chapter 15 chemical kineticsweb.unbc.ca/chemistry/chem101/chem101_ch15_lectures_1_and_2.p… · the...
TRANSCRIPT

Chapter 15 Chemical Kinetics
-for any chemical reaction, there are three questions that are paramount:
- what?- how much?- how fast?

- the first is, in many ways, the most difficult- analytical chemistry allows us to determine
the product of chemical reactions (infrared and NMR spectroscopy, elemental analysis, mass spectroscopy)
- working out the product of chemical reactions is also the most demanding portion of the job
- synthetic chemists develop a certain amount of knowledge that allows them to reasonably guess the results of the reactions
- products generally related to the starting material

- the second question is “thermodynamics”- the extent of a reaction is governed by the
energy difference between the products and the reactants
- the greater the energy difference, the more product should form
- depends upon having a pathway- for example, the organic component of the
human body is thermodynamically unstable relative to carbon dioxide and water but contrary to popular myth people don’t spontaneously burn up!

- the third question deals with “kinetics”- how fast a reaction will proceed is critical
- two demonstrations:
- an iron nail in water
- hydrogen gas (H2) in a balloon

- both reactions are oxidations- iron metal is oxidized to iron (III) - Fe3+
- hydrogen atoms are oxidized to protons – H+
- both are exothermic- the amount of heat given off by either reaction
is about the same (about 800 kJ/mol)
- but they occur at significantly different rates!!- the reaction of oxygen with iron take weeks;
with hydrogen a matter of milliseconds

- Indeed, we can alter the rate of reaction:
- hydrogen in balloon; air outside
- hydrogen and air in balloon
- hydrogen and oxygen in balloon

- reaction of hydrogen with oxygen occurs at the “diffusion limit”
- this means that every time molecules of hydrogen and oxygen collide, the react to give water and recall from the kinetic molecular theory of gases that gas molecules are moving at hundreds of metres per second – typically 1011 collisions per second
- other extreme are reactions that don’t collide very often and have a low probability of success
- the reaction of iron with oxygen depends upon O2 bumping into the surface of the iron and a reaction actually taking place
- (Note: page 615 – combustion and explosions)

Factors that affect reaction rates:- anything that modifies the number of collisions
- temperature – the gas laws tell us that molecules move faster with higher temperatures
- concentration – more molecules means more collisions between reactants per second
- pressure – really just another form of concentration

- physical state – atoms move around in gases and liquids, not in solids
- anything that will allow a collision that is successful
- orientation – molecules must be aligned correctly for reaction to occur
- activation energy – colliding molecules must have sufficient energy to break bonds


15.1 The Rate of a Chemical Reaction
- rate is a measure of how much in how long
- for example,- 75 words per minutes - 60 kilometres per hour- 14 L per 100 kilometres- one problem set per class

- for chemical reactions, we use:
- “moles consumed” or “moles produced” per unit time (typically “seconds”)
why?- moles are a measure of the actual number of
molecules involved in the reaction as a mole represents a specific number of individual molecules

- use units of concentration:
- molarity - moles per litre or mol/litre (M)
- molality - moles per kilogram or mol/kg (m)
- partial pressure - moles per volume (atm)
All are just a measure of moles

Consider the following reaction:
2A + B products
If we had an initial concentration of B equal to 0.014 M and after 26 seconds we had only 0.008 M of B, then:
[B]initial = 0.014M [B]final = 0.008Mtinitial = 0 seconds tfinal = 26 seconds
rate = initial - final concentrations/initial - final time

rate = ([B]o – [B]f)/(to – tf)
= ∆[B] / ∆[t]
= (0.014 – 0.008)/(0-26)
= 0.006 M/-26 s = -2.3 x10-4 M/s

Note:The answer is “negative” in that it is telling us that
B is consumed during the reaction. However, there really isn’t any way for the rate of a reaction to be “negative” – it would be like going -30 kph. Rates should always be reported as a positive number.

If 0.006 M of B is consumed, then the stoichiometry of the reaction tells us that 0.012 M of A is consumed. (2A + B …)
The rate of consumption of [A] must be:
rate = (0.012 M)/(-26 s) = -4.6 x10-4 M/s
So, what is the actual rate of the reaction?

By definition,for the reaction:
aA + bB cC + dD
the reaction rate is:
rate = (-1/a)(∆[A]/∆t) = (-1/b)(∆[B]/∆t) = (1/c)(∆[C]/∆t) = (1/d)(∆[D]/∆t)
Note: for the above example, this means that the rate of the reaction is 2.3 x10-4 M/s


15.2 Measuring Reaction Rates
- measuring “time” is straight forward
- measuring concentration is much more difficult- need a parameter that corresponds to
something observable for the molecules involved in the reaction
- for example, changes in colour or emitted light are frequently used to monitor reactions

- for gases, changes in pressure are directly linked to the number of moles of gas present through the gas laws
- for ionic solutions, conductivity can be used as the conductivity directly corresponds to the number of ions
- for redox reactions, changes in voltage or current can monitor concentration

Any tag will do – provided there is a fixed relationship between the observable property and the molecules of reactant/product present in solution
for example, tritium labeling is often used to monitor biologically interesting molecules as the radiation from the tritium is sensitive to amount of tritium present
page 581 – apparatus for collecting gas from the decomposition of hydrogen peroxide

Having collected concentration and time data, what do we do with it?
time (s) [H2O2] (M)0 2.32
200 2.01400 1.72600 1.49
1200 0.981800 0.623000 0.25
Table 15.1

We can graph this:

we can also calculate specific values:
time (s) [H2O2] (M)0 2.32
200 2.01400 1.72600 1.49
1200 0.981800 0.623000 0.25

Average: the slope of the line joining any two data points is the “average rate” over that intervalnote: not necessarily the “rate of reaction” as it only really works where the relationship between [ ] and time is linear
Instantaneous: the tangent to the curve
Initial: the instantaneous rate at the point of mixing – very difficult to actually measure as it implies a tangent to a point!
So how do we determine the actual reaction rate?


15.3 Effect of Concentration on Reaction Rate: The Rate Law
One of the goals of studying chemical kinetics is to be able to predict the relationship between rates and concentrations at any time during the reaction. This is achieved through a derived equation called “the rate law” or, sometimes, “the rate equation”. (We will use the former.)

For any reaction, such as,aA + bB products
we can express the initial rate asrate = k[A]m[B]n
where ‘m’ and ‘n’ do not necessarily equal the stoichiometric coefficients ‘a’ and ‘b’ (and, indeed, may be ‘zero’ implying that the reaction rate is independent of the concentration of that species!) and ‘k’ is a constant – called ‘the rate constant’

the term ‘order’ is used to describe the relationship between the concentration of a particular species and the reaction rate.
rate = k[A]0 means that the reaction is zero order in [A]
= k[A]1 means that the reaction is first order in [A] (note: we don’t normally write the ‘1’!)
= k[A]2 means that the reaction is second order in [A]
and so on

further, the overall order of a reaction is determined by the sum of the order of each individual concentration
rate = k[A][B] means that the overall order is 2 (or second order) while the order with respect to each reactant is only 1 (or first order)
= k[A]0[B]2 means that the overall order is 2 but the order with respect to ‘A’ is zero, while with respect to ‘B’, the reaction is second order

the effect of the order of a reactant upon a rate is then:
zero order – changing the concentration has no effect on the rate
first order – doubling the concentration doubles the rate- halving the concentration halves the rate
second order – doubling the concentration quadruples the rate- halving the concentration quarters the rate
and so on for higher orders

further, the larger k is, the faster the rate of the reaction for any given set of concentrations
KEY POINTS- the rate law is obtained empirically from the data
that is, the reaction order, rate constant, and rate of reaction are all obtained directly from the measurements
- there is no way to know these just from “looking” at the balanced equation
- the rate law allows us to calculate or express the concentration of a reactant as a function of time

Examples
initial [A] initial rate1.0 M 2.2 x10-2
2.0 M 4.4 x10-2

initial [A] initial rate1.0 M 1.6 x10-3
2.0 M 1.6 x10-3

initial [A] initial rate1.0 M 1.1 x10-2
3.0 M 9.9 x10-2

Calculus and the integrated form of the Rate Law
rates are really just an application of differential calculus
that is, ∆[A] / ∆t = dA/dtwhere dt becomes vanishingly small
that is, for an instantaneous rate, we can express the rate as a derivative and the rate law becomes a differential equation
that is, we can use worked out expressions for differential equations to calculate all of the rate laws


15.4 Zero-Order Reactions
the rate law for a zero order reaction is given byd[A]/dt = -k[A]o = -k
rearranging gives,d[A] = -kdt
and integrating both sides from time zero to time t gives,
∫ d[A] = ∫ -kdt
which equals,[A]t - [A]o = -kt or [A]t = -kt + [A]o

this an equation for a straight line (y = mx + b), where,
- the independent variable (x) is time
- the dependent variable (y) is the concentration of ‘A’ remaining, [A]t
- the slope (m) is –k, the rate constant
- the intercept (b) is the initial concentration of ‘A’, [A]o

Example:The bromination of acetone is a zero order reaction in the presence of excess bromine, giving,
[acetone]t = -kt + [acetone]o
if the initial concentration of acetone equals 0.12 M and the rate constant is 1.8 x10-2 M/s, then how long will it take the acetone to disappear?
(0.0 M) = (-1.8 x10-2 M/s)t + (0.12 M)
t = (0.12 M / 1.8 x10-2 M/s) = 6.7 seconds

What would be the concentration of the acetone remaining after 3 seconds?
[acetone]t = (-1.8 x10-2 M/s)(3 s) + (0.12 M)
= 0.066 M
We can answer any question about the relationship between the rate, the concentration, and the time, provided that we know three out of the four variables.
Book says:- the concentration/time graph is a straight line with a
negative slope

15.5 First-Order Reactions
Overall, a fist order reaction has a rate law that looks something like,
d[A] / dt = -k[A]
which can be re-arranged to:
d[A]/[A] = -kdt
and integrated to give:
ln[A]t - ln[A]o = -kt

which can be rearranged to give the ‘integrated form of the rate law’:
ln[A]t = -kt + ln[A]o
which is the equation of a straight line (y = mx + b) with a slope that gives the rate constant.
Knowing any three variables, we can calculate the fourth.
More importantly, if the concentration of ‘A’ can be measured as a function of time, then a plot of the natural logarithm versus time will give the rate constant

Examples
If the initial concentration of ‘A’ is 0.253 M and the rate constant is 3.8 x10-3 s-1, then how much will be left after 120 seconds?
ln[A]t = -(3.8 x10-3 s-1)(120 s) + ln[0.253]
= -1.8304
[A]t = e-1.8304 = 0.160 M

Or, if the concentration of ‘A’ goes from 0.253 M to 0.1265 M in 180 seconds, what would be the rate constant?
This requires that we rearrange the integrated rate law to give:
-k = (ln[A]t - ln[A]o)/t
substituting in the data gives:
-k = (-2.0675 - -1.3744)/180 = -0.00385 s-1

Note that in the above example, 180 seconds is the time it takes for exactly one half of the reactant to disappear.
This is a special value, called the ‘half-life’ or t½, and it is a constant for a first order reaction.
That is, for a half life of 180 seconds, we have:t [A]0 1.0 M
180 0.5360 0.25540 0.125720 0.0625
etcetera

the half-life of a reaction that is first order can be worked out from our rate law:
ln[A]t = -kt½ + ln[A]o
ln(½[A]o) = -kt½ + ln[A]o
ln(½[A]o) - ln[A]o = -kt½
ln (½[A]o/[A]o) = ln(½) = -0.693 = -kt½
t½ = 0.693/k or k = 0.693/t½

For any first order reaction, the half-life is some constant value – given by 0.693/k. This is a unique feature of first order reactions and has some interesting implications. For example, for the radioactive decay of the elements, the half-life tells us that the material will be around for a very long time as each half-life only destroys one half of the remaining amount.
(see Table 15.4)

Note: many reactions involving gases are ‘first order’ reactions but in these cases, we can use ‘partial pressure’ of the gas as a substitute for concentration. Recall that PV = nRT, so P =(n/V)RT where (n/V) is a measure of moles per unit volume – so PA = [A] (at a constant T). Hence, we can speak about the half-life of gases in the atmosphere or the time it takes for the ozone hole to repair itself.
Another example is the uptake of oxygen by Hemoglobin in the blood, which has a complicated expression for the fraction of blood that is oxygenated:
f = KPo2.8/(1 + KPo
2.8)
which can be used to determine the rate of oxygenation of the blood.


15.6 Second Order Reactions
In this case, d[A]/dt = -k[A]2
which, rearranged, gives:
d[A]/[A]2 = -kdt
and integration gives:
1/[A]t - 1/[A]o = -kt
or 1/[A]t = -kt + 1/[A]o
which, again, is the equation of a straight line.

we can also work out an expression for the half-life of a second order reaction:
1/([A]o/2) = -kt½ + 1/[A]o
2/[A]o = -kt½ + 1/[A]o
2/[A]o - 1/[A]o = -kt½
kt½ = 1/[A]o
and t½ = 1/k[A]o or k = 1/([A]ot½)
Note that this means that t½ depends upon the initial concentration of the reacting species – it will increase as the reaction proceeds. The half-life is not a constant.

Pseudo-first order kinetics- there are two types of second order rate laws that one encounters:
rate = k[A]2 or rate = k[A][B]
- the first can only be monitored as a second order reaction
- the second can be modified to give a “pseudo-first order reaction”
- mathematically, it is far easier to deal with a first order reaction than a second order reaction

Pseudo-first order conditions are created by holding one of the reactants at a constant concentration – which is not really possible as, by definition, a reactant is something that takes part in a reaction!!
Instead, kineticists cheat and hold the concentration “almost” constant.
ex. – in book – a reaction involving water- the concentration of water is always 55.5 M and a reaction that consumes a small amount of water will not change this
hence, rate = k[A][H2O] can be re-written as:rate = k’[A] where k’ = k[55.5M]

more practically speaking,a ratio of 100:1 is more usually sufficient to ensure pseudo-first order conditions:
consider, A + B products
if [A] = 1 and [B] = 100, then [A]o = 1 and [A]f = 0but [B]o = 100 and [B]f = 99
so, the concentration of ‘B’ remains at roughly 100 during the entire reaction and we can write:
rate = k’[A] where k’ = k[B]this has practical advantages when trying to measure the rate of a reaction


15.7 Reaction Kinetics: A Summary
To calculate a rate of reaction when the rate law is know, use the expression: rate = k[A]n[B]m….
To determine a rate of reaction when the rate law is not given, use the integrated form of the rate law and plot the data to obtain a straight line.
To determine the order of a reaction, with respect to the reactants:
- calculate the ratio of rates to the ratio of reactants for two data points
- use the logarithms of the ratios

Order Rate Law Integrated Line Half-lifeForm
0 d[A]/dt = k [A]t = -kt + [A]0 [A] vs. time [A]0/2k
1 d[A]/dt = k[A] ln[A]t = -kt + ln[A]0 ln[A] vs. time 0.693/k
2 d[A]/dt = k[A]2 1/[A]t = -kt + 1/[A]0 1/[A] vs. time 1/k[A]0

Figure 15-7 – page 596


15.8 Theoretical Models for Chemical Kinetics
- when considering a reaction, the rate law tells us what is going on overall – but not what is happening to each molecule- this is the mechanism of the reaction and for many reactions, our understanding of mechanistic questions lead us to be able to predict what is likely to occur and why
- however, it is important to remember that mechanisms are speculations based on the ‘best’ data that we have

Collision Theory
- this approach models all chemical reactions on the basis of the number of collisions that two reactants experience and on how successful those collisions are
- the number of collisions can be worked out on the basis of the gas laws and the Kinetic Molecular Theory (gas phase collisions ~1030 s-1; solution ~1012 s-1)
- but only a fraction are successful at yielding products.- the question is – “how do we measure this?”

- the collision theory equates success to kinetic energy (or “activation energy”)
“the activation energy is the minimum energy above the average kinetic energy that molecules must bring to their collisions for a chemical reaction to occur”
for a gas phase reaction, a Boltzmann distribution applies:
Fig. 15-8
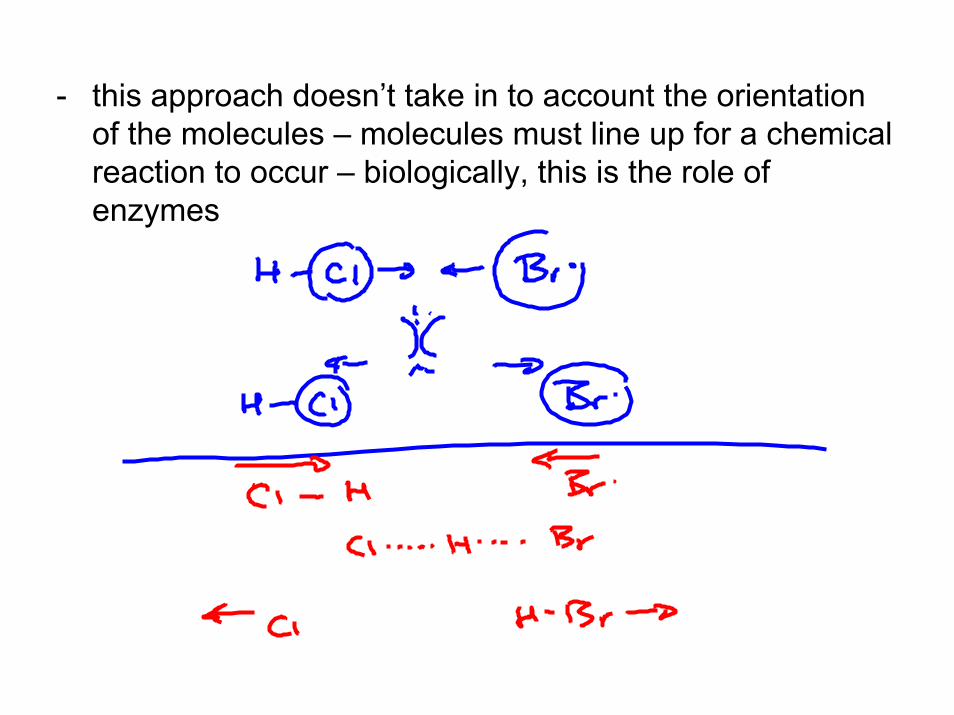
- this approach doesn’t take in to account the orientation of the molecules – molecules must line up for a chemical reaction to occur – biologically, this is the role of enzymes

Transition State Theory- Henry Eyring proposed a model for a chemical reaction in which the molecule or reactants proceed through some intermediate species called “the transition state”
(more accurately, Erying proposed that in going from reactants to products, the molecules must follow a potential energy surface that is continuous)
- Consider the reaction of HCl and Br.

- the reaction proceeds through an “activated complex” which is an intermediary species
- the book says that it is “hypothetical” which is not true –it is not observable but it is not hypothetical
- it is important to note that this approach to viewing a reaction means that all reactions are reversible as any small forward step across the potential energy surface can be reversed – this is known as the “principal of micro-reversibility”
- but for some reactions, it is hard to imagine them being reversed!

- a reaction profile:

15.9 The Effects of Temperature on Reaction Rates
In 1889, Svante Arrhenius determined the relationship between temperature and rate constant:
k = A e(-Eact/RT)
or: ln k = (-Eact/RT) + lnA
note that this is in the form of a straight line with the independent variable being 1/T and the slope (-Eact/R)

In this case, the gas constant is expressed in terms of energy (Joules) rather than volume and pressure (litre.atm):
- R = 8.3145 J/mol.K
This is equivalent to the gas constant used in the gas laws (0.082057 L.atm/mol.K) but I will leave it to you to demonstrate that!

A plot of the natural logarithm of the rate constant against temperature should give a straight line and is the best method of determining the activation energy for a reaction.
But it not always possible to plot the data and the second best method is to use the "Arrhenius Equation":
ln (k2/k1) = (Eact/R)(1/T1 - 1/T2)

- knowing (k1, T1) and (k2, T2), we can calculate Eact
- knowing Eact and (k1, T1), we can calculate k for any temperature or the temperature for any value of 'k'
note: this equation has the same form as the Clausius-Clapeyron equation. Why? Vapour pressure is a rate process - it depends upon the rate that molecules can overcome the activation barrier at the surface of a liquid. This activation barrier is the "surface tension".
An example: T (K) k288 0.052298 0.101308 0.184318 0.332

T (K) k 1/T ln k288 0.052 0.0034722 -2.9545298 0.101 ► 0.0033557 -2.2926308 0.184 0.0032470 -1.6928318 0.332 0.0031447 -1.1026
plotted, using linear regression, this data gives us a straight line with a slope = -5640.6 and an intercept = 16.63
from our equation for a line, ln k = (-Eact/RT) + lnA, we know that the slope is -Eact/R and therefore,
Eact = -(slope)(R) = -(-5640.6)(8.3145 J/mol.K)= 46,900 J/mol or 46.9 kJ/mol

note that if we had simply used the data points in pairs, we would have arrived at different answers:
re-arranging the Arrhenius Equation, we get:
Eact = [R(ln(k2/k1)]/[1/T1 - 1/T2]
using points 1 and 2 gives,
Eact = [(8.3145)(ln(0.101/0.052)]/[1/288 - 1/298]= 47.4 kJ/mol

using points 2 and 3 gives,
Eact = [(8.3145)(ln(0.184/0.101)]/[1/298 - 1/308]= 45.8 kJ/mol
using points 3 and 4 gives,
Eact = [(8.3145)(ln(0.332/0.184)]/[1/308 - 1/318]= 48.1 kJ/mol
- all of which are close to the correct value but not as good as graphing the data set

Another example,if a reaction has a rate constant of 4.36 at 25°C, and an activation energy of 23.2 kJ/mol, what will be the rate of reaction at 50°C?
ln (k2/k1) = (Eact/R)(1/T1 - 1/T2)k1 = 4.36; T1 = 298.15 K; T2 = 323.15 K; Eact = 23,200 J/mol
ln(k2/4.36) = (23,200/8.3145)(1/298.15 - 1/323.15)= 0.72405
ln k2 - ln(4.36) = 0.72405ln k2 = 0.72405 +ln(4.36) = 2.19652
k2 = e2.19652 = 8.99

At what temperature will the above reaction double its rate compared to 25°C? 50°C?
ln (k2/k1) = (Eact/R)(1/T1 - 1/T2)k2/k1 = 2; T1 = 298.15 K; Eact = 23,200 J/mol; T2 = ?
ln 2 = 0.693 = (23,200/8.3145)(1/298.15 - 1/T2)0.693 = 9.35907 - 2790.4/T2
2790.4/T2 = 9.35907 - 0.693 = 8.66607
T2 = 2790.4/8.66607 = 322 K or 49°C

ln (k2/k1) = (Eact/R)(1/T1 - 1/T2)k2/k1 = 2; T1 = 323.15 K; Eact = 23,200 J/mol; T2 = ?
ln 2 = 0.693 = (23,200/8.3145)(1/323.15 - 1/T2)0.693 = 8.63502 - 2790.4/T2
2790.4/T2 = 8.63502 - 0.693 = 7.94202
T2 = 2790.4/7.94202 = 351 K or 78°C
Important to always write out all of the known values and the unknowns - and ask "is the answer reasonable??"

15.10 Reaction Mechanisms
A reaction mechanism is a step-by-step detailed description of a chemical reaction. Each individual step is a single elementary process or reaction. That is, any step that significantly alters a molecule's energy or geometry or produces a new molecule is an elementary step.
Mechanisms are theoretical constructs that must be consistent with the observed data.- all of the elementary steps must add up to the overall reaction

and- must account for the observed rate law
A mechanism should also account for by-products of the reaction.
Only the reactions up to and including the rate determining step are included in the rate law. And, strictly speaking, it is only the reactions up to and including the rate determining step that we can know anything about!

Elementary Processes
1) unimolecular - involving only a single molecule
bimolecular - involving two molecules
termolecular - involving three molecules at the same time - very, very rare
2) the exponents for the concentration terms in a rate law (the order of the reaction) are the stoichiometric factors for an elementary process

i.e. A + B → products
rate = k[A][B] - second order overall and first order in each of 'A' and 'B'
2A + B → products
rate = k[A]2[B] - third order overall, while second order in 'A'
3) Elementary processes may written as reversible reactions (written as equilibria)

4) if a compound is produced in one reaction and consumed in another, then it is not part of the rate law or overall reaction.
More importantly, we can assume that the change in the concentration of any intermediate species with time is essentially 'zero'.
i.e. A + B → CC → products
d[C]/dt = 0 - generally, it is consumed as fast as it is produced

5) The slowest elementary process is the "rate determining step"
- may be the first, last, or anything in between
And, only elementary processes up to and including the rate determining step have any effect on the rate law.

for example, a mechanism with a slow step followed by a fast step:
Overall stoichiometry: 2A + B → products
But the reaction rate law is: rate = k[A][B]
How do we reconcile this?
Postulate a mechanism with two steps and the first step is the slow step.

Step 1 A + B → C
Step 2 C + A → products
if k1 < k2 then k1 is the rate determining step and the observed rate law is that for the first elementary step.
Hence, rate = k1[A][B]
But overall, Step 1 and Step 2 still add up to the overall reaction: 2A + B → products

the book gives the example of the reaction of hydrogen with iodochloride:
H2 + ICl → HI + HClHI + ICl → I2 + HCl
overall: H2 + 2ICl → I2 + 2HCl
rate = k1[H2][ICl]
- chemically speaking, the step with the highest activation barrier is the rate determining step and in this case, it is the first one

Mechanism with a fast equilibrium followed by a slow step
- this is important in biochemistry/biology as many enzymes and other reactions proceed through an association followed by reaction
- in generic terms,A + B C fast
C → products slow
rate should be = k3[C]but 'C' is just an intermediate and not part of the overall reaction - we can use the 'steady state approximation'

the concentration of 'C' doesn't really change over time and is effectively equal to 'zero'
hence, d[C]/dt ≈ 0
and d[C]/dt = k1[A][B] - k2[C] - k3[C] ≈ 0
therefore, k1[A][B] = (k2 + k3)[C]
giving [C] = k1[A][B]/(k2 + k3)
and the overall rate = k1k3[A][B]/(k2 + k3)

now, if k2 > k3, then this simplifies to (k1k3/k2)[A][B] and we can re-write (k1/k2) as "Keq"
then the rate of reaction is simply: rate = k3Keq[A][B]
- this is a fairly common reaction type
- pre-equilibration must happen for most reactions in solution as the reactant must diffuse together before reacting

Chain reactions:initiation: Br2 → 2Br.
propogation: Br. + H2 → HBr + H.
H. + Br2 → HBr + Br.
termination: H. + HBr → H2 + Br.
Br. + Br. → Br2
in this case, d[HBr]/dt = k[H2][Br2]½ / (1 + k'[H2]/[Br2])
where k = 2k2(k1/k5)½ and k' = k4/k3

Enzyme Kineticsbook deals with this under 'catalysis'
notation: E = enzyme; S = substrate; ES = substrate/enzyme
complex
mechanism: E + S ESES → products
where the rate of production of products is:d[products]/dt = k2[ES]

note that the steady state approximation comes into play and the rate of production of the complex is essentially the rate of destruction, hence,
k1[E][S] = (k-1 + k2)[ES]
similar to our pre-equilibrium expression but here we must take into account the total concentration of the enzyme (Eo) which is composed of both free ('E') and complexed('ES') enzyme:
[Eo] = [E] + [ES]
or: [E] = [Eo] - [ES]

which gives us,k1([Eo] - [ES])[S] = (k-1 + k2)[ES]
and rearranging gives,k1[Eo][S] = ((k-1 + k2) + k1[S])[ES]
or: [ES] = k1[Eo][S] / ((k-1 + k2) + k1[S])
and finally,
d[products]/dt = k1k2[Eo][S] / ((k-1 + k2) + k1[S])

if we divide through by (k1/k1) then:
d[products]/dt = (k2k1/k1)[Eo][S] / ((k-1 + k2)/k1 + k1/k1[S])
and if we let Km = (k-1 + k2)/k1
then the rate = k2[Eo][S] / (Km + [S])
This is a very special mechanism and rate law. It is called "Michaelis-Menton Kinetics" and is critical in biological systems where enzyme saturation kinetics can be observed.



15.11 Catalysis
a substance that undergoes no net change during a chemical reaction but affects the rate of a reaction is called a 'catalyst'
all sorts of substances act as 'catalysts'- acid or H+ found in organic chemistry- Superoxide Dismutase - biological catalyst for the
destruction of the superoxide ion- shards of pottery - aluminosilicates - for making
sulphuric acid

General picture is that catalysts work by forming a complex with the reactants which alters the energy profile of the reaction:

Types of catalysts:
Homogeneous - everything is in the same phaseHeterogeneous - reactants and catalysts are in different
phases
Biological - for example, nitrogenase
N2 + 8H+ + 8e- → 2NH3 + H2
- all enzymes are catalysts!