clin neuro mnd davidson06
DESCRIPTION
Medical college lectures: neurology 5th year.TRANSCRIPT

Motor neuron diseaseMND:ALS
Dr. Mohammad Shaikhani.

Lou Gehrig’s Disease
• A well-known baseball player.
• He was diagnosed with ALS.
• Within a year, he could not walk or talk.
• He died in 1941.

MND (ALS): • A progressive disorder of unknown cause.• There is degeneration of motor neurons in the spinal cord,cranial
nerve nuclei& pyramidal neurons in the motor cortex. • 5% of cases are familial, AD,the genetic defect on ch 21, the
enzyme involved superoxide dismutase (SOD1). • For the remaining 95%, ? viral infection, trauma, exposure to
toxins , electric shock. • The prevalence 5/100 000.

MND (ALS):
Clinical features: • A combination of lower &upper motor neuron signs without sensory involvement. • The presence of brisk reflexes in wasted fasciculating limb muscles is typical. • In many patients the clinical features are highly suggestive but alternative diagnoses need to be
carefully excluded specially potentially treatable disorders as;• Diabetic amyotrophy.• Spinal disorders .• Multifocal motor neuronopathy. • EMG helps to confirm presence of fasciculation /denervation, specially helpful when pyramidal
features predominate. • Sensory/motor conduction studies are normal but there may be some reduction in amplitude of
action potentials due axon loss. • Spinal imaging / brain scanning may be necessary to exclude focal spinal or cerebral disease.• CSF exam is usually normal, though a slight elevation in protein may be found.
Clinical features:
Usually after 50 years Very uncommon before 30 years Affects males > females
Onset
Clinical features: Symptoms
Limb muscle weakness, cramps, occasionally fasciculation Disturbance of speech/swallowing (dysarthria/dysphagia)

Clinical features: Signs
Signs Signs
Wasting and fasciculation of muscles Weakness of muscles of limbs, tongue, face & palate Pyramidal signs: spasticity, inc tendon reflexes, extensor plantar. External ocular muscles intact. Sphincters remain intact. No objective sensory deficit. No intellectual impairment in most cases.

Clinical features: course
Signs Signs
Symptoms often begin focally in one part & spread gradually but eventually become widespread
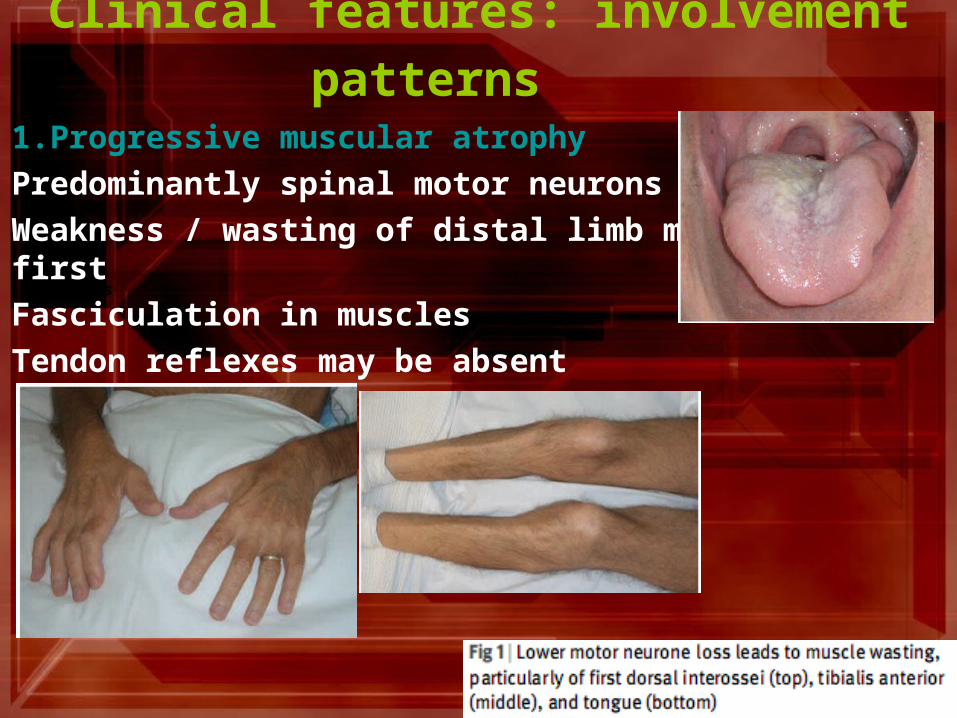
Clinical features: involvement patterns 1.Progressive muscular atrophy
Predominantly spinal motor neurons affected
Weakness / wasting of distal limb muscles at first
Fasciculation in muscles
Tendon reflexes may be absent Signs Signs

Clinical features: involvement patterns
Signs Signs
2.Progressive bulbar palsy
Early involvement of tongue, palate and pharyngeal muscles Dysarthria/dysphagia Wasting / fasciculation of tongue +/- pyramidal signs.

Clinical features: involvement patterns
Signs Signs
3.Amyotrophic lateral sclerosis
Combination of distal & proximal muscle-wasting & weakness, fasciculation Spasticity, exaggerated reflexes, extensor plantars Bulbar & pseudobulbar palsy follow eventually Pyramidal tract features may predominate

DD:
Signs Signs
Management: • The glutamate antagonist, riluzole, shown to have a small effect in prolonging life expectancy
by about 2 month. • It is not clear at which stage of the illness this prolongation occurs, so may not be particularly
helpful. • Other agents such as nerve growth factor show promise. • Psychological / physical support, with help from occupational / speech therapists /
physiotherapists, are essential to maintain the patient's quality of life. • Mechanical aids such as splints, walking aids, wheelchairs / communication devices all help to
reduce handicap. • Feeding by percutaneous gastrostomy may be necessary if bulbar palsy is marked. • Sometimes non-invasive ventilatory support may help distress from weak respiratory muscles
although maintenance ventilation is usually not requested. • Relief of distress in the terminal stages usually requires the use of opiates & sedative drugs.
Management: • The only drug currently available is riluzole (2-amino-6-
[trifluoromethoxy]benzothiazole).• Riluzole blocks glutamic acid release, may slow disease
progression by disrupting glutamate-mediated neurotoxicity. • Administered at 50 mg twice a day,• Riluzole is generally well-tolerated,but may cause nausea ,
general asthenia& methaemoglobinura in overdose.

Management:Future • 1.long-term ceftriaxone slowed the course of disease in a mouse
model, if given in high doses at the onset of the disease, preserved grip strength, slowed weight loss, & increased the overall duration of survival from 122 - 132 days &the mechanism is by modulating glutamate transport.
• 2.Gene therapy • 3. RNA silencing

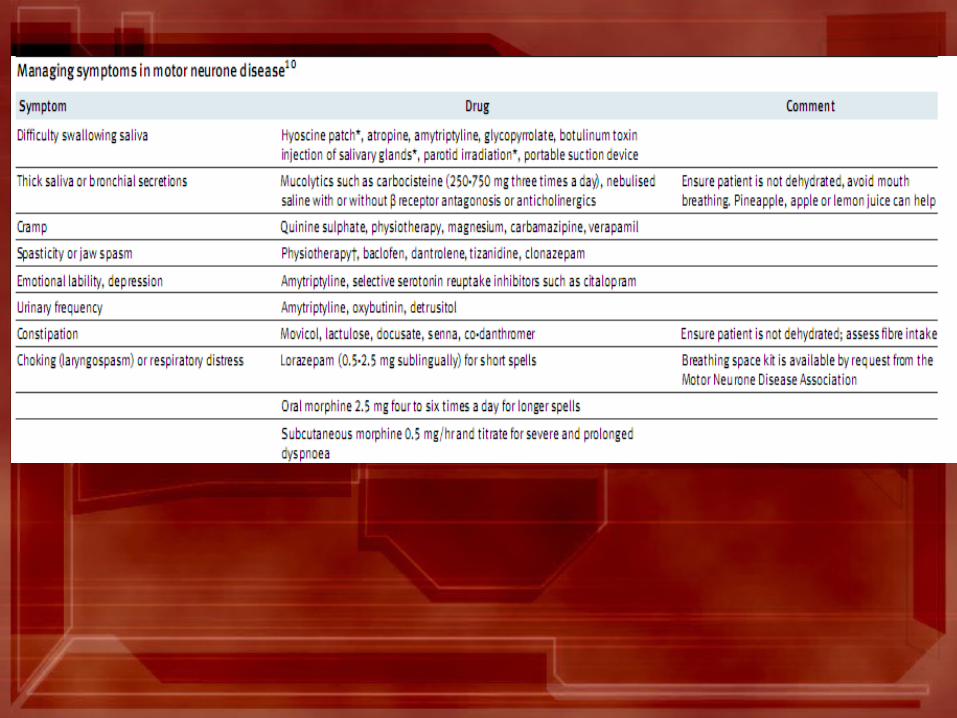

Prognosis: • It is progressive; the mean time from diagnosis to death is 1 year,
with most patients dying within 3-5 years of the onset of symptoms.
• Younger patients & those with early bulbar symptoms tend to show a more rapid course.
• Death is usually from respiratory infection / failure& the complications of immobility.



TYPES OF SPINAL MUSCULAR ATROPHY
Type Onset Inheritance Features Prognosis
Werdnig-Hoffmann
Infancy Autosomal recessive
Severe muscle-wasting/weakness Poor
Kugelberg-Welander
Childhood, adolescence
Autosomal recessive
Proximal weakness and wasting, EMG shows denervation
Slowly progressive disability
Distal forms Early adult life Autosomal dominant
Distal weakness and wasting of hands and feet
Good, seldom disabling
Bulbospinal Adult life, males only
X-linked Facial / bulbar weakness, proximal limb weakness, gynaecomastia
Good