chip-chip: considerations for the design, analysis, and application of genome-wide chromatin...
TRANSCRIPT

www.elsevier.com/locate/ygeno
Genomics 83 (2004) 349–360
Minireview
ChIP-chip: considerations for the design, analysis, and application of
genome-wide chromatin immunoprecipitation experiments
Michael J. Buck and Jason D. Lieb*
Department of Biology and Carolina Center for Genome Sciences, CB 3280, 202 Fordham Hall, University of North Carolina at Chapel Hill,
Chapel Hill, NC 27599-3280, USA
Received 3 October 2003; accepted 12 November 2003
Abstract
Chromatin immunoprecipitation (ChIP) is a well-established procedure used to investigate interactions between proteins and DNA.
Coupled with whole-genome DNA microarrays, ChIPs allow one to determine the entire spectrum of in vivo DNA binding sites for any given
protein. The design and analysis of ChIP-microarray (also called ChIP-chip) experiments differ significantly from the conventions used for
more traditional microarray experiments that measure relative transcript levels. Furthermore, fundamental differences exist between single-
locus ChIP approaches and ChIP-chip experiments, and these differences require new methods of analysis. In this light, we review the design
of DNA microarrays, the selection of controls, the level of repetition required, and other critical parameters for success in the design and
analysis of ChIP-chip experiments, especially those conducted in the context of mammalian or other relatively large genomes.
D 2004 Elsevier Inc. All rights reserved.
Introduction
Interactions between proteins and DNA are fundamental
to life. They mediate transcription, DNA replication, recom-
bination, and DNA repair, all processes that are central to
the biology of every organism. A comprehensive under-
standing of where enzymes and their regulatory proteins
interact with the genome in vivo would greatly increase our
understanding of the mechanism and logic of these critical
cellular events. Over the past several years, advances in
technology have made feasible, in selected organisms, the
goal of cataloging all protein–DNA interactions under a
diverse set of physiological conditions.
Traditional methods of investigation have failed to create
high-resolution, genome-wide maps of the interaction be-
tween a DNA-binding protein and DNA. For example, the
DNA-binding properties of a protein determined by in vitro
oligo selection or gel-shift assays are often poor predictors
of a factor’s actual binding targets in vivo [1]. This is
primarily because transcription factors and other eukaryotic
DNA-binding proteins generally recognize degenerate
motifs of 5 to 10 nucleotides. Even in the simple case of
the yeast genome, a typical transcription factor’s binding
0888-7543/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.ygeno.2003.11.004
* Corresponding author. Fax: +1-919-962-1625.
E-mail address: [email protected] (J.D. Lieb).
site may appear several thousand times. The fact that
consensus DNA binding sites occur far too often in genomic
DNA sequence to provide sufficient specificity has also
frustrated the use of computational approaches to identify
binding sites that are active in vivo. When putative sites of
binding can be identified, methods like DNA footprinting or
ChIP followed by quantitative PCR can be used, but are
applicable only to small segments of hand-chosen genomic
loci. Finally, attempts to determine the genome-wide bio-
logical activity of DNA-binding proteins by measuring
relative transcript level changes in cells lacking the protein
of interest often yield secondary consequences of the
deletion, rather than true primary targets of the regulatory
protein [2,3].
The union of chromatin immunoprecipitation (ChIP) and
whole-genome DNA microarrays (ChIP-chip) circumvents
these limitations by allowing researchers to create high-
resolution genome-wide maps of the in vivo interactions
between DNA-associated proteins and DNA. Currently,
there are about the same number of reviews and book
chapters on ChIP-chip procedures and applications [4–13]
as there are primary papers in the literature [1,14–25]. We
will concentrate on the general considerations for the design
and analysis of ChIP-chip experiments, an area that has not
yet been addressed in detail. A concise review of ChIP-chip
procedures and applications is useful for framing that topic.

enomics 83 (2004) 349–360
An overview of the ChIP-chip experimental procedure
Ranging from yeast to cultured mammalian cells, there
is surprisingly little variation in published ChIP-chip pro-
tocols. Generally, cells are grown under the desired exper-
M.J. Buck, J.D. Lieb / G350
Fig. 1. (A) A summary of the ChIP-chip procedure. See the text for details. (B) Com
and microarray-based experiments. Single-locus experiments use a single internal
the IP, mock IP (or control IP), and input DNA. In microarray experiments, rati
obtained for all other elements, which are termed non-enriched. (C) Global array n
median log2 ratio is equal to 0 for the normalized distribution (blue). (D) The eff
20% of arrayed elements detect five-fold enrichment (log2 STDev = 0.5). The s
average ratios are plotted. The distribution is skewed such that the median log2 ratio
would center the non-enriched population at 0 (green).
imental condition and then fixed with formaldehyde (Fig.
1A). Formaldehyde crosslinks proteins to each other pri-
marily between the e-amino group of lysine residues and
an adjacent peptide bond. Formaldehyde can also form
DNA–protein crosslinks, but only if the DNA is partially
parison of the controls used for single-locus, PCR-based ChIP experiments
control in each sample. The intensity of the target band is compared across
os obtained for enriched elements (boxed in white) are compared to those
ormalization will slide the raw distribution (red) along the x-axis so that the
ect of default normalization on a simulated ChIP-chip experiment in which
imulated experiment was repeated three times, and the distribution of the
of the non-enriched population is at �0.25 (black). The ideal normalization

M.J. Buck, J.D. Lieb / Genom
denatured to expose the –CO–NH moiety at position 1
(N-1) of a guanine or the exocyclic amino groups of an
adenine, guanine, or cytosine. The exact nature of the
crosslinks formed by formaldehyde in chromatin in vivo is
not well characterized, and it is unclear whether the
majority of crosslinks formed are protein–protein or pro-
tein–DNA. In some cases, other crosslinking agents like
dimethyl adipimidate have been used in combination with
formaldehyde [6]. However, formaldehyde remains the
most commonly used fixative because the crosslinks are
heat-reversible, which allows downstream enzymatic treat-
ment of the DNA. After crosslinking, the extract is
sonicated to shear the DNA fragments to the desired size,
usually 1 kb or smaller.
DNA fragments crosslinked to the protein of interest are
enriched in one of three standard ways: immunoprecipita-
tion with a protein-specific antibody, immunoprecipitation
of a tagged protein using an antibody specific to the tag, or
affinity purification using a tag that obviates the need for
antibodies, such as the TAP (tandem affinity purification)
tag [26]. The formaldehyde crosslinks are then reversed and
the DNA is purified. Low DNA yields from the IP reactions
usually make DNA amplification a requirement for DNA
microarray-based detection. Randomly-primed [27] or liga-
tion-mediated PCR-based [28] methods have been most
commonly used, but a recently described linear amplifica-
tion method is likely to give higher fidelity results [29].
Ideally, the IPs can be scaled up economically and ampli-
fication can be avoided.
Enriched DNA is then labeled with a fluorescent mole-
cule such as Cy5 or Alexa 647. The fluorescent molecule
can be introduced directly in the form of a modified
nucleotide [30] or by chemical coupling after the introduc-
tion of an aminoallyl nucleotide derivative [31]. In two-
color array platforms, genomic DNA prepared from IP input
extract is generally used as a reference and similarly
amplified and labeled with a different fluor, such as Cy3
or Alexa 555 [21]. The two probes are then combined and
hybridized to a single DNA microarray. Ideally, to provide a
comprehensive and unbiased survey of protein-DNA inter-
actions, the DNA microarrays used in ChIP experiments
contain elements (deposited DNA fragments) that represent
the entire genome.
The results of the hybridization allow one to identify
which segments of the genome were enriched in the IP.
Since the precise location of each arrayed element is known,
construction of a genome-wide map of in vivo protein–
DNA interactions is possible. The resolution of the method
depends mainly on two factors: the length of the sheared
chromatin enriched by the IP and the length and spacing of
the arrayed DNA elements used to detect the IP-enriched
fragments. Typical yeast experiments achieve a resolution of
about 1 kb, which is sufficient to assign binding to the
regulation of a single gene. Once the bound regulatory
region is identified, the exact binding site can often be
inferred by computational methods [32,33].
Successful applications
The ChIP-chip technique was first applied successfully
to identify binding sites for individual transcription factors
in Saccharomyces cerevisiae [1,15,16]. Later, also in yeast,
a c-Myc epitope protein tagging system was used to map
the genome-wide positions of 106 transcription factors
[17]. Other applications have been reported, including
the study of DNA replication [34], recombination [35],
and chromatin structure [23–25,36]. In these experiments,
microarrays containing f1-kb PCR products representing
ORFs (open reading frames), intergenic regions, or both
were used in conjunction with a two-color experimental
scheme. The PCR products in these arrays were ‘‘tiled’’
across the genome, meaning the PCR products were
directly adjacent to one another along the genome, with
little or no DNA sequence between arrayed elements. The
compact and nonrepetitive nature of the simple genomes
harbored by these model organisms made such an ap-
proach feasible.
Experiments in mammalian systems have proven more
difficult due to the large and repetitive nature of their
genomes. Initial ChIP-chip experiments identified binding
sites for the c-Myc, Max, Gata1, E2F, and Rb transcrip-
tion factors in cultured human cells [18,20–22]. For
practical reasons, the DNA microarrays used in these
pioneering studies represented only a tiny fraction of the
genome. For the c-Myc and Max studies, DNA micro-
arrays were constructed with PCR products spanning the
proximal promoters of 4839 of the approximately 30,000
human genes [18]. The arrayed DNA fragments had an
average size of 900 bp and typically covered a region 650
bp upstream to 250 bp downstream of each gene. In
addition, the arrays contained 729 coding sequences and
221 genomic regions more than 1 kb upstream of a gene.
These arrays were designed to maximize the number of
gene promoters represented while minimizing the number
of arrayed elements. One disadvantage of having one spot
per upstream region is that any interactions occurring
farther than f1 kb away from an arrayed element may
not be detected. A related concern is that the location of
any detected in vivo binding event may not reside directly
in the fragment spotted on the array. The degree of
detected enrichment will correlate inversely with distance
of the binding event from the arrayed element, but this
variation will be impossible to distinguish from variation
produced by other important parameters, such as binding
affinity or site occupancy.
To remedy this shortcoming and ensure that no inter-
actions go undetected, arrays that tile across an entire
regulatory region of particular interest can be designed.
This approach was used to map the Gata-1 transcription
factor to the h-globin locus, by dividing the 75-kb promoter
into 74 segments of approximately 1 kb in length [20]. This
small array was comprehensive, but specific to a single
regulatory region.
ics 83 (2004) 349–360 351

M.J. Buck, J.D. Lieb / Genom352
A third strategy was employed to map the mammalian
transcription factors E2F and Rb. DNA microarrays were
created with 7776 CpG island clones from the UK Genome
Mapping Project Centre’s CGI genomic library [21,22].
CpG islands are short stretches of DNA containing a high
density of nonmethylated CpG dinucleotides and are asso-
ciated with the promoters and the first exon of a gene [37].
Therefore, for studies involving the mapping of transcrip-
tion factors, isolating CpG islands greatly enriches for
regions of potential interest. CpG islands were isolated
through use of an affinity matrix based on the methyl-
CpG binding domain from the chromosomal protein MeCP2
[38]. The clone inserts (0.2–2 kb) were amplified by PCR
before spotting on the array.
This approach reduces the costs associated with order-
ing thousands of primer pairs and potentially provides
unbiased coverage of a large portion of the genome. There
are some trade-offs with this approach. First, at the time
the experiments from Weinmann et al. [21] were per-
formed, the identity of the clones was not known, so spots
that produced interesting results had to be sequenced.
Second, because the identity of the spots was not known,
it was not possible to estimate the level of redundancy or
the degree of coverage prior to embarking on the experi-
ment. Third, the location of any detected in vivo binding
event may not reside directly in the CpG clone spotted on
the array, but instead be up to 2 kb away [11]. Finally,
DNA fragments that are difficult to clone may be under-
represented. Not knowing the above parameters makes it
more difficult to perform a statistical analysis of the results
and could affect interpretation of the data. All of the clones
used for this array have since been sequenced, removing
some of these concerns for that particular set. As is the
case with any array that does not provide complete
coverage, it would be difficult to separate the effects of
distance, binding affinity, or site occupancy on variations
in the observed ratios.
Experimental design and analysis
There are a number of important concerns common to all
DNA microarray experiments. These include the basics of
image acquisition and analysis, background subtraction,
standard normalization algorithms, the need to control for
dye biases, and statistical problems that arise when large
numbers of data points are analyzed. We will not cover these
issues here, since they have been reviewed extensively
elsewhere [39–43].
Among the many hundreds of whole-genome ChIP-chip
experiments that have been performed in yeast, and the few
that have been performed in more complex systems, there is
wide variation in the experimental design, data analysis, and
microarray platforms utilized. What are the factors that one
should consider in choosing the design of a ChIP-chip
experiment?
Which array platform should I choose?
After successfully performing a standard ChIP experi-
ment, a logical next step is to identify comprehensively the
targets of your favorite DNA binding protein or chromatin
component. The first thing to do is choose a DNA micro-
array platform.
There are three main types of DNA microarrays: me-
chanically spotted cDNA or PCR-product arrays, mechan-
ically spotted oligonucleotide arrays, and arrays composed
of oligonucleotides that are synthesized in situ. The most
widely available microarrays contain DNA elements of one
of these types for detecting RNAs transcribed from
expressed genomic regions (or ‘‘ORF arrays’’ for short).
These arrays have traditionally been used for gene expres-
sion studies and are available commercially. The use of ORF
arrays has limited power for ChIP experiments, since most
transcription factor binding sites are located in the intergenic
regions and are therefore not included on these arrays.
Depending on the degree of DNA shearing there may be
enough overlap between immunoprecipitated DNA frag-
ments and the spotted ORF probes to allow identification
of target sites located near an ORF. Experiments in yeast
have found significant enrichment of ORFs when the
neighboring intergenic region is also enriched [15]. In
organisms containing a large number of introns, using
cDNA arrays for ChIP-chip experiments may be trouble-
some. Since introns are spliced away from mRNA, the
arrayed cDNA sequences do not correspond to the linear
sequence of genomic DNA. Therefore, a 1-kb cDNA could
correspond to different fragments of 30 kb of genomic
sequence. Not only will signal be reduced due to a noncon-
tinuous sequence for hybridization, but signal from two
distant binding events could be detected by a single spot.
While this is not a problem in organisms with few introns
like yeast, it does pose a hurdle for mammalian genomes.
The most robust array design for ChIP-chip is one having
contiguous tiled DNA fragments that represent the entire
genome, including the noncoding regions. Whole-genome
tiling arrays consisting of mechanically spotted PCR prod-
ucts have been very useful in organisms with small genomes
like yeast. Two different groups have assembled single
arrays comprising nearly all of the nonrepetitive sequences
of human chromosome 22 (and in one case both 21 and 22),
demonstrating that this can be a practical approach for single
mammalian chromosomes [44,45]. However, mammalian
genomes are 300 times the size of yeast and contain a much
higher proportion of repetitive sequence. Tiling across the
entire genome with small PCR products would require about
3 million DNA spots which with current technology is not
feasible on a single array. Mapped cosmids and BAC clones
have been used to build microarrays [46], and these arrays
could be used to assay ChIP-chip experiments, but the
resolution would be correspondingly low. The optimal length
of arrayed fragments is a balance between the cost of having
many elements and the desire for increased resolution. It is
ics 83 (2004) 349–360

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360 353
important to keep in mind that arrayed elements shorter than
the average size of a sheared chromatin fragment (generally
500–1000 bp) will not increase resolution. Other spotted-
array approaches include tiling individual promoter regions,
using CpG island clones, or representing each of the prox-
imal promoter regions of all known and predicted genes with
one spot each (see Successful applications).
In addition to spotted arrays containing long (>200 bp)
DNA fragments, the use of short- (20–25 bases) or long-
(60–90 bases) oligonucleotide arrays is an attractive possi-
bility. The main advantages would be in avoiding PCR and
mechanical spotting by relying instead on in situ synthesis
from a commercial source and a potential gain in resolution.
These arrays could contain oligonucleotides that tile or are
spaced at regular intervals across a region or genome. There
are no published accounts of the use of such an array for
ChIP-chip experiments, so it is not yet clear that they will
work well for this technique. A major drawback to using
short oligos is potentially poor hybridization to arrayed
elements with low GC content, which are common in non-
coding regions. Selection of a common array hybridization
condition for oligos of widely varying GC content may be
very difficult for mammalian genomes, in which base
composition is highly variable. For this reason, longer oligos
(60–90 bases) are likely to be much more robust in this
context. In addition, if the oligos are not tiled their spacing
will affect target identification. For example, if an oligo is
spaced every 2 kb and the average DNA shear size is 1 kb, a
binding site located 1 kb away from any arrayed element will
exhibit poor enrichment. Again, it would be difficult to
separate the effects of distance from an arrayed element,
binding affinity, or site occupancy on variations in the
observed ratios.
The optimal solution, although still unproven, may be a
tiled, long-oligonucleotide array, which would provide com-
plete coverage and very high resolution for binding-site
identification. A comprehensive comparison of using PCR-
spotted arrays and long- and short-oligonucleotide arrays for
ChIP-chip experiments has not been published. Therefore,
the best array platform for ChIP-chip experiments is not
established. Regardless of whether the arrays are oligo or
amplicon-based, tiling array platforms that provide compre-
hensive coverage may encounter technical problems that will
need to be addressed. These problems include potential
cross-hybridization between homologous genomic regions,
general ‘‘nonspecific’’ cross-hybridization, and the depen-
dence of signal intensity on base composition. Commercial
availability of such long-oligo arrays covering the human
genome may be years away, but custom arrays covering
regions of interest could be synthesized now [47].
Do I really need to use arrays?
The DNA purified from a ChIP experiment can be cloned
and sequenced, providing an alternative to microarray-based
detection [11]. A key advantage to the microarray approach
is that it is able to detect small degrees of relative enrich-
ment genome-wide in a single assay. In contrast, consider
the case in which a 20-fold enrichment of targets is achieved
by IP, and targets represent 1% of all genomic fragments.
If a sequencing approach is chosen, only f17% of all se-
quenced clones would be IP targets at all, and for each
experiment, a very large number of clones would have to be
sequenced to sample the entire IP result with sufficient
coverage to identify targets confidently. This method may
become feasible by devising clever high-throughput
schemes to increase the practical enrichment and decrease
background prior to sequencing. These may include pre-
screening of clones for repetitive elements, modification of
the standard ChIP experiment to include a second IP, or size
selection to limit nonspecific clones and repetitive elements
[11]. In addition to traditional sequencing techniques
(SAGE), commercially available techniques such as mas-
sively parallel signature sequencing can sequence thousands
of cDNA clones simultaneously and be used to sample an
entire ChIP [48]. Sequencing-based approaches could pro-
vide an attractive alternative to array analysis for organisms
with a large genome size.
How many, and what kind of, elements should be on the
array?
How an experiment can be analyzed and interpreted will
be influenced strongly by the number of elements on the
DNA microarray and how many of them correspond to
genomic regions bound by the assayed DNA-binding pro-
tein. In traditional ChIP analysis, specific PCR primers are
used to assay the abundance of a suspected target relative to a
standard genomic fragment that is thought to be non-
enriched by the IP (Fig. 1B). Therefore, all measurements
regarding the degree of enrichment for a tested genomic
region are made relative to a single control fragment.
In contrast, when utilizing a DNA microarray to analyze
IP enrichment, no predetermined single standard is gener-
ally used. All arrayed elements reporting nonenrichment are
used as controls. The elements that will report a nonenriched
result are not assumed beforehand, but are determined after
the experiment is performed. Therefore, for any given
genomic region, data regarding the degree of enrichment
obtained with DNA microarrays are measured relative only
to regions represented by other arrayed elements. This has
the very powerful advantage of allowing interpretation of
experimental results without any knowledge whatsoever of
a protein’s distribution prior to the experiment. It also
eliminates reliance on a single internal control for the
interpretation of results. While some suspected binding sites
(positive controls) are likely to be known prior to the
experiment, it is often difficult to select regions that are
definitely not bound for use as negative controls. Regions
that are not suspected to contain binding sites, for example
ORFs in the case of transcription factors, have been shown
to be enriched in ChIP-chip experiments [1].

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360354
Using a pool of arrayed elements to measure relative
enrichment has some interesting consequences. For exam-
ple, in a hypothetical experiment in which all of the arrayed
elements represent binding targets (as might occur for a
general chromatin factor, even if whole-genome arrays are
used), there will be little or no measurable enrichment of
any particular element relative to any other. The array
readout will appear as if no enrichment was achieved and
the results will be uninterpretable, even though the IP was
successful. More commonly, this situation could arise if too
many ‘‘candidate’’ targets for a transcription factor are used
to create a small array designed specifically for confirma-
tion of suspected targets. This would be the equivalent of, in
a traditional ChIP experiment, unwittingly choosing an
internal standard that represents a genomic fragment
enriched in the IP. In either case, the danger of using an
array rich in targets is that subtle variations in relative
binding among bona fide targets could easily be misinter-
preted as being ‘‘bound’’ or ‘‘unbound’’, with that error
propagating to biological interpretation. Although it is
currently impossible to predict in vivo binding sites accu-
rately prior to performing an experiment, it is very impor-
tant to include intentionally a large number of elements on
the array that are not predicted to be targets. These spots
will not act as ‘‘controls’’ in the traditional sense, since
some of them may in fact be bound. Instead they will
provide a pool of arrayed elements that are likely to detect
background (nonenriched DNA fragments), which will
provide a baseline that can be used for comparison to detect
IP-enriched fragments.
In cases in which a large percentage of arrayed elements
are IP-enriched, the potential for misinterpretation of the
data is increased. This is due to the difficulty in normalizing
ratios in ChIP-chip experiments such that a consistent,
meaningful number is produced for each arrayed element
and experiments can be compared across replicates. Most
global normalization techniques used for gene expression
experiments assume that approximately equal numbers of
arrayed elements detect up- and down-regulated transcripts,
with most transcripts assayed remaining unchanged [49,50].
To determine relative enrichment or depletion of an RNA
message, the median of the ratios for the entire population of
arrayed elements is set to 1 (0 in log2 space), by multiplying
the intensity values of one of the two channels by a constant
for linear (median ratio) or fitting to a line for nonlinear
(Lowess) approaches. In effect, this slides the entire distri-
bution of ratios forward or back along the x axis (Fig. 1C).
However, the assumptions used for this normalization are
explicitly untrue in a ChIP-chip experiment. First, there is
no basis for assuming that any particular genomic fragment
will be specifically depleted in a ChIP experiment. Instead,
there will be two populations of fragments: IP-enriched
genomic fragments and the remaining genomic DNA that
is not IP-enriched. This, coupled with the general use of
total genomic DNA as a reference for ChIP-chip experi-
ments (this is the denominator in the ratio), causes the ratios
obtained in a typical IP experiment to be distributed
asymmetrically about the median. Second, there is no way
to predict accurately how many genomic fragments will be
IP-enriched, so it is difficult to predict how unbalanced the
distribution of data will be. Third, it is difficult to predict
how the ratios of the IP-enriched fragments will behave. For
example, will there be a discrete set of binding targets that
are all enriched to the same degree, creating an easily
discernable class of relatively high ratios? Or will the factor
be bound to some targets more frequently or strongly than
others, creating a continuum of IP-enriched ratios that fades
into noise? This combination of uncertainties makes it
difficult to model how ratios obtained from an IP experi-
ment should be distributed. Even advanced techniques that
select rank-invariant elements to use for normalization fail
on highly skewed data [51].
It is certain, however, that as the percentage of arrayed
elements representing IP-enriched DNA fragments increases,
the log2 median of ratios for the nonenriched population will
not be zero after normalization using the common techniques
(Lowess, rank-invariant selection, or median-ratio normali-
zation). Instead, a negative median will be observed for the
nonenriched class (Fig. 1C). In simulations in which 20% of
the arrayed elements report fivefold enrichment, the log2median of the nonenriched population is centered at �0.25
when normalized with the median-ratio approach, possibly
causing some elements detecting IP-enriched DNA frag-
ments to report log2 ratios less than 0 (STDev = 0.5, average
of three simulations was used). Therefore, if a large percent-
age of arrayed elements represent DNA-binding targets, a
different normalization or analysis technique may be needed
(see Data analysis, below).
There are two ways around this problem. The first is to
select negative controls before the experiment and to use
these for normalization (see above). The second is to try to
distinguish the enriched and nonenriched populations com-
putationally from the raw data and then to use the non-
enriched population for normalization. Rank-invariant
techniques select elements on array whose raw intensity
ranks do not change (in either of the two channels if
performing a two-color experiment). While this approach
works when a low proportion of the arrayed elements are
enriched (<10%), it fails as the percentage of enrichment
increases [51]. The rank-invariant selection schemes used
for expression arrays have not yet been tuned specifically
for use with ChIP-chip data [51,52].
What types of control experiments are best?
It is important to distinguish the function of a control from
that of a hybridization reference. A hybridization reference in
ChIP-chip experiments is a common DNA sample, usually
the sheared genomic DNA from the experimental organism,
that is used as the basis for comparison for each IP exper-
iment. By hybridizing every experiment with a common
reference, accurate ratio measurements can be obtained, and

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360 355
different experiments can be compared more easily. On the
other hand, a control experiment should detect experimental
variation caused by nonbiological sources, including sample
handling, differential PCR amplification, differential label-
ing, or nonspecific antibody interactions. The best control,
when available, is a cell lacking the IP epitope but otherwise
isogenic, such that there is no target for the antibody to bind
specifically. This type of control corrects for sample han-
dling, preferential amplification, labeling biases, and non-
specific antibody interactions. In experiments using an
epitope-tagged protein, this can be achieved easily by using
a cell line lacking the tagged protein.
In many cases the ideal control will not be available and
a mock IP should be performed. In a mock IP experiment,
the protocol is repeated exactly but the antibody is omitted,
or an unrelated antibody for which there is no corresponding
epitope is used, for example, anti-GFP in an unmodified cell
line. Mock IPs control for sample handling, labeling biases,
and preferential amplification, but not for nonspecific anti-
body interactions. A control experiment should never be
used as a reference for an IP experiment, since ideally the
perfect control experiment would be devoid of DNA.
How many times should a ChIP-chip experiment be
repeated?
The high cost of performing DNA-microarray experi-
ments has forced investigators to make difficult choices
about how many times an experiment should be repeated.
The number of times a ChIP-chip experiment needs to be
repeated depends on the fold-enrichment achieved and
experimental variance, two measurements that change with
each combination of antibody, epitope, and DNA micro-
array platform. The variance of an experiment is specific
to each experiment and is hard to model and generalize.
Therefore, there is no ‘‘gold standard’’ for the degree of
repetition. Published experiments have generally achieved
enrichment rates between two- and eightfold (log2 ratios
of 1 to 3) [1,15–20]. Many published ChIP-chip experi-
ments are performed in triplicate, which even in the best
case should be considered the lower limit for reliable
measurements.
The number of replicates required to predict binding
accurately can be estimated from simulations and published
data. For targets with eightfold or higher enrichment, as few
as three replicates may produce reliable site determination
[1,17]. Assuming constant variance, as the enrichment drops,
the number of replicates needs to be increased. Increasing the
measured fold enrichment will reduce the number of repli-
cates required for a ChIP-chip experiment. Enrichment can
be increased by using more specific antibodies, improving
the wash conditions in the IP, improving the specificity of
elutions, reiterating IP steps before the isolation of DNA, or
using shorter sheared chromatin fragments.
Some types of experiments are more likely to exhibit
lower relative enrichment rates than others. For example,
several factors lead to low enrichment rates in whole-
genome ChIPs designed to map the location of specific
histone modifications [24]. First, the number of targets is
potentially very high, which reduces the number of spots
against which a ratio can be measured. Second, the density
of targets may be high, which when coupled with random
shearing may increase the ‘‘baseline’’ against which targets
are measured and make it difficult to resolve adjacent
interactions. Third, the number of sites in the genome in
which a modification can take place is much higher than the
number of arrayed elements. For example, a histone mod-
ification may occur in only a portion of a given genomic
region represented by an arrayed element, or it may occur
many times. The enrichment observed could therefore be a
function of the proportion of the genomic fragment harbor-
ing the modification, rather than the presence or absence of
a single factor. In these types of experiments, in which a
large percentage of the genome (>40%) is enriched, it is
difficult to determine confidently if a specific site is
enriched above background. However, it may be easier
for the experimenter to determine if a group of fragments is
enriched compared to another group (for example ORFs vs
intergenic regions) [23].
In repetitions, what should change, and what should stay
the same?
In most cases, the goal of repeating an experiment is to
determine which parts of the signal represent biological
meaning. One unintended consequence of repeating an
experiment could be to fix variation attributable to some
aspect of the experimental protocol. This is always undesir-
able unless one is troubleshooting a specific problem. To
reduce the likelihood of fixing an artifact, in our opinion
each repetition should assay a completely independent
biological sample, and the experimenter should attempt to
change as many of the seemingly irrelevant variables as
possible. Variables that are good to change with each
repetition include date of the experiment, date of hybridiza-
tion, array batch (or print) used, buffers and other common
reagents used, fluorescent dye combinations, hybridization
chamber type used, scanner used, etc. This way, the values
fixed by the repetition are more likely to be due to biological
state, rather than to systematic error. Technical replicates,
which consist of hybridizing the same biological sample
independently, can of course be useful. For example, label-
ing samples in fluor reverse pairs and combining those data
has been shown to increase power in microarray expression
experiments [42].
Three methods to consider for data analysis
Median percentile rank
One way to avoid many of the previously discussed
problems associated with ratio normalization in ChIP-chip
experiments is to use ranks instead of ratios. The rank of an

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360356
element is simply the position of that element in a list sorted
by ratio in descending order. Ranks are useful because the
magnitude and scale of the actual ratios obtained in any
given experiment become irrelevant; what matters is their
rank order. Most normalization methods do not affect the
rank order of ratios in two-color microarray experiments or
the rank of intensity values in one-color experiments. Rank
methods are most useful when reported ratios vary widely
from experiment to experiment, but the rank order of ratios
is consistent between experiments. In the median percentile
rank method, the percentile rank of the ratio reported by
each element is determined. The percentile rank of a number
x is defined by how many numbers in a given population are
less than x. For example, if 70% of the members of a
population are less than x, the percentile rank of x is 0.7, or
70%. Then, across all replicate experiments, the median
percentile rank for each spot is determined. In an ideal
control experiment in which no genomic fragments were
enriched preferentially, the percentile rank for each spot on a
given array will be a random number between 0 and 1, since
the rank of the spot is due only to noise. Across many
replicates, the medians of the percentile rank values for all
spots will have a normal distribution bounded by 0 and 1,
with a peak at 0.5, or the 50th percentile. With an increasing
number of replicates, the accumulation of values around 0.5
will become increasingly pronounced (Fig. 2A). In contrast,
when a simulated experiment assuming a fourfold IP en-
richment of genomic fragments corresponding to 10% of the
arrayed elements was repeated five times, a bimodal distri-
bution of median rank values was observed (Fig. 2B). This
bimodal distribution results from consistent enrichment of
specific fragments in each of the replicated IP experiments.
The median percentile rank at the trough of the bimodal
distribution is generally selected as a conservative cutoff for
defining targets. This is a very powerful method, because it
allows one to select cutoffs from the distributions of the data
alone, without making any assumptions.
The median percentile rank approach is particularly
useful for identifying targets when more than approximately
4% of the total elements on the array report IP enrichment
[1,15], but is less effective for analysis of proteins with
fewer targets. To analyze the genomic distribution of pro-
teins with fewer DNA-binding sites, a larger number of
repetitions would have to be performed to produce a
bimodal distribution of median ranks. Another significant
disadvantage of this simple method is the potential loss of
amplitude information that is present in the ratio measure-
ments. To capture that information, the single-array error
model or a sliding-window approach may be used.
The single-array error model
The single-array error model was developed to analyze
traditional RNA-based microarray experiments [53] and has
been adapted for ChIP-chip analysis [16,18]. This method
addresses two concerns when combining replicates from
microarray experiments: Do replicates have equal overall
variance, and does every arrayed element report values with
equal measurement error (uncertainty)?
Experimental replicates that have a different overall
variance have different probabilities of outlying events
occurring by chance. For example, in two populations with
average values of 0, if one replicate had a variance of 0.5
and another 1, a measurement of greater than 1 in both
experiments would occur 7.8 and 16% of the time by
chance, respectively. If these replicates were combined
without correcting for differences in the variance, the first
replicate with greater variance would dominate the proper-
ties of the combined dataset. Therefore to combine these
two replicates accurately their variances must be normal-
ized or weighted appropriately. The single-array error
model allows replicate experiments to be averaged with
suitable weight (Fig. 2C).
It has been demonstrated that measurements with low-
intensity signals have a higher relative uncertainty than
measurements with higher intensity signals [53]. As the
intensity in either channel approaches the background signal
it becomes difficult to distinguish true hybridization signal
from nonspecific background. To correct for this increased
uncertainty the single-array error model down-weights
arrayed elements reporting signal close to noise, and those
reporting signal much greater than noise are given increased
weight. Fig. 2D shows a comparison of weighted log ratios
created by the single-array error model and a standard log2ratio as a function of intensity in each channel. The weights
are calculated through the use of a statistic called ‘‘X’’,
which is computed for each measurement on every array.
The distribution of X for each array is normally distributed
with equal variance. A normal or Gaussian distribution is
important because the mean and standard deviation can be
used to estimate the probability of a chance event. For
example, 95% of all the data points will be found within 2
standard deviations of the mean, and p values can be
calculated when datasets from replicate experiments are
combined.
When the number of enriched spots is greater than 5%,
this approach is inaccurate and needs be adjusted, because
the distribution is skewed by a large number of arrayed
elements with a high intensity in one channel. The distri-
bution of X will no longer be normal, and determining the
probability due to random events becomes inaccurate. To
correct this problem, Li et al. [18] suggested that the
nonenriched distribution may be estimated from the nega-
tive half of the X value distribution (where X = 0 is the
reflection point). These values on the left half of the normal
distribution can be ‘‘flipped’’ to estimate the positive X
values on the other half of the distribution. While this
adjustment will work when the percentage of enriched spots
is low (<10%), inappropriate normalization will cause the
true reflection point to be a negative value. Consequently,
the single-array error model should be used to analyze only
datasets containing a low percentage (<10%) of enriched
elements.
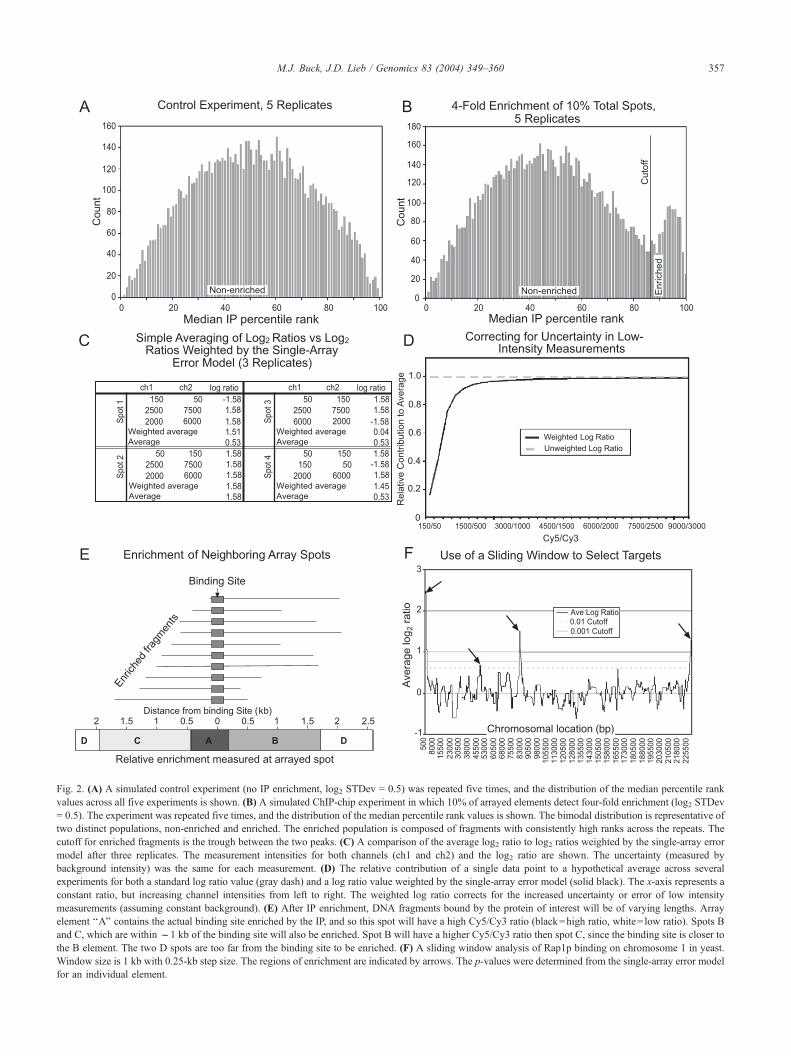
Fig. 2. (A) A simulated control experiment (no IP enrichment, log2 STDev = 0.5) was repeated five times, and the distribution of the median percentile rank
values across all five experiments is shown. (B) A simulated ChIP-chip experiment in which 10% of arrayed elements detect four-fold enrichment (log2 STDev
= 0.5). The experiment was repeated five times, and the distribution of the median percentile rank values is shown. The bimodal distribution is representative of
two distinct populations, non-enriched and enriched. The enriched population is composed of fragments with consistently high ranks across the repeats. The
cutoff for enriched fragments is the trough between the two peaks. (C) A comparison of the average log2 ratio to log2 ratios weighted by the single-array error
model after three replicates. The measurement intensities for both channels (ch1 and ch2) and the log2 ratio are shown. The uncertainty (measured by
background intensity) was the same for each measurement. (D) The relative contribution of a single data point to a hypothetical average across several
experiments for both a standard log ratio value (gray dash) and a log ratio value weighted by the single-array error model (solid black). The x-axis represents a
constant ratio, but increasing channel intensities from left to right. The weighted log ratio corrects for the increased uncertainty or error of low intensity
measurements (assuming constant background). (E) After IP enrichment, DNA fragments bound by the protein of interest will be of varying lengths. Array
element ‘‘A’’ contains the actual binding site enriched by the IP, and so this spot will have a high Cy5/Cy3 ratio (black=high ratio, white= low ratio). Spots B
and C, which are withinf1 kb of the binding site will also be enriched. Spot B will have a higher Cy5/Cy3 ratio then spot C, since the binding site is closer to
the B element. The two D spots are too far from the binding site to be enriched. (F) A sliding window analysis of Rap1p binding on chromosome 1 in yeast.
Window size is 1 kb with 0.25-kb step size. The regions of enrichment are indicated by arrows. The p-values were determined from the single-array error model
for an individual element.
M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360 357

enomics 83 (2004) 349–360
A sliding-window approach
In contrast to mRNA microarray experiments, in which
each arrayed element usually measures the abundance of one
mRNA species, in ChIP-chip experiments each element
measures the abundance of a population of fragments of
assorted lengths due to chromatin shearing (Fig. 2E). There-
fore, arrayed elements representing genomic regions 1 to 2
kb downstream or upstream of the binding site will also
detect enrichment. This effect produces a peak over several
arrayed elements containing genomically adjacent DNA.
This is nonrandom behavior that is not expected from
spuriously high ratio measurements. One can take advantage
of this fact and use it as an independent confirmation of
enrichment for a given genomic region.
When using tiled arrays containing short DNA frag-
ments, several neighboring genomic elements will identify
each protein–DNA interaction. If chromatin is sheared
randomly to an average size of 1 kb in a ChIP experiment,
at least a 2-kb region of the genome surrounding the actual
site of protein–DNA interaction will be enriched. To take
advantage of this unique property of ChIP-chip experiments,
a simple but powerful sliding-window approach has been
developed to characterize binding sites for transcription
factors when using full-genome arrays in yeast (Fig. 2F).
With this approach, a window of 1 kb is slid across a region
or chromosome, and the average log2 ratio of any arrayed
elements that fall within that window is determined. The
window is moved downstream 0.25 kb, and then the
calculation is repeated iteratively for the entire length of
chromosome. This sliding average will identify binding sites
as peaks. The height of peaks caused by spuriously high
ratios will be reduced, since the probability of a neighboring
genomic element also having a high ratio is extremely low.
In addition, a confidence value for each peak can be
assigned based on the number of independent arrayed
elements used to construct the peak. The utility of this
approach does not depend on the absolute number of
targets, but on the density of their distribution. It is
appropriate for detecting any number of targets that are
distributed with a frequency less than approximately three
times the average sheared chromatin size. For example, if
the average sheared chromatin size were 1 kb, this method
would be useful for the detection of any protein predicted
to be spaced at intervals of at least 3 kb. A drawback to
this approach is that it requires high-resolution tiling
arrays.
M.J. Buck, J.D. Lieb / G358
Future applications and challenges
Arrays designed specifically for the ChIP-chip technique
should be developed and utilized. Ideally, arrays should be
designed with short DNA fragments (f0.5 kb) of equal
lengths that are tiled for continuous genomic regions (short
element tiling, or SET, arrays). Use of SET arrays with
ChIP-chip experiments derived from sheared chromatin of
f1 kb should allow for enrichment of the binding site and
at least two neighboring regions, which can be used to
confirm the core binding location. The ratio between the
log2 ratios for the upstream and downstream regions should
be proportional to the distance from the center of the
binding site. In theory, this would allow the center of
binding to be predicted, to the base pair, from the raw data
(Fig. 2E).
Aside from technical advances, which will undoubtedly
allow more accurate and precise determinations of DNA–
protein interactions, simply incorporating ChIP-chip experi-
ments into the standard molecular biology toolbox will
result in a flood of functional data. Time-course experiments
to determine binding order, recruitment relationships, and
codependencies have already been carried out [17]. While
most experiments to date have been performed in culture on
cell lines, bacteria, or yeast, future experiments will include
ChIPs from developing tissues, organs, or cancer biopsies
[54]. Across all of biology, the ChIP-chip platform will be
critical in elucidating the function of genomes and the
proteins they encode.
References
[1] J.D. Lieb, X. Liu, D. Botstein, P.O. Brown, Promoter-specific binding
of Rap1 revealed by genome-wide maps of protein–DNA association,
Nat. Genet. 28 (2001) 327–334.
[2] A. Wagner, Estimating coarse gene network structure from large-scale
gene perturbation data, Genome Res. 12 (2002) 309–315.
[3] T.R. Hughes, M.J. Marton, A.R. Jones, C.J. Roberts, R. Stoughton,
C.D. Armour, H.A. Bennett, E. Coffey, H. Dai, Y.D. He, M.J. Kidd,
A.M. King, M.R. Meyer, D. Slade, P.Y. Lum, S.B. Stepaniants, D.D.
Shoemaker, D. Gachotte, K. Chakraburtty, J. Simon, M. Bard, S.H.
Friend, Functional discovery via a compendium of expression pro-
files, Cell 102 (2000) 109–126.
[4] K.D. Johnson, E.H. Bresnick, Dissecting long-range transcriptional
mechanisms by chromatin immunoprecipitation, Methods 26 (2002)
27–36.
[5] M.H. Kuo, C.D. Allis, In vivo cross-linking and immunoprecipitation
for studying dynamic protein:DNA associations in a chromatin envi-
ronment, Methods 19 (1999) 425–433.
[6] S.K. Kurdistani, M. Grunstein, In vivo protein–protein and protein–
DNA crosslinking for genomewide binding microarray, Methods 31
(2003) 90–95.
[7] B. Nal, E. Mohr, P. Ferrier, Location analysis of DNA-bound proteins
at the whole-genome level: untangling transcriptional regulatory net-
works, Bioessays 23 (2001) 473–476.
[8] V. Orlando, Mapping chromosomal proteins in vivo by formaldehyde-
crosslinked-chromatin immunoprecipitation, Trends Biochem. Sci. 25
(2000) 99–104.
[9] D. Robyr, M. Grunstein, Genomewide histone acetylation microar-
rays, Methods 31 (2003) 83–89.
[10] V.A. Spencer, J.M. Sun, L. Li, J.R. Davie, Chromatin immunopreci-
pitation: a tool for studying histone acetylation and transcription fac-
tor binding, Methods 31 (2003) 67–75.
[11] A.S. Weinmann, P.J. Farnham, Identification of unknown target genes
of human transcription factors using chromatin immunoprecipitation,
Methods 26 (2002) 37–47.
[12] J. Wells, P.J. Farnham, Characterizing transcription factor binding
sites using formaldehyde crosslinking and immunoprecipitation,
Methods 26 (2002) 48–56.

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360 359
[13] J.D. Lieb, Genome-wide mapping of protein–DNA interactions by
chromatin immunoprecipitation and DNA microarray hybridization,
Methods Mol. Biol. 224 (2003) 99–109.
[14] S.K. Kurdistani, D. Robyr, S. Tavazoie, M. Grunstein, Genome-wide
binding map of the histone deacetylase Rpd3 in yeast, Nat. Genet. 31
(2002) 248–254.
[15] V.R. Iyer, C.E. Horak, C.S. Scafe, D. Botstein, M. Snyder, P.O.
Brown, Genomic binding sites of the yeast cell-cycle transcription
factors SBF and MBF, Nature 409 (2001) 533–538.
[16] B. Ren, F. Robert, J.J. Wyrick, O. Aparicio, E.G. Jennings, I. Simon,
J. Zeitlinger, J. Schreiber, N. Hannett, E. Kanin, T.L. Volkert, C.J.
Wilson, S.P. Bell, R.A. Young, Genome-wide location and function
of DNA binding proteins, Science 290 (2000) 2306–2309.
[17] T.I. Lee, N.J. Rinaldi, F. Robert, D.T. Odom, Z. Bar-Joseph, G.K.
Gerber, N.M. Hannett, C.T. Harbison, C.M. Thompson, I. Simon, J.
Zeitlinger, E.G. Jennings, H.L. Murray, D.B. Gordon, B. Ren, J.J.
Wyrick, J.B. Tagne, T.L. Volkert, E. Fraenkel, D.K. Gifford, R.A.
Young, Transcriptional regulatory networks in Saccharomyces cere-
visiae, Science 298 (2002) 799–804.
[18] Z. Li, S. Van Calcar, C. Qu, W.K. Cavenee, M.Q. Zhang, B. Ren, A
global transcriptional regulatory role for c-Myc in Burkitt’s lympho-
ma cells, Proc. Natl. Acad. Sci. USA 100 (2003) 8164–8169.
[19] C.E. Horak, N.M. Luscombe, J. Qian, P. Bertone, S. Piccirrillo, M.
Gerstein, M. Snyder, Complex transcriptional circuitry at the G1/S
transition in Saccharomyces cerevisiae, Genes Dev. 16 (2002)
3017–3033.
[20] C.E. Horak, M.C. Mahajan, N.M. Luscombe, M. Gerstein, S.M.
Weissman, M. Snyder, GATA-1 binding sites mapped in the beta-
globin locus by using mammalian chIp-chip analysis, Proc. Natl.
Acad. Sci. USA 99 (2002) 2924–2929.
[21] A.S. Weinmann, P.S. Yan, M.J. Oberley, T.H. Huang, P.J. Farnham,
Isolating human transcription factor targets by coupling chromatin
immunoprecipitation and CpG island microarray analysis, Genes
Dev. 16 (2002) 235–244.
[22] J. Wells, P.S. Yan, M. Cechvala, T. Huang, P.J. Farnham, Identifica-
tion of novel pRb binding sites using CpG microarrays suggests that
E2F recruits pRb to specific genomic sites during S phase, Oncogene
22 (2003) 1445–1460.
[23] P.L. Nagy, M.L. Cleary, P.O. Brown, J.D. Lieb, Genomewide demar-
cation of RNA polymerase II transcription units revealed by physical
fractionation of chromatin, Proc. Natl. Acad. Sci. USA 100 (2003)
6364–6369.
[24] B.E. Bernstein, E.L. Humphrey, R.L. Erlich, R. Schneider, P. Bou-
man, J.S. Liu, T. Kouzarides, S.L. Schreiber, Methylation of histone
H3 Lys 4 in coding regions of active genes, Proc. Natl. Acad. Sci.
USA 99 (2002) 8695–8700.
[25] H.H. Ng, F. Robert, R.A. Young, K. Struhl, Genome-wide location
and regulated recruitment of the RSC nucleosome-remodeling com-
plex, Genes Dev. 16 (2002) 806–819.
[26] O. Puig, F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-
Nilsson, M. Wilm, B. Seraphin, The tandem affinity purification
(TAP) method: a general procedure of protein complex purification,
Methods 24 (2001) 218–229.
[27] S.K. Bohlander, R. Espinosa III, M.M. Le Beau, J.D. Rowley,
M.O. Diaz, A method for the rapid sequence-independent amplifica-
tion of microdissected chromosomal material, Genomics 13 (1992)
1322–1324.
[28] P.R. Mueller, B. Wold, In vivo footprinting of a muscle specific
enhancer by ligation mediated PCR, Science 246 (1989) 780–786.
[29] C.L. Liu, S.L. Schreiber, B.E. Bernstein, Development and validation
of a T7 based linear amplification for genomic DNA, BMC Genom. 4
(2003) 19.
[30] D.J. Duggan, M. Bittner, Y. Chen, P. Meltzer, J.M. Trent, Expres-
sion profiling using cDNA microarrays, Nat. Genet. 21 (1999)
10–14.
[31] C.C. Xiang, O.A. Kozhich, M. Chen, J.M. Inman, Q.N. Phan,
Y. Chen, M.J. Brownstein, Amine-modified random primers to
label probes for DNA microarrays, Nat. Biotechnol. 20 (2002)
738–742.
[32] X.S. Liu, D.L. Brutlag, J.S. Liu, An algorithm for finding protein–
DNA binding sites with applications to chromatin-immunoprecipita-
tion microarray experiments, Nat. Biotechnol. 20 (2002) 835–839.
[33] X. Liu, D.L. Brutlag, J.S. Liu, BioProspector: discovering conserved
DNA motifs in upstream regulatory regions of co-expressed genes,
Pac. Symp. (2001) 127–138.
[34] J.J. Wyrick, J.G. Aparicio, T. Chen, J.D. Barnett, E.G. Jennings, R.A.
Young, S.P. Bell, O.M. Aparicio, Genome-wide distribution of ORC
and MCM proteins in S. cerevisiae: high-resolution mapping of rep-
lication origins, Science 294 (2001) 2357–2360.
[35] J.L. Gerton, J. DeRisi, R. Shroff, M. Lichten, P.O. Brown, T.D. Petes,
Inaugural article: global mapping of meiotic recombination hotspots
and coldspots in the yeast Saccharomyces cerevisiae, Proc. Natl.
Acad. Sci. USA 97 (2000) 11383–11390.
[36] D. Robyr, Y. Suka, I. Xenarios, S.K. Kurdistani, A. Wang, N. Suka, M.
Grunstein, Microarray deacetylation maps determine genome-wide
functions for yeast histone deacetylases, Cell 109 (2002) 437–446.
[37] F. Antequera, A. Bird, Number of CpG islands and genes in human
and mouse, Proc. Natl. Acad. Sci. USA 90 (1993) 11995–11999.
[38] S.H. Cross, J.A. Charlton, X. Nan, A.P. Bird, Purification of CpG
islands using a methylated DNA binding column, Nat. Genet. 6
(1994) 236–244.
[39] Y. Moreau, S. Aerts, B. De Moor, B. De Strooper, M. Dabrowski,
Comparison and meta-analysis of microarray data: from the bench to
the computer desk, Trends Genet. 19 (2003) 570–577.
[40] Y.F. Leung, D. Cavalieri, Fundamentals of cDNA microarray data
analysis, Trends Genet. 19 (2003) 649–659.
[41] J. Quackenbush, Microarray data normalization and transformation,
Nat. Genet. 32 Suppl. (2002) 496–501.
[42] Y.D. He, H. Dai, E.E. Schadt, G. Cavet, S.W. Edwards, S.B. Stepa-
niants, S. Duenwald, R. Kleinhanz, A.R. Jones, D.D. Shoemaker,
R.B. Stoughton, Microarray standard data set and figures of merit
for comparing data processing methods and experiment designs, Bio-
informatics 19 (2003) 956–965.
[43] N. Kaminski, N. Friedman, Practical approaches to analyzing results
of microarray experiments, Am. J. Respir. Cell Mol. Biol. 27 (2002)
125–132.
[44] J.L. Rinn, G. Euskirchen, P. Bertone, R. Martone, N.M. Luscombe, S.
Hartman, P.M. Harrison, F.K. Nelson, P. Miller, M. Gerstein, S.
Weissman, M. Snyder, The transcriptional activity of human chromo-
some 22, Genes Dev. 17 (2003) 529–540.
[45] P. Kapranov, S.E. Cawley, J. Drenkow, S. Bekiranov, R.L. Strausberg,
S.P. Fodor, T.R. Gingeras, Large-scale transcriptional activity in chro-
mosomes 21 and 22, Science 296 (2002) 916–919.
[46] A.M. Snijders, N. Nowak, R. Segraves, S. Blackwood, N. Brown,
J. Conroy, G. Hamilton, A.K. Hindle, B. Huey, K. Kimura, S. Law,
K. Myambo, J. Palmer, B. Ylstra, J.P. Yue, J.W. Gray, A.N. Jain,
D. Pinkel, D.G. Albertson, Assembly of microarrays for genome-
wide measurement of DNA copy number, Nat. Genet. 29 (2001)
263–264.
[47] E.F. Nuwaysir, W. Huang, T.J. Albert, J. Singh, K. Nuwaysir, A. Pitas,
T. Richmond, T. Gorski, J.P. Berg, J. Ballin, M. McCormick, J. Nor-
ton, T. Pollock, T. Sumwalt, L. Butcher, D. Porter, M. Molla, C. Hall,
F. Blattner, M.R. Sussman, R.L. Wallace, F. Cerrina, R.D. Green,
Gene expression analysis using oligonucleotide arrays produced by
maskless photolithography, Genome Res. 12 (2002) 1749–1755.
[48] S. Brenner, M. Johnson, J. Bridgham, G. Golda, D.H. Lloyd, D.
Johnson, S. Luo, S. McCurdy, M. Foy, M. Ewan, R. Roth, D. George,
S. Eletr, G. Albrecht, E. Vermaas, S.R. Williams, K. Moon, T. Bur-
cham, M. Pallas, R.B. DuBridge, J. Kirchner, K. Fearon, J. Mao, K.
Corcoran, Gene expression analysis by massively parallel signature
sequencing (MPSS) on microbead arrays, Nat. Biotechnol. 18 (2000)
630–634.
[49] C.Workman, L.J. Jensen, H. Jarmer, R. Berka, L. Gautier, H.B. Nielser,
H.H. Saxild, C. Nielsen, S. Brunak, S. Knudsen, A new non-linear

M.J. Buck, J.D. Lieb / Genomics 83 (2004) 349–360360
normalization method for reducing variability in DNA microarray ex-
periments, Genome Biol. 3 (2002) (research 0048.1–0048.16).
[50] Y.H. Yang, S. Dudoit, P. Luu, D.M. Lin, V. Peng, J. Ngai, T.P. Speed,
Normalization for cDNA microarray data: a robust composite method
addressing single and multiple slide systematic variation, Nucleic
Acids Res. 30 (2002) e15.
[51] G.C. Tseng, M.K. Oh, L. Rohlin, J.C. Liao, W.H. Wong, Issues in
cDNA microarray analysis: quality filtering, channel normalization,
models of variations and assessment of gene effects, Nucleic Acids
Res. 29 (2001) 2549–2557.
[52] E.E. Schadt, C. Li, B. Ellis, W.H. Wong, Feature extraction and
normalization algorithms for high-density oligonucleotide gene ex-
pression array data, J. Cell. Biochem. Suppl. Suppl. 37 (2001)
120–125.
[53] C.J. Roberts, B. Nelson, M.J. Marton, R. Stoughton, M.R. Meyer,
H.A. Bennett, Y.D. He, H. Dai, W.L. Walker, T.R. Hughes, M. Tyers,
C. Boone, S.H. Friend, Signaling and circuitry of multiple MAPK
pathways revealed by a matrix of global gene expression profiles,
Science 287 (2000) 873–880.
[54] E.C. Forsberg, K.M. Downs, E.H. Bresnick, Direct interaction of NF-
E2 with hypersensitive site 2 of the beta-globin locus control region in
living cells, Blood 96 (2000) 334–339.