calcium-activated chloride channels in cystic...
TRANSCRIPT

UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Calcium-activated Chloride Channels in Cystic Fibrosis
JOANA RAQUEL DELGADO MARTINS
DOUTORAMENTO EM BIOQUÍMICA
(Especialidade: Genética Molecular)
2011


UNIVERSIDADE DE LISBOA
FACULDADE DE CIÊNCIAS
DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA
Calcium-activated Chloride Channels in Cystic Fibrosis
JOANA RAQUEL DELGADO MARTINS
Tese co-orientada pela Prof. Doutora Margarida D. Amaral e pelo
Prof. Doutor Karl Kunzelmann
DOUTORAMENTO EM BIOQUÍMICA
(Especialidade: Genética Molecular)
2011


Joana Raquel Delgado Martins foi bolseira de
doutoramento da Fundação para a Ciência e Tecnologia do
Ministério da Ciência, Tecnologia e Ensino Superior
SFRH / BD / 28663 / 2006


De acordo com o disposto no artigo 40° do Regulamento de Estudos
Pós-Graduados da Universidade de Lisboa, Deliberação n°961/2003, publicada
no Diário da República – IIa Série, n° 153 de 5 de Julho de 2003, foram
incluídos nesta tese resultados dos artigos abaixo indicados:
Ousingsawat, J., Martins, J. R., Schreiber, R., Rock, J. R., Harfe, B. D.,
Kunzelmann, K. Loss of TMEM16A causes a defect in epithelial Ca2+-
dependent chloride transport. The Journal of Biological Chemistry 284, 28698-
703 (2009).
Martins, J. R., Kongsuphol, P., Almaça, J., AlDehni, F., Clarke, L.,
Schreiber, R., Amaral, M.D., Kunzelmann, K. F508del-CFTR increases the
intracellular Ca2+ signalling that causes enhanced calcium-dependent Cl-
conductance in cystic fibrosis (2010) (Submitted to AJRCMB)
No cumprimento do disposto na referida deliberação, a autora esclarece
serem da sua inteira responsabilidade, excepto quando referido em contrário, a
execução das experiências que permitiram a elaboração dos resultados
apresentados assim como a interpretação e discussão dos mesmos. Os
resultados obtidos por outros autores foram incluídos com autorização dos
mesmos para facilitar a compreensão dos trabalhos e estão assinalados nas
respectivas figuras.

Outros artigos publicados em revistas internacionais contendo resultados
obtidos durante o doutoramento:
Spitzner, M., Martins, J. R., Barro Soria, R., Ousingsawat, J., Scheidt, K.,
Schreiber, R., and Kunzelmann, K. Eag1 and Bestrophin 1 are up-regulated in
fast-growing colonic cancer cells. The Journal of Biological Chemistry 283,
7421-8 (2008).
Aldehni, F., Spitzner, M., Martins, J. R., Schreiber, R. and Kunzelmann,
K. Bestrophin 1 promotes epithelial-to-mesenchymal transition of renal
collecting duct cells. Journal of the American Society of Nephrology : JASN 20,
1556-64 (2009).
Schreiber, R., Uliyakina, I., Kongsuphol, P., Warth, R., Mirza, M., Martins,
J. R. and Kunzelmann K. Expression and function of epithelial anoctamins. The
Journal of Biological Chemistry 285, 7838-45 (2010).
Kunzelmann, K.; Kongsuphol, P.; Chootip, K.; Toledo, C.; Martins, J. R.;
Almaca, J.; Tian, Y.; Witzgall, R.; Ousingsawat, J.; Schreiber, R. Role of the
Ca2+-activated Cl- channels bestrophin and anoctamin in epithelial cells.
Biological chemistry 392, 125-34 (2011).

Preface
i
PREFACE
Cystic Fibrosis (CF) is a life-threatening genetic disorder that primarily
affects Caucasians. The disease is dominated by chronic bacterial infections
resulting from the excessive build-up of a thick mucus in the respiratory system.
Progressive loss of lung function and its ultimate failure is the main cause of
mortality. Other symptoms include pancreatic insufficiency, male infertility and
high sweat electrolytes. Life expectancy of patients suffering from CF is about
37 years of age. In the late 1980s the joint efforts of three independent
laboratories unveiled that the gene coding for the Cystic Fibrosis
Transmembrane Conductance Regulator (CFTR) Cl- channel was the cause of
CF when dysfunctional.
CFTR activity as a Cl- channel is crucial for proper function and ionic
transport of all epithelia, including those of the airways, intestinal tract (including
pancreatic and bile ducts) as well as sweat glands and the male reproductive
system. CFTR protein is a cAMP-dependent and phosphorylation-regulated
chloride (Cl-) channel which, in healthy tissues, is present in the apical
membrane of epithelial cells. In the majority of CF patients CFTR bears a
mutation (F508del) that causes its intracellular retention at the endoplasmic
reticulum (ER), thus preventing it from reaching the plasma membrane of
epithelial cells.
As there is up to now no cure for the disease, most current therapies aim
to alleviate and minimize CF symptoms. However, several drug-discovery
efforts have been attempted to find pharmacological agents capable of
correcting F508del-CFTR intracellular mislocation and potentiating the function
of rescued mutant channels. Although CFTR main function is undoubtedly to
transport Cl- ions, this channel is also a key player in the physiology of epithelial
tissues. Indeed, the complex regulation of ionic transport in tissues affected by
CF compromises other players of which CFTR has been shown to be a key
regulator. Nevertheless, alternatively to the cAMP second messenger pathway

Preface
ii
that leads to CFTR activation, Ca2+ is also able to trigger a generally transient
Cl- conductance in a variety of tissues. It is believed that by stimulating
alternative pathways that enable Cl- secretion, and are thus capable of
replacing at least this aspect of CFTR function in epithelia, the basic CF defect
might be overcome. In this regard, Ca2+-activated Cl- channels (CaCCs) appear
as obvious targets to be stimulated. Moreover, it has been repeatedly reported
that in tissues where CFTR is absent or defective, an enhanced Ca2+-
dependent Cl- secretion is observed, prompting the hypothesis that the
regulation of the two pathways might not be independent. Regardless that the
Ca2+-dependent Cl- conductance has been well-known for more than twenty
years, the molecular identity of CaCC has been considerably controversial.
Multiple proteins have been suggested to account for the Ca2+-dependent Cl-
currents (ICaCC) triggered upon rise in the intracellular Ca2+ concentration
([Ca2+]i) but a consistent candidate has just recently been found.
The objective of this work was to investigate the role and contribution of
two proteins described as CaCCs that could be potentially activated in a CF
scenario. When this doctoral work started, significant experimental data pointed
to bestrophin 1 as the major CaCC candidate, but during its course ANO1 was
definitely identified as the major CaCC. The focus of the present work was thus
both bestrophin 1 (Best 1) and anoctamin 1 (Ano 1 or TMEM16A). The impact
of these proteins in the homeostasis and function of epithelial tissues known to
be affected by CF was assessed here through biochemical and
electrophysiological techniques. The observations found in this study contribute
to a better understanding of how CaCC activity modulates (and is also
modulated) in CF. It also gives new perspectives in using alternative
approaches to overcome the CF basic defect (by the so-called "bypass
therapies ") to the ultimate benefit of CF patients.

Acknowledgments/Agradecimentos
iii
ACKNOWLEDGMENTS/AGRADECIMENTOS
À Professora Margarida Amaral, por me ter dado a excelente
oportunidade de trabalhar no seu laboratório. Agradeço a supervisão e o
entusiasmo sempre presentes, que foram essenciais na realização deste
trabalho.
I would like to thank Karl Kunzelmann for accepting me in his group in
Regensburg and for introducing me to the fascinating field of physiology when I
first arrived in 2006. I am grateful for his support and guidance, as well as for
his inspiring passion for science.
Ao ministério da Ciência e Ensino Superior e à Fundação para a Ciência
e a Tecnologia, por terem possibilitado a realização deste trabalho, através da
concessão da bolsa de doutoramento de que fui recipiente.
A number of scientists have contributed to this work in so many ways. To
Eva Sammels and Jason Rock for essential work. Thank you for making this
thesis more meaningful. A special thanks to Barbara Reich, for her kindness
and patience while helping me with experiments.
A PhD is a challenging test of determination and persistence, and I have
been privileged enough to be surrounded by exceptional people, both in Lisbon
and Regensburg.
I thank everyone in the CF lab in Lisbon: à Marta, por toda a tua alegria e
pela confiança em mim. Marisa, sempre uma fonte de energia e boa
disposição. Filipa, Anabela e Ana Carina por todo o apoio e pela referência que
foram principalmente no início do meu doutoramento. Toby for introducing me
to protein purification, Luka and Shehrazade for your contributions to this work
and for your friendship. Joana, Inna, Luisa Alessio e Mário pelas pessoas
extraordinárias que são. André, (Prof.) Carlos, Carla, Simão e Francisco um
muito obrigada!

Acknowledgments/Agradecimentos
iv
To everyone I met in the in Regensburg: Brigitte, Julia, Agnes, Tini, Tina,
Rene, Nesty, Melanie and Myriam, thank you for your priceless help and
friendship. To Ji and Fadi, for helping me with a number of experiments and for
making science even more fun! Aim and Diana, with whom I shared so many
moments of my life, thank you for your friendship and for being there for me
(also for the invaluable contributions to this work). I am grateful to Prof. Rainer
Schreiber, for sharing his vast knowledge and for all his help and patience.
I am very grateful to everyone who accepted reading and who helped me
revising this thesis.
A David, simplemente por todo. Gracias por el apoyo, sugestiones y el
tiempo dedicado a esta tesis. A Miriam, por la ayuda en la revisión de esta
tesis. Aos amigos de sempre, principalmente Telma, Mariana, Célia e Letícia
por me conseguirem surpreender sempre. Obrigada, por apesar de longe,
andarem sempre por perto.
Aos meus pais. Pelo amor, força e inesgotável confiança em mim e sem
a qual esta tese não teria sido possível. Porque tudo vos devo, e porque nunca
vos poderei agradecer o suficiente, é também vossa esta tese.

Table of contents
v
TABLE OF CONTENTS
PREFACE ................................................................................................................ I
ACKNOWLEDGMENTS/AGRADECIMENTOS .................................................................. III
SUMMARY .............................................................................................................IX
RESUMO.............................................................................................................. XIII
ABBREVIATIONS .................................................................................................. XVII
I GENERAL INTRODUCTION .................................................................................. 3
1 CYSTIC FIBROSIS ............................................................................................. 3
1.1 FROM EARLY REPORTS TO GENE IDENTIFICATION ..................................................... 3
1.2 CLINICAL EXPRESSION ........................................................................................ 4
2 CFTR ............................................................................................................ 6
2.1 FROM GENE TO PROTEIN: THE GENETICS OF CYSTIC FIBROSIS .................................... 6
2.2 CFTR PROTEIN .................................................................................................. 8
2.3 F508DEL-CFTR PROTEIN................................................................................... 11
2.4 CFTR FUNCTIONS IN EPITHELIA ........................................................................... 12
3 CA2+-ACTIVATED CL- CHANNELS .................................................................... 20
3.1 CA2+ VS. CAMP DEPENDENT CL- CONDUCTANCE IN CF ...................................... 20
3.2 PROPERTIES OF CA2+-ACTIVATED CL CHANNELS .................................................. 22
3.3 MOLECULAR IDENTITY OF CACC ....................................................................... 27
4 CF RESCUING STRATEGIES .............................................................................. 39
4.1 RESCUING OF CFTR ......................................................................................... 39
4.2 IDENTIFYING MODIFIER GENES AS TARGETS ........................................................... 40
4.3 BYPASS APPROACHES ...................................................................................... 40
II OBJECTIVES OF THE PRESENT WORK ................................................................... 45

Table of contents
vi
III MATERIALS AND METHODS.............................................................................. 49
1 MOLECULAR BIOLOGY ................................................................................... 49
1.1 PLASMIDS, CDNAS AND SIRNAS ....................................................................... 49
1.2 SITE-DIRECTED MUTAGENESIS ............................................................................ 49
1.3 COMPETENT BACTERIA PREPARATION ................................................................. 50
1.4 TRANSFORMATION AND ISOLATION OF PLASMID DNA .......................................... 51
1.5 PLASMID DNA EXTRACTION AND PRELIMINARY ANALYSIS BY PCR TO ASSESS FOR THE
PRESENCE OF THE RESPECTIVE INSERT ............................................................................. 52
1.6 SEQUENCING OF NOVEL PLASMIDS .................................................................... 52
1.7 RNA ISOLATION, CDNA AND REAL TIME RT-PCR IN CFBE CELLS .......................... 52
1.8 RNA ISOLATION, CDNA AND REAL TIME RT-PCR IN NASAL CELLS: ......................... 53
2 CELL CULTURE, TRANSFECTIONS AND PROTEIN ANALYSIS ....................................... 55
2.1 MAMMALIAN CELL LINES ................................................................................... 55
2.2 CELL CULTURE CONDITIONS .............................................................................. 56
2.3 TRANSFECTIONS ............................................................................................... 56
2.4 IMMUNOCYTOCHEMISTRY.................................................................................. 57
2.5 WESTERN BLOT ................................................................................................ 57
2.6 CO-IMMUNOPRECIPITATION .............................................................................. 58
3 FUNCTIONAL ANALYSIS ................................................................................... 59
3.1 IODIDE QUENCHING ......................................................................................... 59
3.2 MEASUREMENT OF THE INTRACELLULAR CA2+ CONCENTRATION ............................. 60
3.3 CRNA AND DOUBLE ELECTRODE VOLTAGE CLAMP: .............................................. 61
4 ANIMALS ..................................................................................................... 62
4.1 ANIMALS AND TISSUE PREPARATION .................................................................... 62
4.2 MEASUREMENT OF MUCOCILIARY CLEARANCE ................................................... 62

Table of contents
vii
4.3 USSING CHAMBER ............................................................................................ 63
IV RESULTS AND DISCUSSION ............................................................................... 67
CHAPTER 1. LOSS OF ANO 1 CAUSES A DEFECT IN EPITHELIAL CA2+-DEPENDENT CL-
TRANSPORT .................................................................................................. 67
1 ABSTRACT .................................................................................................... 67
2 INTRODUCTION ............................................................................................. 67
3 RESULTS AND DISCUSSION ............................................................................... 68
3.1 DEFECTIVE CA2+-DEPENDENT CL- SECRETION IN AIRWAYS OF ANO 1 -/- ANIMALS .... 68
3.2 ANO 1 IS IMPORTANT FOR MUCOCILIARY CLEARANCE IN MURINE TRACHEA ........... 72
4 SUPPLEMENTARY DATA ................................................................................... 75
CHAPTER 2. F508DEL-CFTR INCREASES THE INTRACELLULAR CA2+ SIGNALLING THAT
CAUSES ENHANCED CA2+-DEPENDENT CL- CONDUCTANCE IN CYSTIC FIBROSIS .............. 77
1 ABSTRACT .................................................................................................... 77
2 INTRODUCTION ............................................................................................. 77
3 RESULTS ....................................................................................................... 80
3.1 CELLULAR LOCALIZATION OF ANO 1 AND BESTROPHIN .......................................... 80
3.2 EXPRESSION OF ANO 1 AND BESTROPHIN 1 IN CF AND NON-CF AIRWAY EPITHELIAL
CELLS AND EFFECTS OF LPS AND CF-SPUTUM ................................................................. 81
3.3 F508DEL-CFTR CHANGES INTRACELLULAR CA2+ SIGNALLING ................................ 84
3.4 ER-TRAPPED F508DEL-CFTR FACILITATES CA2+ MOVEMENTS BY ACTING AS A CL-
COUNTER-ION CHANNEL ............................................................................................. 88
3.5 EXPRESSION OF IRBIT ANTAGONIZES ENHANCED CA2+ SIGNALS IN XENOPUS OOCYTES: .
..................................................................................................................... 89
4 DISCUSSION ................................................................................................. 92
4.1 INFLAMMATION IN CF....................................................................................... 92

Table of contents
viii
4.2 ENHANCED CA2+-DEPENDENT ACTIVATION OF ANO 1 .......................................... 93
4.3 F508DEL-CFTR CONTROLS INTRACELLULAR CA2+ SIGNALLING ............................... 93
4.4 ENHANCED CA2+ SIGNALLING, PROLIFERATION AND THE ROLE OF IRBIT .................. 94
5 SUPPLEMENTARY DATA ................................................................................... 95
V GENERAL DISCUSSION AND FUTURE PERSPECTIVES ............................................ 103
VI REFERENCES ............................................................................................... 113

Summary
ix
SUMMARY
Proper ion transport is crucial for maintenance, hydration and also
protection against infection of epithelial tissues. The dysfunction of the Cystic
Fibrosis Transmembrane Conductance Regulator (CFTR) Cl- channel results in
Cystic Fibrosis (CF), the most common genetic disease among Caucasians.
CFTR and Ca2+-activated Cl- channels (CaCCs) are main Cl- channels present
in the respiratory epithelia and are stimulated by cAMP and Ca2+, respectively.
Data demonstrating upregulated Ca2+ activated Cl- currents (ICaCC) when CFTR
is absent or defective have been well described for long and are of remarkable
interest for further studies and potential therapeutic approaches to CF.
In the first part of this doctoral work, we used Anoctamin 1 (Ano 1) null
mice to look into the role of CaCCs in the airways, so as to elucidate the
potential contribution of this alternative Cl- channel to tissue homeostasis.
Ussing chamber measurements were made on freshly isolated tracheas from
newborn Ano -/- and Ano +/+ mice. Application of the muscarinic M3 receptor
agonist carbachol (CCH) or apical stimulation of purinergic receptors through
UTP and ATP was performed to elicit Ca2+-activated Cl- secretion. In Ano 1 +/+
mice, CCH induced Cl- secretion was sensitive to the CaCCs inhibitor niflumic
acid and also to Ca2+ depletion from the endoplasmic reticulum (ER). On the
other hand, in Ano 1 -/- animals Cl- secretion was completely abolished when
using CCH as an agonist. Additionally, both UTP and ATP evoked a transient
Cl- secretion in Ano 1 +/+ tracheas, but had only small effects on Ano 1 -/-
tracheas. We also indirectly evaluated the efficiency of mucociliary transport in
the airways upon cholinergic stimulation by measuring the movement of
polystyrene microspheres over time on fleshly isolated tracheas. Particle
transport was activated in Ano 1 +/+ tracheas by stimulation with CCH, while on
Ano 1 -/- tissues a lack of cholinergic mucociliary clearance was observed.
Altogether, our results demonstrate that in the absence of Ano 1 both Cl-
secretion and mucociliary clearance are impaired, pointing towards a critical
role of Ano 1 in the airways. Furthermore, we speculate that the novel insights

Summary
x
in the function of Ano 1 provide the basis for novel treatments in CF (the so-
called "bypass therapies"), being Ano 1 an alternative conductor of Cl- when
CFTR is absent or defective.
In the second part of this work we aimed to investigate the link between
CFTR and CaCCs. We first compared the mRNA levels of membrane localized
Ano 1, as well as those of Bestrophin 1, in freshly isolated nasal cells from
F508del-homozygous CF patients and non-CF healthy controls. We also
compared the mRNA levels of these channels in cell lines stably expressing
wild type (wt) or F508del-CFTR. In both cases no increase in transcript levels of
neither Ano 1 nor Best 1 was found in the F508del-CFTR cells. It is thus unlikely
that changes in ion channel expression account for enhanced CaCC activity in
native CF airway epithelial cells. Exposure to lipopolysaccharides (LPS) only
slightly increased Ano 1 and Best 1 mRNA levels, both in wt- and F508del-
CFTR cells. Taken together, the enhanced Ca2+-activated Cl- conductance
observed in airway epithelial cells expressing F508del-CFTR is not explained
neither by an increase in transcripts of known CaCCs nor exposure to
proinflammatory agents such as LPS. We next examined whether F508del-
CFTR has an effect on receptor-mediated Ca2+ signalling. Our results showed
that the presence of F508del-CFTR augments receptor mediated Ca2+
signalling, comparatively to the effects observed in the presence of wt-CFTR.
To elucidate the possible mechanism by which ER-localized F508del-CFTR
augments intracellular Ca2+ signals, we hypothesized that F508del-CFTR
trapped in the ER could affect receptor-mediated Ca2+ release from ER Ca2+
stores. Our results showed that not only UTP-induced increase in intracellular
calcium concentration ([Ca2+]i) was largely Cl- dependent but also that the
presence of F508del-CFTR in the ER may affect intracellular Ca2+ signalling by
acting as a Cl- counter-ion channel, thereby facilitating Ca2+ movement across
the ER membrane.
Altogether, these results give new insights into the fine tuning of ion
transport in the airways. Moreover, they contribute to a better understanding of

Summary
xi
the pathophysiological basis of CF disease, giving new perspectives to bypass
therapies for Cystic Fibrosis.
Keywords: Cystic Fibrosis; CFTR; CaCC; Ano 1; Bestrophin 1; counter
ion channel; Ca2+ signalling; Ca2+-activated Cl- currents; airways.


Resumo
xiii
RESUMO
A Fibrose Quística (FQ) é uma doença genética recessiva fatal que
afecta aproximadamente 70,000 doentes em todo o mundo. A população
Caucasiana é a mais atingida com uma frequência de 1 em cada 2000-4500
nascimentos. A principal causa de morte decorre da doença pulmonar crónica
que afecta os doentes, que se caracteriza por recorrentes infecções
bacterianas nas vias respiratórias e que são principal causa da deterioração
progressiva da função pulmonar. Nos pulmões de doentes com FQ, ocorre um
desequilíbrio iónico do líquido que reveste as vias respiratórias designado ASL,
(do inglês, airway surface liquid) levando à acumulação de um muco espesso.
Esta alteração da viscosidade e de outras propriedades do muco impede o
eficiente transporte mucociliar, o qual facilita a colonização por bactérias,
principalmente Pseudomonas aeruginosa. As recorrentes infecções bacterianas
e o processo inflamatório que daí resulta, geram um ciclo vicioso que conduz à
perda progressiva da função pulmonar e na morte precoce do doente. Um dos
meios de diagnóstico mais usados, mesmo após a descoberta do defeito
molecular responsável pela doença, continua a ser a medição da concentração
de sais no suor (esta está elevada em doentes que sofrem de FQ). Outros
sintomas da FQ, incluem nomeadamente, insuficiência pancreática, meconium
ileus (ileo meconial), diabetes e infertilidade masculina.
De acordo com os dados mais recentes disponíveis, a esperança média
de vida dos doentes é de aproximadamente 37 anos de vida. O constante
aumento da esperança média de vida reflecte as consideráveis melhoras no
tratamento e avanços no conhecimento da doença, nomeadamente desde a
descoberta do gene que está mutado na FQ que codifica para a proteína CFTR
(do inglês, Cystic Fibrosis Transmembrane Conductance Regulator) no final
dos anos 80 do século XX e a demonstração da sua função como canal
transportador de iões cloreto (Cl-). Com efeito, em 1989, os esforços conjuntos
de três laboratórios independentes provaram que a disfunção deste canal de
Cl- era responsável pela doença. Actualmente é amplamente aceite que a

Resumo
xiv
proteína CFTR é essencial para a manutenção do correcto transporte iónico
nas vias aéreas, tracto gastrointestinal (incluindo os ductos pancreáticos e
biliares) assim como nos ductos das glândulas sudoríparas e também no tracto
reprodutivo masculino. A proteína CFTR é um canal de Cl- dependente do
cAMP regulado por fosforilação dependente da PKA (do inglês, protein kinase
A) e que, em indivíduos saudáveis, se localiza na membrana apical das células
epiteliais. Apesar de já terem sido identificadas e descritas mais de 1800
mutações em doentes FQ, a maioria (~90%) apresenta a mutação F508del
pelo menos num alelo. Esta mutação impede a proteína CFTR de ser
correctamente processada até à membrana apical das células epiteliais, sendo
retida intracelularmente a nível do retículo endoplasmático (RE).
Até à data, esta doença é incurável, estando os esforços clínicos
concentrados no alívio e minimização dos sintomas dos doentes. Nestes 22
anos que decorreram desde a descoberta do gene, a comunidade científica
tem também concentrado esforços na descoberta e desenvolvimento de drogas
capazes de corrigir o princípio básico da FQ. A droga ideal seria capaz de não
só corrigir a localização errada da proteína mutada, mas também de potenciar
a sua função.
Apesar da principal função da proteína CFTR ser inquestionavelmente
transportar iões Cl-, este canal é também um factor chave na fisiologia e
correcto funcionamento dos tecidos epiteliais. Com efeito, a complexa
regulação do transporte iónico nos tecidos e órgãos afectados pela FQ inclui
outros intervenientes, os quais foram demonstrados ser regulados pela
proteína CFTR. Porém, para além da sinalização celular dependente de cAMP,
de que depende a activação da CFTR, também a alteração das concentrações
intracelulares de cálcio é capaz de activar o transporte de Cl- em vários tecidos.
Propõe-se assim que, através da estimulação do transporte de Cl- por vias
alternativas às da proteína CFTR, se poderá corrigir pelo menos este aspecto
da FQ, pela compensação do deficiente transporte de Cl- nos tecidos
afectados, numa abordagem terapêutica designada por terapia de "bypass.

Resumo
xv
Neste contexto, os canais de Cl- activados por Ca2+ (CaCCs) são proteínas
extremamente relevantes.
Vários estudos demonstraram que, em tecidos onde não há expressão
de CFTR ou onde esta proteína está mutada, há uma maior secreção de Cl-
dependente de Ca2+. Estas observações levaram à formulação da hipótese de
que a regulação das duas vias poderá acontecer dum modo coordenado.
Apesar deste tipo de correntes de Cl- activadas por Ca2+ (ICaCC) serem
conhecidas há mais de 20 anos, a identidade molecular do CaCC é ainda hoje
debatida. Várias proteínas têm sido sugeridas para explicar a ICaCC, mas
apenas recentemente o candidato consistente foi encontrado. Quando foi
iniciado este trabalho doutoral, a bestrophin 1 (Best 1) era o mais forte
candidato a ser o CaCC. No entanto, enquanto os estudos apresentados nesta
tese decorriam, três laboratórios independentes finalmente identificaram a
proteína anoctamin 1 (Ano 1) como um componente do canal de Cl- activado
por Ca2+ usando diferentes estratégias.
Os estudos apresentados nesta tese tiveram como objectivo investigar o
papel e a contribuição de duas proteínas descritas como CaCCs na Fibrose
Quística: bestrophin 1 (Best 1) e anoctamin 1 (Ano 1). O impacto dessas
proteínas na homeostase e função dos tecidos epiteliais afetados pela FQ foi
estudado por meio de técnicas bioquímicas e eletrofisiológicas.
Na primeira parte deste trabalho utilizou-se um modelo animal de
ratinho/murganho nulo para a proteína Ano1 para investigar a contribuição
deste canal na fisiologia das vias aéreas. Os resultados obtidos deram-nos
novas perspectivas relativamente à contribuição deste canal na doença
pulmonar na FQ, nomeadamente para a secreção de Cl- e manutenção de um
eficiente transporte mucociliar.
A segunda parte desta investigação focou-se na contribuição dos CaCCs
na FQ, nomeadamente na elucidação do mecanismo pelo qual é
frequentemente observado um aumento de ICaCC em doentes FQ. A expressão

Resumo
xvi
das proteínas Ano1 e Bestrophin1, dois candidatos a CaCCs, foi analisada em
linhas celulares que expressam estavelmente a proteína CFTR normal (wild-
type) ou a versão mutada (F508del-CFTR) que fica retida no RE. Os nossos
resultados mostraram que nenhuma das duas proteínas (Ano 1 e Best 1)
apresenta uma maior expressão a nível dos respectivos transcritos nas células
F508del-CFTR. Assim, concluímos que não são as alterações na expressão de
CaCCs que justificam o aumento de ICaCC frequentemente observado em
doentes FQ. Outra hipótese que investigámos foi a possibilidade de ser a
sinalização de Ca2+ que se encontra aumentada alterada em FQ, devido à
expressão da proteína F508del-CFTR no RE. Tal, facilitaria o transporte de
Ca2+ pois, funcionando como um contra-ião, permitiria o simultâneo transporte
de Cl-.
As observações incluídas neste estudo contribuem para uma melhor
compreensão do modo como a actividade dos CaCCs pode modular (e também
é modulada) na Fibrose Quística. Além disso, os resultados aqui incluídos dão
novas perspectivas para possíveis abordagens alternativas para superar o
defeito básico da FQ (terapias de "bypass"), para benefício ultimo dos doentes
FQ.

Abbreviations
xvii
ABBREVIATIONS
Ohm
[Ca2+]i Intracellular calcium concentration
[Cl]i Intracellular chloride concentration
ABC ATP-binding cassette
Ado Adenosine
ADVIRC Autosomal dominant vitreoretinochoroidopathy
Ano 1 Anoctamin 1
ASL Airway surface liquid
ATP Adenosine triphosphate
AVMD Adult-onset vitelliform macular degeneration
Best 1 Bestrophin 1
BHK Baby hamster kidney (cells)
BSA Bovine serum albumin
C terminus Carboxyl terminus
CaCC(s) Ca2+-activated Cl- channel(s)
CaM Calmodulin
CaMKII Calmodulin-dependent protein kinase II
cAMP Cyclic adenosine 3',5'-monophosphate
CB(U)AVD Congenital bi(u)nilateral absence of the vas deferens
CBAVD Congenital bilateral absence of the vas deferens
CCH Carbachol
CF Cystic fibrosis
CFTR Cystic fibrosis transmembrane conductance regulator
CO2 Carbon dioxide
COPII Coated protein II
cRNA Complementary ribonucleic acid
DAPI 4',6'-diamidino-2-phenylindole
DIDS 4,4′-diisothio-cyanostilbene-2,2′-disulfonic acid
DOX Doxycycline

Abbreviations
xviii
DTT Dithiothreitol
ENaC Epithelial sodium (Na+) channel
Endo H Endoglycosidase H
ER Endoplasmic reticulum
ER-QC Endoplasmic Reticulum Quality Control
FBS Fetal bovine serum
Fors Forskolin
G conductance,
GFP Green fluorescent protein
Gte Transepithelial conductance (G)
HBE Human bronchial epithelial (cells)
hBest 1 Human bestrophin 1
HEPES 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
HRP Horseradish peroxidise
I Current
I/F IBMX/Forskolin
IBMX 3-isobutyl-1-methylxanthine
ICaCC Ca2+-activated Cl- (I) current
IP3 Inositol 1,4,5-trisphosphate
IRBIT Inositol-1,4,5-trisphosphate receptors binding protein released
with IP3
Isc Short-circuit current (I)
KO Knockout
LPS Lipopolysaccharides
mAChR Muscarinic acetylcholine receptor
MCC Mucociliary clearance
MSD Membrane spanning domain
N terminus Amino terminus
NBD1 Nucleotide binding domain 1
NHE Na+/H+ exchanger
NHERF Na+/H+ exchange regulatory factor

Abbreviations
xix
NMDG N-methyl-D-glucamine
PCL Periciliary layer
PKA (cAMP-dependent) Protein kinase A
PKC Protein kinase C
PLC Phospholipase C
PTC(s) premature stop codon(s)
Po Open probability
RD Regulatory domain
RPE Retinal pigment epithelial (cells)
RT Reverse transcriptase
Rte Transepithelial resistance
S Siemens
siRNA Small interference ribonucleic acid
SK channel Small conductance Ca2+-activated K+ channel
TM Transmembrane segments
U Unit(s)
UTP Uridine triphosphate
V Volt
Vc Membrane voltage
VGCC Voltage-gated Ca2+ channels
Vte Transepithelial voltage
WB Western blot
wt wild-type
x Xenopus


Part I. GENERAL INTRODUCTION


General Introduction
3
I GENERAL INTRODUCTION
1 CYSTIC FIBROSIS
1.1 FROM EARLY REPORTS TO GENE IDENTIFICATION
Cystic fibrosis (CF) is the most common autosomal recessive fatal
genetic disease among Caucasians, where it has a frequency of one in 2500-
4000 live births. Among Northern European, 1 in 25 individuals are
asymptomatic carriers1. Recent statistical studies indicate that about 25,000
children and adults are affected by the disease only in the United States2, and
about 70,000 worldwide.
CF was first described as a clinical syndrome in the late 1930s by Dr.
Dorothy Andersen3. She later went on to discover the autosomal recessive
inheritance patterns of CF with the help of her colleague Richard Hodges4.
The disease was first described and named “cystic fibrosis of the
pancreas”, based on the destruction of pancreatic exocrine function in affected
patients. However, in the early 1940s, the term “mukoviszidosis” (a German
designation which means “thickened mucus”) was introduced in Switzerland by
Dr Sydney Faber5, broadening the spectrum of the disease beyond this organ.
In the 1950s, Dr. Paul Di Sant'Agnese and collaborators observed that
CF patients have an increase salt content in their sweat6. This later gave birth to
the sweat test, which is still the most common method of diagnosing CF
patients. This major clinical breakthrough allowed an accurate diagnosis from
then on.
Further studies in the early 1980s indicated that every organ impaired by
CF showed a malfunction of the epithelial tissue. Furthermore, it was also
shown that epithelia of CF patients were relatively impermeable to
chloride (Cl-)7. These findings, which were confirmed by several laboratories

Part I
4
worldwide, led to the hypothesis that a defective Cl- channel in the epithelium
accounted for the respiratory failure and that this abnormality could explain the
other clinical manifestations of CF. A major effort was then carried out by
numerous scientists in an attempt to identify the gene responsible for the
disease.
It was in 1989 that finally the joint efforts of three laboratories led to the
cloning of the CF gene. The protein was named “cystic fibrosis transmembrane
conductance regulator” (CFTR)8-10, since the predicted protein sequence did not
resemble that of any other known ion channel. Further proof that this was in fact
the gene responsible for CF came with the detection of the in frame deletion of
three nucleotides that led to the removal of phenylalanine 508 from the coding
region. This mutation was detected on the majority of CF chromosomes and
present in a number of families affected by CF11,12. Later it was shown that
CFTR did indeed function as a Cl- channel13-15. This led to the confirmation that
CF is caused by a defect affecting the transport of Cl- across the epithelial
tissues.
However, as initially demonstrated by Knowles and collaborators16, CF
respiratory epithelium also presents an enhanced sodium (Na+) absorption.
Since water follows the movement of salt transport across the epithelia, some
authors also describe CF as resulting from enhanced water reabsorption and
the consequent increased dehydration and thickness of the mucus in the
respiratory epithelium of CF patients.
1.2 CLINICAL EXPRESSION
With the exception of sweat ducts, the recurring symptom in all organs is
the obstruction of passages by mucus, particularly in the gastrointestinal,
hepatobiliary, reproductive and respiratory tracts.
Pancreatic insufficiency is observed in ~85% of CF patients, as a
consequence of the obstruction of the pancreatic ducts and subsequent
scarring (or fibrosis) of the pancreas. A form of intestinal obstruction called

General Introduction
5
meconium ileus is found in some CF newborns, a condition which can be fatal if
not treated by surgery. Liver disease may also occur throughout life1. In adult
CF patients, male infertility is extensive, mostly due to the congenital bi or
unilateral absence of the vas deferens (CBAVD or CUAVD), while female
patients also show reduced fertility17,18. Most of these complications are
controlled by vitamin supplements, oral pancreatic enzymes, special diets and
anti-inflammatory medication19.
In the lung, however, the inability to clear the excessively viscous mucus
has much more severe consequences namely repetitive airway infections which
lead to deterioration of pulmonary function and is thus the main cause of death
of most CF patients20. Indeed, the thickness of the mucus, not only enhances
bacterial adherence, but also leads to a deficient mucociliary clearance, thus
promoting recurrent infections and consequent exacerbated inflammatory
response. Altogether, these events contribute to progressive respiratory
deficiency and ultimately to death. Lung transplantation is currently the only
definitive treatment for advanced CF. Double-lung or heart-lung transplant is
usually required, having a 55% rate of survival (3 years following the
transplant)19.
It is unquestionable that since the cloning of the CF gene by Riordan et
al.10, there has been immense progress in elucidating the molecular and cellular
pathophysiology of CF. The average life-span of CF patients is steadily
increasing, being currently of about 37 years of age, rising from 32 in 20002.
Nevertheless, most current therapies are still restricted to alleviating symptoms
of the disease and thus, life expectancy and quality of life, although largely
improved, are still limited for CF patients21.
It has thus become clear that correcting the basic defect of CF is
essential to overcome the disease. Initially, great effort and hope was put into
gene therapy. Unfortunately, this approach faced major hurdles related to the
technical implementation of an effective strategy and has not, to date, been
successfully delivered. The use of pharmacological tools to rescue CFTR itself

Part I
6
or bypass strategies manipulating non-CFTR channels are now considered
better and faster routes towards the correction of the basic CF defect22 and will
be further addressed in Section 4 of this introduction.
2 CFTR
2.1 FROM GENE TO PROTEIN: THE GENETICS OF CYSTIC FIBROSIS
The CFTR gene or ABCC7 is a large (~190 kb) gene located on the long
arm of chromosome 7, band 31-32 (7q31-q32). It contains 27 exons that after
splicing result in an mRNA of about 6.2 kb that directs the synthesis of a protein
with 1480 aa residues10. To date, more than 1800 mutations have been
reported to the Cystic Fibrosis Gene Analysis Consortium
(http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html). These mutations can
be classified based on the cellular/molecular mechanism by which they cause
the disease. This classification is also useful for the development of appropriate
pharmacological restoring tools, as mutations in the same class will likely
require a similar therapeutic approach. Mutations of the CFTR gene have been
divided into five classes according to the distinct defects they cause in the
synthesis, trafficking or function of the CFTR protein23,24 (see Figure I. 1).
Figure I. 1 - Classes of CFTR mutations. (Normal) CFTR protein in the plasma
membrane of cells functioning as a Cl- channel; (I) class I mutation: prevents translation;
(II) class II: defective processing; (III) class III: defective regulation; (IV) class IV: defective
conductance; (V) class V: reduced synthesis. See text for further details.
Normal I II III IV V

General Introduction
7
Briefly, Class I mutations result in premature termination of CFTR mRNA
translation in the ribosome with the consequent generation of truncated forms of
CFTR protein, being thus, full-length CFTR absent. Class I mutants are due to
the presence of nonsense mutations, i.e., those leading to premature stop
codons (PTCs), but also due to frameshift and aberrant splicing, mutations
which also result in PTCs. Class II mutations lead to degradation of the protein
within the ER and little or no functional protein reaches the cell membrane.
Mutants in this class include F508del11 and A561E25. The disease severity
correlates with the amount of correctly processed protein that is able to reach
the apical membrane. Class III mutants are located at the cell membrane but
are unable to function as a cAMP-activated Cl- channel (G1244E-CFTR and
G551D-CFTR26). Class IV mutants are also correctly located but have altered
conductance or gating properties causing a reduced Cl- flux through the CFTR
channel. Class V mutants cause a reduction in the levels of normal (functional)
CFTR often due to alternative splicing or impaired membrane recycling.
Although most mutations are rare, the deletion of a single phenylalanine
residue at position 508 (F508del) in exon 10 is the most prevalent disease-
causing mutation, occurring on approximately 70% of all CF chromosomes
worldwide and is associated with a severe phenotype. This mutation is widely
studied with a great effort in restoring CFTR function9.
Despite the possibility, to some extent, of establishing genotype-
phenotype correlations, i.e., the different mutations causing CF allow for
phenotypic predictions regarding the sweat ducts, pancreas and the
reproductive system, correlations concerning the lung are not straightforward27.
For instance, patients who are homozygous for the F508del mutation exhibit a
wide range of severity and rate of development of lung disease.
This scenario clearly reflects the multiplicity of events that occur between
the gene defects and the ultimate clinical phenotype of respiratory insufficiency,
constituting the “CF pathogenesis cascade”22. However, environmental

Part I
8
interactions and the genetic background of the host may also contribute
substantially to the severity of CF lung disease28.
2.2 CFTR PROTEIN
2.2.1 Structure
The CFTR protein is included in one of the largest
families encoded in the human genome – the ATP-Binding Cassette (ABC)
transporter superfamily. This family comprises more than 50 members which
are mostly involved in the active transport of substrates across cell membranes
and is present in a wide range of organisms, from bacteria to man29. A
distinctive feature of these proteins is a highly conserved adenosine 5’-
triphosphate (ATP) binding domain, or “cassette” that provides the appropriate
environment for binding and hydrolysis of ATP. ATP sustains the transport of a
variety of substrates, mostly against a concentration gradient30. Each ABC
transporter is usually specific for a given substrate, being amino acids, proteins,
sugars, lipids and a variety of structurally unrelated drugs the most common
substrates. However, CFTR is functionally distinct from other ABC transporters
as it enables bidirectional permeation of anions, rather than vectorial transport
of solutes31.
The structure of CFTR resembles the one of most ABC transporters and
includes various membrane associated domains. Two membrane-spanning
domains (MSDs) consist of 6 highly hydrophobic transmembrane segments
(TM1-12) that compromise the pore of the channel and anchor the protein at the
MSD1MSD2
MSD1MSD2
Figure I. 2 – Schematic representation of the
structure of CFTR protein. (A) CFTR structure with two
NBDs (NBD1 and NBD2), two MSDs (MSD1 and MSD2),
and a Regulatory Domain (RD). Adapted from Jonh
Hopkins Cystic Fibrosis Centre
(http://www.hopkinscf.org/main/whatiscf/science.ht
ml)297
General Introduction
9
apical membrane of epithelial cells32. Furthermore, two highly conserved
nucleotide binding domains (NBDs), where ATP binds and is hydrolysed, are
peripherally located at the cytoplasmic side of the membrane. Remarkably,
CFTR is the only member of the ABC family with a large regulatory domain
(RD) linking the two halves of the protein where multiple phosphorylation
occurs. Phosphorylation of the RD by cAMP-dependent protein kinase A (PKA),
possibly allows ATP binding to the NBDs, which then dimerize. This event is
later transmitted into the TMs, which alter their conformation, allowing the
opening of the channel pore and passive flow of ions. Moreover, the open
probability (Po) of the channel is controlled by the extent of RD phosphorylation
at multiple sites. This property also reflects the balance between protein kinases
and the phosphatases acting at these sites33.
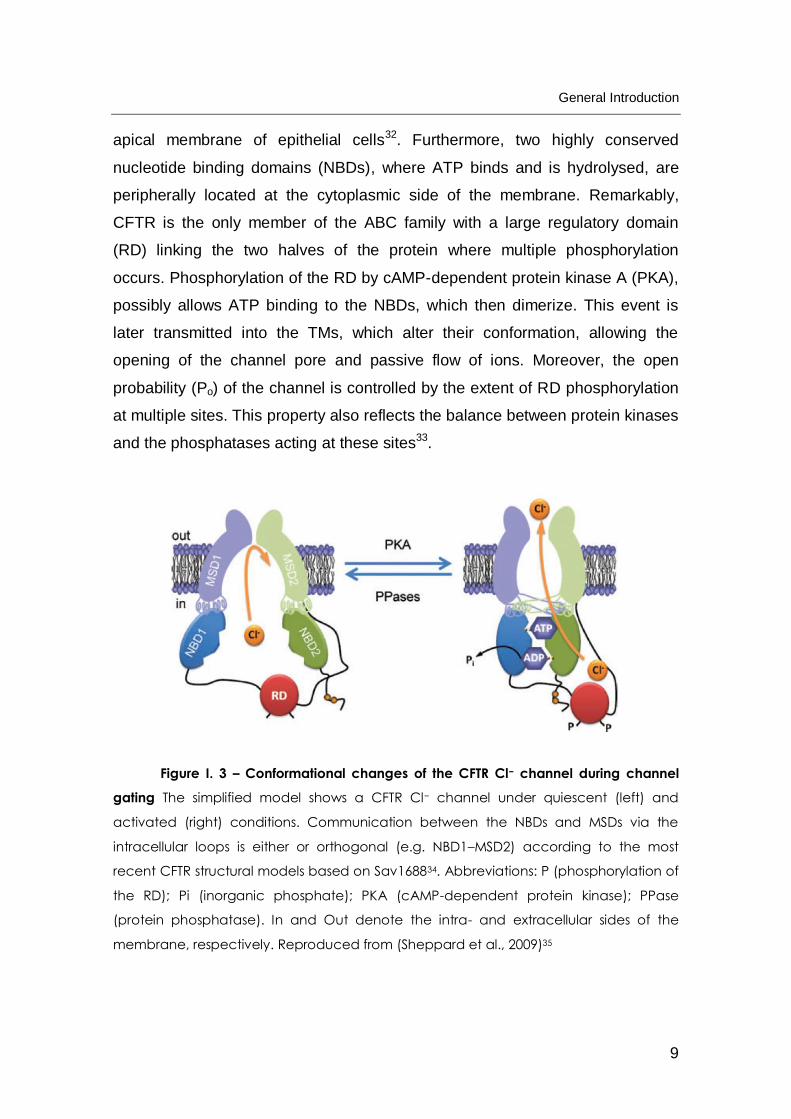
Figure I. 3 – Conformational changes of the CFTR Cl− channel during channel
gating The simplified model shows a CFTR Cl− channel under quiescent (left) and
activated (right) conditions. Communication between the NBDs and MSDs via the
intracellular loops is either or orthogonal (e.g. NBD1–MSD2) according to the most
recent CFTR structural models based on Sav168834. Abbreviations: P (phosphorylation of
the RD); Pi (inorganic phosphate); PKA (cAMP-dependent protein kinase); PPase
(protein phosphatase). In and Out denote the intra- and extracellular sides of the
membrane, respectively. Reproduced from (Sheppard et al., 2009)35

Part I
10
2.2.2 From Biogenesis, through Processing to Degradation
The CFTR protein is an integral membrane glycoprotein located at the
apical membrane of epithelial cells. As other membrane proteins, CFTR is co-
translationally inserted into the membrane of the endoplasmic reticulum (ER),
where it is N-glycosilated. CFTR then follows the general route for export of
proteins to the cell membrane through the secretory pathway. As it proceeds,
the protein undergoes a maturation process which leads to its fully-mature
form11.
The assembly of the various domains into a mature tertiary structure is
mediated by specialized cellular machinery that assists folding and prevents
aggregation of folding intermediates36. The stringent quality control (QC)
mechanism operating at the ER (ERQC), which is capable of discriminating
normally folded from abnormally folded proteins, ensures that only correctly
folded proteins exit the ER and undergo Golgi maturation. The ERQC thus
prevents misfolded CFTR from exiting the ER compartment and is responsible
for sending it for degradation via the cytosolic ubiquitin/proteasome pathway
(UPP)37.
Further progress of CFTR along the secretory pathway is accomplished
by its incorporation into coated protein II (COPII) vesicles and transport to the
Golgi, for which ER retention motifs of the protein need to be masked38,39.
Transport of CFTR protein from the trans-Golgi to the plasma membrane (PM)
can occur indirectly via transcytosis from the basolateral membrane from
recycling endosomes, or directly to the apical membrane by Golgi-derived
vesicles undergoing anterograde traffic40. Once at the plasma membrane, the
amount of CFTR protein at the surface is finely regulated. It can be recycled
and removed from the cell surface by clathrin-mediated endocytosis through
trafficking signal embedded in the primary sequence of CFTR. Furthermore, the
protein can be recycled back to the plasma membrane directly, or through
recycling endosomes. Damaged protein is targeted for lysosomal degradation41.

General Introduction
11
2.3 F508DEL-CFTR PROTEIN
F508del mutation accounts for most of CF cases worldwide, being thus
one of the most widely studied CFTR mutations. It is thus critical to understand
the effects of this mutation in CFTR processing as well as its impact on cellular
homeostasis. CFTR undergoes a fairly complex biosynthetic pathway, which is
in fact rather inefficient. Depending on the cell type, only about 25% to 60% of
pre-cursor wt-CFTR protein is correctly processed and achieves the fully
mature, ER-export competent form42-44. The folding of the F508del-CFTR
mutant into a native conformation is an even less efficient process45. Not only
the mutant protein is trapped in the ER but it is also rapidly recognized and
targeted for proteasomal degradation by the ER-QC11,46,47. As a result, very little
or no F508del-CFTR, depending on the cell type, reaches the cellular
membrane. Furthermore, the F508del-CFTR protein capable of reaching the
membrane is not only thermodynamically unstable, but is also unable to present
normal channel activity, being rapidly endocytosed and degradated34,48,49.
The F508del mutation not only decreases the folding yield and transitions
between the intermediate states in the folding of the domain where it is located -
NBD1, but also prevents interaction of its surface with the rest of the protein,
namely intracellular loop 4 (IL4) in TMD234. This ultimately results in a misfolded
conformation that does not correspond to the higher degree of structure
required to be exported from the ER via COPII-coated vesicles.
Some authors suggest that the altered conformation of F508del-CFTR
exposes targeting motifs which are recognized by the cell ERQC, a situation
that does not occur in the correctly folded wt-CFTR protein. Indeed, CFTR has
four arginine-framed tripeptides (AFTs) along its polypeptide chain, which act as
negative (ER retention) export signals. In practical terms, this means that when
the AFTs are hidden in wt-CFTR, the protein is allowed to transit to the Golgi. If
these signals are exposed in the abnormal conformation of F508del-CFTR, the
protein cannot further mature through the Golgi50. Additionally, F508del-CFTR
may turn inaccessible short di-acidic ER export motifs that are required for

Part I
12
transport out of the ER. These have been proposed to be involved in the
transport of some membrane proteins through the ER exiting sites (ERES),
functioning as positive ER export signal motifs51,52. Although classified as a
trafficking mutant, F508del leads primarily to a protein folding defect with altered
protein trafficking53.
Soon after the identification of the CFTR gene, the F508del mutant was
demonstrated to be “rescuable”. Indeed, it was shown that by lowering the
incubation temperature of cell cultures from 37ºC to 26ºC, partial rescue of the
mutant protein to the PM could be achieved54. Furthermore, its defective
processing and reduced PM localization can also be overcome when
expressing the F508del-CFTR molecule in Xenopus oocytes55, or when highly
overexpressed in mammalian cells49, thus enabling measurable regulated
chloride-channel activity in transfected cells.
2.4 CFTR FUNCTIONS IN EPITHELIA
2.4.1 CFTR as a Cl- channel
The majority of ABC transporters use the energy of ATP hydrolysis to
pump a wide spectrum of substrates through diverse transmembrane pathways.
In contrast, CFTR forms a pathway for passive anion flow that is gated by
cycles of ATP binding and possibly hydrolysis by the NBDs35.
Since the primary structure of CFTR was first elucidated, three
possibilities arose to account for the protein function. It was postulated that it
could be an ATP- driven transporter, a regulator of a separate channel molecule
which would contain the channel “pore” (hence its designation), and thirdly that
CFTR was itself a regulated Cl- channel. Despite the regulatory activity of CFTR
over a variety of other channels and transporters there is compelling proof
indicating that CFTR itself mediates a characteristic epithelial anion
conductance14,56-58.

General Introduction
13
The function and cooperation of the various domains of CFTR confer to
the channel several distinguishing characteristics: i) a small single-channel
conductance (6-10 pS)15; ii) a linear current-voltage (I-V) relationship;
iii) selectivity for anions over cations; iv) anion permeability sequence of
Br- > Cl- >I-; v) time- and voltage-independent gating behaviour; vi) activity
regulated by cAMP-dependent phosphorylation and by intracellular
nucleotides59.
2.4.2 Regulator of Ion Transport across Epithelia
As outlined previously, the role of CFTR in epithelial physiology extends
beyond its function as a Cl- channel, namely as a regulator of other ion
channels and pathways relevant for CF disease.
2.4.2.1 CF airway: Effect on mucociliary transport
Figure I. 4 – Human ciliated airway epithelium (A) Scanning electron micrograph
of human ciliated airway epithelium (HAE) showing cilia and mucus on the luminal
surface. (B) Histological cross-section of HAE showing pseudo-stratified, columnar
airway epithelium with ciliated (cc) and mucin-secreting cells (mc) and basal epithelial
cells (bc). From (Zhang L. et al. 2009)60
The airway surface liquid (ASL) protecting the epithelium lining the
airways (see Figure I. 4) is generally described as a biphasic layer composed of
a periciliary layer (PCL) where the cilia beat, and a more superficial gel layer

Part I
14
that constitutes an efficient barrier against micro-organisms (mucus). The
regulation of ASL in the respiratory epithelia is ensured by both electrolyte
absorptive and secretory pathways, which take place in the surface epithelium
and submucosal glands, respectively61.
On one hand, salt secretion by the submucosal glands forces water to
exit towards the airways lumen, ensuring proper hydration of the epithelia. This
process is essential for efficient mucociliary clearance (MCC), as well as for
mucus exiting from the glands. On the other hand, the hydration of the PCL
under homeostatic conditions is mainly regulated by luminal/basolateral water
transport coupled to active transepithelial Na+ transport. The accompanying
absorptive movement of Cl− and water occurs both through cellular and
paracellular pathways. Under resting conditions, there is no Cl− flux in normal
human airways and the active ion transport is dominated by Na+ absorption
from lumen to submucosal space, while a net secretion is present when the
epithelia is exposed to secretagogues. In CF, both pathways are affected as net
absorption of electrolytes is simultaneously enhanced in the CF surface
epithelium, and secretion by the submucosal glands is inhibited, further
dehydrating the epithelia.
The fine regulation of ion and water transport in the airways epithelium is
thus vital in lung defence62. In this regard, two competing theories have been
proposed to link abnormalities in CF airway epithelial functions to abnormal ion
and water composition of airway mucus or ASL: the “Hypotonic [low
salt]/defensin” and the “Isotonic volume transport/mucus clearance”63 (see
Figure I. 5).

General Introduction
15
Figure I. 5 - Models Explaining ion and water regulation In the Airway Epithelium
in CF. (A) Model depicting relative surface areas of distal and proximal airway regions.
On the left is the depiction of the mechanical (mucus) clearance hypothesis, with the
epithelium controlling the volume of the PCL as the critical element mediating efficient
mucus transport. On the right is shown the chemical shield hypothesis that predicts that
the airway epithelium absorbs salt but not water from ASL to form a "low salt" ASL to
promote antimicrobial activities of defensins. (B) Cl- transport in a normal airway
epithelial cell (C) Isotonic/volume model, where dysfunction of CFTR leads to
hyperabsorption mainly of Na+, decreasing osmotic pressure and consequently
dehydrating the ASL. (D) The hypotonic model, dysfunction of CFTR leads to diminished
absorption of counter-ions, in turn favouring accumulation of salt in the ASL. Adapted
from (Boucher, 2004)28 and (Rowe et al., 2005)21.
The isotonic “low volume” hypothesis focuses on the role of airways MCC. In
this hypothesis, the airway epithelium normally regulates the volume of liquid in
the mucus by isotonic transport to control the efficiency of the mucociliary
transport. In CF, as CFTR is absent from the membrane, epithelial Na+ channel
(ENaC) is no longer inhibited; which leads to an increased isotonic

Part I
16
hyperabsorption of ASL64,65 and consequent decrease in liquid volume at the
surface of the airway epithelium. Further studies suggest an isotonic
composition of normal and CF mucus and a decrease in the fluid volume of CF
airway mucus. According to these authors, a reduction in mucus hydration and
MCC in CF is responsible for the inflammation and infection that lead to
disease66,67.
In contrast, the hypotonic model68-70 postulates that normal airway
epithelium surface absorbs salt but not water (presumably as a consequence of
putative airway epithelial water impermeability or surface forces) to generate a
hypotonic (low salt) ASL. This theory relies on the ability of the superficial
epithelium to regulate the ionic composition, rather than the volume, of ASL. In
CF patients the inability to absorb Cl− through “Cl− impermeable” airways would
consequently render the ASL iso- or hypertonic. Along this line, it has been
postulated that the gene defect in CF leads to a breach in innate immunity.
Studies have shown that altered salt concentration in ASL affects bacterial
survival in the abnormal milieu of the CF airways, where the activity of
antibacterial proteins and peptides (notably defensins) is inhibited69,71,72.
Lowering of the NaCl concentration (≤ 50 mM NaCl) is thought to be sufficient to
activate defensins and create an antimicrobial “shield” on airway surfaces. It
has also been suggested that c-AMP- stimulated bicarbonate (HCO3-) secretion
is mediated via CFTR and that impaired bicarbonate secretion in CF may
induce an abnormally low pH which might also decrease antimicrobial functions
as well as mucus properties73. Others suggest that the absence of functional
CFTR in CF tissues affects the glycoconjugates on the cell surface of epithelial
cells74. These alterations may be crucial for pathogen recognition, ultimately
aggravating CF lung disease75,76.
The resolution of the salt-fluid controversy has major implications in CF
therapeutic strategies. Whether the optimal strategy is to reduce the salt
concentration of the airway liquid in order to increase the innate defence

General Introduction
17
mechanisms or add more liquid to the airway surface in order to improve
mucociliary clearance is still under debate77.
2.4.2.2 Mucus and Bicarbonate Transport
Besides Cl- transport, CFTR has also been implicated in a number of
other functions, namely in HCO3- transport in the lungs, gastrointestinal tract
and pancreas78-81. Although CFTR functions primarily as a Cl- ion channel and
exhibits a low permeability to HCO3- ions82, this permeation has important
implications. HCO3- is involved in several cellular functions, including acting as a
pH buffer and thus enhancing the solubility of many proteins.
It is a long standing controversy whether the abnormally viscous
pathogenic mucus in CF is due to aberrant synthetic processes occurring
intracellularly83,84, or to extracellular events that compromise the environment
where high molecular weight glycoproteins (notably mucins) are produced and
exocytosed85-87.
Some studies have proposed a putative role for CFTR in regulating β-
adrenergic dependent stimulation of mucin secretion88. Other studies have
suggested that over-expression or altered post-translational modification of
certain mucins in CFTR defective cells increases the propensity for bacterial
infection of CF airways77.
Furthermore, it is argued that the abnormal mucus release in CF would
be a consequence of deficient CFTR dependent HCO3- transport in the affected
organs, contributing to the unique “mukoviszidosis” used to describe this
disease in earlier years89. It is thus hypothesized that CFTR permeability to
HCO3- can affect the pH of luminal electrolyte and fluid environment of some
epithelia and consequently affect mucus swelling and hydration during its
exocytosis. Mucins are known to be tightly packed in sub-apical granules which
are exocytosed in response to stimuli. It has been reported that absence of
CFTR increases exocytosis of mucous granules90. Additionally, it has been

Part I
18
suggested that mucins show a tendency to remain compact due to lowered pH
in the extracellular fluid matrix in CF tissues91, thereby augmenting the viscosity
of the ASL. Furthermore, recent results show that HCO3- impedes mucus gel
aggregation and increases the diffusivity of exocytosed mucins most likely by
chelating Ca2+ 89.
2.4.2.3 Regulator of other ion channels
In addition to its function as a Cl- channel, CFTR is frequently involved in
the regulation of a number of other ion channels. As previously outlined, in the
lung, as well as in other epithelial tissues from CF patients, there is a well
recognized hyperabsorption of Na+ mediated by the ENaC92,93. The functional
relationship between CFTR and ENaC is thereby biologically relevant as it
dictates the capacity to clear bacteria and other harmful agents from the lungs
by regulating water levels and composition of ASL28.
Several mechanisms have been proposed to explain this long standing
observation, and how CFTR might downregulate ENaC activity in these tissues.
For instance, some studies showed that the activation of wt-CFTR, in both
recombinant and natives cells94-96, negatively regulates ENaC97-99. Subsequent
studies showed that Cl- currents could account for inhibition of ENaC,
independently of wt-CFTR expression100. Other data suggest that it is the
absence of CFTR from the plasma membrane that leads to hyperactivity of
ENaC101-103. Recently, it was proposed that while ENaC is protected from
proteolytic cleavage, and consequently stimulation of open probability (Po),
when associated with wt-CFTR, F508del-CFTR protein fails provide similar
protection104. Other authors support the hypothesis that wt-CFTR can regulate
the functional and surface expression of endogenous ENaC in airway epithelial
cells by altering the trafficking of the Na+ channel105.
Several reports show that a large dynamic signalling complex consisting
of PDZ domain proteins and kinases is involved in the interaction between
these channels. In this context, the Na+/H+ exchanger regulatory factor isoform-

General Introduction
19
1 (NHERF1) is suggested to mediate the interaction between CFTR and ENaC
indirectly, and even facilitate the reciprocal regulation of these channels (CFTR
downregulates ENaC, whereas ENaC activates CFTR). Recent data, however,
seem to indicate that a physical direct interaction occurs between CFTR and
ENaC106.
In addition to ENaC, CFTR has also been correlated to the regulation of
other Cl- channels such as the outwardly rectifying Cl- channels (ORCCs)107 as
well as CaCCs108.
2.4.2.4 Other Functions
CFTR has also been implicated in the control of oxidative stress in the
airways, as it was found to secrete the antioxidant peptide glutathione (GSH) in
its reduced form109. GSH, the main antioxidant secreted in response to
inflammation in the lung, has been known for long to be reduced in ASL of CF
patients110.Later it was shown that CFTR is capable of mediating transport of
GSH, possibly justifying the augmented basal state of inflammation found in CF
lungs109. Recently CFTR was also implicated in the control of the intracellular
reactive oxidative species (ROS) balance, possibly also related to some CFTR-
dependent modulation of GSH concentration111.
Given the multiple functions assigned to CFTR not only as a transporter
but also as a regulator of other molecular entities, a role for CFTR as an
“anchoring platform” at the cell membrane has been proposed. Specialized
“microdomains” anchored to CFTR may group together a number of proteins
that are dynamically regulated by molecular switches, e.g. PDZ-domain
proteins103. These include signalling molecules, kinases, transport proteins,
PDZ-domain-containing proteins, myosin motors, Rab GTPases and SNAREs.
A better understanding of the complete CFTR “interactome” (CFTR-interacting
proteins) in the most physiologically relevant cells, as well as its dynamic
functional relationships, will certainly give new insights into identifying more
targets for CF therapy.

Part I
20
3 CA2+-ACTIVATED CL- CHANNELS
3.1 CA2+
VS. CAMP DEPENDENT CL- CONDUCTANCE IN CF
As already discussed in section 2.4.2.1, ion transport is relevant for
maintenance of airway hydration and protection against infection. Cl - secretion
across the airway epithelium can be stimulated by a number of secretagogues
that activate distinct second messenger transduction mechanisms.
Figure I. 6 Cell models of the mechanism of electrolyte secretion and electrolyte
absorption in the airway epithelia. (A) in secretory cells, Cl- is taken up from the
basolateral (blood) side by the Na+-K+-2Cl- cotransporter. K+ is recycled via basolateral
K+ channels, and Na+ is pumped out of the cell by the Na+-K+-ATPase. Cl- leaves the cell
via luminal (apical) CFTR Cl- channels, and Na+ is secreted via the paracellular shunt. K+
is also secreted to the luminal side via luminal K+ channels. Depending on the tissue,
intracellular cAMP is increased and secretion is activated by adenosine. (B) in
absorptive epithelial cells, Na+ is taken up by luminal epithelial Na+ channels (ENaC). Cl-
is transported via the basolateral shunt and probably via CFTR Cl - channels. Na+ is
pumped out of the cell by the basolateral Na+-K+-ATPase, whereas Cl- and K+ leave the
cell via Cl- and K+ channels, respectively. In cells that coexpress CFTR and ENaC, CFTR
stimulation and/or expression leads to inhibition of ENaC. The Na+-K+-2Cl- cotransporter
is omitted for clarity. Adapted from (Kunzelmann, 2001)61.
In a secretory epithelium, transporters in the basolateral membrane,
namely the Na+-K+-2Cl- (NKCC1) co-transporter, ensure that Cl- accumulates
inside the cell against its electrochemical gradient, thus developing a favourable

General Introduction
21
driving force for Cl- to exit at the apical membrane. Subsequently, when apically
located Cl- channels are stimulated, Cl- flows into the extracellular environment.
In contrast, in sweat glands and absorptive epithelia, the transport
direction is reverted, i.e. both Cl- and Na+ are absorbed by the apical surface of
epithelia (see Figure I. 6B), building up inside the epithelial cell to be secreted
basolaterally upon appropriate stimuli.
In the apical membrane of airway
epithelial cells in addition to CFTR112
CaCCs are thought to play a role in
epithelial Cl- movement, namely in CF,
where CFTR is defective113. When
stimulated with nucleotides such as ATP
or UTP, airway epithelial cells show a
Ca2+-dependent Cl- secretion due to the
activation of Gq-coupled purinergic
receptors (P2Y). Inositol 1,4,5-
trisphosphate (IP3) production is
consequently increased, leading to Ca2+
release from intracellular (ER) stores. This
is known to augment the short-circuit
current (ISC) and the ASL of a murine
tracheal epithelial cell line112. Moreover,
besides extracellular ATP114,115, a large
class of ligands, including histamine116,117 as well the inflammatory mediator
bradykinin118, has been shown to activate CaCC across the apical membrane of
airway epithelia. This cAMP-independent pathway was established soon after
the identification of CFTR, in studies of CF nasal epithelia. For instance, studies
in the Boucher and Welsh laboratories demonstrated that Ca2+ ionophores are
effective Cl− secretagogues in CF tissues113,119,120. In addition, in the airways of
the CFTR -/- mouse, which lacks CFTR, not only is the CaCC pathway
Figure I. 7 Cell model for Cl-
secretion in airways and proximal
colonic epithelial cells. Two major
pathways for salt transport are
ensured by cAMP-activated CFTR
Cl- channels [adenosine (ADO) in
airways, prostaglandin (PGE2) in
the colon] and by CaCC [P2Y2 and
carbachol (CCH) in airways].
Luminal Basolateral
Ca2+
M3 CCH
EP PGE2
P2YATP
IP3/SOC
IP3/SOC
Cl-PKA
A2BADO cAMP
cAMP
CFTR
Na+
2Cl-
K+
K+Cl-CaCC

Part I
22
preserved, but it appears to be up-regulated121. Some authors also speculate
that the very high contribution of CaCCs to ASL homeostasis in the murine
airway epithelium might explain the lack of a lung phenotype in mouse models
of CF122.
In contrast to the characteristic sustained profile of CFTR currents, Ca2+-
activated Cl- secretion is transient, in part due to the lack of activation of
basolateral Cl- uptake by the cAMP-activated NKCC1 cotransporter. The basal
level of the ASL is thus thought to be controlled by CFTR, whereas CaCCs
seem to acutely regulate the height of the ASL layer in response to agonists.
Furthermore, airflow due to normal tidal breathing confers shear stress on
airway surfaces which release ATP. Ecto-5’ nucleotidases rapidly degrade ATP
to adenosine, which in turn also activates CFTR via A2B receptors123.
It is therefore discussed that the airway mucous layer is regulated by a
cross-talk between CFTR and CaCCs108. Since CFTR and CaCCs are both
apical Cl− channels, it is tempting to speculate that activation of CaCCs could
serve as a therapy for CF. Unfortunately, until recently this line of research has
been held back by the lack of specific CaCC activators and by uncertainty about
the molecular identity of these channels.
The identification of anoctamin 1 (Ano1) as an epithelial CaCC
represents a major breakthrough in this context and remarkable discoveries
arose after it was identified (see section 3.3.3).
3.2 PROPERTIES OF CA2+-ACTIVATED CL CHANNELS
The first references to Ca2+ activated Cl- currents (ICaCC) appeared in the
early 1980s, being described in Xenopus oocytes124,125 and salamander
photoreceptor inner segments126. In relation to oocytes, CaCCs play a role in
the fast block of polyspermy by depolarizing the membrane, and thus
preventing additional sperm entry.

General Introduction
23
Up to now CaCCs have been shown to have other multiple relevant
functions in a variety of tissues namely in epithelial secretion127-129, membrane
excitability in cardiac muscle and neurons130,131, olfactory transduction132,133,
modulation of photoreceptor light responses134, regulation of platelet cell
volume135, regulation of vascular tone136, among others.
Moreover, CaCCs may be involved in several human diseases, including
CF. Although defects in the CFTR Cl− channel cause CF, upregulation of a
Ca2+-activated Cl− current in the airways of CFTR knockout mice can
compensate for the lack of CFTR and improve the pathology in this mouse
model121,122. Since such levels of CaCCs upregulation do not exist in humans, it
is tempting to speculate that this is a reason for the exacerbated CF lung
phenotype.
3.2.1 Biophysical properties
Regarding the biophysical properties of CaCCs, three essential hallmarks
characterize them: i) activation by intracellular Ca2+ with half-maximal
concentrations for activation in the submicromolar range, ii) poor ion selectivity
displaying a selectivity sequence similar to that of Eisenman type I (SCN- > I- >
Br- > Cl- > F-)137 and iii) voltage- and time-dependence of typical macroscopic
currents at subsaturating Ca2+ concentrations (<1 µM).
Concerning their activation, either Ca2+ release from intracellular stores
or Ca2+ influx through store-operated Ca2+ channels (SOC) are responsible for
activating the current. Many cell types express a type of Cl− channel that is
activated by cytosolic Ca2+ concentrations ([Ca2+]i) in the range of 0.2–5 μM138.
The single channel conductance reported for CaCCs ranges from 1 to 50 pS139.
Activation can be accomplished either by direct binding of Ca2+ to the
channel, or indirectly through Ca2+-binding proteins or via phosphorylation
through Ca2+/calmodulin-dependent protein kinase II (CaMKII), depending on
the cell type. Along this line, there is strong evidence in the literature for the
existence of several classes of Ca2+-activated Cl- channels that are thus

Part I
24
differentially regulated and may point toward the existence of different molecular
species of CaCCs. This scenario would also account for the wide range of
reported single channel conductances140.
In spite of the low affinity of CaCCs for Ca2+, several lines of evidence
favour the idea that CaCCs are directly gated by Ca2+. For instance, CaCCs can
be stably activated in excised patches from Xenopus oocytes141, hepatocytes142
and in acinar cells from rat mandibular salivary gland143, even in the absence of
ATP. This suggests that activation of the channels does not require
phosphorylation139. In addition, application of peptide inhibitors of calmodulin
(CaM) and CaMKII to rat parotid acinar cells does not prevent Cl- current
activation by ionomycin-mediated [Ca2+]i increase144. Conversely, in the study
by Arreola and co-workers, the same peptides markedly inhibited the Ca2+-
dependent Cl- currents in the human colonic cell line T84. These authors thus
suggested that CaCCs in T84 cells and in parotid acinar cells must be regulated
by a fundamentally different Ca2+-dependent mechanism.
On the other hand, CaMKII dependent gating of CaCCs has also been
observed in other cell types, for e.g. airway cells128, macrovascular endothelial
cells145, T lymphocytes146, human macrophages147, and pancreatic epithelial
cells148. Furthermore, treatment with the calmodulin blockers calmidazolium and
trifluoroperazine decreases Ca2+-activated Cl- currents145. The possibility that
other components, in addition to Ca2+, are required to open the channel is
further reinforced by excised patch experiments where channel activity runs
down quickly after excision140,149.
Finally, the kinetics of Ca2+ activation constitutes the third major
biophysical fingerprint that characterizes CaCCs. Numerous experiments have
shown that when [Ca2+]i is below 1 μM, ICaCC is both voltage and time
dependent, but at higher [Ca2+]i, the voltage and time dependency is lost.

General Introduction
25
3.2.2 Role of Ca2+-activated Cl- channels
When focusing on the physiological role of CaCCs, the outcome of the
activation of the channels greatly depends on membrane potential, Cl- gradient
and also the [Ca2+]i of the cell under consideration. In most cases, the resting
membrane potential is more negative than ECl. Hence, when [Ca2+]i rises, Cl−
exits the cell, resulting in a depolarization of the plasma membrane. In some
cells this depolarization increases the Po of voltage-gated Ca2+ channels
(VGCCs), which results in additional Ca2+ influx and further depolarization. Due
to osmotic forces and the requirement for charge equality, the efflux of Cl− is
also accompanied by water and Na+. On the other hand, if the membrane
potential is more positive than ECl-, opening of CaCCs leads to Cl- entry and
thus to hyperpolarization.
As above mentioned, CaCCs are essential for the blockade of
polyspermy in Xenopus oocyte, by the generation of the so-called fertilization
potential150. This depolarization is initially triggered by the activation of CaCCs,
which is in turn a consequence of the rise of intracellular Ca2+ which follows
fertilization of the oocyte and subsequent Ca2+ release from intracellular stores.
However, in order to avoid polyspermy this depolarization must be sustained.
As Ca2+ release from internal stores mediated by IP3 is not sufficient,
continuous depolarization is ensured by a tightly-coupled mechanism that
assures that once Ca2+ stores are depleted, activation of Ca2+ influx is assured
by stored-operated Ca2+ channels (SOCs), which further contribute to CaCC
stimulation138.
CaCCs are also involved in the regulation of neuronal and cardiac
excitability, where they are presumably implicated in membrane oscillatory
behaviour, repolarisation of the action potential and generation of after-
polarizations. CaCCs are only found in a subset of neurons, which suggests a
specific function of the channels in such cells. In particular, they are present in
neurons from the dorsal root ganglion (DRG) responsible for sensing skin

Part I
26
temperature, touch, muscle tension and pain140. CaCCs in DRG are responsible
for after-depolarisations following action potentials151.
In cardiac myocytes, the activation of CaCCs may either depolarise or
repolarise the membrane potential depending on the Vm and ECl- and its activity
is linked to several cardiac conditions. Sudden cardiac death in German
shepherd dogs is associated with an abnormal transient outward current that
characterizes the initial phase of repolarisation of the cardiac action potential
(Ito)152. Serious cardiac arrhythmias are also thought to be related to CaCC
activity, due to the onset of oscillatory after-potentials during Ca2+ overload.
Activation of Cl- currents are known to cause important changes in action
potential characteristics and thus contribute to the genesis of arrhythmias153. In
this regard, anion substitution or pharmacologic Cl− channel blockade has been
used to protect against reperfusion and ischemia-induced arrhythmias154,155.
ICaCC is often described in single isolated smooth muscle cells from
several vascular beds and nonvascular smooth muscle types. Furthermore, this
type of currents is often activated by the excitatory neurotransmitters (e.g.,
norepinephrine and acetylcholine, in blood vessels and in the trachea,
respectively). Endothelin and histamine are other mediators that contract
smooth muscle in vascular and airway smooth muscle, respectively. Smooth
muscle contraction usually results from changes in membrane potential. Most
smooth muscle cells possess voltage-dependent Ca2+ channels (VDCC) which
permit the entry of Ca2+ into the cells that produce contraction. The resting
potential of the majority of smooth muscle cells lies between -50mV and -70mV,
while VDCC have a higher Po at more depolarized membrane voltages (-60mV
to -40mV). Thus, it is hypothesized that activation of CaCCs depolarises the
membrane and consequently triggers VDDC activity156.
Additionally, the physiological function of CaCCs in the transduction of
odorous stimuli in vertebrates is well established157. In this case, the Cl− efflux
through CaCCs serves as an amplification system of the odorant-activated
current. The particular role of CaCCs in photoreceptors remains unclear. It has

General Introduction
27
been proposed that they could function in membrane potential stabilization by
counteracting the depolarization induced by Ca2+ entry through VGCC.
Both acinar and duct cells of exocrine glands like the pancreas,
lachrymal, parotid, submandibular and sublingual glands are organs where
CaCCs currents are well known and described158,159. In these organs, fluid and
enzymes are secreted in a Ca2+-dependent manner, triggered by the
parasympathetic neurotransmitter acetylcholine. Activation of the muscarinic
receptor (mAChR) leads to the production of IP3 which releases Ca2+ from
internal stores. As a consequence, CaCCs are activated and subsequently Cl−
exits the cell through the apical membrane. The exit of Cl− forces the movement
of Na+ through the parallel pathway accompanied by water, resulting in salty
fluid secretion. CaCCs constitute the last step in the transepithelial movement of
Cl−, which is the net driving force for the whole secretory process140.
3.3 MOLECULAR IDENTITY OF CACC
Although Ca2+ activated Cl- currents have been studied for almost 30
years, the respective molecular entity responsible for the generation of this type
of current has been controversial. One of the drawbacks of this pursuit is related
with the fact that Xenopus oocytes (one of the favourite systems for expression
of cloned ion channels) are not suitable for expression of this channel, due to
their large endogenous Ca2+-activated Cl- currents. Furthermore, most of the
available ion channel inhibitors were unable to specifically differentiate them
from other types of Cl- channels160. Over the years several candidates were put
forward and are briefly summarized below.
3.3.1 CLCA and other candidates
The first Ca2+-activated Cl− channel (CLCA) cDNA candidate clone was
isolated from a bovine tracheal cDNA expression library. This library was
screened with an antibody generated against a purified protein that behaved as
a CaCC when incorporated into artificial lipid bilayers161. Although induction of

Part I
28
Ca2+-dependent Cl- currents in various cell types was showed when they were
transfected with cDNAs encoding various CLCAs, these proteins are no longer
seriously considered as candidates for CaCC due to various inconsistencies
regarding their characteristics as CaCCs162. Furthermore, CLCAs have very
high homology to known cell adhesion proteins and some are soluble, secreted
proteins163. Despite the fact that the first CLCA was cloned nearly 20 years ago,
structure-function analysis has not provided any clear evidence that a CLCA is
even actually a channel. Additionally, there are differences in the biophysical
properties such as Ca2+ and voltage sensitivity, as well as pharmacology
between CLCA currents and native CaCCs. Another argument against CLCAs
being CaCCs is that a number of cell types, namely Ehrlich cells, that express
native CaCCs do not express CLCAs164. In spite of this controversy, some
authors argue that the processes associated with CLCA expression may be
connected to Cl- channel activity, namely by modulating the conductance of
other ion channels163.
The two human genes hTTHY2 and hTTHY3, or Tweety, have also been
suggested to be responsible for the generation of a large Ca2+ regulated Cl-
current (260pS) found, for instance, in spinal neurons and skeletal muscle165,166.
Nevertheless, the biophysical properties of the generated currents have little
relation to the classical CaCC currents, which are in turn rather small.
Moreover, these proteins are not expressed in cells where a Ca2+-activated Cl-
current has been clearly shown.
3.3.2 Bestrophins
When it was first identified as a Ca2+-activated Cl- channel by Sun and
co-workers, human bestrophin 1 (hBest 1) was initially a promising candidate
for the long searched CaCC, or at least part of it167. Mutations in hBest 1 are
related to various types of retinopathies, namely the autosomal dominant Best
vitelliform macular dystrophy (BVMD or Best disease). This disease is
characterized by an abnormal electro-oculogram (EOG), which is in turn thought

General Introduction
29
to be consistent with a loss of Ca2+ activated Cl- channel activity in the
basolateral membrane of RPE cells168.
3.3.2.1 Bestrophin - From gene to protein
The Best Vitelliform Macular Dystrophy (BVMD) is an inherited
autosomal dominant macular degeneration which is caused by mutations of the
BEST1 gene169. In addition, patients with adult-onset vitelliform macular
degeneration (AVMD) and autosomal dominant vitreoretinochoroidopathy
(ADVIRC) have mutations in Best 1170. hBest 1 is encoded by the vitelliform
macular dystrophy type 2 (VMD2) gene located on chromosome 11q12171. The
human genome encodes four bestrophin paralogs numbered sequentially
(bestrophin 1 to 4).
Best 1 is found at high levels in or at close proximity to the basolateral
membrane of retinal pigment epithelial (RPE) cells172. Subsequent work has
also shown expression of bestrophins in a variety of epithelial and non-epithelial
cells, namely of bestrophin 1 and 2 in airways, colon and kidney tissues173.
Bestrophin 2 (Best 2) was also detected in olfactory sensory neurons of both
human and mouse, and was suggested to play a role in the olfactory
transduction as a CaCC174.
3.3.2.2 Are Bestrophins Ca2+-activated Cl- channels?
In addition, bestrophins have been shown to function as Cl- channels
when heterologously expressed, as well as to produce defective Cl− currents in
many disease-causing mutations of hBest 1175-178. Moreover, it was shown that
Best1 knock-down by siRNA causes a decrease of endogenous Ca2+-activated
Cl- currents in different cell types179-181
Nevertheless, reservations have been expressed questioning the role of
bestrophins as the classical CaCCs. For instance, Best 1 knockout (KO) mice
do not have an ocular pathology; the RPE shows normal Cl- conductance as

Part I
30
well as mutations hBest 1 in patients do not strictly correlate with the expected
changes of electrophysiological properties of the RPE. More relevant is still the
fact that Ca2+ activated Cl- currents are still present in different tissues of Best 1
KO mice173,182.
Despite being both gated directly by Ca2+ without the involvement of
kinases and both exhibiting the same anion selectivity sequence, when
comparing more attentively the biophysical characteristics of classical CaCCs
and bestrophins significant differences are observed. For example,
heterologously expressed hBest1 and mbest2 have an apparent affinity for Ca2+
that is 10 times higher than that of CaCCs. Furthermore, the classical voltage-
dependent kinetics and outward rectification of CaCC is not present in hBest 1
or mbest-2177,183. In this sense, it can be argued that native CaCCs have
another subunit that is not present in the expressed homomeric channels. On
the other hand, certain bestrophins also exhibit a Ca2+-independent volume
regulation, which has not been found in classical CaCCs184,185. In addition,
heterologously expressed bestrophins have been activated using high
concentrations of Ca2+, but have never been shown to be activated through
receptor mediated responses that elevate cytosolic Ca2+186.
3.3.2.3 Regulator of other channels and/or Ca2+ signalling?
On the other hand, Best 1 has also been proposed to be a regulator of
voltage-gated L-type Ca2+ channels, rather than a Ca2+-regulated Cl-
channel182,187,188. Namely, the Strauss laboratory demonstrated that transient
overexpression of hBest 1 in RPE-J cell line can influence the properties of L-
type Ca2+ channels187. Subsequent work in collaboration with Marmorstein
showed that Nimodipine, a voltage-gated Ca2+ (Cav) channel blocker,
diminished the amplitude of the light peak (LP), suggesting a role for Cav in its
generation. Experiments with mBest 1 knockout mice (mBest 1 -/-) provided
additional insights in this matter: a greater Ca2+ response evoked by ATP-
stimulation in RPE cells from mbest1 -/- mice was observed, while ICaCC

General Introduction
31
remained unaltered comparatively to wild-type (wt) animals. The authors
proposed that Best 1 was not required for the generation of LP, but would rather
be responsible for antagonizing the LP luminance response, possibly by
inhibiting an increase in [Ca2+]i through modulation of Cav kinetics upon
stimulation182,189. The observation that some patients with mutations in the
BEST1 gene show normal LP in their electro-oculograms further corroborates
these findings168.
Recently, work from our laboratory suggests that Best 1 may act as a
counter-Cl− channel in the ER, which balances negative charges occurring
through release and reuptake of Ca2+190. This hypothesis is in agreement with
observations in other systems, where compensatory fluxes of Cl- out of the ER
facilitate Ca2+ release/uptake191,192.
Figure I. 8 - Putative role of Best 1 in Ca2+ signalling. (A) Stimulation of G-protein
(Gq/11) and phospholipase C (PLC) coupled receptors will increase IP3, induce initial
Ca2+ release from ER-stores and activate best1. This will facilitate Ca2+ release due to its
function as a counter-ion channel. (B) Alternatively best1 may control Ca2+ influx
through direct interaction with ER-located proteins, such as Stim1. Abbreviations -
SERCA: sarcoplasmic endoplasmic reticulum Ca2+ ATPase; Orai1, TRPC1, TRP3:
components of the store-operated Ca2+ influx. Adapted from (Kunzelmann et al.,
2009)186.
Briefly, release of Ca2+ from the ER stores results in accumulation of
negative charges inside this organelle, which in turn hinders further Ca2+
P2Y
2
IP3
ER
PIP2 Gq/11
PLCß,
Ca2+
ATP
Sti
m1
OR
AI-
1
TRP
3/TR
PC
1
Ca2+
SERCA
0 mV
Cl-
Best1
P2Y
2IP3
ER
PIP2 Gq/11
PLCß,
Ca2+
ATP
Stim1
Cl-
Best1
0 mV
A B

Part I
32
release. Efflux of Cl- through a compensatory Cl- channel would then
compensate for the charge build up and allow greater Ca2+ release.
Furthermore, accumulation of Cl- in the cytosol would be prevented by
opening of Cl- channels on the plasma membrane, allowing Cl- efflux from the
cell, thereby allowing complete dissipation of the charge build-up and thus
facilitating further Ca2+ release192.
3.3.2.4 Other physiological roles of bestrophins
Interestingly, results from our laboratory correlate Best 1 and ICaCC
upregulation during dedifferentiation and proliferation of epithelial cells, as well
as in fast growing cancer cells193,194. Best 2 expression has also been found to
be relevant during differentiation and growth of axons and cilia of sensory
neurons195. On the contrary, native epithelial tissues from human airways,
kidney, and colon show very little ICaCC196. It could thus be speculated that Best
1 affects proliferation either directly as a Cl- channel or by controlling
intracellular Ca2+ signalling186.
Interestingly, bestrophins are highly permeable not only to Cl- but also to
HCO-3197 Recently, Best 2 was shown to mediate HCO-
3 transport by goblet
cells in mouse colon, thus affecting colonic mucosa hydration and pH of the
colonic lumen198. Furthermore, the authors found an intriguing down-regulation
of mBest 2 in CFTR –/– mice.
Although the concept that bestrophins produce Ca2+-activated Cl−
currents has been corroborated in a number of other studies175,199, the
possibility that bestrophins may function as multifunctional proteins has not
been ruled out168,186. They would be thus Cl− channels as well as regulators of
other ion channels, such as voltage-gated Ca2+ channels.

General Introduction
33
3.3.3 Anoctamin 1 (Ano 1)
In 2008 three independent laboratories using different strategies
identified anoctamin 1 (Ano 1, NM 018043) as forming Ca2+-activated Cl-
channels200-202. In the Oh laboratory it was demonstrated that Ano1 was a bona
fide Ca2+-activated Cl- channel activated by both intracellular Ca2+ and Ca2+
mobilizing stimuli202. The channel was identified through a bioinformatic search
of channel- or transporter-like genes with multiple transmembrane domains and
multiple isoforms. On the other hand, Caputo and collaborators treated
bronchial epithelial cells with interleukin-4 (IL-4), which is known to increase
ICaCC. They then performed a global differential gene expression analysis to
identify membrane proteins that were regulated by IL-4, where they proved that
Ano 1 was associated with a ICaCC current200. The third group, Schroeder and
co-workers, showed elegantly that xAno 1 was the long searched Xenopus
CaCC201.
The newly identified CaCC was termed anoctamin due to the anion
selectivity of the protein, while the prefix oct stands for its eight transmembrane
domains202. Independent studies with diverse objectives have also identified
Ano 1 and other members of this family. Hence the multiple designations given
to the protein, as TMEM16A, DOG1 (discovered on GIST-1); TAOS2 (tumour
amplified and overexpressed sequence), ORAOV2 (oral cancer
overexpressed). Anoctamins compromise a family with ten identified paralogs in
mice and humans: TMEM16A – H, J and K respectively renamed Ano 1 to 10.
Curiously, Ano 7 (also called D-TMPP and NGEP) is specifically expressed in
prostate and is up-regulated in a number of prostate tumours160.
Since Ano 1 is the accepted designation for TMEM16A (HUGO
nomenclature committee (http://www.genenames.org/index.html)), this term will
be the used in this dissertation to designate TMEM16A.

Part I
34
These discoveries represent an exciting breakthrough which will help
elucidate the complex contribution of CaCCs in multiple systems, namely in the
CF field, which is of high relevance for the purpose of this doctoral work.
3.3.3.1 Discovery of Ano 1 and other Anoctamins
Ano 1 was known before its identification as CaCC, but up to 2008 had
an unknown function. In 2003, by using bioinformatic tools, Katoh and
collaborators described an uncharacterized gene (FLJ10261) in the human
chromosome 11q13, which had the Ano 1 protein as a product. Interestingly,
this region is one of the most frequently amplified regions of the human genome
in cancer and is known to be amplified e.g. in esophageal cancer, bladder
tumours, and breast cancer203. It is suggested that 11q13 amplification may be
driven by a cassette of genes that provide increased growth capacity or
metastatic advantage to cancer cells. Interestingly, Ano 1 null mice were also
generated prior to identification of its function as CaCC. This model was initially
generated to investigate its function during the development of the limb bud and
how it influenced its outgrowth on a cellular level204-206.
Further in silico studies led to the discovery of other members of the Ano
family of proteins207,208. The use of bioinformatic tools also enabled the
identification of GDD1, the gene responsible for gnathodiaphyseal dysplasia209
(characterized by fragility of the long bones and cemento-osseous lesions of the
jaw), as Ano5 or TMEM16E210.
3.3.3.2 Ano 1 Channel Properties
Up to now several “classical” CaCC characteristics have been confirmed
for Ano 1:
- activation through elevated intracellular Ca2+ with an EC50 of 2.6 μm at
-60 mV, which is very close to that reported for classical CaCCs.

General Introduction
35
- replacement of extracellular Cl− with gluconate has also been shown to
abolish the current.
- the current is blocked by low concentrations of established Cl− channel
blockers as diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS), 5-nitro-2-
(3-phenylpropylamino)benzoic acid (NPPB), niflumic acid (NFA) and
tamoxifen.
Nonetheless, the pharmacology of overexpressed Ano 1 is not
quantitatively the same as reported for native CaCCs. While in native tissues
the IC50 values for DIDS are 16–250μm, 22–68μm for NPPB and 2–44μm for
NFA, currents of overexpressed Ano 1 are blocked almost completely by 10μm
DIDS, NPPB, and (NFA)202. Although the xAno 1 current is insensitive to
tamoxifen, as has been found for native CaCCs, mAno 1 is completely blocked
by this compound. Furthermore, the currents generated by expression of xAno
1, mAno 1 and mTMEM16B in Axolotl oocytes have different kinetics, thus
suggesting that different TMEM16s are likely to have different gating
characteristics201. This also suggests that native currents of CaCCs are formed
by other accessory subunits besides Ano 1, possibly other paralogs that may
alter the pharmacology of the channel160. Still, collectively Ano 1 clearly
demonstrates many, if not all, of the characteristics required of a candidate
gene to produce ICaCC.
3.3.3.3 Expression and Localization
Upon the identification of Ano 1 it was also shown that it is expressed in
tissues that are known to express CaCCs, namely: mammary, salivary glands,
bronchiolar epithelial cells, pancreatic acinar cells, proximal kidney tubule
epithelium, retina and dorsal root ganglion sensory neurons200-202. In our
laboratory, real time PCR analysis was performed in a wide spectrum of mouse
tissues and also found predominant expression of Ano 1 in epithelial tissues,
although it is broadly expressed in almost every kind of mouse tissue186.

Part I
36
3.3.3.4 Predicted Structure
Figure I. 9 - Predicted topology of Ano 1. (A) A number of consensus sites for
phosphorylation by kinases are present in the N-terminus, but no binding site for Ca2+
(numbering of amino acids according to splice product a,c,d)186. Three cysteines (at
position 651, 656, 661) are located in the pore forming loop and are accessible to
sulfhydryl reagents. (B) localization of the alternative segments a, b, c, and d.
Inclusion/skipping of segments b, c, and d is due to alternative splicing of exons 6b, 13,
and 15, respectively. Instead, skipping of segment a occurs by usage of an alternative
promoter.211 Adapted from (Kunzelmann et al, 2009)186 and (Ferrera et al., 2009)211
As a member of the anoctamin family of proteins, Ano 1 has eight
transmembrane domains and a p-loop between TM 5 and 6, which is
speculated to be the pore forming region of the channel (Figure I. 9A).
Moreover, this region shows high homology between the different anoctamins
and mutation of conserved positively charged amino acids results in changes in
the ion selectivity of the channel202.
Ano 1 has no obvious E-F hand-like Ca2+-binding sites or other
consensus binding sites for Ca2+. Investigators speculate that another subunit
might confer Ca2+ sensitivity to the channel202 or that the Ca2+ binding site is of
a novel type that is not easily recognized. Despite such lack of obvious Ca2+-
binding sites, multiple consensus sites for different kinases can be found in the
cytoplasmic domains of the protein186. This protein exhibits at least three
alternative spliced exons (b, c and d), (Figure I. 9B) which alter the biophysical
BA

General Introduction
37
properties of the channel and whose splicing is differentially processed among
various organs211. For instance, segment b (exon 6b) and segment c (exon 13)
influence the Ca2+ sensitivity and voltage dependence of Ano 1-dependent
currents.
3.3.3.5 Role in development and disease
Ano 1 as well as Ano 7, were known to be upregulated during cell
proliferation and cancer long before their identification as ion channels212-215.
Ano 1 antibodies are actually used as markers for gastrointestinal stromal
tumours (GIST)215. This is the most common type of mesenchymal tumour
found in the gastrointestinal tract and although no Ano 1 mutations have been
identified in patients216, its function may support tumour progression. This is
consistent with the findings in our laboratory that Best 1 expression and Ca2+
activated Cl− currents are up-regulated during dedifferentiation and proliferation
of epithelial cells, as well as in fast growing cancer cells. Possibly Ano 1 affects
proliferation through its property as a Cl− channel, or by controlling intracellular
Ca2+ signalling186,193,194. The idea that ion channels may play a role in cancer
has been around for some years217-220, but the discovery of a family of anion
channels associated with multiple cancers will definitively give new insights to
the cancer field.
Ano 1 was also found to be one of the candidate genes responsible for
autosomal recessive hearing impairment221.
The production of Ano 1-/- mouse model also allowed more insights into
the role of this protein in mouse development204-206. These animals died soon
after delivery, in the early postnatal phase, possibly as a result of a pronounced
tracheomalacia which probably causes airway collapse. Rock and co-workers
speculated that the defect in the cartilage rings should be secondary to the
deficient embryonic stratification of the embryonic tracheal epithelium, which
may point toward a fascinating functional cross-talk between the surface
epithelium and the submucosal tissue during development186. Data from

Part I
38
developmental biology also showed accumulation of mucous in the lumen of
tracheas of Ano 1-/- mice, pointing towards an important function of Ano 1 for
MCC in mouse airways 222. It is thus reasonable to speculate that, in human
airways, Ano 1 could be an excellent pharmacological target to overcome the
defective CFTR-dependent Cl− secretion seen in CF patients. Nevertheless, the
discrepancies regarding the higher contribution of CaCC in mouse airways,
comparatively to the human tissue 223,224 must be considered (see Figure I. 10).
Figure I. 10 - Contribution of Ca2+-dependent Cl- secretion by Ano 1 in mouse and
human airways. (A) Schematic Cl− secretion induced by purinergic stimulation. Effect of
luminal ATP is much more pronounced in mouse (right) when compared to human (left)
airways. (B) Cell models for electrolyte transport in human (left) and mouse (right)
airways. CFTR is the dominant pathway for luminal Cl− exit in human airways, while Ano 1
is probably the main secretory pathway in mouse airways. (ADE: adenosine).

General Introduction
39
4 CF RESCUING STRATEGIES
4.1 RESCUING OF CFTR
Taking into account the broad spectrum of influence of CFTR in cellular
and organ homeostasis, it is not surprising that CF is a clinically complex
disease. Since conventional therapy for CF is largely symptomatic, researchers
have gained vast knowledge into the CFTR protein over the years, in order to
tackle the disease at its root. However, a cure for CF basic defect has not yet
been achieved. Nevertheless, various rescuing strategies have attempted to
overcome CF and a better understanding of the cellular defects that cause the
disease will certainly assist the search for a cure.
Firstly, great effort has been placed into developing pharmacological
tools capable of rescuing mutant CFTR, depending on the type of mutation
carried by the protein (See section 2.1 - From Gene to Protein: The genetics of
cystic fibrosis). In this sense, “correctors” of mutant CFTR mislocalization, as
well as “potentiators” of CFTR function, have been developed by high-
throughput screening of large compound libraries. The former group of
compounds (CFTR "correctors") aims at either improving F508del-CFTR folding
and/or increasing its traffic to the plasma membrane by favouring the cellular
milieu, possibly through modulation of interacting proteins, such as chaperones.
CFTR "potentiators", on the other hand, seek stimulation of CFTR function
when already at the plasma membrane. In terms of a “mutation-specific” or
“CFTR-repair” therapy “correctors” correspond to compounds that rescue the
trafficking defect of class II mutations; and “potentiators” are compounds that
stimulate the defective Cl- channel activity of class III, IV and V CFTR mutants,
and possibly also of plasma-membrane rescued class II mutants. An ideal
compound for F508del would thus combine both characteristics, acting both as
a corrector and potentiator.

Part I
40
4.2 IDENTIFYING MODIFIER GENES AS TARGETS
Secondly, genes that modify the effect of CF mutations on lung
dysfunction, i.e. “modifier genes”, also appear to be promising targets to unravel
novel therapeutic approaches. Ultimately this search might lead to the
identification of key genes and proteins that may be rational therapeutic targets
for CF. Different approaches have been taken in order to address this search:
"candidate selection" (which mainly include genes that regulate aspects of
innate lung defense and inflammatory cascades), genetic linkage searches,
such as genome-wide association studies (GWAS), and also complementary
proteomic approaches or high-content siRNA screens designed to find proteins
which function as modifiers225,28.
4.3 BYPASS APPROACHES
Thirdly, as CFTR is an important player in epithelial ion transport, the so-
called “bypass” approach consists in stimulating alternative pathways for Cl-
secretion across the apical membrane or modulating channels that are normally
regulated by CFTR, which ultimately also contribute to CF lung
pathogenesis22,226. As previously discussed (see section 2.4.2 Regulator of Ion
Transport across Epithelia) along with impaired cAMP-dependent Cl- secretion,
enhanced Na+ absorption is present in CF airways87. This results in
hyperabsorption of fluid and electrolytes by the surface epithelium, and
consequent reduction of the ASL. In this regard, blockage of ENaC with specific
inhibitors such as amiloride, benzamil or phenamil could be an alternative for
bypassing pharmacotherapy directly targeted at CFTR. Unfortunately the
duration of amiloride action in vivo is very brief (15–30 min), thereby limiting its
therapeutic efficacy227. In addition, CFTR is involved in HCO3- secretion, control
of osmotic water permeability, electroneutral NaCl transport and many other
aspects of epithelial cell physiology; thus, being actually a true conductance
regulator. Thus, it is possible that these other regulatory functions of CFTR may
not be corrected by focussing on just one.

General Introduction
41
Nevertheless, although CF reflects the dependency of epithelial tissues
on sustained Cl- secretion through CFTR, other channels permeable to Cl- are
present in the apical membrane of airway epithelial cells. It is thus believed that
activation of alternative non-CFTR Cl- channels might help to “bypass” the CF
basic defect, at least the lack of Cl- transport in the airways. Indeed, there is
compelling evidence that CFTR is not the only Cl- channel present in the apical
membrane of airway epithelial cells since the existence of an independent Ca2+
-activated apical Cl− current (ICaCC) has been demonstrated112. Ca2+ activated
Cl- channels (CaCCs) are thereby extremely relevant targets to be addressed in
CF113.


Part II. OBJECTIVES OF THE PRESENT WORK


Objectives
45
II OBJECTIVES OF THE PRESENT WORK
The present doctoral work aimed to elucidate the role and contribution of
CaCCs in Cystic Fibrosis. Biochemical and functional approaches were used so
as to explore alternative ways of overcoming CF disease and also to give new
insights into the mechanistic relationship between CFTR and CaCCs. We
started to explore Bestrophins because at the time these were the strongest
CaCC candidates. However, during the course of this doctoral work, three
independent laboratories using different strategies identified Anoctamin 1 (Ano
1, NM 018043) as forming a Ca2+-activated Cl- channel200-202.
Towards this overall goal, this doctoral work was focussed on the
following two specific objectives:
to use the Ano 1 null mice model to further examine the contribution
of this channel in mouse airway function and to gain new insights into
its contribution in CF lung disease.
to assess the contribution of CaCCs (Ano 1 as well as Bestrophin1)
in a CF setting. To this end, first the expression of these two CaCC
candidates was analysed in the presence of wt-CFTR or ER-retained
F508del-CFTR. Then, changes in Ca2+-signalling were explored and
the hypothesis that F508del-CFTR serves as a means for counter ion
transport that enhances Ca2+ signalling and CaCC activity was tested.


Part III. MATERIALS AND METHODS


Materials and Methods
49
III MATERIALS AND METHODS
1 MOLECULAR BIOLOGY
1.1 PLASMIDS, CDNAS AND SIRNAS
The pRK5 vector carrying cDNA for hBest 1 (NM_004183) was kindly
provided by Dr. Hugh Cahill at John Hopkins University, Baltimore, MD, USA167.
cDNA for human Ano 1 (NM_018043) was cloned from 16HBE cells (kindly
provided by Prof. D. Gruenert, CPMRI, San Francisco, CA) and inserted with a
FLAG tag (DYKDDDDK) into pcDNA3.1 V5-His (Invitrogen, Karlsruhe,
Germany) by Dr. Rainer Schreiber at Prof. Karl Kunzelmann’s lab at the
University of Regensburg, Germany. 16HBE cells express a Ano 1 isoform
containing exons a, b, and c according to Caputo et. al.200. PEXPR-IBA103
carrying the cDNA for mouse inositol-1,4,5-trisphosphate receptors binding
protein released with IP3 (IRBIT, NM_145542) was a kind gift from Dr. Humbert
De Smedt (K.U., Leuven, Belgium). All cDNAs were verified by sequencing.
Silencer® Select Validated siRNA (s650) targeted for IRBIT was
designed and purchased from Ambion (Austin, TX, USA).
1.2 SITE-DIRECTED MUTAGENESIS
The QuickChange® mutagenesis kit (Stratagene, LaJolla, CA, USA) was
used to introduce the FLAG epitope (full sequence DYKDDDDK, after aa L54 in
the first putative extracellular loop of hBest1) as well as the N-glycosylation
(Asn-Ala-Thr) site (full sequence GGNATGG, after aa E63 of hBest1) into the
pRK5-Bestrophin1 cDNA. Each reaction was performed by the use of
complementary pairs of custom designed HPLC-purified mutagenic primers
(Thermo Fisher Scientific, Ulm, Germany) shown in Table III. 1.

Part III
50
Table III. 1 - Custom design primers used to introduce FLAG tag and N-
Glycosylation into prk5-Best1 cDNA
Name of the Fw primer Sequence 5’ 3’ of the Fw primer
JR.B1.L54-FLAG CTTTATTTATAGGCTGGCCCTCGATTACAAGGATGACGACGATAAGACGGAAGAACAACAGCTGATG
JR.B1.E63-NGly-1x GAAGAACAACAGCTGATGTTTGAGGGCGGCAACGCCACCGGCGGCAAACTGACTCTGTATTGCGACAGC
Briefly, this procedure uses a supercoiled dsDNA vector with an insert of
interest and two synthetic oligonucleotide primers (reverse complement of each
other), each containing the desired mutation. For each mutant, a PCR reaction
was set up with 2.5 U of pfuTurbo® DNA polymerase (Stratagene, LaJolla, CA,
USA) or KOD Hot Start DNA polymerase (Calbiochem, Novabiochem,
Novagen®, Merck, Darmstadt, Germany) and cycling parameters set according
to manufacturer’s instructions. Amplification was confirmed by agarose gel
electrophoresis and the resultant nick-containing mutant plasmids were
digested with DpnI (Stratagene, LaJolla, CA, USA) (target sequence: 5’-
Gm6ATC – 3’), specific for methylated and hemimethylated DNA. This allows
destruction of template, thus selecting for mutation containing synthesized DNA.
The nicked vector DNA incorporating the desired mutation was then
transformed into XL1-Blue competent cells (see section 1.4, Part III). Production
of each construct was confirmed by sequencing (see section 1.6, Part III).
1.3 COMPETENT BACTERIA PREPARATION
In the present work, the XL1-Blue bacterial strain was used. This strain
has the following genotype: supE44, hsdR17, recA1, endA1, gyrA46, thi relA1,
lac F’[proAB+ lacIq lacZΔM15::Tnt10 (Tetr)] (Stratagene, La Jolla, CA, USA).
Competent bacteria preparation was performed according to the
technique described by Hanrahan228. Briefly, cells were plated on solid LB

Materials and Methods
51
media (2 % (w/v) bactotriptone, 0.5 % (w/v) yeast bactoextract, 10 mM NaCl,
1.5 % (w/v) agar). A single colony was inoculated in liquid media and incubated
at 37°C with constant agitation (220 rpm). Once a concentration of
approximately 4 - 7x107 bacteria/ml (determined by A550 between 0,45-0,55)
was achieved, bacteria were left on ice for 15 min and then centrifuged at 3000
rpm for 15 min at 4 °C. The pellet was resuspended in a third of the initial
volume of RF1 buffer (100 mM RbCl, 50 mM MnCl2, 30 mM KCH3COO pH7.5,
10 mM CaCl2, 15 % (w/v) glycerol, pH 5.8). The suspension was left on ice for
15 min and then centrifuge at 3000 rpm for 15 min at 4 °C. The pellet was
resuspended in 1/12.5 of the initial volume of buffer RF2 (10 mM MOPS, 10 mM
RbCl, 75 mM CaCl2, 15% (w/v) glycerol, pH 6.8). After a further incubation on
ice for 15min, tubes containing 200 μl aliquots were rapidly frozen in liquid
nitrogen and stored at -80 °C.
1.4 TRANSFORMATION AND ISOLATION OF PLASMID DNA
E. coli cells (XL1B strain) were made competent as described above (see
section 1.2, Part III). An aliquot (10 μl) of each mutagenesis reaction sample
(hbest1 FLAG, hbest1 NG) was added to 200 μl of XL1B competent cells. This
mixture was placed on ice for 30 min and then heat-shock (42 ºC) was applied
for 3 min to allow the uptake of the plasmids. The cells were incubated for 1 h at
37 °C with continuous shaking at 250 rpm, after which the mixture was spread
evenly across the surface of fresh LB-agar plates with 100 μg/ml ampicillin
(Amp) using sterile glass beads. Plates were incubated at 37 °C for 12-16 h.
For DNA extraction colonies were picked from each plate and used to
inoculate 3 ml of LB medium supplemented with 100 μg/ml Amp and grown
overnight at 37 °C, with continuous shaking at 250 rpm. DNA was then
extracted and isolated from the bacterial culture using a miniprep kit
(NucleoSpin Plasmid kit, Macherey-Nagel, Düren, Germany) according to
manufacturer instructions. Plasmid DNA yield was quantified with NanoDrop
spectrophotometer (NanoDrop Technologies, Wilmington, USA) and additionally
DNA quality was analyzed by agarose gel electrophoresis.

Part III
52
1.5 PLASMID DNA EXTRACTION AND PRELIMINARY ANALYSIS BY
PCR TO ASSESS FOR THE PRESENCE OF THE RESPECTIVE INSERT
The presence of the desired constructs was tested by polymerase chain
reaction (PCR). The colonies resulting from the mutagenesis were used as a
template in a reaction solution containing, for a final volume of 50 μl, 1x PCR
buffer I (supplied with Taq DNA Polymerase, Invitrogen™, Karlsruhe,
Germany), 1.5mM MgCl2, 10 pmol of each primer, 200 μM each dNTP, 1.5 U
Taq, DNA Polymerase (Invitrogen™). Reactions were carried out using a
Biometra thermocycler (Biometra, Göttingen, Germany). Specificity of PCR
products was confirmed by sizing on agarose gels. The use of appropriate
primers allowed visualization for the presence or absence of the inserted
mutations. This pair of primers allowed amplification of a hbest1 fragment in
addition to the respective tag.
1.6 SEQUENCING OF NOVEL PLASMIDS
The constructs that were positive for the PCR screening were subjected
to sequencing using the ABI Prism® 3100 Genetic Analyzer (Applied
Biosystems) using the BigDye®Terminator v1.1 sequencing kit (Applied
Biosystems, Norwalk, CA, USA).
1.7 RNA ISOLATION, CDNA AND REAL TIME RT-PCR IN CFBE
CELLS
For real-time PCR, total RNA was isolated using NucleoSpin RNA II
columns (Macherey-Nagel, Düren, Germany). 2µg of total RNA were reverse-
transcribed using random primer and RT (Moloney murine leukaemia virus
reverse transcriptase, Promega, Madison, USA). mRNAs encoding hbest1,
hAno 1 and β-actin were evaluated by real-time PCR using 480 Light Cycler
(Roche, Mannheim, Germany) and Quanti Tect SYBR Green PCR Kit (Quiagen,
Hilden, Germany). Each reaction contained 2 μl of Master Mix (including Taq
polymerase, desoxyribonucleotide triphosphates, and SYBR Green buffer), 1

Materials and Methods
53
pm of each primer sense and antisense, and 1 μl of cDNA. After activation of
the Taq polymerase for 15 min at 95 °C cDNA was amplified for 15 s at 95 °C,
20 s at 55 °C, and 20 s at 72 °C, for 50 cycles. The oligonucleotide primers
were designed using the Universal ProbeLibrary Assay Design Centre from
Roche Applied Science (https://www.roche-applied-
science.com/sis/rtpcr/upl/index.jsp ) and are shown below in Table III. 2:
Table III. 2 - Primers used For Real Time RT PCR in CFBE Cells
Target Sequence 5’ 3’ of Fw and Rv Primers; size of amplified product
hAno 1 (NM_018043.4)
5′-CCTCACGGGCTTTGAAGAG-3′ and 5′-CTCCAAGACTCTGGCTTCGT-3′
hBest1 (NM_004183)
5'-TCTTCACGTTCCTGCAGTTCT-3' and 5'-TCCTCTCCAAAGGGGTTGAT-3'
β-actin (NM_001101)
5′-CAACGGCTCCGGCATGTG-3′ and 5′-CTTGCTCTGGGCCTCGTC-3′
1.8 RNA ISOLATION, CDNA AND REAL TIME RT-PCR IN NASAL
CELLS:
Following approved by the Ethical Review Board of the University of
Lisbon and written consent by patients (or their legal representatives), samples
were collected from F508del-homozygous CF through nasal scraping, and RNA
was isolated using the RNeasy Mini Kit (Qiagen), following manufacturer’s
instructions. Concentration and purity was determined by NanoDrop
spectrophotometry (NanoDrop, Thermo Scientific, Wilmington, DE, USA). An
aliquot containing 1ug of each sample of total RNA was taken and stored at -
80ºC. The stored aliquot was later reverse transcribed to cDNA using
Superscript III Reverse Transcriptase (Invitrogen) according to the
manufacturer’s instructions. The primers used in PCR amplification of Ano 1
and GAPDH cDNA were taken from the Harvard Primerbank

Part III
54
(http://pga.mgh.harvard.edu/primerbank/index.html) and are shown in Table III.
3. Suitability of primers for real time quantitative PCR amplification was tested
by amplifying a product from test cDNAs using the chosen primer pairs in a
conventional PCR reaction, and confirming the absence of non-specific PCR
products for all targets by electrophoretic separation of the products in 2%
ethidium bromide stained agarose gels.
Table III. 3 - Primers used For Real Time RT PCR in nasal Cells
Target Sequence 5’ 3’ of Fw and Rv Primers; size of amplified product
hAno 1 5'-GCGGGACGAGGACACTAAAAT-3' and 5'-CTCTGCACAGCACGTTCCA-3';
74 bp
hBest1 5'-ACTCTGTATTGCGACAGCTACATC-3' and 5'-CGGCAGGTTCTCGTACTG-3'; 108 bp
GAPDH 5'-ATGGGGAAGGTGAAGGTCG-3' and 5'-GGGGTCATTGATGGCAACAATA-3'; 108 bp
Real-time PCR was used to quantify the amount of a target sequence in
a cDNA sample. The target sequence was amplified with primer pairs for Ano 1,
hBEST1 and GAPDH shown above (Table III. 3). The accumulation of double
stranded PCR product DNA was monitored in real time once every cycle during
the amplification reaction by measuring fluorescence of the dsDNA-specific
fluorescent dye SYBR Green, whose intensity is proportional to the amount of
PCR product. The signal curves of standards and unknowns, measured in the
same run, were used for quantification. Quantitative real-time PCR experiments
were performed in an ABI 7000 Sequence detection System (Applied
Biosystems). This instrument, and its associated software, allows the
simultaneous measurement of the fluorescence of SYBR Green in all wells of a
96 well plate throughout the progress of a PCR reaction. The 20 µl PCRs
contained 5 µl of a 1:5 dilution of template cDNA, 0.25 µM of each forward and
reverse primer, and 1x SYBR Green PCR master mix (Applied Biosystems,

Materials and Methods
55
Warrington, UK) according to the manufacturer’s instructions. The template
DNA was either cDNA from Human Bronchial Epithelial (HBE) cells (standards)
or nasal cell cDNA (samples). Each 96 well plate run contained a series of
standards (5 standards with successive 5-fold dilutions) in duplicate, and the 5 x
CF and 5 x non-CF unknowns in triplicate, for two targets (Ano 1 or hBEST1
plus the housekeeping gene GAPDH). The cycling protocol was as follows:
initial denaturation for 10 min at 95°C, followed by 40 cycles with 15 s at 95°C,
and 60 s at 60°C. Fluorescence was measured after each extension phase at
60°C for SYBR Green detection. A melting curve analysis with a temperature
gradient from 60 to 95°C was performed following the PCR amplification, to
confirm that only specific products was amplified. The fluorescence data were
evaluated using the dCT method, and the final relative expression levels are
expressed as 2(-dCT) +/- SEM.
Real time RT-PCR data in nasal cells were generated by Luka Clarke, at
Prof. Margarida Amaral’s laboratory at the University of Lisbon.
2 CELL CULTURE, TRANSFECTIONS AND PROTEIN ANALYSIS
2.1 MAMMALIAN CELL LINES
Cystic Fibrosis Bronchial Epithelial (CFBE) cells stably expressing wt-
CFTR or F508del-CFTR229 were a generous gift from Dr. J.P. Clancy (University
of Alabama at Birmingham, Birmingham, Alabama)
The Baby Hamster Kidney (BHK) cell line is a quasidiploid established
line of Syrian hamster cells, descendent from a clone of an unusually rapidly
growing primary culture of new-born hamster kidney tissue230.
Fisher Rat Thyroid (FRTL) cell line is a diplod and differentiated
established line of Fisher rat cells, descended from a individual epithelial colony
from a pool of thyroid glands pooled from 6-week-old littermates of the NIH
Fischer 344 inbred strain of rats231. This cell line is generally abbreviated to FRT
cell line.

Part III
56
Fluorescently labelled A549 cells expressing wt-CFTR or F508del-CFTR
after induction with doxycycline (DOX) were created in Prof. Amaral’s
laboratory. For this, wt-CFTR and F508del-CFTR were fused in the N-terminus
to mCherry, a fluorescent protein obtained from DsRed by changing the
chromophore environment232. Additionally, a FLAG tag (octapeptide:
DYKDDDDK) was inserted by site-directed mutagenesis (see section 1.2, Part
III) in the fourth extracellular loop of CFTR. By recombination reactions, this
construct was then inserted into the destination vectors with a TetON DOX-
sensitive promoter.
2.2 CELL CULTURE CONDITIONS
CFBE cells were grown in Minimum Essential Medium (MEM)
supplemented with 2 mm glutamine. The cells were seeded on fibronectin
(Invitrogen)/collagen (Vitrogen 100, Angiotech BioMaterials, Palo Alto, CA)-
coated glass coverslips.
Baby hamster kidney (BHK) and Fisher Rat Thyroid (FRT) cells were
cultured in a 1:1 mixture of Dulbecco's modified Eagle's Medium and Ham's F-
12 nutrient medium (DMEM/F12), A549 cells expressing wt-CFTR or F508del-
CFTR were grown in Dulbecco's modified Eagle's medium (DMEM). Expression
of wt-CFTR or F508del-CFTR was induced with 1µg/ml doxycycline (Sigma-
Aldrich, Taufkirchen, Germany) 24h prior to the experiment. All cell lines were
grown at 5% CO2 and 37°C and supplemented with 100 units/ml penicillin, 100
μg/ml streptomycin and 10% fetal bovine serum (FBS), except for the BHK and
FRT cells which were supplemented with only 5% (v/v) FBS instead.
2.3 TRANSFECTIONS
Lipofectamine™2000 Transfection Reagent (Invitrogen, Karlsruhe,
Germany) or FuGENE HD Transfection Reagent (Roche, Mannheim, Germany)
were used for the transfections according to the manufacture’s guidelines. All
experiments were performed 48h to 72h after transfection.

Materials and Methods
57
2.4 IMMUNOCYTOCHEMISTRY
BHK cells transfected with either hbest1-FLAG or Ano 1- FLAG-His were
rinsed twice with cold phosphate-buffered saline (PBS). Non-permeabilized
cells were first incubated with a monoclonal ANTI-FLAG® M2 antibody (Sigma-
Aldrich) for 45 min at 4 ºC. After two washes with PBS the cells (permeabilized
and non-permeabilized) were fixed with 4 % (v/v) formaldehyde in cold PBS for
15 min and washed twice. Permeabilized cells were treated with Triton X-100
(0.25 % w/v) for 20 min and washed twice in PBS. These cells were incubated
with the ANTI-FLAG® M2 antibody for 45 min. Both batches were washed and
incubated with Alexa Fluor 488 conjugated anti-mouse IgG (Invitrogen,
Karlsruhe, Germany) for 45 min at room temperature. Slides were mounted with
Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 4′,6-
diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich) to stain nuclei.
Immunofluorescence staining was observed and recorded either on an
Axioskop fluorescence microscope (Zeiss, Jena, Germany) with Power Gene
810/Probe & CGH software system (Perceptive Scientific Instruments, Chester,
UK) or on a confocal microscope Leica TCS SPE (Leica, Jehna, Germany) and
respective software. No background staining or autofluorescence were
observed with untransfected BHK cells.
2.5 WESTERN BLOT
Protein extracts were prepared using cell lysis buffer (1.5 % (w/v) SDS;
10 % (v/v) glycerol; 0.001 % (w/v) bromophenol blue; 0.5 mM dithiotreitol; 31.25
mM Tris (pH 6.8)). DNA was sheared by sonication or samples were treated
with 1 µl benzonase® endonuclease (5U) (Sigma, Aldrich). Samples were then
quantified through a modified Lowry method. Briefly, 5l of total cell extracts
were diluted in water and proteins were solubilised with 0.015 % (w/v) sodium
deoxycholate. Samples were then precipitated with 7 % (w/v) trichloroacetic
acid and centrifuged at 14000 rpm for 5 min. The supernatant was removed by
suction and the pellet was resuspended in water and incubated with freshly

Part III
58
made solution A (0.625 % (w/v) Na2CO3, 0.1 % (w/v) Na/K tartarate, 0.5 % (w/v)
CuSO4, 1.25 % (w/v) SDS, 100 mM NaOH). Finally, solution B (Folin-Ciocalteau
reagent diluted 5-fold in water) was added to each sample. After an incubation
of 30 min, A750 was measured and protein concentration was determined using
a regression equation established with BSA standards (BioRad, Hercules, CA,
USA) processed in parallel with the samples.
Equal amounts of protein extracts of each sample were digested with
Endoglycosidase H (Endo H) or N-Glycosidase F (PNGase F) (New England
BioLabs, Frankfurt am Main, Germany) at 37ºC overnight according to
manufacture instructions.
The samples were then loaded on 7 % (w/v) mini-gel and separated by
SDS-polyacrylamide gel electrophoreses (PAGE) (Mini-Protean® II, BioRad,
Hercules, CA, USA) and electrotransfered (Mini trans- Blot® BioRad, Hercules,
CA, USA) onto nitrocellulose filters (Schleicher & Schuell, Dassel, Germany).
After blocking with 5 % (w/v) skimmed milk (Molico, Nestle, Vevey, Switzerland)
in PBS containing 0.1 % (v/v) Tween (PBST) for 1 h, the filters were probed with
anti-hbest1 antibody (Davids Biotechnology, Regensburg, Germany) diluted
1:5000 in 3% (w/v) skimmed milk blocking solution overnight (ON) at 4ºC.
Following washing with PBST, filters were incubated with horse radish
peroxidase (HRP)-conjugated anti-rabbit IgG monoclonal antibody diluted
1:3000 in blocking solution. After additional washing in PBST,
chemiluminescent detection was performed using the Super Signal® West Pico
Chemiluminescent Substrate (Pierce, Rockford, IL, USA).
2.6 CO-IMMUNOPRECIPITATION
Co-immunoprecipitation experiments were performed in A549 cells
overexpressing wt-CFTR or F508del-CFTR and in baby hamster kidney (BHK)
cells transiently expressing wt-CFTR or F508del-CFTR. IRBIT was transiently
transfected via adenoviral transduction.

Materials and Methods
59
A549 and BHK cells expressing wt-CFTR or F508del-CFTR as well as
IRBIT were lysed in lysis buffer (50 mM Tris, 0.3 M NaCl, 1% Triton, 0.5mM
dithiothreitol, 0.5mM phenylmethylsulfonyl fluoride, 0.5 mM benzamidine, 5µM
leupeptin, pH 7.5). The lysate (200µg/sample) was cleared by centrifugation (5
min, 10,000 g, 4 °C). Supernatants were incubated for 90 min at 4 °C while
gently mixing (1400 rpm) with antibodies (1.5µg): (i) anti-IRBIT, (ii) anti-CFTR,
(iii) anti-FLAG, (iv) nonspecific mouse IgG (Santa Cruz Biotechnology Inc., sc-
2025), or (v) nonspecific rabbit IgG (Santa Cruz Biotechnology Inc., sc-2027).
Subsequently, r-protein A-TSK-Sepharose resin (50% in lysis buffer) was added
and incubation was continued for another hour. Beads were collected by
centrifugation (20 s, 2000 g) and washed once with Tris-buffered saline (TBS)
supplemented with 0.25% Triton X-100 and 0.1 M LiCl and three times with TBS
supplemented with 0.25% Triton X-100. Finally, the beads were resuspended in
30 µl of NuPAGE loading dye solution sample buffer and heated for 10 min at
75 °C. Supernatants were further analyzed by SDS-PAGE and Western blotting.
Co-imunoprecipitation data for IRBIT and CFTR were obtained by Eva
Sammels at Professor Humbert de Smedt’s laboratory at the Department
Molecular Cell Biology, K.U.Leuven, Leuven, Belgium.
3 FUNCTIONAL ANALYSIS
3.1 IODIDE QUENCHING
Quenching of intracellular fluorescence generated by the iodide sensitive
Enhanced Yellow Fluorescent Protein (EYFP-I152L) was used to measure
anion conductance233. YFPI152L fluorescence was excited at 490 nm using a
polychromatic illumination system for microscopic fluorescence measurement
(TILL Photonics, Gräfelfing, Germany) and the emitted light measured at
535±15 nm with a photomultiplier detector (SF, Zeiss). Quenching of YFP-I152L
fluorescence by I− influx was induced by replacing 5 mM extracellular Cl− by I−.
Images were acquired with a Coolsnap HQ CCD camera (Roper Scientific) and
processed with Metafluor software (Universal Imaging). Cells grown on cover

Part III
60
slips were mounted in a thermostatically controlled imaging chamber
maintained at 37°C. Cells were continuously perfused at 4–5 ml/ min with
Ringer solution. Changes in fluorescence induced by I− are expressed as initial
rates of maximal fluorescence decrease (ΔRF/Δt). For quantitative analysis,
cells with very low or excessively high fluorescence were discarded. The rate of
fluorescence quenching for any given condition was not dependent on the
absolute YFP fluorescence.
3.2 MEASUREMENT OF THE INTRACELLULAR CA2+
CONCENTRATION
Cells were loaded with 2 µM Fura-2/AM (Molecular Probes, Eugene, OR)
in Ringer solution (NaCl 145mM, KH2PO4 0.4mM, K2HPO4 1.6mM, d-glucose
6mM, MgCl2 1mM, Ca-gluconate 1.3mM, pH 7.4) for 1h at room temperature.
Use of the detergent Pluronic F-127 (Molecular Probes, Leiden, Netherlands)
was recommended by the manufacturer, to improve the solubilisation of the
dye. Incubation was carried out in a dark environment, to prevent bleaching of
the dye. Fluorescence was detected in cells perfused continuously at 37 °C,
using an inverted microscope (IMT-2, Olympus, Nuremberg, Germany) and a
high speed polychromator system (VisiChrome, Puchheim, Germany). Fura-2
was excited at 340/380 nm, and emission was recorded between 470 and
550nm using a charge coupled device camera (CoolSnap HQ, Visitron). [Ca2+]i
was calculated from the 340/380 nm fluorescence ratio after background
subtraction.

Materials and Methods
61
The formula used to calculate [Ca2+]i was:
,
where R is the observed fluorescence ratio (340nm/380nm). The values
Rmax and Rmin (maximum and minimum ratios) and the constant Sf2/Sb2
(fluorescence of free and Ca2+-bound Fura-2 at 380 nm) were calculated using
2 µmol/l ionomycin (Calbiochem), 5 µM nigericin, 10 µM monensin (Sigma), and
5 mM EGTA to equilibrate intracellular and extracellular Ca2+ in intact Fura-2-
loaded cells. The dissociation constant for the Fura-2-Ca2+ complex was taken
as 224 nM.
3.3 CRNA AND DOUBLE ELECTRODE VOLTAGE CLAMP:
cDNAs encoding inositol-1,4,5-trisphosphate (IP3) receptor binding
protein released with IP3 (IRBIT), P2Y2-receptors or F508del-CFTR were
linearized in pBluescript with NotI or MluI, and in vitro transcribed using T7, T3,
or SP6 promoter and polymerase (Promega). Oocytes were isolated from adult
Xenopus laevis female frogs and defolliculated by a 45-min treatment with
collagenase (type A, Boehringer, Germany). Subsequently, oocytes were rinsed
and kept at 18 °C in NMDG/ND96 buffer (in mM): NMDG3, 96; HCl, 80; KCl, 2;
CaCl2, 1.8; MgCl2, 1; HEPES, 5; sodium pyruvate, 2.5; pH 7.55), supplemented
with theophylline (0.5 mM) and gentamicin (5 mg/liter). Oocytes were injected
with cRNA (10 ng, 47 nl double-distilled water) encoding IRBIT, P2Y2-receptors,
wt and F508del-CFTR. Water injected oocytes served as controls. 2 - 4 days
after injection, oocytes were impaled with two electrodes (Clark Instruments Ltd,
Salisbury, UK), which had a resistances of < 1 M when filled with 2.7 M KCI.
Using two bath electrodes and a virtual-ground head stage, the voltage drop
across the serial resistance was effectively zero. Membrane currents were
measured by voltage clamping (oocyte clamp amplifier, Warner Instruments
LLC, Hamden CT) in intervals from -60 to +40 mV, in steps of 10 mV, each 1 s.
The bath was continuously perfused at a rate of 5 ml/min. All experiments were
conducted at room temperature (22 °C).

Part III
62
All double electrode voltage clamp data were generated by Patthara
Kongsuphol, at Prof. Karl Kunzelmann’s lab at the University of Regensburg,
Germany.
4 ANIMALS
4.1 ANIMALS AND TISSUE PREPARATION
Animal studies were conducted according to the German laws on
protection of animals. Generation of a null allele of Ano 1 knock-out animals has
been described earlier206. Mice (1–3 days) were sacrificed with Isofluran
(Baxter, Germany). Tracheas were dissected, opened longitudinally on the
opposite side of the cartilage-free zone, and transferred immediately into an ice-
cold buffer solution of the following composition (mM): NaCl 145, KCI 3.8, D-
glucose 5, MgCI2 1, KH2PO4 0,4, K2HPO4 .1,6, Ca-gluconate 1.3, pH 7.4.
4.2 MEASUREMENT OF MUCOCILIARY CLEARANCE
Tracheas isolated from mice (see section 4.1, Part III) were mounted with
insect needles onto extra thick blot paper (Bio-Rad) and transferred into a
water-saturated chamber at 37 °C. The filter paper was perfused with Ringer
solution at a rate of 1 ml/min and at 37 °C. Polystyrene black-dyed
microspheres (diameter 6.51 ± 0.58m, Polybead®, Polyscience Inc.,
Warrington, PA) were washed with Ringer solution (± DIDS 100 µM or ATP 100
µM) and ~10l of particle solution with ~0.5% latex were added onto the
mucosal surface of the trachea. During experiments the serosal side of the
trachea was sequentially perfused under control and stimulated conditions
(±100 µM CCH). Viability of the tracheas was assessed by contraction in
response to CCH stimulation, and those that did not respond were excluded
from the analysis. The particle transport on different conditions was visualized
by recording of images every 10s for 15 min using a Zeiss stereo microscope
Discovery V12 with PlanS objective (1.0x, FWD 81 mm), a Zeiss digital camera
AxioCam ICc1 and the software AxioVision (Release 4.6.3, Zeiss, Göttingen,

Materials and Methods
63
Germany). 5 to 15 moving particles on different regions of the trachea were
selected and the transport was calculated by measurement of the distance
traveled per time (µm/min) using the AxioVision Software (Release 4.6.3, Zeiss,
Göttingen, Germany).
4.3 USSING CHAMBER
After mounting into a perfused micro Ussing chamber, apical and
basolateral surfaces of the epithelium were perfused continuously with buffer
solution at a rate of 5 - 10 ml/min (chamber volume 2 ml). All experiments were
carried out at 37 °C under open circuit conditions. Transepithelial resistance
(Rte) was determined by applying short (1 s) current pulses (I = 0.5 µA) and the
corresponding changes in transepithelial voltage (Vte) and basal Vte were
recorded continuously. Values for the Vte were referred to the serosal side of the
epithelium. The equivalent short-circuit current (Isc) was calculated according to
Ohm’s law from Vte and Rte (Isc = Vte / Rte).
All using chamber data were generated by Jiraporn Ousingsawat, at Prof.
Karl Kunzelmann’s lab at the University of Regensburg, Germany.


Part IV. RESULTS AND DISCUSSION


Loss of Ano 1 Causes a Defect in Epithelial Ca2+
-dependent Cl- Transport
67
IV RESULTS AND DISCUSSION
Chapter 1. LOSS OF ANO 1 CAUSES A DEFECT IN EPITHELIAL
CA2+-DEPENDENT CL- TRANSPORT
Work included in:
Ousingsawat, J., Martins, J. R., Schreiber, R., Rock, J. R., Harfe, B. D., and Kunzelmann, K. Loss of Ano 1 causes a defect in epithelial Ca
2+-
dependent chloride transport.
The Journal of Biological Chemistry 284, 28698-703. (2009)
1 ABSTRACT
Molecular identification of the Ca2+-dependent Cl- channel Ano 1
provided a fundamental step in understanding Ca2+-dependent Cl- secretion in
epithelia. Ano 1 is an intrinsic constituent of Ca2+-dependent Cl- channels in
cultured epithelia, but its physiological role in native epithelial tissues remains
largely obscure. Here, we demonstrate that Cl- secretion in native epithelia
activated by Ca2+-dependent agonists is missing in mice lacking expression of
Ano 1. Ca2+-dependent Cl- transport was missing or largely reduced in isolated
tracheal of Ano 1 -/- animals. Measurement of particle transport on the surface
of tracheas ex vivo indicated largely reduced mucociliary clearance in Ano 1 -/-
mice. These results clearly demonstrate the broad physiological role of Ano 1-/-
for Ca2+-dependent Cl- secretion and provide the basis for novel treatments in
cystic fibrosis.
2 INTRODUCTION
Electrolyte secretion in epithelial tissues is based on the major second
messenger pathways cAMP and Ca2+, which activate the cystic fibrosis
transmembrane conductance regulator (CFTR) Cl- channels and Ca2+-
dependent Cl- channels, respectively10,129,140. CFTR conducts Cl- in epithelial
cells of airways, intestine, and the ducts of pancreas and sweat gland, while

Part IV – Chapter 1
68
Ca2+-dependent Cl- channels secrete Cl- in pancreatic acini and salivary and
sweat glands158,234,235. Controversy exists as to the contribution of these
channels to Cl- secretion in submucosal glands of airways and the relevance for
CF236-238. While cAMP-dependent Cl- secretion by CFTR is well examined,
detailed analysis of epithelial Ca2+-dependent Cl- secretion is hampered by the
lack of a molecular counterpart. Although bestrophins may form Ca2+-
dependent Cl- channels and facilitate Ca2+-dependent Cl- secretion in epithelial
tissues168,179, they are unlikely to form secretory Cl-channels in the apical cell
membrane, because Ca2+-dependent Cl- secretion is still present in epithelia of
mice lacking expression of bestrophin173. Bestrophins may rather have an
intracellular function by facilitating receptor mediated Ca2+ signalling and
activation of membrane localized channels239. With the discovery that Ano 1
produces Ca2+-dependent Cl- currents with biophysical and pharmacological
properties close to those in native epithelial tissues, these proteins are now very
likely candidates for endogenous Ca2+-dependent Cl- channels200-202,222. In
cultured airway epithelial cells, small interfering (si) RNA knockdown of
endogenous Ano 1 largely reduced calcium-dependent chloride secretion200.
However, apart from preliminary studies of airways and salivary glands, the
physiological significance of Ano 1 in native epithelia, particularly in glands, is
unclear202,222.
3 RESULTS AND DISCUSSION
3.1 DEFECTIVE CA2+-DEPENDENT CL
- SECRETION IN AIRWAYS OF
ANO 1 -/- ANIMALS
We examined Ca2+ dependent Cl- secretion in epithelial tissues from wt
mice and from mice lacking expression of Ano 1. As reported earlier, knockout
of Ano 1 in mice leads to death within one month of birth205. Utilizing a micro
Ussing chamber that allows measurements of the transepithelial voltage (Vte) in
tracheas from 3-5 day old pups, we examined Ca2+ dependent Cl- secretion
upon stimulation with the muscarinic M3 receptor agonist carbachol (CCH)240.

Loss of Ano 1 Causes a Defect in Epithelial Ca2+
-dependent Cl- Transport
69
Figure IV.1. 1 - Defective Ca2+-dependent Cl- secretion in airways of Ano 1-/-
animals. A) Ussing chamber recording of the transepithelial voltage (Vte) in isolated
mouse trachea. Carbachol (CCH; 100 µM) activated a transient lumen negative Vte in
tracheas of Ano 1+/+ animals indicating activation of Ca2+-dependent Cl- secretion
(CaCC). CaCC was missing in tracheas of Ano 1-/- mice. B) Summary of the equivalent
short circuit currents (Isc) indicates largely reduced cholinergic Cl- secretion in Ano 1-/-
tracheas. C) Activation of lumen negative Vte and D) short circuit currents by luminal
application of 100 µM UTP in tracheas of Ano 1+/+ mice, which was reduced in Ano 1-/-
tracheas. E) Activation of lumen negative Vte and F) short circuit currents by luminal
-8
-6
-4
-2
0
Vte
Ano1+/+
CCH CCH
2 min
Ano1 -/-A B
C D
-4
-3
-2
-1
0UTP UTP
2 min
0
20
40
60
80
I sc-C
CH
(
A/c
m2) (12)
#
(13)
Ano1-/-
0
20
40
60
80
I sc-U
TP (
A/c
m2)
(15)
(6)#
E F
-4
-3
-2
-1
0ATP
2 min
ATP
0
20
40
60
80
I sc-A
TP (
A/c
m2) (13)
(6)#
Ano1+/+
Ano1+/+ Ano1 -/-
Ano1+/+ Ano1 -/-
Ano1-/-
Ano1+/+
Ano1-/-
Ano1+/+

Part IV – Chapter 1
70
application of 100 µM ATP in tracheas of Ano 1+/+ mice, which was reduced in Ano 1-/-
tracheas. Mean ± SEM, (n) = number of tissues measured, # significant difference when
compared to Ano 1+/+. [Data obtained by Jiraporn Ousingsawat at the Institut für
Physiologie, Universität Regensburg, Germany; included in this thesis with permission]
Basolateral application of CCH induced a negative voltage deflection,
indicating activation of transient Cl- secretion, probably by submucosal glands of
tracheas from Ano 1+/+ pups (Figure IV.1. 1A, B). Cl- secretion was inhibited by
niflumic acid (NFA) and by Ca2+ depletion from the ER, using the Ca2+-pump
inhibitor cyclopiazonic acid (CPA) (Figure IV.1. 2A,B). Thus Ca2+-dependent Cl-
secretion in murine neonatal tracheas is similar to that in adult tracheas but of
much smaller amplitude240. However, Cl- secretion was missing in Ano 1-/-
animals (Figure IV.1. 1A,B). Cl- secretion in wt animals was followed by a
transient K+ secretion (positive voltage deflection) that was present in both +/+
and -/- animals (Figure IV.1. 1A). Thus Ca2+-dependent Cl- secretion was
almost absent in Ano 1-/- animals, while K+ secretion was unaffected.
Stimulation of apical purinergic receptors by UTP increased a transient Cl-
secretion in +/+ tracheas, but had only small effects on -/- tracheas (Figure IV.1.
1C, D). A similar phenotype was observed when P2Y2 receptors were
stimulated with ATP (Figure IV.1. 1E, F).

Loss of Ano 1 Causes a Defect in Epithelial Ca2+
-dependent Cl- Transport
71
Figure IV.1. 2 - Residual Chloride secretion in Ano 1-/- airways is due to CFTR.
A) Transepithelial voltage and effect of carbachol (CCH, 100 µM, black bar) measured
in a trachea of an Ano 1+/+ mouse in the absence and presence of niflumic acid
(NFA, 100 µM). B) Summary of the equivalent short circuit currents (Isc) indicates
inhibition of cholinergic Cl- secretion in Ano 1+/+ tracheas by NFA and cyclopiazonic
acid (CPA; 100 µM). C,D) Effect of the CFTR inhibitor-172 (CFTRinh-172, 5 µM) on UTP- and
ATP-activated Cl- secretion in tracheas of Ano 1+/+ and Ano 1-/- animals. ATP
activated Cl- currents are largely reduced in -/- tracheas. Residual Cl- secretion is
inhibited significantly by the CFTRinh-172. E) ATP-activated Cl- secretion is inhibited by 4,4′-
diisothio-cyanostilbene-2,2′-disulfonic acid (DIDS, 100 µM) in tracheas of Ano 1+/+ but
not Ano 1-/- animals. F) Amiloride (Amil) sensitive Na+ absorption and CFTR-dependent
Cl- secretion activated by 3-isobutyl-1-methylxanthine (IBMX, 100 µM) and forskolin (Fors,
2 µM) were not different between Ano 1+/+ and Ano 1-/- tracheas. Mean ± SEM, (n) =
number of tissues measured, * significant effects of inhibitors (paired t-test), # significant
CD
FE
A B
-4
-3
-2
-1
0V
te (
mV
)
3 min
CCH
NFA
0
10
20
30
40
I sc U
TP (
A/c
m2) (9)
(5)
con
#
CFTRinh-172
0
20
40
60
I sc-A
TP (
A/c
m2) (5)
(4)
con
#
CFTRinh-172
0
20
40
60
I sc-A
TP (
A/c
m2)
(5)
(6)*
Ano1 +/+ Ano1 -/-
con
DIDS
0
20
40
60
80
I s
c (
A/c
m2)
Amil
(9)
IBMX/Fors
(11)
-/-
(5)
(5)
+/+ -/-+/+
0
20
40
60
80
100
I sc-C
CH
(
A/c
m2)
(6-12)
*
*
con
NFA
CPA
*
*
Ano1 +/+ Ano1 -/- Ano1 +/+ Ano1 -/-

Part IV – Chapter 1
72
difference when compared to Ano 1-/-. [Data obtained by Jiraporn Ousingsawat at
the Institut für Physiologie, Universität Regensburg, Germany; included in this thesis with
permission].
The small increase in Cl- secretion by purinergic stimulation of -/-
tracheas is probably due to activation of CFTR241, as the specific inhibitor
CFTRinh-172 inhibited the remaining short circuit current (Isc) (Figure IV.1. 2C,
D). Moreover, other members of the TMEM16-family of proteins are expressed
in mouse trachea, which may also contribute to the remaining Ca2+ activated Cl-
conductance222. In contrast to CFTRinh-172, the CaCC-inhibitor 4,4'-diisothio-
cyanostilbene-2,2'-disulfonic acid (DIDS) inhibited Cl- transport only in wt
animals (Figure IV.1. 2E). Moreover, unlike Ca2+ activated Cl- channels,
amiloride sensitive Na+ absorption and CFTR-dependent Cl- secretion were not
disturbed in Ano 1 null mice (Figure IV.1. 2F).
3.2 ANO 1 IS IMPORTANT FOR MUCOCILIARY CLEARANCE IN
MURINE TRACHEA
Attenuated Cl- secretion in neonatal tracheas could be due to low levels
of expression of Ano 1. In fact, in situ hybridization showed very little signal in
the surface epithelium of neonatal trachea, whereas expression was clearly
detectable in cells of the submucosal glands (Figure IV.1. 3A). No signal was
obtained for the sense probe. Submucosal cells appear to be the major target
for cholinergic stimulation of airway secretion242.
Figure IV.1. 3 A - In situ hybridization of Ano 1 in neonatal Ano 1+/+ trachea.
mRNA for Ano 1 was expressed at low levels in the surface epithelium (left panel) but

Loss of Ano 1 Causes a Defect in Epithelial Ca2+
-dependent Cl- Transport
73
was detectable in submucosal glands (left panel). Bar = 200 µm. No signal was
detected by hybridization with a sense probe (right panel, bar = 50 µm). [Data
obtained by Jason R. Rock at the Department of Molecular Genetics and Microbiology,
University of Florida, Gainesville, Florida 32610; included in this thesis with permission]
Figure IV.1. 3 B-D - Lack of cholinergic airway gland secretion compromises
MCC. B) Tracking of black polystyrene microsphere movement on the surface of a
neonatal Ano 1+/+ trachea in a humidified chamber before and after stimulation with
carbachol (100 µM; CCH). C) Diagram showing particle transport under control

Part IV – Chapter 1
74
conditions and increase after stimulation with CCH. Bar = 1 mm. D) Summary of particle
transport on tracheas of wt (+/+) and Ano 1 -/- animals. Particle transport in -/- tracheas
was neither increased by cholinergic stimulation with carbachol (100 µM) nor inhibited
by DIDS (100 µM). Mean ± SEM, (n) = number of tissues measured. *indicates
significance (unpaired t-test, (P < 0.05)).
As mucociliary transport in the airways depends on cholinergic Cl-
secretion, we examined transport of black polystyrene microspheres on freshly
isolated tracheas in a humidified chamber, as a measure for mucociliary
transport. Mucociliary particle transport was activated in +/+ tracheas by
stimulation with CCH, and was inhibited by the Ano 1 inhibitor DIDS (Figure
IV.1. 3B, D). In contrast, neither cholinergic stimulation nor the Ano 1 inhibitor
DIDS had any effects on particle transport observed in -/- tracheas, indicating
lack of cholinergic mucociliary clearance in Ano 1 -/- mice. Moreover, as
neonatal mouse tracheas show relatively small effects of purinergic agonists
(ATP and UTP) on Cl- secretion (Figure IV.1. 1C-F), stimulation by ATP had
little effects on particle transport in airways of both +/+ and -/- (supplementary
Fig. 1).
Lack of cholinergically stimulated clearance may contribute to the high
lethality of Ano 1-null mice. These results suggest Ano 1 as an important factor
for the control of mucociliary clearance in human airways as suggested in a
previous report222 .

Loss of Ano 1 Causes a Defect in Epithelial Ca2+
-dependent Cl- Transport
75
4 SUPPLEMENTARY DATA
Supplementary Fig.IV.1. 1 - Summary of particle transport on tracheas of wt (+/+)
and Ano 1 -/- animals. Particle transport in +/+ and -/- tracheas was not increased by
purinergic stimulation with ATP (100 µM). Mean ± SEM, (n) = number of tissues measured.
0
300
600
900
1200
pa
rtic
le t
ran
sp
ort
(m
/min
) (7)
(7)
con
ATP
Ano1 -/-Ano1 +/+


Ca2+
signalling in CF epithelial cells
77
Chapter 2. F508DEL-CFTR INCREASES THE INTRACELLULAR
CA2+ SIGNALLING THAT CAUSES ENHANCED CA2+-DEPENDENT
CL- CONDUCTANCE IN CYSTIC FIBROSIS
Manuscript submitted to AJRCMB with minor alterations
1 ABSTRACT
In many cells, an increase in intracellular calcium ([Ca2+]i) activates a
Ca2+-dependent chloride (Cl-) conductance (ICaCC). ICaCC is enhanced in cystic
fibrosis (CF) epithelial cells, lacking Cl- transport mediated by the CF
transmembrane conductance regulator (CFTR). The underlying mechanisms
remain, however, entirely unclear. Here, we show that in freshly isolated nasal
epithelial cells of F508del-homozygous CF patients, expression of the recently
identified Ca2+-activated Cl- channels (CaCC) Ano 1 and bestrophin 1 were
unchanged and can therefore not be the reason for enhanced Cl - conductance
in CF. However, calcium signalling was strongly enhanced after induction of
expression of F508del-CFTR, which is unable to exit the endoplasmic reticulum
(ER). Since receptor-mediated [Ca2+]i increase was largely Cl- dependent, we
suggested that F508del-CFTR functions as an ER chloride counter ion channel
for Ca2+ movement. This was confirmed by expression of a F508del/G551D-
CFTR double mutant, which had no effects on [Ca2+]i. Moreover, F508del-CFTR
could serve as a scavenger for inositol-1,4,5-trisphosphate [IP3] receptor
binding protein released with IP3 (IRBIT). These data explain how ER-localized
F508del-CFTR controls intracellular Ca2+ signalling.
2 INTRODUCTION
There is a well described relationship between the cystic fibrosis
transmembrane conductance regulator (CFTR) and Ca2+-activated Cl- channels
(CaCCs). Enhanced Ca2+ activated Cl- conductance has been detected in the
airways of cystic fibrosis (CF) patients114,223. This finding was confirmed in

Part IV – Chapter 2
78
mouse airways and cultured airway epithelial cells121,243. Other studies have
demonstrated that CFTR has an inhibitory effect on CaCCs244,245, reinforcing a
major role for CFTR as a channel regulator246. Yet, the mechanism for
enhanced Ca2+-dependent Cl- secretion in CF airways remains to be elucidated.
Previous studies demonstrated that CF airway epithelial cells, grown as
polarized primary cultures, exhibit an expansion of the endoplasmic reticulum
(ER) compartment, which correlates with enhanced Ca2+-dependent activation
of a luminal Cl- conductance247.
Pro-inflammatory pathways which appear to be constitutively active in CF
airways, may be the cause of the observed Ca2+i mobilisation from ER luminal
Ca2+ stores and augmented Ca2+-activated Cl- conductance. However, it is
currently under debate whether the upregulation of pro-inflammatory and NFκB
pathways in CF airway epithelial cells leading to those ER/Ca2+ effects, is due
to exogenous infection or this is an intrinsic property of the CF cell. Indeed, it
might result from misfolded F508del-CFTR, causing an unfolded protein
response (UPR) and ER-stress248. Evidence favouring the former against the
latter possibility includes: i) firstly, the increased ER mass and ER-derived Ca2+i
signals revert to normal in primary cultures of F508del-CF bronchial epithelial
cells maintained long term (30-40 days) in the absence of luminal
infection/inflammation249; ii) secondly, primary cultures of non-CF cells exhibit
the same ER/Ca2+ effects as CF cells, when exposed to the supernatant
purulent material (SMM) collected from lungs of CF patients249; iii) thirdly, the
results indicating that F508del-CFTR causes ER stress and activates the
UPR248 were obtained in cells overexpressing F508del-CFTR and hence do not
apply to the physiological situation; iv) moreover, although SMM triggers the
UPR250 typical UPR was found to be absent in primary cultures and cell lines of
CF airway epithelial cells without stimulation250-252; v) finally, patients with other,
non-F508del mutants which do no cause ER retention also have enhanced
CaCCs223. Thus, the current view is that the ER expansion observed in CF cells
is, in fact, an acquired epithelial response to chronic airway
infection/inflammation. Still, the ER/Ca2+ effects in CF cells may be accentuated

Ca2+
signalling in CF epithelial cells
79
by the absence of wt-CFTR expression in epithelial cells247,251,253,254. However,
how CFTR plays a role in such events leading to enhanced Ca2+ signalling
remains unclear.
Two Cl- channels have been shown to contribute to the Ca2+-dependent
Cl- conductance in the airways186. Although expressed at low levels only,
bestrophin 1 (Best 1) facilitates Ca2+ activated Cl- conductance in airway
epithelial cells173. Airway epithelial cells from Best 1 knockout mice have a
reduced ATP-dependent, i.e. Ca2+ activated Cl- conductance173. Although Best
1 has been identified as a bona fide Ca2+-activated Cl- channel186, it is unlikely
that this channel is forming the luminal Cl- channel in airways, since it was
detected in the ER rather than in the plasma membrane in the present and in
previous studies129. Moreover, mBest1-knockout mice are still able to secrete
Cl- upon stimulation with ATP, albeit at reduced levels255. Recent data may
explain these findings since we found that ER-localized Best 1 facilitates
receptor mediated intracellular Ca2+ signalling, probably by serving as a Cl-
counter ion channel in the ER190.
Ano 1 was recently identified as the luminal membrane localized Ca2+-
activated Cl- channel. Ano 1 (anoctamin 1) is a member of a family of 10
homologous proteins that exist as various splicing variants that are abundantly
expressed in epithelial and non-epithelial tissues186,200,202,211. By analyzing Cl-
transport in Ano 1 null mice, we demonstrated that Ano 1 is essential for Ca2+-
dependent Cl- secretion as well as MCC of mouse airways222,256. Therefore, in
the present study, we investigated whether expression of Best 1 and Ano 1 is
different in CF and non-CF epithelial cells, and also whether the expression of
these two CaCCs changes upon exposure to bacterial lipopolysaccharides
(LPS). Although we were unable to detect significant differences in expression
of Ano 1 and Best 1, we could identify the mechanism for increased receptor
mediated Ca2+ signalling in F508del-CFTR expressing cells.

Part IV – Chapter 2
80
3 RESULTS
3.1 CELLULAR LOCALIZATION OF ANO 1 AND BESTROPHIN
Two different types of Ca2+-activated Cl- channels (CaCC) have been
reported to be relevant for Ca2+ dependent Cl- secretion in epithelia. Recently, a
member of the family of anoctamin proteins, Ano 1, has been identified as a
CaCC showing all typical features previously described for these channels
endogenously expressed193,200.
Figure IV.2. 1 - Cellular localization of Ano 1 and bestrophin 1. (A) Expression of
Ano 1 in BHK cells. Non-permeabilized (left panel) and permeabilized (right panel) cells
were exposed to the Flag antibody, which were both equally stained (green
fluorescence). Blue = DAPI. (B) Expression of bestrophin 1 in BHK cells. Non-
permeabilized (left panel) and permeabilized (right panel) cells were exposed to the
Flag antibody, but only permeabilized cells were stained (green). Blue = DAPI. Bar = 20
µm.

Ca2+
signalling in CF epithelial cells
81
This protein is clearly membrane localized when expressed in baby
hamster ovary (BHK) cells, as demonstrated by lack of fluorescence labelling of
a putative extracellularly accessible Flag-tag (Figure IV.2. 1A). Ano 1 is a major
constituent of CaCC and is activated directly by Ca2+ or indirectly through
associated proteins 193,200. Bestrophin 1 (Best 1) expression has also been
demonstrated, both in mouse and human airway epithelial cells, to correlate
with Ca2+-dependent Cl- secretion 173. However, in contrast to Ano 1, Best 1
was found in an intracellular compartment of BHK cells (Figure IV.2. 1B).
Moreover, by introducing glycosylation sites, we demonstrate that Best 1
remains in a core-glycosylated form (Supplementary Fig.IV.2. 1), as shown by
incubation of cell lysates with N-glycosidase F or Endo H which similarly
produced only a single band of lower mobility, indicating that the low-mobility
form is Endo H- sensitive and hence ER-specific. These data reinforce that
indeed Best 1 does not move out of the ER and therefore does not reach the
cell membrane.
The present result is in complete agreement with recent findings, which
locate Best 1 in the ER, where it augments intracellular Ca2+ signalling due to its
function as a counter-ion channel190. In contrast, Ano 1 is a strictly membrane
localized Ca2+-activated Cl- channel that forms the luminal exit pathway for Cl-
186. However, both ion channels contribute to Ca2+-dependent Cl- secretion
since activation of CaCC in Calu3 cells was inhibited by down-regulation of
expression of Ano 1 and Best 1 using siRNA (Supplementary Fig.IV.2. 2), also
as shown earlier 186.
3.2 EXPRESSION OF ANO 1 AND BESTROPHIN 1 IN CF AND NON-CF
AIRWAY EPITHELIAL CELLS AND EFFECTS OF LPS AND CF-SPUTUM
Since enhanced Ca2+-activated Cl- conductance has been reported
earlier in CF nasal epithelial cells 223, we investigated here whether this is due
to increased transcript levels of either Ano 1 and/ or Best 1. To this end, we
quantitatively analyzed by real-time qRT-PCR levels of transcripts from these
two genes in freshly isolated nasal epithelial cells from non-CF healthy

Part IV – Chapter 2
82
individuals and patients homozygous for F508del-CFTR, as well as in isogenic
human airway epithelial cell lines expressing wt- or F508del-CFTR.
Figure IV.2. 2 - Expression of Ano 1 and bestrophin 1 in CF and non-CF airway
epithelial cells and effect of LPS. A) Real time RT-PCR analysis of Ano 1-expression in
freshly isolated nasal epithelial cells from non-CF volunteers and patients homozygous
for F508del-CFTR. B) Real time RT-PCR analysis of bestrophin 1-expression in freshly
isolated nasal epithelial cells from non-CF volunteers and patients homozygous for
F508del-CFTR. C,D) Real time RT-PCR analysis of expression of Ano 1 and bestrophin 1 in
0
0.001
0.002
0.003
0.004
0.005
Re
lative
exp
ressio
no
f A
no
1
(5)
(5)
0
1E-005
2E-005
3E-005
4E-005
5E-005
Re
lative
exp
ressio
no
f h
Be
st
1
(5)
(5)BA
C D
0
40
80
120
160
200
Re
lative
exp
ressio
n
of
An
o 1
/
actin
(%
)
#
(7)(7)
0
40
80
120
160
200
Re
lative
exp
ressio
n
of
hB
est
1 /
a
ctin
(%
)
(7)(5)
E F
-80
-40
0
40
80
A
no
1in
du
ce
d b
y L
PS
(%
)
(7)(7)
-80
-40
0
40
80
h
Be
st
1in
du
ce
d b
y L
PS
(%
)
(7)(5)
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR

Ca2+
signalling in CF epithelial cells
83
CFBE cells stably expressing wt-CFTR or F508del-CFTR. E) Real time RT-PCR analysis of
expression of Ano 1 and bestrophin 1 in CFBE cells stably expressing wt-CFTR or F508del-
CFTR. Change of expression (%) induced by incubation of the cells with LPS (10 µg/ml,
ON). Mean ± SEM, (n) = number of experiments. # significant difference between wt-
CFTR and F508del-CFTR or control and LPS-treatment (unpaired t-test). [Data in panel
(A) obtained by Luka Clarke, At the Faculty of Sciences of the University of Lisbon,
Lisbon, Portugal; included in this thesis with permission]
In native nasal cells, transcripts for Ano 1 were abundant in contrast to
those from Best 1, which were expressed at very low levels. However, these
analyses did not evidence significant differences between CF and non-CF cells
in the levels of transcripts from both ion channels neither in native cells (Figure
IV.2. 2A, B) nor in cell lines (Figure IV.2. 2C, D). It is, thus unlikely that changes
in ion channel expression (Figure IV.2. 2A-D) account for enhanced CaCC
activity in native CF airway epithelial cells.
We further examined whether overnight exposure to lipopolysaccharides
(LPS; 10 µg/ml) would alter levels of Ano 1 or Best 1 transcripts in wt- and
F508del-CFTR CFBE cells. Analysis of Ano 1 and Best 1 transcripts by real
time qRT-PCR demonstrated only slightly increased expression of Ano 1 and
Best 1 under these conditions (Figure IV.2. 2E, F). The same results were
obtained when cells were exposed to supernatant purulent material (SMM)
collected from lungs of CF patients (10 μl in 2ml for 16h) (data not shown).
Taken together, enhanced Ca2+ activated Cl- conductance observed in airway
epithelial cells expressing F508del-CFTR is not explained by an increase in
transcripts of known CaCCs, although previously CF cells were clearly
demonstrated to possess Ca2+-activated Cl- transport, as shown by
fluorescence-quenching of I- sensitive YFP-I152L 233. While CFBE/wt-CFTR
cells did not show activation of CaCC by stimulation with the purinergic agonist
UTP (100 µM), activation of CaCC was readily detectable in CFBE/F508del-
CFTR cells (Supplementary Fig.IV.2. 3 A, B). As expected, substantial
activation of a CFTR Cl--conductance by adenosine (100 µM) was observed in
CFBE/CFTR but not in CFBE/F508del-CFTR cells (Supplementary Fig.IV.2. 3

Part IV – Chapter 2
84
A, B). Moreover, exposure to LPS (10 µg/ml ON) only slightly enhanced
activation of CaCC by UTP in both CFBE/wt-CFTR and CFBE/F508del-CFTR
cells (Supplementary Fig.IV.2. 3 C, D). Thus neither the presence of F508del-
CFTR nor exposure to proinflammatory agents such as LPS increases ion
channel expression.
3.3 F508DEL-CFTR CHANGES INTRACELLULAR CA2+
SIGNALLING
Since expression of Ca2+ activated Cl- channels was not different in cells
expressing F508del-CFTR, we examined whether F508del-CFTR has an effect
on receptor-mediated Ca2+ signalling. An increase in agonist-mediated [Ca2+]i
signals in human CF airway epithelia has been already reported earlier 247.
Figure IV.2. 3 - Ca2+ signalling in CFBE/wt-CFTR and CFBE/F508del-CFTR and
effect of LPS. A) Original recordings of intracellular Ca2+ ([Ca2+]i) increase in CFBE/wt-
CFTR (upper panel) and CFBE/F508del-CFTR (lower panel) cells induced by stimulation
5 min
400
nM
UTP
A
UTP
0
400
800
1200
[Ca
2+] i (
nM
)
# *
*
* #
con
peak
plateau
B (192) (148)
F508del-CFTR
wt-CFTR
-80
-40
0
40
80
[C
a2+] i
UT
P L
PS
(%
)
Ccon
peak
plateau
##
F508del-CFTR
wt-CFTR
F508del-CFTR
wt-CFTR
(65) (138)
D

Ca2+
signalling in CF epithelial cells
85
with UTP (100 µM). B) Summary of baseline [Ca2+]i and UTP induced increase peak and
plateau [Ca2+]i after stimulation with UTP of CFBE/wt-CFTR and CFBE/F508del-CFTR cells.
C) Change (%) of baseline, peak and plateau [Ca2+]i after incubation of CFBE/wt-CFTR
and CFBE/F508del-CFTR cells with LPS (10 µg/ml, ON). Mean ± SEM, (n) = number of cells
measured. . *significant effects of UTP (paired t-test). # significant difference between
wt-CFTR and F508del-CFTR or control and LPS-treatment (unpaired t-test).
In the present study, intracellular Ca2+ was enhanced by UTP stimulation
(100 µM) in both CFBE/wt-CFTR and CFBE/F508del-CFTR cells (Figure IV.2.
3). We found that UTP-induced increases in both peak and plateau [Ca2+]i were
significantly enhanced in CFBE/F508del-CFTR cells, while baseline [Ca2+]i was
identical in both CFBE/wt-CFTR and CFBE/F508del-CFTR cells (Figure IV.2.
3A-C). We also examined whether exposure to LPS (10 µg/ml, overnight)
further increases intracellular Ca2+ signals in both CFBE/wt-CFTR and
CFBE/F508del-CFTR cells, but only found small and inconsistent changes in
both cell lines (Figure IV.2. 3D). These data therefore suggest that the presence
of F508del-CFTR, but not exposure to LPS, augments receptor mediated Ca2+
signalling.
To further examine how F508del-CFTR changes intracellular Ca2+
signalling, we used inducible airway epithelial (A549) cell lines225, which stably
express wt-CFTR or F508del-CFTR under an inducible (Tet-ON) promoter
(Figure IV.2. 4A and Figure IV.2. 5A). We measured UTP (100 µM) activated
Ca2+ transients in wt-CFTR A549 cells under non-induced (no expression) and
induced (expression of wt-CFTR) conditions, and found no increase in [Ca2+]i
but rather a significant decrease in both peak and plateau (Figure IV.2. 4C). In
contrast, induction of expression of F508del-CFTR significantly increased Ca2+
signals elicited by stimulation with UTP (Figure IV.2. 5B, C). Thus, expression of
ER-localized F508del-CFTR clearly augments intracellular Ca2+ signals elicited
by stimulation of GTP-coupled membrane receptors.

Part IV – Chapter 2
86
Figure IV.2. 4 - : Induction of expression of wt-CFTR does not augment Ca2+
signalling. A) Expression of wt-CFTR in non-induced (left upper) and induced (right
upper) A549 cells. B) Original recordings of intracellular Ca2+ [Ca2+]i signals elicited by
UTP (100 µM) in non-induced (no wt-CFTR, left) and induced (wt-CFTR, right) A549 cells.
C) Summary of baseline [Ca2+]i (con) and peak and plateau [Ca2+]i after stimulation
with UTP. *significant effects of UTP (paired t-test). # significant difference between + wt-
CFTR and - wt-CFTR (unpaired t-test).

Ca2+
signalling in CF epithelial cells
87
Figure IV.2. 5 - Induction of expression of F508del-CFTR augments Ca2+ signalling.
A) Expression of F508del-CFTR in non-induced (left upper) and induced (right upper)
A549 cells. B) Original recordings of intracellular Ca2+ ([Ca2+]i signals elicited by UTP (100
µM) in non-induced (no F508del-CFTR, left) and induced (F508del-CFTR, right) A549 cells.
C) Summary of baseline [Ca2+]i (con) and peak and plateau [Ca2+]i after stimulation
with UTP. Bar = 20 µm. Mean ± SEM, (n) = number of cells measured. *significant effects
of UTP (paired t-test). # significant difference between –F508del-CFTR and +F508del-
CFTR or control and LPS-treatment (unpaired t-test).
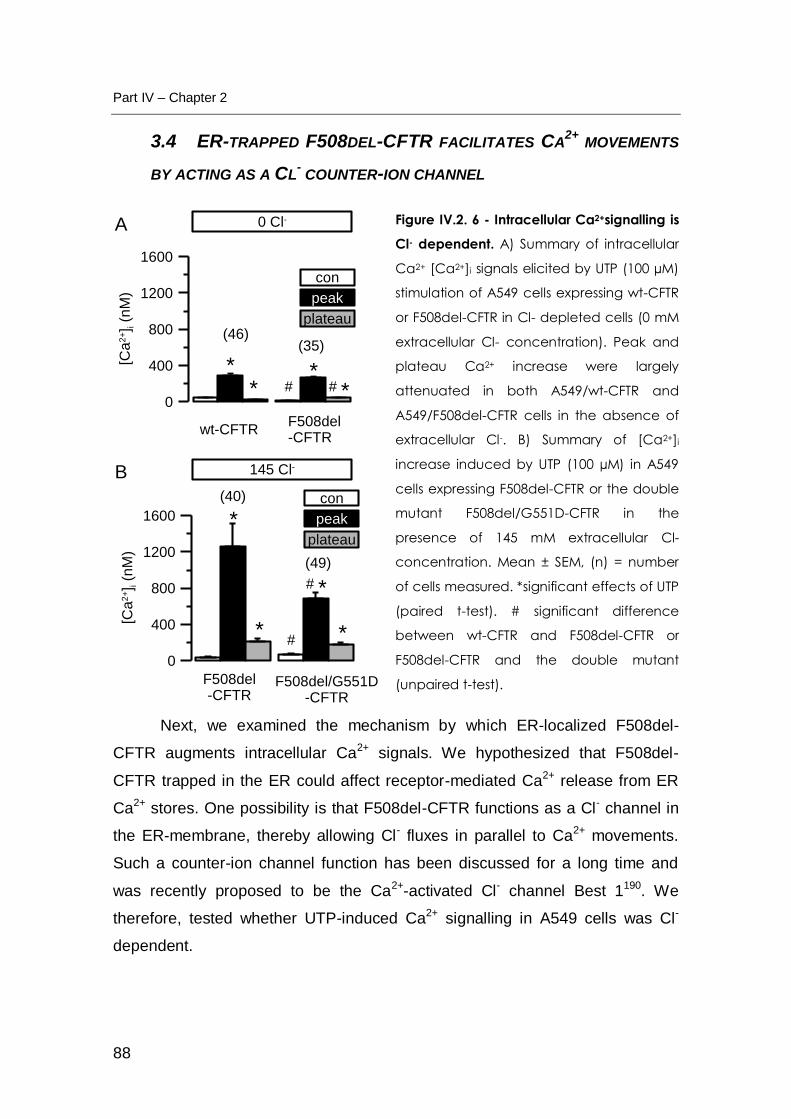
Part IV – Chapter 2
88
3.4 ER-TRAPPED F508DEL-CFTR FACILITATES CA2+
MOVEMENTS
BY ACTING AS A CL- COUNTER-ION CHANNEL
Figure IV.2. 6 - Intracellular Ca2+signalling is
Cl- dependent. A) Summary of intracellular
Ca2+ [Ca2+]i signals elicited by UTP (100 µM)
stimulation of A549 cells expressing wt-CFTR
or F508del-CFTR in Cl- depleted cells (0 mM
extracellular Cl- concentration). Peak and
plateau Ca2+ increase were largely
attenuated in both A549/wt-CFTR and
A549/F508del-CFTR cells in the absence of
extracellular Cl-. B) Summary of [Ca2+]i
increase induced by UTP (100 µM) in A549
cells expressing F508del-CFTR or the double
mutant F508del/G551D-CFTR in the
presence of 145 mM extracellular Cl-
concentration. Mean ± SEM, (n) = number
of cells measured. *significant effects of UTP
(paired t-test). # significant difference
between wt-CFTR and F508del-CFTR or
F508del-CFTR and the double mutant
(unpaired t-test).
Next, we examined the mechanism by which ER-localized F508del-
CFTR augments intracellular Ca2+ signals. We hypothesized that F508del-
CFTR trapped in the ER could affect receptor-mediated Ca2+ release from ER
Ca2+ stores. One possibility is that F508del-CFTR functions as a Cl- channel in
the ER-membrane, thereby allowing Cl- fluxes in parallel to Ca2+ movements.
Such a counter-ion channel function has been discussed for a long time and
was recently proposed to be the Ca2+-activated Cl- channel Best 1190. We
therefore, tested whether UTP-induced Ca2+ signalling in A549 cells was Cl-
dependent.
0
400
800
1200
1600
[Ca
2+] i
(nM
)
con
peak
plateau(46)
(35)
F508del-CFTR
wt-CFTR
# # **
**
0
400
800
1200
1600
[Ca
2+] i
(nM
)
(40)
(49)
F508del/G551D-CFTR
F508del-CFTR
#
# *
*
*
*
con
peak
plateau
145 Cl-
0 Cl-A
B

Ca2+
signalling in CF epithelial cells
89
To that end, we measured Ca2+i transients in induced A549/wt-CFTR and
A549/F508del-CFTR cells in Cl--depleted cells (0 mM extracellular Cl-; (Figure
IV.2. 6A). UTP-induced increase in [Ca2+]i was largely reduced in the absence
of extracellular Cl- and when compared to Ca2+ increase in physiological Ringer
(145 mM extracellular Cl-) solution (Figure IV.2. 3B). Notably, attenuation of
Ca2+ transients in Cl--free solution was more pronounced in F508del-CFTR
expressing cells (Figure IV.2. 3B and Figure IV.2. 6A) and increased Ca2+
signals in F508del-CFTR expressing cells were no longer observed under Cl-
free conditions (Figure IV.2. 6A). In order to further examine whether the ability
of F508del-CFTR to produce a Cl- conductance in the ER-membrane may
influence the Ca2+ signal, we expressed the double mutant F508del/G551D-
CFTR that i) does not traffic to the cell membrane but remains in the ER
(caused by F508del) and ii) does not operate as a Cl- channel due to defective
channel gating (caused by G551D). In fact, A549 cells expressing the double
mutant F508del/G551D-CFTR cells did no longer produce enhanced Ca2+
signals (Figure IV.2. 6B). These results suggest that the presence of F508del-
CFTR in the ER may affect intracellular Ca2+ signalling by acting as a Cl-
counter-ion channel, thereby facilitating Ca2+ movement across the ER
membrane.
3.5 EXPRESSION OF IRBIT ANTAGONIZES ENHANCED CA2+
SIGNALS IN XENOPUS OOCYTES:
It was recently reported, that the inositol-1,4,5-trisphosphate [IP3]
receptor binding protein released with IP3 (IRBIT) binds to CFTR when
released from the IP3-receptor upon binding of IP3257. Because F508del-CFTR
accumulates in the ER being thus in close proximity to the IP3-receptor, we
speculated that F508del-CF may compete with IP3-receptors for binding to
IRBIT258. Thus reduced binding of IRBIT to the IP3-receptor would enhance
agonist-induced Ca2+ release from the ER. To test this hypothesis, we made
use of the expression system in Xenopus oocytes since it allows parallel
expression of several proteins.

Part IV – Chapter 2
90
Figure IV.2. 7 - Potential role of IRBIT for enhanced Ca2+ activated Cl-
conductance in F508del-CFTR expressing oocytes. A) Original recording of whole cell
Cl- currents activated by UTP (100 µM) in P2Y2-receptor expressing Xenopus oocytes. B)
Original recording of whole cell Cl- currents activated by UTP in P2Y2-receptor and wt-
CFTR coexpressing Xenopus oocytes. C) Original recording of whole cell Cl- currents
activated by UTP in P2Y2-receptor and F505del-CFTR coexpressing Xenopus oocytes. D)
Summary of calculated relative whole cell conductances activated by UTP in the
absence or presence of coexpressed IRBIT. Mean ± SEM, (n) = number of cells
measured. # significant difference between different batches (unpaired t-test). [Data
obtained by Patthara Kongsuphol, at the Institut für Physiologie, Universität Regensburg,
Germany; included in this thesis with permission]
0
100
200
300
400
Re
lative
G
(16) (12)
F508del-CFTR
IRB
IT
wt-CFTR
(6)
(6)(15)
(15)
#
#
##
IRB
IT
IRB
IT
-2000
0
2000
4000
6000
I (n
A)
-2000
0
2000
4000
6000
I (n
A)
-2000
0
2000
4000
6000
I (n
A)
1 min
H2O wt-CFTR
H2O
UTP UTP
UTP
A
C
B
D
F508del-CFTR

Ca2+
signalling in CF epithelial cells
91
When P2Y2-receptors were expressed in oocytes, stimulation by UTP
(100 µM) activated endogenous Ca2+-dependent Ano 1 channels and produced
a transient and outwardly rectifying whole-cell Cl- current (Figure IV.2. 7A).
Notably, co-expression of wt-CFTR with P2Y2-receptors slightly, but
significantly augmented the Ca2+-activated Cl- current, while co-expression of
F508del-CFTR induced a significantly larger Ca2+-activated Cl- current (Figure
IV.2. 7B-D). Additional co-expression of IRBIT had little effects on Ca2+-
activated currents in wt-CFTR-expressing cells, but dramatically inhibited UTP-
induced currents in F508del-CFTR co-expressing cells (Figure IV.2. 7D). These
results would be in agreement with the idea that F508del-CFTR accumulating in
the ER may bind IRBIT thereby facilitating agonist-induced IP3 binding to the
IP3 receptor and further increase in intracellular Ca2+, or it could simply be an
non-specific antagonistic effect of IRBIT. To examine binding of IRBIT to CFTR
we performed co-immunoprecipitation experiments in two different cell lines:
A549 cells stably expressing either wt-CFTR or F508del-CFTR, and baby
hamster kidney (BHK) cells transiently expressing wt-CFTR or F508del-CFTR.
Moreover, two different co-immunoprecipitation protocols 257,259 were used and
either IRBIT or CFTR were immunoprecipitated. Under no conditions could we
observe coimmunoprecipitation of the two proteins, as reported by others257.
Therefore we have no evidence that IRBIT binds directly to CFTR when
expressed in these two different cell lines (Supplementary Fig.IV.2. 4)
Supplement 4). Taken together, the present results suggest that enhanced
Ca2+-activated Cl- conductance in CF epithelial cells is due to augmented
intracellular Ca2+ signalling, caused by ER-localized F508del-CFTR. F508del-
CFTR may probably function as an ER-located counter-ion channel which
facilitates Ca2+ release from ER-stores.

Part IV – Chapter 2
92
4 DISCUSSION
4.1 INFLAMMATION IN CF
Although CFTR is known for more than 20 years, it remains enigmatic
how abnormalities in CFTR can cause chronic and persistent pulmonary
inflammation. There is agreement that loss of functional CFTR results in
activation of neutrophils that produce large amounts of proteases and reactive
oxygen species (ROS). These changes are associated with reduced MCC of
airways to get rid of bacteria, and induction of hyperinflammatory responses in
CF airways. The NF-B pathway and Ca2+ mobilization in airway epithelial cells
are believed to be of key importance for lung inflammation, through release of
mediators such as interleukin-8 that participate in further recruitment and
activation of neutrophils, modulation of apoptosis, and loss of epithelial barrier
integrity260.
Several lines of evidence suggest that CFTR mutations, most importantly
F508del-CFTR itself can produce a pro-inflammatory milieu in the airways that
precedes infection. Thus F508del-CFTR has been demonstrated to accumulate
in the ER and to trigger a stress response leading to NFB activation and IL8
production253,261,262. Another study found that inhibition of CFTR in airway
epithelial cells mimicked the CF-typical inflammatory profile with increase in
nuclear NFB and IL-8 secretion254, while others showed that expression of
functional CFTR on the cell surface negatively regulates NFκB mediated innate
immune response263.
This issue, however, is controversial since other in vitro studies did not
detect an intrinsic hyper-inflammatory phenotype in CF cells and demonstrated
increased secretion of pro-inflammatory cytokines by CF airway cells only after
exposure to pathogenic bacteria247,251,264. Moreover, recent data from the
newborn CF pig models evidenced no signs of intrinsic inflammation265.
However, Ribeiro and collaborators demonstrated expansion of an ER

Ca2+
signalling in CF epithelial cells
93
compartment close to the luminal membrane of chronically infected and
inflamed airway epithelia, like those affected by CF247. Large luminal ER pools
lead to enhanced apical Ca2+ signalling triggered by stimulation of purinergic
P2Y2 receptors, which explains the augmented Ca2+-activated Cl- secretion
observed in CF airways. In contrast to these studies we were not able to detect
a significant effect of the major bacterial component LPS or purulent sputum
from CF patients on intracellular Ca2+ signalling and expression of the Ca2+
activated Cl- channels bestrophin 1 or Ano 1.
4.2 ENHANCED CA2+-DEPENDENT ACTIVATION OF ANO 1
Interestingly, pro-inflammatory interleukins have been shown to stimulate
both Ca2+-activated Cl- and SK4 K+ channels266. Galietta and colleagues
actually identified Ano 1 as a Ca2+ activated Cl- channel because Ca2+-activated
Cl- secretion in bronchial epithelial cells was upregulated by IL4200,266. Very
similar the Ca2+ -dependent Cl- channel bestrophin 1 (Best 1) was found to be
upregulated during renal inflammation and post-damage repair190,193. However,
one key finding of the present study was that upregulation of Best 1 or Ano 1
was only minimal in F508del-CFTR expressing cells, and was only little
enhanced upon exposure of the cells to bacterial LPS. This suggests that
although IL-4 and other cytokines can affect transepithelial ion transport in the
human bronchial epithelium by increasing expression of Ano 1, this does not
explain the direct link between F508del-CFTR and CaCC in CF airway epithelial
cells.
4.3 F508DEL-CFTR CONTROLS INTRACELLULAR CA2+
SIGNALLING
The fact that retention of misfolded F508del-CFTR leads to imbalance in
Ca2+ homeostasis is a well recognized fact, however, the reasons for the Ca2+
increase are poorly understood261. Our present data show for the first time a
direct link between F508del-CFTR and intracellular Ca2+ signalling, thereby
connecting F508del-CFTR to enhanced Ca2+-dependent Cl- secretion in CF. It
has been suggested that Ca2+-store depletion might affect chaperone function

Part IV – Chapter 2
94
(notably calnexin) and rescue F508del-CFTR to the cell membrane, but these
data could not be reproduced by other groups267. Moreover, disrupting the
interaction with ER chaperones causes major impairment of cell-surface
expression of wt-CFTR and does not rescue F508del-CFTR268-270
However, our data would explain for the first time why, conversely, Ca2+
signalling is affected by F508del-CFTR, as this defective, but still partially active
CFTR channel may provide a Cl- conductance in the ER-membrane and thus
facilitate Ca2+ movement by allowing transport of the counter-ion Cl-. Notably,
F508del-CFTR, although defective, is still able to generate some Cl-
conductance, as shown earlier55. The concept of a counter-ion channel in the
ER to balance negative charges occurring through Ca2+ release and re-uptake
into the ER-store has long been proposed271. In the sarcoplasmic reticulum
(SR), Cl- channels play an essential role in excitation-contraction coupling, by
balancing charge movement during Ca2+ release and reuptake272,273. This is
also known from airway smooth muscle cells. SR-localized Cl- channels in the
SR membrane allow for neutralization of electrostatic charges that would
otherwise build up during Ca2+ movement274. Thus SR-localized Cl- channels
support Ca2+ signalling and muscle contraction: Blockage of these Cl- channels
might be an effective way to inhibit airway smooth muscle hyperresponsiveness
observed in asthma275. Also for Best 1, a function as a counter-ion channel in
the ER of epithelial cells has been proposed recently190.
4.4 ENHANCED CA2+
SIGNALLING, PROLIFERATION AND THE ROLE
OF IRBIT
Our present data also provide an explanation for the enhanced
proliferation observed for CF epithelial cells. It has been shown earlier that cell
proliferation in bronchial epithelium and submucosal glands of CF patients is
much increased when compared to non-CF cells 276. The high proliferation rate
of CF airway epithelial cells has been explained by the chronic inflammatory
process that takes place in CF airways. However, it is also observed under in
vitro conditions and in the absence of exogenous proinflammatory factors277.

Ca2+
signalling in CF epithelial cells
95
Hyperproliferation of CF epithelial cells with sustained overexpression of IL-8
and matrix metalloproteinase along with reduced terminal differentiation has
been well documented in a sophisticated humanized airway xenograft model,
which recapitulates as close as technically possible how these processes
physiologically occur in the human airway epithelium277. A change in
intracellular Ca2+ signalling, in particular a change in amplitude or frequency of
Ca2+ oscillations in F508del-CFTR expressing cells, would explain such
proliferative activity and the delay in cellular differentiation.
Finally, the present experiments also demonstrate a functional
interference of CFTR with the IP3-receptor binding protein IRBIT. IRBIT has
been shown to suppress the activity of IP3 receptors by competing with IP3 for
a common binding site258. A recent report showed that IRBIT coordinates
epithelial fluid and HCO3- secretion due to stimulation of the Na+/HCO3
- co-
transporter and CFTR257. However, in contrast to that study we were unable to
coimmunoprecipitate CFTR and IRBIT here. Therefore propose that F508del-
CFTR affects Ca2+ signalling in an IRBIT and Cl- dependent manner, due to its
ability to operate as an ER-trapped ion channel.
5 SUPPLEMENTARY DATA
Supplementary Fig.IV.2. 1 - Core
glycosylation of bestrophin 1. Western blot
analysis of bestrophin 1 overexpressed in BHK
cells before (lane 1) and after incubation of
the cell lysates with N-glycosidase F (PNGase,
lane 2), or Endo H (lane 3). Both PNGase and
Endo H produce one single band of lower
mobility suggesting core glycosylation of
bestrophin 1.
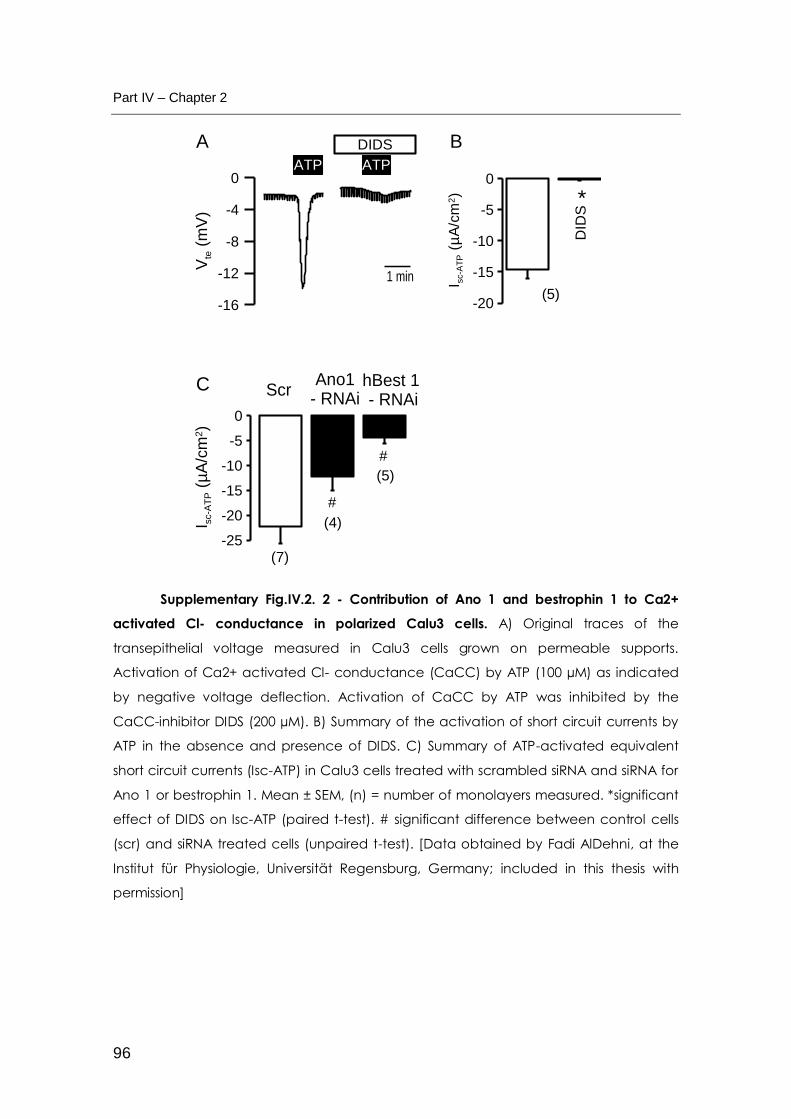
Part IV – Chapter 2
96
Supplementary Fig.IV.2. 2 - Contribution of Ano 1 and bestrophin 1 to Ca2+
activated Cl- conductance in polarized Calu3 cells. A) Original traces of the
transepithelial voltage measured in Calu3 cells grown on permeable supports.
Activation of Ca2+ activated Cl- conductance (CaCC) by ATP (100 µM) as indicated
by negative voltage deflection. Activation of CaCC by ATP was inhibited by the
CaCC-inhibitor DIDS (200 µM). B) Summary of the activation of short circuit currents by
ATP in the absence and presence of DIDS. C) Summary of ATP-activated equivalent
short circuit currents (Isc-ATP) in Calu3 cells treated with scrambled siRNA and siRNA for
Ano 1 or bestrophin 1. Mean ± SEM, (n) = number of monolayers measured. *significant
effect of DIDS on Isc-ATP (paired t-test). # significant difference between control cells
(scr) and siRNA treated cells (unpaired t-test). [Data obtained by Fadi AlDehni, at the
Institut für Physiologie, Universität Regensburg, Germany; included in this thesis with
permission]
-16
-12
-8
-4
0
Vte (
mV
)
DIDS
1 min
-20
-15
-10
-5
0
I sc-A
TP (
µA
/cm
2)
(5)
*
-25
-20
-15
-10
-5
0
I sc-A
TP (
µA
/cm
2)
(5)
hBest 1 - RNAi
#
ScrAno1
- RNAi
#
(4)
(7)
A B
C
ATP ATP
DID
S

Ca2+
signalling in CF epithelial cells
97
Supplementary Fig.IV.2. 3 - Anion conductances in CFBE/wt-CFTR and
CFBE/F508del-CFTR cells. A) Original traces of fluorescence intensity measured in
CFBE/wt-CFTR and CFBE/F508del-CFTR cells. Activation of Ca2+ dependent chloride
conductance (CaCC) by stimulation with UTP (100 µM) was measured by I- quenching
of I- sensitive protein YFP-I152L. Initial slopes of YFP fluorescence quenching by I- influx
correlates to the size of chloride conductance. CaCC was activated by UTP in
CFBE/F508del-CFTR but not in CFBE/wt-CFTR cells. In contrast, activation of CFTR-
conductance by adenosine (100 µM) was observed in CFBE/wt-CFTR but not in
CFBE/F508del-CFTR cells. B) Summary of I- influx measured in CFBE/wt-CFTR and
CFBE/F508del-CFTR cells. C) Activation of CaCC by UTP (100 µM) in CFBE/wt-CFTR and
CFBE/F508del-CFTR cells exposed to LPS (10 µg/ml, ON). D) Change of I- influx (%)
induced by incubation of the cells with LPS (10 µg/ml, ON). Mean ± SEM, (n) = number
of cells measured. *significant effects of UTP and adenosine respectively (paired t-test).
F508del
wt
A B
0
0.1
0.2
0.3
0.4
I- u
pta
ke (
F/s
)
* # *
#
I-
I-UTP
I- Ado
wt-CFTRF508del-CFTR
(73-75) (50-51)
C
0
40
80
120
160
I-
upta
ke L
PS
(%
)
D
##
(45-54)
(39-41)
*
5 min
20
re
l. u
nits
F508del - LPS
wt - LPS
wt-CFTRF508del-CFTR

Part IV – Chapter 2
98
# significant difference between wt-CFTR and F508del-CFTR or control and LPS-
treatment (unpaired t-test).
Supplementary Fig.IV.2. 4 - Lack of co-immunoprecipitation of IRBIT and CFTR.
A,B) WB of wt-CFTR (A) and F508del-CFTR (B) in stably expressing A549 cells. wt-CFTR
and F508del-CFTR could not be co-immunoprecipitated with IRBIT in A549 cells. C,D)
Western blot of IRBIT in A549 cells stably expressing wt-CFTR (C) and F508del-CFTR (D).
IRBIT could not be co-immunoprecipitated with CFTR in A549 cells. E,F) Western blot of
wt-CFTR (E) and F508del-CFTR (F) in overexpressing BHK cells. wt-CFTR and F508del-CFTR
could not be co-immunoprecipitated with IRBIT in BHK cells. G,H) Western blot of IRBIT in
BHK cells overexpressing wt-CFTR (G) and F508del-CFTR (H). IRBIT could not be co-
25 M
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
wtCFTR
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
F508del-CFTR
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
IRBIT
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
IRBIT
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
A B
C D
25 M
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
wtCFTR
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
F508del-CFTR
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
IRBIT
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
IRBIT
Inpu
t
IP a
nti-I
gG ra
bbit
IP a
nti-I
RBIT
IP a
nti-I
gG m
ouse
IP a
nti-C
FTR
E F
G H

Ca2+
signalling in CF epithelial cells
99
immunoprecipitated with CFTR in BHK cells. [Data obtained by Eva Sammels at Professor
Humbert de Smedt’s laboratory at the Department Molecular Cell Biology, K.U.Leuven,
Leuven, Belgium; included in this thesis with permission]


Part V. GENERAL DISCUSSION AND FUTURE
PERSPECTIVES


General Discussion and Future Perspectives
103
V GENERAL DISCUSSION AND FUTURE PERSPECTIVES
The identification of the CFTR gene in 1989 represented a major
hallmark in the CF field and, at the time compromised a great hope in a rapid
discovery of an ultimate cure for CF. Despite the major advances in
comprehension of the molecular basis of the disease, unfortunately a cure
cannot yet be offered to patients suffering from CF. F508del, the most common
mutation present in at least one allele of patients, presents a folding defect and
is consequently retained in the ER and prematurely targeted for degradation.
Only recently small molecules capable of restoring F508del-CFTR folding
and trafficking defects have started to hit the clinical setting, by entering Phase
2 and Phase 3 clinical trials. For instance, VX-809 is a corrector that recently
completed a Phase 2a trial in patients with F508del mutation, in order to test its
safety in a clinical situation278. Another drug presently under trial is potentiator
VX-770 and is capable of stimulating CFTR function at the cell membrane. VX-
770 is currently being evaluated in a Phase III registration program that is
comprised of three different clinical trials in order to determine the safety,
effectiveness and acceptability for approval279.
Although CFTR is clearly a key player in epithelial cell physiology,
attempts have been made regarding the restoration of Cl- secretion in epithelia
independently of CFTR. The so-called “by-pass” therapeutic approach takes
advantage of manipulating other entities present at the membrane of epithelial
cells in order to ultimately enhance Cl- transport and overall balance electrolyte
transport. Moreover, it is also suggested that CFTR might coordinate Cl-
secretion by either acting on other Cl- channels on the apical membrane of
epithelial cells, or indirectly by altering signalling pathways that ultimately result
in an enhanced CFTR independent Cl- secretion.
In fact, several reports demonstrated that an alternative source of Cl -
besides CFTR exists in the apical membrane of epithelial cells. For instance,
simultaneously to the finding that CF was probably cause by impaired cAMP

Part V
104
dependent Cl- channel activity and/or regulation, Ca2+-dependent response was
found to be intact in CF280-282. At the time, this finding was interpreted as a
confirmation that a defective Cl- channel regulation was impaired in CF, rather
than the transport itself280. Subsequent studies by the Boucher group confirmed
that only Ca2+-dependent Cl- secretory mechanisms were functional in freshly
excised CF nasal epithelia113. Interestingly, other studies by the same group
and later also by Mall and co-workers in human CF tissue demonstrated that not
only the Ca2+-mediated Cl- secretory pathway was functional, but also seems to
be upregulated114,223.
Further clues were given by the various CF mouse models generated,
where the investigators were confronted with a puzzling scenario: although the
gastrointestinal phenotype could be recapitulated in the various models,
alterations in ion transport, which lead to the devastating lung disease in CF
patients, appeared to be largely absent122,283. It was initially suggested that the
young aged and the semisterile barrier environment in which the animals were
kept prevented manifestation of the airway pathology seen in humans.
However, KO animals over two years old and kept out of the barrier facility for
over 1 year failed to exhibit lung disease283. A more detailed analysis of murine
airways revealed that there appears to be little or no CFTR expressed284, being
the Cl- secretory activity of this tissue dominated by the alternative non-CFTR
Ca2+-dependent Cl- channels. This also reinforced the idea that alternative
sources of Cl- are capable of overcoming the lack of CFTR in epithelia and
would thereby be excellent targets for further investigation.
While the molecular entity responsible for cAMP dependent Cl- transport
- CFTR - is now known for more than 20 years, the molecular counterpart of
CaCCs was only very recently revealed. Almost simultaneously, data generated
in three independent laboratories, supported the statement that Ano 1 was the
channel responsible for evoking Ca2+-dependent and receptor-mediated Cl-
currents in epithelia. In addition an Ano 1-/- mouse model had been previously
developed as this protein had been also found to be important for development.

General Discussion and Future Perspectives
105
Given this exciting discovery, in this doctoral work we aimed to look at
the particular role played by Ano 1 in Ca2+-dependent Cl- secretion in epithelia
affected by CF.
To this end we compared the electrophysiological properties of tracheas
from Ano 1 +/+ and Ano 1 -/- mice. We initially proved that Cl- secretion elicited
by increase in intracellular Ca2+ due to cholinergic or purinergic stimulation in wt
animals was sensitive to known CaCCs inhibitors, as well as to depletion of
Ca2+ stores. We then compared ICaCC in Ano 1 +/+ and Ano 1 -/- littermates and
found that both cholinergic and purinergic stimulation of Ca2+-dependent Cl-
secretion was indeed largely impaired in Ano 1 -/- tracheas. On the other hand,
neither CFTR activity elicited by IBMX/Forsk nor ENaC amiloride-sensitive
currents was changed in Ano 1 -/- pups. From these experiments, we concluded
that in the lower airways of these animals Ano 1 is the channel mainly
responsible for Ca2+-dependent Cl- secretion. Furthermore, we observed that
the absence of this channel has no impact on either ENaC or on CFTR cAMP-
dependent activity.
Intriguingly, we also observed that the remaining Cl- secretion elicited by
purinergic activation in Ano 1 -/- tracheas was due to CFTR activity, while no
CFTR inhibitor sensitive current was found in the Ano 1 +/+ littermates. The
conventional paradigm is that CFTR is activated through cAMP and protein
kinase A (PKA) such as beta-adrenergic agonists, whereas CaCCs are
activated by Ca2+ agonists like UTP. However, recently CFTR has also been
reported to be activated through additional mechanisms (namely ATP)
suggesting a cross-talk between the Ca2+ and cAMP pathways. For instance,
CFTR dependent Cl- conductance is accomplished through activation of G
proteins in native sweat duct285,286. Furthermore, stimulation of purinergic P2Y2
receptors activates CFTR dependent Cl− currents in CFTR-expressing oocytes,
airway epithelial cells241 and rat submandibular glands287. Moreover, Namkung
and co-workers showed that in well-differentiated primary human bronchial cell
cultures Cl- currents elicited by Ca2+ agonists are mediated by CFTR through a

Part V
106
mechanism involving Ca2+ activation of adenylyl cyclase I (AC1) and cAMP/PKA
signalling288. The authors suggest that compartmentalized AC1-CFTR
association is responsible for Ca2+ / cAMP cross-talk. Our results suggest that
purinergic stimulation of CFTR is present in Ano 1 -/- animals.
Next, we assessed the impact of the activity of CaCC in the maintenance
of proper airways MCC. Stimulation of airways secretion was indirectly
evaluated through analysis of the mucociliary transport of black microspheres in
tracheas of newborn animals. We observed that mucociliary transport that
followed cholinergic stimulation was abolished in Ano 1 -/- comparatively to Ano
+/+ animals. Additionally, MCC was inhibited in the presence of the CaCC
inhibitor DIDS in Ano +/+ mice.
We concluded that the lack of Cl- secretion in Ano 1 null animals and in
Ano 1 inhibited tracheas correlates with a deficient MCC in the analysed
tissues. We therefore, speculate that in mice lower airways, a CF-like
phenotype can be recapitulated by impairment of Ca2+-activated Cl- secretion
through Ano 1, as maintenance of proper ASL and MCC in airways seems to be
Ano 1-dependent.
It is important to bear in mind that animals lacking Ano 1 fail to thrive
during postnatal life, dying within the first nine days of after birth. In addition, the
Ano 1 -/- animals do not gain weight at the same rate as their littermates,
although milk was observed in their stomachs. Phenotypic and physiological
observations on these animals are thus conditioned not only by deletion of Ano
1 but also by the young age of the pups as well as debilitated health condition.
It is also pertinent to observe that the congenital cartilaginous defects
presented by Ano 1 -/- mice are similar (although more severe) to those
described in tracheas from CFTR-/- mice222. Intriguingly, the recently generated
CF pig model shows that neonatal CFTR-/- pigs have reduced tracheal calibre
and circularity. The authors of this study suggest that loss of CFTR produces
tracheal abnormalities that begin early in life, even before the onset of

General Discussion and Future Perspectives
107
inflammation and infection289. These results may reflect a common Cl- secretory
defect mediated by molecularly distinct Cl- channels. Moreover, tracheas from
Ano 1-/- animals exhibit significant, neonatal, lumenal mucus accumulation222,
further supporting our findings that Ano 1 stimulation is responsible, to a great
extent, for maintenance of normal airways hydration. All in all, these results
indicate that stimulation of Ca2+ activated Cl- secretion in CF airways might
alleviate the defect in airways hydration. Furthermore, we speculate that this
approach might also be beneficial for younger patients, as deficiencies in Cl -
transport seem to have a great impact early in airways development.
Another aim of the current doctoral work was to better understand the
mechanisms underlying the long-standing observation that in CF airway
epithelial cells, in parallel with the lack of CFTR-mediated Cl- transport there is
an increased Ca2+-dependent Cl- conductance. Although this has been
repeatedly recognized, the mechanism behind this observation is poorly
understood. Some studies have demonstrated that CFTR has an inhibitory
effect on CaCCs244, while others correlate the findings to altered an Ca2+
signalling pathway present in CF. Previous studies by Ribeiro and collaborators
have shown that inflamed CF airway epithelial cells exhibit an expansion of the
endoplasmic reticulum (ER) compartment, which correlate with the enhanced
Ca2+-dependent activation of a luminal Cl- conductance247. In fact previous work
from our group has also shown that ICaCC is enhanced in allergic airways
inflammation290.
Interestingly, other studies from our group correlated the upregulation of
the Cl- and Ca2+ modulator Best1 and Ca2+ activated Cl− currents (ICaCC) during
dedifferentiation and proliferation of epithelial cells, as well as in fast growing
cancer cells193,194. It was also found that during renal inflammation in LPS-
treated mice or after unilateral urethral obstruction, Best1 was transiently
upregulated. On the other hand, during collecting duct (CD) polarization and
thus terminally differentiated, cell proliferation, ICaCC, and Best1 expression were

Part V
108
remarkably suppressed. These results suggested a negative role for Best1 and
ICaCC in cell proliferation and in tissue repair.
Given the findings on the essential role played by Ano 1 in airways as
CaCC previously presented in this doctoral work, we further looked into CaCC
in CF. We started by comparing the levels of transcripts of Ano 1 and also
Bestrophin1 in both freshly isolated and cultured cells expressing wt or F508del-
CFTR. No upregulation of either Ano 1 or Best 1 transcripts was found in the
cell systems analysed. From this, we concluded that changes in transcriptional
levels of CaCC do not account for the differences observed in CF.
We further hypothesized that altered airway environment resulting from
infection and inflammation present in CF can impact on the innate defense
responses of the epithelia and also on CaCC expression or activity. In this
context we further tested weather exposure to pro inflammatory LPS would
result in altered transcription levels of Ano 1 and Best1. Our studies led to the
observation that neither differential expression of F508del or wt-CFTR alone,
nor inflammation let to increased transcripts of recently identified CaCCs Ano 1
or Best1. From this, we concluded that differential expression of CaCCs would
not account for the enhanced Ca2+ dependent Cl- secretion observed in CF.
The next step was to look into the impact of differential expression of wt
or F508del-CFTR in the Ca2+ homeostasis. Our experiments showed that Ca2+
signalling was strongly enhanced upon of expression of ER-retained F508del-
CFTR. We also found that this UTP mediated [Ca2+]i increase was largely Cl-
dependent. Given this we suggested that F508del-CFTR functioned as an ER
Cl- counter ion channel for Ca2+ movement.
Activity of ER localized wt and F508del-CFTR was reported by Pasyk
and Foskett soon after the identification of CFTR 291. The authors demonstrated
that F508del-CFTR is a functional cAMP-regulated Cl- channel in native ER
membrane of mammalian cells.

General Discussion and Future Perspectives
109
Here, we speculate that basal activity of ER-retained F508del-CFTR
and/or purinergic stimulation of the mutant channels is sufficient to provide an
enhanced counter ion movement for Ca2+. This was confirmed by expression of
a in cis F508del/G551D-CFTR double mutant, which abolished the observed
increase on [Ca2+]I signalling due to expression of F508del-CFTR. We also
tested the hypothesis that F508del-CFTR may serve as a scavenger for inositol-
1,4,5-trisphosphate [IP3] receptor binding protein released with IP3 (IRBIT).
Although no interaction of CFTR and IRBIT could be demonstrated we found
that enhanced CaCC in F508del-CFTR expressing cells was abolished by co-
expression of IRBIT.
Thereby we propose that ER activity of the mutant F508del-CFTR
controls intracellular Ca2+ signalling accounting for enhanced ICaCC and not
changes in Ano 1 or Best1 expression.
Cell proliferation in bronchial epithelium and submucosal glands of CF
patients is increased when compared to non-CF cells276. Taking into
consideration these observations we hypothesize that the increase in Ca2+
signalling and subsequent enhanced CaCC activity result in increased
proliferation observed in CF cells. Furthermore, we speculate that activators of
ICaCC, and in particular of Ano 1, might be beneficial for improvement of lung
function of CF patients, not only through increase of MCC, but also by possibly
favouring tissue repair during inflammation. Conversely, further studies are
crucial for evaluation of a possible therapeutically application of Ano 1
activators, as they could have potential adverse effects, namely in cell
proliferation and fibrosis.
Despite the beneficial aspects of these findings, one must consider that
Ano 1 was initially found to be associated with a number of distinct cancers,
namely head and neck squamous cell carcinoma (HNSCC). Recent studies
have shown that although Ano 1 amplification and expression does not directly
affect cell proliferation, it affects cell properties linked to metastasis, such as
attachment, spreading, and invasion292.

Part V
110
In addition, as suggested previously, Ca2+ activated Cl- conductance
seems to dominate the mouse airways comparatively to CFTR, having a much
smaller contribution in human tissues. Hence, it is important to evaluate the real
participation of Ano 1 in human tissues and if this activation is indeed sufficient
for improving CF in the various organs affected.
Indeed stimulation of purinergic receptors with modified and more stable
synthetic ATP molecules has been one of the strategies adopted to overcome
CF before the identification of Ano 1. Among the most promising new therapies
targeted at increasing mucosal hydration on the surface of the airways,
denufosol tetrasodium is a novel second-generation, metabolically stable,
selective P2Y(2) receptor agonist currently in Phase 3 clinical development293.
However, results from this trial have led to the decision that this compound will
not proceed into further trials (Communication at the NACF Conference, Oct
2010).
Moreover, there are some reports that purinergic stimulation also results
in ENaC inhibition294. As a consequence, drugs acting as purinergic agonist
would have both the potential of increasing secretion of Cl- and attenuating Na+
absorption. In particular it has been shown that direct application of a
membrane-permeant analog of Ins(3,4,5,6)P3 (IP3), INO-4995, to human nasal
airway epithelia reduced the amiloride-inhibitable basal short-circuit current
(Isc)227. In concert such a strategy could help to restore ASL volume in CF
individuals. From our studies a direct regulation of ENaC through CaCC is not
evident since the amiloride sensitive Na+ absorption and CFTR-dependent Cl-
secretion were not disturbed in Ano 1 null mice.
In addition to stimulation of P2Y(2) receptors, drug development would
also benefit from a better understanding of how to prevent inhibition of CaCCs
by inositol 3,4,5,6-tetrakisphosphate (Ins(3,4,5,6)P4 or IP4). As a consequence,
the classically observed transient activity of CaCC could be prolonged and a
sustained activity of the channels would be achieved. This would better

General Discussion and Future Perspectives
111
resemble the activity of CFTR in native tissues and allow further
pharmacological intervention295.
All in all the results presented in this thesis provide valuable insights not
only into the role of Ano 1 as the long searched CaCC in the airways, but also in
the mechanisms Ca2+ signalling in CF. Our results also imply that the F508del
mutation might have an additional impact in CF disease. Besides the
impairment of Cl- secretion, this mutant also perturbs Ca2+ signalling, further
disturbing cellular homeostasis and possibly contributing for the severe
phenotype of F508del-homozygous CF patients.
FINAL REMARKS AND FUTURE DIRECTIONS
The studies presented in this dissertation had the overall goal of
providing new insights in the role and contribution of Ca2+ activated Cl- channels
in CF. From our studies, we concluded that Ano 1 contributes to airway Cl-
secretion and thus for the maintenance of a proper clearance. Ano 1 channels
thereby compromising the long-searched CaCC in airways and are thus a very
promising therapeutical target for CF.
Secondly, we found that neither F508del-CFTR nor LPS treatment
upregulated Ano 1 or Best1 transcript levels, thereby not accounting for the
enhanced Ca2+-activated Cl- secretion reported in CF. Furthermore, we found
and that F508del-CFTR mutant protein trapped in the ER affects Ca2+ signalling
alone, possibly by acting as a Cl- counter-ion channel and in this manner
facilitating Ca2+ movement over the ER membrane.
An obvious follow-up to the present work would be:
1) to evaluate the contribution of Ano 1 in human airways potentially by
using more specific activators that have been developed in the
meantime.

Part V
112
2) Moreover, it is important to understand the consequences of potential
increasing the activity of Ano 1. Studies in a knock-in Ano 1 mouse
model would be a valuable tool for investigating this mater.
3) Furthermore, given the postnatal lethality associated with the deletion
of Ano 1, the generation of a conditional allele of this protein together
with a pulmonary epithelium-specific Cre296 mouse would enable
further studies and possibly give new insights into the contribution of
Ano 1 both the clinic and for basic science.

References
113
VI REFERENCES
1. Collins, F.S. Cystic fibrosis: molecular biology and therapeutic implications. Science 256,
774-9 (1992).
2. Cystic Fibrosis Foundation Patient Registry 2008 Annual Report. (2008).at http://www.cff.org/UploadedFiles/research/ClinicalResearch/2008-Patient-Registry-Report.pdf
3. Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac Disease: a clinical and pathologic study. Archives of Pediatrics Adolescent Medicine 56, 344-399
(1938).
4. Andersen, D.H. & Hodges, R.G. Celiac Syndrome: V. Genetics of Cystic Fibrosis of the Pancreas with a Consideration of Etiology. Archives of Pediatrics Adolescent Medicine 72, 62-80 (1946).
5. Farber, S., Shwachman, H. & Maddock, C.L. Pancreatic function and disease in early life. I. Pancreatic enzyme activity and the celiac syndrome. The Journal of Clinical Investigation 22, 827-38 (1943).
6. Di Santʼagnese, P.A. et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 12, 549-63
(1953).
7. Quinton, P.M. Missing Cl conductance in cystic fibrosis. The American Journal of Physiology 251, C649-52 (1986).
8. Rommens, J.M. et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059-65 (1989).
9. Kerem, B.-sheva et al. Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073-80 (1989).
10. Riordan, J.R. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066-73 (1989).
11. Cheng, S.H. et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827-34 (1990).
12. Cutting, G.R. et al. A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein. Nature 346, 366-9 (1990).
13. Kartner, N. et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell 64, 681-91 (1991).
14. Bear, C.E. et al. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68, 809-18 (1992).
15. Welsh, M.J. et al. Cystic fibrosis transmembrane conductance regulator: a chloride channel with novel regulation. Neuron 8, 821-9 (1992).

Part V
114
16. Knowles, M. et al. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221, 1067-1070 (1983).
17. Chillón, M. et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. The New England Journal of Medicine 332, 1475-80 (1995).
18. Davies, J.C., Alton, E.W.F.W. & Bush, A. Cystic fibrosis. BMJ 335, 1255-9 (2007).
19. McPhee, S.J., Papadakis, M. & Rabow, M.W. Current Medical Diagnosis and Treatment 2011. 1808(McGraw-Hill Medical: 2010).
20. Zielenski, J. & Tsui, L.C. Cystic fibrosis: genotypic and phenotypic variations. Annual Review of Genetics 29, 777-807 (1995).
21. Rowe, S.M., Miller, S. & Sorscher, E.J. Cystic fibrosis. The New England Journal of Medicine 352, 1992-2001 (2005).
22. Amaral, M.D. & Kunzelmann, K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends in Pharmacological Sciences 28, 334-41 (2007).
23. Welsh, M.J. & Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73, 1251-4 (1993).
24. Tsui, L.C. The spectrum of cystic fibrosis mutations. Trends in Genetics 8, 392-8 (1992).
25. Mendes, F. et al. Unusually common cystic fibrosis mutation in Portugal encodes a misprocessed protein. Biochemical and Biophysical Research Communications 311,
665-671 (2003).
26. Parad, R.B. Heterogeneity of phenotype in two cystic fibrosis patients homozygous for the CFTR exon 11 mutation G551D. Journal of Medical Genetics 33, 711-3 (1996).
27. Kerem, E. et al. The relation between genotype and phenotype in cystic fibrosis - analysis of the most common mutation (delta F508). The New England Journal of Medicine 323, 1517-22 (1990).
28. Boucher, R.C. New concepts of the pathogenesis of cystic fibrosis lung disease. European Respiratory Journal 23, 146-158 (2004).
29. Hughes, A.L. Evolution of the ATP-binding-cassette transmembrane transporters of vertebrates. Molecular Biology and Evolution 11, 899-910 (1994).
30. Higgins, C.F. ABC transporters: from microorganisms to man. Annual Review of Cell Biology 8, 67-113 (1992).
31. Riordan, J.R. Assembly of functional CFTR chloride channels. Annual Review of Physiology 67, 701-18 (2005).
32. Chang, X.B. et al. Mapping of cystic fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. The Journal of Biological Chemistry 269, 18572-5 (1994).

References
115
33. Riordan, J.R. CFTR function and prospects for therapy. Annual Review of Biochemistry 77, 701-26 (2008).
34. Serohijos, A.W.R. et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proceedings of the National Academy of Sciences of the United States of America 105, 3256-61
(2008).
35. Hwang, T.-C. & Sheppard, D.N. Gating of the CFTR Cl- channel by ATP-driven
nucleotide-binding domain dimerisation. The Journal of Physiology 587, 2151-61 (2009).
36. Skach, W.R. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney international 57, 825-31 (2000).
37. Amaral, M.D. Processing of CFTR: traversing the cellular maze--how much CFTR needs to go through to avoid cystic fibrosis? Pediatric pulmonology 39, 479-91 (2005).
38. Wang, X. et al. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. The Journal of Cell Biology 167, 65-74
(2004).
39. Bannykh, S.I. et al. Traffic pattern of cystic fibrosis transmembrane regulator through the early exocytic pathway. Traffic 1, 852-70 (2000).
40. Kleizen, B., Braakman, I. & Jonge, H.R. de Regulated trafficking of the CFTR chloride channel. European Journal of Cell Biology 79, 544-56 (2000).
41. Ameen, N., Silvis, M. & Bradbury, N.A. Endocytic trafficking of CFTR in health and disease. Journal of Cystic Fibrosis 6, 1-14 (2007).
42. Amaral, M.D. Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. Journal of Inherited Metabolic Disease 29, 477-87 (2006).
43. Benharouga, M., Sharma, M. & Lukacs, G.L. CFTR folding and maturation in cells. Methods in molecular medicine 70, 229-43 (2002).
44. Varga, K. et al. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. The Journal of Biological Chemistry 279, 22578-84 (2004).
45. Qu, B.H. & Thomas, P.J. Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. The Journal of Biological Chemistry 271, 7261-4 (1996).
46. Lukacs, G.L. et al. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. The EMBO Journal 13, 6076-86 (1994).
47. Ward, C.L., Omura, S. & Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121-7 (1995).
48. Sharma, M. et al. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. The Journal of Biological Chemistry 276, 8942-50 (2001).

Part V
116
49. Dalemans, W. et al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526-8 (1991).
50. Chang, X.B. et al. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Molecular Cell 4, 137-42 (1999).
51. Riordan, J.R. Cystic fibrosis as a disease of misprocessing of the cystic fibrosis transmembrane conductance regulator glycoprotein. American Journal of Human Genetics 64, 1499-504 (1999).
52. Lewis, H. a et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. The EMBO Journal 23, 282-93 (2004).
53. Qu, B.H., Strickland, E.H. & Thomas, P.J. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. The Journal of Biological Chemistry 272, 15739-44 (1997).
54. Denning, G.M. et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761-4 (1992).
55. Drumm, M.L. et al. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 254, 1797-9 (1991).
56. Anderson, M.P. et al. Generation of cAMP-activated chloride currents by expression of CFTR. Science 251, 679-82 (1991).
57. Bear, C.E. et al. Cl- channel activity in Xenopus oocytes expressing the cystic fibrosis
gene. The Journal of Biological Chemistry 266, 19142-5 (1991).
58. Anderson, M.P. et al. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253, 202-5 (1991).
59. Sheppard, D.N. & Welsh, M.J. Structure and function of the CFTR chloride channel. Physiological Reviews 79, S23-45 (1999).
60. Zhang, L. et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS biology 7, e1000155
(2009).
61. Kunzelmann, K. CFTR: interacting with everything? News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society 16, 167-70 (2001).
62. Phillips, J. Bidirectional Transepithelial Water Transport: Measurement and Governing Mechanisms. Biophysical Journal 76, 869-877 (1999).
63. Matsui, H. et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95, 1005-15
(1998).
64. Matsui, H. et al. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. The Journal of Clinical Investigation 105, 1419-27 (2000).

References
117
65. Boucher, R.C. et al. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal
rate and response to adenylate cyclase activation. The Journal of Clinical Investigation 78, 1245-52 (1986).
66. Verkman, A.S. Lung disease in cystic fibrosis: is airway surface liquid composition abnormal? American Journal of Physiology. Lung Cellular and Molecular Physiology 281, L306-8 (2001).
67. Tarran, R. et al. The CF Salt ControversyIn Vivo Observations and Therapeutic Approaches. Molecular Cell 8, 149-158 (2001).
68. Quinton, P.M. Viscosity versus composition in airway pathology. American Journal of Respiratory and Critical Care Medicine 149, 6-7 (1994).
69. Smith, J.J. et al. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229-36 (1996).
70. Zabner, J. et al. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Molecular Cell 2, 397-403 (1998).
71. Bals, R. et al. Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. The Journal of Clinical Investigation 103, 1113-7
(1999).
72. Wine, J.J. The genesis of cystic fibrosis lung disease. The Journal of Clinical Investigation 103, 309-12 (1999).
73. Ballard, S.T. et al. CFTR involvement in chloride, bicarbonate, and liquid secretion by airway submucosal glands. The American Journal of Physiology 277, L694-9 (1999).
74. Dosanjh, A. et al. Heterologous expression of delta F508 CFTR results in decreased sialylation of membrane glycoconjugates. The American Journal of Physiology 266,
C360-6 (1994).
75. Barasch, J. & Al-Awqati, Q. Defective acidification of the biosynthetic pathway in cystic fibrosis. Journal of Cell Science. Supplement 17, 229-33 (1993).
76. Pier, G.B., Grout, M. & Zaidi, T.S. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proceedings of the National Academy of Sciences of the United States of America 94,
12088-93 (1997).
77. Puchelle, E., Bajolet, O. & Abély, M. Airway mucus in cystic fibrosis. Paediatric respiratory reviews 3, 115-9 (2002).
78. Park, H.W. et al. Dynamic regulation of CFTR bicarbonate permeability by Cl-i and its
role in pancreatic bicarbonate secretion. Gastroenterology 139, 620-31 (2010).
79. Hug, M.J., Tamada, T. & Bridges, R.J. CFTR and bicarbonate secretion by [correction of to] epithelial cells. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society 18, 38-42 (2003).

Part V
118
80. Reddy, M.M. & Quinton, P.M. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature 423, 756-60 (2003).
81. Clarke, L.L., Stien, X. & Walker, N.M. Intestinal bicarbonate secretion in cystic fibrosis mice. Journal of the Pancreas 2, 263-7 (2001).
82. Poulsen, J.H. et al. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proceedings of the National Academy of Sciences of the United States of America 91, 5340-4 (1994).
83. Shori, D.K. et al. Altered sialyl- and fucosyl-linkage on mucins in cystic fibrosis patients promotes formation of the sialyl-Lewis X determinant on salivary MUC-5B and MUC-7. Pflügers Archiv : European Journal of Physiology 443 Suppl , S55-61 (2001).
84. Mohapatra, N.K. et al. Alteration of sulfation of glycoconjugates, but not sulfate transport and intracellular inorganic sulfate content in cystic fibrosis airway epithelial cells. Pediatric Research 38, 42-8 (1995).
85. Garcia, M.A.S., Yang, N. & Quinton, P.M. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. The Journal of Clinical Investigation 119, 2613-22 (2009).
86. Coakley, R.D. et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proceedings of the National Academy of Sciences of the United States of America 100, 16083-8 (2003).
87. Donaldson, S.H. & Boucher, R.C. Update on pathogenesis of cystic fibrosis lung disease. Current Opinion in Pulmonary medicine 9, 486-91 (2003).
88. McPherson, M. a & Dormer, R.L. Cystic fibrosis gene and mucin secretion. Lancet 343, 7
(1994).
89. Chen, E.Y.-T. et al. A New Role for Bicarbonate in Mucus Formation. American Journal of Physiology. Lung Cellular and Molecular Physiology (2010).
90. Kuver, R. et al. Absence of CFTR is associated with pleiotropic effects on mucins in mouse gallbladder epithelial cells. American Journal of Physiology. Gastrointestinal and Liver Physiology 291, G1148-54 (2006).
91. Kreda, S.M. et al. Coordinated release of nucleotides and mucin from human airway epithelial Calu-3 cells. The Journal of Physiology 584, 245-59 (2007).
92. Boucher, R.C. et al. Evidence for reduced Cl- and increased Na
+ permeability in cystic
fibrosis human primary cell cultures. The Journal of Physiology 405, 77-103 (1988).
93. Knowles, M.R. et al. Abnormal respiratory epithelial ion transport in cystic fibrosis. Clinics in Chest Medicine 7, 285-97 (1986).
94. Letz, B. & Korbmacher, C. cAMP stimulates CFTR-like Cl- channels and inhibits
amiloride-sensitive Na+ channels in mouse CCD cells. The American Journal of
Physiology 272, C657-66 (1997).

References
119
95. Mall, M. et al. CFTR-mediated inhibition of epithelial Na+ conductance in human colon is
defective in cystic fibrosis. The American Journal of Physiology 277, G709-16 (1999).
96. Mall, M. et al. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis
transmembrane conductance regulator in normal but not in cystic fibrosis airways. The Journal of Clinical Investigation 102, 15-21 (1998).
97. Stutts, M.J. et al. CFTR as a cAMP-dependent regulator of sodium channels. Science 269, 847-50 (1995).
98. Mall, M. et al. Wild type but not deltaF508 CFTR inhibits Na+ conductance when
coexpressed in Xenopus oocytes. FEBS Letters 381, 47-52 (1996).
99. Stutts, M.J., Rossier, B.C. & Boucher, R.C. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. The Journal of Biological Chemistry 272, 14037-40 (1997).
100. König, J. et al. The cystic fibrosis transmembrane conductance regulator (CFTR) inhibits ENaC through an increase in the intracellular Cl
- concentration. EMBO reports 2, 1047-
51 (2001).
101. Suaud, L. et al. Genistein restores functional interactions between Delta F508-CFTR and ENaC in Xenopus oocytes. The Journal of Biological Chemistry 277, 8928-33 (2002).
102. Suaud, L. et al. Regulatory interactions of N1303K-CFTR and ENaC in Xenopus oocytes: evidence that chloride transport is not necessary for inhibition of ENaC. American Journal of Physiology. Cell Physiology 292, C1553-61 (2007).
103. Guggino, W.B. & Stanton, B.A. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nature reviews. Molecular cell biology 7, 426-36 (2006).
104. Gentzsch, M. et al. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na
+ channel. The Journal of Biological Chemistry
285, 32227-32232 (2010).
105. Rubenstein, R.C. et al. Regulation of endogenous ENaC functional expression by CFTR and deltaF508-CFTR in airway epithelial cells. American Journal of Physiology. Lung Cellular and Molecular Physiology (2010).
106. Berdiev, B.K. et al. Molecular proximity of cystic fibrosis transmembrane conductance regulator and epithelial sodium channel assessed by fluorescence resonance energy transfer. The Journal of Bological Chemistry 282, 36481-8 (2007).
107. Schwiebert, E.M. et al. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 81, 1063-73 (1995).
108. Tarran, R. et al. Regulation of murine airway surface liquid volume by CFTR and Ca2+
-activated Cl
- conductances. The Journal of General Physiology 120, 407-18 (2002).
109. Kogan, I. et al. CFTR directly mediates nucleotide-regulated glutathione flux. The EMBO Journal 22, 1981-9 (2003).

Part V
120
110. Roum, J.H. et al. Systemic deficiency of glutathione in cystic fibrosis. Journal of Applied Physiology 75, 2419-24 (1993).
111. lʼHoste, S. et al. CFTR mediates apoptotic volume decrease and cell death by controlling glutathione efflux and ROS production in cultured mice proximal tubules. American Journal of Physiology. Renal Physiology 298, F435-53 (2010).
112. Gabriel, S.E. et al. Permeabilization via the P2X7 purinoreceptor reveals the presence of a Ca
2+-activated Cl
- conductance in the apical membrane of murine tracheal epithelial
cells. The Journal of Biological Chemistry 275, 35028-33 (2000).
113. Boucher, R.C. et al. Chloride secretory response of cystic fibrosis human airway epithelia. Preservation of calcium but not protein kinase C- and A-dependent mechanisms. The Journal of Clinical Investigation 84, 1424-31 (1989).
114. Knowles, M.R., Clarke, L.L. & Boucher, R.C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. The New England Journal of Medicine 325, 533-8 (1991).
115. Mason, S.J., Paradiso, A.M. & Boucher, R.C. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. British Journal of Pharmacology 103, 1649-56 (1991).
116. Noah, T.L. et al. The response of a human bronchial epithelial cell line to histamine: intracellular calcium changes and extracellular release of inflammatory mediators. American Journal of Respiratory Cell and Molecular Biology 5, 484-92 (1991).
117. Clarke, L.L., Paradiso, a M. & Boucher, R.C. Histamine-induced Cl- secretion in human
nasal epithelium: responses of apical and basolateral membranes. The American Journal of Physiology 263, C1190-9 (1992).
118. Clarke, L.L. et al. Effects of bradykinin on Na+ and Cl
- transport in human nasal
epithelium. The American Journal of Physiology 262, C644-55 (1992).
119. Willumsen, N.J. & Boucher, R.C. Activation of an apical Cl- conductance by Ca
2+
ionophores in cystic fibrosis airway epithelia. The American Journal of Physiology 256,
C226-33 (1989).
120. Anderson, M.P. & Welsh, M.J. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proceedings of the National Academy of Sciences of the United States of America 88, 6003-7 (1991).
121. Grubb, B.R., Vick, R.N. & Boucher, R.C. Hyperabsorption of Na+ and raised Ca
2+-
mediated Cl- secretion in nasal epithelia of CF mice. The American Journal of Physiology
266, C1478-83 (1994).
122. Clarke, L.L. et al. Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in Cftr(-/-) mice. Proceedings of the National Academy of Sciences of the United States of America 91,
479-83 (1994).
123. Tarran, R. et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. The Journal of Biological Chemistry 280, 35751-9 (2005).

References
121
124. Miledi, R. A calcium-dependent transient outward current in Xenopus laevis oocytes. Proceedings of the Royal Society of London. Series B, Containing papers of a Biological character. Royal Society (Great Britain) 215, 491-7 (1982).
125. Barish, M.E. A transient calcium-dependent chloride current in the immature Xenopus oocyte. The Journal of Physiology 342, 309-25 (1983).
126. Bader, C.R., Bertrand, D. & Schwartz, E.A. Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. The Journal of Physiology 331, 253-84 (1982).
127. Gray, M. a et al. Chloride channels and cystic fibrosis of the pancreas. Bioscience reports 15, 531-41 (1995).
128. Wagner, J.A. et al. Activation of chloride channels in normal and cystic fibrosis airway epithelial cells by multifunctional calcium/calmodulin-dependent protein kinase. Nature 349, 793-6 (1991).
129. Kunzelmann, K. et al. Calcium-dependent chloride conductance in epithelia: is there a contribution by Bestrophin? Pflügers Archiv : European Journal of Physiology 454, 879-
89 (2007).
130. André, S. et al. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. Journal of neurophysiology 90,
3764-73 (2003).
131. Guo, D. et al. Calcium-activated chloride current contributes to action potential alternations in left ventricular hypertrophy rabbit. American Journal of Physiology. Heart and Circulatory Physiology 295, H97-H104 (2008).
132. Kurahashi, T. & Yau, K.W. Olfactory transduction. Tale of an unusual chloride current. Current Biology 4, 256-8 (1994).
133. Matthews, H.R. & Reisert, J. Calcium, the two-faced messenger of olfactory transduction and adaptation. Current Opinion in Neurobiology 13, 469-75 (2003).
134. Barnes, S. & Deschênes, M.C. Contribution of Ca and Ca-activated Cl channels to regenerative depolarization and membrane bistability of cone photoreceptors. Journal of neurophysiology 68, 745-55 (1992).
135. Fine, B.P., Marques, E.S. & Hansen, K.A. Calcium-activated sodium and chloride fluxes modulate platelet volume: role of Ca
2+ stores. The American Journal of Physiology 267,
C1435-41 (1994).
136. Nelson, M.T. et al. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. The Journal of Physiology 502 ( Pt 2, 259-64 (1997).
137. Wright, E.M. & Diamond, J.M. Anion selectivity in biological systems. Physiological Reviews 57, 109-56 (1977).
138. Hartzell, H.C. Activation of different Cl currents in Xenopus oocytes by Ca liberated from stores and by capacitative Ca influx. The Journal of General Physiology 108, 157-75
(1996).

Part V
122
139. Kuruma, a & Hartzell, H.C. Bimodal control of a Ca2+
-activated Cl- channel by different
Ca2+
signals. The Journal of General Physiology 115, 59-80 (2000).
140. Hartzell, C., Putzier, I. & Arreola, J. Calcium-activated chloride channels. Annual Review of Physiology 67, 719-58 (2005).
141. Gomez-Hernandez, J.M., Stühmer, W. & Parekh, A.B. Calcium dependence and distribution of calcium-activated chloride channels in Xenopus oocytes. The Journal of Physiology 502.3, 569-74 (1997).
142. Koumi, S., Sato, R. & Aramaki, T. Characterization of the calcium-activated chloride channel in isolated guinea-pig hepatocytes. The Journal of General Physiology 104, 357-
73 (1994).
143. Martin, D.K. Small conductance chloride channels in acinar cells from the rat mandibular salivary gland are directly controlled by a G-protein. Biochemical and Biophysical Research Communications 192, 1266-73 (1993).
144. Arreola, J., Melvin, J.E. & Begenisich, T. Differences in regulation of Ca2+
-activated Cl-
channels in colonic and parotid secretory cells. The American Journal of Physiology 274,
C161-6 (1998).
145. Nilius, B. et al. Kinetic and pharmacological properties of the calcium-activated chloride-current in macrovascular endothelial cells. Cell Calcium 22, 53-63 (1997).
146. Nishimoto, I. et al. Regulation of Cl- channels by multifunctional CaM kinase. Neuron 6,
547-55 (1991).
147. Holevinsky, K.O., Jow, F. & Nelson, D.J. Elevation in intracellular calcium activates both chloride and proton currents in human macrophages. The Journal of Membrane Biology 140, 13-30 (1994).
148. Chao, A.C. et al. Calcium- and CaMKII-dependent chloride secretion induced by the microsomal Ca
2+-ATPase inhibitor 2,5-di-(tert-butyl)-1,4-hydroquinone in cystic fibrosis
pancreatic epithelial cells. The Journal of Clinical Investigation 96, 1794-801 (1995).
149. Reisert, J. et al. The Ca-activated Cl channel and its control in rat olfactory receptor neurons. The Journal of General Physiology 122, 349-63 (2003).
150. Webb, D.J. & Nuccitelli, R. Fertilization potential and electrical properties of the Xenopus laevis egg. Developmental Biology 107, 395-406 (1985).
151. Mayer, M.L. A calcium-activated chloride current generates the after-depolarization of rat sensory neurones in culture. The Journal of Physiology 364, 217-39 (1985).
152. Freeman, L.C. et al. Decreased density of Ito in left ventricular myocytes from German shepherd dogs with inherited arrhythmias. Journal of Cardiovascular Electrophysiology 8, 872-83 (1997).
153. Hiraoka, M. et al. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovascular Research 40, 23-33 (1998).

References
123
154. Ridley, P.D. & Curtis, M.J. Anion manipulation: a new antiarrhythmic approach. Action of substitution of chloride with nitrate on ischemia- and reperfusion-induced ventricular fibrillation and contractile function. Circulation Research 70, 617-32 (1992).
155. Tanaka, H. et al. Use of chloride blockers: a novel approach for cardioprotection against ischemia-reperfusion damage. The Journal of Pharmacology and Experimental Therapeutics 278, 854-61 (1996).
156. Large, W.A. & Wang, Q. Characteristics and physiological role of the Ca2+
-activated Cl-
conductance in smooth muscle. The American Journal of Physiology 271, C435-54
(1996).
157. Frings, S., Reuter, D. & Kleene, S.J. Neuronal Ca2+
-activated Cl- channels--homing in
on an elusive channel species. Progress in Neurobiology 60, 247-89 (2000).
158. Jentsch, T.J. et al. Molecular structure and physiological function of chloride channels. Physiological Reviews 82, 503-68 (2002).
159. Kidd, J.F. & Thorn, P. Intracellular Ca2+
and Cl- channel activation in secretory cells.
Annual Review of Physiology 62, 493-513 (2000).
160. Hartzell, H.C. et al. Anoctamin/TMEM16 family members are Ca2+
-activated Cl-
channels. The Journal of Physiology 587, 2127-39 (2009).
161. Cunningham, S. a et al. Cloning of an epithelial chloride channel from bovine trachea. The Journal of Biological Chemistry 270, 31016-26 (1995).
162. Eggermont, J. Calcium-activated chloride channels: (un)known, (un)loved? Proceedings of the American Thoracic Society 1, 22-7 (2004).
163. Loewen, M.E. & Forsyth, G.W. Structure and function of CLCA proteins. Physiological Reviews 85, 1061-92 (2005).
164. Papassotiriou, J. et al. Ca2+
-activated Cl- channels in Ehrlich ascites tumor cells are
distinct from mCLCA1, 2 and 3. Pflügers Archiv : European Journal of Physiology 442,
273-9 (2001).
165. Suzuki, M. & Mizuno, A. A novel human Cl(-) channel family related to Drosophila flightless locus. The Journal of Biological Chemistry 279, 22461-8 (2004).
166. Suzuki, M. The Drosophila tweety family: molecular candidates for large-conductance Ca
2+-activated Cl
- channels. Experimental Physiology 91, 141-7 (2006).
167. Sun, H. et al. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proceedings of the National Academy of Sciences of the United States of America 99, 4008-13 (2002).
168. Hartzell, H.C. et al. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiological Reviews 88, 639-
72 (2008).
169. Petrukhin, K. et al. Identification of the gene responsible for Best macular dystrophy. Nature genetics 19, 241-7 (1998).

Part V
124
170. Krämer, F. et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. European Journal of Human Genetics 8, 286-92
(2000).
171. Marmorstein, A.D. & Kinnick, T.R. Focus on molecules: bestrophin (best-1). Experimental Eye Research 85, 423-4 (2007).
172. Marmorstein, A.D. et al. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proceedings of the National Academy of Sciences of the United States of America 97, 12758-63 (2000).
173. Barro-Soria, R., Schreiber, R. & Kunzelmann, K. Bestrophin 1 and 2 are components of the Ca
2+ activated Cl
- conductance in mouse airways. Biochimica et biophysica acta
1783, 1993-2000 (2008).
174. Pifferi, S. et al. Bestrophin-2 is a candidate calcium-activated chloride channel involved in olfactory transduction. Proceedings of the National Academy of Sciences of the United States of America 103, 12929-34 (2006).
175. OʼDriscoll, K.E. et al. Functional properties of murine bestrophin 1 channel. Biochemical and Biophysical Research Communications 384, 476-81 (2009).
176. Yu, K., Cui, Y. & Hartzell, H.C. The bestrophin mutation A243V, linked to adult-onset vitelliform macular dystrophy, impairs its chloride channel function. Investigative Ophthalmology & Visual Science 47, 4956-61 (2006).
177. Qu, Z. et al. Two bestrophins cloned from Xenopus laevis oocytes express Ca2+
-activated Cl
- currents. The Journal of Biological Chemistry 278, 49563-72 (2003).
178. Tsunenari, T. et al. Structure-function analysis of the bestrophin family of anion channels. The Journal of Biological Chemistry 278, 41114-25 (2003).
179. Barro Soria, R. et al. Bestrophin-1 enables Ca2+
-activated Cl- conductance in epithelia.
The Journal of Biological Chemistry 284, 29405-12 (2009).
180. Chien, L.-ting & Hartzell, H.C. Drosophila bestrophin-1 chloride current is dually regulated by calcium and cell volume. The Journal of General Physiology 130, 513-24
(2007).
181. Matchkov, V.V. et al. Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circulation Research 103, 864-72 (2008).
182. Marmorstein, L.Y. et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1). The Journal of General Physiology 127, 577-89 (2006).
183. Qu, Z., Fischmeister, R. & Hartzell, C. Mouse bestrophin-2 is a bona fide Cl- channel:
identification of a residue important in anion binding and conduction. The Journal of General Physiology 123, 327-40 (2004).

References
125
184. Fischmeister, R. & Hartzell, H.C. Volume sensitivity of the bestrophin family of chloride channels. The Journal of Physiology 562, 477-91 (2005).
185. Chien, L.-ting & Hartzell, H.C. Rescue of volume-regulated anion current by bestrophin mutants with altered charge selectivity. The Journal of General Physiology 132, 537-46
(2008).
186. Kunzelmann, K. et al. Bestrophin and TMEM16-Ca2+
activated Cl- channels with different
functions. Cell Calcium 46, 233-41 (2009).
187. Rosenthal, R. et al. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca
2+ channels in retinal pigment epithelial cells. The
FASEB Journal 20, 178-80 (2006).
188. Reichhart, N. et al. Effect of bestrophin-1 on L-type Ca2+
channel activity depends on the Ca
2+ channel beta-subunit. Experimental Eye Research (2010).
189. Wu, J. et al. Voltage-dependent calcium channel CaV1.3 subunits regulate the light peak of the electroretinogram. Journal of neurophysiology 97, 3731-5 (2007).
190. Barro-Soria, R. et al. ER-localized bestrophin 1 activates Ca2+
-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflügers Archiv : European Journal of Physiology 459, 485-97 (2010).
191. Neussert, R. et al. The presence of bestrophin-1 modulates the Ca2+
recruitment from Ca
2+ stores in the ER. Pflügers Archiv : European Journal of Physiology 460, 163-75
(2010).
192. Janssen, L.J. Ionic mechanisms and Ca2+
regulation in airway smooth muscle contraction: do the data contradict dogma? American Journal of Physiology. Lung Cellular and Molecular Physiology 282, L1161-78 (2002).
193. Aldehni, F. et al. Bestrophin 1 promotes epithelial-to-mesenchymal transition of renal collecting duct cells. Journal of the American Society of Nephrology : JASN 20, 1556-64
(2009).
194. Spitzner, M. et al. Eag1 and Bestrophin 1 are up-regulated in fast-growing colonic cancer cells. The Journal of Biological Chemistry 283, 7421-8 (2008).
195. Klimmeck, D. et al. Bestrophin 2: an anion channel associated with neurogenesis in chemosensory systems. The Journal of comparative neurology 515, 585-99 (2009).
196. Kunzelmann, K. & Mall, M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiological Reviews 82, 245-89 (2002).
197. Qu, Z. & Hartzell, H.C. Bestrophin Cl-
channels are highly permeable to HCO3-. American Journal of Physiology. Cell Physiology 294, C1371-7 (2008).
198. Yu, K. et al. Bestrophin-2 mediates bicarbonate transport by goblet cells in mouse colon. The Journal of Clinical Investigation 120, 1722-35 (2010).
199. Kranjc, A. et al. Regulation of bestrophins by Ca2+
: a theoretical and experimental study. PloS one 4, e4672 (2009).

Part V
126
200. Caputo, A. et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590-4 (2008).
201. Schroeder, B.C. et al. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019-29 (2008).
202. Yang, Y.D. et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210-5 (2008).
203. Katoh, M. & Katoh, M. FLJ10261 gene, located within the CCND1-EMS1 locus on human chromosome 11q13, encodes the eight-transmembrane protein homologous to C12orf3, C11orf25 and FLJ34272 gene products. International Journal of Oncology 22,
1375-81 (2003).
204. Rock, J.R. et al. Identification of genes expressed in the mouse limb using a novel ZPA microarray approach. Gene expression patterns : GEP 8, 19-26 (2007).
205. Rock, J.R. & Harfe, B.D. Expression of TMEM16 paralogs during murine embryogenesis. Developmental Dynamics 237, 2566-74 (2008).
206. Rock, J.R., Futtner, C.R. & Harfe, B.D. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Developmental Biology 321,
141-9 (2008).
207. Katoh, M. & Katoh, M. Identification and characterization of TMEM16E and TMEM16F genes in silico. International Journal of Oncology 24, 1345-9 (2004).
208. Katoh, M. & Katoh, M. Identification and characterization of TMEM16H gene in silico. International Journal of Molecular Medicine 15, 353-8 (2005).
209. Tsutsumi, S. et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). American Journal of Human Genetics 74,
1255-61 (2004).
210. Katoh, M. & Katoh, M. GDD1 is identical to TMEM16E, a member of the TMEM16 family. American Journal of Human Genetics 75, 927-8; author reply 928-9 (2004).
211. Ferrera, L. et al. Regulation of TMEM16A chloride channel properties by alternative splicing. The Journal of Biological Chemistry 284, 33360-8 (2009).
212. Kiessling, A. et al. D-TMPP: a novel androgen-regulated gene preferentially expressed in prostate and prostate cancer that is the first characterized member of an eukaryotic gene family. The Prostate 64, 387-400 (2005).
213. Carneiro, A. et al. Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC cancer 8, 98 (2008).
214. Carles, A. et al. Head and neck squamous cell carcinoma transcriptome analysis by comprehensive validated differential display. Oncogene 25, 1821-31 (2006).
215. Espinosa, I. et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. The American Journal of Surgical Pathology 32, 210-8 (2008).

References
127
216. Miwa, S. et al. Mutation assay of the novel gene DOG1 in gastrointestinal stromal tumors (GISTs). Journal of Gastroenterology 43, 531-7 (2008).
217. Manoury, B., Tamuleviciute, A. & Tammaro, P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. The Journal of Physiology 588, 2305-14 (2010).
218. Pardo, L.A. Voltage-gated potassium channels in cell proliferation. Physiology 19, 285-
92 (2004).
219. Koehl, G.E. et al. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APC(Min/+) mice. Oncogene 29, 1553-60 (2010).
220. Kunzelmann, K. Ion channels and cancer. The Journal of Membrane Biology 205, 159-
73 (2005).
221. Kalay, E. et al. A novel locus for autosomal recessive nonsyndromic hearing impairment, DFNB63, maps to chromosome 11q13.2-q13.4. Journal of Molecular Medicine 85, 397-
404 (2007).
222. Rock, J.R. et al. Transmembrane protein 16A (TMEM16A) is a Ca2+
-regulated Cl-
secretory channel in mouse airways. The Journal of Biological Chemistry 284, 14875-80
(2009).
223. Mall, M. et al. Modulation of Ca2+
-activated Cl- secretion by basolateral K
+ channels in
human normal and cystic fibrosis airway epithelia. Pediatric research 53, 608-18 (2003).
224. Kunzelmann, K., Schreiber, R. & Boucherot, A. Mechanisms of the inhibition of epithelial Na
+ channels by CFTR and purinergic stimulation. Kidney international 60, 455-61
(2001).
225. Almaça, J. et al. Functional Genomics Assays to Study CFTR Traffic and ENaC Function. Diagnosis and Protocols, Volume II: Methods and Resources to Understand Cystic Fibrosis (2011).
226. Tarran, R., Button, B. & Boucher, R.C. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annual Review of Physiology 68, 543-61
(2006).
227. Moody, M. et al. Inositol polyphosphate derivative inhibits Na+ transport and improves
fluid dynamics in cystic fibrosis airway epithelia. American Journal of Physiology. Cell Physiology 289, C512-20 (2005).
228. Glover, D.M. DNA cloning: a practical approach, Volume 1. DNA cloning: a practical approach (Oxford University Press: 1995).
229. Bebok, Z. et al. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. The Journal of Physiology 569, 601-15 (2005).
230. Stoker, M. & Macpherson, I. Syrian hamster fibroblast cell line BHK21 and its derivates. Nature 203, 1355-7 (1964).

Part V
128
231. Ambesi-Impiombato, F.S. Culture of Hormone-Dependent Functional Epithelial Cells from Rat Thyroids. Proceedings of the National Academy of Sciences 77, 3455-3459
(1980).
232. Shu, X. et al. Novel chromophores and buried charges control color in mFruits. Biochemistry 45, 9639-47 (2006).
233. Galietta, L.J.V., Haggie, P.M. & Verkman, A. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Letters 499, 220–224 (2001).
234. Melvin, J.E. et al. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annual review of physiology 67, 445-69 (2005).
235. Petersen, O.H. & Tepikin, A.V. Polarized calcium signaling in exocrine gland cells. Annual Review of Physiology 70, 273-99 (2008).
236. Boucher, R.C. Regulation of airway surface liquid volume by human airway epithelia. Pflügers Archiv : European Journal of Physiology 445, 495-8 (2003).
237. Choi, J.Y. et al. Synergistic airway gland mucus secretion in response to vasoactive intestinal peptide and carbachol is lost in cystic fibrosis. The Journal of Clinical Investigation 117, 3118-27 (2007).
238. Sato, K. & Sato, F. Defective beta adrenergic response of cystic fibrosis sweat glands in vivo and in vitro. The Journal of Clinical Investigation 73, 1763-71 (1984).
239. Milenkovic, V.M. et al. Functional assembly and purinergic activation of bestrophins. Pflügers Archiv : European Journal of Physiology 458, 431-41 (2009).
240. Kunzelmann, K., Schreiber, R. & Cook, D. Mechanisms for the inhibition of amiloride-sensitive Na
+ absorption by extracellular nucleotides in mouse trachea. Pflügers Archiv :
European Journal of Physiology 444, 220-6 (2002).
241. Faria, D., Schreiber, R. & Kunzelmann, K. CFTR is activated through stimulation of purinergic P2Y2 receptors. Pflügers Archiv : European Journal of Physiology 457, 1373-80 (2009).
242. Wine, J.J. Parasympathetic control of airway submucosal glands: central reflexes and the airway intrinsic nervous system. Autonomic Neuroscience 133, 35–54 (2007).
243. Paradiso, a M., Ribeiro, C.M. & Boucher, R.C. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. The Journal of General Physiology 117, 53-
67 (2001).
244. Wei, L. et al. Interaction between calcium-activated chloride channels and the cystic fibrosis transmembrane conductance regulator. Pflügers Archiv : European Journal of Physiology 438, 635-41 (1999).
245. Kunzelmann, K. et al. The cystic fibrosis transmembrane conductance regulator attenuates the endogenous Ca
2+ activated Cl
- conductance of Xenopus oocytes.
Pflügers Archiv : European Journal of Physiology 435, 178-81 (1997).

References
129
246. Kunzelmann, K. The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Reviews of Physiology, Biochemistry and Pharmacology 137, 1-70 (1999).
247. Ribeiro, C.M.P. et al. Chronic airway infection/inflammation induces a Ca2+
i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. The Journal of Biological Chemistry 280, 17798-806 (2005).
248. Bartoszewski, R. et al. Activation of the unfolded protein response by deltaF508 CFTR. American Journal of Respiratory and Critical Care Medicine 39, 448-57 (2008).
249. Ribeiro, C.M.P. The role of intracellular calcium signals in inflammatory responses of polarised cystic fibrosis human airway epithelia. Drugs in R&D 7, 17-31 (2006).
250. Martino, M.E.B. et al. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+
store expansion is mediated by X-box binding protein-1. The Journal of Biological Chemistry 284, 14904-13 (2009).
251. Hybiske, K. et al. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca
2+ of airway epithelia.
American Journal of Physiology. Lung Cellular and Molecular Physiology 293, L1250-60
(2007).
252. Nanua, S. et al. Absence of typical unfolded protein response in primary cultured cystic fibrosis airway epithelial cells. Biochemical and Biophysical Research Communications 343, 135-43 (2006).
253. Antigny, F. et al. Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium 43, 175-83 (2008).
254. Perez, A. et al. CFTR inhibition mimics the cystic fibrosis inflammatory profile. American Journal of Physiology. Lung Cellular and Molecular Physiology 292, L383-95 (2007).
255. Barro-Soria, R., Schreiber, R. & Kunzelmann, K. Bestrophin 1 and 2 are components of the Ca
2+ activated Cl
- conductance in mouse airways. Biochimica et biophysica acta
1783, 1993-2000 (2008).
256. Ousingsawat, J. et al. Loss of TMEM16A causes a defect in epithelial Ca2+
-dependent chloride transport. The Journal of Biological Chemistry 284, 28698-703 (2009).
257. Yang, D. et al. IRBIT coordinates epithelial fluid and HCO3- secretion by stimulating the
transporters pNBC1 and CFTR in the murine pancreatic duct. The Journal of Clinical Investigation 119, 193-202 (2009).
258. Ando, H. et al. IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Molecular Cell 22, 795-806 (2006).
259. Sammels, E. et al. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+
release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. The Journal of Biological Chemistry 285, 18794-805 (2010).
260. Jacquot, J. et al. Airway epithelial cell inflammatory signalling in cystic fibrosis. The International Journal of Biochemistry & Cell Biology 40, 1703-15 (2008).

Part V
130
261. Rottner, M. et al. Exaggerated apoptosis and NF-kappaB activation in pancreatic and tracheal cystic fibrosis cells. The FASEB Journal 21, 2939-48 (2007).
262. Koehler, D.R. et al. Lung inflammation as a therapeutic target in cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology 31, 377-81 (2004).
263. Vij, N., Mazur, S. & Zeitlin, P.L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PloS one 4, e4664 (2009).
264. Becker, M.N. et al. Cytokine secretion by cystic fibrosis airway epithelial cells. American Journal of Respiratory and Critical Care Medicine 169, 645-53 (2004).
265. Rogan, M.P. et al. Pigs and humans with cystic fibrosis have reduced insulin-like growth factor 1 (IGF1) levels at birth. Proceedings of the National Academy of Sciences of the United States of America 107, 20571-5 (2010).
266. Galietta, L.J.V. et al. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. Journal of Immunology 168, 839-45 (2002).
267. Grubb, B.R. et al. SERCA pump inhibitors do not correct biosynthetic arrest of deltaF508 CFTR in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 34,
355-63 (2006).
268. Rosser, M.F.N. et al. Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Molecular Biology of the Cell 19, 4570-9 (2008).
269. Glozman, R. et al. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. The Journal of Cell Biology 184, 847-62
(2009).
270. Farinha, C.M. & Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Molecular and Cellular Biology 25,
5242-52 (2005).
271. Berridge, M.J. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235-49 (2002).
272. Kourie, J.I. ATP-sensitive voltage- and calcium-dependent chloride channels in sarcoplasmic reticulum vesicles from rabbit skeletal muscle. The Journal of Membrane Biology 157, 39-51 (1997).
273. Coonan, J.R. & Lamb, G.D. Effect of chloride on Ca2+
release from the sarcoplasmic reticulum of mechanically skinned skeletal muscle fibres. Pflügers Archiv : European Journal of Physiology 435, 720-30 (1998).
274. Hirota, S. et al. Intracellular Cl- fluxes play a novel role in Ca
2+ handling in airway smooth
muscle. American Journal of Physiology. Lung Cellular and Molecular Physiology 290,
L1146-53 (2006).
275. Janssen, L.J. Asthma therapy: how far have we come, why did we fail and where should we go next? The European Respiratory Journal 33, 11-20 (2009).

References
131
276. Leigh, M.W. et al. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. American Journal of Respiratory Cell and Molecular Biology 12,
605-12 (1995).
277. Hajj, R. et al. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. The Journal of Pathology 211, 340-50 (2007).
278. Cystic Fibrosis Foundation - FAQs About VX-809. at http://www.cff.org/research/ClinicalResearch/FAQs/VX-809/
279. Cystic Fibrosis Foundation - FAQs About VX-770 and VX-809. at http://www.cff.org/research/ClinicalResearch/FAQs/VX-770/
280. Case, M. Physiology. Chloride ions and cystic fibrosis. Nature 322, 407
281. Frizzell, R. a, Rechkemmer, G. & Shoemaker, R.L. Altered regulation of airway epithelial cell chloride channels in cystic fibrosis. Science 233, 558-60 (1986).
282. Welsh, M.J. & Liedtke, C.M. Chloride and potassium channels in cystic fibrosis airway epithelia. Nature 322, 467-70 (1986).
283. Grubb, B.R. & Boucher, R.C. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiological Reviews 79, S193-214 (1999).
284. Zeiher, B.G. et al. A mouse model for the delta F508 allele of cystic fibrosis. The Journal of Clinical Investigation 96, 2051-64 (1995).
285. Reddy, M.M., Sun, D. & Quinton, P.M. Apical heterotrimeric g-proteins activate CFTR in the native sweat duct. The Journal of Membrane Biology 179, 51-61 (2001).
286. Reddy, M.M. & Quinton, P.M. cAMP-independent phosphorylation activation of CFTR by G proteins in native human sweat duct. American Journal of Physiology. Cell Physiology 280, C604-13 (2001).
287. Ishibashi, K., Okamura, K. & Yamazaki, J. Involvement of apical P2Y2 receptor-regulated CFTR activity in muscarinic stimulation of Cl(-) reabsorption in rat submandibular gland. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 294, R1729-36 (2008).
288. Namkung, W., Finkbeiner, W.E. & Verkman, A.S. CFTR-adenylyl cyclase I association responsible for UTP activation of CFTR in well-differentiated primary human bronchial cell cultures. Molecular Biology of the Cell 21, 2639-48 (2010).
289. Meyerholz, D.K. et al. Loss of CFTR Function Produces Abnormalities in Tracheal Development in Neonatal Pigs and Young Children. American Journal of Respiratory and Critical Care Medicine 1-60 (2010).
290. Schreiber, R., Castrop, H. & Kunzelmann, K. Allergen-induced airway hyperresponsiveness is absent in ecto-5ʼ-nucleotidase (CD73)-deficient mice. Pflügers Archiv : European Journal of Physiology 457, 431-40 (2008).

Part V
132
291. Pasyk, E.A. & Foskett, J.K. Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl
- channel is functional when retained in endoplasmic reticulum
of mammalian cells. The Journal of Biological Chemistry 270, 12347-50 (1995).
292. Ayoub, C. et al. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. British Journal of Cancer 103, 715-26 (2010).
293. Kellerman, D. et al. Denufosol: a review of studies with inhaled P2Y(2) agonists that led to Phase 3. Pulmonary Pharmacology & Therapeutics 21, 600-7 (2008).
294. Kunzelmann, K. & Mall, M. Pharmacotherapy of the ion transport defect in cystic fibrosis: role of purinergic receptor agonists and other potential therapeutics. American Journal of Respiratory Medicine : Drugs, Devices, and Other Interventions 2, 299-309 (2003).
295. Shears, S.B. Can intervention in inositol phosphate signalling pathways improve therapy for cystic fibrosis? Expert Opinion on Therapeutic targets 9, 1307-17 (2005).
296. Perl, A.-K.T. et al. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proceedings of the National Academy of Sciences of the United States of America 99, 10482-7 (2002).
297. Johns Hopkins CF Center | What is CF? at http://www.hopkinscf.org/main/whatiscf/index.html