characterization of the c-terminal dna-binding/dna endonuclease region of a group ii intron-encoded...
TRANSCRIPT

Characterization of the C-Terminal DNA-binding/DNAEndonuclease Region of a Group II Intron-encoded Protein
Joseph San Filippo and Alan M. Lambowitz*
Department of Chemistryand Biochemistryand Section of MolecularGenetics and MicrobiologySchool of Biological SciencesInstitute for Cellularand Molecular BiologyUniversity of Texas at AustinMBB2. 234BA, 2500 SpeedwayAustin, TX 78712, USA
Group II intron retrohoming occurs by a mechanism in which the intronRNA reverse splices directly into one strand of a double-stranded DNAtarget site, while the intron-encoded reverse transcriptase uses a C-termi-nal DNA endonuclease activity to cleave the opposite strand and thenuses the cleaved 30 end as a primer for reverse transcription of the insertedintron RNA. Here, we characterized the C-terminal DNA-binding/DNAendonuclease region of the LtrA protein encoded by the Lactococcus lactisLl.LtrB intron. This C-terminal region consists of an upstream segmentthat contributes to DNA binding, followed by a DNA endonucleasedomain that contains conserved sequence motifs characteristic ofH–N–H DNA endonucleases, interspersed with two pairs of conservedcysteine residues. Atomic emission spectroscopy of wild-type and mutantLtrA proteins showed that the DNA endonuclease domain contains asingle tightly bound Mg2þ ion at the H–N–H active site. Although theconserved cysteine residue pairs could potentially bind Zn2þ, the purifiedLtrA protein is active despite the presence of only sub-stoichiometricamounts of Zn2þ, and the addition of exogenous Zn2þ inhibits the DNAendonuclease activity. Multiple sequence alignments identified featuresof the DNA-binding region and DNA endonuclease domain that areconserved in LtrA and related group II intron proteins, and their func-tional importance was demonstrated by unigenic evolution analysis andbiochemical assays of mutant LtrA protein with alterations in key aminoacid residues. Notably, deletion of the DNA endonuclease domain ormutations in its conserved sequence motifs strongly inhibit reverse tran-scriptase activity, as well as bottom-strand cleavage, while retainingother activities of the LtrA protein. A UV-cross-linking assay showedthat these DNA endonuclease domain mutations do not block DNAprimer binding and thus likely inhibit reverse transcriptase activity eitherby affecting the positioning of the primer or the conformation of thereverse transcriptase domain.
q 2002 Elsevier Science Ltd. All rights reserved
Keywords: DNA endonuclease; DNA–protein interaction; metalloenzyme;retrotransposon; ribozyme*Corresponding author
Introduction
Mobile group II introns, found in bacteria andeukaryotic organelles, are catalytic RNAs (“ribo-
zymes”) that encode a reverse transcriptase (RT),which functions in conjunction with the intronRNA to promote mobility.1,2 The best characterizedmobile group II introns are the yeast mtDNA aI1and aI2 introns and the Lactococcus lactis Ll.LtrBintron. These introns insert site-specifically at theunoccupied exon junction in an intronless allele, aprocess termed retrohoming, and also transpose toectopic sites that resemble the normal homing siteat low frequency. Retrohoming occurs by a novelmechanism in which the excised intron RNA usesits ribozyme activity to reverse splice directly into
0022-2836/02/$ - see front matter q 2002 Elsevier Science Ltd. All rights reserved
E-mail address of the corresponding author:[email protected]
Abbreviations used: DTT, dithiothreitol; ICP,inductively coupled plasma; IEP, intron-encoded protein;IPTG, isopropyl-1-thio-b-D-galactopyranoside; LTR, longterminal repeat; mt, mitochondrial; ORF, open readingframe; RNP, ribonucleoprotein; RT, reverse transcriptase;TPRT, target DNA-primed reverse transcription.
doi:10.1016/S0022-2836(02)01147-6 available online at http://www.idealibrary.com onBw
J. Mol. Biol. (2002) 324, 933–951

one strand of a DNA target site, while the intron-encoded RT uses a C-terminal DNA endonucleasedomain to cleave the opposite strand and thenuses the 30 end of the cleaved strand as a primerfor target DNA-primed reverse transcription(TPRT) of the inserted intron RNA.3 – 9 Recentstudies have shown that variations of this mecha-nism are also used for the retrotransposition ofgroup II introns to ectopic sites,10 – 13 and for theretrohoming of a bacterial group II intron thatlacks the conserved endonuclease domain.14 Inthese cases, the intron RNA reverse splices into adouble-stranded or transiently single-strandedDNA target site and is reverse transcribed byusing a primer generated either by non-specificcleavage of the opposite strand or via DNAreplication.
The group II intron-encoded RTs that mediateretrohoming and retrotransposition are multi-functional and contain several conserved domainsassociated with different activities. In the yeastmtDNA and L. lactis Ll.LtrB introns, these are anRT domain, with an N-terminal region Z charac-teristic of the RTs of non-LTR-retroelements,domain X associated with RNA splicing ormaturase activity, and a C-terminal DNA bind-ing/DNA endonuclease region (Figure 1).1,2,15,16
The latter region includes a conserved DNA endo-nuclease domain with amino acid residuesequence motifs characteristic of the H–N–Hfamily of DNA endonucleases, interspersed withtwo pairs of cysteine residues that nominally fitthe consensus CX2 – 4CXNCX2 –4C for a major classof zinc fingers.17 – 19 Because of the conservedcysteine residues, the DNA endonuclease domainhas been referred to as the zinc domain. Thegroup II intron-encoded RTs are related bothstructurally and functionally to those encoded bynon-long terminal repeat (non-LTR) retrotrans-posons, which however, are associated with twoother types of DNA endonuclease domains usedto generate the primer for related TPRT mecha-nisms—an AP endonuclease domain for LINEelements, and a domain related to type II or IIsrestriction enzymes for R2 elements.20 – 23 This situ-
ation suggests a scenario in which the RT domainsof different retroelements became associated withdifferent types of DNA endonuclease domains,enabling similar integration mechanisms.2,3,23,24
The basic outline of the TPRT mechanism usedby the yeast mtDNA and L. lactis Ll.LtrB introns isnow known.1,2 First, the intron-encoded protein(IEP) promotes RNA splicing (maturase activity)by helping fold the intron RNA into the cata-lytically active structure, and it remains bound tothe excised intron lariat RNA to form a ribonucleo-protein (RNP) complex that mediates intronmobility. The group II intron RNPs recognizerelatively large DNA target sites, extending from20 to 25 bps upstream of the intron-insertionsite in the 50 exon to 10 bps downstream in the 30
exon, with both the IEP and base pairing ofthe intron RNA contributing to DNA target siterecognition.10,25 – 28 According to the modelsuggested by DNA footprinting and modification-interference experiments with the lactococcalintron, the IEP first recognizes a small number ofspecific bases in the distal 50-exon region of theDNA target site via major groove interactions.28
These base interactions, bolstered by phosphate-backbone interactions along one face of the DNAhelix, trigger local DNA unwinding, enablingthe intron RNA to base pair to the adjacent 14- to16-nt region of the DNA target site (the EBS/IBSand d–d0 interactions) and then reverse splice intothe top or sense strand. Bottom-strand cleavageoccurs after a lag and requires additional inter-actions between the IEP and the 30 exon, whichpresumably position the C-terminal DNA endo-nuclease domain for cleavage between positionsþ9 and þ10. After bottom-strand cleavage, theIEP must reposition so that the RT domain can usethe 30 end of the cleaved strand as a primer forTPRT of the inserted intron RNA. To completemobility, the cDNA copy of the intron RNA isintegrated into the recipient DNA by cellularrecombination or repair mechanisms.7 – 9
The C-terminal DNA binding/DNA endo-nuclease region of group II IEPs is relatively unex-plored. Sequence comparisons and functional
Figure 1. Map of the LtrA protein encoded by the L. lactis Ll.LtrB group II intron. The RT domain contains conservedsequence motifs I–VII, along with an upstream region Z characteristic of the RTs of non-LTR retroelements.1,2,15,16
Domain X is a putative RNA binding domain, which is required for RNA splicing (“maturase”) activity and is locatedin a position corresponding to the thumb and connecting domains of retroviral RTs.16 Following domain X are theDNA-binding region (D) and DNA endonuclease domain (En). Key amino acid residues are indicated below, as arethe boundaries of the DConEn and DD/ConEn mutants.
934 Group II Intron Protein DNA Endonuclease Region

analyses show that this region consists of a rela-tively unconserved segment downstream ofdomain X that contributes to DNA binding,followed by the C-terminal DNA endonucleasedomain, which contains the conserved H–N–Hand cysteine residue motifs.4,25,28 Deletion of theconserved DNA endonuclease domain abolishesbottom-strand cleavage, but the truncated proteinretains RNA splicing activity and the ability tosupport reverse splicing of the intron RNA intodouble-stranded DNA target sites.4,6 A furthertruncation that deletes the upstream variableregion abolishes stable DNA binding and reversesplicing into double-stranded DNA target sites,but the protein again retains RNA splicing activity,as well as a small amount of reverse splicingactivity with single-stranded DNA target sites(,10% wild-type).4,25,28 The putative DNA-bindingregion is thought to interact with the distal 50-exonregion of the DNA target site to promote unwind-ing of double-stranded DNA substrates andalso makes a major contribution to the binding ofsingle-stranded DNA.25 It is not clear whether thisregion is a separate domain, a linker betweendomain X and the DNA endonuclease domain, oran extension of the DNA endonuclease domain,and it remains possible that other regions of theIEP also contribute to DNA binding.
Here, we analyzed the C-terminal DNA-bindingregion and DNA endonuclease domain of theLtrA protein encoded by the L. lactis Ll.LtrB intron.We identified conserved features of both that arerequired for efficient mobility. Our results suggestthat the DNA endonuclease domain contains asingle catalytically essential Mg2þ ion coordinatedat the H–N–H active site and may function as apositive effector of RT activity.
Results
Multiple sequence alignments
Figure 2 shows multiple sequence alignments ofthe C-terminal region of the LtrA protein (i.e. theregion downstream of domain X) with othergroup II IEPs and related H–N–H DNA endo-nucleases. The latter include a subset of group Iintron homing endonucleases, proteins belongingto the EndoVII-McrA superfamily, and bacterialcolicins and pyocins.29,30 On the basis of phylo-genetic analysis, the group II IEPs have been classi-fied into mitochondrial, chloroplast, and bacteriallineages, with the diverse bacterial lineage beingdivided into a four subclasses (A–D).31 Althoughof bacterial origin, the LtrA protein is closelyrelated to the fungal mtDNA group II IEPsand falls within the mitochondrial lineage.31,32
Consequently, the primary alignment was madefor LtrA with six representative proteins of thatlineage. The group II IEPs belonging to otherlineages and related proteins were then alignedwith the proteins of the mitochondrial lineage.
Amino acid residues with .75% identity in theseven proteins of the mitochondrial lineage areshaded black, while those with .50% similarityare shaded gray. The predicted secondary structureof the LtrA protein is shown above the alignments.
First, the alignments illustrate the upstreamvariable and downstream conserved regionscharacteristic of the C termini of group II IEPs.Without structural data, the boundaries of dif-ferent domains cannot be known with certainty.Assuming that domain X ends after the lastconserved amino acid residue, LtrAs variableDNA-binding region extends from amino acid resi-due 487 to 542, while the conserved endonucleasedomain begins with the first conserved cysteineresidue pair at position 543 and extends essentiallyto the end of the protein. Aligned as shown, theDNA-binding regions of the group II IEPs containno invariant or highly conserved amino acid resi-dues, but do show conservation of similar aminoacid residues at a number of positions, includingan upstream cluster of basic amino acid residuesand a downstream cluster that lies within a pre-dicted a-helix (amino acid residues 523–538).This predicted a-helical region was found in17/18 of the aligned group II IEPs (the exceptionwas the Podospora anserina mt group II IEP), and itis the site of an extensively studied yeast aI2mutant, P714T (boxed in Figure 2), which isdeficient in DNA endonuclease activity, but haselevated RT activity with endogenous mt RNAtemplates.33 – 35 The predicted a-helical region inLtrA and the other group II IEPs is preceded by arelatively long region for which secondary struc-ture could not be predicted using the PSI or PHDprotein folding programs.36,37 A BLAST searchwith the variable region of LtrA (amino acid resi-dues 491–542) found no matches in the GenBankrelease 123.0 data base, including other group IIIEPs.38
The DNA endonuclease domain of LtrA contains27 of 57 amino acid residues that are identical orsimilar to the other proteins of the mitochondriallineage, including several conserved histidineresidues and the two pairs of conserved cysteineresidues (denoted CX2C/1 and CX2C/2). Thisdomain aligns well with the H–N–H domains ofgroup I IEPs, EndoVII-McrA proteins, and bacterialcolicin and pyocins. As noted previously, theconserved cysteine residue pairs are present in theEndoVII-McrA proteins, but absent in group Iintron homing endonucleases and colicins.18,19,29,30
The predicted secondary structure of theH–N–H region of the LtrA protein consists of ab-strand, which contains the first histidine residueof the H–N–H motif, followed by a short a-helix,an extended loop with a second a-helix, followedby a C-terminal a-helix, which contains both thedistal conserved cysteine residue pair and thesecond histidine residue of the H–N–H motif.The conserved asparagine residue of the H–N–Hmotif is replaced in LtrA by a functionally equiva-lent glutamine residue, which is found in the
Group II Intron Protein DNA Endonuclease Region 935
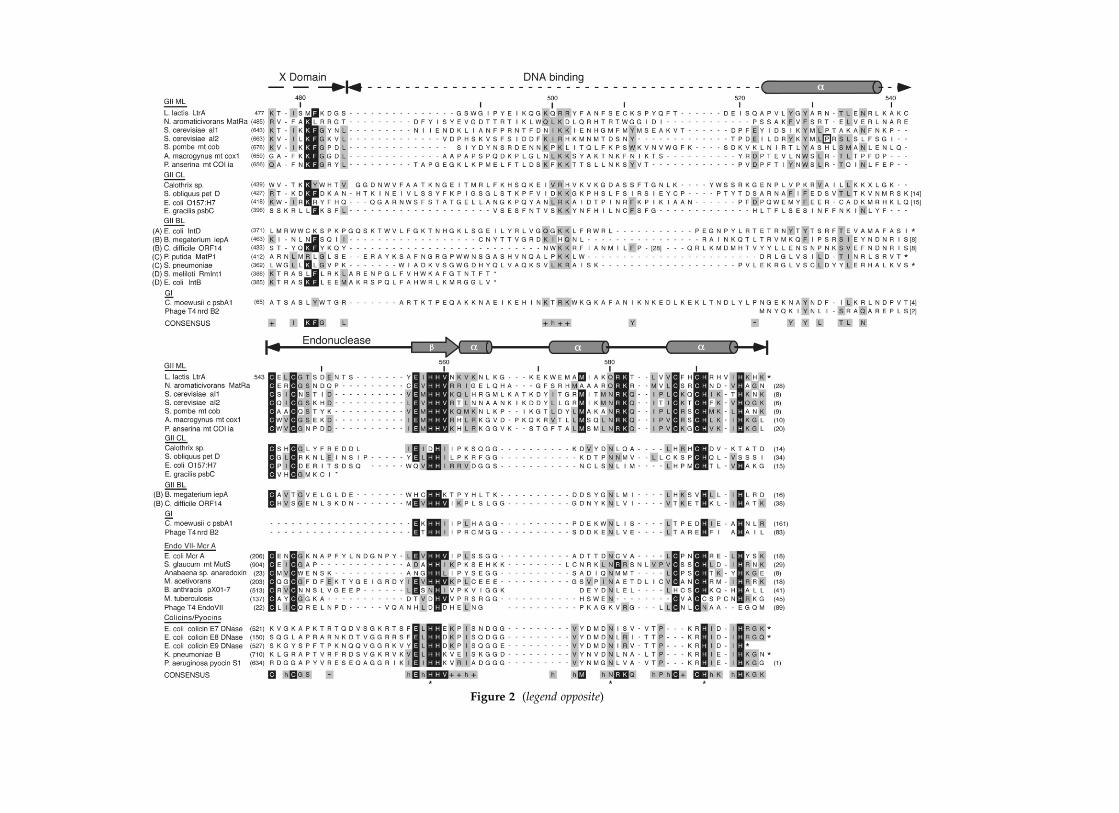
Figure 2 (legend opposite)

predicted loop region downstream of the seconda-helix. The same predicted b/a secondary struc-ture for the conserved H–N–H active-site residueswas found for all group II IEPs in the alignmentand matched the experimentally determined struc-ture for the corresponding regions of colicin E7and E9 DNA endonucleases, as well as phage T4endonuclease VII (see below).
The group II IEPs of the chloroplast lineage andbacterial class B are similar to those of the mito-chondrial lineage in that domain X is followedby a variable region, which includes a conservedcluster of basic amino acid residue and predicteda-helical region, and in most cases an H–N–Hdomain. Interestingly, however, while the H–N–Hmotif is conserved in these proteins, the conservedcysteine residue motifs have diverged, with thealigned chloroplast-lineage proteins retaining onlythree conserved cysteine residues, and the bacterialclass B proteins retaining only one conservedcysteine residue. The proteins shown for bacterialclasses A and C lack the H–N–H domain, but stillretain C-terminal extensions, which again includean upstream cluster of basic residues and adownstream predicted a-helix. Finally, the bac-terial class D proteins, the Sinorhizobium melilotiRmInt1 and Escherichia coli IntB IEPs, lack theH–N–H domain, but still retain a short (20 aaresidue) C-terminal extension after domain X. ThisC-terminal extension is conserved between thetwo class D proteins, but does not appearto be related to that in LtrA. The evolutionarysignificance of these findings is discussed below(see Discussion).
Detailed comparison of LtrA with colicin E9and phage T4 endonuclease VII
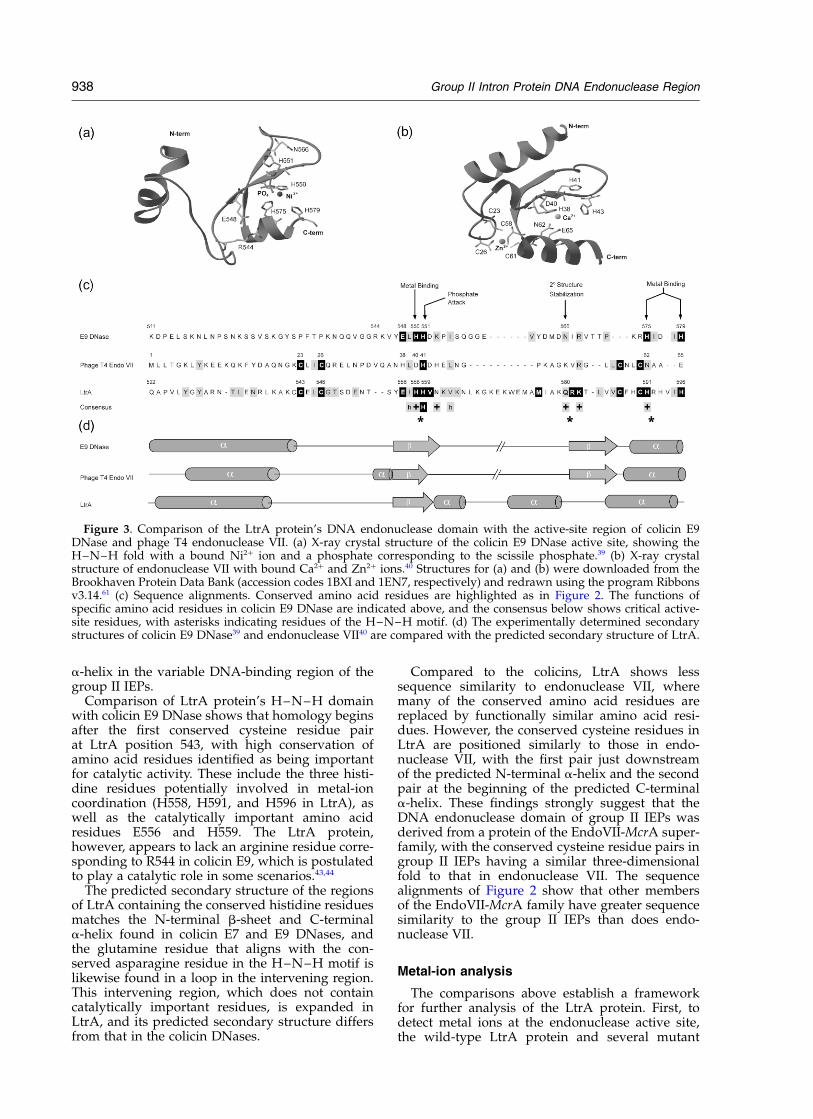
We next compared the DNA endonucleasedomain of LtrA with those of related proteins for
which detailed structural and mechanistic infor-mation is available. Figure 3(a) and (b) show theX-ray crystal structures of colicin E9 DNase andphage T4 endonuclease VII, a Holliday junctionresolvase that is a member of the EndoVII-McrAsuperfamily.39,40 The structures of these enzymes,as well as that of colicin E7 DNase41 show a verysimilar metal ion-dependent nuclease fold consist-ing of a central core of twisted b-strands with aC-terminal a-helix. In the colicin X-ray crystalstructures, the endonuclease active site contains adivalent metal ion, Zn2þ in colicin E7 and Ni2þ incolicin E9, coordinated by two histidine residues,which are part of the H–N–H motif (H550 andH575 in colicin E9), with a third histidine residueinvolved in Zn2þ coordination in colicin E7, butless so in Ni2þ coordination in colicin E9 (colicinE9 H579).41 – 44 Recent studies of colicin E9 DNAsshow that Mg2þ, which is required for double-stranded DNA cleavage, is the physiologicallyrelevant divalent cation bound at the active site.44
Additional conserved residues in colicin E9DNase, which are required for Mg2þ-dependentcleavage activity are R453, R544, E548, andH551.41 – 44 The conserved asparagine residue of theH–N–H motif (N566) is in a loop region, where ithelps stabilize the higher-order structure of thedomain.39 The endonuclease VII structure showssimilarly positioned positively charged residues inthe N-terminal b-strand and C-terminal a-helixinvolved in coordinating the active-site divalentmetal ion, which is Ca2þ in the crystallized form ofthe enzyme.40 The two pairs of conserved cysteineresidues, which are missing in the colicin DNases,are interspersed with the active-site residues andcoordinate a single Zn2þ atom, which may berequired to stabilize the metal-dependent nucleasefold.30,40,45 Notably, the colicin E7 and E9 DNasesand endonuclease VII all have an upstreama-helix positioned similarly to the predicted
Figure 2. Multiple sequence alignments. The C-terminal region of LtrA was aligned with other group II IEPs andrelated domains of other proteins, including group I IEPs (GI); EndoVII-McrA proteins, and bacterial colicins andpyocins. The group II IEPs are classified into mitochondrial (ML), chloroplast (CL), and bacterial (BL) lineages, withthe bacterial lineage divided into subclasses A–D.31 Proteins were aligned using MacVector 6.5.1 (Oxford MolecularLtd., Madison, WI), followed by manual refinement. Amino acid residues conserved in the mitochondrial lineage ofgroup II IEPs are highlighted: black, .75% identity; gray, .50% similarity. Similar amino acid residues are definedbased on the BLOSUM 55.50 matrix as follows: (R, K . H); (H, Q, N); (D, E); (I, L, M, V); (S, T); (Y, F, W).60 Asterisksindicate amino acid residues comprising the H–N–H motif. The predicted secondary structure of the LtrA proteinbased on the PHD folding prediction program36 is shown above the alignments, and a consensus sequence based onthe alignment of mitochondrial lineage group II IEPs is shown below, with þ and 2 indicating positively and nega-tively charged amino acid residues, respectively, and “h” indicating a hydrophobic residue. The site of the P714Tmutation in the yeast aI2 protein is boxed. Accession numbers are: L. lactis LtrA (AAB06503), N. aromaticivoransMatRa (AAD03884), Saccharomyces cerevisae aI1 (CAA24071), S. cerevisae aI2 (CAA24060), S. pombe mt cob (P05511),A. macrogynus mt cox1 (AAC49235), P. anserina mt COI ia (CAA38781), Calothrix sp. (CAA50529), S. obliquus pet D(P19593), E. coli O157:H7 (BAA31792), E. gracilis psbC (QQEGC4), E. coli IntD (BAA22286), B. megaterium iepA(BAA82060), C. difficile ORF 14 (CAA67199), P. putida MatP1 (AAD16434), S. pneumoniae (AAC38715), S. melilotiRmInt1 (CAA72334), E. coli IntB (CAA54637), C. moewusii c psbA1 (P09753), Phage T4 nrd B2 (P32283), E. coli McrA(P24200), S. glaucum mt MutS (AAC16386), Anabaena sp. anaredoxin (Q44141), M. acetivorans (AAM06859), B. anthracispX01-7 (AAD32311), M. tuberculosis (AAK46845), Phage T4 EndoVII (AAD42477), E. coli colicin E7 DNase (Q47112),E. coli colicin E8 DNase (P09882), E. coli colicin E9 DNase (P09883), K. pneumoniae B (AAD39262), P. aeruginosa pyocinS1 (BAA02203).
Group II Intron Protein DNA Endonuclease Region 937
a-helix in the variable DNA-binding region of thegroup II IEPs.
Comparison of LtrA protein’s H–N–H domainwith colicin E9 DNase shows that homology beginsafter the first conserved cysteine residue pairat LtrA position 543, with high conservation ofamino acid residues identified as being importantfor catalytic activity. These include the three histi-dine residues potentially involved in metal-ioncoordination (H558, H591, and H596 in LtrA), aswell as the catalytically important amino acidresidues E556 and H559. The LtrA protein,however, appears to lack an arginine residue corre-sponding to R544 in colicin E9, which is postulatedto play a catalytic role in some scenarios.43,44
The predicted secondary structure of the regionsof LtrA containing the conserved histidine residuesmatches the N-terminal b-sheet and C-terminala-helix found in colicin E7 and E9 DNases, andthe glutamine residue that aligns with the con-served asparagine residue in the H–N–H motif islikewise found in a loop in the intervening region.This intervening region, which does not containcatalytically important residues, is expanded inLtrA, and its predicted secondary structure differsfrom that in the colicin DNases.
Compared to the colicins, LtrA shows lesssequence similarity to endonuclease VII, wheremany of the conserved amino acid residues arereplaced by functionally similar amino acid resi-dues. However, the conserved cysteine residues inLtrA are positioned similarly to those in endo-nuclease VII, with the first pair just downstreamof the predicted N-terminal a-helix and the secondpair at the beginning of the predicted C-terminala-helix. These findings strongly suggest that theDNA endonuclease domain of group II IEPs wasderived from a protein of the EndoVII-McrA super-family, with the conserved cysteine residue pairs ingroup II IEPs having a similar three-dimensionalfold to that in endonuclease VII. The sequencealignments of Figure 2 show that other membersof the EndoVII-McrA family have greater sequencesimilarity to the group II IEPs than does endo-nuclease VII.
Metal-ion analysis
The comparisons above establish a frameworkfor further analysis of the LtrA protein. First, todetect metal ions at the endonuclease active site,the wild-type LtrA protein and several mutant
Figure 3. Comparison of the LtrA protein’s DNA endonuclease domain with the active-site region of colicin E9DNase and phage T4 endonuclease VII. (a) X-ray crystal structure of the colicin E9 DNase active site, showing theH–N–H fold with a bound Ni2þ ion and a phosphate corresponding to the scissile phosphate.39 (b) X-ray crystalstructure of endonuclease VII with bound Ca2þ and Zn2þ ions.40 Structures for (a) and (b) were downloaded from theBrookhaven Protein Data Bank (accession codes 1BXI and 1EN7, respectively) and redrawn using the program Ribbonsv3.14.61 (c) Sequence alignments. Conserved amino acid residues are highlighted as in Figure 2. The functions ofspecific amino acid residues in colicin E9 DNase are indicated above, and the consensus below shows critical active-site residues, with asterisks indicating residues of the H–N–H motif. (d) The experimentally determined secondarystructures of colicin E9 DNase39 and endonuclease VII40 are compared with the predicted secondary structure of LtrA.
938 Group II Intron Protein DNA Endonuclease Region

derivatives were analyzed by inductively coupledplasma (ICP) atomic emission spectroscopy.46 Forthese experiments, the LtrA protein, which hadbeen dialyzed against buffer containing 0.1 mMEDTA, was further purified by chromatographyon a HiTrap SP ion-exchange column to removeany loosely bound metal ions and residual dithio-threitol (DTT; see Materials and Methods). Weverified that this additional step did not signifi-cantly decrease bottom-strand cleavage activity ofthe LtrA protein after reconstitution into RNPparticles, although the protein did appear to showsignificantly decreased stability after prolongedincubation (.20 minutes; not shown).
As summarized in Table 1, the wild-type LtrAprotein preparation contains an approximately 1:1molar ratio of Mg2þ and 0.2 mol/mol Zn2þ. Bycontrast, the DConEn mutant, which has a C-termi-nal truncation that deletes the conserved endo-nuclease domain (see Figure 1), contains greatlydecreased amounts of Mg2þ (0.2 mol/mol), alongwith low, possibly further reduced amounts ofZn2þ (0.1 mol/mol). These findings indicate thatthe Mg2þ detected in the assay is tightly associatedwith the DNA endonuclease domain. AdditionalMg2þ ions expected to be present at the RT activesite are presumably bound less tightly and lostduring purification. The LtrA protein preparationsprior to the additional ion-exchange chroma-tography contained only slightly higher amountsof Zn2þ (0.3(^0.1) mol/mol).
To test the role of the H–N–H motif in metal-ioncoordination, we constructed the mutant H591A,which has alanine residue-substituted for a con-served histidine residue putatively involved inmetal coordination, based on the comparison tocolicin E9 DNase (see Figure 3).39 This mutationdecreased the bound Mg2þ to near backgroundlevels, and it may also slightly decrease the amountof Zn2þ. By contrast, the mutation CX2C/1 ! A,which has alanine residues substituted for bothcysteine residues of the first conserved cysteineresidue pair, did not significantly affect the amountof bound Mg2þ and also retained the same lowamount of Zn2þ. This finding indicates that thecysteine residue motif does not itself bind Mg2þ,
nor is it essential to maintain the structure of theMg2þ-binding fold. Neither the wild-type LtrAprotein nor any of the mutants analyzed containeddetectable Mn2þ or Ni2þ (not shown). Consideredtogether, our results indicate that Mg2þ isthe predominant metal-ion coordinated at theH–N–H active site. The results are consistentwith the possibility that LtrA contains substoichio-metric amounts of Zn2þ, but do not distinguishwhether it is bound specifically or non-specifically.
Effect of exogenous Zn21 on DNAendonuclease activity
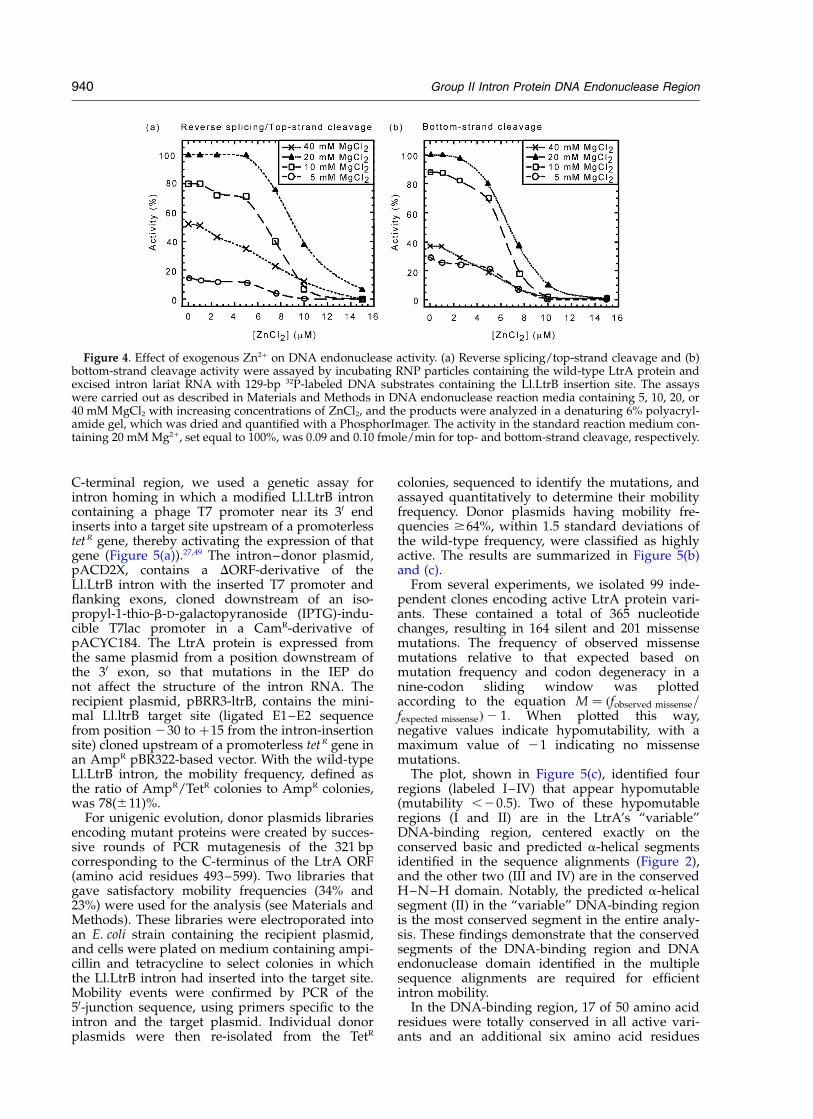
To further investigate the Zn2þ requirement ofthe H–N–H domain, we carried out DNA endo-nuclease assays at different Mg2þ concentrations(5, 10, 20 and 40 mM) in the presence of increasingconcentrations of Zn2þ (Figure 4). In the absence ofZn2þ, both the bottom-strand cleavage and reversesplicing/top-strand cleavage reactions were opti-mal at 20 mM MgCl2. Addition of increasingconcentrations of ZnCl2 progressively inhibitedboth reactions, with the K1/2 ¼ 6.8 mM for bottom-strand cleavage and K1/2 ¼ 7.0–9.5 mM for reversesplicing/top-strand cleavage at all Mg2þ concen-trations tested. The finding that the K1/2 forbottom-strand cleavage is independent of Mg2þ
concentration indicates that exogenous Zn2þ doesnot competitively inhibit the reaction by displacingMg2þ from the active site, and instead inhibitsDNA endonuclease activity by binding to a secondsite. Similar Zn2þ inhibition was also found forLtrA protein that had been further purified by ionexchange chromatography (data not shown).Control experiments showed that 100 mM Zn2þ
does not affect LtrA-promoted RNA splicing ofthe Ll.LtrB intron, indicating that the Zn2þ inhi-bition of DNA endonuclease activity is not due toinhibition of the ribozyme activity of the intronRNA or impaired RNA-protein interaction (notshown). Although the Zn2þ inhibition could bemediated by coordination with the conservedcysteine residue pairs, low amounts of Zn2þ alsoinhibit colicin E9 DNase, which does not containconserved cysteine residue pairs.42 In this case, theZn2þ appears to act by causing changes in proteinstructure, which destabilize the DNA endo-nuclease domain. We conclude that Zn2þ is notrequired for DNA endonuclease activity.
Unigenic evolution analysis
To systematically identify essential and non-essential features of LtrA’s C-terminal DNA-bind-ing/DNA endonuclease region, we used a methodtermed unigenic evolution.47,48 In this method, agenetic screen is used to identify functional vari-ants in a library of mutant proteins generated bymutagenic PCR. The ratio of missense to silentmutations is then analyzed statistically to identifyconserved (hypomutagenic) and variable (hyper-mutagenic) regions. For analysis of LtrA’s
Table 1. Metal-ion analysis
Mg2þ Zn2þ
LtrA 0.9 ^ 0.3 0.2 ^ 0.1DConEn 0.2 ^ 0.1 0.1 ^ 0.1H591A 0.2 ^ 0.1 0.1 ^ 0.1CX2C/1 1.2 ^ 0.1 0.1 ^ 0.1
The Table summarizes molar ratios of Mg2þ and Zn2þ ionsassociated with wild-type and mutant LtrA proteins, deter-mined by ICP atomic emission spectroscopy. Values for thewild-type protein were 6 to 15-fold above background (dialysisbuffer) for Mg2þ and ~2-fold above background for Zn2þ. Back-grounds were 5–40 ng/ml for both Mg2þ and Zn2þ. NeitherMn2þ nor Ni2þ were detected in the same protein preparations.Data are the mean ^SD for three independent preparations ofeach protein.
Group II Intron Protein DNA Endonuclease Region 939
C-terminal region, we used a genetic assay forintron homing in which a modified Ll.LtrB introncontaining a phage T7 promoter near its 30 endinserts into a target site upstream of a promoterlesstet R gene, thereby activating the expression of thatgene (Figure 5(a)).27,49 The intron–donor plasmid,pACD2X, contains a DORF-derivative of theLl.LtrB intron with the inserted T7 promoter andflanking exons, cloned downstream of an iso-propyl-1-thio-b-D-galactopyranoside (IPTG)-indu-cible T7lac promoter in a CamR-derivative ofpACYC184. The LtrA protein is expressed fromthe same plasmid from a position downstream ofthe 30 exon, so that mutations in the IEP donot affect the structure of the intron RNA. Therecipient plasmid, pBRR3-ltrB, contains the mini-mal Ll.ltrB target site (ligated E1–E2 sequencefrom position 230 to þ15 from the intron-insertionsite) cloned upstream of a promoterless tet R gene inan AmpR pBR322-based vector. With the wild-typeLl.LtrB intron, the mobility frequency, defined asthe ratio of AmpR/TetR colonies to AmpR colonies,was 78(^11)%.
For unigenic evolution, donor plasmids librariesencoding mutant proteins were created by succes-sive rounds of PCR mutagenesis of the 321 bpcorresponding to the C-terminus of the LtrA ORF(amino acid residues 493–599). Two libraries thatgave satisfactory mobility frequencies (34% and23%) were used for the analysis (see Materials andMethods). These libraries were electroporated intoan E. coli strain containing the recipient plasmid,and cells were plated on medium containing ampi-cillin and tetracycline to select colonies in whichthe Ll.LtrB intron had inserted into the target site.Mobility events were confirmed by PCR of the50-junction sequence, using primers specific to theintron and the target plasmid. Individual donorplasmids were then re-isolated from the TetR
colonies, sequenced to identify the mutations, andassayed quantitatively to determine their mobilityfrequency. Donor plasmids having mobility fre-quencies $64%, within 1.5 standard deviations ofthe wild-type frequency, were classified as highlyactive. The results are summarized in Figure 5(b)and (c).
From several experiments, we isolated 99 inde-pendent clones encoding active LtrA protein vari-ants. These contained a total of 365 nucleotidechanges, resulting in 164 silent and 201 missensemutations. The frequency of observed missensemutations relative to that expected based onmutation frequency and codon degeneracy in anine-codon sliding window was plottedaccording to the equation M ¼ ðfobserved missense=fexpected missenseÞ2 1: When plotted this way,negative values indicate hypomutability, with amaximum value of 21 indicating no missensemutations.
The plot, shown in Figure 5(c), identified fourregions (labeled I–IV) that appear hypomutable(mutability ,20.5). Two of these hypomutableregions (I and II) are in the LtrA’s “variable”DNA-binding region, centered exactly on theconserved basic and predicted a-helical segmentsidentified in the sequence alignments (Figure 2),and the other two (III and IV) are in the conservedH–N–H domain. Notably, the predicted a-helicalsegment (II) in the “variable” DNA-binding regionis the most conserved segment in the entire analy-sis. These findings demonstrate that the conservedsegments of the DNA-binding region and DNAendonuclease domain identified in the multiplesequence alignments are required for efficientintron mobility.
In the DNA-binding region, 17 of 50 amino acidresidues were totally conserved in all active vari-ants and an additional six amino acid residues
Figure 4. Effect of exogenous Zn2þ on DNA endonuclease activity. (a) Reverse splicing/top-strand cleavage and (b)bottom-strand cleavage activity were assayed by incubating RNP particles containing the wild-type LtrA protein andexcised intron lariat RNA with 129-bp 32P-labeled DNA substrates containing the Ll.LtrB insertion site. The assayswere carried out as described in Materials and Methods in DNA endonuclease reaction media containing 5, 10, 20, or40 mM MgCl2 with increasing concentrations of ZnCl2, and the products were analyzed in a denaturing 6% polyacryl-amide gel, which was dried and quantified with a PhosphorImager. The activity in the standard reaction medium con-taining 20 mM Mg2þ, set equal to 100%, was 0.09 and 0.10 fmole/min for top- and bottom-strand cleavage, respectively.
940 Group II Intron Protein DNA Endonuclease Region
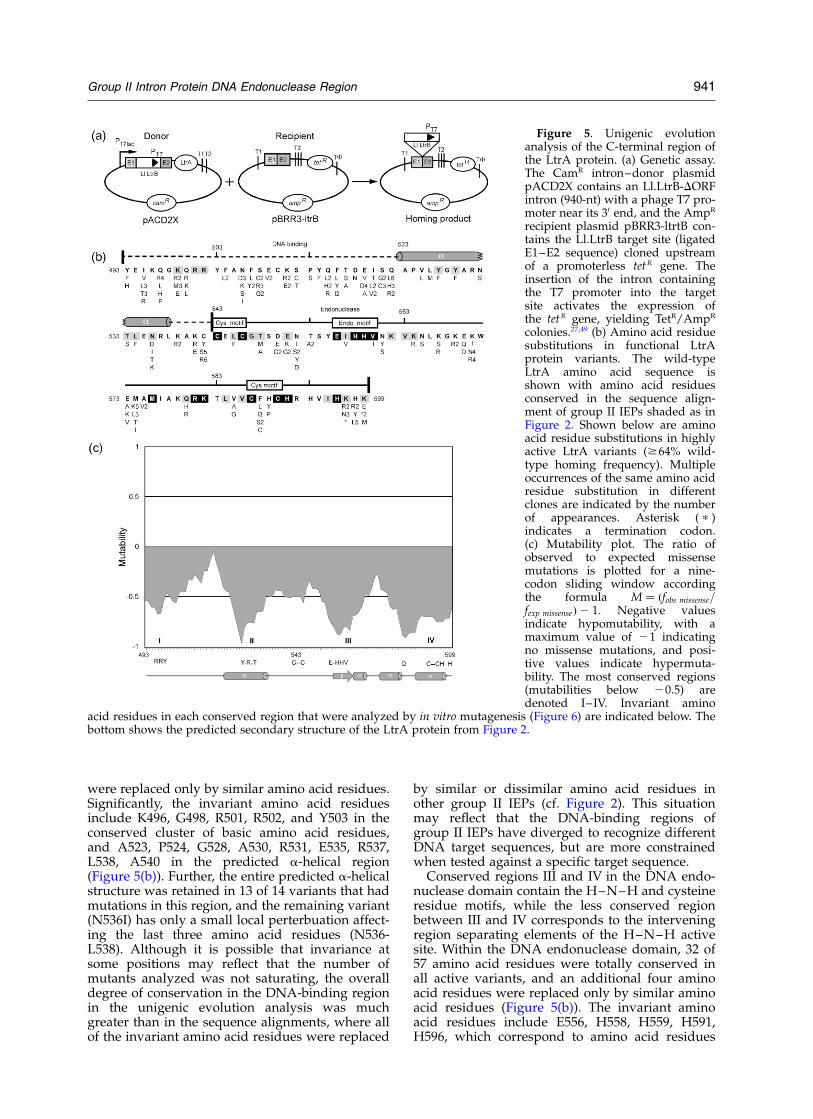
were replaced only by similar amino acid residues.Significantly, the invariant amino acid residuesinclude K496, G498, R501, R502, and Y503 in theconserved cluster of basic amino acid residues,and A523, P524, G528, A530, R531, E535, R537,L538, A540 in the predicted a-helical region(Figure 5(b)). Further, the entire predicted a-helicalstructure was retained in 13 of 14 variants that hadmutations in this region, and the remaining variant(N536I) has only a small local perterbuation affect-ing the last three amino acid residues (N536-L538). Although it is possible that invariance atsome positions may reflect that the number ofmutants analyzed was not saturating, the overalldegree of conservation in the DNA-binding regionin the unigenic evolution analysis was muchgreater than in the sequence alignments, where allof the invariant amino acid residues were replaced
by similar or dissimilar amino acid residues inother group II IEPs (cf. Figure 2). This situationmay reflect that the DNA-binding regions ofgroup II IEPs have diverged to recognize differentDNA target sequences, but are more constrainedwhen tested against a specific target sequence.
Conserved regions III and IV in the DNA endo-nuclease domain contain the H–N–H and cysteineresidue motifs, while the less conserved regionbetween III and IV corresponds to the interveningregion separating elements of the H–N–H activesite. Within the DNA endonuclease domain, 32 of57 amino acid residues were totally conserved inall active variants, and an additional four aminoacid residues were replaced only by similar aminoacid residues (Figure 5(b)). The invariant aminoacid residues include E556, H558, H559, H591,H596, which correspond to amino acid residues
Figure 5. Unigenic evolutionanalysis of the C-terminal region ofthe LtrA protein. (a) Genetic assay.The CamR intron–donor plasmidpACD2X contains an Ll.LtrB-DORFintron (940-nt) with a phage T7 pro-moter near its 30 end, and the AmpR
recipient plasmid pBRR3-ltrtB con-tains the Ll.LtrB target site (ligatedE1–E2 sequence) cloned upstreamof a promoterless tetR gene. Theinsertion of the intron containingthe T7 promoter into the targetsite activates the expression ofthe tetR gene, yielding TetR/AmpR
colonies.27,49 (b) Amino acid residuesubstitutions in functional LtrAprotein variants. The wild-typeLtrA amino acid sequence isshown with amino acid residuesconserved in the sequence align-ment of group II IEPs shaded as inFigure 2. Shown below are aminoacid residue substitutions in highlyactive LtrA variants ($64% wild-type homing frequency). Multipleoccurrences of the same amino acidresidue substitution in differentclones are indicated by the numberof appearances. Asterisk ( p )indicates a termination codon.(c) Mutability plot. The ratio ofobserved to expected missensemutations is plotted for a nine-codon sliding window accordingthe formula M ¼ ðfobs missense=fexp missenseÞ2 1: Negative valuesindicate hypomutability, with amaximum value of 21 indicatingno missense mutations, and posi-tive values indicate hypermuta-bility. The most conserved regions(mutabilities below 20.5) aredenoted I–IV. Invariant amino
acid residues in each conserved region that were analyzed by in vitro mutagenesis (Figure 6) are indicated below. Thebottom shows the predicted secondary structure of the LtrA protein from Figure 2.
Group II Intron Protein DNA Endonuclease Region 941
found to be catalytically essential in H–N–H DNAendonucleases (see above); the two pairs of con-served cysteine residues (C543, C546, C587, C590);and M576, R581, and K582, which were found tobe highly conserved in other group II IEPs in thesequence alignments (see Figure 2).
Biochemical analysis
To investigate the function of different segmentsof LtrA’s C-terminal region, we constructedmutants with alanine residues substituted for keyconserved residues amino acid residues identifiedin the unigenic evolution analysis. The purifiedproteins were assayed for RT activity and reconsti-tuted into RNP particles with in vitro-synthesizedlariat RNA to assay reverse splicing and DNAendonuclease activity. The purified LtrA protein
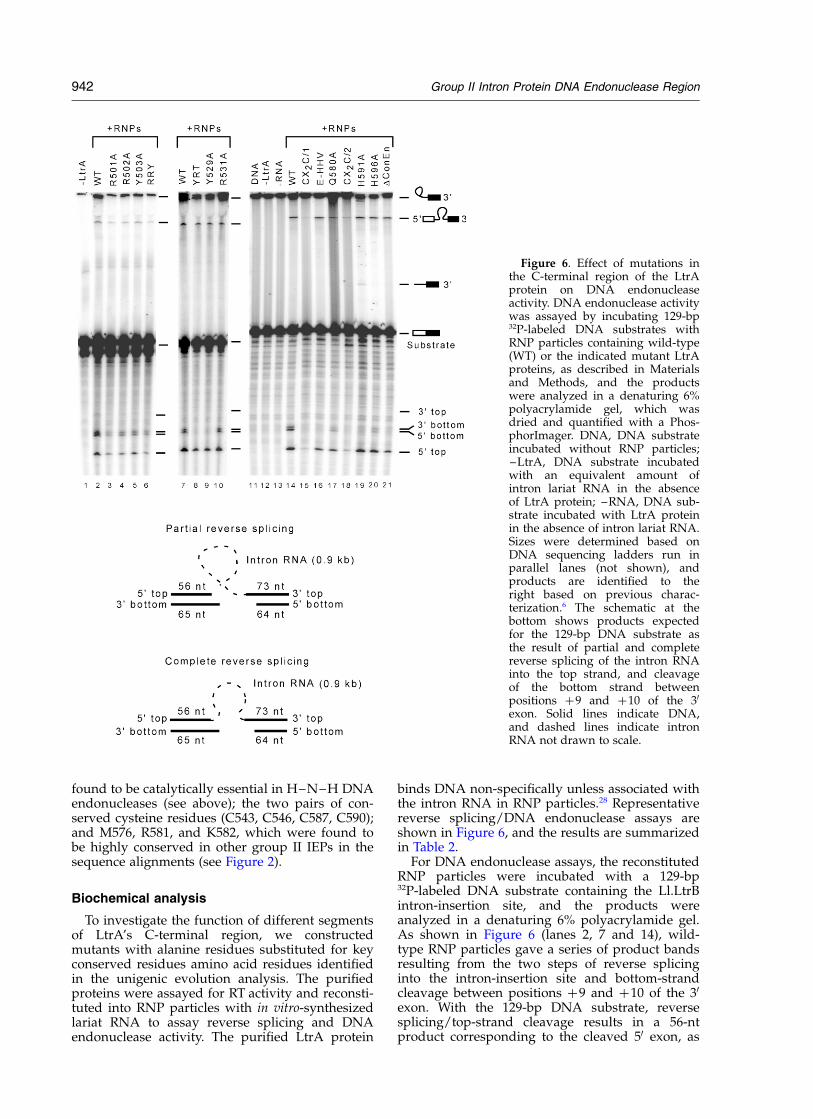
binds DNA non-specifically unless associated withthe intron RNA in RNP particles.28 Representativereverse splicing/DNA endonuclease assays areshown in Figure 6, and the results are summarizedin Table 2.
For DNA endonuclease assays, the reconstitutedRNP particles were incubated with a 129-bp32P-labeled DNA substrate containing the Ll.LtrBintron-insertion site, and the products wereanalyzed in a denaturing 6% polyacrylamide gel.As shown in Figure 6 (lanes 2, 7 and 14), wild-type RNP particles gave a series of product bandsresulting from the two steps of reverse splicinginto the intron-insertion site and bottom-strandcleavage between positions þ9 and þ10 of the 30
exon. With the 129-bp DNA substrate, reversesplicing/top-strand cleavage results in a 56-ntproduct corresponding to the cleaved 50 exon, as
Figure 6. Effect of mutations inthe C-terminal region of the LtrAprotein on DNA endonucleaseactivity. DNA endonuclease activitywas assayed by incubating 129-bp32P-labeled DNA substrates withRNP particles containing wild-type(WT) or the indicated mutant LtrAproteins, as described in Materialsand Methods, and the productswere analyzed in a denaturing 6%polyacrylamide gel, which wasdried and quantified with a Phos-phorImager. DNA, DNA substrateincubated without RNP particles;–LtrA, DNA substrate incubatedwith an equivalent amount ofintron lariat RNA in the absenceof LtrA protein; –RNA, DNA sub-strate incubated with LtrA proteinin the absence of intron lariat RNA.Sizes were determined based onDNA sequencing ladders run inparallel lanes (not shown), andproducts are identified to theright based on previous charac-terization.6 The schematic at thebottom shows products expectedfor the 129-bp DNA substrate asthe result of partial and completereverse splicing of the intron RNAinto the top strand, and cleavageof the bottom strand betweenpositions þ9 and þ10 of the 30
exon. Solid lines indicate DNA,and dashed lines indicate intronRNA not drawn to scale.
942 Group II Intron Protein DNA Endonuclease Region

well as higher molecular weight products resultingfrom partial and complete reverse splicing, whilebottom-strand cleavage after position E2 þ 9results in two closely spaced DNA fragments of 65and 64 nts (see schematic at the bottom of Figure6). As noted previously, the first step of reversesplicing, the attachment of intron lariat RNA tothe 30 exon, is the predominant product in vitro inthe absence of dNTPs, and partially degradedreverse spliced products result from nicking of theintron RNA.6
Analysis of mutations in the DNA-binding region
In the DNA-binding region, we mutated two setsof amino acid residues that were conserved in boththe sequence alignments and unigenic evolutionanalysis: R501, R502, and Y503 in the basic region,and Y529, R531, and T533 in the predicted a-helicalregion (Figure 6, lanes 1–10, and Table 2). Themutant RRY ! A in which R501, R502, and Y503were all changed to alanine residues substantiallyinhibited both reverse splicing and DNA endo-nuclease activity (13(^7)% and 12(^5)% wild-type activity, respectively). Of the three aminoacid residues, the most specific effect on DNAendonuclease activity was seen with the mutantR502A, which had 26(^8) and 32(^6)% wild-typereverse splicing and bottom-strand cleavage,respectively, while retaining 83% wild-type RTactivity. The other single mutants as well as thetriple mutant RRY showed a greater reduction in
RT activity (30–46% wild-type; Table 2). Theseresults are consistent with a reduction in DNA-binding activity more or less equally affectingboth reverse splicing and bottom-strand cleavage,with partial inhibition reflecting redundant inter-actions that contribute to DNA binding.
In the predicted a-helical region, the mutantY-R-T ! A in which Y529, R531, and T533 arereplaced by alanine residues showed reducedreverse splicing/top-strand cleavage activity(59(^5)% wild-type) and no detectable bottom-strand cleavage activity. The single point mutationY529A also inhibited reverse splicing/top strandcleavage activity (42(^11)% wild-type) andabolished bottom-strand cleavage. By contrast, themutation R531A only moderately affected bothtop and bottom strand cleavage (74(^17)% and66(^28)% wild-type, respectively), and T533A hadwild-type levels of both activities (not shown).Thus, Y529 appears to be the most critical of thethree amino acid residues. The pattern of inhibitionfor the Y-R-T ! A and Y529A mutations is consist-ent with disruption of interactions that contributeto both top- and bottom-strand cleavage. Thestronger inhibition of bottom-strand cleavagecould reflect more severe disruption of 30-exoninteractions that are required specifically forbottom-strand cleavage, or disruption of inter-actions with the 50 exon that then mispositionthe DNA endonuclease domain for bottom-strandcleavage. Like the yeast aI2 P714T mutant, whichhas a single amino acid residue change in thesame region, the LtrA mutants Y-R-T ! A, Y529A,and R531A have high RT activity assayed withpoly(rA)/oligo(dT)18 (Table 2). In other assaysusing an in vitro transcript containing the Ll.LtrBintron and flanking exons, with an annealed 30
exon primer E2 þ 10 mimicking that normallyused during TPRT, the Y-R-T mutant had only,50% wild-type RT activity, possibly reflectingmispositioning of the enzyme or DNA primer. Wecould not, however, mimic the primer promiscuitycharacteristic of the yeast aI2 P714T mutant (notshown).34,35
Analysis of mutations in the DNAendonuclease domain
In the DNA endonuclease domain, we con-structed a series of mutants to identify elements ofthe H–N–H active site and assess the function ofthe conserved cysteine residue pairs (Figure 6,lanes 15–21, and Table 2). The previously analyzedDConEn mutant, which lacks the DNA endo-nuclease domain, was included for comparison.The DConEn RNP particles still carry out reversesplicing/top-strand cleavage, but show no detect-able bottom-strand cleavage in agreement withprevious results.6 The mutant E-HHV !A, whichhas alanine residues substituted for the E556,H558 (the first residue of the H–N–H motif),H559, and V560 showed no detectable bottom-strand cleavage, but retained substantial reverse
Table 2. Biochemical activities of wild-type and mutantLtrA proteins
RTReverse splicing/top
strandBottomstrand
WT 100 100 100R501A 34 ^ 10 34 ^ 22 27 ^ 16R502A 83 ^ 25 26 ^ 8 32 ^ 6Y503A 46 ^ 3 109 ^ 21 102 ^ 37RRY 30 ^ 8 13 ^ 7 12 ^ 5YRT 117 ^ 10 59 ^ 5 0Y529A 169 ^ 35 42 ^ 11 0R531A 157 ^ 22 74 ^ 17 66 ^ 28CX2C/1 1 ^ 0 19 ^ 2 0E-HHV 3 ^ 2 86 ^ 5 0Q580A 56 ^ 9 67 ^ 28 70 ^ 32CX2C/2 1 ^ 0 16 ^ 5 0H591A 64 ^ 27 90 ^ 20 2 ^ 2H596A 69 ^ 23 97 ^ 32 24 ^ 9DConEn 2 ^ 2 35 ^ 16 0
RT activity was assayed by incorporation of [a-32P] dTTPusing poly(rA)/oligo(dT)18 as substrate. Reverse splicing/top-strand cleavage and bottom strand cleavage activity wereassayed using both internally labeled and 50-labeled 129-bpDNA substrates, as described in Materials and Methods.Activities were calculated from the proportion of total countsin product bands in each lane and are expressed as percent ofwild-type RNP particles assayed in parallel. Values are themean ^ SD for three independent assays using at least twodifferent protein preparations in each case.
Group II Intron Protein DNA Endonuclease Region 943

splicing/top-strand cleavage activity (86(^5)%wild-type). The mutant H591A, in the second histi-dine residue of the H–N–H motif, had ,2% wild-type bottom-strand cleavage activity, combinedwith high reverse splicing/top-strand cleavageactivity (90(^ )20%). This histidine residue is puta-tively involved in metal-ion coordination, and weshowed above that the H591A mutation stronglydecreased the amount of tightly bound Mg2þ (seeTable 1). Mutation of the neighboring conservedhistidine residue H596A, also putatively involvedin metal-ion coordination, again substantiallyinhibited bottom-strand cleavage (24(^9)% wild-type activity), while the mutations H593A andH598A had no effect on top- or bottom-strandcleavage (not shown). Neither H593 nor H598 areevolutionarily conserved, and in the unigenicevolution analysis truncated LtrA proteins lackingH598 were efficiently mobile (Figure 5). Mutationsin the conserved cysteine residue pairs C543, C546(CX2C/1 ! A) and C587, C590 (CX2C/2 ! A),completely abolished bottom-strand cleavageactivity, while leaving some reverse splicingactivity (19(^2)% and 16(^5)% wild-type activity),as found previously for the analogous yeast aI2mutants.4
In contrast to the large effects of mutating con-served His and Cys residues, mutation of Q580,equivalent to “N” of the conserved H–N–Hmotif, had smaller effects on reverse splicing andbottom-strand cleavage in vitro (67(^28)% and70(^32)% of wild-type activity, respectively, inmultiple experiments). The corresponding aspara-gine residue in colicin E9 DNase is not part of theendonuclease catalytic center, but plays a role instabilizing the higher-order structure of thedomain.43,44 In the unigenic evolution analysis, thisglutamine residue was not invariant, but wasreplaced preferentially by a positively chargedresidue (arginine or histidine residue; Figure 5(b)).The relatively small effect of alanine residue substi-tution at this position could reflect that other inter-actions are largely sufficient to stabilize the domainstructure. Together, these results indicate that theconserved histidine residues are essential forcatalytic activity, as in colicin E7 and E9 DNases,and that the conserved cysteine residues arealso required, presumably to maintain the activeprotein structure.
Mutations in the DNA endonuclease domainstrongly inhibit RT activity
Previous studies with yeast aI2 intron showedthat deletion of the conserved endonucleasedomain or mutations in the conserved H–N–H orcysteine residue motifs strongly inhibit RT activity,while leaving substantial RNA splicing and reversesplicing activity, suggesting a structural or func-tional connection between the DNA endonucleaseand RT activities.34 Similarly, for the LtrA protein,deletion of the conserved DNA endonucleasedomain or multiple mutations in the conserved
histidine and cysteine residues inhibited RTactivity to ,5% of the wild-type level (CX2C/1 ! A, E-HHV !A, CX2C/2 ! A, and DConEn;Table 2). The point mutations H591A and H596A,which more specifically affect the metal ion-binding fold, also moderately inhibited the RTactivity (64% and 69%, wild-type, respectively).All the conserved histidine and cysteine residuemutants still have reverse splicing/top strandcleavage activity, and other experiments showedthat the DConEn and E-HHV !A mutants retainwild-type splicing activity, suggesting that theoverall conformation of the protein is not grosslyaltered (data not shown). Thus, the mutations inthe conserved DNA endonuclease domain appearto affect the RT activity specifically.
Effect of C-terminal mutations on DNAprimer binding
The DNA endonuclease domain is required togenerate the primer for initiation of reverse tran-scription and must therefore be in contact with the30-exon region of the DNA substrate at some pointduring the mobility reactions. Thus, one possibleexplanation for the inhibition of RT activity wasthat the DNA endonuclease mutations prevent thebinding of the DNA primer required for initiationof reverse transcription.34 To test this hypothesis,we adapted a previously developed RT assay inwhich an Ll.LtrB RNA containing the intron andflanking exon sequences is annealed to a 20-merDNA primer E2 þ 10, whose 30 end correspondsto that of the cleaved bottom strand normallyused as the primer for TPRT.6,50 To assay primerbinding, the primer was synthesized with a singleBrdU at the position corresponding to E3 þ 13,enabling efficient UV-cross-linking to the LtrAprotein.
Figure 7(a) shows initial control assays inwhich LtrA was incubated with the Ll.LtrB:BrdU/E2 þ 10 substrate in the presence of 32P-dTTP anddATP, enabling labeling of the primer by 3-ntextension before encountering the first C-residuein the Ll.LtrB RNA template. After UV-irradiationand RNase-digestion of the Ll.LtrB RNA template,LtrA protein containing cross-linked 32P-labeledprimer was detected by SDS-PAGE, followed byautoradiography. The results showed that wild-type (WT) protein and those mutants that retainsubstantial RT activity (H591A and Y-R-T ! A)incorporated 32P-dTTP leading to labeling of thecross-linked E2 þ 10 oligonucleotide, whereasmutants with alterations in the DNA endonucleasedomain that inhibit RT activity (DConEn, CX2C/1 ! A, E-HHV !A) or a mutation in the con-served YADD motif in the RT domain (DD2)showed little or no labeling in the assay. Controlsshowed that cross-linking of the labeled oligo-nucleotide was not observed in the absence of theRNA template or UV irradiation (lanes 1–4). Inaddition, the cross-linked DNA oligonucleotidewas insensitive to DNase treatment, which
944 Group II Intron Protein DNA Endonuclease Region

degraded unbound DNA primer (not shown),suggesting that the cross-linked primer is protectedby the LtrA protein.
To assay primer binding, the experiment wasrepeated now using a 50-labeled BrdU/E2 þ 10primer in the absence of dNTPs. As shown inFigure 7(b), in addition to wild-type LtrA protein,nearly all of the mutant proteins were cross-linkedto the 32P-labeled BrdU/E2 þ 10 primer, includingthose lacking RT activity. As above, the cross-linking was not observed unless the labeled oligo-nucleotide was annealed to the Ll.LtrB RNAtemplate, indicating that it is not due to non-specific interaction of the primer with the LtrAprotein. Further, the cross-linked primer was pro-tected from DNase I digestion, consistent withresults for HIV-1 RT, where 18 nt of primer DNAstrand is protected from hydroxyl radical cleavageby binding at the RT active site.51 The single mutantthat failed to cross-link the labeled oligonucleotide,CX2C/1 ! A, has alterations in the first conservedcysteine residue pair that could either directly
affect primer binding or result in an unfavorableconformational change in the protein. We notethat the first conserved cysteine residue pair isretained in the DConEn mutant, which is capableof primer binding in this assay. Together, thesefindings suggest that the inhibition of RT activityby DNA endonuclease domain mutations is notdue to the inability to bind the primer, a functionpresumably carried out by the RT domain. It ispossible that the DNA endonuclease domainmutations affect the positioning of the primer orinhibit the RT activity by affecting the structure ofthe RT domain.
Discussion
Here, we characterized the C-terminal DNA-binding/DNA endonuclease region of the L. lactisLtrA protein, the first detailed analysis of thisregion in any group II IEP. First, we identifiedtwo functionally important segments of the
Figure 7. UV-cross-linking assayof DNA primer binding by wild-type and mutant LtrA proteins.The RT substrates, shown schematic-ally above each panel, consist ofin vitro transcript Ll.LtrB, with a902-nt Ll.LtrB–DORF intron andflanking exons, annealed to DNAprimer BrdU/E2 þ 10. The primeris complementary to 30 exon pos-itions E2 þ 10 to þ29, with BrdUincorporated in place of the T-resi-due at position þ13. In (a), theLl.LtrB RNA template annealedwith unlabeled DNA primer wasincubated with WT or the indicatedmutant LtrA proteins plus32P-dTTP and dATP, to permit label-ing by incorporation of threedNTPs before encountering thefirst C-residue in the RNA template.The samples were then UV-cross-linked, incubated with RNaseA þ T1 to digest the Ll.LtrB RNA,and analyzed in SDS/9% polyacryl-amide gel, which was driedand autoradiographed. In (b), theLl.LtrB RNA template annealedwith 50-labeled primer was incu-bated with wild-type and mutantLtrA proteins (150 nM) in theabsence of dNTPs. The sampleswere UV-cross-linked and pro-cessed as above. In each case, halfof the sample was treated withDNase I prior to gel analysis.–RNA, LtrA protein and DNAprimer incubated in the absenceof Ll.LtrB RNA template; –UV,parallel sample without UV-cross-
linking. Positions of molecular weight markers (BioRad Broad Range) are indicated to the left, and the position of theLtrA protein with cross-linked 32P-labeled DNA primer is indicated by the arrow to the right.
Group II Intron Protein DNA Endonuclease Region 945

DNA-binding region, one containing a cluster ofbasic amino acid residues and the other containinga predicted a-helix. These features were found inrelated group II IEPs in multiple sequence align-ments, and their functional importance in LtrAwas demonstrated by unigenic evolution analysisand by biochemical assays of mutants with altera-tions in key amino acid residues. The predicteda-helical region is also the site of a previouslystudied yeast aI2 mutation P714T, which inhibitsboth top- and bottom-strand cleavage (25% and6% wild-type, respectively).3 Notably, the con-straints on the amino acid sequence in the DNA-binding region were much greater in the unigenicevolution analysis of LtrA than in naturally occur-ring group II IEPs, where all of the invariantamino acid residues are replaced by similar or dis-similar amino acid residues in some proteins. Thissituation may reflect that the DNA-binding regionhas diverged to recognize different targetsequences in different group II IEPs, but is moreconstrained when tested against its own specifictarget sequence.
We find that the DNA endonuclease domain ofthe LtrA protein contains a metal ion-dependentnuclease fold homologous to that of colicin E7 andE9 DNases and phage T4 endonuclease VII, aHolliday junction resolvase. The sequence align-ments show that LtrA contains most of the aminoacid residues found to be important for catalyticactivity of colicin E9 DNase, and these are presentin a predicted secondary structure context thatmatches the colicin E9 X-ray crystal structure.Further, the catalytically important amino acidresidues are highly constrained in the unigenicevolution analysis, and mutations at key positionsstrongly inhibit bottom-strand cleavage, whileleaving substantial reverse splicing/top-strandcleavage.
ICP atomic emission spectroscopy showed thatLtrA contains a single tightly bound Mg2þ ion,which is associated with the H–N–H endo-nuclease domain, as judged by its disappearancein the C-terminal truncation mutant DConEn(Table 1). Further, the mutation H591A, whichaffects a histidine residue shown to function inmetal-ion coordination in the colicin E7 and E9DNases,39,41 – 44 strongly decreased the amount ofbound Mg2þ and specifically inhibited bottom-strand cleavage (Tables 1 and 2; Figure 6). Thesefindings indicate that that Mg2þ is the predominantactive-site metal ion and that it is coordinatedsimilarly to the metal ions in the colicin DNases.
The conserved cysteine residue pairs found inthe DNA endonuclease domains of group II IEPsare not present in the colicin DNases, but arefound in the related EndoVII-McrA proteins.30 TheX-ray crystal structure of phage T4 endonucleaseVII shows that the two cysteine residue pairs areinterspersed with active-site elements, but cometogether to coordinate a single Zn2þ.40 Further,mutations in the conserved cysteine residue pairsresult in loss of bound Zn2þ, which is correlated
with loss of junction-resolving activity.45 The con-served cysteine residue pairs in the LtrA proteinalign with those in endonuclease VII and arefound in a predicted secondary structure contextsimilar to that in the endonuclease VII crystalstructure. Moreover, the conserved cysteine resi-due pairs were invariant in the unigenic evolutionof active LtrA variants, and alanine-substitutionmutations in either cysteine residue pair abolishedbottom-strand cleavage, while leaving significanttop-strand cleavage. Together, these results suggestthat the conserved cysteine residue pairs in LtrA,like those in endonuclease VII, play a critical rolein maintaining the structure of the DNA endo-nuclease domain.
In contrast to endonuclease VII, we find that thepurified LtrA protein preparations contain onlysubstoichiometric amounts of Zn2þ (0.2 mol/mol),and the addition of exogenous Zn2þ inhibits bothreverse splicing and bottom-strand cleavageactivity. The Zn2þ inhibition is not competitivewith respect to Mg2þ, suggesting that Zn2þ doesnot readily exchange for Mg2þ at the active siteand instead inhibits the DNA endonucleaseactivity by binding specifically or nonspecificallyto one or more secondary sites. Although it is pos-sible that Zn2þ is lost during purification of theLtrA protein, we find no indication of selectiveinactivation of the DNA endonuclease activitycompared to other activities of the protein.
Surprisingly, in addition to inhibiting DNAendonuclease activity, deletion of the conservedendonuclease domain or mutations in its con-served histidine and cysteine residue motifsstrongly inhibit RT activity. These mutations leavesubstantial reverse splicing/top strand cleavageand RNA splicing activity, suggesting that they donot result in gross structural alterations in theprotein. Similar observations were made pre-viously for the yeast aI2 protein.4,34 UV-cross-link-ing experiments showed that the DNA endo-nuclease domain mutants remain capable of DNAprimer binding, which is presumably a function ofthe RT domain. Thus, the inhibition of RT activitycould reflect either that the primer is notpositioned correctly or that mutations in the DNAendonuclease domain affect the structure of theRT domain. The latter situation would be analo-gous to that for HIV-1 RT, where the RT and associ-ated RNase H activities are interdependent, withmutations in one domain frequently affecting theother activity, sometimes to a greater extent.52
Finally, with respect to evolution, our resultssuggest that the most likely progenitor of thegroup II IEP DNA endonuclease domain was aprotein related to the EndoVII-McrA superfamily,which contains an H–N–H-like metal ion-bindingfold with two pairs of conserved cysteine residues,as well as an upstream a-helix positioned similarlyto the predicted a-helix in the DNA-binding regionof group II IEPs.30,40 These features of the C-termi-nal region are conserved in group II IEPs of themitochondrial lineage. By contrast, group II IEPs
946 Group II Intron Protein DNA Endonuclease Region

of the chloroplast lineage and bacterial class B typi-cally contain the conserved H–N–H motif, butonly subsets of the conserved cysteine residues,while bacterial classes A, C, and D completelylack the DNA endonuclease domain. Notably,group II IEPs of the bacterial classes A and C,which lack the DNA endonuclease domain,still have a C-terminal extension downstream ofdomain X, which includes a cluster of basic aminoacid residues and predicted a-helix, shown hereto be functionally important features of the DNA-binding region of LtrA. This situation may reflectthat the DNA-binding region and DNA endo-nuclease domain were acquired together fromthe same EndoVII-McrA-type protein, followed bydivergence or loss of the DNA endonucleasedomain in different lineages, presumably becausethe DNA endonuclease activity is deleterious tothe host.16,31
The bacterial class D proteins RmInt1 and EcIntBdiffer in having only a short (20 aa) C-terminalextension, which appears unrelated to the C-termi-nal regions of other group II IEPs (see Figure 2).The short C-terminal extension in bacterial classD proteins could be a primordial or remnantDNA-binding region, an extension of domain X,or simply a non-functional extension. Notably, theRmInt1 IEP recognizes specific DNA target sitesequences in both the 50 exon proximal to the IBS2sequence and in the 30 exon, suggesting that it hassome (albeit more limited) DNA-binding functionssimilar to those of LtrA.53 Bacterial subclass Cintrons, whose IEPs also lack the DNA endo-nuclease domain but may retain the C-terminalDNA-binding region, appear to insert preferen-tially after rho-independent transcription termi-nators, implying a somewhat different mode ofDNA target site recognition.54 – 56 It will be of inter-est to determine whether the specific DNA-bindingfunctions of these other group II IEPs are associ-ated with the same or different regions of theprotein as those in LtrA.
Materials and Methods
E. coli strains and growth conditions
E. coli strains were BL21(DE3) for LtrA proteinexpression, HMS174(DE3) for group II intron mobilityassays and unigenic evolution, and DH10B for libraryconstruction and cloning. Bacteria were grown in LB orSOB media, with antibiotics added at the followingconcentrations: ampicillin, 100 mg/ml; chloramphenicol,25 mg/ml; tetracycline, 25 mg/ml.
Recombinant plasmids and construction of LtrAprotein mutants
pIMP-1P contains the LtrA ORF cloned behind the tacpromoter in pCYB2 (New England Biolabs, Beverly,MA), with the C-terminus of the ORF fused in-frame toa cassette containing the Saccharomyces cerevisiae VMA1intein and the Bacillus circulans chitin-binding domain.57
pET11-LtrASE8, used in the construction of LtrA pro-tein mutants, contains a modified LtrA ORF, with silentmutations that introduce useful restriction sites, cloneddownstream of the phage T7 promoter in pET-11a(New England Biolabs; Hongwen Ma and A.M.L.,unpublished).
pGMDORF-T7 contains a 902-nt DORF derivative ofthe Ll.LtrB intron and flanking exons cloned down-stream of the phage T7 promoter in pBSSK 2 (Strata-gene, La Jolla, CA). It was derived from pGMDORF bysubstituting the T7 for the T3 promoter.57
pLHS contains a 70-nt sequence corresponding to theligated exon 1 and 2 sequence of the ltrB gene fromposition 235 to þ35 from the intron-insertion site clonedin pBKS þ .6
pACD2, the intron donor plasmid used in mobilityassays, contains a 940-nt Ll.LtrB-DORF intron with a T7promoter inserted in intron domain IV. The intron andflanking exons are cloned downstream of an IPTG-indu-cible T7lac promoter in a CamR pACYC184-derivative,with the LtrA protein expressed from a position down-stream of the 30 exon.27,49 pACD2X is a derivative ofpACD2 that contains a Xho I site inserted 8-bp down-stream of the LtrA ORF termination codon to facilitateconstruction of mutant libraries.
pBRR3-ltrB, the recipient plasmid used for geneticassays, contains the minimal Ll.LtrB target site (ligatedE1–E2 sequence from positions 230 to þ15 from theintron-insertion site) cloned upstream of a promoterlesstetR gene in an AmpR pBR322-derivative.27,49
LtrA mutants were constructed by a two-step PCRusing Vent polymerase, with desired modifications intro-duced via one of the primers.58 The first PCR usedpET11-LtrASE8 as template with a 50 primer (33–45 nts)containing the mutation, and 30 primer DD4 (50-GTAGG-GAGGTACCG CCTTGTTC), complementary to a vectorsequence downstream of the LtrA ORF. The initial PCRproduct was gel-purified and used as the 30 primer in asecond PCR with 50 primer P8A (50-TGGGGGATCCCGT-ATGAGATAAAGC), which overlaps the Bam HI site atthe beginning of LtrA’s C-terminal region. The resultingPCR product was gel-purified and digested with Bam HIand Xma I, generating a 328-bp fragment, which wasswapped for the corresponding fragment of pET11-LtrASE8. The modified C-terminal region was sequencedcompletely, using primer P7A (50-TGAACTCCGCGGG-ATTTGTAATTACTAC), which corresponds to asequence 169-bp upstream of the Bam HI site. The1799-bp Nde I/Xma I fragment containing the modifiedLtrA ORF was then swapped for the correspondingwild-type fragment of the expression construct pIMP-1P(see above).
In vitro transcription
Ll.LtrB RNA was transcribed from Bam HI-digestedpGMDORF-T7, using a T7 MEGAscript kit (Ambion,Austin, TX). It consists of a 207-nt 50 exon, the 902-ntDORF Ll.LtrB intron, and a 95-nt 30 exon.
Sequence alignments
Protein sequences were identified by a BLAST 2.1.2search using the C-terminal region of the LtrA protein(amino acid residues 491–599) as the query.38 Thesequences were aligned using the MacVector 6.5.1ClustalW alignment program, followed by manualrefinement.
Group II Intron Protein DNA Endonuclease Region 947

Purification of the LtrA protein
Wild-type and mutant LtrA proteins expressed frompIMP-1P or its mutant derivatives were purified onchitin affinity columns using the intein-based IMPACTpurification system (New England Biolabs).57 Thepurified proteins were dialyzed at 4 8C in column buffercontaining 0.5 M NaCl, 50 mM Tris–HCl, pH 8.0, and50% (v/v) glycerol, with 0.1 mM EDTA added to removefree or loosely bound cations. For atomic emission spec-troscopy, the proteins were purified further at 4 8Cusing a HiTrap SP ion-exchange column (AmershamPharmacia Biotech, Piscataway, NJ) to remove residualcations and DTT. Protein concentrations were deter-mined by Bradford assays (BioRad, Hercules, CA)against an LtrA protein standard, whose concentrationwas determined by A205 nm and A280 nm, using an extinc-tion coefficient calculated from the amino acidsequence.57 The dialyzed proteins remained active formonths at 270 8C.
ICP atomic emission spectroscopy
Proteins were analyzed by ICP atomic emissionspectroscopy46 to determine the amount of bound cations(Huffman Laboratories, Inc., Golden, CO†).
Intron mobility assays and unigenic evolution
Intron mobility was assayed by using a genetic systemin which a modified Ll.LtrB intron containing a phage T7promoter inserted near its 30 end inserts into a target siteupstream of a promoterless tetR gene, thereby activatingthe expression of that gene.27,49 For mobility assays, theintron donor plasmid pACD2X or donor plasmidlibraries with random PCR-induced mutations in LtrA’sC-terminal region were electroporated into E. coliHMS174(DE3) harboring the recipient plasmid pBRR3-ltrB, which contains the Ll.LtrB insertion site clonedupstream of the promoterless tetR gene. Cells weregrown overnight at 37 8C in LB medium containingchloramphenicol and ampicillin, induced with 100 mMIPTG for 1 h at 37 8C, and then plated on LB containingampicillin and tetracycline to select TetR/AmpR coloniesin which the intron had inserted into the target site.27
For unigenic evolution analysis, LtrA variants thatsupport efficient intron mobility in the above assaywere isolated from donor plasmid libraries containingrandom mutations introduced into LtrAs C-terminalregion by mutagenic PCR.59 To construct the libraries,PCR was carried out in reaction medium containing fiveunits of Taq polymerase (Invitrogen, Carlsbad, CA),7 mM MgCl2, 0.5 mM MnCl2, 0.2 mM dGTP, 0.2 mMdATP, 1 mM dTTP, and 1 mM dCTP, with 20 fmolof pACD2X template and 30 pmol each of 50
primer ZnBamHS (50-TGGAAGTGGTTCGTGGGGG-ATCC), which overlaps the Bam HI site at the beginningof the C-terminal region, and 30 primer ZnXhoAS(50-CGAGAACGGGTGCTCGAGATATCTCA), whichoverlaps the Xho I site at the end of the LtrA ORF.Reaction conditions were 25 cycles of 948 for one minute,458 for one minute, and extension at 728 for two minutes.The 372-bp PCR product was gel-purified, digested withBam HI and Xho I, and swapped for the correspondingfragment of intron donor plasmid pACD2X. The result-ing library was then used as the template for the next
round of mutagenic PCR, thereby creating libraries withincreasing numbers of mutations. The two donor plas-mid libraries used for the unigenic evolution analysishad an average of 3.9 and 5.6 nucleotide substitutions,corresponding to 2.5 and 3.2 amino acid residue substi-tutions per 321 nucleotide residues. Higher mutationfrequencies resulted in intron donor plasmids librarieswith very low homing frequencies (,0.5%).
After selection of TetR/AmpR colonies and confir-mation of correct intron insertion by colony PCR acrossthe 50-junction, donor plasmids were reisolated by trans-forming mini-prepped DNA into HMS174(DE3) andplating onto LB medium plus chloramphenicol. Thereisolated donor plasmids were sequenced using primerP7A (see above) and then retested in mobility assays todetermine mobility frequencies for individual LtrAvariants.27 LtrA variants identified as supporting .64%wild-type mobility assayed in parallel were classifiedas highly active and were used to calculate mutabilityvalues according to the equation M ¼ ðfobserved missense=fexpected missenseÞ2 1; where fobserved missense is the observedfrequency of missense mutations, and fexpected missense isthe expected frequency. The latter was calculated foreach codon based on the probability of a single nucleo-tide change producing a silent or missense mutation,correcting for the transition/transversion ratio (56%/44%) for the variants selected from the library. Themutability values were calculated for a nine-codonsliding window and plotted against the amino acid resi-due position at the center of the window. Negativevalues indicate hypomutability, with a maximum valueof 21 indicating no missense mutations, and positivevalues indicate hypermutability.
Biochemical assays
RT activity was assayed by polymerization of [a-32P]-dTTP (3000 Ci/mmol; New England Nuclear, BostonMA) into high molecular material retained on DE81paper, using either poly(rA)/oligo(dT)18 or in vitro-tran-script Ll.LtrB RNA with an annealed DNA primerE2 þ 10 (complementary to 30-exon positions þ10 toþ29) in the presence of 200 mM of the other dNTPs.50
Reverse splicing and DNA endonuclease activitieswere assayed by incubating group II intron RNP par-ticles with small 32P-labeled DNA substrates generatedby PCR.57 The RNP particles were reconstituted byincubating 1.6 pmol of gel-purified Ll.LtrB lariat RNA,generated by self-splicing of Ll.LtrB RNA, with 1.4 pmolof purified LtrA protein in 4 ml of 50 mM MgCl2 for tenminutes at room temperature. The DNA substrates weregel-purified, 129-bp double-stranded DNAs containingthe ligated exon junction of the ltrB gene, synthesizedby PCR of pLHS using the primers SK (50-CGCTCTAGA-ACTAGTGGATC) and KS (50-TCGAGGTCGACGG-TATC). The 50-labeled substrates were generated by PCRwith 50-labeled top- or bottom-strand primer, and inter-nally labeled DNA substrates were generated by PCR inthe presence of [a-32P]dTTP.6 For the assays, the32P-labeled DNA substrate (1.5 nM; 150,000 cpm) wasincubated with reconstituted RNP particles (4 ml;A260 nm ¼ 0.025 units) for 15–20 minutes at 37 8C in 20 mlof reaction medium containing 10 mM KCl, 20 mMMgCl2, 50 mM Tris–HCl (pH 7.5), with the MgCl2 intro-duced with the reconstituted RNP particles.57 The reac-tions were terminated by extraction with phenol-chloroform-isoamyl alochol (25:24:1), and the productswere ethanol precipitated and analyzed in a denaturing† http://www.huffmanlabs.com/
948 Group II Intron Protein DNA Endonuclease Region

6% (w/v) polyacrylamide gel, which was dried andquantified using a PhosphorImager.
UV-cross-linking assay of DNA primer binding
UV cross-linking assays were carried out with the RTsubstrate consisting of Ll.LtrB RNA plus annealed DNAprimer E2 þ 10 (see above), with BrdU substituted forthe T-residue at E2 þ 13.50 The substrate was generatedby heating 150 nM primer with 150 nM Ll.LtrB RNAto 90 8C for two minutes, then cooling to 37 8C. In oneset of assays, the DNA primer was 50-labeled with[g-32P]ATP (3000 Ci/mmol; New England Nuclear) andT4 polynucleotide kinase (New England Biolabs), andin the second set, the primer was unlabeled, and thereaction mixture contained 10 mCi [a-32P]dTTP (3000 Ci/mmol; New England Nuclear) and 230 mM dATP topermit labeling by 3-nt primer extension before encoun-tering the first C-residue in the template. For the assays,the substrate was incubated with wild-type or mutantLtrA proteins (150 nM) in 72 ml of reaction medium(450 mM NaCl, 5 mM MgCl2, 40 mM Tris–HCl (pH 7.5),100 mg/ml bovine serum albumin, 5 mM DTT) for tenminutes at room temperature and then cross-linked byirradiation with a 240-nm UV lamp for two minutes.The samples were digested with RNase (4 mg RNase Aþ5 units RNase T1; Roche, Indianapolis, IN) ^ DNase I(7.5 units; FPLC-purified; Amersham) for 2 h at 37 8C,and analyzed in SDS-9% (w/v) polyacrylamide gel,which was dried and scanned with a PhosphorImagerto detect LtrA protein cross-linked to radioactive primer.
Acknowledgements
We thank Georg Mohr, Roland Saldanha, NicolasToro, Junhua Zhao, and Jin Zhong for commentson the manuscript. This work was supported byNIH grant GM37949.
References
1. Lambowitz, A. M., Caprara, M. G., Zimmerly, S. &Perlman, P. S. (1999). Group I and group II ribozymesas RNPs: clues to the past and guides to the future.In The RNA World (Gesteland, R. F., Cech, T. R. &Atkins, J. F., eds), 2nd edit., pp. 451–485, Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
2. Belfort, M., Derbyshire, V., Parker, M. M., Cousineau,B. & Lambowitz, A. M. (2002). Mobile introns:pathways and proteins. In Mobile DNA II (Craig,N. L., Craigie, R., Gellert, M. & Lambowitz, A. M.,eds), pp. 761–783, ASM Press Publishers, Washing-ton, DC.
3. Zimmerly, S., Guo, H., Perlman, P. S. & Lambowitz,A. M. (1995). Group II intron mobility occurs bytarget DNA-primed reverse transcription. Cell, 82,545–554.
4. Zimmerly, S., Guo, H., Eskes, R., Yang, J., Perlman,P. S. & Lambowitz, A. M. (1995). A group II intronRNA is a catalytic component of a DNA endonu-clease involved in intron mobility. Cell, 83, 529–538.
5. Yang, J., Zimmerly, S., Perlman, P. S. & Lambowitz,A. M. (1996). Efficient integration of an intron RNA
into double-stranded DNA by reverse splicing.Nature, 381, 332–335.
6. Matsuura, M., Saldanha, R., Ma, H., Wank, H., Yang,J., Mohr, G. et al. (1997). A bacterial group II intronencoding reverse transcriptase, maturase, and DNAendonuclease activities: biochemical demonstrationof maturase activity and insertion of new geneticinformation within the intron. Genes Dev. 11,2910–2924.
7. Eskes, R., Yang, J., Lambowitz, A. M. & Perlman, P. S.(1997). Mobility of yeast mitochondrial group IIintrons: engineering a new site specificity and retro-homing via full reverse splicing. Cell, 88, 865–874.
8. Cousineau, B., Smith, D., Lawrence-Cavanagh, S.,Mueller, J. E., Yang, J., Mills, D. et al. (1998). Retro-homing of a bacterial group II intron: mobility viacomplete reverse splicing, independent of homolo-gous DNA recombination. Cell, 94, 451–462.
9. Eskes, R., Liu, L., Ma, H., Chao, M., Dickson, L.,Lambowitz, A. M. & Perlman, P. S. (2000). Multiplehoming pathways used by yeast mitochondrialgroup II introns. Mol. Cell Biol. 20, 8432–8446.
10. Yang, J., Mohr, G., Perlman, P. S. & Lambowitz, A. M.(1998). Group II intron mobility in yeast mito-chondria: target DNA-primed reverse transcriptionactivity of aI1 and reverse splicing into DNA trans-position sites in vitro. J. Mol. Biol. 282, 505–523.
11. Martinez-Abarca, F. & Toro, N. (2000). RecA-inde-pendent ectopic transposition in vivo of a bacterialgroup II intron. Nucl. Acids Res. 28, 4397–4402.
12. Dickson, L., Huang, H.-R., Liu, L., Matsuura, M.,Lambowitz, A. M. & Perlman, P. S. (2001). Retro-transposition of a yeast group II intron occurs byreverse splicing directly into ectopic DNA sites.Proc. Natl Acad. Sci. USA, 98, 13207–13212.
13. Ichiyanagi, K., Beauregard, A., Lawrence, S., Smith,D., Cousineau, B. & Belfort, M. (2002). Retrotrans-position of the Ll.ltrB group II intron proceeds pre-dominantly via reverse splicing into DNA targets.Mol. Micro. In press.
14. Munoz-Adelantado, E., Martinez-Abarca, F., Garcia-Rodriguez, F. M., San Filippo, J., Lambowitz, A. M.,Toro, N. (2002). Mobility of the Sinorhizobium melilotigroup II intron RmInt1: evidence for reverse splicinginto single-stranded DNA. J. Mol. Biol., submitted forpublication.
15. Michel, F. & Lang, B. F. (1985). Mitochondrial class IIintrons encode proteins related to the reverse tran-scriptases of retroviruses. Nature, 316, 641–643.
16. Mohr, G., Perlman, P. S. & Lambowitz, A. M. (1993).Evolutionary relationships among group II intron-encoded proteins and identification of a conserveddomain that may be related to maturase function.Nucl. Acids Res. 21, 4991–4997.
17. Klug, A. & Schwabe, J. W. R. (1995). Zinc fingers.FASEB J. 9, 597–604.
18. Gorbalenya, A. E. (1994). Self-splicing group I andgroup II introns encode homologous (putative)DNA endonucleases of a new family. Protein Sci. 3,1117–1120.
19. Shub, D. A., Goodrich-Blair, H. & Eddy, S. R. (1994).Amino acid sequence motif of group I intron endo-nucleases is conserved in open reading frames ofgroup II introns. Trends Biochem. Sci. 19, 402–404.
20. Martin, F., Maranon, C., Olivares, M., Alonso, C. &Lopez, M. C. (1995). Characterization of a non-longterminal repeat retrotransposon cDNA (L1Tc) fromTrypanosoma cruzi: homology of the first ORF with
Group II Intron Protein DNA Endonuclease Region 949

the ape family of DNA repair enzymes. J. Mol. Biol.247, 49–59.
21. Feng, Q., Moran, J. V., Kazazian, H. H., Jr & Boeke,J. D. (1996). Human L1 retrotransposon encodes aconserved endonuclease required for retrotrans-position. Cell, 87, 905–916.
22. Yang, J., Malik, H. S. & Eickbush, T. H. (1999). Identi-fication of the endonuclease domain encoded by R2and other site-specific, non-long terminal repeatretrotransposable elements. Proc. Natl Acad. Sci.USA, 96, 7847–7852.
23. Eickbush, T. H. & Malik, H. S. (2002). Origin andevolution of retrotransposons. In Mobile DNA II(Craig, N. L., Craigie, R., Gellert, M. & Lambowitz,A. M., eds), pp. 1111–1144, ASM Press Publishers,Washington, DC.
24. Martinez-Abarca, F., Garcia-Rodriguez, F. M. & Toro,N. (2000). Homing of a bacterial group II intron withan intron-encoded protein lacking a recognizableendonuclease domain. Mol. Micro. 35, 1405–1412.
25. Guo, H., Zimmerly, S., Perlman, P. S. & Lambowitz,A. M. (1997). Group II intron endonucleases useboth RNA and protein subunits for recognition ofspecific sequences in double-stranded DNA. EMBOJ. 16, 6835–6848.
26. Mohr, G., Smith, D., Belfort, M. & Lambowitz, A. M.(2000). Rules for DNA target site recognition by alactococcal group II intron enable retargeting of theintron to specific DNA sequences. Gene Dev. 14,559–573.
27. Guo, H., Karberg, M., Long, M., Jones, J. P., III,Sullenger, B. & Lambowitz, A. M. (2000). Group IIintrons designed to insert into therapeutically rele-vant DNA target sites in human cells. Science, 289,452–457.
28. Singh, N. N. & Lambowitz, A. M. (2001). Interactionof a group II intron ribonucleoprotein endonucleasewith its DNA target site investigated by DNA foot-printing and modification interference. J. Mol. Biol.309, 361–386.
29. Dalgaard, J. Z., Klar, A. J., Moser, M. J., Holley, W. R.,Chatterjee, A. & Mian, I. S. (1997). Statistical model-ing and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an inteinthat encodes a site-specific endonuclease of theH–N–H family. Nucl. Acids Res. 25, 4626–4638.
30. Aravind, L., Makarova, K. S. & Koonin, E. V. (2000).Holliday junction resolvases and related nucleases:identification of new families, phyletic distributionand evolutionary trajectories. Nucl. Acids Res. 28,3417–3432.
31. Zimmerly, S., Hausner, G. & Wu, X. (2001). Phylo-genetic relationships among group II intron ORFs.Nucl. Acids Res. 29, 1238–1250.
32. Mills, D. A., McKay, L. L. & Dunny, G. M. (1996).Splicing of a group II intron involved in the conjuga-tive transfer of pRS01 in lactococci. J. Bacteriol. 178,3531–3538.
33. Kennell, J. C., Moran, J. V., Perlman, P. S., Butow,R. A. & Lambowitz, A. M. (1993). Reverse transcrip-tase activity associated with maturase-encodinggroup II introns in yeast mitochondria. Cell, 73,133–146.
34. Zimmerly, S., Moran, J. V., Perlman, P. S. & Lambow-itz, A. M. (1999). Group II intron reverse transcrip-tase in yeast mitochondria. Stabilization andregulation of reverse transcriptase activity by theintron RNA. J. Mol. Biol. 289, 473–490.
35. Morozova, T., Seo, W. & Zimmerly, S. (2002).Non-cognate template usage and alternative primingby a group II intron-encoded reverse transcriptase.J. Mol. Biol. 315, 951–963.
36. Rost, B. (1996). PHD: predicting one-dimensionalprotein structure by profile based neural networks.Methods Enzymol. 26, 525–539.
37. Jones, D. T. (1999). Protein secondary structure pre-diction based on position-specific scoring matrices.J. Mol. Biol. 292, 195–202.
38. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang,J., Zhang, Z., Miller, W. & Lipman, D. J. (1997).Gapped BLAST and PSI-BLAST: a new generation ofprotein database search programs. Nucl. Acids Res.25, 3389–3402.
39. Kleanthous, C., Kuhlmann, U. C., Pommer, A. J.,Ferguson, N., Radford, S. E., Moore, G. R. et al.(1999). Structural and mechanistic basis of immunitytoward endonuclease colicins. Nature Struct. Biol. 6,243–252.
40. Raaijmakers, H., Vix, O., Toro, I., Golz, S., Kemper, B.& Suck, D. (1999). X-ray structure of T4 endonucleaseVII: a DNA junction resolvase with novel foldand unusual domain-swapped dimer architecture.EMBO J. 18, 1447–1458.
41. Ko, T., Liao, C., Ku, W., Chak, K. & Yuan, H. S.(1999). The crystal structure of the DNase domain ofcolicin E7 in complex with its inhibitor Im7 protein.Structure, 7, 91–102.
42. Pommer, A. J., Kuhlmann, U. C., Cooper, A.,Hemmings, A. M., Moore, G. R., James, R. &Kleanthous, C. (1999). Homing in on the role oftransition metals in the H–N–H motif of colicinendonuclease. J. Biol. Chem. 274, 27153–27160.
43. Pommer, A. J., Cal, S., Keeble, A. H., Walker, D.,Evans, S. J., Kuhlmann, U. C. et al. (2001). Mechanismand cleavage specificity of the H–N–H endo-nuclease colicin E9. J. Mol. Biol. 314, 735–749.
44. Walker, D. C., Georgiou, T., Pommer, A. J., Walker,D., Moore, G. R., Kleanthous, C. & James, R. (2002).Mutagenic scan of the H–N–H motif of colicin E9:implications for the mechanistic enzymology ofcolicins, homing enzymes and apoptotic endo-nucleases. Nucl. Acids Res. 30, 3225–3234.
45. Giraud-Panis, M.-J. E., Duckett, D. R. & Lilley, D. M.J. (1995). The modular character of a DNA junction-resolving enzyme: a zinc-binding motif in bacterio-phage T4 endonuclease VII. J. Mol. Biol. 252, 596–610.
46. Dipietro, E. S., Bashor, M. M., Stroud, P. E., Smarr,B. J., Burgess, B. J., Turner, W. E. & Neese, J. W.(1988). Comparison of an inductively coupledplasma-atomic emission spectrometry method forthe determination of calcium, magnesium, sodium,potassium, copper and zinc with atomic absorptionspectroscopy and flame photometry methods. Sci.Total Environ. 74, 249–262.
47. Deminoff, S. J., Tornow, J. & Santangelo, G. M. (1995).Unigenic evolution: a novel genetic method localizesa putative leucine zipper that mediates dimerizationof the Saccharomyces cerevisae regulator Gcr1p.Genetics, 141, 1263–1274.
48. Friedman, K. L. & Cech, T. R. (1999). Essential func-tions of amino-terminal domains in the yeasttelomerase catalytic subunit revealed by selectionfor viable mutants. Genes Dev. 13, 2863–2874.
49. Karberg, M., Guo, H., Zhong, J., Coon, R., Perutka, J.& Lambowitz, A. M. (2001). Group II introns as con-trollable gene targeting vectors for genetic manipu-lation of bacteria. Nature Biotech. 19, 1162–1167.
950 Group II Intron Protein DNA Endonuclease Region

50. Wank, H., San Filippo, J., Singh, R. N., Matsuura, M.& Lambowitz, A. M. (1999). A reverse transcrip-tase/maturase promotes splicing by binding at itsown coding segment in a group II intron RNA. Mol.Cell, 4, 239–250.
51. Metzger, W., Hermann, T., Schatz, O., LeGrice, S. F. J.& Heumann, H. (1993). Hydroxyl radical footprintanalysis of human immunodeficiency virus reversetranscriptase template primer complexes. Proc. NatlAcad. Sci. USA, 90, 5909–5913.
52. Ding, J., Hughes, S. H. & Arnold, E. A. (1997). Pro-tein-nucleic acid interactions and DNA conformationin a complex of human immunodeficiency virus type1 reverse transcriptase with a double-stranded DNAtemplate-primer. Biopolymers, 44, 125–138.
53. Jimenez-Zurdo, J. I., Garcia-Rodriguez, F. M.,Barrientos-Duran, A. & Toro, N. (2002). DNA target-site requirements for homing in vivo of a bacterialgroup II intron encoding a protein lacking the DNAendonuclease domain. J. Mol. Biol., submitted forpublication.
54. Yeo, C. C., Tham, J. M., Yap, M. W. & Poh, C. L.(1997). Group II intron from Pseudomonas alcaligenesNCIB 9867 (P25X): entrapment in plasmid RP4 andsequence analysis. Microbiology, 143, 2833–2840.
55. Granlund, M., Michel, F. & Norgren, M. (2001).Mutually exclusive distribution of IS1548 andGBSi1, an active group II intron identified in humanisolates of group B streptococci. J. Bacteriol. 183,2560–2569.
56. Dai, L. & Zimmerly, S. (2002). Compilation andanalysis of group II intron insertions in bacterialgenomes: evidence for retroelement behavior. Nucl.Acids Res. 30, 1091–1102.
57. Saldanha, R., Chen, B., Wank, H., Matsuura, M.,Edwards, J. & Lambowitz, A. M. (1999). RNA andprotein catalysis in group II intron splicing andmobility reactions using purified components.Biochemistry, 38, 9069–9083.
58. Sarkar, G. & Sommer, S. S. (1990). The “megaprimer”method of site-directed mutagenesis. BioTechniques,8, 404–407.
59. Cadwell, R. C. & Joyce, G. F. (1992). Randomizationof genes by PCR mutagenesis. PCR Methods Appl. 2,28–33.
60. Henikoff, S. & Henikoff, J. G. (1992). Amino acidsubstitution matrices from protein blocks. Proc. NatlAcad. Sci. USA, 89, 10915–10919.
61. Carson, M. (1991). Ribbons 2.0. J. Appl. Crystallog. 24,958–961.
Edited by M. Belfort
(Received 25 June 2002; received in revised form 9 October 2002; accepted 9 October 2002)
Group II Intron Protein DNA Endonuclease Region 951