bruton's tyrosine kinase inhibitors prevent therapeutic ...lymphoma, chronic lymphocytic...
TRANSCRIPT

Cancer Biology and Signal Transduction
Bruton's Tyrosine Kinase Inhibitors PreventTherapeutic Escape in Breast Cancer CellsXianhui Wang1, Jason Wong1, Christopher J. Sevinsky1, Leila Kokabee1,2,Faiza Khan1, Yan Sun1, and Douglas S. Conklin1
Abstract
We have reported that a novel isoform of BTK (BTK-C)expressed in breast cancer protects these cells from apoptosis. Inthis study, we show that recently developed inhibitors of BTK,such as ibrutinib (PCI-32765), AVL-292, and CGI-1746, reducebreast cancer cell survival and prevent drug-resistant clones fromarising. Ibrutinib treatment impacts HER2þ breast cancer cellviability at lower concentrations than the established breastcancer therapeutic lapatinib. In addition to inhibiting BTK, ibru-tinib, but not AVL-292 and CGI-1746, efficiently blocks theactivation of EGFR, HER2, ErbB3, and ErbB4. Consequently, theactivation of AKT and ERK signaling pathways are also blockedleading to a G1–S cell-cycle delay and increased apoptosis. Impor-tantly, inhibition of BTK prevents activation of the AKT signalingpathway by NRG or EGF that has been shown to promote growth
factor–driven lapatinib resistance in HER2þ breast cancer cells.HER2þbreast cancer cell proliferation is blockedby ibrutinib evenin the presence of these factors. AVL-292, which has no effect onEGFR family activation, prevents NRG- and EGF-dependentgrowth factor–driven resistance to lapatinib in HER2þ breastcancer cells. In vivo, ibrutinib inhibits HER2þ xenograft tumorgrowth. Consistent with this, immunofluorescence analysis ofxenograft tumors shows that ibrutinib reduces the phosphoryla-tion of HER2, BTK, Akt, and Erk and histone H3 and increasescleaved caspase-3 signals. As BTK-C and HER2 are often coex-pressed in human breast cancers, these observations indicatethat BTK-C is a potential therapeutic target and that ibrutinibcould be an effective drug especially for HER2þ breast cancer.Mol Cancer Ther; 15(9); 2198–208. �2016 AACR.
IntroductionBruton's tyrosine kinase (BTK) belongs to the TEC family of
cytoplasmic tyrosine kinases (1). BTK was identified in 1993 as anovel nonreceptor protein tyrosine kinase that is mutated in X-linked agammaglobulinaemia (XLA; refs. 2, 3). It is predominant-ly expressed in hematopoietic cells including erythroid progeni-tors and myeloid cells (4) and is established as a critical regulatorof the B-cell receptor (BCR) signaling, development, differentia-tion, and survival (5–7). Because of its role in BCR signaling–induced proliferation, BTK has emerged as a novel target for thetreatment of rheumatoid arthritis and other immune diseases.Recent studies have focused on the essential role of BTK in manyB-cell leukemias and lymphomas (8, 9) which provides a ratio-nale for targeting the kinase in these malignancies. BTK inhibitorsincluding ibrutinib (10), AVL-292 (11), and CGI-1746 (12)developed as immunosuppressants have been used in clinicaltrials for blood malignancies (13). Recently, ibrutinib (Imbru-vica) gained FDA approval for the treatment of mantle cell
lymphoma, chronic lymphocytic leukemia, and Waldenstr€ommacroglobulinemia (14). An alternate isoform of BTK, BTK-C,was identified as a novel survival factor for breast cancer cells in alarge-scale loss-of-function analysis of human tyrosine kinasesusing an RNA interference library (15). This study showed thatalthough BTK-C is expressed at relatively low levels in severalhuman breast cancer cell lines and tumor tissues, it provides anessential function protecting breast cancer cells from apoptosis.
It has long been appreciated that HER2 is overexpressed oramplified in tumors of about 20% of patients with early-stagebreast cancer and confers an increased disease recurrence and aworse prognosis (16). HER2-directed therapies including trastu-zumab, pertuzumab, ado-trastuzumab, and lapatinib have beenused in clinic and have significantly improved the outlook forpatients with HER2þ breast cancer (17). However, a significantproportion of these patients still relapses and succumbs to theirdisease (18). Therefore, new classes of drugs are needed, especiallyfor HER2þ advanced-stage breast cancer and those that havedeveloped resistance to current therapies.
Here we describe the effects of treating breast cancer cells withrecently developed BTK inhibitors. We find that among the newinhibitors, ibrutinib is particularly effective at inhibiting breastcancer cell growth in vitro and in vivo. Ibrutinib inhibits BTK and allmembers of the ERBB family of receptor tyrosine kinasesmaking itespecially effective at reducing HER2þ breast cancer cell growthand survival. Ibrutinib induces a G1–S arrest and apoptosis inthese cells. We also show that ibrutinib's effects on HER2þ breastcancer cells are not mitigated by NRG1 or EGF stimulation, asoccurs with lapatinib. As the expression of BTK-C and HER2 ispositively correlated in surgical specimens of humanbreast cancertissues, these results indicate that ibrutinib is a potential therapyfor this solid tumor type.
1Cancer Research Center and Department of Biomedical Sciences,State University of New York, University at Albany, Rensselaer, NewYork. 2Department of Molecular Medicine, Pasteur Institute of Iran,Tehran, Iran.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Douglas S. Conklin, Cancer Research Center, Depart-ment of Biomedical Sciences, University at Albany, State University of NewYork,CRCRoom308,OneDiscoveryDrive, Rensselaer, NY 12144. Phone: 518-591-7154;Fax: 518-591-7151; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-15-0813
�2016 American Association for Cancer Research.
MolecularCancerTherapeutics
Mol Cancer Ther; 15(9) September 20162198

Materials and MethodsCell culture and chemicals
Breast cancer and MCF-10A cell lines were obtained from theATCC in 2011. All cell lines were cultured in DMEM (Hyclone)supplemented with 10% FBS (Hyclone) and 100 U/mL of peni-cillin–streptomycin (CellGro), except for MCF-10A cells culturedas indicated in ref. (15). All cell lines were authenticated inMarch2016 by the SUNY-Albany Center for Functional GenomicsMolecular Core Facility using a short tandem repeat method(Promega GenePrint 10 system). BTK inhibitors were suppliedas follows. Ibrutinib was purchased from ChemieTek, AVL-292was purchased from MedKoo Biosciences, CGI-1764 was pur-chased fromAxonMedchem. Recombinant human-b1 andMatri-gel were purchased from R&D Systems. Src inhibitor saracatinibwas purchased from Selleckchem. LY294002 was purchased fromCell Signaling Technology.
Cell viability assaysFor live cell counts, cells were grown on 96-well plates, fixed
with 4% formaldehyde, and counterstained with Hoechst 33342.Images of cells were acquired using an In Cell Analyzer 1000 (GEHealthcare) high-content imaging system. At least 30 fields wereimaged per well. Statistics were performed using the In CellInvestigator 3.4 image analysis software (GE Healthcare).
Three-dimensional (3D) cell culture was performed asdescribed previously (19). BT474 cells were propagated inDMEMwith 10% FBS. Single cells in medium containing 5% Matrigelwere seeded at a density of 5� 104 cells/cm2 on aMatrigel-coatedwell. The topmediumwith 5%Matrigel and ibrutinib or lapatinibwas changed every 3 days. 1 mmol/L ethidium bromide was usedto stain cell death during 3D culture.
Cellswere cultured in6-well plates for indicatednumberofdays.Cells were fixed with 3.7% paraformaldehyde for 10 minutes andwashed with PBS. After washing, cells were stained with 0.05%crystal violet for 30minutes and washed with tap water. Methanol(0.5 mL) was added to the dry plates to solubilize the dye. Crystalviolet staining was measured by reading OD 540 nm of eachsample. Tumorspheres were counted under low magnification.
Cell-cycle analysisCells were cultured in 6-well plates and treated with vehicle,
lapatinib, or ibrutinib at the concentration as indicated for 16hours. After trypsinization, cells were collected and washedwith cold PBS. Cells were fixed with 70% ethanol, washed,and stained with PI/Triton X-100 staining solution (0.1% TritonX-100, 2 mg/mL PI, and 0.2 mg/mL DNAse-free RNAse) for30 minutes. Samples were analyzed by flow cytometry.
ImmunofluorescenceHuman breast cancer tissue sections (BR10010b, US Biomax,
Inc.) were baked for 1 hour at 62�C, subjected to serial alcoholtreatments for rehydration and microwaved in 0.01 mol/L sodi-um citrate for 20 minutes for antigen retrieval. The sections wereserum blocked for 30 minutes, incubated overnight at 4�C withprimary antibodies in PBS and subsequently with Cy5-labeledsecondary antibodies for 60minutes, andnucleiwere stainedwithHoechst 33342. The stained sectionsweremountedwith anti-fadesolution for microscopy. The two by two tables for human datawere analyzed by Fisher exact test. Significance was determined bythe alpha level of 0.05.
ImmunoblottingImmunoblotting was performed essentially as described pre-
viously. Equal amounts of proteins were used. Antibodies usedwere anti-EGFR (1:1,000), anti-HER2 (1:1,000), anti-ERBB3,ERBB4 antibody, anti-BTK, anti-AKT, ERK, anti-PLCg1, PLCg2and anti-PARP (1:1,000, Cell Signaling Technology), anti-Flagantibody (1:1,000, Sigma), anti-rabbit IgG-HRP, and mouseIgG-HRP (1:5,000, Jackson ImmunoResearch; ref. 20).
Apoptosis assayApoptotic cells were identified using an Alexa Fluor 488
Annexin V Apoptosis Kit (Invitrogen). Cells were treated withlapatinib or ibrutinib for 24 hours. Cells were trypsinized andwashed with cold PBS and resuspended in 1� Annexin-bindingbuffer to 1� 106 cells/mL. Alexa Fluor 488 Annexin V (5 mL) and100 mg/mL PI (1 mL) was added to each 100 mL of cell suspension.Cells were incubated for 15 minutes at room temperature. Afterthe incubation, 400 mL 1� Annexin-binding buffer was added,mixed gently, and the sample kept on ice. Samples were analyzedon a BD LSR II Flow Cytometer (BD Biosciences). The data wereanalyzed using the FlowJo software package (Treestar Inc.).
Animal experimentsNOD/SCIDmicewere purchased fromThe Jackson Laboratory.
All mouse procedures were approved by the Animal Care andUseCommittees of The StateUniversity ofNewYork at Albany (SUNYAlbany, Albany, NY) and performed in accordance with institu-tional policies. For xenograft tumor studies, 1 � 106 SKBR3 cellswere suspended in 50-mL Matrigel (BD Biosciences) diluted 1:2withDMEMand injected intomammary fat pad. Treatment beganwhen tumors were palpable. Ibrutinib was given 6 mg/kg/day or12mg/kg/day orally in a vehicle of 1%DMSO/30% polyethyleneglycol/1% Tween 80. Lapatinib was given 75 mg/kg/day or37.5mg/kg/day orally. The tumor volume inmm3 was calculatedby the formula: volume ¼ (width)2 � length/2 every 7 days.
ResultsActivity of BTK inhibitors in human breast cancer cells
In a functional genomic screen, a novel BTK isoform has beenidentified as a gene whose expression protects breast cancer cellsfromapoptosis (15). In addition to genetic evidence, the early BTKinhibitor LFM-A13 was shown to increase apoptosis levels inbreast cancer cells. The recently developed BTK inhibitors, includ-ing ibrutinib, AVL-292, and CGI-1746, are more potent, morespecific, and more useful clinically compared with LFM-A13(12, 21, 22). In 2013, ibrutinib was approved by the FDA fortreatment of B cell malignancies (23, 24). As a first step inexploring the potential clinical utility of the recently developedBTK inhibitors, we performed cell growth assays to determine theeffect of these inhibitors on breast cancer cells. We find thatibrutinib results in decreased cell number in breast cancer cellsMCF7, SKBR3, and MDA-MB-231, but not in MCF-10A cells (Fig.1A). These results are consistent with our previous findings (15).Surprisingly, we observed that HER2þ breast SKBR3 cancer cellsare more sensitive to ibrutinib (1 mmol/L) than nontumorigenic,luminal, or triple-negative cell lines (Fig. 1A). Ibrutinib reducescell numbers by more than 80% at 3 days. To extend our findingsto another HER2þ breast cancer cell, we tested the effect ofibrutinib on BT474 cell survival. The result shows that ibrutinibdecreases HER2þ breast cancer cell growth by 20%–30% at a
BTK Inhibitors and Drug Resistance in Breast Cancer
www.aacrjournals.org Mol Cancer Ther; 15(9) September 2016 2199
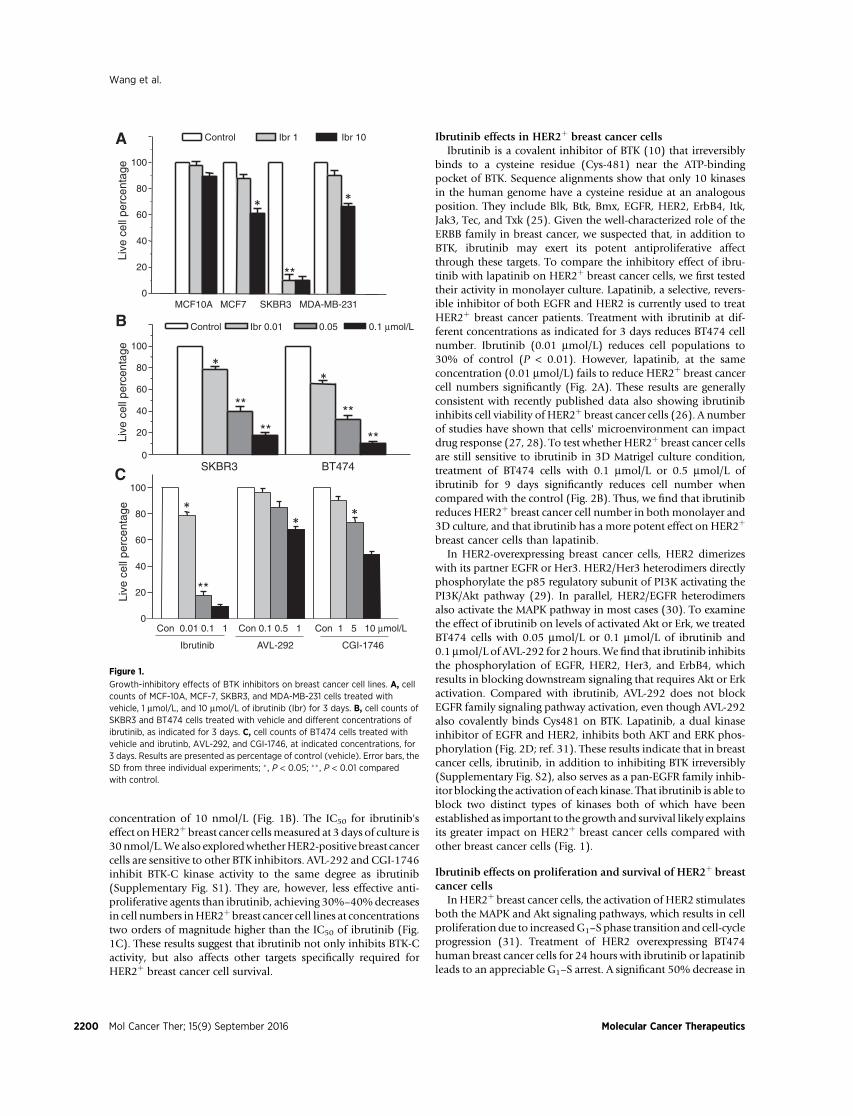
concentration of 10 nmol/L (Fig. 1B). The IC50 for ibrutinib'seffect onHER2þ breast cancer cellsmeasured at 3 days of culture is30nmol/L.We also exploredwhetherHER2-positive breast cancercells are sensitive to other BTK inhibitors. AVL-292 and CGI-1746inhibit BTK-C kinase activity to the same degree as ibrutinib(Supplementary Fig. S1). They are, however, less effective anti-proliferative agents than ibrutinib, achieving 30%–40%decreasesin cell numbers inHER2þbreast cancer cell lines at concentrationstwo orders of magnitude higher than the IC50 of ibrutinib (Fig.1C). These results suggest that ibrutinib not only inhibits BTK-Cactivity, but also affects other targets specifically required forHER2þ breast cancer cell survival.
Ibrutinib effects in HER2þ breast cancer cellsIbrutinib is a covalent inhibitor of BTK (10) that irreversibly
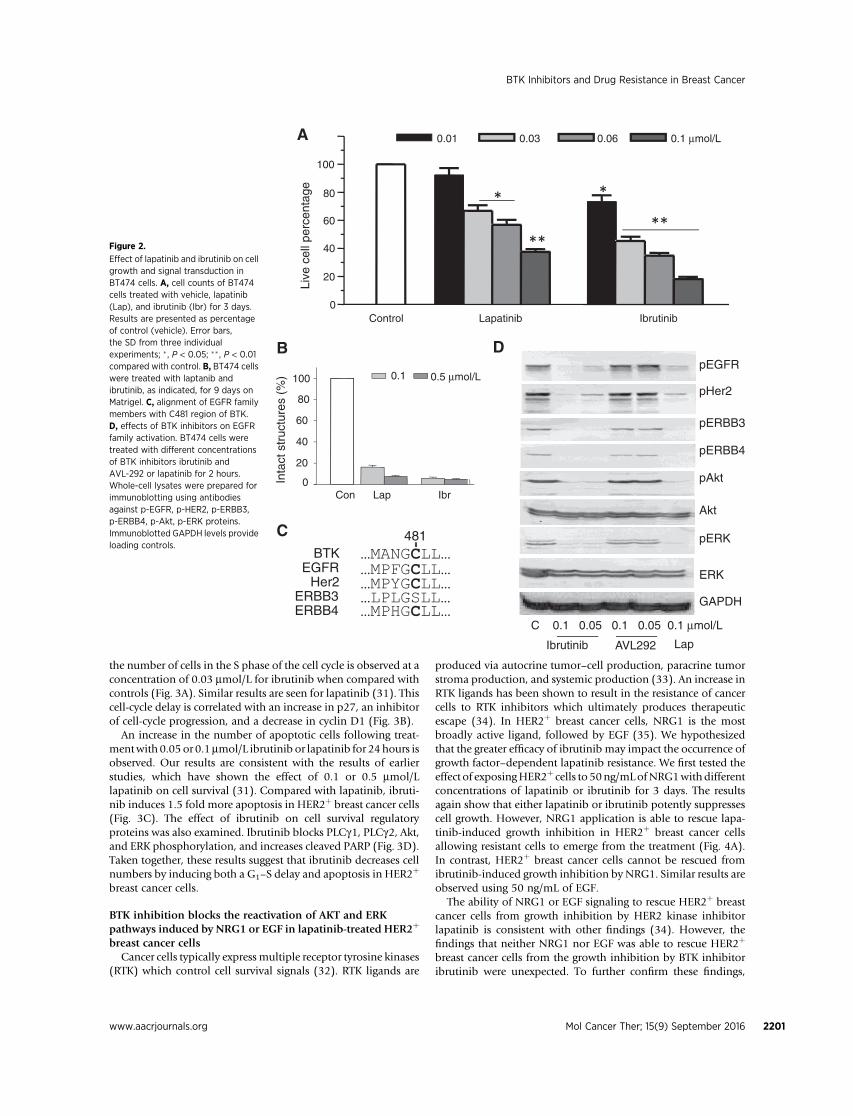
binds to a cysteine residue (Cys-481) near the ATP-bindingpocket of BTK. Sequence alignments show that only 10 kinasesin the human genome have a cysteine residue at an analogousposition. They include Blk, Btk, Bmx, EGFR, HER2, ErbB4, Itk,Jak3, Tec, and Txk (25). Given the well-characterized role of theERBB family in breast cancer, we suspected that, in addition toBTK, ibrutinib may exert its potent antiproliferative affectthrough these targets. To compare the inhibitory effect of ibru-tinib with lapatinib on HER2þ breast cancer cells, we first testedtheir activity in monolayer culture. Lapatinib, a selective, revers-ible inhibitor of both EGFR and HER2 is currently used to treatHER2þ breast cancer patients. Treatment with ibrutinib at dif-ferent concentrations as indicated for 3 days reduces BT474 cellnumber. Ibrutinib (0.01 mmol/L) reduces cell populations to30% of control (P < 0.01). However, lapatinib, at the sameconcentration (0.01 mmol/L) fails to reduce HER2þ breast cancercell numbers significantly (Fig. 2A). These results are generallyconsistent with recently published data also showing ibrutinibinhibits cell viability of HER2þ breast cancer cells (26). A numberof studies have shown that cells' microenvironment can impactdrug response (27, 28). To test whether HER2þ breast cancer cellsare still sensitive to ibrutinib in 3D Matrigel culture condition,treatment of BT474 cells with 0.1 mmol/L or 0.5 mmol/L ofibrutinib for 9 days significantly reduces cell number whencompared with the control (Fig. 2B). Thus, we find that ibrutinibreduces HER2þ breast cancer cell number in both monolayer and3D culture, and that ibrutinib has a more potent effect on HER2þ
breast cancer cells than lapatinib.In HER2-overexpressing breast cancer cells, HER2 dimerizes
with its partner EGFR or Her3. HER2/Her3 heterodimers directlyphosphorylate the p85 regulatory subunit of PI3K activating thePI3K/Akt pathway (29). In parallel, HER2/EGFR heterodimersalso activate the MAPK pathway in most cases (30). To examinethe effect of ibrutinib on levels of activated Akt or Erk, we treatedBT474 cells with 0.05 mmol/L or 0.1 mmol/L of ibrutinib and0.1 mmol/L of AVL-292 for 2 hours.We find that ibrutinib inhibitsthe phosphorylation of EGFR, HER2, Her3, and ErbB4, whichresults in blocking downstream signaling that requires Akt or Erkactivation. Compared with ibrutinib, AVL-292 does not blockEGFR family signaling pathway activation, even though AVL-292also covalently binds Cys481 on BTK. Lapatinib, a dual kinaseinhibitor of EGFR and HER2, inhibits both AKT and ERK phos-phorylation (Fig. 2D; ref. 31). These results indicate that in breastcancer cells, ibrutinib, in addition to inhibiting BTK irreversibly(Supplementary Fig. S2), also serves as a pan-EGFR family inhib-itor blocking the activation of each kinase. That ibrutinib is able toblock two distinct types of kinases both of which have beenestablished as important to the growth and survival likely explainsits greater impact on HER2þ breast cancer cells compared withother breast cancer cells (Fig. 1).
Ibrutinib effects on proliferation and survival of HER2þ breastcancer cells
In HER2þ breast cancer cells, the activation of HER2 stimulatesboth the MAPK and Akt signaling pathways, which results in cellproliferation due to increasedG1–S phase transition and cell-cycleprogression (31). Treatment of HER2 overexpressing BT474human breast cancer cells for 24 hours with ibrutinib or lapatinibleads to an appreciable G1–S arrest. A significant 50% decrease in
**
****
Control Ibr 0.01 0.05 0.1 μmol/L
SKBR3 BT474
Live
cel
l per
cent
age
**
**
Con 0.01 0.1 1 Con 0.1 0.5 1 Con 1 5 10 μmol/L
Ibrutinib AVL-292 CGI-1746
Live
cel
l per
cent
age
**
** *
0
20
40
60
80
100
0
20
40
60
80
100
0
20
40
60
80
100
Control Ibr 1 Ibr 10
MCF10A MCF7 SKBR3 MDA-MB-231
Live
cel
l per
cent
age
**
**
A
B
C
Figure 1.
Growth-inhibitory effects of BTK inhibitors on breast cancer cell lines. A, cellcounts of MCF-10A, MCF-7, SKBR3, and MDA-MB-231 cells treated withvehicle, 1 mmol/L, and 10 mmol/L of ibrutinib (Ibr) for 3 days. B, cell counts ofSKBR3 and BT474 cells treated with vehicle and different concentrations ofibrutinib, as indicated for 3 days. C, cell counts of BT474 cells treated withvehicle and ibrutinb, AVL-292, and CGI-1746, at indicated concentrations, for3 days. Results are presented as percentage of control (vehicle). Error bars, theSD from three individual experiments; � , P < 0.05; �� , P < 0.01 comparedwith control.
Wang et al.
Mol Cancer Ther; 15(9) September 2016 Molecular Cancer Therapeutics2200
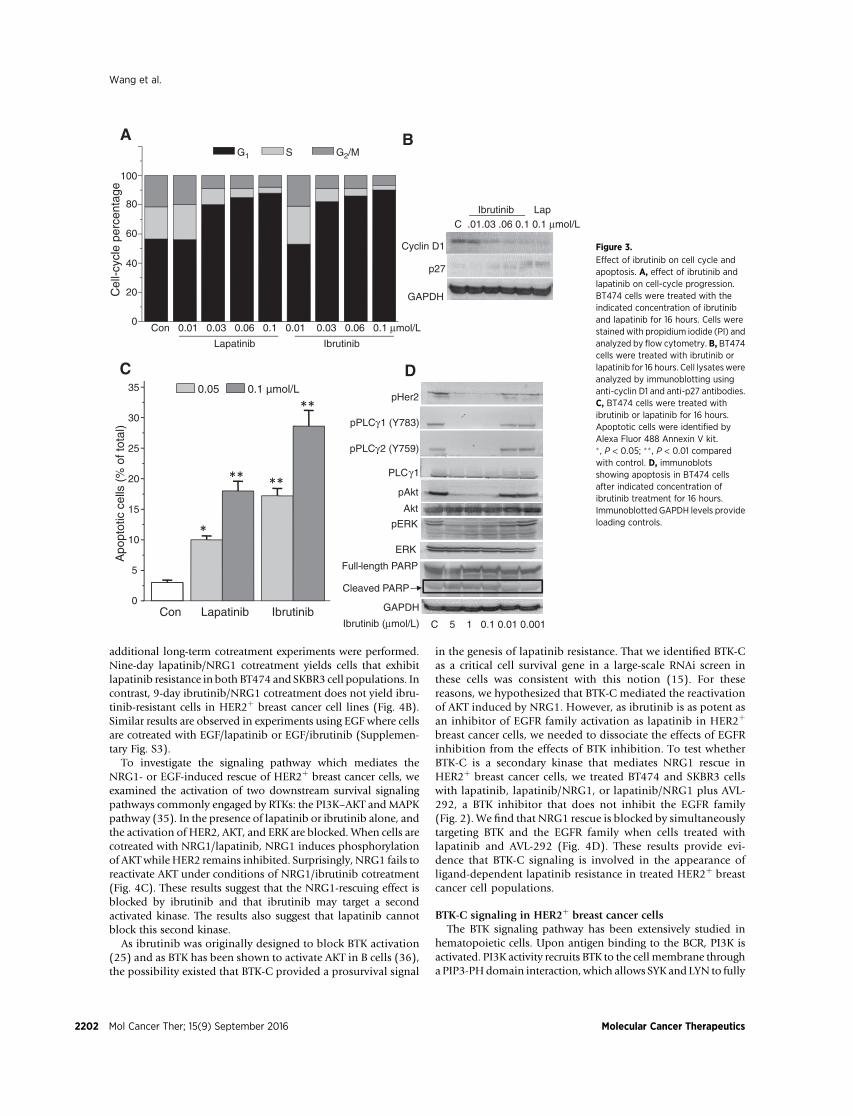
the number of cells in the S phase of the cell cycle is observed at aconcentration of 0.03 mmol/L for ibrutinib when compared withcontrols (Fig. 3A). Similar results are seen for lapatinib (31). Thiscell-cycle delay is correlated with an increase in p27, an inhibitorof cell-cycle progression, and a decrease in cyclin D1 (Fig. 3B).
An increase in the number of apoptotic cells following treat-mentwith 0.05 or 0.1mmol/L ibrutinib or lapatinib for 24hours isobserved. Our results are consistent with the results of earlierstudies, which have shown the effect of 0.1 or 0.5 mmol/Llapatinib on cell survival (31). Compared with lapatinib, ibruti-nib induces 1.5 fold more apoptosis in HER2þ breast cancer cells(Fig. 3C). The effect of ibrutinib on cell survival regulatoryproteins was also examined. Ibrutinib blocks PLCg1, PLCg2, Akt,and ERK phosphorylation, and increases cleaved PARP (Fig. 3D).Taken together, these results suggest that ibrutinib decreases cellnumbers by inducing both a G1–S delay and apoptosis in HER2þ
breast cancer cells.
BTK inhibition blocks the reactivation of AKT and ERKpathways induced by NRG1 or EGF in lapatinib-treated HER2þ
breast cancer cellsCancer cells typically express multiple receptor tyrosine kinases
(RTK) which control cell survival signals (32). RTK ligands are
produced via autocrine tumor–cell production, paracrine tumorstroma production, and systemic production (33). An increase inRTK ligands has been shown to result in the resistance of cancercells to RTK inhibitors which ultimately produces therapeuticescape (34). In HER2þ breast cancer cells, NRG1 is the mostbroadly active ligand, followed by EGF (35). We hypothesizedthat the greater efficacy of ibrutinib may impact the occurrence ofgrowth factor–dependent lapatinib resistance. We first tested theeffect of exposingHER2þ cells to 50ng/mLofNRG1withdifferentconcentrations of lapatinib or ibrutinib for 3 days. The resultsagain show that either lapatinib or ibrutinib potently suppressescell growth. However, NRG1 application is able to rescue lapa-tinib-induced growth inhibition in HER2þ breast cancer cellsallowing resistant cells to emerge from the treatment (Fig. 4A).In contrast, HER2þ breast cancer cells cannot be rescued fromibrutinib-induced growth inhibition by NRG1. Similar results areobserved using 50 ng/mL of EGF.
The ability of NRG1 or EGF signaling to rescue HER2þ breastcancer cells from growth inhibition by HER2 kinase inhibitorlapatinib is consistent with other findings (34). However, thefindings that neither NRG1 nor EGF was able to rescue HER2þ
breast cancer cells from the growth inhibition by BTK inhibitoribrutinib were unexpected. To further confirm these findings,
…MANGCLL……MPFGCLL……MPYGCLL……MPHGCLL…
EGFRHer2
ERBB3ERBB4
BTK
…LPLGSLL…
481
0
20
40
60
80
100 0.1 0.5 μmol/L
Con Lap Ibr
Inta
ct s
truc
ture
s (%
)
B
C
DpEGFR
pERBB3
ERK
C 0.1 0.05 0.1 0.05 0.1 μmol/L
Ibrutinib AVL292 Lap
pHer2
pERBB4
pAkt
Akt
pERK
GAPDH
* *
**
0
20
60
80
100
0.01 0.03 0.06 0.1 μmol/L
Control Lapatinib IbrutinibLi
ve c
ell p
erce
ntag
e
**
A
40Figure 2.
Effect of lapatinib and ibrutinib on cellgrowth and signal transduction inBT474 cells. A, cell counts of BT474cells treated with vehicle, lapatinib(Lap), and ibrutinib (Ibr) for 3 days.Results are presented as percentageof control (vehicle). Error bars,the SD from three individualexperiments; � , P < 0.05; �� , P < 0.01compared with control. B, BT474 cellswere treated with laptanib andibrutinib, as indicated, for 9 days onMatrigel. C, alignment of EGFR familymembers with C481 region of BTK.D, effects of BTK inhibitors on EGFRfamily activation. BT474 cells weretreated with different concentrationsof BTK inhibitors ibrutinib andAVL-292 or lapatinib for 2 hours.Whole-cell lysates were prepared forimmunoblotting using antibodiesagainst p-EGFR, p-HER2, p-ERBB3,p-ERBB4, p-Akt, p-ERK proteins.Immunoblotted GAPDH levels provideloading controls.
BTK Inhibitors and Drug Resistance in Breast Cancer
www.aacrjournals.org Mol Cancer Ther; 15(9) September 2016 2201
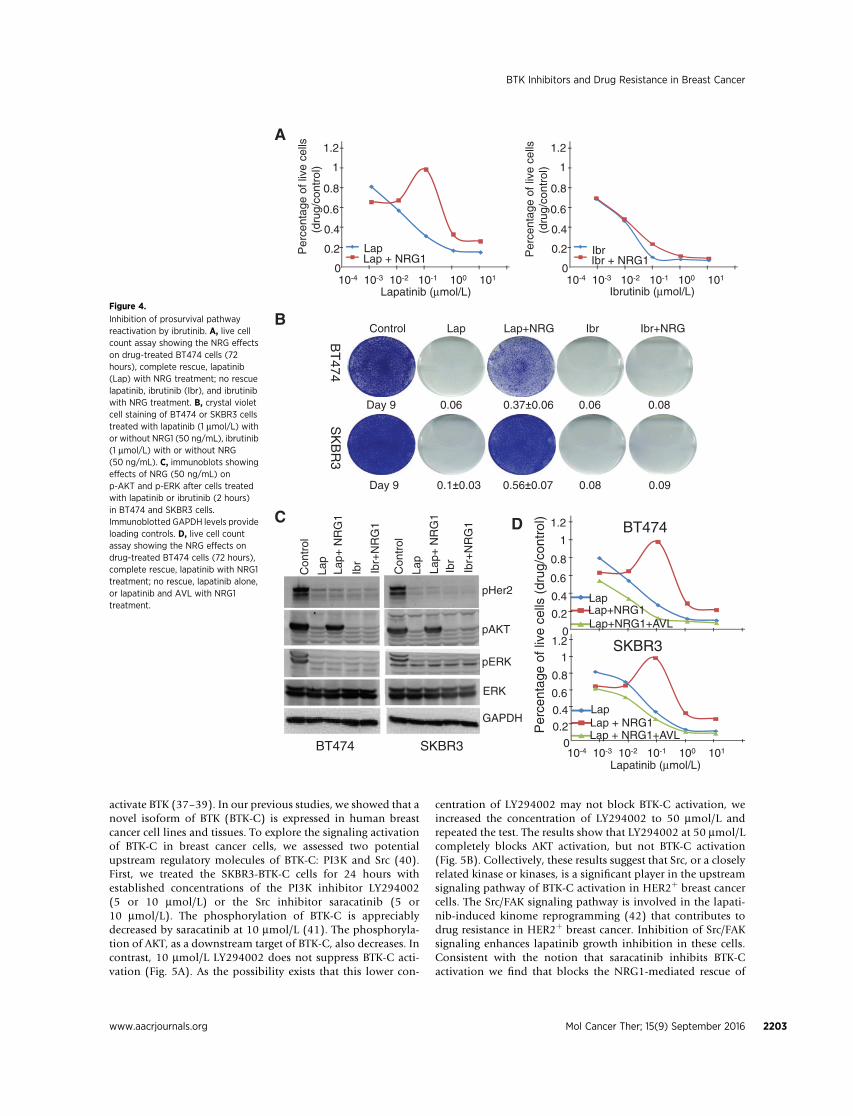
additional long-term cotreatment experiments were performed.Nine-day lapatinib/NRG1 cotreatment yields cells that exhibitlapatinib resistance in both BT474 and SKBR3 cell populations. Incontrast, 9-day ibrutinib/NRG1 cotreatment does not yield ibru-tinib-resistant cells in HER2þ breast cancer cell lines (Fig. 4B).Similar results are observed in experiments using EGF where cellsare cotreated with EGF/lapatinib or EGF/ibrutinib (Supplemen-tary Fig. S3).
To investigate the signaling pathway which mediates theNRG1- or EGF-induced rescue of HER2þ breast cancer cells, weexamined the activation of two downstream survival signalingpathways commonly engaged by RTKs: the PI3K–AKT andMAPKpathway (35). In the presence of lapatinib or ibrutinib alone, andthe activation of HER2, AKT, and ERK are blocked. When cells arecotreated with NRG1/lapatinib, NRG1 induces phosphorylationof AKTwhile HER2 remains inhibited. Surprisingly, NRG1 fails toreactivate AKT under conditions of NRG1/ibrutinib cotreatment(Fig. 4C). These results suggest that the NRG1-rescuing effect isblocked by ibrutinib and that ibrutinib may target a secondactivated kinase. The results also suggest that lapatinib cannotblock this second kinase.
As ibrutinib was originally designed to block BTK activation(25) and as BTK has been shown to activate AKT in B cells (36),the possibility existed that BTK-C provided a prosurvival signal
in the genesis of lapatinib resistance. That we identified BTK-Cas a critical cell survival gene in a large-scale RNAi screen inthese cells was consistent with this notion (15). For thesereasons, we hypothesized that BTK-C mediated the reactivationof AKT induced by NRG1. However, as ibrutinib is as potent asan inhibitor of EGFR family activation as lapatinib in HER2þ
breast cancer cells, we needed to dissociate the effects of EGFRinhibition from the effects of BTK inhibition. To test whetherBTK-C is a secondary kinase that mediates NRG1 rescue inHER2þ breast cancer cells, we treated BT474 and SKBR3 cellswith lapatinib, lapatinib/NRG1, or lapatinib/NRG1 plus AVL-292, a BTK inhibitor that does not inhibit the EGFR family(Fig. 2). We find that NRG1 rescue is blocked by simultaneouslytargeting BTK and the EGFR family when cells treated withlapatinib and AVL-292 (Fig. 4D). These results provide evi-dence that BTK-C signaling is involved in the appearance ofligand-dependent lapatinib resistance in treated HER2þ breastcancer cell populations.
BTK-C signaling in HER2þ breast cancer cellsThe BTK signaling pathway has been extensively studied in
hematopoietic cells. Upon antigen binding to the BCR, PI3K isactivated. PI3K activity recruits BTK to the cell membrane througha PIP3-PHdomain interaction, which allows SYK and LYN to fully
0
5
10
15
20
25
30
35 0.05 0.1 µmol/L
Apo
ptot
ic c
ells
(%
of t
otal
)
Con Lapatinib Ibrutinib
*
** **
**
A B
C
0
20
40
60
80
100
G1 S G2/M
Cel
l-cyc
le p
erce
ntag
e
Con 0.01 0.03 0.06 0.1 0.01 0.03 0.06 0.1 μmol/L
Lapatinib Ibrutinib
Cyclin D1
p27
GAPDH
C .01.03 .06 0.1 0.1 μmol/LIbrutinib Lap
D
pPLCγ1 (Y783)
pPLCγ2 (Y759)
PLCγ1
ERK
Full-length PARP
Cleaved PARP
C 5 1 0.1 0.01 0.001 Ibrutinib (μmol/L)
pHer2
pAkt
pERK
GAPDH
Akt
Figure 3.
Effect of ibrutinib on cell cycle andapoptosis. A, effect of ibrutinib andlapatinib on cell-cycle progression.BT474 cells were treated with theindicated concentration of ibrutiniband lapatinib for 16 hours. Cells werestained with propidium iodide (PI) andanalyzed by flow cytometry. B, BT474cells were treated with ibrutinib orlapatinib for 16 hours. Cell lysateswereanalyzed by immunoblotting usinganti-cyclin D1 and anti-p27 antibodies.C, BT474 cells were treated withibrutinib or lapatinib for 16 hours.Apoptotic cells were identified byAlexa Fluor 488 Annexin V kit.� , P < 0.05; �� , P < 0.01 comparedwith control. D, immunoblotsshowing apoptosis in BT474 cellsafter indicated concentration ofibrutinib treatment for 16 hours.Immunoblotted GAPDH levels provideloading controls.
Wang et al.
Mol Cancer Ther; 15(9) September 2016 Molecular Cancer Therapeutics2202
activate BTK (37–39). In our previous studies, we showed that anovel isoform of BTK (BTK-C) is expressed in human breastcancer cell lines and tissues. To explore the signaling activationof BTK-C in breast cancer cells, we assessed two potentialupstream regulatory molecules of BTK-C: PI3K and Src (40).First, we treated the SKBR3-BTK-C cells for 24 hours withestablished concentrations of the PI3K inhibitor LY294002(5 or 10 mmol/L) or the Src inhibitor saracatinib (5 or10 mmol/L). The phosphorylation of BTK-C is appreciablydecreased by saracatinib at 10 mmol/L (41). The phosphoryla-tion of AKT, as a downstream target of BTK-C, also decreases. Incontrast, 10 mmol/L LY294002 does not suppress BTK-C acti-vation (Fig. 5A). As the possibility exists that this lower con-
centration of LY294002 may not block BTK-C activation, weincreased the concentration of LY294002 to 50 mmol/L andrepeated the test. The results show that LY294002 at 50 mmol/Lcompletely blocks AKT activation, but not BTK-C activation(Fig. 5B). Collectively, these results suggest that Src, or a closelyrelated kinase or kinases, is a significant player in the upstreamsignaling pathway of BTK-C activation in HER2þ breast cancercells. The Src/FAK signaling pathway is involved in the lapati-nib-induced kinome reprogramming (42) that contributes todrug resistance in HER2þ breast cancer. Inhibition of Src/FAKsignaling enhances lapatinib growth inhibition in these cells.Consistent with the notion that saracatinib inhibits BTK-Cactivation we find that blocks the NRG1-mediated rescue of
Control Lap Lap+NRG Ibr Ibr+NRG
BT
474S
KB
R3
Day 9 0.06 0.37 0.06 0.06 0.08
Day 9 0.1 0.03 0.56 0.07 0.08 0.09
Lapatinib (μmol/L)10-4 10-3 10-2 10-1 100 101
0
0.2
0.4
0.6
0.8
1
1.2
LapLap + NRG1
Per
cent
age
of li
ve c
ells
(dru
g/co
ntro
l)
10-4 10-3 10-2 10-1 100 101 0
0.2
0.4
0.6
0.8
1
1.2
IbrIbr + NRG1
Per
cent
age
of li
ve c
ells
(dru
g/co
ntro
l)
Ibrutinib (μmol/L)
Lapatinib (μmol/L)10-4 10-3 10-2 10-1 100 101
0
0.2
0.4
0.6
0.8
1
1.2
LapLap + NRG1P
erce
ntag
e of
live
cel
ls (
drug
/con
trol
)
Lap + NRG1+AVL
SKBR30
0.2
0.4
0.6
0.8
1
1.2
LapLap+NRG1Lap+NRG1+AVL
BT474
A
B
C D
pHer2
pAKT
pERK
ERK
Con
trol
Lap
Lap+
NR
G1
Ibr
Ibr+
NR
G1
Con
trol
Lap
Lap+
NR
G1
Ibr
Ibr+
NR
G1
BT474 SKBR3
GAPDH
Figure 4.
Inhibition of prosurvival pathwayreactivation by ibrutinib. A, live cellcount assay showing the NRG effectson drug-treated BT474 cells (72hours), complete rescue, lapatinib(Lap) with NRG treatment; no rescuelapatinib, ibrutinib (Ibr), and ibrutinibwith NRG treatment. B, crystal violetcell staining of BT474 or SKBR3 cellstreated with lapatinib (1 mmol/L) withor without NRG1 (50 ng/mL), ibrutinib(1 mmol/L) with or without NRG(50 ng/mL). C, immunoblots showingeffects of NRG (50 ng/mL) onp-AKT and p-ERK after cells treatedwith lapatinib or ibrutinib (2 hours)in BT474 and SKBR3 cells.Immunoblotted GAPDH levels provideloading controls. D, live cell countassay showing the NRG effects ondrug-treated BT474 cells (72 hours),complete rescue, lapatinib with NRG1treatment; no rescue, lapatinib alone,or lapatinib and AVL with NRG1treatment.
BTK Inhibitors and Drug Resistance in Breast Cancer
www.aacrjournals.org Mol Cancer Ther; 15(9) September 2016 2203
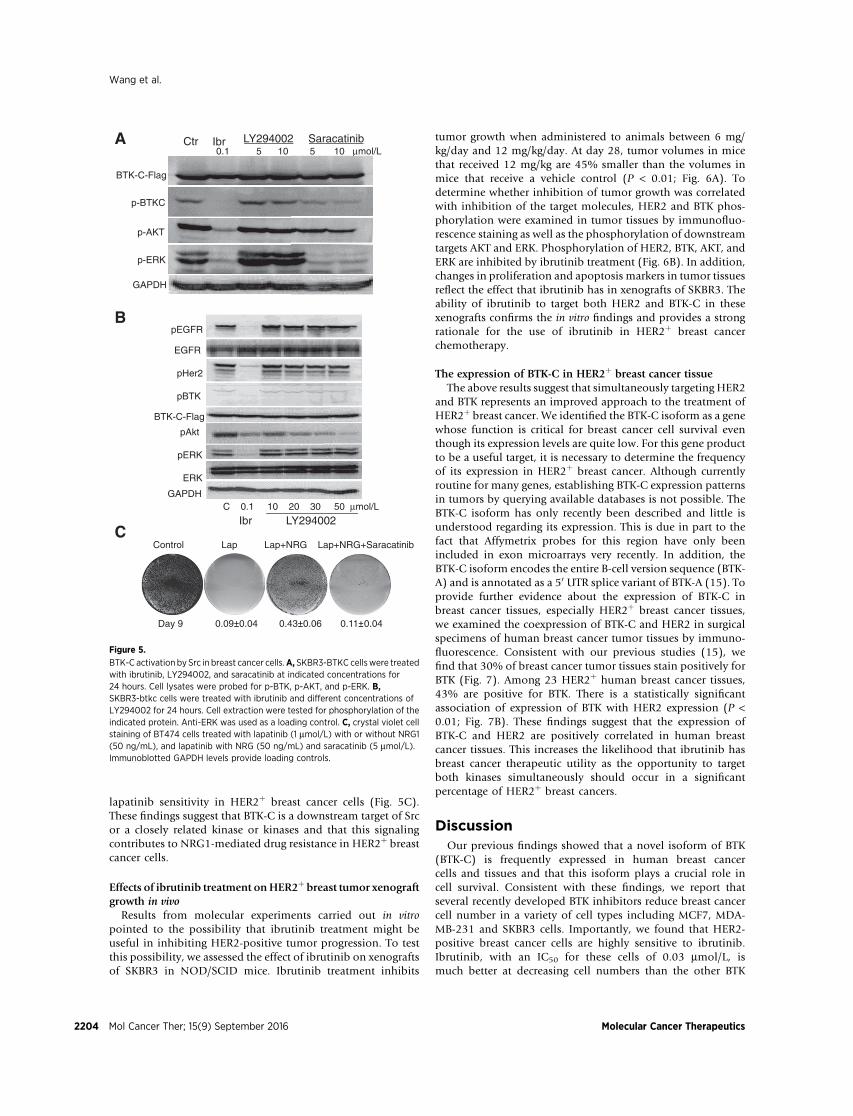
lapatinib sensitivity in HER2þ breast cancer cells (Fig. 5C).These findings suggest that BTK-C is a downstream target of Srcor a closely related kinase or kinases and that this signalingcontributes to NRG1-mediated drug resistance in HER2þ breastcancer cells.
Effects of ibrutinib treatment onHER2þ breast tumor xenograftgrowth in vivo
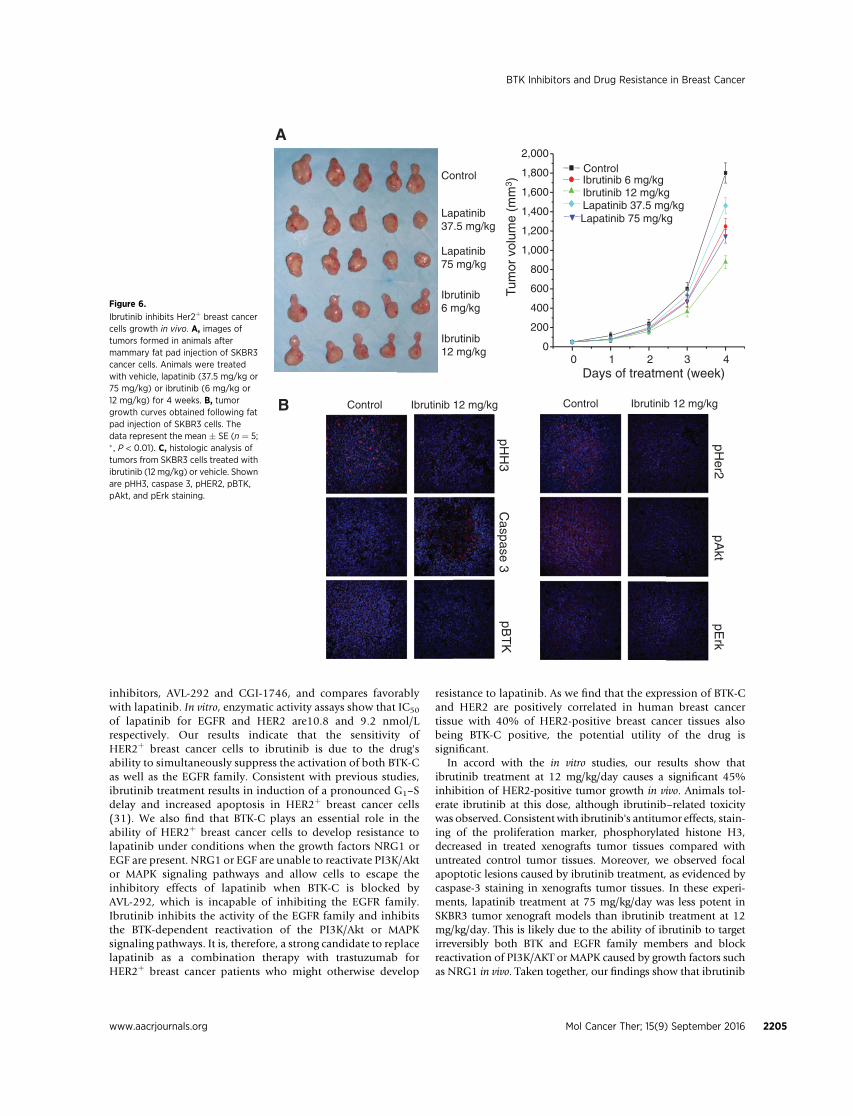
Results from molecular experiments carried out in vitropointed to the possibility that ibrutinib treatment might beuseful in inhibiting HER2-positive tumor progression. To testthis possibility, we assessed the effect of ibrutinib on xenograftsof SKBR3 in NOD/SCID mice. Ibrutinib treatment inhibits
tumor growth when administered to animals between 6 mg/kg/day and 12 mg/kg/day. At day 28, tumor volumes in micethat received 12 mg/kg are 45% smaller than the volumes inmice that receive a vehicle control (P < 0.01; Fig. 6A). Todetermine whether inhibition of tumor growth was correlatedwith inhibition of the target molecules, HER2 and BTK phos-phorylation were examined in tumor tissues by immunofluo-rescence staining as well as the phosphorylation of downstreamtargets AKT and ERK. Phosphorylation of HER2, BTK, AKT, andERK are inhibited by ibrutinib treatment (Fig. 6B). In addition,changes in proliferation and apoptosis markers in tumor tissuesreflect the effect that ibrutinib has in xenografts of SKBR3. Theability of ibrutinib to target both HER2 and BTK-C in thesexenografts confirms the in vitro findings and provides a strongrationale for the use of ibrutinib in HER2þ breast cancerchemotherapy.
The expression of BTK-C in HER2þ breast cancer tissueThe above results suggest that simultaneously targeting HER2
and BTK represents an improved approach to the treatment ofHER2þ breast cancer. We identified the BTK-C isoform as a genewhose function is critical for breast cancer cell survival eventhough its expression levels are quite low. For this gene productto be a useful target, it is necessary to determine the frequencyof its expression in HER2þ breast cancer. Although currentlyroutine for many genes, establishing BTK-C expression patternsin tumors by querying available databases is not possible. TheBTK-C isoform has only recently been described and little isunderstood regarding its expression. This is due in part to thefact that Affymetrix probes for this region have only beenincluded in exon microarrays very recently. In addition, theBTK-C isoform encodes the entire B-cell version sequence (BTK-A) and is annotated as a 50 UTR splice variant of BTK-A (15). Toprovide further evidence about the expression of BTK-C inbreast cancer tissues, especially HER2þ breast cancer tissues,we examined the coexpression of BTK-C and HER2 in surgicalspecimens of human breast cancer tumor tissues by immuno-fluorescence. Consistent with our previous studies (15), wefind that 30% of breast cancer tumor tissues stain positively forBTK (Fig. 7). Among 23 HER2þ human breast cancer tissues,43% are positive for BTK. There is a statistically significantassociation of expression of BTK with HER2 expression (P <0.01; Fig. 7B). These findings suggest that the expression ofBTK-C and HER2 are positively correlated in human breastcancer tissues. This increases the likelihood that ibrutinib hasbreast cancer therapeutic utility as the opportunity to targetboth kinases simultaneously should occur in a significantpercentage of HER2þ breast cancers.
DiscussionOur previous findings showed that a novel isoform of BTK
(BTK-C) is frequently expressed in human breast cancercells and tissues and that this isoform plays a crucial role incell survival. Consistent with these findings, we report thatseveral recently developed BTK inhibitors reduce breast cancercell number in a variety of cell types including MCF7, MDA-MB-231 and SKBR3 cells. Importantly, we found that HER2-positive breast cancer cells are highly sensitive to ibrutinib.Ibrutinib, with an IC50 for these cells of 0.03 mmol/L, ismuch better at decreasing cell numbers than the other BTK
A
B
BTK-C-Flag
p-BTKC
p-AKT
p-ERK
0.1 5 10 5 10 μmol/LSaracatinibLY294002IbrCtr
μμ
GAPDH
GAPDH
EGFR
pHer2
pBTK
pAkt
pERK
ERK
C 0.1 10 20 30 50 μmol/L
Ibr LY294002
pEGFR
BTK-C-Flag
CControl Lap Lap+NRG Lap+NRG+Saracatinib
Day 9 0.09 0.04 0.43 0.06 0.11 0.04
Figure 5.
BTK-C activation by Src in breast cancer cells.A, SKBR3-BTKC cellswere treatedwith ibrutinib, LY294002, and saracatinib at indicated concentrations for24 hours. Cell lysates were probed for p-BTK, p-AKT, and p-ERK. B,SKBR3-btkc cells were treated with ibrutinib and different concentrations ofLY294002 for 24 hours. Cell extraction were tested for phosphorylation of theindicated protein. Anti-ERK was used as a loading control. C, crystal violet cellstaining of BT474 cells treated with lapatinib (1 mmol/L) with or without NRG1(50 ng/mL), and lapatinib with NRG (50 ng/mL) and saracatinib (5 mmol/L).Immunoblotted GAPDH levels provide loading controls.
Wang et al.
Mol Cancer Ther; 15(9) September 2016 Molecular Cancer Therapeutics2204
inhibitors, AVL-292 and CGI-1746, and compares favorablywith lapatinib. In vitro, enzymatic activity assays show that IC50
of lapatinib for EGFR and HER2 are10.8 and 9.2 nmol/Lrespectively. Our results indicate that the sensitivity ofHER2þ breast cancer cells to ibrutinib is due to the drug'sability to simultaneously suppress the activation of both BTK-Cas well as the EGFR family. Consistent with previous studies,ibrutinib treatment results in induction of a pronounced G1–Sdelay and increased apoptosis in HER2þ breast cancer cells(31). We also find that BTK-C plays an essential role in theability of HER2þ breast cancer cells to develop resistance tolapatinib under conditions when the growth factors NRG1 orEGF are present. NRG1 or EGF are unable to reactivate PI3K/Aktor MAPK signaling pathways and allow cells to escape theinhibitory effects of lapatinib when BTK-C is blocked byAVL-292, which is incapable of inhibiting the EGFR family.Ibrutinib inhibits the activity of the EGFR family and inhibitsthe BTK-dependent reactivation of the PI3K/Akt or MAPKsignaling pathways. It is, therefore, a strong candidate to replacelapatinib as a combination therapy with trastuzumab forHER2þ breast cancer patients who might otherwise develop
resistance to lapatinib. As we find that the expression of BTK-Cand HER2 are positively correlated in human breast cancertissue with 40% of HER2-positive breast cancer tissues alsobeing BTK-C positive, the potential utility of the drug issignificant.
In accord with the in vitro studies, our results show thatibrutinib treatment at 12 mg/kg/day causes a significant 45%inhibition of HER2-positive tumor growth in vivo. Animals tol-erate ibrutinib at this dose, although ibrutinib–related toxicitywas observed. Consistent with ibrutinib's antitumor effects, stain-ing of the proliferation marker, phosphorylated histone H3,decreased in treated xenografts tumor tissues compared withuntreated control tumor tissues. Moreover, we observed focalapoptotic lesions caused by ibrutinib treatment, as evidenced bycaspase-3 staining in xenografts tumor tissues. In these experi-ments, lapatinib treatment at 75 mg/kg/day was less potent inSKBR3 tumor xenograft models than ibrutinib treatment at 12mg/kg/day. This is likely due to the ability of ibrutinib to targetirreversibly both BTK and EGFR family members and blockreactivation of PI3K/AKT or MAPK caused by growth factors suchas NRG1 in vivo. Taken together, our findings show that ibrutinib
Control
Lapatinib37.5 mg/kg
Lapatinib75 mg/kg
Ibrutinib6 mg/kg
Ibrutinib12 mg/kg
0 1 2 3 40
200
400
600
800
1,000
1,200
1,400
1,600
1,800
2,000ControlIbrutinib 6 mg/kgIbrutinib 12 mg/kg
Lapatinib 75 mg/kgLapatinib 37.5 mg/kg
Tum
or v
olum
e (m
m3 )
Days of treatment (week)
Control Ibrutinib 12 mg/kg
pHH
3C
aspase 3pB
TK
Control Ibrutinib 12 mg/kg
pHer2
pAkt
pErk
A
B
Figure 6.
Ibrutinib inhibits Her2þ breast cancercells growth in vivo. A, images oftumors formed in animals aftermammary fat pad injection of SKBR3cancer cells. Animals were treatedwith vehicle, lapatinib (37.5 mg/kg or75 mg/kg) or ibrutinib (6 mg/kg or12 mg/kg) for 4 weeks. B, tumorgrowth curves obtained following fatpad injection of SKBR3 cells. Thedata represent the mean � SE (n ¼ 5;� , P < 0.01). C, histologic analysis oftumors from SKBR3 cells treated withibrutinib (12 mg/kg) or vehicle. Shownare pHH3, caspase 3, pHER2, pBTK,pAkt, and pErk staining.
BTK Inhibitors and Drug Resistance in Breast Cancer
www.aacrjournals.org Mol Cancer Ther; 15(9) September 2016 2205

actively blocks BTK and EGFR family activation efficiently inhi-biting HER2þ breast cancer growth in vivo. As a result, this studysupports the use of ibrutinib as a HER2þ breast cancer treatmentand indicates that targeting BTK-C with recently developed BTKinhibitors such as ibrutinib or AVL-292 may avert the develop-ment of drug resistance in breast cancer patients.
The advent of HER2-directed therapy has significantlyimproved the prognosis of patients with metastatic and earlyHER2þ breast cancer. Currently, there are two classes of HER2-targeting drugs used in the clinic. Trastuzumab, trastuzumab-DM1, and pertuzumab are antibody-based drugs, whereaslapatinib is a small molecule that reversibly inhibits bothEGFR and HER2, blocking the PI3K/Akt and MAPK pathways.Lapatinib is the only small compound used to treat HER2þ
breast cancer that has metastasized to the brain due to itsability to cross the blood–brain barrier. Despite this recentprogress, acquired resistance to HER2-directed therapy stillresults in relapse and progression of HER2þ disease. Theprimary mechanism of resistance to lapatinib stems fromincreased levels of EGFR or HER3 ligands such as NRG1 orEGF in the tumor microenvironment which reactivate thePI3K/Akt and MAPK signaling pathways that are blocked bythe drug (34). The activation of the signaling pathway down-stream of EGFR family members other than HER2 is dependenton heterodimerization of the EGFR family member triggeredby NRG1 or EGF binding to the extracellular ligand-bindingdomain. Increased expression of HER2 at the cell membrane
leads to constitutive signaling of downstream pathways: PI3K/Akt and the Ras/Raf/MEK/MAPK, which are involved in regu-lating cell growth, survival, differentiation, migration, andmetastasis. Reactivation of these pathways allows cells toescape from the antiproliferative and antiapoptotic effects ofthe HER2 inhibitors.
Ibrutinib was the first reported covalent inhibitor of BTK (25).As an orally bioavailable, selective and irreversible inhibitor,ibrutinib has been undergoing multiple clinical trials targetingtreatment of various B-cell malignancies and has shown prom-ising clinical efficacy (10). On the basis of sequence alignments,10 kinases in the human genome have an orthologous cysteineresidue (25). The EGFR family includes EGFR/Her1, HER2, Her3,and Her4 all of which are involved in several aspects of tumor-igenesis. These kinases include EGFR, HER2, and Her4 covalentlybind to the thiol group of Cys481 in the ATP pocket region of BTK(position Cys515 of BTK-C). In vitro, enzymatic activity assaysshow that and have IC50 of ibrutinib for each enzyme in thenanomolar range BTK, EGFR,HER2 andHer4 are 0.5, 5.6, 9.4, and0.6 nmol/L respectively. In addition, ibrutinib also inhibits ITK,an essential enzyme in Th2 T cells, which shifts the balancebetween Th1 and Th2 T cells and potentially enhances antitumorimmune responses (43). Given the relevance of this spectrum oftargets, it is therefore not surprising that ibrutinib is effective indecreasing the proliferation and increasing apoptosis in HER2þ
breast cancer cells.In summary, this work demonstrates that BTK-C is a novel
therapeutic target for breast cancer, and that current recentlydeveloped BTK inhibitors may be useful in the treatment ofHER2þ breast cancers. On the basis of its efficacy in breast cancercells, its ability to reduce xenograft growth and its effects on EGFRfamily members, tests of ibrutinib used in a neoadjuvant oradjuvant setting with trastuzumab or other chemotherapeuticcompounds for HER2þ breast cancer is warranted. Moreover, asBTK inhibition abolishes the ability of either NRG1 or EGF toreactivate Akt or Erk in HER2þ breast cancer cells, drugs such asAVL-292might also be useful in suppressing therapeutic escape inthese cancers.
Disclosure of Potential Conflicts of InterestX.Wang, L. Kokabee, andD.S. Conklin are currently inventors on the patents
but do not have ownership interest at this juncture. No potential conflicts ofinterest were disclosed by the other authors.
DisclaimerSome of the data in this study have been used to support one or more patent
applications.
Authors' ContributionsConception and design: X. Wang, D.S. ConklinDevelopment of methodology: X. Wang, J. Wong, L. KokabeeAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): X. Wang, J. Wong, L. Kokabee, D.S. ConklinAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): X. Wang, L. Kokabee, Y. Sun, D.S. ConklinWriting, review, and/or revision of themanuscript: X.Wang, J. Wong, F. Khan,Y. Sun, D.S. ConklinAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): X. Wang, J. Wong, L. Kokabee, F. Khan, Y. Sun,D.S. ConklinStudy supervision: X. Wang, D.S. ConklinOther (conception and design of IHC experiment): C.J. Sevinsky
Her2 expressionPosi�ve Nega�ve
Posi�ve 10 8 18
Nega�ve 13 19 32 BTK-C
Total 23 27 50 <0.01
TotalP
Her2
Case 1 2 3A
B
Figure 7.
Positive correlation between BTK and HER2 expression in surgical specimens ofhuman breast cancer tissue.A,Her2þ and negative surgical specimens of humanbreast tumors (BR10010b) were subject to stain BTK and HER2. The datarepresent 50 of breast tumor specimens. Case 1 and case 2 represent positivestaining BTK-C, case 1 and case 3 represent positive staining HER2. B, summaryof thepositive correlation. The correlation between the overexpression ofBTK-Cand HER2 was analyzed by Fisher exact test.
Wang et al.
Mol Cancer Ther; 15(9) September 2016 Molecular Cancer Therapeutics2206

AcknowledgmentsThe authors thank members of the Conklin laboratory and the University at
Albany Laboratory Animal Care Facility for help with experimentation andcritical reading of the manuscript.
Grant SupportThis work was supported by NCI R01CA136658 and a Technology Accel-
erator Fund Award from The Research Foundation for the State University ofNew York (to D.S. Conklin).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received October 8, 2015; revised April 12, 2016; accepted May 19, 2016;published OnlineFirst June 2, 2016.
References1. Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M.
The Tec family of cytoplasmic tyrosine kinases: mammalian Btk,Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 2001;23:436–46.
2. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al.Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993;72:279–90.
3. Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. Thegene involved in X-linked agammaglobulinaemia is a member of the srcfamily of protein-tyrosine kinases. Nature 1993;361:226–33.
4. Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, et al. Bruton'styrosine kinase (Btk): function, regulation, and transformationwith specialemphasis on the PH domain. Immunol Rev 2009;228:58–73.
5. Aoki Y, Isselbacher KJ, Pillai S. Bruton tyrosine kinase is tyrosine phos-phorylated and activated in pre-B lymphocytes and receptor-ligated B cells.Proc Natl Acad Sci U S A 1994;91:10606–9.
6. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al.Defective B cell development and function in Btk-deficientmice. Immunity1995;3:283–99.
7. Kawakami Y, Yao L, Miura T, Tsukada S, Witte ON, Kawakami T. Tyrosinephosphorylation and activation of Bruton tyrosine kinase upon Fc epsilonRI cross-linking. Mol Cell Biol 1994;14:5108–13.
8. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cellmalignancies. Nat Rev Cancer 2014;14:219–32.
9. Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cellmalignancy. Int Rev Immunol 2012;31:119–32.
10. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al.The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activationand is efficacious inmodels of autoimmune disease and B-cellmalignancy.Proc Natl Acad Sci U S A 2010;107:13075–80.
11. Robak T, Robak E. Tyrosine kinase inhibitors as potential drugs for B-celllymphoid malignancies and autoimmune disorders. Expert Opin InvestDrugs 2012;21:921–47.
12. Di Paolo JA,Huang T, BalazsM, Barbosa J, Barck KH, Bravo BJ, et al. SpecificBtk inhibition suppresses B cell- and myeloid cell-mediated arthritis. NatChem Biol 2011;7:41–50.
13. Vassilev AO, Uckun FM. Therapeutic potential of inhibiting Bruton'styrosine kinase, (BTK). Curr Pharm Des 2004;10:1757–66.
14. Kim ES, Dhillon S. Ibrutinib: a review of its use in patients with mantlecell lymphoma or chronic lymphocytic leukaemia. Drugs 2015;75:769–76.
15. Eifert C, Wang X, Kokabee L, Kourtidis A, Jain R, Gerdes MJ, et al. A novelisoform of the B cell tyrosine kinase BTK protects breast cancer cells fromapoptosis. Genes Chromosomes Cancer 2013;52:961–75.
16. Brown-Glaberman U, Dayao Z, Royce M. HER2-targeted therapy forearly-stage breast cancer: a comprehensive review. Oncology 2014;28:281–9.
17. De Placido S, Pronzato P. Treatment options in HRþ/HER2� advancedbreast cancer patients pretreated with nonsteroidal aromatase inhibi-tors: what does current evidence tell us? Future Oncol 2015;11:975–81.
18. Figueroa-Magalhaes MC, Jelovac D, Connolly RM, Wolff AC. Treatment ofHER2-positive breast cancer. Breast 2014;23:128–36.
19. Wang X, Sun Y, Wong J, Conklin DS. PPARgamma maintains ERBB2-positive breast cancer stem cells. Oncogene 2013;32:5512–21.
20. Wang X, Zhao J. KLF8 transcription factor participates in oncogenictransformation. Oncogene 2007;26:456–61.
21. Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S,et al. Bruton tyrosine kinase represents a promising therapeutic target fortreatment of chronic lymphocytic leukemia and is effectively targeted byPCI-32765. Blood 2011;117:6287–96.
22. Burger JA. Bruton's tyrosine kinase (BTK) inhibitors in clinical trials. CurrHematol Malig Rep 2014;9:44–9.
23. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al.Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia.N Engl J Med 2013;369:32–42.
24. O'Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA,et al. Ibrutinib as initial therapy for elderly patients with chro-nic lymphocytic leukaemia or small lymphocytic lymphoma: anopen-label, multicentre, phase 1b/2 trial. Lancet Oncol 2014;15:48–58.
25. Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al.Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase.ChemMedChem 2007;2:58–61.
26. Grabinski N, Ewald F. Ibrutinib (ImbruvicaTM) potently inhibits ErbBreceptor phosphorylation and cell viability of ErbB2-positive breast cancercells. Invest New Drugs 2014;32:1096–104.
27. Lovitt CJ, Shelper TB, Avery VM. Evaluation of chemotherapeutics in athree-dimensional breast cancer model. J Cancer Res Clin Oncol 2015;141:951–9.
28. Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathwayactivation and response of breast cancer cells to HER2-targeting agents isdependent strongly on the 3D microenvironment. Breast Cancer Res Treat2010;122:35–43.
29. Eroglu Z, Tagawa T, Somlo G. Human epidermal growth factor receptorfamily-targeted therapies in the treatment of HER2-overexpressing breastcancer. Oncologist 2014;19:135–50.
30. Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB,Golas J, et al. Antitumor activity of HKI-272, an orally active,irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res 2004;64:3958–65.
31. Konecny GE, PegramMD, Venkatesan N, Finn R, Yang G, RahmehM, et al.Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res2006;66:1630–9.
32. Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, et al. Akt-RSK-S6 kinasesignaling networks activated by oncogenic receptor tyrosine kinases. SciSignal 2010;3:ra64.
33. Zhang W, Huang P. Cancer-stromal interactions: role in cell survival,metabolism and drug sensitivity. Cancer Biol Ther 2011;11:150–6.
34. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al.Widespreadpotential for growth-factor-driven resistance to anticancer kinase inhibi-tors. Nature 2012;487:505–9.
35. Grant S,Qiao L,Dent P. Roles of ERBB family receptor tyrosine kinases, anddownstream signaling pathways, in the control of cell growth and survival.Front Biosci 2002;7:d376–89.
36. Lindvall J, Islam TC. Interaction of Btk and Akt in B cell signaling. BiochemBiophys Res Commun 2002;293:1319–26.
37. Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, et al.Amplification of B cell antigen receptor signaling by a Syk/ITAM positivefeedback loop. Mol Cell 2002;10:1057–69.
38. Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domainand phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J BiolChem 2001;276:16201–6.
www.aacrjournals.org Mol Cancer Ther; 15(9) September 2016 2207
BTK Inhibitors and Drug Resistance in Breast Cancer

39. Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, ScharenbergA, et al. BTK regulates PtdIns-4,5-P2 synthesis: importancefor calcium signaling and PI3K activity. Immunity 2003;19:669–78.
40. Li Z, Wahl MI, Eguinoa A, Stephens LR, Hawkins PT, Witte ON.Phosphatidylinositol 3-kinase-gamma activates Bruton's tyrosinekinase in concert with Src family kinases. Proc Natl Acad Sci U S A1997;94:13820–5.
41. Bruyere C, Abeloos L, Lamoral-Theys D, Senetta R, Mathieu V, LeMercier M, et al. Temozolomide modifies caveolin-1 expression in
experimental malignant gliomas in vitro and in vivo. Transl Oncol2011;4:92–100.
42. Stuhlmiller TJ,Miller SM, Zawistowski JS,NakamuraK, BeltranAS,DuncanJS, et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep2015;11:390–404.
43. Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Ther-apeutic antitumor immunity by checkpoint blockade is enhanced byibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A2015;112:E966–72.
Mol Cancer Ther; 15(9) September 2016 Molecular Cancer Therapeutics2208
Wang et al.