cancer research chemotherapy-induced genotoxic … genotoxic stress promotes sensitivity to natural...
TRANSCRIPT

Micr
CheSenEna
Jason
Abst
Intro
Innfense(1). Naof recoincludcoatedcludeand cerelatecytokiref. 2)they frologiccriminintensThe
toxicit
AuthorSunnybProgramResearUnivers
Note:Resear
CorresUniversAvenueext. 33
doi: 10
©2010
Cance7102
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
Canceresearch
oenvironment and Immunology
motherapy-Induced Genotoxic Stress Promotessitivity to Natural Killer Cell Cytotoxicity by
R
bling Missing-Self Recognition
H. Fine1, Peter Chen1, Aruz Mesci1, David S.J. Allan1, Stephan Gasser2, David H. Raulet3, and James R. Carlyle1
ractNat
the balectin-gandsby NKinfecteis rapiproteiNotabgands
frequenty and a la
s' Affiliationrook Reseame, Natioch Laboraity of Califo
Supplemench Online (
ponding Auity of Toro, Toronto,82; Fax: 416
.1158/0008-
American A
r Res; 70(
ural killer (NK) cells can recognize and kill tumor cells lacking “self” markers, such as class I MHC, butsis for this recognition is not completely understood. NKR-P1 receptors are members of the C-typerelated NK receptor superfamily that are conserved from rodents to humans. Identification of Clr li-for the NKR-P1 receptors has facilitated functional analysis of MHC-independent target cell recognitioncells. One receptor-ligand pair, NKR-P1B:Clr-b, can mediate “missing-self” recognition of tumor andd cells, but the role of this axis in sensing stressed cells remains unknown. Here, we show that Clr-bdly downregulated in cells undergoing genotoxic and cellular stress at the level of both RNA and surfacen. Stress-mediated loss of Clr-b on leukemia cells enhanced cytotoxicity mediated by NKR-P1B+ NK cells.ly, Clr-b downregulation was coordinated functionally with stress-mediated upregulation of NKG2D li-(but not class I MHC). Our findings highlight a unique role for the MHC-independent NKR-P1B:Clr-b
missing-self axis in recognition of stressed cells, and provide evidence of two independent levels of Clr-b reg-ulation in stressed cells. Cancer Res; 70(18); 7102–13. ©2010 AACR.
led toMHC-nitionspecifstimuMHC-tion bhibitioaloneregulaInd
NKG2latedrecognby a bwhichnume
duction
ate immunity constitutes an important front-line de-and barrier to infectious disease and malignanciestural killer (NK) cells are innate lymphocytes capablegnizing and eliminating a wide variety of target cells,ing transformed, infected, transplanted, antibody-, and stressed cells (2, 3). NK effector mechanisms in-cellular cytotoxicity mediated by perforin, granzymes,ll surface molecules (e.g., FasL, tumor necrosis factor–d apoptosis-inducing ligand), and rapid secretion ofnes (e.g., IFN-γ, tumor necrosis factor-α, chemokines;. Although NK cells are tolerant to normal “self” cells,equently respond to abnormal cells undergoing path-alterations. How NK cells mediate self–nonself dis-ation at the molecular level remains the focus ofe research.
correlation between heightened NK cyto-ck of MHC-I expression on tumor cell lines
modereceptunderrecognThe
cells tderstochemoreplicNKG2ed inway,NKG2divers
s: 1Department of Immunology, University of Toronto,rch Institute, Toronto, Ontario, Canada; 2Immunologynal University of Singapore, Singapore; and 3Cancertory, Department of Molecular and Cell Biology,rnia, Berkeley, California
tary data for this article are available at Cancerhttp://cancerres.aacrjournals.org).
thor: James R. Carlyle, Department of Immunology,nto, Sunnybrook Research Institute, 2075 BayviewOntario, Canada M4N3M5. Phone: 416-480-6100,-480-5703; E-mail: [email protected].
5472.CAN-10-1316
ssociation for Cancer Research.
18) September 15, 2010
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
the hypothesis that NK cells sense the absence of selfI markers on target cells. Termed “missing-self” recog-(4), this pathway is governed by inhibitory receptorsic for self MHC-I molecules that normally overridelatory NK-target interactions. In turn, the loss ofI molecules upon malignant transformation or infec-ecomes sufficient to trigger NK cytotoxicity via disin-n of effector function (5). However, this mechanismis insufficient to explain the complex outcomes thatte NK-target interplay.eed, other potent stimulatory NK receptors such asD recognize “induced-self” ligands dynamically upregu-during stress responses. Thus, a contemporary dual-ition model proposes that NK cell function is regulatedalance of stimulatory and inhibitory receptor signals,in turn are determined by target cell modulation ofrous cognate ligands (3, 6). This more target-centricl highlights the need to understand how both NK cellors and their cognate ligands are modulated in real timepathologic conditions to fully appreciate complex NKition events in vivo.underlying molecular basis behind the ability of NKo recognize “stressed” cells remains incompletely un-od. Recent evidence indicates that cells exposed totherapeutic agents, genotoxic stimuli, or stalled DNAation cycles dynamically upregulate stimulatoryDL (7). This induced-self ligand modulation is mediat-part through the DNA damage response (DDR) path-in particular involving ATR, ATM, and Chk1 (8).
DL upregulation has been observed in response toe genotoxic stimuli (including aphidicolin, cisplatin,for Cancer Research on January 31, 2013org

5-fluoras heaT cellsadditiDNAMHowestressIn a
dent iincludrecepof ClrClr-b ihematMoreolatedClr-bstancethe ronism(seventsa progloss oresponor imfectedto beupreguIn t
on ClrinducpromoscriptsmolecmentsNKG2feine athe ATtion wmainethe ATloss odentlypressiof Clr-of thethe domediatheseself rethe RN
Mate
ChemAph
5-fluo
tin-A,α-amachasedchaseda Gamperfordissoltions
Mice,C14
Ameridentlyin ouras desblastskillerbred atainedsuppleunits/IL-2 dand Msion omarkeCD11cless thcitatiofreezinat all tmaintCell r1 weesencecells wplatestreatm
FlowAll
purchantimpreviofusionReseaStreptwerestainelibur f
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.a
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
ouracil, γ-irradiation, and UV radiation; ref. 9), as wellt shock responses (10), antigen-mediated activation of(11–13), and dysregulation of Dicer expression (14). In
on, ligands for other stimulatory NK receptors, such as-1 (CD226), seem to be similarly upregulated (15).ver, the modulation of inhibitory NK ligands duringresponses remains undocumented to date.ddition to MHC-I molecules, several MHC-indepen-nhibitory ligands have recently been characterized,ing Clr-b (Clec2d), the ligand for the NKR-P1B (Klrb2)tor (3, 16–20). Importantly, the expression pattern-b closely resembles that of classic MHC-I antigens.s widely expressed on normal hematopoietic and non-opoietic cells and is frequently lost on tumor cells (16).ver, like MHC-I, Clr-b expression is rapidly downregu-following cytomegalovirus (CMV) infection (21). Thus,seems to be lost under numerous pathologic circum-s, and might function as an inhibitory rheostat inutine detection of abnormal cells. Although the mecha-) governing Clr-b regulation remain to be elucidated,leading to transformation or infection seem to initiaterammed cellular response pathway culminating in thef Clr-b at the cell surface (17, 21). This intrinsic hostse pathway differs from the extrinsic CTL-selectedmunoevasin-induced loss of MHC-I on tumor or in-cells, and thus, might share common elements knowninvolved in the DDR pathway responsible for NKG2DLlation.his report, we investigated the influence of cell stress-b expression. Various chemotherapeutic agents anders of genotoxic or physiologic stress were found tote a rapid functional downregulation of Clr-b tran-and cell surface protein. Notably, MHC-I cell surface
ules were not substantially altered by similar treat-(or actually increased). Extending previous work,
DL upregulation was found to be blocked by both caf-nd short hairpin RNA (shRNA)-mediated silencing ofR and Chk1 pathway (9). However, Clr-b downregula-as differentially affected by caffeine treatment and re-d largely unaffected by shRNA-mediated knockdown ofR/Chk1 and ATM/Chk2 pathways. This suggests thatf Clr-b following genotoxic stress may occur indepen-of the classic DDR pathway. Interestingly, ectopic ex-on of Clr-b transcripts prevented stress-mediated lossb surface protein. Moreover, pharmacologic inhibitionubiquitin-proteasome degradation pathway uncoupledwnregulation of Clr-b surface protein from the stress-ted loss of endogenous Clr-b transcripts. Collectively,results show that genotoxic stress promotes missing-
gulation of the NK inhibitory ligand, Clr-b, at both showgatingiodideUntresolven
BWZ
A and surface protein levels.
rials and Methods
icals and chemotherapeutic agents
idicolin, bleomycin, cisplatin, camptothecin, etoposide,rouracil, phleomycin, roscovitine, Scriptaid, trichosta-BWtors w
acrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
tunicamycin, 5-aza-2′-deoxycytidine, caffeine, H2O2,nitin, actinomycin-D, and cycloheximide were pur-from Sigma-Aldrich. MG132 and lactacystin were pur-from Calbiochem. γ-Irradiation was performed usingmacell-1000 Cs-source irradiator. UV-C irradiation wasmed using a XL-1000 Spectrolinker. Chemicals wereved in DMSO, water, or PBS, according to the instruc-of the manufacturer.
cell lines, and tissue culture98 and NIH3T3 cells were purchased directly fromcan Type Culture Collection and validated indepen-using our own in-house stocks. MNK-1 was generatedlaboratory from primary mouse fetal thymic NK cells,cribed previously (22). Primary mouse embryonic fibro-(MEF) and interleukin (IL)-2 lymphokine-activated(LAK) cells were generated from B6 and CD-1 micend maintained in our own facilities. Cells were main-in complete medium (DMEM-HG plus 10% FCS andments); MNK-1 cells were maintained in rhIL-2 (300mL of Proleukin; Novartis) and routinely tested forependence (22). All cell lines (C1498, NIH3T3, MNK-1,EF) were authenticated by flow cytometry for expres-f MHC-I and CD45 alleles, Clr-b, and various lineagers (including NK1.1, CD3, CD4, CD8, CD19, CD11b,, and Gr-1). Cell lines were maintained in culture foran 6 months (frequently 1–2 months) following resus-n from stocks within one passage of expansion prior tog. Fibroblasts were maintained in a subconfluent stateimes to prevent the formation of foci, and all cells wereained in logarithmic growth phase prior to treatment.esponses were independently validated followingk of culture in Normocin (Invivogen) to ensure the ab-of Mycoplasma. For most treatments, 2 × 105 to 5 × 105
ere seeded in 2 mL of complete medium in six-welland dosed for 24 hours, with or without 1-hour pre-ents with caffeine, MG132, or lactacystin.
cytometric analysiscommercial monoclonal antibodies (mAb) wereased from BD PharMingen or eBioscience. Biotinylatedouse Clr-b mAb (4A6 mAb; ref. 16) was generatedusly. NKG2DL was visualized using NKG2D/hIgGprotein (23) plus goat anti-hIgG-PE (Jackson Immuno-rch). MHC-I levels were analyzed using anti-KbDb mAb.avidin-R-phycoerythrin or streptavidin-allophycocyaninused as secondary reagents (Invitrogen). Cells wered as described (22), then analyzed using a BD FACSCa-low cytometer and FlowJo software (TreeStar). All plotscells gated for viable cells, as determined by live-cellby forward scatter, side scatter, and lack of propidiumuptake; viability for all experiments was >80% to 90%.ated control stains show analysis of cells treated witht alone.
reporter assay
Z.36 reporter cells (24) expressing CD3ζ-fusion recep-ere described previously (16). For cell-based assays, 105Cancer Res; 70(18) September 15, 2010 7103
for Cancer Research on January 31, 2013org

stimulcocultanalys
RevertranscTot
PureLtranscprimewas p35 cycfor 30
PrimeCAAA3′; G3R-5′-TqRT
ROX (for 3760 secanalysCCCCTGTC
ureendC14ponss.tedfloww u), trm
graygen14
icate. Rresependent experiments.
Fine et al.
Cance7104
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
ator cells were treated for 24 hours, washed, and thenured overnight with 5 × 104 BWZ reporter cells prior tois of β-galactosidase activities (16).
se transcription-PCR/quantitative reverseription-PCRal RNA was prepared using either Trizol or theink RNA Mini Kit (Invitrogen). RNA was reverse-ribed using the Super-Script III Kit and oligo-dTr (Invitrogen). Reverse transcription-PCR (RT-PCR)erformed using a PTC-240G Tetrad (Bio-Rad) for
les (Clr-b) or 28 cycles (G3PDH), as follows: 94°Cseconds, 53°C for 30 seconds, and 72°C for 60 seconds.CAGCTAGC
r Res; 70(18) September 15, 2010
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
rs used were Clrb (624 bp), F-5′-ATGTGTGTCA-GGCTTCC-3′, R-5′-CTAGGAAGGAAAAAAAGGAGTTTG-PDH (452 bp), F-5′-ACCACAGTCCATGCCATCAC-3′,CCACCACCCTGTTGCTGTA-3′.-PCR was performed using iTaq SYBR Green withBio-Rad) on an ABI Prism 7000 (Applied Biosystems)cycles, as follows: 95°C for 30 seconds and 61°C foronds. Products were confirmed by dissociation curveis. Primers used were mClrb (120 bp), F-5′-CATCT-TGAGTCTTGTG-3′ , R-5′-ACTGGGATCTGTTC-TTTG-3′ ; G3PDH (120 bp), F-5′-GACTTCAA‐
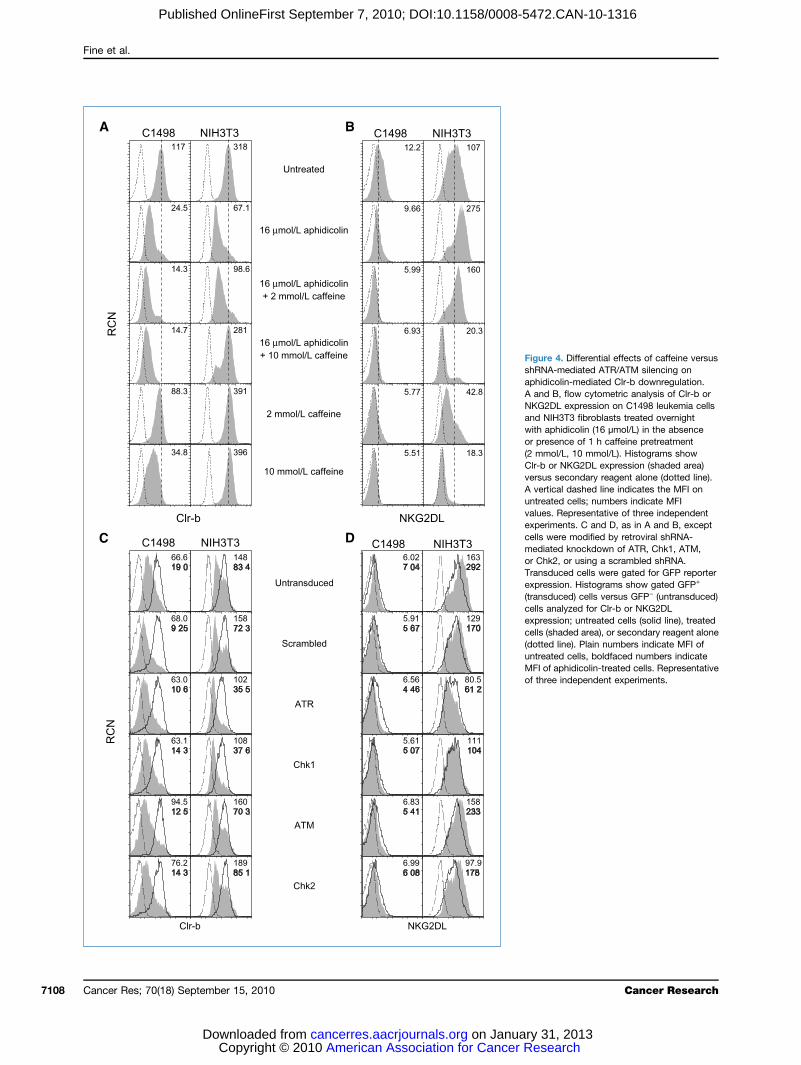
FigdeponresstretreabysholinewithbyreaB, Cindin ARepind
AACTCCCACTCTT-3′ , R-5′CGTATTC-3′.
for Cancer Research on January 31, 2013org
1. Rapid and dose-ent downregulation of Clr-b98 leukemia cells inse to genotoxic and cellularA, C1498 cells wereas indicated and analyzedcytometry. Histogramsntreated cells (thick blackeated cells (thin solid lines,aximal treatment indicatedshading), or secondary
t alone (dotted line).98 cells were treated asd for 24 h, then analyzed asCN, relative cell number.entative of three
-CACCCTGTTGCTG-
Cancer Research

51Cr-rLAK
spleno2,000 ufor 5 t
RetroThe
pMSCbasedLTR-dized aas desbefore
Resu
RapidrespoWe
on mo
rendecytoto(16). Htransfthe reClr-beventcell lintoxic(16). FclassicAs p
kemialine; rlevelswith cduceClr-bof Clr
Figuretreatedlevels o
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.a
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
elease cytotoxicity assayseffector cells were prepared from bonemarrow cells andcytes grown in complete medium containing 1,000 tonits/mL of rhIL-2 plus 15 ng/mL of mIL-15 (Peprotech)o 6 days. Lysis assays were performed as described (16).
viral transductionfull-length Clr-b coding sequence was subcloned intoV2.2-CMV-IRES-GFP. Retroviral shRNA constructson the pMND-Banshee vector (which contains anriven GFP reporter gene) were previously character-nd validated (9). Stable transductants were generatedcribed (16), sorted for GFP, and expanded for 5 daysanalysis.
lts
and dose-dependent loss of cell surface Clr-b innse to diverse genotoxic agents
have previously shown that Clr-b is broadly expressed the mas indicated in Fig. 1, and analyzed by flow cytometry. Bar graphs indicate thef Clr-b, NKG2DL, and MHC-I, relative to untreated control cells. Error bars indic
acrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
rs transformed target cells more susceptible to NKxicity via NKR-P1B–mediated missing-self recognitionowever, the underlying basis for the loss of Clr-b duringormation remains unclear (3). To gain better insight intogulation of Clr-b expression, we investigated whetherlevels are influenced by genotoxic stress, an initiatingin transformation (9). To this end, several Clr-b+ mousees were screened for their responses to various geno-agents, then evaluated for surface expression of Clr-bor comparison, we also monitored the expression ofMHC-I molecules and NKG2DL (23).reviously shown (16), C1498 cells (NKT-like acute leu-; ref. 25), MNK-1 cells (IL-2–dependent thymic pre-NKef. 22), NIH3T3 fibroblasts, and MEF cells express highof cell surface Clr-b. Strikingly, treatment of these cellshemotherapeutic agents and chemicals known to in-genotoxic stress promoted a rapid downregulation ofsurface protein (Fig. 1; Supplementary Fig. S1A). Loss-b expression was a rapid and dynamic event, and
agnitude of downregulation was controlled in a dose-st normal cells, yet frequently lost on tumor cells; this and time-dependent manner (Fig. 1A). To investigate distinct
2. Clr-b downregulation on four different cell lines in response to genotoxic and cellular stress. C1498, NIH3T3, MNK-1, and B6 strain MEF cells were
fold up/downregulation in median fluorescence intensity (MFI)ate SDs of three experiments.Cancer Res; 70(18) September 15, 2010 7105
for Cancer Research on January 31, 2013org

modesresponagentrespon
promomacol
Fine et al.
Cance7106
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
of cellular stress, Clr-b modulation was evaluated inse to a variety of chemotherapeutic and genotoxic
s, as well as agents that induce alternative stress Fig. Sr Res; 70(18) September 15, 2010
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
ted Clr-b downregulation in four cell lines at phar-ogic doses within 24 hours (Fig. 1B; Supplementary
1A). Interestingly, analogous treatments had similarluding (a) camptothecin
urediatotoxctioss.splyzeR-PogrgenomeG2DressadedSOond). Crnigμmcoviomyr-re-1 o98ts incenEMet (Z−R-PtedstimomyZ.Cmalwn.s wicateitu,Z.Presependent experiments.
ses. Several but not all genotoxic agents efficiently effects on Clr-b expression, inc
FigmecytfunstreandanaNKhistreacytNKexp(sh(DMseclineove(20rosble51CCDC14Ploper± StargBWNKpla(−),ionBWNorshocellindin sBWRepind
for Cancer Research on January 31, 2013org
3. Loss of NKR-P1B–ed inhibition of NKicity and Clr-b ligandn in response to genotoxicA, CD-1 strain bone marrowlenic LAK cultures wered by flow cytometry for1B expression (shadedam) versus secondaryt alone (dotted line). B, flowtric analysis of Clr-b,L, and MHC-I (KbDb)ion on treated C1498 cellsarea), untreated cellsalone; solid line), orary reagent alone (dotted1498 cells were treatedht (24 h) with aphidicolinol/L), cisplatin (10 μmol/L),tine (10 μmol/L), orcin (40 μg/mL). C, standardlease cytotoxicity assay ofr B6 LAK cells versustarget cells treated as in B.dicate mean of triplicatetage of specific lysis valuesfor the indicated effector/E:T) ratios. D, top,parental cells or BWZ.CD3ζ/1B reporter cells wereovernight in medium aloneulated using PMA +cin (P/I), or mixed withlr-b stimulator cells (Clr-b).ized OD595–655 values areBottom, C1498 stimulatorere treated with thed concentrations of agentsthen incubated with1B reporter cells overnight.entative of three
Cancer Research

and etin-Aγ-irradtion ation bextentIn c
theseMHC-creasetheseceptibgeneitsuch apropeor NKsistentreatmplatinitor), band 2;the reon NK
GenotcytotorecogLos
wouldobservcells wassaysel acu(26, 27of Clr-ligandcontroNKR-PCD-1 athe massaysNKR-Pweaklref. 28BecauexpresalloweallelespecifavailabAs
spleniNKR-Por cispwhereunaltewith rtreated
upreguagentsmediacytototo aphregulapromomediadicolinroscoincreamentsCD-1and Laphidiloss orecognto theThis sNKR-PtargetcisplatTo s
tion inBWZ.3P1B futo assetor ceresponwhereulatorgenotoovernicells.functiresponsurfacbut noligandother(data
PotenstressPre
aphidiATR/Cmor ceATM/gulati(9). Thway incaffeinupreglin treblock
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.a
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
toposide (topoisomerase-I/II inhibitors); (b) trichosta-and Scriptaid (histone deacetylase inhibitors); and (c)iation, bleomycin, and phleomycin (ionizing radia-nd radiomimetic agents). This highlights a correla-etween similar stress response pathways and theof Clr-b downregulation.ontrast, NKG2DL was frequently upregulated underconditions (Supplementary Fig. S1B; ref. 9), whereasI surface levels remained largely unaffected (or in-d somewhat; Supplementary Fig. S1C). Collectively,combined responses are expected to enhance the sus-ility of target cells to NK cytotoxicity. Because hetero-y in responses existed among the cells tested, factorss transformation state, cell type, or other cell-intrinsicrties might influence the degree or magnitude of Clr-bG2DL modulation. Notably, Clr-b was strongly and con-tly downregulated on several cell lines in response toent with aphidicolin (DNA replication inhibitor), cis-(DNA crosslinking agent), and roscovitine (CDK inhib-ut not bleomycin (DNA-cleaving radiomimetic; Figs. 1Supplementary Fig. S1A). Therefore, we tested whethersponses to these agents had functional consequencesrecognition.
oxic stress enhances susceptibility to NKxicity and diminishes NKR-P1B–mediatednition of Clr-b ligands of surface Clr-b on cells undergoing genotoxic stressbe expected to enhance NK cytotoxicity, as previouslyed for tumor cells (16). To address this, treated targetere analyzed in standard 51Cr-release cytotoxicity. C1498 targets were used because they represent amod-te leukemia cell line used broadly in cytotoxicity assays), they are one of few tumor lines that express high levelsb (16), and they modulate Clr-b but not other known NKs in response to several genotoxic agents. To furtherl for receptor specificity, we took advantage of known1B allelic polymorphisms between two mouse strains,nd B6, which have been previously shown to influenceagnitude of Clr-b–dependent inhibition in cytotoxicity: CD-1 strain LAK cells are strongly inhibited via the1BCD-1 allele, whereas B6-strain LAK cells are onlyy inhibited via the NKR-P1BB6 allele (a.k.a., NKR-P1D;), in response to target cells expressing Clr-b (16, 29).se the NKR-P1B receptor is known to exhibit variegatedsion on NK subsets (3, 29), the use of CD-1 strain LAKd us to monitor surface expression of the NKR-P1BCD-1
(via PK136 mAb; refs. 3, 16, 22, 28), whereas mAbsic for the NKR-P1BB6 allele are not commerciallyle (18, 29).shown in Fig. 3A, both bone marrow–derived andc LAK cells from CD-1 mice express high levels of1B. Furthermore, C1498 targets treated with aphidicolinlatin possess strongly reduced levels of surface Clr-b,as NKG2DL and MHC-I surface levels remain largelyred (Figs. 1, 2, and 3B). In contrast, C1498 cells treated
oscovitine lose Clr-b and induce NKG2DL, whereas cellswith bleomycin maintain Clr-b levels and moderatelycells; rregula
acrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
late NKG2DL. As expected, treatment with genotoxicrendered C1498 targets more sensitive to cytotoxicityted by CD-1 strain LAK (Fig. 3C). Moreover, targetxicity was increased to a greater extent in responseidicolin, cisplatin, or roscovitine (which strongly down-te Clr-b), compared with bleomycin (which does notte loss of Clr-b). On the other hand, cytotoxicityted by B6-strain LAK was minimally affected by aphi-or cisplatin whereas cytotoxicity was increased by
vitine or bleomycin, consistent with the moderatese in NKG2DL expression observed in the latter treat-. Importantly, known receptor polymorphisms affectingversus B6 LAK function are limited to the NKR-P1By49 receptors, yet MHC-I levels do not change uponcolin or cisplatin treatment. This rules out a role forf MHC-I ligands or induction of stimulatory ligandsized by nonpolymorphic NK receptors in contributingaugmented cytotoxicity observed for CD-1 strain LAK.trongly argues that enhanced cytotoxicity mediated by1B+ LAK is due to loss of Clr-b expression on C1498s exposed to the genotoxic agents, aphidicolin andin.pecifically address the loss of surface Clr-b ligand func-isolation, BWZ reporter assays were performed. Here,6 reporter cells (24) expressing a chimeric CD3ζ/NKR-sion-receptor (BWZ.CD3ζ/P1B cells; ref. 16) were usedss NKR-P1B–dependent recognition of treated stimula-lls. As shown in Fig. 3D, BWZ.CD3ζ/P1B reporter cellsd specifically to stimulator cells expressing Clr-b ligand,as BWZ− parental cells fail to respond. Next, C1498 stim-cells were incubated in situ for 24 hours with variousxic agents in 96-well plates, followed by washing andght incubation with BWZ− or BWZ.CD3ζ/P1B reporterImportantly, these results confirm that Clr-b ligandon is lost in a dose-dependent manner, yet only inse to genotoxic agents that downregulate Clr-b celle expression (i.e., aphidicolin, cisplatin, roscovitine,t bleomycin; Fig. 3D; Figs. 1 and 2). This loss of cognatefunction was independently confirmed with three
cell lines treated with the same four genotoxic agentsnot shown).
tial involvement of the ATM/ATR pathways in-mediated Clr-b downregulationvious studies showed a role for the DDR pathway incolin-induced NKG2DL upregulation on fibroblasts (viahk1), as well as constitutive NKG2DL expression on tu-lls (via ATM; refs. 8, 9). In these studies, inhibition of theATR pathways using caffeine abrogated NKG2DL upre-on on fibroblasts in response to aphidicolin treatmenterefore, we investigated the involvement of this path-stress-mediated Clr-b downregulation. Interestingly,e blocked both Clr-b downregulation and NKG2DLulation on NIH3T3 fibroblasts in response to aphidico-atment (Fig. 4A and B). However, caffeine did notaphidicolin-mediated loss of Clr-b on C1498 leukemia
ather, caffeine treatment alone promoted Clr-b down-tion, and the combination of caffeine and aphidicolinCancer Res; 70(18) September 15, 2010 7107
for Cancer Research on January 31, 2013org
ureNAidicnd BG2DNIH
h appresmo
-b osuserticreatues.erimls wdiateChk2nsduressnsduls anressls (shttedreatI ofhree independent experiments.
Fine et al.
Cance7108
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
FigshRaphA aNKandwitor(2 mClrverA vuntvalexpcelmeorTraexp(tracelexpcel(dountMFof t
r Res; 70(18) September 15, 2010
American Association for Cancer Rese Copyright © 2010 on January 31cancerres.aacrjournals.orgDownloaded from
4. Differential effects of caffeine versus-mediated ATR/ATM silencing onolin-mediated Clr-b downregulation., flow cytometric analysis of Clr-b orL expression on C1498 leukemia cells3T3 fibroblasts treated overnighthidicolin (16 μmol/L) in the absenceence of 1 h caffeine pretreatmentl/L, 10 mmol/L). Histograms showr NKG2DL expression (shaded area)secondary reagent alone (dotted line).al dashed line indicates the MFI oned cells; numbers indicate MFIRepresentative of three independentents. C and D, as in A and B, exceptere modified by retroviral shRNA-d knockdown of ATR, Chk1, ATM,, or using a scrambled shRNA.ced cells were gated for GFP reporterion. Histograms show gated GFP+
ced) cells versus GFP− (untransduced)alyzed for Clr-b or NKG2DLion; untreated cells (solid line), treatedaded area), or secondary reagent aloneline). Plain numbers indicate MFI ofed cells, boldfaced numbers indicateaphidicolin-treated cells. Representative
Cancer Research
arch, 2013

promotutiveupregand NfindinviouslinducemighttumorBec
of ATshRNAATM,moduExtendupregshRNAor Chktrast,mediaor C14silencgulatiexpresCol
cannoof theregulaloss ovia a nways (by thebleomtion. Iof p53any inATM/stressturn,mediacells)(36), sDNA-P33, 37)at the
GenotransIt w
the trainfectiregulalevel oized. Tlyzedthe enscriptsstress.
FiguretranscrClr-b dtranscruntreatserial cquantitB, flowClr-b pRetrovifull-lengreportearea) vevalues. A vertical dashed line indicates the median Clr-b expression onuntreated cells. Representative of three independent experiments.
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.aacrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
ted a more striking loss of Clr-b. As a control, consti-NKG2DL expression and stress-mediated NKG2DL
ulation were abrogated on both C1498 leukemia cellsIH3T3 fibroblasts by caffeine (Fig. 4A and B). Thesegs are consistent with differential effects observed pre-y: The ATR pathway seems to regulate aphidicolin-d NKG2DL on fibroblasts, whereas the ATM pathwayregulate constitutive NKG2DL expression on somecells (8, 9).ause caffeine exhibits pleiotropic effects independentR/ATM inhibition (30–33), previously characterizedvectors (9) were used to specifically silence the
ATR, Chk1, and Chk2 gene products, and then Clr-blation on C1498 and NIH3T3 cells was reassessed.ing previous findings (9), aphidicolin-induced NKG2DLulation on NIH3T3 fibroblasts could be blocked by-mediated knockdown of Chk1 or ATR, but not ATM2 (Fig. 4C andD; see also Supplementary Fig. S2). In con-none of the shRNA constructs prevented aphidicolin-ted Clr-b downregulation on either NIH3T3 fibroblasts98 cells. In fact, ATR/Chk1 silencing (but not ATM/Chk2ing) modestly enhanced aphidicolin-mediated downre-on of Clr-b, and somewhat lowered constitutive Clr-bsion, on NIH3T3 fibroblasts (Fig. 4C and D).lectively, although redundancy in the DDR pathwayt be excluded, the failure of shRNA-mediated silencingse kinases to inhibit aphidicolin-mediated Clr-b down-tion (on both NIH3T3 and C1498 cells), suggests thatf Clr-b in response to genotoxic stress might occurovel mechanism, independent of the ATR/ATM path-10). The lack of ATM involvement is further supportedfailure of γ-irradiation and the radiomimetic drugs,ycin and phleomycin, to promote Clr-b downregula-n addition, studies using p53−/− MEF and inhibitorsfunction (such as pifithrin-α; ref. 34) failed to revealvolvement of p53 (a downstream effector of theATR-mediated DDR signaling pathways; ref. 35) on-mediated Clr-b downregulation (data not shown). Inthe partial ability of caffeine to abrogate aphidicolin-ted Clr-b downregulation (on NIH3T3 but not C1498might be due to effects unrelated to the DDR pathwayuch as cell cycle blockade (31), or inhibition of PI3K,K, PAK1, or other caffeine-sensitive kinases (30, 32,. To gain further insight, we examined Clr-b expressiontranscript level.
toxic stress-mediated loss of Clr-b occurs at thecript levelas previously shown that Clr-b is rapidly lost at bothnscript and surface protein levels in response to CMVon (21). However, it is not known whether Clr-b down-tion following genotoxic stress occurs at the transcriptr whether the surface protein is being actively internal-o address this question, Clr-b transcripts were ana-by semiquantitative RT-PCR using primers spanningtire coding sequence. As shown in Fig. 5A, Clr-b tran-
were rapidly lost in C1498 cells undergoing genotoxic5. Genotoxic stress promotes a loss of endogenous Clr-bipts, whereas ectopic expression of Clr-b transcripts abrogatesownregulation. A, semiquantitative RT-PCR analysis of Clr-bipt expression (full-length coding sequence) in C1498 cells eithered or treated with cisplatin (5 μmol/L) as indicated (using 3-foldDNA dilutions). Inverse ethidium bromide gel images andative signal intensities (using Quantity One software) are shown.cytometric analysis of endogenous or ectopically overexpressedrotein on C1498 cells following treatment as indicated.ral C1498 transductants overexpressing a CMV promoter–driventh Clr-b coding sequence were gated according to IRES-GFPr gene expression. Histograms show Clr-b expression (shadedrsus secondary reagent alone (dotted line); numbers indicate MFI
Moreover, the loss of Clr-b transcripts following stress
Cancer Res; 70(18) September 15, 2010 7109
for Cancer Research on January 31, 2013org
inductat theobservfectionof endTo
sequender thvectorsessedtransc
mediaClr-bby traat theNot
portedtransfNKG2slation
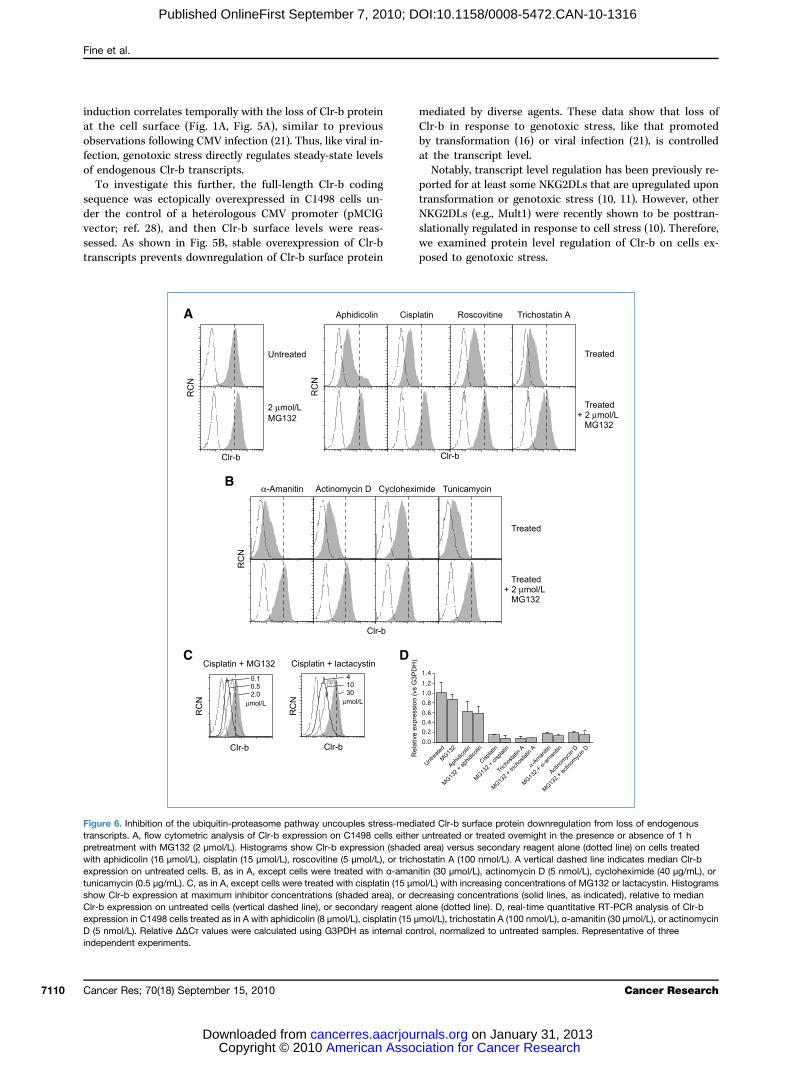
Figuretranscrpretreawith apexpresstunicamshow CClr-b eexpressD (5 nmindepen
Fine et al.
Cance7110
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
ion correlates temporally with the loss of Clr-b proteincell surface (Fig. 1A, Fig. 5A), similar to previousations following CMV infection (21). Thus, like viral in-, genotoxic stress directly regulates steady-state levelsogenous Clr-b transcripts.investigate this further, the full-length Clr-b codingce was ectopically overexpressed in C1498 cells un-e control of a heterologous CMV promoter (pMCIG; ref. 28), and then Clr-b surface levels were reas-
. As shown in Fig. 5B, stable overexpression of Clr-b we exol/L). Relative ΔΔCτ values were calculated using G3PDH as internal control, nodent experiments.
r Res; 70(18) September 15, 2010
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
ted by diverse agents. These data show that loss ofin response to genotoxic stress, like that promotednsformation (16) or viral infection (21), is controlledtranscript level.ably, transcript level regulation has been previously re-for at least some NKG2DLs that are upregulated upon
ormation or genotoxic stress (10, 11). However, otherDLs (e.g., Mult1) were recently shown to be posttran-ally regulated in response to cell stress (10). Therefore,
amined protein level regulation of Clr-b on cells ex-ripts prevents downregulation of Clr-b surface protein posed to genotoxic stress.
6. Inhibition of the ubiquitin-proteasome pathway uncouples stress-mediated Clr-b surface protein downregulation from loss of endogenousipts. A, flow cytometric analysis of Clr-b expression on C1498 cells either untreated or treated overnight in the presence or absence of 1 htment with MG132 (2 μmol/L). Histograms show Clr-b expression (shaded area) versus secondary reagent alone (dotted line) on cells treatedhidicolin (16 μmol/L), cisplatin (15 μmol/L), roscovitine (5 μmol/L), or trichostatin A (100 nmol/L). A vertical dashed line indicates median Clr-bion on untreated cells. B, as in A, except cells were treated with α-amanitin (30 μmol/L), actinomycin D (5 nmol/L), cycloheximide (40 μg/mL), orycin (0.5 μg/mL). C, as in A, except cells were treated with cisplatin (15 μmol/L) with increasing concentrations of MG132 or lactacystin. Histogramslr-b expression at maximum inhibitor concentrations (shaded area), or decreasing concentrations (solid lines, as indicated), relative to medianxpression on untreated cells (vertical dashed line), or secondary reagent alone (dotted line). D, real-time quantitative RT-PCR analysis of Clr-bion in C1498 cells treated as in A with aphidicolin (8 μmol/L), cisplatin (15 μmol/L), trichostatin A (100 nmol/L), α-amanitin (30 μmol/L), or actinomycin
rmalized to untreated samples. Representative of three
Cancer Research
for Cancer Research on January 31, 2013org

RegulgenotAlth
level, ibe pofollowthe Clcouldresiduplemeubiquiexpres(38–41dationstresstor ofInte
gulatioincludand Uslightlface eMG13surfacscriptias α-a(Fig. 6reticuBec
of bloincluder prowas alactacin a dusingImporhad nfollowtional(Fig. 6scriptsNon
essarilof Clr-the acgatesin genOne smal trnated(44). Inacidifimediathesean imwherecontro
in Clr-b transcript levels might rapidly influence Clr-b surfaceexpres
Discu
It hthe NlatedeffectNK ceshowregulanumeto promay isponsing thstress(21), aturesSim
gulatiwas uin resirradiato beand eretentThesedepenstresseHow
regulamechamoterscriptposttrtriguinthat tand rembedzymetranscadenyor immallysequeage sithe otregulatranslsuchThus,untranClr-b–
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.a
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
ation of cell surface Clr-b protein followingoxic stressough Clr-b expression is regulated at the transcriptt is still possible that Clr-b surface protein might alsosttranslationally internalized, either constitutively oring genotoxic stress (10). Consistent with this possibility,r-b cytoplasmic tail contains a number of motifs thataffect its cell surface expression, including several lysinees that may serve as substrates for ubiquitination (Sup-ntary Fig. S3). Indeed, several viruses have evolved E3tin ligases that posttranslationally regulate cell surfacesion of MHC-I, B7 family members, ICAM-1, or NKG2DL). A potential role for the ubiquitin-proteasome degra-pathway in Clr-b downregulation following genotoxicwas investigated using MG132, a pharmacologic inhibi-the 26S proteasome.restingly, MG132 pretreatment abrogated Clr-b downre-n following treatment of C1498 cells with diverse agents,ing aphidicolin, cisplatin, roscovitine, trichostatin-A,V-C irradiation, whereas MG132 treatment aloney (but consistently) increased steady-state Clr-b sur-xpression (Fig. 6A; data not shown). Furthermore,2 administration also blocked downregulation ofe Clr-b in response to generalized inhibitors of tran-on, translation, and posttranslational processing, suchmanitin, actinomycin-D, cycloheximide, and tunicamycinB), the latter of which also promotes endoplasmiclum-associated stress.ause MG132 can exert pleiotropic effects independentcking ubiquitin-dependent proteasomal degradation,ing an ability to inhibit calpains and cathepsins, anoth-teasomal inhibitor of greater specificity, lactacystin,lso used (42). As shown in Fig. 6C, both MG132 andystin prevented stress-mediated Clr-b downregulationose-dependent manner. Similar results were observedNIH3T3 fibroblasts and MEF cells (data not shown).tantly, the administration of proteasomal inhibitorso influence on the downregulation of Clr-b transcriptsing administration of genotoxic agents or transcrip-inhibitors, as revealed by quantitative real-time RT-PCRD). This shows that downregulation of Clr-b tran-can be uncoupled from a loss of Clr-b surface protein.etheless, the MG132 and lactacystin data do not nec-y implicate the proteasome itself in direct degradationb. Pharmacologic inhibition of the proteasome leads tocumulation of nondegraded polyubiquitinated aggre-and a rapid depletion of free ubiquitin levels, resultingeral impairment of ubiquitin-dependent processes (43).uch proteasome-independent process, the endolysoso-afficking pathway, targets endocytosed monoubiquiti-proteins for recycling and/or lysosomal degradationterestingly, chloroquine, an inhibitor of endolysosomalcation (45), also impaired aphidicolin- and cisplatin-ted Clr-b downregulation (data not shown). Collectively,results suggest that ubiquitin-dependent processes playportant role in normal Clr-b cell surface turnover,
as stress-mediated Clr-b downregulation is ultimatelylled at the transcript level. Therefore, any perturbationfuturein resp
acrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
sion.
ssion
as been previously shown that stimulatory ligands forKG2D and DNAM-1 immunoreceptors are upregu-by the DDR pathway following genotoxic stress, anthat enhances the susceptibility of target cells toll–mediated lysis (9, 15). Here, for the first time, wethat genotoxic stress promotes the functional down-tion of an inhibitory NK ligand, Clr-b. The ability ofrous distinct chemotherapeutic and genotoxic agentsmote Clr-b downregulation suggests that cell stressnitiate a conserved and programmed cellular re-e governing Clr-b expression. Thus, pathways govern-e cellular response to transformation (16), genotoxic(an initiating event in transformation), viral infectionnd other pathologic states might share common fea-(46, 47).ilar to the findings reported here for Clr-b downre-on, cell surface expression of the NKG2DL, Mult1,pregulated independently of the ATR/ATM pathwayponse to cell stress induced by heat shock and UVtion (10). Surface expression of Mult1 was showndependent on inhibition of the ubiquitin-proteasomendolysosomal degradation pathways responsible forion of the protein within normal, nonstressed cells.findings highlight a potentially conserved ubiquitin-dent mechanism for Clr-b downregulation underd versus nonstressed conditions.Clr-b transcript and surface protein levels might be
ted requires further investigation. One possiblenism is via the direct control of Clrb (Clec2d) pro-activity and the production of nascent Clr-b tran-s. A second potential mechanism is through theanscriptional regulation of mRNA stability. This in-g possibility was recently suggested by the findinghe rodent Clr-b mRNAs (i.e., the mouse Clec2d8at Clec2d11 gene products; refs. 17, 21) possess anded autocatalytic discontinuous hammerhead ribo-sequence (48). Thus, cleavage of steady-state Clr-bripts by the internal ribozyme upstream of the poly-lation site could promote the loss of mRNA stabilitypair protein translation. Whether protein factors nor-antagonize autocatalysis mediated by the ribozymence, or whether cofactors are recruited to the cleav-te upon genotoxic stress, requires further testing. Onher hand, preexisting Clr-b transcripts may also beted at the level of mRNA stability and/or proteination by microRNA-mediated silencing mechanisms,as that observed for NKG2DL expression (49, 50).Clec2d promoter activity, elements within the Clr-bslated regions, and endogenous or pathogen-encodedspecific microRNAs are all relevant targets to warrant
studies on the missing-self control of Clr-b expressiononse to pathologic processes (16, 17, 21).Cancer Res; 70(18) September 15, 2010 7111
for Cancer Research on January 31, 2013org

In cremaiprogration inknowltransftumorinflueninsighligandrecogn
Disclo
No p
Ackn
ThePflückeC. Kirkh
Grant
HFSPAward E
Refe1. Be
842. Lan3. Me
pro4. Ka
H-fen
5. Macel
6. Ra(NK
7. Gamu
8. Gacel
9. GawaNa
10. NicNK20
11. CeAnNKsus
12. Li JneNK
13. Rabla200
14. Tanda233
15. Soregcelity35
16. CaMiskill
17. Carec
18. IizuGeof
Fine et al.
Cance7112
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
onclusion, how NK cells recognize abnormal targetsns incompletely understood. Understanding how ammed cellular response mediates Clr-b downregula-real time during genotoxic stress will enhance our
edge of the events that lead to innate recognition oformed and infected cells, how malignant NK-resistants evolve, and how chemotherapeutic agents mightce NK recognition of normal and tumor cells. Futuret into this MHC-independent mode of missing-self
modulation will affect our views of NK cell–mediatedtor AwaAwardand CIH
Theof pageaccorda
ition.
sure of Potential Conflicts of InterestRece
ka K, Naidenko OV, Plougastel BF, Fremont DH, Yokoyama WM.netically linked C-type lectin-related ligands for the NKRP1 familynatural killer cell receptors. Nat Immunol 2003;4:801–7.
19. PlofamCd
20. Zhtyp14
21. Voimmu
22. CaJCJ I
23. Cedutor
24. Sahyb
25. Lapreph
26. Kasp35
27. RytarJ E
28. Castr20
29. Aupremi
30. Blotheag
31. Cothe(AT
32. Heofpa
33. Gafeice
34. KoitoSc
35. Sa
r Res; 70(18) September 15, 2010
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
owledgments
authors thank Drs. A. Martin, C.J. Guidos, T.H. Watts, J.C. Zúñiga-r, and L. Horan for helpful comments and suggestions, andam for assistance with p53 genotyping and tissue culture treatments.
Support
Career Development Award CDA-0037/2005, MRI Early ResearcherRA-07-04-071, CIHR Operating Grant FRN-74754, CIHR New Investiga-rd, and BWF Investigator in the Pathogenesis of Infectious Disease(J.R. Carlyle); NSERC PGS-D Award (J.H. Fine); OGS Award (P. Chen);R Vanier Award (A. Mesci).costs of publication of this article were defrayed in part by the paymentcharges. This article must therefore be hereby marked advertisement innce with 18 U.S.C. Section 1734 solely to indicate this fact.
ived 04/14/2010; revised 06/22/2010; accepted 07/15/2010; published
otential conflicts of interest were d isclosed. OnlineFirst 09/07/2010.rencesutler B. Innate immunity: an overview. Mol Immunol 2004;40:5–59.ier LL. NK cell recognition. Annu Rev Immunol 2005;23:225–74.sci A, Ljutic B, Makrigiannis AP, Carlyle JR. NKR-P1 biology: fromtotype to missing self. Immunol Res 2006;35:13–26.rre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of2-deficient lymphoma variants suggests alternative immune de-ce strategy. Nature 1986;319:675–8.cFarlane AW, Campbell KS. Signal transduction in natural killerls. Curr Top Microbiol Immunol 2006;298:23–57.ulet DH. Missing self recognition and self tolerance of natural killer) cells. Semin Immunol 2006;18:145–50.sser S, Raulet DH. The DNA damage response arouses the im-ne system. Cancer Res 2006;66:3959–62.sser S, Raulet DH. Activation and self-tolerance of natural killerls. Immunol Rev 2006;214:130–42.sser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage path-y regulates innate immune system ligands of the NKG2D receptor.ture 2005;436:1186–90.e TJ, Coscoy L, Raulet DH. Posttranslational regulation of theG2D ligand Mult1 in response to cell stress. J Exp Med 2009;6:287–98.rboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A.tigen-activated human T lymphocytes express cell-surfaceG2D ligands via an ATM/ATR-dependent mechanism and becomeceptible to autologous NK- cell lysis. Blood 2007;110:606–15., Rabinovich BA, Hurren R, Cosman D, Miller RG. Survival versusglect: redefining thymocyte subsets based on expression ofG2D ligand(s) and MHC class I. Eur J Immunol 2005;35:439–48.binovich BA, Shannon J, Su RC, Miller RG. Stress renders T cellsts sensitive to killing by activated syngeneic NK cells. J Immunol0;165:2390–7.g KF, Ren H, Cao J, et al. Decreased Dicer expression elicits DNA
mage and up-regulation of MICA and MICB. J Cell Biol 2008;182:–9.riani A, Zingoni A, Cerboni C, et al. ATM-ATR-dependent up-ulation of DNAM-1 and NKG2D ligands on multiple myelomals by therapeutic agents results in enhanced NK cell susceptibil-and is associated with a senescent phenotype. Blood 2009;113:03–11.rlyle JR, Jamieson AM, Gasser S, Clingan CS, Arase H, Raulet DH.sing self-recognition of Ocil/Clr-b by inhibitory NKR-P1 naturaler cell receptors. Proc Natl Acad Sci U S A 2004;101:3527–32.rlyle JR, Mesci A, Fine JH, et al. Evolution of the Ly49 and Nkrp1ognition systems. Semin Immunol 2008;20:321–30.
ugastel B, Dubbelde C, Yokoyama WM. Cloning of Clr, a newily of lectin-like genes localized between mouse Nkrp1a and69. Immunogenetics 2001;53:209–14.ou H, Kartsogiannis V, Hu YS, et al. A novel osteoblast-derived C-e lectin that inhibits osteoclast formation. J Biol Chem 2001;276:916–23.igt S, Mesci A, Ettinger J, et al. Cytomegalovirus evasion of innatemunity by subversion of the NKR-P1B:Clr-b missing-self axis. Im-nity 2007;26:617–27.rlyle JR, Martin A, Mehra A, Attisano L, Tsui FW, Zuniga-Pflucker. Mouse NKR-P1B, a novel NK1.1 antigen with inhibitory function.mmunol 1999;162:5917–23.rwenka A, Bakker AB, McClanahan T, et al. Retinoic acid early in-cible genes define a ligand family for the activating NKG2D recep-in mice. Immunity 2000;12:721–7.nderson S, Shastri N. LacZ inducible, antigen/MHC-specific T cellrids. Int Immunol 1994;6:369–76.
Belle JL, Truitt RL. Characterization of a murine NKT cell tumorviously described as an acute myelogenous leukemia. Leuk Lym-oma 2002;43:1637–44.rlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigenecificity of Ly-49+ IL-2-activated natural killer cells. Nature 1992;8:66–70.an JC, Niemi EC, Nakamura MC, Seaman WE. NKR-P1A is aget-specific receptor that activates natural killer cell cytotoxicity.xp Med 1995;181:1911–5.rlyle JR, Mesci A, Ljutic B, et al. Molecular and genetic basis forain-dependent NK1.1 alloreactivity of mouse NK cells. J Immunol06;176:7511–24.st JG, Gays F, Mickiewicz KM, Buchanan E, Brooks CG. The ex-ssion and function of the NKRP1 receptor family in C57BL/6ce. J Immunol 2009;183:106–16.ck WD, Merkle D, Meek K, Lees-Miller SP. Selective inhibition ofDNA-dependent protein kinase (DNA-PK) by the radiosensitizing
ent caffeine. Nucleic Acids Res 2004;32:1967–72.rtez D. Caffeine inhibits checkpoint responses without inhibitingataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-relatedR) protein kinases. J Biol Chem 2003;278:37139–45.Z, Ma WY, Hashimoto T, Bode AM, Yang CS, Dong Z. Inductionapoptosis by caffeine is mediated by the p53, Bax, and caspase 3thways. Cancer Res 2003;63:4396–401.brielli B, Chau YQ, Giles N, Harding A, Stevens F, Beamish H. Caf-ne promotes apoptosis in mitotic spindle checkpoint-arrestedlls. J Biol Chem 2007;282:6954–64.marov PG, Komarova EA, Kondratov RV, et al. A chemical inhib-
r of p53 that protects mice from the side effects of cancer therapy.ience 1999;285:1733–7.ncar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. MolecularCancer Research
for Cancer Research on January 31, 2013org

mepo
36. Sakin199
37. FoDirphkin
38. CaE3
39. Coensis12
40. CoICA20
41. Thandpro105
42. Leecel
43. Mimbiq
byrepsp
44. Botei39
45. Ohlysag
46. LuviruATlyti
47. WiCein20
48. Mametur
49. Stesy37
Cell Stress Promotes Missing-Self Recognition by NK Cells
www.a
Published OnlineFirst September 7, 2010; DOI:10.1158/0008-5472.CAN-10-1316
chanisms of mammalian DNA repair and the DNA damage check-ints. Annu Rev Biochem 2004;73:39–85.rkaria JN, Busby EC, Tibbetts RS, et al. Inhibition of ATM and ATRase activities by the radiosensitizing agent, caffeine. Cancer Res9;59:4375–82.
ukas LC, Daniele N, Ktori C, Anderson KE, Jensen J, Shepherd PR.ect effects of caffeine and theophylline on p110 δ and other phos-oinositide 3-kinases. Differential effects on lipid kinase and proteinase activities. J Biol Chem 2002;277:37124–30.dwell K, Coscoy L. Ubiquitination on nonlysine residues by a viralubiquitin ligase. Science 2005;309:127–30.scoy L, Sanchez DJ, Ganem D. A novel class of herpesvirus-coded membrane-bound E3 ubiquitin ligases regulates endocyto-of proteins involved in immune recognition. J Cell Biol 2001;155:65–73.scoy L, Ganem D. A viral protein that selectively downregulatesM-1 and B7-2 and modulates T cell costimulation. J Clin Invest
01;107:1599–606.omas M, Boname JM, Field S, et al. Down-regulation of NKG2DNKp80 ligands by Kaposi's sarcoma-associated herpesvirus K5tects against NK cell cytotoxicity. Proc Natl Acad Sci U S A 2008;:1656–61.DH, Goldberg AL. Proteasome inhibitors: valuable new tools for
l biologists. Trends Cell Biol 1998;8:397–403.naugh EG, Chen HY, Davie JR, Celis JE, Neckers L. Rapid deu-
uitination of nucleosomal histones in human tumor cells caused
50. StestrNK
acrjournals.org
American Association Copyright © 2010 cancerres.aacrjournals.Downloaded from
proteasome inhibitors and stress response inducers: effects onlication, transcription, translation, and the cellular stress re-onse. Biochemistry 1997;36:14418–29.nifacino JS, Traub LM. Signals for sorting of transmembrane pro-ns to endosomes and lysosomes. Annu Rev Biochem 2003;72:5–447.kuma S, Poole B. Fluorescence probe measurement of the intra-osomal pH in living cells and the perturbation of pH by variousents. Proc Natl Acad Sci U S A 1978;75:3327–31.o MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalo-s disrupts both ataxia telangiectasia mutated protein (ATM)- andM-Rad3-related kinase-mediated DNA damage responses duringc infection. J Virol 2007;81:1934–50.ebusch L, Neuwirth A, Grabenhenrich L, Voigt S, Hagemeier C.ll cycle-independent expression of immediate-early gene 3 resultsG1 and G2 arrest in murine cytomegalovirus-infected cells. J Virol08;82:10188–98.rtick M, Horan LH, Noller HF, Scott WG. A discontinuous ham-rhead ribozyme embedded in a mammalian messenger RNA. Na-e 2008;454:899–902.rn-Ginossar N, Elefant N, Zimmermann A, et al. Host immunestem gene targeting by a viral miRNA. Science 2007;317:6–81.rn-Ginossar N, Gur C, Biton M, et al. Human microRNAs regulateess-induced immune responses mediated by the receptor
G2D. Nat Immunol 2008;9:1065–73.Cancer Res; 70(18) September 15, 2010 7113
for Cancer Research on January 31, 2013org