chapter 2 amperometric glucose...
TRANSCRIPT

14
CHAPTER 2
AMPEROMETRIC GLUCOSE BIOSENSORS: PAST,
PRESENT AND FUTURE
This chapter is about the past, present and future of technology development of glucose
biosensors.
2.1 Historical Perspectives of Glucose Sensors
Foundations of biosensor and its technological advancements thereof, was laid down by father
of the biosensor, Professor Leland C Clark Jnr., in 1956 with advent of Clark oxygen
electrode. The first biosensor dates back to 1962, when Clark and Lyons of the Cincinnati
Children’s Hospital proposed the first enzyme based electrode to measure blood glucose[26].
The underlying principle being monitoring the oxygen consumption, during the oxidation of
glucose, by glucose oxidase (GOx) entrapped between semipermeable membranes over an
oxygen electrode. Since the pioneering work of Clark and Lyons, although a variety of
techniques and methodologies focusing on improvements of signal transduction and
immobilizations of the biomolecule for glucose biosensor are reported, still it has changed
little in principle over several years.
The electrochemical and colorimetric glucose biosensor developed alongside. First
colorimetric biosensor was launched by Dextrostix in 1965 in the form of blood glucose-
sensing strip based on colorimetric detection of hydrogen peroxide produced during the
oxidation of glucose by glucose oxidase [27]. This was closely followed by first functional
electrochemical biosensor by Updike and Hicks in 1967 [28] for glucose and potentiometric
biosensor for urea by Guilbault and Montalvo in 1969 [29]. Yellow Spring Instrument (YSI)
Company in 1975, launched the first Model 23 YSI glucose analyzer, for direct estimation of
glucose levels in blood. 1974-75 marked a turning point in biosensor history with the
proposed usage of - thermal transducers in enzymatic biosensors [30] and bacteria in place of
enzymes for measurement of alcohol. Later, in 1980 an optical biosensor using alcohol
oxidase enzyme was reported for alcohol detection [31]. Since then different biomolecules –
enzymes, microorganism, DNA, antigen/antibody etc. have been used as bioreceptor element.
Hence, based on the type of biomolecule used biosensor is classified as enzymatic biosensor,

15
microbial or whole cell biosensor, DNA biosensor and immunosensor. In addition to this,
depending on the type of transducer used biosensors are further classified as amperometric,
potentiometric, optical, piezoelectric, calorimetric biosensors. Electrochemical biosensors in
general, and electrochemical glucose biosensor in particular have been studied extensively
presenting various technological advancements leading to improvement in biosensor
parameters like selectivity, response time, stability etc [32]. These technological
developments are discussed in the following sections.
2.2 Technological advancements of electrochemical glucose biosensors
Based on the technology improvement of electrochemical glucose biosensors, three
generations of glucose biosensors have been reported [33].
2.2.1 First-generation of Glucose Biosensors
The first generation glucose biosensors estimated glucose concentration in the sample based
on hydrogen peroxide production by glucose oxidase (GOx) utilizing dissolved oxygen as
given below
A negative potential is applied to the Pt working electrode for a reductive detection of the
oxygen consumption as
The key point of above reaction lies in the redox center of the GOx (FAD) which performs
the function of the initial electron acceptor. The interaction of glucose molecule with flavin
adenine dinucleotiede (FAD) of GOx results in its reduction.
The rejuvenation of the cofactor of enzyme GOx occurs in the presence of molecular oxygen,
resulting in the formation of hydrogen peroxide (H2O2) as,
GOx
Glucose + O2 Gluconic acid + H2O2
O2 + 4H+ + 4e
- 2H2O
Glucose + GOx (FAD) Gluconate + GOx (FADH2)
GOx (FADH2) + O2 GOx (FAD) + H2O2

16
Thus, the rate of reduction of oxygen is directly proportional to the glucose concentration that
is enumerated by either measuring the reduced oxygen concentration or increased
concentration of hydrogen peroxide.
Hydrogen peroxide thus produced as a byproduct is oxidized at platinum (Pt) anode. The
electrons transferred are recognized by electrode and thus the number of electrons transferred
is directly proportional to the number of glucose molecules present.
This glucose biosensing technology of Clark was transferred to Yellow Spring Instrument
Company and on the same principle they launched the first commercial glucose biosensor in
market (Model 23A YSI analyzer) for the direct measurement of glucose in 1975. The usage
of the most expensive metal platinum for fabrication of this electrode restricted the biosensor
to clinical laboratories only.
Major drawbacks of first generation glucose biosensor:
Interference from electroactive species present in blood, such as uric acid, ascorbic
acid and other constituents of blood, at the high operational potential (+0.6V) required
for amperometric measurement of hydrogen peroxide. This limits the high selectivity
of the analyzer and results in inaccurate measurements of glucose concentration.
Oxygen deficit – Sensors involving natural oxygen as the electron acceptor due to
presence of oxidase enzyme, generally face errors resulting from fluctuations in
oxygen tension due to the limited solubility of oxygen in biological fluids. This
reduces the linear range of the biosensor.
Number of approaches have been suggested for addressing this problem, Joseph Wang and
group introduced a biosensor with high oxygen solubility based on a fluorocarbon (Kel-F oil)
pasting liquid [34]. Thus the internal flux of oxygen supports the reaction catalyzed by the
enzyme, even in the absence of oxygen in glucose solution. Other approach was proposed by
Gough’s group, in which they designed a two dimensional cylindrical electrode in which
diffusion of glucose is allowed only from one direction while the oxygen is allowed to diffuse
from both directions into the region where enzyme is immobilized [35, 36]. The above
strategy was achieved by developing a two-dimensional sensor design containing a cylindrical
H2O2 O2 + 2H+ + 2e
-

17
gel with GOx and covering the outer part with a silicone rubber tube which does not allow
glucose but is highly permeable to oxygen.
2.2.2 Second Generation glucose biosensor
Search for an alternative for the natural oxygen acting as an electron acceptor in first
generation biosensors lead the electrochemists to second generation biosensors. Use of
synthetic electron mediators opens up new horizons in the field of biosensors. The synthetic
electron mediators eliminated the need of oxygen for recording the electron transfer at the
electrode surface overcoming the drawbacks of limited oxygen pressure observed in first
generation biosensor. Moreover, the lower redox potential of chosen mediators (-0.1V vs
Ag/AgCl for Pursian Blue) results in no interference from other electro active species such as
ascorbic acid and uric acid. Optimal applied potential for eliminating interference was found
to be between 0.0V to 0.2V. In addition to overcoming the above mentioned two drawbacks,
usage of mediators also ensured faster rate of shuttling of electrons from the redox center of
the enzyme to the surface of the electrode.
Electron transfer rates are affected by the structure of the enzyme and hence accessibility of
the active site. The active center of GOx, the flavin adenine dinucleotide (FAD), is buried
inside a deep pocket between the two subunits of the dimeric enzyme. Thus, the direct
electron transfer from Glucose-reduced GOx(red) to metal electrodes is not facilitated because
of the appreciably large distance between GOx redox centers and the electrode surface (>12-
17Aº), resulting in a much retarded diffusion controlled electron transfer rates.
Mechanism of action of mediators can be explained as:
Glucose from the bulk solution diffuses to the enzyme active site and is converted to gluconic
acid. The electrons released during the above conversion are picked by the mediator and is
reduced; finally at the applied potential oxidation of mediator releases electrons that are
transferred to the electrode. Role of mediators in facilitating electron transfer is further
explained by set of equations given below,

18
where, SM(red) and SM(ox) represents the reduced and oxidized forms of synthetic mediator,
respectively. As represented in above equations the reduction of SM(ox) to form SM(red)
facilitates the reoxidation of reduced form of GOx (FADH2). Further oxidation of SM(red) at
the electrode surface regenerating SM(ox) and two electrons. The number of electron
transferred to the electrode is proportional to the glucose concentration. Some of the common
synthetic electron mediators, which have been used to increase the electron transfer rate or
performance of the sensor, are listed in Table 2.1.
Table 2.1 List of synthetic mediators and their redox potential.
Enzyme Synthetic Mediator Redox potential(Versus
SCE) (mV)
Glucose oxidase Vinyl ferrocene 250
[Fe(CN)6]4- 180
Indigo Disulfonate 188
Methylene blue 217
1,1- dimethyl ferrocene 100
[Ru(CN)6]4- 685
TCNQ 127
Ferrocene carboxylic acid 275
Ferrocene carboxaldehyde 518
TTF 300
Benzyl viologen 370
Hydroxy methyl ferrocene 185
Ferrocene 165
Glucose dehydrogenase N-ethyl phenazene -172
Ferrocene carboxylic acid 275
TMPD -10
1,1-dimethyl ferrocene 100
The characteristics features leading to enhanced usage of mediators are
a low redox potential which helps in avoiding interfering current from coexisting
electro active species leading to false measurements,
their low molecular weight and insoluble nature which allows them to effectively
diffuse without complexing,
high stability in both reduced and oxidized forms and,
low toxicity.

19
All the above mentioned unique qualities lead to an improved linear response and thus a
prolonged lifetime of the biosensor, since the deactivation of the enzyme due to production of
hydrogen peroxide is eliminated. Unfortunately, usage of mediators have their own associated
problems which hinders the successful performance of the biosensors.
Major drawbacks of using mediators in second generation glucose biosensor:
High competition between synthetic mediator and oxygen: Although the
probability of reaction between synthetic mediators with the active center of GOx(red)
occurs at a faster rate than oxygen, still the possibility of competition for oxidation of
the reduced GOx by dissolved oxygen with the synthetic mediator is highly likely,
thus resulting in the accumulation of hydrogen peroxide near the electrode surface
leading to reduced bioactivity of enzyme and biosensor response.
Interference: The possibility of oxidation of coexisting electro active species such as
ascorbate even at low applied potential not only affects the accuracy of the sensor but
also enhances the chances of reaction of the synthetic mediator with interfering
species. Thus leading to further inaccurate or false measurements.
Stability of synthetic mediator near electrode surface: Small size and highly
diffusive nature of synthetic mediators poses problem of leaching of mediator from
intermediate region between enzyme and electrode surface. This limits their use in
applications where continuous operation of biosensor is required to avoid mediator
leaching.
During eighties strategies other than incorporation of synthetic electron mediators [37] to
facilitate electron transfer between the GOx redox center and the electrode surface were also
introduced, such as the concept of wired enzymes [38]. Wired enzymes involved redox
hydrogels (redox ions/mediators immobilized on to hydrogels) acting as electrical wires for
conducting the electron from GOx active center to the electrode surface [39].
2.2.3 Nanomaterials: A better platform for biosensor fabrication
The unique properties of nanostructures have been exploited to achieve parameters like fast
response time, high sensitivities, low detection limits, wide range linearity and low power
requirements necessary for highly precise and defined analyte sensing. The high sensitivity
(196nA/mM) and wide linear range (0.2-20 mM) demonstrated by glucose biosensor based on
modified sol-gel composite at the surface of a basal plane pyrolytic graphite electrode

20
decorated with MWCNT presented by Abdollah Salimi et al. shows the promising behavior of
nanomaterial as compared to the previous hydrogel or membrane based biosensors [40].
Glucose oxidase immobilized on gold nanowires based biosensor that could detect glucose in
just 8 seconds as opposed to few minutes of earlier membrane based biosensors was proposed
by Yashuang Lu et al [41]. The high conductivity and biocompatible behavior of elemental
gold at nanoscale makes them a potential platform form immobilization of Glucose oxidase.
Glucose biosensor based on gold nanoparticles proposed by Sylvain Thibault et al., represents
an extremely efficient system allowing even lower detection of glucose concentrations
(0.37mM) with wide linear range [42].
2.2.4 Third-Generation Glucose Biosensors
In order to avoid complications offered by synthetic or natural mediators in second generation
biosensors, a lot of work is being done for finding new strategies for direct electron transfer
between the electrode and active center of enzyme. This led to development of highly
selective and sensitive third-generation biosensors. However, there are only few reports in the
literature concerning the direct electron transfer (DET) between active center of GOx and
electrode surface, although DET for many enzymes have been achieved [43-45] by
immobilizing them within the thin films with different modifications. The intrinsic barrier to
electron flow is the globular structure of GOx with the active site, containing FAD/FADH2
redox cofactor, buried deep inside a cavity of ~13A◦ is a major hinderance for direct electron
transfer in case of thin film or hydrogels based electrodes. Unsuccessful efforts to obtain
direct electron transfer of GOx at conventional electrodes led to exploration of new electrode
materials. In the year 1987, Albery, Cranston and Bartlett suggested incorporation of organic
conducting salt electrodes in order to avoid protein denaturation and fast direct electron
transfer. These conducting salts can be modified into single crystals, as pressed pellet or a
paste with graphite powder in order to prepare electrode. The conducting organic salts like
tetracyanoquinodimethane (TCNQ) and tetrathiafulvane (TTF), have proved to be useful for
the above application [37, 46]. Different researchers exploited these materials in different
ways to achieve high sensitivities. A third generation glucose sensor based on the growing
tree-shaped crystal structure of TTF-TCNQ was proposed by Khan et al [47]. The reduced
distance between the enzyme active center and electrode and immobilization of enzyme in
correct orientation at the electrode surface allowed direct oxidation of the enzyme at a low
applied potential of 0.1 V, athough no explanation for direct oxidation of obtained results

21
were provided by the authors. Cenas and Kulys [48] presented number of arguments against
the direct electron transfer presented by Palmisano et al in a glucose biosensor fabricated
using growing TTF-TCNQ tree-like crystals through an anti-interference layer of a
nonconducting polypyrrole film [49]. Further, number of mediatorless glucose biosensors
based on different materials like polypyrrole system, oxidized boron-doped diamond
electrodes etc were proposed [50].
Efforts to achieve DET using nanoparticles of different types and size were not very fruitful,
however, SWCNTs and MWCNTs were found to be good candidate. SWCNTs immobilized
vertically on the electrode surface provide suitable orientation for enzyme immobilization and
establishing connection between electrode surface and deeply buried active site [51, 52]. This
enables electron transfer over much longer distances of approximately 150 nm in shorter time
(few seconds) while a diffusion based electron transfer over length scales greater than 8-17A◦
results in much longer time of few minutes. Depending on the length of CNTs and efficient
connectivity with redox center the interfacial electron transfer rate varies, e.g. for 50 nm long
CNTs it is 42s-1
[53] while in another report with PLL-SWCNT-GOx electrode with 23 nm
long SWCNT much higher electron transfer rates of 70-100 s-1
were observed [54]. The
distance between the electrode surface is responsible for the large over potential requirement,
i.e. potential greater than the thermodynamic redox potential of the enzyme. To decrease the
working potential, better connectivity leading to DET is desired. This not just improves the
electron transfer rates but also takes care of the problems of interference from electroactive
species. Recently, research efforts are directed at achieving the same. Although substantial
progress has been made on the electronic coupling of GOx, further improvements in the
charge transport between its FAD redox center and electrodes are desired.
2.3 Glucose Biosensors: Research Efforts 1962-2012
More than 80,000 research articles related to various biosensors have been published since
1962. Out of which ~10% (>8020) of the papers are related to glucose biosensors alone and
greater than 66% of the glucose biosensors are enzymatic glucose biosensors. Recent interest
in nanomaterials is evident from the fact that ~80% of the reported glucose biosensors
research exploit the properties of nanomaterials for improved biosensing. Among
electrochemical, optical, piezoelectric and impedimetric glucose biosensors, amperometric
glucose biosensors (>92%) are most widely studied ones while optical glucose biosensors

22
contributes ~5% and potentiometric being only 2.5%. There are only two research articles on
piezoelectric and one on impedimetric glucose biosensors (see Figure 2.1). Probable reason
behind the above statistics being the ease of fabrication and cost effectivenesss of
amperometric biosensor. Table 2.2 below shows biosensor performance characteristics in
chronological order of various biosensors developed till date.
However, the research ideas are not effectively translated into product as evident from
comparatively much lower number of patents filed (see Table 2.3). Table 2.3 shows the
number of glucose biosensor patents filed and granted by different patent offices – US Patent
Office (USPTO), European Patent Office (EPO) and other countries patent offices (Others).
Table 2.3: Number of patents for glucose biosensor.
The commercial availability of the glucose biosensors confirms the dominating behavior of
the device (> 90% of commercial biosensors are glucose biosensors) in the biosensor market.
Immobilization matrices USPTO EPO Others
Membrane based 306 290 141
Hydrogel based 105 29 94
Nanomaterials based 52 9 49
Application in fermentation
industry
8 5 12
Figure 2.1: Percentage distribution of reported research articles based on
different types of glucose biosensors

23
The above table shows that out of approximately 1000 patents filed in the field only 25
glucose biosensor patents are applied in the field of fermentation industry. Thus more than
95% of the consumer market is occupied by blood glucose monitoring devices and just 3% of
the available technology is applied in the fermentation industry.
Table 2.2: Performance characteristics of amperometric glucose biosensors in chronological order.
Type of support for
Immobilization
Sensitivity Interference Detectio
n limit
Linear
range
Respons
e time
Stability Reference
GOx was immob. on
graphite followed by
adsorption of N-methyl-
phenazinium ion (PMS+)
- Low
interference
from
Galactose and
Mannose
0.5, to
150 μm
2 mM 20-60 s Aleast 9
months
Gunilla
Jönsson et
al., (1985)
[55]
GOx was incorporated into
polypyrrole films that were
electrochemically
deposited on PE.
- - - - 20-40 s 21 days Nicola C.
Foulds et
al., (1986)
[56]
Cellulose acetate, GOx
(crosslinked with
glutaraldehyde) and
polyurethane are placed on
surface of central platinum
wire surrounded by a
stainless steel tubing
- - - 500 mg/dl 100 sec 6 days Kerner W
et al.,
(1988) [57]
Polysiloxanes are used for
interaction between GOx &
CPE
- None by
thiocynates
- 16-71 mM <10 s 2 years
(20%)
Paul D.
Hale et al.,
(1991)
[58]
Incorporation of GOx into
graphite paste modified
with
tetracyanoquinodimethane
(TCNQ)
- - 0.5 mM 5-50mM 15-50
sec
35 days PC Pandey
et al.,
(1992) [59]
Immobilizing GOx and
coating Nafion membrane
7.1 +/- 0.5
nA/mM
- - 0.5-40 mM 30 s 6 days Jianzhong
Zhu et al.,

24
(1994) [8]
Glutaraldehyde & BSA is
used for crosslinking GOx
on CPE
- Interference
from ascorbic
acid, uric acid
and
paracetamol
- 2–20 mM 60-120 s 6 days Miloslav
Pravda et
al., (1995)
[60]
Poly (o-aminophenol) film - - - 0.001-1.0
mM
More
than 4
sec
30 days
(30%
reductio
n)
Z.Zhang et
al.(1996)[6
1]
Sol−gel organic−inorganic
hybrid material was used
for immob. of GOx
600 nA
mmol-1
L-1
Interference by
L-Ascorbate
- 0 to 16 mM 11s 5
months
Bingquan
Wang et
al., (1998)
[62]
Immobilization of GOx
into poly(o-
phenylenediamine) (POPD)
on Pt electrode. Additional
layer of Prussian blue (PB)
was also placed.
0.2 to 0.7
μA mM−1
cm−2
Diminished
ascorbate
interference
- 8 to 14 mM 4 to 8 s - R
Garjonyte
et al.,
(1999) [63]
GOx and poly(p-
phenylenediamine) (poly-
PPD) were coimmobilized
at the surface of a platinum
microdisk electrode
160 μA
cm-2
mM-1
No
interefernce to
ascorbic acid,
uric acid &
cysteine
- 5.0 x 10-5
to
3.0 x 10-3
M
<2 s 2 month Xu Jing-
Juan et al.,
(2000) [64]
3- APTES, Nafion®, GOX,
and perfluorocarbon
polymer (PFCP)
2.21
nA/mM
No
interference
- 2.8 to 167
mM
- 66 days T.
Matsumoto
et al.,
(2001) [65]
Polymerization of p- 5.2 × 10–3
No 10–4
M 2.5 x 10-4
to <2 s 90 days Jing-Juan

25
chlorophenol (4-CP) at a Pt
electrode.
nA L mol–1
interference 1.5 x 10-2
mol/ L
Xu et al.,
(2001) [66]
Co-electrodeposition of a
poly(vinylimidazole)
complex of
[Os(bpy)2Cl]+/2+ (PVI-
Os).
349
nA/mM
No
interference
upto 2 mM
glucose by
ascorbic acid
0.03mM 0 - 30 mM 5 s 64 hours
(50%)
Junjei Fei
et al.,
(2003) [67]
GOx was immob. into a
copper dispersed sol-gel
derived ceramic-
graphite composite
- No
interference to
ascorbic acid
& uric acid.
1.8 × 10-
5
M
4.0×10-5
to 5.6×10-3
M
6-9 s 60 days
(78%)
D. Ravi
Shankaran,
et al.,
(2003) [68]
GOx was immob. into a
sol-gel composite at the
surface of a basal plane
pyrolytic graphite electrode
modified with MWCNT.
196
nA/mM
- 50 μM 0.2-20 mM <5 s 3 weeks. Abdollah
Salimi et
al., (2004)
[40]
Adsorption of GOx at the
platinum nanoparticle-
modified CNT
91mA M-
1cm
-2
Weak
interference of
Ascorbic acid
and Uric acid
- 0.1-
13.5mM
5 s 22 days
(73.5 %)
Hao Tang
et al.,
(2004) [69]
Polyelectrolyte & chitosan
was used to immob. GOx
on Pt electrode
21 mA M−1
cm−2
No
interference
10 μM 0.05–15
mM
<8 s 3
months
(65%)
Minghui
Yang et al.,
(2004) [70]
Microdeposition of
PEDOT/PSS and GODon
ITO-glass
6.43 μA
M−1
cm−2
- - 60 mM - - L. Setti et
al., (2005)
[71]
GOx was immobilized in
sol–gel chitosan/silica
hybrid composite film
420 nA
mM–1
No
interference
8.0×10–6
M
5.0×10–5
to
2.6×10–2
M
10 s 60 days Xue-Cai
Tan et al.,
(2005) [72]
GOx and the electrocatalyst
cobalt phthalocyanine were
mixed with the carbon ink.
1170 nA
mM−1
- 0.025
mM
0.025–2
mM
- - Eric
Crouch
(2005) [73]
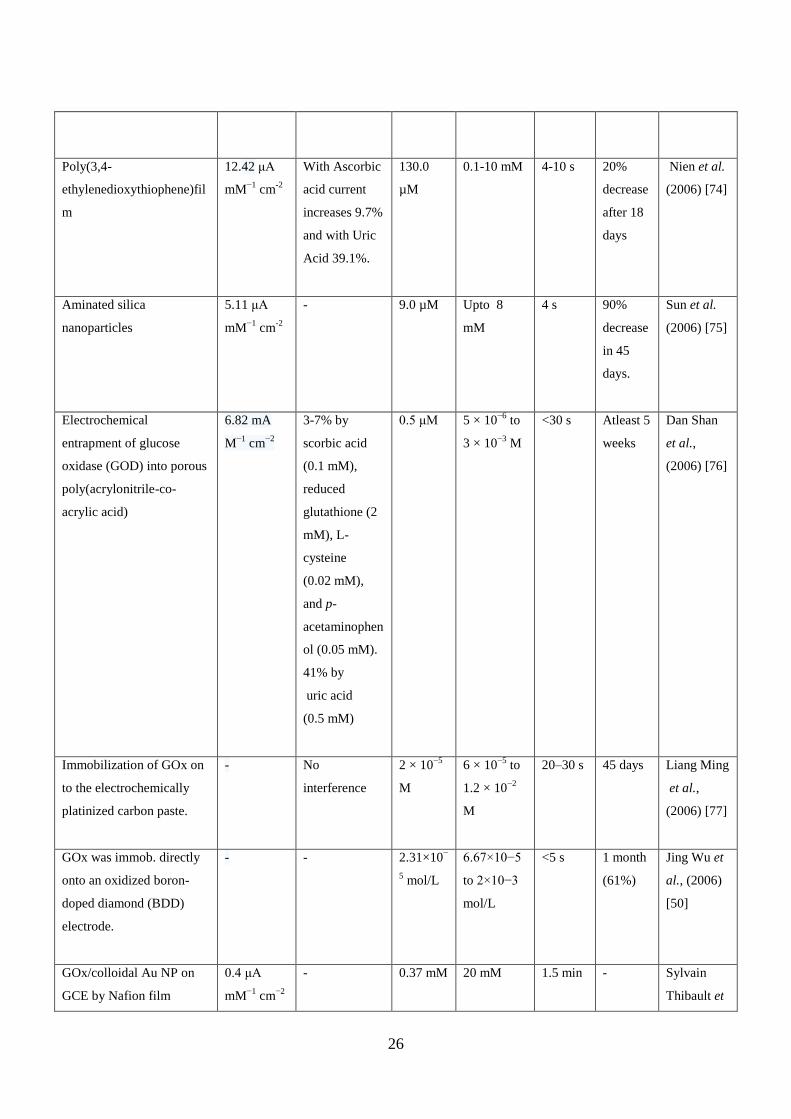
26
Poly(3,4-
ethylenedioxythiophene)fil
m
12.42 μA
mM−1
cm-2
With Ascorbic
acid current
increases 9.7%
and with Uric
Acid 39.1%.
130.0
µM
0.1-10 mM 4-10 s 20%
decrease
after 18
days
Nien et al.
(2006) [74]
Aminated silica
nanoparticles
5.11 μA
mM−1
cm-2
- 9.0 µM Upto 8
mM
4 s 90%
decrease
in 45
days.
Sun et al.
(2006) [75]
Electrochemical
entrapment of glucose
oxidase (GOD) into porous
poly(acrylonitrile-co-
acrylic acid)
6.82 mA
M−1
cm−2
3-7% by
scorbic acid
(0.1 mM),
reduced
glutathione (2
mM), L-
cysteine
(0.02 mM),
and p-
acetaminophen
ol (0.05 mM).
41% by
uric acid
(0.5 mM)
0.5 μM 5 × 10−6
to
3 × 10−3
M
<30 s Atleast 5
weeks
Dan Shan
et al.,
(2006) [76]
Immobilization of GOx on
to the electrochemically
platinized carbon paste.
- No
interference
2 × 10−5
M
6 × 10−5
to
1.2 × 10−2
M
20–30 s 45 days Liang Ming
et al.,
(2006) [77]
GOx was immob. directly
onto an oxidized boron-
doped diamond (BDD)
electrode.
- - 2.31×10−
5 mol/L
6.67×10−5
to 2×10−3
mol/L
<5 s 1 month
(61%)
Jing Wu et
al., (2006)
[50]
GOx/colloidal Au NP on
GCE by Nafion film
0.4 μA
mM−1
cm−2
- 0.37 mM 20 mM 1.5 min - Sylvain
Thibault et

27
al., (2008)
[42]
GOx is immobilized on
CNT surface by assembling
polydiallyldimethylammon
ium chloride (PDDA) layer
- No
interference
7mM 15 μM to 6
mM
- - Guodong
Liu et al.,
(2006) [78]
Polypyrrole (PPy),
functionalized cMWNT,
and GOx
95 nA
mM−1
- - 4 mM - 8 s Yu-Chen
Tsai et al.,
(2006) [79]
Au nanowires-Chitosan
was immob. on GCE
- - 5×10−6
M
10−5
-2×10-
2M
< 8 s 1 month
(85%)
Yashuang
Lu et al,
(2007) [41]
GOx
was immob.on
Pt/sulfonated-
MWCNT/GCE
0.56
μA/mM
- - 6.4
mM
- - H.J. Wang
et al.,
(2007) [80]
PAA, MWCNTs,
cysteamine and GNp,
respectively, followed by
the adsorption of GOD on
Pt electrode
(GOD/GNp/MWCNTs/Pt
electrode)
2.527
μA/mM
No
interference
6.7 μM 0.1–10 mM
glucose
<7 s 7 S Bao-Yan
Wu et al.,
(2007) [81]
Calcium carbonate
nanoparticles was used for
immob. of GOx.
58.1 mA
cm−2
M−1
3-7% by
scorbic acid
(0.1 mM),
reduced
glutathione (2
mM), L-
cysteine
(0.02 mM),
and p-
acetaminophen
ol (0.05 mM).
0.1 μM 0.001–12
mM
6 s 120 days
(86%)
Dan Shan
et al.,
(2007) [82]

28
39% by
uric acid
(0.5 mM)
―Unprotected‖ Pt
nanoclusters mixed with
the nanoscale SiO2
particles
3.85 μA
mM−1
- 1.5 μM 0.27 to 4.08
mM
- - Haipeng
Yang
(2007) [83]
Micro-patterned Prussian
blue (PB) and ferrocene
modified glucose oxidase
covered by a thin Nafion
membrane
- Slight
interference by
Ascorbic acid
on anodic
detection
75 µM 0.1 to 50
mM
1-6 min 1 week
(60%)
Na Zhang
et al.,
(2007) [84]
Layered double hydroxides
(LDHs)
60 mA M−1
cm−2
Negligible 3 μM 6.7 × 10−6
-
3.86 ×10−4
M
5 s - Dan Shan
et al.(2007)
[85]
Exfoliated Graphite
Nanoplatelets Nafion
membrane
14.17 µA
cm-2
mM-1
56.8% for
0.1mM
Ascorbic acid
and 125% for
0.2 mM uric
acid
interference
10 µM upto 6 mM 5
seconds
Stability
for 7
days.
Jue Lu et
al.(2007)
[86]
Amino functionalized
Multi-wall carbon
nanotubes (MWNTs)
7.46 μA
mM−1
cm-2
Minimum
interference.
8.0 µM - <10 s - Sun et al.
(2008) [87]
Single Walled Carbon
Nanohorns
15.0 μA
mM−1
cm-2
Can avoid the
commonly
coexisted
interference.
6.0 µM 0-6 mM - - Liu et al.
(2008) [88]
Layer-by-layer (LBL) self-
assembling of chitosan and
glucose oxidase (GOD) on
a Prussian blue film was
developed
- Low
interference
3.1 μM 6 μM to 1.6
mM
10 s 4 weeks
(73%)
Bing Yin et
al., (2008)
[89]

29
Gold nanoelectrode array
was fabricated by template-
assisted electrodeposition
on general electrodes
1.52 mA
mM-1
cm-2
- 5 x 10-6
M
1 x 10-6
to 1
x 10-2
M
8 s 3
months
(80%)
Yanyan Liu
et al.,
(2009) [90]
Composite material based
on layered double
hydroxides (LDHs) and
chitosan (CHT)
62.6 mA
M−1
cm−2
Weak
interference
0.1 μM 1 × 10−6
to
3 × 10−3
M
5 s 60 days
(70%)
Qiaofang
Shi et al.,
(2008) [91]
Immobilizing glucose
oxidase (GOD) in a titania
sol-gel film, which was
prepared by a vapor
deposition method, on a
Prussian Blue (PB)-
modified electrode
12.74 μA
cm-2
mM-1
For ascorbic
acid, current
decreased
about 3%, &
for Uric Acid
and cysteine
the current
increased 0.2
and
1.2%,
respectively
5 μM 0.02 to 15
mM
< 10 s 3
months
(91%)
Ruping
Liang et
al., (2008)
[92]
Adsorption of
GOx on an AuNPs–
AgCL@polyaniline
(PANI) core-shell
nanocomposites on GCE
- 3, 2.4 and
1.8% for
ascorbic acid,
uric acid &
cysteine.
4 pM 4–34 pM - 2 weeks
(80%)
Wei Yan et
al., (2008)
[93]
Graphite nanoplatelets
(xGnPs) decorated with Pt
and Pd nanoparticles was
used
61.5 ± 0.6
μA mM-1
cm-2
Interference by
ascorbic acid
and uric acid
1 μM 20 mM 2 s 1 week Jue Lu et
al., (2008)
[94]
Encapsulating GOx in the
Nafion– single-walled
carbon
nanohorns(SWCNHs)
composite film
1.06
μA/mM
No
interference of
L-lactate,
glutathione, L-
cysteine, and
p-
aminophenol,
6 μM 0 to 6.0
mM
- - Xiaoqing
Liu et al.,
(2008) [88]

30
L-ascorbate
acid
GOx with
chitosan-AuNP on gold-
Prussian Blue (Au-PB)
nanoparticles (GCE).
9.5 µA M-1
cm-2
- - 3 mM 10 s - Juozas
Kulys et
al.(2008)
[95]
Poly (3,4-
ethylenedioxythiophene) /
Prussian blue bilayer and
multi-walled carbon
nanotubes
2.67 μA
mM−1
cm-2
- - 1–10 mM - 18%
decrease
in 30
days
Chiu et al.
(2009) [96]
Gold nanorods/cellulose
acetate composite film
8.4 μA
mM−1
cm-2
- 20 µM 0.03-
2.2 mM
- 20%
decrease
in 30
days.
Ren et al.
(2009) [97]
Entrapping GOx onto the
inner wall of highly
ordered polyaniline
nanotubes (nanoPANi)
97.18 ±
4.62 μA
mM−1
cm−2
No
interference
0.3 ± 0.1
μM
0.01−5.5
mM
3 s 2 weeks
(91%)
Ziyi Wang
et al.,
(2009) [98]
Alginate (Alg)/layered
double hydroxides (LDHs)
organic-inorganic
composite film
68.9
µA/mM/cm
0- 3.6% by
Ascorbic acid
(0.1 mM), Uric
acid (0.5 mM),
Glutathone
reduced (2
mM), L-
cysteine (0.02
mM), p-
acetaminophen
ol (0.05 mM)
4 × 10−5
M
1.6 × 10−5
−
2 × 10−3
M
10 s 28 days
(87%)
Shou-Nian
Ding et al.,
(2009) [99]
Graphene
- - - 2-14 mM - - Shan et al.
(2009)
[100]

31
Silver nanoparticles/carbon
nanotubes/chitosan film
135.9
µA mM-1
20µM for uric
acid and 8µM
for ascorbic
acid
0.1µM 0.5-50 µM - 9%
decrease
in 10
days and
20% in
about 40
days
Jiehua lin
et al.
(2009)
[101]
Poly (3,4-
ethylenedioxythiophene) /
Prussian blue bilayer and
multi-walled carbon
nanotubes
2.67 μA
cm−2
mM−1
.
- - 1–10 mM - 18%
decrease
in 30
days
Jing-Yang
Chiu et al.
(2009) [96]
GOX is immobilized onto
the CNT/Pt nanosphere.
70
μA/mM/cm
2
- 380 nM 1 μM to
0.75 mM
8 s - Jonathan C.
Claussen
(2010)
[102]
GOx was immob. thin
films of chitosan
containing nanocomposites
of graphene and gold
nanoparticles (AuNPs) at a
gold electrode
0.55μAmM
−1
- 180 μM 2 to 10 mM
and from 2
to 14 mM
- 15 days
(4.6%
reductio
n)
Changshen
g Shan et
al., (2010)
[103]
Utilizes CNTs
electrochemically
decorated with platinum
(Pt) nanospheres to sense
glucose
70
µA/mM/cm
- 380 nM 1 µM to
0.75 mM
8 s - Jonathan C.
Claussen et
al., (2010)
[102]
Silicon dioxide coated
magnetic nanoparticle
decorated multiwalled
carbon nanotubes
(Fe3O4@SiO2/MWNTs)
on a glassy carbon
electrode (GCE)
58.9μA/m
M cm2
Weak
interference by
ascorbic acid
and uric acid
800 nM 1 μM to 30
mM
- - Tessy
Theres
Baby et al.,
(2010) [11]

32
Immobilization of GOx on
nanoflake-like SnS2
modified GCE
7.6 ± 0.5
mA M⁻¹
cm⁻2
None by Uric
acid, and
Ascorbic acid
1.0 ×
10⁻⁵ M
2.5 × 10⁻⁵
M to 1.1 ×
10⁻³ M
8 s 20 days
(8%
reductio
n)
Zhanjun
Yang et al.,
(2011)
[104]
Electrodepositing chitosan
(CS)-glucose
oxidase(GOD)
biocomposite onto the
stainless steel needle
electrode (SSN electrode)
modified by Pt–Pb
nanoparticles (Pt–Pb/SSN
electrode)
0.4485
μA/mM
- - 0.03 to 9
mM
15 s - Meiqing
Guo et al.,
(2011)
[105]
p-tert-
butylthiacalix[4]arene
tetra-amine (TC4TA) is
used for immob. of GOx.
10.2 mA
M−1
cm−2
Weak
interference
20 μM 0.08–10
mM
5 s 20%
decrease
in 30
days
Ming Chen
et al.,
(2011)
[106]
AuNPs and MWCNT
nanocomposite materials
were constructed by
alternate self assembly of
thiol functionalized
MWCNTs and AuNPs,
19.27 μA
mM−1
cm−2
Weak for
ascorbic acid,
uric acid and
acetaminophen
2.3 μM 20 μM to
10 mM
3 s 1 week
(95.4%)
Peng Si et
al., (2011)
[9]
GOx was immob. on
polyaniline-
polyvinylsulphonate
(Pani-Pvs) via the
entrapment technique
- 15% and 25%
for ascorbic
acid & uric
acid.
1.0 ×
10−7
M
1.0 ×
10−7–1.0 ×
10−5 M
200 s 40 days
(80.6%)
Fatma
Arslan
et al.,
(2011)
[107]
Pt nanoparticle
homogeneously decorated
on polyaniline (Pani)-
wrapped boron nitride
nanotubes (BNNTs),
19.02 mA
M–1
cm–2
Negligible for
Ascorbic acid
and Uric acid
0.18 μM 0.01 to 5.5
mM
3 s 40 days
(95%)
Jianmin
Wu et al.,
(2011)
[108]
Palladium 31.2 µA Significant 0.2 µM 0.001-1.0 - 20% Zeng et

33
nanoparticle/chitosan-
grafted graphene
nanocomposites
cm-2
mM-1
interference mM decrease
in 21
days.
al.(2011)
[109]
Immobilizing the PtPd-
MWCNTs catalysts in a
Nafion film on a glassy
carbon electrode.
112 μA
mM−1
cm−2
Negligible
interference
0.031
mM
0.062–
14.07 mM
5 s 28 days
(85%)
Kuan-Jung
Chen et al.,
(2012) [14]
Pt nanoparticles-chitosan
composite film (PtNPs-CS)
- Insignificant
interference by
ascorbic acid,
threonine,
L-cysteine,
uric acid
0.4 μM 1.2 μM to
4.0 mM
<5 s 1 month
(89.6%)
Jingjing Li
et al.,
(2012)
[110]