characterization of proteins - isu public homepage …duahn/teaching/biomodulation and...
TRANSCRIPT

Characterization of Proteins

Determination of the Sizes of Proteins
• Sedimentation analysis
• Centrifugation• Gel filtration• SDS PAGE

Differential Precipitation
• Precipitation:
• The process of formation of a solid that was previously held in solution.
– NH4
SO4
– Polyethylene glycol are added to a protein solution
• A precipitate forms and separated from the solution after centrifugation.
• If the concentrations of NH4
SO4
or polyethylene glycol are increased, more precipitate forms.
3

Centrifugation
• Zonal centrifugation: Mixture to be separated is layered on top of a gradient (e.g.
sucrose or ficoll) increasing concentration down the tube ‐
can be continuous or
discontinuous (layers)
‐
provides gravitational stability as different species move down
tube at different
rates forming separate bands.
– Species are separated by differences in SEDIMENTATION COEFFICIENT
(S)
=
Rate
of movement down tube/Centrifugal force
– S is increased for particle of LARGER MASS
(because sedimenting force a M(1‐vr)
– S is also increased for MORE COMPACT STRUCTURES of equal particle mass
(frictional coefficient is less)

Zonal ultracentrifugation

Centrifugation
• Isopycnic (equal density) centrifugation: Molecules separated on
EQUILIBRIUM POSITION, NOT by RATES of sedimentation.
Each molecule floats or sinks to position where density equals density of
solution (e.g. CsCl gradient for nucleic acid separation).

Gel Filtration Chromatography
7

Size‐Exclusion Chromatography• Separation of
proteins based on
kinetics of moving
through the available
space • Larger proteins have
less space than
smaller molecules• Proteins larger than
matrix elute in void
volume (1 exchange
of volume outside
beads)• Proteins smaller than
matrix partition in
and out of beads• Pore size in beads is
not uniform• Also some surface
interaction with
beads

Gel Filtration Elution Volumes as a Function of Molecular Weight

Gel Electrophoresis

SDS Gel Electrophoresis
• Tris‐glycine buffer• 10% SDS

Protein Size Determination by SDS Polyacrylamide Gel Electrophoresis
+ electrode
- electrode

Protein Detection in Electrophoresis Gel
• Protein detection– Coomassie blue
– Sypro: green, ruby, Red, orange
– Cybergreen– Silver staining
coomassie brilliant blue A595

Detection of Amino Acids, Peptides, and Proteins
• UV• Biuret Reaction• Ninhydrin Reagent • Fluorescarmine
• Ortho‐phthalaldehyde• Coomassie Brilliant Blue
• Silver Staining

Absorption spectra of Trp & Tyr
Beer’s law: A = εcl. Used to estimate protein concentration

Biuret Reaction
• Primarily measure peptide bonds
• Cupric acid react with the peptide groups of proteins and form a cupric ion complex
• Generate purplish‐violet color with Amax
at 540 nm
• Lowry method
• Low sensitivity

Ninhydrin Reaction
• The most widely used reagents for amino acids, peptides, and proteins
• Reacts with amino groups
• The major step is oxidative deamination of an amino acid to CO2, NH3, and an aldehyde conatining one less carbon atom than the original amino acid
• Ninhydrin is simultaneously reduced to ninhydrindantin
• Ninhydrindantin reacts with NH3 and produce purple color (Amax
570 nm)
• Destroy amino acid or peptide and the material detected cannot be used for further characterization

Fluorescarmine
• React with amino group
• Produce fluorescent compounds
• Sensitive• The amino group reacted can be recovered by
hydrolysis and used for further analysis

Ortho‐phthalaldehyde
• React with amino group in the presence of mercaptoethanol
• Forms a fluorescent reaction product
• Sensitive• As Ninhydrin and fluorescarmin, it is not
specific for protein

Circular Dichroism Spectroscopy of Polypeptides
Adapted fromT. E. Creighton, ProteinsW.H.Freeman,1984

Protein Characterization
• Characterization of proteins and peptides involves three different processes:
1.
Determining the Amino Acid Composition
• Involves finding out the amino acids that make up the protein and their number.
2.
Determining the Amino Acid Sequence
• Involves finding out the sequence of amino acids of the proteins in their order.
3.
Determining the Molecular mass of the Protein
21

• The peptide is first hydrolyzed into its constituent amino acids by heating it in 6M HCl at 110ºC for 24‐72 hrs.
The R groups remain intact, except for:
• Trp –
indole ring damaged
• Asn, Gln – converted to Asp, Glu
• Gly does not react
22
Determination of Amino Acid Composition

Amino acid analysis
• The amino acids can be derivatized with ninhydrin or o‐phthalaldehyde to make fluorescent derivatives that are
easy to detect.
• These are chromatographed by reverse‐phase HPLC (high‐ pressure liquid chromatography).
– The characteristic retention times are used to identify the amino acids.
– The fluorescence level can be quantified to determine the amount of that amino acid.

Amino acid analysis.

Protein Sequencing Strategy
1)
Purify protein
2)
Cleave disulfides – react with:A)
reducing agent followed by alkylating agent1)
DTT
2)
β‐ME
B)
performic acid


Determination of Primary Structure 1
• Hydrolyze peptides with hot 6M HCl.– Identify AA and % of each.– Usually done by chromatography
• Identify the N‐term and C‐term AAs– C‐term via carboxypeptidase
– N‐term via Sanger’s Reagent (DNFB) • Fluoro‐2,4‐dinitrobenzene and dansyl chloride • Phenylisothiocyanate (Edman degradation)
5P2-27

Determination of Primary Structure 2
• Selectively fragment large proteins into smaller ones.
– Tripsin: cleave to leave Arg or Lys as C‐term AA
– Chymotrypsin: cleave to leave Tyr, Trp or Phe as C‐term AA
– Cyanogen bromide cleaves at internal Met leaving Met as C‐term homoserine lactone
5P2-28

Determine AA sequence of peptides
• Edman’s reagent:– First, phenylisothiocyanate reacts with the terminal amino
group to form a phenylthiocarbamoyl derivative.– This residue cyclizes under acidic conditions to give a
phenylthiohydantoin (PTH)‐amino acid and a peptide shortened by one amino acid residue.
– This PTH‐amino acid is identified by HPLC.– The amino acid composition of the shortened peptide can
be compared with the original peptide.
5P2-29

30

Sanger’s reagent
• Use fluoro‐2,4‐dinitrobenzene and dansyl chloride
• For N‐terminus amino acid
• Fluoro‐2,4‐dinitrobenzene reacts with an amino group and forms dinitrophenyl peptide
• Dansyl chloride reacts with dinitrophenyl peptide and form dansyl peptide
• Acid hydrolysis and analysis using HPLC


Standard Run on 19 PTH AAs

Residue 1 = Leu

Residue 2 = Ile

Protein Sequencing Strategy
• Reassemble sequence through overlaps of peptides created by different means
• Map disulfides by cleaving protein into peptides before
disulfide bond cleavage.
• After purification of disulfide‐linked peptides and cleavage of their disulfide bonds, sequencing of the
peptides should reveal which cysteines are linked in disulfide bonds.

Det. Primary Structure
• A twelve AA peptide was hydrolyzed.• Trypsin hydrolysis:
– Leu‐Ser‐Tyr‐Gly‐Ile‐Arg
– Thr‐Ala‐Met‐Phe‐Val‐Lys
• Chymotrypsin hydrolysis
–Val‐Lys‐Leu‐Ser‐Tyr
–Gly‐Ile‐Arg
– Thr‐Ala‐Met‐Phe
• Deduce the AA sequence5P2-37
One isC-term
Lys is internal!

Det. Primary Structure
• Tr Leu‐Ser‐Tyr‐Gly‐Ile‐Arg• Ct
Gly‐Ile‐Arg
• Ct Val‐Lys‐Leu‐Ser‐Tyr• Tr Thr‐Ala‐Met‐Phe‐Val‐Lys
• Ct Thr‐Ala‐Met‐Phe
• The complete sequence is:
• Thr‐Ala‐Met‐Phe‐Val‐Lys‐Leu‐Ser‐Tyr‐Gly‐Ile‐Arg
5P2-38
Keeping in mind the N-term AA and overlaping the sequences properly gives:

Protein Identification
• 2D‐GE + MALDI‐MS
– Peptide Mass Fingerprinting (PMF)
• 2D‐GE + MS‐MS
– MS Peptide Sequencing/Fragment Ion Searching
• Multidimensional LC + MS‐MS
– ICAT Methods (isotope labelling)
– MudPIT (Multidimensional Protein Ident. Tech.)
• 1D‐GE + LC + MS‐MS
• De Novo Peptide Sequencing

Isoelectric point (pI)
• Separation by charge:
4
5
6
78
9
10
Stable pH gradient
High pH: protein is negatively charged
Low pH:Protein is positively charged
At the isolectric point the protein has no net charge and therefore no longer migrates in the electric field.

2D‐gel technique example

Advantages vs. Disadvantages
• Good resolution of proteins
• Detection of posttranslational modifications
• Not for hydrophobic proteins
• Limited by pH range • Not easy for low abundant
proteins• Analysis and quantification
are difficult

2D - LC/MS/MS
Study protein complexes without gel electrophoresis
Peptides all bind to cation exchange column
Peptides are separated by hydrophobicity on reverse phase column
Successive elution with increasing salt gradients separates peptides by charge
Complex mixture is simplified prior to MS/MS by 2D LC
(trypsin)

A simple definition of a Mass Spectrometer
• A Mass Spectrometer is an analytical instrument that can separate charged molecules according to their mass–to–charge
ratio. • The mass spectrometer can answer the
questions “what is in the sample”
(qualitative structural information) and “how much is
present”
(quantitative determination) for a very wide range of samples at high sensitivity

Mass Spectrometer
• Works by ionizing molecules and then sorting and identifying the ions according to their mass‐to‐
charge (m/z) ratios.
• Two key components
– ion source, which generates the ions– mass analyzer, which sorts the ions

Mass Spectrometer Schematic
Inlet Ion Source
MassFilter Detector Data
System
High Vacuum SystemTurbo pumpsDiffusion pumpsRough pumpsRotary pumps
Sample PlateTargetHPLCGCSolids probe
MALDIESIIonSprayFABLSIMSEI/CI
TOFQuadrupoleIon TrapMag. SectorFTMS
Microch plateElectron Mult.Hybrid Detec.
PC’sUNIXMac

Ion Sources
• Electrospray ionization (ESI)
• Atmospheric pressure chemical ionization
(APCI)
• Atmospheric pressure photoionization (APPI)
• Matrix Assisted Laser Desorption Ionization (MALDI)

High voltage appliedto metal sheath (~4 kV)
Sample Inlet Nozzle(Lower Voltage)
Charged droplets
+++ ++
+
+++ ++
+
+++ ++
+ +++
+++ +++
+++ ++
++
++
+
++++++
+++
MH+
MH3+
MH2+
Pressure = 1 atm Inner tube diam. = 100 um
Sample in solution
N2
N2 gas
Partial vacuum
Electrospray ionization:
Ion Sources make ions from sample molecules (Ions are easier to detect than neutral molecules.)
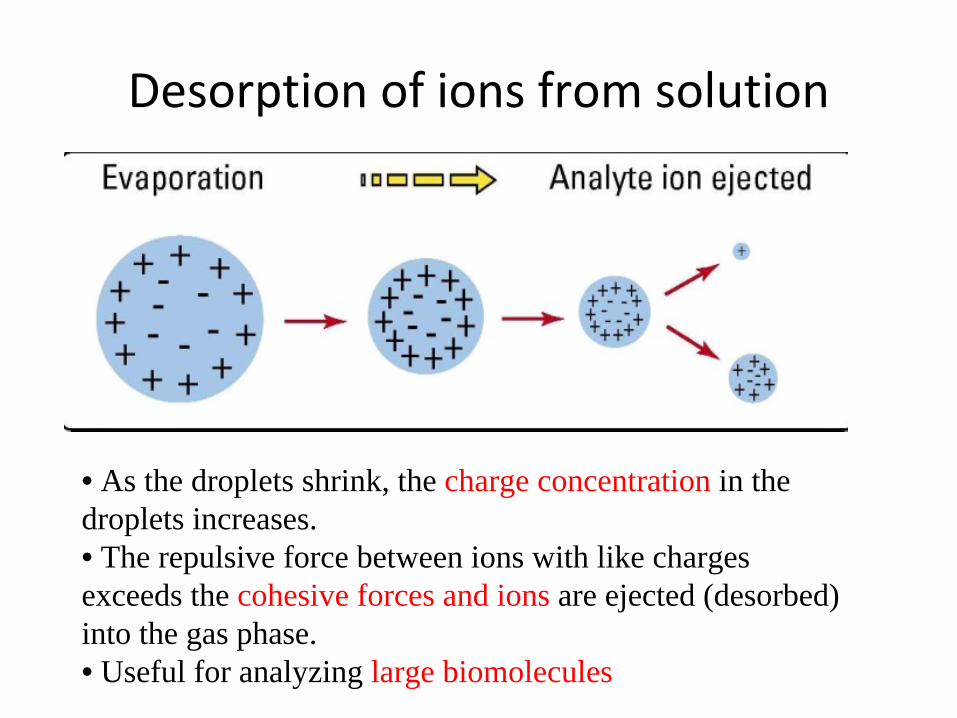
Desorption of ions from solution
• As the droplets shrink, the charge concentration in thedroplets increases.• The repulsive force between ions with like charges exceeds the cohesive forces and ions are ejected (desorbed) into the gas phase. • Useful for analyzing large biomolecules
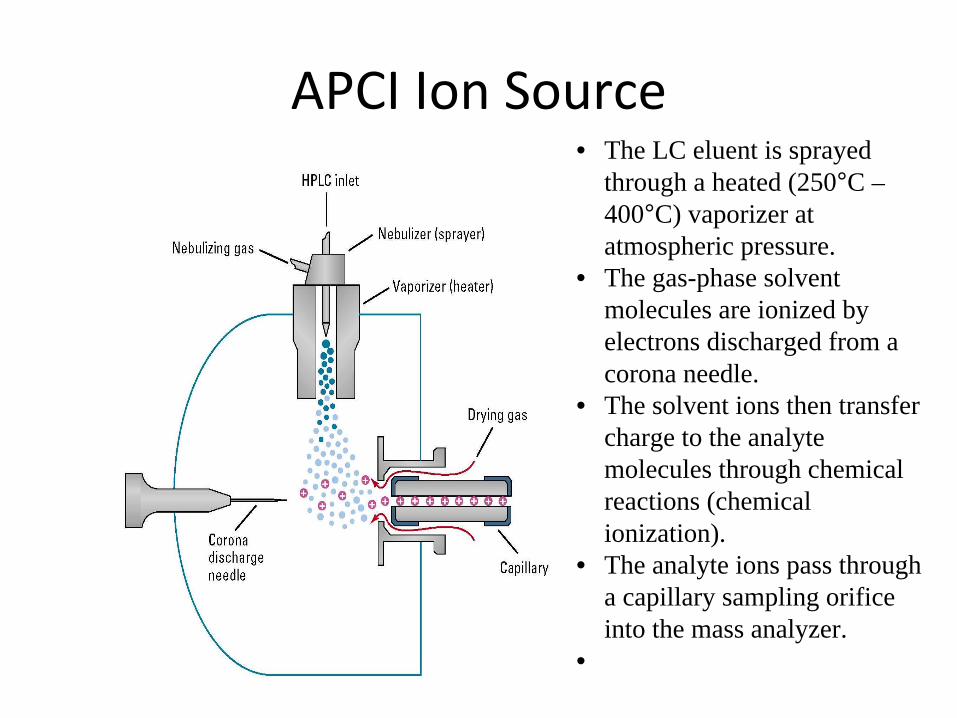
APCI Ion Source• The LC eluent is sprayed
through a heated (250°C – 400°C) vaporizer at atmospheric pressure.
• The gas-phase solvent molecules are ionized by electrons discharged from a corona needle.
• The solvent ions then transfer charge to the analyte molecules through chemical reactions (chemical ionization).
• The analyte ions pass through a capillary sampling orifice into the mass analyzer.
•

APPI Ion Source
• Avaporizer converts the LC eluent to the gas phase.
• A discharge lamp generates photons in a narrow range of ionization energies.
• The energy ionizes analyte molecules while minimizing the ionization of solvent molecules.
• The resulting ions pass through a capillary sampling orifice into the mass analyzer.

MALDI Ionization
++
+
+
---
++
+
+
----+ +
Analyte
Matrix
Laser
+++
• Absorption of UV radiation by chromophoric matrix and
ionization of matrix
• Dissociation of matrix, phase change to super‐compressed gas,
charge transfer to analyte molecule
• Expansion of matrix at supersonic velocity, analyte trapped in
expanding matrix plume (explosion/”popping”)
+
+
+

Mass Analyzers
• Quadrupole• Time‐of‐flight
• Ion trap• Fourier transform‐ion cyclotrone resonance
(FT‐ICR)
• Mass Spectrometers separate ions according to their mass‐to‐charge (m/z) ratios

Quadrupole Mass Analyzer
• Uses a combination of radio frequency and DC
• voltages to operate as a mass filter.
• Has four parallel metal rods.
• Lets one mass pass through at a time. Can scan through all masses or sit at one fixed mass.

Principle of Time of Flight Analyzer
• A uniform electromagnetic force is applied to all ions at the same time, causing them to accelerate down a flight tube.
• Lighter ions travel faster and arrive at the detector first, so the mass-to-charge ratios of the ions are determined by their arrival times.
• Time-of-flight mass analyzers have a wide mass range and can be very accurate in their mass measurements.

Ion Trap
• Consists of a circular ring electrode plus two end caps
that together form a chamber.
• Ions entering the chamber are “trapped”
there by
electromagnetic fields.• Scanning field can eject ions
of specific m/z • Advantages
– MS/MS/MS….. – High sensitivity full scan
MS/MS

Fourier transform‐ion cyclotron resonance (FT‐ICR) Mass Analyzer
• Ions entering a chamber are trapped in circular orbits by powerful electrical and magnetic fields.
• When excited by a radio-frequency (RF) electrical field, the ions generate a time- dependent current.
• This current is converted by Fourier transform into orbital frequencies of the ions which correspond to their mass-to- charge ratios.
• Can perform multiple stages of mass spectrometry without additional mass analyzers.
• Have a wide mass range and excellent mass resolution.
• Very expensive mass analyzers.

What is MS/MS?
MS/MS means using two mass analyzers (combined in one instrument) to select an analyte (ion) from a mixture, then generate fragments from it to give structural information.
Ion source MS-2MS-1
Mixture of ions
Single ion
Fragments

What is MS/MS?
MS/MS+
+
+ +
+
1 peptide selected for
MS/MS
The masses of all the pieces give an MS/MS
spectrum
Peptidemixture
Have only masses to start

Interpretation of an MSMS spectrum to derive structural information is analogous to solving a puzzle
+
+
+ +
+
Use the fragment ion masses as specific pieces of the puzzle to help piece the intact molecule back together

Tandem Mass Spectrometry
• Different MS‐MS configurations
– Quadrupole‐quadrupole (low energy)– Magnetic sector‐quadrupole (high energy)
– Quadrupole‐time‐of‐flight (low energy)
– Time‐of‐flight‐time‐of‐flight (low energy)
• Fragmentation experiments may also be performed on single analyzer instruments such as ion trap instruments
and TOF instruments equipped with post‐source decay

Tandem MS

Fragmentation in MS/MS produces sequence‐specific fragment ions

Mass Spectra
• The peaks in the mass spectrum:– Prefix – Fragments with neutral losses
(‐H2
O, ‐NH3
)– Noise and missing peaks.
G V D L K
mas s0
57 Da = ‘G’ 99 Da = ‘V’LK D V G
and Suffix Fragments.
D
H2O

Protein Identification with MS/MS
G V D L K
mas s0
Inte
nsity
mas s0
MS/MSPeptide Identification:

Peptide sequencing by MS/MSPeptide sequencing by MS/MS
(Standing 2003 Curr. Opin. Struct. Biol. 13, 595-601)

Protein Identification by Tandem Mass Spectrometry
SSeeqquueennccee
S#: 1708 RT: 54.47 AV: 1 NL: 5.27E6T: + c d Full ms2 638.00 [ 165.00 - 1925.00]
200 400 600 800 1000 1200 1400 1600 1800 2000m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
850.3
687.3
588.1
851.4425.0
949.4
326.0524.9
589.2
1048.6397.1226.91049.6489.1
629.0
MS/MS instrumentMS/MS instrument
Database search•Sequestde Novo interpretation•Sherenga

Advantages vs. Disadvantages
• Determination of MW and aa. Sequence
• Detection of posttranslational modifications
• High-throughput capability
• High capital costs• Requires sequence
databases for analysis