associated with pulmonary fibrosis, and
recurrent infections quantitative deficiency of dense granules leading
to mild-moderate bleeding diathesis


Chediak-Higashi
described by Beguez Cesar in 1943, Steinbrinck in 1948, Chédiak in 1952, and Higashi in 1954
AR; abnormal microtubule formation and giant lysozomal granules are present in phagocytes and melanocytes
No degranulation/chemotaxis = recurrent bacterial infections
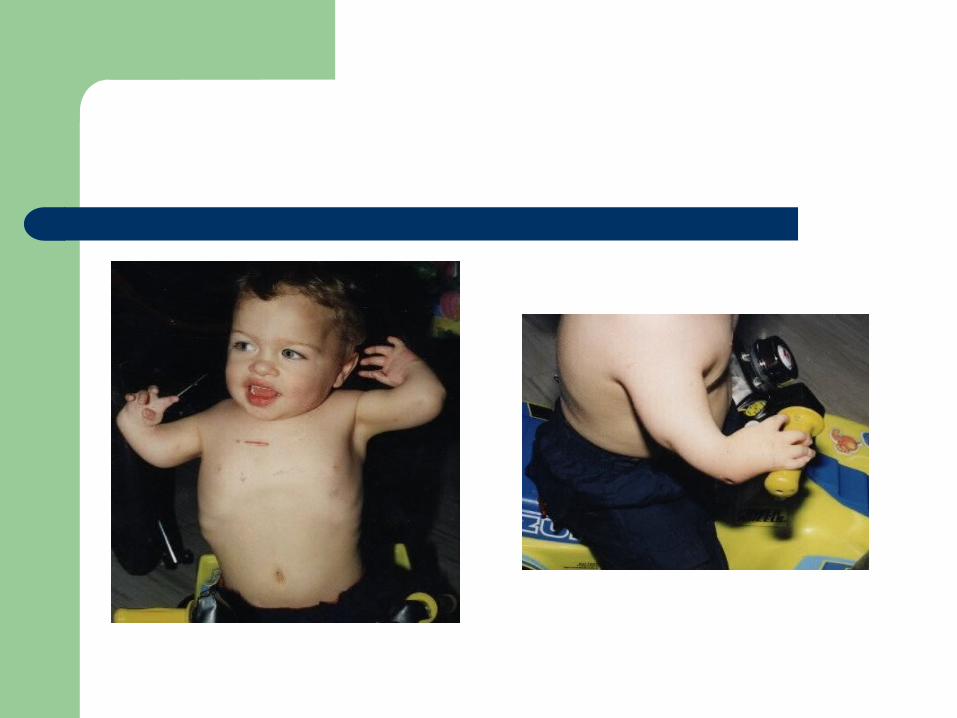
Partial oculocutaneous albinism Dense-body granules decreased/absent
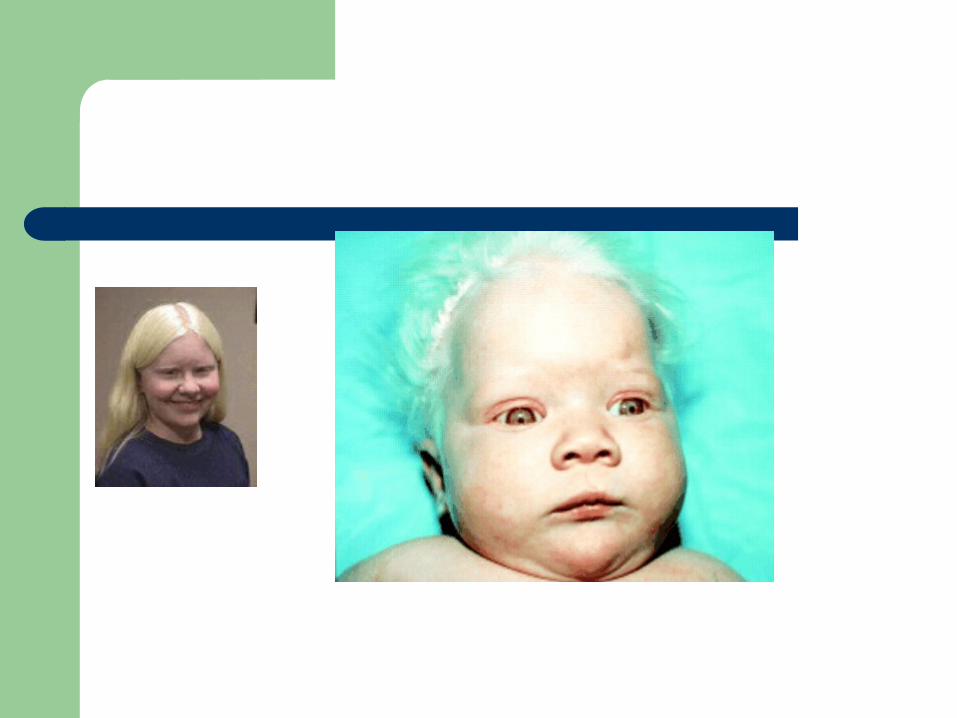


Thrombocytopenia with absent radius (TAR)
First described in 1951 AR, characterized by absent radii,
thrombocytopenia (with storage pool defect), and other abnormalities of the skeletal, GI, cardiovascular system
Etiology unclear Hemorrhage is the major cause of mortality PX is good if survive the two years

Typical Lab Findings– Usually normal platelet
count– Morphology is variable– Platelet aggregation
shows primary wave but absence of secondary wave when stimulated with ADP, epinephrine, arrachidonic acid
– Platelet aggregation with thrombin is usually normal (overrides need for granule release)
– Ristocetin agglutination is normal
Diagnosis – Measure the whole platelet
ATP/ADP ratioNormal: 2.5:1ADP storage pool deficiency
increased >3:1– Often associated with disorders
affecting granules in other cellsChediak-Higashi SyndromeHermansky-Pudlak SyndromeWiskott-Aldrich Syndrome
– Platelets are small with decreased number of both alpha and dense granules

Diagnosis
Platelet aggregation studies may show diminished response to low concentration collagen
ADP and epinephrine show diminished second wave response
Ristocetin shows normal aggregation EM: lack of dense bodies Increased ATP:ADP ratio within platelets


Alpha granule deficiency
Alpha storage pool deficiency, Gray Platelet Syndrome
First described by Raccuglia in 1971 Normal platelets contain approximately 50
granules (PF4, beta-thromboglobulin, PDGF, fibrinogen, vWF, Factor V, fibronectin)
Patients lack granules, present with lifelong, mild to moderate mucocutaneous bleeding

Diagnosis
Prolonged bleeding time, mild thrombocytopenia
A granular, large “gray” platelets on peripheral smear
Aggregation studies: decreased to absent response to collagen
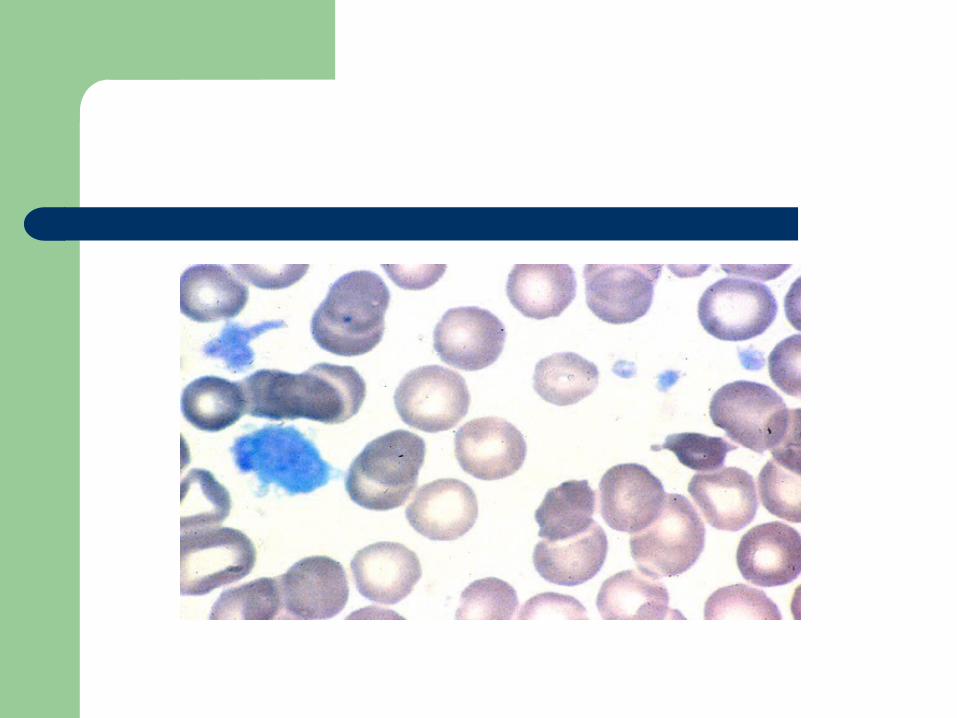

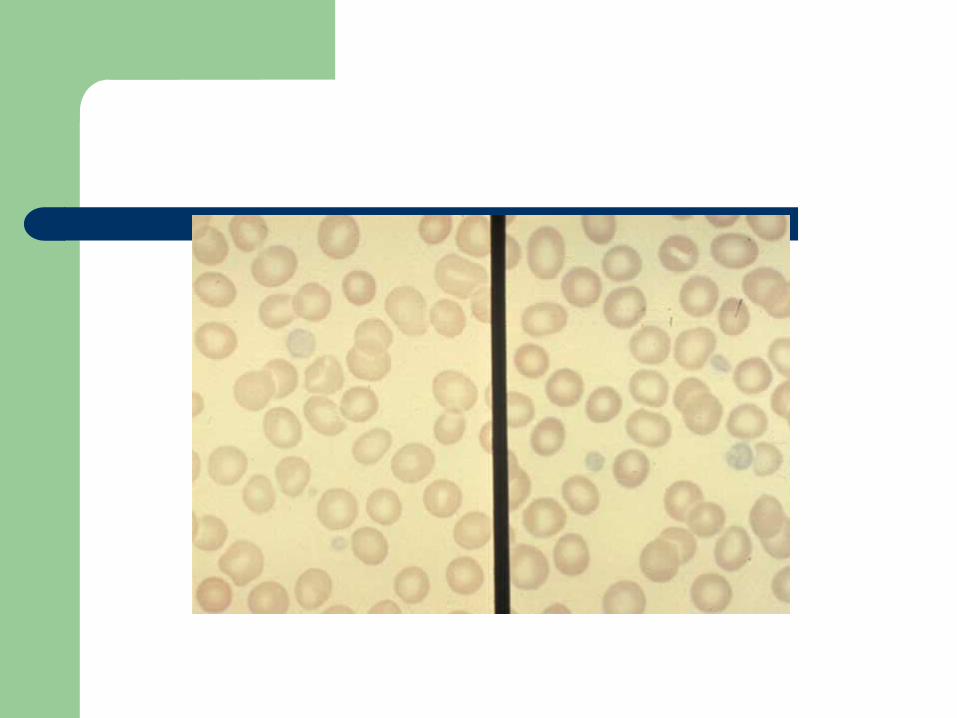

Summary
Morphology and role of the platelet in primary hemostasis
Adhesion: GP1b-V-IX; Bernard-Soulier; aggregates with everything but Ristocetin
Activation (Secretion): dense body deficiency (associated syndromes), alpha granule deficiency
Aggregation: GPIIb-IIIa; Glanzmann’s; no aggregation except for Ristocetin

Differential diagnosisTable. The clinical distinction between disorders of vessels and platelets and disorders of blood coagulation
Fingdings Disorders of platelet or vessels
Disorders of coagulation
Petechiae Characteristic Rare
Deep dissecting hematomas
Rare Characteristic
Superficial ecchymoses Characteristic; usually small and multiple
Common; uaually large and solitary
Hemarthrosis Rare Characteristic
Delayed bleeding Rare Common
Bleeding from superficial cuts and scratches
Persistent; often profuse
Minimal
Sex of patient Relatively more common in females
80-90% of hereditary forms occur only in males
Positive family history Rare Common


Clinical Cases

Nail bed - Hematoma
•Red
•Blue/Gr
•Brown
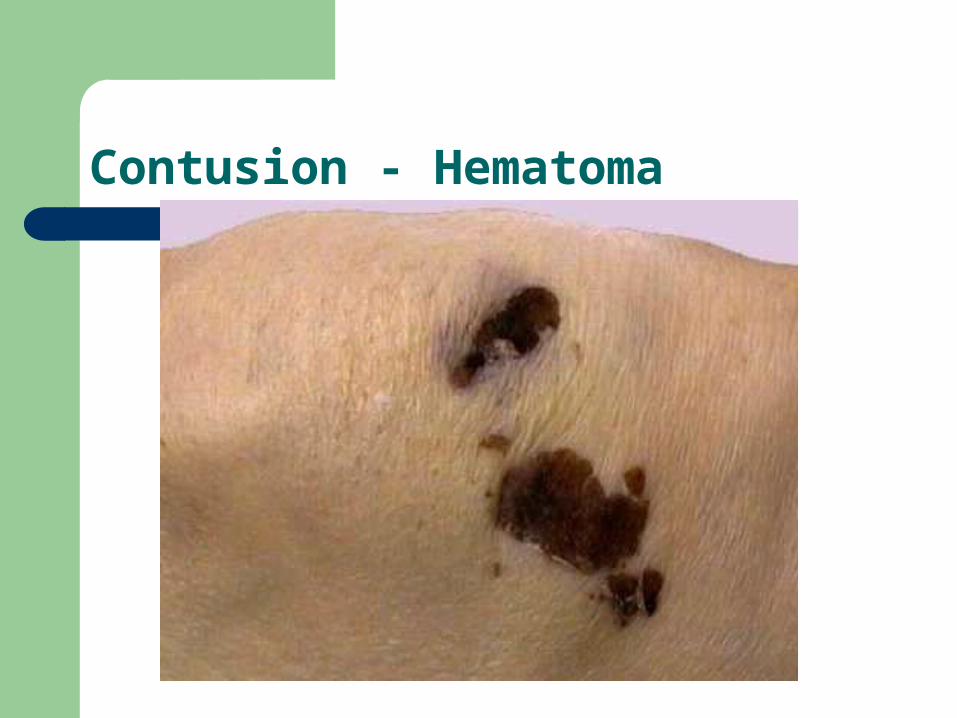
Contusion - Hematoma

Laceration - Trauma


Petechiae & Echymoses -Plt

Petechiae & Echymoses -Plt
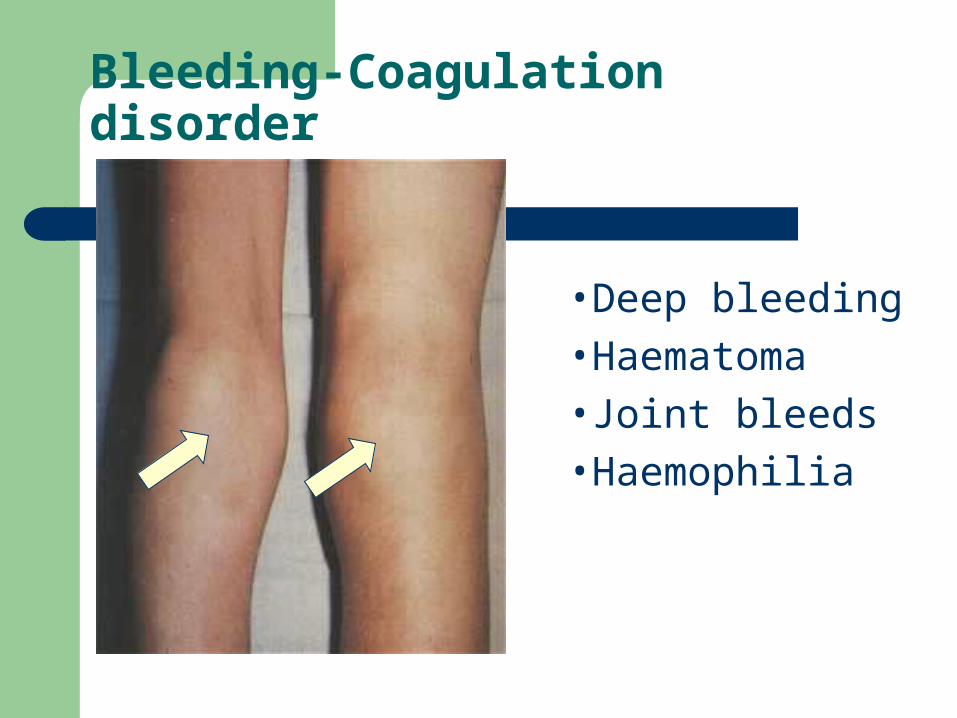
Bleeding-Coagulation disorder
•Deep bleeding
•Haematoma
•Joint bleeds
•Haemophilia

Jaundice
Leptospirosis - Weil’s disease:

Sub Conjuctival HaemorrhageLow PLT

Dengue – Hemorrhagic fever Plt

Bleeding disorders
Vascular abnormalities
Platelet disordersClotting factorabnormalities
DIC

Hereditary Type
The genetic defect can either be the failure of synthesis of one of the proteins or the production of a malfunctioning or abnormal molecule.
Quantitative vs Qualitative But in both of these genetic defects they will
result in slowing down and ineffective production of fibrin.

LAB Screening Tests
Lab screening tests are based on the length of time that it takes a clot to form in plasma.
So screening tests does not differentiate between qualitative or quantitative defects!

CRM + Vs CRM-
CRM is the abbreviation of Cross-Reacting Material Any protein has a functional activity and an immunologic
characteristics. Abnormal protein is functionally ineffective, but is
recognized immunologically.Is called CRM +
No protein (deficient) has no function and also will not be recognized immunologically.
Is called CRM -

Types of Bleeding Disorders
Hemophilia A (factor VIII deficiency)
Hemophilia B (factor IX deficiency)
von Willebrand Disease (vWD)
Other
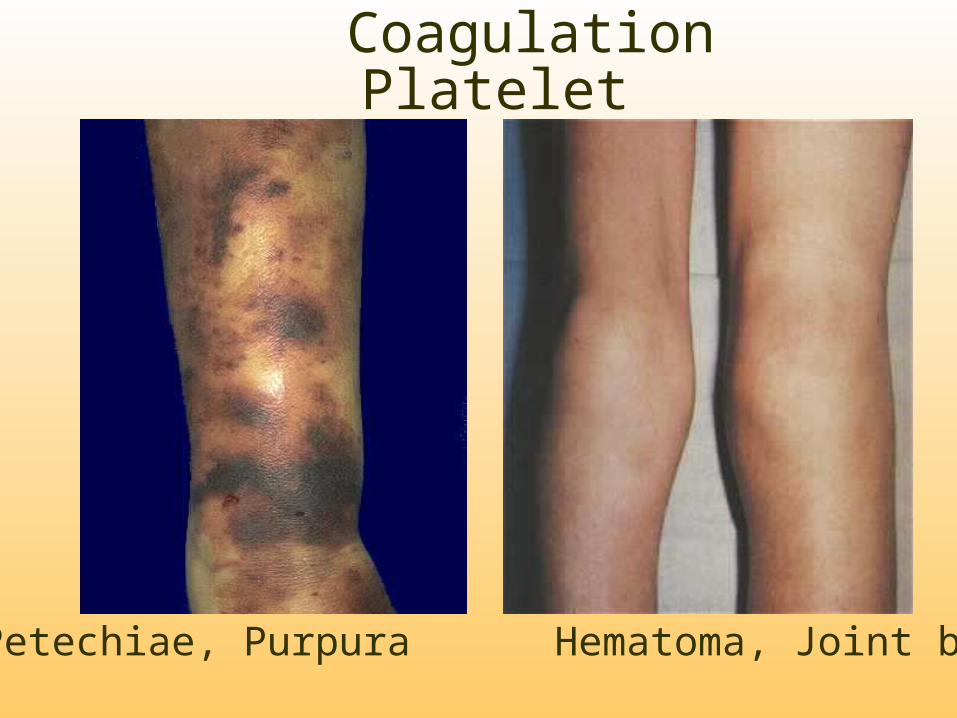
Coagulation Platelet
Petechiae, Purpura Hematoma, Joint bl.

What is Hemophilia?
Hemophilia is an inherited bleeding disorder in which there is a deficiency or lack of factor VIII (hemophilia A) or factor IX (hemophilia B)

Hemophilia A and BHemophilia A Hemophilia B
Coagulation factor deficiency Factor VIII Factor IX
Inheritance X-linked X-linkedrecessive recessive
Incidence 1/10,000 males 1/50,000 males
Severity Related to factor level<1% - Severe - spontaneous bleeding
1-5% - Moderate - bleeding with mild injury 5-25% - Mild - bleeding with surgery or trauma
Complications Soft tissue bleeding

Inheritance of Hemophilia
Hemophilia A and B are X-linked recessive disorders
Hemophilia is typically expressed in males and carried by females
Severity level is consistent between family members
~30 % of cases of hemophilia are new mutations

Genetics of Hemophilia A
The gene F8 is located in the X chromosome, at Xq28, near telomeric region.
It consists of 26 exons, and 25 introns. The F8 gene spans 186 Kb. Mature RNA is 8.8 Kb. The F8 protein is 2351 amino acid. The leader sequence is 19 a.a. So the real protein= 2351-19= 2332 aa.

Genetics of Hemophilia A
Half of hemophilia A patients have no detectable factor VIII;
about 5% have normal levels of dysfunctional factor VIII as protein and are termed CRM+
whereas the rest (45%) have plasma factor VIII Ag protein reduced to an extent roughly comparable to the level of factor VIII:C activity and are designated CRM-.

Detection of Hemophilia
Family history Symptoms
– Bruising– Bleeding with circumcision– Muscle, joint, or soft tissue bleeding
Hemostatic challenges– Surgery– Dental work– Trauma, accidents
Laboratory testing

Screening Tests in Secondary Hemostasis Defects
PT is prolonged or (test extrinsic pathway) APTT is prolonged or (test intrinsic pathway) Both are prolonged. Platelets are normal in count and function. TT (thrombin time): prolonged in disorders of
fibrinogen. If any test is abnormal of these screening tests,
additional testing may resolve the disorder>>>>

Additional Testing
Specific Factor Assays. Fibrinogen Level D-Dimer FDP’s Antithrombin Level The list continues to expand………….

Degrees of Severity of Hemophilia
Normal factor VIII or IX level = 50-150%
Mild hemophilia– factor VIII or IX level = 6-50%
Moderate hemophilia– factor VIII or IX level = 1-5%
Severe hemophilia– factor VIII or IX level = >1%

Hemophilia Prevalence
Hemophilia A; 1 in 5000 population – coagulation factor VIII deficiency
Hemophilia B; 1 in 30000 population – coagulation factor IX deficiency
Hemophilia A is six-fold more prevalent than hemophilia B.

Types of Bleeds
Joint bleeding - hemarthrosis
Muscle hemorrhage
Soft tissue
Life threatening-bleeding
Other

Hemarthrosis

Joint or Muscle Bleeding
Symptoms– Tingling or bubbling sensation– Stiffness– Warmth– Pain– Unusual limb position

Life-Threatening Bleeding
Head / Intracranial– Nausea, vomiting, headache, drowsiness, confusion, visual
changes, loss of consciousness
Neck and Throat– Pain, swelling, difficulty breathing/swallowing
Abdominal / GI– Pain, tenderness, swelling, blood in the stools
Iliopsoas Muscle– Back pain, abdominal pain, thigh tingling/numbness, decreased
hip range of motion

Other Bleeding Episodes
Mouth bleeding
Nose bleeding
Scrapes and/or minor cuts
Menorrhagia

Complications of Bleeding
Flexion contractures
Joint arthritis / arthropathy
Chronic pain
Muscle atrophy
Compartment syndrome
Neurologic impairment

Hemophilia “General Consideration”
There is a strong correlation between residual clotting factor level and severity of bleeding symptoms.
Spontaneous bleeding is only seen in severe disease.
Affected males with severe disease are generally diagnosed by the age of one year.
Even where there is no prior family history of hemophilia, sporadic cases caused by new mutations, which are responsible for 1/3 of cases.
In males with mild hemophilia, it is not unusual for the disorder to not be diagnosed until middle age, possibly following prolonged bleeding at surgery.

Treatment of Hemophilia
Replacement of missing clotting protein– On demand– Prophylaxis
DDAVP / Stimate Antifibrinolytic Agents
– Amicar Supportive measures
– Icing– Immobilization– Rest

Factor VIII Concentrate
Intravenous infusion– IV push– Continuous infusion
Dose varies depending on type of bleeding– Ranges from 20-50+ units/kg. body weight
Half-life 8-12 hours Each unit infused raises serum factor VIII
level by 2 %

Factor IX Concentrate
Intravenous infusion– IV push– Continuous infusion
Dose varies depending on type of bleeding – Ranges from 20-100+ units/kg. body weight
Half-life 12-24 hours Each unit infused raises serum factor IX level
by 1%

History of Clotting Factor Concentrates
Prior to 1950: whole blood
1952: Hemophilia A distinguished from B
1950-1960: FFP and Cryoprecipitate
Early 1970s: Commercial plasma-derived factor concentrates
Mid-late 1970’s: Home infusion practices
1981: First AIDS death in the Hemophilia community

History of Clotting Factor Concentrates (cont’d)
Mid-1983: Factor concentrates heat treated for hepatitis
1985: All products heat treated for viral inactivation
1987: Monoclonal factor concentrates
1992: Recombinant factor VIII
1994: Recombinant factor IX-albumin free
2001: 2nd generation recombinant factor VIII

Infusions of Factor Concentrates
Verify product with physician order. Dose may be +/- 10% ordered. Do not waste factor even if the dose is not exactly
what is ordered. Reconstitute factor per package insert. Infusion rate per package insert or pharmacy
instructions. Document lot number, expiration date, time of
infusion, and exact dose given in units.

Prophylaxis
Scheduled infusions of factor concentrates to prevent most bleeding
Frequency: 2 to 3 times weekly to keep trough factor VIII or IX levels at 2-3%
Types– primary prophylaxis– secondary prophylaxis
Use of IVAD necessary in some patients

DDAVP (Desmopressin acetate)
Synthetic vasopressinMethod of action -
– release of stores from endothelial cells raising factor VIII and vWD serum levels
Administration -– Intravenous– Subcutaneously– Nasally (Stimate)
Side effects

Stimate
How supplied– 1.5 mg./ ml (NOT to be confused with DDAVP nasal spray
for nocturnal enuresis) – 2.5 ml bottle - delivers 25 doses of 150 mcg.
Dosing– Every 24-48 hours prn– >50 kg. body weight - 1 spray (150 mcg.)– >50 kg. body weight - 2 sprays (300 mcg.)

Amicar(epsilon amino caproic acid)
AntifibrinolyticUses
– Mucocutaneous bleeding
Dosing: 50 - 100 mg./kg. q. 6 hoursSide effectsContraindications
– Hematuria

Complications of Treatment
Inhibitors/Antibody developmentHepatitis AHepatitis BHepatitis CHIV

Inhibitors
Definition – IgG antibody to infused factor VIII or IX
concentrates, which occurs after exposure to the extraneous VIII or IX protein.
Prevalence– 20-30% of patients with severe hemophilia A– 1-4% of patients with severe hemophilia B

Hepatitis
Hepatitis A- small risk of transmission– Vaccination recommended
Hepatitis B - no transmissions since 1985– Vaccination recommended
Hepatitis C - no transmissions since 1990– ~90% of patients receiving factor concentrates prior to 1985
are HCV antibody positive

Human Immunodeficiency Virus
No transmissions of HIV through factor concentrates since 1985 due to viral inactivation procedures
HIV seropositive rate -– 69.6% of patients with severe hemophilia A receiving
factor concentrates prior to 1985
– 48.6% of patients with severe hemophilia B receiving factor concentrates prior to 1985

Nursing Considerations
Factor replacement to be given on time Laboratory monitoring Increase metabolic states will increase factor
requirements Factor coverage for invasive procedures Document - infusions, response to treatment Avoid NSAIDS Utilize Hemophilia Center staff for questions /
problems

Psychosocial Issues
GuiltChallenge of hospitalizationsControl issuesFinancial / insurance challengesFeeling different / unable to do certain
activities Counseling needs

Hemophilia Treatment Center Team Members
Patient / Family
Hematologist
Nurse
Social Worker
Physical Therapist
Orthopedist
Primary Care
Infectious Disease
Genetics
Pharmacy
Dental
Hepatology

Role of Hemophilia Treatment Centers
State-of-the-art medical treatment for persons with hemophilia through the life span
Education Research Outreach Model of comprehensive care for chronic
disease

von Willebrand’s Disease

Outline
vWF– Structure– Location– Function
vWD– History– Clinical manifestations – Categories– Diagnosis– Treatment

vWD
Family of bleeding disorders Caused by a deficiency or an abnormality of
von Willebrand Factor

vWF
VWF gene : short arm of chromosome 12– VWF gene is expressed in endothelial cells and
megakaryocytes vWF is produced as a propeptide which is extensively modified to
produce mature vWF– Two vWF monomers bind through disulfide bonds to form
dimers– Multiple dimers combine to form vWF multimers

vWF Production
Vascular endothelial cells Megakaryocytes Most vWF is secreted Some vWF is stored
– Weibel-Palade bodies in endothelial cells
– Alpha granules of platelets
Constitutive and stimulus-induced pathways
Release stimuli (EC)– Thrombin– Histamine– Fibrin– C5b-9 (complement
membrane attack complex)
Release stimuli (platelets)– Thrombin– ADP– Collagen

vWF Function
Adhesion– Mediates the adhesion
of platelets to sites of vascular injury (subendothelium)
Links exposed collagen to platelets
– Mediates platelet to platelet interaction
Binds GPIb and GPIIb-IIIa on activated platelets
Stabilizes the hemostatic plug against shear forces

vW Factor Functions in Hemostasis
Carrier protein for Factor VIII (FVIII)– Protects FVIII from proteolytic degradation– Localizes FVIII to the site of vascular injury– Hemophilia A: absence of FVIII

vWD History
1931: Erik von Willebrand described novel bleeding disorder– Hereditary
pseudohemophilia– Prolonged BT and
normal platelet count– Mucosal bleeding – Both sexes affected
1950s: Prolonged BT associated with reduced FVIII
1970s: Discovery of vWF 1980s: vWF gene cloned

Frequency
Most frequent inherited bleeding disorder– Estimated that 1% of the population has vWD– Very wide range of clinical manifestations– Clinically significant vWD : 125 persons per million
population– Severe disease is found in approximately 0.5-5
persons per million population Autosomal inheritance pattern
– Males and females are affected equally

vWD Classification
Disease is due to either a quantitative deficiency of vWF or to functional deficiencies of vWF– Due to vWF role as carrier protein for FVIII,
inadequate amount of vWF or improperly functioning vWF can lead to a resultant decrease in the available amount of FVIII

vWD Classification
3 major subclasses– Type I: Partial quantitative deficiency of vWF
Mild-moderate disease 70%
– Type II: Qualitative deficiency of vWF Mild to moderate disease 25%
– Type III: Total or near total deficiency of vWF Severe disease 5%
Additional subclass– Acquired vWD

Clinical Manifestations
Most with the disease have few or no symptoms
For most with symptoms, it is a mild manageable bleeding disorder with clinically severe hemorrhage only with trauma or surgery
Types II and III: Bleeding episodes may be severe and potentially life threatening
Disease may be more pronounced in females because of menorrhagia
Bleeding often exacerbated by the ingestion of aspirin
Severity of symptoms tends to decrease with age due to increasing amounts of vWF

Clinical Manifestations
Epistaxis 60% Easy bruising / hematomas 40% Menorrhagia 35% Gingival bleeding 35% GI bleeding 10% Dental extractions 50% Trauma/wounds 35% Post-partum 25% Post-operative 20%

vWD Type I
Mild to moderate disease Mild quantitative deficiency of vWF
– vWF is functionally normal Usually autosomal dominant
– Penetrance may vary dramatically in a single family

vWD Type 2
Usually autosomal dominant
Type 2A – Lack high and
intermediate molecular weight multimers
Type 2B– Multimers bind platelets
excessively Increased clearance
of platelets from the circulation
– Lack high molecular weight multimers
Type 2C– Recessive – High molecular weight vWF
multimers is reduced – Individual multimers are
qualitatively abnormal Type 2M
– Decreased vWF activity– vWF antigen, FVIII, and
multimer analysis are found to be within reference range
Type 2N– Markedly decreased affinity
of vWF for FVIII Results in FVIII levels
reduced to usually around 5% of the reference range.

vWD Type III
Recessive disorder vWF protein is virtually undetectable
– Absence of vWF causes a secondary deficiency of FVIII and a subsequent severe combined defect in blood clotting and platelet adhesion

Acquired vWD
First described in 1970's fewer than 300 cases reported Usually encountered in adults with no personal or family
bleeding history Laboratory work-up most consistent with Type II vWD Mechanisms
– Autoantibodies to vWF– Absorption of HMW vWF multimers to tumors and activated
cells– Increased proteolysis of vWF– Defective synthesis and release of vWF from cellular
compartments Myeloproliferative disorders, lymphoproliferative disorders,
monoclonal gammopathies, CVD, and following certain infections

vWD Screening
PT aPTT (Bleeding time)

vWD: aPTT and PT
aPTT– Mildly prolonged in approximately 50% of patients
with vWD Normal PTT does not rule out vWD
– Prolongation is secondary to low levels of FVIII PT
– Usually within reference ranges Prolongations of both the PT and the aPTT signal a
problem with acquisition of a proper specimen or a disorder other than or in addition to vWD

vWD and Bleeding Time
Historically, bleeding time is a test used to help diagnose vWD– Lacks sensitivity and specificity– Subject to wide variation– Not currently recommended for making the
diagnosis of vWD

vWD Diagnostic Difficulties
vWF levels vary greatly– Physiologic stress– Estrogens– Vasopressin– Growth hormone– Adrenergic stimuli
vWF levels may be normal intermittently in patients with vWD– Measurements should be repeated to confirm abnormal
results– Repeating tests at intervals of more than 2 weeks is
advisable to confirm or definitively exclude the diagnosis, optimally at a time remote from hemorrhagic events, pregnancy, infections, and strenuous exercise
vWF levels vary with blood type

vWD Diagnosis
Ristocetin– Good for evaluating vWF function, – Results are difficult to standardize – Method
Induces vWF binding to GP1b on platelets Ristocetin co-factor activity: measures agglutination of
metabolically inactive platelets RIPA: metabolically active platelets Aggregometer is used to measure the rate of aggregation
vWF Antigen– Quantitative immunoassay or an ELISA using an antibody to
vWF Discrepancy between the vWF:Ag value and RCoF activity
suggests a qualitative defect – Should be further investigated by characterization of the vWF
multimeric distribution

Additional Assays
Multimer analysis PFA-100 closure time
– Screens platelet function in whole blood
– Prolonged in vWD, except Type 2N
FVIII activity assay

vWD Treatment
DDAVP Cryoprecipitate FVIII concentrate

vWD and DDAVP
Treatment of choice for vWD type I– Synthetic analogue of the antidiuretic hormone
vasopressin– Maximal rise of vWF and FVIII is observed in 30-60
minutes– Typical maximal rise is 2- to 4-fold for vWF and 3- to 6-
fold for FVIII– Hemostatic levels of both factors are usually maintained
for at least 6 hours– Effective for some forms of Type 2 vWD
May cause thrombocytopenia in Type 2b– Ineffective for vWD Type 3

Factor VIII Concentrates
Alphanate and Humate P Concentrates are purified to reduce the risk of
blood-borne disease Contain a near-normal complement of high
molecular weight vWF multimers

vWD Treatment
Platelet transfusions– May be helpful with vWD refractory to other therapies
Cryoprecipitate– Fraction of human plasma– Contains both FVIII and vWF– Medical and Scientific Advisory council of the National
Hemophilia Foundation no longer recommends this treatment method due to its associated risks of infection
FFP– An additional drawback of fresh frozen plasma is the large
infusion volume required

Disseminated Intravascular Coagulation

DIC
An acquired syndrome characterized by systemic intravascular coagulation
Coagulation is always the initial event.
Most morbidity and mortality depends on extent of intravascular thrombosis
Multiple causes
6
ThrombosisThrombosis
FibrinFibrin
Red Blood CellRed Blood Cell
PlateletPlatelet
WWW. Coumadin.com

DIC
An acquired syndrome characterized by systemic intravascular coagulation
Coagulation is always the initial event
SYSTEMIC ACTIVATION OF COAGULATION
Intravascular deposition of
fibrin
Depletion of platelets and coagulation
factors
Thrombosis of small and
midsize vesselsBleeding
Organ failure DEATHDEATH

Pathophysiology of DIC
Activation of Blood Coagulation Suppression of Physiologic Anticoagulant
Pathways Impaired Fibrinolysis Cytokines

Pathophysiology of DIC
Activation of Blood Coagulation– Tissue factor/factor VIIa mediated thrombin generation
via the extrinsic pathway complex activates factor IX and X
– TF endothelial cells monocytes Extravascular:
– lung– kidney– epithelial cells

Pathophysiology of DIC
Suppression of Physiologic Anticoagulant Pathways– reduced antithrombin III levels – reduced activity of the protein C-protein S system– Insufficient regulation of tissue factor activity by
tissue factor pathway inhibitor (TFPI)inhibits TF/FVIIa/Fxa complex activity

Pathophysiology of DIC
Impaired Fibrinolysis– relatively suppressed at time of maximal activation
of coagulation due to increased plasminogen activator inhibitor type 1

Pathophysiology of DIC - Cytokines
Cytokines– IL-6, and IL-1 mediates coagulation activation in DIC– TNF-
mediates dysregulation of physiologic anticoagulant pathways and fibrinolysis
modulates IL-6 activity– IL-10 may modulate the activation of coagulation
CoagulationInflamation

Diagnosis of DIC
Presence of disease associated with DIC Appropriate clinical setting
– Clinical evidence of thrombosis, hemorrhage or both.
Laboratory studies– no single test is accurate– serial test are more helpful than single test

Conditions Associated With DIC
Malignancy– Leukemia– Metastatic disease
Cardiovascular– Post cardiac arrest– Acute MI– Prosthetic devices
Hypothermia/Hyperthermia
Pulmonary– ARDS/RDS– Pulmonary
embolism
Severe acidosis Severe anoxia Collagen vascular
disease Anaphylaxis

Conditions Associated With DIC
Infectious/Septicemia– Bacterial
Gm - / Gm +
– Viral CMV Varicella Hepatitis
– Fungal
Intravascular hemolysis Acute Liver Disease
Tissue Injury– trauma– extensive surgery– tissue necrosis– head trauma
Obstetric– Amniotic fluid emboli– Placental abruption– Eclampsia– Missed abortion

Clinical Manifestations of DIC
ORGAN ISCHEMIC HEMOR.Skin Pur. Fulminans
GangreneAcral cyanosis
PetechiaeEchymosisOozing
CNS Delirium/ComaInfarcts
Intracranialbleeding
Renal Oliguria/AzotemiaCortical Necrosis
Hematuria
Cardiovascular MyocardialDysfxn
Pulmonary Dyspnea/HypoxiaInfarct
Hemorrhagiclung
GIEndocrine
Ulcers, InfarctsAdrenal infarcts
Massivehemorrhage.
Ischemic Findingsare earliest!
Bleeding is the most obvious
clinical finding
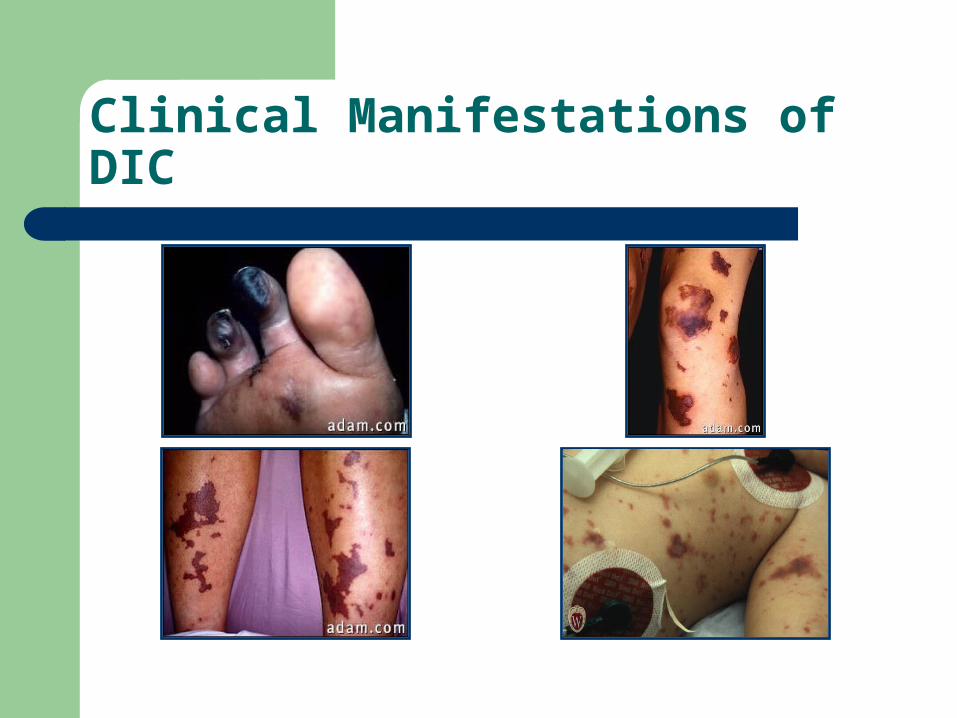
Clinical Manifestations of DIC

Microscopic findings in DIC
Fragments Schistocytes Paucity of platelets

Laboratory Tests Used in DIC
D-dimer* Antithrombin III* F. 1+2* Fibrinopeptide A* Platelet factor 4* Fibrin Degradation
Prod Platelet count Protamine test
Thrombin time Fibrinogen Prothrombin time Activated PTT Protamine test Reptilase time Coagulation factor levels
*Most reliable test

Laboratory diagnosis
Thrombocytopenia– plat count >100,000 or rapidly declining
Prolonged clotting times (PT, APTT) Presence of Fibrin degradation products or positive
D-dimer Low levels of coagulation inhibitors
– AT III, protein C
Low levels of coagulation factors– Factors V,VIII,X,XIII
Fibrinogen levels not useful diagnostically

Differential Diagnosis
Severe liver failure Vitamin K deficiency Liver disease Thrombotic thrombocytopenic purpura Congenital abnormalities of fibrinogen HELLP syndrome

Treatment of DIC
Stop the triggering process .– The only proven treatment!
Supportive therapy No specific treatments
– Plasma and platelet substitution therapy– Anticoagulants– Physiologic coagulation inhibitors

Plasma therapy
Indications– Active bleeding– Patient requiring invasive procedures– Patient at high risk for bleeding complications
Prophylactic therapy has no proven benefit. Cons: Fresh frozen plasma(FFP):
– provides clotting factors, fibrinogen, inhibitors, and platelets in balanced amounts.
– Usual dose is 10-15 ml/kg

Platelet therapy
Indications– Active bleeding– Patient requiring invasive procedures– Patient at high risk for bleeding complications
Platelets– approximate dose 1 unit/10kg

Blood
Replaced as needed to maintain adequate oxygen delivery.– Blood loss due to bleeding– RBC destruction (hemolysis)

Coagulation Inhibitor Therapy
Antithrombin III Protein C concentrate Tissue Factor Pathway Inhibitor (TFPI) Heparin

The major inhibitor of the coagulation cascade– Levels are decreased in DIC.– Anticoagulant and antiinflammatory properties
Therapeutic goal is to achieve supranormal levels of ATIII (>125-150%).
– Experimental data indicated a beneficial effect in preventing or attenuating DIC in septic shock
reduced DIC scores, DIC duration, and some improvement in organ function
– Clinical trials have shown laboratory evidence of attenuation of DIC and trends toward improved outcomes.
– A clear benefit has not been established in clinical trials.
Antithrombin III

Protein C Concentrates
Inhibits Factor Va, VIIa and PAI-1 in conjunction with thrombomodulin.
Protein S is a cofactor Therapeutic use in DIC is experimental and is based on
studies that show:– Patients with congenital deficiency are prone to thromboembolic
disease.– Protein C levels are low in DIC due to sepsis.– Levels correlate with outcome.– Clinical trials show significantly decreased morbidity and
mortality in DIC due to sepsis.

Tissue Factor Pathway Inhibitor
Tissue factor is expressed on endothelial cells and macrophages
TFPI complexes with TF, Factor VIIa,and Factor Xa to inhibit generation of thrombin from prothrombin
TF inhibition may also have antiinflammatory effects Clinical studies using recombinant TFPI are
promising.

Heparin
Use is very controversial. Data is mixed. May be indicated in patients with clinical
evidence of fibrin deposition or significant thrombosis.
Generally contraindicated in patients with significant bleeding and CNS insults.
Dosing and route of administration varies. Requires normal levels of ATIII.

Antifibrinolytic Therapy
Rarely indicated in DIC– Fibrinolysis is needed to clear thrombi from the micro
circulation.– Use can lead to fatal disseminated thrombosis.
May be indicated for life threatening bleeding under the following conditions:
– bleeding has not responded to other therapies and:– laboratory evidence of overwhelming fibrinolysis.– evidence that the intravascular coagulation has ceased.
Agents: tranexamic acid, EACA

Summary
DIC is a syndrome characterized systemic intravascular coagulation.
Coagulation is the initial event and the extent of intravascular thrombosis has the greatest impact on morbidity and mortality.
Important link between inflammation and coagulation. Morbidity and mortality remain high. The only proven treatment is reversal or control
of the underlying cause.