can orbital-free density functional theory simulate molecules? · the journal of chemical physics...
TRANSCRIPT

Can orbital-free density functional theory simulate molecules?Junchao Xia, Chen Huang, Ilgyou Shin, and Emily A. Carter Citation: J. Chem. Phys. 136, 084102 (2012); doi: 10.1063/1.3685604 View online: http://dx.doi.org/10.1063/1.3685604 View Table of Contents: http://jcp.aip.org/resource/1/JCPSA6/v136/i8 Published by the American Institute of Physics. Additional information on J. Chem. Phys.Journal Homepage: http://jcp.aip.org/ Journal Information: http://jcp.aip.org/about/about_the_journal Top downloads: http://jcp.aip.org/features/most_downloaded Information for Authors: http://jcp.aip.org/authors

THE JOURNAL OF CHEMICAL PHYSICS 136, 084102 (2012)
Can orbital-free density functional theory simulate molecules?Junchao Xia,1 Chen Huang,2 Ilgyou Shin,3 and Emily A. Carter1,4,a)
1Department of Mechanical and Aerospace Engineering, Princeton University,Princeton, New Jersey 08544, USA2Department of Physics, Princeton University, Princeton, New Jersey 08544, USA3Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA4Program in Applied and Computational Mathematics Princeton University, Princeton, New Jersey 08544,USA and the Gerhard R. Andlinger Center for Energy and the Environment, Princeton University, Princeton,New Jersey 08544, USA
(Received 27 November 2011; accepted 26 January 2012; published online 22 February 2012)
Orbital-free density functional theory (OFDFT), with its attractive linearly scaling computation costand low prefactor, is one of the most powerful first principles methods for simulating large systems(∼104–106 atoms). However, approximating the electron kinetic energy with density functionalslimits the accuracy and generality of OFDFT compared to Kohn-Sham density functional theory(KSDFT). In this work, we test whether the Huang-Carter (HC) kinetic energy density functional(KEDF), which contains the physics to properly describe covalently bonded semiconductor materi-als, can also be used to describe covalent bonds in molecules. In particular, we calculate a variety ofhomonuclear diatomic molecules with the HC functional within OFDFT. The OFDFT bond dissoci-ation energy, equilibrium bond length, and vibrational frequency of these dimers are in remarkablygood agreement with benchmark KSDFT results, given the lack of orbitals in the calculation. Wevary the two parameters λ (controlling the reduced density gradient contribution to the nonlocal ker-nel) and β (the exponent of the density in the nonlocal term) present in the HC KEDF and findthat the optimal λ correlates with the magnitude of the highest occupied molecular orbital - lowestunoccupied molecular orbital energy gap. Although the HC KEDF represents a significant improve-ment over previous KEDFs in describing covalent systems, deficiencies still exist. Despite the sim-ilar overall shape of the KSDFT and OFDFT ground state electron densities, the electron densitywithin the bonding region is still quite different. Furthermore, OFDFT is not yet able to give rea-sonable description of magnetic states. The energy orderings of the triplet and singlet states of Si2
and Al family dimers are not consistent with KSDFT or experimental results and the spin polariza-tion distributions also differ widely between the two theories. © 2012 American Institute of Physics.[http://dx.doi.org/10.1063/1.3685604]
I. INTRODUCTION
Among first principles quantum mechanics methods forstudying molecular and material properties, the most widelyemployed one today is density functional theory (DFT), thecornerstone of which was established by Hohenberg andKohn.1 One DFT scheme in particular, Kohn-Sham DFT(KSDFT),2 has become one of the most powerful methodsavailable due to its ability to deliver quite accurate predic-tions at a reasonable computational cost. However, the orbitalorthonormalization and k-point sampling required in KSDFTmakes the computation scale as the cube of the system size N,often with a large prefactor. Although a number of linear scal-ing KSDFT algorithms exist,3–8 they generally only becomelinear scaling above ∼100 atoms due to a large algorithmicprefactor and are usually applicable only for nonmetallic sys-tems. Even though a couple of metallic/semi-metallic systemshave been studied with linear scaling KSDFT,8 a consider-able buffer size is required to reach an accuracy comparableto conventional KSDFT methods. As a result, the prefactorof the linear scaling algorithm can become very large, which
a)Author to whom correspondence should be addressed. Electronic mail:[email protected].
is proportional to the KSDFT computation cost in each sub-domain. The prefactor will be even larger if k-point samplingis needed when periodic boundary conditions are applied tosub-domains. As a result, in practice it is still prohibitive tocarry out KSDFT simulations to answer interesting scientificquestions if the sample size is much beyond a few hundredatoms, even with access to supercomputing power.
An alternative DFT scheme, orbital-free DFT (OFDFT),9
demonstrates promising competence in simulating muchlarger numbers of atoms.10–13 Because the only variable inOFDFT is the density distribution of electrons, the numberof degrees of freedom is reduced from 3N (in the case of Norbitals) to only 3 (for the electron density). This tremen-dous simplification allows the computation to scale quasi-linearly with the system size (O(NlnN)). In contrast to thelinear scaling KSDFT methods, OFDFT has a much smallerprefactor,10 and shows excellent accuracy and efficiency whensimulating metallic systems.10–15 OFDFT simulations of sam-ple sizes containing thousands and even 104 atoms are nowroutine,11, 14, 15 with benchmark calculations performed formore than 106 atoms, on a modest number of processors.10
As usual, however, a tradeoff exists between efficiencyand accuracy. Despite its attractive quasilinear scaling cost,
0021-9606/2012/136(8)/084102/13/$30.00 © 2012 American Institute of Physics136, 084102-1

084102-2 Xia et al. J. Chem. Phys. 136, 084102 (2012)
OFDFT is thus far less accurate than and not as generallyapplicable as KSDFT. The inaccuracy compared to KSDFToriginates from two sources. Unlike KSDFT in which orbital-based nonlocal pseudopotentials (NLPSs) can be employed toaccount for the interaction between electrons and ions (nucleiplus their core electrons),16–18 the lack of orbitals in OFDFTmeans the only option is to use a local pseudopotential (LPS),which provides much less flexibility. However, some LPSshave demonstrated excellent accuracy and transferability,19–21
even for a transition metal,22 when compared to NLPSs, sug-gesting that inaccuracies in the electron-ion term in principlecan be overcome for OFDFT. Instead, the major source of er-ror in OFDFT lies in describing the non-interacting electronkinetic energy in terms of only the electron density, using a ki-netic energy density functional (KEDF). Although the Hohen-berg and Kohn theorems1 proved the existence of a universalKEDF, they do not offer any specific details for constructingit. The exact form of the KEDF remains unknown, althoughmany approximations to it have been proposed through years.The Thomas-Fermi (TF) KEDF,23–25 which of course pre-ceded modern DFT, was the first “naïve” attempt but it is onlyexact for the non-interacting uniform electron gas and fails topredict any atomic shell structure or chemical bonding.26, 27
Inclusion of the von Weizsäcker (vW) KEDF (Refs. 28–30)(the TFλvW model) improves the TF model, but still is notaccurate for most systems. Other local or semilocal KEDFmodels, containing higher order derivatives of the density,were proposed later on but offered little improvement.31, 32
In recent decades, several nonlocal KEDFs, such as theChacón-Alvarellos-Tarazona,33–35 Wang-Teter (WT),36 andWang-Govind-Carter (WGC) (Ref. 37) KEDFs have beenproposed, which are all based on linear response theory. TheseKEDFs exhibit greatly improved accuracy compared to lo-cal or semilocal KEDFs, and demonstrated accuracy com-parable to KSDFT for nearly-free-electron-like main groupmetals.38–40 However, constructing an accurate KEDF for sys-tems other than main group metals remained elusive. For ex-ample, the aforementioned nonlocal KEDFs are mostly in-adequate for studying covalently bonded materials,41 wherevalence electrons are more localized and the linear responsebehavior is rather different from that in metallic systems.42
Very recently, the Huang-Carter (HC) KEDF was pro-posed for semiconductor materials based on the dielectricresponse of semiconductors. It exhibited remarkable accu-racy and transferability in calculating properties of silicon andgroup III/V semiconductors.43 This motivated us to considerif this HC KEDF could improve the generality of OFDFTsuch that other types of systems could be accurately treated. Inparticular, because the HC KEDF is able to treat the covalentbonds in crystals, we sought to test the validity of OFDFTin treating covalent bonds in molecules. Here we focus onhomonuclear diatomics (dimers) so as to study purely cova-lent bonds, though one example of a heteronuclear diatomicis also provided.
Despite their seeming simplicity, quantitative treatmentof dimers remains a huge challenge for OFDFT. To ourknowledge, very few studies44 have been done on moleculedissociation using self-consistent OFDFT calculations. Somehave employed Hartree-Fock or KSDFT densities to evaluate
non-self-consistent OFDFT bond energies in small moleculessuch as F2 and the CH4 molecule.45–47 All of them, how-ever, demonstrated unsatisfactory OFDFT results due to in-accurate KEDFs such as those based on a gradient expansion.On the other hand, the nonlocal KEDFs proposed for nearly-free-electron-like metals based on the perturbed uniform elec-tron gas are also not appropriate for describing molecules.A simple dimer is even more difficult for OFDFT to treatthan semiconductor crystals, because multiple covalent bondscan be involved and spin-polarized calculations are also re-quired for the open-shell ground states of some dimers. Inthe present work, a variety of dimers are calculated utilizingOFDFT with the HC KEDF. Via comparison with KSDFT re-sults, we demonstrate that OFDFT can treat dimer moleculesremarkably well given the lack of a wavefunction, althoughOFDFT clearly still has remaining defects to be rectified.
The rest of this paper is structured as follows. InSecs. II and III, the formalisms and numerical details aregiven. Then results for properties of different kinds of dimersare presented, including equilibrium bond lengths, bond dis-sociation energies, and vibrational frequencies. Predictionsmade with the HC and other semilocal and nonlocal KEDFsare compared to KSDFT benchmarks. Ground state densitydistributions are also analyzed and compared with KSDFTdensities. The sensitivity of the results to the parameters inthe HC KEDF is also presented and discussed. We end witha discussion of prospects for future improvements.
II. FORMALISM
The Hohenberg-Kohn theorems1 state that the electronictotal energy can be expressed as a functional of the electrondensity alone:
E[n] = Ts[n] + J [n] + Exc[n] + Eext [n], (1)
where n is the total electron density, Ts is the non-interactingelectron kinetic energy, J is the Hartree electron repulsionenergy, and Exc is the exchange-correlation energy. The lastterm, Eext, is the energy related to external fields, such as ionic(or nuclear) potentials. This Hohenberg-Kohn energy func-tional in Eq. (1) is precisely the OFDFT energy functional.
A number of the dimers have ground states that are open-shell multiplets. To treat them, we must extend the OFDFTformalism to allow for spin polarization. The total OFDFTenergy functional including spin polarization is simply writ-ten as
E[nup, ndown] = Ts[nup, ndown] + J [nup, ndown]
+Exc[nup, ndown] + Eext [nup, ndown],
(2)
where nup and ndown are the densities of spin up and spindown electrons, respectively. The Hartree and external en-ergies generally remain the same as in the spin-unpolarizedformalism, though local spin-dependent electron-ion poten-tials could be used.48 The exchange-correlation energy canbe evaluated within local spin density approximation (LSDA)(Refs. 49–52) or spin-polarized generalized gradient approx-imation (GGA) functionals.53 The non-interacting kinetic

084102-3 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
energy is evaluated through the equation,54
Ts[nup, ndown] = 1
2Ts[2nup] + 1
2Ts[2ndown]. (3)
The total energy is variationally minimized subject to the con-straint that the total number of electrons remains fixed. Theproblem of finding a minimum with a constraint can be con-verted to a problem of finding a stationary point by means ofLagrange multipliers:
δ
{E[n] − μ
(∫n(r)dr − N
)}= 0, (4)
where N is the total electron number and μ is the chemicalpotential. For spin-polarized and fixed magnetization calcula-tions, we simply generalize to
δ
{E[nup, ndown] − μup
(∫nup(r)dr − Nup
)
−μdown
(∫ndown(r)dr − Ndown
)}= 0, (5)
where Nup and Ndown are the spin up and spin down electronnumbers, and μup and μdown are the chemical potentials forspin up and spin down electrons, respectively. Our optimiza-tion algorithm is implemented similarly to that described inRef. 55, except that in each iteration the optimization direc-tions θα and θβ are simultaneously optimized to minimizethe total energy via a two-dimensional conjugate gradientsearch instead of the approximation introduced in the origi-nal paper.55
III. NUMERICAL DETAILS
The OFDFT calculations are performed with a modifiedversion of our PROFESS 2.0 code56, 57 and the benchmark KS-DFT calculations are carried out with the ABINIT code.58
In both OFDFT and KSDFT calculations, bulk-derived lo-cal pseudopotentials (BLPSs) are used. For all local densityapproximation (LDA) BLPSs except that of lithium, we em-ploy previously reported ones,21, 43 while the lithium LDABLPS and all GGA BLPSs were generated as described inthe literature;21 details are given in the Appendix. For theexchange-correlation functionals, the Perdew-Zunger (PZ)(Refs. 49, 50, and 52) form of the LSDA and the Perdew-Burke-Ernzerhof (PBE) (Ref. 53) form of the GGA is em-ployed in all calculations.
As mentioned earlier, our focus is on testing the HCKEDF for use in molecular simulations. The HC KEDF pos-sesses a similar form to some previous nonlocal KEDFs:36, 37
Ts[n] = TTF[n] + TvW[n] + TNL[n], (6)
TTF[n] = CTF
∫n(r)5/3dr, (7)
TvW[n] = 1
8
∫ ∇n(r) · ∇n(r)
n(r)dr, (8)
TNL[n] = C
∫∫n(r)8/3−βω(r, r′)n(r′)βdrdr′, (9)
where TTF is the TF KEDF with CTF
= 3/10(3π2)2/3; TvW is the vW KEDF, and TNL is thenonlocal term, which includes a single-density-dependentkernel:
ω(r, r′) = ω[ξ (r, r′)|r − r′|], (10)
ξ (r, r′) = kF (r)(1 + λs(r)2), (11)
s(r) = |∇n(r)|n(r)4/3
, (12)
−βηω̃(η)′ + (5 − 3β)βω̃(η) = 5
3[F (η) − 3η2 − 1], (13)
where kF is the Fermi wave vector, kF(r) = (3π2n(r))1/3.The kernel can be solved for numerically in reciprocal spaceaccording to Eq. (13), where η is a dimensionless momen-tum vector, η = q/(2kF). For the parameters λ and β in theHC KEDF, selected values in the literature43 (λ = 0, 0.01,and 0.01177, β = 0.65 and 0.7143) are employed. For somedimers, the parameters are tuned slightly to achieve betterresults. We also compare the accuracy of the HC KEDF tothe TF1/5vW,29, 59 TF1/9vW,31 WGC,37 and WT (Ref. 36)KEDFs. When using WGC and WT KEDFs, an average elec-tron density needs to be specified, which is not well definedfor molecules. Here we first obtain the ground state densityfrom KSDFT calculations at the equilibrium bond length ofeach dimer. The average density is then determined by av-eraging the total electron density in the bonding region be-tween nuclei, where the density is greater than 10−2 a.u. Thekinetic energy cutoff for the plane-wave basis is selected sothat the total energy is converged to within 1 meV/atom.KSDFT calculations all employ a 900 eV kinetic energy cut-off and OFDFT calculations use a kinetic energy cutoff of1600 eV. In each total energy vs. bond length curve, the ini-tial guess for the wavefunctions in KSDFT calculations is setto be the default in the ABINIT code for the first bond length,usually 1.8 Å. Upon increase of the bond length, for each ofthe subsequent bond length calculations, the initial guess istaken as the resulting wavefunctions from the previous (onestep smaller) bond length calculation. In OFDFT calculations,the initial guess of the density is always taken as the uniformaverage density of the system (defined as discussed above).
To test the quality of OFDFT for treating diatomicmolecules, different kinds of dimers including Al2, Ga2, In2,Si2, P2, As2, Sb2, Li2, and Mg2 are considered. A heteronu-clear diatomic molecule, AlP, is also examined. For each ofthem, the equilibrium bond length (re), vibrational frequency(ωe) and bond dissociation energy (D0) is computed and thencompared with KSDFT results. In both OFDFT and KSDFTcalculations, the periodic cell is set to be 20 × 10 × 10 Å, withthe dimer in the center of the cell aligned in the longest direc-tion, which guarantees the distance between nearest images islarger than 10 Å for all bond lengths considered. In generating

084102-4 Xia et al. J. Chem. Phys. 136, 084102 (2012)
the total energy versus bond length curves, the dimer is keptin the center while the distance between two atoms is varied.The magnetization is fixed to be 0 in the singlet calculationsand 2 in the triplet calculations. Around the total energy mini-mum, a ±0.01 Å region is employed to perform quadratic fit-ting so as to calculate the equilibrium bond length re and thevibrational frequency ωe, which is then used to calculate thezero-point energy, �E0 = hωe/4π . To calculate the bond dis-sociation energy, the difference between the total energies atthe equilibrium bond length and the dissociation limit (bondlength equal to 10 Å) is first computed, and then we subtractthe zero-point energy to obtain the D0 values reported. Singleatom energies are also calculated, with a 10 × 10 × 10 Å pe-riodic cell. The resultant energy is then multiplied by two toobtain the full dissociation limit energy.
IV. RESULTS AND DISCUSSION
A. Energy analysis
Table I lists results for Al2, Ga2, and In2, for which thetriplet state is the ground state for all three dimers; the ex-act ground state electron configuration has been under debatefor a long time, with two candidates 3�u and 3 g
− nearlydegenerate.60–72 Some recent experiments and calculations fa-vor 3�u as the ground state.62–65, 69, 70 Here we obtained a3 g
− ground state for Al2 and Ga2 with both LSDA and spin-polarized GGA exchange-correlation functionals, consistentwith some previous KS-DFT calculations.66, 68 For In2, LSDAcalculations predict a 3�u ground state while spin-polarizedGGA calculations give a 3 g
− ground state. The table alsoincludes singlet state results for comparison. In the electronconfiguration of the singlet state, one spin up and one spin
TABLE I. OFDFT, KSDFT, and experimental bond dissociation energies(D0), equilibrium bond lengths (re), and vibrational frequencies (ωe) for Al2,Ga2, and In2 in singlet (Ms = 0) and triplet (Ms = 1) states. λ and β arethe parameters in the HC KEDF (see text). The optimal λ = 0 for all threemolecules. OFDFT values are listed first while KSDFT values are listed inparentheses.
D0 re ωe
Dimer β (eV) (Å) (cm−1)
Expt. Al2 (Ms = 1) . . . 1.55a 2.466a 350a
Ga2 (Ms = 1) . . . 1.40a 180b
In2 (Ms = 1) . . . 1.01a 118b
LDA Al2 (Ms = 0) 0.51 1.72 (1.74) 2.498 (2.473) 285 (346)Al2 (Ms = 1) 0.7143 1.70 (2.00) 2.489 (2.464) 324 (351)Ga2 (Ms = 0) 0.51 1.67 (1.69) 2.329 (2.323) 208 (212)Ga2 (Ms = 1) 0.7143 1.64 (1.96) 2.312 (2.312) 211 (216)In2 (Ms = 0) 0.51 1.64 (1.64) 2.669 (2.644) 139 (154)In2 (Ms = 1) 0.7143 1.65 (1.87) 2.661 (2.633) 148 (157)
GGA Al2 (Ms = 0) 0.51 1.50 (1.49) 2.543 (2.497) 309 (328)Al2 (Ms = 1) 0.7143 1.49 (1.80) 2.519 (2.490) 321 (343)Ga2 (Ms = 0) 0.51 1.29 (1.28) 2.447 (2.422) 165 (192)Ga2 (Ms = 1) 0.7143 1.25 (1.60) 2.431 (2.411) 175 (193)In2 (Ms = 0) 0.7143 1.15 (1.18) 2.775 (3.131) 102 (89)In2 (Ms = 1) 0.7143 1.05 (1.31) 2.798 (3.080) 100 (94)
aReference 71.bReference 72.
down electron occupies each of the π -orbitals, respectively,resulting in an open-shell singlet. However, due to the singledeterminant nature of KSDFT, it actually cannot be consid-ered as a true 1�g state.
The parameters in the HC KEDF in Table I were ad-justed to generate reasonable D0 values, while re changes lit-tle with these parameter alterations. For these three dimers,the HC KEDF parameters can be adjusted such that OFDFTD0 results are in remarkably good agreement with the KS-DFT benchmarks. In all cases, λ = 0 gave the best results,while β = 0.51 or 0.7143 were optimal. For singlet states, theD0 difference between OFDFT and KSDFT is usually lessthan 0.03 eV, while for triplet states, the difference is a littlelarger, about 0.3 eV. The re values computed by OFDFT arevery close to the KSDFT values, with deviations usually lessthan 0.05 Å (except for the GGA results of In2). The OFDFTvibrational frequencies are also in a reasonable range aroundthe KSDFT values. We observe the usual overbinding by LDAin both KSDFT and OFDFT results: LDA generally predictslarger D0, smaller re, and larger ωe than GGA does. Remark-ably, OFDFT-GGA predicts D0, re, and ωe within 0.15 eV,0.05 Å, and 30 cm−1 of experiment, respectively.
To put this level of agreement in perspective, Table IIcompares the ability of different KEDFs used within OFDFTto predict D0. TF KEDF results are not listed because (as ex-pected) it did not produce bonding for any dimer. Also, WGCKEDF results could not be obtained because the evaluation ofthe WGC KEDF via Taylor expansion for highly fluctuatingdensities makes the calculation unstable and hard to converge.Although all the other KEDFs examined predict bound states,the TFλvW KEDFs greatly underbind the molecules while theWT KEDF greatly overbinds them. The HC KEDF is the onlyone that can obtain near quantitative agreement with KSDFTfor covalent bond energies, albeit without a universal param-eter set (optimal λ or β varies somewhat).
Despite this achievement, Fig. 1 reveals the first clue ofremaining flaws in our formalism. The KSDFT and OFDFTpotential energy curves are compared in Fig. 1 for the twodifferent spin states of Al2. KSDFT (black and red squares)predicts the triplet state to be lower in energy than the sin-glet state, consistent with experiment. OFDFT, independentof KEDF used, predicts the reverse energy ordering, with the
TABLE II. D0 values of Al2, Ga2, In2, and Si2 in singlet (Ms = 0) andtriplet (Ms = 1) states calculated by KSDFT, OFDFT with HC, TFλvW(λ = 1/5 and 1/9) and WT KEDFs. All values are in eV. LDA exchange-correlation is used.
OFDFT OFDFT OFDFT OFDFTDimer KSDFT (HC) (TF1/5vW) (TF1/9vW) (WT)
Al2 (Ms = 0) 1.74 1.72 0.73 0.36 7.99Al2 (Ms = 1) 2.00 1.70 0.64 0.29 13.44Ga2 (Ms = 0) 1.69 1.67 0.63 0.27 10.24Ga2 (Ms = 1) 1.96 1.64 0.52 0.21 16.73In2 (Ms = 0) 1.64 1.64 0.63 0.29 6.66In2 (Ms = 1) 1.87 1.65 0.56 0.24 12.60Si2 (Ms = 0) 4.59 4.86 0.84 0.33 13.82Si2 (Ms = 1) 4.66 4.66 0.73 0.26 28.11

084102-5 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
FIG. 1. Total energy vs. bond length (r) curves for singlet and triplet Al2from KSDFT and OFDFT with the TF1/9vW KEDF and the HC KEDF (λ= 0 and β = 0.65). All calculations use the LDA exchange-correlation func-tional. The KSDFT curves exhibit a slight jump just beyond re when the en-ergy ordering of the σ - and π -MOs flips, while the OFDFT curves are smootheverywhere.
triplet state much higher in energy than the singlet state. Herewe used for both spin states a value of β intermediate betweenthe two optimal values in the HC KEDF. Adjusting β over alarge range (0.51–1.0) did not change the ordering of the twospin states. Other KEDFs such as TF1/9vW produces similarenergy orderings, as shown in Fig. 1. The origin of this inac-curacy will be discussed further when analyzing the electrondensity distribution in Sec. IV B.
The situation is quite similar for Si2. The ground state isalso a triplet (3 g
−) (Ref. 71) state. Table III displays the pre-dicted properties for Si2 and Fig. 2 provides the correspond-ing potential energy curves. Again, the OFDFT HC KEDFand KSDFT D0, re, and ωe values match reasonably well butOFDFT still incorrectly predicts the ground state to be sin-glet instead of triplet. Nevertheless, Table II reveals that theHC KEDF is the best among the KEDFs, with TFλvW againgreatly underbinding and WT vastly overbinding.
More attention could be paid to the choice of parametersin the HC KEDF for the Al family dimers and Si2. Wesee that zero λ is optimal for the Al family dimers whilenon-zero λ is optimal for Si dimer. In the HC KEDF paper,43
zero λ was found to be optimal for metallic systems, such
TABLE III. OFDFT, KSDFT, and experimental D0, re, and ωe for Si2 insinglet (Ms = 0) and triplet (Ms = 1) states. OFDFT values are listed firstwhile KSDFT values are listed in parentheses. λ = 0.01, β = 0.65 is used inthe HC KEDF.
D0 re ωe
Dimer (eV) (Å) (cm−1)
Expt. Si2 (Ms = 1) 3.21a 2.246a 510.98a
LDA Si2 (Ms = 0) 4.86 (4.59) 2.246 (2.284) 513 (501)Si2 (Ms = 1) 4.66 (4.66) 2.259 (2.277) 486 (509)
GGA Si2 (Ms = 0) 4.48 (3.93) 2.283 (2.309) 469 (489)Si2 (Ms = 1) 4.30 (4.15) 2.296 (2.303) 487 (492)
aReference 71.
FIG. 2. Total energy vs. bond length curves for singlet and triplet Si2 fromKSDFT and OFDFT with TF1/9vW KEDF and HC KEDF (λ = 0.01 and β
= 0.65). All calculations use the LDA exchange correlation functional.
as face-centered-cubic (fcc) bulk Al, while non-zero λ isoptimal for covalent systems, such as bulk cubic diamond(CD) Si. As it turns out, the optimal parameter set for Si2,λ = 0.01 and β = 0.65, is the same as that for bulk CDSi.43 The coincidence is interesting although these dimersare molecular rather than bulk crystals. We will return to theeffects of parameters λ and β in Sec. IV C.
Table IV shows predicted properties of P2, As2, and Sb2,which all form triply bonded singlet (1 g
+) ground states.For dimers of this family, although re does not change muchwhen the HC KEDF parameters are adjusted, zero or small λ
yields too small Do and ωe values compared to KSDFT. TheOFDFT values increase with λ, however even with λ equalto 0.03, OFDFT still underbinds these dimers, producingsmaller D0, larger re, and smaller ωe than KSDFT. Furtherincreases of λ offer little improvement, with OFDFT D0
values ∼2 eV smaller than KSDFT values. OFDFT re valuesare ∼0.2–0.3 Å larger than those of KSDFT and the vibra-tional frequencies are smaller in OFDFT than in KSDFT by∼100–200 cm−1. Despite the lack of quantitative agreement
TABLE IV. OFDFT, KSDFT, and experimental D0, re, and ωe for P2, As2,and Sb2. OFDFT values are listed first while KSDFT values are listed inparentheses. λ = 0.03, β = 0.51 is used in the HC KEDF.
D0 re ωe
Dimer (eV) (Å) (cm−1)
Expt. P2 5.03a 1.89a 781a
As2 3.96a 2.10a 430a
Sb2 3.09a 2.34a 270a
LDA P2 7.12 (9.54) 2.138 (1.942) 628 (790)As2 6.12 (8.35) 2.247 (2.032) 352 (459)Sb2 4.71 (6.65) 2.710 (2.431) 212 (283)
GGA P2 6.74 (9.38) 2.124 (1.915) 622 (812)As2 5.49 (7.62) 2.324 (2.084) 327 (425)Sb2 4.57 (5.92) 2.706 (2.459) 213 (261)
aReference 71.

084102-6 Xia et al. J. Chem. Phys. 136, 084102 (2012)
TABLE V. D0 values of P2, As2, Sb2, and Li2 calculated by KSDFT,OFDFT with HC, TFλvW (λ = 1/5 and 1/9) and WT KEDFs. All valuesare in eV. LDA exchange-correlation is used.
OFDFT OFDFT OFDFT OFDFTDimer KSDFT (HC) (TF1/5vW) (TF1/9vW) (WT)
P2 9.54 7.12 0.94 0.39 15.80As2 8.35 6.12 0.66 0.20 12.91Sb2 6.65 4.71 0.71 0.31 4.73Li2 1.45 0.97 0.33 0.24 1.61
with KSDFT, the covalent bonding physics contained in theHC KEDF yields far superior results to those provided byTFλvW and WT KEDFs, as seen in Table V. However, thetriple bond character is clearly more challenging to capture inOFDFT than the single or double bonds of group III and IVdimers.
Finally, we consider Li2 and Mg2 with results listed in Ta-ble VI. OFDFT fortuitously predicts a D0 in excellent agree-ment with experiment for Li2, but it is ∼30% smaller thanKSDFT, which it is meant to approximate. The bond lengthsare overestimated while the vibrational frequencies are under-estimated for Li2 within OFDFT, with respect to both KSDFTand experiment. For Mg2, OFDFT predicts a much larger D0
than KSDFT and experiment, which is actually not surpris-ing. Since each of the Mg atoms has two 3s electrons andforms a closed-shell structure, the Mg dimer only forms a vander Waals bond. The poor prediction of OFDFT for Mg2 isexpected because the HC KEDF is not designed to describethe van der Waals interaction. Furthermore, it is known thatKSDFT with LDA or GGA does not treat the van der Waalsinteraction accurately, which is reflected in the significant dif-ference between the KSDFT and experimental values.
The trends in PBE GGA D0 and re for all dimers exceptMg2 are summarized in Fig. 3. Mg2 is not included sincethe van der Waals interaction is not our focus here. We ob-serve that OFDFT generally produces similar trends for D0
as KSDFT. The D0 values for Al, Ga, In, and Si dimers arereasonably close to the KSDFT ones, independent of spinstate, while OFDFT predicts smaller D0 for Li, P, As, and Sbdimers. For re, OFDFT values fluctuate around the KSDFTvalues, but the deviation is not large, at most ∼0.3 Å off theKSDFT values.
We note that KSDFT has difficulty describing the dissoci-ation limit, where the molecular orbitals formed by p atomic
FIG. 3. The general trend of D0 and re values from OFDFT and KSDFT.(a) singlet state (Ms = 0); (b) triplet state (Ms = 1), all calculations use thePBE GGA exchange-correlation functional.
states, are nearly degenerate in energy. The single determi-nant description in KSDFT cannot treat this kind of multi-reference case properly. This can be seen in the large differ-ence between KSDFT and experimental D0 values, especiallyfor the group V family dimers. Instead, single atom calcula-tions can be carried out and their total energies (multipliedby two) could be considered as the total energy at the fullydissociated limit. The corrected D0 values computed in thisway are listed in Table VII. The results from OFDFT calcu-lated in the same way are also listed. We see that for Li2 andthe group III family dimers, OFDFT results are still in rathergood agreement with the KSDFT and experimental values,while OFDFT predicts much larger D0 values for Si2 and the
TABLE VI. OFDFT, KSDFT, and experimental D0, re, and ωe for Li2 and Mg2. OFDFT values are listed first whileKSDFT values are listed in parentheses.
D0 re ωe
Dimer Parameters (eV) (Å) (cm−1)
Li2 Expt. . . . 1.05a 2.67a 351a
LDA λ = 0.01, β = 0.7143 0.97 (1.45) 3.125 (2.781) 237 (314)GGA λ = 0.01, β = 0.7143 0.93 (1.42) 3.103 (2.775) 267 (314)
Mg2 Expt. . . . 0.05a 3.89a 51a
LDA λ = 0, β = 0.65 1.43 (0.21) 2.740 (3.405) 224 (114)GGA λ = 0, β = 0.65 1.28 (0.15) 2.807 (3.493) 223 (110)
aReference 71.
084102-7 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
TABLE VII. Experimental, OFDFT, and KSDFT D0 values calculated using the two single atom dissociation limit;see text for details. All units are in eV. The HC KEDF with optimal λ and β values is used in OFDFT calculations.
Dimer OFDFT_LDA OFDFT_GGA KSDFT_LDA KSDFT_GGA Expt.
Al2 (Ms = 0) 2.61 2.30 1.76 1.50 . . .Al2 (Ms = 1) 1.70 1.49 1.96 1.66 1.55a
Ga2 (Ms = 0) 2.69 1.81 1.71 1.29 . . .Ga2 (Ms = 1) 1.64 1.27 1.92 1.45 1.4a
In2 (Ms = 0) 2.38 1.82 1.66 1.20 . . .In2 (Ms = 1) 1.65 1.05 1.84 1.23 1.01a
Si2 (Ms = 0) 8.34 7.68 3.87 3.43 . . .Si2 (Ms = 1) 7.30 6.72 4.05 3.61 3.21a
P2 15.89 15.29 6.31 5.57 5.03a
As2 14.34 13.10 5.23 4.02 3.96a
Sb2 10.92 10.14 4.02 2.83 3.09a
Li2 1.10 0.92 0.95 0.71 1.05a
Mg2 1.43 1.28 0.21 0.14 0.05a
aReference 71.
group V family dimers. This is because the HC KEDF is stillnot accurate enough to describe open-shell multiplets, as al-ready demonstrated by the incorrect energy ordering betweensinglet and triplet states. The total energy computed for a sin-gle open-shell multiplet atom is inaccurate, leading to poorD0 values, especially for Si atom (Ms = 1) and the group Vfamily atoms (Ms = 3/2). It is well known that single atomcalculations pose a real challenge for OFDFT calculations.73
For now, we do not focus on the accuracy of OFDFT in calcu-lating single open-shell atoms. Instead, OFDFT D0 values arecalculated only from the potential energy curves at the dimerdissociation limit (10 Å) and then compared to the consistentKSDFT values.
Besides homonuclear diatomic molecules, one heteronu-clear diatomic molecule, AlP, is also calculated and resultsare shown in Table VIII. It has been found in experiments andcomputations that two triplet states, 3 − and 3� are nearlydegenerate with 3 − slightly lower than 3�.74, 75 Here weonly report the triplet ground state, which is found to be a3� state. For the singlet state, similar to the group III dimers,KSDFT tries to describe an open-shell singlet state, but itis extremely difficult to converge because the required two-determinant description is missing in the KSDFT formalism.Through slight tuning of the parameters in the HC KEDF, rea-sonably good D0, re, and ωe could also be obtained compared
TABLE VIII. OFDFT, KSDFT, and experimental D0, re, and ωe for AlP(Ms = 1). λ = 0.003, β = 0.65 is used in the HC KEDF OFDFT calculations.
D0 re ωe
Method (eV) (Å) (cm−1)
Expt. . . . 2.20a . . . . . .
LDA KSDFT 2.92 2.233 539OFDFT_HC 3.02 2.341 454
OFDFT_TF1/5vW 0.75 2.642 232OFDFT_TF1/9vW 0.32 2.833 160
GGA KSDFT 2.57 2.235 530OFDFT_HC 2.73 2.366 410
OFDFT_TF1/5vW 0.51 2.724 126OFDFT_TF1/9vW 0.23 3.294 148
aReference 71.
to the KSDFT values. The HC KEDF results again are far su-perior to the other KEDFs.
B. Electron density analysis
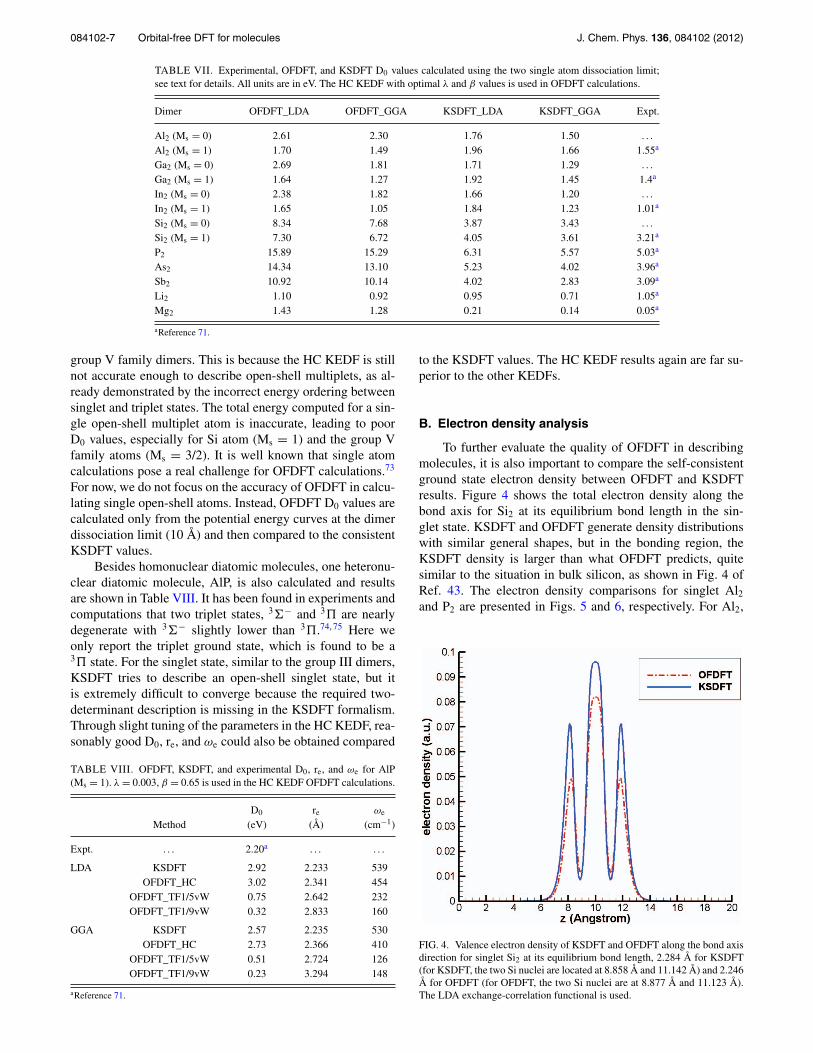
To further evaluate the quality of OFDFT in describingmolecules, it is also important to compare the self-consistentground state electron density between OFDFT and KSDFTresults. Figure 4 shows the total electron density along thebond axis for Si2 at its equilibrium bond length in the sin-glet state. KSDFT and OFDFT generate density distributionswith similar general shapes, but in the bonding region, theKSDFT density is larger than what OFDFT predicts, quitesimilar to the situation in bulk silicon, as shown in Fig. 4 ofRef. 43. The electron density comparisons for singlet Al2and P2 are presented in Figs. 5 and 6, respectively. For Al2,
FIG. 4. Valence electron density of KSDFT and OFDFT along the bond axisdirection for singlet Si2 at its equilibrium bond length, 2.284 Å for KSDFT(for KSDFT, the two Si nuclei are located at 8.858 Å and 11.142 Å) and 2.246Å for OFDFT (for OFDFT, the two Si nuclei are at 8.877 Å and 11.123 Å).The LDA exchange-correlation functional is used.

084102-8 Xia et al. J. Chem. Phys. 136, 084102 (2012)
FIG. 5. Valence electron density of KSDFT and OFDFT along the bond axisdirection for singlet Al2 at its equilibrium bond length, 2.473 Å for KSDFT(the two Al nuclei are at 8.764 Å and 11.237 Å) and 2.498 Å for OFDFT (thetwo Al nuclei are at 8.751 Å and 11.249 Å). The LDA exchange-correlationfunctional is used.
OFDFT has larger density than KSDFT in the bonding re-gion. The OFDFT density in the bonding region is increasedhere because a smaller λ (λ = 0) is used in the Al dimer calcu-lations. Generally, a larger λ lowers the OFDFT density in thebonding region, as will be discussed in Sec. IV C. This effectis further illustrated in the electron density comparison for P2,where KSDFT predicts a much larger density than OFDFT inthe bonding region.
A similar density comparison can also be carried out forthe triplet states of the dimers. Figure 7 demonstrates the spinup and spin down electron density for triplet Si2. KSDFThas larger density in the bonding region for both spin up
FIG. 6. Valence electron density of KSDFT and OFDFT along the bond axisdirection for singlet P2 at its equilibrium bond length, 1.942 Å for KSDFT(the two P nuclei are at 9.029 Å and 10.971 Å) and 2.138 Å for OFDFT (thetwo P nuclei are at 8.931 Å and 11.069 Å). The LDA exchange-correlationfunctional is used.
FIG. 7. (a) spin up and (b) spin down valence electron densities of KSDFTand OFDFT along the bond axis direction for triplet Si2 at its equilibriumbond length, 2.277 Å for KSDFT (the two Si nuclei are at 8.862 Å and 11.139Å) and 2.259 Å for OFDFT (the two Si nuclei are at 8.871 Å and 11.130 Å).The LDA exchange-correlation functional is used.
and spin down electrons. Unlike KSDFT, OFDFT does nothave equivalent densities in both spin channels. Althoughthe general shapes look similar in KSDFT and OFDFT, thediscrepancy between the two can be seen most clearly bycomparing another important dimensionless variable, thespin polarization ζ , which is calculated as
ζ (r) = nup(r) − ndown(r)
nup(r) + ndown(r). (14)
In Fig. 8, isosurfaces of different ζ values for triplet Si2 aredisplayed such that we may compare how the isosurfacesevolve with decreasing ζ . In KSDFT, when ζ is large, theisosurface has a donut-like shape around the bond axis. Thismakes complete sense because the extra two spin up electronscomprising the triplet occupy two degenerate, orthogonalπ -orbitals. As the ζ value decreases, the isosurface expands.However, in OFDFT, the shape of the isosurface is entirely

084102-9 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
FIG. 8. Spin polarization ζ iso-surfaces for triplet Si2: KSDFT results for(a) ζ = 0.7, (b) ζ = 0.4, and (c) ζ = 0.15. OFDFT results for (d) ζ = 0.7,(e) ζ = 0.4, and (f) ζ = 0.15. All calculations employ the LDA exchange-correlation functional.
different. Furthermore, the evolving trend is also reversed.The isosurface is large when ζ is large and shrinks when ζ
reduced. The inaccurate behavior of OFDFT for triplet statecalculations is reflected not only through the incorrect energyordering between singlet and triplet dimers, but also throughthe totally different spin polarization distributions from theKSDFT results. This is likely the origin of the incorrect orderof spin states and clearly requires further advances in theLPSs and KEDFs to remedy.
C. Effects of the parameters
By tuning the HC KEDF parameters λ and β, the abso-lute energy curve may shift upward or downward, as shown inFig. 9. Generally, increasing λ moves the whole curve upwardwhile increasing β moves the curve downward. This behavioris consistent for all of the dimers, as demonstrated in Fig.10, where the absolute total energies for all dimers at theirequilibrium bond lengths are plotted. For every parameter setshown, the curve is shifted such that the OFDFT energy ofGa2 matches the KSDFT value for Ga2. The shifted curvesalmost overlap with each other, which implies that despitedifferent absolute energy values with different parameter sets,the general trend of the absolute energies of each dimer at itsequilibrium bond length is similar, both for singlet and tripletcalculations.
FIG. 9. Total energy vs. bond length curves of KSDFT and OFDFT withdifferent HC KEDF parameter values. (a) Si2 in its singlet state; (b) Si2in its triplet state; and (c) P2. All calculations employ the LDA exchange-correlation functional.
The behavior displayed in Figs. 9 and 10 indicates thatthere could be a universal parameter set that could producea total energy at re close to the KSDFT values for everydimer. However, as mentioned earlier, the parameter choice(especially λ) greatly affects the D0 and ωe values, as wellas the ground state electron density distribution. Table IXshows results for Si2 using different parameter values in theHC KEDF with the LDA exchange-correlation functional. Al-though changing parameters has only a small effect on re (dif-fering less than 0.1 Å), D0 and ωe are much more sensitive tothe parameter choice, particularly between zero and non-zeroλ parameter sets. Non-zero λ tends to produce a deep well,leading to large D0 and ωe, while zero λ predicts a shallow

084102-10 Xia et al. J. Chem. Phys. 136, 084102 (2012)
FIG. 10. Absolute total energy of each dimer at its equilibrium bond length.OFDFT curves are shifted to guarantee that energy values of Ga2 matcheswith the KSDFT results. (a) singlet state dimers; (b) triplet state dimers. Allcalculations employ the LDA exchange-correlation functional.
TABLE IX. D0, re, and ωe results of KSDFT and OFDFT with differentparameter values for Si2. The LDA exchange-correlation functional is used.
D0 re ωe
Method (eV) (Å) (cm−1)
SingletKS 4.59 2.284 501OFDFTλ = 0, β = 0.65 1.80 2.307 377λ = 0, β = 0.7143 1.78 2.296 395λ = 0.01, β = 0.65 4.86 2.246 513λ = 0.01, β = 0.7143 4.94 2.235 521λ = 0.01177, β = 0.65 5.01 2.247 518λ = 0.01177, β = 0.7143 5.10 2.236 527TripletKS 4.66 2.277 509OFDFTλ = 0, β = 0.65 1.66 2.321 391λ = 0, β = 0.7143 1.64 2.310 380λ = 0.01, β = 0.65 4.66 2.259 486λ = 0.01, β = 0.7143 4.74 2.248 485λ = 0.01177, β = 0.65 4.82 2.260 512λ = 0.01177, β = 0.7143 4.91 2.249 472
FIG. 11. (a) spin up and (b) spin down electron density for triplet Al2 at itsequilibrium bond length with different λ values in the HC KEDF. β is fixedto be 0.7143. The LDA exchange-correlation functional is used.
well, which can also be seen in Fig. 9. Generally, the largerλ is, the larger D0 and ωe will be. Increasing the other pa-rameter, β, also slightly raises the D0 values, but much lessdramatically.
Besides D0, re, and ωe values, we also analyze the effectof changing the HC KEDF parameters on the electron densitydistributions, as shown in Fig. 11 for triplet Al2 with differ-ent λ values. As mentioned in Sec. IV B, the reason why thedensity comparison between OFDFT and KSDFT has contra-dictory behavior for Si2 versus Al2 is the different parametersets employed for these two dimers. For Al2, the OFDFT den-sity with λ equal to zero is larger than the KSDFT density inthe bonding region. If λ is increased, the OFDFT density inthe bonding region decreases gradually for both spin channelsas shown in Fig. 11. Although it is possible to select a param-eter set to produce a ground state electron density distributionclose to the KSDFT one, an accurate D0 or ωe would not beguaranteed. This indicates that the current KEDF is still notgeneral enough to predict good self-consistent energies andelectron densities simultaneously.

084102-11 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
TABLE X. Optimal parameter sets of OFDFT for all dimers with the LDAexchange-correlation functional.
Dimer Best λ Best β
Ms = 0Al 0 0.51Ga 0 0.51In 0 0.51Si 0.01 0.65P 0.03 0.51Sb 0.03 0.51As 0.03 0.51Li 0.01 0.7143
Ms = 1Si 0.01 0.65Al 0 0.7143Ga 0 0.7143In 0 0.7143
Finally, reasonable parameters for different dimers areanalyzed and suggested. Since satisfactory energy and den-sity results cannot be guaranteed at the same time, here theparameters are tuned mainly based on D0 values. The opti-mal parameter sets for each dimer are listed in Table X. Inorder to analyze the trend in those parameters, we revisit therelationship between the parameter λ and the static dielectricconstant.43 In the HC KEDF, if we recover the original non-local form of s(r),43
s(r) =[n(r) − n(r′)
|r − r′|]
1
n(r)4/3, (15)
and assume s(r) is very small so that we can perform a first-order Taylor expansion for the nonlocal term, then
TNL[n] = C
∫∫n(r)8/3−βω(kF (r)|r − r′|)n(r′)βdrdr′
+C
∫∫n(r)8/3−βω′|kF |r−r′|kF (r)λ
[n(r)−n(r′)
|r−r′|]2
× |r − r′|n(r)8/3
n(r′)βdrdr′. (16)
Inserting Eq. (16) together with the TF and vW terms intoEq. (11) or Eq. (12) of Ref. 43 and carrying out the functionalderivative, we see that the TF and the vW KEDFs and thefirst term on the right-hand side of Eq. (16) do not contributeto the 1/|r − r′| or 1/q2 term, while one can show that thelast term on the right-hand side of the Eq. (16) generates a
term proportional to 1/|r − r′| or 1/q2 in the reciprocal space.Consequently, an approximate relation between λ and staticdielectric constant can be obtained:
λ ∝ A
ε̃(0) − 1, (17)
where A is related to the kernel derivative, the density distri-bution, and β. This equation implies that the larger the staticdielectric constant, the smaller the magnitude of λ. The signof A could not be universally determined, but in practice pos-itive λ usually improves the results both for bulk materialsand molecules. Physically, this relation also makes completesense. For a metal with an infinitely large dielectric constant,λ would be zero and the kernel would return to a similar formas those in the WT or WGC KEDFs, whereas for a semicon-ductor or insulator with a finite dielectric constant, λ wouldbe non-zero and consequently the gradient term would be ex-plicitly included in the kernel.
For molecules, the static dielectric constant is not welldefined. However, the relationship derived above can be gen-eralized at least conceptually. Usually in semiconductors orinsulators, the smaller the band gap is, the larger the staticdielectric constant is (as noted above, metals with zero bandgap have infinite dielectric constant). As a result, we may ob-tain an approximate relation that the smaller the band gap,the smaller λ should be. In molecules, the band gap can betaken as the energy difference between the highest occupiedmolecular orbital (HOMO) and the lowest unoccupied molec-ular orbital (LUMO). Although strictly speaking the HOMO-LUMO energy gap in KSDFT is not equal to the true energygap, it usually can give a rough estimate of the true gap. TheHOMO-LUMO gap energies from KSDFT calculations areshown in Table XI. For the group III family dimers, we knowthat the optimal λ is zero, and the HOMO-LUMO gap is theenergy difference between the σ and π orbitals, which is verysmall. For Si2, the optimal λ equals 0.01, and the gap is theenergy difference between the π bonding and π* antibond-ing orbitals, which is significantly larger, as listed in the ta-ble. The situation is similar for the group V family dimerswith the optimal λ equal to 0.03, and even larger HOMO-LUMO gaps. Thus the trend for the HOMO-LUMO gap isqualitatively consistent with the above relation. The smallerthe HOMO-LUMO gap, the smaller λ is. In general, larger λ
is suggested to treat systems with large HOMO-LUMO gapswhile zero or small λ is appropriate for systems with smallHOMO-LUMO gaps.
TABLE XI. Energy differences �ε between the HOMO and the LUMO in KSDFT calculations of each dimer at its re.The LDA exchange-correlation functional is used.
Al2 Ga2 In2 Si2Dimers (Ms = 0) (Ms = 0) (Ms = 0) (Ms = 0) P2 As2 Sb2 Li2
�ε (eV) 0.21 0.21 0.11 2.50 3.70 3.33 2.37 1.59Dimers Al2 Ga2 In2 Si2
(Ms = 1) (Ms = 1) (Ms = 1) (Ms = 1)�ε (eV) 0.20 0.23 0.13 2.37

084102-12 Xia et al. J. Chem. Phys. 136, 084102 (2012)
V. CONCLUSIONS
In this work, a variety of diatomic molecules were cal-culated by OFDFT with the HC KEDF to test its ability todescribe covalent bonds in molecules as well as semiconduc-tor crystals. By adjusting the two HC KEDF parameters, λ
and β, OFDFT can generate reasonably quantitative bond dis-sociation energies, equilibrium bond lengths, and vibrationalfrequencies compared to KSDFT, a vast improvement overother KEDFs such as TFλvW or the WT KEDFs. However,for those dimers with triplet ground states, OFDFT predictsthe singlet state to have lower energy, which is not consistentwith KSDFT and experiment. This failure is further reflectedby comparing ground state electron densities. OFDFT yieldsa similar overall electron density distribution to KSDFT, butthe density mismatch in the bonding region is still significant.Furthermore, OFDFT produces a significantly different spinpolarization distribution from the KSDFT one, which is likelythe reason for the incorrect energy ordering between differ-ent spin states. Finally, the effects of parameters on proper-ties were analyzed. In terms of absolute energies, larger λ andsmaller β tend to raise the total energy, but the relative trendof total energy for each dimer at its equilibrium bond length issimilar and consistent with KSDFT results. The λ value alsoinfluences the density distribution. Increasing λ tends to lowerthe electron density in the bonding region. Although by tuningλ it is possible to make the OFDFT density match reasonablywell with the KSDFT one, the current KEDF cannot guar-antee a good electron density and bond dissociation energy atthe same time. Unlike the equilibrium bond length which doesnot change much when adjusting parameters, the bond disso-ciation energy is very sensitive to the λ value. Zero and smallλ generates shallow wells and small D0 values, while large λ
predicts deep wells and large D0 values. Finally, the optimalvalue of λ was qualitatively related to a molecule’s HOMO-LUMO gap. Zero or small λ is suggested for molecules withsmall HOMO-LUMO gaps and large λ is better for moleculeswith large HOMO-LUMO gaps.
Although the HC KEDF can predict quite good resultsfor D0, re, and ωe for diatomic molecules, it still producesconsiderable errors, such as significant inaccuracy in describ-ing spin state ordering and the related mismatch in the elec-tron spin densities. Further research is needed to improve theKEDF and potentially the LPS to make OFDFT a completelyreliable and general quantum mechanics simulation tool.
ACKNOWLEDGMENTS
We are grateful for support from the Office of Naval Re-search and the National Science Foundation (NSF).
APPENDIX: CONSTRUCTION AND TESTINGOF NEW BLPSs
For both KSDFT and OFDFT calculations, BLPSs(Ref. 21) are employed with the PZ (Refs. 49 and 50) LDAand the PBE (Ref. 53) GGA exchange-correlation function-als. For all LDA BLPSs except for Li, we use previously re-ported ones.21, 43 The Li LDA BLPS and all GGA BLPSs areconstructed with the same inversion method described in the
literature.21 For each element, target electron densities for fcc,body-centered-cubic (bcc), simple cubic (sc), and CD crys-tal structures are obtained by carrying out KSDFT calcula-tions with Troullier-Martins NLPSs (Ref. 18) generated bythe FHI98 code76 with default cutoff radii. The local termis selected as the d channel for Li, Mg, Al, Si, P, As, andSb, and the s channel for Ga and In. The inversion proce-dure described in Ref. 21 is carried out by a modified versionof ABINIT.58 During the construction of the BLPSs, Fermi-Dirac smearing with a smearing width equal to 0.1 eV isused and the plane-wave basis kinetic energy cutoff is set to1600 eV. The k-point mesh used during the BLPS construc-tion is 20 × 20 × 20 per unit cell for all structures. The num-ber of atoms per unit cell is as follows: one atom for fcc, bcc,sc, and body-centered tetragonal In (Ref. 78) structures, twoatoms for α-As,79 α-Sb,80 CD, and hexagonal closest-packedstructures, three atoms for 9R Li,81 eight atoms for α-Ga,82
and A17 P.83 Equilibrium structures are relaxed with KSDFTwith a force threshold of 5 × 10−5 hartree/bohr and a stressthreshold of 5 × 10−7 hartree/bohr.3 The structures are thenexpanded and compressed by 2% to obtain total energy ver-sus volume points, which are fit to Murnaghan’s equation ofstate77 to calculate bulk moduli. The total energy differencebetween different phases at their equilibrium volumes is thephase energy difference.
Two parameters in the BLPS construction, the value ofthe non-Coulombic part of the BLPS at q = 0 in reciprocalspace and the position beyond which the BLPS recovers theCoulomb tail in real space, are adjusted so as to reproducethe equilibrium volumes and bulk moduli calculated byKSDFT with the NLPS, for the experimental ground statephase of each element. The BLPSs are further tested foreach element with different crystal structures other than itsground state phase. In the test calculations, 900 eV kineticenergy cutoffs and 20 × 20 × 20 k-point meshes in each unitcell are employed. Fermi-Dirac smearing with a smearingwidth of 0.1 eV is used for metallic solids and no smearingis used for insulators. Comparisons of results obtained usingthe NLPSs versus the BLPSs are listed in the supplementalmaterial Tables S1-S9.84 The new BLPSs are also plotted inthe supplemental information Figures S1-S3.84 The BLPSresults are in good agreement with the NLPS results overall.Except for the CD structure of Li, the BLPS bulk moduliare generally reasonably close to the NLPS values. For equi-librium volumes, the difference between BLPS and NLPSpredictions is quite small for all structures and elements.Finally, the energy orderings predicted by BLPSs exactlyreproduce the NLPS results, and the phase energy differencesalso generally agree well with the NLPS results, except insome cases such as fcc Si. The ground state of Ga is predictedby the NLPS and the BLPS to be fcc instead of α-Ga, whichmay be due to the lack of a nonlinear core correction.85 Thefcc structure is predicted to be the ground state of Li, but theenergy differences between fcc, bcc, and the experimentallyobserved 9R structure are very small, less than 2 meV, whichis certainly within the KSDFT LDA or GGA uncertainty.
1P. Hohenberg and W. Kohn, Phys. Rev. 136, B864 (1964).2W. Kohn and L. J. Sham, Phys. Rev. 140, A1133 (1965).

084102-13 Orbital-free DFT for molecules J. Chem. Phys. 136, 084102 (2012)
3W. Kohn, Phys. Rev. Lett. 76, 3168 (1996).4S. Goedecker, Rev. Mod. Phys. 71, 1085 (1999).5S. Ismail-Beigi and T. A. Arias, Phys. Rev. Lett. 82, 2127 (1999).6S. Y. Wu and C. S. Jayanthi, Phys. Rep. 358, 1 (2002).7T. A. Arias, Rev. Mod. Phys. 71, 267 (1999).8F. Shimojo, R. K. Kalia, A. Nakano, and P. Vashishta, Phys. Rev. B 77,085103 (2008).
9Y. A. Wang and E. A. Carter, in Theoretical Methods in Condensed PhaseChemistry, edited by S. D. Schwartz (Kluwer, Dordrecht, 2000), p. 117.
10L. Hung and E. A. Carter, Chem. Phys. Lett. 475, 163 (2009).11I. Shin, A. Ramasubramaniam, C. Huang, L. Hung, and E. A. Carter,
Philos. Mag. 89, 3195 (2009).12Q. Peng, X. Zhang, L. Hung, E. A. Carter, and G. Lu, Phys. Rev. B 78,
054118 (2008).13L. Hung and E. A. Carter, J. Phys. Chem. 115, 6269 (2011).14L. Hung and E. A. Carter, Modell. Simul. Mater. Sci. Eng. 19, 045002
(2011).15I. Shin and E. A. Carter, Modell. Simul. Mater. Sci. Eng. 20, 015006
(2011).16D. R. Hamann, M. Schlüter and C. Chang, Phys. Rev. Lett. 43, 1494 (1979).17L. Kleinman and D. M. Bylander, Phys. Rev. Lett. 48, 1425 (1982).18N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 (1991).19B. Zhou, Y. A. Wang, and E. A. Carter, Phys. Rev. B 69, 125109 (2004).20S. Watson, B. J. Jesson, E. A. Carter, and P. A. Madden, Europhys. Lett.
41, 37 (1998).21C. Huang and E. A. Carter, Phys. Chem. Chem. Phys. 10, 7109 (2008).22B. Zhou and E. A. Carter, J. Chem. Phys. 122, 184108 (2005).23L. H. Thomas, Proc. Cambridge Philos. Soc. 23, 542 (1927).24E. Fermi, Rend. Accad. Naz. Lincei 6, 602 (1927).25E. Fermi, Z. Phys. 48, 73 (1928).26E. Teller, Rev. Mod. Phys. 34, 627 (1962).27E. H. Lieb and B. Simon, Adv. Math. 23, 22 (1977).28C. F. v. Weizsäcker, Z. Phys. 96, 431 (1935).29K. Yonei and Y. Tomishima, J. Phys. Soc. Jpn. 20, 1051 (1965).30M. Levy, J. P. Perdew and V. Sahni, Phys. Rev. A 30, 2745 (1984).31C. H. Hodges, Can. J. Phys. 51, 1428 (1973).32D. R. Murphy, Phys. Rev. A 24, 1682 (1981).33E. Chacón, J. E. Alvarellos, and P. Tarazona, Phys. Rev. B 32, 7868 (1985).34P. García-González, J. E. Alvarellos, and E. Chacón, Phys. Rev. B 53, 9509
(1996).35P. García-González, J. E. Alvarellos, and E. Chacón, Phys. Rev. B 57, 4857
(1998).36L. W. Wang and M. P. Teter, Phys. Rev. B 45, 13196 (1992).37Y. A. Wang, N. Govind, and E. A. Carter, Phys. Rev. B 60, 16350 (1999);
64(E), 089903 (2001).38E. Smargiassi and P. A. Madden, Phys. Rev. B 51, 117 (1995).39K. M. Carling and E. A. Carter, Modell. Simul. Mater. Sci. Eng. 11, 339
(2003).40G. Ho, M. T. Ong, K. J. Caspersen, and E. A. Carter, Phys. Chem. Chem.
Phys. 9, 4951 (2007).41B. Zhou, V. L. Ligneres, and E. A. Carter, J. Chem. Phys. 122, 044103
(2005).42R. M. Pick, M. H. Cohen, and R. M. Martin, Phys. Rev. B 1, 910 (1970).43C. Huang and E. A. Carter, Phys. Rev. B 81, 045206 (2010).44K. Yonei, J. Phys. Soc. Jpn. 31, 882 (1971).45J. P. Perdew, M. Levy, G. S. Painter, S. Wei, and J. B. Lagowski, Phys. Rev.
B 37, 838 (1988).46S. Iyengar, M. Ernzerhof, S. N. Maximoff, and G. E. Scuseria, Phys. Rev.
A 63, 052508 (2001).
47L. A. Constantin and A. Ruzsinszky, Phys. Rev. B 79, 115117 (2009).48V. Cocula, C. J. Pickard, and E. A. Carter, J. Chem. Phys. 123, 214101
(2005).49D. M. Ceperley and B. J. Alder, Phys. Rev. Lett. 45, 566 (1980).50J. P. Perdew and A. Zunger, Phys. Rev. B 23, 5048 (1981).51J. P. Perdew and Y. Wang, Phys. Rev. B 45, 13244 (1992).52U. von Barth and L. Hedin,J. Phys. C 5, 1629 (1972).53J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
(1996).54G. L. Oliver and J. P. Perdew, Phys. Rev. A 20, 397 (1979).55H. Jiang and W. Yang, J. Chem. Phys. 121, 2030 (2004).56G. Ho, V. L. Ligneres, and E. A. Carter, Comput. Phys. Commun. 179, 839
(2008).57L. Hung, C. Huang, I. Shin, G. Ho, V. L. Ligneres, and E. A. Carter, Com-
put. Phys. Commun. 181, 2208 (2010).58X. Gonze, J. M. Beuken, R. Caracas, F. Detraux, M. Fuchs, G. M.
Rignanese, L. Sindic, M. Verstraete, G. Zerah, F. Jollet, M. Torrent, A. Roy,M. Mikami, P. Ghosez, J.-Y. Raty, and D. C. Allan, Comput. Mater. Sci. 25,478 (2002).
59Y. Tomishima and K. Yonei, J. Phys. Soc. Jpn. 21, 142 (1966).60D. S. Ginter, M. L. Ginter, and K. K. Innes, Astrophys. J. 139, 365 (1964).61T. H. Upton, J. Phys. Chem. 90, 754 (1986).62C. W. Bauschlicher, H. Partridge, S. R. Langhoff, P. R. Taylor, and
S. P. Walch, J. Chem. Phys. 86, 7007 (1987).63M. F. Cai, T. P. Dzugan, and V. E. Bondybey, Chem. Phys. Lett. 155, 430
(1989).64H. J. Himmel and B. Gaertner, Chem.-Eur. J. 10, 5936 (2004).65X. Tan and P. J. Dagdigian, J. Phys. Chem. A 107, 2642 (2003).66B. Song and P. Cao, J. Chem. Phys. 123, 144312 (2005).67K. K Das, J. Phys. B 30, 803 (1997).68Y. Zhao, W. Xu, Q. Li, Y. Xie, and H. F. Schaefer III, J. Phys. Chem. A
108, 7448 (2004).69G. Balducci, G. Gigli, and G. Meloni, J. Chem. Phys. 109, 4384 (1998).70K. Balasubramanian and J. Li, J. Chem. Phys. 88, 4979 (1988).71K. Huber and G. Herzberg, Constants of Diatomic Molecule (Van Nostrand
Reinhold, New York, 1979).72F. W. Froben, W. Schulze, and U. Kloss, Chem. Phys. Lett. 99, 500
(1983).73R. G. Parr and W. Yang, Density Functional Theory of Atoms and
Molecules (Oxford University Press, New York, 1989).74H. Gomez, T. R. Taylor, Y. Zhao, and D. M. Neumark, J. Chem. Phys. 117,
8644 (2002).75Z. Gan, D. J. Grant, R. J. Harrison, and D. A. Dixon, J. Chem. Phys. 125,
124311 (2006).76M. Fuchs and M. Scheffler, Comput. Phys. Commun. 119, 67 (1999).77F. D. Murnaghan, Proc. Natl. Acad. Sci. U.S.A. 30, 244 (1944).78Smithells Metals Reference Book, edited by E. A. Brandes and G. B. Brook,
7th ed. (Elsevier, New York, 1998).79L. F. Mattheiss, D. R. Hamann, and W. Weber, Phys. Rev. B 34, 2190
(1986).80L. M. Falicov and P. J. Lin, Phys. Rev. 141, 562 (1966).81A. W. Overhauser, Phys. Rev. Lett. 53, 64 (1984).82M. Bernasconi, G. L. Chiarotti, and E. Tosatti, Phys. Rev. B 52, 9988
(1995).83A. Brown and S. Rundqvist, Acta Crystallogr. 19, 684 (1965).84See supplementary material at http://dx.doi.org/10.1063/1.3685604 for the
comparison of bulk properties predicted by BLPSs and NLPSs for eachelement, as well as plots of the newly constructed BLPSs.
85S. G. Louie, S. Froyen, and M. L. Cohen, Phys. Rev. B 26, 1738 (1982).